
Competitive Adsorption of Proteins and Low-molecular-weight Surfactants Computer Simulation and...
23
Advances in Colloid and Interface Science 107 (2004) 27–49 0001-8686/04/$ - see front matter 2003 Elsevier B.V. All rights reserved. doi:10.1016/j.cis.2003.08.003 Competitive adsorption of proteins and low-molecular-weight surfactants: computer simulation and microscopic imaging Luis A. Pugnaloni , Eric Dickinson *, Rammile Ettelaie , Alan R. Mackie , Peter J. Wilde a a, a b b Procter Department of Food Science, University of Leeds, Leeds LS2 9JT, UK a Food Materials Science Division, Institute of Food Research, Norwich Laboratory, Norwich Research Park, Colney NR4 7UA, UK b Abstract Proteins and low-molecular-weight (LMW) surfactants are used in the food industry as emulsifying (and foaming) ingredients and as stabilizers. These attributes are related to their ability to adsorb at fluid–fluid (and gas–fluid) interfaces lowering the interfacial (and surface) tension of liquids. Hence, the study of the properties of adsorbed layers of these molecules can be expected to lead to a better understanding of their effect on food products. Direct proof of the validity of mesoscopic models of systems of proteins and LMW surfactants can only be achieved by quantitative theoretical predictions being tested against both macroscopic and mesoscopic experiments. Computer simulation constitutes one of the few available tools to predict mathematically the behaviour of models of realistic complexity. Furthermore, experimental techniques such as atomic force microscopy (AFM) now allow high resolution imaging of these systems, providing the mesoscopic scale measurements to compare with the simulations. In this review, we bring together a number of related findings that have been generated at this mesoscopic level over the past few years. A useful simple model consisting of spherical particles interacting via bonded and unbonded forces is described, and the derived computer simulation results are compared against those from the imaging experiments. Special attention is paid to the adsorption of binary mixtures of proteins, mixtures of LMW surfactants, and also proteinqsurfactant mixed systems. We believe that further development of these mathematically well-defined physical models is necessary in order to achieve a proper understanding of the key physico–chemical processes involved. 2003 Elsevier B.V. All rights reserved. Keywords: Proteins; Surfactants; Competitive adsorption; Emulsion and foam stability; Atomic force microscopy; Brownian dynamics simulation; Phase separation Contents 1. Introduction ......................................................................................................................................... 28 1.1. The background ............................................................................................................................. 28 1.2. Computer simulation ...................................................................................................................... 29 1.3. A simple model for proteins and surfactants at interfaces ................................................................... 31 1.4. Microscopy at interfaces ................................................................................................................. 33 1.5. Phase separation at interfaces .......................................................................................................... 34 2. Adsorption of one-component systems .................................................................................................... 36 2.1. Adsorption of LMW surfactants ...................................................................................................... 36 2.2. Adsorption of globular proteins ....................................................................................................... 37 3. Competitive adsorption of mixed LMW surfactants ................................................................................. 39 4. Competitive adsorption of mixed proteins ............................................................................................... 41 5. Competitive adsorption of proteins and LMW surfactants ......................................................................... 44 *Corresponding author. Tel.: q44-113-3432956; fax: q44-113-3432982. E-mail address: [email protected] (E. Dickinson).
-
Upload
rayito-de-luz -
Category
Documents
-
view
219 -
download
1
description
proteins
Transcript of Competitive Adsorption of Proteins and Low-molecular-weight Surfactants Computer Simulation and...

Advances in Colloid and Interface Science 107(2004) 27–49
0001-8686/04/$ - see front matter� 2003 Elsevier B.V. All rights reserved.doi:10.1016/j.cis.2003.08.003
Competitive adsorption of proteins and low-molecular-weight surfactants:computer simulation and microscopic imaging
Luis A. Pugnaloni , Eric Dickinson *, Rammile Ettelaie , Alan R. Mackie , Peter J. Wildea a, a b b
Procter Department of Food Science, University of Leeds, Leeds LS2 9JT, UKa
Food Materials Science Division, Institute of Food Research, Norwich Laboratory, Norwich Research Park, Colney NR4 7UA, UKb
Abstract
Proteins and low-molecular-weight(LMW) surfactants are used in the food industry as emulsifying(and foaming) ingredientsand as stabilizers. These attributes are related to their ability to adsorb at fluid–fluid(and gas–fluid) interfaces lowering theinterfacial (and surface) tension of liquids. Hence, the study of the properties of adsorbed layers of these molecules can beexpected to lead to a better understanding of their effect on food products. Direct proof of the validity of mesoscopic models ofsystems of proteins and LMW surfactants can only be achieved by quantitative theoretical predictions being tested against bothmacroscopic and mesoscopic experiments. Computer simulation constitutes one of the few available tools to predict mathematicallythe behaviour of models of realistic complexity. Furthermore, experimental techniques such as atomic force microscopy(AFM)now allow high resolution imaging of these systems, providing the mesoscopic scale measurements to compare with thesimulations. In this review, we bring together a number of related findings that have been generated at this mesoscopic level overthe past few years. A useful simple model consisting of spherical particles interacting via bonded and unbonded forces isdescribed, and the derived computer simulation results are compared against those from the imaging experiments. Special attentionis paid to the adsorption of binary mixtures of proteins, mixtures of LMW surfactants, and also proteinqsurfactant mixed systems.We believe that further development of these mathematically well-defined physical models is necessary in order to achieve aproper understanding of the key physico–chemical processes involved.� 2003 Elsevier B.V. All rights reserved.
Keywords: Proteins; Surfactants; Competitive adsorption; Emulsion and foam stability; Atomic force microscopy; Brownian dynamics simulation;Phase separation
Contents
1. Introduction ......................................................................................................................................... 281.1. The background............................................................................................................................. 281.2. Computer simulation...................................................................................................................... 291.3. A simple model for proteins and surfactants at interfaces................................................................... 311.4. Microscopy at interfaces................................................................................................................. 331.5. Phase separation at interfaces.......................................................................................................... 34
2. Adsorption of one-component systems.................................................................................................... 362.1. Adsorption of LMW surfactants...................................................................................................... 362.2. Adsorption of globular proteins....................................................................................................... 37
3. Competitive adsorption of mixed LMW surfactants................................................................................. 394. Competitive adsorption of mixed proteins............................................................................................... 415. Competitive adsorption of proteins and LMW surfactants......................................................................... 44
*Corresponding author. Tel.:q44-113-3432956; fax:q44-113-3432982.E-mail address: [email protected](E. Dickinson).

28 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
6. Concluding remarks.............................................................................................................................. 47Acknowledgements................................................................................................................................... 48References............................................................................................................................................... 48
Fig. 1. Schematic representation of surface-active species.(a) Proteinand surfactant molecules.(b) Representation of the proteins and sur-factants as particles according to the model presented in this paper.
1. Introduction
1.1. The background
Proteins and low-molecular-weight(LMW) surfac-tants are key components of many foodstuffsw1x. Somedairy products, for example ice cream, contain bothproteins and LMW surfactants in their formulation. Bothtypes of molecules can adsorb at fluid interfaces, reduc-ing the interfacial tension and so facilitating the forma-tion of emulsions and foams and providing stability todroplets and bubbles. However, their molecular proper-ties are very differentw2x.LMW surfactants are small molecules each consisting
of a hydrophilic head group and one or several hydro-phobic tails. When such molecules reach an air–wateror oil–water interface, they tend to adsorb by arrangingthe hydrophobic tails within the non-aqueous phase andthe hydrophilic head in the water phase(see Fig. 1a).LMW surfactants are very mobile and they are particu-larly efficient at reducing the interfacial tension. As aresult, they rapidly coat the newly created oil–water andair–water interface during emulsification and foaming.Proteins are high-molecular-weight molecules each
consisting of a chain of amino acids. As there are polar,non-polar and ionic amino acids, proteins contain amixture of hydrophilic and hydrophobic groups. Inaqueous solution, a protein molecule will tend to fold
in a coil-like structure in order to expose the mosthydrophilic groups to the water and hide the mosthydrophobic segments in the centre of the coil(see Fig.1a). However, when a protein molecule reaches an air–water or oil–water interface, the molecule will partiallyunfold orientating its hydrophobic groups towards thenon-aqueous phase(Fig. 1a). Proteins are very slow atdiffusing and adsorbing as compared with LMW surfac-tants; and they do not normally lower the interfacialtension so efficiently. However, proteins form thickprotective layers at the surface of oil droplets and gasbubbles which, under appropriate conditions, can preventcoalescence after an emulsion or foam has been formedthereby conferring long-term stability to the system.When a mixture of LMW surfactants or a mixture of
proteins or a surfactantqprotein mixed system isexposed to an interface, the different species competeto adsorb and lower the interfacial tensionw3x. Duringthe equilibration of the interface—this may take fromseconds in the case of pure LMW surfactants to up toseveral hours for protein-containing systems—the mol-ecules adsorb and desorb dynamically. If one of thecomponents of the mixture is presented to the interfacefirst, then the second component will tend to replace theadsorbed molecules of the first type partially or totallydepending on the relative surface-activity of the speciesand their mutual cross interaction. Interestingly, in somecases, the mixture does not adsorb homogeneously overthe surface. On the contrary, interfacial regions rich inone or the other species are present either during theequilibration process or in metastable states.It is a common practice in the study of such systems
(and in general of most physico–chemical systems) togenerate a mesoscopic model of the molecules involvedin order to explain the behaviour of the system in aqualitative fashion. These kinds of models, which areoften not very precisely defined, take into account a fewessential characteristics of the molecules under study—such as molecular weight, flexibility, hydrophilic–lipo-philic balance, presence of reactive groups, and chargedistribution—instead of the detailed chemical structure.This provides a useful way of elucidating the importantfactors affecting a particular phenomenon. However, themajority of these models are expressed in a qualitativelanguage during the interpretation of experimentalresults, but if these types of models are amenable to beexpressed in a physico–mathematical language describ-ing quantitatively the molecules and their interactions,the calculation of the behaviour of an ensemble ofmolecules can be made by solving the basic equations

29L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
of motion through the use of a suitable computersimulation algorithmw4,5x.Atomic force microscopy(AFM) is a powerful exper-
imental tool to probe very small length scales on asurface w6x, hence the usefulness of this technique toinspect the structure of adsorbed protein films at inter-faces w7x. It is particularly interesting that with AFMwe are able to reach resolutions almost down to molec-ular length scales. Therefore, any mesoscopic model canbe assessed at the ultimate level of description, i.e. downto the small-scale structures formed by the moleculesthemselves. Additionally, fluorescence microscopy pro-vides a method to distinguish different species in aprotein mixturew8x. This can be particularly useful, forexample, if one is interested in studying the homogeneityof the layer formed at the interface.In this paper, we review a number of experiments and
computer simulations of model systems that have beencarried out in recent years. We pay special attention tothe description of the structure of the interface. Wedescribe a general but simple mesoscopic model foradsorbing proteins and surfactants, and several examplesof computer simulation results on the competitiveadsorption of these types of molecules are shown.Atomic force microscopy and fluorescence microscopyexperiments are presented and compared with the sim-ulation results. Additionally, we present some rheologi-cal properties of adsorbed protein layers that can beextracted from the simulations.
1.2. Computer simulation
In this section we give a concise overview of themost common computer simulation techniques relevantto this topic. We recommend the reader to refer to thebook by Allen and Tildesleyw5x for a thorough intro-duction to the subject.Every physico–chemical system comprises a number
of basic interacting entities. Depending on the intendedlevel of description, these entities can be atoms, mole-cules or molecular complexes. We give the nameparti-cle to any of these basic entities. A theoretical modelfor these particles consists in a mathematical definitionof the way they interact with each other and withexternal forces. When such a model is complex, as isusually the case when dealing with complex fluids, thephysical equations that describe the behaviour of asystem composed of thesemodel particles are unattain-able analytically. In a few cases, further approximationscan be used to simplify the equations. Mainly, however,a numerical solution of the set of fundamental physicalequations is normally the practical alternative. Thisnumerical approach is referred to as ‘computer simula-tion’. The two basic techniques for performing computersimulations consist in either solving the equations ofmotion of the involved entities(particles) that form the
system—this is termed molecular dynamics(MD) sim-ulation—or in generating ensembles of representativeconfigurations of the system at random—what is calledMonte Carlo(MC) simulation.It is important to mention here that there exist other
numerical techniques to study the properties of differentmolecular models.Self-consistent field theory for exam-ple, is a very useful technique to study theequilibriumproperties of molecular systems(see for example Ref.w9–12x). However, such types of studies are not com-puter simulations in the sense that the fundamentalphysical equations are solved indirectly. Inself-consis-tent field theory, the unknown function to be calculatednumerically is the density distribution of molecularsegments at thermodynamic equilibrium rather than thepositions andyor velocities of each segment.In a classical MD simulation, Newton’s equations are
solved for a set of particles. The computation involvesthe solution of a system ofN coupled differentialequations(one for each particle) of the form
2Nd r tŽ .i
extw z w zx | x |m sF r t q F r t ,r t , (1)Ž . Ž . Ž .i i i i,j i jy ~ y ~82dt j/i
wherem is the mass of particlei, r (t) is its position ati i
time t, and andF are the forces applied oni byextFi i,j
any external field and by particlej, respectively. Ofcourse, solution of such a system of differential equa-tions requires initial conditions for the positions of theparticles r (ts0) and their velocitiesv (ts0). Then,i i
any appropriate finite difference technique can be usedto solve the equations numerically and so obtain theposition r (t) and velocitiesv (t) of the particles ati i
discrete intervals of timeDt. Once the trajectories of theparticles have been calculated for a long enough periodof time—millions of units ofDt, typically—the macro-scopic properties(specific heat, viscosity, etc.) andmesoscopic properties(structure factor, clustering, etc.)can be extracted through the appropriate time averages.Classical MC simulation relies on the ensemble sta-
tistical theory. According to this theory, an average overall possible configurations of the system is equivalentto an average over the configurations that the systemvisits during its time evolution, as long as it is inthermodynamic equilibrium. Therefore, a representativeset of MC configurations can be generated at randomand then used to obtain mesoscopic and macroscopicaverages. The simulations carried out under the MCscheme are normally simpler and more efficient thanthe ones based on MD. However, since the real trajec-tories of the particles are not calculated, no directdynamic information can be extracted from MC simu-lations. Thus, only equilibrium properties of the systemsof interest can be studied via MC simulation. Unfortu-nately, many of the most interesting properties of realsurface-active molecules are related to their slow dynam-

30 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
ics, which evolves over periods of hours, days or evenweeks, without reaching proper thermodynamicequilibrium.There is also a third, more coarse-grained simulation
approach, namely Brownian dynamics(BD). In studyingsystems of surface-active molecules(and complex fluidsin general), a mixture of solvent molecules(typicallywater) and surface-active molecules(proteins or LMWsurfactants) has to be simulated. Normally, such solu-tions contain a few surface-active molecule for everytens of million of water molecules. Therefore, it is clearthat the simulation of a few hundred surface-activemolecules by the MD or MC approach would involvethe calculation of trajectories or configurations of aprohibitively large number of solvent molecules. How-ever, if we are not actually interested in the behaviourof the solvent, it is more sensible to avoid such a wasteof computing resources by focusing just on the surface-active species alone, and including the solvent contri-bution as an effective external perturbation. This is thebasis of Brownian dynamics.In a BD simulation, as in MD, the equations of
motion of the particles are solved numerically. However,no solvent particles are included explicitly. Instead,every other particle is subjected to two external forcesthat mimic the effect of the solvent on them. These twoforces are the drag force and the random buffeting thatgenerates the Brownian motion of the solute. In otherwords, BD is concerned with the numerical solution ofthe Langevin equation. For an isolated spherical particlethe drag force is taken asy3p h s v , whereh is thei
solvent viscosity,s is the particle diameter, andv isi
the particle velocity. The random buffeting is simulatedthrough a random Gaussian distribution with zero meanand variance 2k Ty(3p h s), which is consistent withB
the fluctuation–dissipation theorem and leads to Ein-stein’s relation for the diffusion coefficientwD sk Ty0 B
(3p h s)x. Consequently, we have to solve a system ofN coupled differential equations—one for each surface-active particle—of the form
2Nd r tŽ .i
extw z w zx | x |m sF r t q F r t ,r tŽ . Ž . Ž .i i i i,j i jy ~ y ~82dt j/i
dr tŽ .iRy3phs qF t . (2)Ž .idt
Here, represents the random force. Again, as inRF tŽ .i
MD simulation, finite difference algorithms are used tosolve these equations numerically.An important point has to be made with respect to all
types of computer simulation. Because the systemsstudied are invariably much smaller than real systems,an unrealistically large proportion of particles are closeto the edges of the system boundaries. Therefore, one
expects that the presence of any container wall shouldaffect significantly the behaviour of the entire simulatedsystem. A way to lessen this effect is to use periodicboundaries conditions. That is, we allow particles onone side of the system to interact with particles on theopposite side, as if copies of the virtual container wereplaced all around the system to create borders of thesame nature as the system itself. Within the samescheme, a particle that crosses the boundaries of thesystem (say by diffusion) is introduced through theopposite edge to create a smooth motion across theborder. Although this computational trick is very usefuland efficient, it cannot eliminate other small-size effects:for example, in the calculation of long-ranged particle–particle forces or in the analysis of density fluctuationslarger than the basic simulation system size. In practice,the only way of ensuring that size effects do notinfluence a particular result is to perform independentsimulations with different system sizes and then extrap-olate the results to infinitely big systems.It is appropriate also here to mention briefly the non-
equilibrium simulation techniques used to analyse theresponse of a system to an external perturbationw5,13x.It is the case that the linear response of the system tosmall perturbations can be obtained from an equilibriumMD or BD simulation through the fluctuation–dissipa-tion theoremw5x. However, non-equilibrium simulationtechniques require less computer effort to obtain asimilar degree of accuracy, and they also allow us tostudy the response of the system to large perturbations,where the response is likely to be highly non-linear.The response of the system to an external perturbation
can be measured through the interparticle stress tensors. For a pairwise-additive interaction it is given by theKirkwood formula w14x
N Ny11s srk Td y r F , (3)ab B ab aij bij88V j)i is1
whereV is the volume of the system. Here,a and bindicate the different Cartesian components of the stresstensors, the interparticle distancer , and the interpar-ij
ticle force F , respectively. Normally, the macroscopicij
rheological quantities of interest are related to thesymmetric part ofs, i.e.,
1s s s qs . (4)Ž .ab ab ba2
Shear flow can be imposed on a simulated ensembleof particles in much the same fashion as for a realsystem in the laboratory. The most widely used methodinvolves the creation of a shear flow profile by theaddition of a position and time-dependent external forcequantified by the affine straing (t) applied on thexy-xy

31L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
plane (or any other appropriate plane). The periodicboundary conditions have then to be modified to accountfor a linear flow profile across the infinite replicasw15x.For a sine wave input at frequencyfsvy2p, we have
g t sg sin vt , (5)Ž . Ž .xy 0
and the time domain response is
s tŽ .xysG9 v sin v t qG0 v cosv t , (6)Ž . Ž . Ž . Ž .
g0
whereG9(v) andG0(v) are the storage and loss moduli,respectively. These can be extracted by integrating overa sufficiently large integral number of shear cyclesn as
2npv vG9 v s s t sin vt dt, (7)Ž . Ž . Ž .xy|npg 00
2npv vG0 v s s t cosvt dt. (8)Ž . Ž . Ž .xy|npg 00
Several initial cycles of the oscillation have to bediscarded from the analysis so as to ensure that only thesteady state response to the perturbation is accountedfor.The dilatational rheology of a system can be extracted
in a completely analogous fashion to the shear rheologyw16,17x. In this case, the system is subjected to a smalloscillatory change in volume. This is achieved bycreating an extensional flow profile on application of aposition- and time-dependent external force quantifiedby the dilatational strain-rate. Again, the periodic bound-ary conditions have to be adapted in order to accountfor a volume-changing system. After an integral numberof dilatational cycles, the dilatational storage modulus´9(v) and loss modulus0(v) can be extracted throughequations analogous to Eqs.(7) and (8). Additionally,we can investigate large deformation-dilatational rheol-ogy by compressing(expanding) the system at a con-stant strain-rate down to(up to) few times the originalsize and analysing the response of the system thoughthe stress tensorw18x.It is worth mentioning here that in what follows we
concentrate on interfacial systems. Therefore, the rheo-logical properties discussed above should be adapted tointerfacial shear and interfacial dilatational quantities byusing the appropriate interfacial stress tensor. A detaileddiscussion and application of interfacial shear and dila-tational rheology in computer simulation can be foundin Ref. w17x.
1.3. A simple model for proteins and surfactants atinterfaces
In order to study the competitive adsorption of differ-ent surface-active components, a simple simulation
approach has been developed by Wijmans and Dickinsonw18,19x. The model is a straightforward variation of aprevious one designed to describe interacting proteinsystems in the bulk(i.e. far from interfaces) w20x. In itssimplest version, the model consists of two types ofspherical particles(of diameterss ands ) interacting1 2
via a steeply repulsive spherical core potential:
36B EsC Ff r s´ , (9)Ž .c ijD Grij
where ,s ands are the diameters ofss1y2 s qsŽ .i j i j
the interacting particles,r s±r yr ± is the centre-to-ij i j
centre distance of the particle pair, and´ is an energyparameter that is set equal tok T. As usual,k is theB B
Boltzmann constant andT is the absolute temperature.All quantities are then expressed in units of´, s and1
for energy, length and time, respectively, withys m y´1 1
s andm being the diameter and mass of the particles1 1
of type 1.To mimic adsorption, each particle of typea (as1
or 2) interacts with an external potential, acting in thez-direction:
af z sŽ .s i
36 36S B E B E B Es s s1 1 a aC F´ q´ zy Fzi cD GB E B Es s 2C F C Fa aC F C Fs qwy zy s qwq zyŽ . Ž .1 i 1 iT D D GG D D GG2 2
(10)
U
B E B E B Es s s1 1 a36 36 aC F´ q´ zy )zi cT D GB E B Es s 2C F C Fa aa aC F C Fs qwy z y s qwq z yŽ . Ž .1 c 1 cD D GG D D GGV 2 2
This potential has a square-well-like shape with oneinfinite wall and one finite wall. The infinite wallprevents particles from escaping to the phase in whichthey are not soluble—typically air or oil in the case ofproteins. Conversely, the finite wall allows for inter-change of particles between the interface and the phasewhere they dissolve—typically an aqueous solution inan experimental setup. It is important to note that somesurfactants can also dissolve in a non-aqueous medium.The adaptation of Eq.(10) to account for oil-solublesurfactants is straightforward but will not be consideredhere. The parameterw in Eq. (10) defines a minimumwidth for the potential well, and it is usually set to0.05s . Then, should be greater thanw. The param-az1 c
eter defines simultaneously the total width of theazcpotential well and the particle adsorption energy foreach type, namely w18x. A particlea a a aE 'f z yf 0Ž . Ž .ads s c s
i of typea is said to be adsorbed if . Notice thatazFzi c
the potential representing the presence of the interfaceis positioned in such a way that particles adsorb by‘touching’ the interface, which is atzs0. Therefore,particles of different sizes adsorbed at the interface will

32 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
not have their centres on the samez-plane, as exempli-fied in Fig. 1b.In order to account for the intermolecular cross-
linking behaviour of some proteins, the adsorbing par-ticles can also interact through flexible bonds(see Fig.1b). Nodes are created on the nominal surface of thespherical particles(s y2 from the centre). The bondi
interaction acts along the straight line that joins thecorresponding nodes, and it depends on the node-to-node distanceb only:ij
S0 b Fbij 1
T 2B Eb ybij 1C Ff b s ´ b -b Fb . (11)Ž .B ij B 1 ij maxUD Gb0
2B Eb ybT max 1C F´ b )bB ij maxD GV b0
We note that, as these forces do not need to operatein purely radial directions, they can give rise to torquesacting on each particle. Consequently, a rotational con-tribution to the equation of motion has to be applied inaddition to Eq.(1) or Eq. (2).The interparticle bonds can be created or destroyed
through different protocols. Normally, a bond would becreated with the given probability when two parti-abPBcles—one of typea and one of typeb—approach withina distanceb . Initially, nodes are created on the line that1
joins the particle centres. After bond formation, thenodes that define the ends of the bond are fixed withinthe corresponding particle reference system. That is, thenodes remain fixed at the initial position on the surfaceof each particle. If the particle moves such that thelength of a bond exceedsb , the bond is deemed tomax
be broken. By settingb s`, irreversible(unbreaka-max
ble) bonds can be simulated.In some cases, to allow for various kinds of repulsive
and attractive interactions, an additional non-bindingforce between the particles can be introduced. Thesimplest case is dictated by the interaction pair potential
abS B Er yrc ijab abT C F´ r FrUB ij cabUab D Gr ysf r s , (12)cŽ .UB ij Tab
V0 r )rij c
where defines the range of the interaction,ab abr ´c UB
accounts for its strength, and again ;ss1y2 (s qs )a b
s ands are the diameters of the interacting particles.a b
The indicesa andb indicate the type of particlei andj, respectively.Adding together all the above contributions, the force
acting on particlei of typea at time is given byt
aw w zx |F t sy=r f z tŽ . Ž .ii s iy ~x
y
abw z w z w zzx | x | x |q f r t qf b t qf r t , (13)Ž . Ž . Ž .c ij B ij UB ijy ~ y ~ y ~|8
~j/i
whereb identifies the corresponding type for particlej.The two terms on the right hand side of Eq.(13)correspond, respectively, to the two first terms on theright hand side of either Eq.(1) or Eq. (2). The torquearound the centre of particlei is
w zx |t t s yb ==bf b t . (14)Ž . Ž .ji j B ijy ~8
j
This last sum runs over all the bonds of particlei, whereb is the position of nodej with respect to the centre ofj
the particle.The system is not homogeneous in thez-direction
since the interfacial external potential depends on thez-coordinate of the particles. Therefore, the periodicboundary conditions can be applied in thex- and y-directions only. Particles cannot escape in the negativez-direction(upwards in Fig. 1b), as they are trapped bythe infinite wall of the interface, but they can moveaway (and become ‘lost’) in the positive z-direction(downwards in Fig. 1b). In order to avoid this effect,which would otherwise not be present if periodic bound-ary conditions were applicable in thez-direction, anadditional ‘wall’ needs to be added at the givenz-position. One way of achieving that is by introducingan external potential of the form:
36B Esaa C Ff z s´ , (15)Ž .w iD Gz yzw i
wherez is the position of the restricting ‘wall’. In thisw
way particles initially placed betweenzs0 and zszwcan only move within this range in thez-direction.The properties of the present model can, in principle,
be studied by means of MD, MC or BD. However, thefirst two techniques would require, in principle, theintroduction of an extra species in the system to accountfor the solvent. Therefore, BD simulations are normallymore efficient, as they introduce the solvent effect byjust assuming a bulk solvent viscosity,h. In this case,the unit of time is generally changed to a more conven-ient scale: this is the average time taken for a particleto diffuse a distance equal to its radius in an infinitelydiluted system, i.e.
2 3sy2Ž . ps ht s s . (16)r 6D 8k T0 B
The model defined so far consists of a number of

33L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
parameters that characterize the system. These parame-ters are: the sizes of the particless and s , the1 2
corresponding adsorption energies and (deter-1 2E Eads ads
mined by and , respectively), the maximum length1 2z zc c
of the bondsb , the strength of the bonds defined bymax
, the equilibrium length of the bondsb , the2´ ybB 0 1
reaction probabilities , the strengths of11 22 12P , P andPB B B
the long-range forces defined by and,11 22 12´ , ´ and´UB UB UB
finally, the ranges of the non-bonded forces. By selecting different sets of parameters,11 22 12r , r andrc c c
we are able to model various sorts of systems like binarymixtures of proteins, binary mixtures of LMW surfac-tants or proteinqLMW surfactant mixtures. In latersections, we show how this can be done and how resultsfrom the simulated systems compare to some recentexperimental results from AFM and fluorescencemicroscopy.It is worth emphasising that the model presented in
this section does not consider the internal structure andflexibility of the molecules. This is particularly inade-quate for very flexible protein molecules, as the modelcannot account for the unfolding of the molecule uponadsorption. However, some globular proteins such asb-lactoglobulin may be reasonably well represented by themodel at mesoscopic resolution, since unfolding takesplace over long time scales, especially with stronglyinteracting proteins. A molecular dynamics simulationof the entire structure of a singleb-casein moleculeadsorbing to an oil–water interface over a time periodof 1 ns has been carried out recentlyw21x. Naturally, thecomputational cost of simulating an entire protein filmusing this scheme for experimentally significant timescales is prohibitive for today’s computer resources.
1.4. Microscopy at interfaces
As has already been stated, interfacial films formedby the adsorption of surface-active species to a gas–liquid (or liquid–liquid) interface can be simulated byBD. In order to make a comparison between the patternsformed by these simulations and experimental systemswe require methods of probing similar length scales.This is complicated by the fact that the film sits at aliquid boundary, making it inherently unstable. Theproblem of imaging such interfacial films can beapproached in two different ways. Either the sample canbe viewed non-invasively in-situ using optical tech-niques or the interfacial film can be transferred, unchan-ged, onto a solid support where it becomes amenable toimaging at high resolution by probe microscopy. Bothapproaches have advantages and disadvantages, whichmake them useful under different circumstances.The foremost probe microscopy is atomic force
microscopy (AFM), which uses a sharpened siliconnitride cantilever to scan over the surface of the sample.In its normal mode of operation the deflection of the
cantilever is maintained at a predetermined value as thescan progresses in order to keep the forces actingbetween tip and sample constant. By monitoring themovements of the scanner that are required to maintainconstant deflection of the cantilever, the sample topog-raphy is revealed as a three-dimensional image. AFMhas the advantage over electron microscopy of beingable to operate in liquids at ambient temperatures andpressures, and so it is an ideal instrument for biologicalstudies. Its resolution is limited not by diffraction butby the sharpness of the tip. In practice, AFM canroutinely achieve molecular resolution and in idealcircumstances sub-molecular resolutionw22x. However,in order to obtain these high levels of resolution, thesample needs to be mounted on an atomically flat solid–substrate such as mica. This is achieved by a techniqueknown as Langmuir–Blodgett(LB) transfer where thesolid support is dipped, at low speed through theinterface into the sub-phase, and then removed. Thetransfer of the interfacial film is controlled by the surfaceproperties of the substrate. With a hydrophilic substratethe contact angle between the liquid and the substratedictates that the film will only transfer on the upwardstroke, removing a single section of interfacial film.Although the film may be altered during the transfer,comparisons with Brewster angle microscopyw23x andspecially designed in-situ AFM experimentsw24x haveshown the same behaviour and morphology as thoseusing the LB transfer technique. Despite the high spatialresolution, because the technique is essentially topo-graphical, it cannot be used to distinguish betweenmolecules with only small differences. Most proteinmolecules for example look very similar to one anotherin AFM images. In Section 5, we give some examplesof AFM images of LB films drawn from fluid–fluidinterfaces where mixtures of proteins and LMW surfac-tants were adsorbed competitively.Optical techniques that can interrogate an interfacial
film in-situ such as Brewster angle microscopy(BAM)suffer from limited resolution(typically 1mm). Despitethe limited spatial resolution of BAM it can be extremelysensitive to surface composition. The two requirementsfor obtaining good BAM images are phase regions ofsufficient size, typically tens of microns, and significantvariation of the optical properties between the twophases. For example, one common use of BAM is tolook at the complex interfacial phase behaviour of lipidsw25x. In such systems small changes in the orientation(tilt) of the molecule between liquid-expanded andliquid-condensed phases are readily apparent if thedifferent domains are large enough. The limited spatialresolution means that this technique is not suited tolooking at the formation of structures in protein filmsor mixed proteinqLMW surfactant films unless(oruntil) the phase separated regions become sufficientlylarge. BAM is certainly not suitable for looking at phase

34 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
separation in mixed protein systems because of thesimilarity in optical properties.Although neither of the two techniques discussed
above can distinguish between different proteins, the useof a combination of LB transfer to a transparent solid–substrate and fluorescence microscopy can be used toprovide images with a resolution of a few hundrednanometres—which is the diffraction limit for opticalmethods. In this method proteins are covalently linkedto various fluorescent probes. The labelled protein solu-tions are then used to form an interfacial film that istransferred to a solid support for imaging. An additionaldisadvantage of this approach is the effect the fluores-cent moiety might impart to the functionality of theprotein. Examples from two groups who have usedvariations on this technique are given in Section 4.
1.5. Phase separation at interfaces
When surface-active molecules adsorb at an interfacethey achieve a higher local concentration than in thecorresponding bulk solution. Consequently, the pairinteractions between the molecules become more impor-tant at the interface in determining the overall behaviourof a mixture of surface-active components than they doin solution. One of the most interesting features of moreconcentrated mixtures is the greater likelihood of phaseseparation. It seems relevant, therefore, to speculate onsituations under which surface phase separation mightoccur. In the following, we discuss the possible casesthat may arise in this respect during the adsorption ofbinary mixtures of surface-active molecules.A crucial factor in determining the possible occur-
rence of phase separation at an interface relates to thetime scale over which the interface equilibrates with itscorresponding bulk solution. For highly non-solublesurfactants or high-molecular-weight proteins, present onsurfaces, such processes can be particularly sloww26x.Thus, it is easy to encounter situations where theduration of an experiment is far shorter than the neces-sary equilibration times. In such cases, the overallcomposition of the interface remains virtually constantduring the experiment, having initially been set by theamount of each surface-active species that is introducedonto the interface. For this very slow exchange dynam-ics, then, the interfacial region may be considered as aseparate phase isolated from the bulk. Associated withsuch an interface, covered by a binary mixture of twodifferent surface active molecules, is an excess freeenergy per unit areaf (G , G ), whereG andG denotes 1 2 1 2
the coverage of each species in the mix. It must benoted that in general, should phase separation occur ata certain range of values ofG andG , then f (G , G )1 2 s 1 2
is not experimentally accessible in this range. Rather,its behaviour has to be inferred, albeit qualitatively, froma suitable theoretical model; a situation not dissimilar
to bulk systems. Outside this range, however, informa-tion regardingf (G , G ) is obtained from experimentals 1 2
data which, for example, might involve surface pressureand interfacial tension measurements. The phase sepa-ration behaviour at the interface is dictated by the formof f (G , G ). In particular, if the matrix of seconds 1 2
derivatives (≠f y≠G ≠G ) (Hessian matrix) is positives i j
definite everywhere, then the interface remains stableagainst fluctuations in composition at all values ofcoverage and will show no tendency for surface phaseseparation. For an interface to begin to exhibit suchbehaviour, the Hessian matrix must cease to be positivedefinite at least over a certain range ofG andG . In1 2
this range of values of surface coverage, the mechanismof phase separation is spinodal decomposition, whileoutside the range phase separation can occur through anucleation and growth process.From a more physical point of view, conditions that
lead to the ‘convex’ form off (G , G ) and the onsets 1 2
of phase separation at air–water interfaces, are onlyrealised if certain degree of unfavourable interactionsare present between the two surface-active species orbetween these and the water molecules. While theformer set of interactions can arise in certain circum-stances, the latter are highly unlikely. Even for highlyinsoluble surfactants at the interfaces, it is the polar orionic hydrophilic sections of the molecules that remainin the aqueous phase. Such sections have a strongpreference to be in contact with the solvent moleculesand in the absence of the hydrophobic parts would, bythemselves, be soluble in water. Thus, for amphiphilicmolecules at air–water interfaces, the only remaininginteraction that can realistically lead to phase separationbehaviour is that existing between the two surface-activespecies. This in turn is dominated by the lateral inter-actions between the hydrophobic parts of the two dif-ferent sets of molecules. For high-molecular-weightsynthetic co-polymers, consisting of large hydrophilicand hydrophobic blocks, the required unfavourable inter-actions are relatively easy to obtain. Small incompati-bilities between different monomers, comprising thehydrophobic parts of the two species, become far moreprominent because of the substantial size of the inter-acting blocks concerned, as well as the relatively higherconcentration of the molecules on the interface. Achiev-ing the same level of unfavourable interactions betweenLMW surfactant species is more difficult and requiresthe hydrophobic parts of the two sets of molecules tobe chemically very different. An interesting example ofsuch a system, which has received some attention in theliterature (see for example Ref.w27x), involves binarymixtures of fluorocarbon–hydrocarbon-based insolublesurfactants placed on air–water interfaces.In contrast to synthetic block co-polymers, proteins
are made from a variety of hydrophilic and hydrophobicamino acids that tend to be more uniformly distributed

35L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
along the length of the chains. In other words, suchmolecules rarely possess large blocks, which happen toconsist almost entirely of the same strand of amino acidmonomers. Furthermore, the same variety of amino acidgroups is to be found in different protein species. Thus,even if a certain degree of incompatibility betweendifferent hydrophobic amino acid types exists, the struc-ture and composition of proteins, as discussed above,makes the possibility of large enough direct unfavoura-ble lateral interactions between such molecules unlikely.At first sight then, the above discussion suggests that
mixtures of different proteins should not exhibit inter-facial phase separation behaviour, at least not in thestrict thermodynamic sense. Any observed patternsresembling onset of phase separation are to be interpret-ed as transients, which should decay out over sufficientlylong periods of time. However, in a previous publicationw51x, we have argued that this picture might not alwaysbe true. Protein molecules often contain groups, whichcan associate or even form inter-molecular bonds. Anextreme and rather well studied casew28x involvesproteins which contain thiol groups(–SH), as say withb-lactoglobulin. In bulk solutions, such groups arehidden from each other within the interior of themolecules. However, on the air–water interface,b-lactoglobulin molecules unfold and expose these reactivegroups, thus giving rise to the possibility of formationof covalent bonds between the chains. Though in thecase of covalent bonds these bonds tend to be irrevers-ible, in other circumstances weaker bonds(e.g. hydrogenbonding) might form, which can break and reform. Ithas been speculatedw51x that the presence of such bondsbetween one set of proteins, but not the other, canprovide an effective attraction, which can overcome theentropy of mixing, causing formation of separate phaseson the interface. Certainly, examples of such behaviourhave been found in bulk solutions for water solublepolymers. For example, incorporation of a few smallhydrophobic groups, comprising no more than 2% wywof the polymer to polyethylene glycol(PEG) chains, isknown to cause phase separation in solutions consistingof mixtures of such modified and identical non-modifiedPEG moleculesw29x. Both resulting phases have beenreported to be clear solutions, with roughly the sameamount of PEG present in each. However, the viscositiesof the two phases are found to be five orders ofmagnitude differentw54x. This is thought to be due tothe presence of predominately modified PEG in onephase and that of non-modified chains in the other. Thehydrophobic groups on the modified chains are knownto associate reversiblyw30x in water, forming spanningnetworks that lead to the observed high viscosity. It hasbeen shown theoretically that such association results ina net attraction between modified PEG molecules suffi-cient to explain the observed behaviour of such mixturesw54x. As discussed in the later section, following the
presentation of a number of simulation results, it isproposed that a similar mechanism can operate betweenmixtures of two different proteins residing on interfaces.However, an important point to emphasise is that suchassociation or bonds have to be transient to allowsufficient mobility on the interface. Permanent bondslead to a system that becomes kinetically trapped,arresting the subsequent phase separation dynamics.While bonds have to be weak enough to be reversible,they have to nevertheless have sufficient strength to leadto enough effective attraction to induce phase separation.An important question is, therefore, whether these twoopposing requirements can simultaneously be met inreal or even in model simulated systems. We shallattempt to provide some clues to the answer in Section4.Our discussion so far has been entirely concerned
with systems where the dynamic of exchange betweenthe bulk and the interface is assumed to be very slow.It is also useful to briefly consider the opposite casewhere the bulk solution and the interface are at equilib-rium. For such systems, just as before, there is an excessfree energy per unit areaf (G , G ), arising from thes 1 2
presence of the interface. However, unlike the situationdiscussed above, the overall composition of the interfa-cial layer is no longer constrained. Since molecules ofboth species can freely exchange with the bulk, it isappropriate now to consider the interfacial thermody-namic potential(per unit area), i.e.
b bA G ,G sf G ,G ym G ym G (17)Ž . Ž .s 1 2 s 1 2 1 1 2 2
where is the chemical potential of the speciesi inbmi
the bulk solution. The composition of the interface isdetermined by the values ofG andG , which happen1 2
to minimise the function defined in Eq.(17). At thislowest value,A is also equal to the interfacial tensions
for the interfacew31x. In most circumstances one expectsa single global minimum for the function in Eq.(17).Nevertheless, one might argue that for some particularset of values of bulk concentration of the two surface-active species,A (G , G ) might develop two minima,s 1 2
at different interfacial compositions, which just happento have the same minimum value. In principle then, twosuch phases can co-exist on the interface. In practice,however, achieving this situation is highly unlikely. Firstof all, the situation will only arise at some preciselyrelated values of bulk concentration of the two species.Controlling the amount of two sets of molecules to sucha degree would be difficult. Even if such a situationcould be realised, there are no constraints on the overallcomposition of the interface. Therefore, in time, one ofthe phases will tend to dominate at the expense of theother. This will be determined by the kinetic of adsorp-tion of the two species and the initial conditions of theexperiment.

36 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 2. Adsorption of spheres to an initially clean interface for variousadsorption energies form BD simulations.(a) Occupied area fractionof the interfacef as a function of reduced timetyt . The bold linea
r
corresponds to the theoretical prediction for a diffusion-limitedadsorption.(b) Adsorption isotherms, i.e.f vs. bulk volume fractiona
f at equilibrium. The continuous lines are fitted according to theb
Volmer isotherm withKs35, 250 and 1100 forE s6.00(h), 8.94ads
(n), and 13.29(s), respectively.
Our motivation, in discussing these cases involvingfree exchange of surface-active species with the bulk, isto emphasise the importance ofirreversible adsorptionas a prerequisite to having true phase separation pro-cesses at the surface. We finish this section by drawingattention to a situation intermediate between the twocases discussed above, where one of the surface-activespecies is irreversibly adsorbed, whilst the other is inequilibrium with the bulk solution. This is an interestingpossibility that can arise when a high-molecular-weightprotein is very slowly displaced by relatively solubleLMW surfactant molecules, adsorbing from bulk. Onceagain the simulation results(presented in Section 5) canprovide some clues as to the likelihood of phase sepa-ration behaviour of such systems.
2. Adsorption of one-component systems
To understand the simulations of the competitiveadsorption of proteins and LMW surfactants we havefirst to consider the behaviour of each separate type ofmolecule adsorbing to an interface. Although no AFMimaging can be done for pure surfactant layers—mainlybecause of the high mobility of these molecules—someimages have been obtained forb-lactoglobulin adsorbedat oil–water interfacesw3x. Furthermore, interfacial rhe-ological properties of adsorbed proteins have been inves-tigated extensively in the laboratoryw32,33x and also incomputer simulationsw18,17x. In the following twosections we describe separately the main features ofsimulated adsorption of pure LMW surfactants and pureproteins.
2.1. Adsorption of LMW surfactants
When an oil–water or air–water interface is presentedto a LMW surfactant dissolved in one of the phases, itsamphiphilic character drives a surface adsorption pro-cess. Although there is an entropy loss(mainly due tothe loss of a degree of freedom in the direction perpen-dicular to the interface), the orientation of the surfactantmolecules at the interface(with the hydrophobic tailprotruding into the hydrophobic phase and the hydro-philic group into water) reduces the free energy of thesystem as a whole. There is a binding energy permolecule of, say, a fewk T. Therefore, surfactant mol-B
ecules tend to accommodate themselves preferentially atthe interface thereby reducing the interfacial tensiong.Surfactant molecules are relatively small and so theydiffuse rapidly to and from the interface. After a shortperiod of time a dynamical equilibrium state is reached,where the number of molecules adsorbing at the inter-face at any time equals the number of moleculesdesorbing back into the bulk phase, driven by thermalmotion. Following equilibration, the concentration ofthe adsorbate molecules at the interface is constant and
so is the interfacial tensiong. Dynamical interfacialtension measurementsw35x often provide useful infor-mation regarding the equilibration process in suchsystems.The concentration of highly idealized spherical adsor-
bate species at the interface, obtained by BD, is shownin Fig. 2 as a function of time and bulk concentrationf . The surface concentration is expressed as areab
fraction—the fraction of the interfacial area covered bythe adsorbate molecules. ForN adsorbed spherical mol-ecules of diameters at an interface of area , the areaAfraction is f 'p Ns y(4A). The simulation corre-a 2
sponds to the model described in Section 1.3, but withjust a single type of particle used in order to simulatepure surfactant adsorption. In this case, the only parti-cle–particle interaction involved is the repulsive spher-ical core potential f (r ). Particles were initiallyc ij
randomly distributed in bulk. In Fig. 2a, the time

37L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 3. Interfacial structure of a close-packed protein monolayer from simulation(a) and AFM (b). The AFM image corresponds to ab-lactoglobulin Langmuir–Blodgett film at the oil–water interface(100=100 nm).
evolution of the concentration at the interface is dis-played for various adsorption energiesE . For highads
values ofE —where every molecule that collides withads
the interface is ‘irreversibly’ adsorbed, and so theprocess is strictly diffusion-limited—the initial part ofthe curves agrees with experimentw34x and diffusiontheory w35x in that the area fractionf increases asa
. In Fig. 2b, a set of adsorption isotherms isD typy 0
shownw18x. The area fraction is plotted against the bulkvolume fraction of the particles. The fitted curvescorrespond to Volmer isothermsw36x,
a aw zf f bexp sKf , (18)x |2 a 2 aps y4yf ps y4yfy ~
with K an adjustable constant. As expected, for a givenbulk volume fraction, the equilibrium area fraction atthe interface increases for increasing adsorption energy.Equally, for a given adsorption energy, the equilibriumarea fraction increases with increasing bulkconcentration.We should point out here that our model surfactant
molecules are not strictly amphiphilic in the sense thatthey do not associate into micelles, and they interactwith each other only through a repulsive core potential.The surfactant attributes captured by our model are thesmall molecular size and the tendency to adsorb revers-ibly at the interface through the moderately low adsorp-tion energy per molecule.
2.2. Adsorption of globular proteins
Proteins, like LMW surfactants, can also adsorb tointerfaces lowering the interfacial tension. Since themolecular mass is typically 100 times that of LMWsurfactants, they diffuse more slowly to the interface.
Protein chains are normally highly folded in solutionwith the hydrophobic amino acids tending to reside inthe centre of the globule—away from water—while thehydrophilic amino acids tend to be at the globule surface.When a protein adsorbate reaches an oil–water or air–water interface, it partially unfolds exposing its hydro-phobic groups to the non-aqueous phase. In that waythe protein reduces the surface free energy of the system,and the binding energy per molecule is much higherthan for surfactant adsorbates. Therefore, proteins arealso surface-active molecules, although LMW surfac-tants can typically reduce the interfacial tension to alarger extent. This is due to the simpler structure andsmaller size of LMW surfactants, which allows them tobe arranged more efficiently at the interface.(In Section3, we will show how important this size effect can befor mixed systems.)Many globular proteins can also form chemical and
physical linkages with other protein molecules. Whenproteins adsorb, they reach higher local concentrationsthan in the bulk, and so the protein–protein interactionsplay a more important role. Moreover, when they unfold,a large number of chemical groups on the proteinmolecule are exposed to other neighbouring molecules.As a consequence, the formation of a protein–proteinlinkage is a very common event in globular proteinfilms. Indeed, many of these films behave like two-dimensional gels, held together by a cross-linked net-work of bonds, some strong(permanent) and someweak(transient).In Fig. 3, we show the interfacial structure of a
simulated layer of protein-like spherical particlesadsorbed at an interface(a) together with a AFM imageof a realb-lactoglobulin monolayer adsorbed at the oil–water interface(b). As in the previous section, particlesof a single species are used in simulating the modelusing a BD algorithm. However, this time, the adsorbate

38 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 4. Interfacial shear rheology of a cross-linked monolayer of par-ticles (reproduced from Ref.w17x). (a) Normalised stress time cor-relation functionc (t)yc (0) for a two-dimensional system(the areas s
fraction is f s0.31) and a three-dimensional system(the volumea
fraction isf s0.05) as a function of reduced timetyt . (b) Storagebr
and loss moduliG9(d) and G0(j) vs. (reduced) shear frequencyft . The maximum applied strain isg s0.05, which is in the linearr 0
regime.
spheres are allowed to interact via the formation ofbonds wsee Eq.(11)x in addition to the core repulsivepotential. The particles adsorb to the interface with ahigh adsorption energyE s44.0, and the bond para-ads
meters are: s1.0, b s1.0, b s0.1 and b s0.3.B 1 0 max
Bonds are created with probabilityP s1.0 when a pairB
of particles approaches to withinb , and destroyed when1
the bond length exceedsb . However, as proteins aremax
much more reactive at the interface than in bulk solution,no bond is created unless both approaching particles arefully adsorbed. All the adsorbate spheres(4000 in total)are initially placed at random in the simulation box(L sL s40, L s6). Soon after the simulation starts,x y z
the spheres pack the interface and develop the network-like structure shown in Fig. 3a. As we can see, thisstructure compares rather well(in terms of the length-scales of heterogeneities) with the experimental one,obtained using AFM imaging ofb-lactoglobulin at theoil–water interface(Fig. 3b).Detailed studies of the rheological properties of these
types of simulated films have been carried outw18,17x.However, definitive comparisons with experimentalresults are still awaited(for a review on experimentaltechniques see Ref.w37x). The small-deformation shearrheology of a monolayer of bond-forming spheres showssimilar characteristics to the shear rheology of an anal-ogous bulk system. The main difference is that the stresstime correlation function, defined asC t sŽ .s
, decays more slowly in the¯ ¯N MN s 0 s t y rk TŽ . Ž . Ž .ab ab B
two-dimensional case, perhaps not surprisingly giventhe more restricted topology of a two-dimensional net-work. In Fig. 4, we show the stress time correlationfunction (a), and the storage and loss moduli as afunction of the frequency of the applied strain(b). Theresults correspond to a system withNs1000 particlesirreversibly adsorbed(i.e. E s`, which correspondsads
to z s`). After adsorption (f s0.31), particlesac
formed bonds withP s10 , assuming bond parame-y3B
ters ´ s1.0, b s1.0, b s0.1 andb s1.0. Once aB 1 0 max
stable structure had been formed, the bonding probabilitywas set to zero and the rheological properties of theinterface determined.The slow decay of the stress time correlation function
(Fig. 4a), as compared with a similar three-dimensionalsystem, indicates that the interfacial viscosity—whichcorresponds to zero frequency shear—is rather high.This is to be expected since at the interface the particleconcentration is normally much higher than in bulksolution, and as already mentioned, the particles havealso a more restricted mobility in the two-dimensionalspace of the interface. As far as the storage and lossmoduli are concerned(Fig. 3b), the same qualitativefeatures are seen in the interfacial and bulk gel-likeprotein systems. Frequency-dependent storage and lossmoduli have been measuredw38,39x, but no direct
quantitative comparison with simulated data is possiblebecause of the somewhat arbitrary choice of the para-meters in the simulation model.The small-deformation dilatational rheology of this
same system has been also studiedw17x. In Fig. 5a, weshow the storage and loss dilatational moduli based onisotropic expansion and compression of the interface.Again, the gel-like behaviour of the film is clear sincethe storage modulus builds up with increasing frequencywhile the loss modulus vanishes at relatively low fre-quencies. This information can be obtained experimen-tally by using, for example, a pendant-bubble dynamictensiometer where a protein-coated bubble is subjectedto volume oscillations that induce surface area changes(see for example Ref.w40x).In Fig. 5b, the large-deformation surface dilatational
rheology of a simulated protein film is represented asstress vs. timew18x. The model film(Ns1000 particlesadsorbed withf s0.63 and bond parameterss1.0,a
B
b s0.1, b s0.1 andb s`) is compressed in thex-1 0 max

39L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 5. Interfacial dilatational rheology of a cross-linked monolayerof particles.(a) Storage and loss dilatational moduli´9(d) and´0(j)vs. dilatational frequencyft for the same system as in Fig. 4(repro-r
duced from Ref.w17x). The maximum relative increase of the inter-facial area (isotropic compression and expansion) during theoscillation was 0.05.(b) Interfacial stress responses (normal com-xx
ponent of the stress tensor parallel to the direction of compressionand expansion) of a system with area fractionf s0.63 (reproduceda
from Ref. w18x). From t s0 to t s0.5 the interface is compressed* *
(up to 50%) and it is re-expanded betweent s0.5 and t s1.0.* *
Curves a–c correspond to adsorption energiesE s2.66, 6.00 andads
8.94 k T, respectively. The strain-rate is . Curve d corre-y10.283tB r
sponds to the same system as b, but for a strain-rate of .y10.0283tr
direction up to roughly half of its initial surface areaand re-expanded back to its original size. The compres-sion and expansion at constant strain-rate is achieved˙by rescaling the position of the particles so that thelength of the simulation box in thex-direction varieswith time as . In addition, the positions˙L t sL exp´tŽ . Ž .x x,0
of the particles are rescaled in thez-direction accordinglyto maintain the system at constant volume. The timescale in Fig. 5b has been adjusted so thatt s0.5*
corresponds to the time at which the maximum com-pression is reached andt s1.0 corresponds to the time*
at which the system is returned back to its originalshape. Therefore, results corresponding to differentstrain-rates are presented with different time scales.Curves a–c are for different adsorption energies, andcurve d is the same adsorption energy as b but at alower strain-rate. As we can see, the stress becomesnegative during the compression phase(i.e. the interfa-cial tension decreases), and it is greater in absolute
value for the systems with higher particle adsorptionenergies. This is what we expect: particles need to beremoved from the interface in order to relax the filmstress, and this is harder to achieve with stronglyadsorbing particles. The minimum of the curves isassociated with the point at which many of the particlesstart to desorb to release interfacial pressure and an‘orogenic’ mechanism has been suggested to producethis processw7x. Comparing curves b and d in Fig. 5b,it becomes clear that a low strain-rate allows the proteinfilm to rearrange its internal structure at the interfacemore efficiently leading to a lower stress in absoluteterms. Recently developed instrumentsw41x might beable to produce experimental results in situations com-parable to those studied here by computer simulation.To conclude this section, we should emphasise that
these simulations on large-deformation dilatational rhe-ology were carried out with irreversible protein–proteinbonds(b s`) and no bonds were allowed to formmax
during the compression–expansion process. Therefore,the topology of the bond network was restricted toremain unaltered during the deformation process. In Fig.6, we show a compressed and a re-expanded proteinfilm where bonds were allowed to break(b s1.0s)max
and new bonds were allowed to form(P s10 ). Aftery3B
re-expansion, the film presents cracks due to the bondsbroken during the rearrangement of the internal structureof the film. The restrictions imposed by the newlyformed bonds make the film protrude into the bulk afterre-expansion because many of the particles cannot thenre-adsorb. This is in contrast with the previous simula-tions where the film re-adsorbed fully after expansionw18x.
3. Competitive adsorption of mixed LMW surfactants
When a mixture of two different LMW surfactants(or any mixture of surface-active species) is allowed toadsorb at an interface, the two kinds of molecules haveto ‘compete’ for space: hence, the name ‘competitiveadsorption.’ Naively, we might expect the adsorbatewith the larger value ofE to dominate the interface ifads
the mixture bulk concentration ratio is approximately1:1. For species with equal binding capacity for thesurface, it would be expected that the one present athigher bulk concentration would predominate at theinterface. However, the molecule–molecule interactionsalso play an important role in determining the finalcomposition at the interface.There is, however, another equally important factor—
regardless of inter-molecular interactions—affecting thecomposition of the mixture at the interface: the sizeratio of the species. It has been shownw42x by MDsimulation that small spheres can displace bigger onesfrom an interface even if their adsorption energies areidentical. This effect is explained from the thermodyn-

40 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 6. Snapshot of a compressed interfacial film after a large number of extra bonds have been formed(a), and the same system after re-expansion(b). In each case a top-view and a side-view are given. Reproduced from Ref.w18x.
Fig. 7. Displacement of a monolayer of adsorbed spheres by smallerparticles. Number of adsorbed big spheresN as a function of time.b
The 1000 initially adsorbed spheres(adsorption energy 6.0k T) areB
of unit diameter. Displacement is produced by 2000 small spheres(ofsame adsorption energy as big ones) of diameter 0.3(e), 0.5 (h),0.7 (n), and 0.9(s).
amical point of view, by the Braun–Le Chatelier’sˆprinciple applied to surfacesw43x. In fact, this can alsobe the case for small spheres having lower adsorptionenergy than the big spheres. In Fig. 7, we show somesimulated data illustrating this behaviour. Using themodel described in Section 1.3, we explore the displace-ment of big spheres by smaller ones. One thousandparticles of type 1(with unit diameter) are initiallyplaced at the interface. Then, twice this number ofsmaller particles(diameter 0.3, 0.5, 0.7 or 0.9) is addedto the bulk, beneath the interface. The particles interactwith each other only via the repulsive core interactionpotential, and both species have the same adsorptionenergy ( ). Fig. 7 shows the time-1 2E sE s6.0k Tads ads B
dependence of the number of particles of type 1 as theyare displaced by particles of type 2 during the equilibra-tion process. Results for various sizes of type 2 particlesare compared. As we can see, smaller particles promotea more efficient displacement. This is because thenumber density at the interface can be greatly enhancedby adsorbing several small particles in the space requiredfor adsorbing a big particle. As the adsorption energy isidentical for both species, the more particles of any typethat the interface contains, the lower is the free energyof the system. The actual number of small adsorbatesthat can be placed in the same space as a big adsorbategrows as the square of the diameter ratio. Therefore, ifthe size ratio is large enough, even small adsorbateswith low adsorption energies(LMW surfactants) canefficiently displace big particles that have a high affinityfor the interface(even proteins). This effect is further
enhanced by the non-ideal entropy of mixing of non-equally-sized molecules as demonstrated by Lucassen-Reyndersw44x by recognising that the total Gibbs freeenergy of the adsorbed mixture is expressed moreappropriately as a function of the area fraction ratherthan the mole fraction of the molecules. Of course, theequilibrium adsorbed amount of type 1 particles ulti-mately depends not only on the size ratio, but also onthe bulk concentration of both species, and also theintermolecular interactions. We do not attempt here to

41L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
analyse the thermodynamic properties of the model sincewe are more interested in the non-equilibrium propertiesof the initial stages of the competitive adsorption.We conclude this section with a comment on the
likelihood of lateral phase separation in this kind ofsystem. Surfactants are in general small molecules andtheir adsorption energies are of the order of just a fewk T w37x. Therefore, surfactant molecules adsorb andB
desorb from liquid–liquid interfaces in a relatively rapidmanner, so promoting a constant exchange of particleswith the bulk phase. As we have discussed in Section1.5, a mixture of adsorbates with such a property isunlikely to form different co-existing phases at theinterface even if their cross interaction favours de-mixing.
4. Competitive adsorption of mixed proteins
The competitive adsorption of sticky hard spheres hasbeen studied in the framework of the Percus–Yevickintegral equation theory, both in generalw45x and inrelation to protein mixturesw46x. The model systemw47xconsists of a binary mixture of particles interactingthrough a hard sphere particle–particle potential and asticky surface–particle potential. It has been shownw46xthat for binary mixture of equally sized adsorbing hardspheres with no extra interactions, the isotherms calcu-lated from the Percus–Yevick theory reproduce qualita-tively the experimental results obtained for thecompetitive adsorption ofa -caseinyb-casein mixturess1
in oil-in-water emulsions. It is worth mentioning thatthis type of integral equation theory applies properlyjust to systems at equilibrium. Fortunately, mixtures ofthe disordered proteinsa -casein andb-casein come tos1
equilibrium fairly rapidly—unlike most globular pro-teins—and so the theoretical assumption is realistic inthis case.In relation to the kinetics of the competitive adsorp-
tion of proteins, there has been a significant amount ofwork done on the diffusion equation and its relationshipto surface tension measurements(for a review see Ref.w35x). As far as the sticky surface model is concerned,the assumption that the protein–protein interaction iswell represented by a hard sphere potential may bereasonably reliable only at low or moderate ionicstrength where the protein molecules do not aggregateas a result of electrostatic screening of the attractiveinteractions. Unfortunately, theoretical studies thatinclude protein–protein stickiness in addition to pro-tein–surface stickinessw48x have not provided a simpleexpression for the isotherms.Recently, there have been experimental reportsw49,50x
of high-resolution microscopic images of mixed proteinfilms. Binary mixtures of proteins labelled with differentfluorescent dyes have been adsorbed from bulk aqueoussolution to the air–water interface. After transferring
each adsorbed film onto an appropriate substrate, sepa-rate images of each species could be recorded byilluminating the sample with different wavelengths oflight. The results of some of these experiments appearto be contradictory, however, as phase separation of thecomponents after ageing has been seen in some casesw8,49x, but not in othersw50x. In any case, it is clearthat the viscoelastic character of the films kineticallylimits the molecular mobility at the interface and henceany thermodynamically driven structural rearrange-ments. As a result, lateral phase separation, if it occurs,may typically take place only after several days ofsurface ageing. However, when strong intermolecularinteractions are present, the system can be kineticallytrapped, so that interfacial phase separation cannot occurat all during normal experimental time-scales.In order to model the dynamic behaviour of this type
of system, some over-damped BD simulations have beenperformedw51x. The assembly contains 2000 particlesof two types (i.e. 4000 particles altogether). Particleswere initially distributed at random in a prism-like boxof sides L =L =L (40=40=6). The value of thex y z
parameterz in the interfacial potential, Eq.(2), was setc
to 0.15s. This implies an adsorption energy of 44.0k TBfor both kinds of particles. As the simulation evolves,the individual particles reaching the interface are trappedby the strong external force field and so they remainadsorbed essentially irreversibly(no spontaneousdesorption over the simulation time-scale). However,they can readily move in the plane of the interface. Thestrong adsorption energy here is essential in order toavoid complications of desorption during the simulation,and hence to set the composition of the interface onceit is packed with particles. Otherwise, phase separationis not to be expected, as pointed out previously(Section1.5).The model particles of type 1 can interact through
flexible bonds as described by Eq.(11). In order tomimic the effect of the interfacial unfolding behaviourof real proteinsw33x, we allow these particles to formbonds only when they are located at the interface—reflecting the fact that macromolecular reorganization atthe interface generally enhances greatly the probabilityof reacting groups becoming exposed to neighbouringproteins molecules.In Fig. 8, the simulated structure of the interface is
illustrated by representing type 1 particles as lightspheres and type 2 particles as dark spheres. In the setof top-view pictures(a)–(c), various different types ofinteractions have been adopted in addition to the repul-sive hard-core spherical potential. While pictures(a)–(c) include the adsorbed particles only, picture(d)shows a side view of the entire system(displaying bothadsorbed and non-adsorbed particles) corresponding to(c). Picture (a) refers to a system where the type 1particles can form elastic bonds once they reach the

42 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 8. Two-dimensional patterns formed by mixtures of model spherical particles as they adsorb to a planar interface. Parts of the replicas of thecentral simulation box are also displayed. Dark particles do not form bonds. In(a), light particles form irreversible bonds. In(b), light particlesform reversible bonds. In(c), neither dark nor light particles form bonds but an effective repulsion has been introduced between unlike particles.A three-dimensional profile of the system in(c) is also shown(d). Reproduced from Ref.w51x.
interface. These bonds are irreversible(i.e. b s`)max
and moderately inflexible(´ s3.0k T)—a case thatb B
might for instance represent the situation where inter-molecular covalent disulfide bonds are formed betweenadsorbedb-lactoglobulin moleculesw52x. After a shorttime, corresponding to the time necessary to saturate theinterface with particles, the interfacial structure becomestrapped in a particular configuration, like the one shownin Fig. 8a. It now no longer evolves with time. Thebond-forming type 1 particles build a two-dimensionalnetwork, which resembles a gel-like structure with porescontaining the type 2 particles. Although regions rich inone or other of the species can be seen in picture(a),these are of the order of a few molecules in size. Areduction in free energy would be achieved if the systemwere to become phase-separated. However, the rigidityof the interfacial network traps the system kinetically,preventing the structure from becoming more heteroge-neous. This picture(a) seems to agree rather well withfluorescence microscopy experiments onb-lactoglobu-linyb-caseinw50x where a ‘perfectly mixed’ system wasobserved after three days of ageing. In Fig. 9a, wepresent a fluorescence image of a mixedb-casein(lightcolour)qb-lactoglobulin(dark colour) film after 2 days
of ageing showing no evidence of surface phaseseparation.In picture(b) of Fig. 8 we see the result of a similar
kind of simulation to the one displayed in picture(a),but, in this new example, the bonds were allowed tobreak and reform(b 50.3s), and they were alsomax
more flexible (´ s1.0k T), corresponding to lessb B
strong, ‘physical’ association, as opposed to ‘chemical’bonds in(a). Because of these changes, the more mobiletype 1 particles at the surface can now rearrange moreefficiently and it is clear from picture(b) that the systemis indeed phase separating. In the initial stages, thesystem builds a network similar to the one shown in(a). However, the reversible character of the bondingallows the surface structure to rearrange, leading to morecompact type 1 clusters that become increasingly dis-connected. The ageing of this system would be expectedeventually to lead to a proper macroscopic phase-separated structure—but the simulations become prohib-itively expensive at these later stages because thediffusion of a big type 1 cluster through a dense type 2environment is inevitably very slow. This type of sce-nario is normally referred to as ‘transient gelation’, andit has been well documented in bulk systemsw53x. Based

43L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 9. Fluorescence images of mixed protein films adsorbed to air–water interfaces.(a) b-casein(light colour)yb-lactoglobulin(dark colour)after 2 days of ageing(reproduced from Ref.w50x). The image is 63=48 mm and it presents a homogeneous grey texture.(b) a-lactalbumin(light colour)yb-casein(dark colour) after 4 days of ageing(reprinted with permission from Ref.w49x Copyright 2001 American ChemicalSociety). The image is 1200mm across and it shows ana-lactalbumin-rich oval region.
Fig. 10. Fluorescence scanning near-field optical micrograph for aphase-separated poly(octadecyl methacrylate)ypoly(isobutyl methac-rylate) binary mixture at the air–water interface(reprinted with per-mission from Ref.w56x Copyright 2001 American Chemical Society).The light coloured areas correspond to the poly(octadecyl methacry-late) molecules.
on the experimental evidence, phase separation of thischaracter might well be encountered in soy 11Syb-casein,a-lactalbuminyb-caseinw49x, and bovine serumalbuminyb-casein mixturesw8x. It has also been dem-onstrated elsewherew54,55x that phase separationbetween two very similar polymeric species can occurin the bulk if the molecules of one of the species canself-associate. In Fig. 9b is a fluorescence imagew49xof a mixeda-lactalbumin(light colour)qb-casein(darkcolour) film after 4 days of ageing showing ana-lactalbumin-rich oval region. Interestingly, these typesof interfacial phase-separated structures do not appearfor this system when investigated at low bulk concen-trationsw49x.An example of a simulated layer of two non-bond-
forming species is shown in picture(c) of Fig. 8. Inthis case, we have ‘switched off’ the formation of bondsbetween type 1 particles, but instead have included arepulsive interaction between the unlike species( 12´ sUB
). Additionally, the simulation box122.0k T andr s1.2sB c
is enlarged in thez-direction (40=40=18) to reducethe bulk volume fraction of the particles and so to avoidpossible bulk immiscibility. We find that the interfacialregion phase separates very quickly through what seemsto be the spinodal decomposition mechanism. This effectis seen soon after the interface is packed with particles,in sharp contrast to the case depicted in picture(b), forwhich much longer runs were required in order toobserve the nucleation of clusters of type 1 particles.As we have discussed in Section 1.5, rapid phaseseparation is commonly seen in binary polymer mixturesrather than in protein mixtures since much higherdegrees of incompatibility are normally achieved in theformer case. Fig. 10 shows an experimental example ofa phase-separating polymer mixture at the air–waterinterfacew56x.By studying experimental adsorption isotherms,
incompatibility in interfacial mixing of some pairs ofproteins has been inferredw57x. That is, the actualadsorption isotherm for a mixture of proteins can be
compared against the theoretical Langmuir isotherm fora non-interacting mixture. Then, the extent of the devi-ation can be used to provide a measure of the degree of‘cross interaction’ present in the system. This measurehas been suggestedw57x as being an indicator of thedegree of(im)miscibility of the system. However, thismight not actually be the case. For example, if theproteins of the unlike species cross-link strongly to eachother at the surface, they could form a very well mixedsystem w58x, even though the strong cross interactionwould lead to a large deviation of the actual adsorptionisotherm from that corresponding to a non-interactingmixture. A further complicating fact is that true ther-modynamic equilibrium might be extremely hard toachieve even after long periods of ageing in these highly

44 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
concentrated and highly interacting protein mixtures.Therefore, the incipient phase separation may not actu-ally be seen in some experiments, because the particularpreparation procedure of the system leads to a kineticallytrapped configuration, as discussed above.
5. Competitive adsorption of proteins and LMWsurfactants
When a solution containing protein and LMW surfac-tant is adsorbed, the surfactant is likely to prevail at theinterface, after equilibration, if both species are presentat high enough bulk concentrationsw37,52,59x. This isdue to the fact that LMW surfactants are much smallerin size than proteins, and so they can reduce theinterfacial tension more efficiently by adsorbing a muchlarger number of molecules within the same surface area(see the discussion in Section 3 about the relativeparticle size effect on competitive adsorption of LMWsurfactants). However, during the adsorption of the twospecies, the protein molecules do partially adsorb at theinterface in the very initial stages of the processw60x .1
Soon after, however, an increasing number of smallsurfactant molecules adsorb at the interface and displacethe protein molecules, eventually removing all of theinitial protein film. A similar sort of behaviour occursif the surfactant is introduced into the sub-phase afterthe protein film has developed at the interface.It is important to mention that there exists an addi-
tional mechanism for the displacement of a protein filmby surfactant molecules. Water-soluble surfactant mole-cules can bind to protein molecules both by electrostaticinteraction(if the surfactant is ionic) and by hydropho-bic interaction involving their hydrophobic tails andhydrophobic groups of the proteinw35,37x. If the hydro-philic–lipophilic balance is larger for the protein–sur-factant complex than for the protein molecule on itsown, a solubilization mechanism will facilitate the dis-placement of some of the protein from the monolayeron the interfacew52x.Although many experiments have been devised to
study the properties of protein films being displaced byLMW surfactants, it is only recently that the first imagesof these films have been obtained by means of theatomic force microscopew7x. The outcome of theseexperiments has been very valuable, providing evidenceagainst the common assumption in many theoretical
Although the diffusion coefficient is lower for proteins, they1
occupy a larger area when they reach the interface. Indeed, duringthe very initial stages of adsorption the surface concentration increaseswith time as G(t)s2 c (D typ) , with c the molar bulk1y2
0 0 0
concentrationw35x. Therefore the area fraction occupied by theparticles increases asf (t)sp G(t) s y4;s . Hence, the biggera 2 3y2
the particles are the more surface they initially cover at the interfacefor a given bulk concentration. This effect is very much enhanced byunfolding in the case of proteins, since they can diffuse faster in thefolded configuration and occupy a larger area after unfolding.
approaches(see for example Ref.w35x), namely that thepreferential adsorption of the surfactant occurs in ahomogeneous way throughout the interface. The mainstructural features of the displacement of milk proteinsby LMW surfactant have been identified in this workw7x. In Fig. 11, we show AFM images ofb-casein(pictures a–c) and b-lactoglobulin (picture d) filmsdisplaced by the non-ionic surfactant Tween20. Afterthe protein film has formed, small pools of surfactantdevelop in the defects of the protein monolayer(picturea). Then, these surfactant domains grow with time andwith increasing surfactant bulk concentration, increasingthe interfacial pressure and compressing the protein film(picture b). Beyond a critical surface pressure the proteinfilm starts buckling into the bulk phase and consequentlyincreasing the film thickness. As surfactant moleculescontinue adsorbing, the surfactant domains start to coa-lesce and parts of the protein film detach from theinterface (picture c). Finally, a continuous surfactantphase develops at the interface leaving just a fewdisconnected protein areas adsorbed, which eventuallyalso detach from the interface.The compression mechanism that promotes the local
thickening of the protein layer has led to the descriptionof the competitive displacement process as ‘orogenicdisplacement’ by analogy with the geological process ofmountain formation. The regions of the interface coveredwith protein behave like a protein film subjected tomechanical compression(see Section 2.2) under theinfluence of the growing surfactant domains.Fig. 11d corresponds to an intermediate state of the
displacement process for ab-lactoglobulinyTween20mixture. From this AFM image, we see that althoughthe general process described above applies to mostproteins, individual protein films can show very differentstructures. Whereas the disordered proteinb-caseinallows the formation of nearly circular surfactantdomains, the globular proteinb-lactoglobulin developsvery irregular gaps. The main reason for the irregularshaped domains is the gel-like structure of theb-lactoglobulin films created by a network of disulfidebondsw52x, which can support local stresses, leading toa fracture-like disruption of the film. The structuresencountered depend not only on the type of protein(disordered or globular, bond-forming or weakly-inter-acting), but also on the type of surfactant(ionic or non-ionic, water-soluble or oil-soluble) w61x and the natureof the fluid interface(air–water or oil–water) w3x. InFig. 12, for example, we can note the different structuresobtained during displacement ofb-casein by Tween20at the air–water and oil–water interfaces. It has beensuggestedw3x that theb-casein network may be strongerat the oil–water interface than at the air–water interface,which makes it more likely to present fracture-typedisruptions as the surfactant domains grow at the oil–water interface. The study of the displacement of mixed

45L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 11. Atomic force micrographs of spread protein films displaced by adsorbing surfactant Tween20:(a)–(c) b-casein films for increasingsurface pressure( ) as the surfactant adsorbs;(d) b-lactoglobulin at intermediate displacement.(a) 1.6=1.6 mm, Ps15.9 mNym.Psg ygH O2
(b) 6.4=6.4 mm, Ps16.7 mNym. (c) 6.4=6.4 mm, Ps19.2 mNym. (d) 3.2=3.2 mm, Ps21.8 mNym. Similar results were obtained whenthe protein was co-adsorbed with the surfactant instead of spread on the water surface. Reproduced from Ref.w7x.
Fig. 12. Atomic force micrographs of spreadb-casein films displaced by adsorbing Tween20:(a) b-casein film at the air–water interface; 6.4=6.4mm, Ps19.2 mNym (reproduced from Ref.w7x); (b) b-casein film at the oil–water interface; 6.4=6.4mm,Ps30.5 mNym (reproduced fromRef. w3x).
protein films by surfactants has shownw50x structuresintermediate between those obtained with the corre-sponding pure protein films.
An attempt has been made to study these proteinqLMW surfactant systems by computer simulationw19,62x using the general model presented in Section

46 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 13. Interfacial structure of a simulated protein film displaced by surfactant as seen from below(the bulk phase). The big light spheresrepresent the protein-like molecules and the small dark spheres represent the surfactant. The images are labelled in chronological order from theinsertion of the surfactant beneath the interface(a) to the intermediate stages of the displacement(d). Only those surfactant molecules adsorbeddirectly at the interface are displayed. By contrast, all the protein molecules are drawn, and so many of them are actually protruding into the bulksolution. The central simulation box plus a part of the replicas are shown to give a better idea of the spatial texture of the system.
1.3. In Fig. 13, we show a typical result from a BDsimulation. The model system consists of two types ofparticles: protein-like particles represented as large lightspheres(type 1) and the surfactant-like particles repre-sented as small dark spheres(type 2). Proteins aremodelled as bond-forming particles(´ s1.0k T, b sB B 0
0.1s , b s0.1s , b s0.4s and ) with a11P s1.01 1 1 max 1 B
relatively low adsorption energy( ). Sur-1E s2.66k Tads B
factants, in contrast, are modelled as small particles(s s0.5s ) that do not form bonds and have a rela-2 1
tively high adsorption energy( ). As2E s29.5k Tads B
already mentioned, real surfactant molecules normallyhave lower adsorption energies than proteins, and themain reason for their great effectiveness in reducing theinterfacial tension is the more efficient coverage of theinterface due to their smaller size. However, in asimulation, the use of very small surfactant moleculesis prohibitively expensive in terms of computationalresources. The smaller the surfactants the more mole-cules we need to introduce in the system in order fillthe space between the big protein molecules. Using ahigher adsorption energy for the simulated surfactantmolecules is a pragmatic way to overcome the problem
of using not such small surfactant molecules but stillmaking them more efficient in competition for theinterface. It is worth mentioning that we compare theresults of these simulations with experiments onb-lactoglobulinqTween20 mixtures. In this case, wherethe surfactant molecules are reasonably big, the diameterratio between the two species is approximately 3 if weassume size is proportional to the cube root of molecularweight. Therefore, the diameter ratio of 2 used in thesimulations is not too unrealistic with respect to thisparticular system.The simulation of the competitive displacement of
globular protein by surfactant is carried out as follow.Firstly, 1000 type 1 particles(the proteins) are placedat the interface( ) and they are allowed toaf s0.641
cross-link to form a gel-like film. Then, after equilibra-tion, 2000 type 2 particles(the surfactants) are intro-duced beneath the interface. The type 2 particles areconstrained by a reflecting wall located atz53.5s . In1
this way we avoid the problem of the surfactant diffusingaway from the interface and ‘diluting’ the bulk solution.In Fig. 13, we can see how the surfactant moleculesadsorb in the gaps between the proteins. Then, these
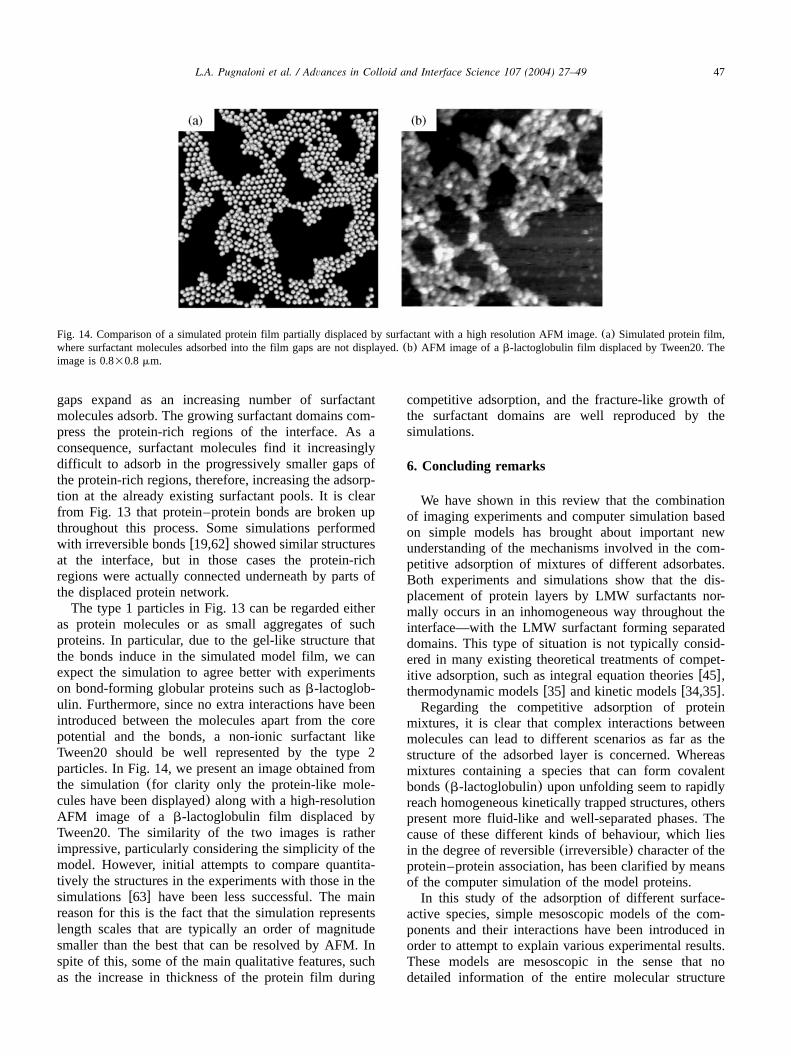
47L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
Fig. 14. Comparison of a simulated protein film partially displaced by surfactant with a high resolution AFM image.(a) Simulated protein film,where surfactant molecules adsorbed into the film gaps are not displayed.(b) AFM image of ab-lactoglobulin film displaced by Tween20. Theimage is 0.8=0.8mm.
gaps expand as an increasing number of surfactantmolecules adsorb. The growing surfactant domains com-press the protein-rich regions of the interface. As aconsequence, surfactant molecules find it increasinglydifficult to adsorb in the progressively smaller gaps ofthe protein-rich regions, therefore, increasing the adsorp-tion at the already existing surfactant pools. It is clearfrom Fig. 13 that protein–protein bonds are broken upthroughout this process. Some simulations performedwith irreversible bondsw19,62x showed similar structuresat the interface, but in those cases the protein-richregions were actually connected underneath by parts ofthe displaced protein network.The type 1 particles in Fig. 13 can be regarded either
as protein molecules or as small aggregates of suchproteins. In particular, due to the gel-like structure thatthe bonds induce in the simulated model film, we canexpect the simulation to agree better with experimentson bond-forming globular proteins such asb-lactoglob-ulin. Furthermore, since no extra interactions have beenintroduced between the molecules apart from the corepotential and the bonds, a non-ionic surfactant likeTween20 should be well represented by the type 2particles. In Fig. 14, we present an image obtained fromthe simulation(for clarity only the protein-like mole-cules have been displayed) along with a high-resolutionAFM image of a b-lactoglobulin film displaced byTween20. The similarity of the two images is ratherimpressive, particularly considering the simplicity of themodel. However, initial attempts to compare quantita-tively the structures in the experiments with those in thesimulationsw63x have been less successful. The mainreason for this is the fact that the simulation representslength scales that are typically an order of magnitudesmaller than the best that can be resolved by AFM. Inspite of this, some of the main qualitative features, suchas the increase in thickness of the protein film during
competitive adsorption, and the fracture-like growth ofthe surfactant domains are well reproduced by thesimulations.
6. Concluding remarks
We have shown in this review that the combinationof imaging experiments and computer simulation basedon simple models has brought about important newunderstanding of the mechanisms involved in the com-petitive adsorption of mixtures of different adsorbates.Both experiments and simulations show that the dis-placement of protein layers by LMW surfactants nor-mally occurs in an inhomogeneous way throughout theinterface—with the LMW surfactant forming separateddomains. This type of situation is not typically consid-ered in many existing theoretical treatments of compet-itive adsorption, such as integral equation theoriesw45x,thermodynamic modelsw35x and kinetic modelsw34,35x.Regarding the competitive adsorption of protein
mixtures, it is clear that complex interactions betweenmolecules can lead to different scenarios as far as thestructure of the adsorbed layer is concerned. Whereasmixtures containing a species that can form covalentbonds(b-lactoglobulin) upon unfolding seem to rapidlyreach homogeneous kinetically trapped structures, otherspresent more fluid-like and well-separated phases. Thecause of these different kinds of behaviour, which liesin the degree of reversible(irreversible) character of theprotein–protein association, has been clarified by meansof the computer simulation of the model proteins.In this study of the adsorption of different surface-
active species, simple mesoscopic models of the com-ponents and their interactions have been introduced inorder to attempt to explain various experimental results.These models are mesoscopic in the sense that nodetailed information of the entire molecular structure

48 L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
(atom-by-atom) is considered. Clearly, further develop-ment of theoretical models for these types of adsorbateis needed to allow for surfactant micellization andprotein–surfactant complexation. However, we believethat the model adsorbates should still remain mesoscopicin character. The reason for this is two-fold. Firstly, theway we want to describe these generic systems is byusing handy uncomplicated models of the molecules. Atthis stage, it seems worth putting these types of mesos-copic model to the test, rather than incorporating highlydetailed chemical structures that might hide the keyfeatures that determine the generic behaviour of thesystems under study. Secondly, the computer resourcesrequired to simulate over a long time-scale even justone protein molecule at the level of its atomic structure,not to mention an entire adsorbed protein layer, is atpresent unavailable to us, and is likely to remain so forseveral years to come.
Acknowledgments
This research was supported by the BBSRC(UK).Computing was done on the Leeds Grid Node 1 facility,funded under the 2001 HEFCE Science Research Invest-ment Fund initiative at the University of Leeds, whichis one of the partners in the White Rose Grid project.
References
w1x E. Dickinson, An Introduction to Food Colloids, OxfordUniversity Press, Oxford, 1992.
w2x P.J. Wilde, Curr. Opin. Colloid Interface Sci. 5(2000) 176.w3x A.R. Mackie, A.P. Gunning, P.J. Wilde, V. Morris, Langmuir
16 (2000) 2242.w4x E. Dickinson, D.J. McClements, Advances in Food Colloids,
Blackie, Glasgow, 1995, Chapter 4.w5x M.P. Allen, D.J. Tildesley, Computer Simulation of Liquids,
Clarendon Press, Oxford, 1987.w6x V.J. Morris, A.R. Kirby, A.P. Gunning, Atomic Force Micros-
copy for Biologists, Imperial College Press, London, 1999.w7x A.R. Mackie, A.P. Gunning, P.J. Wilde, V.J. Morris, J. Colloid
Interface Sci. 210(1999) 157.w8x T. Sengupta, S. Damodaran, J. Colloid Interface Sci. 229
(2000) 21.w9x F. Drolet, G.H. Fredrickson, Phys. Rev. Lett. 83(1999) 4317.
w10x R. Mezzenga, G.H. Fredrickson, Macromolecules 36(2003)4457.
w11x E. Dickinson, D.S. Horne, V.J. Pinfield, F.A.M. Leermakers, J.Chem. Soc. Faraday Trans. 93(1997) 425.
w12x E. Dickinson, V.J. Pinfield, D.S. Horne, F.A.M. Leermakers, J.Chem. Soc. Faraday Trans. 93(1997) 1785.
w13x P.B. Visscher, P.J. Mitchell, D.M. Heyes, J. Rheol. 38(1994)465.
w14x M. Doi, S.F. Edwards, The Theory of Polymer Dynamics,Oxford University Press, New York, 1986, Chapter 3.
w15x A.W. Lees, S.F. Edwards, J. Phys. C 5(1972) 1921.w16x M. Whittle, E. Dickinson, J. Chem. Soc. Faraday Trans. 94
(1998) 2453.w17x C.M. Wijmans, E. Dickinson, Langmuir 14(1998) 7278.w18x C.M. Wijmans, E. Dickinson, Phys. Chem. Chem. Phys. 1
(1999) 2141.
w19x C.M. Wijmans, E. Dickinson, Langmuir 15(1999) 8344.w20x M. Whittle, E. Dickinson, Mol. Phys. 90(1997) 739.w21x M.M. Cassiano, J.A.G. Areas, J. Mol. Struct.(Theochem) 539
(2001) 279.w22x A. Philippsen, W. Im, A. Engel, T. Schirmer, B. Roux, D.J.
Muller, Biophys. J. 82(2002) 1667.¨w23x A.R. Mackie, A.P. Gunning, M.J. Ridout, P.J. Wilde, J.R.
Patino, Biomacromolecules 2(2001) 1001.w24x A.P. Gunning, A.R. Mackie, P.J. Wilde, V.J. Morris, Langmuir
15 (1999) 4636.w25x C.M. Rosetti, R.G. Oliveira, B. Maggio, Langmuir 19(2003)
377.w26x E. Dickinson, in: S.E. Harding, S.E. Hill, J.R. Mitchell(Eds.),
Biopolymer Mixtures, Nottingham University Press, Notting-ham, 1995, p. 349.
w27x A. Garciaa, P. Creux, J. Lachaise, R.S. Schechter, J. ColloidInterface Sci. 261(2003) 233.
w28x E. Dickinson, D.J. McClements, Advances in Food Colloids,Blackie, Glasgow, 1995, Chapter 2.
w29x T. Annable, R. Ettelaie, J. Chim. Phys. 93(1996) 899.w30x A. Yekta, B. Xu, J. Duhamel, H. Adiwidjaja, M.A. Winnik,
Macromolecules 28(1995) 956.w31x R.J. Hunter, Foundation of Colloid Science, volume 1, Clar-
endon Press, Oxford, 1987.w32x B.S. Murray, in: R. Miller, D. Mobius(Eds.), Proteins at¨
Liquid Interfaces, Elsevier, Amsterdam, 1998, p. 179.w33x B.S. Murray, E. Dickinson, Food Sci. Technol. Int.(Jpn) 2
(1996) 131.w34x C.H. Chang, E.I. Franses, Colloids Surf. A 100(1995) 1.w35x R. Miller, V.B. Fainerman, A.V. Makievski, J. Kragel, D.O.¨
Grigoriev, V.N. Kazakov, et al., Adv. Colloid Interface Sci. 86(2000) 39.
w36x J. Lyklema, Fundamentals of Interface and Colloid Science,volume 2, Academic Press, London, 1995, Chapter 1.
w37x M.A. Bos, T. van Vliet, Adv. Colloid Interface Sci. 91(2001)437.
w38x G.B. Bantchev, D.K. Schwartz, Langmuir 19(2003) 2673.w39x J. Benjamins, F. van Voorst Vader, Colloids Surf. 65(1992)
161.w40x L.G. Cascao-Pereira, O. Theodoly, H.W. Blanch, C.J. Radke,˜ `
Langmuir 19(2003) 2349.w41x D.B. Jones, A.P.J. Middelberg, Langmuir 18(2002) 5585.w42x E. Dickinson, E.G. Pelan, J. Chem. Soc. Faraday Trans. 89
(1993) 3435.w43x P. Joos, G. Serrien, J. Colloid Interface Sci. 145(1991) 291.w44x E.H. Lucassen-Reynders, Colloids Surf. A 91(1994) 79.w45x J.W. Perram, E.R. Smith, Chem. Phys. Lett. 39(1976) 328.w46x E. Dickinson, J. Chem. Soc. Faraday Trans. 88(1992) 3561.w47x R.J. Baxter, J. Chem. Phys. 49(1968) 2770.w48x D.Y.C. Chan, B.A. Pailthorpe, J.S. McCaskill, D.J. Mitchell,
B.W. Ninham, J. Colloid Interface Sci. 17(1979) 27.w49x T. Sengupta, S. Damodaran, J. Agric. Food Chem. 49(2001)
3087.w50x A.R. Mackie, A.P. Gunning, M.J. Ridout, P.J. Wilde, V.J.
Morris, Langmuir 17(2001) 6593.w51x L.A. Pugnaloni, R. Ettelaie, E. Dickinson, Langmuir 19(2003)
1923.w52x E. Dickinson, Colloids Surf. B 15(1999) 161.w53x H. Tanaka, J. Phys: Condens. Matter 12(2000) R207.w54x T. Annable, R. Ettelaie, Macromolecules 27(1994) 5616.w55x M. Tsianou, K. Thuresson, L. Piculell, Colloid Polym. Sci. 279
(2001) 340.w56x H. Aoki, S. Ito, J. Phys. Chem. B 105(2001) 4558.w57x L. Razumovsky, S. Damodaran, J. Agric. Food Chem. 49
(2001) 3080.

49L.A. Pugnaloni et al. / Advances in Colloid and Interface Science 107 (2004) 27–49
w58x E. Dickinson, in: S.E. Harding, S.E. Hill, J.R. Mitchell(Eds.),Biopolymer Mixtures, Nottingham University Press, Notting-ham, 1995, p. 349.
w59x J. Kragel, R. Wustneck, F. Husband, P.J. Wilde, A.V. Makiev-¨ ¨ski, D.O. Grigoriev, J.B. Li, Colloids Surf. B 12(1999) 399.
w60x A.R. Mackie, A.P. Gunning, P.J. Wilde, V.J. Morris, Langmuir16 (2000) 8176.
w61x A.P. Gunning, Encyclopedia of Surface and Colloid Science,Marcel Dekker, New York, 2002, p. 3858.
w62x L.A. Pugnaloni, R. Ettelaie, E. Dickinson, Colloids Surf. B 31(2003) 149.
w63x A.R. Mackie, A.P. Gunning, L.A. Pugnaloni, E. Dickinson, P.J.Wilde, V.J. Morris, Langmuir 19(2003) 6032.


















