
Adsorption of surf act ants and polymers at the …ps24/PDFs/Adsorption of Surfactants and...1....
24
\; A COLLOIDS AND SURFACES Colloids and Surfa.:es A: Physicochemical and EngineeringAspects 123-124(1997)491-513 ELSEVIER Adsorption of surf act antsand polymers at the solid-liquid interface P. Somasundaran *, S. Krishnakumar Langmuir Center for Colloids and Interfaces, CohDnbia University, New York. NY 10027, USA Received 30 July 1996; accepted 6 August 1996 Abstract Surfactants, polymers and their mixtures are widely used in several important industrial processes such as dispersion-flocculation. ftotation, dewatering, paints and pigments and oil recovery. Many types of surfactants (anionic, cationic or non-ionic) and polymers (charged and otherwise) are used individually or in combination to achieve desired surface modifications. The adsorption of these molecules at the solid-liquid interface depends on several factors suchas the nature of the substrate, solvent, adsorbatespecies, the presence of secondary competing-coo~ erative species,temperature and even mode of mixing. The nature of the adsorbed layer determines the surface modification achieved and this in turn dependson the adsorption mechanisms and the conditions prevailing during and after adsorption. Several spectroscopic techniques such as ftuorescence,electron spin resonance and Raman spectroscopy have been employed to investigate the structure of various adsorbed layers and to obtain correlations with observed changesin the dispersion properties. In this paper, we review our previous work on adsorption of various ionic and non-ionic surfactants, polymers and their mixtures and mechanisms involved. in aqueous and non-aqueousmedia. C 1997 Published by Elsevier Science B. V. f~~flj;$~ ~""~-.,..."v..,. ~~~iXf§~f; ~.~,:".~"'";""~ ~t~f[}Ji':1 s : .. Keywords: Adsorption; Polymers; Solid-liquid interface; Surfactants 1. Introduction I . Adsorption of molecules on solids from solution is important in controlling a variety of interfacial processes such as mineral flotation, flocculation- dis~rsion, blood clotting, self-assembly and enhanced oil recovery. Adsorption results from energetically favorable interactions between the solid adsorbate and the solute species and is often a complex processsinceit can be influenced by all solid, solvent and solute componentsof the system. Several interactions suchas electrostaticattraction, covalent bonding, hydrogen bonding or non-polar interactions between the adsorbate and the adsor- bate species, and lateral interaction between the ad$orbed species as wen as their desolvation can contribute to the adsorption and desorption processes. Adsorption can be considered to be a process of selective partitioning of the adsorbate species to the interfacein preference to the bulk and is the result of interactions of suchspecies with the surface species on the solid. The interactions responsible for adsorption can be either physical or chemical in nature.Adsorptioncan be broadly classified into two categories, physical adsorption and chemical adsorption, depending on the n~ture of the forcesinvolved [I]. Physical adsorption is usuany weak and reversible and involves sman . Corresponding author. 0927-1157/97/$17.0001997 Published by ElsevierScience B. V. All rights reserved. PII 80927-7757(96)03829-0 ~ ~-:j
Transcript of Adsorption of surf act ants and polymers at the …ps24/PDFs/Adsorption of Surfactants and...1....

\;
ACOLLOIDSANDSURFACES
Colloids and Surfa.:esA: Physicochemical and Engineering Aspects 123-124(1997)491-513ELSEVIER
Adsorption of surf act ants and polymers at the solid-liquidinterface
P. Somasundaran *, S. KrishnakumarLangmuir Center for Colloids and Interfaces, CohDnbia University, New York. NY 10027, USA
Received 30 July 1996; accepted 6 August 1996
Abstract
Surfactants, polymers and their mixtures are widely used in several important industrial processes such asdispersion-flocculation. ftotation, dewatering, paints and pigments and oil recovery. Many types of surfactants(anionic, cationic or non-ionic) and polymers (charged and otherwise) are used individually or in combination toachieve desired surface modifications. The adsorption of these molecules at the solid-liquid interface depends onseveral factors such as the nature of the substrate, solvent, adsorbate species, the presence of secondary competing-coo~erative species, temperature and even mode of mixing. The nature of the adsorbed layer determines the surfacemodification achieved and this in turn depends on the adsorption mechanisms and the conditions prevailing duringand after adsorption. Several spectroscopic techniques such as ftuorescence, electron spin resonance and Ramanspectroscopy have been employed to investigate the structure of various adsorbed layers and to obtain correlationswith observed changes in the dispersion properties. In this paper, we review our previous work on adsorption ofvarious ionic and non-ionic surfactants, polymers and their mixtures and mechanisms involved. in aqueous andnon-aqueous media. C 1997 Published by Elsevier Science B. V.
f~~flj;$~~""~-.,..."v..,.~~~iXf§~f;~.~,:".~"'";""~
~t~f[}Ji':1s:..
Keywords: Adsorption; Polymers; Solid-liquid interface; Surfactants
1. IntroductionI.
Adsorption of molecules on solids from solutionis important in controlling a variety of interfacialprocesses such as mineral flotation, flocculation-dis~rsion, blood clotting, self-assembly andenhanced oil recovery. Adsorption results fromenergetically favorable interactions between thesolid adsorbate and the solute species and is oftena complex process since it can be influenced by allsolid, solvent and solute components of the system.Several interactions such as electrostatic attraction,covalent bonding, hydrogen bonding or non-polar
interactions between the adsorbate and the adsor-bate species, and lateral interaction between thead$orbed species as wen as their desolvation cancontribute to the adsorption and desorptionprocesses.
Adsorption can be considered to be a processof selective partitioning of the adsorbate speciesto the interface in preference to the bulk and isthe result of interactions of such species with thesurface species on the solid. The interactionsresponsible for adsorption can be either physicalor chemical in nature. Adsorption can be broadlyclassified into two categories, physical adsorptionand chemical adsorption, depending on the n~tureof the forces involved [I]. Physical adsorption isusuany weak and reversible and involves sman. Corresponding author.
0927-1157/97/$17.0001997 Published by Elsevier Science B. V. All rights reserved.PII 80927-7757(96)03829-0
~~-:j

P. Somasundaran, S. Krishnakrmlar / Colloids Surfaces A: Physicochem. £IIg. Aspects 123-124 (1997) 491-513492
where r (mol g-l) is the adsorption density,Cc (moll-i) and Cj (moll-i) are the initial andfinal concentrations of the adsorbate, V (I) is thevolume of solution and W (g) is the mass of theadsorbent.
The net driving force for adsorption &G' can beconsidered to be the sUm of a number of contribut-ing forces:&Gads 0 = &Gelec ° + &Gcbem 0 + &Gc -c °
+&Gc-.O&GHo+&GHzoo+... (3)where &Geiec 0 is the electrostatic interaction term,&Gchcm ° is the chemical term due to covalent
bonding, &Go-c° is the lateral interaction term due.to the cohesive chain-chain interaction amongadsorbed long chain surfactant species, &G~o isdue to interaction between hydrocarbon chainsand hydrophobic sites on the solid, &GHo is thehydrogen bonding term, and &GHzoo is the solva-tion or desolvation term resulting from hydrationof the adsorbate or any other species from theinterface during adsorption. For each surfactant-solid-solvent system, several of the above termscan be significant depending on the mineral andthe surfactant type, surfactant concentration,background electrolyte and solvent temperature.For non-metallic minerals, electrostatic attractionand lateral interaction effects are considered to bethe major factors determining adsorption while,for salt-type minerals such as calcite and sulfidessuch as galena, the chemical term often becomessignificant. In organic liquids the electrostaticforces are minimal and adsorption depends on thehydrophobic and hydrophilic interactions amongthe c9nstituents.
t~~'~*ji~~:;'i;
energy changes. van der Waals forces and electro-static forces are primarily responsible for physicaladsorption which is also characterized by a highrate of adsorption and formation of multilayers[2]. Chemical adsorption occurs through covalentbonding between the adsorbate and the surfacespecies on the solid. Chemical adsorption normallyinvolves an activation stage and is characterizedby relatively high energy changes and a low rateof adsorption. Such adsorption is usually strongand irreversible and is limited to a monolayer. Adistinction between physical and chemical adsorp-tion can usually be made from the temperaturedependence of the adsorption process. In the caseof physical adsorption the adsorption generallydecreases with temperature while in the case ofchemisorption it increases with temperature.However, it must be noted that the distinctionbetween physical and chemical adsorption is arbi-trary and in many cases an intermediate characterof adsorption is encountered. In some cases suchas adsorption of gases on metal surfaces, phy-sisorption may take place initially and may befollowed by adsorbent-adsorbate reactions, result-ing in chemisorption [3].
Adsorption isotherms are commonly used todescribe adsorption processes and these representa functional relationship between the amountadsorbed and the activity of the adsorbate at aconstant temperature. The adsorption density,which is the amount of adsorbate removed fromsolution to the interface, can be expressed as [4]
(1)
where rj is the adsorption density in the plane c5,which is at the distance of closest approach ofcounterions to the surface, r is the effective radiusof the adsorbed ions, C (mol ml-1) is the bulkconcentration of the adsorbate, R is the gas con-stant, T is the absolute temperature and ~Gads° isthe standard free energy of adsorption. In practice,however, the adsorption density is measured asdepletion of the adsorbate from the solution:
2. Surfactant adsorption in aqueous media
2.1.. Ionic surfactants
A typical Somasundaran-Feurstenau typeadsorption isotherm of charged surfactants onoppositely charged surfaces is shown in Fig. I [5]where the adsorption of negatively charged sodiumdOdecylsulfate (SDS) on positively charged alu-mina is shown. This isotherm is characterized by
v(2)r-(Cc-G)w
~ Ki~i~~~i~:;~,
~
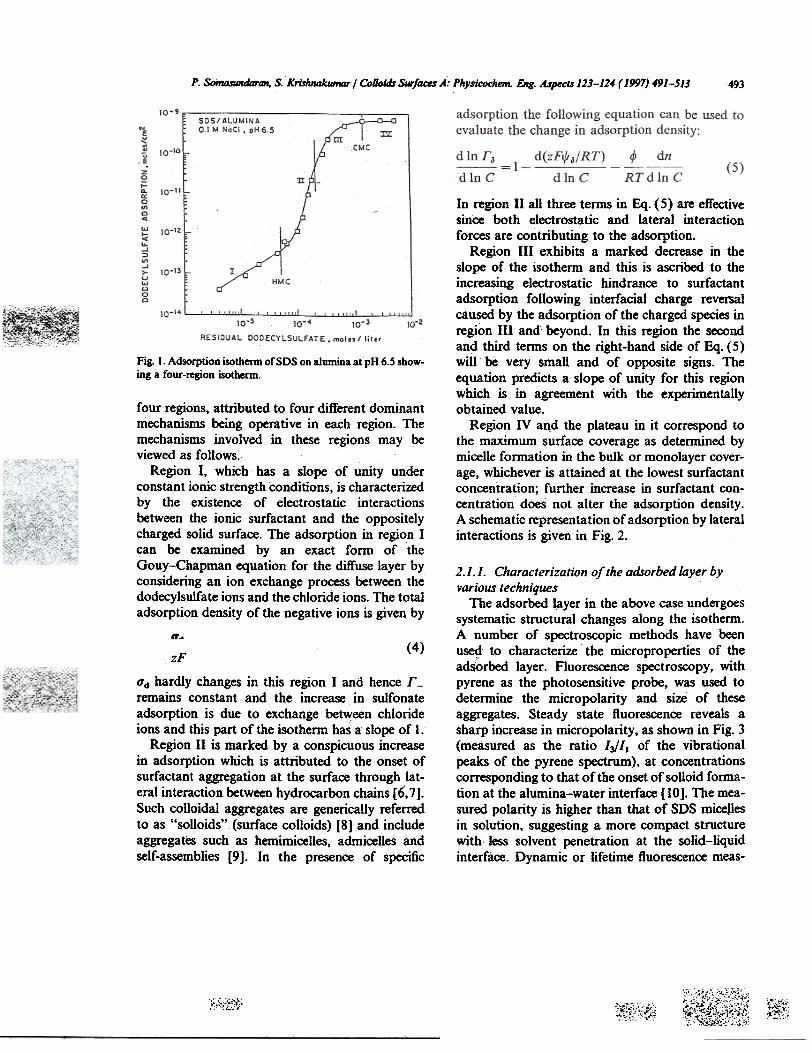
P. SoInD.SIIIIda1iU1, S. Krishnakumar I Colloidr Surfaces A: Physicochem. EIIg. Aspects 123-114 (]997) 49]-5]3 493
Fig. I. Adsorption isotherm of SDS on alumina atpH 6.S show-ing a four-region isothenn.
In region II all three tenDS in Eq. (5) are effectivesince both electrostatic and lateral interactionforces are contributing to the adsorption.
Region III exhibits a marked decrease in theslope of the isothernt and this is ascribed to theincreasing electrostatic hindrance to surfactantadsorption following interfacial charge reversalcaused by the adsorption of the charged species inregion III and beyond. In this region the secondand third ternts on the right-hand side of Eq. (5)will be very small and of op~site signs. Theequation predicts a slope of unity for this regionwhich is in agreement with the experimentallyobtained value.
Region IV aqd the plateau in it correspond tothe maximum surface coverage as detennined bymicelle formation in the bulk or monolayer cover-age, whichever is attained at the lowest surfactantconcentration; further increase in surfactant con-centration does not alter the adsorption density.A schematic representation of adsorption by lateralinteractions is given in Fig. 2.
four regions,' attributed to four different dominantmechanisms being operative in each region. Themechanisms involved in these regions may beviewed as follows.
Region I, which has a slope of unity underconstant ionic strength conditions, is characterizedby the existence of electrostatic interactionsbetween the ionic surfactant and the oppositelycharged solid surface. The adsorption in region Ican be examined by an exact form of theGouy-Chapman equation for the diffuse layer byconsidering an ion exchange process between thedodecylsulfate ions and the chloride ions. The totaladsorption density of the negative ions is given by
2.1.1. Characterization of the adsorbed layer byvarious techniques
The adsorbed (ayer in the above case undergoessystematic structural changes along the isothenn.A number of spectroscopic methods have been~ to characterize the microproperties of theadsorbed layer. Fluorescence spectroscopy, withpyrene as the photosensitive probe, was used todetemtine the micropolarity and size of theseaggregates. Steady state fluorescence reveals asharp increase in micropolarity, as shown in Fig. 3(measured as the ratio lJi/1 of the vibrationalpeaks of the pyrene spectrum), at concentrationscorresponding to that of the onset of solloid forma-tion at the alumina-water interface [10]. The mea-sured polarity is higher than that of SDS miceJIesin solution, suggesting a more compact structurewith Jess solvent penetration at the solid-liquidinterface. Dynamic or lifetime fluorescence meas-
~.-r_= (4)zF
O"d hardly changes in this region I and hence r -remains constant and the increase in sulfonateadsorption is due to exchange between chlorideions and this part of the isotherm has a slope of 1.
Region II is marked by a conspicuous increasein ads:Qrption which is attributed to the onset ofsurfactant aggregation at the surface through lat-eral interaction between hydrocarbon chains [6,7].Such colloidal aggregates are generically referredto as "soUoids" (surface colloids) [8] and includeaggregates such as hemimicelles, admiceUes andself-assemblies [9]. In the presence of specific
~
.;~,~~;~~')i ~~'i?J~:'~;~1

P. Soma.SIDrdaran; s. Kris1lllakJlmar I Colloidr SlII'faces -4: Physicochem. Eng. Aspecu 123-124 ( 1991) 491-513494
12:
m~... ..-0' ...
~
+ + + +
~
- - n+ + +
+ + + +~
ee~.+ + +- -..7
Fig. 2. Schematic ~resentation of the correlation of surfa~ charge andtbe growth of aggregates: (a) reverse orientation model; (b)bilayer model.
aggregates rather than the formation of new aggre-gates due to lack of positive adsorption sites. Suchadsorption is possible because of the hydrophobicinteraction between the hydrocarbon tails of thesurf~ctant molecules already adsorbed and theadsorbing molecules. The new molecules adsorbingat the solid-liquid interface can be expected toorient with the ionic head towards the water sincethe solid particles possess a net negative chargesimilar to that of the surfactant under theseconditions.
Additional information was obtained by con-ducting electron spin resonance (ESR) studies byco-iidsorbihg a paramagnetic probe, doxyl stearicacid, together with the surfactant [12,13].Information on micropolarity and microviscO$itycan be obtained by measuring the hyperfine split-ting constant AN and the rotational correlationtime 'reo The latter is the time required for acomplete rotation of the nitroxide radical aboutits axis [14]. The surfactant adsorption itself Wasfound to be not significantly affected by the pres-ence of small quantities of the probe. The chan~esin the ESR spectrum on surfactant adsorptionhave been used to quantify the microviscosity of
urements were used [11] to estimate the size of theadsorbed aggregates. The aggregates in region IIappear to be of relatively uniform size (120-160)while in region III there is a marked growth in theaggregate size (above 250). In region II, the surfaceis not fully covered and enough positive sitesremain as adsorption sites. The transition fromregion II to III corresponds to the isoelectric point(IEP) of the mineral, and adsorption in region IIIis proposed to occur through the growth of existing
~~~~ ~~i]~l~~;*~ ~~R~j~@~l~ ~~~~~~~
~~:rii:.~{~,..,

P. Soma.nmdaran, s. Krishnakwnar I CoOoids SlIrjaces A: Physicochem. Eng. Aspects 123-124 ( 1997) 491-513495
the adsorbed layer. The hyperfine splitting con-stants of 16-doxyl stearic acid measured in dodecylsulfonate solloids (hemimicelles) (15.0 G) areindicative of a less polar environment in compari-son with its value for water (16.0 G) and SDSmicelles ( 15.6 G), corroborating the resultsobtained from the fluorescence studies. Similarly,microviscosities estimated from Tc measurementsand calibrated against Tc measured in etha-nol-glycerol mixtures give reasonably high micro-viscosity values for the solloids (Fig. 4). When theposition of the nitroxide group was varied from 5to 12 to 16 along the stearic acid chain, it experi-enced a different viscosity within the solloid. Theseobservations may be explained by assuming amodel for the adsorption of the probe in whichthe carboxylate group is bound to the aluminasurface (Fig. 5). Such a model would require usto attribute greater mobility for the nitroxidemoiety near the SDS-H2O interface (as in the 16-doxyl stearic acid case) and less mobility for the5-doxyl case. This work is the first reported indica-tion of variations inmicroviscosity within a surfac-tant solloid as estimated by any known technique.
In addition to these two techniques. excited stateresonance Raman spectroscopy, using Tris(2,2'-bipyridyl) ruthenium(lI) chloride, Ru(bpy)i+, asa reporter molecule [15], was employed to studythe nature of the solloids. Significant perturbationsin the vibrational spectrum of this molecule wereobserved and correlated with the forDlation andmicrostructure of the solloids in this case.
2.1.2. Effect of functional group of surfactants onadrorption
The structure of the adsorbed layer depends onthe packing of the molecules which in turn dependson the mutual repulsion and steric constraintsamong adsorbate species [16]. Adsorption iS0-therms of 4Cll 3,S-paraxylene sulfonate (Para-I),4Cll 2,5-paraxylene sulfonate (Para-2) and 4Cll2,4-metaxylenesulfonate (Meta) on alumina fromwater are shown in Fig. 6. Average aggregationnumbers of the solloids increase from 17 to 76with increase in adsorption in all three cases. Theaggregation numbers of the two paraxylene sulfo-nates are similar throughout the range studied..However, at higher adsorption densities, the aggre-~~t~i;
~.
,~xw~i=z0
~w~~0U-I~Z0
~'0~
Fig. 4. Comparison of ESR spectra of 16-doxyl stearic acid in solloids, micelles and ethanol-glycero1 mixtures and correspondingrotational correlation times.
:;!Jf~'1l~ @~~ ~~~

496 P. s s. ~ I Co1JDi4s Swf-.4: ~ EII8.~ I2J-124 ( 1991) 491-$11
a:nN-~
0-X
N ICO.OOEu
"
...:i
a.S) 1.00, 1(00.00
RESIDUAL SULFONATE CONC.. k~ol/m3x 105
IO.cr. 100.m
Fag. 6. Adsorption of alkylxylene sulfooates on alumina with the average ~tion numbers as determined by dynamic: ftuores:euce.
gation number of the metaxylenesulfonate is lowerthan that of the paraxylenesulfonates. This sug-gests higher steric hirtdrance to the packing of thesurfactant molecules in the soUoids of the metaxy-lenesulfonate. Microca1orimetric studies have
shown that, at low adsorption densities, theadsorption is enthalpy driven while at higheradsorption densities the en tropic tenD becomesdominant [17]. At higher adsorption densities theadsorption entropy is higher for the paraxylene
:1;~';;A..:
->-..--cnzw0
z0-.-a..«0cn0~
10.00
1.00
O.

P. SOnIa$WIdaran, S. KrUhnakumar I Colloids Surfaces .4: Physicochem. Eng. Aspects 123-124 ( 1997) 491-513 497
sulfonates, indicating tighter packing of moleculesin the soUoids. On the basis of this, the effect ofthe change in position of functional groups on thearomatic ring of the alkylxylene sulfonates onadsorption can be attributed to the steric con-straints on the packing of surfactant molecules intheir aggregates..
2.2. Adsorption of non-ionic surfactants
The adsorption of non-ionic ethoxylated surfac-tants has become of much interest recently owingto their potential applications in processes such asdetergency, cosmetics and enhanced oft recovery.The adsorption of non-ionic surfactants differsfrom that of ionic surfactants largely because ofthe absence of electrostatic interactions. Non-ionicethoxylated alcohols exhibit strong adsorption onsilica but I)ot on some other minerals such asalumina. Since hydrogen bonding is relatively weakin comparison with electrostatic and chemicalbonding, the nature of the water structure at thesolid-liquid interface will be of particular impor-tance for the adsorption of non-ionics. The lackof adsorption, for example, on certain mineralssuch as alumina is speculated to be due to the factthat thesunactant molecules are unable to disruptthe rigid water layer surrounding the substrate.
The adsorption of dodecyloxyheptaethoxyethylalcohol (CI2EOs) alcohol on silica [18] displays anisothenn similar in shape to the isothenn of dode-cylsulfate on alumina (Fig. 7). The absence ofelectrostatic repulsion between the adsorbed non-ionic species results in this case in a higher slopefor the hemimicellar region and absence of regionIII. Adsorption of this type of surfactant dependson the degree of ethoxylation as well as the lengthof the alkyl chain [19] (Figs. 8 and 9). At constantchain length, the extent of adsoI;"Ption at lowconcentrations is greater for surfactants withhigher degree of ethoxylation. However, the pla-teau adsorption is higher for the sunactants withlower degree of ethoxylation. A linear relationshipis obtained when the parking area at plateauadsorption is plotted as a function of the ethoxyla-tion number. This yields a parking areaper-OCH2CH2 segment of9.2 A2, suggesting directadsorption of the ethylene oXide' chains on the
silica surface. On the contrary, the alkyl chainlength affects only the onset of plateau adsorption.This is in line with the decrease in critical micelleconcentration (CMC) with increase in hydro-carbon chain length. On the basis of these observa-tions hydrogen bOnding is proposed to be thedriving force for adsorption at low concentrationswhile the higher uptake of longer EO surfactantsat such concentrations is due to the cumulativehydrogen bonding interactions of the EO chainswith the hydroxylated silica surface. In contrast,at higher concentrations hydrophobicchain-chaininteractions become more significant as evidencedby the progressive increase in slope of the adsoxp-tion isotherm with a decrease in EO number (thesmaller the EO number the lesser. is the sterichindrance to chain-chain interactions by theethoxyl groups).
A new class of sugar-based non-ionic surfactantshas attracted more attention recently owing tothe fact that they are easily biodegradable.Information on adsorption of such surfactants onsolid substrates is however very limited in litera-ture. Smith et al. [23] measured adsorption ofthree alkyl polYglucosides on titanium dioxide.They postulated that the hydroxyl groups on thesurfactants are slightly acidic in nature and canhydrogen bond with the basic OH groups on thetitania particles. Results obtained in our laborato-ries [20] show that ~o-dodecyl maltoside adsorbsstrongly on alumina and hematite but not on silica(Fig. 10). Also, molecular parking area calcula-tions based on the adsorption plateau suggest theexistence of surfactant bilayers on the solid.
2.). Adsorption of surfactant mixtures
Surfactants are generally used as mixtures toserve different purposes. A typical feature of theadsorption of ionic-non-ionic surfactant mixturesand oppositely charged ionic surfactant mixturesis the synergistic interaction at the interface as wellas in solution. Adsorption of non-ionic ethoxylatedalcohols on alumina is negligible, but it can beenhanced by several orders of magnitude by co-adsorption of an anionic surfactant. Similarly,anionic surfactants do not adsorb on negativelycharged. silica. but substantial adsorption can be
~~4
"~,,:,.'
~?~~ ~~;~~;~t~~FiW~r;~~;~ti;~~'~"I',r~..~1:!.:-'~¥W""~',-"'~'~
~!i~']1~:"!;;;;";:::~:,:,,:.;[";::;:..!,,
:.,;;~ ~~
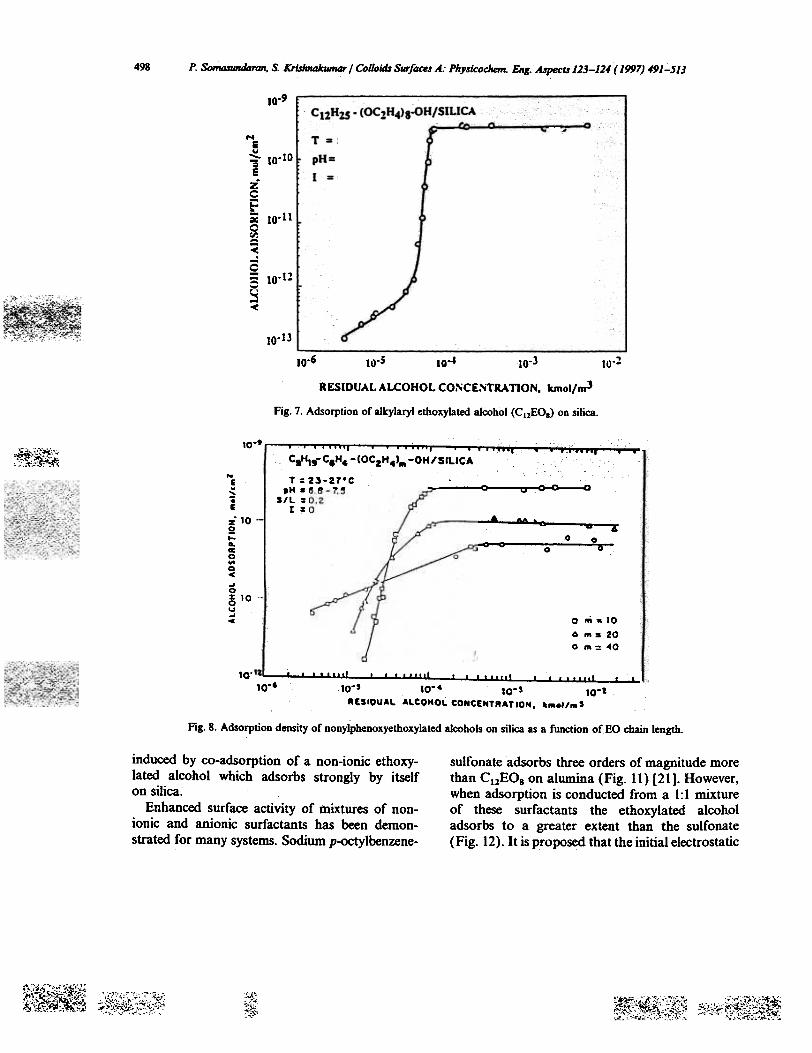
498 P. Somtl.fundaran, S. Krishnakumar / Colloids Surfaa.r.4.. Phy.ricochem. Eng.A.rpect.r 123-:124 ( 1997) 491-513
10.9
CUHzs. (OCzH4>a-OH/SILICA
T ~SOoC
pHa 6.9I ~ O.03M N.CI
NE..
- 10.10
iZc
F= 10.110~-:
'<.
c
-=: 10.12~~-<
10.13
10.6 10.5 10-3 10.2IO~
RESIDUAl. ALCOHOL CO~CE.'jTRA 110N, kmol/mJ
Fig. 7. Adsorption of alkylaryi ethoxylated alcohol (CuEO.) on silica.
10.'
CsHt9-C,H
T:23-27.C -L~' I,H : '.8 -7.55/1.. : 0.2
1:0
-- -.~.~":"'": ';~~;~f~
c~ 0 w a ~
/' -:', " ~,
N
E~..~
z' 10.'~2.....~0~0~...0S 10.11
U....c 0 ,,; .10
4 '" = 200 '" = 40
I10'-_"""'" ,. , ,.,!10.t 10.' 10- 4 10.' 10.1
AUIOUA\. A'-COHO\. CONCENTRATION. '",.,1-'
Fig. 8. Adsorption density of nonylphenoxyethoxylated alcohols on silica as a function of EO chain length.
sulfonate adsorbs three orders of magnitude morethan C12EOs on alumina (Fig. II) [21]. However,when adsorption is conducted from a I: I mixtureof these surfactants the ethoxylated alcoholadsorbs to a greater extent than the sulfonate(Fig. 12). It is proposed that the initial electrostatic
induced by co-adsorption of a non-ionic ethoxy-tatoo alcohol which adsorbs strongly by itselfon silica.
Enhanced surface activity of mixtures of non-ionic and anionic surfactants has been demon-strated for many systems. Sodium p-octylbenzene-
~" ~',.,"..;~.:~
I""""'-':"'~'"~~O;-~'J"
~'~' "~!,J~""."..."i;.~, "'.~
~\~::;,*:~:, -, ".oc ~f~;;~;i~;'~~~ ~~ ~~~~i~~!~~ ~4~~~~~~:::~f'f;

P. Soma.tIIIIdaran. S. Kr;silllaklUllar / CQIloids Surface3 A.: Physicochem. Enf. ASpecU 113-124 ( 1997) 491-511 499
lxlO'S,
e lxlO"'=-0
.!b
~ lxlO.7
lxlO"
lxlO"
Fig. 10. Adsorption of Il-dodecyl-maltoside on different solids.
adsorption of the sulfonate provides a sufficientnumber of hydrophobic sites for solloid-typeadsorption of the ethoxylated alcohol. As a resultof the synergism the overall adsorption of thesulfonate is also enhanced and the concentrationof solloid formation is lowered by more than anorder of magnitude. The presence of the non-ionic
surfactant between the sulfonate ions in the sol-loids enhances the sulfonate adsorption by reduc-ing the electrostatic repulsion between the 'ionicsulfonate head groups- Similar results have beenobtained with SDS and C12EOs- Fluorescenceprobing of the adsorbed layer conducted in thiscase [22] revealed that the micropolarity of the
',"'ijf':~;~i Si(,'
~::i';:;~~.~rf~~~;c~~:

P. ~ s. Kri.shnDkJDIIa; / Colloids surfaces.4..' Physicochem. Eng. ~ts 123-:-124 (1997) 491-513soo
Fig. II. AdsOrption of Crbenzene sulfonate and CuEO.alcohol on alumina.
aggregates. Another interesting observation is theconstancy of the micropolarity suggesting that theaggregates do not change much with concen-tration. ~ic fluorescence estimation of therelative aggregation numbers in these ~temsreveals a higher aggregation number .than for pureSDS. Clearly the reduction in electrostatic repul-sion is an im~rtant reason for the observedincrease in aggregation num~r (Fig. 13). Table Ishows the aggregation number of each surfactantin the mixed layer. At low adsorption densities thenumber of C1zEOs molecules is lower than that ofSDS suggesting that the C1zEOs aggregationdepends on SDS adsorption. As the adsorptionin~, the lateral chain interactions become thedominant mechanism and the numbers of SDSand CuEOs in the aggregates become more orless equal.
Similar synergistic effects have also beenobserved in the case of adsorption of cationic-non-ionic surfactant mixtures at the alumina-waterinterface [23]. The adsorption of mixtures of thecationic tetradecyl trirnethyl ammonium chloride(TTAC) and non-ionic pentadecylethoxylatednonyl phenol (NP-15) was studied at several ratios.ft~
~
~~z~~
5~~
1fT' 10-' Ir 10-3 10-2 10-1
RE.'If.DfIAL S1JRFACTANT roNC kmnl m3
Fig. 12. Adsorption of c.-~ne sulfonate and C11EO.alcohol from their mixture at an initial compo~tion ofSO mol. ~ sulfonate-SO mol. % alcohol.
SDS-C12EOs adsorbed layer is higher than thatof the adsorbed layer of pure SDS, but similar tothat of SDS-CuEOs micelles in solution. Thissuggests that the ethoxyl chain is the key factordetermining the polarity of the adsorbed laye~
~!i~if~i~~~
~~,:,.,..;f%~; ~

P. Soma.nIndanm, S. Krishnakwrrar I OJIloids Surfaces A: PhysicOchem. £IIg. AsPects 123-124 (1997) 491-$13 SOl
Table IVariation in surfactant aggregation number in the mixed adsorbed layer of sodium d6decylsulfate and dod«Yioxyheptaethoxyethylalcohol
Surfactant:pyrene
(adsorbed/layer)ko(ns-l)
Total adsorption
(mol m-~kc(ns-t)
N-SDS
~
CuEO, Total
1.1 X 10-123 X 10-13.2 X 10-16.2 X 10-11.9 X 10-6
151173179187194
0.0150.0130.0100.00790.0071
0.18
0.0550.0150.0110.0055
0.680.770.791.600.86
6172,.828S
4661677783
10713314..128168
The NP-15 does not adsorb on alumina by itselfpossibly because the ethylene oxide groups arestrongly hydrated in solution and the adsorptionforce is not strong enough to cause displacementof these water molecules. As expected, the cationicTfAC does adsorb strongly on alumina at pH 10and exhibits an isotherm typical of that of ionicsurfactant adsorption on an oppositely chargedsurface. This adsorption changes significantly inthe presence of the non-ionic surfactant NP-15.As in the previous case, solloid aggregation occursat lower TfAC concentrations as the NP-15 con-centration is increased (Fig. 14). Interestingly, atrelatively high NP-15 amounts (ratio 1:4) no evi-dence of solloid formation is observed. Theincrease in TfAC adsorption at low concentrations
is attributed to charge screening by the non-ionicsurfactant which reduces the electrostatic repulsionbetween the cationic head groups. Plateau adsorp-tion of TI'AC decreases owing to the competitionof the bulky nonyl phenol with TI'AC for adsorp-tionsites (Fig. 15). The adsorption density ofNP-15 from its mixtures with TI'AC reveals enhance-ment of adsorption of NP-15 by lTAC due tosolubilization in the lTAC surface aggregates.Calculation of the monomer concentrations of theindividual surfactants performed using the regularsolution approximation revealed the adsorption oflTAC from the mixtures to correspond to itsmonomer activity. However, a similar correlationdoes not exist for the non-ionic surfactant and onecan conclude that NP-15 adsorption depends notonly on its monomer activity but also on the
4xlO"
rU~1
fp-o--oIxIO"
1x10-1
IxIO"
{
4/.~ 0
6° OJ rII.J,;.~- '/ O
0 _-A .,.-, "6,..0'0/
,0,.6 ~/
i~~c0°,"Q<
~!:
Fig. IS. Adsorption of NP-15 on alumina in the prcsence ofvarying amounts of TTAC at pH 10; IS, 0.03 M Naa.
6..~ .. . .txl0txtO" txl0-5 lxtO" txl0-J txlO-1 Sxl0-2
tTAC ~ COMenIr8IiOlL kmoVm J
Fig. 14. Adsorption of TfAC in the p~ce and absence ofNP-1S at pH 10; IS, 0.03 M NaCL
~:i~~ ~~!~;j~j~...co""

P. SomaII8Idaran. s. Krishnokumar I CoUoids Swfacn.4.: Physici>CMm. &g. Aspects 123-114(1997) 491-513SO2
proposed to be due to additional adsorption ofCuEOs in reverse orientation. At low concen-trations, the chain-chain interactions between theadjacently adsorbed SDS and CuEOs moleculesprovide additional energy gain for the adsorptionprocess. Such hydrophobic interactions at theinterface will result in the formation of the hydro-phobic microdomains into which the hydrocarbonchains of additional CuEOs molecules can becomeincorpOrated as a second layer. This is schemati-cally shown in Fig. 17. The evolution of such anadsorbed layer clearly changes the wetting beha-vior of the particle surface.
3. Surfactants in non-aqueous media
Surfactant adsorption in non-aqueous media isimportant in various technological processes suchas ceramic and magnetic tape, ink, paint andpigment manufacture. Adsorption in this case isusually governed by the polarity differentialbetween the solvent, solute and substrate and thiscan be related to the acid-base characteristics ofthese components.
adsorption of1TAC and the adsorbed layer struc-ture. It is to be noted that the regular solutiontheory failed to predict the change in even thebulk monomer concentration ofNP-IS.
Fig. 16 shows the adsorption of NP-lS when1TAC is pre-adsorbed on alumina. In this casealso NP-lS adsorbs synergistically because of theadsorbed TTAC and the NP-lS adsorptionincreases with 1TAC adsorption density. It is alsoobserved that once the lTAC forms hemimicellesat the alumina-water interface there is no furthereffect of increasing lTAC adsorption density onthe NP-lS adsorption. It should also be noted thatNP-lS adsorption density in this case is lower thanthat from mixtures, leading to the inference thatNP-IS adsorption depends to a large extent on thestructure of the adsorbed layer. When it is presentin mixtures, NP-lS can adsorb as the lTAC layerevolves but, when the 1TAC is pre-adsorbed,adsorption is more difficult as it should change the1TAC adsorbed layer first and this may involve alarge activation energy. .
Both SOS and C12EOs adsorb on kaolinite bythemselves. Adsorption of C12EOs on kaolinite isinfluenced by the presence of anionic SOS. Theplateau C12EOs adsorption in all cases is higherfrom its mixtures with SOS than in its absence[24J. These higher plateau adsorption values are
:~::;::'~~~.~;,~:
3.1. Ionic surfactants on oxide surfaces
The density of the surface hydroxyls determinesthe relative basicities of many minerals and thiscan be inferred from the IEPs of the minerals inaqueous solutions (the lower the IEP the moreacidic is the mineral). Alumina has a high surfacehydroxyl density (10-15 OH nm-~, a high IEP
L////////////7//// ///.////
(a) (b) (c)
rig. 17. Schematic repr~tation of the adsorption mechanismsfor SDS'-CuEO. mixtures on kaolinite.
Fig. 16. Effect oflTAC order of addition on NP-15 adsorptionon alumina; varying amounts of pre-adsorbed lTAC and NP-15 added together with lTAC.
f\~~:
~~.~~~1; ~ "',h::O-c-:t+.
;f(~t1:

P. Soma3IDIdaran. S. Krislurakumar I Colloids Surfaces .4.: Physicochem.Eng. A¥ctsJ23-124 ( 1997) 491-513 SO3
NE""0E
.0-x.;::--;;c
t)
"g
c.2""Q....0~
"g-<
(8.5) and is a basic oxide. On the contrary, silicahas a lower hydroxyl density (3-40Hnm-~, alow IEP (less than 2.5) and is an acidic oxide. Thedensity of the hydroxyl groups and hence thebasicity of the surface can be controlled also bydehydroxylation of the surface by extended heatingat elevated temperatures.
Fig.18 shows the adsorption isotherms ofAerosol-OT (AOT) on alumina and silica fromcyclohexane [25]. It can be seen that the anionicsurfactant has a greater affinity for the basic oxidethan for the acidic oxide. The situation is reversedin the case of the adsorption of the cationicsurfactant dimethyl dodecylamine (DDA). DDAadsorbs more on acidic silica than on alumina(Fig. 19). Calculations based on a plateau adsorp-tion value of3 x 10-6mol m-2 on alumina give aparking area of about O.55nm-2 for the AOTmolecule. This is in goOd agreement with thepublished values for AOT parking areas at thewater-xylene and water-isooctane [26] interfacessuggesting that it adsorbs at the alumina-cyclohex-ane interface as a monolayer in an orientationperpendicular to the adsorbent with the hydro-carbon chain extending into the solution. Fig. 18also shows the adsorption of AOT on dehydroxy-lated alumina. Dehydroxylation, in this case, is
Fig. 19. Adsorption of cationic DDA on different solids.
achieved by heating the alumina at 900°C for 48 hand verified by the disappearance of the OHvibration bands at 3800 cm-1 from the IRspectrum of the alumina sample. Dehydroxylationincreases the acidity of the surface and hencereduces the adso~tion of the anionic AOT. It isclear from these resultS that AOT adsorbs throughinteractions with the hydroxyl groups on the oxidesurface. These results also suggest that for a givensurfactant the relative acidity of the oxide surfaceis one of the major factors that determine theextent of interaction.
3.2. Adsorption on graphite
'{tie adsorption isothenn of AOT on graphitefrom cyclohexane follows a very interesting patternas shown in Fig. 20 [27]. The adsorption increasessharply in the initial part suggesting high affinityof the surfactant for the solid at low concen-trations. The adsorption then appears to reach aplateau and calculations based on apparent mono-layer coverage at this level yields a parking areaof approximately 1.03 nm2 (AOT molecule)-l.This molecular parking area corresponds to anAOT molecule adsorbing flatly on the solid-liq~idinterface. At higher AOT concentrations, above10-2mol1-1, a further sharp increase in the
0 5 10 15 20 25 30,Residual Concentration x 10 . moil'
35
Fig. 18. Adsorption isotheml of OT on different solids fromcyclobexane.
~
}*~::~:!;.;~:~~~t~~,

SO4 P. Soma.fIIIIdaran; s. Kri.JhnakuInQr I Colloidf SlIT/aces A: Physicochem.EIIg. Aspecu 123-124 (1997) 491-51J
to form aggregates at the interface as shown in~ig. 21(b). This leads to a sharp increase inadsorption density as well as a decrease in settlingrate due to the disappearance of the interparticleaggregation observed at low surface coverages.Formation of such reverse hemimicelle-like aggre-gates at the interface has been reported previouslyfor the adsorption of 1-decanol on graphitizedcarbon black from non-polar solvents [28].
"E
"0E
.00-
~..c."0C
.C?-
o...0III
"0.
3.3. Surfactant desorption and effect of solventI,;,.
0While adsorption studies yield infonnation on
the interactions responsible for adsOrption, desorp-tion studies give infonnation regarding the revers-ibility of the adsorption and hence a qualitativehandle on the strength of the solute-solid inter-actions. Fig. 22 shows data for the desorption ofAOT, pre-adsorbed on alumina from cyclohexane,into solvents of different polarities. It can be seenthat as the solvent polarity is increased the amountof surfactant desorbed rises sharply above a certainvalue of the dielectric constant. As the solventpolarity increases, .the surfactant interacts morewith the solvent with a concomitant reduction inthe surfactant-solid interaction. This increase insolvent-surfactant interaction with increasingpolarity is also evident from the data for aggrega-tion of AOT in different solvents [29]. Aggregationwas observed in solvents of low polarities and theextent of aggregation decreased as the polarity ofthe solvent was increased as a result of increasedinteraction of the surfactant with the solvent. Most
c
smol/IResidual Concentration x 10
Fia- 20. Adsorption of AOT on graphite from cyclohexane.
.:.:~,;.~~-:::.H,.~..,~
:;~~:I;~"'I;
absorbed amount occurs and this reaches to about5 times the initial plateau value at abOut 3 x 10-2moll-1.
At low surface coverages the polar head groupof the flatly adsorbing AOT is exposed to thesolvent with which it is not compatible. In such acase the AOT molecules, as shown in Fig. 21 (a),can fonn inte~article aggregates that would effec-tively create a polar microdomain to shield thehead groups from the solvent. Suchan interparticleaggregation can account for the increased settlingrate at low coverages. As the AOT concentrationis increased the adsorbed molecules are proposed
A B
Fig. 21. Schematic representation of adsorption of AOT on graphite (a) at ww concentrations by interparticle aggregation and (b)at high concentration~ through intraparticle aggregation.
~.~~ ;;:~q?l[:Aoi:,~,:~t~.

P. SOma"rmdaran, s. Krishnakwrrar / Colloids Surfaces A: Ph~hem. Eng. Aspects 123-124 ( 1997) 491-513 50S
c0
:J~..
a40
0
N
Fig. 22. Desorption of AOT pre-adsorbed on alumina insolvents of different polarities.
~:
~~:~~_.!~";'~~-";'-i; --..-~~1!;"t.:;,~j
. "'~"'#~'--~,-~~ -""'-~::i'-,.. . ,~.~~?,.".~
~~~m~j~i~
interestingly, beyond a certain solvent polarity theaffinity of the surfactant for the oxide surfacestarts to increase again as indicated by the decreasein the desorbed amount. At higher solvent polari-ties, the hydrocarbon chain of the surfactant isless compatible with the solvent and tends to formaggregate structures essential to remove the hydro-carbon parts from the bulk solvent. This can beaccomplished via micelle formation in solution oradsorption at a less polar interface. Evidently thelatter mechanism is favored in this case.
Studies with different oxides (Fig. 23) revealedsimilar trends with the curves shifting to the rightin the low dielectric constant regime and movingto lower desorption values in the higher dielectricconstant region as the acidity of the oxide isincreased. The acidity of the oxides [30] increasesin the order alumina < hematite < rutile < silicaand the observed shifts also follow the same trend.This demonstrates the effect of the nature of thesolid on the solvent influence on adsorption.
A semiquantitative description of this adsorp-tion process was. carried out using interactionparameters to characterize the mutual interactionsbetween the solid, surfactant and solvent. Forsolvents the interaction parameters are the totalsolubility parameters as defined by Hansen [31].The interaction parameters for surfactants have
been estimated in some cases by considering themas supercooled liquids. Alternatively, these can betheoretically calculated from their structural for-mulae using the group theory approach [32]. Forsolid surfaces the interaction parameter can beestimated using a procedure similar to that usedfor polymers wherein the polymer is characterizedby the center of its volume of solubility in a three-dimensional plot of the interaction parameters ofvarious solvents [33]. The net adsorption energyE, is the affinity of the solute j for the solid surfaces less the affinity of the solvent i for the surfaceand the affinity of the solute for the solvent [34]:
Et=jsE-isE-ljE (6)
A similar approach is adopted to develop ageneralized model for surfactant adsorption in thiscase. Simple algebraic differences between theinteraction parameters are taken as being propor-tional to the strength of their mutual interaction.The larger the absolute value of bj-bjo the weakeris the interaction between the two species, j and j:
net adsorption = fObsolid - blOlvent 1,lblOlute
-baotventl,lblOlJd -bIOIuteD (7)
These three binary interactions can be combinedin a number of different ways. The simplest caseis to take a linear combination of these terms asshown below
berr=abs(AlblOlid -blOlftDtl +B)blOlute
-bIOlVeDtl- qblOlid -bso'uteD (8)
where beff is the effective interaction parameterand A, B, C are numerical constants and willd~pend on specific system interactions such aselectrostatic forces or lateral interaction amongadsorbed molecules. It can be seen from the earlierdiscussion that for good adsOrption this sumshould be large or in other words the larger thevalue of berr the greater is the adsorption tendency.Conversely, one can argue that a low value ofberr will favor surfactant partitioning into the bulksolvent. Eq. (8) has been tested for a number ofdifferent surfactant-solvent-solid systems [35].
Thus adsorption in non-polar media is governedby the relative interactions of the systemcomponents as described by their interaction
'~."", i;c' ".
"l'~ :~~~~~f~:~~~; ~.!;~

SO6 P. Somasulrdal'lJllo S. KrishnakIImar I Colloids Surfaces A:Physicoclrem. £fig. A.rpect.J 123-124 (1997) 491-513
100 0 Alumina. Suicaa Hematite. Rullle80
c0
:::a-s..001to
Q
~
~
40
20
00 10 30 -40 50 60
Dieleclric Constant
70 eo
Fig. 23. Desorption of AOT from different solids into solvents of different polarities.
parameters. -The adsorption of ionic surfactantson hydrophilic substrates proceeds to a monolayerand there is no further evidence of lateral inter-actions or aggregation among the adsorbed mole-cules. However, when the substrate is hydrophilicreverse solloids can form on the surface owing tothe interactions among the polar head groups ofthe adsorptive molecules.
4. Polymers in aqueous media
Polymers can exist in different confonnationsboth in the solution and in the adsorbed state.Adsorption of polymeric materials onto solid sur-faces can be quite different from that of smallmolecules in that the polymer adsorption is greatlyinfluenced by the multifunctional groups that itpossesses [36]. This stems from the widely varyingsizes and configurations available for the polymer.In addition macromolecules usually possess manyfunctional groups each having a potential toadsorb on one or more given surfaces.
measuring the depletion of the polymer concen-tration in the solution after contact with the 'solid.Linear flexible macromolecules can assumedifferent conformations at the interface and alsoexhibit varying degrees of attachment to the sur-face [37]. At the solid-liquid interface the macro-molecules usually prefer a conformation thatallows for maximum segment-surface contact.Since the attachment of one segment will increasethe probability of neighboring segments to beadsorbed and the number of functional groups permolecule is large, multiple bonding between thepolymer and the surface is favored. The result isnormally an interfacial conformation consisting ofsequepces of adsorbed segments ("trains")alternating with free three-dimensional "loops"extending away from the surface and with thechain terminating at either end in two free dangling"tails" [38] (Fig. 24).
Adsorption causes a loss of entropy of the poly-mer. Therefore, for adsorption to be favorable, theinteraction energy per segment-surface attachmentmust be large. For polyelectrolytes, the major driv-ing force for adsorption is the electrostatic attrac-tion. To investigate this the adsorption ofpolyacrylic acid (PAA) on Sumitomo AKP aluminawas studied under various pH conditions. Fig. 25shows the effect of pH on the maximum adsorption
4.1. Adsorption ofpo/ye/ectro/ytes
Adsorption isothenns of polymers are deter-mined similarly to those for surfactants. i.e. by
~~,~j\i:~~~~l~-;!""'"~O;" j.".,
:;;:::.\,~: :~m;~ ,-~ ,,""';t.""!,;:'f~-'~'
~..~.;;;,r!-P'-"~'.~'

P. SomaSIInJaran, s. Krishnakllmar f CoUoids Sllr/aces.4.: PhysicOchem. Eng. Aspects 121-124 (1997) 491-513SO1
lOO~S~ .60
---TAIL40
"'\
~.~-..'..r~"'\:NS
"9\>20
E
..OJ 0
i~ .20N
Fig. 24. Schematic representation of an ~rbed polymer at aplane interface showing the presence of trains, loops and tails.
-40
.a
-80
Fig. 26. Variation in , potential of alumina suspensions withadsorbed PM.
Another important factor affecting polymeradsorption is the polymer molecular weight {3S].For non-porous substrates where adsorption isrestricted only by the extent of accessible surface,the general observation is that adsorption increaseswith increase in chain length. This is observed inthe case ofPAA adsorption on non-porousAKPSOalumina as shown in Fig. 27 and is expectedbecause the number of polymer-segment bondsincreases with chain length of the polymer mole.;
Fig. 25. Effect of pH on the adsorption of PAA on alumina.I
0.9density of PAA on alumina [39]. As can be seen,there is a decrease in the adsorption density withan increase in pH until the IEP of alumina isreached. Fig. 26 shows the variation in { potentialof alumina suspensions in the presence and absenceof adsorbed PAA. As can be seen clearly, theadsorption of the n~tively charged polymerdecreases the { potential significantly and alsolowers the ffiP of the alumina. These results suggestthat electrostatic forces are predominantly responsi-ble for the adsorption ofPAA on alumma. However,even under conditions where the surface is highlynegatively charged (pH 10) and the polymer highlyionized there is some adsorption observed. Thismay be attributed to the fact that at pH 10 thereare still some positive sites on the alumina surfacewhich can anchor some polymer segments.
. ~~~_. ,,"- - - - --A-.:~""'-. -&~~- - - -0
0.8
0.7
~~0.6F.:S 0.5.~a 0.4
~<
~
D SOOO 0 1 SOOOO fj. 8000000.3
0.2
0.1
00 SOO 1000 1500
Residual COGcealralioG, ppm2000
Fig. 27. Effect of PAA molecular weight on its adsorption onnon-porous AKPSO alumina.
~M~,~~f
V(~~~~ri::If~:j~~~,-,~.,~t;.".,;:i~;.~.!".;~",~:"
.~f~)r~;;;,~~;~'
..

.. P. Som4rImdaran. s. KrishnOkUma'l CoHoids Surfaces..4: P":)';tiCocJ.~ Eng.A.spects 123-114 (1997) 491-513
depending on the pH. The rationale behind theuse of this technique is the observation that theextent of excimer fomlation which depends onthe interaction of an excited state pyrene of thepolymer pendant group with another pyrene groupin the ground state has a direct bearing on thepolymer conformation. This may be understOodwith reference to Fig. 30 which shows that. at lowpH, there is a better probability for intramolecularexcimer formation between pyrene groups resultingfrom a favorable coiled confomlation. Similarly,a low probability for the excimer fomlation athigh pH may be understood as a consequence ofthe repulsion between the highly ionized carb-oxylate groups in the pol~er and the subsequentstretching of the polymer chain. This difference isreflected in the nature of their fluorescence spectraas seen in Fig. 31. Also, these studies have demon-strated that the PAA adsorbed in the stretchedfoml on alumina at high pH could not becomecoiled because the conformation could not bealtered by lowering the pH subsequently (Fig. 32)[41]. In contrast, the polymer adsorbed in the
cule. However, the adsorption density of PAA onthe porous Linde alumina decreases with increasein molecular weight (Fig. 28). This is attributedto the exclusion of the higher molecular weightpolymers from a large fraction of the pores whichare accessible only to the smaller molecules.
4.1.1. Polymer conformation by spectroscopictechniques
PAA can exist in different conformationsdepending on the solvent, pH, and ionic strengthconditions (Fig. 29) [40]. Such a flexibility alsoinfluences its adsorption characteristics on solidsand in turn affects subsequent suspension behavior.Using a fluorescent labelled polymer and by moni-toring the extent of excimer formation it wasshown that the polymer at the interface could havea stretched or coiled conformation at the interface
~
"".~ ' -.~ ~ ~
\.ow ~H Hlq.. P"
Fig.30. Schematic representation or the correlation or theextent or excimer rOmlation and intrastrand coiling or pyrene-
1abe\ed(»AA.
Fig. 28. Effect of PAA molecular weight on its adsorption onporous Linde alumina. MOI1~
.~
i.sc9
:1~.~~~
~
~.9-~~~~~ -++++--+
(a) pH 4.4+- - - -+--(b) pH 10.5
350 400 SSG 600450 500
Wavelength (om)
Fig. 29. Schematic representation of PAA conformation underdifferent pH conditions.
Fig. 31. Fluo~ emjssion spectra of adsorbed polymer attwo pH values.
:~~,. :;::~;:~:;.:
~ .":t:,.:;., "". "..:.iY~
"""¥~... "'~-""""~:

P. SQma3widarall, S. Krlshnakwnar I Colloids Surfaces .4: Physicochem. Eng. Aspects 123-124 (1997) 491-513 SO9
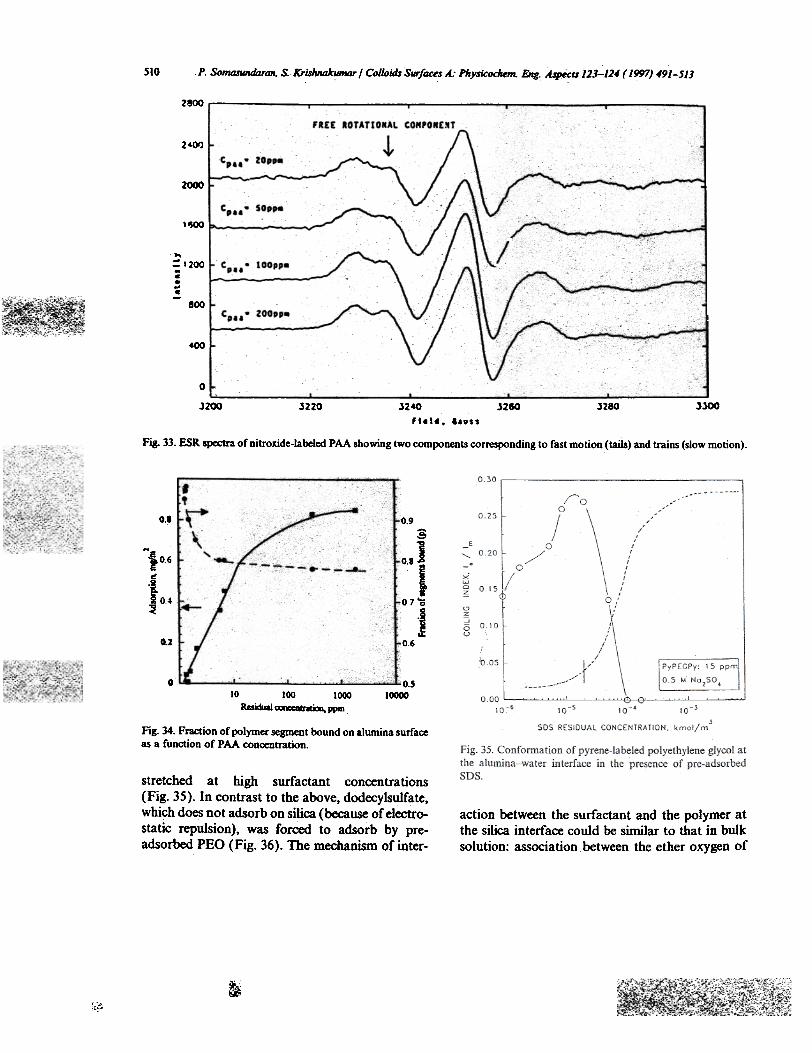
(Fig. 33). These spectra are composed of twodistinct components: non-restricted ("free rota-tional"), corresponding to the parts of polymermolecules in solution ("loops" and "tails"), andrestricted ("frozen"), corresponding to the partsof polymer molecules directly bound or in veryclosed contact with solid surfaces. The relativeincrease in the peak at 3235 G in the series ofspectra collected at increasing polymer concen-tration reveals an increase in non-restrictedspectral component. This suggests that with anincrease in the number of polymer molecules atthe solid-liquid interface, a larger and larger frac-tion of the polymer molecules are pushed awayfrom the solid surface. Using computer simulation,we decomposed the observed spectra and calcu-lated the ratio of spectral components. The fractionof segment bound (relative number of "trains")together with the adsorption isotherm of polymerare given in Fig. 34. It can be seen that the numberof free polymer segments (loops and .tails) is at amaximum at the polymer adsorption plateau, indi-cating inteimolecular repulsion of the polymermolecules at high packing density.
a) k.w ph
~ "(9-
b) b;lhph
"'!o -- ~D ~~---++++
~+ + + ...aIiDt/1OiuIioa
,T
Iow~ hiaIIph. - .-- -"-'--""-~---'-~ -~
+ + + .OIpaJIded/adIOrbed
suoaabiDdilll
B E. . . .coiIcd/adoorbed~biJIdiDa
~~,~~1~~~
billl ph low phC .-:;..~- -~- F
~ ~ ~~~/-
partiaUy expandedfadaorbcd apaoded/odJOrbcd
Fig. 32. Schematic representation of the adsorption process ofpyrene-Jabeled PAA on alumina. A) At low pH polymer iscoiled in solution which leads to B) adsorption in coiled formC) Subsequent raising of the pH causes some expansion of thepolymer D) Polymer at high pH in solution is extended andbinds E) strongly to the surface in this conformation F)Subsequent lowering of pH does not aUow for sufficientintrastrand interactions for coiling to occur.
5. Adsorption of polymer-surfactant mixtures
coiled form at low pH did stretch out when thepH was increased.
4.1.2. Determination of bound fraction of theadsorbed polymer using electron spin resonance
spectroscopyESR spectroscopy is a powerful analytical tool,
which can provide infomlation on the mobility ofspecific molecules and thus fluidity of the microdo-main environment. In our study, PAA withattached nitroxyl groups (molar ratio 1:100) wasused to investigate polymer conformation on solidsurfaces [42]. The number of polymer segmentsbound to the surface ("trains") can be estimatedfrom the complex ESR spectra obtained fromlabeled polymer adsorbed on alumina surface
Mixtures of polymers and surfactants are widelyused in various commercial fonnulations sincethese mixtures possess special interfacial and col-loidal properties. Adsorption of polymer-surfac-tant complexes has been studied and they are alsoknown to have very interesting surface-modifyingprop;erties. For example, it has been shown thatintetactions between a cationic polymer andan j anionic surfactant promote the flotation of
anionic quartz.In one study using polyethylene oxide (PEO)
and SDS, the former of which nonnally does notadsorb on alumina, was induced to adsorb by pre-adsorbed SDS [43]. There was near completeadsorption of the polymer once the solloid awe-gates of the surfactant formed at the interface.Interestingly, the conformation (as observed cbyfluorescence emission) of .the co-adsorbed PEOwas markedly influenced as a result of its inter-action with SDS with the polymer being completely
!~fiE1
..~.~ .
~~;.~~
510 P. SomaslDldarQll, S.Krishnak umar I ColJoidsSlir/aces A: Physicodlem. Eng, Aspects 123-124 ( 1997) 491-513
2800
,.££ .OTATrOaAL CO"'Oa£!lT
! I2400
C,..- 20".2000
c,..- SOp,.=:=. ~:.-/1~ .~~~/ ~- - -
~ c . 100"./~L~-~~~':'::-~~ ---". ~
/I
..:: I 200...-c
-800 c . ZOOpp.p..
~
600
0
J200 JJOO3220 32.0F'.14, "UII
3260 J280
Fig. 33. ESR spectra ofnitroxide-labeled PAA showing two components corresponding to fast motion (tails) and trains (slow motion).
..
f.~:' \
0.1
"':'0.6ec
104
----.,..;
0.9~
]0.8 .
I07'-0
t...
0.6
,4-
0.1
0 0.'1000010 100 1000
ResidUIi --.:aIIr8!icm, ppm
Fig. 34. Fr8(:tion of polymer ~ent bound on alumina surfaceas a function of PAA concentration.
stretched at high surfactant concentrations(Fig. 35). In contrast to the above, dOdecylsulfate,which does not adsorb on silica (because of electro-static repulsion), was forced to adsorb by pre-adsorbed PEa (Fig. 36). The mechanism of inter-
action between the surfactant and the polymer atthe silica interface could be similar to that in bulksolution: association between the ether oxygen of
~',,~

P. Soma.nIndaran, s. KrlshllakWrlar / Colloids Surfaces A: Physkochem &g. AspeCU 12J-124 (1997) 491-51J Sit
0.00007
.0.00G0e
the binding of 80S to pre-adsorbed PEa, suggest~ing that the adsorbing tendency is reduced oncethe complex forms in solution. There was, how~ever, no effect of the pre-adsorbed PEa amounton the SDS uptake at the silica interface. In factat higher pre-adsorbed PEO levels there was somedesorption of PEa as the SOS adsorbs. Theseresults suggest that there is a minimum amount ofPEO required to activate the uptake of SDS andthis amount is lower than that required for inter-actions in solution. Thus, the surface is seen insome way to activate the polymer-surfactant inter-action. Oearly various types of polymer-surfac-tant interactions can be designed to achievedesirable interfacial properties.
Er.I;
G 20.00005 $IUCA: 25 m Iv
"I PREAOSORBEO no 4 "'V/v.. I pH - 8.5~ 0.00004 r-0- It r~ 0.00003,~ l
t~ 0.00002:'
, i.
ii /A
h'0.00001
0.000001
Fig. 36. Adsorption or 50s on silica with pre-adsorbed PEO.
6. Summary and conclusionsPEa wifu the surfactant head group. Such bindingoccurs ina narrow concentration range and satura-tion adsorption is reached near the CMC of thesurfactant. The sharp increase in adsorption isattributed to the strong interaction between thehydrocarbon chains of the surfactant moleculesthat are anchored on the polymer chain ratherthan on the silica surface. It is to be noted thatthere was a measurable difference in the adsorptionof dodecylsulfate depending on whether adsorp-tion took place from a bulk solution of PEa andSDS or from SDS solution onto pre-adsorbedPEa (Fig. 37). Adsorption of SDS fromPEO-SDS mixtures was consistently lower than
9;*,~}}~~:~~f~~ '"",:.-.r~'~k ,,,i:~~~'! }l~;~s
;.~.;Lc,.,~.,,~
If~~'r~;ij~..~0-
f
Adsorption of surfactants and polymers at thesolid-liquid interface depends on the nature of thesurfactant or polymer, the solvent and 1he sub-strate. In addition, system properties such as tem-perature and even system history can be expectedto affect adsorption significantly.
Ionic surfactants adsorbing on oppositelycharged surfaces exhibit a typical four-region iso-therm. At low concentrations the adsorption ismainly by ion exchange (electrostatic interaction).At higher concentrations lateral interactionsamong the adsorbed molecules causes a sharp risein adsorption which plateaus out once the CMCof the surfactant is reached. In some cases, at highconcentrations a second layer of surfactant canad$orb with a reverse orientation.
iNon-ionic surfactants adsorb primarily throughhydration or hydrogen bond interactions and thisdepends on the hydration characteristics of thesubstrate. In this case the lateral interactions aremuch stronger owing to the absence of electrostaticrepulsion between the surfactant head groups andthis leads to a sharp increase in the adsorptioncorresponding to solloid formation. The adsorp-tion of non-ionic surfactants can be greatlyenhanced by the presence of ionic surfactants andvice vet'Sa. Experiments have shown that non-ionicand ionic surfactants when used in mixtures can~ forced to adsorb on substrates on which they
Fig. 37. Adsorption of SDS onto silica from PEo-SDS mix.tures showing effect of mode .of addition.
~~~

512 P. Soma.rImdara1I, S. KrirIIIIQk1mIar I Colloids SurfacesA-: Physicochenlo Eng.AsJi:ects l1J-124 ( 1997) 491-513
dency of polymers and surfactants to interactin solution at low concentrations via interactionsamong the hydrophobic moieties. In the case ofmixed systems, the order of addition is found tobe an important parameter determining the equi-librium adsorption..
Acknowledgment
The authors wish to acknowledge NationalScience Foundation (NSF-CfS-93 I 1940),Engineering Research Center, University ofFlorida, Environmental Protection Agency(RR823301-O1-O) for financial support.
References
iJ:!#
do not exhibit much adsorption by themselves.The synergistic effect of these mixtures on adsorp-tion is hypothesized to be due to reduction incharge repulsion leading to better packing of theionic surfactants while for the non-ionic surfac-tants the increase in adsorption is due to theirsolubilization in the hydrophobic microdomainsformed by the ionic surfactants. While adsorptionof simple components can be expressed in termsof the properties of the components and the solid,the adsorption of mixtures cannot yet be describedtheoretically. In fact interactions even in bulksolution do not follow, for example, the regularsolution theory in many cases.
In organic media, adsorption depends on therelative acid-base characteristics of the solute,solvent and substrate. Adsorption of ionic surfac-tants on oxide surfaces proceeds to a monolayerand there is no evidence of lateral interactionbetween the adsorbed molecules. However, onhydrophobic surfaces, interactions are possibleamong the polar groups of the adsorbed moleculesleading to the formation of reverse solloids and aconcomitant increase in adsorption density some-what resembling solloid formation in aqueousmedia.
Polymer adsorption in aqueous media is con-trolled by the polymer charge, molecular weight,solvent, solution conditions (pH, ionic strength)and porosity of the substrate. Polyelectrolyteadsorption proceeds mainly by electrostatic inter-actions while non-ionic PEO adsorbs by hydrogenbonding interactions similarly to the non-ionicsurfactants. However, the adsorbed layer structureis very different in the case of polymers from thatof the surfactants. A polymer can have variousconfigurations at the interface and this has beenstudied for the case of PAA on alumina by fluores-cence and ESRspectroscopy. The adsorbed layerin this case can be thought of as a fuZzy polymerlayer with trains, loops and tails. Also, it is clearthat the reorientation of the polymer molecules atthe interface takes place in response to changes inthe surrounding environment such as changingpH in the case of PAA on alumina. Polymer-surfactant mixtures exhibit synergism in adsorp-tion akin to ionic-non-ionic surfactant mixtures.The origin of this synergism lies. in the ten-
[ I] A. W. Adamson, Physical Chemistry of Surfaces, Wiley,New York, 4th edn., 1982.
[2] G.D. Parfitt, C.H. Rochester, Adsorption of small mole.cules, in: G.D. Parfitt. C.H. Rochester (Eds.), Adsorptionfrom Solution at the Solid/liquid Interface, AcademicPress,New York:, 1983, pp. 3-48.
[3] A Zangwill, in: Physics at Surfaces, Cambridge UniversityPress, New York:, 1988, Chapter 14.
[4] E.D. Goddard, P. Somasundaran, Croat. Chem. Acta 48(1976) 451
[5) P. Somasundaran, D.W. Feurstenau, J. Phys. Chem. 70(1966)90
[6] A.M. Gaudin, D.W. Feurstenau, Min. Eng. 7 (1955) 66[7] P. SoD1asundaran, D.W. Feurstenau, J. Phys. Chem. 70
(1966) 90[8] P. Somasundaran, J.T. Kunjappu, Colloids Surf. 37
(1~89) 245[9) JJ{. Harwell, D. Bitting, Langmuir 3 (1987) 500
[10] P. Somasundaran, P. Chandar, NJ. Turro, Colloids Surf.20 (1986) 145
[II] P. Chandar, P. Somasundaran, NJ. Turro, J. ColloidInterface Sci. 117 (1987) 31
[12] K.C. Waterman, NJ. Turro, P. Chandar, P.Somasundaran, J. Phys. Chem. 90 (1986) 6829
[13] P. Chandar, P. Somasundaran, K.C. Waterman, NJ.Turro, I. Phys. Chem. 91 (1986) ISO
[14] I.H. Wertz, I.R. Bolton, Electron Spin Resonance-Elementary Theory and Practical Applications, Chapmanand HaD, New York, 1986.
[IS) P. Somasundaran, J.T. Kunjappu, Langmuir 5 (1989) 215[16] A Sivakumar, P. Somasundaran, Adsorption of alkylXy-
lenesuJfonates on alumina: a ft~orescence probe study,Langmuir 10 (1994) 131
~':~;[~.~;f;
fiE~s

P. Somao\1mdaraII; S. Krislmakuntar I Colloids Surfaces A: PhysicocMm. Eng. Aspects 12J-124 ( 1997) 491-513 513
(31] C.M. Hansen, J. Paint Technol. 39 (1967) 104,505.(32] P.A. Small, J. Appl. Chern. 3 (1953) 71(33] K.V.s.N. Raju, M. Yaseen, Langmuir 8 (1992) 43(34] M.J. Mcguire, I.H. Suffet, J, Am. Waterworks Assoc. 70
(1978)621(35] S. Krishnakumar, Doctoral Thesis, Columbia UniVersity,
1995.(36] H. Morawctx, Macromolecules in Solution, Wiley, New
York. 1975.(37] D.H. Napper, Polymeric Stabilization of Colloidal
Dispersions, Conoid Science Monograph, Academic Press,London, 1983.
(38] T. Sato, R. Ruch, Stabilization of Colloidal Dispersionsby Polymer Adsorption, Dekker, New York, 1990.
(39] B. Markovic, Doctoral ThesiS, University of Zagreb, 1996.(40] K.s. Arora, NJ. Turro, J. Polym. Sci. 25 (1987) 243(41] P. Chandar, P. Somasundaran, K.C. Waterman. NJ.
Turro, Langmuir 3 (1987) 298(42] x. Yu, Doctoral Thesis, Columbia University, 1995.(43] C. Maltesh, P. Somasundaran, J. Colloid Interface Sci. 153
(I) (1992) 298
[17] A. Sivakumar, P. SOmaSUDdaran, S. Thach. Colloids Surf.70 (1993) 69
[18] E. Fu, Doctoral Thesis, Columbia UniVersity, 1987.[19] P.Somasundaran, E. Snell, Q. Xu, J. Colloid Interf~ Sci.
144 (1) (1991) 159[20] P. Somasundaran, L. Zhang, unpublished results.[21] P. Somasundaran, E. Fu,Q.Xu, Langmuir (1992) 1065.[22] Q. Xu, Doctoral Thesis, Columbia University, 1992.[23] L. Huang. C. Maltesh, P. Somasundaran, J. Conoid
Interf~ Science 177 (1996) 222[24] Q. Xu, P. Somasundaran, Miner. Metall. Process. 9
(1992) 29[25) S. Krisbnakumar, P. Somasundaran. Langmuir 10
(1994) 2786[26] A.N. Maitra, H.F. Eicke, J. Pbys.Chem. 85 (1981) 2687[27] S. Krishnakumar, P. Somasundaran, Colloids Surf., to
be published.[28] T.Gu,B.-Y. Zhu, Colloids Surf. 46 (1990) 81[29] S. Krlshnakumar,P. Somasundaran, J. Colloid Interf~
Sci. 162 (1994) 425[30] E. Koksal, R.. Ramachandran, P. Somasundaran, C.
Maltesb, Powder Technol. 62 (1990) 253
~
~



















