
Chronic Constrictive Pericarditis: Pending Issues
86
Indian Heart J 2003; 55: 305–309 Chronic Constrictive Pericarditis: Pending Issues SS Kothari, Ambuj Roy, VK Bahl Department of Cardiology, All India Institute of Medical Sciences, New Delhi C hronic constrictive pericarditis (CCP) is a well characterized clinical entity. 1–3 With advances in the understanding of hemodynamics, Doppler echocardio- graphy, and cardiac cross-sectional imaging, the diagnosis of CCP has been straightforward. However, it often masquerades as chronic liver disease, cardiomyopathy, unexplained heart failure, etc. Furthermore, there is a remarkable paucity of information regarding the pathogenesis, and the optimal management of patients with CCP. In this article, we provide a brief perspective on the recent advances in CCP, and highlight the need for more evidence-based data regarding a disease that is still common in India. Etiology Identifying the etiology of CCP has been notoriously difficult. The vast majority of patients with idiopathic CCP are suspected to have had tuberculosis in the past. While proven tubercular pericarditis may present with dense fibrosis without any direct evidence of tuberculosis, such fibrosis may follow other etiologies of CCP as well. The reported prevalence of idiopathic CCP has varied from 24% to 61% in Indian studies, 4–6 depending on the criteria used to diagnose tubercular CCP. Tuberculosis remains a common cause of CCP in developing countries, with a reported incidence of 38%–83%. 4,7–9 The etiology of CCP in the western world has now undergone a significant change, with tuberculosis reported in only 0%–1% of cases in some recent series. 10,11 The leading identifiable causes of CCP are following cardiac surgery and radiation therapy, besides viral pericarditis. 10–12 The incidence of CCP complicating cardiac surgery is between 0.025% and 0.15% of patients undergoing open heart surgery. 13 However, the lack of reports of post-surgical and post- radiation CCP from India is remarkable. An informal inquiry in the five major Indian medical college hospitals did not yield cases of CCP following cardiac surgery. Possibly such cases are being missed. Pathogenesis The precise pathogenesis of CCP remains unknown, and is scantily investigated. CCP may result from the progression of an acute pericarditis from a dry stage through an effusive, absorptive, and constrictive phase sequentially; or it may result from a smouldering fibrosis with no previous history of an acute pericarditis (as occurs in the majority of patients). The factors responsible for the resolution of inflammation or its progression to severe fibrosis remain unknown. For example, in nearly half the patients with tubercular pericardial effusion resolution occurs without constriction, while the rest develop CCP despite adequate and similar antitubercular therapy. 14 The virtual absence of CCP following rheumatic fever, 15 and its low incidence in tubercular pericarditis in HIV-positive patients are noteworthy. 16 Little research has been done to unravel the inflammatory repertoire of the pericardial tissue. 17 The role of cytokines in tubercular pericardial effusion has been investigated recently. It appears that tubercular pericarditis is a hypersensitivity reaction to antigens such as tuberculoproteins. The increased production of interferon- gamma, tumor necrosis factor-alpha, and interleukin-1 and interleukin-2 in tubercular pericardial effusion suggest that the inflammation is orchestrated by T-helper-1 lymphocytes. 18 T-lymphocytes and activated macrophages seem to play an important role in granuloma formation and fibrosis. 19 However, the exact mechanism has not been elucidated. Similarly, the mechanisms of constriction in postoperative patients, in patients with collagen vascular disease, or those with the rare but interesting hereditary disease mulibreynanism remain unexplored and unexplained. Mechanism of Fluid Retention In general, patients with CCP retain more water and sodium than patients with myocardial disease. The ascites in CCP is out of proportion to edema, and also occurs earlier than peripheral edema, a sequence opposite to that seen with other causes of congestive heart failure (CHF). The cause of this “ascites precox” has never been satisfactorily explained. It could simply be a reflection of very high right atrial pressures compared to other causes Editorial Correspondence: Professor VK Bahl, Department of Cardiology, All India Institute of Medical Sciences, New Delhi 110029 e-mail: [email protected] EDITORIAl-New.p65 11/19/2003, 11:23 PM 305
Transcript of Chronic Constrictive Pericarditis: Pending Issues

Indian Heart J 2003; 55: 305–309 Kothari et al. Chronic Constrictive Pericarditis 305
Chronic Constrictive Pericarditis: Pending Issues
SS Kothari, Ambuj Roy, VK BahlDepartment of Cardiology, All India Institute of Medical Sciences, New Delhi
Chronic constrictive pericarditis (CCP) is a wellcharacterized clinical entity.1–3 With advances in the
understanding of hemodynamics, Doppler echocardio-graphy, and cardiac cross-sectional imaging, the diagnosisof CCP has been straightforward. However, it oftenmasquerades as chronic liver disease, cardiomyopathy,unexplained heart failure, etc. Furthermore, there is aremarkable paucity of information regarding thepathogenesis, and the optimal management of patientswith CCP. In this article, we provide a brief perspective onthe recent advances in CCP, and highlight the need for moreevidence-based data regarding a disease that is still commonin India.
Etiology
Identifying the etiology of CCP has been notoriouslydifficult. The vast majority of patients with idiopathic CCPare suspected to have had tuberculosis in the past. Whileproven tubercular pericarditis may present with densefibrosis without any direct evidence of tuberculosis, suchfibrosis may follow other etiologies of CCP as well. Thereported prevalence of idiopathic CCP has varied from 24%to 61% in Indian studies,4–6 depending on the criteria usedto diagnose tubercular CCP. Tuberculosis remains acommon cause of CCP in developing countries, with areported incidence of 38%–83%.4,7–9 The etiology of CCPin the western world has now undergone a significantchange, with tuberculosis reported in only 0%–1% of casesin some recent series.10,11 The leading identifiable causes ofCCP are following cardiac surgery and radiation therapy,besides viral pericarditis.10–12 The incidence of CCPcomplicating cardiac surgery is between 0.025% and0.15% of patients undergoing open heart surgery.13
However, the lack of reports of post-surgical and post-radiation CCP from India is remarkable. An informalinquiry in the five major Indian medical college hospitalsdid not yield cases of CCP following cardiac surgery. Possiblysuch cases are being missed.
Pathogenesis
The precise pathogenesis of CCP remains unknown, and isscantily investigated. CCP may result from the progressionof an acute pericarditis from a dry stage through an effusive,absorptive, and constrictive phase sequentially; or it mayresult from a smouldering fibrosis with no previous historyof an acute pericarditis (as occurs in the majority ofpatients). The factors responsible for the resolution ofinflammation or its progression to severe fibrosis remainunknown. For example, in nearly half the patients withtubercular pericardial effusion resolution occurs withoutconstriction, while the rest develop CCP despite adequateand similar antitubercular therapy.14 The virtual absenceof CCP following rheumatic fever,15 and its low incidencein tubercular pericarditis in HIV-positive patients arenoteworthy.16 Little research has been done to unravel theinflammatory repertoire of the pericardial tissue.17 The roleof cytokines in tubercular pericardial effusion has beeninvestigated recently. It appears that tubercular pericarditisis a hypersensitivity reaction to antigens such astuberculoproteins. The increased production of interferon-gamma, tumor necrosis factor-alpha, and interleukin-1 andinterleukin-2 in tubercular pericardial effusion suggest thatthe inflammation is orchestrated by T-helper-1lymphocytes.18 T-lymphocytes and activated macrophagesseem to play an important role in granuloma formationand fibrosis.19 However, the exact mechanism has not beenelucidated. Similarly, the mechanisms of constriction inpostoperative patients, in patients with collagen vasculardisease, or those with the rare but interesting hereditarydisease mulibreynanism remain unexplored andunexplained.
Mechanism of Fluid Retention
In general, patients with CCP retain more water andsodium than patients with myocardial disease. The ascitesin CCP is out of proportion to edema, and also occursearlier than peripheral edema, a sequence opposite to thatseen with other causes of congestive heart failure (CHF).The cause of this “ascites precox” has never beensatisfactorily explained. It could simply be a reflection ofvery high right atrial pressures compared to other causes
Editorial
Correspondence: Professor VK Bahl, Department of Cardiology,All India Institute of Medical Sciences, New Delhi 110029e-mail: [email protected]
EDITORIAl-New.p65 11/19/2003, 11:23 PM305

306 Kothari et al. Chronic Constrictive Pericarditis Indian Heart J 2003; 55: 305–309
of CHF. Increased venous pressures, hypoalbuminemiaresulting from protein-losing enteropathy, cardiaccirrhosis, increased capillary permeability, and impedanceto lymph flow have been suggested but appear to beinadequate explanations.
The mechanisms of fluid retention in CCP have beenexplored in a systematic fashion in only one study.20 In 16patients with untreated CCP, it was found that themagnitude and mechanisms of sodium and water retentiondiffered somewhat from a similar congestion resulting fromdecreased cardiac output due to myocardial failure. For acomparable reduction in cardiac output, volume retentionwas higher and vascular resistance was lower in patientswith CCP compared to those with myocardial disease.20 Theactivation of the renin–angiotensin–aldosterone andsympathetic systems were found to be similar in CCP andother causes of CHF.20 The atrial natriuretic peptide (ANP)levels in patients with CCP were five times higher than thosein normal controls, but were only one-third of those seenin myocardial disease.20,21 The ANP levels in CCP are lessthan what might be expected on the basis of right atrialpressures alone. This may be primarily due to less distensibleatria caused by the constrictive process in CCP, as ANPrelease is mediated by atrial stretch. The greater salt andwater retention in CCP could be due to smaller increase inANP levels. The ANP hypothesis has been suggested toexplain the lack of pulmonary edema in CCP andtamponade despite very high central pressures.22 ANP isshown to increase capillary hydraulic conductivity, andenhance transcapillary fluid movement.22 To the best of ourknowledge, ANP in restricted cardiomyopathy (RCM) hasnot been studied systematically.
Interestingly, other neurohumoral or autonomic effectsmay also be important. In CCP, severe autonomicdysfunction was reported in all segments of the autonomicnervous system.23 In contrast, autonomic dysfunction wasmuch less in patients with RCM, and involved theparasympathetic efferent pathway.24 Thus, mechanismsother than ANP may also be operative.
Prevention of Constrictive Pericarditis
Nonsteroidal anti-inflammatory drugs, colchicines, andsteroids may reduce the chances of CCP by preventingrecurrence of pericarditis. However, the data on this aspectare scanty and inconclusive. Limited information isavailable regarding even the commonly occurringtubercular pericarditis. The oft-quoted South African trial25
in 240 patients of tubercular pericarditis reported asignificant reduction in the need for repeat pericardio-centesis in patients randomized to receive steroids for 11
weeks. But steroids did not reduce the need forpericardiectomy at 24 months.25 The study has beencriticized as it did not include all-cause mortality, did notanalyze data on the basis of intention-to-treat, and excludedpatients not adhering to the protocol. A re-analysis of thistrial by Ntsekhe et al.26 also did not give definite answers.In the other trial of steroids in patients with establishedCCP, 2 of the 53 patients (4%) treated with prednisoloneand 7 of the 61 patients treated with a placebo (11%) diedfrom pericarditis, and 11 (21%) and 18 (30%), respectively,required pericardiectomy. However, these differences werenot statistically significant.27
Corticosteroids could have an adverse impact on thealready compromised immune status of patients with HIVand tubercular pericarditis. However, in a recent trial of58 patients, steroids led to a significant reduction in all-cause mortality in HIV-positive patients with tubercularpericarditis on 18-month follow-up.16 Interestingly, a re-analysis of the trial data failed to show the benefit ofsteroids.26 Thus, there is still a need for large, multicentric,prospective controlled trials to accurately assess the benefitof adjuvant steroids in tubercular pericarditis.
Role of pericardial drainage: Usually, large effusionsthat are unresponsive to treatment, unexplained effusionsor effusions that last for longer than 3 months warrantpericardiocentesis. Such pericardiocentesis is curative inhalf the cases.1 Whether routine drainage of pericardialeffusion is helpful in preventing the occurrence of CCP hasrarely been investigated.25,28 Strang et al.25 randomizedpatients with tubercular pericarditis to open drainage, orno drainage, and followed them up for up to 24 months toassess their outcome. Open pericardial drainage did notdecrease mortality or improve clinical outcome at follow-up. The routine drainage of pericardial fluid in tubercularpericarditis does not seem to be useful, especially in areaswith a high prevalence. It may be useful in areas wheretuberculosis is an uncommon cause of pericarditis as it mayhelp to obtain pericardial fluid for examination. In a studyof 71 patients with large pericardial effusion (>2 cm onechocardiography) of different etiologies, routine drainagedid not yield significant diagnostic information, and wasnot useful in preventing the occurrence of cardiactamponade or CCP.28
The efficacy of pericardiocentesis in preventing CCP inhemorrhagic effusion has not been tested specifically. Inpurulent pericarditis, adequate drainage and fibrinolyticagents seem to reduce the occurrence of CCP, but againthe data are limited.29 In one study of 6 children withpyopericardium, instillation of intrapericardialstreptokinase at a dose of 10 000 to 15 000 U/kg twice
EDITORIAl-New.p65 11/19/2003, 11:23 PM306

Indian Heart J 2003; 55: 305–309 Kothari et al. Chronic Constrictive Pericarditis 307
daily for a mean of 6 days led to the resolution ofpericarditis, and freedom from CCP on follow-up.29
Diagnosis of CCP
Hemodynamics: The hemodynamic criteria to diagnoseCCP, viz. elevated diastolic pressures, typical pressurewaveforms, and equilibration of pressure of all fourchambers in diastole, have been discussed widely, yet thesemay not be diagnostic in individual cases. In one study, one-fourth of the patients could not be identified by these criteriaof equalization of the right and left ventricular diastolicpressures within 5 mmHg, pulmonary artery systolicpressure less than 50 mmHg, and right ventricular end-diastolic pressure more than one-third of the rightventricular systolic pressure.30 Evidence of dissociation ofintracardiac and intrathoracic pressures, and the presenceof ventricular interdependence (i.e. reciprocal respiratoryvariation in right ventricular/left ventricular pressures)have improved the accuracy. Ventricular interdependencehad 100% sensitivity and 95% specificity for distinguishingCCP from RCM.31 However, in real-world situations severalpatient-related or other factors complicate theinterpretation of laboratory data. The presence of localizedconstriction, associated myocardial dysfunction, othervalve disease, obesity, obstructive airway disease, previousinfarction, and other co-morbidities may influence theirmeasurement. For example, a disproportionately elevatedpulmonary artery wedge pressure was seen in patients withmitral valve disease,32 and even evidence of ventricularinterdependece was reportedly lacking in a patient with alocalized constriction.33
Doppler echocardiography: Doppler echocardiographicparameters have been a very valuable addition to thediagnosis of CCP, and in its differentiation from RCM. Theinitial study that suggested a cut-off of 25% respiratoryvariation in mitral valve inflow velocities was based on thedata from only 7 patients with CCP.34 Subsequently, a largerstudy found that 12% of patients did not show diagnosticchanges in these echocardiographic parameters.35 Severalcaveats need to be considered in individual cases. Forexample, a very high preload may be one reason for theabsence of respiratory variation,36 and a reduction inpreload may be required to bring out the typical pattern,or fluid challenge may be required to diagnose patients withoccult CCP.37 In addition, irregular breathing, irregularheart rate, and short diastolic periods resulting fromtachycardia may cause difficulty in the interpretation ofrespiratory variation of Doppler velocities. The additionaluse of tissue Doppler imaging (TDI) may further enhance
the diagnostic utility of echocardiography. Reduced mitralannular velocity on TDI was reported in RCM, whereasthese parameters were normal in patients with CCP in theinitial reports.38,39
Cross-sectional imaging: The other commonly usedinvestigation for the diagnosis of CCP is cross-sectionalimaging with computerized tomography (CT) and magneticresonance imaging (MRI). The diagnostic hallmark of thesetests is the presence of pericardial thickening with orwithout calcification. While a minimal amount ofpericardial calcification is better detected by CT scan, MRIprovides better soft tissue characterization.40 Pericardialthickening of more than 3 mm is usually taken asabnormal; the usual thickening of the pericardium being1–2 mm. The actual thickness of the pericardium is 0.4–1mm, and this discrepancy between MRI and pathologicalmeasurement is most likely due to a combination of volumeaveraging, chemical shift artifact, motion artifact, and theinclusion of a small amount of pericardial fluid in MRImeasurements.40 Though commonly used, the data on thediagnostic utility of CT/MRI in CCP are not robust. In onesmall study of 29 patients, pericardial thickening on MRIwas found to be 88% sensitive and 100% specific fordetecting CCP.41
It is important to look for additional findings suggestiveof CCP, such as dilated inferior vena cava (IVC), enlargedatria, tubular-shaped ventricles, ascites and pleural effusionon CT/MRI images. Myocardial tagging has also beensuggested as a new method of mitral/tricuspid annularmovement. As normal ventricles contract, there is aslippage between the myocardium and pericardium;however, once pericardial adhesions develop, this slippageis absent and the tag lines passing through the myocardiumand pericardium are not deformed during the cardiaccycle.42 The presence of abnormal diastolic motion of theseptum on cine MRI may also be a useful finding to diagnoseCCP, and to distinguish it from RCM.43
Thus, no single test in isolation should be considereddiagnostic of CCP.
CCP with Normal Pericardial Thickness
It is well known that pericardial thickening may be presentwithout CCP; however, it is not as well recognized that CCPmay occur with normal thickness of the pericardium,which was reported in 18% of 143 patients in a recentlypublished series from the Mayo Clinic. These patients mostcommonly had post-surgical and post-radiation CCP.12
Their clinical characteristics were similar to patients withCCP with a thickened pericardium, and pericardiectomy
EDITORIAl-New.p65 11/19/2003, 11:23 PM307

308 Kothari et al. Chronic Constrictive Pericarditis Indian Heart J 2003; 55: 305–309
was very effective in relieving the symptoms. Microscopyof the excised pericardium was abnormal in all the patients,and included the presence of focal fibrosis, focal calcificationor inflammation. Thus, pericardiectomy should not bedenied to patients with typical clinical and hemodynamicfeatures of CCP but with a normal pericardial thickness onimaging.
Transient CCP
It is conceivable that occasionally, the inflammatoryexudates causing clinical CCP may resolve with no featuresof constriction on follow-up.44,45 Such an event occurredin 15% of patients with tubercular pericarditis in one reportfrom South Korea.46 The resolution occurred within 2months in the majority of patients. Perhaps transient CCPcould occur even more often with purulent pericarditis.However, decisions regarding the management of thesepatients should be based on the overall clinical profile.
Surgical Aspects
Pericardiectomy is the definitive treatment for CCP but itstiming, surgical approach, and preoperative stabilizationneed careful consideration. Various approaches topericardiectomy, i.e. median sternotomy, lateralthoracotomy and bilateral thoracotomy with or withoutthe use of cardiopulmonary bypass, and anterior or totalremoval of the pericardium have been described dependingon patient population or personal preferences. The surgicalmortality of pericardiectomy continues to be high, and hasbeen generally reported to be 6%–12%4,10,47.A subgroup ofpatients with calcific CCP had a mortality of 19% in a recentseries.48 The negative predictors of survival afterpericardiectomy include NYHA class IV, low-voltage ECGcomplex, markedly increased atrial pressure, associatedorgan failure, and post-radiation CCP.10,46,47,49
The optimal timing of pericardiectomy is important.Pericardiectomy is unwarranted too early or too late in thecourse of the disease.1 Long-standing CCP may lead tomyocardial atrophy. Medical therapy may be better in somepatients with CCP having adverse risk factors in whom thesurgical mortality approaches 30%–40%.1 In purulentpericarditis, it is important not to intervene in the subacutestage when a plane of cleavage has not developed clearly.50
The occurrence of low cardiac output following CCP is oftena reflection of the chronicity of CCP and associatedmyocardial atrophy. Acute cardiac dilatation and failurefollowing pericardiectomy may occur unpredictably,51 andis not well characterized. Preoperative inotropes for 48
hours,47 and preoperative digitalization are often used toreduce the chances of postoperative heart failure but theireffectiveness is not clear. The improvement followingpericardiectomy may be rapid but, at times, occurs over afew weeks.1 However, residual CCP or recurrence of CCPdue to epicardial constriction remains a persistent problem.Multiple incisions into the fibrous epicardium whileprotecting the myocardium and coronary arteries are usefulin relieving the constriction (waffle procedure).52 In patientswith extensive calcific plaques, where large plaques do notpermit the development of cleavage planes, wedge incisionsthat reach up to the epicardium help release constriction.47
More recently, ultrasonic decalcification has been used intough calcific lesions.48
Conclusion
CCP continues to intrigue and engage clinicians despiteadvances in knowledge about the disease. No single test ordiagnostic finding should be considered pathognomic ofCCP. A combination of clinical and investigative resultsshould be thoughtfully analyzed to prevent misdiagnosis.Further research is required to understand thepathogenesis, prevention, and optimal treatment of CCP.
References
1. Hoit BD. Management of effusive and constrictive pericardial heartdisease. Circulation 2002; 105: 2939–2942
2. Myers RB, Spodick DH. Constrictive pericarditis: clinical andpathophysiologic characteristics. Am Heart J 1999; 138: 219–232
3. Nishimura RA. Constrictive pericarditis in the modern era: adiagnostic dilemma. Heart 2001; 86: 619–623
4. Bashi VV, John S, Ravikumar E, Jairaj PS, Shyamsunder K,Krishnaswami S. Early and late results of pericardiectomy in 118cases of constrictive pericarditis. Thorax 1988; 43: 637–641
5. Anand SS, Saini VK, Wahi PL. Constrictive pericarditis. Dis Chest1965; 47: 291–295
6. Bhayana JN, Prusty S, Singhal VS, Gupta MP, Sharma SR, PadmavatiS. Surgical treatment of constrictive pericarditis. Indian Heart J 1971;23: 205–211
7. Pedreira Perez M, Virgos Lamela A, Crespo Mancebo FJ, CervantesJL, Fernandez de la Reguera G, Barragan Garcia R. [40 years’experience in the surgical treatment of constrictive pericarditis]. ArchInst Cardiol Mex 1987; 57: 363–373
8. Raffa H, Mosieri J. Constrictive pericarditis in Saudi Arabia. East AfrMed J 1990; 67: 609–613
9. Arsan S, Mercan S, Sarigul A, Atasoy S, Demircin M, Dogan R, et al.Long-term experience with pericardiectomy: analysis of 105consecutive patients. Thorac Cardiovasc Surg 1994; 42: 340–344
10. Ling LH, Oh JK, Schaff HV, Danielson GK, Mahoney DW, Seward JB,et al. Constrictive pericarditis in the modern era: evolving clinicalspectrum and impact on outcome after pericardiectomy. Circulation1999; 100: 1380–1386
11. Oh KY, Shimizu M, Edwards WD, Tazelaar HD, Danielson GK. Surgicalpathology of the parietal pericardium: a study of 344 cases (1993–1999). Cardiovasc Pathol 2001; 10: 157–168
EDITORIAl-New.p65 11/19/2003, 11:23 PM308

Indian Heart J 2003; 55: 305–309 Kothari et al. Chronic Constrictive Pericarditis 309
12. Talreja DR, Edwards WD, Danielson GK, Schaff HV, Tajik AJ, TazelaarHD, et al. Constrictive pericarditis in 26 patients with histologicallynormal pericardial thickness. Circulation 2003; 108: 1852–1857
13. McCaughan BC, Schaff HV, Piehler JM, Danielson GK, Orszulak TA,Puga FJ, et al. Early and late results of pericardiectomy for constrictivepericarditis. J Thorac Cardiovasc Surg 1985; 89: 340–350
14. Hageman JH, D’esopo ND, Glenn WW. Tuberculosis of thepericardium. A long-term analysis of forty-four proved cases. N EnglJ Med 1964; 270: 327–332
15. Przybojewski JZ. Rheumatic constrictive pericarditis. A case reportand review of the literature. S Afr Med J 1981; 5919: 682–686
16. Hakim JG, Ternouth I, Mushangi E, Siziya S, Robertson V, Malin A.Double blind randomised placebo controlled trial of adjunctiveprednisolone in the treatment of effusive tuberculous pericarditis inHIV seropositive patients. Heart 2000; 84: 183–188
17. Leak LV, Ferrans VJ, Cohen SR, Eidbo EE, Jones M. Animal model ofacute pericarditis and its progression to pericardial fibrosis andadhesions: ultrastructural studies. Am J Anat 1987; 180: 373–390
18. Burgess LJ, Reuter H, Carstens ME, Taljaard JJ, Doubell AF. Cytokineproduction in patients with tuberculous pericarditis. Int J Tuberc LungDis 2002; 6: 439–446
19. Sheffield EA. The pathology of tuberculosis. In: Davis PDO (ed).Clinical tuberculosis. London: Chapman and Hall Medical; 1994.pp. 44–54
20. Anand IS, Ferrari R, Kalra GS, Wahi PL, Poole-Wilson PA, Harris PC.Pathogenesis of edema in constrictive pericarditis. Circulation 1991;83: 1880–1887
21. Anand IS, Ferrari R, Kalra GS, Wahi PL, Poole-Wilson PA, Harris PC.Edema of cardiac origin. Studies of body water and sodium, renalfunction, hemodynamic indexes, and plasma hormones in untreatedcongestive cardiac failure. Circulation 1989; 80: 299–305
22. Spodick D. Low atrial natriuretic factor levels and absent pulmonaryedema in pericardial compression of the heart. Am J Cardiol 1989;63: 1271–1272
23. Singh M, Anand IS, Kalra G, Bali HK, Wahi PL. Autonomic functionsin chronic constrictive pericarditis before and after pericardiectomy[Abstr]. Circulation 1989; 80; (Suppl II): 667
24. Singh M, Juneja R, Bali HK, Varma JS. Autonomic functions inrestrictive cardiomyopathy and constrictive pericarditis: acomparison. Am Heart J 1998; 136: 443–448
25. Strang JIG, Kakaza HHS, Gibson DG, Girling DJ, Nunn AJ, Fox W.Controlled clinical trial of complete open surgical drainage and ofprednisolone in treatment of tuberculous pericardial effusion inTranskei. Lancet 1988; ii: 759–764
26. Ntsekhe M, Wiysonge C, Volmink JA, Commerford PJ, Mayosi BM.Adjuvant corticosteroids for tuberculous pericarditis: promising, butnot proven. QJM 2003; 96: 593–599
27. Strang JIG, Kakaza HHS, Gibson DG, Girling DJ, Nunn AJ, Fox W.Controlled trial of prednisolone as adjuvant in the treatment oftuberculous constrictive pericarditis. Lancet 1987; 2: 1418–1422
28. Merce J, Sagrista-Sauleda J, Permanyer-Miralda G, Soler-Soler J.Should pericardial drainage be performed routinely in patients whohave a large pericardial effusion without tamponade? Am J Med1998; 105: 106–109
29. Juneja R, Kothari SS, Saxena A, Sharma R, Joshi A. Intrapericardialstreptokinase in purulent pericarditis. Arch Dis Child 1999; 80:275–277
30. Vaitkus PT, Kussmaul WG. Constrictive pericarditis versus restrictivecardiomyopathy: a reappraisal and update of diagnostic criteria. AmHeart J 1991; 122: 1431–1441
31. Hurrell DG, Nishimura RA, Higano ST, Appleton CP, Danielson GK,Holmes DR Jr, et al. Value of dynamic respiratory changes in left andright ventricular pressures for the diagnosis of constrictivepericarditis. Circulation 1996; 93: 2007–2013
32. Kothari SS, Narula J, Tandon R, Shrivastava S. Cardiac compressionwith mitral stenosis: a haemodynamic challenge. Int J Cardiol 1993;39: 216–218
33. Hasuda T, Satoh T, Yamada N, Sakamaki F, Kyotani S, Nakanishi N,et al. A case of constrictive pericarditis with local thickening of thepericardium without manifest ventricular interdependence.Cardiology 1999; 92: 214–216
34. Hatle LK, Appleton CP, Popp RL. Differentiation of constrictivepericarditis and restrictive cardiomyopathy by Dopplerechocardiography. Circulation 1989; 79: 357–370
35. Oh JK, Hatle LK, Seward JB, Danielson GK, Schaff HV, Reeder GS, etal. Diagnostic role of Doppler echocardiography in constrictivepericarditis. J Am Coll Cardiol 1994; 23: 154–162
36. Oh JK, Tajik AJ, Appleton CP, Hatle LK, Nishimura RA, Seward JB.Preload reduction to unmask the characteristic Doppler features ofconstrictive pericarditis. A new observation. Circulation 1997 18;95;796–799
37. Abdalla IA, Murray RD, Lee JC, White RD, Thomas JD, Klein AL. Doesrapid volume loading during transesophageal echocardiographydif ferentiate constrictive pericarditis from restrictivecardiomyopathy? Echocardiography 2002; 19: 125–134
38. Garcia MJ, Rodriguez L, Ares M, Griffin BP, Thomas JD, Klein AL.Dif ferentiation of constrictive pericarditis from restrictivecardiomyopathy: assessment of left ventricular diastolic velocities inlongitudinal axis by Doppler tissue imaging. J Am Coll Cardiol 1996;27: 108–114
39. Ha JW, Oh JK, Ommen SR, Ling LH, Tajik AJ. Diagnostic value ofmitral annular velocity for constrictive pericarditis in the absence ofrespiratory variation in mitral inflow velocity. J Am Soc Echocardiogr2002; 15: 1468–1471
40. Glockner JF. Imaging of pericardial disease. Magn Reson Imaging ClinN Am 2003; 11: 149–162
41. Masui T, Finck S, Higgins CB. Constrictive pericarditis and restrictivecardiomyopathy: evaluation with MR imaging. Radiology 1992; 182:369–373
42. Kojima S, Yamada N, Goto Y. Diagnosis of constrictive pericarditis bytagged cine magnetic resonance imaging. N Engl J Med 1999; 341:373–374
43. Giorgi B, Mollet NR, Dymarkowski S, Rademakers FE, Bogaert J.Clinically suspected constrictive pericarditis: MR imaging assessmentof ventricular septal motion and configuration in patients andhealthy subjects. Radiology 2003; 228: 417–424
44. Sagrista-Sauleda J, Permanyer-Miralda G, Candell-Riera J, Angel J,Soler-Soler J. Transient cardiac constriction: an unrecognized patternof evolution in effusive acute idiopathic pericarditis. Am J Cardiol1987; 59: 961–966
45. Oh JK, Hatle LK, Mulvagh SL, Tajik AJ. Transient constrictivepericarditis: diagnosis by two-dimensional Doppler echocardio-graphy. Mayo Clin Proc 1993; 68: 1158–1164
46. Yang HS, Song JK, Song JM, Kang DH, Lee CW, Nam GB, et al. Clinicalcharacteristics of constrictive pericarditis diagnosed by echo-Dopplertechnique in Korea. J Korean Med Sci 2001; 16: 558–566
47. Ufuk Y, Kestelli M, Yilik L, Ergunes K, Kanlioglu N, Emrecan B, et al.Recent surgical experience in chronic constrictive pericarditis. TexHeart Inst J 2003; 30: 27–30
48. Ling LH, Oh JK, Breen JF, Schaff HV, Danielson GK, Mahoney DW, etal. Calcific constrictive pericarditis: is it still with us? Ann Intern Med2000; 132: 444–450. Erratum in : Arch Intern Med 2003; 133: 659
49. Tirilomis T, Unverdorben S, von der Emde J. Pericardectomy forchronic constrictive pericarditis: risks and outcome. Eur J CardiothoracSurg 1994; 8: 487–492
50. Gibson DG. Pericardial disease. In: Weatherall DJ, Ledingham JGG,Warell DA (eds). The Oxford textbook of medicine. Oxford: OxfordUniversity Press; 1989. pp. 13.304–13.312.
51. Sunday R, Robinson LA, Bosek V. Low cardiac output complicatingpericardiectomy for pericardial tamponade. Ann Thorac Surg 1999;67: 228–231
52. Heimbecker RO, Smith D, Shimizu S, Kestle J. Surgical technique forthe management of constrictive epicarditis complicating constrictivepericarditis (the waffle procedure). Ann Thorac Surg 1983; 36:605–606
EDITORIAl-New.p65 11/19/2003, 11:23 PM309

310 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
Prudent Diet and Preventive Nutrition From Pediatricsto Geriatrics: Current Knowledge and Practical
Recommendations
Enas A Enas, A Senthilkumar, Hancy Chennikkara, Marc A BjurlinCoronary Artery Disease in Asian Indians (CADI) Research Foundation, and University of Illinois, Chicago, USA
man is what he eats” (German proverb). Foodprovides not only the essential nutrients for life but
also other bioactive compounds for the promotion of healthand the prevention of disease.1–3 The results of 50 years ofintensive worldwide research support the conclusion thatdiet is the major environmental cause of atherosclerosisand cardiovascular diseases (CVD), especially in geneticallysusceptible individuals.4 A high-caloric diet, combined withlimited physical activity, contributes to dyslipidemia, insulinresistance, diabetes, and obesity. All these abnormalitiesincrease the risk of CVD. Over the past few decades, theprevalence of obesity has doubled in adults, and quadrupledin teenagers in the USA. A similar pattern is emerging inIndia, where an epidemic of coronary artery disease (CAD)and diabetes is under way, with no signs of a downturn.Whereas the rates of CAD have declined by 60% in the US,the rates have increased by 300% in India over the past 30years.5 The public and physicians are constantly bombardedwith confusing and conflicting dietary advice. This reviewanalyzes the important recent developments in the fieldsof diet and nutrition for the prevention and treatment ofCVD and diabetes, with particular attention to AsianIndians.
Facts and Myths about Cholesterol, Fats, and Meats
The modern understanding of the role of nutrition in heartdisease began in 1903 when Anitschkow and Chalatowfound that a diet of meat, milk, and egg producedatherosclerosis in rabbits. A decade later, serum totalcholesterol (TC) level was found to be the agent responsible.Contrary to common belief, the contribution of dietarycholesterol to serum TC is small (<10 mg/dl). The averageadult on a western diet consumes about 300 mg ofcholesterol daily, which is about the size of 3 toothpicks,and hardly 3 cal. Nonetheless, high intakes of dietarycholesterol increase the number of circulating low-density
lipoprotein (LDL) particles.6 Dietary cholesterol is foundonly in the animal kingdom; 3 oz of beef, lamb, or porkcontains 75 mg of cholesterol. Most of the cholesterol inpoultry is in the skin, and some in dark meat. One cup ofmilk has 33 mg, 2 egg yolks have 560 mg, and 100 g ofbrain has 2000 mg of cholesterol. One hundred grams ofshrimp contain about 150 mg of cholesterol but <1 g ofsaturated fat. The recommended dietary intake ofcholesterol and various types of fat is given in Table 1.1,6–12
The contribution of dietary saturated fat to serum TC isvery large—10 times greater than that of dietarycholesterol. Fats are substances consisting of a combinationof fatty acids, which are classified as saturated (SAFA),monounsaturated (MUFA), polyunsaturated (PUFA), andtransunsaturated (TRAFA), depending on the location andnumber of double bonds.13 It is not often appreciated thatthe quality of the fat is more important than the quantityof fat consumed. The National Cholesterol EducationProgram (NCEP) recommends an intake of total fat of 25%–35%, MUFA up to 20%, PUFA up to 10%, and SAFA <7%of the total energy14 (Table 1). Although many affluentAsian Indians consume 50% of energy from fat, the averageconsumption is about half this amount (20%–25% of theenergy). Increasing the MUFA intake to 20%, and total fatintake to 35% of the energy appears to be appropriate forAsian Indians because of the beneficial effects on high-density lipoprotein (HDL) and triglycerides (TG) . The NCEPdietary guidelines for PUFA and SAFA seem appropriate forAsian Indians without any modification.
Saturated fatty acids, the arch villain ofatherosclerosis: Excessive consumption of SAFA is theprincipal dietary culprit contributing to elevated serumTC level, which is the primary determinant ofatherosclerosis.15,16 Differences in CAD mortality worldwideare explained by differences in SAFA intake and theresulting serum TC levels in 40 countries, except for France,Finland, and India.16–18 Intake of SAFA suppresses the LDL-receptor activity and decreases the clearance of LDL fromthe circulation, resulting in a marked elevation of its level.19
Review Article
Correspondence: Dr Enas A Enas, CADI Research Foundation, 1935, GreenTrails, Lisle IL, 60523 USA. e-mail: [email protected]
“A
IHJ-584-03.p65 11/19/2003, 11:27 PM310
Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 311
SAFA raises the serum TC level thrice as much as PUFA,and MUFA lowers it. For example, substitution of 20% ofthe daily energy intake of carbohydrate by SAFA increasesthe TC level by 30 mg/dl, whereas PUFA and MUFA lower itby 10 mg/dl.13 Most of this increase is due to an increase inLDL. Although some increase in HDL also occurs, it is notsufficient to offset the atherogenicity and thrombogenicityresulting from marked elevation of LDL.6,20
Our diet contains SAFA of different chain lengths withvarying atherogenic properties. According to their chainlengths, SAFA can be classified as short chain (4:0–6:0),medium chain (8:0–10:0), long chain (12:0–18:0), andvery long chain (20:0–24:0) fatty acids. Stearic acid(C18:0) is desaturated to oleic acid soon after its absorption,and hence does not raise the TC level.21,22 Therefore, its useneed not be restricted and, in fact, it can be recommended.23
SAFA with chain lengths of 12–16 have the mostcholesterol-raising properties.24 These are lauric acid(C12:0), myristic acid (C14:0), and palmitic acid (C16:0).These 3 fatty acids account for only 25%–30% of the totaldietary fat but 60%–70% of SAFAs in western diets.24
Palmitic acid is the most common fatty acid in the humandiet, and the principal SAFA in both animal fats and palmoil. In a study conducted in a metabolic ward, 40% ofenergy as palmitic acid raised the TC by 25 mg/dl v. 15 mg/dl with lauric acid.21 Myristic acid is the most powerfulcholesterol-raising SAFA, and increases the TC level 50%more than palmitic acid. Replacement of 20% of energyfrom carbohydrate with myristic acid raises the blood TClevel by 46 mg/dl, compared to 30 mg/dl with palmitic acid,and 20 mg/dl with lauric acid.25 Most of the rise in the TClevel is due to an increase in LDL, the respective contributionfrom HDL being 16 mg/dl, 8 mg/d and 12 mg/dl.25 Themajor sources of myristic acid are butter and tropical oils(Table 2).6,20–25 The TC-raising potential of lauric acid is 33%less than that of palmitic acid, and it is the principal SAFAin coconut and palm kernel oils, both containing 48%.23–25
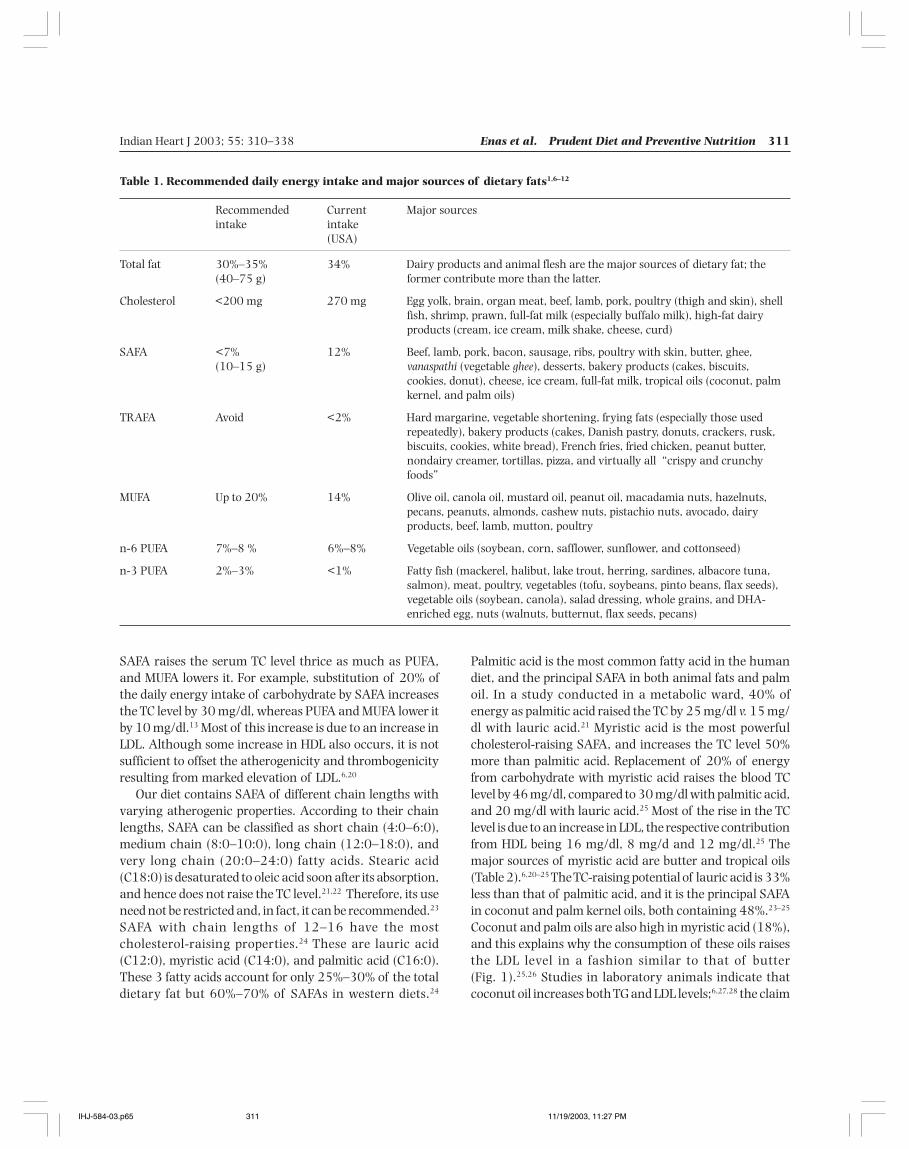
Coconut and palm oils are also high in myristic acid (18%),and this explains why the consumption of these oils raisesthe LDL level in a fashion similar to that of butter(Fig. 1).25,26 Studies in laboratory animals indicate thatcoconut oil increases both TG and LDL levels;6,27,28 the claim
Table 1. Recommended daily energy intake and major sources of dietary fats1,6–12
Recommended Current Major sourcesintake intake
(USA)
Total fat 30%–35% 34% Dairy products and animal flesh are the major sources of dietary fat; the(40–75 g) former contribute more than the latter.
Cholesterol <200 mg 270 mg Egg yolk, brain, organ meat, beef, lamb, pork, poultry (thigh and skin), shellfish, shrimp, prawn, full-fat milk (especially buffalo milk), high-fat dairyproducts (cream, ice cream, milk shake, cheese, curd)
SAFA <7% 12% Beef, lamb, pork, bacon, sausage, ribs, poultry with skin, butter, ghee,(10–15 g) vanaspathi (vegetable ghee), desserts, bakery products (cakes, biscuits,
cookies, donut), cheese, ice cream, full-fat milk, tropical oils (coconut, palmkernel, and palm oils)
TRAFA Avoid <2% Hard margarine, vegetable shortening, frying fats (especially those usedrepeatedly), bakery products (cakes, Danish pastry, donuts, crackers, rusk,biscuits, cookies, white bread), French fries, fried chicken, peanut butter,nondairy creamer, tortillas, pizza, and virtually all “crispy and crunchyfoods”
MUFA Up to 20% 14% Olive oil, canola oil, mustard oil, peanut oil, macadamia nuts, hazelnuts,pecans, peanuts, almonds, cashew nuts, pistachio nuts, avocado, dairyproducts, beef, lamb, mutton, poultry
n-6 PUFA 7%–8 % 6%–8% Vegetable oils (soybean, corn, safflower, sunflower, and cottonseed)
n-3 PUFA 2%–3% <1% Fatty fish (mackerel, halibut, lake trout, herring, sardines, albacore tuna,salmon), meat, poultry, vegetables (tofu, soybeans, pinto beans, flax seeds),vegetable oils (soybean, canola), salad dressing, whole grains, and DHA-enriched egg, nuts (walnuts, butternut, flax seeds, pecans)
IHJ-584-03.p65 11/19/2003, 11:27 PM311
312 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
that lauric acid does not raise TC is not supported by scientificdata.21,23 Recent studies have shown that caprylic acid (C:8)and capric acid (C:10) raise the LDL level to about 50% thatof palmitic acid, and raise the TG level.6,23–25 Coconut oilcontains 14% of these two cholesterol-raising SAFA.
Replacing 5% of the daily energy intake of SAFA withMUFA and PUFA could reduce the risk of CAD by 42%.29
Therefore, substituting MUFA and PUFA for SAFA andTRAFA is more effective in lowering the risk of CAD than
simply reducing the total amount of fat.30 Since 1970, thetotal fat intake decreased from 42% to 34%, and SAFA from18% to 12% in the USA, as a result of nationwide changesin dietary habits.2,31 This change in dietary fat intake isprimarily responsible for the decrease in serum TC level from220 to 200 mg/dl in the US population. This decrease inTC level is principally responsible for the dramatic reductionin CAD, during a period when the rates of obesity anddiabetes doubled in Americans.32
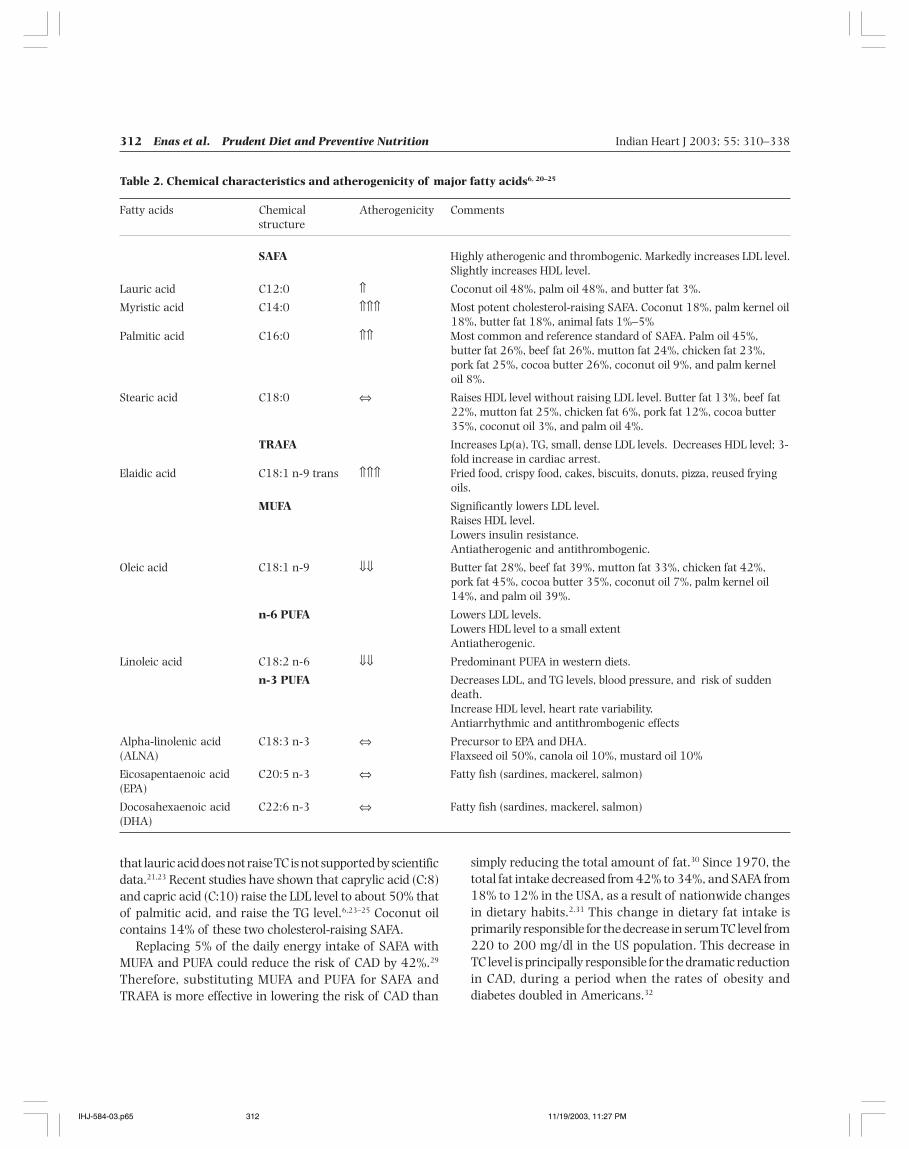
Table 2. Chemical characteristics and atherogenicity of major fatty acids6, 20–25
Fatty acids Chemical Atherogenicity Commentsstructure
SAFA Highly atherogenic and thrombogenic. Markedly increases LDL level.Slightly increases HDL level.
Lauric acid C12:0 ⇑ Coconut oil 48%, palm oil 48%, and butter fat 3%.
Myristic acid C14:0 ⇑⇑⇑ Most potent cholesterol-raising SAFA. Coconut 18%, palm kernel oil18%, butter fat 18%, animal fats 1%–5%
Palmitic acid C16:0 ⇑⇑ Most common and reference standard of SAFA. Palm oil 45%,butter fat 26%, beef fat 26%, mutton fat 24%, chicken fat 23%,pork fat 25%, cocoa butter 26%, coconut oil 9%, and palm kerneloil 8%.
Stearic acid C18:0 ⇔ Raises HDL level without raising LDL level. Butter fat 13%, beef fat22%, mutton fat 25%, chicken fat 6%, pork fat 12%, cocoa butter35%, coconut oil 3%, and palm oil 4%.
TRAFA Increases Lp(a), TG, small, dense LDL levels. Decreases HDL level; 3-fold increase in cardiac arrest.
Elaidic acid C18:1 n-9 trans ⇑⇑⇑ Fried food, crispy food, cakes, biscuits, donuts, pizza, reused fryingoils.
MUFA Significantly lowers LDL level.Raises HDL level.Lowers insulin resistance.Antiatherogenic and antithrombogenic.
Oleic acid C18:1 n-9 ⇓⇓ Butter fat 28%, beef fat 39%, mutton fat 33%, chicken fat 42%,pork fat 45%, cocoa butter 35%, coconut oil 7%, palm kernel oil14%, and palm oil 39%.
n-6 PUFA Lowers LDL levels.Lowers HDL level to a small extentAntiatherogenic.
Linoleic acid C18:2 n-6 ⇓⇓ Predominant PUFA in western diets.
n-3 PUFA Decreases LDL, and TG levels, blood pressure, and risk of suddendeath.Increase HDL level, heart rate variability.Antiarrhythmic and antithrombogenic effects
Alpha-linolenic acid C18:3 n-3 ⇔ Precursor to EPA and DHA.(ALNA) Flaxseed oil 50%, canola oil 10%, mustard oil 10%
Eicosapentaenoic acid C20:5 n-3 ⇔ Fatty fish (sardines, mackerel, salmon)(EPA)
Docosahexaenoic acid C22:6 n-3 ⇔ Fatty fish (sardines, mackerel, salmon)(DHA)
IHJ-584-03.p65 11/19/2003, 11:27 PM312

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 313
Transfatty acids (TRAFA)—the hardened fat thathardens arteries fast: TRAFA is formed during the partialhydrogenation of vegetable oils, a process that converts oilsinto solid or semisolid fats for subsequent use in foodproducts. This process not only improves the texture andfirmness but also markedly increases the shelf-life of foodby minimizing oxidative spoilage.33 Elaidic acid (n-9 trans18:1) is the principal TRAFA, although several other transisomers are also formed.23 Such oils are used in commercialbaked goods, and for cooking in most fast-food chains inwestern countries.34 Perhaps an equally important andoften neglected cause of TRAFA formation is thespontaneous hydrogenation of vegetable oils during deep-frying.35 Very small amounts of TRAFA are also found inbeef and dairy products (Table 1).
Consumption of TRAFA has a greater adverse effect onlipoproteins than that of SAFA.36 Whereas both SAFA andTRAFA increase LDL levels considerably, TRAFA alsodecreases HDL levels, thereby increasing the TC/HDL ratio,the single best lipid-related risk factor for CAD.37 Replacing9% of calories from SAFA with TRAFA results in a 20%decrease in HDL level.38 Other important adverse effects ofTRAFA consumption include increases in lipoprotein(a)(Lp[a]), TG, and small, dense LDL levels,34,38–44 as well asincreased platelet aggregation, endothelial dysfunction,and sudden death.34 TRAFA are stronger predictors of CADand diabetes than SAFA and carbohydrates.1,45–47 In theNurses’ Health Study, women in the highest versus thelowest quartile of TRAFA consumption had a 50% higherrisk of CAD.48 It is estimated that a substitution of 2% ofcalories from TRAFA with MUFA and PUFA results in a 53%reduction in CAD risk—a risk double that of substitutionof calories from SAFA.29 TRAFA consumption also markedlyincreases the postprandial insulin response in diabeticpatients.49 Replacing 2% of energy from TRAFA with PUFA
would lead to a 40% reduction in diabetes.45 SAFA caloriesshould not be replaced by TRAFA calories; doing so is likejumping from the frying pan to the deep-fat fryer.50
The average consumption of TRAFA in the USA andEurope is low (<2% of energy or 11–27 g/day).51,52
However, TRAFA accounts for about 5% of fat in Americandiets, and 5% of fat stored in adipose tissue.33,50,53 Buttercontains 60% SAFA, whereas stick margarine contains16% TRAFA. The tub or soft margarine contains only 2 gof TRAFA per 15 ml. Therefore, the fat-spread of choiceremains soft margarine;54 olive oil may be an even bettersubstitute. Although many margarines and shorteningspreviously contained up to 50% of TRAFA, in most westerncountries, these products currently have a low TRAFAcontent due to recent manufacturing changes.47 However,frying fats used in fast-food outlets still contain over 30%of TRAFA. French fries sold in these outlets provide 7–8 gof TRAFA per portion. About one-third of TRAFA in thewestern diet comes from French fries, fried chicken, pizza,and cookies.
The TRAFA consumption is likely to be high in AsianIndians because deep-frying is a favorite mode of cookingat home as well as in restaurants. Deep-frying is associatedwith spontaneous hydrogenation and TRAFA formation,and repeated re-use of oils previously used for deep-fryingmay further increase the TRAFA content. These practicesappear to be the norm rather than the exception, and maybe of enormous public health importance, especially withregard to elevated Lp(a) levels, and high rates of CAD inthis population. There is an urgent need to ascertain anddisseminate the TRAFA content of vanaspathi (vegetableghee) and frying oils used in India. As of today, we are notaware of any industrial manufacturing changes aimed atlowering the TRAFA content of Indian foods, as has beendone in western countries.
MUFA, the good fat that raises the good cholesterol:Diets high in MUFA (oleic acid C18:1) make LDL resistantto oxidation, restore LDL-receptor activity, and markedlylower LDL levels. Substitution of 20% of energy fromcarbohydrates with MUFA decreases TC by 10 mg/dl. Thereduction in TC is 3-fold higher when MUFA replaces SAFA.For example, TC decreases by 40 mg/dl when 20% of energyfrom SAFA is replaced with MUFA.13, 25 The effect on small,dense LDL is even greater. Other beneficial effects of MUFAinclude the favorable influence on blood pressure,endothelial activation, inflammation, and thrombogenesis.
A higher intake of MUFA lowers insulin resistanceand diabetes, unlike SAFA and TRAFA, which increaseit.45,49,55–60 Consumption of MUFA offers the unique
Fig. 1. Predicted change in HDL and LDL when all fat in Dutch diet is replacedby a particular oil.
IHJ-584-03.p65 11/19/2003, 11:27 PM313

314 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
advantage of effectively lowering LDL levels withoutlowering HDL or raising TG levels. Individuals with low HDLlevels have a high risk of CAD.61,62 Subjects with high TG,especially those with the metabolic syndrome and diabetes,are highly sensitive to the TG-raising effects of a highcarbohydrate load. A high carbohydrate diet is associatedwith highly atherogenic, small, dense LDL particles, whilehigh-fat diets are associated with less atherogenic, buoyantLDL particles. Thus, replacing SAFA with MUFA is moreeffective in preventing CAD than reducing the total fatintake, especially in Asian Indians, a population with highrates of prevalence of the metabolic syndrome and diabetes.The NCEP III has recommended up to 20% of total caloriesfrom MUFA (Table 1). This recommendation seemsparticularly appropriate for Asian Indians.32
In Mediterranean countries, the high intake of MUFAin the form of olive oil is inversely related to CAD.15 TheNurses’ Health Study and other studies of almost 300 000Americans showed that a diet rich in MUFA in the form ofcanola oil also reduces the risk of CAD.29,63,64 Contrary tocommon belief, energy-controlled, high-MUFA diets do notpromote weight gain, and are more acceptable than low-fat diets for weight loss in obese subjects. The addition ofMUFA should be at the expense of SAFA and carbohydrates.Since all fats are high in calories (9 cal/g), failure to decreasethe energy from carbohydrates and SAFA would invariablyresult in weight gain, and mitigate most of the beneficialeffects of MUFA.
Meat and dairy products, which are also rich in SAFA,provide most of the MUFA in western diets. Olive oil andcanola oil are good sources of MUFA (Table 3),65 canola oilappears to be even better as it contains less SAFA and morePUFA, especially alpha-linolenic acid (ALNA). Mustard oilis high in MUFA but also high in erucic acid, which is knownto have toxic effects on the heart. Canola oil is geneticallyengineered mustard oil without erucic acid. Nuts andavocado are excellent sources of MUFA and arerecommended, provided the quantity is no more than 50–100 g/day.66 Groundnut (peanut) products are a rich sourceof MUFA; they are inexpensive and widely available inIndia.67
PUFA, another healthy substitute for SAFA: There are2 series of PUFA that are deemed essential. Linoleic acid(C18:2 n-6) is the predominant omega-6 or n-6 PUFA. Thepredominant (parent) omega-3 or n-3 PUFA is linolenic acid(18:3 n-3).23 Linoleic acid increases the fecal excretion ofsteroids, and inhibits the hepatic synthesis of apo B-containing lipoproteins. Replacing SAFA with PUFAreverses the suppression of LDL-receptor activity bycholesterol-raising SAFA (similar to that of MUFA).6,68
Substituting 20% of energy from SAFA with PUFAdecreases the TC level by 40 mg/dl. Most of the reductionis in LDL, and the number of apo B particles.23,25 PUFA doesnot raise the TG level, and sometimes lowers it.69 The twoundesirable effects of PUFA are increased susceptibility forperoxidation, and lowering of the HDL level.10,70–72 HDLlevels are reduced by about 1% for every 2% of MUFA orSAFA energy substituted with PUFA.6,70–72
The substitution of PUFA for SAFA calories has played amajor role in reducing TC levels and CAD in the USA. TheCAD mortality rate declined by 60% in the past 3 decadesin the USA.73 About a third of the decline in CAD rates isattributed to a 6%–8% decrease in the serum TC level inthe population; this, in turn, was due to an increase in theconsumption of PUFA from 3% to 6%, and a decrease inSAFA consumption from 16% to 12% of the energy. Theimportance of PUFA is further underscored by the markeddifferences in PUFA consumption, which parallel the 4-folddifference in CAD rates between France and Finland.16
Vegetable oils, such as soybean, corn, safflower, sunflower,and cottonseed, are the primary sources of n-6 PUFA(Table 1). Their average consumption in the western diet is6%–8% of energy, (17 g/day for men, and 12 g/day forwomen).74
Contrary to previous fears, n-6 PUFA do not antagonizethe anti-inflammatory effects of n-3 PUFA nor do they raisethe risks of breast, colorectal, or prostate cancer in
Table 3. MUFA, PUFA, and SAFA content (%) in 100 g ofvarious cooking oils65
MUFA PUFA SAFA
Sunflower oil, high oleic (>70%) 84 4 10Safflower oil, high oleic (>70%) 75 14 6Olive oil 74 8 14Almond oil 70 17 8Mustard oil 59 21 12Canola oil 59 30 7Cod liver oil 47 23 23Peanut oil 46 32 17Sunflower oil, linoleic (<60%) 45 40 10Sesame oil 40 42 14Rice bran oil 39 35 20Palm oil 37 9 49Cocoa butter 33 3 60Corn oil 24 59 13Soybean oil 23 58 14Walnut oil 23 63 9Sunflower oil, linoleic (>60%) 20 66 10Cottonseed oil 18 52 26Safflower oil, linoleic (>70%) 14 75 6Palm kernel oil 11 2 82Coconut oil 6 2 92
IHJ-584-03.p65 11/19/2003, 11:27 PM314

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 315
humans.75,76 However, a very high n-6 PUFA to n-3 PUFAratio may increase the thrombogenicity through increasedproduction of arachidonic acid and thromboxane A
2. This
is because linoleic and linolenic acids use the same set ofenzymes for desaturation and chain elongation.77 An n-6PUFA to n-3 PUFA ratio of 3:1 appears to be optimum.78–80
Japan, which has one of the highest rates of fishconsumption, has recently changed the recommendationof this ratio from 4:1 to 2:1;81 this ratio may be advisablefor vegetarians).9
Fish, a tasty way to prevent sudden death: Fish do notdie from myocardial infarction (MI), and populations thatconsume large amounts of marine foods have a lowprevalence of CVD death.82–99 Replacing high-fat meat withfish is also associated with a decreased risk of CAD. Theresults of several large studies show that one or two fishmeals per week are associated with a 30%–50% reductionin sudden death.84,95,98 In a meta-analysis of 11 prospectivestudies involving 116 764 individuals, fish consumptionwas inversely related to CAD death. This report suggeststhat 40–60 g/day of fish consumption is optimal, andresults in a 40%–60% risk reduction.100 Greater intake hasno additional benefits, and suggests a threshold effect.101,102
However, a recent large study of 5103 women with diabetesshowed a dose–response relationship. Consumption of fish1–3 times per month was associated with a 40% riskreduction, and a 64% risk reduction was seen among thosewho consumed fish >5 times per week.102,103 The benefit isseen in people with and without prior heart disease.104
These benefits persist as long as the fish consumption iscontinued.
Fish is a tasty food that contains many essentialnutrients, such as selenium, iodine, vitamin D, and n-3PUFA.97,105 The beneficial effects of fish are largely mediatedthrough n-3 PUFA, which displace arachidonic acid fromplatelet phospholipid stores, thereby reducing the availablesubstrate for thromboxane A
2 synthesis.106,107 The principal
ef fects of n-3 PUFA are antithrombogenic andantiarrhythmic, whereas that of n-6 PUFA isantiatherogenic.108–117 The serum levels of n-3 PUFA areinversely related to sudden death.66,82,89,93,95,118–121 Theconsumption of n-3 PUFA decreases blood pressure, andhomocysteine level, increases HDL level, and improveshemostatic factors (Table 4).10,75,122–132 A 30%–50%reduction in TG can be achieved by taking 3–5 g/day of n-3 PUFA.130
The major n-3 PUFA in fish oils are eicosapentaenoicacid (EPA) (20:5 n-3) and docosahexaenoic acid (DHA)(22:6 n-3); together they constitute 26% of fish oil fattyacids.23 The benefits from n-3 PUFA are greater with DHA
and EPA found in fatty fish, shellfish, and marine mammalsthan with ALNA found in canola oil, soybean oil, andwalnut.108 It is important to distinguish between lean andfatty fish for cardioprotection, because the content of n-3PUFA is highest in fatty fish.119 Fatty fish, such as mackerel,sardine, and salmon, are widely available and inexpensive.Heating is associated with significant loss of n-3 PUFA.9
Frying fish is associated with an even greater loss of EPAand DHA, and may be particularly harmful if fried inSAFA.133 The current intake of DHA and EPA is only 200mg/day, and needs to be increased 5-fold to meet the dietarygoals.11
Both plant-based (ALNA), and fish-based (EPA and DHA)supplements have shown benefits in secondaryprevention.89,90,134,135 In one such trial of 605 French menrecovering from an MI, there was a 70% reduction in totaland cardiac death during a follow-up of 27 months in thosewho received an experimental “Mediterranean diet” usingcanola oil-based margarine, enriched with n-3 PUFA.136 Inanother large randomized study of 11 324 survivors of arecent MI, there was a 20% reduction in total deaths, 30%reduction in CVD deaths, and 45% reduction in suddendeaths among those who received n-3 PUFA 1g/day.90 Thetotality of the data suggest that n-3 PUFA can be consideredas the best antiarrhythmic agent and antifibrillatorytreatment.106,117,133,135 Cardiologists and their patientsshould pay serious attention to this new paradigm in thediet–heart hypothesis, and increase the intake of fish andfish oil.133,137
The amount of n-3 PUFA necessary for cardioprotectionis surprisingly low. The current recommendation is to take2–3 fish meals per week (200–300 g/week of fish). A less
Table 4. Omega-3 fatty acids and CVD10,75,122–132
• Decrease the risk of ventricular fibrillation and sudden deathIncrease cell membrane PUFAFavorably alter cardiac ion channel function and actionpotentialDecrease the ventricular fibrillation thresholdIncrease heart rate variability
• Decrease the risk of stroke and MI• Decrease platelet reactivity, aggregability, and the risk of
thrombosis• Reduce monocyte reactivity• Reduce inflammatory cytokines and response• Improve endothelial function• Reduce the expression of vascular adhesion molecules• Markedly lower the TG and remnant lipoprotein levels• Decrease the growth of atherosclerotic plaques• Decrease homocysteine levels• Improve insulin sensitivity and reduce the risk of diabetes• Slightly lower blood pressure
IHJ-584-03.p65 11/19/2003, 11:27 PM315

316 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
attractive alternative is to consume 1000 mg/day of n-3PUFA (contained in 3000 mg of fish oil capsules).10,11,74,96,133
The current intake of n-3 PUFA in the US is 1600 mg/dayor 0.7% of the calories, which is about half therecommendation.11 Fish is more beneficial than fish oil, butthe latter may be required in most patients with CAD toobtain the required amount of n-3 PUFA.90 Patients withCAD should consume about 1800 mg/day of n-3 PUFA(DHA and EPA) as the best insurance against sudden death.
Alpha-Linoleic acid (ALNA)—the n-3 PUFA of theplant kingdom: There is no DHA and EHA in a vegetariandiet. Vegetarians derive their n-3 PUFA almost exclusivelyfrom ALNA, which is also the major type of n-3 PUFA inomnivores.138 There is increasing evidence for thecardioprotective effects of ALNA, albeit less than EPA andDHA.139 In a large study involving 43 700 men, increasedintake of ALNA reduced the risk of MI by 60%.64 A similarrisk reduction was also observed in the Nurses’ HealthStudy and Multiple Risk Factor Intervention Trial(MRFIT).93,140 Some vegetable oils are high in ALNA(flaxseed oil 50%, canola oil 10%, mustard oil 10%, soybeanoil 7%) while others are low (groundnut oil <0.5%).141
Walnuts are a rich source of ALNA; small concentrationsare found in green leafy vegetables, corn oil, almonds,hazelnuts, cereals, pulses, millets, and spices.78,142 Walnutsand canola oil account for most of the ALNA in the westerndiets.78,101 The recommended intake of ALNA is 2% ofenergy but the current intake in the USA is 0.6% ofenergy.11,74
ALNA is readily converted to EPA, and more slowly toDHA; the latter being the major component of phospholipidmembranes of the brain and retina.78 The beneficial effectsof ALNA are less than half that of DHA and EPA, becausethe conversion of ALNA to the more active longer-chainmetabolites is inefficient: <5%–10% for EPA, and 2%–5%for DHA.80,141,143 This explains why vegetarians have lowerlevels of n-3 PUFA than omnivores, and also higher plateletaggregability.77 Since the biological effects of plant n-3PUFA are significantly lower than marine n-3 PUFA, therequirements may be higher (3% of energy) for vegetariansthan for nonvegetarians.9,74,77
Protein: Americans eat 80–90 g/day of protein, which istwice the daily requirement, and most of this comes frommeat, which is also high in SAFA. Up to 25% of daily energyfrom protein (but not more than 100 g/day) is permissibleif the major source of protein is plant-based. Nuts areimportant sources of plant protein along with soy, bran,beans, and legumes. Substituting protein for carbohydratesincreases HDL and lowers TG levels.144,145 In a meta-analysis
of 38 controlled human clinical trials, consumption of soyprotein (47 g/day) was associated with a significant 13%decrease in LDL, 10% decrease in TG, and a 2% increase inHDL levels.146 This led to FDA approval for the use of foodlabels for the health claim that soy protein can reduce therisk of heart disease.
Meat: Although meat contains a significant amount ofSAFA, almost half the SAFA is stearic acid, which does notraise TC levels. In addition, meat contains up to 45% ofcholesterol-lowering MUFA. Furthermore, lean meat hasmuch less SAFA than fatty cuts of meat (Table 5).6,147 Leanbeef is an excellent source of protein and MUFA, and hasless SAFA than chicken (dark meat); 6 oz of lean beefcontains 3.0 g of SAFA v. a chicken thigh which contains5.2 g of SAFA (the term loin or round signifies lean meatwhereas prime or rib signifies fat cuts with very high SAFAin the USA). Chicken and lean beef (not fatty meat) havesimilar ef fects on plasma lipoproteins, and areinterchangeable in a healthy diet.30,148,149
Glycemic Load: A Potent Predictor of theMetabolic Syndrome and Diabetes
The source, nature, and amount of carbohydrates have aprofound influence on postprandial glycemia, which in turnis directly associated with the risk of CAD in patients withdiabetes.6,150,151 Foods containing the same amount ofcarbohydrate (carbohydrate exchange) may have up to a5-fold difference in glycemic impact, depending on thedifferences in the digestion and absorption.152,153 Theglycemic index is an extension of the fiber hypothesis, andwas proposed in 1981 as a physiological system for theclassification of carbohydrate-containing foods.154,155
Table 5. Atherogenic, antiatherogenic, and neutral fattyacids in selected meats and cooking oils6,147
Fat Total Cholesterol- Stearic Oleic acid:SAFA raising acid: cholesterol-
(%) SAFA cholesterol lowering (%) neutral MUFA (%)
SAFA (%)
Beef fat 51 29 22 39Mutton fat 54 29 25 33Chicken fat 30 24 6 42Pork fat 39 27 12 45Butter fat 66 53 13 28Cocoa butter 61 26 35 35Coconut oil 92 89 3 7Palm kernel oil 74 71 3 14Palm oil 50 46 4 39
IHJ-584-03.p65 11/19/2003, 11:27 PM316

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 317
Carbohydrate classified by glycemic index, in contrast to itstraditional classification as either simple or complex, is abetter predictor of CAD in epidemiological studies.156 Theglycemic index is a scientific measure of the glycemicresponse to various foods, and is obtained from publishedfood tables. The hierarchy of the glycemic index begins withbeans, lentils, rice, spaghetti, potatoes, white bread (withrefined flour), and refined grain cereals.150 A high glycemicindex indicates a lower quality of carbohydrate associatedwith low HDL levels, and low rates of satiety. 157,158 Fruits,nonstarchy vegetables, parboiled rice, and legumes have alow glycemic index.159 The glycemic index of potato is102%, white bread 100%, whereas that of apple is 55%,and broccoli 13%. Glycemia observed after consuming driedpeas is only one-third that of an equivalent amount ofpotatoes. Since peas are also high in fiber, theirconsumption needs to be encouraged, especially in patientswith diabetes.153,160
Glycemic load is the product of the glycemic value ofthe food and its carbohydrate content (per serving) dividedby 100. For example, carrot has a high glycemic index buta low glycemic load (Table 6).152,161 The overall daily dietaryglycemic load is calculated by adding the glycemic loads ofall the different foods consumed in a given day. Accordingly,the glycemic load can be decreased by reducing the amountof carbohydrate intake and/or by consuming foods with alow glycemic index.162 In addition to the quality andquantity of carbohydrates consumed, the glycemic load alsorepresents diet-induced insulin demand.163,164 PAI-1 levelsare significantly increased with high glycemic load, anddecreased with low glycemic load.165
Dietary carbohydrates drive TG much more than dietaryfat.6 A high glycemic load produces only mild incrementsin TG levels in individuals with normal TG levels butmarked elevation in those with fasting lipemia and/orobesity.6,166–168 A low HDL level is a strong risk factor forCAD, even when the TC level is not elevated.169,170 A highglycemic load produces a low HDL, particularly whensubstituted for MUFA or PUFA.23,157,166,171–179 In a prospectivestudy of 75 521 women followed up for 10 years, those inthe highest quintile of glycemic load had double the risk ofCAD after adjustment for age, smoking status, total energyintake, and other risk factors (p<0.0001).156
More importantly, a glycemic load promotes diabetes,especially in those with insulin resistance.156,161,180–183
(Fig. 2)183 This is particularly true for refined carbohydrates,sweets, white bread, and potatoes.45,156,183,184 Thus, a highglycemic load may be considered a risk factor of equalimportance as high SAFA diet in precipitating diabetes. Alow glycemic load can reduce insulin secretion in patients
Table 6. Glycemic index of common foods152,161
Glycemic Serving Carbohydrate Glycemicindex size per serving load
Basmati rice 58 150 g 38 22Brown rice (South India) 50 150 g 33 16Parboiled rice (Canada) 48 150 g 36 26White rice (Uncle Ben’s) 45 150 g 36 16Curry rice (Japan) 67 150 g 61 41Jasmine rice 109 150 g 42 46White bread 70 2 slices 30 21Uppuma 18 150 g 33 6Upittu 68 150 g 42 28Chapati 76 60 g 30 23Dosai 77 150 g 39 30Idli 77 250 g 52 40Poori 70 150 g 41 28Pongal 68 250 g 52 35Millet/ragi 104 70 g 50 52Barley 43 150 g 37 16Tapioca 70 250 g 18 12Kellogg’s cornflakes 81 30 g 26 24Milk, full-fat 27 250 g 12 3Yogurt 36 200 g 9 3Orange juice(reconstituted USA) 57 250 ml 26 15Pineapple 59 120 g 13 7Plums 39 120 g 12 5Prunes 29 60 g 33 10Raisins 64 60 g 44 28Cantaloupe 65 120 g 6 4Plantain, green 38 120 g 21 8Banana, unripe 70 120 g 45 31Banana 53 170 g 25 13Strawberry jam 51 30 g 20 10Laddu 27 50 g 31 8Black-eyed beans 42 150 g 30 13Chickpeas 10 150 g 30 3Kidney beans (rajmah) 13 150 g 25 3Lentils 30 150 g 17 5Lima beans 32 150 g 30 10Pinto beans 39 150 g 26 10Soy beans 15 150 g 6 1Sweet corn 59 80 g 18 11Green peas 39 80 g 7 3Carrot 47 80 g 6 3Beet root 64 80 g 7 5Potato 85 150 g 30 26Split peas 32 150 g 19 6Snicker bar 55 60 g 35 19Pizza Hut supreme
pan pizza 36 100 g 24 9Coca Cola 58 250 g 26 14Instant noodles 47 180 g 40 19Macaroni 45 180 g 49 22Spaghetti 68 220 g 27 19
IHJ-584-03.p65 11/19/2003, 11:27 PM317

318 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
with type 2 diabetes, decrease insulin requirements in type1 diabetes, and improve glycemic control in both types ofdiabetes. The incremental benefit from low glycemic loadis similar to that offered by pharmacological agents thatalso target postprandial hyperglycemia, such as alpha-glycosidase inhibitors.185,186 The benefit of low glycemic loadon the development of diabetes is similar to MUFA, PUFA,whole grains, fiber, fruits, and vegetables.
Whole Grains: The Foundation of Healthy Food
Whole grains have been the staple food worldwide forcenturies, especially among vegetarians.187,188 Whole grainand legume consumption not only decreases blood sugarand insulin resistance but also prevents the developmentof diabetes, particularly in people with the metabolicsyndrome.185,186 Whole-grain products are a good sourceof fiber, minerals, as well as several vitamins, includingvitamins B and E. In a 12-year follow-up of 42 898 men,the risk of developing diabetes was 42% lower in those withthe highest intake of whole grains. The risk was reducedby 52% in those who also engaged in physical activity, and87% in those who also had a low BMI.189 The risk reductionwas attributed to higher intakes of cereal fiber andmagnesium. Intake of whole-grain cereal is inverselyassociated with hypertension, CAD, stroke, and CVDmortality190,191 (Table 7).192–206 In another study, 25% –30%reduction in stroke was observed with the intakeof whole grains—similar in magnitude to that ofstatins.206–208 In sharp contrast, intake of refined grainsincreases the risk of diabetes, stroke and CVD.191,205–212
These prospective data highlight the importanceof distinguishing whole-grain from refined-graincereals in the prevention of CVD and diabetes.209 Efforts
Fig. 2. Risk of diabetes in 65 173 US women during 6 years of follow-up:influence of glycemic load and fiber
Table 7. CVD risk reduction demonstrated with selectedfood groups192–206
Author CVD risk reduction (%)
Fruits and vegetables (15%–48%)Bazzano et al.192 25Liu et al.193 15Joshipura et al.194 30Joshipura et al.195 20Gaziano et al.196 48Knekt et al.197 35
Nuts (19%–48%)Albert et al.198 48Ellsworth et al.199 19Hu et al.200 35Fraser et al.201 38Fraser et al.202 40Jiang et al.203 21 (diabetes)
Whole grains (32%–44%)Jacobs et al.204 33Fraser et al.201 44Liu et al.205 32Liu et al.206 32 (stroke)
should be made to replace refined-grain with whole-grainfoods.189
A whole-grain food includes all the edible parts of thegrain: the bran, the germ, and the endosperm.213 Grindingor milling, using modern technology, leads to the loss ofmany beneficial micronutrients, antioxidants, minerals,phytochemicals, fiber, and much of the germ.214 As a result,refined grain products are devoid of most vitamins andessential fatty acids, and contain more starch.215 Becauseof the loss of bran and pulverization of the endosperm,refined grains are digested and absorbed rapidly, resultingin a large increase in the levels of blood sugar and insulin.215
The common grains consumed in the West include wheat,oats, rye, rice, barley, and corn.213 In the USA, rye bread isan important source of whole grain consumption, andresults in a lower glucose response than white bread.152,212
Whole-grain, ready-to-eat cereal contains >25% wholegrain content by weight.189 The recommended intake is atleast 6 servings of grain (but not more than 11) with atleast 3 being whole grains. The current intake of wholegrains is less than half a serving/day or 15% of the grainintake. Only 2% of the 150 lb of wheat flour consumed percapita in the USA is whole-grain flour.216 Commonlyconsumed refined grain foods include white rice (idli, dosa),refined wheat and flour (white bread), pancakes, cakes,sweet rolls, English muffins, muffins, waffles, rolls, biscuits,pizza, and refined-grain ready-to-eat breakfast cereal, andtheir use should be minimized.
IHJ-584-03.p65 11/19/2003, 11:27 PM318

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 319
Nuts: A Wholesome Food and Powerhouse ofHealthy Fats and Nutrients
Extensive studies during the past decade have transformedthe image of nuts from fattening snacks to a wholesomeand heart-healthy food to be consumed daily.198–202,215 Nutsare rich sources of protein, antioxidants, fiber, vitamins andminerals (especially potassium and magnesium). Nuts yield5%–10% fiber, and 12%–25% protein. The consumptionof nuts is also associated with a reduced risk of CAD inseveral studies.198–202,217,218 Yet, nuts are not generallyrecommended as snacks because of their high fat content.Although nuts contain 45%–80% fat, most of the fats arethe highly beneficial MUFA and PUFA (Table 8).65
Nuts, particularly almonds, significantly improve lipidprofiles because of the high fiber and MUFA component.The dose–response effects of almonds were compared withlow-SAFA (<5% energy), whole-wheat muffins used as thecontrol diet in a randomized crossover study involving 27dyslipidemic men and women. Three isoenergeticsupplements each (mean 423 kcal/day; 22% of energy)were consumed for 1 month. The supplement consisted offull-dose almonds (73 g/day), half-dose almonds plus half-dose muffins, and full-dose muffins. Full-dose almondsproduced a highly significant decrease in the Lp(a) level(8%), LDL:HDL ratio (8%), and oxidized LDL (14%)compared to the control diet.219 A 9% decrease in the LDLlevel occurred with 73 g/day of almonds, and 4% decreasewith 37 g/day (handful) of almonds. This result translatesto a 1% reduction in LDL for every 7 g/day of almonds, andis consistent with other studies.220,221 More importantly,there was no difference in body weight between the almondand muffin diet.222 Nuts are energy-dense, and contain
160–200 cal/oz. It cannot be overemphasized that energyfrom nuts should replace the unhealthy calories from SAFAand refined grains to prevent weight gain.
Consumption of other nuts (except coconuts) is equallybeneficial. For example, a 10% reduction in the LDL levelcan be achieved by the daily consumption of 40 g ofwalnuts, peanuts or pistachios, 70 g almonds, 100 gmacadamia nuts, and 110 g of pecans.223–230 Nuts are aseffective as increasing physical activity and trimmingcalories to increase HDL levels. Adding 2 oz or 60 g of nutsto a diet is a delicious way to decrease the TC/HDL ratioand CAD risk.8,14,23,231,232 Nuts also improve insulinsensitivity and prevent diabetes.233 In a prospective studyof 83 818 women, 3206 new cases of type 2 diabetes wereobserved during a follow-up of 16 years.203 Consumptionof nuts was inversely associated with the risk of type 2diabetes after adjustment for age, body mass index (BMI),physical activity, smoking, alcohol use, and dietary factors(total calories, fat calories, and fiber). The risk of diabeteswas reduced by 27% in those who consumed >5 oz/weekof nuts or peanut butter compared to those who almostnever ate these products.203 The proscription of nuts canno longer be justified. In fact, regular nut consumption asreplacement for refined grains and high-fat meats isstrongly recommended.161,234
Fruits and Vegetables, the Natural Way to ConsumeAntioxidants and Flavonoids
Fruits and vegetables are rich in a myriad of nutrients andphytochemicals, including fiber, vitamins B and C,antioxidants, potassium, and flavonoids.190,215
Phytochemicals are bioactive nonnutrient plant
Table 8. MUFA, PUFA, and SAFA content (in g) in tree nuts and fruit (all nuts are dry roasted without salt except coconut)65
Calories per 100 g Fat content per 100 g MUFA PUFA SAFA
NutsMacadamia nuts 718 76 59 1 12Hazelnuts 646 62 47 8 5Pecans 710 74 44 21 6Almonds 597 53 34 13 4Cashew nuts 574 46 27 8 9Pistachio nuts 570 46 24 14 6Walnuts 607 57 13 37 4Flax seed 492 34 7 22 3Coconut meat, creamed 684 69 3 1 61Coconut meat, sweetened, shredded 501 35 2 0 31
FruitsAvocados, California 177 17 11 2 3Olives 115 11 8 1 1Avocados, Florida 112 9 5 1 2
IHJ-584-03.p65 11/19/2003, 11:27 PM319

320 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
compounds linked to a reduced risk of chronic diseases.Fruits and vegetables decrease blood pressure,homocysteine, and cancer, especially that of the GItract.211,235,236 Since fruits and vegetables are rich inpotassium, their liberal intake is recommended for theprevention and treatment of hypertension.237 Good sourcesof potassium include bananas, oranges, beans, fish, anddairy products. While you can get an overdose of potassiumfrom pills, you cannot get an overdose of potassium fromfood. Moreover, dietary supplements do not have the healthbenefits associated with a diet rich in fruits and vegetables.For example, the antioxidant value of 100 g of apple isequivalent to 1500 mg of vitamin C.3
Several large studies, including one comprising 84 000women and 42 000 men, have shown a significant inverseassociation between the consumption of fruits andvegetables and CVD mortality190,194,195 (Table 8). Therelationship is particularly strong with vitamin C-richfruits, green leafy vegetables, and carotenoid vegetables(carrots, broccoli, spinach, lettuce, tomatoes, and yellowsquash).192–196,238,239 Consuming fruits and vegetables (3times/day compared with <1 time/day was associated witha 27% lower incidence of stroke, a 42% lower strokemortality, a 24% lower CAD mortality, a 27% lower CVDmortality, and a 15% lower all-cause mortality afteradjustment for standard CVD risk factors.192 In sharpcontrast, consumption of potatoes and French fries increasethe risk of CAD and stroke.152,215
The landmark study of the Dietary Approaches to StopHypertension (DASH)240 has yielded tremendous insightsinto the benefits of increased intakes of various types offruits and vegetables. The DASH diet is rich in vegetables,fruits, and low-fat dairy products (9 servings of fruits andvegetable combined per day).240 As compared with thecontrol diet with a high sodium, the DASH diet with a lowsodium intake led to a decrease in systolic blood pressureof 7 mmHg in normotensive individuals, and 11.5 mmHgin hypertensive individuals. The benefits of the DASH dieton lipoprotein levels were equally spectacular, with an 11mg/dl decrease in LDL and a 4 mg/dl increase in HDL levelswithout significant effects on TG levels. Men had a greaterreduction in LDL level than women, with no differencebetween Whites and Blacks. These results suggest that theDASH diet is likely to reduce the risk of CAD and can berecommended as an overall eating plan.241 The currentintake of fruits and vegetables is 3 servings/day each in theUSA; only 23% consume the recommended 5 servings/dayeach.242 The DASH diet is feasible in the real world, unlikethe array of drastic diets which are impossible to continuefor more than a few months.240
Flavonoids: Flavonoids are secondary metabolites thatplants use to attract pollinators, repel predators, and tocolor flowers, leaves, and fruits.243 Important biologicaleffects of flavonoids include the scavenging of oxygen-derived free radicals, inhibition of LDL oxidation, increasein HDL levels, and protection against CVD and severalchronic diseases.244–248 The beneficial effects of thesenatural products on health were known long before thediscovery of flavonoids. The major sources of flavonoids arevegetables (onions, kale, broccoli), fruits (apples, grapes,berries), olive oil, and beverages such as tea andwine.244,248,249 Other sources include grains, bark, roots,stems, and flowers. Flavonoids present in red wine couldbe partly responsible for the low CAD mortality seen in redwine drinkers (“French Paradox”). Red wine is the majorsource of flavonoid in France and Italy (40%), onions andapples in Finland, and olive oil in Greece.250 The strong tasteof extra-virgin olive oil is partly caused by the abundanceof flavonoids.
Antioxidants: Oxidative modification of LDL acceleratesatherosclerosis whereas dietary antioxidants prevent LDLoxidation. These antioxidants include vitamin C, vitaminE, beta-carotene, selenium, flavonoids, magnesium, andMUFA.251 It is worth emphasizing that vitamin pills are nosubstitute for a healthy diet. Although an earlier studysuggested some benefits from antioxidant vitaminsupplementation, several subsequent studies involvingmore than 100 000 patients have consistently failed todemonstrate any benefit. More recent studies suggest thatpossible harm may outweigh the benefit of thesevitamins.252–254 In a recent study, the use of vitamins E andC reduced the lipid-lowering efficacy of statins and niacinby 50%. More importantly, the clinical event reduction waslowered from 90% to 60%.255 The current scientificevidence does not support any protective role of vitaminsE, C, and beta-carotene supplements; their use only createsa diversion away from proven therapies.256 The USPreventive Service Task Force (USPSTF) recommendsagainst the use of beta-carotene supplements.257 It is worthnoting that the oxidative modification of LDL continues tobe relevant, and people should obtain their antioxidantvitamins from food sources. (However, folic acid fortificationis recommended in women who are pregnant or mightbecome pregnant.)
Non-nutritive Food Adjuncts
Fiber: The term dietary fiber was coined to describe theplant cell wall removed during the refining process.258
Dietary fiber improves coagulation, fibrinolysis, insulin
IHJ-584-03.p65 11/19/2003, 11:27 PM320

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 321
sensitivity, LDL, and blood pressure levels.259–265 Fiber isparticularly concentrated in bran. Insoluble fiber shortensthe intestinal transit, resulting in less time for carbohydrateabsorption.266 Soluble (viscous) fiber, such as beta-glucan,which is found in oat bran, delays gastric emptying, andslows the absorption and digestion of carbohydrates. Theseprocesses lead to a slower release of glucose into thecirculation, resulting in a reduced demand forinsulin.187,189,267 An intake of 16 g of total fiber is associatedwith a 12% decrease in CAD risk.268 FDA has permittedcardiovascular health claims to be made by the industryfor 2 viscous fibers, beta-glucan and psyllium.269 Psylliumsupplementation significantly lowers TC and LDL levels; itis safe and well tolerated.270
The benefit of whole grains appears to be mediatedprimarily through the greater intake of fiber, and is greaterwith cereal fiber than vegetable or fruit fiber.212,263,264,271,272
Approximately one-fourth of the fiber provided by cerealsources is water soluble.268 Cereal fiber consumption isassociated with a 21% lower risk of incident CVD, and 30%lower risk of diabetes.164,188,259,273 (Fig. 2). Cereal fiberconsumption may reduce the risk of CVD via thesubstitution effect, replacing the intake of other foodshaving potentially detrimental effects. In addition to cerealgrains, legumes are also excellent sources of water-solubledietary fiber. Half a cup of cooked beans contains, on anaverage, 6 g of total fiber and 2 g of soluble fiber.274
The current ADA recommendation for a healthy diet isto consume 25 g/day of fiber with about one-third fromsoluble fiber. In one study, type 2 diabetics consuming 50g/day of fiber (25 g soluble, 25 g insoluble) lowered theblood sugar by 13 mg/dl, plasma TC by 7%, and TG levelsby 10%.275 Thus, a high intake of dietary fiber, above thelevel recommended by the ADA, particularly of the solubletype, improves glycemic control, insulin levels, and plasmalipid concentrations in patients with type 2 diabetes.275
Plant sterols and stanols: Plant sterols and stanols arestructural analogues of cholesterol. Low-fat plant stanol-containing margarines lower plasma LDL levels (by as muchas 12%) in those with hypercholesterolemia by suppressingcholesterol absorption.276–279 In one randomized controlledstudy, the reduction in LDL with such supplements wassimilar to 20 mg of lovastatin (30% with statin v. 28.6%with diet).280 Various plant supplements have been shownto reduce LDL by 40% (Table 9).146,219,265,280–283
Spices: Plants have the capacity to synthesize a diversearray of chemicals. Spices are aromatic vegetablesubstances, the significant function of which is foodseasoning rather than nutrition. Typically, spices are the
Table 9. A portfolio of dietary factors useful for LDLreduction146,219,265,280–283
Decrease in LDL(%)
SAFA intake <7% 10Dietary cholesterol intake <200 mg 5Body weight 5 kg 5Plant sterols/stanols 1–3 g/day 5Soy protein 25 g/day 5Nuts (almonds) 50 g/day 5Viscous fiber intake 5–10 g/day 5Total LDL reduction Full portfolio 40
dried aromatic parts of plants, generally the seeds, berries,roots, pods, and sometimes leaves, that mainly grow intropical countries. Common spices include turmeric,paprika, saffron, cinnamon, nutmeg, red and black pepper.In contrast, herbs used in cooking are typically composedof leaves and stems.284
Caffeine: Caffeine is found in coffee, tea, soft drinks,chocolate, and some nuts. Finland has one of the highestrates of per capita coffee drinking (13 kg/year).285 In aprospective study of 20 179 Finnish adults, coffee drinkingwas not associated with an increased risk of MI. However,consumption of large quantities of boiled unfiltered coffeeraises cholesterol and homocysteine levels.285–287 In anexperiment involving 10 volunteers, who consumed theequivalent lipid content of 6–7 cups of boiled unfilteredcoffee daily for 6 weeks, the LDL levels increased by 33 mg/dl.288–290 These data suggest that daily consumption of 1–2cups of coffee is safe with no particular health benefits orrisks.
Tea: Tea, the most widely consumed beverage in the worldother than water, has been associated with lowercardiovascular risk.291–295 Unlike coffee, tea consumption isassociated with a substantial reduction in LDL levels. Tea isrich in flavonoids. Green tea contains catechins, whereasblack tea, formed from the polymerization of catechins,contains theaflavins.291 In one recent study, theaflavin-enriched green tea extract reduced the LDL level by 16%.296
Tea is the major source of flavonoid intake in Japan (>80%);the Japanese consume an estimated 7 cups/day of teacompared to half a cup/day in the USA. Adding milk totea, as is common in the UK and India, abolishes thebeneficial effect of tea.297
Alcohol: Moderate intake of alcohol (one drink a day forwomen and 2 drinks a day for men) may decrease the riskof CAD.298 Recently, it has been shown that only one drinkper week is enough to provide cardiac protection (45 ml ofspirits or 350 ml of beer or 120 ml of wine); the
IHJ-584-03.p65 11/19/2003, 11:27 PM321

322 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
cardioprotection is similar for beer, wine, whiskey, brandy,vodka, rum, and drinks in equivalent amounts.299,300 Morethan 2 drinks per day does not provide any additionalprotection and, in fact, the net effect may be harmful untilthe age of 45 years in men and 55 years in women.301 Likecarbohydrates, consumption of large quantities of alcoholraises TG levels.6,302 Other dangers of excessive alcoholconsumption includes alcohol dependence, liver disease,high blood pressure, obesity, stroke, traffic accidents,spousal abuse, suicide, and breast and other cancers. Giventhese risks, the American Heart Association cautionspeople against increasing their alcohol intake or startingto drink if they do not already do so.
Weight Gain and Weight Loss Diets
Excess calories and obesity: Diets of any type containingmore energy than needed or expended will lead to obesityand dyslipidemia.303 A calorie is a calorie whether it comesfrom carbohydrates, fat, or protein. Excess calories of anykind will eventually be converted by insulin to body fat.304
A common misconception is that dietary fat of any kind isfattening, while low-fat and high-protein diets haveslimming properties. It is absolutely vital that bothphysicians and the public understand that it is the excesscalories and not diet composition that causes weightgain.304–309 There is no evidence of weight gain with a highMUFA diet, compared with a high carbohydrate diet, underisoenergetic conditions.310,311
Obesity is not only a reflection of overnutrition but alsoan important contributor to the mass dyslipidemia seen inIndia and western countries.6 Obesity in general isaccompanied by the increased production of apo B and adecrease in the HDL levels.6,312 Humans have a limitedcapacity to store energy as carbohydrates. Whencarbohydrate intake exceeds storage and oxidationcapacities, the excess is converted to fat by de novolipogenesis that leads to high TG levels.313 This process isincreased several-fold in people with the metabolicsyndrome which, if left untreated, leads to overt diabetes(25-fold risk).312 Body fatness and not lean body mass isthe principal determinant of diabetes and prediabetes.314
At a given BMI, Asian Indians have 7%–10% higher bodyfat; accordingly, BMI <23 is termed optimum; BMI 23–25overweight, and >25 obese in Asian Indians. Likewise, theoptimum waist circumference is lower in Asian Indiansthan Whites with a cut-off <90 cm in men and <80 cm inwomen.315 Although obesity and dyslipidemia areuncommon in less affluent societies, some individuals maybe excessively sensitive to caloric excess.6
Fast foods rapidly produce plaques. The averageAmerican gained 9 lb in the past decade. A third ofvegetable taken in the USA are either French fries or potatochips.305 In one study, overweight subjects who consumedfairly large amounts of sucrose (28% of energy), mostly asbeverages, had increased energy intake, body weight, fatmass, and blood pressure after 10 weeks. These effects werenot observed in a similar group of subjects who consumedartificial sweeteners.316 Restricting the dietary cholesterolcan achieve a 3% reduction in TC level, whereas losingweight from trimming extra calories can reduce LDL by 5%to 20%.8
Weight loss: The recipe for effective weight loss is acombination of motivation, physical activity and caloricrestriction; maintenance of weight loss is a balance betweencaloric intake, and physical activity, with life-longadherence. Each pound of body fat contains 3500 cal.Therefore, a person who consumes 500 cal less than hespends each day can lose 1 lb of fat a week. Any higherweight loss is due to a more severe caloric restriction orwater loss rather than fat loss. The minimum caloric intakein a medically unsupervised weight loss diet is 1500 cal/day for men, and 1200 cal/day for women. Superior long-term participation and adherence is observed in a high-fatdiet rather than a low-fat diet (35% v. 20%), especially inwestern cultures.309 The greater success rate is due to higherpalatability of the high-fat diet provided by mixed nuts andlean meat.309 Furthermore, the long-term outcome of areduced-fat diet consumed ad libitum for weight control isdismal. In one study, compared with the control group,weight decreased in the reduced-fat diet group significantlyby 3 kg in 1 year but diminished to an insignificant 1 kg by5 years.317 Until more information becomes available, “theprudent diet,” which is a balanced diet, is the one to followfor young and old alike.318
Very low fat diet: Some experts have argued for a verylow-fat diet (<10%).319 Since these diets are not high-protein diets (like the Atkins diet), they are in reality veryhigh in carbohydrate. High-carbohydrate diets (theMacrobiotic diet) increase insulin resistance and induce themetabolic syndrome. In controlled trials, low-fat, high-carbohydrate diets decreased HDL levels. Replacing 10%of energy from SAFA with carbohydrate lowers the HDLlevels by 5 mg/dl, even when the carbohydrate consumedis complex.62,173,320,321 There is also a marked increase in TGlevel, which makes LDL small, dense, and moredangerous.173,320–322 The ef fect is strongest whencarbohydrates replace SAFA but is also seen whencarbohydrates replace MUFA and PUFA. The effect is seen
IHJ-584-03.p65 11/19/2003, 11:27 PM322

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 323
in both short- and long-term trials, and is therefore not atransient phenomenon. Therefore, replacement of SAFAmust be achieved through increasing MUFA and not bycarbohydrates. The adverse effects of high-carbohydratediets (high glycemic load) in the metabolic syndrome anddiabetes have not received due attention, especially in theIndian literature. The recommended carbohydrate intakeis <50% of calories in people with the metabolic syndromeor diabetes (NCEP).
The allure and dangers of very low-carbohydrate,high-protein diets: High-protein diets that are extremelylow in carbohydrates are touted as a new strategy forsuccessful weight loss by many.323 Most such diets contain<10% carbohydrates, 25%–35% protein, and 55%–65%fat. Because the protein is provided mainly by animalsources, these diets are high in SAFA and cholesterol. Thus,these diets are truly high-fat diets masquerading as high-protein diets. Advocates of this diet often promote seriousmisconceptions about carbohydrates, insulin resistance,ketosis, and fat burning as the mechanisms of action forweight loss. To avoid excess load on the kidneys, the totalprotein intake should not exceed 100 g/day.324 Moreimportantly, the body has an obligatory requirement forglucose of about 100 g/day, largely determined by themetabolic demands of the brain.324,325
In randomized studies, the extent of weight loss wassmall (4 kg), and adherence to the diet was low.326,327 Inone study, although a low-carbohydrate diet produced a 4%greater weight loss at 6 months than did the conventionaldiet, the differences did not persist at 1 year. Furthermore,adherence was poor, and attrition was high in both thehigh- and low-carbohydrate groups. Longer and largerstudies are required to determine the long-term safety andefficacy of low-carbohydrate, high-protein, high-fat diets.326
Two recent studies have provided insight into high-proteindiets; the initial weight loss is due to fluid loss and ketosis-induced appetite suppression. The monotony of this dietalso results in involuntary caloric restriction.326,327
The beneficial ef fects on blood lipids and insulinresistance are due to the weight loss, and not the change incaloric composition. Such diets increase LDL but decreaseTG levels, in sharp contrast to high-carbohydrate diets,which increase TG, and decrease HDL levels. Althoughthese diets may not be harmful for most healthy people overa short period of time, there are no long-term scientificstudies to support their overall efficacy and safety. Markedlyatherogenic profiles have also been reported in childrenwith ketogenic diets. At 6 months, the high-fat ketogenicdiet significantly increased plasma LDL levels by 50 mg/dl,
triglycerides 58 mg/dl, apo B 49 mg/dl, and non-HDLcholesterol 63 mg/dl. The mean HDL-cholesterol levelsdecreased significantly.328 These lipid abnormalities inchildren are more than likely to translate into a high riskof heart disease as young adults.
High-protein diets also do not provide the variety of foodsneeded to continue the diet on a long-term basis. High-protein diets are not recommended, and are perhapsdangerous because they restrict most healthful foods thatprovide essential nutrients, especially fruits and vegetables.Individuals who follow these diets are therefore at risk forcompromised vitamin and mineral intake, as well aspotential cardiac, renal, bone, and liver abnormalitiesoverall.324 The consumption of a very low-carbohydrate dietfor 6 weeks delivers a high acid load to the kidney, increasesthe risk of stone formation, decreases body calcium, andmay increase the risk of bone loss and fractures.329 A high-protein diet is the ultimate antithesis of the prudent diet. Itis important to realize that diets are not for 6 weeks, 6months or 6 years, but for a lifetime. Although most quick-fix diets have a short-term success rate >90%, the long-term failure rate is 100%.
Healthy and Contaminated Vegetarian Diets
Omnivores or nonvegetarians outnumber vegetarians 10to 1 in western cultures. Vegetarians include vegans whodo not consume any animal products, ovo-vegetarians whoconsume egg, lacto-vegetarians who consume milk, ovo-lacto-vegetarians who consume egg and milk, and semi-lacto-vegetarians who eat small amounts of meat (<1 time/week). Ironically, most self-defined vegetarians in westerncountries consume red meat and poultry, albeitinfrequently, and in very small quantities. In a recentsurvey, only 1% of self-reported vegetarians did not eat meatin the USA, whereas about 6% of Americans who do notconsume any meat did not identify themselves asvegetarians.330–332
Western vegetarians generally consume a healthier dietthan omnivores; healthy foods such as soy, nuts, legumesand vegetables replace meat.333 They generally have twicethe fish consumption of nonvegetarians.330 This is not thecase with Indian vegetarians who shun fish. US vegetarianseat more whole-grain products, dark green and deep yellowvegetables, whole-wheat bread, brown rice, soy milk, tofu,meat substitutes, legumes, lentils, garbanzos, walnuts, andpecans.330 However, they eat the same amount of food asomnivores (1000 kg/year) but are usually thinner.334 Ahealthy vegetarian diet is characterized by more frequent
IHJ-584-03.p65 11/19/2003, 11:27 PM323

324 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
consumption of fruits and vegetables, whole grains,legumes and nuts, resulting in higher intakes of dietaryfiber, antioxidants and phytochemicals.335 Thus avegetarian diet contains a portfolio of natural products thatcan improve both the carbohydrate and lipid abnormalitiesin diabetes.187
Vegetarians eat about two-thirds of SAFA, and one-halfof cholesterol as omnivores; vegans consume one-half ofSAFA and no cholesterol.9,336 Cholesterol levels amongwestern vegetarians are 15–25 mg/dl lower thanomnivores.337–340 Vegans have very low levels of LDL.341,342
Nuts, viscous fibers (from oats and barley), soy proteins,and plant sterols in vegetarian diets improve serum lipidlevels.337 Furthermore, substituting soy or other vegetableproteins for animal proteins reduces the risk of developingnephropathy in type 2 diabetes.
With the exception of tropical oils, calories from plantsources are negatively correlated with CAD mortality,whereas calories from animal sources are positivelycorrelated.16 Olive oil, fresh fruits, and vegetables areprotective against heart disease, and seem to play a greaterrole in the French paradox than wine.16 Greaterconsumption of whole milk and other animal productswere important contributors to Finland having the highestrates of CAD.16 In a prospective study of 4671 vegetariansand 6225 nonvegetarians, followed up for 10–12 years,BMI, TC, and CAD mortality was substantially lower amongvegetarians than in the nonvegetarians. Other studies alsosuggest a protective effect of vegetarianism against manydiseases. Vegetarians in western countries, but not in India,enjoy remarkably good health, exemplified by low rates ofobesity,334,343 diabetes,344 CAD345–347 and cancer,337 and a 3–6 year increase in life expectancy.333,348 It is not clearwhether this is due to abstinence from meat or to a greaterconsumption of heart-healthy food.349
Indian vegetarianism, a form of “contaminatedvegetarianism”: Most Asian Indians are lacto-ovo-vegetarians, unlike western vegetarians. About 50% ofAsian Indians are vegetarians, but their lipoprotein levels,and rates of diabetes and CAD are no different from thoseof nonvegetarians.350,351 This phenomenon is due tocontaminated vegetarianism, wherein vegetarians manageto consume excessive amounts of SAFA and TRAFA. In theCADI study, Asian Indian physicians in the USA followed aheart-healthy diet, with 32% energy from total fat, and 8%from SAFA, which is the recommendation by the NCEP.350
This appears to be an exception rather than the rule. In aCanadian study, Asian Indians consumed more fried foodsand high-fat dairy products, such as full-fat milk than White
Canadians.352 Although the intake of fat is 20%–25%energy in most Asian countries, many affluent AsianIndians consume >50% of their calories from fat.
Indian vegetarians consume liberal amounts of bakeryproducts, butter, ghee, cheese, ice cream, curd, and otherdairy products to overcompensate for not using meat.Contrary to popular belief, dairy products are the majorsource of SAFA, even in the western diet. It is worthhighlighting that SAFA intake from high-fat dairy productsincreases LDL levels 3 times as much as it raises the HDLlevel.353 Meat is expensive, and consumed in very smallquantities by Indian omnivores because of cultural andfinancial reasons. This is in sharp contrast to an annualper capita consumption of 124 kg meat and 23 kg fish byAmericans.354 Prolonged cooking of vegetables, as ispractised in India, virtually destroys every nutrient beforeit is consumed. A major problem overlooked in the Indiandiet is the high glycemic load, resulting in high TG and lowHDL levels.355 There appears to be a threshold forcarbohydrate consumption with an intake >280 g/dayoften resulting in atherogenic dyslipidemia.350
Deep-frying and reuse of frying oil: Deep-frying, acommon form of cooking among Asian Indians, isassociated with spontaneous hydrogenation, and theformation of TRAFA. Reuse of oil used for deep-frying hasbeen shown to produce endothelial dysfunction.356
Repeated reuse of such oil is exceedingly common amongAsian Indians.357 HDL inhibits LDL oxidation primarilythrough its paraxonase activity; reuse of frying oil reducesparaxonase activity, and thus reduces the ability of HDL toprevent LDL oxidation.356–360 Fats that have been heated forprolonged periods in air contain many dangerous productsfrom oxidation and breakdown of lipids. These compoundsinclude hydroxy peroxides, aldehydes, polymers, hydroxyfatty acids, hydroperoxy epoxides, and hydroperoxyalkenals.361 In one study, fast-food restaurant cooking oil,just before the weekly change, was compared to unused oil.The repeatedly used oil had 4 times higher peroxide levels,7 times higher carbonyl levels, and 17 times higher levelsof acids.357
Ghee: Ghee is one of the most important sources of dietaryfat and a common cooking medium.362,363 Use of ghee fordeep-frying is considered gourmet among Asian Indians.Ghee or clarified butter is anhydrous milk fat, and is rich inMUFA (32%) and SAFA (62%), most of which arecholesterol-raising (myristic acid 17%, palmitic acid 26%).It is perhaps more harmful than butter due to the addedpresence of cholesterol oxides, which are generated during
IHJ-584-03.p65 11/19/2003, 11:27 PM324

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 325
its preparation by prolonged heating of butter.362–364 Liberaldietary exposure to cholesterol oxides from ghee is a likelycontributor to the high frequency of CAD among AsianIndians.364 There are conflicting data on the risk of heartdisease with ghee.365,366 We are unaware of any biologicalexplanation as to why Asian Indians can be immune to theunfavorable effects of butter and/or ghee. In addition to milkghee, vegetable ghee (vanaspathi) is also immensely popularin Indian cooking, which exerts similar adverse effectsthrough its high TRAFA content.
Tropical oils: The term tropical oils refers to coconut, palmkernel, and palm oils. These oils contain a very highpercentage of SAFA, unlike other vegetable oils such asrapeseed oil (mustard oil), sesame oil, and rice bran oil,which are low in SAFA and high in MUFA (Table 3). Tropicaloils are more atherogenic and thrombogenic than muttonand beef fat; the latter contains <5% myristic acidcompared to 18% in coconut and palm kernel oils.104 Infact, these oils contain more TC-raising SAFA than animalfats—coconut oil 89%, palm kernel oil 71%, and palmoil 46% compared to <30% for butter fat, beef fat, porkfat, and chicken fat (Table 5).6,147 Tropical oils accountfor <2% of energy (<4 g/day) in the USA. but 25%or more in many other countries.147,367 Tropical oils arealso found in commercially baked cakes, biscuits, cookies,and “snack foods”. In Mauritius, a regulated change in theSAFA content by substituting soybean oil for palm oilresulted in a dramatic 32 mg/dl fall in TC level, andunderscores the crucial role of cooking oils in populationlevels of TC.368
Coconut oil: Coconut oil contains mostly cholesterol-raising SAFA (8% caprylic, 6% capric, 45% lauric, 17%myristic, and 8% palmitic acid).369 Rabbits fed a commercialchow diet containing 0.5% cholesterol and 14% coconutoil developed more severe dyslipidemia and atherosclerosisthan rabbits fed the same diet containing olive oil insteadof coconut oil. The average plasma TC level was 2-fold, andTG level 20-fold higher in the coconut oil-fed rabbits thanin the olive oil-fed rabbits.28 Cox et al.369,370 have reportedthe cholesterol-raising effects of coconut oil to be similarto that of butter. In a comparative study of diets rich inbeef fat versus coconut oil, the plasma TC, LDL, and HDLresponses were lower with beef fat than coconut oil,commensurate with the lower proportion of cholesterol-raising SAFA in beef (29%) than coconut oil (89%)371 (Table5). A Malaysian study in which 22% of the energy intakewas substituted with coconut oil found an increase of 40mg/dl in TC, 29 mg/dl in LDL, 36 mg/dl in TG, and 4 mg/dlin HDL levels.372 The impact on LDL and HDL by using
various fats as the sole source of fat in a Dutch populationis shown in Fig. 1. Note the marked increase in LDL incontrast to HDL with the use of coconut oil.
Kerala, renowned for the universal and liberal consump-tion of coconut milk and oil, not only has the highest levelof TC in India, but also the highest rate of CAD.373 Theproportion of subjects with high TC (>239 mg/dl) in Keralais almost double that of the USA. (32% v. 18%).374 This isin sharp contrast to the Japanese among whom only 6%have high TC.375 In Sri Lanka, which also has a very highrate of CAD, about 80% of the fat in the habitual diet comesfrom coconut.134,374,376
Consumers need to be educated about the atherogenicand antiatherogenic effects of various cooking oils, as wellas animal and vegetable ghee. There is little awareness, andeven controversy, about the atherogenic effects of certainfoods and oils, especially in regions where the production,sale, and consumption of such oils have a profound impacton the regional economy.
Prudent Diet for All Ages and the EntirePopulation
The traditional Mediterranean diet is characterized byabundant plant foods (vegetables, breads, pastas, beans,nuts, and seeds). Fresh fruit is the typical daily dessert, andolive oil is used as the principal source of fat. Dairy products(principally cheese and yogurt), fish, and poultry areconsumed in low-to-moderate amounts. Red meat and eggare consumed in low amounts (0–4 eggs weekly). Wine isconsumed in low-to-moderate amounts, normally withmeals. This diet is typically high in total fat (35%–45%) butlow in SAFA (7%–8% of energy). Greater adherence to thetraditional Mediterranean diet is associated with asignificant reduction in total mortality.377 The 6 beneficialcomponents of this diet have recently been elucidated. Theyare vegetables, legumes, whole-grain cereal, fish, fruit, andnuts, which form the basis for the “prudent diet”377
(Table 10).199–203,225–227,231–236
According to the new paradigm, dietary pattern ratherthan individual nutrients appears to be more important.Recent research suggests the existence of a food synergy inwhich the beneficial effects of healthy foods are magnifiedwhen several different types of foods are consumed.213 Huet al. have developed the concept of “prudent diet” (modifiedfrom the Mediterranean diet).30,378–380 The “prudent diet”has a higher intake of vegetables, fruits, legumes, wholegrains, fish, and poultry, whereas the “western diet” ischaracterized by a higher intake of red meat, processedmeat, refined grains, sweets, desserts, French fries, and
IHJ-584-03.p65 11/19/2003, 11:27 PM325

326 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
high-fat dairy products.378,380 The “prudent diet” isassociated with a 24% decreased risk of CVD compared toa 46% increased risk with the western diet.215,380
Consumers are bombarded on a daily basis with theBabel of nutritional breakthroughs.381 Food companiesadvertize their products, nutrition researchers publicizetheir latest results, and the media are more interestedin a controversial story than in scientific facts.30 Trivialreports are often publicized as major breakthroughs bythe media, and cause confusion among consumers. It isdifficult for most journalists and consumers to tell thedifference between a major research finding and a trivialreport.381
The dangers of the current western diet and thecontaminated vegetarian diet, and the remarkable benefitsof the prudent diet need to be disseminated amongcardiologists, physicians, and the public. This diet can besustained lifelong but needs to be adapted to Indianingredients and cooking methods. Several countries havedeveloped dietary guidelines to reduce nutritionalinformation anarchy. The Indian consensus on the prudentdiet should incorporate scientific facts, and the culturalpreferences appropriate for different parts of India. Such
information needs to be adopted by the scientificcommunity, and adapted by the food industry.
Current Knowledge on Preventive andTherapeutic Nutrition
Randomized, controlled clinical trials, meta-analysis, andsystematic reviews are considered the ultimate tests of thebenefits of therapeutic interventions. Such reviews haveshown a 24% reduction in major coronary events in dietarytrials lasting >2 years.382 The TC/HDL ratio is the single bestlipid predictor of CVD. This ratio is determined by 3 partlyopposing dietary factors—the proportion of energy fromSAFA, which raises TC; the proportion of energy from totalfat, which raises HDL; and the excess in total energy intake,which produces obesity and secondarily lowers HDL.20 Thegreatest reduction in CVD risk is achieved by LDL-loweringby reducing SAFA intake. Decreasing SAFA intake is bestaccomplished by reducing the intake of high-fat dairyproducts, and increasing fiber-rich foods. A dietincorporating lean beef, skinless chicken, and fatty fish hasbeen shown to improve the lipid profile by 5%–10%.383
The preferred replacement for SAFA is MUFA or PUFA
Table 10. The benefits and sources of selected heart-healthy foods199–203, 225–227, 231–236
Category Benefits Sources/preferred foods
Whole grains Good source of fiber, minerals, and Whole wheat, oats, rye, barley, corn, brown ricevitamins E and B and wild rice
Delay gastric emptying and blunts postprandial Whole-grain oatmeal, whole-grain, insulin response pumpernickel, rye or dark bread
Improve insulin sensitivity Whole-grain, ready-to-eat cereal⇓ Insulin secretion Cooked cereal⇓ Insulin resistance, and diabetes PastaImprove lipids (⇑ HDL) Popcorn⇓ Hypertension Bran⇓ CVD incidence and CVD mortality Buckwheat, cracked wheat, wheat germ
Fruits and vegetables Rich in fiber, flavonoids, and antioxidants Green leafy vegetables⇓ Blood pressure Carotenoid vegetables (carrots, broccoli, spinach,⇓ LDL lettuce, tomato, and yellow squash)⇑ HDL⇓ Homocysteine Citrus (vitamin C-rich) fruits⇓ CVD and mortality Temperate fruits (cantaloupe, apple, pear)⇓ Cancer, especially GI tract Tropical fruits (mango, papaya, pineapple,⇓ All-cause mortality jackfruit, guava, banana)
Viscous fiber ⇓ LDL Cereal grains (oats—beta glucan)⇓ Postprandial glycemia Legumes, Psyllium (Metamucil)
Soy protein ⇓ LDL Soy bean
Plant sterols and stanols ⇓ LDL Low-fat plant stanols containing margarines
Nuts ⇓ LDL See Table 6⇑ HDL
IHJ-584-03.p65 11/19/2003, 11:27 PM326

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 327
and not carbohydrates (Table 11). Replacing SAFA withcarbohydrates decreases the LDL levels but makes LDLsmall, dense, and more dangerous by increasing the TGlevels.30 Substituting carbohydrates with MUFA decreasesthe LDL level, and increases the HDL level. PUFA and MUFAincrease insulin sensitivity, and decrease the risk of type 2diabetes.45,384–386 Substantial evidence indicates that dietsusing MUFA and PUFA as the predominant form of dietaryfat, an abundance of fruits and vegetables, and adequaten-3 fatty acids can offer significant protection against CAD,stroke and diabetes (Table 8). Adequate consumption offruits and vegetables provides most of the necessaryantioxidants, and are preferable to dietary supplements inthe form of pills. Replacing a high glycemic with a lowglycemic index, and reducing the glycemic load can reducethe risk of diabetes161 (Table 6).
Nuts, once deemed unhealthy because of their high fatcontent, have become an important part of diets designedto control weight, lower blood pressure and cholesterol, andachieve secondary prevention of CAD, besides addingvariety, texture and flavor to dishes.145,211,215,387 Unless abeneficial effect is clearly demonstrated by well-designedscientific studies, the liberal use of butter, ghee, palm oil,
and coconut (oil and milk) should be discouraged. However,in diets with a negligible intake of fish, meat, milk, and dairyfat, the modest use (<7% of energy) of such oils may bepreferable to no fat at all.
Practical Recommendations
Better food habits can help reduce the risk of diabetes, MI,stroke, and death. A healthy eating plan means choosingthe right foods to eat, and preparing them in a healthy way.A healthy diet involves a decrease in the use of refinedgrains, tropical oils, egg yolks, animal, dairy, andhydrogenated fats, and an increase in the consumption ofwhole grains, vegetables, nuts, legumes, and fruits.7
Increasing the MUFA intake up to 20% of energy, as areplacement for SAFA and carbohydrates, may help preventand treat the metabolic syndrome, diabetes, and CVD. Sucha strategy can also significantly reduce the need for lipid-optimizing drugs. Since meat contains one-third MUFA andone-third cholesterol–neutral stearic acid, its consumptioncan also be incorporated into a healthy diet, provided leancuts are used, and the quantity limited to 150 g/day.6
Practical recommendations on diet are given in Table 12.
Table 11. Summary of current knowledge on diet
• Quality of fat is more important than the quantity of fat consumed.• SAFA is the principal determinant of elevated LDL levels. Reduction in energy intake from SAFA is the cornerstone of dietary
modification. Dairy products provide more SAFA than meat, even in western diets.• Contrary to common belief, MUFA and PUFA can significantly lower LDL levels. Replacement of 20% of energy from SAFA with
MUFA or PUFA decreases TC level by 40 mg/dl.• The atherogenicity and thrombogenicity of tropical oils are several times higher than meat.• Increase in LDL level from lean meat is no higher than chicken, and need not be eliminated from a healthy diet.• n-3 PUFA found in fatty fish is antithrombotic, antiarrhythmic, and prevents sudden death.• The adverse effects from TRAFA consumption are greater than those from SAFA, because of increase in LDL and Lp(a) levels, and
decrease in HDL level.• Both quality (glycemic index) and quantity (glycemic load) of carbohydrates are important determinants of insulin resistance and
the metabolic syndrome. A high glycemic index or load portends an inferior quality of carbohydrates.• Although carbohydrates do not raise LDL levels, a high glycemic load is the major determinant of postprandial lipemia.• Vegetarian diet is unhealthy if it contains large amounts of SAFA and TRAFA.• Nuts, fruits, vegetables and whole grains can each reduce the risk of CVD by 15%–45%.• Nuts are wholesome and nutritious food.• The risk from alcohol outweighs the benefit in men <45 years and women <55 years of age.• Drinking coffee does not increase the risk of heart disease but consumption of large amounts of unfiltered coffee can increase TC
levels.• Tea is rich in antioxidants and flavonoids, and is associated with a reduced risk of CAD; however, most of the beneficial effects are
neutralized with the addition of milk.• A calorie is a calorie, whether derived from carbohydrates, protein or fat.• Obesity is a reflection of overnutrition (caloric excess), and a major cause of dyslipidemia.• Caloric excess, irrespective of the composition of food, can raise LDL and lower HDL levels as a secondary effect of obesity.• Weight reduction seen in high protein diets is due to the involuntary caloric restriction from the monotony of the diet rather than
the diet composition.
IHJ-584-03.p65 11/19/2003, 11:27 PM327

328 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
Conclusions
People eat specific foods because of their taste, easyavailability and affordability, but are often unaware of thehealth benefits and risks. Dietary modifications remain thecornerstone of both the treatment and prevention ofdiabetes and CVD, the twin epidemics of the twenty-firstcentury.234,388–391 A prudent diet together with regularphysical activity, avoidance of smoking, and maintenanceof a healthy body weight may prevent the majority ofdiabetes and CVD in the Indian population.30 Aggressivedietary interventions may reduce CVD events to a similarmagnitude as that achieved with statins. Compared withmedical or surgical interventions, nutritional interventionis low-risk, low-cost, and readily available.259 A variety ofwhole grains, not refined grains, as well as various types offruits and vegetables should be the main form ofcarbohydrates.242 Prolonged cooking of vegetables shouldbe avoided. It is important to realize that the vegetarian dietis healthy only when it is low in SAFA, and the predominantenergy is from foods with a low glycemic index.215 The bestway to counter the perils of contaminated vegetarianismis by substituting full-fat dairy products with low-fat dairyproducts. Cooking oils containing high SAFA should bereplaced with those containing high MUFA. Deep-frying,especially with previously used oils, should be discouraged.Nuts are healthy, wholesome foods, and their use shouldbe encouraged as a replacement for unhealthy calories.
A diet rich in fish has multiple benefits, including raisingHDL, and lowering TG levels, and preventing suddendeath.383 Consumption of fish is preferable to taking a largenumber of fish oil capsules. There is increasing evidencethat dietary and lifestyle modifications begun in childhoodare likely to have benefits later in life.392 Therefore, thesedietary guidelines are applicable to all Asian Indians >2years of age, and not just those with diabetes or heartdisease.
References
1. Kris-Etherton P, Daniels SR, Eckel RH, Engler M, Howard BV, KraussRM, et al. Summary of the scientific conference on dietary fattyacids and cardiovascular health: conference summary from thenutrition committee of the American Heart Association. Circulation2001; 103: 1034–1039
2. Wylie-Rosett J. Fat substitutes and health: an advisory from theNutrition Committee of the American Heart Association.Circulation 2002; 105: 2800–2804
3. Liu RH. Health benefits of fruit and vegetables are from additiveand synergistic combinations of phytochemicals. Am J Clin Nutr2003; 78 (Suppl): 517S–520S
4. Connor WE. The decisive influence of diet on the progression andreversibility of coronary heart disease. Am J Clin Nutr 1996; 64:253–254
5. Enas EA. Coronary artery disease epidemic in Indians: a cause foralarm and call for action. J Indian Med Assoc 2000; 98: 694–695,697–702
6. Grundy SM, Denke MA. Dietary influences on serum lipids andlipoproteins. J Lipid Res 1990; 31: 1149–1172
7. Schaefer EJ. Lipoproteins, nutrition, and heart disease. Am J ClinNutr 2002; 75: 191–212
Table 12. Dietary recommendations
• A minimum carbohydrate intake of 100 g/day is necessary but should not exceed more than 300 g/day.• Indians with or predisposed to the metabolic syndrome and diabetes should limit carbohydrate intake to <50% of energy.• Most carbohydrate calories should be from whole grains and low glycemic index foods. A total fat intake of 30%–35% is preferable
to a very high carbohydrate diet.• Reduce SAFA intake to <7% of the daily energy (10 g for women and 15 g for men), and cholesterol intake to <200 mg/day.• Substitute excess SAFA and carbohydrate calories with MUFA (≤20%) and PUFA (≤10%).• Substitute full-fat with low-fat milk and dairy products.• Choose lean meat or skinless poultry, and limit the amount to <150 g/day.• Minimize the intake of TRAFA by avoiding fried or crispy foods.• Use cooking oils with beneficial effects on lipids and avoid deep-frying, especially with previously used oils.• Consume 2–3 fish meals (200–300 g) per week; avoid frying to maintain the benefits.• Those who are unable to consume fish may take the equivalent of 1g/day of EPA and DHA (3g of fish oil); 2 g/day is needed for
those with heart disease or high TG.• Protein intake, up to 25% of energy, is permissible if most of the protein is from plant sources.• Control caloric intake to achieve and maintain optimum weight and waistline.• Increase physical activity to >60 min every day.• Reduce intake of salt to <6 g/day.• Eat a variety of foods, including whole grains, nuts, legumes, fruits, and vegetables.• Consume nuts, up to 60 g/day, as a substitute for unhealthy foods.• Increase intake of fruits to >5 servings/day (500 g/day), and use whole fruits instead of juices.• Increase intake of vegetables to >5 servings/day (500 g/day), and avoid prolonged cooking of vegetables.• Limit alcohol intake to 1–6 drinks/week.
IHJ-584-03.p65 11/19/2003, 11:27 PM328

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 329
8. Denke MA. Dietary prescriptions to control dyslipidemias.Circulation 2002; 105: 132–135
9. Davis BC, Kris-Etherton PM. Achieving optimal essential fatty acidstatus in vegetarians: current knowledge and practicalimplications. Am J Clin Nutr 2003; 78 (Suppl): 640S–646S
10. Kris-Etherton PM, Harris WS, Appel LJ; AHA Nutrition Committee.American Heart Association. Omega-3 fatty acids andcardiovascular disease: new recommendations from the AmericanHeart Association. Arterioscler Thromb Vasc Biol 2003; 23:151–152
11. Harper CR, Jacobson TA. The fats of life: the role of omega-3 fattyacids in the prevention of coronary heart disease. Arch Intern Med2001; 161: 2185–2192
12. Jonnalagadda S, Mustard V, Champagne C. Margarine and plasmacholesterol—a perspective for dieticians on trans fatty acids in thediet. Mosby-Year Book Inc. 1995; 2: 9–16
13. Enas EA. Cooking oil, cholesterol and CAD: facts and myths. IndianHeart J 1996; 48: 423–427
14. NCEP III. Third Report of the National Cholestrol EducationProgram (NCEP) Adult Treatment Panel III: National Institute ofHealth; 2002
15. Verschuren WM, Jacobs DR, Bloemberg BP, Kromhout D, MenottiA, Aravanis C, et al. Serum total cholesterol and long-termcoronary heart disease mortality in different cultures. Twenty-fiveyear follow-up of the seven countries study. JAMA 1995; 274:131–136
16. Artaud-Wild SM, Connor SL, Sexton G, Connor WE. Differences incoronary mortality can be explained by differences in cholesteroland saturated fat intakes in 40 countries but not in France andFinland. A paradox. Circulation 1993; 88: 2771–2779
17. Aravanis C, Corcondilas A, Dontas AS, Lekos D, Keys A. Coronaryheart disease in seven countries. IX. The Greek islands of Crete andCorfu. Circulation 1970; 41 (Suppl): 88–100
18. Kimura N, Keys A. Coronary heart disease in seven countries. X.Rural southern Japan. Circulation 1970; 41 (Suppl): I101–112
19. Schafer EJ, Lichtenstein A, Lamon-Fava S, McNamara JR, OrdovasJM. Lipoproteins, nutrition, aging, and atherosclerosis. Am J ClinNutr 1995; 61 (Suppl): 726S–740S
20. Knuiman JT, West CE, Katan MB, Hautvast JG. Total cholesteroland high density lipoprotein cholesterol levels in populationsdiffering in fat and carbohydrate intake. Arteriosclerosis 1987; 7:612–619
21. Denke MA, Grundy SM. Comparison of effects of lauric acid andpalmitic acid on plasma lipids and lipoproteins. Am J Clin Nutr1992; 56: 895–898
22. Bonanome A, Grundy SM. Effect of dietary stearic acid on plasmacholesterol and lipoprotein levels. N Engl J Med 1988; 318:1244–1248
23. Grundy SM. Comparison of monounsaturated fatty acids andcarbohydrates for lowering plasma cholesterol. N Engl J Med 1986;314: 745–748
24. Renaud S, Delorgel M. Dietary lipids and their relation to ischemicheart disease from epidemiology to prevention. J Int Med 1989;225 (Suppl): 39–46
25. Katan MB, Zock PL, Mensink RP. Dietary oils, serum lipoproteins,and coronary heart disease. Am J Clin Nutr 1995; 61 (Suppl):1368S–1373S
26. Mann JI. The role of nutritional modifications in the prevention ofmacrovascular complications of diabetes. Diabetes 1997; 46(Suppl): S125–S130
27. Hostmark AT, Spydevold O, Eilertsen E. Plasma lipid concentrationand liver output of lipoproteins in rats fed coconut fat or sunfloweroil. Artery 1980; 7: 367–383
28. Van Heek M, Zilversmit DB. Evidence for an inverse relation betweenplasma triglyceride and aortic cholesterol in the coconut oil/
cholesterol-fed rabbit. Atherosclerosis 1988; 71: 185–19229. Hu FB, Stampfer MJ, Manson JE, Rimm E, Colditz GA, Rosner BA,
et al. Dietary fat intake and the risk of coronary heart disease inwomen. N Engl J Med 1997; 337: 1491–1499
30. Hu FB, Willett WC. Optimal diets for prevention of coronary heartdisease. JAMA 2002; 288: 2569–2578
31. Ernst ND, Sempos CT, Briefel RR, Clark MB. Consistency betweenUS dietary fat intake and serum total cholesterol concentrations:the National Health and Nutrition Examination Surveys. Am J ClinNutr 1997; 66 (Suppl): 965S–972S
32. Willet WC. Polyunsaturated fat and risk of cancer. BMJ 1995; 311:1239–1240
33. Byers T. Hardened fats, hardened arteries? N Engl J Med 1997; 337:1544–1545
34. Lemaitre RN, King IB, Raghunathan TE, Pearce RM, Weinmann S,Knopp RH, et al. Cell membrane trans-fatty acids and the risk ofprimary cardiac arrest. Circulation 2002; 105: 697–701
35. Katz AM. Trans-fatty acids and sudden cardiac death. Circulation2002; 105: 669–671
36. Zock PL, Katan MB. Trans fatty acids, lipoproteins, and coronaryrisk. Can J Physiol Pharmacol 1997; 75: 211–216
37. Ascherio A, Hennekens CH, Buring JE, Master C, Stampfer MJ,Willett WC. Trans-fatty acids intake and risk of myocardialinfarction. Circulation 1994; 89: 94–101
38. de Roos NM, Bots ML, Katan MB. Replacement of dietary saturatedfatty acids by trans fatty acids lowers serum HDL cholesterol andimpairs endothelial function in healthy men and women.Arterioscler Thromb Vasc Biol 2001; 21: 1233–1237
39. Mauger JF, Lichtenstein AH, Ausman LM, Jalbert SM, JauhiainenM, Ehnholm C, et al. Ef fect of dif ferent forms of dietaryhydrogenated fats on LDL particle size. Am J Clin Nutr 2003; 78:370–375
40. Willet WC, Ascherio A. Response to the international life sciencesinstitutes report on trans fatty acids. Am J Clin Nutr 1995; 62:524–526
41. Lichtenstein AH, Ausman LM, Jalbert SM, Schaefer EJ. Effects ofdifferent forms of dietary hydrogenated fats on serum lipoproteincholesterol levels [Erratum in N Engl J Med 1999; 341: 856]. NEngl J Med 1999; 340: 1933–1940
42. Mensink RP, Zock PL, Katan MB, Hornstra G. Effect of dietary cisand trans fatty acids on serum lipoprotein [a] levels in humans. JLipid Res 1992; 33: 1493–1501
43. Judd JT, Baer DJ, Clevidence BA, Muesing RA, Chen SC, WeststrateJA, et al. Effects of margarine compared with those of butter onblood lipid profiles related to cardiovascular disease risk factors innormolipemic adults fed controlled diets. Am J Clin Nutr 1998; 68:768–777
44. Aro A, Jauhiainen M, Partanen R, Salminen I, Mutanen M. Stearicacid, trans fatty acids, and dairy fat: effects on serum andlipoprotein lipids, apolipoproteins, lipoprotein(a), and lipid transferproteins in healthy subjects. Am J Clin Nutr 1997; 65: 1419–1426
45. Salmeron J, Hu FB, Manson JE, Stampfer MJ, Colditz GA, Rimm EB,et al. Dietary fat intake and risk of type 2 diabetes in women. Am JClin Nutr 2001; 73: 1019–1026
46. Ascherio A, Katan MB, Zock PL, Stampfer MJ, Willett WC. Transfatty acids and coronary heart disease. N Engl J Med 1999; 340:1994–1998
47. Oomen CM, Ocke MC, Feskens EJ, van Erp-Baart MA, Kok FJ,Kromhout D. Association between trans fatty acid intake and10-year risk of coronary heart disease in the Zutphen ElderlyStudy: a prospective population-based study. Lancet 2001; 357:746–751
48. Willett WC, Stampfer MJ, Manson JE, Colditz GA, Speizer FE, RosnerBA, et al. Intake of trans fatty acids and risk of coronary heartdisease among women. Lancet 1993; 341: 581–585
IHJ-584-03.p65 11/19/2003, 11:27 PM329

330 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
49. Christiansen E, Schnider S, Palmvig B, Tauber-Lassen E, PedersenO. Intake of a diet high in trans monounsaturated fatty acids orsaturated fatty acids. Effects on postprandial insulinemia andglycemia in obese patients with NIDDM. Diabetes Care 1997; 20:881–887
50. Byers T. Body weight and mortality. N Engl J Med 1995; 333:723–724
51. Enig MG, Atal S, Keeney M, Sampugna J. Isomeric trans fatty acidsin the U.S. diet. [Erratum in J Am Coll Nutr 1991; 10: 514]. J AmColl Nutr 1990; 9: 471–486
52. Hulshof KF, van Erp-Baart MA, Anttolainen M, Becker W, ChurchSM, Couet C, et al. Intake of fatty acids in western Europe withemphasis on trans fatty acids: the TRANSFAIR Study. Eur J ClinNutr 1999; 53: 143–157
53. Trans fatty acids and coronary heart disease risk. Report of theexpert panel on trans fatty acids and coronary heart disease. Am JClin Nutr 1995; 62: 655S–708S; discussion 518–526
54. Denke MA, Adams-Huet B, Nguyen AT. Individual cholesterolvariation in response to a margarine- or butter-based diet: a studyin families. JAMA 2000; 284: 2740–2747
55. Parillo M, Rivellese AA, Ciardullo AV, Capaldo B, Giacco A,Genovese S, et al. A high-monounsaturated-fat/low-carbohydratediet improves peripheral insulin sensitivity in non-insulin-dependent diabetic patients. Metabolism 1992; 41: 1373–1378
56. Vessby B, Aro A, Skarfors E, Berglund L, Salminen I, Lithell H. Therisk to develop NIDDM is related to the fatty acid composition ofthe serum cholesterol esters. Diabetes 1994; 43: 1353–1357
57. Vessby B, Unsitupa M, Hermansen K, Riccardia G, Rivellese AA,Tapsell LC, et al.; KANWU Study. Substituting dietary saturatedfor monounsaturated fat impairs insulin sensitivity in healthy menand women: the KANWU Study. Diabetologia 2001; 44: 312–319
58. Heine RJ, Mulder C, Popp-Snijders C, van der Meer J, van der VeenEA. Linoleic-acid-enriched diet: long-term effects on serumlipoprotein and apolipoprotein concentrations and insulinsensitivity in noninsulin-dependent diabetic patients. Am J Clin Nutr1989; 49: 448–456
59. Feskens EJ, Kromhout D. Habitual dietary intake and glucosetolerance in euglycaemic men: the Zutphen Study. Int J Epidemiol1990; 19: 953–959
60. Marshall JA, Hoag S, Shetterly S, Hamman RF. Dietary fat predictsconversion from impaired glucose tolerance to NIDDM. The SanLuis Valley Diabetes Study. Diabetes Care 1994; 17: 50–56
61. Kukita H, Imamura Y, Hamada M, Joh T, Kokubu T. Plasma lipidsand lipoproteins in Japanese male patients with coronary arterydisease and in their relatives. Atherosclerosis 1982; 42: 21–29
62. Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipidsand lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb1992; 12: 911–919
63. Hu FB, Willett WC. Diet and coronary heart disease: findings fromthe Nurses’ Health Study and Health Professionals’ Follow-upStudy. J Nutr Health Aging 2001; 5: 132–138
64. Ascherio A, Rimm EB, Giovannucci EL, Spiegelman D, StampferM, Willet WC. Dietary fat and the risk of coronary heart disease inmen: cohort follow up study in the United States. BMJ 1996; 313:84–90
65. USDA nutrient database for standard reference. Accessed October6, 2003. Available from: URL: http://www.nal.usda.gov/fnic/cgi-bin; www.nal.usda.gov/fnic/cgi-bin
66. Hu FB, Stampfer MJ. Nut consumption and risk of coronary heartdisease: a review of epidemiologic evidence. Curr Atheroscler Rep1999; 1: 204–209
67. Ghafoorunissa. Fats in Indian diets and their nutritional and healthimplications. Lipids 1996; 31 (Suppl): S287–S291
68. Lichtenstein AH, Ausman LM, Carrasco W, Jenner JL, Gualtieri LJ,Goldin BR, et al. Effects of canola, corn, and olive oils on fasting
and postprandial plasma lipoproteins in humans as part of aNational Cholesterol Education Program Step 2 diet. ArteriosclerThromb 1993; 13: 1533–1542
69. Chait A, Onitiri A, Nicoll A, Rabaya E, Davies J, Lewis B. Reductionof serum triglyceride levels by polyunsaturated fat. Studies on themode of action and on very low density lipoprotein composition.Atherosclerosis 1974; 20: 347–364
70. Mattson FH, Grundy SM. Comparison of effects of dietarysaturated, monounsaturated, and polyunsaturated fatty acids onplasma lipids and lipoproteins in man. J Lipid Res 1985; 26:194–202
71. Vega GL, Groszek E, Wolf R, Grundy SM. Influence ofpolyunsaturated fats on composition of plasma lipoproteins andapolipoproteins. J Lipid Res 1982; 23: 811–822
72. Jackson RL, Kashyap ML, Barnhart RL, Allen C, Hogg E, Glueck CJ.Influence of polyunsaturated and saturated fats on plasma lipidsand lipoproteins in man. Am J Clin Nutr 1984; 39: 589–597
73. Johnson CL, Rifkind BM, Sempos CT, Cassoll MD, Bachorik PS,Briefel RR, et al. Declining serum total cholesterol levels amongUS adults. The National Health and Nutrition ExaminationSurveys. JAMA 1993; 269: 3002–3008
74. Simopoulos AP, Leaf A, Salem N. Workshop on the essentiality ofand recommended dietary intakes for n-6 and n-3 fatty acids.Bethesda, MD: National Institutes of Health; 1999
75. Pischon T, Hankinson SE, Hotamisligil GS, Rifai N, Willett WC,Rimm EB. Habitual dietary intake of n-3 and n-6 fatty acids inrelation to inflammatory markers among US men and women.Circulation 2003; 108: 155–160
76. Zock PL, Katan MB. Linoleic acid intake and cancer risk: a reviewand meta-analysis. Am J Clin Nutr 1998; 68: 142–153
77. Li D, Sinclair A, Wilson A, Nakkote S, Kelly F, Abedin L, et al. Effectof dietary alpha-linolenic acid on thrombotic risk factors invegetarian men. Am J Clin Nutr 1999; 69: 872–882
78. Connor WE. Alpha-linolenic acid in health and disease. Am J ClinNutr 1999; 69: 827–828
79. Djousse L, Pankow JS, Eckfeldt JH, Folom AR, Hopkins PN, ProvinceMA, et al. Relation between dietary linolenic acid and coronaryartery disease in the National Heart, Lung, and Blood InstituteFamily Heart Study. Am J Clin Nutr 2001; 74: 612–619
80. Gerster H. Can adults adequately convert alpha-linolenic acid (18:3n-3) to eicosapentaenoic acid (20: 5n-3) and docosahexaenoicacid (22: 6n-3)? Int J Vitam Nutr Res 1998; 68: 159–173
81. Simopoulos AP, Leaf A, Salem N Jr. Workshop statement on theessentiality of and recommended dietary intakes for omega-6 andomega-3 fatty acids. Prostaglandins Leukot Essent Fatty Acids 2000;63: 119–121
82. Bang HO, Dyerberg J, Hjoorne N. The composition of food consumedby Greenland Eskimos. Acta Med Scand 1976; 200: 69–73
83. Kromhout D, Bosschieter EB, de Lezenne Coulander C. The inverserelation between fish consumption and 20-year mortality fromcoronary heart disease. N Engl J Med 1985; 312: 1205–1209
84. Albert CM, Hennekens CH, O’Donnell CJ, Ajani UA, Carey VJ, WillettWC, et al. Fish consumption and risk of sudden cardiac death.JAMA 1998; 279: 23–28
85. Hojo N, Fukushima T, Isobe A, Gao T, Shiwaku K, Ishida K, et al.Effect of serum fatty acid composition on coronary atherosclerosisin Japan. Int J Cardiol 1998; 66: 31–38
86. Tavani A, Pelucchi C, Negri E, Bertuzzi M, La Vecchia C. n-3polyunsaturated fatty acids, fish, and nonfatal acute myocardialinfarction. Circulation 2001; 104: 2269–2272
87. Marckmann P. Fishing for heart protection. Am J Clin Nutr 2003;78: 1–2
88. Marckmann P, Gronbaek M. Fish consumption and coronary heartdisease mortality. A systematic review of prospective cohort studies.Eur J Clin Nutr 1999; 53: 585–590
IHJ-584-03.p65 11/19/2003, 11:27 PM330

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 331
89. Burr ML, Fehily AM, Gilbert JF, Rogers S, Holliday RM, SweetnamPM, et al. Effects of changes in fat, fish, and fibre intakes on deathand myocardial reinfarction: diet and reinfarction trial (DART).Lancet 1989; 2: 757–761
90. GISSI. Dietary supplementation with n-3 polyunsaturated fattyacids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenzanell’Infarto miocardico. Lancet 1999; 354: 447–455
91. Rissanen T, Voutilainen S, Nyyssonen K, Lakka TA, Salonen JT. Fishoil-derived fatty acids, docosahexaenoic acid and docosapentaenoicacid, and the risk of acute coronary events: the Kuopio ischaemicheart disease risk factor study. Circulation 2000; 102: 2677–2679
92. Daviglus ML, Stamler J, Orencia A, Dyer AR, Liu K, Greenland P, etal. Fish consumption and the 30-year risk of fatal myocardialinfarction. N Engl J Med 1997; 336: 1046–1053
93. Dolecek TA. Epidemiological evidence of relationships betweendietary polyunsaturated fatty acids and mortality in the multiplerisk factor intervention trial. Proc Soc Exp Biol Med 1992; 200: 177–182
94. Rodriguez BL, Sharp DS, Abbott RD, Burchfiel CM, Masaki K, ChyouPH, et al. Fish intake may limit the increase in risk of coronaryheart disease morbidity and mortality among heavy smokers. TheHonolulu Heart Program. Circulation 1996; 94: 952–956
95. Siscovick DS, Raghunathan TE, King I, Weinmann S, WicklundKG, Albright J, et al. Dietary intake and cell membrane levels oflong-chain n-3 polyunsaturated fatty acids and the risk of primarycardiac arrest. JAMA 1995; 274: 1363–1367
96. Leaf A, Kang JX, Xiao YF, Billman GE. Clinical prevention of suddencardiac death by n-3 polyunsaturated fatty acids and mechanismof prevention of arrhythmias by n-3 fish oils. Circulation 2003;107: 2646–2652
97. Harris WS. Fish oils and plasma lipid and lipoprotein metabolismin humans: a critical review. J Lipid Res 1989; 30: 785–807
98. He K, Rimm EB, Merchant A, Rosner BA, Stampfer MJ, Willett WC,et al. Fish consumption and risk of stroke in men. JAMA 2002;288: 3130–3136
99. Hu FB, Stampfer MJ, Manson JE, Aschrio A, Colditz GA, Speizer FE,et al. Dietary saturated fats and their food sources in relation tothe risk of coronary heart disease in women. Am J Clin Nutr 1999;70: 1001–1008
100. Bucher HC, Hengstler P, Schindler C, Meier G. N-3 polyunsaturatedfatty acids in coronary heart disease: a meta-analysis ofrandomized controlled trials. Am J Med 2002; 112: 298–304
101. Siscovick DS, Lemaitre RN, Mozaffarian D. The fish story: a diet–heart hypothesis with clinical implications: n-3 polyunsaturatedfatty acids, myocardial vulnerability, and sudden death. Circulation2003; 107: 2632–2634
102. Hu FB, Bronner L, Willett WC, Stampfer MJ, Rexrode KM, AlbertCM, et al. Fish and omega-3 fatty acid intake and risk of coronaryheart disease in women. JAMA 2002; 287: 1815–1821
103. Hu FB, Cho EC, Rexrode KM, Albert CM, Manson JE. Fish and long-chain omega-3 fatty acid intake and risk of coronary heart diseaseand total mortality in diabetic women. Circulation 2003; 107:1852–1857
104. Ulbricht TL, Southgate DA. Coronary heart disease: seven dietaryfactors. Lancet 1991; 338: 985–992
105. Adler AJ, Holub BJ. Effect of garlic and fish-oil supplementation onserum lipid and lipoprotein concentrations in hypercholesterolemicmen. Am J Clin Nutr 1997; 65: 445–450
106. De Caterina R, Madonna R, Zucchi R, La Rovere MT.Antiarrhythmic effects of omega-3 fatty acids: from epidemiologyto bedside. Am Heart J 2003; 146: 420–430
107. Axelrod L, Camuso J, Williams E, Kleinman K, Briones E, SchoenfeldD. Effects of a small quantity of omega-3 fatty acids oncardiovascular risk factors in NIDDM. A randomized, prospective,
double-blind, controlled study. Diabetes Care 1994; 17: 37–44108. Dewailly EE, Blanchet C, Gingras S, Lemieux S, Sauve L, Bergeron
J, et al. Relations between n-3 fatty acid status and cardiovasculardisease risk factors among Quebecers. Am J Clin Nutr 2001; 74:603–611
109. Kang JX, Leaf A. Prevention of fatal cardiac arrhythmias bypolyunsaturated fatty acids. Am J Clin Nutr 2000; 71 (Suppl):202S–207S
110. Leaf A, Kang JX, Xiao YF, Billman GE, Voskuyl RA. Theantiarrhythmic and anticonvulsant effects of dietary N-3 fattyacids. J Membr Biol 1999; 172: 1–11
111. Leaf A, Kang JX, Xiao YF, Billman GE. n-3 fatty acids in theprevention of cardiac arrhythmias. Lipids 1999; 34 (Suppl):S187–S189
112. McLennan PL, Abeywardena MY, Charnock JS. Dietary fish oilprevents ventricular fibrillation following coronary arteryocclusion and reperfusion. Am Heart J 1988; 116: 709–717
113. Sellmayer A, Witzgall H, Lorenz RL, Weber PC. Effects of dietaryfish oil on ventricular premature complexes. Am J Cardiol 1995;76: 974–977
114. Leaf A. The electrophysiologic basis for the antiarrhythmic andanticonvulsant effects of n-3 polyunsaturated fatty acids: heartand brain. Lipids 2001; 36 (Suppl): S107–S110
115. Fauchier L, Babuty D, Cosnay P, Fauchier JP. Prognostic value ofheart rate variability for sudden death and major arrhythmicevents in patients with idiopathic dilated cardiomyopathy. J Am CollCardiol 1999; 33: 1203–1207
116. Christensen JH, Korup E, Aaroe J, Toft E, Moller J, Rasmussen K, etal. Fish consumption, n-3 fatty acids in cell membranes, and heartrate variability in survivors of myocardial infarction with leftventricular dysfunction. Am J Cardiol 1997; 79: 1670–1673
117. Christensen JH, Skou HA, Fog L, Hansen V, Vesterlund T, DyerbergJ, et al. Marine n-3 fatty acids, wine intake, and heart rate variabilityin patients referred for coronary angiography. Circulation 2001;103: 651–657
118. Kromhout D. Fish consumption and sudden cardiac death. JAMA1998; 279: 65–66
119. Oomen CM, Feskens EJ, Rasanen L. Fish consumption and coronaryheart disease mortality in Finland, Italy, and The Netherlands. AmJ Epidemiol 2000; 151: 999–1006
120. Lemaitre RN, King IB, Mozaffarian D, Kuller LH, Tracy RP, SiscovickDS. n-3 polyunsaturated fatty acids, fatal ischemic heartdisease, and nonfatal myocardial infarction in older adults:the Cardiovascular Health Study. Am J Clin Nutr 2003; 77:319–325
121. Luostarinen R, Boberg M, Saldeen T. Fatty acid composition in totalphospholipids of human coronary arteries in sudden cardiac death.Atherosclerosis 1993; 99: 187–193
122. Forsyth JS, Willatts P, Agostoni C, Bissenden J, Casaer P, Boehm G.Long chain polyunsaturated fatty acid supplementation in infantformula and blood pressure in later childhood: follow up of arandomised controlled trial. BMJ 2003; 326: 953
123. Pauletto P, Pauto M, Angeli MT, Pessina AC, Munhambo A, Bittolo-Bon G, et al. Blood pressure, serum lipids, and fatty acids inpopulations on a lake-fish diet or on a vegetarian diet in Tanzania.Lipids 1996; 31(Suppl): S309–S312
124. Pauletto P, Puato M, Caroli MG, Casiglia E, Munhambo AE,Cazzolato G, et al. Blood pressure and atherogenic lipoproteinprofiles of fish-diet and vegetarian villagers in Tanzania: theLugalawa study. Lancet 1996; 348: 784–788
125. Chan DC, Watts GF, Mori TA, Barrett PH, Redgrave TG, Beilin LJ.Randomized controlled trial of the effect of n-3 fatty acidsupplementation on the metabolism of apolipoprotein B-100 andchylomicron remnants in men with visceral obesity. Am J Clin Nutr2003; 77: 300–307
IHJ-584-03.p65 11/19/2003, 11:27 PM331

332 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
126. Nestel PJ, Connor WE, Reardon MF, Connor S, Wong S, Boston R.Suppression by diets rich in fish oil of very low density lipoproteinproduction in man. J Clin Invest 1984; 74: 82–89
127. Failor RA, Childs MT, Bierman EL. The effects of omega 3 andomega 6 fatty acid-enriched diets on plasma lipoproteins andapoproteins in familial combined hyperlipidemia. Metabolism 1988;37: 1021–1028
128. Parks JS, Wilson MD, Johnson FL, Rudel LL. Fish oil decreaseshepatic cholesteryl ester secretion but not apoB secretion in Africangreen monkeys. J Lipid Res 1989; 30: 1535–1544
129. Shahar E, Folsom AR, Wu KK, Dennis BH, Shimakawa T, ConlanMG, et al. Associations of fish intake and dietary n-3polyunsaturated fatty acids with a hypocoagulable profile. TheAtherosclerosis Risk in Communities (ARIC) Study. ArteriosclerThromb 1993; 13: 1205–1212
130. Dunstan DW, Mori TA, Puddey IB, Beilin LJ, Burke V, Morton AR,et al. The independent and combined effects of aerobic exercise anddietary fish intake on serum lipids and glycemic control in NIDDM.A randomized controlled study. Diabetes Care 1997; 20: 913–921
131. Appel LJ, Miller ER 3rd, Seidler AJ, Whelton PK. Doessupplementation of diet with ‘fish oil’ reduce blood pressure? Ameta-analysis of controlled clinical trials. Arch Intern Med 1993;153: 1429–1438
132. Schmidt EB. The potential of omega-3 fatty acids to reduce CAD.Proceedings of the conference on omega-3 fatty acid in nutritionand vascular biology. Houston, Texas 1994. pp. 208–211
133. Mozaffarian D, Lemaitre RN, Kuller LH, Burke GL, Tracy RP,Siscovick DS; Cardiovascular Health Study. Cardiac benefits of fishconsumption may depend on the type of fish meal consumed: theCardiovascular Health Study. Circulation 2003; 107: 1372–1377
134. Yancy WS Jr, Westman EC, French PA, Califf RM. Diets and clinicalcoronary events: the truth is out there. Circulation 2003; 107:10–16
135. Marchioli R, Barzi F, Bomba E, Chieffo C, Di Gregorio D, Di MascioR, et al. Early protection against sudden death by n-3polyunsaturated fatty acids after myocardial infarction: time-course analysis of the results of the Gruppo Italiano per lo Studiodella Sopravvivenza nell’Infarto Miocardico (GISSI)-Prevenzione.Circulation 2002; 105: 1897–1903
136. de Lorgeril M, Salen P, Martin JL, Monjaud I, Delaye J, Mamelle N.Mediterranean diet, traditional risk factors, and the rate ofcardiovascular complications after myocardial infarction: finalreport of the Lyon Diet Heart Study. Circulation 1999; 99:779–785
137. Zock PL, Kromhout D. [Nutrition and health—fish fatty acidsagainst fatal coronary heart disease.] Ned Tijdschr Geneeskd 2002;146: 2229–2233
138. Leaf A. Dietary prevention of coronary heart disease: the Lyon DietHeart Study. Circulation 1999; 99: 733–735
139. Erkkila AT, Lehto S, Pyorala K, Uusitupa MI. n-3 Fatty acids and5-y risks of death and cardiovascular disease events in patientswith coronary artery disease. Am J Clin Nutr 2003; 78: 65–71
140. Hu FB , Stampfer MJ, Manson JE, Rimm EB, Wolk A, Colditz GA, etal. Dietary intake of alpha-linolenic acid and risk of fatal ischemicheart disease among women. Am J Clin Nutr 1999; 69: 890–897
141. Ghafoorunissa. Requirements of dietary fats to meet nutritionalneeds and prevent the risk of atherosclerosis—an Indianperspective. Indian J Med Res 1998; 108: 191–202
142. Kris-Etherton PM, Taylor DS, Yu-Poth S. Polyunsaturated fattyacids in the food chain in the United States. Am J Clin Nutr 2000;71: 179S–188S
143. Burdge GC, Wootton SA. Conversion of alpha-linolenic acid toeicosapentaenoic, docosapentaenoic and docosahexaenoic acidsin young women. Br J Nutr 2002; 88: 411–420
144. Wolfe BM. Potential role of raising dietary protein intake for
reducing risk of atherosclerosis. Can J Cardiol 1995; 11 (Suppl):127G–131G
145. Hu FB, Stampfer MJ, Manson JE, Rimm E, Colditz GA, Speizer FE, etal. Dietary protein and risk of ischemic heart disease in women.Am J Clin Nutr 1999; 70: 221–227
146. Anderson JW, Johnstone BM, Cook-Newell ME. Meta-analysis ofthe effects of soy protein intake on serum lipids. N Engl J Med 1995;333: 276–282
147. Saturated fatty acids in vegetable oils. Council on Scientific Affairs.JAMA 1990; 263: 693–695
148. Watts GF, Ahmed W, Quiney J. Effective lipid lowering dietsincluding lean meat. Br Med J (Clin Res Ed) 1988; 296: 235–237
149. Scott LW, Dunn JK, Pownall HJ, Branchi DJ, McMann MC, Herd JA,et al. Effects of beef and chicken consumption on plasma lipid levelsin hypercholesterolemic men. Arch Intern Med 1994; 154:1261–1267
150. Bornet FR, Costagliola D, Rizkalla SW. Insulinemic and glycemicindexes of six starch-rich foods taken alone and in a mixed mealby type 2 diabetics. Am J Clin Nutr 1987; 45: 588–595
151. Bonora E, Muggeo M. Postprandial blood glucose as a risk factorfor cardiovascular disease in type II diabetes: the epidemiologicalevidence. Diabetologia 2001; 44: 2107–2114
152. Foster-Powell K, Holt SH, Brand-Miller JC. International table ofglycemic index and glycemic load values: 2002. Am J Clin Nutr2002; 76: 5–56
153. Jenkins DJ, Ghafari H, Wolever TM, Taylor RH, Jenkins AL, BarkerH, et al. Relationship between rate of digestion of foods and post-prandial glycaemia. Diabetologia 1982; 22: 450–455
154. Jenkins DJ, Wolever TM, Taylor RH, Barker H, Fielden H, Baldwin J,et al. Glycemic index of foods: a physiological basis for carbohydrateexchange. Am J Clin Nutr 1981; 34: 362–366
155. Jenkins DJ, Kendall CW, Augustin LS, Franceschi S, Hamidi M, MarcC, et al. Glycemic index: overview of implications in health anddisease. Am J Clin Nutr 2002; 76: 266S–273S
156. Liu S, Willett WC, Stampfer MJ, Hu FB, Franz M, Sampson L, et al.A prospective study of dietary glycemic load, carbohydrate intake,and risk of coronary heart disease in US women. Am J Clin Nutr2000; 71: 1455–1461
157. Ford ES, Liu S. Glycemic index and serum high-density lipoproteincholesterol concentration among US adults. Arch Intern Med 2001;161: 572–576
158. Brand-Miller JC, Holt SH, Pawlak DB, McMillan J. Glycemic indexand obesity. Am J Clin Nutr 2002; 76: 281S–285S
159. Ludwig DS. The glycemic index: physiological mechanisms relatingto obesity, diabetes, and cardiovascular disease. JAMA 2002; 287:2414–2423
160. Schafer G, Schenk U, Ritzel U, Ramadori G, Leonhardt U.Comparison of the effects of dried peas with those of potatoes inmixed meals on postprandial glucose and insulin concentrationsin patients with type 2 diabetes. Am J Clin Nutr 2003; 78: 99–103
161. Willett W, Manson J, Liu S. Glycemic index, glycemic load, and riskof type 2 diabetes. Am J Clin Nutr 2002; 76: 274S–280S
162. Wolever TM, Mehling C. Long-term effect of varying the source oramount of dietary carbohydrate on postprandial plasma glucose,insulin, triacylglycerol, and free fatty acid concentrations insubjects with impaired glucose tolerance. Am J Clin Nutr 2003; 77:612–621
163. Wolever TM, Jenkins DJ, Jenkins AL, Josse RG. The glycemic index: methodology and clinical implications. Am J Clin Nutr1991; 54: 846–854
164. Stevens J, Ahn K, Juhaeri, Houston D, Steffan L, Couper D. Dietaryfiber intake and glycemic index and incidence of diabetes inAfrican–American and white adults: the ARIC study. Diabetes Care2002; 25: 1715–1721
165. Jarvi AE, Karlstrom BE, Granfeldt YE, Bjorck IE, Asp NG, Vessby
IHJ-584-03.p65 11/19/2003, 11:27 PM332

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 333
BO. Improved glycemic control and lipid profile and normalizedfibrinolytic activity on a low-glycemic index diet in type 2 diabeticpatients. Diabetes Care 1999; 22: 10–18
166. Liu S, Manson JE, Stampfer MJ, Holmes MD, Hu FB, Hankinson SE,et al. Dietary glycemic load assessed by food-frequencyquestionnaire in relation to plasma high-density-lipoproteincholesterol and fasting plasma triacylglycerols in postmenopausalwomen. Am J Clin Nutr 2001; 73: 560–566
167. Melish J, Le NA, Ginsberg H, Steinberg D, Brown WV. Dissociationof apoprotein B and triglyceride production in very-low-densitylipoproteins. Am J Physiol 1980; 239: E354–E362
168. Sane T, Nikkila EA. Very low density lipoprotein triglyceridemetabolism in relatives of hypertriglyceridemic probands. Evidencefor genetic control of triglyceride removal. Arteriosclerosis 1988;8: 217–226
169. Goldbourt U, Yaari S. Cholesterol and coronary heart diseasemortality. A 23-year follow-up study of 9902 men in Israel.Arteriosclerosis 1990; 10: 512–519
170. Goldbourt U, Holtzman E, Neufeld HN. Total and high densitylipoprotein cholesterol in the serum and risk of mortality: evidenceof a threshold effect. Br Med J (Clin Res Ed) 1985; 290: 1239–1243
171. Knuiman JT, West CE, Burema J. Serum total and high densitylipoprotein cholesterol concentrations and body mass index in adultmen from 13 countries. Am J Epidemiol 1982; 116: 631–642
172. Grundy SM, Florentin L, Nix D, Whelan MF. Comparison ofmonounsaturated fatty acids and carbohydrates for reducing raisedlevels of plasma cholesterol in man. Am J Clin Nutr 1988; 47:965–969
173. Mensink RP, Katan MB. Effect of monounsaturated fatty acidsversus complex carbohydrates on high-density lipoproteins inhealthy men and women. Lancet 1987; 1: 122–125
174. Kesteloot H, Huang DX, Yang XS, Claes J, Rosseneu M, Geboers J, etal. Serum lipids in the People’s Republic of China. Comparison ofWestern and Eastern populations. Arteriosclerosis 1985; 5:427–433
175. Frost G, Leeds AA, Dore CJ, Madeiros S, Brading S, Dornhorst A.Glycaemic index as a determinant of serum HDL-cholesterolconcentration. Lancet 1999; 353: 1045–1048
176. Jackson RL, Yates MT, McNerney CA, Kashyap ML. Diet and HDLmetabolism: high carbohydrate vs. high fat diets. Adv Exp Med Biol1987; 210: 165–172
177. Brussaard JH, Katan MB, Groot PH, Havekes LM, Hautvast JG.Serum lipoproteins of healthy persons fed a low-fat diet ora polyunsaturated fat diet for three months. A comparison oftwo cholesterol-lowering diets. Atherosclerosis 1982; 42: 205–219
178. Kuusi T, Ehnholm C, Huttunen JK. Concentration and compositionof serum lipoproteins during a low-fat diet at two levels ofpolyunsaturated fat. J Lipid Res 1985; 26: 360–367
179. Jones DY, Judd JT, Taylor PR, Campbell WS, Nair PP. Influenceof caloric contribution and saturation of dietary fat on plasmalipids in premenopausal women. Am J Clin Nutr 1987; 45:1451–1456
180. Jeppesen J, Schaaf P, Jones C, Zhou MY, Chen YD, Reaven GM. Effectsof low-fat, high-carbohydrate diets on risk factors for ischemicheart disease in postmenopausal women. Am J Clin Nutr 1997;65: 1027–1033
181. Mann JI. Diet and risk of coronary heart disease and type 2 diabetes.Lancet 2002; 360: 783–789
182. Salmeron J, Ascherio A, Rimm EB, et al. Dietary fiber, glycemic load,and risk of NIDDM in men. Diabetes Care 1997; 20: 545–550
183. Salmeron J, Manson JE, Stampfer MJ, Colditz GA, Wing AL, WillettWC. Dietary fiber, glycemic load, and risk of non-insulin-dependentdiabetes mellitus in women. JAMA 1997; 277: 472–477
184. Howard BV, Wylie-Rosett J. Sugar and cardiovascular disease: astatement for healthcare professionals from the Committee on
Nutrition of the Council on Nutrition, Physical Activity, andMetabolism of the American Heart Association. Circulation 2002;106: 523–527
185. Pereira MA, Jacobs DR Jr, Pins JJ, et al. Effect of whole grains oninsulin sensitivity in overweight hyperinsulinemic adults. Am J ClinNutr 2002; 75: 848–855
186. Bazzano LA, He J, Ogden LG, Loria C, Vupputuri S, Myers L, et al.Legume consumption and risk of coronary heart disease in US menand women: NHANES I Epidemiologic Follow-up Study. Arch InternMed 2001; 161: 2573–2578
187. Jenkins DJ, Kendall CW, Marchie A, Jenkins AL, Augustin LS, LudwW, et al. Type 2 diabetes and the vegetarian diet. Am J Clin Nutr2003; 78 (Suppl): 610S–616S
188. Liu S. Whole-grain foods, dietary fiber, and type 2 diabetes:searching for a kernel of truth. Am J Clin Nutr 2003; 77: 527–529
189. Fung TT, Hu FB, Pereira MA, Liu S, Stampfer MJ, Colditz GA, et al.Whole-grain intake and the risk of type 2 diabetes: a prospectivestudy in men. Am J Clin Nutr 2002; 76: 535–540
190. Steffen LM, Jacobs DR Jr, Stevens J, Shahar E, Carithers T, FolsomAR. Associations of whole-grain, refined-grain, and fruit andvegetable consumption with risks of all-cause mortality andincident coronary artery disease and ischemic stroke: theAtherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr2003; 78: 383–390
191. Liu S, Manson JE, Stampfer MJ, Hu FB, Giovannucci E, Colditz GA,et al. A prospective study of whole-grain intake and risk of type 2diabetes mellitus in US women. Am J Public Health 2000; 90:1409–1415
192. Bazzano LA, He J, Ogden LG, Loria CM, Vupputuri S, Myers L, et al.Fruit and vegetable intake and risk of cardiovascular disease in USadults: the first National Health and Nutrition Examination SurveyEpidemiologic Follow-up Study. Am J Clin Nutr 2002; 76: 93–99
193. Liu S, Lee IM, Ajani U, Cole SR, Buring JE, Manson JE. ; Physicians’Health Study Intake of vegetables rich in carotenoids and risk ofcoronary heart disease in men: The Physicians’ Health Study. Int JEpidemiol 2001; 30: 130–135
194. Joshipura KJ, Ascherio A, Manson JE, Stampfer MJ, Rimm EB,Speizer FE, et al. Fruit and vegetable intake in relation to risk ofischemic stroke. JAMA 1999; 282: 1233–1239
195. Joshipura KJ, Hu FB, Manson JE, Stamfer MJ, Rimm EB, Speizer FE,et al. The effect of fruit and vegetable intake on risk for coronaryheart disease. Ann Intern Med 2001; 134: 1106–1114
196. Gaziano JM, Manson JE, Branch LG, Colditz GA, Willett WC, BuringJE. A prospective study of consumption of carotenoids in fruits andvegetables and decreased cardiovascular mortality in the elderly.Ann Epidemiol 1995; 5: 255–260
197. Knekt P, Reunanen A, Jarvinen R, Seppanen R, Heliovaara M,Aromaa A. Antioxidant vitamin intake and coronary mortality ina longitudinal population study. Am J Epidemiol 1994; 139:1180–1189
198. Albert CM, Gaziano JM, Willett WC, Manson JE. Nut consumptionand decreased risk of sudden cardiac death in the Physicians’Health Study. Arch Intern Med 2002; 162: 1382–1387
199. Ellsworth JL, Kushi LH, Folsom AR. Frequent nut intake and riskof death from coronary heart disease and all causes inpostmenopausal women: the Iowa Women’s Health Study. NutrMetab Cardiovasc Dis 2001; 11: 372–377
200. Hu FB, Stampfer M, Manson J. Frequent nut consumption and riskof coronary heart disease in women: prospective cohort study. BMJ1998; 317: 1341–1345
201. Fraser GE, Sabate J, Beeson WL, Strahan TM. A possible protectiveeffect of nut consumption on risk of coronary heart disease.The Adventist Health Study. Arch Intern Med 1992; 152:1416–1424
202. Fraser GE, Shavlik D. Risk factors for all-cause and coronary heart
IHJ-584-03.p65 11/19/2003, 11:27 PM333

334 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
disease mortality in the oldest-old. The Adventist Health Study. ArchIntern Med 1997; 157: 2249–2258
203. Jiang R, Manson JE, Stampfer MJ, Liu S, Willett WC, Hu FB. Nutand peanut butter consumption and risk of type 2 diabetes inwomen. JAMA 2002; 288: 2554–2560
204. Jacobs DR Jr, Meyer KA, Kushi LH, Folsom AR. Whole-grain intakemay reduce the risk of ischemic heart disease death inpostmenopausal women: the Iowa Women’s Health Study. Am JClin Nutr 1998; 68: 248–257
205. Liu S, Stampfer MJ, Hu FB, Giovannucci E, Rimm E, Manson JE, etal. Whole-grain consumption and risk of coronary heart disease:results from the Nurses’ Health Study. Am J Clin Nutr 1999; 70:412–419
206. Liu S, Manson JE, Stampfer MJ, Rexrode KM, Hu FB, Rimm EB, etal. Whole grain consumption and risk of ischemic stroke in women:a prospective study. JAMA 2000; 284: 1534–1540
207. Jacobs DR Jr, Meyer KA, Kushi LH, Folsom AR. Whole-grain intakemay reduce the risk of ischemic heart disease death inpostmenopausal women: the Iowa Women’s Health Study. Am JClin Nutr 1998; 68: 248–257
208. Jacobs DR Jr, Meyer HE, Solvoll K. Reduced mortality among wholegrain bread eaters in men and women in the Norwegian CountyStudy. Eur J Clin Nutr 2001; 55: 137–143
209. Liu S, Sesso HD, Manson JE, Willett WC, Buring JE. Is intake ofbreakfast cereals related to total and cause-specific mortality inmen? Am J Clin Nutr 2003; 77: 594–599
210. Meyer KA, Kushi LH, Jacobs DR Jr, Slavin J, Sellers TA, Folsom AR.Carbohydrates, dietary fiber, and incident type 2 diabetes in olderwomen. Am J Clin Nutr 2000; 71: 921–930
211. Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, SacksFM, et al. A clinical trial of the effects of dietary patterns on bloodpressure. DASH Collaborative Research Group. N Engl J Med 1997;336: 1117–1124
212. Montonen J, Knekt P, Jarvinen R, Aromaa A, Reunanen A. Whole-grain and fiber intake and the incidence of type 2 diabetes. Am JClin Nutr 2003; 77: 622–629
213. Jacobs DR Jr, Steffen LM. Nutrients, foods, and dietary patterns asexposures in research: a framework for food synergy. Am J Clin Nutr2003; 78 (Suppl): 508S–513S
214. Kushi LH, Lenart EB, Willett WC. Health implications ofMediterranean diets in light of contemporary knowledge. 1. Plantfoods and dairy products. Am J Clin Nutr 1995; 61: 1407S–1415S
215. Hu FB. Plant-based foods and prevention of cardiovascular disease:an overview. Am J Clin Nutr 2003; 78 (Suppl): 544S–551S
216. Cleveland LE, Moshfegh AJ, Albertson AM, Goldman JD. Dietaryintake of whole grains. J Am Coll Nutr 2000; 19 (Suppl):331S–338S
217. Barnard ND, Scialli AR, Bertron P, Hurlock D, Edmonds K, Talev L.Effectiveness of a low-fat vegetarian diet in altering serum lipids inhealthy premenopausal women. Am J Cardiol 2000; 85: 969–972
218. Fraser GE. Nut consumption, lipids, and risk of a coronary event.Clin Cardiol 1999; 22 (Suppl): III11– III15
219. Jenkins DJ, Kendall CW, Marchie A, Parker TL, Connelly PW,Qian W, et al. Dose response of almonds on coronary heart diseaserisk factors: blood lipids, oxidized low-density lipoproteins,lipoprotein (a), homocysteine, and pulmonary nitric oxide: arandomized, controlled, crossover trial. Circulation 2002; 106:1327–1332
220. Spiller GA, Jenkins DA, Bosello O, Gates JE, Cragen LN, Bruce B.Nuts and plasma lipids: an almond-based diet lowers LDL-C whilepreserving HDL-C. J Am Coll Nutr 1998; 17: 285–290
221. Abbey M, Noakes M, Belling GB, Nestel PJ. Partial replacement ofsaturated fatty acids with almonds or walnuts lowers total plasmacholesterol and low-density-lipoprotein cholesterol. Am J Clin Nutr1994; 59: 995–999
222. Sabate J. Nut consumption and body weight. Am J Clin Nutr 2003;78 (Suppl): 647S–650S
223. Almario RU, Vonghavaravat V, Wong R, Kasim-Karakas SE. Effectsof walnut consumption on plasma fatty acids and lipoproteins incombined hyperlipidemia. Am J Clin Nutr 2001; 74: 72–79
224. Curb JD, Wergowske G, Dobbs JC, Abbott RD, Huang B. Serum lipideffects of a high-monounsaturated fat diet based on macadamianuts. Arch Intern Med 2000; 160: 1154–1158
225. Sabate J, Fraser GE, Burke K, Knutsen SF, Bennett H, Lindsted KD.Effects of walnuts on serum lipid levels and blood pressure innormal men. N Engl J Med 1993; 328: 603–607
226. Edwards K, Kwaw I, Matud J, Kurtz I. Effect of pistachio nuts onserum lipid levels in patients with moderate hypercholesterolemia.J Am Coll Nutr 1999; 18: 229–232
227. Morgan WA, Clayshulte BJ. Pecans lower low-density lipoproteincholesterol in people with normal lipid levels. J Am Diet Assoc 2000;100: 312–318
228. O’Byrne DJ, Knauft DA, Shireman RB. Low fat-monounsaturatedrich diets containing high-oleic peanuts improve serum lipoproteinprofiles. Lipids 1997; 32: 687–695
229. Zambon D, Sabate J, Munoz S, Campero B, Casals E, Merlos M, etal. Substituting walnuts for monounsaturated fat improves theserum lipid profile of hypercholesterolemic men and women. Arandomized crossover trial. Ann Intern Med 2000; 132: 538–546
230. Rajaram S, Burke K, Connell B, Myint T, Sabate J. Amonounsaturated fatty acid-rich pecan-enriched diet favorablyalters the serum lipid profile of healthy men and women. J Nutr2001; 131: 2275–2279
231. Garg A, Bonanome A, Grundy SM, Zhang ZJ, Unger RH.Comparison of a high-carbohydrate diet with a high-monounsaturated-fat diet in patients with non-insulin-dependentdiabetes mellitus. N Engl J Med 1988; 319: 829–834
232. Kris-Etherton PM, Zhao G, Binkoski AE, Coval SM, Etherton TD.The effects of nuts on coronary heart disease risk. Nutr Rev 2001;59: 103–111
233. Lovejoy JC, Most MM, Lefevre M, Greenway FL, Rood JC. Effect ofdiets enriched in almonds on insulin action and serum lipids inadults with normal glucose tolerance or type 2 diabetes. Am J ClinNutr 2002; 76: 1000–1006
234. van Dam RM, Willett WC, Rimm EB, Stampfer MJ, Hu FB. Dietaryfat and meat intake in relation to risk of type 2 diabetes in men.Diabetes Care 2002; 25: 417–424
235. Appel LJ, Miller ER 3rd, Jee SH, Stolzenberg-Solomon R, Lin PH,Erlinger T, et al. Effect of dietary patterns on serum homocysteine:results of a randomized, controlled feeding study. Circulation 2000;102: 852–857
236. Key TJ, Allen NE, Spencer EA, Travis RC. The effect of diet on riskof cancer. Lancet 2002; 360: 861–868
237. Whelton PK, He J, Cutler JA, Brancati FL, Appel LJ, Follmann D, etal. Effects of oral potassium on blood pressure. Meta-analysis ofrandomized controlled clinical trials. JAMA 1997; 277:1624–1632
238. Gillman MW, Cupples L, Gagnon D. Protective effects of friuts andvegetables on development of stroke in men. JAMA 1995; 273:1113 –1117
239. Liu S, Manson JE, Lee IM, Cole SR, Hennekens CH, Willett WC, etal. Fruit and vegetable intake and risk of cardiovascular disease:the Women’s Health Study. Am J Clin Nutr 2000; 72: 922–928
240. Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D,et al; DASH-Sodium Collaborative Research Group. Effects on bloodpressure of reduced dietary sodium and the Dietary Approachesto Stop Hypertension (DASH) diet. DASH-Sodium CollaborativeResearch Group. N Engl J Med 2001; 344: 3–10
241. Obarzanek E, Sacks FM, Vollmer WM, Bray GA, Miller ER 3rd, LinPH, et al; DASH-Research Group Effects on blood lipids of a blood
IHJ-584-03.p65 11/19/2003, 11:27 PM334

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 335
pressure-lowering diet: the Dietary Approaches to StopHypertension (DASH) Trial. Am J Clin Nutr 2001; 74: 80–89
242. Li R, Serdula M, Bland S, Mokdad A, Bowman B, Nelson D. Trendsin fruit and vegetable consumption among adults in 16 US states:Behavioral Risk Factor Surveillance System, 1990–1996. Am JPublic Health 2000; 90: 777–781
243. Katan MB. Flavonoids and heart disease. Am J Clin Nutr 1997; 65:1542–1543
244. Nijveldt RJ, van Nood E, van Hoorn DE, Boelens PG, van Norren K,van Leeuwen PA. Flavonoids: a review of probable mechanisms ofaction and potential applications. Am J Clin Nutr 2001; 74: 418–425
245. Hollman PC, Hertog MG, Katan MB, Role of dietary flavonoids inprotection against cancer and coronary heart disease. Biochem SocTrans 1996; 24: 785–789
246. Knekt P, Jarvinen R, Reunanen A, Maatela J. Flavonoid intake andcoronary mortality in Finland: a cohort study. BMJ 1996; 312:478–481
247. Keli SO, Hertog MG, Feskens EJ, Kromhout D. Dietary flavonoids,antioxidant vitamins, and incidence of stroke: the Zutphen study.Arch Intern Med 1996; 156: 637–642
248. Knekt P, Kumpulainen J, Jarvinen R, Rissanen H, Heliovaara M,Reunanen A, et al. Flavonoid intake and risk of chronic diseases.Am J Clin Nutr 2002; 76: 560–568
249. Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D.Dietary antioxidant flavonoids and risk of coronary heart disease:the Zutphen Elderly Study. Lancet 1993; 342: 1007–1011
250. Hertog MG, Kromhout D, Aravanis C, Blackburn H, Buzina R,Fidanza F, et al. Flavonoid intake and long-term risk of coronaryheart disease and cancer in the seven countries study. Arch InternMed 1995; 155: 381–386
251. Jha P, Flather M, Lonn E, Farkouh M, Yusuf S. The antioxidantvitamins and cardiovascular disease. A critical review ofepidemiologic and clinical trial data. Ann Intern Med 1995; 123:860–872
252. Stephens NG, Parsons A, Schofield PM, Kelly F, Cheeseman K,Mitchinson MJ. Randomised controlled trial of vitamin E in patientswith coronary disease: Cambridge Heart Antioxidant Study(CHAOS). Lancet 1996; 347: 781–786
253. Brown BG, Zhao XQ, Chait A, Fisher LD, Cheung MC, Morse JS, etal. Simvastatin and niacin, antioxidant vitamins, or thecombination for the prevention of coronary disease. N Engl J Med2001; 345: 1583–1592
254. Heart Protection Study Collaborative Group. MRC/BHF HeartProtection Study of antioxidant vitamin supplementation in20,536 high-risk individuals: a randomised placebo-controlledtrial. Lancet 2002; 360: 23–33
255. Brown BG, Cheung MC, Lee AC, Zhao XQ, Chait A. Antioxidantvitamins and lipid therapy: end of a long romance? ArteriosclerThromb Vasc Biol 2002; 22: 1535–1546
256. Gotto AM. Antioxidants, statins, and atherosclerosis. J Am CollCardiol 2003; 41: 1205–1210
257. U.S. Preventive Services Task Force. Routine vitaminsupplementation to prevent cancer and cardiovascular disease:recommendations and rationale. Ann Intern Med 2003; 139:51–55
258. Burkitt D, Trowell H. Refined carbohydrate foods and disease: someimplications of dietary fiber. London: Academic Press 1975
259. Mozaffarian D, Kumanyika SK, Lemaitre RN, Olson JL, Burke GL,Siscovick DS. Cereal, fruit, and vegetable fiber intake and the riskof cardiovascular disease in elderly individuals. JAMA 2003; 289:1659–1666
260. Anderson JW. Dietary fiber prevents carbohydrate-inducedhypertriglyceridemia. Curr Atheroscler Rep 2000; 2: 536–541
261. Burke V, Hodgson JM, Beilin LJ, Giangiulioi N, Rogers P, Puddey IB.
Dietary protein and soluble fiber reduce ambulatory blood pressurein treated hypertensives. Hypertension 2001; 38: 821–826
262. Pereira MA, Pins JJ. Dietary fiber and cardiovascular disease:experimental and epidemiologic advances. Curr Atheroscler Rep2000; 2: 494–502
263. Kushi LH, Meyer KA, Jacobs DR Jr. Cereals, legumes, and chronicdisease risk reduction: evidence from epidemiologic studies. Am JClin Nutr 1999; 70 (Suppl): 451S–458S
264. Wolk A , Manson JE, Stampfer MJ, Coldtiz GA, Hu FB, Speizer FE, etal. Long-term intake of dietary fiber and decreased risk of coronaryheart disease among women. JAMA 1999; 281: 1998–2004
265. Brown L, Rosner B, Willett WW, Sacks FM. Cholesterol-loweringeffects of dietary fiber: a meta-analysis. Am J Clin Nutr 1999; 69:30–42
266. Slavin JL, Martini MC, Jacobs DR Jr, Marquart L. Plausiblemechanisms for the protectiveness of whole grains. Am J Clin Nutr1999; 70: 459S–463S
267. Anderson JW, Bryant CA. Dietary fiber: diabetes and obesity. Am JGastroenterol 1986; 81: 898–906
268. Bazzano LA, He J, Ogden LG, Loria CM, Whelton PK; NationalHealth and Nutrition Examination Survey I Epidemidogic Follow-up Study. Dietary fiber intake and reduced risk of coronary heartdisease in US men and women: the National Health and NutritionExamination Survey I Epidemiologic Follow-up Study. Arch InternMed 2003; 163: 1897–1904
269. Ripsin CM, Keenan JM, Jacobs DR Jr, Elmer PJ, Welch RR, Van HornL, et al. Oat products and lipid lowering. A meta-analysis. JAMA1992; 267: 3317–3325
270. Anderson JW, Allgood LD, Lawrence A, Altringer LA, Jerdack GR,Hengehold DA, et al. Cholesterol-lowering effects of psyllium intakeadjunctive to diet therapy in men and women withhypercholesterolemia: meta-analysis of 8 controlled trials. Am JClin Nutr 2000; 71: 472–479
271. Rimm EB, Ascherio A, Giovannucci E, Spiegelman D, Stampfer MJ,Willett WC. Vegetable, fruit, and cereal fiber intake and risk ofcoronary heart disease among men. JAMA 1996; 275: 447–451
272. Pietinen P, Rimm E, Korhonen P, Hartman AM, Willett WC, AlbanesD, et al. Intake of dietary fiber and risk of coronary heart diseasein a cohort of Finnish men. The Alpha-Tocopherol, Beta-CaroteneCancer Prevention Study. Circulation 1996; 94: 2720–2727
273. Liu S, Buring JE, Sesso HD, Rimm EB, Willett WC, Manson JE. Aprospective study of dietary fiber intake and risk of cardiovasculardisease among women. J Am Coll Cardiol 2002; 39: 49–56
274. Van Horn L. Fiber, lipids, and coronary heart disease. A statementfor healthcare professionals from the Nutrition Committee,American Heart Association. Circulation 1997; 95: 2701–2704
275. Chandalia M, Garg A, Lutjohann D, von Bergmann K, Grundy SM,Brinkley LJ. Beneficial effects of high dietary fiber intake in patientswith type 2 diabetes mellitus. N Engl J Med 2000; 342: 1392–1398
276. Vanstone CA, Raeini-Sarjaz M, Parsons WE, Jones PJ. Unesterifiedplant sterols and stanols lower LDL-cholesterol concentrationsequivalently in hypercholesterolemic persons. Am J Clin Nutr 2002;76: 1272–1278
277. Hallikainen MA, Uusitupa MI. Effects of 2 low-fat stanol ester-containing margarines on serum cholesterol concentrations as partof a low-fat diet in hypercholesterolemic subjects. Am J Clin Nutr1999; 69: 403–410
278. Gylling H, Radhakrishnan R, Miettinen TA. Reduction of serumcholesterol in postmenopausal women with previous myocardialinfarction and cholesterol malabsorption induced by dietarysitostanol ester margarine: women and dietary sitostanol.Circulation 1997; 96: 4226–4231
279. Blair SN, Capuzzi DM, Gottlieb SO, Nguyen T, Morgan JM, CaterNB. Incremental reduction of serum total cholesterol and low-density lipoprotein cholesterol with the addition of plant stanol
IHJ-584-03.p65 11/19/2003, 11:27 PM335

336 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
ester-containing spread to statin therapy. Am J Cardiol 2000; 86:46–52
280. Jenkins DJ, Kendall CW, Marchie A, Faulkner DA, Wong JM, deSouza R, et al. Effects of a dietary portfolio of cholesterol-loweringfoods vs lovastatin on serum lipids and C-reactive protein. JAMA2003; 290: 502–510
281. Jenkins DJ, Kendall CW, Vuksan V. Viscous fibers, health claims,and strategies to reduce cardiovascular disease risk [Editorial;Comment]. Am J Clin Nutr 2000; 71: 401–402
282. Nguyen TT. The cholesterol-lowering action of plant stanol esters.J Nutr 1999; 129: 2109–2112
283. Olson BH, Anderson SM, Becker MP, Anderson JW, HunninghakeDB, Jenkins DJ, et al. Psyllium-enriched cereals lower blood totalcholesterol and LDL cholesterol, but not HDL cholesterol, inhypercholesterolemic adults: results of a meta-analysis. J Nutr1997; 127: 1973–1980
284. Lampe JW. Spicing up a vegetarian diet: chemopreventive effectsof phytochemicals. Am J Clin Nutr 2003; 78 (Suppl): 579S–583S
285. Kleemola P, Jousilahti P, Pietinen P, Vartiainen E, Tuomilehto J.Coffee consumption and the risk of coronary heart disease anddeath. Arch Intern Med 2000; 160: 3393–3400
286. Thelle DS. Coffee, tea and coronary heart disease. Curr Opin Lipidol1995; 6: 25–27
287. Thelle DS, Arnesen E, Forde OH. The Tromso heart study. Doescoffee raise serum cholesterol? N Engl J Med 1983; 308:1454–1457
288. Zock PL, Katan MB, Merkus MP, van Dusseldorp M, Harryvan JL.Ef fect of a lipid-rich fraction from boiled coffee on serumcholesterol. Lancet 1990; 335: 1235 –1237
289. Olthof MR, Hollman PC, Zock PL, Katan MB. Consumption of highdoses of chlorogenic acid, present in coffee, or of black tea increasesplasma total homocysteine concentrations in humans. Am J ClinNutr 2001; 73: 532–538
290. Mennen LI, de Courcy GP, Guilland JC, Ducros V, Bertrais S, NicolasJP, et al. Homocysteine, cardiovascular disease risk factors, andhabitual diet in the French Supplementation with AntioxidantVitamins and Minerals Study. Am J Clin Nutr 2002; 76: 1279–1289
291. Kris-Etherton PM, Keen CL. Evidence that the antioxidantflavonoids in tea and cocoa are beneficial for cardiovascular health.Curr Opin Lipidol 2002; 13: 41–49
292. Sesso HD, Gaziano JM, Buring JE, Hennekens CH. Coffee and teaintake and the risk of myocardial infarction. Am J Epidemiol 1999;149: 162–167
293. Geleijnse JM, Launer LJ, Hofman A, Pols HA, Witteman JC. Teaflavonoids may protect against atherosclerosis: the RotterdamStudy. Arch Intern Med 1999; 159: 2170–2174
294. Mukamal KJ, Maclure M, Muller JE, Sherwood JB, Mittleman MA.Tea consumption and mortality after acute myocardial infarction.Circulation 2002; 105: 2476–2481
295. Peter U, Poole C, Arab L. Does tea affect cardiovascular disease? Ameta-analysis. Am J Epidemiol 2001; 154: 495–503
296. Maron DJ, Lu GP, Cai NS, Wu ZG, Li YH, Chen H, et al. Cholesterol-lowering effect of a theaflavin-enriched green tea extract: arandomized controlled trial. Arch Intern Med 2003; 163:1448–1453
297. Hertog MG, Sweetnam PM, Fehily AM, Elwood PC, Kromhout D.Antioxidant flavonols and ischemic heart disease in a Welshpopulation of men: the Caerphilly Study. Am J Clin Nutr 1997; 65:1489–1494
298. Gaziano JM, Gaziano TA, Glynn RJ, Sesso HD, Ajari UA, StampferMJ, et al. Light-to-moderate alcohol consumption and mortalityin the Physicians’ Health Study enrollment cohort. J Am Coll Cardiol2000; 35: 96–105
299. Berger K, Ajani UA, Kase CS, Gaziano JM, Buring JE, Glynn RJ, etal. Light-to-moderate alcohol consumption and risk of stroke
among U.S. male physicians. N Engl J Med 1999; 341: 1557–1564300. Di Castelnuovo A, Rotondo S, Iacoviello L, Donati MB, De Gaetano
G. Meta-analysis of wine and beer consumption in relation tovascular risk. Circulation 2002; 105: 2836–2844
301. Thadhani R, Camargo CA Jr, Stampfer MJ, Curhan GC, Willett WC,Rimm EB. Prospective study of moderate alcohol consumption andrisk of hypertension in young women. Arch Intern Med 2002; 162:569–574
302. Crouse JR, Grundy SM. Effects of alcohol on plasma lipoproteinsand cholesterol and triglyceride metabolism in man. J Lipid Res1984; 25: 486–496
303. Krauss RM, Eckel RH, Howard B, Appel LJ, Daniels SR, DeckelbaumRJ, et al. AHA Dietary Guidelines: revision 2000: a statement forhealthcare professionals from the Nutrition Committee of theAmerican Heart Association. Circulation 2000; 102: 2284–2299
304. Freedman MR, King J, Kennedy E. Popular diets: a scientific review.Obes Res 2001; 9 (Suppl): 1S–40S
305. Ornish D. Dean Ornish, MD: a conversation with the editor.Interview by William Clifford Roberts, MD. Am J Cardiol 2002; 90:271–298
306. Kennedy ET, Bowman SA, Spence JT, Freedman M, King J. Populardiets: correlation to health, nutrition, and obesity. J Am Diet Assoc2001; 101: 411–420
307. Zambon A, Sartore G, Passera D, Francini-Pesenti F, Bassi A, BassoC, et al. Effects of hypocaloric dietary treatment enriched in oleicacid on LDL and HDL subclass distribution in mildly obese women.J Intern Med 1999; 246: 191–201
308. Heilbronn LK, Noakes M, Clifton PM. Effect of energy restriction,weight loss, and diet composition on plasma lipids and glucose inpatients with type 2 diabetes. Diabetes Care 1999; 22: 889–895
309. McManus K, Antinoro L, Sacks F. A randomized controlled trial ofa moderate-fat, low-energy diet compared with a low fat, low-energy diet for weight loss in overweight adults. Int J Obes RelatMetab Disord 2001; 25: 1503–1511
310. Shah M, Garg A. High-fat and high-carbohydrate diets and energybalance. Diabetes Care 1996; 19: 1142–1152
311. Seidell JC. Dietary fat and obesity: an epidemiologic perspective.Am J Clin Nutr 1998; 67: 546S–550S
312. Sattar N, Gaw A, Scherbakova O, Ford I, O’Reilly DS, Haffner SM,et al. Metabolic syndrome with and without C-reactive protein asa predictor of coronary heart disease and diabetes in the West ofScotland Coronary Prevention Study. Circulation 2003; 108:414–419
313. Schwarz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novolipogenesis in normoinsulinemic and hyperinsulinemic subjectsconsuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr 2003; 77: 43–50
314. Edelstein SL, Knowler WC, Bain RP, Andres R, Barrett-Cornor EL,Dowse GK, et al. Predictors of progression from impaired glucosetolerance to NIDDM: an analysis of six prospective studies. Diabetes1997; 46: 701–710
315. Illustrated Guide to heart disease in Asian Indians. Accessed on10 Sept 2003. Available from: URL: “http: /www.cadiresearch.com;www.cadiresearch.com.
316. Raben A, Vasilaras TH, Moller AC, Astrup A. Sucrose comparedwith artificial sweeteners: different effects on ad libitum food intakeand body weight after 10 wk of supplementation in overweightsubjects. Am J Clin Nutr 2002; 76: 721–729
317. Swinburn BA, Metcalf PA, Ley SJ. Long-term (5-year) effects of areduced-fat diet intervention in individuals with glucoseintolerance. Diabetes Care 2001; 24: 619–624
318. Bonow RO, Eckel RH. Diet, obesity, and cardiovascular risk. N EnglJ Med 2003; 348: 2057–2058
319. Ornish D. Low-fat diets. N Engl J Med 1998; 338: 127–129320. Katan MB. Effect of low-fat diets on plasma high-density lipoprotein
IHJ-584-03.p65 11/19/2003, 11:27 PM336

Indian Heart J 2003; 55: 310–338 Enas et al. Prudent Diet and Preventive Nutrition 337
concentrations. Am J Clin Nutr 1998; 67: 573S–576S321. Mensink RP, Katan MB. Effect of a diet enriched with
monounsaturated or polyunsaturated fatty acids on levels of low-density and high-density lipoprotein cholesterol in healthy womenand men. N Engl J Med 1989; 321: 436–441
322. Ginsberg H, Olefsky JM, Kimmerling G, Crapo P, Reaven GM.Induction of hypertriglyceridemia by a low-fat diet. J Clin EndocrinolMetab 1976; 42: 729–735
323. Atkins RC, Ornish D, Wadden T. Low-carb, low-fat diet gurus faceoff. Interview by Joan Stephenson. JAMA 2003; 289: 1767–1768,1773
324. St Jeor ST, Howard BV, Prewitt TE, Bovee V, Bazzarre T, Eckel RH;Nutrition Committee of the Council on Nutrition, Physical Activity,and Metasodium of the American Heart Association. Dietaryprotein and weight reduction: a statement for healthcareprofessionals from the Nutrition Committee of the Council onNutrition, Physical Activity, and Metabolism of the American HeartAssociation. Circulation 2001; 104: 1869–1874
325. Cahill GF Jr. Starvation in man. N Engl J Med 1970; 282: 668–675326. Foster GD, Wyatt HR, Hill JO, McGuckin BG, Brill C, Mohammed
BS, et al. A randomized trial of a low-carbohydrate diet for obesity.N Engl J Med 2003; 348: 2082–2090
327. Samaha FF, Iqbal N, Seshadri P, Chicano KL, Daily DA, McGrory J,et al. A low-carbohydrate as compared with a low-fat diet in severeobesity. N Engl J Med 2003; 348: 2074–2081
328. Kwiterovich PO Jr, Vining EP, Pyzik P, Skolasky R Jr, Freeman JM.Effect of a high-fat ketogenic diet on plasma levels of lipids,lipoproteins, and apolipoproteins in children. JAMA 2003; 290:912–920
329. Reddy ST, Wang CY, Sakhaee K, Brinkley L, Pak CY. Effect of low-carbohydrate high-protein diets on acid–base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis 2002;40: 265–274
330. Haddad EH, Tanzman JS. What do vegetarians in the United Stateseat? Am J Clin Nutr 2003; 78 (Suppl): 626S–632S
331. Singh RB, Sharma JP, Rastogi V, Raghuvanshi RS, Moshiri M, VermaSP, et al. Prevalence of coronary artery disease and coronary riskfactors in rural and urban populations of north India. Eur Heart J1997; 18: 1728–1735
332. Kant AK, Block G, Schatzkin A, Ziegler RG, Nestle M. Dietarydiversity in the US population, NHANES II, 1976–1980. J Am DietAssoc 1991; 91: 1526–1531
333. Singh PN, Sabate J, Fraser GE. Does low meat consumption increaselife expectancy in humans? Am J Clin Nutr 2003; 78 (Suppl): 526S–532S
334. Appleby PN, Thorogood M, Mann JI, Key TJ. Low body mass indexin non-meat eaters: the possible roles of animal fat, dietary fibreand alcohol. Int J Obes Relat Metab Disord 1998; 22: 454–460
335. Rajaram S. The effect of vegetarian diet, plant foods, andphytochemicals on hemostasis and thrombosis. Am J Clin Nutr2003; 78 (Suppl): 552S–558S
336. Janelle KC, Barr SI. Nutrient intakes and eating behavior scores ofvegetarian and nonvegetarian women. J Am Diet Assoc 1995; 95:180–186, 189, quiz 187–188
337. Key TJ, Fraser GE, Thorogood M, Appleby PN, Bernal V, Reeves G,et al. Mortality in vegetarians and non-vegetarians: a collaborativeanalysis of 8300 deaths among 76,000 men and women in fiveprospective studies. Public Health Nutr 1998; 1: 33–41
338. Burr ML, Bates CJ, Fehily AM, St Leger AS. Plasma cholesterol andblood pressure in vegetarians. J Hum Nutr 1981; 35: 437–441
339. Thorogood M, Carter R, Benfield L, McPherson K, Mann JI. Plasmalipids and lipoprotein cholesterol concentrations in people withdifferent diets in Britain. Br Med J (Clin Res Ed) 1987; 295: 351–353
340. Fisher M, Levine PH, Weiner B, Ockere IS, Johnson B, Johnson UH,
et al. The effect of vegetarian diets on plasma lipid and plateletlevels. Arch Intern Med 1986; 146: 1193–1197
341. Jenkins DJ, Kendall CW, Popovich DG, Vidgen E, Mehling CC,Vuksan V, et al. Effect of a very-high-fiber vegetable, fruit, and nutdiet on serum lipids and colonic function. Metabolism 2001; 50:494–503
342. Sacks FM, Castelli WP, Donner A, Kass EH. Plasma lipids andlipoproteins in vegetarians and controls. N Engl J Med 1975; 292:1148–1151
343. Singh PN, Lindsted KD. Body mass and 26-year risk of mortalityfrom specific diseases among women who never smoked.Epidemiology 1998; 9: 246–254
344. Snowdon DA, Phillips RL. Does a vegetarian diet reduce theoccurrence of diabetes? Am J Public Health 1985; 75: 507–512
345. Snowdon DA, Phillips RL, Fraser GE. Meat consumption and fatalischemic heart disease. Prev Med 1984; 13: 490–500
346. Fraser GE, Lindsted KD, Beeson WL. Effect of risk factor values onlifetime risk of and age at first coronary event. The Adventist HealthStudy. Am J Epidemiol 1995; 142: 746–758
347. Thorogood M, Mann J, Appleby P, McPherson K. Risk of death fromcancer and ischaemic heart disease in meat and non-meat eaters.BMJ 1994; 308: 1667–1670
348. Key TJ, Appleby PN, Davey GK, Allen NE, Spencer EA, Travis RC.Mortality in British vegetarians: review and preliminary resultsfrom EPIC-Oxford. Am J Clin Nutr 2003; 78 (Suppl): 533S–538S
349. Burr ML, Butland BK. Heart disease in British vegetarians. Am JClin Nutr 1988; 48: 830–832
350. Yagalla MV, Hoerr SL, Song WO, Enas E, Garg A. Relationship ofdiet, abdominal obesity, and physical activity to plasma lipoproteinlevels in Asian Indian physicians residing in the United States. JAm Diet Assoc 1996; 96: 257–261
351. Enas EA, Garg A, Davidson MA, Nair VM, Huet BA, Yusuf S.Coronary heart disease and its risk factors in first-generationimmigrant Asian Indians to the United States of America. IndianHeart J 1996; 48: 343–353
352. Enas EA, Yusuf S. Third meeting of the International WorkingGroup on Coronary Artery Disease in South Asians. 29 March1998, Atlanta, USA. Indian Heart J 1999; 51: 99–103
353. Sacks FM, Ornish D, Rosner B, McLanahan S, Castelli WP, KassEH. Plasma lipoprotein levels in vegetarians. The effect of ingestionof fats from dairy products. JAMA 1985; 254: 1337–41
354. Pimentel D, Pimentel M. Sustainability of meat-based and plant-based diets and the environment. Am J Clin Nutr 2003; 78 (Suppl):660S–663S
355. Abbasi F, McLaughlin T, Lamendola C, Kim HS, Tanaka A, Wang T,et al. High carbohydrate diets, triglyceride-rich lipoproteins, andcoronary heart disease risk. Am J Cardiol 2000; 85: 45–48
356. Williams MJ, Sutherland WH, McCormick MP, de Jong SA, WalkerRJ, Wilkins GT. Impaired endothelial function following a meal richin used cooking fat. J Am Coll Cardiol 1999; 33: 1050–1055
357. Sutherland WH, Walker RJ, de Jong SA, van Rij AM, Phillips V,Walker HL. Reduced postprandial serum paraoxonase activity aftera meal rich in used cooking fat. Arterioscler Thromb Vasc Biol 1999;19: 1340–1347
358. Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, FogelmanAM, et al. Protective effect of high density lipoprotein associatedparaoxonase. Inhibition of the biological activity of minimallyoxidized low density lipoprotein. J Clin Invest 1995; 96: 2882–2891
359. Witztum JL. The oxidation hypothesis of atherosclerosis. Lancet1994; 344: 793–795
360. Steinberg D. Lewis A. Conner Memorial Lecture. Oxidativemodification of LDL and atherogenesis. Circulation 1997; 95:1062–1071
361. Thompson JA, May WA, Paulose MM, Peterson RJ, Chang SSChemical reactions involved in the deep-fat frying of foods. VII.
IHJ-584-03.p65 11/19/2003, 11:27 PM337

338 Enas et al. Prudent Diet and Preventive Nutrition Indian Heart J 2003; 55: 310–338
Identification of volatile decomposition products of trilinolein. JAm Oil Chem Soc 1978; 55: 897–901
362. Vijaya Kumar M, Sambaiah K, Lokesh BR. The anhydrous milk fat,ghee, lowers serum prostaglandins and secretion of leukotrienesby rat peritoneal macrophages. Prostaglandins Leukot Essent FattyAcids 1999; 61: 249–254
363. Simmons D, Williams R. Dietary practices among Europeans anddifferent South Asian groups in Coventry. Br J Nutr 1997; 78:5–14
364. Jacobson MS. Cholesterol oxides in Indian ghee: possible cause ofunexplained high risk of atherosclerosis in Indian immigrantpopulations. Lancet 1987; 2: 656–658
365. Gupta R, Prakash H. Association of dietary ghee intake withcoronary heart disease and risk factor prevalence in rural males. JIndian Med Assoc 1997; 95: 67–9, 83
366. Lip GY, Malik I, Luscombe C, McCarry M, Beevers G. Dietary fatpurchasing habits in whites, blacks and Asian peoples in England–implications for heart disease prevention. Int J Cardiol 1995; 48:287–293
367. Mendis S, Samarajeewa U, Thattil RO. Coconut fat and serumlipoproteins: effects of partial replacement with unsaturated fats.Br J Nutr 2001; 85: 583–589
368. Dowse GK, Gareeboo H, Alberti KG, Zimmet P, Tuomilehto J, PurranA, et al. Changes in population cholesterol doncentrations andother cardiovascular risk factor levels after five years of the non-communicable disease intervention programme in Mauritius.Mauritius Non-communicable Disease Study Group. BMJ 1995;311: 1255–1259
369. Cox C, Mann J, Sutherland W, Chisholm A, Skeaff M. Effects ofcoconut oil, butter, and safflower oil on lipids and lipoproteins inpersons with moderately elevated cholesterol levels. J Lipid Res1995; 36: 1787–1795
370. Cox C, Sutherland W, Mann J, de Jong S, Chisholm A, Skeaff M.Effects of dietary coconut oil, butter and safflower oil on plasmalipids, lipoproteins and lathosterol levels. Eur J Clin Nutr 1998; 52:650–654
371. Reiser R, Probstfield JL, Silvers A, Scott LW, Shorney ML, WoodRD, et al. Plasma lipid and lipoprotein response of humans to beeffat, coconut oil and safflower oil. Am J Clin Nutr 1985; 42:190–197
372. Ng TK Hayes KC, Dewitt GF, Jagathesan M, Satgunasingam N, OngAS, et al. Dietary palmitic and oleic acids exerts similar effects onserum cholesterol and lipoprotein profiles in normocholesterolemicmen and women. J Am Coll Nutr 1992; 11: 383–390
373. Kutty VR, Balakrishnan KG, Jayasree AK, Thomas J. Prevalence ofcoronary heart disease in the rural population ofThiruvananthapuram district, Kerala, India. Int J Cardiol 1993;39: 59–70
374. Joseph A, Kutty VR, Soman CR. High risk for coronary heart diseasein Thiruvananthapuram city: a study of serum lipids and otherrisk factors. Indian Heart J 2000; 52: 29–35
375. Burchfiel CM, Abbott R, Curb S, Rodriguez B, Yano K. Distributionand correlates of lipids and lipoproteins in elderly Japanese-American men: the Honolulu Heart Program. Artherioscler ThrombVasc Biol 1996; 16: 1356–1364
376. Mendis S. Cardiovascular disease in Sri Lanka: Sri Lanka: Ministry ofHealth, 1998
377. Trichopoulou A, Costacou T, Bamia C, Trichopoulos D. Adherenceto a Mediterranean diet and survival in a Greek population. N EnglJ Med 2003; 348: 2599–2608
378. Hu FB, Rimm EB, Stampfer MJ, Ascherio A, Spiegelman D, WillettWC. Prospective study of major dietary patterns and risk ofcoronary heart disease in men. Am J Clin Nutr 2000; 72:912–921
379. Hu FB. Dietary pattern analysis: a new direction in nutritionalepidemiology. Curr Opin Lipidol 2002; 13: 3–9
380. Fung TT, Willett WC, Stampfer MJ, Manson JE, Hu FB. Dietarypatterns and the risk of coronary heart disease in women. ArchIntern Med 2001; 161: 1857–1862
381. Truswell S. Practical and realistic approaches to healthier dietmodifications. Am J Clin Nutr 1998; 67(Suppl): 583S–590S
382. Hooper L, Summerbell CD, Higgins JP, Thompson RL, Capps NE,Smith GD, et al. Dietary fat intake and prevention of cardiovasculardisease: systematic review. BMJ 2001; 322: 757–763
383. Beauchesne-Rondeau E, Gascon A, Bergeron J, Jacques H. Plasmalipids and lipoproteins in hypercholesterolemic men fed a lipid-lowering diet containing lean beef, lean fish, or poultry. Am J ClinNutr 2003; 77: 587–593
384. Meyer KA, Kushi LH, Jacobs DR Jr, Folsom AR. Dietary fat andincidence of type 2 diabetes in older Iowa women. Diabetes Care2001; 24: 1528–1535
385. Hu FB, van Dam RM, Liu S. Diet and risk of type II diabetes: therole of types of fat and carbohydrate. Diabetologia 2001; 44:805–817
386. Summers LK, Fielding BA, Bradshaw HA, Ilic V, Beysen C, ClarkML, et al. Substituting dietary saturated fat with polyunsaturatedfat changes abdominal fat distribution and improves insulinsensitivity. Diabetologia 2002; 45: 369–377
387. de Lorgeril M, Renaud S, Mamelle N, Salen P, Martin JL, MonjaudI, et al. Mediterranean alpha-linolenic acid-rich diet in secondaryprevention of coronary heart disease. Lancet 1994; 343:1454–1459
388. Ros E. Dietary cis-monounsaturated fatty acids and metaboliccontrol in type 2 diabetes. Am J Clin Nutr 2003; 78 (Suppl):617S–625S
389. Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H,Ilanne-Parikka P, et al. Prevention of type 2 diabetes mellitus bychanges in lifestyle among subjects with impaired glucosetolerance. N Engl J Med 2001; 344: 1343–1350
390. Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, etal. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women.N Engl J Med 2001; 345: 790–797
391. Franz MJ, Bantle JP, Beebe CA, Brunzell JD, Chiasson JL, Garg A, etal; American Diabetes Association. Evidence-based nutritionprinciples and recommendations for the treatment and preventionof diabetes and related complications. Diabetes Care 2003; 26(Suppl): S51–S61
392. Deckelbaum RJ, Fisher EA, Winston M, Kumanyika S, Lauer RM,Pi-Sunyer FX, et al. Summary of a scientific conference onpreventive nutrition: pediatrics to geriatrics. Circulation 1999; 100:450–456
IHJ-584-03.p65 11/19/2003, 11:27 PM338

Indian Heart J 2003; 55: 339–343 Lateef et al. Displacement of the Precordial Leads and ECG 339
Vertical Displacement of the Precordial Leads AltersElectrocardiographic Morphology
Fatimah Lateef, Narayan Nimbkar, Zhuang Kun Da, Foo Rui MinDepartment of Emergency Medicine, Singapore General Hospital, Singapore,
Uniformed Services University of Health Sciences Bethesda, Maryland, USA; National University of Singapore
Electrocardiography (ECG), introduced by Einthoven in1902, is the graphical display of electrical potential
differences of an electric field originating in the heart andrecorded on the body surface. It represents a uniquetechnology that is safe, simple, and reproducible.1 AccurateECG interpretation assumes that technical standards areadhered to during the acquisition and recording of tracings.A number of technical factors may alter the quality ofrecorded ECG. Some of these are patient-related, some areoperator dependent, and others relate to the equipmentutilized for recording.2
The exact placement of electrodes for ECG depends onthe interpretation and skill of the person performing it (e.g.nurses, technicians, medical students). Placement ofprecordial electrodes for the recording of a 12-lead ECG issubject to variation. Technical variability represents thelargest source of error for variations in the amplitude and
waveform of the chest lead ECG. Precordial leads arecommonly placed either too high, too low or horizontallydisplaced from their anatomically defined sites. Personsrecording ECGs should ensure that the chest leads areplaced in the proper positions, and electrodes have good skincontact to minimize artefacts. Incorrect placement maylead to false diagnosis of infarction or cause alterations inthe ECG interpretation.3,4 In practice, the effect of thedisplacement on individual ECGs is unknown.
The present study was done to assess if verticaldisplacement of electrodes af fects the waveform ofprecordial leads of the ECG in healthy adult volunteers.
Methods
This was a clinical experimental investigation. FollowingECGs in supine position were performed in healthy, adultvolunteers, with no history of ischemic heart disease (IHD).
(i) a standard 12-lead ECG with the usual accepted,surface precordial lead placement;
Original Article
Correspondence: Fatimah Lateef, Department of Emergency Medicine,Singapore General Hospital, Outram Road, Singapore 169608.e-mail: [email protected]
Background: The present study was undertaken to assess whether the vertical displacement of electrodesaffects the waveforms of precordial leads.Methods and Results: Two hundred forty healthy, adult volunteers had a standard 12-lead electrocardiogram,a 12-lead electrocardiogram with the precordial leads displaced 2 cm cranially, and another with the precordialleads displaced 2 cm caudally from the standard positions. All the three sets of electrocardiograms were visuallycompared, and changes noted. One hundred twenty male and 120 female volunteers, 20–68 years of age, wereanalyzed. Fifty-four males (45.0%) and 2 females (1.7%) showed no difference between the 3 sets ofelectrocardiograms, while 66 males (55.0%) and 118 females (98.3%) had some changes. R wave amplitudechanges were noted in 63 male (52.5%) and 111 female (92.5%) volunteers; S wave amplitude changes wereseen in 59 males (49.2%) and 99 females (82.5%); T wave changes in 5 males (4.2%) and 3 females (2.5%); STsegment changes in 1 male (0.8%) and none of the females; and QRS morphologic changes in 1 male (0.8%)and 12 females (10.0%).Conclusions: Precordial electrocardiographic waveform changes were seen with the vertical displacement ofthe precordial leads. This will have implications on the interpretation of serial electrocardiograms. Healthcareproviders should take into consideration this deviation when interpreting serial ECGs. (Indian Heart J 2003;55: 339–343)
Key Words: Electrocardiography, Precordial leads, Lead placement
IHJ-455-03.p65 11/19/2003, 11:24 PM339
340 Lateef et al. Displacement of the Precordial Leads and ECG Indian Heart J 2003; 55: 339–343
(ii) a 12-lead ECG, as in (i), but with all the precordial leads(i.e. V
1 to V
6) shifted 2.0 cm cranially; and
(iii) a 12-lead ECG as in (i), but with all the precordial leadsshifted 2.0 cm caudally.
Measurements were done with a standard ruler. All thethree ECGs for each volunteer were visually compared, andthe differences documented (e.g. R and S wave amplitudechanges, changes in the QRS morphology, ST segmentchanges, T wave morphology and appearance/disappearance of Q waves). T wave changes refer to eitheran upright, inverted or flattened T wave. ST segmentchanges may represent an isoelectric ST segment, or STelevation, or depression. The morphological changes wouldrefer to changes such as the appearance of a bundle branchblock pattern.
The standard 12-lead ECG for each person was used asthe control against which the other two ECGs, which haddeliberate alterations in the positions of the precordial leads,were compared. When the tracings with the displacedelectrodes did not resemble the standard ECG tracings, thevariations were confirmed by repeating the tracings.
The study was approved by the hospital ethicscommittee.
Results
There were a total of 240 volunteers, 20–68 years of age.One hundred and twenty were males and 120 females.Fifty-four males (45.0%) and 2 females (1.7%) showed nodifferences when their three sets of ECGs were compared,while 66 males (55.0%) and 118 females (98.3%) had somechanges noted. R wave amplitude changes were noted in63 male (52.5%) and 111 female (92.5%) volunteers; Swave changes in 59 males (49.2%) and 99 females (82.5%)(Figs 1a–c); T wave changes in 5 males (4.2%) and 3 females(2.5%) (Fig. 2a–c); ST segment changes in 1 male (0.8%)and none of the females; and QRS morphological changesin 1 male (0.8%) and 12 females (10.0%) (Figs 2a–e)(Table 1). There was no Q wave appearance ordisappearance noted in the group studied. Table 1demonstrates the confidence intervals at p=0.01.
R and S wave changes were mainly amplitude changes(i.e. the height of the R wave and depth of the S wave). Therange of R wave amplitude change was from +3 mm to+15 mm (positive indicates above the isoelectric baseline),while that for S was –2 mm to –15 mm (negative indicatesbelow the isoelectric baseline) (Figs 1a–c). All the volunteerswho had S wave changes also had R wave changes.
Analysis of the changes showed that these werecommonest in leads V
1, V
2, and V
3, irrespective of whether
the displacement was cranial or caudal (Tables 2 and 3).
Fig. 1a. Normal position: amplitude of the R wave in V3 (+27 mm).
Fig. 1b. 2 cm cranial: amplitude of the R wave in V3 (+9 mm).
Fig. 1c. 2 cm caudal: amplitude of the R wave in V3 (+10 mm).
Changes were not observed in the ramaining precordialleads, namely leads V
4, V
5, and V
6.
As the volunteers did not have any history ofcardiovascular problems, none of them presented with anycardiorespiratory symptoms. There was one volunteer whowas detected to have left ventricular hypertrophy on ECG,which was repeated for reconfirmation. She wassubsequently offered assessment by a cardiologist as anoutpatient.
Discussion
There are numerous potential clinical uses of the 12-lead
IHJ-455-03.p65 11/19/2003, 11:24 PM340
Indian Heart J 2003; 55: 339–343 Lateef et al. Displacement of the Precordial Leads and ECG 341
Fig. 2c. 2 cm caudal: QRS complex in V1 shows RS pattern with T waveinversion in V3.
Table 1. Changes observed when comparing the 3electrocardiograms
Changes Number of Number ofobserved males (%) females (%)
None 54 (45.0) 2 (1.7)CI: 33.87–56.65 CI: 0.33–8.07
Changes 66 (55.0) 118 (98.3)CI: 43.35–66.13 CI: 91.3–99.67
R wave amplitude 63 (52.5) 111 (92.5)CI: 40.94–63.80 CI: 83.85–96.70
S wave amplitude 59 (49.2) 99 (82.5)CI: 37.77–60.65 CI: 71.93–89.66
T wave 5 (4.2) 3 (2.5)QRS morphological 1 (0.8) 12 (10.0)ST segment 1 (0.8) 0
CI: confidence intervalNote: Some patients had more than one category of change (e.g. patientA had both R wave and S wave amplitude changes), therefore, the totalnumber of changes may be more than the number of volunteers.For male volunteers, T wave, QRS morphology, and ST segment changeswhen taken together, the CI at p=0.01 was 33.87%–56.65%.For female volunteers, the CI at p=0.01 was 6.64%–22.29%.
Table 2. Detailed changes among male volunteers
Changes Leads Cranial Caudaldisplacement displacement
R wave V1
6 9V
229 34
V3
42 38
S wave V1
0 0V
219 22
V3
40 37
T wave V1
0 1V
23 1
V3
3 3
QRS complex V1
1 0
ST segment V1
0 0V
21 0
V3 0 0
Note: Some volunteers showed changes in more than one lead, and alsochanges in either one or both of the cranial and caudal displacement leads.For these reasons, the numbers may not tally, and may be more than thetotal number of male volunteers.
Fig. 2a. Normal position: QRS complex in V1 showing an RS pattern with Twave inversion in V3.
Fig. 2b. 2 cm cranial: QRS complex in V1 showing an RSR' pattern with uprightT wave in V3.
Fig. 2d. Enlargement of Fig. 2a (normal) V1: RS pattern
Fig. 2e. Enlargement of Fig. 2b (2cm cranial) V1: RSR' pattern
ECG. The ECG may reflect changes associated with primaryand secondary myocardial processes, metabolic andelectrolyte abnormalities, as well as toxic or therapeuticeffects of drugs or devices. ECGs are composed of a numberof waveforms, each with its own sensitivity and specificity,
IHJ-455-03.p65 11/19/2003, 11:24 PM341

342 Lateef et al. Displacement of the Precordial Leads and ECG Indian Heart J 2003; 55: 339–343
and each influenced by a variety of pathologic andpathophysiologic factors. For example, although STsegment and T wave changes are the commonest and mostsensitive, these changes are also the least specific.3–5
There is continuing uncertainty about standardizedprocedures for the placement of ECG chest electrodes.Technical variability represents the largest source of errorfor short-term variations in amplitude and waveforms ofthe chest lead ECG. There is uncertainty about locating themid-clavicular line and, thus, the position of V
4,
particularly in obese persons, and females. Appropriateselection of the position for chest leads may help inremoving the inconsistencies in ECG interpretation andselection criteria for therapy, thus improving comparabilityof treatment results.6
Amplitude changes are not uncommon. Previous studieson modified positions of the limb leads have shownconsiderable amplitude and waveform changes in thefrontal plane ECG.7,8
The R wave (QRS complex) reflects the depolarizationphase of the ventricles. It has been shown that in thosewith coronary artery disease/ischemic heart disease, thereis often poor progression of the R wave as one moves acrossthe precordial leads and approaches V
6.9
As for extrasystoles, Michaelides et al.10 in a study usingangiographic confirmation, showed that the R wave of theextrasystoles is positively correlated with the number ofobstructed coronary arteries, Barnhill et al.11 studied R andS waves (terminal portion of the QRS complex) in patients
with myocardial infarction (MI), and concluded that thesewere commonly affected, and very sensitive to changes andvariations.
Feldman et al.,12 in a study that examined R waveamplitude and left ventricle chamber size, concluded thatthere is a direct and dynamic correlation between the twofactors. Chamber size and distance from the left ventricleto leads V
5 and V
6 were major determinants of the R wave
amplitude. R and S wave amplitudes are also important indiagnostic criteria such as left ventricular hypertrophy(LVH).
The morphology of the ECG depicts the resultantelectrical vector in 3 dimensions, with time being the fourthdimension. Hence, it is natural that the morphology of theECG is known to be affected by the position of the heart,position of the unipolar leads in relation to the heart, aswell as the pathway between the heart and unipolarelectrodes. For example, with alteration in position of theprecordial electrodes, the current may have to pass througha different, more circuitous route, if there is nonconductivetissue encountered along the more direct pathway. Otherfactors that have been shown to affect ECG morphologyinclude body position,13 and age.14 There have been somestudies on ethnicity, and its effect on ECGs.15–16 However,most of these studies are small and may be applicable tothe local, ethnic population only.
It has often been thought that with changes in theplacement of leads, there will be ST segment alterationsaffecting our interpretation of MI (i.e. ST segmentelevation), especially when comparing serial ECGs.17 In thisstudy, there was only one male patient (0.8%) in whom STsegment change was noted. This assumption may not beas common as previously thought.
There is intra-individual precordial voltage variation inserial ECGs. It is necessary to improve reproducibility andprecision in the performance of ECGs. There is also a needto emphasize how electrode placement is taught to nurses,as well as their accountability for this duty.18,19
A new device known as the precordial ECG BELT, usinga 6-lead precordial application, has been used in studies tocompare its accuracy with standard precordial leadapplication. Results have indicated disagreement betweenthe two methods.20 The most obvious weakness of the ECGBELT was the inability to obtain a reading with a stablebaseline. The findings suggest that the ECG BELT is notadequate for clinical application in its current form.20
Another alternative may be a device with less surfaceconnections, such as the EASI ECG. This device uses only 5surface connections to derive the 12-lead ECG. Perhaps withless connections, there would be less placement error and
Table 3. Detailed changes among female volunteers
Changes Leads Cranial Caudaldisplacement displacement
R wave V1
4 0V
256 46
V3
71 73
S wave V1
1 3V
227 32
V3
71 64
T wave V1
1 0V
21 2
V3
1 1
QRS complex V1
2 3V
27 4
V3
3 5
Note: Some volunteers showed changes in more than one lead, and alsochanges in either one or both of the cranial and caudal displacement leads.For these reasons, the numbers may not tally, and may be more than thetotal number of female volunteers.
IHJ-455-03.p65 11/19/2003, 11:24 PM342

Indian Heart J 2003; 55: 339–343 Lateef et al. Displacement of the Precordial Leads and ECG 343
variations. The EASI system has been favorably comparedwith the standard 12-lead ECG.
Therefore, to reduce inconsistencies, the practice ofmarking precordial lead positions for short-term serial ECGsshould be encouraged. Physicians should also be morecognizant of ECG changes caused by precordial leaddisplacement when comparing serial ECGs.
This study has shown that variations do exist amongnormal individuals. It is the first time this has been provenobjectively on normal individuals. This suggests the needfor standardization, or the use of a device to assist in leadplacement, which can ensure accuracy, reproducibility, andobjectivity.
References
1. Einthoven W. [Weiteresinber das elektrokardiogramm]. Arch GesamtePhysiol 1908; 172: 517–520
2. Fisch C. Evolution of the clinical electrocardiogram. J Am Coll Cardiol1989; 14: 1127–1138
3. Friedberg CK, Zager A. “Non-specific” ST and T wave changes. ClinAll Round 1961; 23: 655–661
4. Hurst JW. Images in cardiovascular medicine. “Switched” precordialleads. Circulation 2000; 101: 2870–2871
5. Kadish AH, Buxton AE, Kennedy HL, Knight BP, Mason JW, SchugerCD, et al. ACC/AHA clinical competence statement on electro-cardiography and ambulatory electrocardiography. A report ofthe ACC/AHA/ACP-ASIM Task Force on Clinical Competence(ACC/AHA Committee to Develop a Clinical Competence Statementon Electrocardiography and Ambulatory Electrocardiography).J Am Coll Cardiol 2001; 38: 2091–2100
6. Rautaharju PM, Park L, Rautaharju FS, Crow R. A standardizedprocedure for locating and documenting ECG chest electrodepositions: consideration of the effect of breast tissue on ECGamplitudes in women. J Electrocardiol 1998; 31: 17–29
7. Rautaharju PM, Prineas RJ, Crow RS, Seale D, Furberg C. The effect
of modified limb electrode positions on electrocardiographic waveamplitudes. J Electrocardiol 1980; 13: 109–113
8. Takuma K, Hori S, Sasaki J, Shinozawa Y, Yoshikawa T, Hand S, et al.An alternative limb lead system for electrocardiographs in emergencypatients. Am J Emerg Med 1995; 13: 514–517
9. Chia BL. Ischaemic heart disease. In: Clinical electrocardiography.3rd ed. World Scientific Publishers. pp. 11-37
10. Michaelides AP, Vyssoulis GP, Paraskevas PG, Skouros CG, TsiamisEG, Toutouzas PK. Significance of R wave changes in exercise-induced supraventricular extrasystoles. Angiographic correlates. JElectrocardiol 1993; 26: 197–206
11. Barnhill JE 3rd, Tendera M, Cade H, Campbell WB, Smith RF.Depolarization changes early in the course of myocardial infarction:significance of changes in the terminal portion of the QRS complex.J Am Coll Cardiol 1989; 14: 143–149
12. Feldman T, Borow RM, Neumann A, Lang RM, Childers RW. Relationof electrocardiographic R-wave amplitude to changes in leftventricular chamber size and position in normal subjects. Am J Cardiol1985; 55: 1168–1174
13. Adams MG, Drew BJ. Body position effects on the ECG: implicationsfor ischemia monitoring. J Electrocardiol 1997; 30: 285–291
14. Bachman S, Sparrow D, Smith LK. Effects of aging on theelectrocardiogram. Am J Cardiol 1981; 48: 513–516
15. Mansi IA, Nash IS. Ethnic differences in the ST segment of theelectrocardiogram: a comparative study among six ethnic groups.Am J Emerg Med 2001; 19: 541–554
16. Chen CY, Chiang BN, Macfarlane PW. Normal limits of theelectrocardiogram in a Chinese population. J Electrocardiol 1989; 22:1–15
17. Herman MV, Ingram DA, Levy JA, Cook JR, Athans RJ. Variability ofelectrocardiographic precordial lead placement: a method to improveaccuracy and reliability. Clin Cardiol 1991; 14: 469–476
18. Bupp JE, Dunger M, Lawrence C, Wingate S. Placement of cardiacelectrodes: written, simulated, and actual accuracy. Am J Crit Care1997; 6: 457–462
19. McConnell EA. Applying cardiac monitor electrodes. Nursing 1998;28: 26
20. Bell SJ, Clifton J, Pease J, Greenfield JC, Leggett S, Maynard C, et al:The evaluation of a precordial ECG BELT: technologist satisfactionand accuracy of recording. J Electrocardiol 2001; 34: 155–159
IHJ-455-03.p65 11/19/2003, 11:24 PM343

344 Shrivastava et al. Coronary Calcium and CAD Indian Heart J 2003; 55: 344–348
Coronary Calcium and Coronary Artery Disease:An Indian Perspective
Sameer Shrivastava, Vinayak Agrawal, Ravi R Kasliwal, Dhanraj R Jangid,Ashok Sen, Atul Verma, Naresh Trehan
Departments of Cardiology and Nuclear Medicine, Escorts Heart Institute and Research Centre, New Delhi
Calcification is closely associated with atheromatousplaque, and is a recognized marker for coronary artery
disease.1 Until recently, it was believed that calcium presentin the atheromatous plaques is in the form of calciumphosphate, precipitated in a passive manner. However,recent findings indicate that calcification is an activeprocess and calcium is deposited as hydroxyapatite, similarto that in active bone formation.2 It is known that theamount of calcium in the coronary arteries is better
correlated with the amount of atherosclerosis in differentindividuals and, to a lesser extent, with the segments ofthe coronary tree in the same individual. It is not knownwhether the quantity of calcium tracks the quantity ofatherosclerosis over time in the same individual. There arenumerous articles on the correlation of coronary calciumwith angiographically proven coronary artery disease(CAD); however, there are no such large studies from India.This study was aimed at using helical computedtomography (CT) scan for calculating coronary calciumscores (CCSs) in the Indian population.
Original Article
Background: Coronary artery calcification is a part of the development of atherosclerosis. It occurs exclusivelyin atherosclerotic arteries and can be used as a noninvasive marker of coronary atherosclerosis. As there is nolarge-scale study on coronary calcium score reported in the Indian population till date, this study was undertakenfor calculating the score in Indians at intermediate-to-high risk of coronary artery disease, and to correlate itwith angiographically proven coronary artery disease.Methods and Results: A total of 388 consecutive patients who underwent coronary calcium scoring andcoronary angiography were included in the study. Calcium scoring was performed based on a modification ofthe Agatston Score using a high-speed computed tomography scanner (GE® CT/i scanner). Coronary calciumscores were correlated with the presence or absence of significant coronary artery disease (defined as ≥70%stenosis of at least one major epicardial coronary artery) on angiography. Out of 388 patients who underwentcoronary angiography, 298 were found to have significant coronary artery disease. Mean coronary calciumscore was significantly higher in patients with angiographically proven coronary artery disease (226.7±65.2)as compared to those who had normal angiograms (20.29±56.7; p value<0.0001). All the 72 patients whohad a score >400 had an abnormal angiogram (sensitivity 23.1%, specificity 100%, positive predictive value100%, and negative predictive value 24.1%). On the other hand, among the patients who had a score >0, 298were found to have abnormal angiograms, while 16 had normal angiograms (sensitivity 95.5%, specificity78.9%, positive predictive value 94.9%, and negative predictive value 81.1%).Conclusions: Detection of coronary calcium score by a helical computed tomography scanner is a useful toolfor predicting the presence of significant coronary artery disease in intermediate-to-high risk patients. Anabsolute score of 0 has a high negative predictive value for the presence of coronary artery disease, and mayobviate the need to perform coronary angiogram in intermediate-risk patients. On the other extreme, score>400 is highly predictive of the presence of coronary artery disease, and virtually confirms the presence ofsignificant coronary artery disease in intermediate-to-high risk patients. (Indian Heart J 2003; 55:344–348)
Key Words: Coronary artery disease, Coronary calcium score, Computed tomography
Correspondence: Dr Vinayak Agrawal, 103, Siddhartha Enclave, NewDelhi. e-mail: [email protected]
IHJ-478-03.p65 11/19/2003, 11:25 PM344

Indian Heart J 2003; 55: 344–348 Shrivastava et al. Coronary Calcium and CAD 345
Methods
The study included 388 consecutive patients whounderwent coronary calcium scoring as well as coronaryangiography. These included patients who had presentedwith either typical symptoms suggestive of ischemic heartdisease (with or without a positive stress test), or atypicalsymptoms with a positive stress test for reversiblemyocardial ischemia, putting these patients into a pretestprobability of intermediate-to-high risk category. All thepatients underwent detailed clinical evaluation,biochemical tests, coronary calcium scoring, and coronaryangiography. Biochemical tests included fasting and post-prandial blood sugar, and fasting lipid profile.
Assessement of CCSs was done by using a high-speedCT scanner (GE® CT/i scanner). Scores were computergenerated using an advanced software “smart-scorecalcium” adapting a modification based on Agatston Score.Total CCSs thus obtained were correlated with the presenceor absence of significant CAD. Significant CAD was definedas ≥70% stenosis of at least one of the major epicardialcoronary arteries on coronary angiography. Informedconsent was obtained from the entire patient populationenrolled in the study. The study was approved by thehospital ethics committee.
Statistical analysis : The data obtained were maintainedon a Microsoft excel spreadsheet. The differences in variousparameters between the different groups were assessed byusing Chi-square and t tests, as indicated. Sensitivity,specificity, positive predictive value (PPV), and negativepredictive value (NPV) for dif ferent risk scores werecalculated. A p value <0.05 was considered significant. Allthe statistical analyses were done on SPSS 10.0 forWindows.
Results
A total of 388 consecutive patients underwent coronarycalcium scoring and coronary angiograms to rule outsignificant CAD. Three hundred twenty-five patients(83.8%) were male while 63 (16.2%) were female. Themean age of the patients was 52.3±11.3 years (range 15–78 years). Out of these patients, 76.2% were less than 60years of age, and 23.8% were more than 60 years of age.For the purpose of analysis, patients were divided into fourgroups according to CCS: 0, 1–100, 101–400, and >400.There was no statistically significant difference in theprevalence of various conventional cardiovascular riskfactors in different calcium score groups (Tables 1 and 2).The presence or absence of significant CAD on coronary
angiography in different calcium score groups is presentedin Table 3. Sensitivity, specificity, PPV, and NPV for theprediction of significant CAD were calculated for threedifferent calcium scores, namely >0, >100, and >400(Table 4). All the 72 patients who had CCSs >400 hadabnormal angiograms, yielding a sensitivity of 23.1%,specificity 100%, PPV 100%, and NPV 24.1%. Twohundred thirty-six patients with CCS>100 had abnormalangiograms, with only 4 patients having normalangiogram (sensitivity 75.6%, specificity 94.7%, PPV98.3%, and NPV 51.3%). Out of 314 patients who had
Table 1. Incidence of various risk factors in relation to thecalcium score
Risk factor Calcium score p value
≤100 101–400 >400(n=148) (n=168) (n=72)
Age (years) 47.2±10.7 51.3±10.6 49.2±10.1 nsMale (%) 86.5 80.4 86.1 nsHypertension (%) 12.5 11.1 14.7 nsDiabetes (%) 3.8 5.6 8.8 nsFamily history (%) 11.5 11.1 8.8 nsSmoking (%) 5.4 3.7 2.9 ns
ns: not statistically significant
Table 2. Correlation of calcium score with lipid profile
Lipid profile (mg/dl) Calcium score p value
≤100 101–400 >400(n=148) (n=168) (n=72)
Total cholesterol 204±45 220±57 216±10 nsTriglycerides 220±47 181±41 129±33 nsHigh-density 42±11 44±9 41±13 ns
lipoproteinsLow-density 125±37 142±47 140±18 ns
lipoproteinsVery low-density 40±9 34±8 25±7 ns
lipoprotein
ns: not statistically significant
Table 3. Coronary calcium score correlation with coronaryangiography findings
Calcium scores>400 101–400 1–100 0
(n=72) (n=168) (n=74) (n=74)
Abnormal angiogram 72 164 62 14Normal angiogram 0 4 12 60
IHJ-478-03.p65 11/19/2003, 11:25 PM345

346 Shrivastava et al. Coronary Calcium and CAD Indian Heart J 2003; 55: 344–348
CCSs >0, 298 were found to have abnormal angiogramswhile 16 patients had normal angiograms (sensitivity95.5%, specificity 78.9%, PPV 94.9%, and NPV 81.1%).
Discussion
Quantification of calcium in the coronary arteries has beenshown to reflect the total atherosclerotic plaque burden.3,4
This quantification can be determined in a straightforwardmanner by using advanced high-speed CT scans includinghelical CT scans, multi-slice CT scans, and electron-beamCT (EBCT) scans. In several studies, detection andquantification of coronary artery calcification with EBCTin suspected CAD patients has been compared with widelyavailable conventional helical CT scan. Although imagesof the heart from conventional CT scans may suffer fromcardiac motion artifacts, calcium scoring with spiral ormulti-slice CT scanners has the potential to identify patientswith CAD with an accuracy similar to that of EBCT scan.5–7
Protocols with prospective or retrospective electrocardio-graphy triggering can be used, which appear to yield resultssimilar to Agatston CCS with EBCT scans, despite technicaldifferences.8
Calcification is not found in normal coronary arteries.The atherosclerotic process starts in the second decade oflife, when calcium deposition begins. The calciumdeposition increases with age, and with progression ofatherosclerotic lesions. Coronary artery calcification istemporally related to vascular inflammation and the demiseof lipid-laden macrophages.9,10 The exact mechanism isunclear, and may be due to the deposition of extracellularcalcium from dying cells, or the formation of bone-likestructure by calcifying cells.11,12 Since calcium is depositedonly in the atherosclerotic plaques and not in the normalvessels, and since atherosclerosis is a diffuse process, a highcoronary calcium burden reflects the presence of moreextensive coronary atherosclerosis (Fig. 1). Hence, higherCCSs are associated with the presence of significant CAD
and future risk of adverse coronary events. Moreover,contrary to popular belief, coronary calcium depositionmay represent a type of plaque instability, namely plaquerupture. It was earlier believed that calcified areas areunlikely to be associated with the sites of plaque rupture.13
However, the stiffened calcific area can induce stress at thejunction of the calcified and noncalcified sections, whichis a common site of plaque rupture.14 Burke et al.15 foundthat, contrary to expectations, the degree of calcificationwas greatest for acute and healed plaque ruptures, and leastfor plaque erosion. It has been suggested that the presenceof calcium within a heterogeneous, fat-laden plaquerenders the plaque more susceptible to stress cracking, ascompared to a plaque that contains no calcium.16
However, calcium deposition as detected by coronarycalcium scoring does not correlate with the site and severityof luminal narrowing on coronary angiography. Theobservation that coronary plaque burden is poorlycorrelated with luminal narrowing has been confirmed bystudies using intravascular ultrasound (IVUS), and can beexplained by the process of vascular remodeling.17–19 As theatherosclerotic plaque accumulates in the coronary arterywall, there is an outward remodeling leading to thepreservation of luminal size.20 However, the likelihood ofplaque rupture leading to acute MI is not decreased andmay even be increased. Thus, the apparent lower specificityof CCS for angiographic obstruction in some studies mayin fact be due to the inability of angiography to detectextraluminal plaque.21 However, an increased CCS wouldmean the likelihood of significant CAD elsewhere or atanother site.
Our data also suggest that in symptomatic patients, themere presence of calcium is a very sensitive index for thepresence of significant CAD somewhere in the coronaryarteries. Any CCS >0 has a high sensitivity and reasonablygood specificity, with good PPV to predict the presence ofobstructive CAD. There have been numerous studies that
Table 4. Sensitivity, specificity, positive predictive value,and negative predictive value of different calcium scoresfor predicting the presence or absence of significantcoronary artery disease
Calcium scores
>400 >100 0
Sensitivity 23.1 75.6 95.5Specificity 100 94.7 78.9Positive predictive value 100 98.3 94.9Negative predictive value 24.1 51.3 81.1
Fig. 1. Normal and high calcium score images.
Zero Calcium Socre High Calcium Socre
IHJ-478-03.p65 11/19/2003, 11:25 PM346

Indian Heart J 2003; 55: 344–348 Shrivastava et al. Coronary Calcium and CAD 347
combined to obtain complementary information.35 Anegative (<0) calcification generally excludes obstructiveCAD. Hence, this technique can be used to evaluate patientswith atypical chest pain. If the CCS is zero, the source ofpain is unlikely to be due to CAD. If the CCS is very high(>400), the chances of the presence of obstructive CADare steep.
Since coronary calcium scoring is a screening tool, atechnique that has a very high sensitivity for the predictionof obstructive CAD is desirable. In our study, which involvedintermediate-to-high risk patients, a CCS >0 had a veryhigh sensitivity for obstructive CAD with fairly highspecificity. There was a progressive increase in PPV withincreasing CCS, suggesting that high CCSs are increasinglyassociated with obstructive CAD. Thus, a CCS of zero maybe used to obviate the need for coronary angiography inintermediate-risk patients and in selected high-risk patients,when coronary angiography is not the preferred methoddue to concomitant co-morbid conditions (such as renaldysfunction, terminal illnesses, etc.). In such patients, ahigh CCS, particularly >400, virtually confirms thepresence of significant CAD, and can help in devisingfurther management strategies.
Limitations: Our study suffered from certain limitations.It involved only those individuals who were at intermediate-to-high risk of having significant CAD. Patients at low riskof CAD could not be included because of obvious ethicalreasons. Hence, there was a definite selection bias that couldnot have been avoided.
References
1. Rifkin RD, Parisi AF, Folland E. Coronary calcification in the diagnosisof coronary artery disease. Am J Cardiol 1979; 44: 141–147
2. Doherty TM, Detrano RC. Coronary artery calcification as an activeprocess: a new perspective on an old problem. Calcif Tissue Int 1994;94: 1597–1604
3. Rumberger JA, Simons DB, Fitzpatrick LA, Sheedy PF, Schwartz RS.Coronary artery calcium area by electron-beam computedtomography and coronary atherosclerotic plaque area. Ahistopathologic correlative study. Circulation 1995; 92: 2157–2162
4. Sangiorgi G, Rumberger JA, Severson A, Edwards WD, Gregoire J,Fitzpatrick LA, et al. Arterial calcification and not lumen stenosis ishighly correlated with atherosclerotic plaque burden in humans: ahistologic study of 723 coronary artery segments usingnondecalcifying methodology. J Am Coll Cardiol 1998; 31: 126–133
5. Becker CR, Knez A, Jakobs TF, Aydemir S, Becker A, Schoepf UJ, et al.Detection and quantification of coronary artery calcification withelectron-beam and conventional CT. Eur Radiol 1999; 9: 620–624
6. Carr JJ, Crouse JR 3rd, Goff DC Jr, D’Agostino RB Jr, Peterson NP,Burke GL. Evaluation of subsecond gated helical CT for quantificationof coronary artery calcium and comparison with electron beam CT.AJR Am J Roentgenol 2000; 174: 915–921
7. Knez A, Becker C, Becker A, Leber A, White C, Reiser M, et al.Determination of coronary calcium with multi-slice spiral computed
have proved coronary calcium scoring to be an excellentpredictor of the presence of angiographically provenobstructive CAD, and future risk of adverse cardiovascularevents. In a large study conducted by Budoff et al.22 in 710symptomatic subjects with a mean age of 56 years,comparing EBCT scan with coronary angiography, thesensitivity, specificity, and NPV were 95%, 44%, and 84%,respectively. Breen et al.23 and Kaufmann et al.24
demonstrated the relationship between CCS and coronaryartery lumen obstruction as assessed by coronaryangiography. Kaufmann et al.24 studied 160 symptomaticindividuals 23–59 years of age and compared CCS withcoronary artery stenosis. Sensitivity, specificity, and accuracyranged from 81% to 86%. Several other groups25–30 haveconfirmed the association of increased CCS with increasedpresence and severity of CAD.
He et al.31 studied nearly 400 asymptomatic patientsusing EBCT scan and SPECT, and showed that none of thepatients with a CCS <10 had an abnormal SPECT. Out ofof these, 2.6% of patients with CCS 10–100, 11.3% witha CCS of 101–399, and 46% with CCS>400 had a SPECTshowing ischemia. This high sensitivity and low specificityprobably relates to the fact that only 20% of theatherosclerotic plaque is calcified. However, the absence ofcalcification seems to indicate a lack of significant luminalstenosis.
In the Rotterdam Coronary Calcification Study,32 astrong and graded association was found between coronarycalcification and MI. In a similar study of patients withsuspected CAD, CT scan showed a 65% sensitivity, and 87%specificity for coronary artery calcification.33 According tothe American Heart Association Expert Consensusdocument on the use of EBCT for the diagnosis andprognosis of CAD,34 a meta-analysis of the relationshipbetween CAD and calcium prevalence in patientsundergoing EBCT and cardiac catheterization to determinethe diagnostic accuracy of EBCT in catheterized patientsdemonstrated a high sensitivity of EBCT for CAD, a muchlower specificity and an overall predictive accuracy of 70%in typical CAD patient populations. The test has proven tohave a predictive accuracy approximately equivalent to thealternative methods for diagnosing CAD, but was not foundto be superior to alternative noninvasive tests such as SPECTimaging. A positive calcium score might be valuable indetermining whether a patient who appears to be atintermediate risk of CAD is actually at high risk. Conversely,a low or absent calcium score may also prove useful indetermining a low likelihood of developing CAD. Theabsence of coronary calcification identifies patients withfalse-positive exercise treadmill testing, and the two can be
IHJ-478-03.p65 11/19/2003, 11:25 PM347

348 Shrivastava et al. Coronary Calcium and CAD Indian Heart J 2003; 55: 344–348
tomography: a comparative study with electron-beam CT. Int JCardiovasc Imaging 2002; 18: 295–303
8. Schmermund A, Mohlenkamp S, Erbel R. The latest on the calciumstory. Am J Cardiol 2002; 90: 12L–14L
9. Stary HC. The sequence of cell and matrix changes in atheroscleroticlesions of coronary arteries in the first forty years of life. Eur Heart J1990; 11 (Suppl): 3–19
10. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull WJr, et al. A definition of advanced types of atherosclerotic lesions anda histological classification of atherosclerosis. A report from theCommittee on Vascular Lesions of the Council on Arteriosclerosis,American Heart Association. Circulation 1995; 92: 1355–1374
11. Parhami F, Bostrom K, Watson K, Demer LL. Role of molecularregulation in vascular calcification. J Atheroscler Thromb 1996; 3:90–94
12. Watson KE, Abrolat ML, Malone LL, Hoeg JM, Doherty T, Detrano R,et al. Active serum vitamin D levels are inversely correlated withcoronary calcification. Circulation 1997; 96: 1755–1760
13. Cheng GC, Loree HM, Kamm RD, Fishbein MC, Lee RT. Distributionof circumferential stress in ruptured and stable atheroscleroticlesions. A structural analysis with histopathological correlation.Circulation 1993; 87: 1179–1187
14. Demer LL, Watson KE, Bostrom K. Mechanism of calcification inatherosclerosis. Trends Cardiovasc Med 1994; 4: 45–49
15. Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R.Coronary risk factors and plaque morphology in men with coronarydisease who died suddenly. N Engl J Med 1997; 336: 1276–1282
16. Schwarz U, Buzello M, Ritz E, Stein G, Raabe G, Wiest G, et al.Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol Dial Transplant 2000; 15: 218–223
17. Klingensmith JD, Vince DG, Kuban BD, Shekhar R, Tuzcu EM, NissenSE, et al. Assessment of coronary compensatory enlargement bythree-dimensional intravascular ultrasound. Int J Card Imaging 2000;16: 87–98
18. Nissen SE. Shortcomings of coronary angiography and theirimplications in clinical practice. Cleve Clin J Med 1999; 66: 479–485
19. Nissen SE, Yock P. Intravascular ultrasound: novel pathophysiologicalinsights and current clinical applications. Circulation 2001; 103:604–616
20. Fishbein MC, Siegel RJ. How big are coronary atherosclerotic plaquesthat rupture? Circulation 1996; 94: 2662–2666
21. Baumgart D, Schmermund A, Goerge G, Haude M, Ge J, Adamzik M,et al. Comparison of electron beam computed tomography withintracoronary ultrasound and coronary angiography for detectionof coronary atherosclerosis. J Am Coll Cardiol 1997; 30: 57–64
22. Budoff MJ, Georgiou D, Brody A, Agatston AS, Kennedy J, WolfkielC, et al. Ultrafast computed tomography as a diagnostic modality inthe detection of coronary artery disease: a multicenter study.Circulation 1996; 93: 898–904
23. Breen JF, Sheedy PF 2nd, Schwartz RS, Stanson AW, Kaufmann RB,Moll PP, et al. Coronary artery calcification detected with ultrafast
CT as an indication of coronary artery disease. Radiology 1992; 185:435–439
24. Kaufmann RB, Peyser PA, Sheedy PF, Rumberger JA, Schwartz RS.Quantification of coronary artery calcium by electron beamcomputed tomography for determination of severity of angiographiccoronary artery disease in younger patients. J Am Coll Cardiol 1995;25: 626–632
25. Bielak LF, Kaufmann RB, Moll PP, McCollough CH, Schwartz RS,Sheedy PF 2nd. Small lesions in the heart identified at electron beamCT: calcification or noise? Radiology 1994; 192: 631–636
26. Rumberger JA, Schwartz RS, Simons DB, Sheedy PF 3rd, EdwardsWD, Fitzpatrick LA. Relation of coronary calcium determined byelectron beam computed tomography and lumen narrowingdetermined by autopsy. Am J Cardiol 1994; 73: 1169–1173
27. Agatston AS, Janowitz WR, Kaplan G, Gasso J, Hildner F, ViamonteM Jr. Ultrafast computed tomography-detected coronary calciumreflects the angiographic extent of coronary arterial atherosclerosis.Am J Cardiol 1994; 74: 1272–1274
28. Kajinami K, Seki H, Takekoshi N, Mabuchi H. Noninvasive predictionof coronary atherosclerosis by quantification of coronary arterycalcification using electron beam computed tomography:comparison with electrocardiographic and thallium exercise stresstest results. J Am Coll Cardiol 1995; 26: 1209–1221
29. Stanford W, Travis ME, Thompson BH, Reiners TJ, Hasson RR,Winniford MD. Electron-beam computed tomographic detection ofcoronary calcification in patients undergoing percutaneoustransluminal coronary angioplasty: predictability of restenosis. Apreliminary report. Am J Card Imaging 1995; 9: 257–260
30. Budoff MJ, Georgiou D, Brody A, Agatston AS, Kennedy J, WolfkielC, et al. Ultrafast computed tomography as a diagnostic modality inthe detection of coronary artery disease: a multicenter study.Circulation 1996; 93: 898–904
31. He ZX, Hedrick TD, Pratt CM, Verani MS, Aquino V, Roberts R, et al.Severity of coronary artery calcification by electron beam computedtomography predicts silent myocardial ischemia. Circulation 2000;101: 244–251
32. Vliegenthart R, Oudkerk M, Song B, van der Kuip DA, Hofman A,Witteman JC. Coronary calcification detected by electron-beamcomputed tomography and myocardial infarction. The RotterdamCoronary Calcification Study. Eur Heart J 2002; 23: 1596–1603
33. Masuda Y, Naito S, Aoyagi Y, Yamada Z, Uda T, Morooka N, et al.Coronary artery calcification detected by CT: clinical significance andangiographic correlates. Angiology 1990; 41: 1037–1047
34. O’Rourke RA, Brundage BH, Froelicher VF, Greenland P, Grundy SM,Hachamovitch R, et al. American College of Cardiology/AmericanHeart Association Expert Consensus Document on electron-beamcomputed tomography for the diagnosis and prognosis of coronaryartery disease. J Am Coll Cardiol 2000; 36: 326–340
35. Lamont DH, Budoff MJ, Shavelle DM, Shavelle R, Brundage BH, HagarJM. Coronary calcium scanning adds incremental value to patientswith positive stress tests. Am Heart J 2002; 143: 861–867
IHJ-478-03.p65 11/19/2003, 11:25 PM348

Indian Heart J 2003; 55: 349–353 Malhotra et al. Prehospital Delay in Treatment of AMI 349
Prehospital Delay in Patients Hospitalized WithAcute Myocardial Infarction in the Emergency Unit of a
North Indian Tertiary Care Hospital
S Malhotra, M Gupta, KK Chandra, A Grover, P PandhiDepartments of Pharmacology and Cardiology, Post Graduate Institute of Medical Education and Research, Chandigarh
Acute myocardial infarction (AMI) results fromprolonged myocardial ischemia, precipitated in most
cases by an occlusive coronary thrombus at the site of apre-existing atherosclerotic plaque. It is a major cause ofmortality and morbidity in the general population.Currently, the prevalence of coronary artery disease (CAD)in the age group of 40–50 years in urban India is 4-foldhigher than in the United States1 (10% v. 2.5%).
Numerous studies have demonstrated the benefits ofthrombolytic therapy for patients with evolving AMI.2 Thusattention has shifted towards efforts to increase the use ofthis therapy. However, the benefits of thrombolytic therapyare greater in the first few hours after the onset ofsymptoms.3 Hence, patient-associated delay in therecognition of symptoms of heart attack has been identifiedas a major obstacle to the widespread use of thrombolytictherapy.4 The duration of prehospital delay includes the
time required to recognize the symptoms, decision to seekcare, arrange transportation, travel to the hospital, and thetime required for diagnosis.5
A large number of factors have been attributed toprehospital delay; these include sociodemographic,behavioral, clinical, and situational constraints.6 The aimof the present study was to examine the extent of, andfactors associated with, delay in seeking medical care(usually thrombolytic therapy) in patients with AMI.
Methods
The study was conducted in patients presenting with AMIadmitted to the medical emergency or coronary care unitof the Nehru Hospital, Post Graduate Institute of MedicalEducation and Research, Chandigarh from December 2001to June 2002. One hundred four consecutive patients ofMI were interviewed using a pre-designed proforma. Thechief constituents of the questionnaire included thedemographic characteristics of the patient, nature and timeof onset of symptoms, pre-existing illness and treatment
Original Article
Background: Prompt treatment of patients presenting with acute myocardial infarction decreases the incidenceof death from early arrhythmia, and maximizes the potential benefit of thrombolytic therapy. Prehospital delayhas been identified as a major obstacle to the widespread use of thrombolytic therapy. The aim of the presentstudy was to examine the extent of, and factors associated with, delay in seeking medical care (usuallythrombolytic therapy) in patients with acute myocardial infarction.Methods and Results: The study was conducted in patients visiting the medical emergency unit of the NehruHospital, Post Graduate Institute of Medical Education and Research, Chandigarh. A total of 104 patientsdiagnosed with acute myocardial infarction were interviewed using a pre-designed proforma. Pain-to-door, anddoor-to-drug times, were the main outcome measures. The corrected mean (SEM) and median (range) pain-to-door times were 8.5 (0.8) hours and 5.2 (0.5–24) hours, respectively. Out of 104 patients, 38 did not receivethrombolytic therapy. In those who did not receive thrombolytic therapy, prior therapy at local health centers,lack of knowledge of symptoms, and transportation problems were the main reasons for hospital delay. Themean (SEM) and median (range) of door-to-drug times were 1.2 (0.1) hours and 1 (0.2–3.5) hours, respectively.(Indian Heart J 2003; 55: 349–353)
Key Words: Prehospital delay, Thrombolytic therapy, Acute myocardial infarction
Correspondence: Dr (Mrs) P Pandhi, Department of Pharmacology, PostGraduate Institute of Medical Education and Research, Chandigarh 160012.e-mail: [email protected]
IHJ-470-03.p65 11/19/2003, 11:25 PM349

350 Malhotra et al. Prehospital Delay in Treatment of AMI Indian Heart J 2003; 55: 349–353
received, and any previous knowledge about symptoms ofheart attack. The patients were then asked about the timeat which they presented to the emergency unit and receivedmedical care. The main outcome measures included: (i)pain-to-door time (time interval between the onset ofsymptoms suggestive of AMI and arrival in the emergencyunit); and (ii) door-to-drug time (time interval betweenarrival in the emergency unit and administration ofthrombolytic therapy).
Statistical analysis: The data were expressed as mean(±SEM) and median. To examine the differences betweendifferent delay groups for continuous variables, t tests andanalysis of variance (ANOVA) were used. A value of p<0.05was considered statistically significant.
Results
Of the 104 consecutive MI patients studied, 66 receivedthrombolytic therapy. We initially examined the associationbetween demographic and clinical factors, and durationof prehospital delay (Table 1). The average and mediandelay times increased with advancing age. Men did notexperience significant delay as compared to women. Also,delay was not significant in patients with respect to thelocation and type of MI. Patients with a history of anginaexhibited prolonged delay compared with patients withoutangina (13.4 v. 3.2 hours). However, patients with ahistory of diabetes and hypertension did not experiencesignificant delays as compared with patients without theseconditions. Patients seeking treatment from localphysicians delayed longer than those who camestraightaway to the emergency unit (16.1 v. 6 hours) (Fig.1). We also found that patients who did not know thesymptoms of heart attack delayed longer than those whohad a good knowledge (Fig. 1).
Pain–door–drug times: The mean (SEM) and median(range) pain-to-door times were 13 (2.1) hours and 5.2(0.5–12) hours, respectively. The corrected (delay equals24 hours for those delaying 24 hours or more) mean (SEM),and median (range) pain-to-door times were 8.5 (0.8) hoursand 5.2 (0.5–24) hours, respectively (Fig. 2).
Fig. 1. Effect of treatment order and patients’ knowledge of disease symptomson prehospital delay.
Fig. 2. Prevalence of prehospital delay (pain-to-door time in patients with acutemyocardial infarction).
The mean (SEM) and median (range) of door-to-drugtimes were 1.2 (0.1) hours and 1 (0.2–3.5) hours,respectively (Table 2).
Prehospital delay was the cause for 30 of the 38 patients(79%) not receiving any thrombolytic therapy. In theremaining 8 patients, the causes for not receivingthrombolysis were: unaffordability of urokinase (3),unaffordability of streptokinase (1), associatedcontraindications for thrombolysis (2), and already receivedthrombolysis (2) (Fig. 3). Prior therapy at local healthcareservices, lack of knowledge of symptoms, andtransportation problems were the main reasons forprehospital delay in nonthrombolyzed patients.
Fig. 3. Reasons for not receiving thrombolytic therapy.
Prior local treatment None No Yes
Treatment color Knowledge of disease symptoms
Preh
ospi
tal d
elay
[cor
rect
ed m
ean
(SEM
) hou
rs]
Prehospital delay
Unaffordability of urokinase
Unaffordability of streptokinase
Associated contraindications forthrombolysis
Already thrombolized
IHJ-470-03.p65 11/19/2003, 11:25 PM350
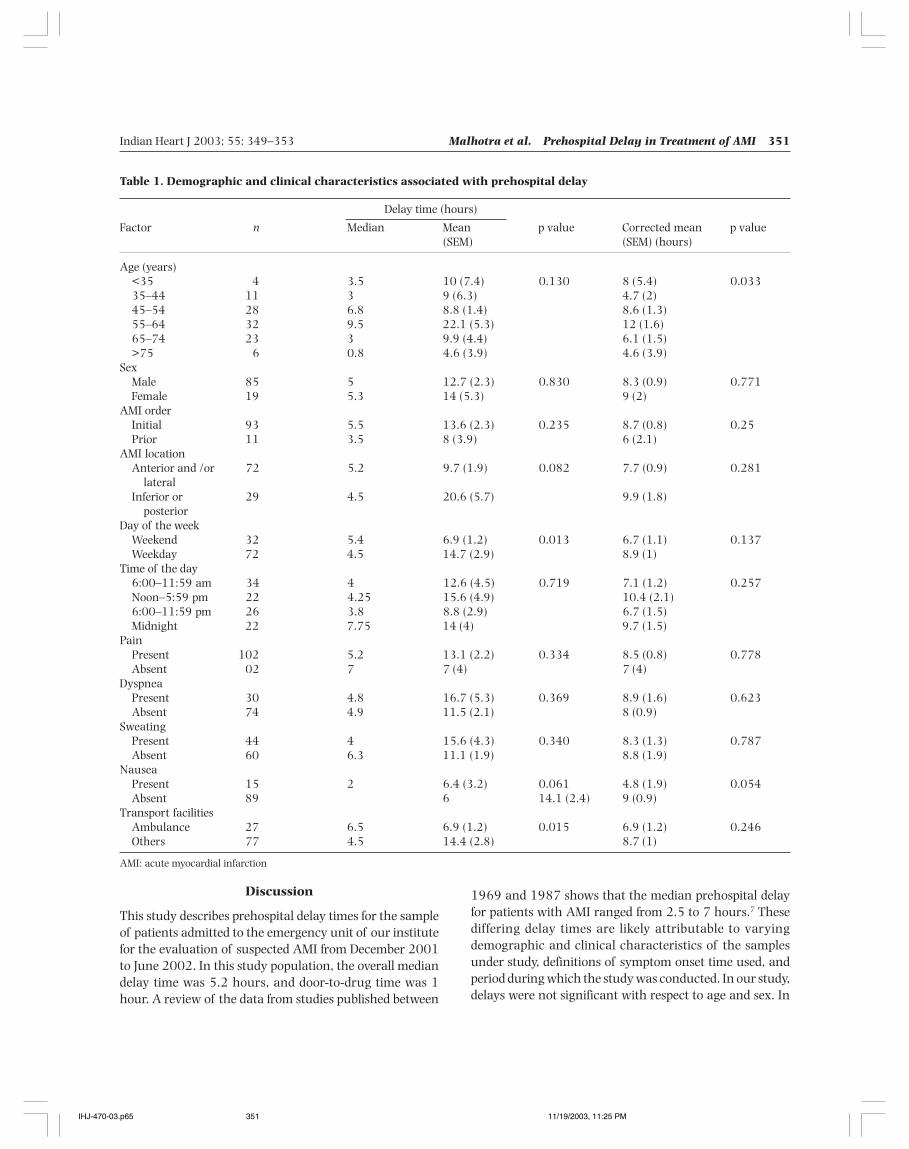
Indian Heart J 2003; 55: 349–353 Malhotra et al. Prehospital Delay in Treatment of AMI 351
Discussion
This study describes prehospital delay times for the sampleof patients admitted to the emergency unit of our institutefor the evaluation of suspected AMI from December 2001to June 2002. In this study population, the overall mediandelay time was 5.2 hours, and door-to-drug time was 1hour. A review of the data from studies published between
1969 and 1987 shows that the median prehospital delayfor patients with AMI ranged from 2.5 to 7 hours.7 Thesediffering delay times are likely attributable to varyingdemographic and clinical characteristics of the samplesunder study, definitions of symptom onset time used, andperiod during which the study was conducted. In our study,delays were not significant with respect to age and sex. In
Table 1. Demographic and clinical characteristics associated with prehospital delay
Delay time (hours)
Factor n Median Mean p value Corrected mean p value(SEM) (SEM) (hours)
Age (years)<35 4 3.5 10 (7.4) 0.130 8 (5.4) 0.03335–44 11 3 9 (6.3) 4.7 (2)45–54 28 6.8 8.8 (1.4) 8.6 (1.3)55–64 32 9.5 22.1 (5.3) 12 (1.6)65–74 23 3 9.9 (4.4) 6.1 (1.5)>75 6 0.8 4.6 (3.9) 4.6 (3.9)
SexMale 85 5 12.7 (2.3) 0.830 8.3 (0.9) 0.771Female 19 5.3 14 (5.3) 9 (2)
AMI orderInitial 93 5.5 13.6 (2.3) 0.235 8.7 (0.8) 0.25Prior 11 3.5 8 (3.9) 6 (2.1)
AMI locationAnterior and /or 72 5.2 9.7 (1.9) 0.082 7.7 (0.9) 0.281
lateralInferior or 29 4.5 20.6 (5.7) 9.9 (1.8)
posteriorDay of the week
Weekend 32 5.4 6.9 (1.2) 0.013 6.7 (1.1) 0.137Weekday 72 4.5 14.7 (2.9) 8.9 (1)
Time of the day6:00–11:59 am 34 4 12.6 (4.5) 0.719 7.1 (1.2) 0.257Noon–5:59 pm 22 4.25 15.6 (4.9) 10.4 (2.1)6:00–11:59 pm 26 3.8 8.8 (2.9) 6.7 (1.5)Midnight 22 7.75 14 (4) 9.7 (1.5)
PainPresent 102 5.2 13.1 (2.2) 0.334 8.5 (0.8) 0.778Absent 02 7 7 (4) 7 (4)
DyspneaPresent 30 4.8 16.7 (5.3) 0.369 8.9 (1.6) 0.623Absent 74 4.9 11.5 (2.1) 8 (0.9)
SweatingPresent 44 4 15.6 (4.3) 0.340 8.3 (1.3) 0.787Absent 60 6.3 11.1 (1.9) 8.8 (1.9)
NauseaPresent 15 2 6.4 (3.2) 0.061 4.8 (1.9) 0.054Absent 89 6 14.1 (2.4) 9 (0.9)
Transport facilitiesAmbulance 27 6.5 6.9 (1.2) 0.015 6.9 (1.2) 0.246Others 77 4.5 14.4 (2.8) 8.7 (1)
AMI: acute myocardial infarction
IHJ-470-03.p65 11/19/2003, 11:25 PM351

352 Malhotra et al. Prehospital Delay in Treatment of AMI Indian Heart J 2003; 55: 349–353
Table 2. Duration of prehospital delay and timing ofadministration of thrombolytic therapy in patients withacute myocardial infarction
Pain-to-door Door-to-drug times (hours)time. Duration n Median Mean (SEM) p value*of prehospitaldelay (hours)
<1 6 0.8 1.0 (0.3) 0.6271–1.9 16 0.8 0.1 (0.1)2–3.9 16 0.7 1.2 (0.2)4–5.9 10 1.4 1.4 (0.2)6–11.9 16 1 1.4 (0.3)>12 2 1.5 1.5 (0.5)Total 66 1 1.2 (0.1)
*p value denotes the comparison of mean door-to-drug times in the variousgroups of patients with different durations of prehospital delay.
an ambulance can quickly transport patients directly to thetertiary-care center if necessary. Secondly, generalpractitioners can be formally trained to administerstreptokinase themselves, and deal with the complicationsarising from streptokinase therapy.
In the present study, out of 104 patients, 66 werethrombolyzed. Also, prehospital delay was significantlylonger in the nonthrombolysed patients (corrected mean15 hours v. 4.6 hours). There is overwhelming evidence ofthe beneficial effects provided by reperfusion strategies inpatients with AMI.10 Although various contraindicationsto the administration of thrombolytic therapy presentlyexist, including prolonged prehospital delay, thrombolytictherapy is underutilized in the management of AMI.Findings from the Worcester Heart Attack Study show thatpatients who delayed for more than 6 hours were 6.5 timesless likely to receive thrombolytic agents compared topatients who arrived within 1 hour of the onset of AMI.8
In the present study, out of 38 nonthrombolysed patients,30 presented to the hospital after 6 hours; the remainingeither could not afford thrombolytic therapy or werealready thrombolysed. Another aspect of prehospital delayhighlighted by our study is knowledge regarding heartattack symptoms. Patients who knew about the symptomsof heart attack delayed less than those who were ignorant(2.9 v. 11.9 hours). Thus, educational approaches areessential to reduce the extent of patient delay, and toenhance widespread use of time-dependent managementstrategies in patients hospitalized with AMI.
Hospital-associated delays in administeringthrombolytic therapy: In our study, mean and mediandoor-to-drug times in thrombolysed patients were 1.2 (0.1)hours and 1 (0.2–3.5) hours, respectively. However, aprevious study suggests an association of increasing delaysin thrombolytic therapy with increasing duration of pre-hospital delay. The National Heart Attack Alert Programhas appropriately called attention to reduction of door-to-drug times. However, a few studies have examined therelationship between the extent of prehospital and hospitaldelay with thrombolytic therapy.
Limitations of the study: There are several limitationsof the present study. The effect of prehospital delay ondisease outcomes was not studied. Patients who died of AMIoutside the hospital and those with silent MI were notstudied. Appropriate caveats need to be planned for theinterpretation of study findings, since different delaypatterns may have existed in patients with poorly definedtime of onset of symptoms. The number of patientsincluded in the study was also small. However, our data
contrast, earlier studies suggested that older individualstook longer to access medical care than youngerindividuals.8 This might be due to the still prevalent jointfamily norms in Indian society. The median delay in patientswith a past medical history of ischemic heart disease wasnot significantly different from that in patients without sucha history. Thus, the patient’s behavior was not modified bythe past experience of an acute attack. It is also interestingto note that the majority of patients studied had no historyof angina (100 v. 4), diabetes (93 v. 11), or hypertension(87 v. 17). It could be speculated that patients with theabove diseases were more health conscious, and tookmedicines regularly. Also, prehospital delay was notdifferent in patients with or without the above diseases. Ourresults are contradictory to those of previous studies wherepatients with a history of MI and diabetes delayed longerthan those without the disease.7
Pain was the most common presenting symptom in ourstudy (102 v. 2). However, it was found that presentingsymptoms such as pain, dyspnea, sweating, and nausea arenot related to pain-to-door time. It has been seen that thebehavior of patients with collapse, pain or pathologicalbreathlessness after MI is at an instinctive level, though tosome extent, it may be culturally related.9
In contrast to previous studies, the day of the week andtime of presentation did not affect the prehospital time.Prehospital delay was significantly higher in patients (9.9v. 5.2 hours, p=0.002) who had visited a local practitionerthan those who came straightaway to the emergency unitof the hospital. While the reasons for such a practice maybe speculative, we suggest that this delay in time can bereduced if there is a 24-hour ambulance service with ahotline and facilities for fascimile transmission of ECG. Such
IHJ-470-03.p65 11/19/2003, 11:25 PM352

Indian Heart J 2003; 55: 349–353 Malhotra et al. Prehospital Delay in Treatment of AMI 353
are representative of a typical tertiary care hospital and,therefore, such data can be generalized to other hospitalsas well.
In conclusion, prehospital delay is the main reason fornot receiving thrombolytic therapy. The main factorscontributing to prehospital delay are: poor knowledge ofsymptoms, prior treatment from local practitioners, andunavailability of rapid transport facilities. Targeteducational efforts, and provision of rapid transportationfacilities can help reduce the excessive case fatality ratesattributed to prehospital delay.
References
1. Enas EA, Garg A, Davidson MA, Nair VM, Huet BA, Yusuf S. Coronaryheart disease and its risk factors in first-generation immigrant AsianIndians to the United States of America. Indian Heart J 1996; 48:343–353
2. Effectiveness of intravenous thrombolytic treatment in acutemyocardial infarction. Gruppo Italiano per lo Studio dellaStreptochinasi nell’ Infarto Miocardico (GISSI). Lancet 1986; 1:397–402
3. Second International Study of Infarct Survival (ISIS-2) SteeringCommittee. Intravenous streptokinase given within 0–4 hours of
onset of myocardial infarction reduced mortality in ISIS-2. Lancet1987; 1: 502–508
4. Goldberg RJ, McGovern PG, Guggina T, Savageau J, Rosamond WD,Luepker RV. Prehospital delay in patients with acute coronary heartdisease: concordance between patient interviews and medicalrecords. Am Heart J 1998; 135: 93–99
5. Goff DC Jr, Feldman HA, McGovern PG, Goldberg RJ, Simons- MortonDG, Cornell CE, et al. Prehospital delay in patients hospitalized withheart attack symptoms in the United States: the REACT trial. RapidEarly Action for Coronary Treatment (REACT) Study Group. AmHeart J 1999; 138: 1046–1057
6. Hackett TP, Cassem NH. Factors contributing to delay in respondingto the signs and symptoms of acute myocardial infarction. Am JCardiol 1969; 24: 651–658
7. Yarzebski J, Goldberg RJ, Gore JM, Alpert JS. Temporal trends andfactors associated with extent of delay to hospital arrival in patientswith acute myocardial infarction: the Worcester Heart Attack Study.Am Heart J 1994; 128: 255–263
8. Goldberg RJ, Yarzebski J, Lessard D, Gore JM. Decade-long trends andfactors associated with time to hospital presentation in patients withacute myocardial infarction: the Worcester Heart Attack Study. ArchIntern Med 2000; 160: 3217–3223
9. Rawles JM, Haites NE. Patient and general practitioner delays in acutemyocardial infarction. Br Med J 1988; 196: 882–884
10. Goff DC, Nichoman NZ, Ramsay DJ, et al. A population basedassessment of the use and effectiveness of thrombolytic therapy: theCorpus Christi Heart Project. Ann Epidemiol 1995; 5: 171–178
IHJ-470-03.p65 11/19/2003, 11:25 PM353

354 Roy et al. Mitral Valve Repair for Nonrheumatic Mitral Regurgitation Indian Heart J 2003; 55: 354–357
Mitral Valve Repair for NonrheumaticMitral Regurgitation
Swapnadeep Roy, Shiv Kumar Choudhary, Arkalgud Sampath KumarDepartment of Cardiothoracic and Vascular Surgery, All India Institute of Medical Sciences, New Delhi
Surgical treatment of severe mitral regurgitation (MR)currently involves either mitral valve (MV) repair or
replacement (mechanical prosthesis/bioprosthesis).Although valve replacement has been the widely acceptedmodality, there are numerous complications associatedwith this procedure such as thrombosis, thromboembolism,anticoagulant-related hemorrhage, and bioprostheticdegeneration over a period of 7–15 years, necessitating asecond surgical procedure. Therefore, repair as thepreferred option, whenever possible, is gaining popularityamong cardiac surgeons.
The etiology of the MR may be rheumatic ornonrheumatic. In the latter category, there are numerouscauses such as congenital (often associated with atrialseptal defect), myxomatous degeneration, ischemic, andinfective. While the results of repair for rheumatic valvedisease often have a late phase of deterioration due to
progression of the rheumatic valvulitis, the reported resultsof repair in nonrheumatic cases have generally been moresatisfactory. We report our experience with repair in thissubset of patients.
Methods
From January 1988 to April 2002, 116 patients with severeMR of nonrheumatic etiology underwent MV repair. Theage range of the patients was 2–67 years (mean age 26.4years). Of these patients, 59 were males (50.8%), and 57females (49.2%). The causes of nonrheumatic MR werecongenital in 56 patients (47.4%), myxomatousdegeneration with cusp prolapse in 44 (37.9%), infectiveendocarditis in 7 (6%), and ischemic in 9 (7.7%). Six of thepatients had a primum ASD (partial atrioventricular septaldefect), and 46 had a secundum ASD. An isolated cleft MVwas responsible for the MR in 4 patients.1 In patients withmyxomatous degeneration, there was cusp prolapse withchordal elongation/rupture, predominantly in the posterior
Original Article
Background: We studied the results of mitral valve repair in patients with severe mitral regurgitation ofnonrheumatic etiology.Methods and Results: Between January 1988 and April 2002, 116 patients, of which 59 were male and 57female, with severe mitral regurgitation of nonrheumatic etiology, underwent mitral valve repair using a varietyof techniques. Their mean age was 26.4 years (range 2–67 years). The cause of mitral regurgitation wascongenital in 56 patients, myxomatous in 44, infective endocarditis in 7, and ischemic in 9. Ninety patientswere in preoperative New York Heart Association class III, and 26 in class IV. Reparative procedures includedposterior teflon felt collar annuloplasty (modified Cooley’s) in 80 patients, chordal shortening in 37, cusp excisionin 34, cleft closure in 8, chordal transfer in 6, and neochordae in 3. The early mortality was 3.4% (4 patients).Follow-up ranged from 1 to167 months (mean 47 months), and was 95% complete. There were 2 late deaths(1.7%). Six patients (5.2%) underwent reoperation for severe mitral regurgitation post-repair. Of the remaining104 patients, 90 (86.5%) had no or trivial mitral regurgitation at the last follow-up. Actuarial, reoperation-free, and event-free survival at 130 months was 93%±3.6%, 89.9%±6%, and 69.7%±13.7%, respectively.Ninety-two patients (88.5%) were in New York Heart Association class I at the last follow-up.Conclusions: Mitral valve repair in nonrheumatic mitral regurgitation patients provides satisfactory resultswith current surgical techniques, and is the preferred option in this subset of patients. (Indian Heart J 2003;55: 354–357)
Key Words : Mitral regurgitation, Mitral valve repair, Valvular heart disease
Correspondence: Professor A Sampath Kumar, Department ofCardiothoracic and Vascular Surgery, Cardiothoracic Centre, AIIMS,New Delhi 110029. e-mail: [email protected]
IHJ-507-03.p65 11/19/2003, 11:26 PM354

Indian Heart J 2003; 55: 354–357 Roy et al. Mitral Valve Repair for Nonrheumatic Mitral Regurgitation 355
mitral leaflet. In the ischemic category, we included patientswith chronic ischemia only, in whom the MR was due topapillary muscle dysfunction or chordal elongation/rupture. Associated annular dilatation was present in allthese patients due to left ventricular dilatation. In patientswith infective endocarditis, there were primarily localizedareas of leaflet destruction, and repair was undertaken onlyafter an adequate course of antibiotics (at least 2 weeks).
Seventy-four patients (63.8%) were in sinus rhythmpreoperatively, and 42 in atrial fibrillation (36.2%). Thepredominant presenting symptom was dyspnea onexertion. Ninety patients were in New York HeartAssociation (NYHA) class III, and 26 were in class IV.
Preoperative transthoracic echocardiography wasperformed in all the patients. Cardiac catheterization andcoronary arteriography were performed in patients above40 years of age, or if there was a suspicion of associatedaortic valve disease, or coronary artery disease. The severityof MR was assessed by echocardiography and/orangiography.
Surgical technique: After induction of generalanesthesia, transesophageal echocardiography (TEE) wasperformed in all the patients after 1994. Intraoperatively,the surgical approach was either a mid-sternotomy(n=100) or a right anterolateral thoracotomy (n=16). Thelatter approach was primarily used for cosmetic reasons inyoung females. Before 1996, moderately hypothermic(32 oC) cardiopulmonary bypass (CPB), and since 1996,normothermic perfusion was used in all the patients. Cold-blood cardioplegia and topical ice-slush was used for theprotection of the myocardium. Bicaval cannulation wasused in all the patients. The MV was exposed through astandard incision in the left atrial wall behind the interatrialgroove. In patients with an ASD, the approach was throughthe right atrium. After assessing the morphology, a varietyof reparative techniques were utilized, as described earlier.2–5
The adequacy of repair was confirmed by injecting salineinto the left ventricular cavity and observing the coaptationof the leaflets. At the end of the procedure afterdiscontinuing CPB, TEE was performed in all the patientstreated after 1994 to confirm the adequacy of repair.
The various procedures utilized for the MV repair are asfollows: (i) posterior teflon felt collar annuloplasty (modifiedCooley’s)2 in 80 patients (68.9%); (ii) chordal shortening3
in 37 (31.9%); (iii) cusp excision (quadrangular resectionof posterior mitral leaflet) in 34 (29.3%); (iv) cleft closurein 8 (6.9%); (v) chordal transfer in 6 (5.2%); and (vi)neochordae in 3 (2.6%).
Associated procedures performed in these patients
included the following: (i) aortic valve replacement(prosthetic) in 8 (primarily for aortic regurgitation);(ii) aortic valvuloplasty in 15; (iii) closure of atrial septaldefect in 52; (iv) coronary artery bypass grafting in 5 (3 ofthe other 4 patients with ischemic MR had prior successfulangioplasty of the diseased target vessels; the remainingpatient had scarred myocardium with no reuptake onthallium scan).
Follow-up ranged from 1 to 168 months (mean 47months). The follow-up was 95% complete. At each follow-up visit, a transthoracic echocardiogram (TTE) wasperformed in all these patients to assess the status of theMV repair, besides looking for any associated lesions.Statistical analysis was performed using SPSS software ona Windows operating system. Kaplan–Meier survivalestimates were derived for this group of patients.
Results
All the patients survived the operation. The mean bypasstime was 51 min (range 28–70 min), and the mean aorticcross-clamp time was 37 min (range 21–58 min). Themean intensive care unit stay was 2 days (range 1–6 days),and the mean postoperative ventilation was for a period of8 hours (range 6–72 hours). The mean postoperative staywas 5 days (range 4–9 days).
Early mortality (30-day) was seen in 4 patients (3.4%).In 1 patient, the cause of death was persistent low outputfollowing the repair (preoperatively the patient was incongestive heart failure unresponsive to medicalmanagement). The second patient developed acute renalfailure in the immediate postoperative period andsuccumbed. Two other patients died of persistentintractable ventricular arrhythmias.
In the surviving patients, a pre-discharge TTE wasperformed to evaluate the status of the MV repair. In allthe 112 patients, there was no or mild MR that correlatedwell with the TEE, performed at the end of operation in theoperating room.
There were 2 late deaths (1.7%). Both patients haddeveloped severe left ventricular dysfunction secondary tosevere MR and succumbed before reoperation. Six patients(5.2%) underwent reoperation for severe MR. Of thesepatients, 5 had congestive heart failure, and the sixthpatient was in NYHA class III prior to reoperation. In 5 ofthe 6 patients, the cause of failure of the original repairwas due to suture dehiscence of the annuloplasty, or thoseused for chordal shortening. In one of them, there wasprogression of the original myxomatous degeneration withchordal rupture of originally uninvolved chordae. All the
IHJ-507-03.p65 11/19/2003, 11:26 PM355

356 Roy et al. Mitral Valve Repair for Nonrheumatic Mitral Regurgitation Indian Heart J 2003; 55: 354–357
6 patients had a prosthetic MV replacement, and survived.Of the 104 late survivors, 92 (86.5%) are in NYHA class
I, and 12 in NYHA class II at the last follow-up. Eightypatients (76.9%) are in sinus rhythm, while the rest are inchronic atrial fibrillation (AF) (all of them were also in AFpreoperatively). One of the 104 late survivors (0.9%) hada thromboembolic complication (the patient was in AFpostoperatively). There were no other complications. Ofthose who were reoperated, all have improved to NYHAclass II status.
At the last follow-up, 90 of the 104 surviving patientshave no or trivial MR on echocardiography, 6 have mildMR, and 8 have moderate-to-severe MR.
The actuarial, reoperation-free, and event-free survivalin the group of operative survivors at 130 months was93%±3.6%, 89.9%±6%, and 69.7%±13.6%, respectively(Kaplan–Meir estimates). Freedom from thromboembolicevents at 108 months was 98.6% (Figs 1–3).
Discussion
In India, rheumatic heart disease remains the predominantcause of MR. However, MR due to nonrheumatic causes isnot uncommon.
Repair of the MV has gradually become widely accepted,ever since it was popularized by Carpentier.7 For non-rheumatic severe MR, repair of the MV has shown excellentlong-term durability. The Carpentier group6–8 has reporteda 15-year actuarial survival of 72.4%, with freedom fromreoperation at 15 years being 92.7%. This grouppredominantly used a prosthetic ring in most of theirannuloplasty procedures. Galloway et al.9 reported on theirresults with Carpentier-type MV reconstructions with anoperative mortality of 5%, and a 5-year freedom fromreoperation rate of 94.4%. Actuarial freedom fromthromboembolic complications was 93.9% at 15 years, and96.5% were free from anticoagulant-related hemorrhage.Similar results have been reported by other groups also.Overall, the results are better than those reported withprosthetic MV replacement.10
Our results compare favorably with other reports in theliterature. Our 10-year survival of about 93% with areoperation-free rate of 89% is comparable to most reportedseries. Functional rehabilitation and the absence ofmedication are marked advantages. Preservation of leftventricular function is another advantage.
Conclusion: Repair of the MV rather than its replacementshould be the procedure of choice in patients with non-rheumatic MR.
Probability 100 100 99 99 99 99 97.3 95.3 92.9 92.9at risk 111 103 86 74 69 60 52 41 17 6
Fig. 1. Probability of actuarial survival in patients of mitral valve repair fornonrheumatic mitral regurgitation (Kaplan–Meier estimates).
Probability 100 99 99 97.8 97.8 97.8 97.8 95.6 95.6 89.9at risk 111 103 86 76 70 64 53 42 18 4
Fig. 2. Probability of reoperation-free survival in patients after mitral valverepair for nonrheumatic mitral regurgitation (Kaplan–Meier estimates).
Probability 100 99 98 96.8 96.8 96.8 96.8 90.5 83.7 83.7at risk 111 103 86 77 74 63 52 41 19 6
Fig. 3. Probability of event-free survival in patients after mitral valve repair fornonrheumatic mitral regurgitation (Kaplan–Meier estimates).
IHJ-507-03.p65 11/19/2003, 11:26 PM356

Indian Heart J 2003; 55: 354–357 Roy et al. Mitral Valve Repair for Nonrheumatic Mitral Regurgitation 357
Acknowledgments
The authors acknowledge the help provided by the residentsof the department for the excellent record-keeping thatgreatly facilitated the analysis of results. We would also liketo thank Mr Rajbir of the Department of Biostatistics, forthe statistical analysis.
References
1. Mohanty SR, Choudhary SK, Ramamurthy S, Kumar AS. Isolatedcongenital anterior mitral leaflet cleft: a rare cause of mitralinsufficiency. J Heart Valve Dis 1999; 8: 67–70
2. Kumar AS, Kumar RV, Shrivastava S, Venugopal P, Sood AK, GopinathN. Mitral valve reconstruction. Early results of a modified Cooleytechnique. Tex Heart Inst J 1992; 19: 107–111
3. Kumar AS, Bhan A, Kumar RV, Shrivastava S, Sood AK, Gopinath N.Cusp-level chordal shortening for rheumatic mitral regurgitation.Early results. Tex Heart Inst J 1992; 19: 47–50
4. Kumar AS, Rao PN. Restoration of pliability to the mitral leafletsduring reconstruction. J Heart Valve Dis 1995; 4: 251–253
5. Choudhary SK, Talwar S, Dubey B, Chopra A, Saxena A, Kumar AS.Mitral valve repair in a predominantly rheumatic population. Long-term results. Tex Heart Inst J 2001; 28: 8–15
6. Deloche A, Jebara AV, Relland JY, Chauvaud S, Fabiani JN, Perier P,et al. Valve repair with Carpentier techniques. The second decade. JThorac Cardiovasc Surg 1990; 99: 990–1001
7. Carpentier A. Cardiac valve surgery—the “French correction”. JThorac Cardiovasc Surg 1983; 86: 323–337
8. Carpentier A, Chauvaud S, Fabiani JN, Deloche A, Relland J, LessanaA, et al. Reconstructive surgery of mitral valve incompetence: ten-year appraisal. J Thorac Cardiovasc Surg 1980; 79: 338–348
9. Galloway AC, Colvin SB, Baumann FG, Grossi EA, Ribakove GH,Harty S, et al. A comparison of mitral valve reconstruction withmitral valve replacement: intermediate-term results. Ann Thorac Surg1989; 47: 655–662
10. Sand ME, Naftel DC, Blackstone EH, Kirklin JW, Karp RB. Acomparison of repair and replacement for mitral valve incompetence.J Thorac Cardiovasc Surg 1987; 94: 208–219
IHJ-507-03.p65 11/19/2003, 11:26 PM357

358 Srimannarayana et al. Left Atrial Thrombus in Mitral Stenosis Indian Heart J 2003; 55: 358–361
Prevalence of Left Atrial Thrombus in Rheumatic MitralStenosis With Atrial Fibrillation and its Response to
Anticoagulation: A TransesophagealEchocardiographic Study
J Srimannarayana, RS Varma, S Satheesh, R Anilkumar, J BalachanderDepartment of Cardiology, Jawaharlal Institute of Postgraduate Medical Education and Research, Pondicherry
Rheumatic mitral stenosis with atrial fibrillation (AF) isa common clinical problem in India. It is associated
with a very high risk of embolic cerebrovascular accidents,which is reported to be as much as seventeen times greaterthan in unaffected controls.1
Transesophageal echocardiography (TEE) is wellestablished as the gold standard for detecting thrombi inthe left atrium (LA) and the LA appendage. The sensitivityand specificity of TEE are reported to be 100% and 99%,respectively.2 The semi-invasive nature and safety of the testmake it ideal for serial follow-up of thrombi in the LA bodyand appendage. Though it is known that thrombi arecommon in patients with mitral stenosis and AF, only smallstudies have documented the exact frequency of occurrence
of these thrombi, and their fate on optimal oralanticoagulation.
The objectives of this study were to document thefrequency of occurrence of LA body and appendage clotsin patients with severe mitral stenosis and AF, and to studythe effect of oral anticoagulation on clot dissolution.
Methods
Consecutive patients with severe mitral stenosis and AF,who were being evaluated for percutaneous transvenousmitral commissurotomy (PTMC), underwent TEEevaluation. Patients were excluded from the study if therewas considerable mitral regurgitation or aortic valve disease.A total of 490 patients (343 females [70%], and 147 males[30%]) were included in the study. The mean age of thesepatients was 27.4±8.41 years (range 21–65 years).
Original Article
Correspondence: Professor R Anilkumar, Department of Cardiology,JIPMER, Pondicherry 605006. e-mail: [email protected]
Background: The frequency of occurrence of left atrial thrombi, and the effect of anticoagulation in patientswith rheumatic mitral stenosis and atrial fibrillation is not well established. This study was conducted to evaluatethe occurrence of left atrial body and left atrial appendage clots in patients with rheumatic mitral stenosis andatrial fibrillation, and to document the effect of long-term anticoagulation on clot dissolution.Methods and Results: Consecutive patients with severe rheumatic mitral stenosis and atrial fibrillation wereassessed by transesophageal echocardiography. Those with left atrial body or left atrial appendage clots wereanticoagulated with oral nicoumalone. Transesophageal echocardiography was then repeated in patients onanticoagulation who were on regular follow-up, and in whom percutaneous transvenous mitral commissurotomycould be considered. Of the 490 patients studied, 163 had left atrial body or left atrial appendage clots. A repeattransesophageal echocardiographic examination was done in 50 patients who had optimal anticoagulation fora period of 6 months. Only 2 of the 17 patients who had left atrial body clots had successful clot dissolution afterlong-term anticoagulation, while the left atrial appendage clots disappeared in 31 of 33 patients (p<0.001).Conclusions: Left atrial clots are present in a third of patients with severe rheumatic mitral stenosis and atrialfibrillation. Isolated left atrial appendage clots in patients with rheumatic mitral stenosis and atrial fibrillationcan disappear with long-term anticoagulation, while thrombi that extend into the left atrial body may persistdespite optimal anticoagulation. (Indian Heart J 2003; 55: 358–361)
Key Words: Rheumatic mitral stenosis, Left atrial thrombi, Echocardiography
IHJ-527-03.p65 11/19/2003, 11:26 PM358

Indian Heart J 2003; 55: 358–361 Srimannarayana et al. Left Atrial Thrombus in Mitral Stenosis 359
No clots were detected on repeat TEE in 31 out of 33patients (93.9%) with isolated LA appendage clots (Figs 1and 2). These 31 patients underwent PTMC successfully.Only 2 out of 17 patients (11.8%) with a previous LA bodyclot had no detectable clots. Patients with persistent LA clotswere referred for an open mitral valvotomy andthrombectomy. A comparison between those in whom theclot disappeared, and those in whom the clot persistedrevealed that the location of the LA clot (isolated LAappendage v. LA body) alone was a significant predictor ofthrombus dissolution. There were no statistically significantdifferences in other parameters, such as valve orifice, LAsize, mitral regurgitation, age, gender, and INR valuesbetween these groups.
An ACUSON 128-XP echocardiographic systemequipped with omniplane and biplane transesophagealprobes was used for this study. TEE was performed accordingto the standard procedure after obtaining informed consent.All the patients were put on oral anticoagulation withnicoumalone. The international normalized ratio (INR) wasmaintained between 2.5 and 3.5. Repeat TEE wasperformed after 6 months in patients (i) with LA body orLA appendage clots; (ii) on regular anticoagulation, andfollow-up with INR; and (iii) with valve morphologysuitable for PTMC.
Patients who were very sick (in NYHA class IV), andthose whose valve morphology precluded PTMC wereimmediately referred for appropriate surgery.
Of the patients undergoing repeat TEE, patients with anisolated LA appendage clot (group A) were compared withpatients in whom the thrombus extended into the LA bodyor who had isolated LA body clots (group B). The groupwith successful clot dissolution, and the group withpersistence of the thrombus were also compared.
Continuous variables were expressed as mean valueswith standard deviation, while proportions were presentedas numbers and percentages of the total. The significanceof differences between means was tested by the Student’st test, while that between proportions was tested with theChi-square test. A p value <0.05 was considered significant.
Results
Incidence and location of thrombi: Of the 490 patientswho underwent TEE, LA clots were present in 163 (33.2%).Isolated LA appendage clots were found in 88 patients(18%). Isolated LA body clots or LA appendage clotsextending into the LA body were found in 75 patients(15.3%). LA spontaneous echocardiographic contrast(LASEC) was present in 365 patients (74.5%). None of thepatients had right atrial thrombi.
Resolution of thrombi: TEE was repeated after 6 monthsof optimal oral anticoagulation in 50 out of 163 patients.These patients had maintained their INR in the range of2.5–3.5, and were on regular follow-up. Nearly half thepatients (48%) were above the age of 35 years (range 21–55 years). None of these patients had any other risk factors,such as diabetes mellitus, hypertension or smoking. Acomparison of the baseline characteristics, echocardio-graphic parameters, and mean INR between the group ofpatients with isolated LA appendage clots (n=33) and thegroup with clots extending to the LA body (n=17) is givenin Table 1. There were no statistically significant differencesamong these parameters between the two groups.
Table 1. Comparison of patients who underwent repeatTEE with isolated LA appendage clot versus those withclots extending to the LA cavity
Characteristic Group A Group B p value(n=33) (n=17)
Mean age (years) 33.91±7.82 33.8±9.01 nsMales 10 (30.3%) 9 (52.9%) nsFemales 23 (69.7%) 8 (47.1%)Mean MVO (cm2) 0.9±0.15 0.96±0.14 nsMean MPG (mmHg) 20.15±4.7 22.06±4.51 nsMean LA size (AP diameter, cm) 3.73±0.6 3.71±0.5 nsMitral regurgitation (grade 1/4) 7 (21.2%) 4 (23.5%) nsMean INR 2.87±0.3 2.89±0.23 ns
Group A: patients with isolated LA appendage clot; Group B: patients with clotsextending to the body of the LA or isolated LA body clots; MVO: mitral valve orifice;MPG: mean pressure gradient; AP: anteroposterior; INR: international normalizedratio; ns: not significant; LA: left atrial; TEE: transesophageal echocardiography
Discussion
Conventional transthoracic echocardiography (TTE) has apoor yield in the detection of LA appendage thrombi.3 Theposterior location of the LA in the chest as well as the poor
Fig. 1. Isolated left atrial appendage clot before anticoagulation.
IHJ-527-03.p65 11/19/2003, 11:26 PM359

360 Srimannarayana et al. Left Atrial Thrombus in Mitral Stenosis Indian Heart J 2003; 55: 358–361
visualization of the LA appendage contribute to the lack ofaccuracy of TTE. On the other hand, TEE is an excellentmethod for detecting atrial thrombi, especially those locatedin the LA appendage.
There are few reports on the prevalence of LA body andLA appendage clots in patients with severe mitral stenosisand AF. In a small group of 50 patients with mitral stenosisand AF, Hwang et al.4 observed an LA thrombus in 28patients (56%) by TEE. In another small study of 22 patientswith mitral stenosis and AF, Karatasakis et al.5 observedan LA thrombus in 12 patients (54%). In our study of 490patients, the prevalence was 33.5%. Considering the sizeof the study group, this can be considered a representativefigure for the prevalence of LA thrombi in patients withsevere mitral stenosis and AF. Thus, it can be stated that 1out of every 3 patients with severe mitral stenosis and AFwill have an LA thrombus.
Anticoagulation is conventionally used to reduce the riskof the thromboembolic events associated with AF. Oralanticoagulants act by inhibiting the synthesis of vitaminK-dependent coagulation factors. This prevents newthrombus formation, and promotes adherence andorganization of old thrombi to the surroundingendocardium.6–8 However, recent studies have supportedan alternative hypothesis of benefit that includes not onlythe prevention of new thrombus formation but also theresolution of existing thrombi.9
In situations in which a thrombus is already present,anticoagulation prevents further thrombus extension,thereby facilitating the action of endogenous fibrinolysis.A study conducted by Kandpal et al.10 showed that 41.7%of isolated LA appendage clots resolved in contrast to 12.5%of LA body clots in patients with mitral stenosis after 6months of oral anticoagulation. Resolution of atrial
thrombi after oral anticoagulation in patients withnonvalvar AF has also been examined in several studies.11,12
In the present study, it was seen that thrombi in the LAappendage disappeared on anticoagulation, whereasthrombi in the LA body did not. Success in resolving LAappendage thrombi vis-à-vis LA body thrombi is crucial,given that most clinically encountered thrombi are locatedin the LA appendage. This difference is possibly explainedby the fact that patients with an LA body clot have a higherclot burden, which does not permit dissolution withendogenous fibrinolysis despite optimal anticoagulation. Itis probable that clots localized to the LA appendage, with amuch smaller clot burden, are dissolved by endogenousfibrinolysis, while effective oral anticoagulation preventsfresh thrombus formation.
A study conducted by Murillo et al.13 showed that PTMCcan be performed in patients with an LA appendage clotwith TEE guidance and fluoroscopic control. However, thiscan be fraught with the risk of embolism. Hence, theinterventional cardiologist will always prefer the LA andLA appendage to be free of clots before embarking onballoon mitral valvotomy. Based on our study, it is possibleto evolve a strategy of treatment in patients with severemitral stenosis, AF, and LA thrombus. Patients with an LAthrombus extending well into the LA body can be directlyreferred for surgery, since they are unlikely candidates forclosed procedures. However, patients with isolated LAappendage clot and mitral stenosis can afford to be onanticoagulation for a period of 6 months, and then undergoa repeat TEE, especially if they can be stabilized to NYHAclass II or less. If the clot resolves, as in a good percentageof our subjects, these patients can undergo PTMC.
Some limitations of our study have to be emphasized. Arepeat TEE was done in only about one-third of the patientswith LA clots. The rest of the patients did not undergo arepeat TEE as part of the study because of one or more ofthe following reasons: valve morphology precluded PTMC,NYHA class III or class IV patients who were judged to needearly surgical treatment, suboptimal anticoagulation, andirregular follow-up. Thus, the results of the study shouldbe interpreted with some caution, and a further, large-scalestudy may be necessary to confirm the findings of our study.The effect of anticoagulation on LASEC was not studiedsince the mere presence of LASEC was not considered to bea contraindication for PTMC. Also, it was not practicallypossible to determine the duration of AF in these patients,and correlate it with clot prevalence and resolution.
Conclusions: One-third of patients with severe mitralstenosis and AF have LA body or LA appendage clots.Isolated LA appendage clots in patients with rheumatic
Fig. 2. The same patient as in Fig. 1, showing no left atrial appendagethrombus after anticoagulation for 6 months.
IHJ-527-03.p65 11/19/2003, 11:26 PM360

Indian Heart J 2003; 55: 358–361 Srimannarayana et al. Left Atrial Thrombus in Mitral Stenosis 361
mitral stenosis and AF can disappear with long-termanticoagulation. Such patients may become favorablecandidates for PTMC. Clot dissolution with oralanticoagulation is unlikely in those with an LA body clot.These patients can be referred early for open mitralvalvotomy and thrombectomy.
References
1. Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologicfeatures of chronic atrial fibrillation: the Framingham study. N EnglJ Med 1982; 306: 1018–1022
2. Manning WJ, Weintraub RM, Waksmonski CA, Haering JM, RooneyPS, Maslow AD, et al. Accuracy of transesophageal echo-cardiography for identifying left atrial thrombi. Ann Intern Med 1995;123: 817–822
3. Aschenberg W, Schluter M, Kremer P, Schroder E, Siglow V, BleifeldW. Transesophageal two-dimensional echocardiography for thedetection of left atrial appendage thrombus. J Am Coll Cardiol 1986;7: 163–166
4. Hwang JJ, Chen JJ, Lin SC, Tseng YZ, Kuan P, Lien WP, et al. Diagnosticaccuracy of transesophageal echocardiography for detecting leftatrial thrombi in patients with rheumatic heart disease havingundergone mitral valve operations. Am J Cardiol 1993; 72: 677–681
5. Karatasakis GT, Gotsis AC, Cokkinos DV. Influence of mitralregurgitation on left atrial thrombus and spontaneous
echocardiographic contrast in patients with rheumatic mitral valvedisease. Am J Cardiol 1995; 76: 279–281
6. Goldman M. The management of chronic atrial fibrillation. ProgCardiovasc Dis 1960; 2: 465–479
7. Mancini GB, Goldberg AL. Cardioversion of atrial fibrillation:consideration of embolization, anticoagulation, prophylacticpacemaker, and long-term success. Am Heart J 1982; 104: 617–621
8. Tsai LM, Hung JS, Chen JH, Lin LJ, Fu M. Resolution of left atrialappendage thrombus in mitral stenosis after warfarin therapy. AmHeart J 1991; 121: 1232–1234
9. Collins LJ, Silverman DI, Douglas PS, Manning WJ. Cardioversion ofnonrheumatic atrial fibrillation. Reduced thromboemboliccomplications with 4 weeks of precardioversion anticoagulation arerelated to atrial thrombus resolution. Circulation 1995; 92: 160–163
10. Kandpal B, Garg N, Anand KV, Kapoor A, Sinha N. Role of oralanticoagulation and inoue balloon mitral valvulotomy in presenceof left atrial thrombus: a prospective serial transesophgealechocardiographic study. J Heart Valve Dis 2002; 11: 594–600
11. Corrado G, Tadeo G, Beretta S, Tagliagambe LM, Manzillo GF, SpataM, et al. Atrial thrombi resolution after prolonged anticoagulationin patients with atrial fibrillation. Chest 1999; 115: 140–143
12. Jaber WA, Prior DL, Thamilarasan M, Grimm RA, Thomas JD, KleinAL, et al. Efficacy of anticoagulation in resolving left atrial and leftatrial appendage thrombi: a transesophgeal echocardiographic study.Am Heart J 2000; 140: 150–156
13. Murillo H, Ayala F, Lepe L, Madrid R, Solorio S, Lara A, et al.[Percutaneous mitral commissurotomy in patients with mitral valvestenosis and thrombosis of the left atrium.] Arch Inst Cardiol Mex1999; 69: 134–138
IHJ-527-03.p65 11/19/2003, 11:26 PM361

362 Mishra et al. Percutaneous Balloon Angioplasty of Membranous Obstruction Indian Heart J 2003; 55: 362–364
Percutaneous Balloon Angioplasty of MembranousObstruction of the Inferior Vena Cava
TK Mishra, SN Routray, M Behera, UK Patnaik, C SatapathyDepartment of Cardiology, SCB Medical College, Cuttack, Orissa
Membranous obstruction of the inferior vena cava(MOVC) is relatively common in Asia and Africa.1
Many cases have been reported from India.2 MOVC is oneof the important causes of Budd–Chiari syndrome.Intervention is often necessary, as medical treatment isineffective. The disease should be managed early, becauselong-standing MOVC can result in hepatocellularcarcinoma.1 Till the introduction of balloon dilatation fortreating MOVC cases, surgery was the only option available.Here, we report our experience with balloon angioplastyfor managing these cases.
Methods
Between January 1999 and January 2002, 19 patients whowere clinically diagnosed to have inferior vena cava (IVC)obstruction were taken up for the study. The disease wassuspected when the patients presented with ankle edema,ascites, and prominent veins over the abdomen and back,with the direction of blood flow from below upwards. Aftertaking a careful history, detailed physical examination was
carried out in all the patients. Hematological tests,coagulation profile, liver function tests, and abdominalultrasound with Doppler study of the IVC was undertaken.
After clinical diagnosis, the patients were taken up forinferior vena cavography, and subsequent angioplasty.Catheterization and angiography of the IVC was donethrough the femoral vein with a pigtail catheter.Simultaneous right atrial catheterization was done throughthe antecubital vein for simultaneous measurement of thepressure gradient between the IVC and right atrium (RA).
If the vena cavography demonstrated a completeobstruction, the IVC was first punctured by the stiff end ofa guidewire inserted through a multipurpose catheter.Predilatation of the lesion was initially done with aperipheral angioplasty balloon (3×20 mm) if the Josephballoon could not be negotiated across the site of the lesion.Angioplasty was attempted with the Joseph balloon, whichis commonly used for balloon mitral valvotomy. The size ofthe balloon used varied from 22 to 24 mm, depending uponthe size of the IVC. After the balloon was placed across thelesion, it was inflated and deflated 2–3 times. Repeatvenography was done to show blood flow towards the RA.The success of the procedure was confirmed bydemonstrating reduction or disappearance of the gradientbetween the IVC and RA.
Original Article
Correspondence: Dr SN Routray, Department of Cardiology, Qr. No. 3R-8,Near Cancer Wing, SCB Medical College, Cuttack 753007.e-mail: [email protected]
Background: Membranous obstruction of the inferior vena cava is common in African and Asian countries.Methods and Results: Between January 1999 and January 2002, 19 patients were prospectively studied. Themean age of the patients was 38±6.9 years. All of them had swelling of the abdomen and ankle edema. Fivepatients (26.3%) had jaundice, 9 (47.3%) had hepatomegaly, and 5 (26.3%) splenomegaly. Ultrasonographycould detect the site of obstruction in 18 patients (94.7%). Vena cavography demonstrated obstruction of theinferior vena cava at the level of the diaphragm, with 2 patients (10.5%) having additional intrahepaticobstruction. The mean pressure gradient was 22±3.5 mmHg. Seventeen patients underwent balloon angioplastyusing a Joseph balloon. The procedure was successful in 15 patients (88.2%). The post-angioplasty mean pressuregradient was 5±1.4 mmHg. On follow-up, 3 patients (20%) developed features of restenosis; out of them, 2underwent successful redilatation.Conclusions: Balloon angioplasty of membranous obstruction of the inferior vena cava is feasible with a highsuccess rate, without any rupture of the inferior vena cava. (Indian Heart J 2003; 55: 362–364)
Key Words: Inferior vena cava obstruction, Balloon angioplasty, Percutaneous intervention
IHJ-413-03.p65 11/19/2003, 11:24 PM362

Indian Heart J 2003; 55: 362–364 Mishra et al. Percutaneous Balloon Angioplasty of Membranous Obstruction 363
Results
Nineteen patients were initially diagnosed to have MOVC.The mean age at presentation was 38±6.9 years. The meanduration of illness was 3.5±0.6 years. The male:femaleratio was 16:3. The chief complaints were pain in theabdomen in 15 patients (79%), and anorexia in 18 (94.7%).All the patients complained of swelling of the abdomen andankle edema: 5 (26.3%) had jaundice. In all the patients,the veins of the anterior abdominal wall and back wereprominent, with the direction of blood flow from belowupwards. Nine patients (47.3%) had hepatomegaly, and 5(26.3%) splenomegaly.
The mean hemoglobin concentration was 8±1.3 g/dl.The TLC and platelet count were normal. One patient hada prolonged prothrombin time. Abnormal liver function,as assessed by serum albumin and alanine transferase, waspresent in 5 patients (26.3%).
Ultrasonography and Doppler study of the IVCdemonstrated the site of obstruction in 18 patients (94.7%).Before the ultrasound examination, 5 patients (26.3%)were diagnosed to have cirrhosis of the liver. Inferior venacavography demonstrated the site of obstruction of the IVCat the level of the diaphragm in all the patients. Two patients(10.5%) had an additional obstruction in the hepatic vein;they were referred for surgical intervention. Of theremaining 17 patients selected for angioplasty, 13 (76.4%)had complete obstruction, and 4 (23.6%) had partialobstruction, with a meatus <3 mm. The mean pressuregradient between the IVC and RA was 22±3.5 mmHg.
Fifteen patients (88.2%) underwent successful balloondilatation without any rupture of the IVC. The procedurewas abandoned in 2 patients (11.8%), because of inabilityto cross the lesion. The post-procedure mean gradient insuccessful cases was 5±1.4 mmHg.
All the 15 patients who underwent successful dilatationwere followed up for a mean period of 8 months (range 3–24 months). Three patients (20%) developed recurrenceof symptoms such as ascites. Two of them underwentsuccessful redilatation following the demonstration ofrestenosis in the cavogram. One patient refused a repeatprocedure.
Discussion
Chronic Budd–Chiari syndrome can be due to multiplecauses. The impediment to blood flow can occur anywherefrom the RA to the small radicles of the hepatic vein. MOVCaccounts for 40% cases of Budd–Chiari syndrome inAfrica.3 It is still not known whether the obstructingmembrane is congenital or acquired. Yamamoto et al.4 have
classified MOVC into 3 groups. In group A, there wasincomplete obstruction of the IVC, in group B theobstructing membrane was complete and thin; in group Cthe membrane was complete and thick. Of the 17 patientswho were taken up for angioplasty in our series, themembrane was complete in 13 (76.4%).
The mean age at presentation in our patients was38±6.9 years. The majority of MOVC patients were alsobetween 30–40 years of age in the series reported fromSouth Africa by Simson.1 In our series, jaundice was evidentin 5 cases (26.3%). It was present in 5.8% of patients inthe series reported by Kohli et al.5 The high incidence ofjaundice in our cases was probably due to late presentationof the patients. Ultrasound examination revealed the siteof obstruction in 94.7% of our patients. Kohli et al.5 couldidentify obstruction of the IVC by plain ultrasonography in91% of cases.
In our series, 2 patients (10.5%) had additionalobstruction of the hepatic vein. Combined obstruction hasbeen reported in 7 out of 75 cases studied by Kohli et al.5
Medical treatment is ineffective.6 Previously, surgicaloptions included membranotomy, done by a finger7 or metalbougie,8 or cavoatrial shunts done to bypass theobstruction.
Angioplasty has become the treatment of choice forMOVC. Successful dilatation has been reported in 93.3%–100% of cases from India.5,9 In our series, proceduralsuccess was 88.2%. In all our successful cases, the site ofcomplete obstruction could be punctured by the stiff endof the guidewire. The obstruction can also be pierced byBrockenbrough’s septal puncture needle, which was usedby Kar et al.9 and Patel et al.10 with excellent results.
In earlier studies, either a pulmonary balloon 8 or Inoueballoon11,12 has been used to dilate an obstruction of theIVC. We attempted dilatation using a Joseph balloon withgratifying results. It is safe with a high success rate. Theballoon could be reused several times, which is important,as the majority of our patients are poor.
Restenosis was detected in 20% of our patients duringfollow-up. 14.3% of cases developed restenosis in the seriesby Kohli et al.5 Tyagi et al.,13 however, reported a very highincidence (36.4%) of restenosis.
Conclusions: Membranous obstruction of the IVC can giverise to substantial morbidity because of massive ascites andankle edema. A high index of suspicion is necessary, as thedisease is often confused with cirrhosis of the liver.Ultrasonography with Doppler study of the IVC can bediagnostic, though vena cavography is required forconfirmation. In the majority of patients, obstruction canbe successfully tackled by balloon angioplasty without
IHJ-413-03.p65 11/19/2003, 11:24 PM363

364 Mishra et al. Percutaneous Balloon Angioplasty of Membranous Obstruction Indian Heart J 2003; 55: 362–364
fear of rupture of the IVC. Long-term follow-up of post-angioplasty patients is necessary to further validate thesuccess of the procedure.
References
1. Simson IW. Membranous obstruction of the inferior vena cava andhepatocellular carcinoma in South Africa. Gastroenterology 1982;82: 171–178
2. Victor S, Jayanthi V, Kandaswamy I, Ratnasabapathy A,Madanagopalan N. Retrohepatic cavoatrial bypass for coarctationof inferior vena cava with a polytetrafluoroethylene graft. J ThoracCardiovasc Surg 1986; 91: 99–105
3. Schafer OF, Sorrell MF. Vascular diseases of liver. In: Sleisenger andFordtran’s gastrointestinal and liver diseases. 6th ed. 1998. p. 1189
4. Yamamoto S, Yokoyama Y, Takeshige K, Iwatsuki S. Budd–Chiarisyndrome with obstruction of the inferior vena cava. Gastroenterology1968; 54: 1070–1084
5. Kohli V, Pande GK, Dev V, Reddy KS, Kaul U, Nundy S. Managementof hepatic venous outflow obstruction. Lancet 1993; 342: 718–722
6. Ahn SS, Yellin A, Sheng FC, Colonna JO, Goldstein LI, Busuttil RW.Selective surgical therapy of Budd–Chiari syndrome providessuperior survivor rates than conservative medical management.J Vasc Surg 1987; 5: 28–37
7. Hirooka M, Kimura C. Membranous obstruction of the hepaticportion of the inferior vena cava. Surgical correction and etiologicalstudy. Arch Surg 1970; 100: 656–663
8. Rogers MA, Chesler E, Ou Plesis L, Jeffe N, Jouvert E. Membranousobstruction of the hepatic segment of inferior vena cava. Br J Surg1967; 54: 221–225
9. Kar AK, Mandal M. Percutaneous balloon angioplasty of inferiorvena cava stenosis. Cardiology Today 1999; 5: 221–228
10. Patel T, Shah S, Sanghvi K, Fonseca K. Successful stenting of acomplex inferior vena cava stenosis using a modified sharprecanulization technique. Catheter Cardiovasc Interv 2001; 52:492–495
11. Bahl VK, Bhargava B, Chandra S. Percutaneous balloon dilatationof IVC obstruction using Inoue Catheter. Indian Heart J 1997; 49:427–428
12. Yang XL, Cheng TO, Chen CR. Successful treatment by percutaneousballoon angioplasty of Budd–Chiari syndrome caused bymembranous obstruction of inferior vena cava: 8-year follow-upstudy. J Am Coll Cardiol 1996; 28: 1720–1724
13. Tyagi S, Jain BL, Kumar N, Lahoti D, Arora R. Balloon dilatation ofinferior vena cava stenosis in Budd–Chiari syndrome. J AssocPhysicians India 1996; 44: 378–380
IHJ-413-03.p65 11/19/2003, 11:24 PM364

Indian Heart J 2003; 55: 365–367 Goel et al. Thrombin Injection for Femoral Artery Pseudoaneurysm 365
Sonographically Guided Thrombin Injection for theTreatment of Femoral Artery Pseudoaneurysm
Pravin K Goel, Nitin Modi, Sanjay Saran Baijal, Manoj Kathuria, Surendra K AgrawalDepartments of Cardiology, Radiology, and Cardiovascular and Thoracic Surgery,
Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow
Femoral artery pseudoaneurysm is an uncommon butwell-recognized complication after percutaneous
arterial catheterization. The incidence of pseudoaneurysmformation is reported to be up to 1% after diagnostic studies,and 3.2% after interventional procedures.1,2 Traditionally,these pseudoaneurysms require reparative surgery. In1986, Cope et al. described the technique of percutaneousthrombin injection for small pseudoaneurysm in the iliac,femoral, and popliteal arteries.3 There is, however, limitedexperience with this technique on large aneurysms. Wedescribe a case of a large pseudoaneurysm successfullymanaged with percutaneous intra-aneurysmal injection ofbovine thrombin.
Case Report
A 66-year-old hypertensive, nondiabetic, nonsmoker maleunderwent diagnostic cardiac catheterization through theright femoral artery using 6 F arterial sheath. Five thousandunits of heparin was given during the procedure, and postprocedure, the right lower limb was immobilized for 6hours. After 6 hours, the patient was allowed to carry outhis routine activities and was to be discharged the followingday. However, about 18 hours post procedure, the patientcomplained of severe pain in the right groin with obviousswelling of the thigh. There was no external bleeding fromthe puncture site. Doppler study of the femoral artery
showed a large pseudoaneurysm (10×4×4 cm) with anarrow neck of 0.5 cm. The aneurysm was largely locatedanterior to the femoral artery all along its length, for up toabout 10 cm below the puncture site. Ultrasound-guidedcompression of the neck was tried but was not successful.Considering the large size of the aneurysm,ultrasonographically guided thrombin injection into thepseudoaneurysm was planned.
Technique: After taking informed consent, a completeDoppler assessment of the aneurysm and the patency ofdistal run-off was obtained. The right groin was prepared,draped and an echo-probe placed directly over thepseudoaneurysm. The flow into the aneurysm was assessedby both the color and angio modes on the HP Sonos 5500machine. Obliteration of flow into the aneurysm wasconfirmed by compression of the neck of thepseudoaneurysm with the color mode on. Under localanesthesia, a 22-gauge spinal needle was then passedpercutaneously directly into the pseudoaneurysm cavityunder ultrasonographic guidance. The tip of the needle waspositioned within the center of the cavity, to keep it awayfrom the neck. A small 2 ml syringe containing bovinethrombin (Parke Davis) 1000 U/ml concentration wasattached to the needle and 1 ml injected into the cavity withcontinuing compression of the neck. The echogenicitywithin the aneurysm was seen to increase immediately, buton color Doppler study repeated after 3–4 min, some flowinto the cavity was still seen. Thrombin injection wasrepeated using the same technique and, this time, dense
Brief Report
Correspondence: Dr PK Goel, Department of Cardiology, SGPGIMS, RaeBareli Road, Lucknow 226014. e-mail: [email protected]
The formation of pseudoaneurysm in the femoral artery after cardiac catheterization is a well-recognizedcomplication occurring in 1%–4% of cases. It is traditionally managed surgically and has a high morbidity.Prolonged ultrasound-guided compression of the neck of the pseudoaneurysm, and ultrasound-guided injectionof thrombin into the aneurysm are newer modalities of treatment especially for small aneurysms. We describethe case of a giant pseudoaneurysm of the right femoral artery, post-arteriography, which was successfullymanaged with ultrasonographically guided percutaneous thrombin injection. (Indian Heart J 2003; 55:365–367)
Key Words: Pseudoaneurysm, Femoral artery, Thrombin
IHJ-486-03.p65 11/19/2003, 11:25 PM365

366 Goel et al. Thrombin Injection for Femoral Artery Pseudoaneurysm Indian Heart J 2003; 55: 365–367
occurs in 1%–3% of cases following percutaneous arterialcatheterization.1,2 The treatment options are surgical repair,covered stents, coil embolization or, ultrasound-guidedcompression and direct percutaneous thrombin injection.Operative repair is usually associated with high morbiditysecondary to delayed ambulation and/or wound infection.Fellmeth4 in 1991 described ultrasound-guidedcompression repair. With this treatment, flow into thepseudoaneurysm is halted by applying pressure with theultrasound probe to the neck of the pseudoaneurysm andmaintaining such pressure until spontaneous thrombosisof the aneurysm cavity occurs. This procedure, however, isoften very painful, and has an overall success rate as low as45% to up to 100%, but a recurrence rate of up to 30%.5–7
The technique of ‘clotting aneurysm’ by direct injectionof diluted thrombin was first described in 1986 by Cope etal.3 For the next decade, however, not much attention wasgiven to this technique, and it is only in the past 1–2 yearsthat there has been a resurgence of interest, with multiplestudies showing success rates of 96%–100% (Table1). Theoverall complication rate of this technique is less than 2%and the most dreaded complication is downstreamembolization.18 Samal et al.10 described the technique ofballoon occlusion of the neck of the aneurysm by using acontralateral approach to prevent spillover of thrombin intothe distal circulation in 4 cases. We in our limited
echogenicity developed within seconds. Repeat study after3–4 min showed no flow into the cavity, without anyarterial compression. Good distal run-off on color Dopplerwas confirmed before the end of the procedure. As aprecautionary measure the patient was kept on bed restwith the right lower limb immobilized for another 6 hoursto ensure clotting of any small nonthrombosed lobulationthat might have been left. Repeat Doppler study done after24 and 48 hours showed total thrombosis within thepseudoaneurysm cavity, which by then had also regressedin size, and there was no flow into the aneurysm cavity.
Discussion
With the increasing number of cardiac catheterizationprocedures being performed, the total number ofcases with complications are bound to increase.Pseudoaneurysm formation of the the femoral artery
Table 1. Results from different studies for ultrasound-guided thrombin injection for the obliteration of femoralartery pseudoaneurysm
Number Success Number ofof cases (%) complications
Friedman et al.8 40 98 2Edgerton et al.9 47 94 1Samal et al.10 4 100 0Sheiman et al.11 (Simple) 45 100 0
(Complex) 9 56 0Paulson et al.12 26 96 0Kang et al.13 74 100 0La Perna et al.14 70 94 0Hughes et al.15 9 100 0Sievert et al.16 29 100 0Morrison et al.17 39 100 0Brophy et al.18 15 100 0Lennox et al.19 30 100 0Pezzullo et al.20 23 96 1Tamim et al.21 10 100 0Vermeulen et al.22 8 100 0Taylor et al.23 29 93 0Wixon et al.24 11 100 0Liau et al.25 5 100 0
Figs. 1–6. Ultrasonographic pictures in angio mode demonstrating thrombininjection into the femoral artery pseudoaneurysm and thrombus formation. (a)Origin of the pseudoaneurysm from the femoral artery with a narrow neck seenin the angio mode. (b) Giant pseudoaneurysm and blood flow into the aneurysmseen in the angio mode, (c) Injection needle positioned into the aneurysm whilemaintaining compression on the neck of pseudoaneurysm. (d) Thrombin beinginjected into the aneurysm while continuing pressure on the aneurysmal neck.The injection stream is highlighted in the angio mode with no other flow intoaneurysm during the injection. (e) Formation of post-injection thrombus, i.e.density of aneurysm changed within 3 min. (f) Compression released and still noflow occurring into the aneurysm with the pseudoaneurysm neck seen as a stump.
a b
c d
e f
IHJ-486-03.p65 11/19/2003, 11:25 PM366

Indian Heart J 2003; 55: 365–367 Goel et al. Thrombin Injection for Femoral Artery Pseudoaneurysm 367
experience, however, found that a transient compressiongiven to the proximal femoral artery during injection ofthrombin, and confirmation of no flow into the aneurysmwith color/angio mode, may be as good a method to avoidspillover, which also avoids the need for further invasivehandling of the already damaged site. We do feel, however,that caution must be exercised with AV fistula andaneurysm involving bifurcation of the femoral artery as insuch cases a spillover could occur using ultrasound-guidedcompression alone. Most failures/recurrences wereobserved in patients with complex aneurysms, i.e.multilobed aneurysms. Other possible complications couldinclude allergy to bovine thrombin and the formation of alocal abscess formation. Reviewing the literature, themaximum volume of aneurysmal sac thrombosed by thistechnique has been 55 cm3. Our case, on the other hand,had a giant pseudoaneurysm with a volume of nearly80 cm3 and was successfully managed using 2000 unitsof thrombin, without any complications.
Among the other nonsurgical approaches, it may beworth mentioning the technique of temporary coilplacement in the pseudoaneurysm sac, described recentlyby Trehan et al.26 This technique, however, is more invasivewith a 5 F sheath being placed directly into the aneurysmsac, and a PDA coil (8 mm) kept in place for over 2 hoursbefore its final removal. Experience with this technique islimited to a small aneurysm only.
We conclude that sonographically guided directthrombin injection into the aneurysmal sac is a safe, quick,and ef fective technique for managing a giantpseudoaneurysm at the the puncture site. It is well toleratedby the patient, with low morbidity, and avoids the need forsurgery, and could thus be considered as a first-line therapyin these patients.
References
1. Zahn R, Thoma S, Fromm E, Lotter R, Zander M, Seidl K, et al.Pseudoaneurysm after cardiac catheterization: therapeuticinterventions and their sequelae: experience in 86 patients. CathetCardiovasc Diagn 1997; 40: 9–15
2. Chatterjee T, Do DD, Kaufmann U, Maier F, Meier B. Ultrasound-guided compression repair for treatment of femoral arterypseudoaneurysm: acute and follow-up results. Cathet Cardiovasc Diagn1996; 38: 335–340
3. Cope C, Zeit R. Coagulation of aneurysm by direct percutaneousthrombin injection. AJR Am J Roentogenol 1986; 147: 383–387
4. Fellmeth BD, Roberts AC, Bookstein JJ. Postangiographic femoralartery injuries: nonsurgical repair with US-guided compression.Radiology 1991; 178: 671–675
5. Agarwal R, Agrawal SK, Roubin GS, Berland L, Cox DA, Iyer SS, etal. Clinically guided closure of femoral arterial pseudoaneurysmscomplicating cardiac catheterization and coronary angioplasty.Cathet Cardiovasc Diagn 1993; 30: 96–100
6. Dean S, Olin ZW, Piedmonte MA. Ultrasound-guided compressionclosure of postcatheterisation pseudoaneurysms during concurrent
anticoagulation. J Vasc Surg 1995; 23: 28–357. Cox GS, Young JR, Gray BR, Grubb MW, Hertzer NR. Ultrasound-
guided compression repair of postcatheterization pseudoaneurysms:results of treatment in one hundred cases. J Vasc Surg 1994; 19: 683–686
8. Friedman SG, Pellerito JS, Scher L, Faust G, Burke B, Safa T.Ultrasound-guided thrombin injection is the treatment of choice forfemoral pseudoaneurysms. Arch Surg 2002; 137: 462–464
9. Edgerton JR, Moore DO, Nichols D, Lane BW, Magee MJ, Dewey TM,et al. Obliteration of femoral artery pseudoaneurysm by thrombininjection. Ann Thorac Surg 2002; 74: S1413–S1415
10. Samal AK, White CJ, Collins TJ, Ramee SR, Jenkins JS. Treatment offemoral artery pseudoaneurysm with percutaneous thrombininjection. Catheter Cardiovasc Interv 2001; 53: 259–263
11. Sheiman RG, Brophy DP. Treatment of iatrogenic femoralpseudoaneurysms with percutaneous thrombin injection: experiencein 54 patients. Radiology 2001; 219: 123–127
12. Paulson EK, Sheafor DH, Kliewer MA, Nelson RC, Eisenberg LB,Sebastian MW, et al. Treatment of iatrogenic femoral arterialpseudoaneurysms: comparison of US-guided thrombin injectionwith compression repair. Radiology 2000; 215: 403–408
13. Kang SS, Labropoulos N, Mansour MA, Michelini M, Filling D, BaublyMP, et al. Expanded indications for ultrasound-guided thrombininjection of pseudoaneurysms. J Vasc Surg 2000; 31: 289–298
14. La Perna L, Olin JW, Goines D, Childs MB, Ouriel K. Ultrasound-guidedthrombin injection for the treatment of postcatheterisationpseudoaneurysms. Circulation. 2000; 102: 2391–2395
15. Hughes MJ, McCall JM, Nott DM, Padley SP. Treatment of iatrogenicfemoral artery pseudoaneurysms using ultrasound-guided injectionof thrombin. Clin Radiol 2000; 55: 749–751
16. Sievert H, Baser A, Pfeil W, Fach A, Scherer D, Spies H, et al. [Thetreatment of iatrogenic spurious aneurysm of the femoral artery bydirect thrombin injection.] Dtsch Med Wochenschr 2000; 125: 822–825
17. Morrison SL, Obrand DA, Steinmetz OK, Montreuil B. Treatment offemoral artery pseudoaneurysms with percutaneous thrombininjection. Ann Vasc Surg 2000; 14: 634–639
18. Brophy DP, Sheiman RG, Amatulle P, Akbari CM. Iatrogenic femoralpseudoaneurysms: thrombin injection after failed US-guidedcompression. Radiology 2000; 214: 278–282
19. Lennox AF, Delis KT, Szendro G, Griffin MB, Nicolaides AN, CheshireNJ. Duplex-guided thrombin injection for iatrogenic femoral arterypseudoaneurysm is effective even in anticoagulated patients. Br JSurg 2000; 87: 796–801
20. Pezzullo JA, Dupuy DE, Cronan JJ. Percutaneous injection ofthrombin for the treatment of pseudoaneurysms aftercatheterization: an alternative to sonographically guidedcompression. AJR Am J Roentgenol 2000; 175: 1035–1040
21. Tamim WZ, Arbid EJ, Andrews LS, Arous EJ. Percutaneous inducedthrombosis of iatrogenic femoral pseudoaneurysms followingcatheterization. Ann Vasc Surg 2000; 14: 254–259
22. Vermeulen EG, Umans U, Rijbroek A, Rauwerda JA. Percutaneousduplex-guided thrombin injection for treatment of iatrogenic femoralartery pseudoaneurysms. Eur J Vasc Endovasc Surg 2000; 20: 302–304
23. Taylor BS, Rhee RY, Muluk S, Trachtenberg J, Walters D, Steed DL, etal. Thrombin injection versus compression of femoral arterypseudoaneurysms. J Vasc Surg 1999; 30: 1052–1059
24. Wixon CL, Philpott JM, Bogey WM Jr, Powell CS. Duplex-directedthrombin injection as a method to treat femoral arterypseudoaneurysms. J Am Coll Surg 1998; 187: 464–466
25. Liau CS, Ho FM, Chen MF, Lee YT. Treatment of iatrogenic femoralartery pseudoaneurysm with percutaneous thrombin injection. JVasc Surg 1997; 26: 18–23
26. Trehan VK, Mukhopadhyay S, UmaMahesh CR, Yusuf J, SuryavanshiS, Aora R. Successful closure of iatrogenic femoral arterypseudoaneurysm: a new minimally invasive technique. Indian HeartJ 2002; 54: 702–704
IHJ-486-03.p65 11/19/2003, 11:25 PM367

368 Trehan et al. Stenting of a Septal Perforator Post-MI Indian Heart J 2003; 55: 368–369
Stenting of a Septal Perforator for Post-MyocardialInfarction Angina
Vijay Trehan, Saibal Mukhopadhyay, Umamahesh C Rangasetty, Jamal Yusuf, Mohit D Gupta, UA KaulDepartment of Cardiology, GB Pant Hospital, New Delhi
Acute myocardial infarction (AMI) is usually caused bythe thrombotic occlusion of a major epicardial vessel.
Branch occlusion alone, causing AMI in the absence ofinvolvement of a major epicardial artery, is unusual. Wereport a case in which a patient with anteroseptalmyocardial infarction (MI) and intractable post-MI anginawas found to have isolated 90% obstruction of the firstseptal perforator artery, which was successfully dilated andstented.
Case Report
A 52-year-old male, a known hypertensive on irregulartreatment, presented to us with refractory post-MI angina.He had had an anteroseptal MI 48 hours earlier for whichhe was thrombolized with streptokinase (1.5 million U over1 hour) within 3 hours of the onset of chest pain. Thepatient was taken up for coronary angiography and possiblerevascularization. Coronary angiogram revealed athrombus-containing 90% discrete stenosis of the firstseptal perforator artery just after its origin (Figs 1a and 1b).The left anterior descending coronary artery (LAD) alongwith the other epicardial vessels were normal. The patientwas taken up for angioplasty. He was given a bolus ofabciximab (0.25 mg/kg body weight) followed by aninfusion of 10 µg/min for 12 hours. The lesion in the septalperforator artery was crossed with a Luge Wire (BostonScientific Scimed, Inc., USA), and serially dilated with a2.5×12 mm and 3×13 mm balloon. Following balloon
dilatation, a check angiogram revealed TIMI 2 flow with50% residual stenosis. A mounted stent of 3×13 mm (BxVelocity, Cordis, USA) was deployed at 14 bars, andpersistent TIMI 3 flow was achieved (Fig. 2). There were nocomplications during or after the procedure. The patientbecame asymptomatic and was discharged after 72 hours.On follow-up of nearly a year, the patient is asymptomatic.
Discussion
The interventricular septum (IVS) forms the vital wall forboth the ventricles, and constitutes about one-third of thetotal left ventricular (LV) mass.1 The septal perforator
Brief Report
Correspondence: Dr Umamahesh C Rangasetty, Department ofCardiology, GB Pant Hospital, New Delhi 110002.e-mail: [email protected]
Occlusion of a septal perforator branch alone, without the involvement of the left anterior descending coronaryartery, leading to acute myocardial infarction is unusual. We report a case in which an isolated severely stenoticthrombus-containing first septal artery causing intractable post-myocardial infarction angina was successfullydilated and stented. (Indian Heart J 2003; 55: 368–369)
Key Words: Septal artery, Stents, Myocardial infarction
Fig. 1a. Anteroposterior cranial view showing significant eccentric stenosis inthe first septal perforator artery just after its origin.
IHJ-442-03.p65 11/19/2003, 11:24 PM368

Indian Heart J 2003; 55: 368–369 Trehan et al. Stenting of a Septal Perforator Post-MI 369
case reports of isolated septal artery obstruction.3,4 Therehave also been reports of angioplasty of the septalperforators, usually done in patients with critical stenosisor total occlusion of the LAD.5,6 Recently, there have been acouple of reports of stenting of a septal perforator artery.One was in a post-coronary bypass surgery patient withan occluded LAD,7 while the other was in a patient withmultivessel disease.8 However, to the best of our knowledge,there have been no reports of angioplasty or stenting of anisolated culprit septal artery. The first septal perforator,apart from supplying the IVS, also supplies the bundle ofHis, and in 50% hearts, the atrioventricular node.9 Ourpatient, with refractory post-MI angina due to occlusionof the first septal perforator, was at increased risk ofreinfarction that could have disrupted either the conductionsystem or the IVS, causing its rupture. Unlike the epicardialarteries, septal arteries are subjected to a higher extraneouspressure. Although long-term results of angioplasty of theseptal perforator have not shown increased rates ofrestenosis,5 the behavior of stents in response to a highextrinsic pressure needs to be observed. This situation issimilar to the stenting of myocardial bridges.
References
1. Kaul S. The interventricular septum in health and disease. Am HeartJ 1986; 112: 568–581
2. Banka VS, Agarwal JB, Bodenheimer MM, Helfant RH.Interventricular septal motion: biventricular angiographicassessment of its relative contribution to left and right ventricularcontraction. Circulation 1981; 64: 992–996
3. Nishimura RA, Tajik AJ, Seward JB. Distinctive two dimensionalechocardiographic appearance of septal infarct secondary to isolatedocclusion of first septal perforator artery. Rev Cardiovasc Ultrasound1984; 1: 97–101
4. Plokker HM, Ernest SM, Van Tellingen C, Bruschke AV. Isolatedobstruction of large septal perforators. Am J Cardiol 1988; 62: 142–143
5. Vemuri DN, Kochar GS, Maniet AR, Banka VS. Angioplasty of theseptal perforators: acute outcome and long-term clinical efficacy. AmHeart J 1993; 125: 682–686
6. Comazzi JL, Jang GC, Marsa RJ, Willis WH Jr, Anderson DL, WarehamEE. Percutaneous transluminal angioplasty of a large septal artery.Cathet Cardiovasc Diagn 1983; 9: 181–186
7. Ozdemir M, Timurkaynak T, Cemri M, Boyaci B, Yalcin R, Cengel A,et al. Stenting of the septal perforator coronary artery. J InvasiveCardiol 2001; 13: 694–697
8. Regar E, Kozuma K, Ligthart J, et al. Coronary stent implantation inseptal perforator artery. Jpn Circ J 2000; 64: 802–804
9. Pujadas G (ed). Coronary angiography. New York: McGraw-Hill BookCompany, Inc. 1980; p. 72
Fig. 1b. Left anterior oblique cranial view showing the thrombus in the lesion.
Fig. 2. Angiographic result following stenting of the septal artery.
branches of the LAD2 contribute blood supply to theanterior two-thirds of the IVS. Obstruction of the septalperforators in patients with diffuse coronary artery diseaseis common. However, isolated obstruction of a septalperforator artery is very rare. Till date, there have been 2
IHJ-442-03.p65 11/19/2003, 11:24 PM369

370 Bedi et al. MV Replacement on a Beating Heart Indian Heart J 2003; 55: 370–372
Mitral Valve Replacement on a Beating Heart
Harinder S Bedi, Raman P Singh, Vipin Goel, Purshottam LalMetro Heart Institute, Noida
Cardioplegic techniques carry with them the almostobligatory sequel of some degree of reperfusion injury.1
Any technique that avoids the ischemic component ofcardioplegia and an arrested heart by keeping the heartbeating will go a long way in reducing iatrogenic damageto the heart. While the heart–lung machine can be avoidedin most cases of coronary artery bypass grafting (CABG),2
it is still mandatory in open heart valvular surgery. Wepresent a case of a high-risk mitral valve operationperformed on a beating heart.
Case Report
A 15-year-old girl presented with orthopnea and congestiveheart failure. She was a known case of rheumatic heartdisease with severe mitral regurgitation, moderate mitralstenosis, severe pulmonary artery hypertension, and severefunctional tricuspid regurgitation. The diagnosis wasconfirmed by echocardiography. She did not show muchimprovement with aggressive medical therapy, and wastaken up for emergency surgery. Since she would have beenat a high risk of reperfusion injury if standard techniquesof cardioplegic arrest were used, a technique of on-pumpbeating heart surgery was planned.
The operation was performed through a mediansternotomy. After heparinization, total cardiopulmonarybypass (CPB) was established by aortic and bicavalcannulation. The patient was actively warmed to 36.5°C,and pulsatile flow maintained at the highest pressure thatwas safely possible, keeping the mean systemic pressure
above 60 mmHg. The pump flow was kept at 2.5 L/min/m2,and the FiO2 was kept at 100%.
A retrograde self-inflating coronary sinus cannula(Medtronic DLP, Grand Rapids, MI) was inserted by astandard closed technique. The position was confirmed bypalpation, and the balloon positioned as close to thecoronary sinus ostium as possible to avoid right ventricularmalperfusion. The central perfusion port was connected tothe standard cardioplegic line circuit, and oxygenatedunmodified (noncardioplegic) blood primed into thecannula. With an aortic root vent and maximal venousdrainage, retrograde perfusion was started, andsimultaneously the aorta was cross-clamped. Retrogradeperfusion flow was maintained at 250–300 ml/min withconstant monitoring of the intracoronary sinus pressurevia its integral pressure line, keeping a mean coronary sinuspressure of 40 mmHg. A phosphodiesterase inhibitor—milrinone 0.5 µg/kg/min—was infused in the retrogradeline by a syringe pump to increase the retrograde perfusionflow by inducing vasodilatation. Samples of the bloodentering the coronary sinus and of the blood in the aorticroot vent were taken for analysis and calculation of theoxygen extraction ratio across the myocardium.3 The leftatrium was now opened and mitral valve replacement withpreservation of the posterior mitral apparatus carried outon the beating heart (Figs 1 and 2). At the end of theprocedure, the cardiac cavities were filled with blood (byinflating the lungs) and with warm saline, and the leftatrium was closed. The aortic root vent was kept on suction,the aortic cross-clamp released, and the retrograde coronarysinus cannula removed. The right atrium was opened, and astandard repair (DeVega annuloplasty) of the tricuspid valvecarried out.
Brief Report
We report the case of a patient who needed mitral valve replacement but was at a high risk of myocardial injurywith the conventional technique (cardioplegic arrest on cardiopulmonary bypass). Valve replacement was carriedout on a beating heart on cardiopulmonary bypass by perfusing the heart continuously with oxygenatednoncardioplegic normothermic blood via the coronary sinus. (Indian Heart J 2003; 55: 370–372)
Key Words: Mitral valve replacement, Cardiopulmonary bypass, Beating heart
Correspondence: Dr Harinder Singh Bedi, Chief Cardiac Surgeon andChairman, Cardio-Vascular Surgery, Metro Heart Institute, X-1, Sector 12,Noida, NCR, Delhi. e-mail: [email protected]; [email protected]
IHJ-492-03.p65 11/19/2003, 11:25 PM370
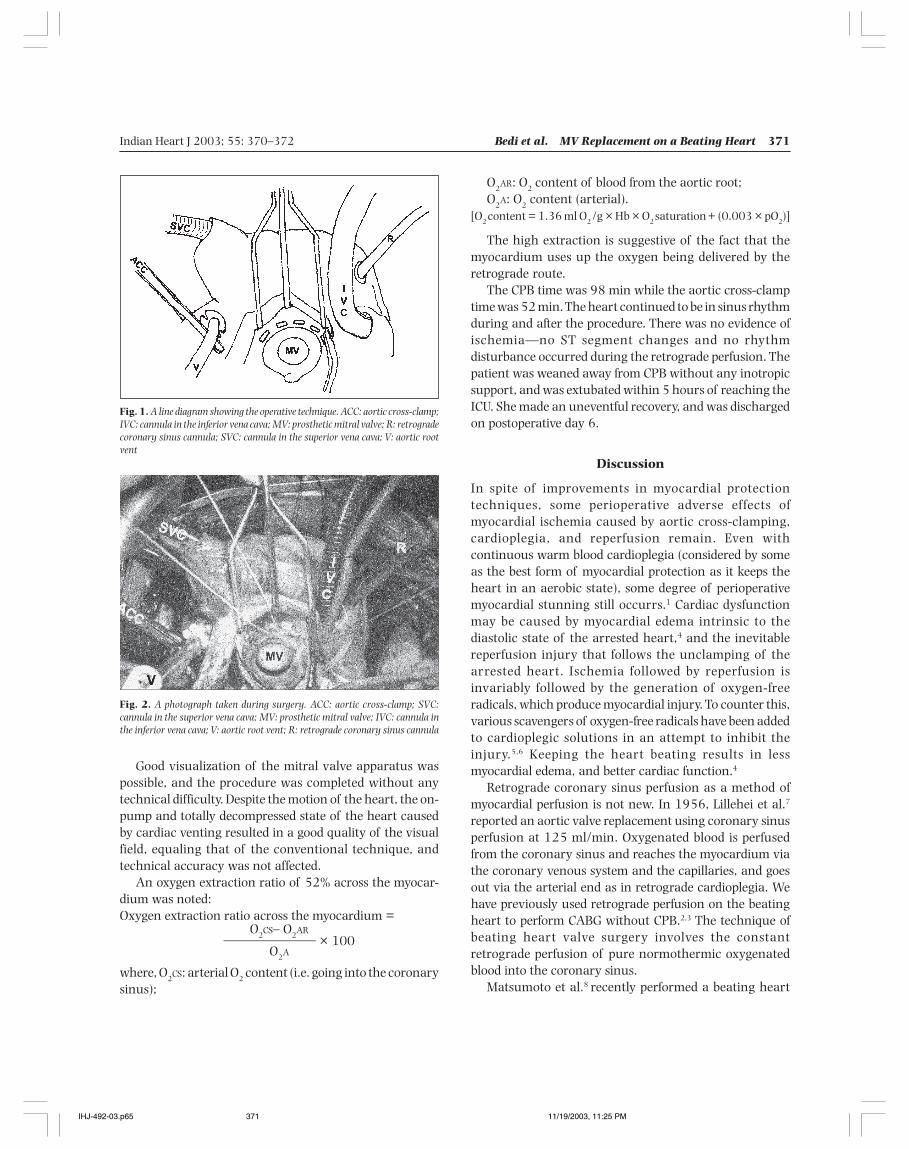
Indian Heart J 2003; 55: 370–372 Bedi et al. MV Replacement on a Beating Heart 371
Good visualization of the mitral valve apparatus waspossible, and the procedure was completed without anytechnical difficulty. Despite the motion of the heart, the on-pump and totally decompressed state of the heart causedby cardiac venting resulted in a good quality of the visualfield, equaling that of the conventional technique, andtechnical accuracy was not affected.
An oxygen extraction ratio of 52% across the myocar-dium was noted:Oxygen extraction ratio across the myocardium =
O2CS– O2AR_________________ × 100O2A
where, O2CS: arterial O2 content (i.e. going into the coronarysinus);
O2AR: O2 content of blood from the aortic root;O2A: O2 content (arterial).
[O2 content = 1.36 ml O2 /g × Hb × O2 saturation + (0.003 × pO2)]
The high extraction is suggestive of the fact that themyocardium uses up the oxygen being delivered by theretrograde route.
The CPB time was 98 min while the aortic cross-clamptime was 52 min. The heart continued to be in sinus rhythmduring and after the procedure. There was no evidence ofischemia—no ST segment changes and no rhythmdisturbance occurred during the retrograde perfusion. Thepatient was weaned away from CPB without any inotropicsupport, and was extubated within 5 hours of reaching theICU. She made an uneventful recovery, and was dischargedon postoperative day 6.
Discussion
In spite of improvements in myocardial protectiontechniques, some perioperative adverse ef fects ofmyocardial ischemia caused by aortic cross-clamping,cardioplegia, and reperfusion remain. Even withcontinuous warm blood cardioplegia (considered by someas the best form of myocardial protection as it keeps theheart in an aerobic state), some degree of perioperativemyocardial stunning still occurrs.1 Cardiac dysfunctionmay be caused by myocardial edema intrinsic to thediastolic state of the arrested heart,4 and the inevitablereperfusion injury that follows the unclamping of thearrested heart. Ischemia followed by reperfusion isinvariably followed by the generation of oxygen-freeradicals, which produce myocardial injury. To counter this,various scavengers of oxygen-free radicals have been addedto cardioplegic solutions in an attempt to inhibit theinjury.5,6 Keeping the heart beating results in lessmyocardial edema, and better cardiac function.4
Retrograde coronary sinus perfusion as a method ofmyocardial perfusion is not new. In 1956, Lillehei et al.7
reported an aortic valve replacement using coronary sinusperfusion at 125 ml/min. Oxygenated blood is perfusedfrom the coronary sinus and reaches the myocardium viathe coronary venous system and the capillaries, and goesout via the arterial end as in retrograde cardioplegia. Wehave previously used retrograde perfusion on the beatingheart to perform CABG without CPB.2,3 The technique ofbeating heart valve surgery involves the constantretrograde perfusion of pure normothermic oxygenatedblood into the coronary sinus.
Matsumoto et al.8 recently performed a beating heart
Fig. 1. A line diagram showing the operative technique. ACC: aortic cross-clamp;IVC: cannula in the inferior vena cava; MV: prosthetic mitral valve; R: retrogradecoronary sinus cannula; SVC: cannula in the superior vena cava; V: aortic rootvent
Fig. 2. A photograph taken during surgery. ACC: aortic cross-clamp; SVC:cannula in the superior vena cava; MV: prosthetic mitral valve; IVC: cannula inthe inferior vena cava; V: aortic root vent; R: retrograde coronary sinus cannula
IHJ-492-03.p65 11/19/2003, 11:25 PM371

372 Bedi et al. MV Replacement on a Beating Heart Indian Heart J 2003; 55: 370–372
valve procedure on 25 patients with good results. Theyshowed, by intraoperative monitoring, that myocardialtissue oxygen saturation (SO2) levels were maintained atthe preoperative physiologic values throughout theprocedure. A good myocardial oxygen consumption, asnoted by us, indicates that the myocardium is in a state ofaerobic metabolism that is maintained by the retrogradecoronary sinus perfusion.
The advantages of the technique are: a perfused beatingheart that does not have to ‘‘work’’ (pump blood); noreperfusion injury (as there is no ischemia); anuninterrupted performance of the operation; a long periodof continuous oxygenated blood delivery that maintains thebeating of the heart; appropriate pH, effective delivery ofsubstrates, and removal of acid metabolites; more uniformoxygenated blood distribution in the presence of anyassociated coronary artery stenosis; testing of the mitralvalve repair, when performed, can be done under the actualphysiologic conditions of a normal left ventricular tone withpreserved three-dimensional architecture of the beatingheart (with conventional techniques, the mitral valve is ina motionless flaccid state that may not accurately reflectits function in the beating heart); avoidance of injury tothe ostia of the coronary arteries; and the possibility ofperforming ablation for atrial fibrillation on the beatingheart. The normothermic state of the blood ensuresmaximal vasodilatation of the cardiac veins, and steady,uniform flow and distribution. Should the flow beinadequate or if there is malperfusion, it will be reflectedby ECG changes (ST segment changes, bradycardia,arrhythmias, etc.), and the flow can be suitably adjusted.The option of converting to the conventional technique—cardiac arrest with hyperkalemic blood cardioplegia— isalways immediately available, if the surgeon is not satisfied
with the beating heart technique.The beating heart technique is thus a useful addition to
the armamentarium of the cardiac surgeon in dealing withpatients undergoing high-risk valve surgery. A comparisonof this procedure versus the arrested heart technique in alarge group of patients is required to judge the superiorityof the beating heart technique.
References
1. Misare BD, Krukenkamp IB, Lazer ZP, Levitsky S. Recovery ofpostischemic contractile function is depressed by antegrade warmcontinuous blood cardioplegia. J Thorac Cardiovasc Surg 1993; 105:37–44
2. Bedi HS, Suri A, Kalkat MS, Sengar BS, Mahajan V, Chawla R, et al.Global myocardial revascularization without cardiopulmonarybypass using innovative techniques for myocardial stabilization andperfusion. Ann Thorac Surg 2000; 69: 156–164
3. Bedi HS. Selective graft and coronary sinus perfusion in off pumpCABG: is it necessary? [Reply]. Ann Thorac Surg 2001; 71: 1070–1072
4. Mehlhorn U, Allen SJ, Adamus DL, Davis KL, Gogola GR, WartersRD. Cardiac surgical conditions induced by beta blockade: effect onmyocardial fluid balance. Ann Thorac Surg 1996; 62: 143–150
5. Vinten-Johansen J, Ronson RS, Thourani VH, Wechsler AS. Surgicalmyocardial protection. In: Gravlee GP, Davis RF, Kurusz M, Utley JR(eds). Cardiopulmonary bypass—principles and practice. 2nd ed.Phliladelphia: Lippincott Williams and Wilkins; 2000. p. 214
6. Kirklin JW, Barratt-Boyes BG. Myocardial management duringcardiac surgery with cardiopulmonary bypass. In: Kirklin JW,Barratt-Boyes BG (eds). Cardiac surgery. Morphology, diagnostic criteria,natural history, techniques, results and indications. 2nd ed. New York:Churchill Livingstone; 1993. p. 129
7. Lillehei CW, DeWall RA, Gott VL, Varco RL. The direct visioncorrection of calcific aortic stenosis by means of a pump oxygenatorand retrograde coronary sinus perfusion. Dis Chest 1956; 30:123–131
8. Matsumoto Y, Watanabe G, Endo M, Sasaki H, Kasashima F, KosugiI. Efficacy and safety of on-pump beating heart surgery for valvulardisease. Ann Thorac Surg 2002; 74: 678–683
IHJ-492-03.p65 11/19/2003, 11:25 PM372

Indian Heart J 2003; 55: 373–375 Radha et al. Catheter Elimination of Accessory Pulmonary Blood Flow 373
Transcatheter Closure of Native Pulmonary Artery for theElimination of Accessory Pulmonary Blood Flow After
Bidirectional Glenn Shunt
Anil Sivadasan Radha, Bhava RJ Kannan, Raman Krishna KumarDivision of Pediatric Cardiology, Amrita Institute of Medical Sciences and Research Centre, Kochi
Bidirectional Glenn shunt (BDGS) is frequentlyperformed as part of a multistaged palliation of
congenital heart disease in patients with a single ventriclephysiology to decrease the volume overload in patients whoare candidates for future Fontan type repair.1,2 It is oftenadvocated as an intermediary step for high-risk patients.3
Traditionally, accessory sources of pulmonary blood flow,such as from the native pulmonary valve, are eliminatedto minimize pulmonary artery (PA) pressure aftercavopulmonary anastomosis (Glenn shunt). Recently,however, there is a growing trend towards preservation ofblood flow through the native pulmonary valve to improvesaturation. However, occasionally patients can developheart failure when blood flow through the nativepulmonary valve is excessive. We report a child whopresented with elevated PA pressures due to excessiveantegrade pulmonary blood flow after a Glenn shunt andthe successful interruption of this additional flow using anAmplatzer duct occluder (ADO).
Case Report
The patient was an 11-year-old boy with severe cyanosis(basal saturation of 65%) and marked limitation of physicalactivity. The anatomic diagnosis was situs solitus, D-loop,levocardia, bilateral superior vena cava (SVC), double outletright ventricle, multiple muscular ventricular septal defects
(VSDs), and severe infundibular and valvar pulmonicstenosis. At the age of 3 years, the child underwent atrialseptectomy with Brock procedure (enlargement of rightventricular outflow in relation to the pulmonary valve).Cardiac catheterization at admission showed elevated rightatrial (mean: 18 mmHg) and PA pressures (29/17 mmHg,mean: 23 mmHg) with a pulmonary-to-systemic blood flowratio of 3:1, and the calculated indexed pulmonary vascularresistance was 0.14 Wood units/m2. He underwent right-sided cavopulmonary anastomosis with ligation of the leftSVC and enlargement of the atrial septal defect. Ligationof the PA could not be done as planned due to denseadhesions in the region of the main pulmonary artery(MPA). His immediate postoperative course was uneventful.On postoperative day 11, it was noticed that he hadprominent neck vein pulsations, and prominent chest wallveins, which were seen to drain from above downwards.Besides, he had an overt right heart failure with ascites andhepatomegaly. Echocardiography showed systolicretrograde pulsatile flow in the right SVC due to excessiveantegrade flow from the right ventricle into the PA.
Informed consent was taken and cardiac catheterizationwas performed under conscious sedation. The femoralvessels and the right subclavian vein were cannulatedpercutaneously. The SVC pressure tracing showed abiphasic wave pattern and was markedly elevated (38/18mmHg, mean 26 mmHg). The PA angiogram showednormal branch pulmonary arteries with rapid wash-off ofthe contrast, and the PA annulus measured 12.5 mm. Thepulmonary valve was crossed in a retrograde fashion witha 0.035" exchange length (300 cm) Roadrunner wire
Brief Report
We report a case where excessive accessory pulmonary blood flow via the native pulmonary valve aftercavopulmonary anastomosis resulted in pulmonary hypertension and heart failure. This flow was successfullyeliminated in the cardiac catheterization laboratory using an Amplazter duct occluder that was placed acrossthe native pulmonary valve. (Indian Heart J 2003; 55: 373–375)
Key Words: Catheter intervention, Amplatzer duct occluder, Cavopulmonary shunt
Correspondence: Dr R Krishna Kumar, Division of Pediatric Cardiology,Amrita Institute of Medical Sciences and Research Centre, Elamakkara PO,Kochi 682026. e-mail: [email protected]
IHJ-502-03.p65 11/19/2003, 11:25 PM373
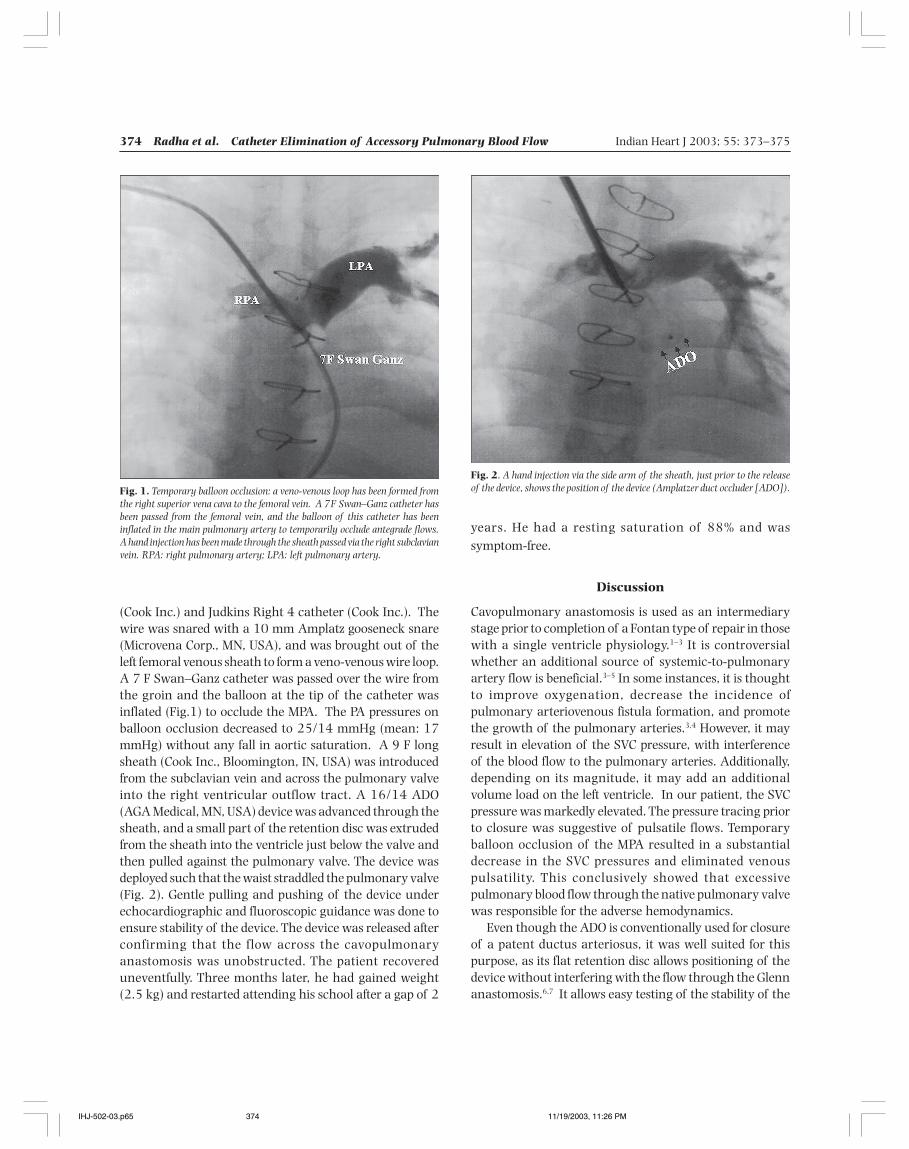
374 Radha et al. Catheter Elimination of Accessory Pulmonary Blood Flow Indian Heart J 2003; 55: 373–375
(Cook Inc.) and Judkins Right 4 catheter (Cook Inc.). Thewire was snared with a 10 mm Amplatz gooseneck snare(Microvena Corp., MN, USA), and was brought out of theleft femoral venous sheath to form a veno-venous wire loop.A 7 F Swan–Ganz catheter was passed over the wire fromthe groin and the balloon at the tip of the catheter wasinflated (Fig.1) to occlude the MPA. The PA pressures onballoon occlusion decreased to 25/14 mmHg (mean: 17mmHg) without any fall in aortic saturation. A 9 F longsheath (Cook Inc., Bloomington, IN, USA) was introducedfrom the subclavian vein and across the pulmonary valveinto the right ventricular outflow tract. A 16/14 ADO(AGA Medical, MN, USA) device was advanced through thesheath, and a small part of the retention disc was extrudedfrom the sheath into the ventricle just below the valve andthen pulled against the pulmonary valve. The device wasdeployed such that the waist straddled the pulmonary valve(Fig. 2). Gentle pulling and pushing of the device underechocardiographic and fluoroscopic guidance was done toensure stability of the device. The device was released afterconfirming that the flow across the cavopulmonaryanastomosis was unobstructed. The patient recovereduneventfully. Three months later, he had gained weight(2.5 kg) and restarted attending his school after a gap of 2
years. He had a resting saturation of 88% and wassymptom-free.
Discussion
Cavopulmonary anastomosis is used as an intermediarystage prior to completion of a Fontan type of repair in thosewith a single ventricle physiology.1–3 It is controversialwhether an additional source of systemic-to-pulmonaryartery flow is beneficial.3–5 In some instances, it is thoughtto improve oxygenation, decrease the incidence ofpulmonary arteriovenous fistula formation, and promotethe growth of the pulmonary arteries.3,4 However, it mayresult in elevation of the SVC pressure, with interferenceof the blood flow to the pulmonary arteries. Additionally,depending on its magnitude, it may add an additionalvolume load on the left ventricle. In our patient, the SVCpressure was markedly elevated. The pressure tracing priorto closure was suggestive of pulsatile flows. Temporaryballoon occlusion of the MPA resulted in a substantialdecrease in the SVC pressures and eliminated venouspulsatility. This conclusively showed that excessivepulmonary blood flow through the native pulmonary valvewas responsible for the adverse hemodynamics.
Even though the ADO is conventionally used for closureof a patent ductus arteriosus, it was well suited for thispurpose, as its flat retention disc allows positioning of thedevice without interfering with the flow through the Glennanastomosis.6,7 It allows easy testing of the stability of the
Fig. 1. Temporary balloon occlusion: a veno-venous loop has been formed fromthe right superior vena cava to the femoral vein. A 7F Swan–Ganz catheter hasbeen passed from the femoral vein, and the balloon of this catheter has beeninflated in the main pulmonary artery to temporarily occlude antegrade flows.A hand injection has been made through the sheath passed via the right subclavianvein. RPA: right pulmonary artery; LPA: left pulmonary artery.
Fig. 2. A hand injection via the side arm of the sheath, just prior to the releaseof the device, shows the position of the device (Amplatzer duct occluder [ADO]).
IHJ-502-03.p65 11/19/2003, 11:26 PM374

Indian Heart J 2003; 55: 373–375 Radha et al. Catheter Elimination of Accessory Pulmonary Blood Flow 375
device before release. We chose an oversized duct occluderinstead of a muscular VSD occluder because it is lessexpensive. The precise positioning of the device such thatit straddles the pulmonary valve is critical. If the device istoo far into the ventricle, it could slip down and getembolized through the systemic circuit. If the device isplaced entirely distal to the pulmonary valve, it can migratedistally and can occlude the branch pulmonary arteries.Because of the absence of a retention disc on the pulmonaryarterial aspect, we chose to oversize the device substantially.
Conclusions: This report demonstrates theunconventional use of an ADO for occlusion of the nativepulmonary valve. This offers an easy and nonsurgicalsolution in this subset of patients who would otherwiseneed repeat surgical procedures. Temporary balloonocclusion is a useful maneuver to ascertain the contributionof accessory pulmonary blood flow so that the benefits ofeliminating this flow can be predicted.
References
1. Mazzera E, Corno A, Picardo S, Di Donato R, Marino B, Costa D, et al.Bidirectional cavopulmonary shunts: clinical applications as stagedor definitive palliation. Ann Thorac Surg 1989; 47: 415–420
2. Allgood NL, Alejos J, Drinkwater DC, Laks H, Williams RG.Effectiveness of the bidirectional Glenn shunt procedure for volumeunloading in the single ventricle patient. Am J Cardiol 1994; 74:834–836
3. Masuda M, Kado H, Shiokawa Y, Fukae K, Suzuki M, Murakami E, etal. Clinical results of the staged Fontan procedure in high-riskpatients. Ann Thorac Surg 1998; 65: 1721–1725
4. Mainwaring RD, Lamberti JJ, Uzark K, Spicer RL. Bidirectional Glenn.Is accessory pulmonary blood flow good or bad? Circulation 1995;92 (Suppl 9): II294–II297
5. Migliavacca F, de Leval MR, Dubini G, Pietrabissa R. A computationalpulsatile model of the bi-directional cavopulmonary anastomosis:the influence of pulmonary forward flow. J Biomech Engineer 1996;11: 520–528
6. Masura J, Walsh KP, Thanopoulous B, Chan C, Bass J, Goussous Y, etal. Catheter closure of moderate- to large-sized patent ductusarteriosus using the new Amplatzer duct occluder: immediate andshort-term results. J Am Coll Cardiol 1998; 31: 878–882
7. Ebeid MR, Masura J, Hijazi ZM. Early experience with the Amplatzerductal occluder for closure of the persistently patent ductusarteriosus. J Interv Cardiol 2001; 14: 33–36
IHJ-502-03.p65 11/19/2003, 11:26 PM375

376 Singh et al. LA–CS Dissociation After RFA for AP Indian Heart J 2003; 55: 376–378
Left Atrial–Coronary Sinus Dissociation Following anAttempt at Radiofrequency Ablation for a Left Lateral
Accessory Pathway
Balbir Singh, Rakesh Sapra, Ripen K Gupta, Tapan Ghose, Rahul Trehan, Upendra KaulDepartment of Cardiology, Batra Hospital and Medical Research Centre, New Delhi
Radiofrequency (RF) ablation for accessory pathways(APs) is safe and effective.1,2 The mapping of APs
involves the use of multipolar electrodes placed at differentsites. Coronary sinus (CS) electrograms assume animportant role (particularly for left-sided pathways), andgenerally represent the sequence of left atrial (LA)activation.3 We report a case of an orthodromic tachycardiausing a concealed left lateral AP in which the activationsequence in CS electrograms reversed following an attemptat RF ablation without altering the AP conduction.
Case Report
A 23-year-old man was referred to our institute forrecurrent episodes of supraventricular tachycardia (SVT)lasting for several hours. He was taken up for anelectrophysiologic study and possible ablation. The AHinterval was 110 ms while the HV interval was 45 ms.Retrograde conduction on ventricular pacing was eccentric,with earliest activation at the distal CS (Fig. 1a). An SVTwith a cycle length (CL) of 330 ms could easily be induced
with a single atrial extrastimulus, with a retrograde atrialactivation sequence similar to that during ventricularpacing (Fig. 1b).
After a trans-septal puncture, a 7 F ablation catheter(Cordis Webster) was introduced to map the left free wallAP along the lateral mitral annulus. During ventricularpacing, a site with the shortest VA interval preceding allother atrial activations was chosen for the application ofRF energy (Fig. 2). During the RF application, it was realizedthat the retrograde activation sequence had changed to oneresembling concentric activation (Fig. 3a).
The RF lesion was completed after 60 s, and thestimulation protocols repeated. An SVT could be inducedagain but with a different activation sequence (proximal tothe distal CS sequence, and earliest activation at the Hisbundle site) (Fig. 3b). On observing this change in the atrialactivation sequence, this tachycardia was thought to beeither an atypical atrioventricular nodal re-entranttachycardia or a slow conducting anteroseptal AP. Adiagnosis of ectopic atrial tachycardia was excluded byoverdrive pacing from the right ventricle. This resulted inentrainment of the tachycardia with the same atrialsequence and, on abrupt termination of pacing, the returnsequence was an A–V electrogram favoring re-entry ratherthan atrial tachycardia.4 However, on mapping with theablation catheter around the mitral annulus, it was soon
Brief Report
Coronary sinus electrograms generally represent the sequence of left atrial activation, and are very helpful inlocalizing and differentiating left lateral accessory pathway-mediated tachycardia from other supraventriculartachycardias. The activation of the coronary sinus from the left atrium occurs through muscle bridges, whichmay be discrete or form an intermingled continuum. These muscle bridges, if disconnected, may dissociate thecoronary sinus from the left atrium, in which case the coronary sinus electrograms do not represent left atrialactivation, and do not help to understand, or may cause misinterpretation of, the mechanism of supraventriculartachycardia. We report one such case of orthodromic supraventricular tachycardia mediated through the leftlateral accessory pathway in which the coronary sinus got dissociated from the left atrium during radiofrequencyablation. (Indian Heart J 2003; 55: 376–378)
Key Words: Left lateral accessory pathway, Coronary sinus, Radiofrequency ablation
Correspondence: Dr Balbir Singh, Senior Consultant, InterventionalCardiology and Cardiac Electrophysiology, Batra Hospital and Medical ResearchCentre, 1, Tughlakabad Institutional Area, Mehrauli Badarpur Road, New Delhi110062.
IHJ-409-02.p65 11/19/2003, 11:24 PM376
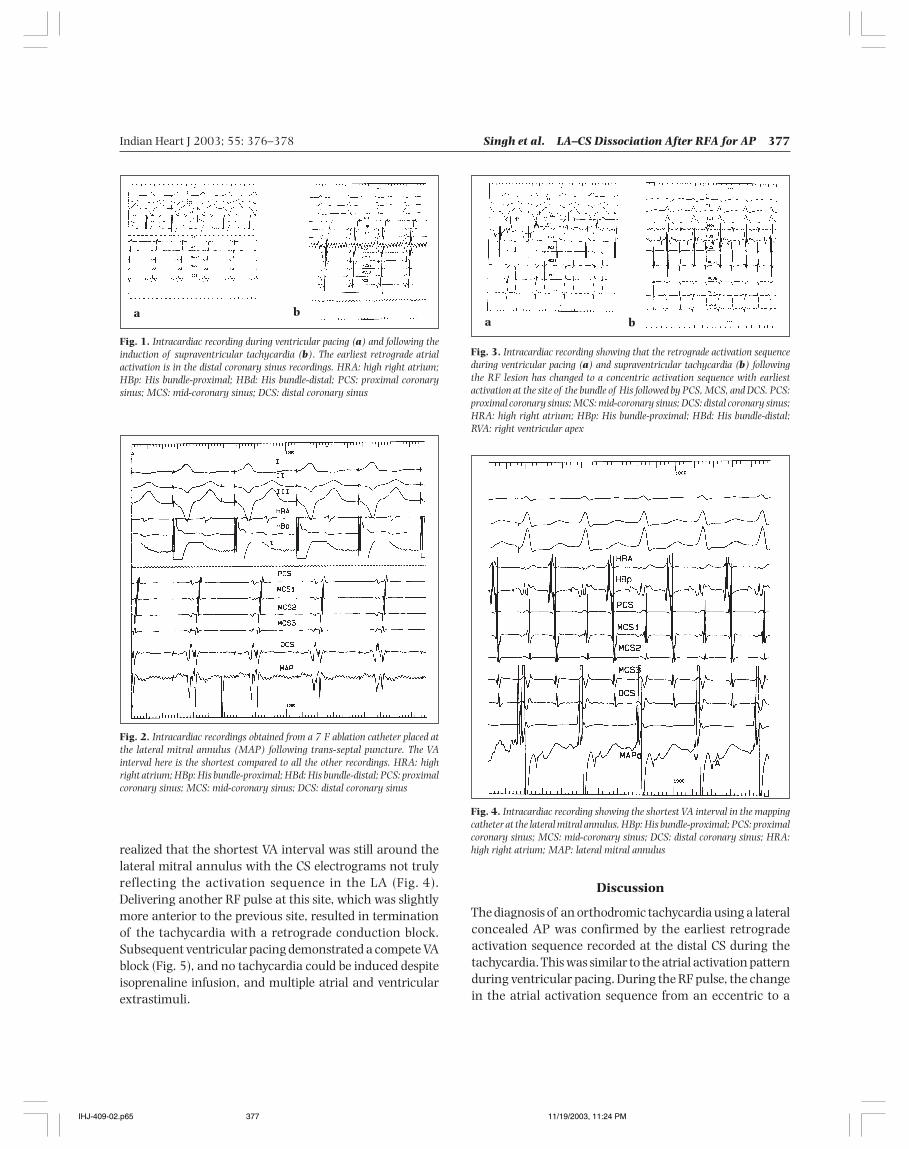
Indian Heart J 2003; 55: 376–378 Singh et al. LA–CS Dissociation After RFA for AP 377
Fig. 1. Intracardiac recording during ventricular pacing (a) and following theinduction of supraventricular tachycardia (b). The earliest retrograde atrialactivation is in the distal coronary sinus recordings. HRA: high right atrium;HBp: His bundle-proximal; HBd: His bundle-distal; PCS: proximal coronarysinus; MCS: mid-coronary sinus; DCS: distal coronary sinus
Fig. 2. Intracardiac recordings obtained from a 7 F ablation catheter placed atthe lateral mitral annulus (MAP) following trans-septal puncture. The VAinterval here is the shortest compared to all the other recordings. HRA: highright atrium; HBp: His bundle-proximal; HBd: His bundle-distal; PCS: proximalcoronary sinus; MCS: mid-coronary sinus; DCS: distal coronary sinus
a ba b
Fig. 3. Intracardiac recording showing that the retrograde activation sequenceduring ventricular pacing (a) and supraventricular tachycardia (b) followingthe RF lesion has changed to a concentric activation sequence with earliestactivation at the site of the bundle of His followed by PCS, MCS, and DCS. PCS:proximal coronary sinus; MCS: mid-coronary sinus; DCS: distal coronary sinus;HRA: high right atrium; HBp: His bundle-proximal; HBd: His bundle-distal;RVA: right ventricular apex
Fig. 4. Intracardiac recording showing the shortest VA interval in the mappingcatheter at the lateral mitral annulus. HBp: His bundle-proximal; PCS: proximalcoronary sinus; MCS: mid-coronary sinus; DCS: distal coronary sinus; HRA:high right atrium; MAP: lateral mitral annulusrealized that the shortest VA interval was still around the
lateral mitral annulus with the CS electrograms not trulyreflecting the activation sequence in the LA (Fig. 4).Delivering another RF pulse at this site, which was slightlymore anterior to the previous site, resulted in terminationof the tachycardia with a retrograde conduction block.Subsequent ventricular pacing demonstrated a compete VAblock (Fig. 5), and no tachycardia could be induced despiteisoprenaline infusion, and multiple atrial and ventricularextrastimuli.
Discussion
The diagnosis of an orthodromic tachycardia using a lateralconcealed AP was confirmed by the earliest retrogradeactivation sequence recorded at the distal CS during thetachycardia. This was similar to the atrial activation patternduring ventricular pacing. During the RF pulse, the changein the atrial activation sequence from an eccentric to a
IHJ-409-02.p65 11/19/2003, 11:24 PM377
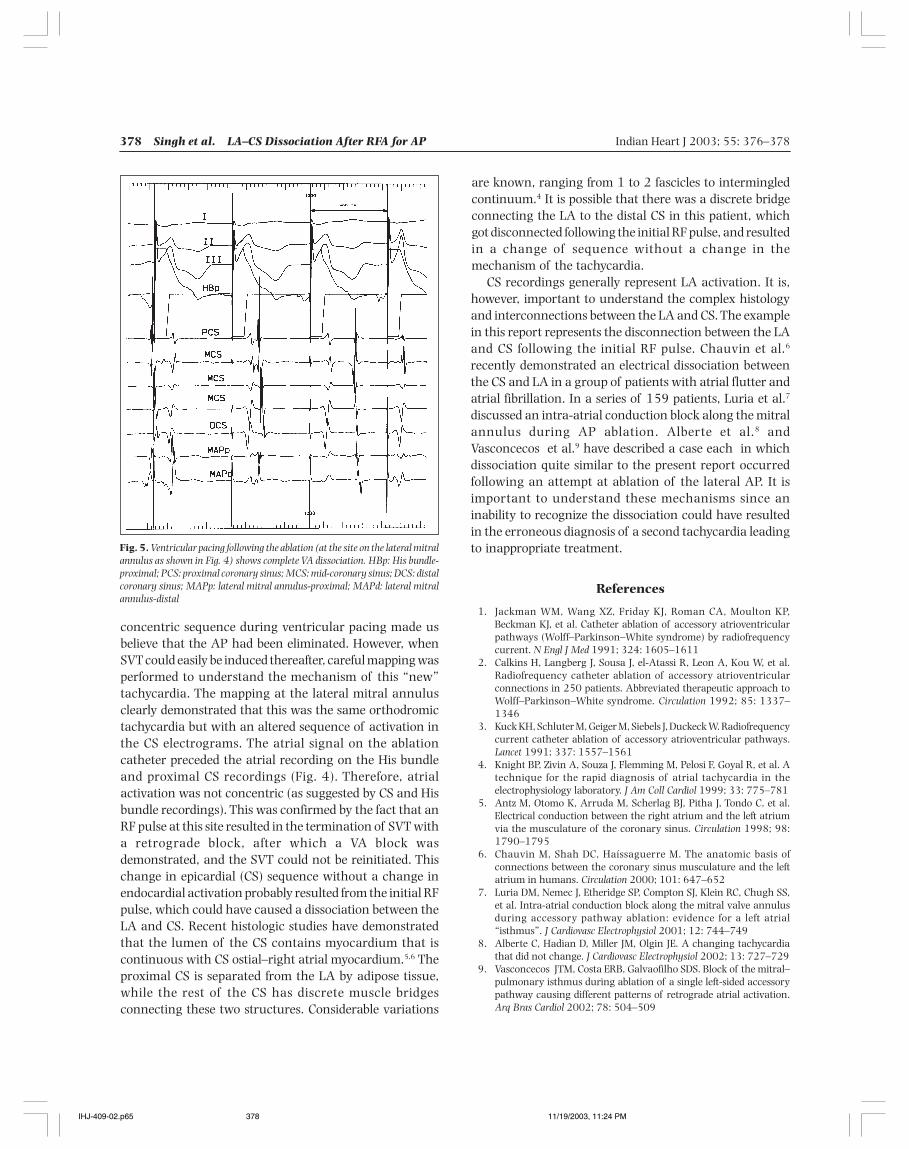
378 Singh et al. LA–CS Dissociation After RFA for AP Indian Heart J 2003; 55: 376–378
concentric sequence during ventricular pacing made usbelieve that the AP had been eliminated. However, whenSVT could easily be induced thereafter, careful mapping wasperformed to understand the mechanism of this “new”tachycardia. The mapping at the lateral mitral annulusclearly demonstrated that this was the same orthodromictachycardia but with an altered sequence of activation inthe CS electrograms. The atrial signal on the ablationcatheter preceded the atrial recording on the His bundleand proximal CS recordings (Fig. 4). Therefore, atrialactivation was not concentric (as suggested by CS and Hisbundle recordings). This was confirmed by the fact that anRF pulse at this site resulted in the termination of SVT witha retrograde block, after which a VA block wasdemonstrated, and the SVT could not be reinitiated. Thischange in epicardial (CS) sequence without a change inendocardial activation probably resulted from the initial RFpulse, which could have caused a dissociation between theLA and CS. Recent histologic studies have demonstratedthat the lumen of the CS contains myocardium that iscontinuous with CS ostial–right atrial myocardium.5,6 Theproximal CS is separated from the LA by adipose tissue,while the rest of the CS has discrete muscle bridgesconnecting these two structures. Considerable variations
are known, ranging from 1 to 2 fascicles to intermingledcontinuum.4 It is possible that there was a discrete bridgeconnecting the LA to the distal CS in this patient, whichgot disconnected following the initial RF pulse, and resultedin a change of sequence without a change in themechanism of the tachycardia.
CS recordings generally represent LA activation. It is,however, important to understand the complex histologyand interconnections between the LA and CS. The examplein this report represents the disconnection between the LAand CS following the initial RF pulse. Chauvin et al.6
recently demonstrated an electrical dissociation betweenthe CS and LA in a group of patients with atrial flutter andatrial fibrillation. In a series of 159 patients, Luria et al.7
discussed an intra-atrial conduction block along the mitralannulus during AP ablation. Alberte et al.8 andVasconcecos et al.9 have described a case each in whichdissociation quite similar to the present report occurredfollowing an attempt at ablation of the lateral AP. It isimportant to understand these mechanisms since aninability to recognize the dissociation could have resultedin the erroneous diagnosis of a second tachycardia leadingto inappropriate treatment.
References
1. Jackman WM, Wang XZ, Friday KJ, Roman CA, Moulton KP,Beckman KJ, et al. Catheter ablation of accessory atrioventricularpathways (Wolff–Parkinson–White syndrome) by radiofrequencycurrent. N Engl J Med 1991; 324: 1605–1611
2. Calkins H, Langberg J, Sousa J, el-Atassi R, Leon A, Kou W, et al.Radiofrequency catheter ablation of accessory atrioventricularconnections in 250 patients. Abbreviated therapeutic approach toWolff–Parkinson–White syndrome. Circulation 1992; 85: 1337–1346
3. Kuck KH, Schluter M, Geiger M, Siebels J, Duckeck W. Radiofrequencycurrent catheter ablation of accessory atrioventricular pathways.Lancet 1991; 337: 1557–1561
4. Knight BP, Zivin A, Souza J, Flemming M, Pelosi F, Goyal R, et al. Atechnique for the rapid diagnosis of atrial tachycardia in theelectrophysiology laboratory. J Am Coll Cardiol 1999; 33: 775–781
5. Antz M, Otomo K, Arruda M, Scherlag BJ, Pitha J, Tondo C, et al.Electrical conduction between the right atrium and the left atriumvia the musculature of the coronary sinus. Circulation 1998; 98:1790–1795
6. Chauvin M, Shah DC, Haíssaguerre M. The anatomic basis ofconnections between the coronary sinus musculature and the leftatrium in humans. Circulation 2000; 101: 647–652
7. Luria DM, Nemec J, Etheridge SP, Compton SJ, Klein RC, Chugh SS,et al. Intra-atrial conduction block along the mitral valve annulusduring accessory pathway ablation: evidence for a left atrial“isthmus”. J Cardiovasc Electrophysiol 2001; 12: 744–749
8. Alberte C, Hadian D, Miller JM, Olgin JE. A changing tachycardiathat did not change. J Cardiovasc Electrophysiol 2002; 13: 727–729
9. Vasconcecos JTM, Costa ERB, Galvaofilho SDS. Block of the mitral–pulmonary isthmus during ablation of a single left-sided accessorypathway causing different patterns of retrograde atrial activation.Arq Bras Cardiol 2002; 78: 504–509
Fig. 5. Ventricular pacing following the ablation (at the site on the lateral mitralannulus as shown in Fig. 4) shows complete VA dissociation. HBp: His bundle-proximal; PCS: proximal coronary sinus; MCS: mid-coronary sinus; DCS: distalcoronary sinus; MAPp: lateral mitral annulus-proximal; MAPd: lateral mitralannulus-distal
IHJ-409-02.p65 11/19/2003, 11:24 PM378

Indian Heart J 2003; 55: 379–381 Chockalingam et al. Left Atrial Appendage Aneurysm 379
Left atrial appendage (LAA) aneurysm is a rarelyencountered congenital defect. Most LAA aneurysms
are diagnosed incidentally. They can result in arrhythmiasor systemic embolization.1 We report a child with massiveLAA aneurysm who presented with effort intolerance andarrhythmias.
Case Report
A previously healthy 12-year-old boy presented with a6-month history of effort intolerance, retrosternal prickingtype of chest discomfort, and palpitations. His NYHAfunctional status had progressed to class III. The pulse,blood pressure, and jugular venous pressure were normal.His cardiac examination revealed normal apical impulsein the left 5th intercostal space. Heart sounds were normalwithout murmurs or gallops. The initial electrocardiogram(ECG) was normal, and when repeated during palpitations,showed narrow QRS nodal re-entrant tachycardia at a rateof 235/min.
Chest X-ray PA view showed cardiomegaly with a CTratio of 64% with straightening of the left heart border. Itshowed filling out of the cardiac bay (Fig. 1). The lateralview X-ray confirmed that the enlarged chamber was theleft atrium (LA). Lung markings were unremarkable.
Transthoracic echocardiography revealed a very large(15×10 cm) echo-free space compressing the left ventricle
(Figs 2, 3). The chamber connected to the LA in the regionof the LAA. Autocontrast was present within the LAAaneurysm due to sluggish flow (Fig. 2). To-and-fro bloodflow was demonstrated using Doppler echo (Fig. 3). Therelative size and location of the aneurysm was delineatedby magnetic resonance imaging (MRI) (Fig. 4). Cardiaccatheterization confirmed normal chamber pressures andsaturations. The LA injection by trans-septal punctureopacified a large chamber adjacent to the left heart (Fig. 5).
The patient was operated on cardiopulmonary bypass.The aneurysm was resected and the histopathology reportconfirmed benign cardiac and fibrous tissue. His symptomsimproved rapidly and he is now doing well 10 months afterthe resection without recurrence of palpitation.
Brief Report
Correspondence: Dr Anand Chockalingam, Madras Medical College andResearch Institute, 9 A Taylors Road, Kilpauk, Chennai 600010.e-mail: [email protected]
Left atrial appendage aneurysm is a rarely reported condition. Symptoms are absent in childhood and diagnosisis usually incidental. Systemic embolization or arrhythmia can bring these cases to medical attention. We reportthe case of a 12-year-old male with massive left atrial appendage aneurysm who presented with effort intoleranceand supraventricular arrhythmia. The diagnosis was made by transthoracic echocardiography. Magneticresonance imaging and left atriogram were also done before surgical resection. (Indian Heart J 2003; 55:379–381)
Key Words: Left atrial appendage, Supraventricular tachycardia, Echocardiography
Massive Left Atrial Appendage Aneurysm Presenting asSupraventricular Tachycardia
Anand Chockalingam, R Alagesan, M Nandakumar, G GnanaveluDepartment of Cardiology, Madras Medical College and Research Institute, Chennai
Fig. 1. Chest X-ray in posteroanterior view showing filling out of the cardiacbay, altering the left cardiac border.
IHJ-547-03.p65 11/19/2003, 11:26 PM379
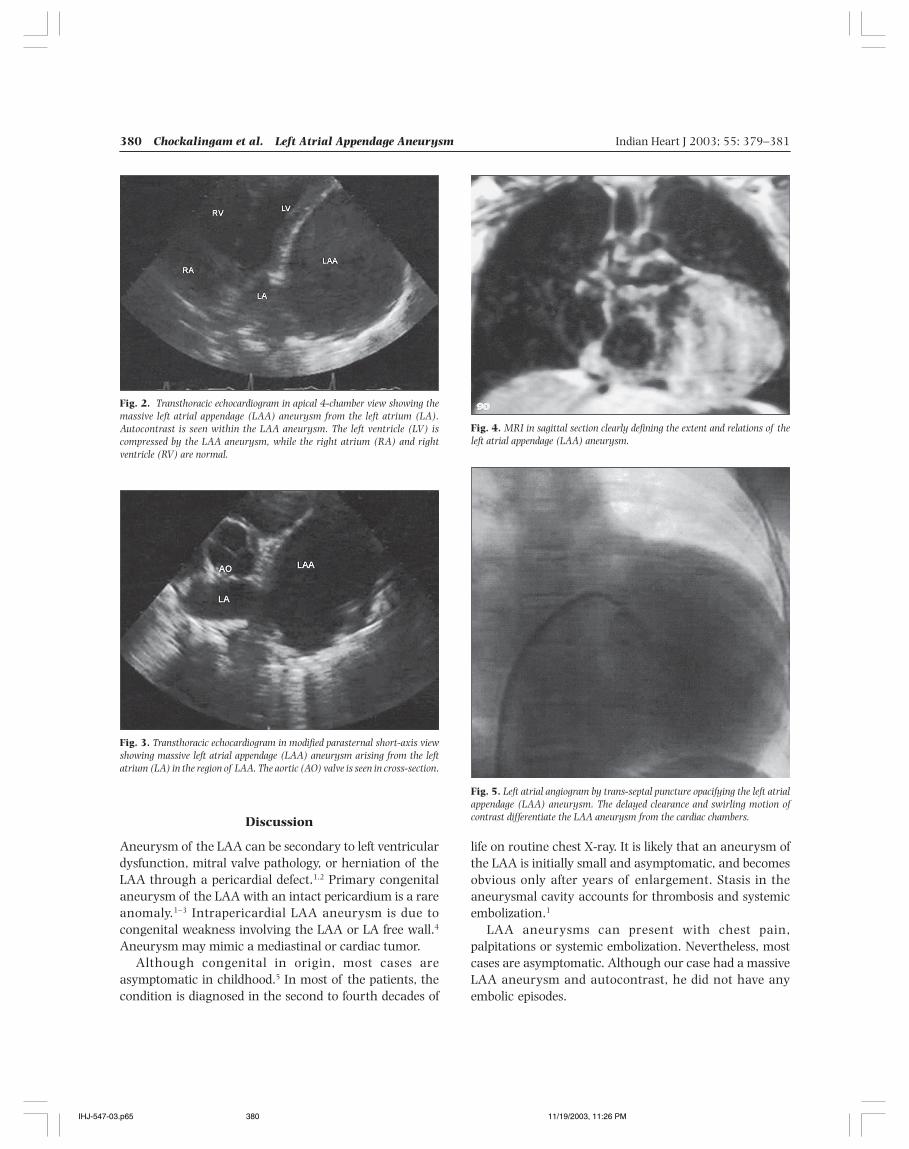
380 Chockalingam et al. Left Atrial Appendage Aneurysm Indian Heart J 2003; 55: 379–381
life on routine chest X-ray. It is likely that an aneurysm ofthe LAA is initially small and asymptomatic, and becomesobvious only after years of enlargement. Stasis in theaneurysmal cavity accounts for thrombosis and systemicembolization.1
LAA aneurysms can present with chest pain,palpitations or systemic embolization. Nevertheless, mostcases are asymptomatic. Although our case had a massiveLAA aneurysm and autocontrast, he did not have anyembolic episodes.
Fig. 2. Transthoracic echocardiogram in apical 4-chamber view showing themassive left atrial appendage (LAA) aneurysm from the left atrium (LA).Autocontrast is seen within the LAA aneurysm. The left ventricle (LV) iscompressed by the LAA aneurysm, while the right atrium (RA) and rightventricle (RV) are normal.
Fig. 3. Transthoracic echocardiogram in modified parasternal short-axis viewshowing massive left atrial appendage (LAA) aneurysm arising from the leftatrium (LA) in the region of LAA. The aortic (AO) valve is seen in cross-section.
Discussion
Aneurysm of the LAA can be secondary to left ventriculardysfunction, mitral valve pathology, or herniation of theLAA through a pericardial defect.1,2 Primary congenitalaneurysm of the LAA with an intact pericardium is a rareanomaly.1–3 Intrapericardial LAA aneurysm is due tocongenital weakness involving the LAA or LA free wall.4
Aneurysm may mimic a mediastinal or cardiac tumor.Although congenital in origin, most cases are
asymptomatic in childhood.5 In most of the patients, thecondition is diagnosed in the second to fourth decades of
Fig. 4. MRI in sagittal section clearly defining the extent and relations of theleft atrial appendage (LAA) aneurysm.
Fig. 5. Left atrial angiogram by trans-septal puncture opacifying the left atrialappendage (LAA) aneurysm. The delayed clearance and swirling motion ofcontrast differentiate the LAA aneurysm from the cardiac chambers.
IHJ-547-03.p65 11/19/2003, 11:26 PM380

Indian Heart J 2003; 55: 379–381 Chockalingam et al. Left Atrial Appendage Aneurysm 381
Physical examination is often unrevealing.6 ECG mayreveal tachyarrhythmia, as in our case. A radiographicdifferential for filling out of the cardiac bay region (Fig. 1)includes partial absence of the pericardium along withatrial herniation through the defect, paracardiac tumorsor cysts, enlarged coronary sinus, aneurysmal dilatationof the LA due to mitral valvular disease, and pulmonaryartery aneurysm.
Echocardiography, radionuclide angiography, CT scan,MRI, and cardiac catheterization have been used toestablish the diagnosis. Angiography requires trans-septalpuncture, and may not detect an LAA aneurysm filled witha thrombus. In our case, the diagnosis was made duringscreening echocardiography. Due to adequatedemonstration of the aneurysm and its origin bytransthoracic windows, transesophageal echocardiography(TEE) was not required.
Foale et al. proposed the following criteria to diagnosecongenital LAA aneurysm:8
(i) origin from an otherwise normal LA; (ii) well-definedcommunication with the LA; (iii) position within thepericardium; and (iv) distortion of the LV free wall by theaneurysm.
Our patient fulfilled all the 4 criteria for the diagnosis of anLAA aneurysm.
Associated anomalies such as atrial septal defect (ASD),persistent left superior vena cava, anomalous pulmonary
venous drainage, and renal artery anomalies have beenreported.9,10 Aneurysmectomy abolishes recurrentarrhythmias and thromboembolism. Thus, surgicalresection, with or without cardiopulmonary bypass, isrecommended even in asymptomatic patients.
References
1. Bramlet DA, Edwards JE. Congenital aneurysm of left atrialappendage. Br Heart J 1981; 45: 97–100
2. Tanabe T, Ishizaka M, Ohta S, Sugie S. Intrapericardial aneurysm ofthe left atrial appendage. Thorax 1980; 35: 151–153
3. Godwin TS, Avager P, Key JA. Intrapericardial aneurysmal dilatationof left atrial appendage. Circulation 1988; 37: 397–400
4. Shaher RM, Alley R, Anis W, Mintzer J. Congenital enlargement ofthe left atrium. J Thorac Cardiovasc Surg 1972; 63: 292–299
5. Behrendt DM, Aberdeen E. Congenital aneurysm of the left atrium.Ann Thorac Surg 1972; 13: 54–59
6. Krueger SK, Ferlic RM, Mooring PK. Left atrial appendage aneurysm:correlation of noninvasive with clinical and surgical findings: reportof a case. Circulation 1975; 52: 732–738
7. Comess KA, Labate DP, Winter JA, Hill AC, Miller DC. Congenital leftatrial appendage aneurysm with intact pericardium: diagnosis bytransesophageal echocardiography. Am Heart J 1990; 120: 992–996
8. Foale RA, Gibson TC, Guyer DE, Gillam L, King MF, Weyman AE.Congenital aneurysms of the left atrium: recognition by cross-sectional echocardiography. Circulation 1982; 66: 1065–1069
9. Parmley LF. Congenital atriomegaly. Circulation 1982; 25: 553–55810. Labarre TR, Stamato NJ, Hwang MH, Jacobs WR, Stephanides L,
Scanlon PJ. Left atrial appendage aneurysm with associatedanomalous pulmonary venous drainage. Am Heart J 1987; 114:1243–1245
IHJ-547-03.p65 11/19/2003, 11:26 PM381
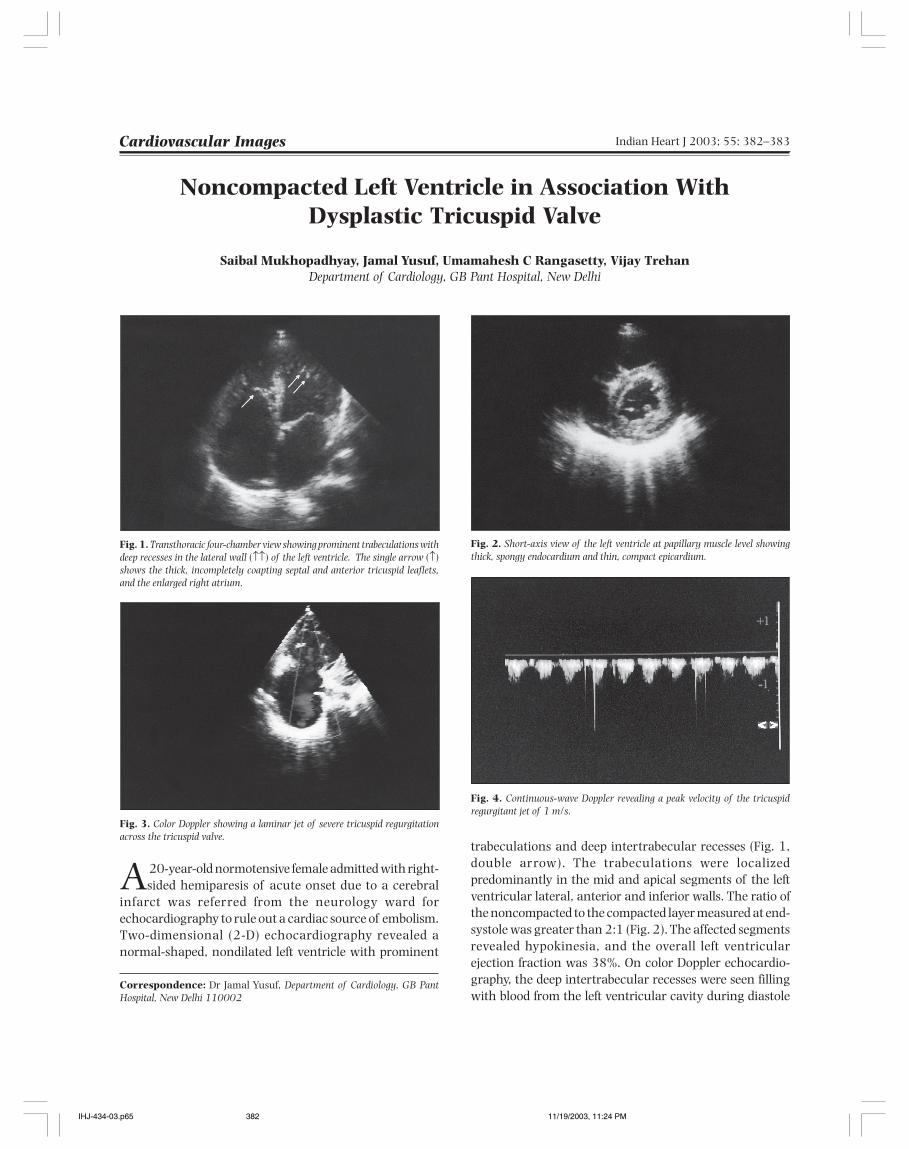
382 Mukhopadhyay et al. Noncompacted LV With Dysplastic Tricuspid Valve Indian Heart J 2003; 55: 382–383
Noncompacted Left Ventricle in Association WithDysplastic Tricuspid Valve
Saibal Mukhopadhyay, Jamal Yusuf, Umamahesh C Rangasetty, Vijay TrehanDepartment of Cardiology, GB Pant Hospital, New Delhi
A 20-year-old normotensive female admitted with right-sided hemiparesis of acute onset due to a cerebral
infarct was referred from the neurology ward forechocardiography to rule out a cardiac source of embolism.Two-dimensional (2-D) echocardiography revealed anormal-shaped, nondilated left ventricle with prominent
Cardiovascular Images
Correspondence: Dr Jamal Yusuf, Department of Cardiology, GB PantHospital, New Delhi 110002
Fig. 1. Transthoracic four-chamber view showing prominent trabeculations withdeep recesses in the lateral wall (↑↑) of the left ventricle. The single arrow (↑)shows the thick, incompletely coapting septal and anterior tricuspid leaflets,and the enlarged right atrium.
Fig. 2. Short-axis view of the left ventricle at papillary muscle level showingthick, spongy endocardium and thin, compact epicardium.
Fig. 3. Color Doppler showing a laminar jet of severe tricuspid regurgitationacross the tricuspid valve.
Fig. 4. Continuous-wave Doppler revealing a peak velocity of the tricuspidregurgitant jet of 1 m/s.
trabeculations and deep intertrabecular recesses (Fig. 1,double arrow). The trabeculations were localizedpredominantly in the mid and apical segments of the leftventricular lateral, anterior and inferior walls. The ratio ofthe noncompacted to the compacted layer measured at end-systole was greater than 2:1 (Fig. 2). The affected segmentsrevealed hypokinesia, and the overall left ventricularejection fraction was 38%. On color Doppler echocardio-graphy, the deep intertrabecular recesses were seen fillingwith blood from the left ventricular cavity during diastole
IHJ-434-03.p65 11/19/2003, 11:24 PM382

Indian Heart J 2003; 55: 382–383 Mukhopadhyay et al. Noncompacted LV With Dysplastic Tricuspid Valve 383
with reversed flow in systole. No thrombus could bedetected in the intertrabecular recesses. The mitral andaortic valves were normal. The right atrium was enlarged(Fig. 1) and the septal and anterior leaflets of the tricuspidvalve appeared thickened with incomplete coaptation insystole (Fig. 1, single arrow). There was no excessive apicaldisplacement of the septal leaflet as seen in Ebstein’sanomaly. Color Doppler echocardiography across thetricuspid valve revealed a laminar jet of severe tricuspidregurgitation (TR) (Fig. 3) with continuous-wave Dopplerrevealing a peak velocity of the regurgitant jet of 1 m/s(Fig. 4). Thus, a diagnosis of dysplastic tricuspid valve wasmade. The right ventricle and pulmonary valve werenormal.
Noncompacted myocardium has been frequentlyreported in patients with congenital left or right ventricularoutflow tract obstruction. In these patients with a “spongymyocardium”, the recesses represent “persisting sinusoids”that fail to regress during ontogenesis due to persistentpressure overload, and communicate with the coronaryarteries.1–3 In contrast, isolated ventricular noncompaction(IVNC) is considered a congenital anomaly due to the arrestof compaction of the loose myocardial meshwork duringfetal ontogenesis,4 when the intertrabecular recessescovered with endothelial cells are filled with blood from theventricular cavity without evidence of communication tothe epicardial coronary artery system.2,5 These recessespredispose to local thrombus formation but are often missedon echocardiography when associated with prominenttrabeculations.6
Noncompacted myocardium and tricuspid valvedysplasia as individual conditions are well described. Ourpatient had all the characteristic echocardiographicfeatures of left ventricular noncompaction as described byJenni et al.4 along with a coexistent congenital anomaly—tricuspid valve dysplasia—a combination not yet reported
in the literature, to the best of our knowledge. Earlydiagnosis of ventricular noncompaction and the correctmanagement of such patients is crucial, as the clinicalmorbidity includes heart failure, caused by progressiveventricular dysfunction, arrhythmias and systemic embolicevents.7
Our patient presented with asymptomatic leftventricular dysfunction detected incidentally onechocardiography, but there was no clinical evidence ofright ventricular systolic dysfunction. The patient was puton medical therapy with a combination of beta-blockersand ACE inhibitors (to arrest negative remodeling and theprogression of heart failure), and oral anticoagulants toprevent the formation of thrombi in the left ventricle.
References
1. Dusek A, Osadal B, Duskova M. Postnatal persistence of spongymyocardium with embryonic blood supply. Arch Pathol 1975; 99:312–317
2. Angelini A, Melacini P, Barbero F. Evolutionary persistence of spongymyocardium in humans [Abstr]. Circulation 1999; 99: 247S
3. Lauer RM, Fink RM, Petry EL, Dunn MI, Deihl AM. Angiographicdemonstration of intramyocardial sinusoids in pulmonary valveatresia with intact ventricular septum and hypoplastic right ventricle.N Engl J Med 1964; 271: 68–72
4. Jenni R, Oechslin E, Schneider J, Jost CA, Kaufmann PA.Echocardiographic and pathoanatomical characteristics of isolatedleft ventricular non-compaction: a step towards classification as adistinct cardiomyopathy. Heart 2001; 86: 666–671
5. Oechslin E, Jost CA, Rojas JR, Kaufmann PA, Jenni R. Long term followup of 34 adults with isolated left ventricular non-compaction: adistinct cardiomyopathy. J Am Coll Cardiol 2000; 36: 493–500
6. Boyd MT, Seward JB, Tajik AJ, Edward WD. Frequency and locationof prominent left ventricular trabeculations at autopsy in 474normal human hearts: implications for evaluation of mural thrombiby two-dimensional echocardiography. J Am Coll Cardiol 1987; 9:323–326
7. Ichida F, Hamamichi Y, Miyawaki T. Clinical features of isolated non-compaction of left ventricle. J Am Coll Cardiol 1999; 34: 233–240
IHJ-434-03.p65 11/19/2003, 11:24 PM383

384 Letters to the Editor Indian Heart J 2003; 55: 384–385
The search for new risk factors that can satisfactorilyexplain the prevalence and severity of coronary heart
disease (CHD) has brought into focus the role of infections,particularly the association of Helicobacter pylori (H. pylori)Chlamydia pneumoniae (C. pneumoniae), and cytomegalo-virus with CHD.1 Although many such studies are availablefrom western countries, studies in the Indian context arelacking. Identification of a causal linkage of any kind ofinfection with CHD can lead to timely intervention withantibiotics, which may decrease the disease burden.
One hundred and seventeen angiographically provenpatients (35–79 years of age) with unstable angina (UA),and 16 patients with chronic stable angina (CSA) werematched with 90 healthy blood donors (20–60 years ofage) free from clinical CHD confirmed by 12-lead restingECG. Fasting serum samples were drawn from these healthycontrols and patients. C. pneumoniae and H. pylori-specificIgG was detected by the ELISA technique.
A total of 59 (58%), 9 (56%), and 48 subjects (53%)were seropositive for H. pylori in the UA, CSA, and controlgroups, respectively. The number of subjects seropositivefor C. pneumoniae in the UA, CSA, and control groups were67 (66%), 15 (94%), and 73(81%), respectively. Nosignificant statistical difference in seropositivity was found
Seropositivity of Chlamydiapneumoniae and Helicobacter pyloriAmong Coronary Heart DiseasePatients and Normal Individuals ina South Indian Population
between the study group and the controls, either for H.pylori or for C. pneumoniae. The prevalence of H. pylorireported in our study is within the range reported in studiesfrom India (40%–74%).2 There has been no report on theseropositivity of C. pneumoniae from India till date. Our studyshowed a high prevalence of C. pneumoniae (70.1% inpatients, and 81% in controls), which is in agreement withthe normal Chinese population (61.5%).3
It has been postulated that in countries with poorsocioeconomic conditions, there may be a link betweenCHD and some undefined environmental exposures,including infections. It is possible that cardiovascular riskincreases with cumulative or earlier exposure to morepathogens or specific potentially atherogenic microbes.4
References
1. Danesh J, Collins R, Peto R. Chronic infections and coronary heartdisease: is there a link? Lancet 1997; 350: 430–436
2. Kate V, Ananthakrishnan N, Ratnakar C, Badrinath S. Anti H. pyloriIgG seroprevalence rates in asymptomatic children and adults fromSouth India. Indian J Med Microbiol 2000; 19: 73–78
3. Ni AP, Lin GY, Yang L, He HY, Huang CW, Liu ZJ, et al. A seroepi-demiologic study of Chlamydia pneumoniae, Chlamydia tranchomatisand Chlamydia psittaci in different populations on the mainland ofChina. Scand J Infect Dis 1996; 28: 553–557
4. Taylor C. Workshop on emerging issues in microbial infections andcardiovascular diseases, October 1998. NIAID Council News 1999;8: 5
Abhijit Chaudhury, D Rajasekhar, SAA Latheef,G Subramanyam
Departments of Microbiology and CardiologySri Venkateswara Institute of Medical Sciences, Tirupati
Letters to the Editor
Letter-to-Editors.p65 11/19/2003, 11:28 PM384

Indian Heart J 2003; 55: 384–385 Letters to the Editor 385
Delayed Tamponade Following theSudden Braking of a SpeedingVehicle
Motor vehicle accidents often cause blunt cardiactrauma with pericardial hemorrhage and rapid
tamponade.1 We describe a patient who presented withbleeding, not to the emergency services, but to thecardiologist.
A 63-year-old hypertensive male on beta-blockerspresented with a 3-day history of dyspnea on exertion andprofound weakness. His symptoms started 1 day after thesudden braking of his car while he was travelling in theback seat without a seat belt. He reported having protectedhimself by holding onto the seat in front of him, and deniedimpact to the chest. He was well for a few hours butdeveloped pain in the back of the neck the same night. Overthe next 3 days, he developed progressive weakness anddyspnea. On examination, his pulse rate was 80 bpm (onatenolol) and the blood pressure 140/90 mmHg with apulsus paradoxus of 10 mmHg. The jugular venouspressure was elevated to 15 mmHg and heart sounds werefaint. The ECG showed a relative decrease in the QRSvoltages compared to the previous recording. A chest X-ray showed an enlarged cardiac silhouette with acardiothoracic ratio of 70%.
The patient's hemoglobin level was 12 g%; it had been14.5 g% during a routine health checkup 3 months earlier.Serum transminases and alkaline phosphatase wereelevated three-fold, as is common in subacute tamponade.
The troponin T concentration was normal (<0.01 ng/ml).Echocardiography revealed a moderate-size pericardialeffusion with collapse of the right atrium, and exaggeratedrespiratory variation in the tricuspid and mitral Dopplerflow velocities. The patient underwent a therapeutictapping of the pericardial fluid. Seven hundred and twentyml of bloody fluid was aspirated via the subxiphoidapproach and a pigtail catheter left inside for 24 hours.There was no recurrence of effusion, and the patient madean uneventful recovery. The fluid had a hemoglobin contentof 10 g%, was sterile on culture and had no malignant cells.The patient was doing well on follow-up at 6 months.
We believe that the bleeding resulted from an injurycaused by the sudden deceleration of the car.2 Subacutetraumatic pericardial bleeds should be managed andfollowed up like other cases of pericardial hemorrhage,including long-term follow-up for the development ofconstrictive pericarditis.2
References
1. Rashid MA, Wikstrom T, Ortenwall P. Cardiac injuries: a ten-yearexperience. Eur J Surg 2000; 166: 18–21
2. Karmy-Jones R, Yen T, Cornejo C. Pericarditis after trauma resultingin delayed cardiac tamponade. Ann Thorac Surg 2000; 74: 239–241
Rajiv Bajaj, Harbans Singh WasirDepartment of Cardiology
Batra HospitalNew Delhi
Letter-to-Editors.p65 11/19/2003, 11:28 PM385

386 Selected Summaries Indian Heart J 2003; 55: 386–389Selected Summaries
Comparison of Carvedilol and Metoprolol on Clinical Outcomes inPatients With Heart Failure in the Carvedilol or Metoprolol
European Trial (COMET): Randomized Controlled Trial
PA Poole-Wilson et al. COMET Investigators. Lancet 2003; 362: 7–13
Summary
COMET was a multicenter, randomized, double-blind,parallel-group trial comparing the effects of carvedilol andmetoprolol on the clinical outcome of patients with chronicheart failure (CHF). The study involved 3029 patients from341 centers in 15 European countries. Symptomatic NYHAclasses II–IV (<5% were class IV) CHF patients receivingboth angiotensin-converting enzyme inhibitors (ACE-I) anddiuretics for at least 2 weeks were included in the study. Allof them had left ventricular (LV) systolic dysfunction asevidenced by ejection fraction (EF) <35% or left ventricularend-diastolic dimension (LV-EDD) >6 cm, and fractionalshortening (FS) <20%. Patients with an unstable medicalcondition, uncontrolled hypertension or recent change intreatment were excluded from the study. Carvedilol (1511patients) was started at 3.125 mg twice daily, and doubledevery 2 weeks to achieve a target dose of 25 mg twice daily.Metoprolol was started at 5 mg twice daily, and stepped upto 50 mg twice daily. The patients were followed up for amean duration of 58±6 months. The baselinecharacteristics were similar in both the groups. The meanage was 62±11 years, one-fifth were female, mean durationof CHF was >40 months, and more than 50% had anischemic etiology. The mean EF was 26%±7%. About 60%of patients were receiving digitalis but very few patientswere receiving angiotensin receptor blockers (ARBs). Theprimary end-point of the study was all-cause mortality.Later, a composite end-point of all-cause mortality or all,cause admission was admitted as a second primary end-point in recognition of its importance, after publication ofthe findings of the MERIT-HF study. The all-cause mortalitywas 17% lower with carvedilol (34% v. 40%; 95% Cl: 0.74–0.93, p=0.0017). The survival advantage was apparentwithin 6 months, and was consistent among all thesubgroups. The composite end-point was also 6% lower inthe carvedilol group, though this was statisticallynonsignificant (74% v. 76%; 95% CI: 0.86–1.02, p=0.122).Sudden cardiac death was the most common mode ofdeath, accounting for 43% of deaths in the carvedilol group,and 44% in the metoprolol group. Circulatory heart failurecontributed to one-third of deaths in both the groups.Three-fourth of the patients in the carvedilol arm reachedthe target dose (mean dose 41.8±14.6 mg), whereas 78%achieved the target dose in the metoprolol group (meandose achieved 85±28.9 mg). However, the reduction inheart rate was greater in the carvedilol group (13.3 beats/min v. 11.7 beats/min; 95% CI: 2.7 to –0.6). The incidenceof side-effects and drug withdrawals (32%) were similar inboth the groups. The authors concluded that carvedilolextends a survival advantage vis-a-vis metoprolol inpatients with symptomatic CHF.
Comments
In patients with symptomatic CHF, the aims of treatmentare two-fold. The first and foremost is to relieve symptomsand second, even more important than the first, is toimprove survival. While diuretics and digoxin are wellknown for the symptom relief they provide in CHF, theyconfer very little, if any, survival advantage. In this context,the only drugs that improve survival are ACE-I, beta-blockers, aldosterone antagonists, and ARBs. However, thebenefit achieved by a combination of these renin–angiotensin–aldosterone system (RAAS) modifying drugsis still controversial. For instance, the VAL-HEFT studyshowed that while a combination of ACE-I with ARB, orbeta-blocker (BB) with ARB was beneficial, the combinationof all three agents (ACE-I + BB + ARB) is actuallydetrimental. The second issue has been the actual drugused. Among the beta-blockers, carvedilol (COPERNICUS,CHRISTMAS, CAPRICORN), metoprolol (MDC, MERIT-HF),and bisoprolol (CIBIS I and II) have all been shown to beuseful. However, which of these beta-blockers, if any, ismore beneficial is still uncertain. Theoretically, metoprololand bisoprolol are both β
1- selective, and should therefore
be more effective. Carvedilol is not only β1- and β
2-active,
but also blocks the α1-adrenergic receptors. However,
carvedilol has several other mechanisms by which it canbe useful in patients with CHF. It increases insulinsensitivity, and has potent antioxidant action, therebyimproving endothelial dysfunction and reducing apoptosis.Indeed, a recent meta-analysis has suggested that the useof carvedilol led to a greater improvement in EF (one of theprime mechanisms for improvement of survival) ascompared to metoprolol. The COMET trial is the first largestudy to compare these two drugs head-on. This studysuggests that carvedilol is superior to metoprolol not onlyin reducing the combined rate of mortality and hospitaladmission but, more importantly, in improving survival (asmuch as 17% over a 5-year period). However, this study isnot without limitations. The mean dose of metoprolol, i.e.85 mg, is much lower than that achieved in the MERIT-HFstudy (>162 mg). Secondly, the salt of metoprolol, i.e.metoprolol tartarate, is also different from metoprololsuccinate (metoprolol CR/XL) used in the MERIT-HF study.Some studies have shown that the tartarate salt may haveat least 30%–35% reduced bioavailability as compared tothe succinate salt. Indeed, the reduction in heart rate andblood pressure was higher in the carvedilol arm, perhapsreflecting inadequate dosage in the metoprolol arm.Another important limitation of the study was that follow-up EF values were not obtained.
IHJ-Selected Summary.p65 11/19/2003, 11:27 PM386

Indian Heart J 2003; 55: 386–389 Selected Summaries 387
Summary
EUROPA was a randomized, double-blind, placebo-controlled, largest-ever trial of the angiotensin-convertingenzyme inhibitor (ACE-I) perindopril, evaluating thereduction of cardiac events in low-risk patients with provencoronary artery disease (CAD) but with no evidence of CHF.It enrolled 13 655 patients ≥18 years of age, anddocumented CAD as evidenced by previous MI, coronaryrevascularization, angiographically proven CAD (>70%stenosis) or positive stress test in men. Patients withevidence of CHF, hypotension/uncontrolled hypertensionor due for PCI, recent use of ACE-I or angiotensin receptorblockers (ARBs), renal insufficiency or elevated serumpotassium (>5.5 mmol/L) were excluded from the study.The primary end-point was combined CVS mortality +nonfatal AMI + survival of sudden cardiac death (SCD).Patients initially received 4 mg perindopril for 4 weeks,increased to 8 mg for another 4 weeks. Patients were thenrandomized to receive either 8 mg perindopril or a placebofor at least 3 years, the dose being reduced to 4 mg if it wasnot tolerated. The mean follow-up was 4.2 years. Overall,baseline characteristics were similar in both the groups. Themean age was 60±9 years, and 85% were male. They weregenerally low-risk CAD patients, 92% on platelet inhibitors,62% on beta-blockers, and 48% on statins. Perindoprilsignificantly reduced the combined end-point from 9.9%in the placebo group to 8% (RR 20%, p=0.0003). Thebeneficial effect became apparent at 1 year of treatment,and was seen in all the sub-groups. Perindopril use wasassociated with a reduction in all the secondary end-pointsas well. Total mortality reduced by 11% (ns), MI decreasedby 23.9% (p=0.001), and CHF decreased by 39.2%(p=0.002). About 50 patients needed to be treated withperindopril over 4 years to prevent one major CVS event,and this benefit was over and above the medications alreadybeing used.
Comments
The HOPE study has already established the role of ACE-I,especially ramipril, in patients with high-risk CAD. In theEUROPA study, perindopril was chosen because of itsantiischemic and antiatherogenic effects, its Effects oncardiovascular remodeling, and its blood pressure-loweringeffect. The baseline characteristics of the EUROPA studywere different from those of the HOPE study. In the HOPE
Efficacy of Perindopril in Reduction of Cardiovascular Events Among Patients WithStable Coronary Artery Disease: Randomized, Double-Blind, Placebo-Controlled,
Multicenter Trial (the EUROPA Study)
KM Fox et al. The EUROPA Trial Investigators. Lancet 2003; 362: 782–788
study, all the patients were >55 years, whereas in theEUROPA study, at least one-third of patients were <55years. In addition, more HOPE patients had diabetesmellitus, hypertension, stroke, and obstructive peripheralvascular disease. This difference is well reflected in theplacebo event rates, where the total mortality in the HOPEstudy versus the EUROPA study was 12.2% v. 7.4%; CVSmortality 8.1% v. 4.4%, and Q wave MI 3.2% v. 2.1%. Withthis background, the results of the EUROPA study were evenmore remarkable and, indeed, may appear to extend theindication of ACE-I to even low-risk CAD patients. However,if we look carefully, even the CAD patients enrolled in thetwo studies were different. In the HOPE trial, only aboutone-third of the patients were post-MI, whereas in theEUROPA trial, nearly two-thirds were post-MI. The role ofACE-I in post-MI is already well established. If we considersubgroup analysis, the benefit in the non-MI patients wasnot statistically significant in the EUROPA study. In otherwords, perindopril is effective in only those patients whohave suffered a prior MI. Thus, these results cannot bedirectly extrapolated to the garden variety of CAD patients;rather, they are more applicable to the post-MI population.Furthermore, another important limitation of the EUROPAstudy is that the ejection fraction was not considered. Sincemost patients in the EUROPA study were post-MI, it is likelythat the mean ejection fraction in this study would be low.The role of ACE-I in patients with a low ejection fraction isagain well established. It can be argued that the effect ofperindopril could be due to its antihypertensive effect.However, the BP reduction was small, 5/2 mmHg, whichcould not account for the large reduction in CVS events. Tobetter understand the mechanism of CVS event reduction,5 substudies of EUROPA are under way. The PERTINENTstudy is evaluating the predictive value of several plasmaand serum markers associated with atherosclerosis, and theeffect of perindopril on these levels. The PERFECT studywill measure blood flow in the brachial artery using B-modeimaging. The PERSPECTIVE study aims to investigate theeffects of perindopril on progression and regression indiabetics, and the PERGENE study will geneticallycharacterize the EUROPA population. The results of thesestudies are expected within a year. Another interestingpoint that has emerged from the EUROPA study is thatmortality reduction was superior in the subgroup ofpatients on beta-blockers. The reduction of events inpatients not on beta-blockers was statistically insignificant.Further studies will be required to clarify this concept.
IHJ-Selected Summary.p65 11/19/2003, 11:27 PM387

388 Selected Summaries Indian Heart J 2003; 55: 386–389
Beneficial Effect of Immediate Stenting After Thrombolysisin Acute Myocardial Infarction
B Scheller et al. SIAM III study group. J Am Coll Cardiol 2003; 42: 634–641
Summary
The Southwest German Interventional Study in Acute MyocardialInfarction (SIAM III) study is a multicenter, randomized,prospective, controlled trial in which immediate stenting after fullthrombolysis was compared with the conservative approach ofdelayed stenting after 2 weeks in patients presenting with acuteSTEMI. The study included patients presenting within 12 hoursof the onset of symptoms with diagnostic electrocardiograms whohad no contraindication to thrombolysis. Angiographic inclusioncriteria were an infarct-related coronary artery >2.5 mm,diameter stenosis of at least 70% or TIMI flow <grade 3. Patientswere recruited from community hospitals located within 34 kmof an interventional center. Reteplase was administered in 2boluses of 10 IU 30 min apart. Patients also received 250 mgaspirin IV, and a bolus of 5000 IU heparin, followed by an infusionadjusted to maintain an activated partial thromboplastin time of50 to 70 ms. During thrombolysis, the patients were randomizedto either immediate or elective stenting. Patients randomized toimmediate stenting were shifted to interventional centers within6 hours where the infarct-related artery was stented; 2 weeks later,a repeat angiography was performed. The second groupunderwent elective angiography and stenting performed at 2weeks. Patients in both the groups were clinically followed up atregular intervals, and repeat angiography was scheduled at 6months. A total of 163 patients were included in the study. GroupI (immediate stenting group) had a significant reduction in thecombined end-point of ischemic events, death, reinfarction, andtarget lesion revascularization at 6 months (25.6% v. 50.6%,p=0.001). This beneficial effect was largely driven by a reductionin recurrent ischemic events (4.9% v. 28.4%, p=0.01). On anintention-to-treat basis, immediate stenting was also associatedwith a reduction in the 6-month combined end-point of death,reinfarction, and target vessel revascularization (27.7% v. 39.8%,p=0.049). Patients with stenting showed an improvement in LVfunction at 2 weeks with further improvement at 6 months,whereas the delayed stenting group (group II) showed nostatistically significant improvement in LV function. Majorbleeding complications occurred in 9.8% of patients whounderwent immediate stenting versus 7.4% in the delayed stentinggroup (p=0.374). There were 5 deaths in group II within 48 hoursof thrombolysis, whereas all the patients in group I survived theacute phase. Immediate stenting after thrombolysis is significantlymore beneficial compared to delayed stenting.
Comments
Thrombolysis results in TIMI-3 flow in only 60% of patients withacute MI. Moreover, around a third of those thrombolyzed haverecurrent ischemia. Numerous studies have shown that primaryPCI is superior to pharmacologic thrombolysis with higherreperfusion rates, and improved event-free survival. The maindrawback remains the immediate availability of centers with thefacility for PCI. Hence, in a real-world situation, a considerabletime delay is usually encountered in transportation of the patient,and mobilization of appropriate resources. As we are aware, time
is all-important in salvage of the myocardium. The ACC/AHArecommends a door-to-balloon time of 90±30 min, but in a day-to-day situation, this is usually not possible. In the NationalRegistry of Myocardial Infarction (NRMI), there was a mediantreatment time delay of >2 hours in almost 90% of patients. Inthe recently published results of the DANAMI-2 study, there wasa lower composite end-point of death, MI or stroke at 30 days inpatients who were transferred from community hospitals totertiary care centers with intervention facilities, compared withpatients receiving on-site thrombolysis (8.5% v. 14.2%, p=0.002).A similar benefit has been seen in the PRAGUE study, as well asthe AIR-PAMI study. Though these trials have shown that the timedelay in PCI is offset by the superiority of the results of angioplastyover thrombolysis, the door-to-balloon time remains important.In the NRMI registry, a door-to-balloon time >2 hours wasassociated with an increase in mortality. Numerous fibrinolysistrials have shown that the time from the onset of symptoms tovessel recanalization is a predictor of myocardial salvage andsurvival. Given the lack of widespread availability of PCI centers,alternative approaches with a combination of pharmacologic andmechanical revascularization methods are being tried. Therationale behind facilitated PCI was to improve outcomes byattempting to achieve earlier reperfusion while awaiting definitiveintervention at a center with PCI facility. This combinationapproach is an attempt to buy time, and widen the therapeuticwindow for revascularization. Moreover, patients have a betterTIMI flow grade and lesser thrombus burden, thus reducing thechances of microvascular embolization, improving lesion focus,and improving the overall results. However, earlier attempts withthis complementary therapy were not encouraging. Data from theThrombolysis and Angioplasty in Myocardial Infarction (TAMI-1) study, and the European Cooperative study group showed nobenefit in survival or ventricular function, and were in factassociated with a higher complication rate. However, thedevelopment of bolus thrombolytics with enhanced fibrinsensitivity, and excellent safety and efficacy profiles, along withimprovement in PCI hardware, including thrombectomy anddistal protection devices, have led to a resurgence in the use offacilitated PCI.
In this trial, the time to angiography was 6.7 hours, exceedingthe currently recommended time. But the setting was a real-worldsituation that would be commonly encountered in practice. In theSIAM III study, over 60% of patients in the immediate stentinggroup had TIMI-3 flow; this figure increased to 98%. Thistranslated into an improved LV function at 2 weeks and 6 months.Despite the long time taken till intervention, there was a clearbenefit, and no increase in the complication rates. In conclusion,this trial has proved the relative superiority of thrombolysisfollowed by immediate stenting, despite the time involved intransfer to the tertiary care center. The use of Gp IIb/IIIaantagonists would further improve the outcome of immediatestenting. In conclusion, immediate stenting after thrombolysis issafe, and improves event-free survival as well as ventricularfunction compared with elective stenting at 2 weeks following MI.
IHJ-Selected Summary.p65 11/19/2003, 11:27 PM388

Indian Heart J 2003; 55: 386–389 Selected Summaries 389
Summary
The SIRIUS investigators conducted a randomized double-blind trial in 1058 patients with a newly diagnosed singlelesion in a native coronary artery, comparing a sirolimus-eluting stent with a standard stent. Although previous trialshave shown a reduction in restenosis rates with sirolimusstents, the patients enrolled were at a lower risk forrestenosis with discrete lesions. This study enrolled patientswith more challenging coronary lesions with the majority(56%) having class B
2 or C lesions (ACC/AHA classi-
fication). The average vessel diameter was 2.8 mm, and themean lesion length was 14.4 mm. There were 533 patientsin the sirolimus stent group, and 525 in the standard stentgroup. The groups were well matched with respect todemographical characteristics, cardiac risk profile,symptomatology, lesion anatomy, and procedural factor.Follow-up angiography (performed in around 85% ofpatients in both groups) showed that the minimal luminaldiameter stenosis, and the late lumen loss in the in-stentzone as well as the in-segment zone were significantly(p=0.001) better in the sirolimus stent group.
There was a higher rate of in-stent restenosis and in-segment stenosis (35.4% and 36.3%, respectively) in thestandard stent group compared with the medicated stentgroup (3.2% and 8.9%, respectively). Intravascularultrasound revealed a reduced neointimal volume in thein-stent zone (4.4 mm3 v. 57.6 mm3, p<0.001). Theprimary end-point of target vessel failure rate (compositeof death from cardiac causes, myocardial infarction, andrepeat revascularization) within 270 days was reduced by58% with sirolimus stents (110 patients v. 56 patients,p<0.001). This reduction was driven largely by a decreasein the frequency of the need for revascularization of thetarget vessel (16.6% v. 4.1%, p<0.001)). Diabetes wassignificantly associated with an increased risk of restenosisalong with reference vessel diameter and lesion length.However, sirolimus stents showed a consistent benefit evenin these high-risk subgroups as compared with standardstents.
Comments
Sirolimus (rapamycin) is a macrolide antifungal withpowerful immunosuppressant properties; it inhibits severalregulators of cell-cycle progression, and the migration ofvascular smooth muscle cells. This unique property led toits use in coronary artery stents to prevent neointimalproliferation. The Randomized Study with the SirolimusEluting Bx Velocity Balloon Expandable Stent (RAVEL)showed a significant reduction in the frequency of in-stentrestenosis (from 26.6% to 0%). The present study enrolledpatients with a higher frequency of cardiac risk factors,such as diabetes and hypercholesterolemia, more complexmorphology, and longer lesions. In the RAVEL study, therewere no type C lesions, and the mean lesion length was9.58±3.25 mm compared with 14.4±5.8 mm in thepresent study. Similarly, only 19% of the patients haddiabetes and 40% dyslipidemia compared with 26% and74% in this study, respectively. Intravascular ultrasoundrevealed a 92% reduction in neointimal volume, a 77%reduction in the rate of target lesion revascularization, andan 85% reduction in the rate of non-QMI. The highfrequency of in-segment restenosis compared to in-stentrestenosis (8.9% v. 3.2%) in the sirolimus stent group wasdue to a higher rate of restenosis at the proximal marginof the stent, and is attributed to balloon injury to the vesselprior to stent deployment. The authors recommend the useof shorter balloons and longer stents that would have noexposed balloon-injured area. The restenotic lesions werefocal compared with diffuse disease with standard stents,and hence more amenable to complex balloon angioplasty.The encouraging results from the trial suggest that specialstents may maintain their superiority over standard stentsin a diverse patient population. As sirolimus-coated stentsmove into general use, and with a longer follow-up, it islikely that the rate of restenosis will be higher. This trialhas shown the beneficial effect of sirolimus-coated stentsin a population at a higher risk, and with more complexdisease than previous studies.
Sirolimus-Eluting Stents Versus Standard Stents in PatientsWith Stenosis in a Native Coronary Artery
Moses et al. SIRIUS Investigators. N Engl J Med 2003; 349: 1315–1323
IHJ-Selected Summary.p65 11/19/2003, 11:27 PM389

Calendar of Conferences Indian Heart J 2003; 55: 390
November 14–15, 2003, 4th Annual Conference ofNuclear Cardiological Society of India, Vellore, IndiaContact: Organizing Secretary
Department of Nuclear MedicineChristian Medical CollegeVellore, Tamil Nadu, IndiaFax: 0416 2232103e-mail: [email protected]
December 4–7, 2003, 55th Annual Conference ofCardiological Society of India, Kolkata, IndiaContact: Dr Asok Kumar Kar, Organizing Secretary
Indian Heart HouseP-60, CIT Road, Scheme VIIM,Kankurgachi, Kolkata 700054, IndiaFax: 033 355 6308e-mail: [email protected]
January 9–11, 2004, Joint Meeting of InternationalSociety for Heart Research and InternationalAcademy of Cardiovascular Sciences, Lucknow, IndiaContact: Professor VK Puri, Organizing Secretary
Department of CardiologyCSM Medical UniversityLucknow, IndiaFax: 0522 225 5830e-mail: [email protected]
May 6–9, 2004, 4th Congress of the Asian PacificSociety of Atherosclerosis and Vascular Disease, Bali,IndonesiaContact: Dr Slamet Suyono, MD
Pacto Convex Ltd, Lagoon Tower Level B-1Jagarta Hilton InternationalJI Gatot Subroto, Jakarta 10270, IndonesiaFax: 62 21 5705798e-mail: [email protected]
November 7–10, 2004, 77th Scientific Session,American Heart Association (AHA), New Orleans,Louisiana, USAContact: American Heart Association
7320 Greenville AvenueDallas TX 75231, USATel: 1 214 373 6300Fax: 1 214 373 3406
November 26–28, 2004, 3rd International Congresson Cardiovascular Disease, Taipei, TaiwanContact: Dr CE Chiang
Taiwan Society of Cardiology7F, No. 27, Min-Chuan, West RoadTaipei 104, TaiwanFax: 886 2 25076180e-mail: [email protected]; [email protected]
November 28–30, 2004, 10th World Congress onClinical Nutrition, ThailandContact: Dr Buncha Ooraikul
or Dr Tapan Basu, Department of Food Scienceand Nutrition, University of Alberta, EdmontonCanada T6G 2MFax: 403 4924821e-mail: [email protected]
May 21–25 2005, 4th International Conference onPreventive Cardiology, Foz do Iguassu, BrazilContact: Dr Mario Maranhao, Chairman, Congress
Internationales SA/Lidy Groot Congress EventsP.O. Box 83005 1080, AA AmsterdamThe NetherlandsFax: 3120 6758236e-mail: [email protected]
Cal of Conference.p65 11/19/2003, 11:22 PM390


















