
As process development and manufacturing (cmc) for biologics development-an overview 26 nov09

By Susan Dana Jones PhD Patricia Seymour and Howard L Levine PhD BioProcess Technology Consultants
THE CURRENT ECONOVllC CRISIS increasing competition and the need to make healthcare more affordable combine to present formidable challenges to todays biopharmaceushy
tical industry By 2015 spending on prescription medicines in the Us alone forecast to be approximately 5450 billion repshyresenting more than 10 of total Us hea lthcare expenditures Therapeutic monoclonal antibodies have now become the dominant component of the biopharmaceutical market with combined 2008 revenues estimated to be nearly $30 billion Continued development of this exciting class of products is expected to continue to drive the overall sales of biopharmashyceutical products in the future
BioProcess Technology Consultants recently published The Development of Therapeutic Monoclonal Antibody Products a comshyprehensive report outlining the complex technical regulatory and strategic Chemistry Manufacturing and Control (CMC) activities necessary to successfully advance new monoclonal antibody products from discovery to First-in-Human clinical trials and the market as quickly and economically as possible This article based in part on the report will summarize the overall costs and timing for development of a new monoclonal antibody product and outline the critical development manushyfacturing quality and regulatory activities required to support the successful filing of an Investigational New Drug (IND) application to initiate human clinical trials
CMC development is one of the most critical and time-
consuming tasks required to enable human clinical testing of a new monoclonal antibody candidate This task includes the construction and testing of a production cell line and manufacshyturing process for production of the product as well as develshyopment of suitable analytical methods to characterize the antishybody and ensure that it is safe and has the desired functional properties The numerous interdependent CMC activities required to advance a monoclonal antibody candidate from discovery to clinical tria ls and the estimated overall costs of these activities are outlined in Table 1 Many of the activities included in Table 1 must be substantially completed before anishymal tOXIcology studies can be performed since these studies must use product that is produced using essentially the same process that will be used to make material for the human clinishycal trials Detailed project planning and management is essenshytial to coordinate these multiple activities and ensure technical and regulatory success
Susan Dana Jones PhD is vice president and senior consultant
of 8ioProcess Technology Consultants Patricia Seymour is a
senior consultant at the firm and Howard L Levine PhD is
the companys founder president and principal consultant Correspondence about this article can be addressed to
sjonesbioprocessconsultantscom
6~CONTRACT PHARMA bull April 20 I 0 wwwcontractpharmacom
Table I Estimated cost of IND-enabling CMC activities release and the use of platform processes for upstream and downstream processing with full process optimization and valshyidation being performed later in development The use of platshyform processes allows the rapid production of monoclonal antishybody products for hwnan clinical evaluation
Activity Estimated Cost
Analytical development and qualification $1050000
Cell line and upstream
process development $600000
Downstream process development and
viral clearance validation $900000
Formulation development $300000
Bulk drug substance manufacturing
including stability studies $3300000
Final drug product manufacturing
including stability studies $550000
IND Preparation (CMC Section) $100000
TOTAL $6800000
Figure 1 provides a useful framework for understanding the interdependencies of the various tasks that must be completed prior to filing an INO and initiating human clinical trials for a monoclonal antibody product Each of the major groups of tasks listed in Figure 1 is generally performed by distinct resources either within a product development company or by a contract service provider The technical and business details of any given monoclonal antibody product development proshygram may r ult m some variance from this typical timeline Some companies have reported CMC development timelines as short as 10 - 13 months but it is more typical for these CMC
development activities to take at least 18 months to complete at a cost of approximatel S7 million
Process Development Activities The first CMC activities that are required for monoclonal antishybody development are the development of suitable analytical methods and the generation of a production cell line for the
Since the CMC activshyities described here are Figure I Timeline for Key Chemistry Manufacturing and Control Tasks for on the critical path to a
Therapeutic Monoclonal Antibody Product Developmentsuccessful INO filing it is essential that they be completed as quickly as possible to facilitate the initiation of human clinical trials A typical time line for CMC
development activities required prior to filing an INO is shown in Figure 1 Unlike other therapeutic recombishynant proteins the comshymonality in molecular structure of monoclonal antibody products enables the use of comshymon platform technoloshygies or processes for both upstream and downstream processshying Therefore the timeshyline shown in Figure 1 is based on the assumpshytion that development is carried out in the conshytext of a reasonably well-established set of analytical methods for product characterizashytion and QC testing and
Months from start of program
1 2 3 4 5 6 7 B 9 10 11 12 13 14 15 16 17 18
1 Analytical Methods Preliminary Development Assays amp Standards -Analyt ical Methods Development
Assay Qualification amp Transfer to QC -Prod ct Characterization
Clinical Reference Standards -2 Upstream Process Expression Vecwr bullCell Line SelectIon amp Opt imization
Cell Banking amp Characterization
Initial Media amp Bioreactor Process Development
3 Downstream Process Purification Process Development
Initial Viral Validation Studies
4 Formulation Development PremiddotFormulat ion Development
Clinical Formulation DevelClpment
5 Drug Substance ManufactUring Process amp Analytical Documentation
PredinicallEnglneenng Drug Substance Production
Phase I (GMP) Drug Substance Production
Preliminary Drug Substance Stability Studies i bull6 Drug Product Manufacturing Clin ical FillFinish Process Development bullProcess amp Analyt ical Documentation bullPreciinica llEngineuroeuroring Drug Product Production bull bull bullPhase I (GMP) Drug Product Production
Prelim inary Drug Product Stability Studies
7 Regulatory IND Filing Preparat ion - CMC Section
IND Filing www_contractpharma_com CONTRACT PHARMA bull April 20 I 0 61
product Development of a suitable genetically-modified engishyneered cell line capable of producing a monoclonal antibody with sufficiently high productivity is one of the most time-conshysuming CMC development activities Using traditional selecshytion methods cell line development can take as long as nine months to complete However many technologies are currentshyly available that improve the speed of cell line development producing cell lines with sufficient specific productivity for manufacturing in as little as three to six months At least some
Meeting the necessary purity levels
is an absolute regulatory requirement
e fJ fr
analytical methods must be in place prior to initiation of cell line development to insure that the right product is produced by the candidate cell lines and the final selected cell line As the development program progresses additional analytical methshyods are normally developed to support celi culture and purifishycation development quality control testing and characteriza-
LYOPHILIZAT ION TEC HNOLOG Y INC Jnipoundj a U f9 Sc ipound ~pound a nd J t hn o(0 JJ
Contract Manufacturing Services with Unparalleled Lyophilization Expertise
Outstanding quality and service is what you have come to expect from OUf development capabilities Now realize the same benefits by preparing your Pbase I and II clinical trial materials at LT
bull Thermal Analysis bull Clinical Matena l Preparation bull Formulation Design bull Quality and Regulatory Support bull Process Engineering bull Technical Services bull Design Space Studies bull On-site Training
Visit our website or call us to discuss a better way to get your product to the clinic
30 INDIAN DRIVEmiddot IVYLAND PA 18974 USA PHONE +1 (215 ) 396-8373 bull FAX +1 ( 215 ) 396-8375 inquirylyo-t com bull www lyotechnology com
62 CONTRACT PHARMA bull April 2~ __
tion of the bulk drug substance and final drug product Following cell line development a suitable cell culture
process that enables production of sufficient quantities of the monoclonal antibody product in an appropriately sized bioreshyactor must be developed During cell culture process developshyment media composition and bioreactor conditions are studied and optimized using a number of screening and factoshyrial experiments to determine the effects of multiple middotariables on growth and viability specific productivity antibody
e a 5
r bull com e eoon ep
yields product consistency and impurity levels Upstream process development ends with the definition of a robust cell culture process that is based on the optimal conditions identishyfied to consistently generate high product yield and quality product homogeneity and minimal amounts of impurities and contaminants
The downstream process consists of a series of recovery and purification steps to enable the antibody to be purified to homogeneity The antibody produced in the upstream process is separated from impurities (substances related to the desired monoclonal antibody product or process that are undesirable in the final product) and potential contaminants (substances that are not intended to be m the process product or intermeshydiates but which may be present and require removal) to levels sufficient for delivery to a patient The development of a suitshyable recovery and purification process for a monoclonal antishybody focuse on two main technical aspects - the effectiveness of the removal of imp unties and contaminants (ie the product purity) and the yield of the product (the percent of the active antibody product present in the bioreactor batch that is recovshyered in the final bulk drug substance) All processes involve tradeoffs between yield and purity so the major overall goal of downstream process development is to insure that the purity is sufficient for the monoclonal antibody product to be safe for its intended use while simultaneously maximizing yield Meeting the necessary purity levels is an absolute regulatory requirement while product yield is primarily an economic conshycern for the company developing the product The purified monoclonal antibody solution resulting from the recovery and purification process is referred to as the bulk drug substance
Other CMC development activities that are essential to enable a successful [ND filing include formulation developshyment and stability testing The antibody must be formulated to ensure that the proper dose of active antibody reaches the proper site of action in the body upon administration to the patient and that the drug product is stable during storage Formulation development should be initiated as soon as prodshyuct is available even product from process development activshyities which may not be fully representative of the final process
wwwcontra~tpharmaco~
To select an optimized formulation the monoclonal antibody product is prepared in a number of different formulations and subjected to accelerated storage conditions Analytical methods that are stability-indicating are used to determine which forshymulations are best for maintaining antibody structure and function Data supporting the structural and functional stabilishyty of an antibody must be generated prior to an 110 filing and the stability program must continue for as long as a specific lot of antibody product is used in the clinic
Scale-up and Manufacturing For First-in-Human Clinical Trials After process development is completed the upstream and downstream process for production of a monoclonal antibody product must be scaled-up to the intended manufac turing scale for production of early-stage clinical trial material Scale-up is required in order to produce sufficient amounts of material for clinical and other needs For the production process scale-up involves careful engineering and testing to nsure that the process performs as expected and the product is not adversely affected Some scale-up activities can be performed in the development laboratories but to reach the final production scale one or more engineering or demonstration batches are often produced at the intended manufacturing sca le These non-GMP batches are often produced in the GMP production suite and material from these studies is often used for preclinishycal GLP animal studies in support of an lND application and to produce a reference standard The production of an engineershying batch (or batches) enables final process optimIzation and resolution of any scale-up or production issues prior to the first batch produced under cGMP
Following production of the bulk drug substance for clinishycal use the antibody must be filled into vials or yringes to generate the final drug product for shipment to the cl inic The manufacture of a monoclonal antibody drug product involves three critical processes - formulation sterili zation and asepshytic filling of the product - shown schema tic lly in Figure 2 In the first step excipients such as buffering agents s tabili zshyers or cryoprotectants are added to the bulk drug substance so lution to ensure the stability of the product in the final conshytainer and the protein concentration is djusted to the desired level for storage and administration The fo rmulated bulk drug substance is sterilized by filtration to remove all bioburshyden from the solution and produce a sterile product for parshyenteral administration This sterile bu lk drug substance is normally aseptically filled into pre-ste rilized vials or syringes although other containers such as IV bags are occashysionally used
Figure 2 Sieps in the Manufacture of a
Monocianal Antibody Drug Product
FonnulacedBulk drug
bulkdrug substance wbS13nce
Sterile bulk drug substance
Drug
product
wwwcontractpharmacom
The first commercial therapeutic monoclonal antibody prodshyuct was approved in 1986 and more than 20 antibody products have been approved since then Since monoclonal antibodies are such a dominant element in todays biopharmaceutical marshyket Significant effort has gone into developing more efficient technologies and tools to enable higher titers better yields in downstream processing and more cost effective manufacturing processes that will impact the cost of goods of commercial prodshyucts Some of these technologies ha ve already been implementshyed in existing commercial antibody manufacturing processes whereas other cutting-edge novel technologies are still finding their way into development programs
The regulatory environment for biopharmaceuticals is conshystantly evolVing For example as recently as 10 years ago most cell culture was performed in the presence of serum Today there are advanced media formulations that enable initial cell transfection cloning selection and culture in the bioreactor in the absence of any serum and removal of serum from processshyes is now expected by the regulatory agenCies The requireshyments for purity and for viral clearance validation have also become more stringent in the past decade These regulatory changes Impac t the cost and time needed to develop manufacshyturing processes for new monoclonal antibody products
The critical CMC development activities for new monoclonshyal antibody produ ts consist of a series of well defined steps which when carefully managed and followed enable compashynies to ad ance new monoclonal antibody product candidates into human clinical trials with a high probability of success These activities normal require approximately 18 months to complete although advances ill all aspects of process developshyment ma y lead to some reductions in this time line in the upcoming years bull
References
Ransohoff TC et al (BioProcess Technology Consultants Inc Acton MA) Cell culture manufacturing capacity trends and
outlook through 20 13 Springfield 01A) Pharmsource
Information Services 2008 Dec 126 p
2 Ransohoff TC Levine HL CapaCity counts Inside Outsourcing
Pharm Exec (Internet] 2009 Nov (Cited 2009 Dec I 1]29( II
Suppl) 18-23 Available from httpwww nxtbookcom
nxtbooksadva nstari ins ideoutsourcinamp-20091 Ii ndexphpl startid= 18
3 The Development of Therapeutic Monoclonal Antibody
Products Levine HL Jagschies G editors Boston (MA) BioProcess Technology Consultants Inc 20 I 0 Feb 332 p
4 Jones SO Technologies to Improve Cell Line Development and Engineering Presented at Cambridge Healthtech Institute Peptalk 2009 Jan I I- 6 Coronado CA
5 Box GE et a l Statistics for experimenters design innovation
and discovery Second ed Hoboken (NJ) John Wiley amp Sons c2005 664 p
CONTRACT PHARMA bull April 20 I 0 63
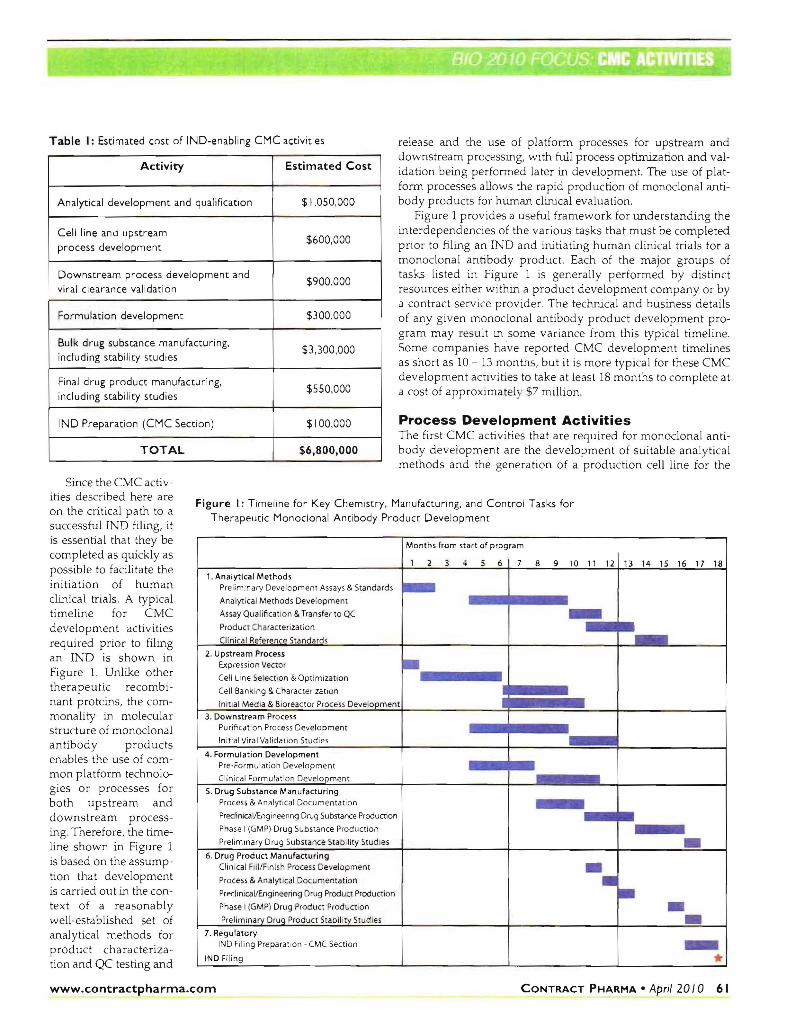
Table I Estimated cost of IND-enabling CMC activities release and the use of platform processes for upstream and downstream processing with full process optimization and valshyidation being performed later in development The use of platshyform processes allows the rapid production of monoclonal antishybody products for hwnan clinical evaluation
Activity Estimated Cost
Analytical development and qualification $1050000
Cell line and upstream
process development $600000
Downstream process development and
viral clearance validation $900000
Formulation development $300000
Bulk drug substance manufacturing
including stability studies $3300000
Final drug product manufacturing
including stability studies $550000
IND Preparation (CMC Section) $100000
TOTAL $6800000
Figure 1 provides a useful framework for understanding the interdependencies of the various tasks that must be completed prior to filing an INO and initiating human clinical trials for a monoclonal antibody product Each of the major groups of tasks listed in Figure 1 is generally performed by distinct resources either within a product development company or by a contract service provider The technical and business details of any given monoclonal antibody product development proshygram may r ult m some variance from this typical timeline Some companies have reported CMC development timelines as short as 10 - 13 months but it is more typical for these CMC
development activities to take at least 18 months to complete at a cost of approximatel S7 million
Process Development Activities The first CMC activities that are required for monoclonal antishybody development are the development of suitable analytical methods and the generation of a production cell line for the
Since the CMC activshyities described here are Figure I Timeline for Key Chemistry Manufacturing and Control Tasks for on the critical path to a
Therapeutic Monoclonal Antibody Product Developmentsuccessful INO filing it is essential that they be completed as quickly as possible to facilitate the initiation of human clinical trials A typical time line for CMC
development activities required prior to filing an INO is shown in Figure 1 Unlike other therapeutic recombishynant proteins the comshymonality in molecular structure of monoclonal antibody products enables the use of comshymon platform technoloshygies or processes for both upstream and downstream processshying Therefore the timeshyline shown in Figure 1 is based on the assumpshytion that development is carried out in the conshytext of a reasonably well-established set of analytical methods for product characterizashytion and QC testing and
Months from start of program
1 2 3 4 5 6 7 B 9 10 11 12 13 14 15 16 17 18
1 Analytical Methods Preliminary Development Assays amp Standards -Analyt ical Methods Development
Assay Qualification amp Transfer to QC -Prod ct Characterization
Clinical Reference Standards -2 Upstream Process Expression Vecwr bullCell Line SelectIon amp Opt imization
Cell Banking amp Characterization
Initial Media amp Bioreactor Process Development
3 Downstream Process Purification Process Development
Initial Viral Validation Studies
4 Formulation Development PremiddotFormulat ion Development
Clinical Formulation DevelClpment
5 Drug Substance ManufactUring Process amp Analytical Documentation
PredinicallEnglneenng Drug Substance Production
Phase I (GMP) Drug Substance Production
Preliminary Drug Substance Stability Studies i bull6 Drug Product Manufacturing Clin ical FillFinish Process Development bullProcess amp Analyt ical Documentation bullPreciinica llEngineuroeuroring Drug Product Production bull bull bullPhase I (GMP) Drug Product Production
Prelim inary Drug Product Stability Studies
7 Regulatory IND Filing Preparat ion - CMC Section
IND Filing www_contractpharma_com CONTRACT PHARMA bull April 20 I 0 61
product Development of a suitable genetically-modified engishyneered cell line capable of producing a monoclonal antibody with sufficiently high productivity is one of the most time-conshysuming CMC development activities Using traditional selecshytion methods cell line development can take as long as nine months to complete However many technologies are currentshyly available that improve the speed of cell line development producing cell lines with sufficient specific productivity for manufacturing in as little as three to six months At least some
Meeting the necessary purity levels
is an absolute regulatory requirement
e fJ fr
analytical methods must be in place prior to initiation of cell line development to insure that the right product is produced by the candidate cell lines and the final selected cell line As the development program progresses additional analytical methshyods are normally developed to support celi culture and purifishycation development quality control testing and characteriza-
LYOPHILIZAT ION TEC HNOLOG Y INC Jnipoundj a U f9 Sc ipound ~pound a nd J t hn o(0 JJ
Contract Manufacturing Services with Unparalleled Lyophilization Expertise
Outstanding quality and service is what you have come to expect from OUf development capabilities Now realize the same benefits by preparing your Pbase I and II clinical trial materials at LT
bull Thermal Analysis bull Clinical Matena l Preparation bull Formulation Design bull Quality and Regulatory Support bull Process Engineering bull Technical Services bull Design Space Studies bull On-site Training
Visit our website or call us to discuss a better way to get your product to the clinic
30 INDIAN DRIVEmiddot IVYLAND PA 18974 USA PHONE +1 (215 ) 396-8373 bull FAX +1 ( 215 ) 396-8375 inquirylyo-t com bull www lyotechnology com
62 CONTRACT PHARMA bull April 2~ __
tion of the bulk drug substance and final drug product Following cell line development a suitable cell culture
process that enables production of sufficient quantities of the monoclonal antibody product in an appropriately sized bioreshyactor must be developed During cell culture process developshyment media composition and bioreactor conditions are studied and optimized using a number of screening and factoshyrial experiments to determine the effects of multiple middotariables on growth and viability specific productivity antibody
e a 5
r bull com e eoon ep
yields product consistency and impurity levels Upstream process development ends with the definition of a robust cell culture process that is based on the optimal conditions identishyfied to consistently generate high product yield and quality product homogeneity and minimal amounts of impurities and contaminants
The downstream process consists of a series of recovery and purification steps to enable the antibody to be purified to homogeneity The antibody produced in the upstream process is separated from impurities (substances related to the desired monoclonal antibody product or process that are undesirable in the final product) and potential contaminants (substances that are not intended to be m the process product or intermeshydiates but which may be present and require removal) to levels sufficient for delivery to a patient The development of a suitshyable recovery and purification process for a monoclonal antishybody focuse on two main technical aspects - the effectiveness of the removal of imp unties and contaminants (ie the product purity) and the yield of the product (the percent of the active antibody product present in the bioreactor batch that is recovshyered in the final bulk drug substance) All processes involve tradeoffs between yield and purity so the major overall goal of downstream process development is to insure that the purity is sufficient for the monoclonal antibody product to be safe for its intended use while simultaneously maximizing yield Meeting the necessary purity levels is an absolute regulatory requirement while product yield is primarily an economic conshycern for the company developing the product The purified monoclonal antibody solution resulting from the recovery and purification process is referred to as the bulk drug substance
Other CMC development activities that are essential to enable a successful [ND filing include formulation developshyment and stability testing The antibody must be formulated to ensure that the proper dose of active antibody reaches the proper site of action in the body upon administration to the patient and that the drug product is stable during storage Formulation development should be initiated as soon as prodshyuct is available even product from process development activshyities which may not be fully representative of the final process
wwwcontra~tpharmaco~
To select an optimized formulation the monoclonal antibody product is prepared in a number of different formulations and subjected to accelerated storage conditions Analytical methods that are stability-indicating are used to determine which forshymulations are best for maintaining antibody structure and function Data supporting the structural and functional stabilishyty of an antibody must be generated prior to an 110 filing and the stability program must continue for as long as a specific lot of antibody product is used in the clinic
Scale-up and Manufacturing For First-in-Human Clinical Trials After process development is completed the upstream and downstream process for production of a monoclonal antibody product must be scaled-up to the intended manufac turing scale for production of early-stage clinical trial material Scale-up is required in order to produce sufficient amounts of material for clinical and other needs For the production process scale-up involves careful engineering and testing to nsure that the process performs as expected and the product is not adversely affected Some scale-up activities can be performed in the development laboratories but to reach the final production scale one or more engineering or demonstration batches are often produced at the intended manufacturing sca le These non-GMP batches are often produced in the GMP production suite and material from these studies is often used for preclinishycal GLP animal studies in support of an lND application and to produce a reference standard The production of an engineershying batch (or batches) enables final process optimIzation and resolution of any scale-up or production issues prior to the first batch produced under cGMP
Following production of the bulk drug substance for clinishycal use the antibody must be filled into vials or yringes to generate the final drug product for shipment to the cl inic The manufacture of a monoclonal antibody drug product involves three critical processes - formulation sterili zation and asepshytic filling of the product - shown schema tic lly in Figure 2 In the first step excipients such as buffering agents s tabili zshyers or cryoprotectants are added to the bulk drug substance so lution to ensure the stability of the product in the final conshytainer and the protein concentration is djusted to the desired level for storage and administration The fo rmulated bulk drug substance is sterilized by filtration to remove all bioburshyden from the solution and produce a sterile product for parshyenteral administration This sterile bu lk drug substance is normally aseptically filled into pre-ste rilized vials or syringes although other containers such as IV bags are occashysionally used
Figure 2 Sieps in the Manufacture of a
Monocianal Antibody Drug Product
FonnulacedBulk drug
bulkdrug substance wbS13nce
Sterile bulk drug substance
Drug
product
wwwcontractpharmacom
The first commercial therapeutic monoclonal antibody prodshyuct was approved in 1986 and more than 20 antibody products have been approved since then Since monoclonal antibodies are such a dominant element in todays biopharmaceutical marshyket Significant effort has gone into developing more efficient technologies and tools to enable higher titers better yields in downstream processing and more cost effective manufacturing processes that will impact the cost of goods of commercial prodshyucts Some of these technologies ha ve already been implementshyed in existing commercial antibody manufacturing processes whereas other cutting-edge novel technologies are still finding their way into development programs
The regulatory environment for biopharmaceuticals is conshystantly evolVing For example as recently as 10 years ago most cell culture was performed in the presence of serum Today there are advanced media formulations that enable initial cell transfection cloning selection and culture in the bioreactor in the absence of any serum and removal of serum from processshyes is now expected by the regulatory agenCies The requireshyments for purity and for viral clearance validation have also become more stringent in the past decade These regulatory changes Impac t the cost and time needed to develop manufacshyturing processes for new monoclonal antibody products
The critical CMC development activities for new monoclonshyal antibody produ ts consist of a series of well defined steps which when carefully managed and followed enable compashynies to ad ance new monoclonal antibody product candidates into human clinical trials with a high probability of success These activities normal require approximately 18 months to complete although advances ill all aspects of process developshyment ma y lead to some reductions in this time line in the upcoming years bull
References
Ransohoff TC et al (BioProcess Technology Consultants Inc Acton MA) Cell culture manufacturing capacity trends and
outlook through 20 13 Springfield 01A) Pharmsource
Information Services 2008 Dec 126 p
2 Ransohoff TC Levine HL CapaCity counts Inside Outsourcing
Pharm Exec (Internet] 2009 Nov (Cited 2009 Dec I 1]29( II
Suppl) 18-23 Available from httpwww nxtbookcom
nxtbooksadva nstari ins ideoutsourcinamp-20091 Ii ndexphpl startid= 18
3 The Development of Therapeutic Monoclonal Antibody
Products Levine HL Jagschies G editors Boston (MA) BioProcess Technology Consultants Inc 20 I 0 Feb 332 p
4 Jones SO Technologies to Improve Cell Line Development and Engineering Presented at Cambridge Healthtech Institute Peptalk 2009 Jan I I- 6 Coronado CA
5 Box GE et a l Statistics for experimenters design innovation
and discovery Second ed Hoboken (NJ) John Wiley amp Sons c2005 664 p
CONTRACT PHARMA bull April 20 I 0 63

product Development of a suitable genetically-modified engishyneered cell line capable of producing a monoclonal antibody with sufficiently high productivity is one of the most time-conshysuming CMC development activities Using traditional selecshytion methods cell line development can take as long as nine months to complete However many technologies are currentshyly available that improve the speed of cell line development producing cell lines with sufficient specific productivity for manufacturing in as little as three to six months At least some
Meeting the necessary purity levels
is an absolute regulatory requirement
e fJ fr
analytical methods must be in place prior to initiation of cell line development to insure that the right product is produced by the candidate cell lines and the final selected cell line As the development program progresses additional analytical methshyods are normally developed to support celi culture and purifishycation development quality control testing and characteriza-
LYOPHILIZAT ION TEC HNOLOG Y INC Jnipoundj a U f9 Sc ipound ~pound a nd J t hn o(0 JJ
Contract Manufacturing Services with Unparalleled Lyophilization Expertise
Outstanding quality and service is what you have come to expect from OUf development capabilities Now realize the same benefits by preparing your Pbase I and II clinical trial materials at LT
bull Thermal Analysis bull Clinical Matena l Preparation bull Formulation Design bull Quality and Regulatory Support bull Process Engineering bull Technical Services bull Design Space Studies bull On-site Training
Visit our website or call us to discuss a better way to get your product to the clinic
30 INDIAN DRIVEmiddot IVYLAND PA 18974 USA PHONE +1 (215 ) 396-8373 bull FAX +1 ( 215 ) 396-8375 inquirylyo-t com bull www lyotechnology com
62 CONTRACT PHARMA bull April 2~ __
tion of the bulk drug substance and final drug product Following cell line development a suitable cell culture
process that enables production of sufficient quantities of the monoclonal antibody product in an appropriately sized bioreshyactor must be developed During cell culture process developshyment media composition and bioreactor conditions are studied and optimized using a number of screening and factoshyrial experiments to determine the effects of multiple middotariables on growth and viability specific productivity antibody
e a 5
r bull com e eoon ep
yields product consistency and impurity levels Upstream process development ends with the definition of a robust cell culture process that is based on the optimal conditions identishyfied to consistently generate high product yield and quality product homogeneity and minimal amounts of impurities and contaminants
The downstream process consists of a series of recovery and purification steps to enable the antibody to be purified to homogeneity The antibody produced in the upstream process is separated from impurities (substances related to the desired monoclonal antibody product or process that are undesirable in the final product) and potential contaminants (substances that are not intended to be m the process product or intermeshydiates but which may be present and require removal) to levels sufficient for delivery to a patient The development of a suitshyable recovery and purification process for a monoclonal antishybody focuse on two main technical aspects - the effectiveness of the removal of imp unties and contaminants (ie the product purity) and the yield of the product (the percent of the active antibody product present in the bioreactor batch that is recovshyered in the final bulk drug substance) All processes involve tradeoffs between yield and purity so the major overall goal of downstream process development is to insure that the purity is sufficient for the monoclonal antibody product to be safe for its intended use while simultaneously maximizing yield Meeting the necessary purity levels is an absolute regulatory requirement while product yield is primarily an economic conshycern for the company developing the product The purified monoclonal antibody solution resulting from the recovery and purification process is referred to as the bulk drug substance
Other CMC development activities that are essential to enable a successful [ND filing include formulation developshyment and stability testing The antibody must be formulated to ensure that the proper dose of active antibody reaches the proper site of action in the body upon administration to the patient and that the drug product is stable during storage Formulation development should be initiated as soon as prodshyuct is available even product from process development activshyities which may not be fully representative of the final process
wwwcontra~tpharmaco~
To select an optimized formulation the monoclonal antibody product is prepared in a number of different formulations and subjected to accelerated storage conditions Analytical methods that are stability-indicating are used to determine which forshymulations are best for maintaining antibody structure and function Data supporting the structural and functional stabilishyty of an antibody must be generated prior to an 110 filing and the stability program must continue for as long as a specific lot of antibody product is used in the clinic
Scale-up and Manufacturing For First-in-Human Clinical Trials After process development is completed the upstream and downstream process for production of a monoclonal antibody product must be scaled-up to the intended manufac turing scale for production of early-stage clinical trial material Scale-up is required in order to produce sufficient amounts of material for clinical and other needs For the production process scale-up involves careful engineering and testing to nsure that the process performs as expected and the product is not adversely affected Some scale-up activities can be performed in the development laboratories but to reach the final production scale one or more engineering or demonstration batches are often produced at the intended manufacturing sca le These non-GMP batches are often produced in the GMP production suite and material from these studies is often used for preclinishycal GLP animal studies in support of an lND application and to produce a reference standard The production of an engineershying batch (or batches) enables final process optimIzation and resolution of any scale-up or production issues prior to the first batch produced under cGMP
Following production of the bulk drug substance for clinishycal use the antibody must be filled into vials or yringes to generate the final drug product for shipment to the cl inic The manufacture of a monoclonal antibody drug product involves three critical processes - formulation sterili zation and asepshytic filling of the product - shown schema tic lly in Figure 2 In the first step excipients such as buffering agents s tabili zshyers or cryoprotectants are added to the bulk drug substance so lution to ensure the stability of the product in the final conshytainer and the protein concentration is djusted to the desired level for storage and administration The fo rmulated bulk drug substance is sterilized by filtration to remove all bioburshyden from the solution and produce a sterile product for parshyenteral administration This sterile bu lk drug substance is normally aseptically filled into pre-ste rilized vials or syringes although other containers such as IV bags are occashysionally used
Figure 2 Sieps in the Manufacture of a
Monocianal Antibody Drug Product
FonnulacedBulk drug
bulkdrug substance wbS13nce
Sterile bulk drug substance
Drug
product
wwwcontractpharmacom
The first commercial therapeutic monoclonal antibody prodshyuct was approved in 1986 and more than 20 antibody products have been approved since then Since monoclonal antibodies are such a dominant element in todays biopharmaceutical marshyket Significant effort has gone into developing more efficient technologies and tools to enable higher titers better yields in downstream processing and more cost effective manufacturing processes that will impact the cost of goods of commercial prodshyucts Some of these technologies ha ve already been implementshyed in existing commercial antibody manufacturing processes whereas other cutting-edge novel technologies are still finding their way into development programs
The regulatory environment for biopharmaceuticals is conshystantly evolVing For example as recently as 10 years ago most cell culture was performed in the presence of serum Today there are advanced media formulations that enable initial cell transfection cloning selection and culture in the bioreactor in the absence of any serum and removal of serum from processshyes is now expected by the regulatory agenCies The requireshyments for purity and for viral clearance validation have also become more stringent in the past decade These regulatory changes Impac t the cost and time needed to develop manufacshyturing processes for new monoclonal antibody products
The critical CMC development activities for new monoclonshyal antibody produ ts consist of a series of well defined steps which when carefully managed and followed enable compashynies to ad ance new monoclonal antibody product candidates into human clinical trials with a high probability of success These activities normal require approximately 18 months to complete although advances ill all aspects of process developshyment ma y lead to some reductions in this time line in the upcoming years bull
References
Ransohoff TC et al (BioProcess Technology Consultants Inc Acton MA) Cell culture manufacturing capacity trends and
outlook through 20 13 Springfield 01A) Pharmsource
Information Services 2008 Dec 126 p
2 Ransohoff TC Levine HL CapaCity counts Inside Outsourcing
Pharm Exec (Internet] 2009 Nov (Cited 2009 Dec I 1]29( II
Suppl) 18-23 Available from httpwww nxtbookcom
nxtbooksadva nstari ins ideoutsourcinamp-20091 Ii ndexphpl startid= 18
3 The Development of Therapeutic Monoclonal Antibody
Products Levine HL Jagschies G editors Boston (MA) BioProcess Technology Consultants Inc 20 I 0 Feb 332 p
4 Jones SO Technologies to Improve Cell Line Development and Engineering Presented at Cambridge Healthtech Institute Peptalk 2009 Jan I I- 6 Coronado CA
5 Box GE et a l Statistics for experimenters design innovation
and discovery Second ed Hoboken (NJ) John Wiley amp Sons c2005 664 p
CONTRACT PHARMA bull April 20 I 0 63

To select an optimized formulation the monoclonal antibody product is prepared in a number of different formulations and subjected to accelerated storage conditions Analytical methods that are stability-indicating are used to determine which forshymulations are best for maintaining antibody structure and function Data supporting the structural and functional stabilishyty of an antibody must be generated prior to an 110 filing and the stability program must continue for as long as a specific lot of antibody product is used in the clinic
Scale-up and Manufacturing For First-in-Human Clinical Trials After process development is completed the upstream and downstream process for production of a monoclonal antibody product must be scaled-up to the intended manufac turing scale for production of early-stage clinical trial material Scale-up is required in order to produce sufficient amounts of material for clinical and other needs For the production process scale-up involves careful engineering and testing to nsure that the process performs as expected and the product is not adversely affected Some scale-up activities can be performed in the development laboratories but to reach the final production scale one or more engineering or demonstration batches are often produced at the intended manufacturing sca le These non-GMP batches are often produced in the GMP production suite and material from these studies is often used for preclinishycal GLP animal studies in support of an lND application and to produce a reference standard The production of an engineershying batch (or batches) enables final process optimIzation and resolution of any scale-up or production issues prior to the first batch produced under cGMP
Following production of the bulk drug substance for clinishycal use the antibody must be filled into vials or yringes to generate the final drug product for shipment to the cl inic The manufacture of a monoclonal antibody drug product involves three critical processes - formulation sterili zation and asepshytic filling of the product - shown schema tic lly in Figure 2 In the first step excipients such as buffering agents s tabili zshyers or cryoprotectants are added to the bulk drug substance so lution to ensure the stability of the product in the final conshytainer and the protein concentration is djusted to the desired level for storage and administration The fo rmulated bulk drug substance is sterilized by filtration to remove all bioburshyden from the solution and produce a sterile product for parshyenteral administration This sterile bu lk drug substance is normally aseptically filled into pre-ste rilized vials or syringes although other containers such as IV bags are occashysionally used
Figure 2 Sieps in the Manufacture of a
Monocianal Antibody Drug Product
FonnulacedBulk drug
bulkdrug substance wbS13nce
Sterile bulk drug substance
Drug
product
wwwcontractpharmacom
The first commercial therapeutic monoclonal antibody prodshyuct was approved in 1986 and more than 20 antibody products have been approved since then Since monoclonal antibodies are such a dominant element in todays biopharmaceutical marshyket Significant effort has gone into developing more efficient technologies and tools to enable higher titers better yields in downstream processing and more cost effective manufacturing processes that will impact the cost of goods of commercial prodshyucts Some of these technologies ha ve already been implementshyed in existing commercial antibody manufacturing processes whereas other cutting-edge novel technologies are still finding their way into development programs
The regulatory environment for biopharmaceuticals is conshystantly evolVing For example as recently as 10 years ago most cell culture was performed in the presence of serum Today there are advanced media formulations that enable initial cell transfection cloning selection and culture in the bioreactor in the absence of any serum and removal of serum from processshyes is now expected by the regulatory agenCies The requireshyments for purity and for viral clearance validation have also become more stringent in the past decade These regulatory changes Impac t the cost and time needed to develop manufacshyturing processes for new monoclonal antibody products
The critical CMC development activities for new monoclonshyal antibody produ ts consist of a series of well defined steps which when carefully managed and followed enable compashynies to ad ance new monoclonal antibody product candidates into human clinical trials with a high probability of success These activities normal require approximately 18 months to complete although advances ill all aspects of process developshyment ma y lead to some reductions in this time line in the upcoming years bull
References
Ransohoff TC et al (BioProcess Technology Consultants Inc Acton MA) Cell culture manufacturing capacity trends and
outlook through 20 13 Springfield 01A) Pharmsource
Information Services 2008 Dec 126 p
2 Ransohoff TC Levine HL CapaCity counts Inside Outsourcing
Pharm Exec (Internet] 2009 Nov (Cited 2009 Dec I 1]29( II
Suppl) 18-23 Available from httpwww nxtbookcom
nxtbooksadva nstari ins ideoutsourcinamp-20091 Ii ndexphpl startid= 18
3 The Development of Therapeutic Monoclonal Antibody
Products Levine HL Jagschies G editors Boston (MA) BioProcess Technology Consultants Inc 20 I 0 Feb 332 p
4 Jones SO Technologies to Improve Cell Line Development and Engineering Presented at Cambridge Healthtech Institute Peptalk 2009 Jan I I- 6 Coronado CA
5 Box GE et a l Statistics for experimenters design innovation
and discovery Second ed Hoboken (NJ) John Wiley amp Sons c2005 664 p
CONTRACT PHARMA bull April 20 I 0 63


















