Bronchial Asthma(Jaypee)
347
-
Upload
india2puppy -
Category
Documents
-
view
75 -
download
10
description
Indian Textbook on Bronchial Asthma by the Jaypee Publishing Company.
Transcript of Bronchial Asthma(Jaypee)


Severe Asthma (Fatal Asthma) 1
BronchialAsthma

BronchialAsthma
D BeheraMD (Medicine) FCCP FNCCP FICP FICA MNAMS (Medicine)
Dip. NBE (Respiratory Medicine)
ProfessorDepartment of Pulmonary Medicine
Postgraduate Institute of Medical Education and ResearchChandigarh (India)
JAYPEE BROTHERSMEDICAL PUBLISHERS (P) LTD
New Delhi
Second Edition

Published by
Jitendar P VijJaypee Brothers Medical Publishers (P) LtdEMCA House, 23/23B Ansari Road, DaryaganjNew Delhi 110 002, IndiaPhones: +91-11-23272143, +91-11-23272703, +91-11-23282021,+91-11-23245672Fax: +91-11-23276490, +91-11-23245683 e-mail: [email protected] our website: www.jaypeebrothers.com
Branches• 202 Batavia Chambers, 8 Kumara Krupa Road
Kumara Park East, Bangalore 560001, Phones: +91-80-22285971,+91-80-22382956, +91-80-30614073 Tele Fax: +91-80-22281761e-mail: [email protected]
• 282 IIIrd Floor, Khaleel Shirazi Estate, Fountain PlazaPantheon Road, Chennai 600008, Phones: +91-44-28262665,+91-44-28269897 Fax: +91-44-28262331e-mail: [email protected]
• 4-2-1067/1-3, Ist Floor, Balaji Building, Ramkote Cross RoadHyderabad 500095, Phones: +91-40-55610020, +91-40-24758498Fax: +91-40-24758499 e-mail: [email protected]
• 1A Indian Mirror Street, Wellington SquareKolkata 700013, Phone: +91-33-22451926 Fax: +91-33-22456075e-mail: [email protected]
• 106 Amit Industrial Estate, 61 Dr SS Rao RoadNear MGM Hospital, Parel, Mumbai 400012Phones: +91-22-24124863, +91-22-24104532, +91-22-30926896Fax: +91-22-24160828 e-mail: [email protected]
Bronchial Asthma
© 2005, D Behera
All rights reserved. No part of this publication should be reproduced, stored in aretrieval system, or transmitted in any form or by any means: electronic, mechanical,photocopying, recording, or otherwise, without the prior written permission of theauthor and the publisher.
This book has been published in good faith that the material provided by author isoriginal. Every effort is made to ensure accuracy of material, but the publisher,printer and author will not be held responsible for any inadvertent error(s). In case ofany dispute, all legal matters are to be settled under Delhi jurisdiction only.
First Edition: 2000Second Edition: 2005
ISBN 81-8061-434-4
Typeset at JPBMP typesetting unitPrinted at Gopsons Papers Ltd, A-14, Sector 60, Noida 201 301, India

Dedicated tothe loving memory of
my distinguished teacherlate Dr SK Malik

Foreword
The prevalence of bronchial asthma, a major public health problem is increasing worldwide.Several studies have demonstrated that there is an increase in morbidity and mortality frombronchial asthma. Over and under treatment of asthma may be responsible for high mortalityrates. Until recently bronchospasm that results from hyperresponsiveness of the airways tomultiplicity of stimuli has been regarded as the main cause of airway dysfunction in asthma.Bronchial asthma is now considered as a chronic inflammatory disease of the airways. Thisrealization that inflammation is the key factor in the pathogenesis of asthma is reflected in thechange in asthma therapy with emphasis on inhaled anti-inflammatory drugs. There aremany controversies in the management of bronchial asthma especially the role ofimmunotherapy. Many new drugs are under development and yet there is no cure for asthma.
In a country like India with different socio-cultural diversities and beliefs, the treatment ofasthma varies and the existence of different systems of medicine in our country complicatesthe treatment issues. Prof D Behera, a renowned Pulmonologist of our country and Professorof Pulmonary Medicine at the Postgraduate Institute of Medical Education and Research,Chandigarh has taken up the challenge of bringing out the updated second edition of hisbook, “Bronchial asthma”. The tremendous response to the first edition of his book is atestimony to the academic excellence of this book. The second edition has 21 chapters includingepidemiology, pathophysiology, clinical presentation, complications, management andvarious guidelines. This revised edition is a comprehensive review of bronchial asthma andprovides practical information for Physicians and Pulmonologists who have to takeappropriate diagnostic and therapeutic decisions in patients with bronchial asthma.I congratulate Dr Behera for his tireless efforts to bring out the second edition of this book.
VALLABHBHAI PATEL CHEST INSTITUTEUNIVERSITY OF DELHI, P.O. BOX NO. 2101
DELHI-110 007, INDIA
Dr. V.K. VijayanMD (Med), Ph D (Med), D Sc, FAMSFCAI, FNCCP (I), FICC, FCCP (USA)Director
Tel. (O) : 91-11-7666180(R) : 91-11-7667027
Fax : 91-11-7667420E-mail : [email protected]
Date: ...........................July 8, 2004
Dr VK VijayanDirector

Preface to the Second Edition
Bronchial asthma is a common respiratory disorder affecting approximately3-5 percent of the population, although there is a wide variation in itsprevalence in the world, even in the same country at different parts. Over theyears our understanding about the disease has changed. One of the majorchanges in our thinking about the pathophysiology of the disease is that thedisease is inflammatory in nature rather than the earlier simplistic view of itbeing a simple bronchospastic disorder. A number of cytokines and mediatorstake part in its causation. Accordingly the approach to management ofasthma has also changed. A number of guidelines have come up in recentyears and there is a constant renewal in some of the concepts. Althoughthere is no guideline for adult Indian patients, the same is given for children.The chapter on bronchial asthma in children is not complete in all aspects,but it will give a brief account of the same for the pulmonary physician. Thisedition has brought out some of these changes. Further, the references areupdated with Vancouver style.
D Behera

Preface to the First Edition
Bronchial asthma is a common disease affecting nearly 3 to 5 percent of thepopulation. Although incidence- and prevalence-wise the disease is not morecommon than tuberculosis in this country, the major difference is its recurringnature with periods of remissions and exacerbation. In some cases life long,and in many cases most of the times, medications with anti-asthma drugswill be required for symptom-free life. This is a major contrast to tuberculosiswhere treatment for 6 to 9 months will cure the disease. Earlier conceptsabout bronchial asthma, that it is a bronchospastic disease, have changed inrecent years, wherein it is proved that it is an inflammatory disease. A widearray of cells with a number of cytokines take active role in the patho-physiology of the disease.
The idea of writing this book came to my mind while I was preparing forthe second edition of my textbook entitled Pulmonary Medicine. I thought achapter on Bronchial Asthma in a textbook may not give sufficient justificationto cover the explosion of recent knowledge acquired about the disease,particularly our understanding of its pathophysiology and approach tomanagement.
D Behera

1. Epidemiology ........................................................................................................................ 1
2. Aetiology ............................................................................................................................... 14
3. Pathophysiology of Bronchial Asthma ............................................................................ 40
4. Pathology .............................................................................................................................. 86
5. Clinical Presentation of Bronchial Asthma ..................................................................... 92
6. Diagnosis of Bronchial Asthma ........................................................................................ 98
7. Prognosis of Bronchial Asthma ...................................................................................... 114
8. Complications of Bronchial Asthma .............................................................................. 117
9. Management of Bronchial Asthma ................................................................................ 127
10. Pharmacologic Management of Asthma ....................................................................... 134
11. Inhalation Therapy ........................................................................................................... 176
12. Therapeutic Approach in Patients with AsthmaI. Chronic Bronchial Asthma ........................................................................................... 183
13. Therapeutic Approach in Patients with AsthmaII. Acute Severe Asthma (SA) ......................................................................................... 208
14. Management of Asthma with Special Problems ......................................................... 235
15. New Treatment Modalities/Newer Drugs for Bronchial Asthma ............................ 247
16. New Guidelines for Asthma Management(Non-pharmacological Management) ............................................................................ 256
17. New Guidelines for Asthma Management (Pharmacological Management) ........ 265
18. New Guidelines for Asthma Management (Acute Asthma) ..................................... 276
19. Alternate Treatments in Asthma .................................................................................... 293
20. Severe Asthma (Fatal Asthma, Refractory Asthma) .................................................... 306
21. Asthma in Children .......................................................................................................... 314
Index ..................................................................................................................................... 337
Contents

DEFINITION
Asthma is a disease whose presence dates back to at least the time of Hippocrates whonoted a condition of ‘deep and heavy breathing’. The Greeks had labelled this condition as“asthma”, the meaning of which was panting. Bronchial asthma is difficult to define since itis not one homogenous condition and because there is no one objective measurement orseries of measurements that can be used to make the diagnosis of asthma. A widelyacceptable definition still remains elusive ever since it was first defined in 1959 by an expertstudy group during the CIBA Foundation Guest Symposium.1 The Global Initiative forAsthma (1995) defines asthma on the basis of its pathogenesis (vide infra). The clinician,immunologist, physiologist, and pathologist all have their own perspective of asthma, andall these perspectives are difficult to merge into a comprehensive definition sufficientlyspecific to exclude other diseases. Earlier definitions were non-specific and therefore thecondition was both under and over-diagnosed.2,3 However, during the past one-decadethere have been major changes in the concepts of pathophysiology of asthma. Whereas thecondition was previously considered as a bronchospastic disorder only, it is now recognisedthat asthma is primarily an inflammatory disease. The current definition incorporates bothof these components and a generally agreed-on working definition of asthma is as follows:4
“Bronchial asthma is a disease characterised by (i) airway obstruction (airway narrowing)that is reversible (but not completely so in some patients) either spontaneously or withtreatment; (ii) airway inflammation; and (iii) airway hyperresponsiveness to a variety ofstimuli”.
Subsequently, the Consensus Report5 describes asthma as a “Chronic inflammatorydisorder of the airways in susceptible individuals, inflammatory symptoms are usuallyassociated with widespread but variable airflow obstruction and an increase in airwayresponse to a variety of stimuli. Obstruction is often reversible, either spontaneously orwith treatment.”
PREVALENCE
The prevalence of asthma is not exactly known. This is because the precise way how todefine asthma in population studies is defined differently. Questionnaires are the mostpractical tools to use in screening population for asthma. Such questionnaires have beenvalidated to assess the ability of individual questions and combination of questions to predictwhich individuals in the population have either clinical diagnoses of asthma or non-specificbronchial hyperreactivity to agents like methacholine or histamine.6 Unfortunately,
Epidemiology
1

2 Bronchial Asthma
physician-diagnosis of asthma and bronchial hyperreactivity are not particularly good “goldstandards” for identifying asthma. While the former can miss milder forms of asthma, thelater is present in many people without asthma.6-8 To avoid these limitations, many studiesnow use questionnaires.6,9-11 In general, questions about “ever having asthma”, “ever havingasthma diagnosed by a physician”, and “having wheezing during the previous 12 months”have been the questions with best sensitivity and specificity for prediction of the flawedgold standards. These questionnaires, of course are being used most often in recent studies.However, earlier surveys will have flaws as mentioned, and the difference prevalence ratesin different studies in the past can be explained in part due to these methodologicaldifficulties. Nonetheless, in many countries, the prevalence of asthma has increased in recentdecades.12,13
The disease has reached epidemic proportions affecting 155 million individuals in theworld. About 15% (one out of seven) of children in United Kingdom wheeze and similarnumber suffers from the related disorders of atopic dermatitis. The prevalence has risenover the past 30 years all over the world particularly in all Westernised societies perhaps asa result of the loss of childhood infections.14 While asthma is one of the less common causesof death, the magnitude of the problem is evident from the fact that during a 10 yearsperiod from 1978 to 1987, there were 1,87,000 deaths in USA, Canada, England, Wales,France, West Germany, and Japan.15,16 Since the definition of asthma was varying, theavailable statistics is viewed with some skepticism. In general, it seems that asthma remainsunder diagnosed especially during childhood. There is some evidence that bronchial asthmais increasing in a number of countries particularly New Zealand, UK and USA.15,17 Anestimated 10 million persons in the USA had asthma. In the general population, asthmaprevalence rates increased 29% from 1980 to 1987.
Bronchial asthma is the most common chronic respiratory disorder among all age groupswith a reported prevalence of 5 to 10%.18 During the last decade, studies from differentcountries keeping appropriate statistics have reported a significant rise in asthma morbidityand mortality.18-28 In the United States, approximately 17 million people have asthma (andasthma related symptoms) account for 10 million missed school days, > 1.5 millionemergency department visits, approximately 500,000 hospitalisations and > 5000 deathsannually. In 1998, the direct and indirect expenditures for the treatment of asthma in theUnited States were approximately $11.3 billion.29 The overall 1988 asthma death rate was1.9/100,000 persons with much lower rates in persons younger than 45 years, risingdramatically with increasing age.18-30 Asthma is the most common chronic disease of childrenin USA.31,32 About 6 million children in the United States have asthma compared to 3.1 millionin 1984, an increase of 80%. Annually, asthma accounts for 12 million primary care visits,1.6 million emergency department visits, 11 million missed school days, 200,000 hospitaladmissions, and 150 paediatric deaths.33 Improved personal behaviour and medical carehave a limited sustained impact on childhood asthma until basic environmental issues aremodified.34 Various other statistics also prove that both asthma and allergic rhinitis haveincreased in recent years. The effect of these disorders on children and adults is considerablein terms of morbidity and lost productivity resulting from the disease and its treatment .35,36
In addition, hospitalisation due to asthma and deaths attributed to asthma are increasing,despite the availability of effective drugs.37 From 1982 to 1992, the overall annual age-adjustedprevalence rate of self reported asthma increased 42% (from 34.7 per 1,000 people to 49.4

Epidemiology 3
per 1,000 people). Even more alarming is the observation that during this period, the overallannual age-adjusted death rate for asthma increased 40%.38
One disadvantage with these statistics is that these are based on informations obtainedby questionnaire and in most cases identical questions were not used at each survey.39
However, from available data, both morbidity and mortality from asthma in New Zealandare amongst the highest in the world.40 A survey of 12-year-old school children carried outin New Zealand and South Wales41 revealed a higher prevalence in the former (17%) thanin the later (12%). New Zealand children were also more likely than the Wales children tohave a history of “wheeze ever” (27% vs. 22%) and wheeze brought on by running (15% vs.10.5%). The sex ratio of asthmatic and wheezy children was very similar in the two countries.The overall prevalence of asthma is estimated at 13.7%, bronchial hyperresponsiveness at13.4%, and atopy at 31.1% in the age range of 13 to 18 years. The prevalence of bronchialhyperresponsiveness in those without asthma symptoms is 3%. Both current asthmasymptoms and bronchial hyperresponsiveness are more common among females. In a studyto determine the prevalence of asthma in cohorts of Finnish young men in the period 1926-1989, Haahtela et al42 found that during 1926-1961 the prevalence was steady at between0.02 and 0.08%. Between 1961 and 1966, however, a continuous, linear rise began, theprevalence increasing from 0.29% in 1966 to 1.79% in 1989, that is, representing a six-foldincrease. The rise is 20 folds compared with that in 1961. Much of this increase appears realand not merely due to an improvement in the methods of diagnosis over these years. Areview of the available published figures for children in United Kingdom revealedprevalence for “wheeze in the previous year” of between 4.9 and 15% and “wheeze ever”between 9.9 and 24.9%. Figures for “asthma ever” varied between 1.2 and 5%.
A simple flow diagram of the natural history of asthma17 based on the prevalence ofchildhood wheeze in Australia is shown in Figure 1.1.
Fig.1:1. Natural history of bronchial asthma in Australian children. Thehatching represents the approximate percentages in each group who areatopic and who have bronchial hyperresponsiveness. The top line indicatesthe group who are atopic and who wheeze while the bottom line representsthose without evidence of (a) allergy; (b) wheeze; (c) and persistent wheeze

4 Bronchial Asthma
The Figure 1.1 also shows the approximate number of people entering adult life withpersistent wheeze. This study showing natural history of asthma is based on the prevalenceof atopy as measured by skin tests and the prevalence of childhood wheeze in Australia. Anumber of studies from around the world show that the prevalence of atopy is between 30-50% in children.43-46 In addition, the number of children who have wheezed at sometime isaround 25-30%.47-49 Most children with persistent wheeze are atopic.50,51 About 7% of thepatients have persistent asthma as reported from Australia by Woolcock et al.18
Adequate prevalence data from most developing countries is not available either for childrenor adults. Although it is a general perception that bronchial asthma is a very common problemin India, apart from tuberculosis, authentic information is not available regarding itsprevalence or incidence. Whatever data is available, it lacks the uniformity of definition,problems of sample size, and analytical methodology used. From different studies, theprevalence of asthma has been reported to be 1.2 to 6.2% in adults in the western world. In a survey of respiratory symptoms in India, the prevalence of asthma has been reportedto be 0.6 and 3.2% in rural and urban women respectively. The same in urban males has been4%.52-55 The prevalence was reported to be 1.76% in an urban population in the mid sixties.56
It was also reported by the same investigators that the prevalence in the morbidity surveys ofgovernment employees and their families in Delhi was 1.8%.56 However, in recent years twostudies from Mumbai and Northern India are available.57,58 The study from Greater Mumbairevealed a prevalence of 3.5% by physician diagnosis and 17% using a very broad definitionincluding those with asymptomatic bronchial reactivity. Prevalence of asthma in Mumbaiwas similar in males and females (3.8 and 3.4% respectively). In the North Indian survey, avalidated questionnaire was used tested against physician—diagnosed asthma and theprevalence in the population was assessed.58 The true population prevalence was reported as3.94% in urban and 3.99% in rural males and 1.27% in both urban and rural females. A recentstudy from Delhi59 estimated the risk of asthma in children to be very high.59 Prevalence ofasthma symptoms in children was determined in the International Study of Asthma andAllergies in Childhood (ISAAC) in the age groups of 6-7 and 13-14 years using a standardisedsample survey.60,61 Prevalence of “ever asthma” varied from 1.8 to 12.4% with an overall figureof 4.5%. The figure of “ever asthma” in 12 months is not strictly same as prevalence of asthmain adults. The overall prevalence of asthma in children of 10-18 years age at Chandigarh was2%, using the same methodology as in adults.58,62
Since morbidity depends, at least partly, on prevalence, the trends should be similar.Other indices of morbidity such as days lost from work and restriction in lifestyle, nocturnaldisturbances with symptoms and hospital admission rates confirm the trends and extent ofproblem due to asthma. It is clear that the most dramatic increase in admission to hospitalshas been in children. All the data collected on the basis of above informations indicatecontinuing extensive morbidity from asthma, although more effective treatment may bemodifying this.
MORTALITY
Statistics for deaths from asthma yield widely variable mortality rates between countries.15
Increasing asthma mortality was first highlighted in the early-mid 1960’s63,64 when therewas a dramatic increase in asthma deaths in England and Wales, Australia and New Zealand.This was most apparent in children 10-14 years, but was also apparent for all age groups,
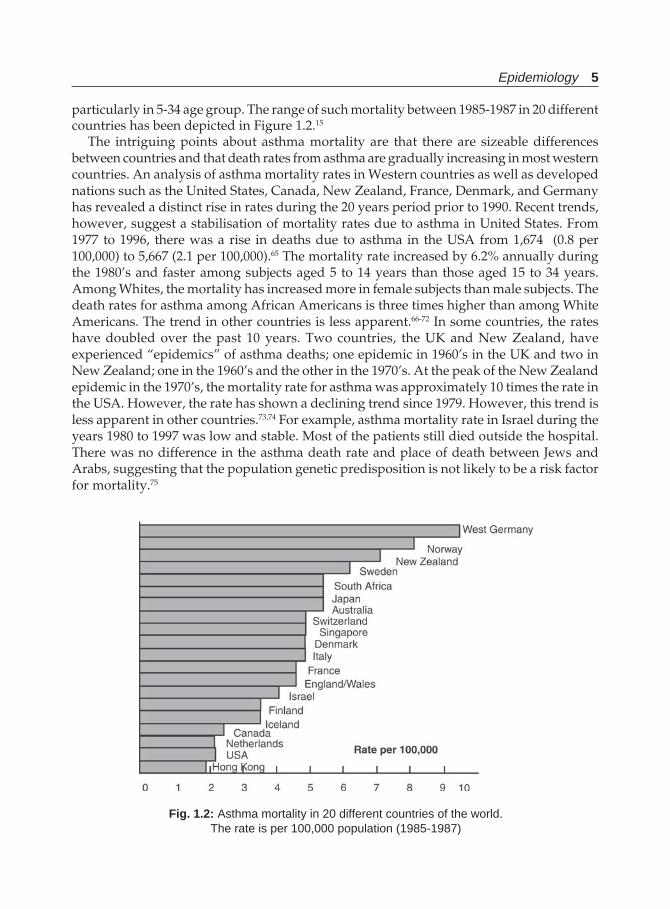
Epidemiology 5
particularly in 5-34 age group. The range of such mortality between 1985-1987 in 20 differentcountries has been depicted in Figure 1.2.15
The intriguing points about asthma mortality are that there are sizeable differencesbetween countries and that death rates from asthma are gradually increasing in most westerncountries. An analysis of asthma mortality rates in Western countries as well as developednations such as the United States, Canada, New Zealand, France, Denmark, and Germanyhas revealed a distinct rise in rates during the 20 years period prior to 1990. Recent trends,however, suggest a stabilisation of mortality rates due to asthma in United States. From1977 to 1996, there was a rise in deaths due to asthma in the USA from 1,674 (0.8 per100,000) to 5,667 (2.1 per 100,000).65 The mortality rate increased by 6.2% annually duringthe 1980’s and faster among subjects aged 5 to 14 years than those aged 15 to 34 years.Among Whites, the mortality has increased more in female subjects than male subjects. Thedeath rates for asthma among African Americans is three times higher than among WhiteAmericans. The trend in other countries is less apparent.66-72 In some countries, the rateshave doubled over the past 10 years. Two countries, the UK and New Zealand, haveexperienced “epidemics” of asthma deaths; one epidemic in 1960’s in the UK and two inNew Zealand; one in the 1960’s and the other in the 1970’s. At the peak of the New Zealandepidemic in the 1970’s, the mortality rate for asthma was approximately 10 times the rate inthe USA. However, the rate has shown a declining trend since 1979. However, this trend isless apparent in other countries.73,74 For example, asthma mortality rate in Israel during theyears 1980 to 1997 was low and stable. Most of the patients still died outside the hospital.There was no difference in the asthma death rate and place of death between Jews andArabs, suggesting that the population genetic predisposition is not likely to be a risk factorfor mortality.75
Fig. 1.2: Asthma mortality in 20 different countries of the world.The rate is per 100,000 population (1985-1987)

6 Bronchial Asthma
All statistics shown are derived from published population and mortality data availablefrom the national statistics centres in each country. West Germany reported over 9 deathsper 100,000 followed by Norway, New Zealand, and Sweden. Netherlands, USA, and HongKong reported asthma mortality rates less than 2/100,000.
The reasons for the trends in mortality due to asthma and for the sizeable differencesbetween countries are not clear.76-79 The increase in mortality in most countries cannot beprimarily due to an increase in the prevalence of asthma as the rise in mortality isdisproportionately greater than that of the prevalence.80 In the last decade, though, thestabilisation of mortality, and even a decrease in mortality, from asthma has been reported.81-84
A number of reasons have been proposed including: (i) Partial contribution from the shiftof International code of death (ICD-8 to ICD-9). Due to this, the term asthmatic bronchitiswas coded as asthma rather than bronchitis; (ii) Shifts in physician diagnosis patterns,especially from bronchitis to asthma in the 0-5 years age group and from COPD to asthmain smokers past middle life. There is clearly some misclassification of asthma deaths withover-reporting over age 50 and under-reporting in the younger age groups; (iii) An increasein the prevalence and or severity of asthma; (iv) Increased diagnosis of asthma; and(v) Adverse drug effects. In the 60’s overuse of adrenaline in Europe and currently the useof fenoterol have been postulated to be contributory to the mortality due to asthma. However,these postulates have not been confirmed.85-89 Other possible contributors are delay in care,poor compliance, lack of access to health care, theophylline toxicity, besides the overuse ofβ-agonists.90-92 Most likely cause of the recent decline in asthma deaths is the more judicioususe of prophylactic treatment, particularly inhaled steroids, as a possible factor.93,94 Raceand socioeconomic status may influence the outcome of an asthma attack.95,96 Hospitaladmission rates for asthma are high and have increased in the last few decades.97,98 However,some patients die before they can receive medical care.98-100 The exact proportion of deathsoccurring outside the hospital and its association with genetic, environmental or socialfactors is not clear. An estimated 15 million persons in the United States have bronchialasthma, and the number is increasing. Although asthma is generally treated in an outpatientbasis, increased hospitalisation rates have been observed. Hospitalisation rates and episodesof asthma have increased in all age groups particularly in boys up to 4 years old.101
Hospitalisation rates are twice as common in African Americans as White Americans.102
Causes for the Increase in Asthma Mortality
Besides the above mentioned reasons, many other causes have been advocated for theincrease in asthma mortality and morbidity and they include allergen exposure, air pollution,medication use, inadequate access to health care, increased incidence of viral infections,and physician management of asthma (Discussed subsequently).
The risk of death due to asthma appears to predominate in large urban areas with highrates of poverty. Risk of hospitalisation for children with asthma is 8.4 times greater inareas with population of lower socioeconomic status and 5.3 times greater in areas with alarger African American population.103 Lower socioeconomic status and African Americanrace are strong risk factors for hospitalisation and mortality from asthma.
NATURAL HISTORY OF BRONCHIAL ASTHMA
Over the last few decades the natural evolution of asthma from childhood to adulthood hasbeen the subject of many reviews and studies and more than 50 such well-designed

Epidemiology 7
publications highlight the subject.104 It was long believed that the prognosis for asthmaoriginating in infancy or childhood was good, and that in most patients the symptoms wouldresolve by the age of puberty. However, a review of literature shows that not all patientsbecome asymptomatic in adulthood. In fact, asthma symptoms persist in 30-80% of adultpatients. Although epidemiological studies have shown a fair chance of either “remission” ora reduction in asthma symptoms between the ages of 10 and 20 years,105-108 and most populationbased and clinical studies have also shown a reduction in asthma symptoms with age, therelapse rates after a symptom-free interval is also high.107,109 It has also been shown that, evenin the absence of asthma symptoms, subjects may still have obstructive lung functions andincreased bronchial hyperresponsiveness.110-116
No definite information is available about the progression of asthma through childhoodand adolescence.117 Martinez118 studied the natural history of wheezing in children aged0-6 years and found that approximately half of the children experienced wheezing illness atsometime during the study period. In some of them wheezing occurred early in life but resolvedby the age of three years (transient early wheezing), some experienced wheezing illness betweenthe ages of three and six years (late onset wheezing) and others had wheezing illness throughoutthe entire study period (persistent wheezing). Different risk factors were associated with theseresults. Children with transient early wheezing had reduced pulmonary function, as measuredby functional residual capacity shortly after birth and before any lower respiratory tractillness had occurred. The risk was also increased in children whose mothers smoked duringpregnancy. Congenitally smaller airways may therefore predispose children to wheezingillness early in life. Children with late and persistent wheezing are more likely to be atopic asassessed by elevated serum IgE levels and skin test reactivity, asthmatic mothers, and theirlung function decreased after the age of one till they are six years of age. This study suggeststhe presence of two distinct wheezing illnesses up to the age of six years. As discussed above,there are varying reports about the persistence/disappearance of asthma symptoms inadolescence. However, some reports suggest that up to 80% of asthmatics may becomeasymptomatic during puberty.119,120 In a cohort study of Australian school children121 testedinitially at the age of 8-10 years and then again at 12-14 years of age, the persistence ofbronchial hyperresponsiveness at 12-14 years of age was found to be related to the severity ofdisease at 8-10 years of age, the atopic status of the child, and parental history of bronchialasthma. Most of the children who had a slight or mild degree of bronchial hyperresponsivenessat 8-10 years of age lost their increased response by the age of 12-14 years. Only 15.4% ofchildren with severe or moderate bronchial hyperresponsiveness at the initial assessmenthad normal levels of bronchial responsiveness at the later assessment. Whether the decline inreported symptoms is real or the result of the children increasingly denying their illness asthey reach puberty remains to be clarified. The reduced bronchial responsiveness may favourthe hypothesis of a real reduction in the activity of the disease or higher doses of the provocativeagents like histamine or methacholine may be needed to provoke hyperresponsiveness inlarger airways of rapidly growing children.
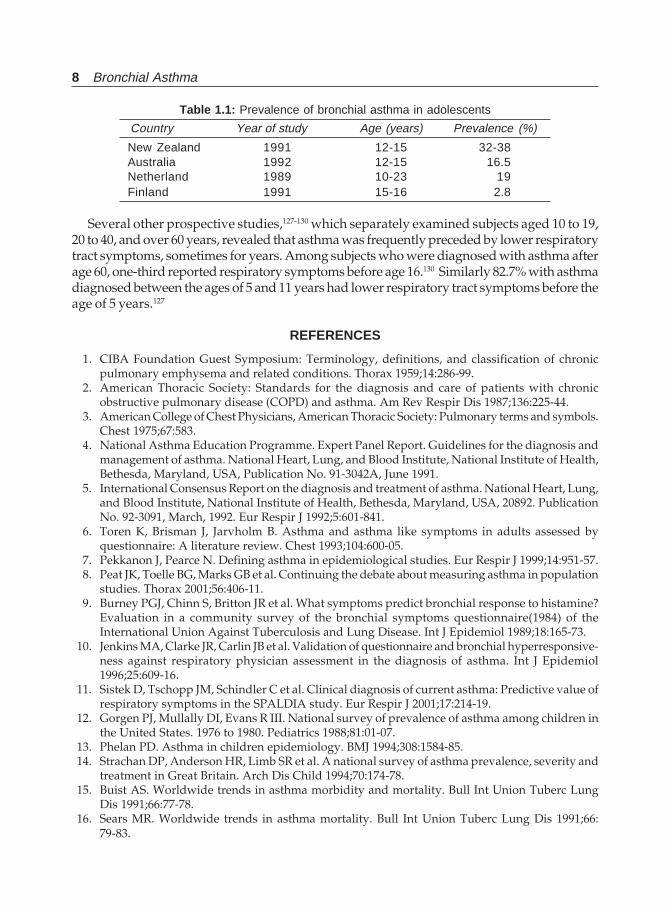
As against the above figures, the prevalence of bronchial asthma in adolescents in 4 differentcountries122 varied from 2.8 to 38% at different ages and is summarised inTable 1.1.123-126 This shows a significant number still will have asthma in adolescence.
8 Bronchial Asthma
Table 1.1: Prevalence of bronchial asthma in adolescents
Country Year of study Age (years) Prevalence (%)
New Zealand 1991 12-15 32-38Australia 1992 12-15 16.5Netherland 1989 10-23 19Finland 1991 15-16 2.8
Several other prospective studies,127-130 which separately examined subjects aged 10 to 19,20 to 40, and over 60 years, revealed that asthma was frequently preceded by lower respiratorytract symptoms, sometimes for years. Among subjects who were diagnosed with asthma afterage 60, one-third reported respiratory symptoms before age 16.130 Similarly 82.7% with asthmadiagnosed between the ages of 5 and 11 years had lower respiratory tract symptoms before theage of 5 years.127
REFERENCES
1. CIBA Foundation Guest Symposium: Terminology, definitions, and classification of chronicpulmonary emphysema and related conditions. Thorax 1959;14:286-99.
2. American Thoracic Society: Standards for the diagnosis and care of patients with chronicobstructive pulmonary disease (COPD) and asthma. Am Rev Respir Dis 1987;136:225-44.
3. American College of Chest Physicians, American Thoracic Society: Pulmonary terms and symbols.Chest 1975;67:583.
4. National Asthma Education Programme. Expert Panel Report. Guidelines for the diagnosis andmanagement of asthma. National Heart, Lung, and Blood Institute, National Institute of Health,Bethesda, Maryland, USA, Publication No. 91-3042A, June 1991.
5. International Consensus Report on the diagnosis and treatment of asthma. National Heart, Lung,and Blood Institute, National Institute of Health, Bethesda, Maryland, USA, 20892. PublicationNo. 92-3091, March, 1992. Eur Respir J 1992;5:601-841.
6. Toren K, Brisman J, Jarvholm B. Asthma and asthma like symptoms in adults assessed byquestionnaire: A literature review. Chest 1993;104:600-05.
7. Pekkanon J, Pearce N. Defining asthma in epidemiological studies. Eur Respir J 1999;14:951-57.8. Peat JK, Toelle BG, Marks GB et al. Continuing the debate about measuring asthma in population
studies. Thorax 2001;56:406-11.9. Burney PGJ, Chinn S, Britton JR et al. What symptoms predict bronchial response to histamine?
Evaluation in a community survey of the bronchial symptoms questionnaire(1984) of theInternational Union Against Tuberculosis and Lung Disease. Int J Epidemiol 1989;18:165-73.
10. Jenkins MA, Clarke JR, Carlin JB et al. Validation of questionnaire and bronchial hyperresponsive-ness against respiratory physician assessment in the diagnosis of asthma. Int J Epidemiol1996;25:609-16.
11. Sistek D, Tschopp JM, Schindler C et al. Clinical diagnosis of current asthma: Predictive value ofrespiratory symptoms in the SPALDIA study. Eur Respir J 2001;17:214-19.
12. Gorgen PJ, Mullally DI, Evans R III. National survey of prevalence of asthma among children inthe United States. 1976 to 1980. Pediatrics 1988;81:01-07.
13. Phelan PD. Asthma in children epidemiology. BMJ 1994;308:1584-85.14. Strachan DP, Anderson HR, Limb SR et al. A national survey of asthma prevalence, severity and
treatment in Great Britain. Arch Dis Child 1994;70:174-78.15. Buist AS. Worldwide trends in asthma morbidity and mortality. Bull Int Union Tuberc Lung
Dis 1991;66:77-78.16. Sears MR. Worldwide trends in asthma mortality. Bull Int Union Tuberc Lung Dis 1991;66:
79-83.

Epidemiology 9
17. Woolcock AJ. Worldwide trends in asthma morbidity and mortality. Explanation of trends. BullInt Union Tuberc Lung Dis 1991;66:85-89.
18. Woolcock AJ, Peat JK, Salome CM et al. Prevalence of bronchial hyperresponsiveness and asthmain a rural adult population. Thorax 1987;42:361-368.
19. Sears MR. International trends in asthma mortality. Allergy Proc 1991;12:155.20. Jackson R, Sears MR, Beaglehole R et al. International trends in asthma mortality:1970 to 1985.
Chest 1988;94:914-18.21. Evans R, Mullally DI, Wilson RW et al. National trends in the morbidity and mortality of asthma
in the US. Chest 1987;91(Suppl 6):65S-74S.22. Sly RM. Mortality from asthma. 1979-1984. J Allergy Clin Immunol 1988;82:705-17.23. Weiss KB, Wagener DK. Changing patterns of asthma mortality: Identifying target populations
at high-risk. JAMA 1990;264:1683-87.24. Gerjen PJ, Weiss KB. Changing patterns of asthma hospitalisation among children; 1979 to 1987.
JAMA 1990;264:1688-92.25. Weiss KB, Gergen PJ, Wagener DK. Breathing better or wheezing worse? The changing
epidemiology of asthma morbidity and mortality. Annu Rev Public Health 1993;14:491-513.26. Whitelaw WA. Asthma deaths. Chest 1991;99:1507-10.27. Mao Y, Semenciw R, Morrison H et al. Increased rates of illness and death from asthma in
Canada. Can Med Assoc J 1987;137:620-24.28. Williams MH. Increasing severity of asthma from 1960-1987. N Engl J Med 1989;320:1015-16.29. Center for Disease Control and Prevention. Forecasted state-specific estimates of self reported
asthma prevalence – United States, 1998;MMWR Morb Mortal Wkly Rep 1998;47:1002-25.30. Ehrlich RI, Bourne DE. Asthma deaths among coloured and white South Africans; 1962-88. Respir
Med 1994;88:195-202.31. Gergen PJ, Mullally DI, Evans R. National survey of prevalence of asthma among children in
the United States 1976 to 1980. Pediatrics 1988;81:01-07.32. Taylor WB, Newacheck PW. Impact of childhood asthma on health. Paediatrics 1992;90:657-62.33. Centers for Disease Control and Prevention. Asthma mortality and hospitalisation among children
and young adults 1980-1983. MMWR Morb Mort Wkly rep 1996;45:350-53.34. Cloutter M, Wakefield D, Hall CB, Bailit H. Childhood asthma in an urban community.
Prevalence, care system, and treatment. Chest 2002;122:1571.35. Anderson HR, Bailey PA, Cooper JS et al. Morbidity and school absence caused by asthma and
wheezing illness. Arch Dis Child 1983;58:777-84.36. Vuurman EFPM, van Vaggel LMa, Uiterwijk MMC et al. Seasonal allergic rhinitis and
antihistaminic effects on children’s learning. Ann Allergy 1993;71:121-26.37. Turkeltaub PC, Gergen PJ. Prevalence of upper and lower respiratory conditions in the US
population by social and environmental factors: Data from the Second National Health andNutrition Examination Survey. 1976 to 1980 (NHANES II). Ann Allergy 1991;67(2 pt 1):147-54.
38. Asthma statistics in the United States from 1982 to 1992. MMWR 1995;43:952-55.39. Costello J. Asthma-the problem and the paradox. Postgrad Med J 1991;67(Suppl 4):S1.40. Shaw RA, Crane J, O’Donnell TV. Asthma symptoms, bronchial hyperresponsiveness and atopy
in a Maori and European population. NZ Med J 1991;104:175.41. Barry DM, Burr ML, Limb ES. Prevalence of asthma among 12 years old children in New Zealand
and South Wales: A comparative survey. Thorax 1991;46:405.42. Haahtela T, Lindholm H, Bjorksten F, Koskenvuo K, Laitinen LA. Prevalence of asthma in Finnish
young men. Br Med J 1990;301:266.43. Hurry VM, Peak JK, Woolcock AJ. Prevalence of respiratory symptoms, bronchial hyperrespo-
nsiveness and atopy in school going children living in the Villawood area of Sydney. Austr NZJ Med 1988;18:745-52.
44. Goodfrey RC, Griffiths M. The prevalence of immediate skin tests to Dermatophagoidespteronyssinus and grass pollen in school children. Clin Allergy 1976;6:79-82.

10 Bronchial Asthma
45. Clifford RD, Howell JB, Radford M, Holgate ST. Association between respiratory symptoms,bronchial response to methacholin, and atopy in two age groups of school children. Arch DisChild 1989;64:1133-39.
46. Burrows B, Lebowitz MD, Barbee RA. Respiratory disorders and allergy skin reactions. AnnIntern Med 1976;84:134-39.
47. Kaplan BA, Masci-Taylor CGN. Asthma and wheezy bronchitis in British National Sample. JAsthma 1987;24:289-96.
48. Schachter EN, Doyle CA, Beck GJ. A prospective study of asthma in a rural community. Chest1984;85:623-30.
49. Sears MR, Jones DT, Holdaway MD et al. Prevalence of bronchial reactivity to inhaled methacholinin New Zealand children. Thorax 1986;41:283-89.
50. McNichol KH, Williams HE. Spectrum of asthma in children-II. Allergic components. Br Med J1973;4:12-16.
51. Van Asperen PP, Kemp AS, Mukhi A. Atopy in infancy predicts the severity of bronchialhyperresponsiveness in later childhood. J Allergy Clin Immunol 1990;85:790-95.
52. Behera D, Jindal SK. Respiratory symptoms in Indian women using domestic cooking fuels.Chest 1991;100:385.
53. Behera D, Malik SK. Chronic respiratory disease in Chandigarh teachers- a follow up study. IndJ Chest Dis All Sci 1987;29:25.
54. Behera D, Malik SK. Chronic respiratory disease and ventilatory function in adult rural Oriyafemales. Lung India 1988;6:127.
55. Jindal, S.K., Bhaskar, BV and Behera, D: Respiratory disease pattern in a large referral hospitalin India. Lung India 1989; 7: 119-21.
56. Viswanathan R, Prasad M, Thakur AK, Sinha SP, Prakash N, Mody RK et al. Epidemiology ofasthma in an urban population; A random survey. J Ind Med Ass 1966;46:480.
57. Chougule R, Shetye VM, Parmer JR et al. Prevalence of respiratory symptoms, bronchialhyperreactivity and asthma in a mega city: Results of the European Community RespiratoryHealth Survey in Mumbai. Am J Respir Crit Care Med 1998;158:547-54.
58. Jindal SK, Gupta D, Aggarwal AN, Jindal RC, Singh V. Study of prevalence of asthma in adultsin North India using a standardised questionnaire. J Asthma 2000;37:345-51.
59. Chhabra SK, Epidemiology of childhood asthma. Indian J Chest Dis Allied SS 1998; 40:179-94.60. The International Study of Asthma and Allergies in Childhood (ISAAC)Steering Committee:
Worldwide variations in the prevalence of symptoms of asthma, allergic rhino conjunctivitis,and atopic eczema: ISAAC. Lancet 1998;351:1225-32.
61. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee:Worldwide variations in the prevalence of symptoms of asthma, and allergies in childhood(ISAAC). Eur Respir J 1998;12:315-35.
62. Jindal SK. Asthma epidemiology in India. Chest2001; 2(Indian Edition):115.63. Speizer FE, Doll R, Heaf P. Observations on recent increase in mortality from asthma. Br Med J
1968;1:335-39.64. Fraser PM, Speizer FE, Water SDM, Doll R, Mann NM. The circumstances preceding death from
asthma in young people in 1968-1969. Br J Dis Chest 1971;65:71-84.65. Sly R. decreases in asthma mortality in the United States. Ann Allergy Asthma Immunol
2000;85:121-27.66. Evans R, Mullally DI, Wilson RW et al. National trends in the morbidity and mortality of asthma
in the US prevalence, hospitalisation, and mortality of asthma over two decades; 1965-1984.Chest 1987;91:65S-74S.
67. Buist AS. Asthma mortality: What have we learnt? J Allergy Clin Immunol 1989;84:275-83.68. Sheffer AI, Buist AS. Proceedings of the asthma mortality task force. J Allergy Clin Immunol
1987;80:361-62.

Epidemiology 11
69. Khanna PM, Linger J. Asthma mortality and hospitalisation among children and young adults;United States, 1980-1993. JAMA 1996;275:1535-37.
70. Buist AS, Vollmer WM. Reflections in the rise in asthma morbidity and mortality. JAMA19990;264:1719-20.
71. Burney P. Asthma deaths in England and Wales 1931-85: Evidence for a true increase in asthmamortality. J Epidemiol Community Health 1988;42:316-20.
72. Weiss KB, Wagener DK. Changing pattern of asthma mortality. JAMA 1990;264:1683-87.73. Salas-Ramirez M, Sagura N, Martinez C. Trends in asthma mortality in Mexico. Bol Oficina
Sanit Panam 1994, 116:298-306.74. Molinari J, Chatkin J. Tendencia da mortalidade por asthma bronnquica no Rio Grande do Sul.
J Pneumonol 1995;21:103-06.75. Picard E, Barmeir M, Schwartz S et al. Rate and place of death from asthma among different
ethnic groups in Israel. National trends 1980 to 1997. Chest 2002;122:1222-27.76. Sears MR, Rea HH, De Boer G et al. Accuracy of certification of death due to asthma—A national
study. Am J Epidemiol 1986;124:1004-11.77. Hunt LW, Mair JE, Laplante JM et al. Causes of death in a population with asthma. Am Rev
Respir Dis. 1989;139:A486.78. Riou B, Barriot P. Accuracy of asthma mortality in France. Chest 1990;97:507-08.79. World Health Organisation. Manual of the international statistical classification of diseases,
injuries and causes of death: Based on the recommendation of the ninth revision conference,1975. WHO, Geneva, 1979, Vol 1.
80. Garrett J, Kolbe J, Richards G et al. Major reduction in asthma morbidity and continued reductionin asthma mortality in New Zealand: What lessons have been learned? Thorax 1995;50:303-11.
81. Sly RM. Changing asthma mortality. Ann Allergy 1994;73:259-68.82. Vergara C, Caraballo L. Asthma mortality in Colombia. Ann Allergy Asthma Immunol 1998;80:55-
60.83. Pearce N, Beasley R, Crane J et al. End of the New Zealand asthma mortality epidemic. Lancet
1995;345:41-44.84. Sly RM, O’Donnell R. Stabilisation of asthma mortality. Ann Allergy Asthma Immunol
1997;48:347-54.85. Stolley PD, Schinnar R. Association between asthma mortality and isoprenol aerosols: A review.
Preventive Med 1978;7:519-38.86. Esdaile JM, Feinstein AR, Horwitz RI. Can general mortality data implicate a therapeutic agent?
Arch Intern Med 1987;147:543-49.87. Crane J, Flatt A, Jackson R et al. Prescribed fenoterol and death from asthma in New Zealand.
Lancet 1989;1:917-22.88. Poole C, Lanes SF, Walker AM et al. Fenoterol and fatal asthma. Lancet 1990;335:920.89. Beasley R, Smith K, Pearce N et al. Trends in asthma mortality in New Zealand, 1908-1986. Med
J Aust 1990;152:570-73.90. Sly RM. Mortality from asthma. J Allergy Clin Immunol 1989;84:421-34.91. Spitzer WO, Suissa S, Ernst P et al. The use of beta-agonists and the risk of death and near-death
from asthma. New Engl J Med 1992;326:501-06.92. Sly RM. O’Donnell R. Regional distribution of deaths from asthma. Ann Allergy 1989;62:347-54.93. Goldman M, Rachmiel M,Gendler M et al. Decrease in asthma mortality rate in Israel from
19991-1995: Is it related to increased use of inhaled corticosteroids? J allergy Clin Immunol2000;105:71-74.
94. Campbell MJ, Gogman GR, Holgate ST et al. Age, specific trends in asthma mortality in Englandand Wales, 1983-1995; Results of an observational study. BMJ 1997;314:1439-41.
95. Respiratory diseases disproportionately affecting minorities. The NHLBI Working Group. Chest1995;108:1380-92.

12 Bronchial Asthma
96. Lang D. Trends in US asthma mortality: Good news and bad news. Ann Allergy Asthma Immunol1997;78:333-36.
97. Gergen PJ, Weiss KB. Changing patterns of asthma hospitalisation among children. 1979 to1987. JAMA 1990;264:1688-92.
98. To T, Dick P, Feldman W et al. A cohort study in childhood asthma admissions and readmissions.Paediatrics 1996;98:191-95.
99. Jones AP, Bentham G. Health service accessibility and death from asthma in 401 local authoritydistricts in England and Wales. 1988-92. Thorax 1997;52:218-22.
100. Capewell S. The continuing rise in emergency admissions. BMJ 1996;312:991-992.101. Vollmer et al. Am Rev Respir Dis 1993;147:347102. Osborne M. Clinical asthma: Will NAEP guidelines help? Pulm Perspectives 1994;11(1):1-3.103. Castro M, Halstead J, Schechtman K et al. Risk factors for asthma morbidity and mortality in a
large metropolitan city. J Asthma 2001;38:625-36.104. Roorda RJ. Prognostic factors for the outcome of childhood asthma in adolescence. Thorax
1996;51(Suppl 1):S7-S12.105. Peckham C, Butler N. A national study of asthma in childhood. J Epidemiol Community Health
1978;32:79-85.106. Anderson HR, Bland JM, Patel S, Pekham C. The natural history of asthma in childhood. J
Epidemiol Community Health 1986;40:121-29.107. Bronniman S, Burros B. A prospective study of the natural history of asthma. Remissions and
relapse rates. Chest 1986;90:480-84.108. Aberg N, Engstrom I. Natural history of allergic diseases in children. Acta Paediatr Scand
1990;79:206-11.109. Radford PG, Hopp RJ, Biven RE et al. Longitudinal changes in bronchial hyperresponsiveness
in asthmatic and previously normal children. Chest 1992;101:624-29.110. Friberg S, Bevegard S, Graff-Lonnevig V, Hallback I. Asthma from childhood to adulthood. A
follow-up study of 20 subjects with special reference to work capacity and pulmonary gasexchange. J Allergy Clin Immunol 1989;84:183-90.
111. Ferguson AC. Persisting airway obstruction in asymptomatic children with asthma with normalpeak expiratory flow rates. J Allergy Clin Immunol 1988;82:19-22.
112. Cooper DM, Cutz E, Levison H. Occult pulmonary abnormalities in asymptomatic asthmaticchildren. Chest 1977;71:361-65.
113. Blackhall M. Ventilatory function in subjects with childhood asthma who have become symptomfree. Arch Dis Child 1970;45:363-65.
114. Cade JF, Pain MCF. Pulmonary function during clinical remission of asthma. How reversible isasthma? Aust NZ J Med 1973;3:545-51.
115. Strachan DP. The prevalence and natural history of wheezing in early childhood. J Royal CollGen Pract 1985;35:182-84.
116. Peat JK. Salome CM, Toelle BG, Bauman A, Woolcock AJ. Reliability of a respiratory historyquestionnaire and effect of mode of administration on classification of asthma in children. Chest1992;102:153-57.
117. von Mutius E. Progression of allergy and asthma through childhood to adolescence. Thorax1996;51(Suppl 1):S3-S6.
118. Martinez FD, Wright AL, Taussig LM et al. Asthma and wheezing in the first six years of life.N Engl J Med 1995;332:133-38.
119. Park Es, Golding J, Carswell F, Stewart-Brown S. Pre-school wheezing and prognosis at 10.Arch Dis Child 1986;61:642-46.
120. Balfour-Lynn. Childhood asthma and puberty. Arch Dis Child 1985;60:231-35.121. Peat JK, Salome CM, Sedgewick CS, Kerrebijn J, Woolcock AJ. A prospective study of bronchial
hyper responsiveness and respiratory symptoms in a population of Australian school children.Clin Exp Allergy 1989;19:299-306.

Epidemiology 13
122. Price JF. Issues in adolescent asthma: What are the needs? Thorax 1996;51(Suppl 1):S13-S17.123. Robson B, Woodman K, Burgess C et al. Prevalence of asthma symptoms among adolescents in
the Wellington region by area and ethnicity. NZ Med J 1993;106:239-41.124. Forero R, Bauman A, Young L, Larkin P. Asthma prevalence and management in Australian
adolescents; results from three community surveys. J adolescent Health 1992;13:707-12.125. Kolnaar B, Beissel E, van-den-Bosch WJ et al. Asthma in adolescents and young adults: Screening
outcome versus diagnosis in general practice. Fam Pract 1994;11:133-40.126. Rimpela AH, Savonius B, Rimpela MK, Haahtela T. Asthma and allergic rhinitis among Finnish
adolescents in 1977-1991. Scand J Soc Med 1995;23:60-65.127. Dodge R, Martinez FD, Cline MG, Lebowitz MD, Burrows B. Early childhood respiratory
symptoms and the subsequent diagnosis of asthma. J Allergy Clin Immunol 1996;95:48-54.128. Dodge R, Burrows B, Lebowitz MD, Cline MG. Antecedent features of children in whom asthma
develops during the second decade of life. J Allergy Clin Immunol 1993;92:744-49.129. Dodge R, Cline MG, Lebowitz MD, Burrows B. Findings before the diagnosis of asthma in young
adults. J Allergy Clin Immunol 1994;94:831-35.130. Burrows B, Lebowitz MD, Barbee RA, Cline MG. Findings before the diagnosis of asthma among
the elderly in a longitudinal study of a general population sample. J Allergy Clin Immunol1991;88:870-77.

14 Bronchial Asthma
A number of factors are responsible either in the causation or exacerbation of bronchialasthma. A brief account of each of these factors will be discussed.
ATOPY AND ALLERGY
The association between asthma and allergy has long been recognised. It has been reportedthat 75-85% of patients with asthma have positive immediate skin reactions to commoninhalant allergens. There are at least 6 major evidences to prove that most asthma in youngpeople is due to exposure to allergens or to sensitisers. They are summarised below.
i. Most people with asthma are atopic, which can be measured by skin tests or withmeasurements of specific IgE. In population studies and in clinical practice, it is clearthat majority of young people are atopic. Furthermore, in most population studies ofasthma, atopy has been found to be the most important single risk factor. House dustmite allergens appear to be the most common one associated with asthma.
ii. Challenge with allergens in atopic asthmatics increases the severity of the disease. Thestimulus is capable of increasing this for days and sometimes for weeks. This impliesthat allergens play a role in maintaining the disease.
iii. Occupational asthma occurs due to allergens and sensitisers. In some healthy people,who are exposed to these agents, sensitisation occurs and is followed by episodic wheeze.Unless the subject is removed from the source, episodic symptoms continue, and withtime become persistent.
iv. It has been shown that subjects with apparently intrinsic asthma (normal skin tests),have higher levels of circulating IgE than the non-asthmatic population.
v. Improvement in the symptomatology occurs on allergen withdrawal, which provesthe causal relationship between the two.
vi. Population studies have clearly demonstrated the association between atopy andasthma.
It has been shown that in Indonesian children, there is less atopy and less asthma.Similarly studies from France have reported a lower prevalence of asthma where mitesare less in number. There is a strong co-relation between allergic sensitisation to commonaeroallergens and the subsequent development of asthma. There is also a strongassociation between allergen exposure in early life and sensitisation to these allergens,although it has not been possible to demonstrate an association between allergenexposure and the development of asthma.1
Aetiology
2

Aetiology 15
Some studies, however, challenged the assumption that childhood asthma is largely ofallergic etiology.2 Pearce et al3 reviewed the epidemiological evidence implicatingaeroallergen exposure in the primary causation of asthma, and concluded that the availabledata do not indicate that aeroallergen exposure is a major risk factor. In a further study,they reported that atopy attributes only 38% to the causation of asthma.2 Some investigatorshave observed a weak and inconsistent association between atopy and asthma prevalence.On the contrary, recent studies suggest that among those reporting wheezing in the previousmonths have a stronger relationship with atopy for those reporting > 12 episodes of wheezingin the past 12 months compared to those reporting 1 to 3 episodes in the last 12 months.4
The proportion of “asthma-ever” attributable to atopy was 33%, while the proportion was89% for those who were attending hospitals (indicating more severe form). Based on thesefindings, it is suggested that atopy contributes more to the frequent or severe asthma thanto mild or infrequent asthma.4 These findings are consistent with other studies. The importantassociation of atopy with childhood asthma is well recognised.5 A review of studies relatingatopy to asthma notes that in cross-sectional studies conducted exclusively or predominantlyin children, the proportion of cases attributable to atopy varied from 25 to 63%, with aweighted mean of 38%.6 Relationship of atopy and severity of asthma is a well-known fact.6
Atopy is also related to degree of bronchial hyperreactivity.7,8 Conversely, in patients havingwheeze in the previous 12 months, bronchial hyperactivity is related to both atopy andmeasures of disease severity such as peak expiratory flow variability.9
Taken together these facts are strong evidence for the role of atopy in asthma. Eventhough not all asthmas are associated with or perpetuated by exposure to common airborneallergens, exposure to these agents plays a major role. Both indoor and outdoor allergenexposures have increased asthma morbidity. People now spend a substantial proportion ofthe time indoors. Most of the responsible allergens are probably prevalent inside the housessince this is where human beings spend most of their lives. The most important onesthroughout the world appear to be the house dust mites, grass pollens, animal proteins, andmoulds. Recent changes in housing styles in many western countries may have led toincreased allergens levels. Houses tend to have less ventilation, making them more humid,and there has been widespread introduction of carpeted floors and pets living in the houses.Whereas house dust mite is the most important and common indoor allergen linked toasthma,10 Outdoor allergens such as grass pollen, soyabean dust and Alternaria alternatehave been specifically linked to severe asthma exacerbations.11,12 There has also been spreadof plants, cockroaches and perhaps mites. Moreover, the climate of a particular area mayfavour the availability of various allergens, which in part may be responsible for thedifference in the prevalence of asthma in various countries. Another important factor is theway the allergen is handled. Pollutions add to the allergenicity of aeroallergens. Thepredominance of these allergens will of course depend upon various factors, particularlylocal. Studies in asthmatics of allergen skin reactivity, IgE antibody levels, and bronchialprovocation have all helped establish the important role of allergens in many asthmaexacerbations.13 Further, reducing the patient’s exposure to allergens can help bring asthmasymptoms under control. A growing number of uncontrolled and controlled studies suggestthat allergen eradication and avoidance measures lead to improvement in bronchialhyperresponsiveness, severity of symptoms, and requirements of asthma medications.14
Recent research suggests that for many allergic disorders associated with aeroallergens,the process of IgE sensitisation begins right early in life while the immune response is still

16 Bronchial Asthma
developing. It has been shown that the level of dust mite allergen present in the homeduring the first year of life is a major factor in determining whether an infant born of anallergic mother who is genetically susceptible, did in fact develop allergy or asthma by thetime they are 11 years of age. Moreover, the density of allergen (per gram of dust) is animportant factor in determining the age of onset of first symptoms. Higher is theconcentration of allergen earlier is the onset of disease.
Allergenic pollens vary at different places. The predominant offending allergen willvary with locality, lifestyle, season, and climate. For example, in Delhi, Amarantus pollen isthe most common offending allergen followed by Cassia siamea, Ricinus, Brassica, Imperata,Prosopis, Cenchrus, Cassia occidentalis, etc.15 Prosopis is the commonest antigenic pollen inBikaner, Lucknow, and Varanasi.16-18 Brassica is the commonest pollen in Bhopal andKanpur.19,20 On the other hand Parthenium is the commonest offending agent in Kolhapur .21
In the United kingdom, 50-75% of atopic asthmatics react to house dust mite, similar numberto grass pollens, 35-55% to cat dander, 10-20% to dog dander, 10-20% to tree pollens,10-15% to moulds, and fewer than 10% to food allergens.13 In contrast, keeping cats as pets,unlike in many western countries is not a common practice in India. Therefore, cat or dogdander allergy may not be that important in this country. On the other hand because oftropical climates, and peculiar habit of storage of food articles, cockroaches grow plenty inthis country. Therefore, these might be an important allergen for people of India.
The importance of allergy is different for different age groups. In infants, allergens playa less important role than other ages and viral respiratory infections are the principal triggers.Although allergic reactions to food can occur in infants, foods are not the common triggers.Studies in children suggest that allergy influences the persistence and severity of asthma. Itis reported by several authors that severity of childhood asthma corelates with the numberof positive immediate skin tests. Children with multiple positive skin tests are more likelyto have daily rather than intermittent symptoms of asthma. The important allergens inchildren after infancy appear to be inhalants. Aeroallergens are important in patients whosedisease has started before the age of 30 years or who are exposed to occupational allergens.Patient can also have allergy for the first time after the age of 30. However, in adults theintensity of allergic skin tests does not appear to be associated with increased severity ofasthma. Food allergies are not common triggers for asthma in adults. The patient may haveaspirin sensitivity, but it has no immunological basis.
Different Allergens (Figs 2.1a to 2.1h)
i. Important outdoor allergens include pollens and moulds.Pollen Particles greater than 10 micron in diameter are usually cleared in the nose andmouth and do not penetrate the lower respiratory tract. However, there are some plants,which produce allergen-containing particles that are less than 10 microns. Ragweedand grass pollination are definitely associated with asthma. Pollen allergy is usuallyseason-related and is more closely linked to hay fever and allergic conjunctivitis.Mould Mould spores are generally smaller than pollen grains and are more likely topenetrate the lower respiratory tract. Mould spores exist primarily outdoors and tendto be seasonal. Some fungi sporulate on warm, dry summer days and others prefer therainy season. The species of the fungus vary with the geographic distribution accordingto climatic conditions.

Aetiology 17
Fig. 2.1a: Dust during cleaning Fig. 2.1b: Pollen
Fig. 2.1e: House dust mite in the bedding Fig. 2.1f: Perfumes
Fig. 2.1c: Smoke Fig. 2.1d: Domestic fuel

18 Bronchial Asthma
ii. Although house dust itself is not an allergen, there are allergic components in it. Themost important ones are mites, animal danders, and cockroaches.House dust mite plays a major role in the causation of asthma, although it does not leaveany immediately perceptible sting or bite. This is the agent most widely implicated inthe pathogenesis and provocation of allergic asthma. They are arachnoids distantlyrelated to ticks and spiders. They are ubiquitous, living in the house dust that providesboth their shelter and food (scales of skin shed by humans). They occur in environmentswith sufficient humidity since they are quite dependent for survival on moisture fromthe atmosphere. Loss of water from the mite body constrains their growth, but mitesare capable of extracting water vapour even from air that is only 50% saturated. Livemites are equipped with suckers at the tips of their legs, which make them difficult toremove by vacuuming. The commonest mite is Dermatophagoides pteronyssinus. Otherspecies may also exist in small numbers. Mite antigen is found throughout the home,wherever human dander, the food for the mite, is found.
High levels are found in mattresses, pillows, carpets, upholstered furnitures, bedcovers, clothes, and soft toys. The principal allergen of the house dust mite is found inits faeces. A gram of dust may contain 1,000 mites and 250,000 faecal pellets. Thesepellets are quite large and 10-40 microns in size, similar to pollen grains and sharesome of the aerodynamic properties with them. Like larger pollen grains, they do noteasily enter the lower respiratory tract and are rapidly cleared from the airway by
Fig. 2.1g: Pets (Animal dander)
Fig. 2.1h: Mould in the wall

Aetiology 19
gravity. Mite antigen is readily demonstrated in the air during cleaning. Some miteallergens may be smaller that may be in the respirable range for the lower respiratorytract. The major allergens of house dust mites are probably digestive enzymes,collectively designated as group I allergens or Der p I, and there are now tests availableto quantitate this.
The improvement of asthma in children residing in high altitude where low humidityconstrains dust mite growth or in patients admitted to the relatively dust-free environ-ment of a hospital14 indicates the contribution of the house dust mite to asthmaexacerbation. A study of children requiring hospitalisation for asthma found that therisk of re-admission was associated with continued exposure to high concentrations ofmite allergen.22-28
Animal allergens Dogs, cats, and other pet animals including rodents are commonlykept in homes. Danders from these animals contribute greatly to the allergeniccomponents of house dust. All warm-blooded pets can cause allergic reactions, includingthe birds and small rodents. Products made from feathers retain the allergens frombird. All breeds of cats produce common allergens, and cat saliva and cat danders arepotent allergens. Dogs also produce common allergens, although minor breeddifferences may exist. For several reasons cat allergen is more likely to cause sensitisationthan that of dogs. The major cat allergen is Fel d I, which is a protein secreted by thecat’s salivary, sebaceous, and lacrimal glands. The protein is very stable and loses noneof its antigenic potency for at least a month. It is coated on to the fur by the usualgrooming, and at the rate cats shed their fur and dander, a reservoir of the antigenrapidly accumulates in household furnishings. In addition, Fel d I, particles are lessthan 2.5 mm in diameter and flake-shaped, making them easily airborne and easilyrespirable. While air filtration can remove some of the allergens, little permanentreduction occurs unless carpets, furnishings, and other reservoirs of coated fur (the catitself) are removed. It takes several months before the concentration of allergens indomestic dust falls after removal of the pet.
A number of epidemiological studies suggest that close contact with a cat or dog invery early infancy reduces subsequent prevalence of allergy and asthma. This may bea consequence of high allergen exposure inducing tolerance.29-31
Cockroach allergen The cockroach appears to be important, particularly in warmerclimates and inner side of the house in cooler climates. Cockroach allergy has beenidentified as an important cause of asthma. This form of asthma—“The cockroachasthma”—is a more severe form of the disease, having perennial symptoms, and highlevels of IgE. Cockroaches produce several allergens, which produce sensitisation.Usually there is exposure to high levels of this allergen at homes. The important domesticspecies are Blattella germanica and Periplaneta American.
Kinds of Allergens
The allergens are Bla g 2 (inactive aspartic protease), Bla g 4 (calycin), Bla g 5 (glutathione– S-transferase), Bla g 6 (troponin), the Group I cross-reactive allergen Bla g 1 and Per a 1,Per a 3 (arylphorin), and Per a 7 (tropomyosin). Although elimination of cockroaches totallyis difficult, development of cockroach allergens as recombinant proteins has led to bettercontrol of this form of asthma.32 Indoor moulds are prominent in environments withincreased humidity. Bathrooms, kitchens, basement areas, and perspiration on pillows are

20 Bronchial Asthma
the common sites of mould growth. Cockroach sensitivity in children has been associatedwith greater symptom frequency and more emergency department visits due toasthma.33-36 Similar observations are made for elderly patients with asthma also.37
Risk Allergens: Responsible for Acute Attacks
Threshold concentrations of allergens that can be regarded as risk factors for acute attacksinclude:
• 10 μg/g dust of group I mite allergen• 8 μg/g dust of Fel d I, the major cat allergen• 10 μg/g dust of Can f I, the major dog allergen• 8 μg/g dust of cockroach allergen
FOOD ALLERGEN AND BREASTFEEDING
In the first 1 or 2 years of life, food sensitivity is an important factor in the development ofallergies. Breastfeeding has been advocated as a method of preventing allergy and asthma.With breastfeeding there is a decreased risk (about 20%) for development of asthma.38 Impactof exclusive breastfeeding in children at 6 years of age has shown that the introduction ofmilk other than breast milk before the age of 4 months of age is a significant risk factor forincreased likelihood of bronchial asthma.39 However, another study has shown an increasedrisk of wheezing, particularly in asthmatic mothers and if the child is also atopic.40
There are some reports that regular consumption of oily fish is associated with a reducedrisk for asthma in children, although subsequent studies have not shown clinical benefits ofsupplemental ω3 fatty acids over a 6 months period.41,42 Further, it has been hypothesised thatdecreased dietary antioxidant vitamin intake is associated with increased asthma.43 Higherconcentrations of vitamin intake are associated with a decreased serum levels if IgE and asignificant decrease of atopy.44 Recent experimental data showed a reduced risk with intakeof lectins (wheat germ agglutinin from whole wheat products).45
INFECTION
It has long been recognised that viral respiratory infections provoke and alter asthmaticresponses. Over 80% of acute asthma exacerbations in school children and about 60% inadults result from viral infections, mostly common cold viruses. These observations havesuggested that viral infections may be intimately involved in the development of asthmaand allergy. The susceptibility of the asthmatic airway to viral inflammation is due topersistent allergic mast cell and eosinophil-derived inflammation stimulates the release ofcytokines such as tumour necrosis factor-alpha, which cause an increase in the expressionof receptors for human respiratory viruses on the airway lining epithelium. In case of mostrhinoviruses, the receptor is an adhesion molecule (intracellular adhesion molecule-1). TheViral respiratory illnesses may produce their effect by causing epithelial damage, producingspecific immunoglobulin IgE antibodies directed against respiratory viral antigens andenhancing mediator release. Once the virus enters the epithelial cells, it replicates andgenerates a wide variety of proinflammatory cytokines, which further enhance eosinophiland mast cell inflammation. Apart from aggravating clinical asthma, viral upper respiratory

Aetiology 21
infections increase airway responsiveness, which may persist for many weeks after theinfection.
Provocateurs of Asthma
The principal infection provocateurs of asthma in childhood during the first 2 years oflife are respiratory syncytial virus (RSV), parainfluenza virus, and rhinovirus. Influenzavirus is much more common in older children and adults. Early hospitalisation for respiratorysyncytial virus, croup, or bronchiolitis is associated with greater airway responsiveness andmore frequent history of wheezing.46 Other microorganisms that can exacerbate bronchialasthma include Mycoplasma pneumoniae. Although bacterial infection i s no t a cause ofsuch exacerbations, it has been reported recently that H.influenzae and other Gram-negativebacteria can synthesise histamine both in vivo and in vitro.47 The presence of this mediatormay contribute to the bronchoconstriction and other effects of histamine that can accompanybronchial infection. Pseudomonas infection in cystic fibrosis is responsible for a hyperreactivityreaction in these patients. A recent study in 101 nonsmoking severe asthmatics showsassociation between accelerated loss of lung function and seropositivity to Chlamydiapneumoniae.48
Interestingly, in recent years it is also observed that some infections are protective ofbronchial asthma. While viral infections can undoubtedly cause deterioration of establishedasthma, viral or bacterial infections during the first three years of life may serve a protectivefunction against the development of allergic diseases. Possibly they evoke a Th1-like protectiveresponse with the generation of IFN-gamma and IL-2. Thus, if multiple infections occur duringthe first few years of life, high concentrations of these Th1 cytokines could inhibit the releaseof Th2 cytokines, thereby tuning the mucosal immune response away from allergensensitisation. This hypothesis is supported from observations from an African study, wherechildren infected with measles during the first year of life had a 63% lesser chance of developingpositive skin tests to common aeroallergens. Similarly repeated vaccination with BCG exerteda protective effect against the development of allergy in young Japanese children. Both measlesand BCG are potent stimulators of the Th1 cytokine response. Another support of this protectiveinfection comes from observations comes from the fact that the increase in asthma and allergywith movement to urban areas may be related to a decrease in early exposure to parasiticinfections. One study from slum are of Caracas, Venezuela showed that antihelminthictreatment causes a decrease in IgE level, but was accompanied by an increase in skin testreactivity to house dust mite. In contrast, in the untreated children, the parasitic colonisationcontinued, IgE levels increased but the dust mite sensitisation fell. It indirectly means thateradication of parasites or reduced opportunities for infection could, in part, explain the ruralto urban differences in the prevalence of allergic diseases. These observations led to the“Hygiene hypothesis” of bronchial asthma. This suggests that early exposure to microbialproducts will switch off allergic responses preventing allergic disorders like asthma.49
Epidemiological studies comparing large populations who have or have not had suchexposures support the hypothesis.50 The hygiene hypothesis explains that allergic diseaseswere prevented by infections in early childhood, transmitted by unhygienic contact witholder siblings or acquired prenatally. Over the past century declining family size, improvedhousehold amenities, and higher standards of personal cleanliness may have resulted inmore atopic diseases.49 It is further proposed that modern vaccinations, fears of germs and

22 Bronchial Asthma
obsession with hygiene are depriving the immune system of input on which it is dependent.Recent data suggest that exposure of young children to older children at home or to childrenat day-care protects against the development of asthma and frequent wheezing later inchildhood. A double blind placebo controlled trial using the probiotic.
Lactobacillus CG, observed a reduced incidence of atopic eczema but no effect on IgE antibodysensitisation, important for bronchial asthma. However, this study has the limitation of smallsample size and early age limit of interpretation.51
DRUGS
About 5 to 20 per cent of adults with asthma will experience severe and even fatal exacer-bations of bronchoconstriction after ingestion of aspirin or certain non-steroidal anti-inflammatory drugs (NSAIDs). These drugs are as follows:52-58
• Aspirin • Ibuprofen • Indomethacin• Piroxicam • Sulindac • Tolmetin• Naproxen • Fenoprofen • Meclofanamate• Mefenamic acid • Diclofenac sodium
The list is not complete and aspirin sensitivity implies cross-reactivity with other non-steroidal medications. The prevalence increases with increasing severity of asthma. In theseindividuals, ingestion of aspirin is followed within 1 to 2 hours by the onset of bronchospasm,which may be accompanied by rhinitis and/or urticaria. An association between aspirinsensitivity in people with asthma and the presence of sinusitis and nasal polyps is oftenstressed. Although there is a statistical relation, many patients with nasal polyps are notaspirin sensitive, and many patients with asthma and aspirin sensitivity have not beenfound to have nasal polyps. It is likely that sinusitis, nasal polyps, and aspirin sensitivity allincrease in prevalence with increasing severity of asthma and they are not causally related.
Although the exact mechanism is not known, it is nonimmunologic and probably dependson inhibition of cyclo-oxygenase. Accordingly, the arachidonic acid metabolism proceeds viathe lipo-oxygenase pathway producing leukotrienes (see pathogenesis). Although the exactpathogenesis of aspirin-induced asthma is unclear, studies have demonstrated thatleukotrienes play an important role in airway narrowing and other signs in these patients.These observations are derived from the fact that urinary LTE4 is two-folds to ten-folds higherin these patients than in aspirin tolerant patients.59-61 Several leukotriene modifiers inhibit theasthma response in oral or inhaled bronchial provocation tests, such as aspirin andnonsteroidal anti-inflammatory drugs,62-64 and improve respiratory function by expandingthe airway in patients with aspirin induced asthma.65 An additional hypothesis for themechanism of aspirin sensitivity suggests that there is increased target organ sensitivity toleukotrienes. The inhibition of cyclo-oxygenase is a property common to all of the drugsproducing this adverse reaction. Although analgesics not inhibiting this enzyme are generallyconsidered to be safe, the most frequently employed alternative, acetaminophen, has beenreported to cause asthma exacerbations in a few aspirin-sensitive patients.
Other drugs that are known to exacerbate asthma include beta-blocker drugs (i.e. propra-nolol and nadolol). Eye drop preparations of this class of drugs also can induce asthma.Recently, inhaled verapamil, a calcium channel blocker, has been reported to induce severebronchospasm in mild asthma.

Aetiology 23
EXERCISE-INDUCED ASTHMA66-71
Exercise-induced asthma’ (EIA) is often used to describe the asthma of persons in whomexercise is the predominant or even the only identified trigger to airflow obstruction. Noavailable data support the concept that exercise-induced asthma represents a distinctpathologic or pathophysiologic entity. Exercise-induced bronchoconstriction is onemanifestation of the asthmatic diathesis. Most, virtually all, people with asthma have airwayhyperirritability that leads to exercise-induced asthma if the provocative stimulus - eucapnicvoluntary hyperventilation- is appropriately intensified. Accordingly, this condition shouldbe anticipated in all asthma patients. For some asthmatics, exercise is the only trigger. Inaddition, most patients in whom exercise is the predominant trigger, will have otheradditional sensitivities that either can be found in the clinical history or will evolve overtime. It is estimated that approximately 40 per cent of children with allergic rhinitis, butwithout clinical asthma, have EIA. This situation probably holds true for adults. UntreatedEIA can limit and disrupt normal life. Although individual episodes of EIA are short lived,the severity and impact is striking.
During short (few minutes) periods of exercise, airways actually dilate. Exercise-inducedasthma is the airway narrowing that occurs minutes after the onset of vigorous activity.Airway narrowing develops within 2-3 minutes after cessation of exercise. It generallyreaches its peak about 5-10 minutes after cessation of activity and usually resolvesspontaneously in the next 30-90 minutes or within a few minutes of administration of aninhaled beta-adrenergic bronchodilator. There are some reports now that a late phase ofEIA exists.72,73 However, this phase is uncommon (EIA is a nonimmunologic form of asthma)and not severe unlike the late phase of allergen-induced asthma. Ambient air conditionsduring the post-exercise period also influence the degree of bronchoconstriction thatdevelops. A rapid change to warm, moist air post-exercise tends to worsen the developmentof airflow obstruction.74 Some patients who engage in continuous, repetitive exercise periods,EIA diminishes or is completely abated during a refractory period that usually lasts 2 hoursafter an exercise challenge. This is referred to as “refractory period”. Because of thisphenomenon, many asthmatic athletes report that a warm-up period of sub-maximal exercisehelps to minimise exercise-induced symptoms.75 During sustained exercise they are oftenable to “work through’ initial respiratory symptoms, i.e. experience resolution of initialsymptoms despite continued exercise.
In contrast to asthma in general, which is characterised by both smooth muscle contractionand airway inflammation, exercise-induced asthma is due mainly to smooth musclecontraction. Therefore some investigators call this as airflow-induced bronchoconstriction(AIB) or exercise-induced bronchospasm (EIB). Although the exact mechanism of asthmais debated, it is generally established that EIA is due to loss of heat or water or both, fromthe lung during exercise resulting from hyperventilation of air that is cooler and dryer thanthat of the bronchial tree. The key aspects of the triggering stimulus are the level of ventilationduring exercise and the temperature/water content of the inspired air.70 The higher theminute ventilation during exercise and the colder and drier the inspired air, the greater isthe stimulus for bronchoconstriction. How this airway cooling causes bronchoconstriction,is not exactly clear. It has been suggested that heat and water loss leads to changes in airwayfluid osmolarity which initiates mediator release that cause constriction in the smoothmuscle. Some investigators believe that airway cooling triggers bronchoconstriction inasthmatic subjects, and a rewarming-induced hyperaemia and oedema results in airway

24 Bronchial Asthma
obstruction. Another hypothesis put forth is that exercise-induced bronchoconstriction resultsfrom an imbalance between two opposing mechanisms: an excitatory pathway stimulated byairway drying and an inhibitory pathway initiated by airway cooling. It is speculated thatcooling attenuates hypocapnia, hypertonic aerosol- and dry air-induced bronchospasm via acold induced reduction in neuronal activity or mediator production and release.
Effects of Exercise
An athlete’s minute ventilation during exercise is determined in part by the workloadundertaken as measured by oxygen consumption and in part by the degree of deconditioningas measured by minute ventilation. Thus, for all asthmatics, regular exercise that improvescardiovascular fitness and thereby increased oxygen extraction from the blood by exercisingmuscle can help reduce exercise-induced bronchoconstriction by lowering the level ofventilation needed during any given exercise task. Rate, depth, and pattern (I:E ratio) ofbreathing at a given level of ventilation during eucapnic voluntary hyperventilation are notimportant determinants of bronchoconstriction.71 To reduce/avoid EIA, avoidance of a cold/dry environment is preferable. Swimming is the preferred exercise for persons with asthmabecause of this mechanism. Other inhaled irritants in the ambient air including high levels ofair pollutants and smoke can also trigger asthma especially during exercise when larger thannormal volumes of these irritants are inhaled.
OCCUPATION AND ASTHMA
Occupational asthma is the commonest industrial lung disease in the developed world withover 400 causes.76-78 It may account for about 10% of adult onset asthma.79 Environmentalagents related to work place have been recognised as the causative agents for respiratorydiseases for many centuries. Bernardino Ramazzini had recognised the importance ofoccupation in the causation of asthma as early as 1713 particularly in grain workers, bakers,millers, sulphur workers, and other occupations. With increased industrialisation, simplechemicals and organic compounds have been used more often with a consequent increase innew respiratory hazards, particularly occupational asthma. Occupational asthma may accountfor about 10% of adult onset asthma.79 It is now the commonest industrial lung disease in thedeveloped world with over 400 causes.80-86
Agents causing occupational asthma are usually encountered in an industrial setting,but it is also possible for persons outside the working area to develop disease aftercontamination of their environment by a point source industrial chemical irritant or allergen.Industries in which asthma occurs include plastics and paint manufacturing, electronics,welding, metal refining, photography, health-related industries, antibiotics and cosmeticmanufacturing, dyeing, forestry, and food processing. Asthma can also result from massivepollution due to transportation accidents or gross contamination of the local environmentby manufacturing industries. It can also occur in more unrecognised ways like materialscontaminating air conditioning system inlets from near by factories, or by contamination ofworkers or of their clothing. Thus the strict definition of occupational asthma as reversibleobstructive airways disease contracted in the work place may underestimate the real extent of theproblem.

Aetiology 25
Prevalence of Asthma in Workers
Although the exact prevalence of occupational asthma is not known and will vary accordingto the setting in which it occurs, on the industrial agent involved, on the intensity of exposure,and on working conditions, industrial hygiene, and engineering factors; it is reported thatbetween 5 to 15% of all cases of asthma in Japan are occupational. Bakers exposed to flourdust develop asthma at a rate of 10-30%, in washing powder industry, up to 60% of theworkers become sensitised to Bacillus subtilis, and in the cotton industry the prevalence ofbyssinosis is 25-29% in workers exposed in the carding process and 10-29% in those exposedin the spinning process. Similarly 5% of the western red cedar workers, 6% of the animalhandlers, 5% of the workers in plastics industry (volatile isocyanates), and 30-50% of thoseworking in the metal industry using soluble platinum salts develop the disease.
Agents capable of inducing occupational asthma can be vapours, gases, aerosols, orparticulate matters and can range from very low molecular weight inorganic chemicals tocomplex organic macromolecules. Some of these agents are shown in Table 2.1.
Table 2.1: Selective agents known to cause occupational asthma
Agents Occupation
1. Natural organic environmental agents.Animal proteins (urine, danders) Laboratory workers/VeterinariansShellfish, egg proteins, pancreatic enzymes papain, amylase Food processingB.subtilis enzymes Detergent factoryPoultry mites, droppings, feathers Poultry farmersFlour grain BakersStorage mites, soyabean, wheat FarmersMidges Fish food manufacturingSilk-worm moths and larvae Silk workersCastor beans, Coffee seeds bean FarmersColophony Electric solderingWood dusts (red cedar, oak,mahogany, etc) Carpenters and Saw mill workersGrain dust (moulds, insects, grain) Shipping workersCotton dust Cotton mill workersStorage mites, fungi, ragweed, pollen Granary workers
2. Organic chemicals.Isocyanates (TDI, MDI, HDI) Plastic and foamAntibiotics, piperazine, methyl dopa ManufacturingDisinfectants Hospital workersParaphenylene diamine Fur dyeingFormaldehyde, ethylene diamine Rubber processingFurfuryl alcohol resin Foundry workersDimethyl ethanolamine toluene di-isocyanate Automobile painting
3. Inorganic chemicals.Platinum salts RefiningNickel salts PlatingCobalt salts Diamond polishingChromium salts Stainless steel weldingAluminium fluoride ManufacturingPersulphate Beauty shopVanadium Refinery workersStainless fumes Welding

26 Bronchial Asthma
Occupational asthma can be mediated by any of the several, mechanisms. They include,reflex vagal bronchoconstriction in response to an irritant effect on specific receptors;inflammatory bronchoconstriction secondary to toxic concentration of gases (nonspecificcomplement activation, neuro-peptide release, disrupted cell membrane releasing arachidonicacid products); direct pharmacological reaction by agents such as organic insecticides(parasympathetic agonists) and beta-adrenergic blocking agents; or by immunologicmechanisms. Some agents also act via alternative path way of complement activation throughan antibody-independent mechanism.
TARTRAZINE AND SULPHITE SENSITIVITY
Tartrazine is a yellow dye commonly employed in food and medications. Beginning in 1958,a number of reports appeared linking this agent with the occurrence of acutebronchoconstriction. The reaction is particularly noted in those with aspirin sensitivity.Although the exact prevalence is not known, there are reports of positive challenge in up to22% of unselected asthma patients and 25-50% of those with aspirin sensitivity. It is not aninhibitor of cyclo-oxygenase. However, the incidence of tartrazine-induced asthma is verylow and perhaps is limited to those rare individuals who appear to have an immunologicallymediated sensitivity to the dye.87
Sulphiting agents88-90 have been used to preserve foods and beverages since ancient times.They maintain the crisp and fresh appearance of the foods, prevent browning, and controlmicrobial growth and spoilage. The agents used include sulphur dioxide as well as thesodium and potassium salts of sulphite, bisulphite, and metabisulphite. All these agentsrelease sulphur dioxide gas under suitable conditions of warmth and acidity. Major sourcesof exposure to sulphites are processed potatoes, shrimp, dried fruits, beer and wine. Anothersource of sulphite exposure for patients with asthma is medication. Sulphites are used toprevent oxidation of beta-adrenergic agonists. For this purpose, sulphites are contained insome nebuliser solutions, injected epinephrine, and injected local anaesthetics containingepinephrine. Except in vary rare individuals with true allergy to sulphites, the amount ofinjected solutions is inconsequential. However, the amount in the nebuliser solutions issufficient to cause paradoxical bronchoconstriction or a blunted bronchodilator responsein these subjects.
Exposure to sulphites, particularly in restaurant salad bars in western countries, or afterdrinking wine or beer, has been reported to be responsible for fatal attacks of asthma andits use has been banned in many countries. Sulphur dioxide released in the mouth andstomach from sulphites has been incriminated as the cause of precipitation of asthma in avast majority of patients. Sulphur dioxide is a known irritant and asthmatics are particularlysusceptible. The levels released from food and beverages may be sufficient to account forthe bronchoconstriction. However, all patients with asthma do not react adversely tosulphites. This may be due to varying extent of inhalation of liberated sulphur dioxide bydifferent patients or there may be a subset of asthmatics, which have low levels of theenzyme sulphite oxidase. These patients will be able to metabolise sulphites to harmlesssulphates. A small number of asthma patients may have true allergy to sulphites, in whoman immediate skin test reactivity can be demonstrated.

Aetiology 27
RHINITIS AND SINUSITIS
A possible relation between sinusitis and activation of asthma has been postulated recently.A high incidence of radiographic evidence of sinusitis on the order of 40 to 60 % has beendemonstrated in asthmatic patients. However, the question is, does this association representan epiphenomenon? There is suggestive clinical evidence that sinusitis not only occurs inassociation with asthma but may also play some role in its pathogenesis. Studies of childrenand adults after medical or surgical therapy indicate that the asthmatic state may improvewith proper management of the underlying sinusitis. It is also likely that nasal and sinuspathology can aggravate asthma, particularly if there is uncontrolled drainage of mucoidor mucopurulent material down the nasopharynx where it can contribute to cough andirritability of larynx. This material may also be aspirated into the lower respiratory tract,especially during sleep. It is also possible, but not proven, that sinus infection may aggravateasthma through reflex mechanisms.91-93
Although historically, it was believed that structurally and functionally there aredifferences within the respiratory tract which have been used as the basis for separating theairway into upper and lower respiratory tracts, it is now being appreciated that allergicrhinitis and bronchial asthma are considered as ‘one airway, and one disease’.94 Theprevalence of asthma and allergic rhinitis is increasing in the general population, and ahigh proportion of new patients have coexisting upper and lower respiratory tract disease.It is estimated that 60 to 70% of patients who have asthma have also coexisting allergicrhinitis. During the past decade with increased understanding, current thinking is emergingthat they should better be described as a continuum of inflammation involving one commonairway. Traditional therapies originally indicated for allergic rhinitis and asthma are beingreassessed to explore their potential utility in both these conditions. Recently, there hasbeen a renewed interest in the role that histamine plays in lower airway disease, and interestin increasing in the theory that leukotrienes, which are more potent inflammatory mediatorsthan histamine, play a role in upper airway disease as well. Because its important role inthe pathogenesis of both airways disease, leukotriene receptor antagonists are recently haveemerged as important therapeutic advances that have potential clinical utility in both asthmaand allergic rhinitis.
GASTRO-OESOPHAGEAL REFLUX (GER)
A number of reports are available in the medical literature on the relationship between gastro-oesophageal reflux (GER) and pulmonary disease. Since the late seventies, numerousinvestigators have reported on epidemiology, mechanisms and clinical trials in an effort topiece together the gastro-oesophageal reflux and asthma. Epidemiological evidence for theassociation suggest that about three-fourth of the asthmatics, independent of the use ofbronchodilators, have acid gastro-oesophageal reflux, increased frequency of reflux episodes,or heart burn, and about 40 per cent have reflux oesophagitis. As early as 1967, Urschel andPaulson reported that of 636 patients scheduled for an operative treatment for GER, 60%also had pulmonary symptoms.95 Since then, many studies have shown a high prevalenceof GER among patients with asthma.96,97 A recent report says that even asthmatics withouthaving reflux symptoms have a high prevalence (62%) of abnormal results for 24-hoursoesophageal tests.98 The simultaneous occurrence of GER and asthma suggests a causal

28 Bronchial Asthma
relationship. The occurrence of GER after bedtime is strongly associated with asthma,respiratory symptoms, and obstructive sleep apnoea syndrome.99
Two separate mechanisms are involved in the gastro-oesophageal reflux and asthmarelationship.99,100
i. Reflex vagal bronchoconstriction occurs secondary to stimulation of sensory nerve fibresin the lower oesophagus. This mechanism is supported by the findings that acid infusionof the oesophagus in asthmatic patients leads to increased airway resistance that rapidlyreverses with antacids and infusion of acid into the lower oesophagus of asthmaticchildren during sleep induces bronchoconstriction.
ii. The second proposed mechanism is micro-aspiration, particularly during sleep. This issupported by the findings of (a) a large vagally mediated increase in airway resistancewith minute quantities of hydrochloric acid infused into the trachea of cats; (b) a highprevalence rate of hiatus hernia and gastro-oesophageal reflux in patients with bronchialasthma and (c) an incidence of gastro-oesophageal reflux in 63 per cent of childrenwith asthma. The prevalence of gastro-oesophageal reflux is increased at least three-folds in both children and adults with bronchial asthma. The evidence for the relationshipalso has gained support from the results of clinical trials.
iii. The partial narrowing or occlusion of the upper airway during sleep, followed by anincrease in intrathoracic pressure, might predispose the patient to nocturnal GER and,consequently, to respiratory symptoms.99 Both medical treatment with antacids andpostural therapy and surgical management of gastro-oesophageal reflux have resultedin improvement of asthma symptoms. However, other studies have not demonstratedsuch a beneficial effect.101-105
Prevalence of gastro-oesophageal reflux in asthmatics can be summarised as follows:106
• 57% of asthmatics have heartburn• 41% of asthmatics note reflux-associated respiratory symptoms• 82% of asthmatics have abnormal oesophageal acid contact times• 43% of asthmatics have oesophagitis• Heartburn is more prevalent in asthmatics over 65 years of age (35%) compared with
asthmatics 18-34 years of age (23%)• Heartburn is associated with a higher rate of future asthma hospitalisation• Subjects reporting nocturnal GER have higher asthma prevalence rates and symptoms
of obstructive sleep apnoea• Proximal oesophageal acid exposure is present in 48% of asthmatics• In children : abnormal oesophageal pH tests are present in 62% and GER is a risk
factor for asthma (OR 1.9).
PSYCHOLOGICAL FACTORS
There has been a great deal of controversy regarding the cause and effect relationship ofasthma and psychological factors. Many patients with asthma acknowledge thatexacerbations are provoked by psychological events, such as shock, bereavement, orexcitement. However, such factors are rarely the dominant cause of disease. Suggestionand hypnosis may have some beneficial effect in modifying the asthmatic reactions.Depression most often associated with asthma may be secondary to a chronic disease. Inrare instances, patients commit suicide. Although the information linking depression and

Aetiology 29
increased death from asthma is derived from clinical reports, the association, however, isstriking. In a review of cases in which children died suddenly and unexpectedly of asthma,there is clinical evidence that the children had expressed despair, hopelessness, a wish todie, and other evidence of depression. Other psychological problems that are documentedas associated with those at increased risk of mortality include alcohol abuse, documenteddepression, recent family loss and disruption, recent unemployment, and schizophrenia.The severe asthmatic attack is very frightening and such patients are understandablyanxious. Occasionally, psychological illness, family disputes or marital disharmony maybe major factors in the aetiology of intractable asthma.107-109
POLLUTION
Pollution with particulate matter adds to the allergenicity of aeroallergens. Passive smokingis known to be a risk factor110 and there is evidence that diesel fumes are associated withincreased allergic responses. Similarly smokers have increased bronchial hyperreactivityto a variety of stimuli. A small increase in allergen exposure will make the airway morereactive, which will result in a large increase in severity and potential deaths. Ozone andother oxidants contained in photochemical smog which occurs in areas of high traffic density,high sunlight and temperature inversions as in Los Angeles and Athens, act as respiratoryirritants and can exacerbate asthma. Similarly other atmospheric pollutants as in highlyindustrialised area containing sulphur dioxide and other smoke particulates can provokeasthma. Indoor air pollution due to cooking fuels such as gas, biomass, and kerosene containoxides of nitrogen and are responsible for increased respiratory symptoms as reported insome studies.111
Other environmental pollutants such as diesel particulates, and noxious gases like ozone,sulphur dioxide, and nitrous oxides may be important in the development in youngchildren.112
Air pollution is partly being incriminated as a possible contributing factor in the recentrise in the prevalence, morbidity and mortality of asthma globally.113 Although recent studieshave not established a direct causal relation of air pollution and bronchial asthma, there isnow substantial evidence that air pollution can contribute significantly to asthma morbidityand mortality. Ambient levels of air pollutants exacerbate mucosal inflammation in asthmaticairways, can affect lung function, and potentiate inhaled response to aeroallergens. Emissionsfrom motor vehicles are a major source of these pollutants. Retrospective analysis of pollutionepisodes in the world history (Meuse Valley, Belgium -1930; Donnora, Pennsylvania-1948;London 1952) have identified a link between respiratory morbidity and mortality and highlevels of sulphur dioxide and black smoke, although these studies were not primarilyfocussed to study the association between asthma and air pollution.114-116 Reports from theTokyo-Yokohama area of Japan where USA soldiers were based, revealed many cases ofasthmatic bronchitis characterised by cough, wheeze, and breathlessness associated witheosinophilia and positive skin prick tests. This area experienced smog as it was highlyindustrialised and surrounded by hills. These individuals experienced relief of theirsymptoms when they moved out to less polluted areas. This entity is known as “Tokyo-Yokohama asthma”. However, since the levels of pollutants were not measured, this couldnot be attributed to any specific pollutant.117-119 Other studies from Yokkaichi, Japan,120
Birmingham, UK,121 Seattle,122 Utah valley,123 Southern Ontario and Toranto124-126 have shown

30 Bronchial Asthma
positive correlations between asthma exacerbations and SO2, ozone, fine particulate matter,and sulphates.127-129 Indoor air pollution is a contributory factor in exacerbation of bronchialasthma.130
Environmental tobacco smoke is important in the development of childhood asthmaand in the worsening of asthma in children and adults.131 The earlier and the greater thedegree of environmental tobacco smoke, the greater the likelihood of asthma developing inchildren. In infants exposed to prenatal and postnatal cigarette smoking, have altered lungfunction.130,132,133 These limitations in lung functions may be secondary to smaller lung size,and less maturity of lungs secondary to in utero lung growth retardation because of persistentexposure of lungs to nicotine.134-136 Increased bronchial responsiveness after birth occurs ininfants exposed to maternal smoking.137 Infants exposed to smoking are at increased risk ofdeveloping asthma later in life.138-146 It is, however, not clear whether increased bronchialreactivity after birth plays a role, if any, in the development of asthma. It is also not clearwhether the increased bronchial reactivity in these infants is purely genetic, or whether it isthe result of lung injury from exposure to cigarette smoke.
Asthmatic smokers have increased hyperresponsiveness to methacholine.147 Asthmaticsmokers have higher sputum total cell and neutrophil numbers and IL-8 concentrationscompared to asthmatic nonsmokers. In contrast, sputum eosinophils and eosinophil-cation-protein levels are higher in nonsmoking than smoking asthmatics, suggesting a normalisingeffect of smoking on the Th1/Th2 balance. Thus upon the eosinophils inflammation, smokinginduces neutrophilic airway inflammation in asthma.147 Further, smoking asthmatics showno improvement in lung function, airway hyperresponsiveness, and sputum eosinophiliaon treatment with steroid inhalation.148 This decreased steroid responsiveness is responsiblefor the faster decline in FEV1 seen in smoking asthmatics.
ENDOCRINAL FACTORS
Although the exact role of hormones in asthma has not been defined, a number of patientscomplain of exacerbation of their symptoms during or preceding menstruation.Retrospective studies suggest that in approximately one-third of women, asthma becomesworse during pregnancy; in one-third, it becomes better; and in one-third, it remainsunchanged. In women in whom asthma becomes worse during pregnancy, peak severityoccurs at 29-36 weeks of gestation. Asthma becomes less severe during the last 4 weeks ofpregnancy. The change in the severity of asthma during pregnancy is sometimes dramaticand tends to be consistent in subsequent pregnancies. Most patients return to a pre-pregnancy level of severity by 3 months of postpartum.149-152 There may be an improvementin airway responsiveness during pregnancy that is greatest in those with the mosthyperresponsive airway initially. It is also reported that improvements in responsivenessare associated with improvements in clinical asthma severity. However, progesterone alonedid not appear to be the sole contributor to these improvements. It is also suggested thatoestrogen plays a role in the pathophysiology of asthma and long-term use and/or highdoses of postmenopausal hormone therapy increase subsequent risk of asthma.153
Several observations have been made on the influence of thyroid hormones on asthma.Hyperthyroidism is accompanied by many manifestations suggesting over stimulation ofthe sympathetic system and this condition is a contraindication for use of β-2 agonists. One,therefore would expect that patients of bronchial asthma, who in addition develop hyper-thyroidism, should either have a decreased requirements of bronchodilators or ameliorationof their symptoms. However, the reverse has been observed. Asthmatics who develop

Aetiology 31
hyperthyroidism, do far worse than euthyroid asthmatics. In some hyperthyroid asthmaticsfollowing treatment of hyperthyroidism, not only asthma improves, but in rare instancesthey become completely asymptomatic. Similar discrepancies have also been observed inhypothyroidism. Various mechanisms such as changes in beta adrenoceptor activity andaltered prostaglandin metabolism have been proposed to explain these observations.Bronchodilator response is impaired in the presence of excess thyroid hormones, whichimproves after euthyroid state is achieved.154
GENETICS AND ASTHMA
Genetic factors play a contributing role in the pathogenesis of asthma.155 There are severalstudies indicating familial aggregation of asthma. It is a frequent clinical observation thatasthma runs in families. Moreover, other atopic conditions like allergic rhinitis and atopicdermatitis are common among the family members of the asthma patients. The concordanceof asthma in monozygotic (MZ) twins is reported to be significantly greater than that indizygotic (DZ) twins. Though the dosage of inhaled antigens and other factors influencethe likelihood of clinical disease, recent family studies suggest that atopy is dominantlyinherited. Molecular genetic linkage studies indicate that the “atopy” gene locus is onchromosome 11.156-159
Cytokines are important components in the pathogenesis of asthma (see below). Thegenes for these cytokines are encoded in a small region in the long arm of chromosome 5and a number of them are coordinately regulated. T cells that differentiate along this routeand preferentially release cytokines of the IL-4 gene cluster are called Th2-like. These Th2-like lymphocytes and their cytokines are over represented in tissue biopsy studies in patientswith allergic diseases. The chromosome 5 contains an IL-4 gene cluster which encodes theallergic cytokines IL-3,4,5,9,13 and GM-CSF (granulocyte macrophage colony-stimulatingfactor). This gene is closely linked to inheritance of an increased IgE response and to increasedbronchial hyperresponsiveness. Further, human genome studies have revealed that allergicdiathesis is linked with a region on the long arm of chromosome 12 which contains thegene encoding interferon-γ) (INF-γ), which is a powerful suppressor of Th2 responses. It isestablished that there is a reciprocal relationship between Th2 and Th1 responses withIL-10 derived from Th2 cells inhibiting Th1 responses while INF-γ generated by Th1 cellsinhibits Th2 responses. It is possible that, in allergic diseases like asthma, there is an increasein the expression of genes, which regulates Th2 cytokines, a decrease in expression of genesthat regulate INF-γ production, or a combination of both.160
The other genetic component that plays an important role is the ability of a susceptibleindividual to recognise an environmental allergen as foreign and starts an allergic immuneresponse. This component operates through the human lymphocyte antigen (HLA, or MHCclass II) molecules HLA-DR, HLA-DP, HLA-DQ, which provide the mechanism for antigenrecognition and presentation to and by T and B lymphocytes.160
Many candidate genes and positional cloning have recently been identified.161 The firstgenome-wide screen for linkages to asthma identifies linkages on chromosomes 4q,6 (nearthe major histocompatibility complex, (MHC), 7,11q containing FcεR1-β, 13q and 16.Linkages have been confirmed to chromosomes 4,11,13 and 16. Suggestive evidences arealso found for linkages and replication for loci on chromosomes 5q, 12q, 19q, and 21q.Different linkages have been reported from different ethnic groups. The loci most consistentlyand robustly identified are on chromosomes 5, 6, 12 and 13.

32 Bronchial Asthma
Various candidate genes those have been implicated in atopy and asthma are summarisedin Table 2.2.162
Several studies have shown that polymorphisms in the β2 adrenergic receptor geneinfluences responsiveness to β-agonists. Similarly polymorphisms in the 5-lipoxygenase geneand the leukotriene C4 synthase gene have been associated with response to medications thattarget leukotriene metabolism. These findings suggest the potential for pharmacogenetictailoring of therapy in individual asthmatic patients.163
Environmental risk factors for development of asthma are summarised in Table 2.3.
Table 2.2: Candidate genes for asthma
Chromosome Gene
1 IL-105 IL-4 promoter
IL-5IL-9IL-12BIL-13GM-CSFCD 14β2 adrenergic receptor
6 TNF-αHuman leukocyte antigens
11 FcεR1-β,CC16
12 Interferon γ13 STAT 514 T-cell receptor α/β complex16 IL-4 receptor (IL-4α)
Table 2:3: Environmental risk factors for the development of bronchial asthma
Allergens Food allergensInhalant allergens
Pollutants Environmental tobacco smokeDiesel particulatesNoxious gases (ozone, SO2, NO2)
Infections ViralRespiratory syncytial virusParainfluenza virusHuman rhinovirus
BacterialMycobacteriumChlamydia Mycoplasma Lactobacillus
Dietary modifications ωωωωω3 fatty acidsVitamins
AntioxidantsLectins

Aetiology 33
REFERENCES
1. Lau S, Illi S, Sommerfeld C et al. Early exposure to house-dust mite and cat allergens anddevelopment of childhood asthma: A cohort study. Multicentre Allergy Study Group. Lancet2000;356:1392-97.
2. Pearce N, Pekkanen J, Beasley R. How much asthma is really attributable to atopy? Thorax1999;54:268-72.
3. Pearce N, Douwes J, Beasley R. Is allergen exposure the primary cause of asthma. Thorax2000;55:424-31.
4. Ponsonby AL, Garenby P, Glagow N, Mullins R, McDonald T, Hurwitz M. Which clinicalsubgroup within the spectrum of child asthma are attributable to atopy? Chest 2002;121:135-42.
5. Host A, Halken S. The role of allergy in childhood asthma. Allergy 2000;55:600-08.6. Weissman DN. Epidemiology of asthma. Severity matters. Chest 2002;121:6-8.7. Burrows B, sears MR, Flannery EM et al. Relations of bronchial responsiveness to allergy skin
test reactivity, lung function, respiratory symptoms, and diagnoses in thirteen-year-old NewZealand children. J Allergy Clin Immunol 1995;95:548-56.
8. Soriano JB, Anto JM, Sunyer J et al. Risk of asthma in the general Spanish population attributableto specific immune response. Int J Epidemiol 1999;28:728-34.
9. Toelle BG, Peat JK, Salome CM et al. Toward a definition of asthma for epidemiology. Am RevRespir Dis 1992;146:633-37.
10. Sporik K, Holgate ST, Platts-mills TA, Cogswell JJ. Exposure to house dust mite allergen (DerpI) and development of asthma in childhood. A prospective study. NEJM 1990; 323:502-07.
11. Anto JM, Sunyer J, Reed CE, et al. Preventing Asthma epidemics due to soyabean by dust-control measures. N Engl J Med 1993; 329:1760-63.
12. O’HallarenMT, Yaginger JW, Offord KP et al. Exposure to an aeroallergen as possible precipitatingfactor in respiratory arrest in young patients with asthma. N Engl J Med 1991;324:359.
13. Colloff MJ, Ayres J, Carswell F, Howarth PH, Merrett TG, Mitchell EB, Walshaw MJ, Warner JO,Warner JA, Woodcock AA. The control of allergens of dust mites and domestic pets: a positionpaper. Clin Exp Allergy. 1992; 22 Suppl 2:1-28.
14. Platts-Mills TA, Tovey ER, Mitchell EB, Moszoro H, Nock P, Wilkins SR. Reduction of bronchialhyperreactivity during prolonged allergen avoidance. Lancet. 1982; 2:675-8.
15. Shivpuri DN, Dua KL. Studies in pollen allergy in Delhi area. Part IV: Clinical investigations.Ind J Med Res 1983;51:68.
16. Gupta KD, Agarwal RG, Sulemani AA. Pollination calendar of allergenic plants of Bikaner(Rajsthan), Part 3; Investigations of pollens. J Asso Phys India 1969;17:225.
17. Agnihotri MS, Singh AB. Observations on pollinosis in Lucknow with special reference tooffending factors. Aspects Allergy Appl Immunol 1971;5:135-45.
18. Jha VK, Sundaramma M, Mishra SK, Joshi M. Clinical study with some airborne pollens anddust as respiratory allergens Around Varanasi. Ind J Chest Dis 1975;10:155.
19. Tiwari UC. Studies in allergenic pollen in Bhopal atmosphere. Aspects Allergy Appl Immunol1978;10:65-72.
20. Mittal OP, Katiyar SK. Results of intradermal tests by various allergens in cases of nasobronchialallergy in Kanpur. Aspects Allergy Appl Immunol 1978;11:188-97.
21. Chaubal PD, Gadve SB. Study of pollen allergy in Kolhapur during monsoon. Ind J Chest Dis1984;26:38-40.
22. Sporik et al. Clin Exp Allergy 1993;23:74023. Nelson HS. The atopic diseases. Ann Allergy 1985;55:441.24. Zimmerman B, Feanny S, Reisman J et al. Allergy in asthma I. The dose relationship of allergy to
severity of childhood asthma. J Allergy Clin Immunol 1988;81:63.25. Martoin AJ, Landau LI, Phelan PD. Natural history of allergy in asthmatic children followed to
adult life. Med J Aust 1981;2:470.

34 Bronchial Asthma
26. Zimmerman B, Chambers C, Forsyth L. Allergy in asthma. II. The highly atopic infant and chronicasthma. J Allergy Clin Immunol 1988;81:71.
27. Inouye T, Tarlo S, Broder I et al. Severity of asthma in skin-test negative and skin-test positivepatients. J Allergy Clin Immunol 1985;75:313,
28. Murray AB, Ferguson AC, Morrison B. The seasonal variation of allergic symptoms induced byhouse dust mites. Ann Allergy 1980;45:347.
29. Remes ST, Castro-Rodriguez JA, Holberg CJ et al. Dog exposure in infancy decreases thesubsequent risk of frequent wheeze but not of atopy. J Allergy Clin Immunol 2001;108:509-15.
30. Hessaelmar B, Aberg N, Aberg B et al. Does early exposure to cat or dog protect against laterallergy development? Clin Exp Allergy 1999;29:611-17.
31. Platts-Mills T, Vaughan J, Squillace S et al. Sensitisation, asthma, and a modified Th2 responsein children exposed to cat allergen: a population based cross-sectional study. Lancet 2001;357:752-56.
32. Arruda LK, Chapman MD. The role of cockroach allergens in asthma. Curr Opinion Pulm Med2001;7:14-19.
33. Platts-Mills T, Vervloet D, Thomas W et al. Indoor allergens and asthma. Report of the thirdInternational workshop. J Allergy Clin Immunol 1997;100:S1-S24.
34. Rosenstreich D, Eggleston P, Kattan M et al. The role of cockroach allergy and exposure tocockroach allergen in causing morbidity among inner-city children with asthma,. New Engl JMed 1997;336:1356-63.
35. Eggleston P, Rosenstreich D, Lynn H et al. Relationship of indoor allergen exposure to skin testsensitivity in inner-city children with asthma. J Allergy Clin Immunol 1998;102:563-70.
36. Gelber L, seltzer I, Bouzoukis J et al. Sensitisation and exposure to indoor allergens as riskfactors for asthma among patients presenting to hospital. Am Rev Respir Dis 1993;147:573-78.
37. Rogers L, Cassino C, Berger KL et al. Asthma in the elderly. Cockroach sensitisation and severityof airway obstruction in elderly nonsmokers. Chest 2002;122:1580-86.
38. Park JK, Li J. Reversing the trend: Reducing the prevalence of asthma. J Allergy Clin Immunol1999;103:1-10.
39. Oddy WH, Holt PG, Sly PD et al. Association between breast-feeding and asthma in 6-year-oldchildren: findings of prospective birth cohort study. BMJ 1999;319:815-19.
40. Wright AL, Holberg CJ, Taussig LM et al. Factors influencing the relation of infant feeding toasthma and recurrent wheeze in childhood. Thorax 2001;56:192-97.
41. Hodge L, Salome CM, Peat JK et al. Consumption of oily fish and childhood asthma risk. Med JAust 1996;164:137-40.
42. Hodge L, Salome CM, Hughes JM et al. Effect of intake of dietary omega –3 and omega-6 fattyacids on severity of asthma in children. Eur Respir J 1998;11:361-65.
43. Britton JR, Pavord I, Richards K et al. Dietary antioxidant vitamins intake and lung function ingeneral population. Am J Respir Crit Care Med 1995;151:1383-87.
44. Fogarty A, Lewis S, Weiss S et al. Dietary vitamin E, IgE concentrations and atopy. Lancet2000;256:1573-74.
45. Watzl B, Neudecker C, Hansch GM et al. Dietary wheat germ agglutinin modulates albumin-induced immune responses in Brown Norway rats. Br J Nutr 20001;85:483-90.
46. Bailey WC, Clark NM, Gotsch AR, et al. Asthma prevention. Chest 1992;102(Suppl):216S-231S.47. Sheinman BD, Devalia JL, Crook SJ, Tabaqchali S. Synthesis of histamine by Haemophilus
influenzae. Br Med J 1986;292:857.48. Ten Brinke A, van Dissel JT, Sterk PJ et al. Persistent airflow limitation in adult onset nonatopic
asthma is associated with serologic evidence of Chlamydia pneumoniae infection. J Allergy ClinImmunol 2001;107:449-54.
49. Holt PG, Sly PD, Bjorksten B. Atopic versus infectious diseases in childhood: A question ofbalance? Pediatr Allergy Immunol 1997;8:53-58.

Aetiology 35
50. Strachan DP. Family size, infection and atopy. The first decade of the “hygiene hypothesis”.Thorax 2000;55(Suppl 1):S2-S10.
51. Kallomaki M, Salminen S, Arvilommi H et al. Probiotics in primary prevention of atopic disease;a randomised placebo-controlled trial. Lancet 2001;357:1076-79.
52. Stevenson DD, Simon RO. Aspirin sensitivity: Respiratory and cutaneous manifestations, In.Middleton E, Reed C, Ellis EF et al (Eds). Allergy Principles and Practice, 3rd Ed, St Luis, CVMosby, 1988.
53. Stevenson SD. Diagnosis, prevention and treatment of adverse reactions to aspirin and NSAIDs.J Allergy Clin Immunol 1984;74:617.
54. Ayers JG, Flemming DM, Whittington RM. Asthma death due to ibuprofen. Lancet. 1987;i:1082.55. Raine JM, Palazzo MG, Kerr JH, Sleight P. Near fatal bronchospasm after oral nadolol in a
young asthmatic and response to ventilation with halothane. Br Med J 19821;282:548.56. McGarigle P, Tribe AE. Eye drop induced asthma. Med J Aust 1985;142:425.57. Barnes PJ. Our changing understanding of asthma. Respiratory Med 1989;83(Suppl A):17.58. Harman E, Hill M, Piper JA, Hendeles L. Inhaled verapamil-induced bronchoconstriction in
mild asthma. Chest 1991;100:17.59. Szceklik A, Stevenson DD. Aspirin induced asthma: Advances in pathogenesis and management.
J Allergy Clin Immunol 1999;104:5-13.60. Stevenson DD, Simon RA. Sensitivity to aspirin and nonsteroidal anti-inflammatory drugs. In:
Middleton E, reed CE, Ellis EF et al, (Eds). Allergy: Principles and Practice. 5th Eds. St Louis,MO, Mosby, 1998;1225-34.
61. Smith CM, Hawksworth RJ, Thien FC et al. Urinary leukotriene E4 in bronchial asthma. EurRespir J 1992;5:692-99.
62. Christie PE, Tagari P, Ford-Hutchinson AW et al. Urinary E4 concentrations increase after aspirinchallenge in aspirin-sensitive asthma patients. Am Rev Respir Dis 1991;143:1025-29.
63. Yamamoto H, Nagata M, Kiramtsu K et al. Inhibition of analgesic-induced asthma by leukotrienereceptor antagonist ONO-1078. Am J Respir Crit Care Med 1994;150:254-57.
64. Obase Y, Shimoda T, Tomari S, et al. Effects of pranlukast on chemical mediators in inducedsputum on provocation tests in atopic and aspirin-intolerant asthmatic patients. Chest2002;121:143-50.
65. Dahlen B, Nizankowska E, Szceklik A, et al. Benefits from adding the 5-lipoxygenase inhibitorzyluton to conventional therapy in aspirin-induced asthmatics. Am J Respir Crit Care Med1998;157:1187-94.
66. Freed AN, Stream CE. Airway cooling: Stimulus specific modulation of airway responsivenessin the canine lung periphery. Eur Respir J 1991;4:568.
67. McFadden ER. Exercise and asthma. N Engl J Med 1987;317:502.68. Anderson SD. Issues in exercise-induced asthma. J Allergy Clin Immunol 1985:76:763.69. Godfrey S. Controversies in pathogenesis of EIA. Eur Respir J 1986;68:81.70. McFadden ER Jr, Ingram RH Jr. Exercise-induced asthma: Observations on the initiating stimulus
(Correction of published data) N Engl J Med. 1984; 311:1127-8.71. Ingenito EP, Pichurko BM, Caeurfur J, Drazen JM, Ingram RH Jr., and Solway J. Breathing pattern
after respiratory heat loss but not bronchoconstrictor response in asthma. Lung 1990; 168:23-34.72. Rubinstein I, Levison H, Slutsky As et al. Immediate and delayed bronchoconstriction after
exercise in patients with asthma. N Engl J Med 1987;317:482-8573. Karjalainen J. Exercise response in 404 young men with asthma: No evidence for a late asthmatic
reaction. Thorax 1991;46:100-04.74. McFadden ER Jr, Kimberly AM, M. Lennes, Strohl KP. Post exertional airway rewarming and
Thermal induced asthma. J Clin Invest 1986;78:18-25.75. Reiff DB, Choudary NB, Pride NB, Ind PW. Effect of prolonged submaximal warm up exercise
on exercise induced asthma. Am Rev Respir Dis 1989; 139:479-84.

36 Bronchial Asthma
76. Ross DJ. Ten-years of the SWORD project. Surveilance of work-related and occupationalrespiratory diseases. Clin Exp Allergy 1999;29:750-53.
77. Hendrick DJ, Burje PS,. Asthma. In. Hendrick DJ, Beckett W, Burge PS, charge A (Eds).Occupational disorders of the lung. Recognition, Management, and prevention. London WBSaunders, 2002:33-76.
78. Banks DE, Wang ML. Occupational asthma: “the big picture”. Occup Med 2000;15:335-58.79. Meredith S, Nordman H. Occupational asthma: Measures of frequency from four countries.
Thorax 1996;51:435-40.80. Banks DE, deSazo RD. An overview of occupational asthma, principles of diagnosis and
management. Immunol Allergy Pract 1985;7:122.81. Brooks SM. Occupational asthma. Chest 1987;87:218S.82. Chan-Yeung M, Lam S. Occupational asthma. Am Rev Respir Dis 1986;133:686.83. Environmental and occupational asthma. Guest Editor Merchant JA. Chest 1990;98:146S.84. Ross DJ. Twenty years of the SWORD project: Surveillance of work-related and occupational
respiratory disease. Clin Expt Allergy 1999;29:750-53.85. Hendrick DJ, Burge PS. Asthma. In. Hendrick DJ, Beckett W, Burge PS, Churg A (Eds).
Occupational disorders of the lung: Recognition, management, and prevention. London WBSaunders, 2002;15:335-58.
86. Banks DE, Wang ML. Occupational asthma: The big picture. Occup Med 2000;15:335-58.87. Stevenson DD, Simon RA, Lumry WR, Mathison DA. Adverse reactions to tartrazine. J Allergy
Clin Immunol 1986;78:182-90.88. Simon RA. Sulfite challenge for the diagnosis of sensitivity. Allergy Proc 1989;10:357-62.89. Bush RK, Taylor SL, Busse W. A critical evaluation of clinical trials in reactions to sulfites. J
Allergy Clin Immunol 1986;78:191-202.90. Taylor SL, Bush RK, Selner JC et al. Sensitivity to sulfited foods among sulfite-sensitive subjects
with asthma. J Allergy Clin Immunol 1988;81:1159-67.91. Slavin RG. Sinusitis—Present state of the art. Allergy Proc 1991;12:163.92. Rachelefsky GS, Katz RM, Siegel SC. Chronic sinus disease with associated reactive airway
disease in children. Paediatrics 1984;73:526.93. Mings R, Friedman WH, Linford PA, Slavin RG. Five year follow-up of the effects of bilateral
intranasal sphino-ethmoidectomy in patients with sinusitis and asthma. Am J Rhinology1987;79:247.
94. Grossman J. One airway, one disease. Chest 1997;111:11S-16S.95. Uschel HC, Paulson DL. Gastroesopgeal reflux and hiatus hernia: Complications and therapy.
Thorax Cardiovascular Surg1967;53:21-32.96. Sontag SJ. Gastro-oesophageal reflux and asthma. Am J Med 1997;103(Suppl 1):84S-90S.97. Harding SM, Sontag SJ. Asthma and gastrooesophageal reflux. Am J Gastroenterol 2000;95
(Suppl):S23-S32.98. Harding SM, Guzzo MR, Richter JE. The prevalence of gastro-oesophageal reflux in asthma
patients without reflux symptoms. Am J Respir Crit Care Med 2000;162:34-39.99. Gislason T, Janson C, Vermeire P et al. Respiratory symptoms and nocturnal gastro-oesophageal
reflux. A population-based study of young adults in three European countries. Chest 2002;121:158-63.
100. Harding SM. Nocturnal asthma: Role of nocturnal gastresophageal reflux. Chronbiol Int1999;16:641-62.
101. Nelson HS. Worsening asthma: is reflux esophagitis to blame? J Rev Respir Med 1990;11:827-44.102. Sontag SJ. Gut feelings about asthma. The burp and the wheeze. Chest 1991;99:1321.103. Mays EE. Intrinsic asthma in adults: Association with gastrooesophageal reflux. JAMA
1976;236:2626.

Aetiology 37
104. Field SK. Sutherland LR. Does medical antireflux therapy improve asthma in asthmatics withgastrooesophageal reflux. Chest 1998;114:275-83.
105. Field SK, Gelfand GAJ, McFadden SD. The effect of antireflux surgery in asthmatics withgastrooesophageal reflux. Chest 1999;116:766-74.
106. Harding SM. Acid reflux and asthma. Curr Opin Pulm Med 2003;9:42-45.107. Miller BD. Depression and asthma: A potentially lethal mixture. J Allergy Clin Immunol
1987;80:481-86.108. Strunk RC. Identification of the fatality-prone subjects with asthma. J Allergy Clint Immunol
1989;83:477-85.109. Rea HH, Scragg R, Jackson R, Beaglehole E, Fenwick J, Southerland D. A case control study of
deaths from asthma. Thorax 1986;41:833-39.110. Gupta D, Aggarwal AN, Kumar R, Jindal SK. Prevalence of bronchial asthma and association
with environmental tobacco exposure in adolescent school children in Chandigarh, North India.Journal of Asthma 2001; 38:501-07.
111. Behera D, Jindal SK. Respiratory symptoms in Indian women using domestic cooking fuels.Chest 1991;100:385.
112. Weiss ST, Tager IB, Schenker M et al. The health effects of involuntary smoking. Am Rev RespirDis 1983;128:933-42.
113. Krishna MT, Chauhan AJ, Frew AJ. Air pollution and asthma: What is the association? ClinAsthma Rev 1997;1:7-12.
114. Department of Health, Committee on the Medical Effects of Air Pollutants. Asthma and OutdoorAir Pollution. HMSO, London 1995.
115. Ministry of Health. Mortality and Morbidity during the London Fog of 1952. HMSO, London1954.
116. Schrenk HH, Heinmann H, Claydon GD, Gafarar WM, Wexler H. Air pollution in Donora PA:Epidemiology of the unusual smog episode of October 1948. Public Health Bulletin No 306.Washington D: Public Health Service 1949.
117. Phleps HW, Koike S. Tokyo-Yokohama asthma. The rapid development of respiratory distresspresumably due air pollution. Am rev Respir Dis 1962;86:55-63.
118. Oshima Y, Ishizaka T, Miyamato T, Kabe J, Makino S. A study of Tokyo-Yokohama asthmaamong Japanese. Am Rev Respir Dis 1964;90:632-34.
119. Tremonti LP. Tokyo-Yokohama asthma. Ann Allergy 1970;28:590-95.120. Kitagawa T. Cause analysis of Yokkaichi asthma episode in Japan. JAPCA 1984;34:743-46.121. Walters S, Griffiths RK, Ayers J. Temporal association between hospital admissions for asthma
in Birmingham and ambient levels of sulphur dioxide and smoke. Thorax 1994;49:79-83.122. Schwartz J, Slater D, Larson TV, Pierson WE, Koenig JQ. Particulate air pollution and hospital
emergency room visits for asthma in Seattle. Am Rev Respir Dis 1993;147:826-31.123. Pope CA. Respiratory disease associated with community air pollution and a steel mill, Utah
Valley. Am J Public Health 1989;79:623-28.124. Bates DV, Sitzo R. Relationship between air pollutant levels and hospital admissions in Southern
Ontario. Can J Public Health 1983;74:117-22.125. Bates DV, Sitzo R. Air pollutant levels and hospital admissions in Southern Ontario: The acid
summer haze effect. Environ Res 1987;43:317-31.126. Bates DV, Sits R. The Ontario air pollution study; Identification of the causal agent. Environ
Health Perspect 1989;79:69-72.127. Committe of the Environmental and occupational health Assembly of the American Thoracic
Society. Health effects of outdoor air pollution. Am J Respir Crit Care Med 1996;153:3-50.128. Pande JN, Bhatta N, Biswas D, Pandey RM, Ahluwalia G, Siddaramaiah NH, Khilnani GC.
Outdoor Air pollution and emergency room visits at a Hospital in Delhi. Indian J Chest DisAllied Sci 2002; 44:13-19.

38 Bronchial Asthma
129. Cunningham J, O’Connor GT, Dockery DW, Speizer FE. Environmental tobacco smoke, wheezing,and asthma in children in 24 communities. Am J Respir Crit Care Med 1996;153:218-24.
130. Martinez FD, Wright AL, Tausig LM et al. Asthma and wheezing in the first six years of life.New Engl J Med 1995;332:133-38.
131. Committee of the Environmental and occupational Health Assembly of the American ThoracicSociety: Health of outdoor air pollution. Am J Respir Crit Care Med 1996;153:3-50.
132. Stick SM, Burton PR, Gurrin L et al. Effect of maternal smoking during pregnancy and a familyhistory of asthma on respiratory function in newborn infants. Lancet 1996;348:1060-64.
133. Tager IB, Ngo L, Hanrahan JP. Maternal smoking during pregnancy: effects on lung functionduring the first 18 months of life. Am J Respir Crit Care Med 1995;152:977-83.
134. Chen MF, Kimizuka G, Wang NS. Human fetal lung changes associated with maternal smokingduring pregnancy. Pediatr Pneumonol 1987;3:51-58.
135. Lieberman E, Torday J, Barbieri R et al. Association of intrauterine cigarette smoke exposurewith indices of fetal lung maturation. Obstet Gynecol 1992;79:564-70.
136. Sheikh S, Goldsmith LJ, Howell L et al. Comparison of lung function in infants exposed tomaternal smoking and in infants with a family history of asthma. Chest 2002;116:52-58.
137. Young S, Le Souef PN, Greelhoed GC et al. The influence of family history of asthma and parentalsmoking on airway responsiveness in early infancy. New Engl J Med 1991;324:1168-73.
138. Infante-Rivard C. Childhood asthma and indoor environmental risk factors. Am J Epidemiol1003;137:834-44.
139. Arshad SH, hide DW. Effects of environmental factors on the development of allergic disordersin infancy. J Allergy Clin Immunol 1992;90:235-41.
140. Weitzman M, Gortmaker S, walker DK et al. Maternal smoking and childhood asthma. Pediatrics1990;85:505-11.
141. Stoddard JJ, Miller T. Impact parental smoking on the prevalence of wheezing respiratory illnessin children. Am J Epidemiol 1995;141:96-102.
142. Lewis S, Richards D, Bynner J et al. Prospective study of risk factors for early and persistentwheezing in childhood. Eur Respir J 1995;8:349-56.
143. Weiss ST. Environmental tobacco smoke and asthma. Chest 1993;104:991-92.144. Martinez FD, Cline M, Burrows B. Increased incidence of asthma in children of smoking mothers.
Pediatrics 1992;89:21-26.145. Ehrlich RL, Tott DD, Jordan E et al. Risk factor for childhood asthma and wheezing: Importance
of maternal and household smoking. Am J Respir Crit Care Med 1996;154:681-85.146. Wright AL, Holberg C, Martinez FD et al. Relationship of parental smoking to wheezing and
non-wheezing lower respiratory tract illness in infancy. J Pediatr 1991;118:207-14.147. Chalmers DW, MacLeod KJ, Thomson L et al. Smoking and airway inflammation in patients
with mild asthma. Chest 2001;120:1917-22.148. Chalmers DW, MacLeod KJ, Little SA et al. Influence of cigarette smoking on inhaled
corticosteroid treatment in mild asthma. Thorax 2002;57:226-30.149. Stenius-Aarniala B, Piirila P, Teramo K. Asthma and pregnancy; a prospective study of 198
pregnancies. Thorax 1988;43:12-18.150. Editorial. Pregnancy and the asthmatic. Respir Med 1991;85:451.151. Schatz M, Harden KM, Forsythe A et al. The course of asthma during pregnancy, postpartum,
and with successive pregnancies: a prospective analysis. J Allergy Clin Immunol 1988;81:509.152. Schatz M, Zeiger RS, Harden KM et al. The safety of inhaled β-agonist bronchodilators during
pregnancy. J Allergy Clin Immunol 1988;82:686.153. Troisi RJ, Speizer FE, Willett WC, trichopoulos D, Rosner B. Menopause, postmenopausal
estrogen preparations, and the risk of adult-onset asthma: A prospective cohort study. Am JRespir Crit Care Med 1995;152:1183-88.

Aetiology 39
154. Behera D, Roy R, Dash RJ, Jindal SK: Airway response to inhaled fenoterol in hyperthyroidpatients before and after treatment. J Asthma 1992; 29:307, 369-74.
155. Meyers DA, Bleecker ER. Approaches to mapping genes for allergy and asthma. Am J Crit CareMed 1995;152:411-13.
156. Hopkin J. Genetics and lung disease. Advances in our understanding of emphysema, cysticfibrosis, and asthma. Br Med J 1991;302:1222.
157. Cookson WOCM, Hopkin JM. Dominant inheritance of atopic immunoglobulin-E responsiveness.Lancet 1988;i:86.
158. Cookson WOCM, Sharp P, Faux J, Hopkin JM. Linkage between immunoglobulin-E responsive-ness underlying asthma and rhinitis and chromosome 11q. Lancet 1989;i:1292.
159. Nieminen MM, Kaprio J, Koskenvuo M. A population-based study of bronchial asthma in adulttwin pairs. Chest 1991;100:70.
160. Holgate ST. Asthma and allergy: Interaction of immunology and environment. PulmonaryPerspectives 1997;14:4-6.
161. Cookson WOC. Asthma genetics. Chest 2002;121:7S-13S.162. Sandford A, Pare P. The genetics of asthma: The important questions. Am J Respir Crit Care
Med 2000;161:S202-S206.163. Joos L, Sandford AJ. Genotype predictors of response to asthma medications. Curr Opin Pulm
Med 2002;8:9-15.

40 Bronchial Asthma
Pathophysiology ofBronchial Asthma
3
Bronchial asthma is a disease characterised by wide variation over short periods of time inthe resistance to flow in the airways. The hallmark of the disease is the airflow obstruction.Most asthma is of allergic origin. In this form, it is viewed as a sum of three features: theearly asthmatic reaction (EAR); the late asthmatic reaction (LAR); and bronchialhyperresponsiveness, with varying contribution from each. The cellular response in LARin non-allergic asthma is similar, but little is known of the underlying aetiology. Threefactors narrow airway caliber to limit the flow:
• Airway smooth muscle contraction;• Gland and epithelial secretions and exudation into the airway lumen; and• Inflammatory oedema and vasodilatation (hyperaemia).
Early Asthmatic Reaction (EAR)
In atopic individuals, bronchial challenge/inhalation of appropriate antigens will elicit anearly response, which is maximum at 15 minutes and characterised by smooth musclecontraction, exudation of plasma, and mucus production. This reaches its peak in aboutthirty minutes and resolves within 90-180 minutes. This early reaction is IgE dependentand is the result of IgE binding to mast cells by its Fc portion and to specific antigens by itsF(ab) portion. When IgE-sensitised mast cells are exposed to antigen against which the IgEmolecule is directed, pre-formed and newly generated mediators are released.1 These can bedetected in the blood as they overflow into the circulation, in bronchoalveolar lavage fluid,and as metabolites in the urine and include histamine, prostaglandin D2, and leukotriene C4from airway mast cells.2 This early response is due to the release of histamine. This reactioncan be prevented by pre-medication with sodium cromoglycate and nedocromil sodium andβ-2 agonists,3 but not with steroids.
Late Asthmatic Reaction (LAR) and Bronchial Hyperreactivity (BHR)
The EAR is followed by a complete or partial recovery period over the next 1 to 2 hours andthen by a further progressive fall in respiratory function, which is maximal between 6 to 12hours. A further recovery occurs by 24 to 36 hours. This response can only be partiallyreversed by β-2 agonist, but pre-medication with cromolyn and corticosteroids inhibits thisresponse. The LAR is also characterised by the release of inflammatory mediators into thesame fluids. However, during this phase there is a striking infiltration of inflammatory

Pathophysiology of Bronchial Asthma 41
cells with activation of these cells which include eosinophils, neutrophils, and lymphocytes.This LAR is thought to be a primary mechanism responsible for airway (bronchial)hyperresponsiveness (BHR). The BHR is an exaggerated bronchoconstriction of smoothmuscles and airway narrowing on exposure to small quantity of non-allergic stimulantthat usually does not provoke such a reaction in normal subjects.4 The BHR usually precedesthe onset of LAR.5 This may last for several days or occasionally weeks.6 The BAL fluidfrom these subjects contains increased eosinophils, eosinophilic cationic protein, CD4+ Tlymphocytes, macrophages, monocytes, basophils, and neutrophils.3,6 The selectiverecruitment of these leucocytes into the airways during the LAR are probably due to therelease of local and circulating messengers, i.e. cytokines from the cells in the airway mucosain relation to allergen exposure with the subsequent effect of recruiting mature and precursorcells from the bone marrow and other sites of leucocyte sequestration.7 Mucosal oedemaand vasodilatation are the important components of airway obstruction during the LARand contraction of airway smooth muscle contribute substantially to the EAR.
It is clear from studies in human and animals that the two phases of bronchoconstrictionresponse to inhaled antigen have distinct characteristics. The immediate response to antigenoccurs before airway inflammation is established histologically, is abolished or attenuatedby prior bronchodilator drugs like β-2 adrenergic agonist, is sensitive to the effects of anti-inflammatory drugs, and is not associated with an increased bronchial hyperreactivity. Incontrast, the LAR is associated with histologic evidence of airway inflammation, is relativelyresistant to bronchodilator drugs, is lessened by corticosteroids, and is associated withbronchial hyperreactivity. Rabbit experiments showed that if they are depleted of neutrophilsand then exposed to inhaled antigen, there will be an immediate bronchoconstrictor response,but there will be no late bronchoconstriction, neither they develop bronchial hyperreactivity.These findings suggest that airway inflammation underlines the bronchial hyperreactivitycharacteristic of LAR. Non-immunologic causes of airway inflammation are also associatedwith the development of bronchial reactivity. Inhaled ozone and viral infections damagethe bronchial epithelium, leading to an inflammatory response in the bronchial walls andbronchial hyperreactivity develops once airway inflammation become evident. Similarlyneutropenic dogs do not develop hyperreactivity after exposure to ozone. All these supportthe hypothesis that inflammatory processes are important in the pathogenesis of bronchialhyperreactivity.8
The results of skin testing of allergic subjects indicate that isolated immediatehypersensitivity reactions occur in about 20% of positive challenges, isolated late phasereactions in 6-14%, and both reactions in 66 to 85%.
Thus, it is apparent that both inflammation and bronchial hyperreactivity are importantto bring about these changes. A series of events including cellular elements, mediators, andneuropeptides in a coordinated manner are responsible for the ultimate airway obstruction.
A number of cells and chemical by-products take part in the pathogenesis of bronchialasthma to bring about changes outlined above. The stimulus/stimuli in a susceptible hoststarts the ball rolling so that a number of cells with their products cause various changesthat are characteristic of bronchial asthma. The understanding and concept of pathogenesisof bronchial asthma has changed considerably over the past decade. Bronchial asthma isnow considered as a heterogeneous disorder with multiple triggers. However, certainfeatures are common to all asthmatics: Airway inflammation and hyperresponsiveness to abroad range of stimuli. It is also known over the last few years that there is a close relationship

42 Bronchial Asthma
between airway inflammation and hyperresponsiveness. Exposure to oxidants, pollutants,viral infections, chemicals, and allergens are all associated with inflammation and theseinflammatory stimuli are associated with airway hyperresponsiveness. Most studies haveshown that airway inflammation precedes hyperresponsiveness and may be a prerequisitefor the development of hyperresponsiveness and clinical bronchospasm. On the other hand,airway inflammation as in purulent bronchitis may be present without hyperresponsivenessor bronchospasm. Thus, airway inflammation, a hallmark of bronchial asthma is of a specificnature that differs from other types of inflammation.
Bronchial asthma is now established as an inflammatory disease of the airways associatedwith inflammatory cell infiltration, epithelial damage, and subepithelial fibrosis. Broncho-alveolar lavage studies from patients with bronchial asthma have shown increased numberof eosinophils, mast cells, epithelial cells, and various humoral and chemical mediators ofasthma.9-15 Histopathological studies also have shown epithelial shedding and influx ofeosinophils into the airway mucosa.16-18 Fresh biopsies from asthmatics of varying severityhave shown epithelial changes, deposition of collagens, and influx of inflammatory cellseven in patients with mild asthma.19 Further, presence of increased number of eosinophilsin the sputum and peripheral blood of patients with bronchial asthma has been known formany years. It is also reported subsequently that eosinophils and mast cells increasequantitatively during exacerbations of asthma. Substantial data also support the role forboth neutrophils and macrophages.20 Specific subtypes of lymphocytes (T-helpers2 [Th2])may orchestrate a unique inflammatory response in the asthmatic lung and may significantlymodulate the function not only of the inflammatory cells, but also non-inflammatory cellswhich include endothelial cells, platelets, sensory nerves, and airway epithelial cells.21 Thesenon-inflammatory cells may contribute to the inflammatory response and may also directlyparticipate in the regulation of normal airway tone. The function of these cells can bemodulated in asthmatics and they may produce mediators with effects on airwayfunction.22-31
INFLAMMATORY CELLS IN ASTHMA
Mast Cells
Mast cells have been recognised since a long time as the main effectors cells in early asthmareaction.32,33 Normal human respiratory tract contains large numbers of mast cells beneaththe bronchial epithelium and alveolar walls. Increased numbers of mast cells and histamine(a product of mast cells) have been found in the bronchoalveolar lavage fluid obtainedfrom patients with bronchial asthma.34,35 These calls are derived from CD34+ cells in thebone marrow.36 Based on the production of proteases, a number of subtypes of mast cellsexist in human beings.37 A large number of biologically active molecules, both preformed,i.e. histamine, proteases38 and newly synthesised,39 are released from the mast cell duringthe allergic reaction when its high affinity, IgE receptors are cross-linked with antigen.40
After immunological activation, some populations of mast cells metabolise arachidonicacid, primarily through the cyclooxygenase pathway to prostaglandin (PGD2), andthromboxane A2, whereas other populations of mast cells metabolize arachidonic acidprimarily through the 5'-lipooxygenase pathway to LTB4 and LTC4 (Fig. 3.1). All mast cellshave secretory granules that contain large amounts of histamine, proteoglycans, heparin,and proteases. These preformed substances are exocytosed from the cell after immunologic

Pathophysiology of Bronchial Asthma 43
activation. It has been shown recently in experimental animals that certain activated mastcells also release transiently a large number of cytokines affect the tissue microenvironmentduring inflammation. These substances are GM-CSF, INF-γ, IL-1, IL-3-6, PAF, transforminggrowth factor, JE, and M1P1. These cytokines are capable of recruiting, priming andactivating other cells involved in inflammation. Through the release of cytokines similar tothose released from TH2 lymphocytes, it is possible that mast cells also play an importantrole in the development of LAR in addition to its primary role in EAR.
It has been suggested that mast cells also possess anti-inflammatory properties throughrelease of heparin and related proteolysis. The tissue damaging properties of cationic proteinmediators released from eosinophils (see later) are neutralised by the highly anionic heparin.It has also been shown that heparin inhibits the increased vascular permeability inducedby a wide range of agonists, can inhibit lymphocyte activation and trafficking and likeglucocorticoids is capable of inhibiting delayed hypersensitivity responses. Thus, it ishypothesised that an imbalance between these inflammatory and anti-inflammatorysubstances will decide the final outcome. However, this has not yet been proven.
Although mast cells are primary cells in EAR through IgE dependent release ofspasmogenic mediators, they also have an important role in LAR as they also produce
Fig. 3.1: Arachidonic acid metabolism and mediator release (LT- Leukotriene)

44 Bronchial Asthma
GM-CSF, interleukins, etc.41 although some other reports indicate that they are less likely to beinvolved in the chronic inflammatory response.42,43
Eosinophils44-47
The importance of eosinophils in the causation of bronchial asthma is evident in view ofextensive tissue, blood and sputum eosinophilia in this disease. Biopsy studies bothpostmortem and during life have shown the presence of excess eosinophils in the bronchialmucosa of these patients. They also play a key role in asthma, and their presence in theairways characterises the inflammation of asthma, which has been termed as “chroniceosinophilic bronchitis”. The number of activated eosinophils is closely related to asthmaseverity and may be associated with epithelial shedding. Their development is dependenton T cell function. The IL-5 specifically stimulates eosinophil differentiation. They havereceptors for IgG, IgA, and IgE on their cell surface.
These cells are able to produce many mediators that are responsible for the disorderedairway function characteristic of asthma. These substances include:
• Platelet activating factor• LTB4
• LTC4• PGE2
• 15-HETE• Oxygen radicals and• Four cytotoxic proteins48-51
i. Major basic protein (MBP)ii. Eosinophil cationic protein (ECP)
iii. Eosinophil-derived neurotoxin (EDN) andiv. Eosinophil peroxidase (EPO).
All these mediators are released by activated eosinophils. The release of these mediatorsresults in bronchoconstriction, epithelial damage, and recruitment and priming of otherinflammatory cells. Eosinophil maturation and priming are under the control of IL-3, IL-4,IL-5, and GM-CSF (Granulocyte macrophage-colony stimulating factor), cytokines releasedfrom a number of cell types in the airways including activated T cells of the TH2 type, andmast cells. Another molecule present in the eosinophils is the Charcot-Leydon crystal proteinthat possesses lysophospholipase activity.
Eosinophils have characteristic granules and granule proteins. The granule is composedof a crystalloid core and a matrix. The above four proteins are present in the granules. Thegenes of these proteins are cloned and the cDNA for MBP specifies the existence of a pro-MBP molecule that is composed of an acid-rich portion and a basic MBP portion. EDN andECP are both ribonucleases. In addition, ECP is a potent helminthotoxin. EPO is a memberof the peroxidase multigene family that is composed of myeloperoxidase, thyroid peroxidase,and lactoperoxidase. The MBP is toxic to respiratory epithelium and is elevated in the sputumof patients with asthma. It has also been shown that MBP is deposited in the damaged areasin the epithelium. Not only MBP, but also ECP and EPO alone, as well as EPO in the presenceof halide and hydrogen peroxidase, damage bronchial epithelium. Experimental studieshave shown that eosinophil proteins, particularly MBP applied to respiratory epitheliumstimulates smooth muscle contraction and also can increase the sensitivity of the smooth

Pathophysiology of Bronchial Asthma 45
muscle to acetylcholine, which suggests that eosinophil is an effector of the changes ofbronchial hyperreactivity in vivo.
Lymphocytes52-70
Although production of IgE by B lymphocytes is well known, the role of T lymphocytes inbronchial asthma had received little attention till recently.52,53 Chronic asthma, at least inpart, represents a form of delayed-type hypersensitivity involving interactions between“activated” lymphocytes and eosinophils. There are a number of evidences to prove thatthese cells play important roles in this disease.
i. T lymphocytes secrete lymphokines, IL-4, and interferon-γ, that closely regulate IgEproduction by B lymphocytes. While IL-4 stimulates, interferon-γ inhibits IgE synthesis.
ii. IL-3, and IL-5, and GM-CSF regulate eosinophil production, and IL-3, and IL-4 areimportant regulators of mast cell and basophil production.
iii. T cells are attracted to the bronchial mucosal surface to the site of inflammation byspecific receptors both on themselves and on the mucosal capillary and endothelialvenules.
iv. Local accumulation of CD8+ cells in early phase reactions recovered in BAL fluidsuggests that the subsequent late phase reaction may be in part, under the control ofT cells. Although CD8+ cells are not a part of this reaction, it has been found thatsubstantial infiltration of CD4 IL-2R+ lymphocytes, and activated (EG2+) eosinophilsoccurs in allergen-induced late phase reaction in atopic subjects. Recently it has beenreported that a high percentage of these CD4+ cells are UCLH-1 or memory cells, thatrespond to recall antigens.
v. Patients with acute severe asthma have activated CD4+ lymphocytes in their blood,the number of which returns to baseline value after successful treatment. The elevationis associated with increased serum concentration of IL-2 soluble receptors and INF-γ.These changes corelate well with the severity of disease.
vi. Corticosteroid resistant asthmatics have chronically activated circulating T cells (IL-2Rand HLA-DR positive).
vii. More recently, direct evidence of T cell involvement in bronchial asthma is acquired bythe study of mucosal biopsy specimens from volunteers. Electron microscopy hasrevealed elevated numbers of activated “irregular” lymphocytes in the bronchialmucosa. There is a significant increase in the number of IL-2R+ (CD25) cells both at thecentral and subsegmental airways.It is now well established that there are two types of T cells.54 They are Th1 and Th2,
divided on the basis of lymphokines they secrete. While the Th1 cells secrete IL-2, interferon-alpha and tumour necrosis factor-beta, the Th2 cells secrete IL-4,IL-5, IL-6, and IL-10, andIL-3 and GM-CSF are secreted by both. While INF-γ inhibits the development of Th2 cells,55
IL-10 inhibits Th1 proliferation.56 Details of such interaction are being discussed above undergenetics and asthma. More recently, another stable phenotype among T-helper clones hasbeen recognised both in mouse and man, which is called Th0. This subtype is characterisedby an ability to generate a large variety of cytokines, including IL-4 and INF-γ, which arecharacteristic of either the Th1 or Th2 subset.
Therefore, activated CD4+ cells are a feature of both active and chronic asthma, more sofor the later, and their presence is associated with actively secreting eosinophils. T cellsmay be directly involved in eosinophil recruitment and activation by secreting various

46 Bronchial Asthma
interleukins which favour the synthesis of IgE and activation of eosinophils and mast cells.There is a preferential expansion of type Th2 T cells secreting IL-3, IL-4, IL-5 and GM-CSF withfewer Th1 cells whose cytokine profile includes IL-2 and interferon-γ. Such a mechanismwould explain the peculiarities of allergic inflammation involving isotype switching to IgEsynthesis, and the preferential recruitment of eosinophils and mast cells.
Lymphokines and various other cytokines that are relevant to airway inflammation inasthma are shown subsequently.57-68 Two important cytokines , that are particularly importantin bronchial asthma are IL-4 and NF-KB.69,70 IL-4 is essential for IgE production. INF-γdiminishes cell processing necessary for IL-4 production.69 Thus, interplay of these cytokineswill decide whether IL-4 producing cells will be produced, and, thus, whether IgE will beproduced in response to various allergic stimuli. The role of transcription-factor NF-KB isemerging recently to play a key role in the pathogenesis of bronchial asthma. It is postulatedthat inflammatory signals activate transcription factors such as NF-KB and this in turn willswitch on the inflammatory genes, which will lead on to the increased expression ofinflammatory proteins. Corticosteroids are potent inhibitors of NF-KB and their anti-inflammatory action is believed to be mediated through this mechanism.70
Thus, it may be summarised that T cell participation is an important event in allergicdiseases and asthma. Th2 cells are more important by the way of production of variouscytokines which are necessary for allergic responses. In contrast, Th1 cells are primarilyresponsible for classic delayed hypersensitivity. Products of Th1 type cells, principallyINF-γ, inhibit or antagonize Th2 effector function. IL-4 induces IgE synthesis, and INF-γ isa strong inhibitor of this process. Such control establishes a model of how IgE can be tightlyregulated in vitro. The Th2 pathway is also involved in regulation of eosinophilia, mast cellactivity and IgE synthesis. The differentiation into Th1 and Th2 cells are again regulated bycytokines. While IL-4 may act directly on the precursor T cell to induce Th2 differentiation,interferon and transforming growth factor-beta TGF-β.71 While T cell sensitisation is animportant factor in the development of IgE production to a particular antigen and T cellsubsets are important in establishing the process of airway hyperresponsiveness.Experimental data have shown that the transfer of antigen-specific IgE, immediate cutaneoushypersensitivity, and increased airway responsiveness may be mediated, depending onthe antigen, by specific Vβ expressing T cell subsets.
While the precise mechanisms by which inflammatory cells are recruited into the lungsare not fully understood, increasingly available evidence suggest that the activation ofantigen-specific CD4+ T cells of the type 2 T-helper (Th2) subset in the lungs, which resultsin IL-5 secretion, plays a major role in asthmatic airway inflammation.72 CD4+ T cellactivation leading to cytokine production and effector function requires two signals fromthe antigen-presenting cell (APC). The first signal is triggered by the interaction betweenantigen-specific T cell receptor and peptide-major histocompatibility complex II complexeson APCs. The second signal or ‘co-stimulatory’ signal is triggered by CD80 (B7-1) and CD86(B7-2) of the APC binding to the CD28 and cytotoxic T lymphocyte antigen (CTLA-4) of theT lymphocytes.73-76 In the absence of co-stimulatory signals, the T cell-dependent immuneresponse is greatly diminished, or even eliminated.77 Thus, costimulatory signals may fulfilla valuable role in T lymphocyte activation, Type 1 T-helper (Th1) or Th2 cell differentiation,and the production of different cytokines.78
CTLA-4 is a second co-stimulatory molecule and is a homologue of CD28. It is expressedonly on activated T cells, binds to accessory molecule B7,79 and mediates T cell-dependent

Pathophysiology of Bronchial Asthma 47
immune response. Signalling through CTLA-4 may down regulate Th1 cell proliferationby inhibiting the production of IL-2 and IL-2 receptor expression.80,81 However, the role ofCTLA-4 remains uncertain, with some studies79 the CTLA-4 might also deliver a positivesignal to Th2 cell activation. Disruption of this delicate balance of immune regulation couldlead to autoimmune diseases or atopic diseases. Therefore, CTLA-4 is considered to beimportant in the development of many of the immunologic and physiologic features ofasthma.
Polymorphisms of the CLTA-4 gene, located on chromosome 2q33, could thus have effectson immune response. Three CTLA-4 genes are known at present.82-86 The CTLA-4 promoter(-318 C/T) T allele may serve as a clinically useful marker of severe asthma. This promoterpolymorphism is associated with asthma severity, but not with asthma, atopy, or bronchialhyperresponsiveness. A significant association is found between severe asthma andbronchial hyperresponsiveness.85
Monocytes and Macrophages
Several findings favour a role for macrophages in bronchial asthma.86-94 Firstly, after in vivoand in vitro contact with specific allergen or non-specific stimulus, alveolar macrophagesfrom asthmatics have been shown to release lysosomal enzymes, prostaglandin (TxB2),leukotrienes, and platelet-activating factor (PAF). They are also able to generate oxygenfree radicals, neutral proteases, and β-glucuronidase after non-specific stimulation. Someof these studies also have shown that macrophages from asthmatics are hyperactive andrelease more lipid-derived mediators than those from the normal subjects.
Secondly, a subpopulation of peripheral blood monocytes and alveolar macrophagesare IgE receptor positive.95,96 Whereas in normal healthy humans, only 5 to 10% of the alveolarmacrophages and 10-15% of the peripheral monocytes are IgE Fc positive, these numbersincrease dramatically in asthmatics. As many as 80% of the monocytes and up to 30% of themacrophages recovered from BAL fluid in mild asthmatics will be IgE receptor positive.The percentage may be still higher in severe forms of asthma. The macrophage IgE receptors(IgE FcR) has a low affinity for IgE compared to that of the mast cell. This lower affinitybinding suggests that IgE immune complexes may be more important in activation of thesecells compared to mast cells and basophils that are sensitised by monomeric IgE, because oftheir greater strength of binding to this FcR.
Thirdly, it has been demonstrated that active macrophages are present at the air-surfaceinterface of human airways as well as in alveoli. Therefore it is possible that these cellsinteract with any inhaled allergen.
Fourthly, macrophages are capable of releasing several potent neutrophil chemotaxins.These include complement fragments, fibronectins, neutrophil attractant/activating protein-1 (IL-8), and LTB4. IL-8 is also chemotactic for lymphocytes. The production of LTB4 frommacrophages is greater on a nanogram per cell basis than other cells. LTB4 and PAF arechemo attractants for eosinophils. Macrophages produce histamine-releasing factor(s) thatinduce the release of histamine from basophils.
Fifthly, macrophage function is altered by lymphokines, such as INF-γ. It is reasonableto hypothesize that this lymphokine and/or others, such as IL-4, may regulate the numberof IgE FcRs on lung macrophages.

48 Bronchial Asthma
Basophils
Basophils are histamine releasing cells in the late phase reaction of asthma unlike mastcells, which release histamine in the early phase reaction. The spontaneous release ofhistamine is quite high by these activated basophils (20-40% of total). This release processhas slow kinetics and is temperature dependent. Various cytokines (IL-1, IL-3, and histaminereleasing factor) and PAF have an up regulatory/stimulatory effect on blood basophils.Any or all of these cytokines could prime the basophils such that they become responsiveto very low concentrations of stimuli or some could directly trigger basophil mediator release.
Epithelial Cells and Adhesion Molecules
The infiltration of inflammatory cells into the airways is dependent on the expression ofadhesion molecules on inflammatory cells and endothelial cells of the bronchial circulation.97
One consequence of inflammation is epithelial injury. Morphological studies have shownthat asthma is associated with epithelial injury. These changes range from minor disruptionof the epithelium with loss of ciliated cells to complete denudation of the epithelium. Thesestructural changes in the epithelial barrier can lead to increased permeability to inhaledallergens, irritants, and inflammatory mediators. In addition, transudation of fluids andreduced clearance of inflammatory substances and respiratory secretions occur withdisruption of epithelial mucociliary mechanisms. The epithelium also participates inmediator release and metabolism.98-101 They have the capacity to produce PGE2, PGF2α,12-and 15-hydroxy eicosatetraenoic acid, GM-CSF, etc. The bronchial hyperresponsivenessin asthma is attributed to the epithelial cell damage. The airway epithelial cells have aprotective role against various tachykinins.
Currently, adhesion molecules are considered to be important in the causation of airwayinflammation, although the specific mechanism is still under investigation.102-109 Adhesionof various inflammatory cells to the bronchial vascular endothelium is a key step in theinitiation and propagation of inflammation. This is effected by the interaction of variousadhesion molecules expressed on endothelial cells, epithelial cells, platelets, and leucocytes.These molecules are specific glycoproteins that are grouped into different families dependingupon their molecular structure. These include integrins, immunoglobulin super gene family(intracellular adhesion molecule-ICAM, vascular cell adhesion molecule-VCAM; plateletendothelial adhesion molecule-PCAM), selectins (E-selectin like ELAM-1, and ECAM-1,P-selectin, L-selectin) and carbohydrates are important for lung inflammation. Expressionof various adhesion molecules is regulated by various mediators of inflammation.
Neutrophils
Although neutrophils are found in large proportions in the bronchial wall andbronchoalveolar lavage fluid in bronchial asthma, it is not clear if they have any definiterole to play in bronchial asthma. However, such neutrophils in bronchial asthma showincreased expression of membrane complement receptors and enhanced toxicity forcomplement coated antigens. They also have ability to alter airway function. These findingsuggest that neutrophils probably participate in inflammation of bronchial asthma.

Pathophysiology of Bronchial Asthma 49
CYTOKINES IN BRONCHIAL ASTHMA
Cytokines are extracellular signalling proteins, usually less than 80 KD in size, and many areglycosylated. They are produced by different cell types. A detailed discussion on the role ofdifferent cytokines is given below.
Various cytokines and their function are shown in Table 3.1.
Table 3.1: Various cytokines, their source and function inthe pathogenesis of bronchial asthma110
Cytokines Origin Function
IL-1 Various cells Increased expression of endothelial adhesion moleculesIL-2 Th2-cells Eosinophil activationIL-3 Th2 cell, mast cells Eosinophil and neutrophil differentiation, activation,
eosinophils and eosinophils survival, eosinophil chemotaxisIL-4 T cell, mast cell IgE synthesis, T cell growth, endothelial adhesionIL-5 T cell, mast cells, Eosinophil differentiation, maturation, activation,
eosinophils, adhesion, priming and chemotaxis,basophil differentiation and priming, cofactor in IgEsynthesis
IL-6 T cells T cell growth factor, eosinophil chemo-attractantIL-8 Monocytes, T cells Neutrophil chemo-attractant and activator,
fibroblasts inhibits IgE synthesisIL-10 T cell Inhibition of Th1 cytokine, stimulates monocytes
productionIL-12 T cells NK cell and T cell growth, IgE synthesis inhibitionIL-13 T cells Critical regulator of allergic responseGM-CSF T cells, mast cells Granulocyte differentiation, activation, survival,chemotaxis macrophages, eosinophils, epithelial cellsIFN-γ T cells Eosinophil and macrophage activationTumour necrosis T cell and macrophage activation,factorPlatelet derived Monocytes, Fibrosis, Th2 cytokine inhibitiongrowth factor (PDGF) Macrophages
INFLAMMATORY MEDIATORS IN ASTHMA
From the foregoing paragraphs it is apparent that a number of mediators released by differentcells are important for various changes observed in bronchial asthma.111-116 They are generatedby recruited cells and resident cells of the airways. These mediators cause contraction ofairway smooth muscle, increased mucus secretion, microvascular leak, further recruitmentand activation of various inflammatory cells, all essential changes in bronchial asthma.
LEUKOTRIENES
Of the many mediators that have been implicated in the asthmatic response, thesulphidopeptide leukotrienes are of interest because they have the potential of involvementin both aspects of the asthma syndrome, i.e. hyperresponsiveness, and inflammation. Theoriginal discovery of a slow-reacting substance was that of smooth muscle contractile activitydistinct from histamine; it was distinguished from histamine on the basis that its effectswere slow in onset and prolonged in duration.117 The subsequent isolation and elucidation

50 Bronchial Asthma
of the structure of slow-reacting substance was identified as cysteinyl leukotrienes (LTC4,LTD4, LTE4) which are synthesised and exported into the microenvironment by a number ofthe above mentioned inflammatory cells, including mast cells and eosinophils.118-127
Furthermore, since plasma leakage is prominent in more severe asthma, it is likely that thevascular endothelium will be exposed to cells capable of donating LTA4. It is well establishedthat the cysteinyl leukotrienes are formed when LTA4 exporting cells, such as polymo-rphonuclear leucocytes (neutrophils and eosinophils) provide LTA4 for effector cells suchas vascular endothelial cells or platelets.
As shown in Figure 3.2 arachidonic acid (AA) released from membrane phospholipidsduring cell activation may be oxidatively metabolised by the enzymes of the cyclooxygenaseor lipooxygenase pathways.109 Arachidonate is presented to the 5-lipooxygenase enzymeby 5-lipooxygenase-activating protein (FLAP).120 This FLAP is a cofactor resident in thenuclear membrane. While cyclooxygenase pathway produces prostaglandins andthromboxane, the 5-lipooxygenase pathway generates 5-hydroperoxy-eicosatetraenoic acid(5-HPETE) or is converted enzymatically to the unstable intermediate LTA4. LTA4 ismetabolised by an epoxide hydrolase to LTB4, or by a glutathion-S-transferase (LTC4synthase) to LTC4.
128 LTC4 is cleaved by glutamyl-transpeptidase to LTD4, which is convertedby a peptidase to LTE4, these enzymes being ubiquitous in the tissues and circulation(Fig. 3.2). LTB4 is a potent chemo attractant for neutrophils, and the sulphidopeptideleukotrienes (LTC4, LTD4, and LTE4) are potent spasmogens for non-vascular smooth muscleand comprise the activity previously known as slow-reacting substance of anaphylaxis(SRS-A).
The leukotrienes have profound biochemical and physiologic effects, even in Pico molarconcentrations. The importance of leukotrienes has been suggested in a wide variety ofdisorders that include hepatorenal syndromes, myocardial ischaemia, and inflammatoryconditions of bowel, skin and joints,122 besides their involvement principally in bronchialasthma.123 These include severe airway obstruction, i.e. bronchoconstriction,129 oedema,130
and increased secretion of bronchial mucus from submucosal gland secretion.131 The most
Fig. 3.2: Synthesis of leukotrienes and their function

Pathophysiology of Bronchial Asthma 51
prominent effect is their ability to mediate airway narrowing in normal subjects as well asin subjects with asthma. The airway obstruction is prolonged compared to that induced byhistamine. LTC4 and LTD4 are approximately 3000 times more potent in contracting theairway compared to histamine in normal subjects. LT4 is also a potent bronchoconstrictoralthough 30-100 times less potent than the above two. LTE4 induces a state of enhancedairway responsiveness in asthmatics, but not in normal subjects. Inhalation of LTE4at dosesthat induce a small but significant contractile response enhances the response to subsequentadministration of inhaled histamine. This enhancement is on the order of a four-fold shiftin the histamine dose-response curve with the effect lasting approximately 24 hours withsmall effects persisting for up to a week. Thus a state of airway hyperresponsiveness ismaintained.
Leukotriene B4 (LTB4) is a potent chemotactic factor and is responsible, in part, for therecruitment of inflammatory cells to the airway and stimulation of secretion of inflammatoryproducts. Their role in the smooth muscle contraction is controversial, although some studiessuggest that they may increase airway smooth muscle responsiveness to subsequentstimulation. This can also modulate the immune response by inhibiting the capacity tomount a delayed hypersensitivity response.132
The cells producing leukotrienes are only macrophages, neutrophils, eosinophils, andmast cells that can synthesise them from the substrate arachidonic acid. However, subsequentenzymes like LTA4 hydrolase, and LTC4 synthase are more broadly distributed includingnon-inflammatory cells, airway epithelial cells and in the lung lining fluids. It is also nowrecognised that synthesis of leukotrienes in the lung may involve a single inflammatorycell type or an interaction between inflammatory and non-inflammatory cells termed“transcellular metabolism”. Some reports suggest that even transcellular metabolism maybe the principal source of LTC4 in the lungs.133,134
These leukotrienes are recovered from nasal lavage fluid after inhalation challenge.Significantly larger quantities are also recovered from the BAL fluid from subjects withsymptomatic asthma. Sulphidopeptide leukotrienes have been detected in the plasma duringasthma attacks. Larger quantities of these substances have been recovered from the urineof asthma patients during acute spontaneous attacks than found in normal subjects.
The recent development and usefulness of leukotriene receptor antagonists and synthesisinhibitors in bronchial asthma further emphasizes the role of these leukotrienes in thepathogenesis of this condition.135-138
Leukotrienes are important in asthma, and leukotriene modifiers modulate antigen-induced asthma. Leukotrienes participate in the pathogenesis of bronchial asthma besidesthe involvement eosinophilic airway inflammation.139 Overproduction of leukotrienes notonly occurs in house dust mite provoked asthma, but also in aspirin induced bronchialasthma, although the mechanisms of such overproduction are different. While in the formerthe overproduction occurs with an antigen-antibody reaction, in aspirin-induced asthma,the overproduction is due to a shift to the 5-lipooxygenase series of the arachidonatecascade.140 Pranleukast, a leukotriene inhibitor suppresses the increased values of sputumeosinophil count and eosinophil cationic protein during house dust mite-induced asthmaare suppressed by further, this drug increases FEV1 that falls during such provocation.140
The role of leukotrienes in the pathogenesis of aspirin-induced asthma comes from the factthat airway narrowing and other signs in these patients are associated with 2-10 fold highervalues of LTE4 in the urine of these patients compared to aspirin tolerant patients.141-143

52 Bronchial Asthma
Further, several leukotriene modifiers inhibit the asthma response in oral or inhaledbronchoprovocation by aspirin and other non-steroidal anti-inflammatory agents144,145 andimprove respiratory function by bronchodilatation.146
Mast Cell Proteases
As much as 70% of the weight of a mast cell consists of proteases that are enzymatically activeat neutral pH. These cells express a complex array of proteases, which consist of serineproteases, tryptases, and chymase. These enzymes regulate neuropeptide regulation in theairways, smooth muscle contraction, and submucosal gland secretion.147-149 Histamine, anothermast cell product has a well-established role in the pathogenesis of asthma. It inducesbronchospasm, increases vascular and epithelial permeability, and increases the mucousglycoprotein secretion.150
Histamine
The role of histamine in the pathogenesis of bronchial asthma is well established for a longtime. Histamine induces bronchoconstriction, increases epithelial and vascular permeability,and increases the secretion of mucus glycoproteins.150 In patients of bronchial asthma, thelevels of histamine are increased in blood and bronchoalveolar lavage fluid.151,152
Prostaglandins
PGD2 and PGF2- A are very potent bronchoconstrictor agents. The former has greaterbronchoconstrictor activity compared to that of the later or histamine.153 Both theseprostaglandins also potentiates the bronchoconstricting activity of histamine andmethacholine.155,156 On the other hand, PGE1 and PGE2 has bronchodilating effect. WhileThromboxane A2 (TXA2) is a bronchoconstrictor, vasoconstrictor, and platelet aggregator,PGI2 is a bronchodilator, vasodilator and prevents platelet aggregation.156,157
Platelet-activating Factor (PAF)
PAF has attracted attention as an important mediator of bronchial asthma.158-161 Recovery ofthis substance from bronchoaveolar lavage fluid in antigen exposed individuals supportssuch a role.162,163 It is an important mediator involved in the bronchial hyperresponsivenessin addition to having action of bronchoconstriction, stimulation of eosinophil and eosinophilaccumulation in the airway, induction of airway microvascular leakage and oedema, andincreased airway secretions and epithelial permeability.
Bradykinin
Bradykinin is another important inflammatory mediator in asthma and asthmatics haveincreased responsiveness to bradykinin,164 and the levels are found to be high in broncho-alveolar lavage fluid from these patients165 The substance causes bronchoconstriction,increases vascular permeability, has vasodilator activity, increases mucus secretion, activatesC-fibre nerve endings, enhances neuropeptide release from sensory nerves, and increasescholinergic reflex.164, 166,167 Bradykinin mediates its effects through BK1 and BK2 receptors,166
although the effects on airways are primarily mediated via BK2 receptors. It also releasestachykinins from airway sensory nerves.

Pathophysiology of Bronchial Asthma 53
CytokinesCytokines are extracellular signalling proteins, usually less than 80 kD in size and manyare glycosylated. They are produced by different cell types involved in cell-to-cell interactions,having an effect on closely adjacent cells, and therefore function in a predominantly paracrinefashion. They may also act at a distance (endocrine) and may have effects on the cell of origin(autocrine). A classification according to function is proposed in Table 3.2.
Table 3.2: Classification of cytokines and cytokine receptors
Cytokines
Pro-inflammatory cytokines IL-1α/β, TNFα/β, IL-6, IL-11, IFN-γ
Cytokines involved in atopy IL-4, IL-13 (promoters); IFN-γ, IL-12 (inhibitors)
Cytokines of eosinophil chemo-attraction IL-2, IL-3, IL-4, IL-5, GM-CSF, RANTES, eotaxin,and activation MCP-3, MCP-4
Th2 cytokines IL-4, IL-5, IL-10, IL-13
Cytokines involved in T cell IL-16, RANTES, MIP-1α/βchemo-attraction
Cytokines of neutrophil chemo-attraction IL-8, IL-1α/β, TNFα/βand activation
Anti-inflammatory cytokines IL-10, IL-4, IL-13, IL-12, IL-1ra
Growth factors PDGF, TGF-β, FGF, EGF, TNF-α, SCF
Cytokine receptorsCytokine receptor super family IL-2Rβ-and γ-chains, IL-4R, IL-3R α-and β-chains,
IL-5 α-and β-chains, IL-6R, gp130, IL-12R,GM-CSFR; soluble forms by alternative splicing(e.g. IL-4R)
Immunoglobulin super family IL-1R, IL-6R, PDGFR, M-CSFR
Protein kinase receptor super family PDGFR, EGFR, FGFR
Interferon receptor super family IFN-α/β receptor, IFN-γ receptor and IL-10 receptor
Never growth factor super family NGFR, TNFR-1(p55), TNFR-II(p75)
Seven-transmembrane G-protein Chemokine receptorscoupled receptor super family
The effects of an individual cytokine may be influenced by other cytokines releasedsimultaneously from the same cell or from target cells following activation by the cytokine,and are mediated by binding to cell surface high-affinity receptors usually present in lownumbers, which can be up regulated with cell activation. The receptors for many cytokineshave been regrouped into super families based on the presence of common homology regions(Table 3.3). Cytokines themselves may induce the expression of receptors which may changethe responsiveness of both source and target cells. Some cytokines may stimulate their ownproduction in an autocrine manner, where as others stimulate the synthesis of differencecytokines that have a feedback stimulatory effect on the first cytokine, resulting in an increasein its effects. The effects of cytokines are summarised in Table 3.3.168

54 Bronchial Asthma
Table 3.3: Summary of effects of cytokines
Cytokines Important cellular and mediator effects
LymphokinesIL-2 • Eosinophilia in vivo
• Growth and differentiation of T cellsIL-3 • Eosinophilia in vivo
• Pluripotent haematopoietic factorIL-4 • ↑ Eosinophil growth
• ↑ Th2; ↓ Th1• ↑ IgE• ↑ Mucin expression and goblet cells
IL-5 • eosinophil maturation• ↓ Apoptosis• ↓ Th2 cells• BHR
IL-13 • Activates eosinophils• ↓ apoptosis• ↑ IgE• ↑ mucin expression and goblet cells
IL-15 • As for IL-2• Growth and differentiation of T cells
IL-16 • Eosinophil migration• Growth factor and chemotaxis of T cells (CD4+)
IL-17 • T cell proliferation• Activates epithelia, endothelial cells, fibroblasts
Pro- inflammatoryIL-1 • ↑ adhesion to vascular endothelium; cosinophil accumulation in vivo
• Growth factor for Th2 cells• B cell growth factor; neutrophil chemo-attractant; T cell and
epithelial activation• BHR
TNF-α • Activation epithelium, endothelium, antigen-presenting cells;monocytes/macrophages
• BHR• ↑ IL-8 from epithelial cells• ↑ MMPs from macrophages
IL-6 • T cell growth factor• B cell growth factor• ↑ IgE
IL-9 • ↑ Activated T cells and IgE from B cells• ↑ Mast cell growth and differentiation• ↑ Mucin expression and goblet cells• Causes eosinophilic inflammation and BHR
IL-11 • B cell growth factor• Activates fibroblast• BHR
GM-CSF • Eosinophil apoptosis and activation; induces release of leukotrienes
Contd...
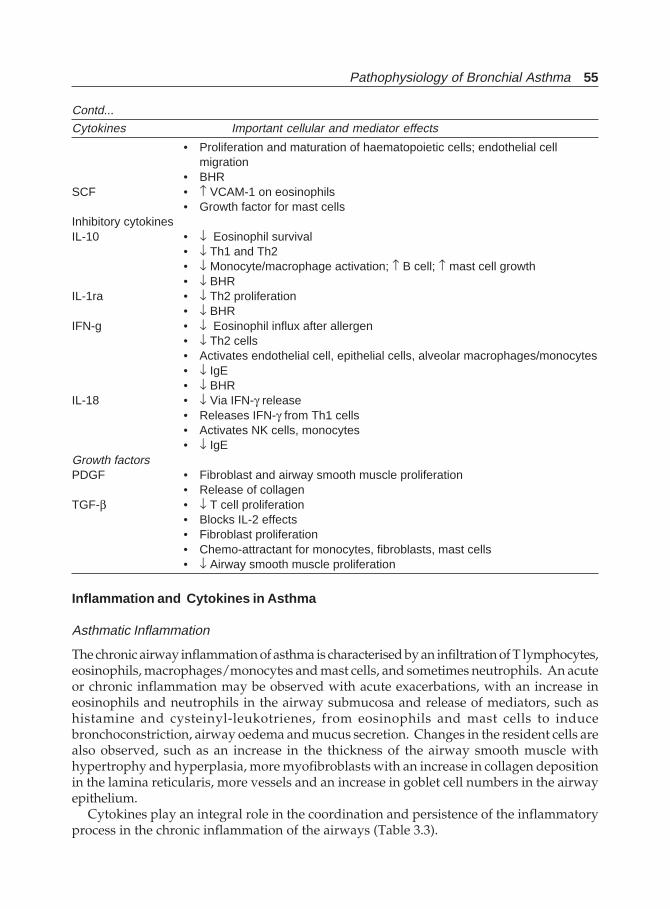
Pathophysiology of Bronchial Asthma 55
• Proliferation and maturation of haematopoietic cells; endothelial cellmigration
• BHRSCF • ↑ VCAM-1 on eosinophils
• Growth factor for mast cellsInhibitory cytokinesIL-10 • ↓ Eosinophil survival
• ↓ Th1 and Th2• ↓ Monocyte/macrophage activation; ↑ B cell; ↑ mast cell growth• ↓ BHR
IL-1ra • ↓ Th2 proliferation• ↓ BHR
IFN-g • ↓ Eosinophil influx after allergen• ↓ Th2 cells• Activates endothelial cell, epithelial cells, alveolar macrophages/monocytes• ↓ IgE• ↓ BHR
IL-18 • ↓ Via IFN-γ release• Releases IFN-γ from Th1 cells• Activates NK cells, monocytes• ↓ IgE
Growth factorsPDGF • Fibroblast and airway smooth muscle proliferation
• Release of collagenTGF-β • ↓ T cell proliferation
• Blocks IL-2 effects• Fibroblast proliferation• Chemo-attractant for monocytes, fibroblasts, mast cells• ↓ Airway smooth muscle proliferation
Inflammation and Cytokines in Asthma
Asthmatic Inflammation
The chronic airway inflammation of asthma is characterised by an infiltration of T lymphocytes,eosinophils, macrophages/monocytes and mast cells, and sometimes neutrophils. An acuteor chronic inflammation may be observed with acute exacerbations, with an increase ineosinophils and neutrophils in the airway submucosa and release of mediators, such ashistamine and cysteinyl-leukotrienes, from eosinophils and mast cells to inducebronchoconstriction, airway oedema and mucus secretion. Changes in the resident cells arealso observed, such as an increase in the thickness of the airway smooth muscle withhypertrophy and hyperplasia, more myofibroblasts with an increase in collagen depositionin the lamina reticularis, more vessels and an increase in goblet cell numbers in the airwayepithelium.
Cytokines play an integral role in the coordination and persistence of the inflammatoryprocess in the chronic inflammation of the airways (Table 3.3).
Contd...
Cytokines Important cellular and mediator effects

56 Bronchial Asthma
Th2-associated Cytokines
CD4+ T lymphocytes of the asthmatic airways express Th2 cytokines including IL-3, IL-4, IL-5, IL-10, IL-13 and GM-CSF. The primary signals that activate Th2 cells may be related to thepresentation of a restricted panel of antigens in the presence of appropriate cytokines. Dendriticcells are ideally suited to being the primary contact between the immune system and externalallergens. Co-stimulatory molecules on the surface of antigen-presenting cells, in particularB7.2/ CD28 interaction, may lead to proliferation of Th2 cells.169 With the expression of IL-4,synthesis of IgE by B lymphocytes on immunoglobulin isotype switching occurs.170 IgEproduced in asthmatic airways binds to FcεRI receptors (high-affinity IgE receptors) on mastcells priming them for activation by antigen. The maturation and expansion of mast cells frombone marrow cells involve growth factors and cytokines such as SCF and IL-3 derived fromstructural cells. Bronchoalveolar mast cells from asthmatics show enhanced release ofmediators such as histamine. Mast cells also elaborate IL-4 and IL-5.171 IL-4 also increases theexpression of an inducible form of the low-affinity receptor for IgE (FcεRII or CD23) on Blymphocytes and macrophages.172 IL-4 drives the differentiation of CD4+ Th precursors toTh2- like cells.
IL-18 is a cytokine with potent interferon (IFN)- γ-inducing activity. It is predominantlyproduced by activated macrophages and synthesised with IL-12 to induce (IFN)- γ synthesisfrom T lymphocytes, promoting differentiation of T cells to the Th1 subsets. The IL-18 levelsare low in the BAL fluid of patients with bronchial asthma. This inherently low levels of IL-18 may be associated with pathogenesis of asthmatic airway inflammation.173
Antigen presentation Cytokines may play an important role in antigen presentation(Fig. 3.3). Airway macrophages are usually poor at antigen presentation and suppressT cell proliferative responses (possible via release of cytokines such as IL-1 receptorantagonist), but in asthma there is reduced suppression after exposure to allergen.174 BothGM-CSF and IFN-γ increase the ability of macrophages to present allergen and expressHLA-DR.175 IL-1 is important in activating T lymphocytes and is an important co-stimulatorof the expansion of Th2 cells after antigen presentation.176 Airway macrophages may be animportant source of first-wave cytokines, such as IL-1, TNF-α and IL-6, which may bereleased on exposure to inhaled allergens via FcεRI receptors. These cytokines, may then acton epithelial cells to release a second wave of cytokines, including GM-CSF, IL-8 and RANTESwhich then leads to influx of secondary cells, such as eosinophils, which themselves mayrelease multiple cytokines.
Eosinophil-associated cytokines The differentiation, migration and pathobiological effectsof eosinophils may occur through the effects of GM-CSF, IL-3, IL-5 and certain chemokinessuch as eotaxin.177,178 IL-5 and eotaxin also induce the mobilisation of eosinophils andeosinophil precursors into the circulation.179 Mature eosinophils may show increase survivalin bronchial tissue.180 Eosinophils themselves may also generate other cytokines such asIL-3, IL-5 and GM-CSF.181
Cytokines such as IL-4 may also exert an important regulatory effect on the expressionof adhesion molecules such as VCAM-1, both on endothelial cells of the bronchial circulationand on airway epithelial cells. IL-1 and TNF-α increase the expression of ICAM-1 in bothvascular endothelium and airway epithelium.182 Cytokines also play an important role inrecruiting inflammatory cells to the airways.
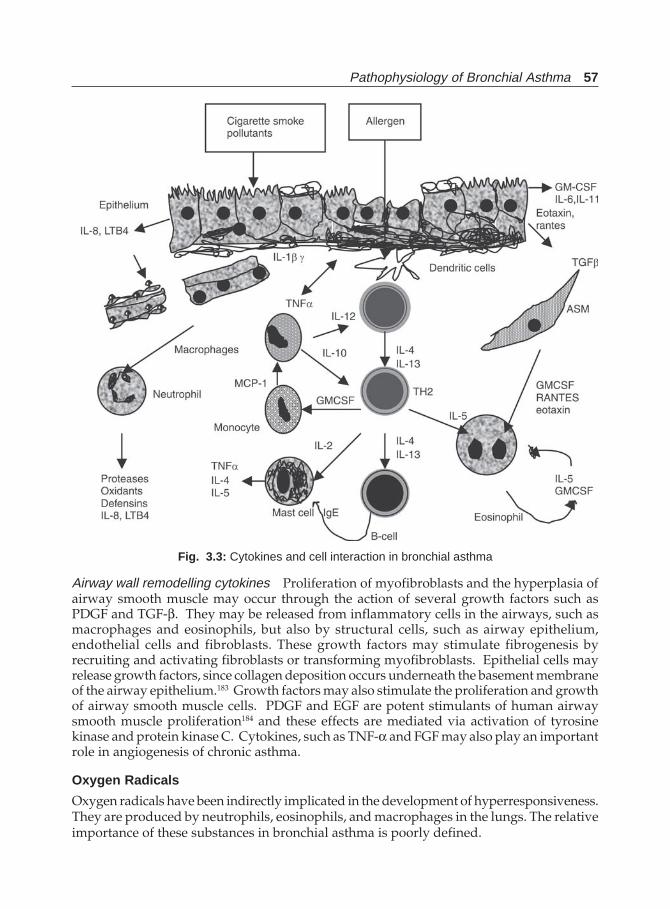
Pathophysiology of Bronchial Asthma 57
Airway wall remodelling cytokines Proliferation of myofibroblasts and the hyperplasia ofairway smooth muscle may occur through the action of several growth factors such asPDGF and TGF-β. They may be released from inflammatory cells in the airways, such asmacrophages and eosinophils, but also by structural cells, such as airway epithelium,endothelial cells and fibroblasts. These growth factors may stimulate fibrogenesis byrecruiting and activating fibroblasts or transforming myofibroblasts. Epithelial cells mayrelease growth factors, since collagen deposition occurs underneath the basement membraneof the airway epithelium.183 Growth factors may also stimulate the proliferation and growthof airway smooth muscle cells. PDGF and EGF are potent stimulants of human airwaysmooth muscle proliferation184 and these effects are mediated via activation of tyrosinekinase and protein kinase C. Cytokines, such as TNF-α and FGF may also play an importantrole in angiogenesis of chronic asthma.
Oxygen Radicals
Oxygen radicals have been indirectly implicated in the development of hyperresponsiveness.They are produced by neutrophils, eosinophils, and macrophages in the lungs. The relativeimportance of these substances in bronchial asthma is poorly defined.
Fig. 3.3: Cytokines and cell interaction in bronchial asthma

58 Bronchial Asthma
Nitric Oxide
Although it is now well established that normal subjects have measurable concentrations ofnitric oxide (NO.) in their expired air, in patients with bronchial asthma the peak or mixedexpired NO are about 50% higher.185-187 Furthermore, compared with normal subjects, theairways of patients with asthma have up regulated expression of type II nitric oxide synthase,NOS.188 Taken together, these findings have led to the speculation that expired concentrationsof NO reflect the inflammatory microenvironment of the asthmatic airway wall.189
Neurotrophins
The neurotrophins are a family of peptides that promote survival, growth, and differentiationof neurons. They may also influence the function of non-neuronal cell types, includingimmune cells. The development and maintenance of asthma are thought to involve nervoussystem and the immune system, but the exact role that the neurotrophins play is unclear.The cellular sources of neurotrophins include mast cells, lymphocytes, macrophages,epithelial cells, smooth muscle cells, and eosinophils. The action of neurotrophin receptorslike Trk (tyrosine kinase) acts possibly act in concert with known immune regulating factorsto modulate the maturation, accumulation, proliferation, and activation of immune cells.Neurotrophins also can modulate afferent nerve function by stimulating the production ofneuropeptides within airway afferent neurons. These neuropeptides may be released fromthe central terminals of airway afferent neurons, which leads to increased autonomic reflexactivity, and increased reactivity in the airways.190
The role of different mediators is summarised in Table 3.4.
Table 3.4: Role of mediators causing pathological changes in asthma
Pathological changes Mediator implicated
Bronchospasm Histamine (H1response)LTC4, LTD4, LTE4Prostaglandins and TXA2BradykininPlatelet activating factorAcetylcholin
Mucosal oedema Histamine (H1response)LTC4, LTD4, LTE4Prostaglandin EBradykininPlatelet activating factor
Cellular infiltration Eosinophil chemotactic factor(airway hyperreactivity) Neutrophil chemotactic factor
HETEsLTB4
Mucus secretion Histamine (H1response)LTC4, LTD4, LTE4Prostaglandins generatingFactor of anaphylaxisProstaglandinsHETEsAcetylcholinMacrophage mucus secretagauge
Desquamation O–2, H2O2, OH–
Proteolytic enzymesBasement membrane thickening O–
2, Proteolytic enzymes

Pathophysiology of Bronchial Asthma 59
NEUROPEPTIDES IN ASTHMA
There is increasing evidence that abnormal neurogenic mechanisms and neuropeptidescontributing in the pathophysiology of bronchial asthma.191-197 Autonomic nerves regulateairways smooth muscle tone, mucous secretion, blood flow, vascular permeability, andmigration and release of inflammatory cells.198,199 Neuropeptides are small amino acidcomponents that are localised to neurons. Originally described in the gastrointestinal tract,neuropeptides were first termed “gut hormones”. Upon their discovery subsequently inbrains, they were termed as “gut-brain hormones”. However, now it is established thatthese peptides are present throughout the body and may be produced by, localised to, cellsother than cells of the nervous system. In the respiratory tract, they are located in neurons,neuroendocrine cells, and inflammatory cells. Neuroendocrine cells are granulated epithelialcells found throughout the conducting airways. They contain a number of peptides, includingcalcitonin, katacalcin, CGRP (calcitonin gene-related peptide), and bombesin. Neuropeptidessuch as VIP (vasoactive intestinal peptide) has been identified in various inflammatorycells including eosinophils, mast cells, and mononuclear and polymorphonuclear leucocytes.Once released these peptides act as either neurotransmitters, hormones, or mediators. Theymodulate airway caliber, vascular tone, mucus secretion, and vascular permeability. Theyare also capable of affecting inflammatory cell function by modulating mediator releaseand chemotactic responses. Their wide spread distribution and different physiological effectsmake neuropeptides excellent candidates to play important roles in asthma.
The neural control of the airways is mediated by three pathways: cholinergic (para-sympathetic); adrenergic (sympathetic); and the nonadrenergic noncholinergic (NANC)pathways.191 The cholinergic nervous system is considered excitatory in nature because itplays an important role in maintaining bronchial smooth muscle tone and in mediatingacute bronchospastic responses. The system consists of vagal afferent fibres in and aroundthe airway lumen that travels to the central nervous system and then terminate in efferentfibres. The later innervate airway smooth muscle. There are three types of pharmacologicallydefined muscarinic receptors, which are important in regulating the smooth muscle tone.The M1 receptor is located in the parasympathetic ganglia and facilitates vagal transmission.The M3 receptors are present in large airways and in some peripheral airways and arelargely responsible for smooth muscle contraction. The M2 receptor functions as anautoreceptor in airway tissue, acting as a feedback-inhibitory receptor to reduce neurotrans-mission. Acetylcholin is the cholinergic messenger. Acetylcholin normally binds to thecholinergic receptor and causes release of cyclic 3',5'-guanosine monophosphate (cyclic-GMP). This causes bronchoconstriction. Cholinergic nerves are the dominant neuralbronchoconstrictor pathways for human lungs. Triggers like sulphur dioxide, prosta-glandins, histamine, and cold air stimulate afferent receptors causing reflex broncho-constriction. Inflammatory mediators like histamine, prostaglandins, and bradykininstimulate irritant receptors and C-fibre endings in the airway leading to a reflexbronchoconstriction.200 Neurotransmitters like TxA2, PGD2, and tachykinins enhance Acetyl-choline release from the postganglionic nerves in the airways. It is suggested that M2
autoreceptors are dysfunctional in bronchial asthma.201
The sympathetic nervous system in the bronchial tree is inhibitory because of its prominentairway relaxant effect. This is mediated by β-receptor stimulation and by cAMP. Adrenergic

60 Bronchial Asthma
fibres represent only a minor component of the total nerve fibres in human airways. Althoughthere is little or no direct sympathetic innervations of human airways, there are many α-and β-adrenergic receptors that are important in regulating bronchomotor tone. Earlier itwas believed the imbalance between cGMP and cAMP production was the underlyingmechanism of bronchial asthma, (Yin-Yang hypothesis).
The neurotransmitters for the NANC nervous system were initially thought to be purinenucleotides, such as adenosine and adenosine triphosphate and accordingly the NANCnerves were termed “purinergic”. However, now it is believed that the neurotransmittersare not purines, but peptides, and thus the nerves are “peptidergic”. Although a number ofneurotransmitters have been identified, only VIP, peptide histidine methionine (PHM),and nitric oxide may be the neurotransmitters of the nonadrenergic inhibitory nervous systemand thus are important endogenous bronchodilators.202,203 They also decrease mucus secretionand manifest anti-inflammatory actions. Deficiency of this system has been postulated tocontribute to the development of bronchial hyperreactivity. Functional deficiencies of thesystem can result from blockade of nonadrenergic pathways at the level of ganglia or nerveendings; from deficiency of airway VIP or PHM receptors, or from enhanced breakdown ofneuropeptides by peptidases released from inflammatory cells in the asthmatic airway. Ithas been demonstrated recently that there is a loss of VIP from pulmonary nerve fibres inasthmatics. Immunoreactive VIP is observed within nerves in more than 90% of lung sectionsfrom normal subjects but is not identified in any lung sections from patients with asthma.However, it is not clear whether it is a primary or secondary event.
Other peptides such as substance P, neurokinin A (substance K, neuromodulin L) andcalcitonin gene-related peptide (CGRP) are believed to be neurotransmitters of thenoncholinergic excitatory system and thus act as endogenous bronchoconstrictors.204-209 Thesepeptides also play a role in regulating mucus production, pulmonary vasomotor tone,mucosal permeability, and inflammatory cell function. A number of substances are knownto release neuropeptides from these nerves include capsaicin (most potent), irritant gases,antigen, and various inflammatory mediators, including histamine, bradykinin, andprostaglandins. These neuropeptides have the remarkable ability to affect multiple cells inthe airways and to provoke many responses including cough, mucus secretion, smoothmuscle contraction, plasma extravasations, and neutrophil adhesion. This series of effectsis termed as “neurogenic inflammation”.210-215 An enzyme neutral endopeptidase (NEP)exists on the surfaces of all lung cells. The enzyme inactivates the neuropeptides limitingtheir concentration. Angiotensin converting enzyme (ACE) also helps in the degradation ofthese neuropeptides. Thus neurogenic inflammatory responses are normally mild andprobably protective in nature. It is proposed that in asthma, a decrease in the normaldegradation process of substance P occurs by NEP or ACE. Cigarette smoke, respiratoryviral infections, and inhalation of industrial pollutant toluene diisocyanate inhibit NEPand exaggerate neurogenic inflammation. In addition, there are reports that there are moresubstance P immunoreactive nerves in the lungs of patients with asthma compared to thatin normal subjects.
Therefore, in addition to the proposed changes in the cholinergic and adrenergic nervoussystems, subjects with asthma have now been revealed to potentially have changes in theirnonadrenergic inhibitory and noncholinergic excitatory nervous system. These changeswill lead to an imbalance in the autonomic nervous system and predispose subjects withasthma towards bronchospasm (Fig. 3.4).

Pathophysiology of Bronchial Asthma 61
It is suggested that abnormal control of the airway is the underlying mechanism ofbronchial hyperreactivity, with a preponderance of excitatory (cholinergic and α-adrenergic)or a deficiency of inhibitory (α-adrenergic) control.
Bronchial Hyperreactivity
Airway hyperresponsiveness to a large number of stimuli is a characteristic feature of asthmain humans. Various components of the tracheobronchial tree might contribute to thisphenomenon, such as smooth muscle, the bronchial epithelium, various neurohumoralmechanisms and the mechanical linkage between the lung parenchyma and the airwaysincluding the baseline airflow obstruction. The degree of responsiveness can be furtherincreased by a series of stimuli associated with inflammation in the periphery of the lung.Such stimuli actually induce an asthmatic state or heighten the vulnerability of asthmatics,making them more prone to overt attacks in response to minor stimuli that would beordinarily tolerated. Depending upon the inciting stimulus, different cells and mediatorsmay be playing a role in producing and perpetuating the inflammatory state and producingfurther increases in responsiveness. The level of airway responsiveness usually correlateswith the clinical severity of asthma and medication requirement. The airways of asthmaticsubjects are 14-fold, 15-fold, 5-fold, 9-fold, and 194-fold more responsive than were theairways of normal subjects to histamine, methacholine, LTC4, LTD4, and LTE4 respectivelyin a direct comparison of the potencies of these substances in six asthmatics and six controls.112
Further, cumulative data suggest that hyperresponsiveness to the leukotrienes may be moremarked in the central rather than the peripheral airways of asthmatic subjects. Bisgardreported that the airways of 8 asthmatic subjects were more responsive to LTD4 than werethose of 9 nonasthmatic controls; the relative differences in potencies between asthmaticand controls were 100- to 1000-fold when measured in terms of FEV1 but they were only 15-fold differences in V30.
216 Similarly Smith et al reported a 30% fall in V30 in response to LTD4
Fig. 3.4: Autonomic imbalance postulated for bronchial asthma

62 Bronchial Asthma
was accompanied by a 60% fall in sGaw in asthmatic subjects but only a 30% fall in sGaw innormal controls.217 In another study Davidson et al reported a 30% fall in V30 induced byinhaled histamine was accompanied by a 10 and 13% fall in FEV1 in asthmatic and normalsubjects, respectively. But when the same individuals inhaled LTE4, a 30% fall in V wasaccompanied by a 17% fall in FEV1 in asthmatic subjects and a 3% fall in FEV1 in normalcontrols.218 While FEV1 represents the central airway function, V30 represents small orperipheral airways function. The interaction of various factors and the pathophysiology ofbronchial asthma is summarised in Figure 3.5.
Beta-adrenergic Receptors (β β β β β AR) and Asthma
β ARs belong to the family of adrenergic receptors that use the endogenous catecholaminesepinephrine and norepinephrine (and, to a lesser extent dopamine) as agonists. Nine differentadrenergic receptor subtypes have been cloned.219 There are three β AR subtypes (β1, β2, β3)and they couple to the stimulatory G-protein, Gs, which results in activation of adenylcyclase and increases in intracellular cAMP. The β2 AR is expressed to some extent in virtuallyevery tissue in the body. In the lung, this is present in epithelium, smooth muscle of bronchiand bronchioles, submucosal glands, the endothelium and smooth muscle of pulmonaryarteries, alveolar walls, immune cells including mast cells, macrophages, eosinophils,neutrophils, and lymphocytes. There are reports that β3AR also regulates bronchial smoothmuscle tone in pharmacological in vivo studies. β2 AR has been studied extensively andthought to have important therapeutic implications. Recent genetic polymorphisms of theβ2 AR have been identified in the population, which may be the basis of a more severe formof the disease or the basis of the heterogeneity of receptor expression and response to beta-agonists observed clinically.220 Some of the important molecular domains that have beenfound to be important for receptor function have also been identified. Although a numberof studies have addressed whether β2 AR are dysfunctional in asthma, there appears to beno consensus in this matter.221 It seems that beta-receptor dysfunction may not be the primarylesion in asthma. Perhaps this occurs as a secondary phenomenon in asthma either becauseof the drugs used and thus acquired or there may be a receptor mutation or polymorphism.
Szentivanyi proposed in 1968 that asthma may be due to an inherited or acquired deficitin β-adrenoceptor function.222 Several lines of evidence suggest that the β2-adrenoceptormay be abnormal in asthma, making the β2-adrenoceptor gene an attractive candidategene in this disease. Administration of β2-adrenoceptor agonists increases airway tone andresponsiveness in patients with asthma.223 Bronchial or tracheal smooth muscle obtained ateither autopsy or surgery from asthmatic patients show a deficit in β-adrenoceptorfunction.224-227 A large number of polymorphisms or point mutations have been describedin the human β2-adrenoceptor gene. A restriction fragment length polymorphism (RFLP)of this gene has been reported using the restriction enzyme Ban I.228 Another biallelicpolymorphism is reported using the restriction enzyme Fnu4HI,229 while subsequentinvestigations reported nine different point mutations within the coding region, four ofwhich result in changes in amino acid residues 16, 27, 34 and 164.230
Moreover, cells transferred with β2-adrenoceptor complimentary DNA containing themutations at amino acid positions 27 or 164 showed altered β-adrenoceptor function. Studieson the distribution of Ban I polymorphisms in South African asthmatics showed the presenceof both these alleles in this group, but the genotypes were found with similar frequencies in

Pathophysiology of Bronchial Asthma 63
allergic and nonallergic subjects. Further studies on sequencing of the β2-adrenoceptor geneidentified nine separate point mutations or polymorphisms, but there was no significantdifference in the frequency of alleles between the asthmatic and nonasthmatic patients.230
Japanese investigation on family members of asthmatics found a higher prevalence of asthmain family members who lacked the 3.1 kb Ban I RFLP, but the findings were not sufficient toexclude genetic linkage to either methacholine responsiveness or allergy.231 Subsequent
Fig. 3.5: Interaction of various factors in the causation of asthma

64 Bronchial Asthma
studies also showed that distribution of these alleles was not different between asthmaticsand nonasthmatics,232 although it was not possible to exclude an association. In a morerecent study to exclude genetic linkage between the β2-adrenoceptor gene and asthma,allergy, and methacholine airway hyperresponsiveness, indicated that these are not linkedto a dominant β2-adrenoceptor gene with strong effect in families with an inherited patternof asthma.233
Nitric Oxide (NO) and Bronchial Asthma185-188
Nitric oxide is synthesised from L-arginine by the enzyme NO synthase (NOS). Two formsof NO is known; iNOS (independent of Ca++) and cNOS (Ca++/calmodiulin-dependent,constitutive form). While the former is induced by TNF-alpha and beta, interferon gamma,endotoxins, interleukin-1 and other cytokines, stimulation of the later occurs throughmediators like bradykinin, histamine, PAF, acetylcholine, and leukotrienes. Thus, it isobvious that NO has the potential to affect a number of cells critical for normal lung functionand NO possibly plays a key role in the pathogenesis of asthma and its inhibitors may beuseful therapeutically to treat asthma.221 Nitric oxide is present in the expired air of healthyindividuals.234 It is a known bronchial smooth muscle relaxant. Thus its level should bereduced in bronchial asthma. But on the contrary, NO is higher in the expired air ofasthmatics,185,235 and epithelial NOS is higher in the epithelial cells in them.236 This impliesthat NO may increase in asthma as a compensatory response to other factors, such as thosethat cause bronchoconstriction or inflammation. Further, elevated NO might exacerbatebronchial obstruction because NO relaxes vascular smooth muscle and thus, vascularengorgement which is an important pathogenetic mechanism. In addition, elevated NOmay result in elevated NO reaction products, such as superoxides, particularly peroxynitrite,which may cause airway damage if excess. However, it is not clear whether elevated NO ispart of the primary pathologic process in asthma or is a compensatory response.
SUMMARY OF EVENTS LEADING TO AIRWAYS INFLAMMATION
The pathogenesis of bronchial asthma is more clearly understood in extrinsic or allergicasthma and is summarised in Figure 3.6.237 Although the terms “intrinsic” and “extrinsic”no longer adequately reflect our knowledge of the clinical syndrome of asthma, recentadvances in the understanding of its pathophysiology indicate that it is a heterogenousdisorder with multiple triggers. There are, however, features, which are virtually commonto all asthmatics. These include airways inflammation and hyperreactivity to a broad rangeof stimuli. The chronic allergic response is a continuous process of IgE generation, mast cellactivation, and eosinophil recruitment. These processes are orchestrated by T lymphocytes.In atopic individuals, T lymphocytes receive an allergen-specific signal from highlyspecialised antigen presenting cells, called dendritic cells, at mucosal surfaces. Presentationof allergen peptides to the T cell usually occurs in local lymphoid tissue along with theessential engagement of co-stimulatory molecules (B7 and CD28) and results in thedifferentiation of the naive T cell to one that generates a range of cytokines which upregulatecells and antibodies involved in the allergic response. CD4+ lymphocytes of the Th2-typeare activated and clonally expand after capture and processing of inhaled allergens likecigarette smoke, house dust mites, pollen, viral infection, fungi, etc. by the dendritic cells
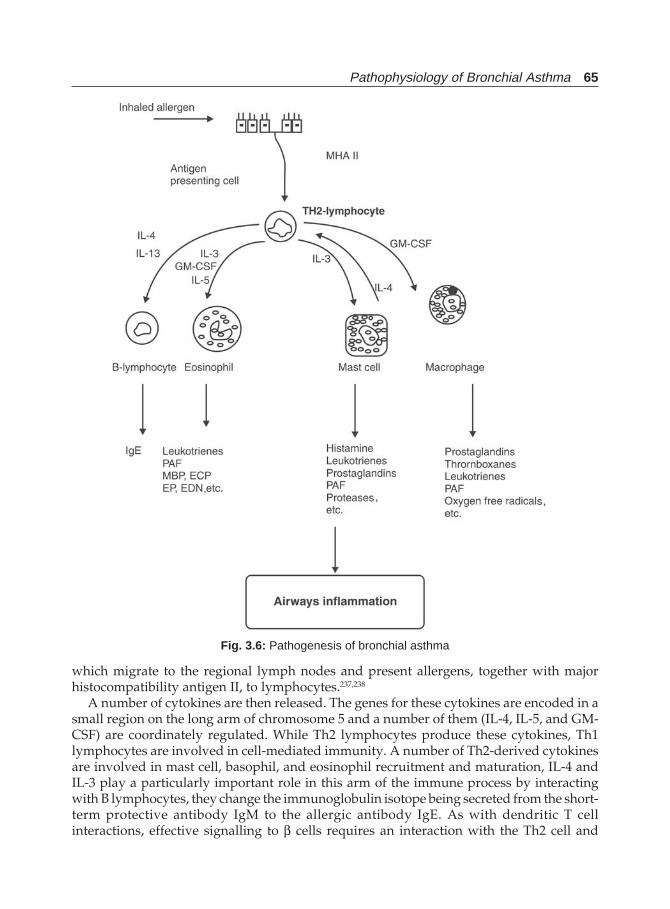
Pathophysiology of Bronchial Asthma 65
Fig. 3.6: Pathogenesis of bronchial asthma
which migrate to the regional lymph nodes and present allergens, together with majorhistocompatibility antigen II, to lymphocytes.237,238
A number of cytokines are then released. The genes for these cytokines are encoded in asmall region on the long arm of chromosome 5 and a number of them (IL-4, IL-5, and GM-CSF) are coordinately regulated. While Th2 lymphocytes produce these cytokines, Th1lymphocytes are involved in cell-mediated immunity. A number of Th2-derived cytokinesare involved in mast cell, basophil, and eosinophil recruitment and maturation, IL-4 andIL-3 play a particularly important role in this arm of the immune process by interactingwith B lymphocytes, they change the immunoglobulin isotope being secreted from the short-term protective antibody IgM to the allergic antibody IgE. As with dendritic T cellinteractions, effective signalling to β cells requires an interaction with the Th2 cell and

66 Bronchial Asthma
involves antigen presentation and engagement of a second set of co-stimulatory molecules(CD40 and its ligand, CD40L). If T and B cell interact in the presence of antigen, IL-4 or IL-13,and co-stimulatory molecules, allergen-specific IgE is generated. If IL-4 or IL-3 is present, butcell-cell contact does not occur, only non-specific IgE is generated. Thus IgE has the importantrole of linking allergen recognition to cell signalling in a variety of cells, which release a rangeof active mediators. IL-4 produced by Th2 lymphocytes ‘fuels’ the inflammatory reactions inthe airways and leads to production of further Th2 lymphocytes and to differentiation andmaturation of IgE producing B lymphocytes. A strong genetic component plays importantrole in the form of an ability of a susceptible individual to recognise an environmental allergenas foreign and mounts an allergic immune response through the human lymphocyte antigen(HLA or MHC class II) molecules. The second component of the gene involves the genesresponsible for cytokine response.
Allergen specific IgE binds to IgE receptors on several inflammatory cell types such aseosinophils, mast cells, and macrophages. High affinity IgE receptors are an important linkbetween the presence of specific antigen in the microenvironment and activation of mast cellsand other cells. Antigen-specific IgE binds to effector cells via specific IgE receptors; whenantigen binds an adequate number of these receptors to initiate receptor clustering, signaltransduction occurs. The molecular nature of the IgE receptor has now been clearly defined;221
it is composed of four chains: an alpha chain, a beta chain, and two gamma chains. While thealpha chain binds IgE, it is thought that the gamma chains are the units that initiatesintracellular signal transduction; however, the specific mechanisms of transduction are notestablished.
The inflammatory cells then release various inflammatory mediators outlined above, whichaccentuates airways’ inflammation including the release of 5-lipooxygenase products andproteases. Leukotrienes along with other products cause bronchoconstriction and otherchanges characteristic of bronchial asthma. Mast cell proteases are also important players inthe inflammatory process. Neutral endopeptidase (NEP) is a major enzyme of importance inlimiting the biologic activity of small peptide mediators such as substance P or neurokinin A.The beta-adrenergic receptor and nitric oxide represent two effector mechanisms that areimportant in modifying the biology of an asthmatic response.
Although smooth muscle constriction can lead to airways obstruction, it is now understoodthat nonmuscular airway obstruction is not less important. The importance of airway wallremodelling with thickening of the airway wall due to infiltration with inflammatory cellsand alteration in the amount and type of collagen deposited in the airway is reflected in theenhanced degree of obstruction that is observed for a given level of smooth muscle activationin the remodelled wall. The wall is also thickened and obstructed due to the engorgement ofthe bronchial blood vessels. Such engorgement could account for a significant component ofasthmatic airway narrowing under certain circumstances. The presence of intraluminal fluidsincluding mucosubstances further obstruct airways and could make it more difficult forindividuals to clear secretions from their airways.
The relationship between airway inflammation and the development of airwayhyperresponsiveness and clinical asthma has been well established during the last decade.Exposure to oxidant pollutants, some chemicals, antigens, and viral respiratory tract infectionsare all associated with inflammatory cell infiltration into the airway and these inflammatorystimuli are also associated with the development of airway hyperresponsiveness. Most studieshave shown that airway inflammation precedes the development of hyperresponsiveness

Pathophysiology of Bronchial Asthma 67
and may be the prerequisite feature necessary for the development of both hyperresponsivenessand clinical bronchospasm. The relationship between airway inflammation, bronchialhyperreactivity and airway obstruction in asthma is shown in Figure 3.7.
Although, approximately one-half of the children with wheezing in infancy and youngchildhood will no longer be wheezing at 6 years of age,239 a different type of observation hasbeen noted in children with wheezing in their bronchoalveolar lavage fluid. Increased numbersof cells and increased neutrophils in BAL samples have been reported in children havingwheezing.240-244 In contrast, BAL eosinophilia is a common finding in adults with asthma.Eosinophilia and elevated IgE levels have also been found in infants who subsequentlydevelop asthma. It is possible that neutrophil-induced inflammation is important in the earlystages of wheezing in infants. It is also possible that this neutrophil response may be aresponse to an unrecognised infection.
While the pathogenesis of occupational asthma, intrinsic asthma and other forms of asthmais less clearly understood, these conditions are thought to involve a cytokine “cascade” similarto that involved in extrinsic or allergic asthma.237
The mechanisms of allergy in causing episodic and chronic asthma are shown inFigure 3.8.
Aspirin Induced Asthma
Patients with bronchial asthma and sensitivity to aspirin (ASA) and other nonsteroidal anti-inflammatory drugs are often corticosteroid-dependent and have the accompanying symptomsof rhinosinusitis, rhinorrhoea, nasal congestion, anosmia, loss of taste, and recurrent severenasal polyposis.245 Upon challenge with aspirin or other cyclooxygenase inhibitors thesepatients have increased cysteinyl leukotriene release as detected in urine,246,247 in nasallavage,248,249 and in bronchial lavage fluids,250 in contrast to aspirin-tolerant subjects. These
Fig. 3.7: Interaction of various factors
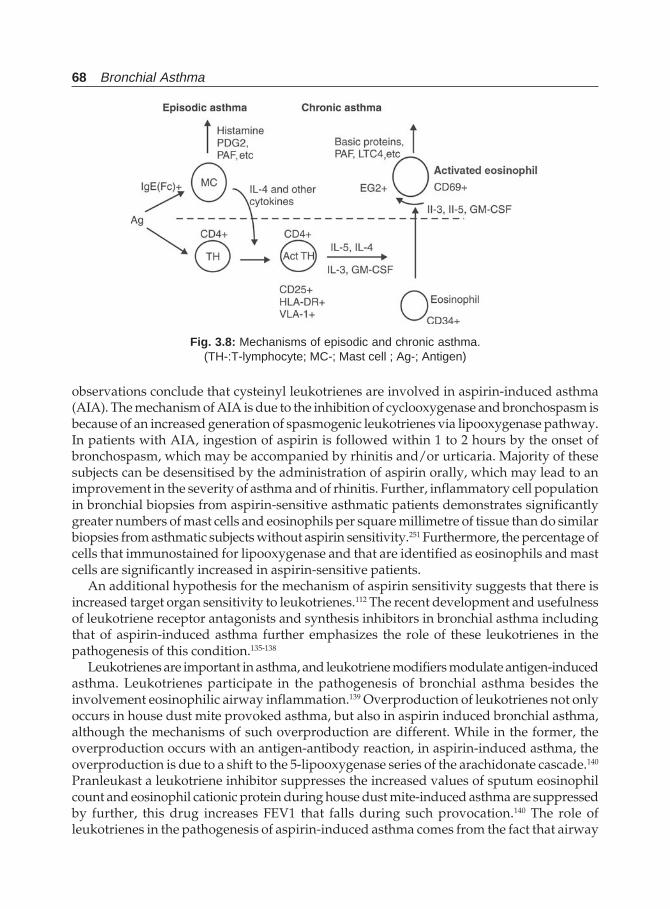
68 Bronchial Asthma
observations conclude that cysteinyl leukotrienes are involved in aspirin-induced asthma(AIA). The mechanism of AIA is due to the inhibition of cyclooxygenase and bronchospasm isbecause of an increased generation of spasmogenic leukotrienes via lipooxygenase pathway.In patients with AIA, ingestion of aspirin is followed within 1 to 2 hours by the onset ofbronchospasm, which may be accompanied by rhinitis and/or urticaria. Majority of thesesubjects can be desensitised by the administration of aspirin orally, which may lead to animprovement in the severity of asthma and of rhinitis. Further, inflammatory cell populationin bronchial biopsies from aspirin-sensitive asthmatic patients demonstrates significantlygreater numbers of mast cells and eosinophils per square millimetre of tissue than do similarbiopsies from asthmatic subjects without aspirin sensitivity.251 Furthermore, the percentage ofcells that immunostained for lipooxygenase and that are identified as eosinophils and mastcells are significantly increased in aspirin-sensitive patients.
An additional hypothesis for the mechanism of aspirin sensitivity suggests that there isincreased target organ sensitivity to leukotrienes.112 The recent development and usefulnessof leukotriene receptor antagonists and synthesis inhibitors in bronchial asthma includingthat of aspirin-induced asthma further emphasizes the role of these leukotrienes in thepathogenesis of this condition.135-138
Leukotrienes are important in asthma, and leukotriene modifiers modulate antigen-inducedasthma. Leukotrienes participate in the pathogenesis of bronchial asthma besides theinvolvement eosinophilic airway inflammation.139 Overproduction of leukotrienes not onlyoccurs in house dust mite provoked asthma, but also in aspirin induced bronchial asthma,although the mechanisms of such overproduction are different. While in the former, theoverproduction occurs with an antigen-antibody reaction, in aspirin-induced asthma, theoverproduction is due to a shift to the 5-lipooxygenase series of the arachidonate cascade.140
Pranleukast a leukotriene inhibitor suppresses the increased values of sputum eosinophilcount and eosinophil cationic protein during house dust mite-induced asthma are suppressedby further, this drug increases FEV1 that falls during such provocation.140 The role ofleukotrienes in the pathogenesis of aspirin-induced asthma comes from the fact that airway
Fig. 3.8: Mechanisms of episodic and chronic asthma.(TH-:T-lymphocyte; MC-; Mast cell ; Ag-; Antigen)

Pathophysiology of Bronchial Asthma 69
narrowing and other signs in these patients are associated with 2-10 fold higher values ofLTE4 in the urine of these patients compared to aspirin tolerant patients.142-144 Further, severalleukotriene modifiers inhibit the asthma response in oral or inhaled bronchoprovocation byaspirin and other non-steroidal anti-inflammatory agents144,145 and improve respiratoryfunction by bronchodilatation.146
Virus-induced Asthma
Viral infections have been considered to play a significant role in the development andconsolidation of obstructive airway disease. This may occur by amplification of the responseto cigarette smoke, induction of steroid resistance,252 enhanced sensitisation to inhaledallergens due to increased permeability and recruitment of dendritic cells,253 or reactivation oflatent but persistent virus due to insufficient T-helper-1-type immune response and/oradministration of corticosteroids.254 Viral respiratory infections increase symptoms of bronchialasthma in many patients.255 Rhinovirus increases airway responsiveness and also promotesthe likelihood of a late allergic reaction to allergen.256,257 Enhanced airway responsivenessand the late allergic reaction persist for weeks beyond the viral infection. Lymphocytes areactivated during incubation with rhinovirus and secrete cytokines, like γ -interferon. Althoughγ-interferon does not have any proinflammatory activity like those of Il-4 and 5, it does affecteosinophil function, including promotion of survival. Furthermore, γ-interferon can augmentbasophil mediator release. Thus, lymphocyte activation by virus may provide a very differentcytokine profile and in this manner selectively enhances inflammation.71,258
Exercise-induced Asthma (EIA)
Exercise-induced asthma is a temporary increase in the airway resistance following vigorousphysical activity. Obstruction to airflow begins soon after cessation of exercise and peaks in5-10 minutes.259 Most patients will recover completely in the next 30-60 minutes, but in fewthis EAR will be followed by a LAR several hours after the initial response subsides.260,261
Two major hypotheses have been put forward to explain the mechanism whereby waterand heat loss by hyperventilation with exercise causes airway narrowing.
i. The EIA is a consequence of thermodynamic events that occur within the tracheobronchialtree during or after hyperventilation that is associated with exercise.262 Because of thishyperventilation during exercise, there is a fall in the airway temperature and respiratorywater loss, i.e. evaporation causes cooling.263 Mouth breathing to meet increased demandof oxygen further aggravates this factor because air bypasses the nasal air-conditioningmechanism. Thus, during re-warming of the airways by reactive hyperaemia of thebronchial circulation with subsequent airway oedema of the bronchial wall during thepost-exercise period.264 Further, the event precipitates bronchoconstriction. The magnitudeof bronchospasm is directly proportional to the heat loss from the respiratory tract requiredto bring the inspired air to alveolar conditions.265 Oedema due to hyperaemia ofmicrocirculation may be the cause of bronchial obstruction developing after exercise. It isalso possible that patients with EIA may have hyperplastic capillary bed that developexaggerated hyperaemia and airway oedema leading on to bronchial obstruction.262
ii. The other mechanism of EIA may be as a result of water loss from mucosal surface andresulting increase in osmolarity of the fluid interface of the mucosal surface in the airways,

70 Bronchial Asthma
which may lead to mast cell and basophil degranulation and precipitating EIA.266,267
Exercise-induced bronchoconstriction, a feature of 70-80% of asthmatics,268 is triggeredby drying of the bronchial epithelium due to airway water loss from the tracheobronchialtree.269-273 During exercise, the ventilation rate increases, and thus the respiratory tractneeds to condition much larger volumes of air over a much shorter time during exercisecompared with rest, and airway dehydration occurs with subsequent exercise-inducedbronchoconstriction. The findings that269 inhaling fully humidified air at body conditionscould prevent exercise-induced bronchoconstriction demonstrated the importance of waterloss from the airway. It is also been recommended swimming as the exercise leasttroublesome to asthmatic patients because of the humidity of the inspired air, a phenomenonthat is supported by comparative studies of diverse sporting activities.274,275
Since mast cell-derived mediators, such as histamine and leukotrienes, may cause not onlyairway smooth-muscle contraction, but also airway oedema, it is possible that both of thesehypotheses are related to the airway narrowing following exercise in asthmatics. Exercise-induced bronchospasm is, at least in part, due to bronchial microvascular phenomena suchas vascular engorgement and plasma leakage that could thicken the mucosa and therebynarrow airway diameters, which could in turn amplify the effects of airway smooth musclecontraction.
Various reports give conflicting results concerning the role of inflammation in EIA.276,277
However, some believe that EIA, to a larger extent, is mediated through the release ofbronchoconstrictor substances from inflammatory cells in the airway wall. Leukotrienes seemto play a particularly important role in this response. This conclusion is arrived fromobservations made in antileukotriene drug studies in EIA.278,279 Similarly antileukotrienes arehelpful in cold air-induced bronchial asthma280 highlighting the role of cold air in causingEIA. Further, eucapnic voluntary hyperventilation manoeuvres designed to simulate exercise-induced bronchoconstriction in the laboratory, demonstrate that airway fluid-loss has a similarbronchoconstrictor effect to histamine.281-284 It is also demonstrated that the release of histamine,a potent bronchoconstrictor, and other pro-inflammatory bronchoconstrictor mediators,including cysteinyl-leukotrienes,285 from mast cells and other airway cells under hyperosmolarconditions.286-288 These findings underline the bronchoconstrictor potential of airwaydehydration. Presence of thermally sensitive neural receptors in the airways of patientssusceptible to EIA may be responsible for bronchoconstriction in response to cold air.267
Recently another hypothesis suggests that increased excessive production of nitric oxideduring exercise289,290 increases airway vascular permeability, that co-relates with the severityof exercise-induced bronchoconstriction in asthmatics. Assessment of albumin flux in airwaylining fluid stimulated by hypertonic saline solution is a sensitive predictor of the severity ofthis phenomenon.291
Occupational Asthma
Bronchial hyperreactivity is a characteristic feature of occupational asthma.292 Specificinhalation challenge tests may induce any of the five types of reactions: (i) isolated early;(ii) isolated late; (iii) biphasic; (iv) continuous; or (v) atypical asthmatic reactions.293 An earlyreaction occurs within a few minutes after an inhalation challenge, reaches maximal intensitywithin 30 minutes, and ends within 60-90 minutes. An isolated late asthma reaction occurs4-6 hours after the challenge, reaches maximal intensity within 8-10 hours, and ends after

Pathophysiology of Bronchial Asthma 71
24-48 hours. A biphasic reaction is an early reaction with spontaneous recovery followed bya late asthma reaction. In a continuous type of asthma reaction there will be no remissionbetween the early and late reactions. Atypical reactions usually start 2 hours after a challengeand last for a few hours.294 Generally, IgE-dependent agents induce isolated early reactions orbiphasic reactions, and IgE-independent agents will induce isolated late, biphasic or atypicalasthma reactions.
Occupational asthma induced by IgE-dependent agents is similar to allergic asthma.295
Most high-molecular-weight compounds (5000 or more daltons) induce asthma by producingspecific IgE antibodies. These molecules such as proteins, glycoproteins and polysaccharidesare usually complete antigens. Some low-molecular-weight molecules (<5000 daltons) likeacid anhydrides and platinum salts act as haptens and induce specific IgE antibodies bycombining with a body protein. The specific reaction between antigen and IgE gives rise to acascade of events that is responsible for the activation of inflammatory cells. Both preformedand newly formed inflammatory mediators are released, and they orchestrate the inflammatoryevents already outlined above. However, for many low-molecular-weight molecules, such asisocyanates, specific IgE antibodies have not been identified or are found only in a smallproportion of cases.296 Presence of these antibodies does not necessarily mean the cause of thedisease, but may be the markers of exposure .297 In addition to IgE-mediated reactions,immunoglobulins of the IgG class, possibly IgG4, may be involved in immediate-type reaction.
T lymphocytes may be directly involved in the inflammatory process.297,298 Pathologic airwaychanges are similar to those in patients with other forms asthma. Some of them includeaccumulation of inflammatory cells, mostly eosinophils, oedema, hypertrophy of smoothmuscle, subepithelial fibrosis, and exudation of fluid and mucus.299-301 An increase in theactivated eosinophils and T lymphocytes has been found in the mucosa and sub-mucosa,and mast cells increase in the epithelium.302 Animal models for pathogenic and immunologicmechanisms of bronchial asthma have also confirmed these observations.303
Some other mechanisms that are responsible for occupational asthma are as follows; (i)reflex vagal bronchoconstriction in response to an irritant-effect on specific receptors; (ii)inflammatory bronchoconstriction secondary to toxic concentrations of gases (nonspecificcomplement activation, neuropeptide release, perturbation of cell membrane releasingarachidonic acid products); (iii) a direct pharmacological reactions by agents like organicinsecticides with anticholinergic activity (parasympathetic agonists) and beta-adrenergicblocking agents; (iv) or immunologic mechanism leading to allergic tissue injury.304
Irritant induced occupational asthma (Reactive airways dysfunction syndrome, RADS) ispersistent asthma and airway hyperresponsiveness, which develops after acute inhalation ofa respiratory irritant in toxic concentrations.305 The onset of respiratory symptoms and thepresence of airway hyperresponsiveness within a few hours of exposure to an identifiableirritant distinguish this entity from hypersensitivity induced occupational asthma. This formof asthma is associated with the workplace, in which wheezing illness starts within 24 hoursor less of a single large exposure to an irritant. The condition is inflammatory, but does notinvolve immunological recognition of the irritant, so that continued low-level exposure to thecausative agent can be tolerated without problem. Bronchial biopsy studies in theseindividuals have shown bronchial epithelial cell injury with desquamation, and bronchialwall inflammation, with infiltration of plasma cells and lymphocytes, but not eosinophils.306-
307 The diagnosis is made by the presence of non-specific responsiveness and a compatiblehistory. The prognosis varies, but most often the condition improves.

72 Bronchial Asthma
Nocturnal Asthma
Diurnal variations (circadian rhythm) is normally seen in healthy normal individuals as wellas in patients with bronchial asthma. Lowest airways function during early in the morningand best during the mid-day and evening has been shown by various investigators.308-310
Frequent occurrence of nocturnal symptoms has been shown in many reports.311-313 Patients ofbronchial asthma show a greater bronchial reactivity at 4 AM when compared to at 4 PM.314 Itis also seen that majority of asthma deaths occur most often at night.315,316
Although mechanisms involved in nocturnal asthma are not clearly understood, multiplefactors seem to be involved. In allergic individuals, allergen exposure during evening hoursinitiates a cascade of events to produce a LAR. Further, exposure to house-dust mite mayprecipitate an EAR, and these factors precipitate bronchoconstriction.317 Lowest levels ofserum adrenaline and cortisol, and highest levels of histamine during night hours could beresponsible for nocturnal episodes in asthmatic individuals318 The BAL fluid recovered frompatients having nocturnal asthma shows greater number of eosinophils and neutrophils at4 AM compared to that at 4 PM. This indirectly suggests worsening of inflammation duringnight319 Increased vagal tone at night or gastro-oesophageal reflux leading to increased vagaltone may further be contributory to increased bronchial reactivity and bronchial asthma atnight. Changes of body temperature, i.e. lowering of temperature, and increased accumulationof secretions in the respiratory tract during sleep may be additional factors.320-321
REFERENCES
1. Barnes PJ, Chung KF, Page CP. Inflammatory mediators in asthma. Pharmacol Rev 1988;40:49-84.
2. Holgate ST, Finnerty JP. Recent advances in the understanding the pathogenesis of asthma andits clinical consequences. Cl J Med 1988;249:5-9.
3. Cockcroft DW, Murdock KY. Comparative effects of inhaled salbutamol, sodium cromoglycate,and beclomethasone dipropionate on allergen-induced early asthmatic response, late asthmaticresponse, and allergen-induced increase in bronchial responsiveness to histamine. J All ClinImmunol 1987;79:734-40.
4. Postuma DS, Koeter GH, de Vries K. Clinical expression of airway hyperreactivity in adults. ClinRev Allergy 1990;7:321-343.
5. Durham SR, Craddock CF, Cookson WOO, Benson MK. Increase in airway responsiveness tohistamine precede allergen-induced late asthmatic response. J allergy Clin Immunol 1988;82:764-70.
6. Metzger WJ, Zavala D, Richardson HB et al. Local allergen challenge and bronchoalveolarlavage of allergic asthmatic lungs: Description of the model and local airway inflammation. Amrev Respir Dis 1987;135:433-40.
7. Holgate ST. Pharmacological modulation of asthma in relation to mechanisms. All Proc1991;12:151-54.
8. Madison JM. Chronic asthma in the adult: Pathogenesis and pharmacotherapy. Seminar RespirMed 1991;12:175-84.
9. Kelly C, Ward C, Stenton CS, Bird G, Hendrick DJ, Walters EH. Number and activity ofinflammatory cells in bronchoalveolar lavage fluid in asthma and their relation to airwayresponsiveness. Thorax 1988;43:684-92.
10. Casale TB, Wood D, Richardson HB et al. Elevated bronchoalveolar lavage fluid histamine levelsin allergic asthmatics are associated with methacholine bronchial hyperresponsiveness. J ClintInvest 1987;79:1197-1203.

Pathophysiology of Bronchial Asthma 73
11. Wardlan AJ, Dunnette S, Gleich GJ, Collins JV, Kay AB. Eosinophils and mast cells in broncho-alveolar lavage with mild asthma: Relationship to bronchial hyperreactivity. Am Rev Respir Dis1983;137:62-69.
12. Liu MC, Blercker ER, Lichtensetein LM et al. Evidence of elevated levels of histamine, prostaglandinD2, and other bronchoconstricting prostaglandins in the airway of subjects with mild asthma.Am Rev Respir Dis 1990;142:126-32.
13. Diaz P, Galleguillos FR, Ganzales MC, Pantin C, Kay AB. Bronchoalveolar lavage in asthma; theeffect of DSCG on leucocyte count, immunoglobulins and complement. J All Clin Immunol1984;74:41-48.
14. Kirby JG, Hargreave FE, Gleich GJ, O’Byrne PM. Bronchoalveolar cell profiles of asthmatic andnon-asthmatic subjects. Am Rev Respir Dis 1987;136:379-83.
15. Flint KC, Leung KBP, Hudspith BN et al. Bronchoalveolar mast cells in extrinsic asthma: Amechanism for initiation of antigen-specific bronchoconstriction. Br Med J 1985;291:923-26.
16. Haahtela T (editorial). The importance of inflammation in early asthma. Respir Med 1995;89:461-62.
17. Glynn AA, Michaels L. Bronchial biopsy in chronic bronchitis and asthma. Thorax 1960;15:142-53.
18. Dunnil MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of thebronchi in normal subjects, in status asthmaticus, in chronic bronchitis and emphysem. Thorax1969;24:176-79.
19. Laitinen LA, Heino M, Laitinen A, Kava T, Haahtela T. Damage of the airway epithelium andbronchial reactivity in patients with asthma. Am Rev Respir Dis 1985;131:599-606.
20. Bigby TD. Inflammatory mediators and asthma. Pulmonary Perspectives 1992;9(4):6-9.21. Robinson et al. N Engl J Med 1992;326:298-304.22. International symposium on airway hyperreactivity. AM Rev Respir Dis 1991;143:S3.23. Johnston SL, Holgate S. The inflammatory response in asthma. Br J Hosp Med 1991;46:84.24. Sheffer AL, Fink JN. Immunopharmacologic update. J Allergy Clin Immunol 1991;88:303.25. Ingram RH Jr. Asthma and airway hyperresponsiveness. Annu Rev Med 1991;42:139.26. Boushey HA, Holtzman MJ, Sheller JR, Nadel JA. Bronchial hyperreactivity. Am Rev Respir Dis
1980;121:1389.27. Busse WW. The role of inflammation in asthma: a new focus. J Respir Dis 1989;10:72.28. Djukanovic R, Roche WR, Wilson JW et al. Mucosal inflammation in asthma. Am Rev Respir Dis
1990;142:434.29. Holgate ST, Beasley R, Twentymen OP. The pathogenesis and significance of bronchial
hyperresponsiveness in airway disease. Clin Sci 1987;73:561.30. Chanez P, Bousquet J, Couret I et al. Increased number of hypodense alveolar macrophages in
patients with bronchial asthma. Am Rev Respir Dis 1991;144:923.31. Page CP. Are mast cells bad? Postgrad Med J 1991;67(Suppl 4):S6.32. Patterson NAM, Wassermann SI, Said JW, Austen KF. Release of chemical mediators from
partially purified human lung mast cells. J Immunol 1976;117:1356-62.33. Kay AB, Austen KF. The IgE mediated release of an eosinophil leucoyte chemotactic factor from
human lungs. J Immunol 1971;107:899-902.34. Tomioka M, Ida S, Shindoh Y, Ishizaka T, Takishima T. Mast cells in bronchoalveolar lumen of
patients with bronchial asthma. Am Rev Respir Dis 1984;129:1000-05.35. Walls AF, Djukanorie R, Walters C et al. Mast cell activation in atopic and intrinsic asthma.
J allergy Clin Immunol 1991;87:215.36. Krishenbaum AS, Goff JP, Kessler SW et al. The effect of IL-3 and stem cell factor on the appearance
of mast cells and basophils from CD-34+ pleuripotent progenitor cells. J Immunol 1992;148:772-77.

74 Bronchial Asthma
37. Drazen JM, Evans JF, Stevens RL, Shipp MA. Inflammatory effector mechanisms in asthma. AmJ Crit Care Med 1995;152:403-07.
38. Cruzen N, Rafferty P, Holgate ST. Effect of a cyclooxygenase inhibitor, flurbiprofen and a H1-histamine receptor antagonist, terfenadine, alone and in combination on allergen inducedbronchoconstriction in man. Thorax 1987;42:946-52.
39. Piacentinii GL, Kaliner M. The potential role of leukotrienes in bronchial asthma. Am Rev RespirDis 1991;143:S96-S99.
40. Casale TB, Wood D, Richerdson HB et al. Direct evidence of a role for mast cells in the pathogenesisof antigen induced bronchoconstriction. J Clin Invest 1987;80:1507-11.
41. Romagnani S, Maggi E, Passonchi P, Macchia D, Piccini MP, Ricci M. Increased numbers ofTh-2 like CD4+ T cells in target organs and in the allergen specific receptor of allergic patients—Possible role of IL-4 produced by non-T cells. Arch Allergy Appl Immunol 1991;94:133-36.
42. Barnes PJ. New concepts in the pathogenesis of bronchial hyperresponsiveness and asthma.J allergy Clin Immunol 1989, 83:1013-26.
43. Mcfadden ER, Gilbert IA. 1992-asthma. N Engl J Med 1992.44. Bousquet J, Chanez P, Lacoste JY et al. Eosinophilic inflammation in asthma. N Engl J Med
1990;323:1033-39.45. Ohashi Y, Matojima S, Fukuda T, Makano S. Airway hyperresponsiveness increased intracellular
spaces of bronchial epithelium, and increased infiltration of eosinophils and lymphocytes inbronchial mucosa in asthma. Am Rev Respir Med 1992;145:1469-76.
46. Dunhil MS. The pathology of asthma with special reference to changes in the bronchial mucosa.J Clin Pathol 1960;13:27-33.
47. Dunhill MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of thebronchi in the normal subjects, in chronic bronchitis, and in emphysema. Thorax 1969;24:176-79.
48. Filley WV, Holley KE, Kephart GM, Gleich GJ. Identification by immunofluorescence ofeosinophil granule major basic protein in lung tissue of patients with bronchial asthma. Lancet1982;ii:11-16.
49. Lam S, LeRice J, Philips D, Chan YM. cellular and protein changes in bronchial lavage fluid afterlate asthmatic reaction in patients with red cedar asthma. J Allergy Clin Immunol 1987;80:44-50.
50. DeMonchey JG, Kauffman HF, Venge P et al. Bronchoalveolar eosinophils during allergeninduced late asthmatic reactions. Am Rev Respir Dis 1985;131:373-76.
51. Sedgwick JB, Calhoun GJ, Kita H, et al. Immediate and late airway response of allergic rhinitispatients to segmental antigen challenge. Characterisation of eosinophils and mast cell mediators.Am Rev Respir Dis 1991;144:1274-81.
52. Hamid Q, Azzawi M, Ying S et al. Expression of mRNA for interleukin 5 in mucosal bronchialbiopsies from asthma. J Clin Invest 1991;87:1541-46.
53. Robinson DS, Homid Q, Ying S et al. Predominant Th2-like bronchoalveolar lavage T-lymphocytepopulation in atopic asthma. N Engl J Med 1992;326:298-304.
54. Mossman TR, Coffman RL. Th1 and Th2 cells; Different patterns of lymphokine secretion leadsto different functional properties. Ann Rev Immunol 1989;7:145-73.
55. Gajewski TF, Joyce J, Pitch FW. Antiproliferative effect of interferon-gamma in immuneregulation. III. differential secretion of Th1 and Th2 murine helper T lymphocyte clone usingrecombinants Il-2 and recombinant IFN-gamma. J Immunol 1989;143:15-22.
56. Fiorentino D, Bond HW, Mossman TR. Two types of mouse T-helper cells. IV. Th2 clones secreta factor that inhibits cytokine production by Th1 clones. J Exp Med 1989;170:2081-95.
57. De Prete G, Maggi E, Parrochi P et al. IL-4 is an essential factor for IgE synthesis induced in vitroby human T cells and their supernatants. J Immunol 1988;140:4193-98.
58. Spits H, Yessel Y, Takelsy Y et al. recombinant interleukin-4 promotes the growth of humanT cells. J Immunol 1987;139:1142-47.
59. Schleimer RP, Sterbinsky SA, Kaiser J et al. IL-4 induces adherence of human eosinophils and

Pathophysiology of Bronchial Asthma 75
basophils but not neutrophils to endothelium; association with expression of VCAM-1. J Immunol1992;148:1086-92.
60. Desermaux P, Janin A, Colombal JF et al. Interleukin-5 messenger RNA expression by eosinophilsin the intestinal mucosa of patients with celiac disease. J Exp Med 1992;175:293-96.
61. Campbell HD, Tucker WQJ, Hort Y et al. Molecular cloning, nucleotide sequence and expressionof the gene encoding human eosinophil differentiation factor (IL-5). Proc Natl Acad Sci (USA)1987;84:6629-33.
62. Saito H, Hatake K, Dvorak AM et al. Selective differentiation and proliferation of haematopoiticcell induced by recombinant human interleukins. Proc Natl Acad Sci (USA) 1988;85:2288-92.
63. Sonoda Y, Arai N, Ogawa M. Humoral regulation of eosinophilopoiesis in vitro; analysis of thetargets of interleukin-3, granulocyte/macrophage colony stimulating factor (GM-CSF), andinterleukin-5. Leukaemia 1989;3:14-18.
64. Walsh GM, Hartnell A, Wardhwa AJ et al. IL-5 enhances the in vitro adhesion of human eosino-phils, but not neutrophils, in a leukocyte integrin (CD11/18) dependant manner. Immunology1990;71:258-65.
65. Lopez AF, Sanderson CJ, Gamble JR, et al. Recombinant human interleukin-5 is a selectiveactivator of human eosinophil function. J Exp Med 1988;167;219-24.
66. Fugisawa T, Abu-ghazaleh R, Kita H, Sanderson CJ, Gleich GJ. Regulatory effect of cytokines oneosinophil degranulating. J Immunol 1990;140;642-46.
67. Schmi R, Wardlaw AJ, Cornwell O et al. Interlukin-5 selectively enhances the chemotactic responseof eosinophils obtained from normal but not from eosinophilic subjects. Blood 1992;79;2952-59.
68. Pene I, Rousset F, Brier F et al. Interleukin-5 enhances IL-4 induced age production by normalhuman B cells; The role of soluble CD 23 antigen. Eur J Immunol 1988;18:929-35.
69. NIH conference. Asthma. Ann Intern Med 1994;121:698-708.70. Barnes PJ. Molecular mechanisms of asthma: Order now emerging from chaos. (Amberson
Lecture). ALA/ATS Daily Bulletin 1996;May 12:1,4.71. Busse WW, Coffman RL, Gelfand EW, Kay AB, Rosenwasser LJ. Mechanisms of persistent
airway inflammation in asthma. A role for T cell and T cell products. Am J Crit Care Med1995;152:388-93.
72. Tsuyuki S, Tsuyuki J., Einsle K et al. Costimulation through B7-2 (CD86) is required for theinduction of a lung mucosal T helper cell 2 (Th2) immune response and altered airwayhyperresponsiveness, J Exp Med 1997;185:1671-1679.
73. Weaver CT, Unanue ER. The costimulatory function of antigen-presenting cells. Immunol Today1990;11:49-55.
74. Schwartz RH. Costimulation of T-lymphocytes: The role of CD28, CTLA-4 and B7/BB1 ininterleukin-2 production and immunotherapy. Cell 1992;71:1065-68.
75. Jenkins MK, Johnson JG. Molecules involved in T cell costimulation. Curr Opin Immunol1993;5:361-67.
76. Jenkins MK. The role of cell division in the induction of clonal anergy. Immunol today 1992;13:69-73.
77. Keane-Myers A, Gause WC, Linsley PS et al. B7-CD28/CTLA-4 costimulatory pathways arerequired for the development of T-helper cell 2-mediated allergic airway responses to inhaledantigens. J Immunol 1997;158:2042-49.
78. Le Gros G, Erb K, Harris N et al. Immunoregulatory networks in asthma. Clin Exp Allergy1999;28:S92-S96.
79. Djukanovic R. The role of costimulation in airway inflammation. Clin Exp Allergy 2000;30:S46-S50.
80. Krummel MF, Allison JP. CTLA-4 engagement inhibits accumulation and cell cycle progressionupon activation of resting T cell. J Exp Med 1996;183:2533-40.
81. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation.J Exp Med 1996;183:2541-50.

76 Bronchial Asthma
82. Nisticio L, Buzzetti R, Pritchard LE. The CTLA-4 gene region of chromosome 2q33 is linked to,and associated with type 1 diabetes. Hum Mol Genet 1996;5:1075-80.
83. Polymeropoulos MH, Xiao H, Rath DS et al. Dinucleotide repeat polymorphism at the humanCTLA-4 gene. Nucleic Acids Res 1991;19:4018.
84. Deichman K, Heinzman A, Bruggenolt E et al. An Mse1 RFLP in the human CTLA-4 promoter.Biochem Biophys Res Commmun 1996;225:817-18.
85. Lee SY, Lee YH, Shin C et al. Association of asthma severity and bronchial hyperresponsivenesswith a polymorphism in the cytotoxic T-lymphocyte antigen-4 gene. Chest 2002;122:171-76.
86. Fels AOS, Cohn ZA. The alveolar macrophage. J Appl Physiol 1986;60:353-69.87. Sibille Y, Reynolds HY. Macrophages and polymorph nuclear neutrophils in lung defense and
injury. Am Rev Respir Dis 1990;141:471-501.88. Takemura R, Werb Z. Secretory products of macrophages and their physiological functions. Am
J Physiol 1984;246:C1-C9.89. Rich EA, Tweardy DJ, Fujiwara H, Ellner J. Spectrum of immunoregulatory functions and
properties of human alveolar macrophages. Am Rev Respir Dis 1987;136:258-65.90. Poulter LW, Power C, Burke C. The relationship between bronchial immunopathology and
hyperresponsiveness in asthma. Eur Respir J 1990;3:792-799.91. Crofton RW, Diesselhoff-den Dulk MMC, van Furth R. The origin, kinetics, and the characteristics
of chuffer cells in normal steady state. J Exp Med 1978;148:1-17.92. Meuret G, Schildkuecht O, Joder P, Senn H. Proliferation activity and bacteriostatic potential of
human blood monocytes, macrophages and pleural effusions, ascites, and of alveolarmacrophages. Blood 1980;40:17-25.
93. Van Furth R, Diesselhoff-den Dulk MMC, The kinetics of promonocytes and monocytes in thebone marrow. J Exp Med 1970;132:813-28.
94. Meuret G. The kinetics of mononuclear phagocytes in man. In: Schmalzl F, Han D, Schafer HE,(Eds). Hematology and Blood transfusion; Berlin: Springer vela; 1981; 27:11-22.
95. Joseph M, Tonnel AB, Capron A, Voisin C. Enzyme release and superoxide anion production byhuman alveolar macrophages stimulated with immunoglobulin E. Clin Exp Immunol 1980;40:416-22.
96. Fuller RW, Morris PK, Richmond R et al. Immunoglobulin E-dependant stimulation of humanalveolar macrophages: Significance in type I hypersensitivity. Clin Exp Immunol 1986;65:416-26.
97. Albelda SM. Endothelial and epithelial cell adhesion molecules. Am J Respir Cell Mol Biol1991;4:195-203.
98. Elia C, Bucca C, Rolla G, Scappaticci E, Cantino D. A freeze fracture study of human bronchialepithelium in normal, bronchitic, and asthmatic subjects. J Sub-microsc Cytol Pathol 1988;20:509-17.
99. Jeffery PK, Wardlaw AJ, Nelson FC, Collins JV, Kay AB. Bronchial biopsies in asthma: An ultra-structural, quantitative study and correlation with hyperreactivity. Am Rev Respir Dis1989;140:1745-53.
100. Churchil L, Chilton FH, Resau JH, Bascom R, Hubbard WC, Proud D. Cyclooxygenase metabolismof endogenous arachidonic acid by cultured human tracheal epithelial cells. Am Rev Respir Dis1989;140:449-59.
101. Frossard N, Rhodenk J, Batrnes PJ. Influence of epithelium removal on guineapig airway responseto tachykinins: Role of end peptidase and cyclooxygenase. J Pharmacol Exp Therap 1989; 248:292-97.
102. Joseph M, Pilewski M, Steven MA. Adhesion molecules in the lung: An overview. Am Rev RespirDis 1993; 148(Suppl):S31-S37.
103. Albelda SM, Buck CA. Integrins and other cell adhesion molecules. FASEB J 1990;4:2868-80.

Pathophysiology of Bronchial Asthma 77
104. Williams AF, Barelay AN. The immunoglobulin super gene family-domains for cell surfacerecognition. Ann Rev Immunol 1988;6:381-405.
105. Lasky LA. Selectins: Interpreters of cell-specific carbohydrate information during inflammation.Science 1992;258:964-69.
106. Berg EL, Robinson MK, Mansson O, Butcher EC, Magnani JL. A carbohydrate domain commonto both Sialyl lea and Sialyl Lax is recognised by endothelial cell adhesion molecule (ELAM-1). BrJ Boil Chem. 1991;266:14869-872.
107. McDonald JA. Receptors for extracellular matrix components. Am J Physiology 1989;257:L331-37.
108. Torsi MF, Stark JM, Smith CW et al. Induction of ICAM-1 expression on human airway epithelialcells by inflammatory cytokines: Effects on neutrophil-epithelial cell adhesion. Am J Respir CellMol Biol 1992;7:214-21.
109. Look DC, Rapp SR, Keller BT, Holtzmzn MJ. Selective induction of intercellular adhesion molecule-1 by interferon-gamma in human airway epithelial cells. Am J physiology 1992;263:L79-l87.
110. Kumar S, Mohan A, Sharma SK, Pande JN. Recent concepts in the pathogenesis of bronchialasthma. Ind J Chest Dis All Sc 1997;39:27-45.
111. Lee TH, Arm J. Leukotrienes. Allergy Proc 1991;12:139-42.112. Brown PH, Crompton GK, Greening AP. Proinflammatory cytokines in acute asthma. Lancet
1991;338:590.113. Milgrom H. Asthma- something old, something new. Postgrad Med J 1991;67(Suppl 4):S13.114. Jansen HM, Neijens HJ. Immunoregulation of asthma. Eur Respir J 1991;4(Suppl 13):3S.115. Vercelli D, Geha RS. Regulation of IgE synthesis in humans: A tale of two signals. J Allergy Clin
Immunol 1991;88:285.116. Wenzel SE, Westcott JY, Smith HR, Larsen GL. Spectrum of prostanoid release after
bronchoalveolar allergen challenge in atopic asthmatics and in control groups. Am Rev RespirDis 1989;139:450.
117. Kellaway C.H, Trethewie R. The liberation of a slow reacting smooth muscle stimulatingsubstance in anaphylaxis. Q J Exp Physiol 1940;30:121-45.
118. Taylor IK. Cysteinyl leukotrienes in asthma: Current state of therapeutic evaluation. Thorax1995;50:1005-10.
119. Samuelsson B. Leukotrienes: Mediators of immediate hypersensitive reactions and inflammation.Science 1983; 220:568-75.
120. Lewis RA, Austen KF. The biologically active leukotrienes. J Clin Invest 1984;73:889-97.121. Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of lipoxygenase pathway.
New Engl J Med 1990;323:645-55.122. Barnes PJ, Chung KF, Page CP. Inflammatory mediators and asthma. Pharmacol Rev 1988;40:
49-84.123. Piacentini GL, Kaliner MA. The potential roles of leukotrienes in bronchial asthma. Am Rev
Respir Dis 1991;143:S96-S99.124. Arm JP, Lee TH. Sulphidopeptide leukotrienes in asthma. Clin Sci 1993;84:501-10.125. Dahlen SE, Hansson G, Hedquist P et al. Allergen challenge of lung tissue from asthmatics
elicits bronchial contraction that co-related with the release of leukotrienes C4, D4, and E4. ProcNatl Acad Sci, USA 1983;80:1712-16.
126. MacGlashan DW, Schleimer RP, Peters SP et al. Generation of leukotrienes by purified humanlung mast cells. J Clin Invest 1982;70:747-51.
127. Weller PF, Lee CW, Foster DW et al. Generation and metabolism of 5-lipoxygenase pathwayleukotrienes by human eosinophils: Predominant production of leukotriene C4. Proc Natl AcadSci USA. 1983;80:7626-30.
128. Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of lipoxygenase pathway.New Engl J Med 1990;323:645-55.

78 Bronchial Asthma
129. Kern R, Smith LJ, Patterson R et al. Characterisation of the airway response to inhaled leukotrieneD4 in normal subjects. Am Rev Respir Dis 1986;133:1127-32.
130. Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4, LTD4,LTE4 and LTB4) in human skin. J Invest Dermatol 1983;80:115-19.
131. Coles SJ, Neill KH, Leid LM et al. Effects of leukotrienes C4 and D4 on glycoprotein and lysozymesecretion by human bronchial mucosa. Prostaglandins 1983;25:155-70.
132. O’Byrne PM, Leikauf GD, Aizwa H et al. Leukotriene B4 induces airway hyperresponsiveness indogs. J Appl Physiol 1985;59:1941-46.
133. Black PN, Fuller RW, Taylor GW et al. Bronchial reactivity is not increased after inhalation ofleukotriene B4 and prostaglandin D2. Br J Clin Pharmacol 1988;25:667P.
134. Bigby T. Inflammatory mediators and asthma. Pulm Perspectivs 1992;9(4):6-9.135. Holgate ST, Bradding P, Sampson AP. Leukotriene antagonists and synthesis inhibitors: New
directions in asthma therapy. J Allergy Clin Immunol 1996;98:1-13.136. Fischer AR, McFadden CA, Frantz R, Awni WM, Cohn J, Drazen JM, Israel E. Effect of chronic
5-lipoxygenase inhibition on airway hyperresponsiveness in asthmatic subjects. Am J RespirCrit Care Med 1995;152:1203-07.
137. Wenzel SE. New approaches anti-inflammatory therapy for asthma. Am J Med 1998;104:287-300.
138. Liu MC, Dube LM, Lancaster J et al. Acute and chronic effects of a 5-lipoxygenase inhibitor inasthma: A 6-month randomised multicentre trial. J Allergy Clin Immunol 1996;98:859-71.
139. Drazen JM, Israel F, O’Byrne PM. Treatment of asthma with drugs modifying the leukotrienepathway. N Engl J Med 1999;340:197-206.
140. Obase Y, Shimoda T, Tamari S et al. Effects of pranlukast on chemical mediators in inducedsputum on provocation tests in atopic and aspirin-intolerant asthmatic patients. Chest 2002;121:143-50.
141. Szczeklik A, Stevenson DD. Aspirin-induced asthma: Advances in pathogenesis and management.J Allergy Clin Immunol 1999;104:5-13.
142. Stevenson DD, Simon RA. Sensitivity to aspirin and nonsteroidal anti-inflammatory drugs: In:Middleton E, Reed CE, Ellis EF et al. (Eds). Allergy: Principles and Practice: 5th Edition. St Louis,MO: Mosby: 1998;1225-34.
143. Smith CM, Hawksworth RJ, Thien FC et al. Urinary leukotriene E4 in bronchial asthma. EurRespir J 1992;5:693-99.
144. Christie PE, Tagari P, Ford-Hutchinson AW et al. Urinary leukotriene E4 concentrations increaseafter aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis 1991;143:1025-29.
145. Yamamoto H, Nagata M, Kraits K et al. Inhibition of analgesic-induced asthma by leukotrienereceptor antagonist ONO-1078. Am J Respir Crit Care Med 1994;150:254-57.
146. Dahlen B, Nizankowska E, Szczeklik A et al. Benefits from adding the 5-lipoxygenase inhibitorzileuton to conventional therapy in aspirin-induced asthmatics. Am J Respir Crit Care Med1998;157:1187-94.
147. Caughey GH. Roles of mast cell tryptase and chymase in airway function. Am J Physiol1989;257:L39-46.
148. Nadel JA. Roles of mast cell proteases in airways. Drugs 1989;37:51-55.149. Drazen JM, Evans JF, Stevens RL, Shipp MA. Inflammatory effector mechanisms in asthma. Am
J Respir Crit Care Med 1995;152:403-07.150. White MV, Slater JE, Kaliner MA. Histamine and asthma. Am Rev Respir Dis 1987;135:1165-76.151. Simon RA, Stevenson DD, Arroyane CM, Tan EM. The relationship of plasma histamine to the
activity of bronchial asthma. J Allergy Clin Immunol 1977;60:312-16.152. Barnes PJ, Ind PW, Brown MJ. Plasma histamine and catecholamines in stable asthmatic subjects.
Clin Sci 1982;62:661-65.

Pathophysiology of Bronchial Asthma 79
153. Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The bronchoconstrictor effect of inhaledprostaglandin D2 in normal and asthmatic men. N Engl J Med 1984;311:209-13.
154. Walters EH, Parrish RW, Bevan C, Smoth AP. Induction of bronchial hyperresponsivity: Evidencefor a role of prostaglandins. Thorax 1981;36:571-74.
155. Fuller RW, Dixon CMS, Dollery CT, Barnes PJ. Prostaglandins D potentiates airway responsesto histamine and methacholine. Am Rev Respir Dis 1986;133:252-54.
156. Henderson WR (Jr). Eicosanoids and platelet activating factor in allergic respiratory disease. AmRev Respir Dis 1991;143:S86-S90.
157. Henderson WR (Jr). Lipid derived and other chemical mediators of inflammation in the lung.J Allergy Clin Immunol 1987;79:543-53.
158. Hozawa S, Haruta Y, Ishioka S, Yamakido M. Effects of a PAF antagonist, Y-24180, on bronchialhyperresponsiveness in patients with asthma. Am J Respir Crit Care Med 1995;152:1198-1202.
159. Evans TW, Chung K, Rogers DF, Barnes PJ. Effect of platelet-activating factor on airway vascularpermeability: possible mechanisms. J Appl Physiol 1987;63:479-84.
160. Wirtz H, Lang M, Sannwald U, Hahn H. Mechanism of platelet-activating factor (PAF)-inducedsecretion of mucus from tracheal submucosal glands in ferrets (abstract). Fed Proc 1986;45:486.
161. Wardlow AJ, Moqbel R, Cornwell O, Kay AB. Platelet-activating factor: A potent chemotacticand chemokinetic factor for human eosinophils. J Clin Invest 1986;78:1701-06.
162. Stenton Sc, Court EN, Kingston WP et al. Platelet-activating factor in bronchoalveolar lavagefluid from asthmatic subjects. Eur Respir J 1990;3:408-13.
163. Nakamura T, Morita Y, Kuriyama M, Ishitara K, Ito K, Miyamoto T. Platelet-activating factor inlate asthmatic responses. Int Arch Allergy Appl Immunol 1987;82:57-61.
164. Simonsson BG, Skoogh BE, Bergh NP, Anderson R, Sredmyr N. In vivo and in vitro effect ofbradykinin on bronchial motor tone in normal subjects and patients with airway obstruction.Respiration 1973;30:378-88.
165. Christiansen SC, Proud D, Sarnoff RB et al. Elevation of tissue kallikrein and kallidin in theairways of asthmatic subjects after endobronchial allergen challenge. Am Rev Respir Dis1992;145:900-05.
166. Polosa R, Holgate ST. Comparative airway response to inhaled bradykinin, kallidin, and (desArg) bradykinin in normal and asthmatic airways. Am Rev Respir Dis 1990;142:1367-71.
167. Regoli D, Barabe J. Pharmacology of bradykinin and related kinins. Pharmacol Rev 1980;32:1-46.
168. Chung KF, Barnes PJ. Cytokines in asthma. Thorax 1999;54:825-57.169. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell co-stimulation. Annu Rev
Immunol 1996;14:233-58.170. Geha RS. Regulation of IgE synthesis in humans. J Allergy Clin Immunol 1992; 90:143-50.171. Bradding P, Roberts JA, Britten KM et al. Interleukin 4, -5, and tumor necrosis factor-a in normal
and asthmatic airways: Evidence for the human mast cell as a source of these cytokines. Am JRespir Cell Mol Biol 1994;10:471-80.
172. Vercelli D, Jabara HH, Lee BW et al. Human recombinant interleukin-4 induces FCeRH/CD23on normal human monocytes. J Exp Med 1988;167:1406-16.
173. Ho LP, Davis M, Denison A et al. Reduced interleukin-18 levels in BAL specimens from patientswith asthma compared to patients with sarcoidosis and healthy control subjects. Chest2002;121:1421-26.
174. Spiteri M, Knight RA, Jeremi JY et al. Alveolar macrophage-induced suppression of T cellhyperresponsiveness in bronchial asthma is reversed by allergen exposure. Eur Respir J1994;7:1431-38.
175. Fischer HG, Frosch S, Reske K et al. Granulocyte-macrophage-colony-stimulating factor activatesmacrophages derived from bone marrow cultures to synthesis of MHC class II molecules and toaugmented antigen presentation function. J Immunol 1988;141:3882-88.

80 Bronchial Asthma
176. Chang TL, Shea CH, Urioste S et al. Heterogenicity of helper/inducer T lymphocytes: Lymphokineproduction and lymphokine responsiveness. J Immunol 1990;145:2803-08.
177. Sanderson CJ, Warren DJ, Strath M. Identification of a lymphokine that stimulates eosinophildifferentiation in vitro. Its relationship to interleukin 3 and functional properties of eosinophilsproduced in cultures. J Exp Med 1985;162:60-74.
178. Hallsworth MP, Litchfield TM, Lee TH. Glucocorticoids inhibit granulocyte-macrophage-colony-stimulating factor and interleukin 5 enhanced in vitro survival of human eosinophils. Immunology1992;75:382-85.
179. Collins PD, Griffith-Johnson DA, Jose PJ et al. Co-operation between interleukin 5 and thechemokine, eotaxin, to induce eosinophil accumulation in vivo. J Exp Med 1995;182:1169-74.
180. Rothenberg ME, Owen WFJ, Siberstein DS. Human eosinophils have prolonged survival, enhancedfunctional properties and become hypodense when exposed to human interleukin.J Clin Invest 1988;81:1986-92.
181. Moqbel R, Hamid Q, Ying S et al. Expression of mRNA and immunoreactivity of the granulocyte/macrophage colony-stimulating factor (GM-CSF) in activated human eosinophils. J Exp Med1991;174:749-52.
182. Tosi MF, Stark JM, Smith CW et al. Induction of ICAM-1 expression on human airway epithelialcells by inflammatory cytokines. Effects on neutrophil-epithelial cell adhesion. Am J Respir CellBiol 1992;7:214-21.
183. Brewster CEP, Howarth PH, Djukanovic R et al. Myofibroblasts and subepithelial fibrosis inbronchial asthma. Am J Respir Cell Mol Biol 1990;3:507-11.
184. Hirst SJ, Barnes PJ, Twort CHL. Quantifying proliferation of cultured human and rabbit airwaysmooth muscle cells in response to serum and platelet-derived growth factor. Am J Respir CellMol Biol 1992;7:574-81.
185. Massaro AF, Mehta S, Lilly CM, Kobazik L, Reilly JJ, Drazen JM. Elevated nitric oxide concentrationsin isolated lower airway gas of asthmatic subjects. Am J Respir Crit Care Med 1996;153:1510-14.
186. Alving K, Weitzberg E, Lundberg JM. Increased amount of nitric oxide in exhaled air of asthmatics.Eur Respir J 1993;6:1368-70.
187. Kharitonov SA, Yates D, Robbins RA et al. Increased nitric oxide in exhaled air of asthmaticpatients. Lancet 1994;343:133-35.
188. Hamid Q, Springall DR, Riveros-Moreno V et al. Induction of nitric oxide synthase in asthma.Lancet 1993;342:1510-13.
189. Ligget SB, Levi R, Metzger H. G-Protein coupled receptors, nitric oxide, and the IgE receptor inasthma. Am J Respir Crit Care Med 1995;152:394-402.
190. Carr MJ, Hunter DD, Undem BJ. Neurotrophins and asthma. Curr Opin Pulm Med 2001;7:1-7.191. Barnes PJ. The third nervous system in the lung: Physiology and clinical perspectives. Thorax
1984;39:561-67.192. Casale TB. Neuropeptides and the lung. J Allergy Clin Immunol 1991;88:1.193. Casale TB, Neuromechanisms of asthma. Ann Allergy 1987;59:391.194. Nadel JA. Neutral endopeptidase modulates neurogenic inflammation. Eur Respir J 1991;4:745.195. Drazen JM, Israel E. Asthma: A solution to half the puzzle. Am Rev Respir Dis 1991;144:743.196. Bleecker ER. Cholinergic and neurogenic mechanisms in obstructive airways disease. Am J Med
1986;81:93.197. Kowalski ML, Diddier A, Kaliner MA. Neurogenic inflammation in the airways. Am Rev Respir
Dis 1989;140:101.198. Barnes PJ. Neural control of human airways in health and disease. Am Rev Respir Dis
1986;134:1289-1314.199. Barnes PJ. Neural control of airway function: New perspectives. Mol Aspects Med 1990;11:351-
423.200. Nadel JA, Barnes PJ, Holtzmzn MJ. Autonomic factors in the hyperreactivity of airway smooth

Pathophysiology of Bronchial Asthma 81
muscle. In: Hand book of physiology: The respiratory system III. American Physiology Society;1986:693-702.
201. Minett PAH, Lammers J, Dixon CMS, McCuska MT, Barnes PJ. A muscarinic agonist inhibitsreflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol 1989;67:2461-65.
202. Palmer JB, Cuss FM, Barnes PJ. VIP and PHM and their role in nonadrenergic inhibitory responsesin isolated human airways. J Appl Physiol 1986;61:1322-28.
203. Belvisi MG, Stretton CD, Verlenden GM, Jacob MH, Barnes PJ. Inhibitory NANC nerves inhuman tracheal smooth muscle involvement of VIP and NO. Am Rev Respir Dis 1991;143:A355.
204. Lundberg JM, Saria A, Lundblad L et al. Bioactive peptide in capsaici sensitive C-fibre afferents ofairways, functional and pathophysiological implications, New York.; Marcel Decker; 1987:417-45.
205. Rogers DF, Belvisi MG, Arsudkij JB, Evans TW, Barnes PJ. Effects and interactions of sensoryneuropeptides on airway microvascular leakage in guineapigs. Br J Pharmacol 1988;95:1109-16.
206. Johnson AR, Aston J, Schulz WW, Erdos EG. Neutral metalloendopeptidases in human lungtissue and cultured cells. Am Rev Respir Dis 1985;132:564-68.
207. Rogers DF, Arsudkij JB, Barnes PJ. Effects of tachykinins on mucus secretion on human bronchiin vitro. Eur J Pharmacol 1989;174:283-86.
208. Mak JCM, Barnes PJ. Autoradiographic localisation of calcitonin gene-related peptide bindingsites in human and guineapig lung. Peptides 1988;9:957-64.
209. Barnes PJ. Neuropeptide and asthma. Am Rev Respir Dis 1991;143:S28-S33.210. Barnes PJ. Neurogenic inflammation and asthma. J Asthma 1992;29:165-80.211. Naline E, Devillier P, Drape G et al. Characterisation of neurokinin effects and receptor selectivity
in human isolated bronchi. Am Rev Respir Dis 1989;140:679-86.212. Barnes PJ, Dewar A, Rogers DF. Human bronchial secretion: effect of substance P, muscarinic
and adrenergic stimulation in vitro. Br J Pharmacol 1986;89:787P.213. Richardson PS, Webber SE. The control of mucus secretion in the airways by peptidergic
mechanisms. Am Rev Respir Dis 1987;136:S72-S76.214. Fuller RW, Maxwell DL, Dixon CMS et al. The effects of substance P on cardiovascular and
respiratory function in human subjects. J Appl Physiol 1987;62:1473-79.215. Sekizawa K, Tamaoki J, Graf PD et al. Enkephalinase inhibitor potentiates mammalian tachykinins-
induced contraction in ferret trachea. J Pharmacol Exp Ther 1987;243:1211-17.216. Bisgaard H, Groth S, Madsen F. Bronchial hyperreactivity to leukotriene D4 and histamine in
exogenous asthma. Br Med J 1985;290:1468.217. Smith LJ, Greenberger PA, Patterson R et al. The effect of inhaled leukotrienes in humans. Am
Rev Respir Dis 1985;131:368.218. Davidson AB, Lee TH, Scanlon PD et al. Bronchoconstrictor effects of leukotriene E4 in normal
and asthmatic subjects. Am rev Respir Dis 1987;135:333.219. Liggett SB, Raymond JR. Pharmacology and molecular biology of adrenergic receptors. In PM
Bouloux, Editor, Catecholamines. WB Saunders, London, 1993;279-306.220. Reishaus E, Innis M, MacIntyre N, Liggett SB. Mutations in the gene encoding for the beta-2
adrenergic receptor in normal and asthmatic subjects. Am J Respir Cell Mol Biol 1993;8:334-39.221. Liggett SB, Levi R, Metzger H. G-protein coupled receptors, nitric oxide, and the IgE receptor in
asthma. Am J Respir Crit Care Med 1995;152:394-402.222. Szentivanyi A. The β-adrenergic theory of the atopic abnormality in bronchial asthma. J Allergy
1968;42:203-32.223. McNeil RS. Effect of α–adrenergic blocking agent propranolol on asthmatics. Lancet 1964;2:
1101-02.224. Goldie RG, Spina D, Henry PJ et al. In vitro responsiveness of human asthmatic bronchus to
carbachol, histamine, badrenoceptor agonists, and theophylline. Br J Clin Pharmacol 1986;22:669-76.

82 Bronchial Asthma
225. Bai TR. Abnormalities in airway smooth muscle in fatal asthma. Am Rev Respir Dis 1990;141:552-57.
226. Bai TR. Abnormalities airway smooth muscle in fatal asthma: a comparison between trachea andbronchus. Am Rev Respir Dis 1991;143:441-43.
227. Cerrina J, Ladurie MLR, Labat C et al. Comparison of human bronchial muscle response tohistamine in vitro with histamine and isoproterinol agonists in vitro. Am Rev Respir Dis 1986;134:57-61.
228. Lentes KU, Berretinni WH, Hoche MR et al. A biallelic DNA polymorphism of the humanβ2-adrenergic receptor detected by Ban I-Adrbr-2. Nucleic Acids Res 1988;16:2359.
229. McQuitty CK, Emala CW, Hirshman CA et al. polymorphism in the human β2-adrenergic receptorgene detected by restriction endonuclear digestion with Fnu4HI. Hum Genet 1994;93:225.
230. Reihsaus E, Innis M, AacIntyre N et al. Mutations in the gene encoding for the β2-adrenergicreceptor in normal and asthmatic subjects. Am J Respir Cell Biol 1993;8:334-39.
231. Ohe M, Munakata M, Hizawa N et al. β2-adrenergic receptor gene restriction fragment lengthpolymorphism and bronchial asthma. Thorax 1995;50:353-59.
232. Taguchi H, Ohe M, Hizawa M. XV International Congress of Allergology and Clinical Immunology(abstract). ACI News 1994;89(Suppl 2):A317.
233. Emala CW, McQuitty CK, Eleff SM et al. Asthma, allergy, and airway hyperresponsiveness arenot linked to the b2-adrenoceptor gene. Chest 2002;121:722-31.
234. Gustafsson LE, Leone AM, Persson MG, Wilklund NP, Moncada S. Endogenous nitric oxide ispresent in the exhaled air of rabbits, guinea pigs, and humans. Biochem Biophysics Res Comm1991;181:852-57.
235. Springall DR, Hamid OA, Buttery LKD et al. Nitric oxide synthase induction in airways ofasthmatic subjects. Am Rev Respir Dis 1993;147:A515.
236. Gaston B, Drazen J, Chee CBE, Wohl MEB, Stamler JS. Expired nitric oxide (NO) concentrationsare elevated in patients with reactive airway disease. Endothelium 1993;1:87-92.
237. Pauwels R. Asthma: Managing the underlying disease. Eur Respir Rev 1994;4:291-94.238. Holt PG. Regulation of antigen-presenting cell function(s) in lung and airway tissue. Eur Respir
J 1993;6:120-29.239. Martinez FD, Wright AL, Taussig LM et al. Asthma and wheezing in the first six years of life.
N Engl J Med 1995;332:133-38.240. Marguet C, jouen-Boedes F, Dean TP et al. Bronchoalveolar cell profiles in children with asthma,
infantile wheezing, chronic cough, or cystic fibrosis. Am J Respir Crit Care Med 1999;159:1533-40.
241. Schellhase DE, Pawcett DD, Schutze GE et al. Clinical utility of flexible bronchoscopy andbronchoalveolar lavage in young children with recurrent wheezing. J Pediatr 1998;132:312-18.
242. Krawiee ME, Westcott JY, Wei Chu H et al. Persistent wheezing in very young children isassociated with lowered respiratory inflammation. Am J Respir Crit Care Med 2001;163:1338-43.
243. Cloutier MM. Neutrophils or eosinophils in young children with wheezing. Chest 2002;122:761-63.
244. Le Bourgeots M, Goncalves M, Le Clincher L et al. Bronchoalveolar cells in children , 5 years oldwith severe recurrent wheezing. Chest 2002;122:791-97.
245. Samter M, Biers RF. Intolerance to aspirin: clinical studies and consideration of its pathogenesis.Ann Intern Med 1968;6:975-83.
246. Christie PE, Tagari P, Ford-Hutchinson AW, et al. Urinary leukotriene E4 concentrations increaseafter aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis 1991;143:1025-29.
247. Knapp HR, Sladek K, Fitzgerald GA. Increased excretion of leukotriene E4 during aspirin-induced asthma. J lab Clin Med 1992;119:48-51.

Pathophysiology of Bronchial Asthma 83
248. Ferreri NR, Howland WC, Stevenson AD, Spiegelberg ML. Release of leukotrienes, prosta-glandins and histamine into nasal secretions of aspirin-sensitive asthmatics during reaction toaspirin. Am Rev Respir Dis 1988;137:847-54.
249. Ortolani C, Mirone C, Fontana A, Folco GC et al. Study of mediators of anaphylaxis in nasalwash fluids after aspirin and sodium meta-bisulphite nasal provocation in intolerant rhiniticpatients. Ann Allergy 1987;59:106-12.
250. Sladek K, Dworksi R, Soja J, et al. Eicosanids in bronchoalveolar lavage fluid of aspirin-tolerantpatients with asthma after aspirin challenge. Am J Respir Crit Care Med 1994;149:940-46.
251. Nasser SMS, Pfister R, Christie PE, Sousa AR, et al. Inflammatory cell populations in bronchialbiopsies from aspirin sensitive asthmatic subjects. Am J Respir Crit Care Med 1996;153:90-96.
252. Hogg JC. Role of latent viral infections in chronic obstructive pulmonary disease and asthma.Am J Respir Crit Care Med 2001;164:S71-S75.
253. Schwarze J, Gelfand EW. Respiratory viral infections as promoters of allergic sensitisation andasthma in animal models. Eur Respir J 2002;19:341-49.
254. ven HL. Role of persistent infection in the control and severity of asthma: focus on Chlamydiapneumoniae. Eur Respir J 2002;19:546-56.
255. Gypear D, Busse WW. Role of virus infection in asthma. Immunol Allergy Clin North Am1993;13:745-68.
256. Lemanske RF, Jr, Dick EC, Swenson CA, Vrtis TF, Busse WW. Rhinovirus upper respiratoryinfection increases airway hyperreactivity and late asthmatic reactions. J Clin Invest 1989;83:1-10.
257. Calhoune WJ, Swenson CA, Dick EA, et al. Experimental rhinovirus-16 infection potentiateshistamine release after antigen bronchoprovocation in allergic subjects. Am Rev Respir Dis1991;144:1267-73.
258. Huftel MA, Swenson CA, Borcherding WR et al. The effect of T cell depletion on enhancedbasophil histamine release after in vitro incubation with live influenza A virus. Am J Respir CellMol Biol 1992;7:434-40.
259. Godfrey S. Exercise-induced asthma. In: Bierman CW, Perlmanlman DS, Ed: Allergic Diseasefrom infancy to adulthood. Philadelphia; WB Saunders Co.; 1988;597-606.
260. Belcher NG, O’Hickey S, Arm JP et al. Pathogenic mechanisms of exercise-induced asthma andthe refractory period. NER Allergy Proc 1988;9:199-201.
261. Lee TH, Nagacura T, Papageoriou N et al. Exercise-induced late asthmatic reaction withneutrophil chemotactic activity. N Engl J Med 1983;308:1502-05.
262. Gilbert IA, Fouke JM, McFadden ER Jr. Heat and water flux in the intrathoracic airways andexercise-induced asthma. J Appl Physiol 1987;631:681-91.
263. Godfrey S. Bronchial challenge by exercise or hypertension. In: spector Sl, Ed: provocativechallenge procedures; background and methodology. New York, Futura Publishing Co; 1989;365-94.
264. McFadden ER. Hypothesis: exercise-induced asthma as a vascular phenomenon. Lancet1990;335:880-82.
265. McFadden ER Jr, Pichurko B, Bowman HF et al. Thermal mapping of the airways in humans.J Appl Physiol 1985;58:564-70.
266. Anderson SD. Is there a unifying hypothesis for exercise induced asthma? J Allergy Clint Immune1984;73:660-65.
267. Anderson SD. Issues in exercise-induced asthma. J Allergy Clint Immune 1985;76:763-72.268. Anderson SD, Silverman M, Godfrey S et al. Exercise-induced asthma – a review. Br J Dis Chest
1975;69:1-39.269. Chen WY, Horton DJ,. Heat and water loss from the airways and exercise-induced asthma.
Respiration 1977;34:305-13.

84 Bronchial Asthma
270. Strauss RH, McFadden ER Jr., Ingram RH Jr et al. Influence of heat and humidity on the airwayobstruction induced by exercise in asthma. J Clin Invest 1978;61:433-40.
271. Strauss RH, McFadden ER Jr., Ingram RH Jr et al. Enhancement of exercise-induced asthma bycold air. New Engl J Med 1977;297:743-47.
272. Deal EC Jr, McFadden ER Jr., Ingram RH Jr et al. Hyperapnea and heat flux: initial reactionsequence in exercise-induced asthma. J Appl Physiol 1979;46:476-83.
273. Anderson SD, Schoefield RE, Follet R et al. Sensitivity to heat and water loss at rest and duringexercise in asthmatic patients. Eur J Respir Dis 1982;63:459-71.
274. Huang SW, Veiga R, Sila U et al. The effects of swimming on asthmatic children, participants ina swimming program in the city of Baltimore. J Asthma 1989;26:117-21.
275. Bar-Or O, Inbar O. Swimming and asthma: Benefits and deleterious effects. Sports Med 1992;14:397-405.
276. Spector SL. Update on exercise-induced asthma. Ann Allergy 1993;71:571-77.277. McFadden ER, Gilbert IA. Exercise-induced asthma. N Engl J Med 1994;330:1362-67.278. Makkar HK, Lau LC, Thomson HW, Binks SM, Holgate ST. The protective effect of inhaled
leukotriene D4 receptor antagonist ICi 204,219 against exercise-induced asthma. Am Rev RespirDis 1993;147:1413-18.
279. Reiss TF, Bronsky E, Hendeles L et al. MK-0476, a potent leukotriene (LT)D4 antagonist, inhibitsexercise-induced bronchoconstriction in asthmatics at the end of a once daily dosing schedule(abstract). Am J Respir Crit Care Med 1995;151:A377.
280. Glass M, Snadder LA, Israel E. Effect of the inhaled LTD4 receptor antagonist, ICI-204,219, oncold-air-induced bronchoconstriction in patients with asthma.(abstract). J Allergy Clin Immunol1994;93:295A.
281. Scharf SM, Heimer D, Walters M. Bronchial challenge with room temperature isocapnichyperventilation: a comparison with histamine challenge. Chest 1985;88:586-93.
282. Rosenthal RR. Simplified eucapnic voluntary hyperventilation challenge. J Allergy Clin Immunol1984;73:676-79.
283. Argyros GJ, Phillips YY, Rayburn DB et al. Water loss without heat flux in exercise-inducedbronchospasm. Am Rev Respir Dis 1993;147:1419-24.
284. Eliasson AH, Phillips YY, Rajagopal KR et al. Sensitivity and specificity of bronchial provocationtesting: an evaluation of 4 techniques in exercise-induced bronchospasm. Chest 1992;102:347-55.
285. Anderson SD, Deviskas E, Smith CM. Exercise-induced asthma: a difference in opinion regardingthe stimulus. Allergy Proc 1989;10:215-16.
286. Eggleston PA, Kagey-Sobotka A, Lichtenstein LM. A comparison of the osmotic activation ofbasophils and human lung mast cells. Am Rev Respir Dis 1987;135:1043-48.
287. Silber G, Proud D, Warner J et al. In vivo release of inflammatory mediators by hyperosmolarsolutions. Am Rev Respir Dis 1988;137:606-12.
288. Moloney E, O’Sullivan S, Hogan T et al. Airway dehydration: A therapeutic target in asthma?Chest 2002;121:1806-11.
289. Barnes PJ, Belvisi MG. Nitric oxide and lung disease. Thorax 1993;48:1034-43.290. Kanazawa H, Hirata K, Yoshikawa J. Role of endogenous nitric oxide in exercise-induced airway
narrowing in patients with bronchial asthma. J Allergy Clin Immunol 2000;106:1081-87.291. Kanazawa H, Asai K, Hirata K, Yoshikawa J. Vascular involvement in exercise-induced airway
narrowing in patients with bronchial asthma. Chest 2002;122:166-70.292. Lam S, Wong R, Chan-Yeung M. Nonspecific bronchial reactivity in occupational asthma. J
Allergy Clin Immunol 1979;63:28-34.293. Peppys J, Hutcheroft BJ. Bronchial provocation tests in etiologic diagnosis and analysis of asthma.
Am Rev Respir Dis 1975;112:829-59.294. Perrin B, Cartier A, Ghezzo H et al. Reassessment of the temporal patterns of bronchial obstruction
after exposure to occupational sensitizing agents. J Allergy Clin Immunol 1991;87:630-39.

Pathophysiology of Bronchial Asthma 85
295. Chan-Yeung M, Malo JL. Occupational asthma. New Engl J Med 1995;333-107-12.296. Mapp CE, Boschetto P, Dal Vecchio L, Maestrelli P, Fabbri LM. Occupational asthma due to
isocyanate. Eur Respir J 1988;1:273-79.297. Frew AJ, Chan H, Dryden P, Salari H, Lam S, Chan-Yeung M. Immunologic studies of the
mechanisms of occupational asthma caused by western red cedar. J Allergy Clin Immunol1993;92:466-78.
298. Kay AB, Corrigan CJ, Frew AJ. The role of cellular immunology in asthma. Eur Respir J1991;13:105s-112s.
299. Kusaka Y, Nakano Y, Shirakawa Y, Morrimoto K. Lymphocyte transformation with cobalt inhard metal asthma. Ind Health 1989;27:155-63.
300. Saetta M, Di Stefano A, Maestrelli P et al. Airway mucosal inflammation in occupational asthmainduced by toluene diisocyanate. Am Rev Respir Dis 1992;145:160-68.
301. Lam S, LeRiche J, Phillips D, Chan-Yeung M. Cellular and protein changes in bronchial lavagefluid after late asthmatic reaction in patients with red cedar asthma. J allergy Clin Immunol1987;80:44-50.
302. Bentley AM, Maestrelli P, Saetta M et al. Activated T lymphocytes and eosinophils in the bronchialmucosa in isocyanate-induced asthma. J Allergy Clin Immunol 1992;89:821-29.
303. Karol MH. Animal models of occupational asthma. Eur Respir J 1994;7:555-68.304. Soto-Aguilar MC, Salvaggio JE. Immunologic aspects of occupational asthma. Seminars Respir
Med 1991;12:185-95.305. Taylor AJN. Respiratory irritants encountered at work. Thorax 1996;51:541-45.306. Brooks SM, Weiss MA, Bernstein IL. Reactive airways dysfunction syndrome (RADS). Persistent
asthma syndrome after high level irritant exposure. Chest 1985;88:376-84.307. Gautrin D, Boulat LP, Boulat M et al. Is reactive airway dysfunction syndrome a variant of
occupational asthma? J Allergy Clin Immunol 1994;93:12-22.308. Staudinger HW, Steinijsnd VW. Theophylline steady-state pharmacokinetics: Recent concepts
and their application in the chrono-therapy of bronchial asthma. In: Lemmer B, Huller H, Ed;Clinical chronopharmacology, Clin Pharmacol 1996;6:136-47.
309. Gupta ML, behera D: Pattern of airflow obstruction in Bronchial Asthma—An observation onHome-Monitoring of Peak Expiratory Flow Rate. J Ass Phy India 1997;45:94-96.
310. Turner-Warwick M. Epidemiology of nocturnal asthma. Am J Med 1988;85:6-8.311. Shah A. Bronchial asthma and sleep disturbances. Ind J Chest Dis All Sc 1997;39:77-79.312. Meijer GG, Oosterhoff Y, Weersink EJM, Postma DS, Gerritsen J, van Aalderen WMC. Nocturnal
dyspnoea: Prevalence in asthmatic children. Eur Respir J 1991;4:523S.313. Martin RJ, Cicutoo LC., Ballard RD. Factors related to the nocturnal worsening of asthma. Am
Rev Respir Dis 1990;141:33-38.314. Hetzel MR, Clark TJH, Branthwaite MA. Asthma: Analysis of sudden deaths and ventilatory
arrests in hospital. Br Med J 1977;1:808-11.315. Robertson CF, Rubinfeld AR, Bowes G. Deaths from asthma in Victoria; A 12-months survey.
Med J Austr 1990;152:511-17.316. Mohiuddin AA, Martin RJ. Circadian basis of the late asthmatic response. Am Rev Respir Dis
1990;142:1153-57.317. Barnes P, Fitzgerald G, Brown M, Dollery C. Nocturnal asthma and changes in circulatory
epinephrine, histamine and cortisol. N Engl J Med 1980;303:263-67.318. Martin RJ, Cicutto LC, Smith HR et al. Airways inflammation in nocturnal asthma. Am Rev
Respir Dis 1991;143:351-57.319. Chen WY, Chai H. Airway cooling and nocturnal asthma. Chest 1982;81;675-800.320. Bush RK. Nocturnal asthma: Mechanisms and the role of theophylline in treatment. Postgrad
Med J 1991;67(Suppl 4):S20.321. Asthma: A nocturnal disease. Proceedings of a symposium. Am J Med 1988;85 (Suppl 1B):2.

86 Bronchial Asthma
Pathology
4
The earlier descriptions of histological changes in bronchial asthma relied on postmortemspecimens taken from people dying in status asthmaticus. Since the 1960s, epithelialshedding and influx of eosinophils into the airway mucosa have been associated withbronchial asthma.1,2 Large segments of the airway from the major bronchi to the peripheryare occluded with a mixture of tenacious secretion containing serum protein mixed withmucus and cellular debris. Crystalline material consisting largely of major basic proteinderived from eosinophil granules (Charcot-Leyden crystals) may be present. There isoedema, dense eosinophilic infiltration, and epithelial denudation in the bronchial wall.Airway samples obtained at open lung biopsy show goblet cell hyperplasia, peribronchialsmooth muscle hypertrophy and apparent basement membrane thickening. Further evidenceof epithelial shedding in asthmatics is provided by the findings of clumps of epithelial cellsin the sputum of such patients during acute attacks. The strips of epithelial cells are calledCurschmann’s spirals. Clumps of cells (Creola bodies) or isolate metaplastic cells arecommon. However, no detailed pathological changes were available in milder forms ofasthma before the use of fibreoptic bronchoscope (Fig 4.1a-c)
Fibreoptic bronchoscopy has helped in sampling the bronchial mucosa as well as thesubmucosa from the subcarinal levels in asthmatics at various stages of their disease. In1985, fresh biopsies were taken from eight asthmatics, with two of them having mild asthma,three having moderate and three with severe asthma.3 All of them showed virtual destructionand shedding of epithelium at the three airway levels studied. This was in contrast toperfectly intact epithelium found in a control subject. The most important observation wasthat epithelial changes and influx of inflammatory cells also existed in the two untreatedpatients who had mild disease both clinically and functionally. The existence of severeinflammatory changes is well known from necropsy studies on patients died of bronchialasthma. However, significant inflammation is also present in early asthma in patients withonly a short duration of symptoms, or with mild disease.4-6 These bronchial mucosal biopsyfindings resulted in surprising results. Biopsies taken from mild asthmatics requiring onlyoccasional bronchodilators, showed them to be always abnormal compared to that fromnonatopic normal individuals. Such changes included the presence of mast cells at variousstages of degranulation, and a wide spread infiltration of eosinophils. Most of the eosinophilsrevealed the ultrastructural features of activation and degranulation. Eosinophils,neutrophils, and mononuclear cells were present in increased numbers in the postcapillaryvenules, and were frequently in close contact with the vascular endothelium.
Another important observation was the presence of apparent thickening of the sub-epithelial basement membrane.7-9 Although the basement membrane is of normal thickness,
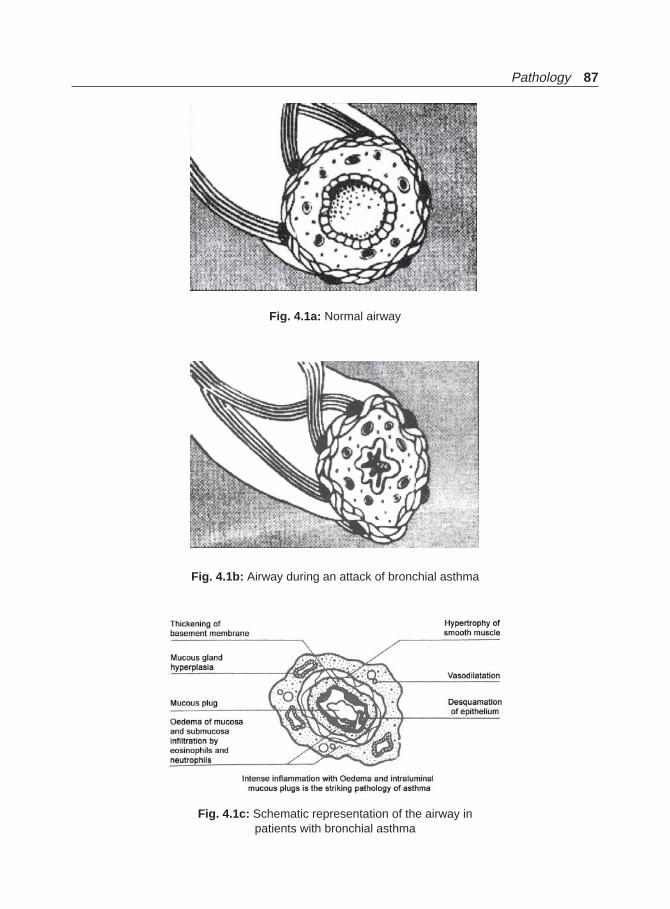
Pathology 87
Fig. 4.1a: Normal airway
Fig. 4.1b: Airway during an attack of bronchial asthma
Fig. 4.1c: Schematic representation of the airway inpatients with bronchial asthma

88 Bronchial Asthma
the subepithelial band consists of dense cross-linked collagen fibrils. Monoclonal antibodystudies suggest that the sub-basement membrane band consists of types III and V collagen,together with fibronectin but not laminin. This suggests the fibroblastic origin of the band.Recent data further revealed an expanded network of subepithelial myofibroblasts with bothcontractile and collagen secreting properties. The number of myofibroblasts correlates withthe degree of subepithelial thickening, suggesting a repair response secondary to chronicinflammation. Extensive collagen deposition within the bronchial mucosa might influencethe mechanical properties of the airways and contribute towards bronchialhyperresponsiveness and irreversible airflow obstruction. The thickness of the reticularbasement is increased even in mild asthma and is correlated with airway obstruction andhyperresponsiveness. It is therefore, suggested that anti-inflammatory treatment with inhaledsteroids should be started in the early stage of bronchial asthma to prevent structural changesfrom occurring in the airway wall.10
Similar changes have been described in the asthmatic airway in childhood. Bronchialbiopsy specimens from children show thickening and hyalinization of the basement membrane.The ciliated epithelial cells showed loss of cilia in some cases. Overactive fibroblasts areconstant findings. There is submucosal infiltration with degranulating mast cells andlymphocytes. Eosinophils are present in some cases.11
AIRWAY REMODELLING
Chronic inflammation in the airways leads to structural changes, including hypertrophy andhyperplasia of airway smooth muscle and thickening of the reticular layer of basementmembrane. This later thickening is due to the deposition of collagen from activatedmyofibroblasts in response to cytokines and growth factors released during the inflammatoryresponse.12 There is extensive deposition of collagen beneath the true basement membrane.Using immunostaining this collagen is identified as predominantly types III and V, but nottype IV (basement membrane)13 collagen. Thus, the increased subepithelial collagen in asthmadoes not represent a thickening of the true basement membrane but rather collagen laid downby fibroblasts with the lamina propria. Although the factor(s) controlling the proliferationand collagen-secreting activities of the myofibroblasts is not known, these structural changesmay underlie the progressive and irreversible airflow obstruction that is seen in patients withpoorly controlled asthma over a period of time.
The remodelling of airways in bronchial asthma involves structural changes in theepithelium, the myofibroblasts, and extracellular matrix including the basement membrane,and smooth muscle. This remodelling process is mainly caused by a complex interaction ofinflammatory cells that are central to the pathogenesis of asthma with structural tissue cells.The inflammatory cells such as eosinophils, T cells, mast cells and macrophages togetherwith structural tissue cells, play important effector role through the release of a number ofcytokines, mediators, and chemokines.
Remodelling of the airways in asthma involves structural changes in the epithelium, themyofibroblasts and extracellular matrix including basement membrane, and smooth muscle.This remodelling process is mainly orchestrated by a complex interaction of inflammatorycells that are central in the pathogenesis of asthma with structural tissue cells. Epithelialinjury plays an important role in asthma airway remodelling.14 An intrinsically aberrantepithelium when injured by toxic mediators, cause the epithelium to be in a chronic state of

Pathology 89
increased injury and repair. Pro-fibrotic stimuli cause subsequent subepithelial basementmembrane and submucosal alterations of collagen, elastin, and smooth muscle fibres. Thisinteraction is called the epithelial mesenchymal trophic unit. Patients of asthma have increasedgoblet cell hyperplasia, and increased stored mucin in the airway epithelium.
Airway inflammation and remodelling contribute significantly to the decline in lungfunction in bronchial asthma. Generally, lung function increases during childhood, levelsoff around 25-35 years of age, and declines after the age of 35 years. However, in asthmaticchildren the observation is different. A girl developing asthma at age of 7 years would have5% reduction in FEV1 by age of 10 years and a 7% deficit by age of 15 years compared withchildren without asthma.15,16 Similar observations are made for adult asthmatics that mayalso have increased decline in lung function during their life.17 This enhanced decline inlung function is present in both sexes and is further enhanced by smoking. On this logic a175 cm tall, nonsmoker, nonasthmatic man had an average FEV1 of 3.05 L, compared withthe FEV1 of 1.99 L for a man of the same age and height who smoked and had asthma.Further, there may be a subset of nonsmoking asthmatics those have an excess overall declinein lung function. This may lead to severe, potentially life-threatening, irreversible airwayobstruction without the presence of emphysema.18
Further, ongoing inflammation results in more severe airway hyperreactivity, and lowerlung function as well as accelerated loss of FEV1.
Airway Pathology during Asthma Remission
Spirometric abnormalities and bronchial hyperresponsiveness to methacholine or cold airchallenge during clinical remission of asthma are often observed.19,20 It is unclear whetherthese functional abnormalities reflect persistent activity of the airways inflammatory processor merely indicates structural changes of the airways as a consequence of childhood asthma.These structural changes (airway remodelling) are probably early events in the course ofthe disease that appear to progress. The process of remodelling leads to thickening of theairway wall.21-24 The exact physiologic consequences of airway wall thickening are notcompletely understood.25 If airway wall thickening is present in subjects in clinical remissionof asthma, it could at least partly account for the functional abnormalities including bronchialhyperreactivity observed during remission. On the other hand, ongoing active airwayinflammation will have substantial impact on the risk of relapse later in life. Therefore,subjects with subclinical airway inflammation could benefit from anti-inflammatorytreatment.26-28 Elevated exhaled nitric oxide (eNO) levels and bronchial hyperreactivity duringclinical remission have been demonstrated recently implying ongoing inflammation.29 Recentstudies have shown that eosinophils, T cells, mast cells, and IL-5 are significantly elevated inthe airway mucosa of subjects with bronchial asthma in remission compared with controlsubjects.13 Also blood eosinophil cell counts were higher in subjects with clinical remission.Blood eosinophil cell counts, exhaled nitric oxide (eNO) levels, and bronchial response toadenosine-5’-monophosphate correlated significantly with the quantity of tissue eosinophils.Significant airway remodelling was found in subjects in clinical remission. Matrixmetalloproteinase-9 concentrations are increased in severe, persistent asthma and followingairway challenge.30 These results indicate ongoing airway inflammation and airwayremodelling in adolescents in clinical remission of atopic asthma. Subclinical airwayinflammation may well determine the risk of an asthma relapse later in life.

90 Bronchial Asthma
REFERENCES
1. Glynn AA, Michaels L. Bronchial biopsy in chronic bronchitis and asthma. Thorax 1960;15:142-53.
2. Dunhill MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of thebronchi in normal subjects, status asthmaticus, in chronic bronchitis and emphysema. Thorax1969;24:176-79.
3. Laitinen LA, Heino M, Laitinen A, Kava T, Haahtela T. Damage of the airway epithelium andbronchial reactivity in patients with asthma. Am Rev Respir Dis 1985;131:599-606.
4. Laitinen LA, Laitinen A, Heino M, Haahtela T. Eosinophilic airway inflammation duringexacerbation of asthma and its treatment with inhaled corticosteroid. Am Rev Respir Dis1992;42:423-27.
5. Laitinen LA, Laitinen A, Haahtela T. Airway mucosal inflammation even in patients with newlydiagnosed asthma. Am Rev Respir Dis 1993;147:697-704.
6. Jeffrey PK, Wardlaw AJ, Nelson FC, Collins JV, Kay AB. Bronchial biopsies in asthma. An ultra-structural quantitative study and correlation with hyperreactivity. Am Rev Respir Dis1989;140:1745-53.
7. Brewster CE, Howarth PH, Djukanovic R et al. Myofibroblast and subepithelial fibrosis inbronchial asthma. Am J Respir Cell Mol Biol 1990;3:507.
8. Reid LM. Workshop on pathology; summary of workshop manuscripts and discussion withrecommendation from the panel. J Allergy Clin Immunol 1987;80(Suppl):403.
9. Cutz E, Levison H, Cooper DM. Ultrastructure of airways in children with asthma. Histo-pathology 1978:2:407.
10. Shiba K, Kasahara K, Nakajima H, Adachi M. Structural changes of the airway wall impairrespiratory function, even in mild asthma. Chest 2002;122:1622-26.
11. Cokugras H, Akcakaya N, Seckin I, Camcroglu Y, Sarmurat M, Aksoy F. Ultrastructuralexamination of bronchial biopsy specimens from children with moderate asthma. Thorax2001;56:25-29.
12. Djuknovie R, Roche WR, Wilson JW et al. Mucosal inflammation in asthma. Am Rev Respir Dis1990;142:437-52.
13. Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics.Lancet 1989;i:520-23.
14. Holgate ST, Davies DE, Lackie PM et al. Epithelial mesenchymal interactions in the pathogenesisof bronchial asthma. J Allergy Clin Immunol 2000;105: 193-204.
15. Weiss ST, Tosteson TD, Segal MR et al. Effect of asthma on pulmonary function in children. Alongitudinal population based study. Am Rev Respir Dis 1‘992;145:58-64.
16. Roorda RJ, Germitsen J, van Aalderen WM et al. Follow up of asthma from childhood toadulthood: Influence of potential childhood risk factors on the outcome of pulmonary functionand bronchial responsiveness in adulthood. J Allergy Clin Immunol 1994;93:575-84.
17. Lange P, Parmer J, Vestbo J et al. A 15-year follow up study of ventilatory function in adultswith asthma. New Engl J Med 1998;339:1194-1200.
18. Nick HT, Hacken T, Postma Ds, Timens W. Airway remodelling and long-term decline in lungfunction in asthma. Curr Opin Pulm Med 2003;9;9-14.
19. Kerrebijn KF, Fioole AC, van Bentveld RD. Lung function in asthmatic children after year ormore without symptom or treatment. Br Med J 1978;I:886-88.
20. Gruber W, Eber E, Steinbrugger B, Modl M, Weinhandle E, Zach MS. Atopy, lung function,and bronchial responsiveness in symptom-free pediatric asthma patients. Eur Respir J1997;10:1041-45.
21. Sears MR. Consequences of long-term inflammation. The natural history of asthma. Clin ChestMed 2000;21:315-29.

Pathology 91
22. Gillis HL, Lutchen KR,. Airway remodelling in asthma amplifies heterogeneities in smoothmuscle shortening causing hyperresponsiveness. J Appl Physiol 1999;86:2001-12.
23. Djukanovic R. Asthma: a disease of inflammation and repair. J Allergy Clin Immunology2000;105:522-26.
24. Vignola AM, Chanez P, Bonsignore G, Godard P, Bousquet J. Structural consequences of airwayinflammation in asthma. J Allergy Clin Immunol 2000;105:514-17.
25. Fahy JV, Corry DB, Boushey HA. Airway inflammation and remodelling in asthma. Curr OpinPulm Med 2000;6:15-20.
26. Hodshino M, Takahashi M, Takai Y, Sim J. Inhaled corticosteroids decrease subepithelial collagendeposition by modulation of the balance between matrix metalloproteinase-9 and tissue inhibitorof metalloproteinase-1 expression in asthma. J Allergy ClinImmunol 1999;104:356-63.
27. Laitinen A, Altraza A, Kampe M, linden M, Virtanen I, Laitinen LA. Tenascin in airway basementmembrane of asthmatics and decreased by an inhaled steroid. Am J Respir Crit Care Med1997;156:951-58.
28. Van den Toorn LM, Prins JB, Overbeek SE, Hoogsteden HC, de Jongste JC. Adolescents in clinicalremission of atopic asthma have elevated exhaled nitric oxide levels and bronchial hyper-responsiveness. Am J Respir Crit Care Med 2000;162;953-57.
29. Van den Toorn LM, Overbeek SE, de Jongste JC, Leman K, Hoogsteden HC, Prins JB. Airwayinflammation is present during clinical remission of atopic asthma. Am J Respir Crit Care Med2001;164:2107-13.
30. Mattos W, Lam S, Russell R et al. Matrix metalloproteinase-9 expression in asthma. Effect ofasthma severity, allergen challenge, and inhaled corticosteroids. Chest 2002;122:1543-52.

92 Bronchial Asthma
Clinical Presentation ofBronchial Asthma
5
The clinical presentations of bronchial asthma are heterogeneous, falling into every agegroup from infancy to old age, and the spectrum of signs and symptoms varies in degree ofseverity from patient to patient, as well as within each patient, over time. Detailed clinicalhistory taking is very important in the clinical diagnosis of bronchial asthma. The usualsymptoms include cough, wheezing, shortness of breath, chest tightness, and modest degreeof sputum production. The sputum is usually white or clear, and the patient may sometimesnotice more solid or greenish streaks in it. Dry cough may be the only manifestation ofasthma in some (cough variant asthma). It is estimated that about 10% of the population,i.e. double the number having overt asthma symptoms experience asthma-like symptoms.1
Conditions known to be associated with bronchial asthma include rhinitis, sinusitis, nasalpolyposis, or atopic dermatitis. Between 60-78% of patients, who have asthma havecoexisting allergic rhinitis.2 Further, allergic rhinitis has been recognised as a risk factor forasthma and between 20-38% of patients who have allergic rhinitis have coexisting asthma.Most patients will complain of the onset of an attack of bronchial asthma following allergicpharyngitis, in the form of sore throat, pain in the throat, itching, sneezing, running nose ora blocked nose. Viral infection of the upper airways is another important preceding eventin many patients.3 The pattern of symptoms may be perennial, seasonal, or perennial withseasonal exacerbations. The symptomatology is generally episodic, although may becontinuous or continuous with acute exacerbations. There is usually a circadian variationwith more nocturnal symptoms.4 These nocturnal attacks wake the patient in early hours ofmorning and the patient feel the need to get out of bed and want to open the window forair. Exacerbation of symptoms, may occur after several minutes of usually unaccustomedexertion, increase in severity over a minute or two and wane over about half an hour. Theprecipitating event (discussed above under etiology) may or may not be evident from history.
The incidence of IgE mediated allergy (allergic rhinitis, atopic dermatitis, hay fever)and bronchial asthma in close relatives is very high. The detailed medical history of thepatient including other allergic disorders and in children history of early life injury to airways(bronchopulmonary dysplasia, history of pulmonary infiltrates, documented pneumonia,viral bronchiolitis, recurrent croup, symptoms of gastro-oesophageal reflux and passiveexposure to smoking) may be rewarding.5 Many other risk factors discussed above likedomestic dust mite, pollens, mould, furred animals, airborne irritants, tobacco smoke, arealso capable of aggravating asthma and are known as asthma triggers as they can provoke

Clinical Presentation of Bronchial Asthma 93
asthma attacks. Other triggers include smoke from domestic cooking fuels, physical activity(running and other exercises), extreme emotional expressions (laughing or crying hard),cold air or weather changes, food additives, cold drinks, and drugs like aspirin. People withasthma may have one or more triggers, and different individuals have different triggers.
Physical examination of chronic asthma (for acute attacks see later) should focus on theupper respiratory tract, the skin and the chest. The findings may reveal the presence ofrhinitis and/or sinusitis in the form of purulent nasal discharge, pale nasal mucosa, postnasaldrip, and nasal polyps. Flexural eczema may indicate the presence of atopic dermatitis. Inchildren, there will be evidence of hyperinflation of the lungs with use of accessory musclesand appearance of hunched shoulders and “pigeon chest”. The intensity of the breath soundsin symptomatic asthma will be reduced and the expiratory phase is prolonged. Presence ofrhonchi is a characteristic finding in asthma and will be present in most patients. However,neither its presence nor its absence will confirm or exclude bronchial asthma. Rhonchi maybe heard in many other conditions including chronic bronchitis, pulmonary oedema,bronchial stenosis, foreign body aspiration, upper airway obstruction, aspiration pneumoniaand pulmonary embolism, etc. It is often said that “all that wheezes is not asthma”.Moreover, wheezing is not a reliable sign of severity. Crepitations are not the findings ofasthma unless there is secondary infection or a complication like allergic bronchopulmonarymycosis.
CLASSIFICATION
Intrinsic and Extrinsic Asthma
Some investigators try to classify bronchial asthma into the intrinsic and extrinsic types.The intrinsic asthma usually has late onset with no history of atopy or allergy and is non-seasonal. The skin test for allergens is usually negative and the serum IgE level is oftennormal. The symptoms are generally severe, they do not respond well to conventionaltherapy and a greater likelihood that the patient will need maintenance oral steroids, towhich the response is dramatic. A great majority of these patients have auto-antibodies tosmooth muscle and among women, thyroid and gastric antibodies and antinuclear factor.Asthma associated with polyarteritis nodosa and aspirin-sensitive bronchial asthma areusually of intrinsic type. However, many do not agree with this classification in view of therecent understanding of the underlying pathogenesis of asthma. Moreover, history of allergyor the responsible allergen is not easy to find out always.
Late Onset Asthma
Late onset asthma is a much used but poorly defined term. The difficulty arises because ofthe lack of appreciation of the difference between truly late onset asthma and asthma that isrecognised late.6,7 This is important because the missed asthmatic patient with long-standingunder treated asthma is more likely to develop irreversible airflow obstruction. Althoughthere is no agreeable definition of this entity, a reasonable definition would be “asthmawith onset of symptoms in adult life in a patient with no pre-existing, persistent respiratorysymptoms”. True onset of asthma is perhaps more common than it was appreciated andmay affect one in 50 of the adult population, assuming a 5% prevalence of asthma overall.Asthma perhaps occurs more frequently in the elderly than is usually appreciated and

94 Bronchial Asthma
may, therefore, be under diagnosed and under treated.8 Although several studies reportthe characteristics of older patients with asthma, few have described patients with onset ofasthma after the age of 65 years. Available studies are limited by small number of patients.9,10
In a recent population based study, the incidence of asthma was found to be more commonin the elderly.11 The age-and sex-adjusted incidence was 95/100,000 at or after the age of65 years.
Late onset asthma has sometimes been equated with intrinsic asthma, but in some patientsthere will be other important causes that must be recognised. Asthma induced by drugs,and occupational asthma may belong to this category. Asthma of adult onset may be thefirst sign of the development of polyarteritis nodosa. Women, who develop adult onsetasthma more often than men, often give a history of asthma beginning at the menopause.Many patients report that their symptoms started after a respiratory tract infection.
Occupational Asthma
Occupational asthma is the most common occupational lung disease in developed worldand accounts for 26-52% of all occupational lung diseases in UK, and Canada.12 About 15%of bronchial asthma are due to occupational exposure as reported from USA.13 About 250agents have been identified that can cause occupational asthma and some of them areindicated in earlier in the section under aetiology. Isocyanates that are widely used in manyindustries are responsible for the most common form of the disease and the prevalence ofisocyanate-induced asthma in exposed workers is close to 10%.14
There can be two categories of asthma related to the workplace. They are: occupationalasthma and work-aggravated asthma. Occupational asthma is characterised by variableairflow limitation, bronchial hyperresponsiveness, or both, due to conditions in particularwork environment, not to stimuli outside the workplace.15 On the other hand, work-aggravated asthma is preexisting or concurrent asthma that is aggravated by irritants orphysical stimuli in the workplace. Occupational asthma may develop in a person withpreexisting asthma or concurrent asthma after workplace exposure. There is usually a latentperiod between the first exposure to the offending agent and the onset of asthma. Thisperiod may vary from a few weeks to over 20 years. Occupational asthma with latencyincludes all instances of immunologic asthma, although the immunologic mechanism hasnot been identified for all agents. The other type of occupational asthma is without a latentperiod and the worker develops symptoms immediately upon working with the samesubstance. This is usually due to exposure to high concentrations of irritant gases, fumes,or chemicals on one or several occasions-reactive airway dysfunction syndrome.16
Exposure is the most important determinant whether occupational asthma develops.Higher the degree of exposure to an agent, higher is the prevalence of occupational asthma.History of atopy and smoking are important determinants to induce occupational asthmathat occurs through an IgE-dependent mechanism. The duration of exposure is not important.About 40% of patients with occupational asthma have symptoms within 2 years of exposureand in 20%, symptoms develop after 10 years of exposure.17 HLA class II alleles are involvedin some cases of isocyanate-induced asthma. The patient usually complains of chestsymptoms after working hours, in the evenings and at nights, but not during workinghours at the onset of the illness. Improvement in symptoms occurs at weekends or duringlonger periods away from work and worsening on return to work suggests but does not

Clinical Presentation of Bronchial Asthma 95
confirm occupational asthma. Runny and itchy eyes and nose and sneezing often accompanyrespiratory symptoms. Peak flow monitoring is important to recognize this problem.
Diagnosis of occupational asthma includes:a. Diagnosis of asthma andb. Establishment of a relation between asthma and work.
The diagnosis of asthma is based on compatible history and the presence of variableairflow obstruction or, in the absence of airflow limitation, the presence of pharmacologicallyinduced bronchial hyperresponsiveness. The number of criteria required to establish therelation to work depends on the purpose for which the diagnosis is made. They are morestringent if the diagnosis is required for medical purposes, and the relation to work shouldbe objectively demonstrated. But for screening examinations in the workplace or for fieldepidemiological surveys, less stringent diagnostic requirements can increase the sensitivityof case detection. An occupational cause should be sought for all new onset asthma inadults. The disease should be suspected in a person exposed at work to agents known tocause occupational asthma. History of both past and current exposures is required to beobtained since previous exposure to such agents may have induced permanent asthma.Such list of agents is available.18 However, the inability to identify an agent should not ruleout the diagnosis of occupational asthma. Detailed assessment of workplace exposure mayhelp determine the specific type of occupational asthma. The history should include specificjob duties and work processes for both the patient and the coworkers. A visit of the site bythe physician may help to understand the work situation better.
The diagnosis should always be confirmed by objective measurements. Various methodsused to diagnose occupational asthma include questionnaire, immunological testing,bronchial responsiveness to methacholin or histamine, measurement of FEV1 before andafter work, peak expiratory flow monitoring, specific inhalation challenges in a hospitallaboratory, and serial FEV1 measurements at work under supervision. There are manyadvantages and disadvantages of all these tests. While questionnaire is simple and sensitive,it has a low specificity. Although immunological tests are simple and sensitive, they canonly be used for high-molecular weight and some low-molecular weight agents. The testonly identifies sensitisation, but not disease. Further, most of the allergens are not availablecommercially.
Nocturnal Asthma
Nocturnal asthma symptoms are frequent and about 39% of asthmatics awaken nightly,and 94% have nocturnal awakenings at least once a month. A number of mechanisms havebeen hypothesised to explain the phenomenon of nocturnal asthma including exposure todust mite allergen, late-phase allergic reactions, effects of posture and sleep stages on airwaytone, gastro-oesophageal reflux, impaired mucociliary clearance, airway cooling, andchanges in circadian rhythms of circulating hormones (adrenaline and steroids). While nosingle mechanism can explain these changes, circadian rhythms may be particularly relevant.Normal airway tone increases during sleep and is magnified in asthmatics. Normally thereis a rhythm city in the lung function parameters with maximum readings between 3-4 PMand the lowest being at 3-4 AM. This is exaggerated in asthmatics. Bronchial responsivenessto histamine and allergen challenge increases during sleep and mast cell mediator release

96 Bronchial Asthma
is enhanced. Circulating eosinophils increase, which may allow their ingress into pulmonarytissue. Together with a decreased plasma catecholamine and cortisol levels all these factorsmay influence airway tone, inflammation, and responsiveness during sleep and producethe observed clinical picture.
The characteristic symptomatology is described above.
PATTERNS OF AIRFLOW OBSTRUCTION IN CHRONIC ASTHMA
Chronic asthma may be classified according to patterns of variations in their airflowobstruction.18
1. “Brittle” asthma2. “The morning dipper”3. The irreversible asthma”
a. A group never achieving a normal peak flow, but showing a reversible component,either spontaneously or after specific drug therapy.
b. A subgroup having a reversible FVC, but irreversible FEV1 and PEFR.c. The “drifter”, having irreversible airflow obstruction gradually improving over
weeks of intensive therapy.
Brittle Asthma
This is a form of intractable and persistent asthma resistant to all conventional therapy.There will be no wheeze at one moment, but gross wheezing may be present over a shortperiod of time. Most often they are misunderstood to have deliberate or emotional asthma.Serial measurements of PEFR show a chaotic pattern, with normal to grossly abnormalpatterns of airflow obstruction. They occur randomly throughout 24 hours. Low readingsmay reverse to normal with small doses of bronchodilators, but stabilisation is difficult.The salient feature of this asthma is their response to sympathomimetic drugs but withoutstabilisation. The patient may be atopic or non-atopic. Cromoglycate and steroid therapywill not be able to stabilize. These patients usually need frequent bronchodilators. However,not all patients are resistant to conventional therapy.
Morning Dippers
These are the patients who have worsening of their symptoms during early hours of thenight and is discussed above. The rhythm city is maintained during the day and reductionoccurs early in the morning. During day times, the patient may be completely normal andstable, so that no abnormality may be detected during the visit to the doctor. In children,the attack is usually worse around 2 AM and in adults it is variable increasing slowly andrapidly from midnight. Waking does not change the attack. It is observed in sleep workersthat the attack is worse towards the end of sleeping hours.
REFERENCES
1. National Asthma Programme in Finland 1994-2004. Ministry of Health and Social Welfare,Helsinki, 1994; quoted in Haahtela T. The importance of inflammation in early asthma. RespiratoryMed 1995;89:461-62.
2. Braman SS, Barrows AA, deCotiis BA et al. Airway hyperresponsiveness in allergic rhinitis: Arisk factor for asthma. Chest 1987;91:671-74.

Clinical Presentation of Bronchial Asthma 97
3. Steinium-Aarniale B. The role of infection in asthma. Chest 1987;91:157S.4. Clark TJV. Diurnal rhythm of asthma. Chest 1987;91:137S.5. Rachelefsky GS, Katz RM, Siegel SC. Chronic sinus disease with associated reactive airway
disease in children. Paediatrics 1984;73:526.6. Ayres JG. Late onset asthma. Br Med J 1990;300:1602.7. Lee HY, Stretton TB. Asthma in the elderly. Br Med J 1972;4:93-95.8. Banerjee DK, Lee GS, Malik SK, et al. Under diagnosis of asthma in the elderly. Br J Dis Chest
1987;81:23-29.9. Burr ML, Charles TJ, Roy K et al. Asthma in the elderly: an epidemiological survey. BMJ
1979;1:1041-44.10. Braman SS, Kaemmerlen JT, Davis SM. Asthma in the elderly: a comparison between patients
with recently acquired and long-standing disease. Am Rev Respir Dis 1991;143:336-40.11. Bauer BA, Reed CE, Yunginger JW, Wollan PC, Silverstein MD. Incidence and outcomes of
asthma in the elderly. A population based study in Rochester, Minnesota. Chest 1997;111:303-10.
12. Chan-Yeung M, Malo JL. Occupational asthma. New Engl J Med 1995;333:107-12.13. Blane P. Occupational asthma in national disability survey. Chest 1987;92:613-17.14. Chan-Yeung M, Malo JL. Epidemiology of occupational asthma. In: Busse WW, Holgate ST
(Eds). Asthma and rhinitis. Boston; Blackwell Scientific Publications. 1995;44-57.15. Bernstein IL, Chan-Yeung M, Malo JL, Bernstein DI. Definition and classification of asthma. In:
Bernstein IL, Chan-Yeung M, Malo JL, Bernstein DI (Eds). Asthma in the workplace. New York:Marcel Dekker, 1993;1-4.
16. Brooks SM, Weiss MA, Bernstein IL. Reactive airway dysfunction syndrome (RADS): persistentasthma syndrome after high level irritant exposures. Chest 1985;88:376-84.
17. Malo JL, Ghezzo H, D’Aquino C, L’Archeveque J, Cartier A, Chan-Yeung M. Natural history ofoccupational asthma; relevance of type of agent and other factors in the rate of development ofsymptoms in affected subjects. J Allergy Clin Immunol 1992;90:937-44.
18. Turner Warwick M. On observing patterns of airflow obstruction in chronic asthma. Br J DisChest 1977;71:73-86.

98 Bronchial Asthma
Diagnosis ofBronchial Asthma
6
The diagnosis of asthma is a clinical one; there is no confirmatory diagnostic blood test,radiographic or histopathological investigation. In some people, the diagnosis can becorroborated by suggestive changes in lung function tests. The clinical diagnosis of asthmais not always simple and the absence of an agreed definition of the disease is a problem,with many descriptions existing. However, while making a diagnosis of bronchial asthmaThe International Consensus Report definition of asthma should be kept in mind whichstates that it is “a chronic inflammatory disorder of the airways…..in susceptible individuals,inflammatory symptoms are usually associated with widespread but variable airflowobstruction and an increase in airway response to a variety of stimuli. Obstruction is oftenreversible, either spontaneously or with treatment”.
Some of the symptoms of asthma are shared with diseases of other systems. Even whenthe symptom of breathlessness is thought to be due to lung disease, there are numerousrelatively common lung diseases and differentiation of an airway disorder needs to bemade from both infections, and pulmonary thromboembolic disease and restrictive lungdisorders. Features of an airway disorder such as cough, wheeze and breathlessness shouldbe corroborated where possible by measurement of airflow limitation. They may be dueeither to a localised airway obstruction (e.g. tumour, foreign body, vocal cord dysfunctionor post-tracheostomy stenosis), or to a generalised problem (such as asthma, chronicobstructive pulmonary disease (COPD), bronchiectasis, cystic fibrosis or obliterativebronchiolitis).
Symptoms of Asthma
To avoid misdiagnosis it is essential to remember that people with asthma may suffer froma variety of symptoms, none of which is specific for asthma:
• Wheeze• Shortness of breath• Chest tightness• CoughThe hallmark of asthma is that these symptoms tend to be:• Variable• Intermittent• Worse at night• Provoked by triggers including exercise

Diagnosis of Bronchial Asthma 99
When cough is the predominant symptom without wheeze, this is often refered to ascough variant asthma.
Signs of Asthma
During exacerbations, the patient will often have wheeze and reduced lung function, eitherreduced peak flow or an obstructive pattern on spirometry. The presence of wheeze (usuallydiffuse, polyphonic, bilateral and particularly expiratory) is a cardinal sign of asthma and,if present, should be documented in clinical notes. Outside acute episodes, there may be noobjective signs of asthma. Patients who present with chronic asthma may have signs ofhyperinflation with or without wheeze.
Additional information which may contribute towards a clinical suspicion of asthmaincludes: personal or family history of asthma or other atopic condition (eczema, allergicrhinitis); worsening of symptoms after exposure to recognised triggers such as pollens,dust, feathered or furry animals, exercise, viral infections, chemicals, and environmentaltobacco smoke; and worsening of symptoms after taking aspirin/non-steroidal anti-inflammatory medication or use of β blockers. A good medical history is enough in most ofthe time to diagnose bronchial asthma. Particular attention should be paid to the precipitatingand/or aggravating factors. Pattern of symptoms may be perennial, seasonal, or perennialwith seasonal exacerbations. The symptoms may also be continuous, episodic, or continuouswith acute exacerbations. The onset, duration, and frequency of symptoms like number ofdays per week or month are also important to note. Day-night (circadian) variation withspecial reference to nocturnal symptoms should be asked for in each case. This should alsoinclude the age of onset and age of diagnosis, progress of the disease, previous and presentevaluation of the disease, treatment, and response to such therapy, living situation withhome age, location, cooling, and heating (central with gas, oil, electric, or kerosene), woodburning fire place, type of domestic cooking fuel used, carpeting, and humidifier. Specialattention should be paid in enquiring about the patient’s living room with particularreference to pillow, bed, floor covering, and dust collectors. Information regarding animalsin home and exposure to cigarette smoke, direct or side stream, is important. The impact ofdisease on the patient including number of emergency department visits, hospitalisation,history of life-threatening acute exacerbations, ventilatory support, requirement of oralsteroid therapy, number of school or work days missed, limitation of activity especiallysports, nocturnal awakening and the effect on growth, development, behaviour, school orwork achievements and lifestyles need to be assessed. Similarly, the impact on family includingdisruption of family life, effect on spouse and children, and economic impact needs assessment.Patient, parental, and spouse knowledge of asthma and belief in the chronicity of asthma andin the efficacy of treatment, ability of the patient and the family to cope with disease, level offamily support and economic resources are helpful in planning out a management programmefor the patient which is to be evaluated at the time of diagnosis. Any precipitating and/oraggravating factors like viral respiratory tract infections,1 exposure to environmental allergens,exposure to occupational chemicals or allergens, impact of environmental changes like movinginto a new home, going on a vacation, and/or alterations in workplace, work process, ormaterial used, exposure to irritants like tobacco smoke, strong odour, air pollutants like ozone,oxides of sulphur, occupational chemicals, vapors, gases and aerosols, emotional expressionslike anger, laughter, frustrations, crying, fear, drugs like aspirin, beta blockers, nonsteroidal

100 Bronchial Asthma
anti-inflammatory drugs, food additives like sulphites, change in weather, particularlyexposure to cold air, exercise, endocrinal factors like menstruation, pregnancy, thyroid disease,etc. are very important to evaluate since they will be very helpful in identifying the possibleagent/factor(s) that care responsible in causing the disease.
Family history of allergic diseases, asthma in close relatives is important pointers. In thepersonal and past history, any previous allergic disease like chronic rhinitis, repeated throatinfections, sneezing, dermatitis, gastrointestinal disturbances, adverse reaction to foods, historyof pulmonary infiltrates, and history of smoking or passive smoking are important points tonote.
Diagnosis of allergy in an asthma patient requires a thorough history taking. For example,if the asthma worsens in certain months and other symptoms of allergy like allergic rhinitis,sneezing, itching, running nose and nasal obstruction occur at the same time, pollens andoutdoor moulds are the responsible allergens. If symptoms appear when visiting a housewhere there are indoor pets or if the symptoms improve when the patient is away from homefor a week or longer, animal dander is the offending agent. Further evidence of animal dandercomes from the fact that eyes may itch and become red after handling the pet. If the pet licks thepatient, a red, itchy welt develops. If symptoms are more where a carpet is being vacuumedand bed making makes asthma worse, most likely mites are the responsible antigens. Mouldallergy is usual if symptoms develop around hay and on being exposed into a dampenvironment. If symptoms are related to certain job activities, either at work and they improvewhen away from work for a few days will indicate occupational asthma.
Physical findings of bronchial asthma are already discussed above. Although recurrentepisodes of cough and wheezing with breathlessness are almost always due to bronchialasthma in both children and adults, there are other causes of airways obstruction leading towheezing. The diagnosis may be little more difficult in children and infants rather in adults.
Laboratory Studies
Spirometry should be undertaken to document severity of airflow obstruction and to establishacute bronchodilator responsiveness for all patients in whom the diagnosis of asthma isbeing considered. All patients suspected to have bronchial Asthma should have spirometrydone at least for initial assessment. However, it is important to use an accurate spirometerand the procedure being done correctly.2 In bronchial asthma, typically one gets an obstructivepattern. Usually there will be a normal vital capacity with either impaired FEV1 or impairedMMEF.3 When the FEV1 is severely reduced with clear evidence of obstruction (FEV1/FVCratio less than 75% predicted), the vital capacity can also be reduced due to severe obstructionalone which prevents all the air to be emptied out during forced expiration. The mid-expiratoryflow rate is useful as a screening test but it is too sensitive to assess the severity of obstruction.The FEV1 is the single best measure of pulmonary function for assessing severity, althoughPEFR when measured accurately correlated well with FEV1. Bronchial asthma has a significantimpact on lung function decline, although not as great as COPD. Decline in FEV1 in patientswith bronchial asthma is significantly influenced by baseline FEV1, disease duration, andFEV1 variability. Moreover, the rate of FEV1 decline seems to increase in younger subjects onlywhen the baseline function is poorer.4
The low FEV1 in bronchial asthma is due to increased resistance because of broncho-constriction and remodelled airway walls. However, recently it is reported that measurement

Diagnosis of Bronchial Asthma 101
of maximum static pleural pressure at different lung volumes showed marked loss of lungrecoil in patients with moderate and severe asthma.5 Loss of this elastic recoil accounted formore than half of the reduction in total maximum airflow in these patients. This low elasticrecoil in patients of asthma is due to long-term corticosteroid therapy, which has knowndetrimental effect on connective tissue, smoking, mechanical fatigue due to the persistentstretch in over inflation, and altered surfactant levels. Further accumulation of inflammatorycells has been reported in the alveolar tissue of these patients.6
While complete spirometry can be done in a laboratory only, the patient can measure thepeak expiratory flow rate (PEFR) himself. Such measurement has many benefits. It provides asimple, quantitative, reproducible measure of airway obstruction that can be obtained usinginexpensive, portable peak flow meters. PEFR has a very good correlation with FEV1. This isalmost analogous to measuring blood pressure with a sphygmomanometer. This simpleobjective measurement of lung function helps detecting early deterioration of lung function.7,8
The most common strategy employed to support a clinical diagnosis of asthma is to demonstratethe presence of an abnormal, short-term variable airflow obstruction. Spontaneous variableairflow obstruction can be assessed by using peak expiratory flow monitoring at home9 ortreatment induced variable airflow obstruction can be assessed in the laboratory by measuringthe bronchodilator response to β2-agonists or the bronchoconstrictor response to short-actingairway smooth muscle spasmogens like methacholine.
Patients with stable asthma should be encouraged to measure their peak expiratory flowrates at least one or two days a week to detect any slow deterioration and to start recording itregularly if they develop a respiratory tract infection, increase in wheeze, or other symptomsof increasing airway obstruction. They should normally measure their PEFR twice daily, onwaking and in the evening, before using a bronchodilator, and perhaps four times a dayduring exacerbations. On each occasion, three readings should be taken and the best recordedgraphically for easy inspection. Patients at increased risk, who are those recently admitted tohospitals with acute asthma, brittle asthmatics, unstable asthma, those requiring varyingdoses of systemic steroids to control their symptoms, and those using home nebulizers shouldrecord their PEFR more often.
Home recordings of PEFR should improve the detection of under treated asthma. Patientsthought to overuse their β2-agonist inhalers may show previously unrecognised nocturnalasthma or pronounced morning dipping. Recordings may also allow unnecessary drugs tobe withdrawn, thus reducing morbidity and cost of treatment. High dose corticosteroidsrequired initially may not be necessary subsequently. Thus in summary, measurement ofPEFR is valuable in medical care settings to:
• Assess the severity of asthma as a basis for making treatment decisions, such as admissionto or release from the hospital or initiation of oral steroids.
• Monitor response to therapy during an acute exacerbation.• Monitor response to chronic therapy and provide objective justification for therapy to
patients.• Diagnose exercise-induced asthma.• Detect asymptomatic deterioration in lung function in the office and intervene before it
becomes more serious.• Monitor degree of airflow obstruction during a series of office visits to assess the overall
success of therapy.

102 Bronchial Asthma
The primary limitation of PEFR measurement is that it is effort dependent and validmeasurements depend upon the patient’s willingness and ability to exhale as hard aspossible. Adequate training and periodic checkups are necessary to verify the accuracy. Inaddition, PEFR measures only large airway function; therefore, patients with mild asthmawhose pathophysiologic abnormalities are linked to the small airways may be underdiagnosed if spirometry, which measures flow rates at low lung volumes (i.e., FEF50,FEF25-75), is not performed.• Other laboratory investigations for bronchial asthma include:• Complete and differential blood counts; chest X-ray (to rule out other causes of airway
obstruction, and to detect associated complications);• Sputum examination and stain for eosinophils (sputum eosinophils are highly characteri-
stic of asthma and neutrophils predominate in bronchitic sputum); nasal secretions andstain for eosinophils (neutrophilic nasal discharge indicates sinusitis); sputum differentialeosinophil count is one of the most useful objective tests in patients with bronchial asthma10
and• Complete pulmonary function studies including flow-volume loops which may reveal the
presence of upper airway obstruction.
Determination of specific IgE antibodies to common inhalant allergens with skin tests orwith in vitro test is useful to find out the role of allergy in the patient’s asthma. Incorporatinga skin prick test using commonly inhaled allergens is a simple, safe, inexpensive, rapid, andmost common way of assessing the contribution of atopy.11 The incidence of positive skinprick test result, at least to one aeroallergen in asthmatic adults residing in the UK, age range18-50 years, was 90%,12 whereas a positive result has been found in 15-40% of normalindividuals.13-15 Inclusion of this test in suspected asthma cases can reduce the cost of thisprocess significantly, and the test can be used as a reliable method to predict the absence ofasthma in young adults.16
OBJECTIVE TESTS
Obstructive airways disease produces a decrease in peak expiratory flow (PEF) and forcedexpiratory volume in one second (FEV1). One or both of these should be measured, but may benormal if the measurement is made between episodes of bronchospasm. If they are repeatedlynormal in the presence of symptoms, then a diagnosis of asthma must be in doubt. Variabilityof PEF and FEV1, either spontaneously over time or in response to therapy is a characteristicfeature of asthma. Although the normal level of diurnal variability is open to question, sequentialmeasurement of PEF may be useful in the diagnosis of asthma. Calculating variability may bedone in one of several ways. A 20% or greater variability in amplitude % best with a minimumchange of at least 60L/min, ideally for three days in a week for two weeks seen over a periodof time, is highly suggestive of asthma.17-23
Many patients with asthma will demonstrate variability below 20%, making this areasonably specific but insensitive diagnostic test. That is, marked variability of peak flowand easily demonstrated reversibility confirms a diagnosis of asthma, but smaller changes donot necessarily exclude the diagnosis.

Diagnosis of Bronchial Asthma 103
Diagnosis of Asthma Using PEF
Amplitude % best = (highest–lowest)/highest × 100Highest PEF = 400 1/minLowest PEF = 300 l/minAmplitude = 400 l/min – 300 l/min = 100 l/minPercentage PEF variability = (400-300)/400 × 100 = 25%
The objective measurements helpful in the diagnosis of asthma include:• > 20% diurnal variation on > 3 days in a week for two weeks (to be maintained in a
diary)• or FEV1 > 15% (and 200 ml) increase after short acting β2-agonist (salbutamol 400 μg
by metered dose inhaler (pMDI) +spacer or 2.5 mg by nebuliser)• or FEV1 > 15% (and 200 ml) increase after trial of steroid tablets (prednisolone 30 mg/
day for 14 days)• or FEV1 > 15% decrease after six minutes of exercise (running)• Histamine or methacholine challenge in difficult cases
Methods for Measuring Reversibility
• An increase after inhalation of a short acting β2-agonist (e.g. salbutamol 400 mg by metereddose inhaler (pMDI) +spacer or 2.5 mg by nebuliser)
• An increase after a trial of steroid tablets (prednisolone 30 mg/day for 14 days)• A decrease after six minutes of exercise, e.g. running. A resting measurement is to be
taken first and then the patient is to be asked to exercise for six minutes, a further readingis to be taken and then every 10 minutes for 30 minutes. As this procedure may rarelyinduce significant asthma, facilities for immediate treatment should be available.Objective tests should be used to try to confirm a diagnosis of asthma before long-term
therapy is started. Each of the above methods can be used, measuring either PEF (a 20%change from baseline and at least 60 l/min) or FEV1 (15% change and at least 200 ml).24
Bronchodilators reduce hyperinflation. Measurements of lung volumes before and afterbronchodilators add sensitivity when examining for bronchodilator responsiveness.25
Other investigations that may be helpful include rhinoscopy, sinus X-ray and broncho-provocation tests,26,27 provocative challenge with occupational allergens and evaluation ofpH for gastro-oesophageal reflux.
Bronchoprovocation Test
Bronchoprovocation test is indicated to assess the airway hyperresponsiveness in the formof increased bronchoconstrictor response to a variety of physical, chemical, or pharma-cological stimuli.28-30 This can better be assessed in a specialised pulmonary testing facilityusing bronchial challenge or provocation techniques. The most commonly employedmethods used to evaluate airway hyperresponsiveness include inhalation provocation withmethacholine or histamine and exercise challenge. During such a test changes in pulmonaryfunction are measured with serial spirometry after inhaling incremental doses of an agonistsuch as methacholine or histamine or after exercise.31 The results are then expressed eitheras the cumulative dose or the concentration of agonist that produces a 20% fall in FEV1(PD20). Methacholine bronchoprovocation testing is frequently used to diagnose airway

104 Bronchial Asthma
hyperresponsiveness and asthma. A > 20% reduction in FEV1 following methacholineadministration is a common parameter used to determine airway hyperresponsiveness. Someobserved that the slope of the decline of FEV1 with increasing dose of methacholine is a betterway of measuring responsiveness because a value can be assigned to all subjects. Alternatively,a > 40% reduction in specific airway conductance (sGaw) can be used to determine airwayhyperresponsiveness.32,33 Regardless of which test is selected, according to the AmericanThoracic Society guidelines, the changes in the test parameter following methacholinechallenge must exceed 2 SDs or coefficients of variation for repeated measures in the sameindividual before a statistically significant change can be established.33 Although either of thetwo measurements is good enough, a substantial number of patients have a reduction inSGaw alone in response to methacholine, and this response is seen in patients with a higherFEF25-75 / FVC ratio.34 Large, central airway obstruction is best detected by SGaw measurements,while both large and small airway narrowing will affect measurements of FEV1.
Methacholine responsiveness is often used to confirm asthma status in patients, and as apredictor of later development of respiratory disease.35,36 It is widely used in epidemiologicalstudies, where a standardised tool for measurements of bronchial responsiveness tomethacholine has been developed to estimate variation in prevalence of increased bronchialresponsiveness and predictors of asthma in different groups.37 Various such predictors arethe FEV1 and symptom status, female sex, smoking, atopy, occupational exposure, andgeographical regions are associated with increased responsiveness. Smaller airways are moreresponsive than larger ones, and the reduction in responsiveness diminishes with eachincrease of lung size.38
Methacholine challenge testing may cause an acute episode of vocal cord adduction andthus, positive results may not reflect underlying reactive airways disease. However, a flatteningor truncation of the inspiratory flow-volume loop after the patient undergoes methacholinetesting is not diagnostic for the presence of inspiratory vocal cord adduction.39
Results of exercise provocation are expressed as the peak fall in FEV1 after exercise.Asthmatics respond to bronchoprovocation with greater degree of airflow obstruction thannormal subjects.40 Other conditions that are associated with an increased bronchial hyper-reactivity include allergic rhinitis, cystic fibrosis, COPD, normal persons after a viral upperrespiratory tract infection or oxidant exposure, and smokers.40,41 Diurnal variation in themeasurement of PEFR is an indirect but clinically useful way of the degree of bronchialhyperreactivity even if there may be some variation.42
Bronchial provocation test is helpful in the differential diagnosis of asthma when therespiratory history, physical findings, and PEFR variations are not adequate to confirm theclinical diagnosis. These situations include cough variant asthma and exercise-induceddyspnoea.28,43
There is no one test or set of tests that should be ordered for every patient. Selection of testsshould be individualised. However, with careful attention to the history, physical examination,and laboratory results, a correct diagnosis of asthma will be made in virtually all instances.
Asthma may be under diagnosed particularly in young children, if they only wheezewhen they have respiratory infections which may be dismissed as wheezy bronchitis,asthmatic bronchitis, bronchitis, or pneumonia. Although recurrent episodes of cough andwheezing are almost always due to asthma in both children and adults, there are other

Diagnosis of Bronchial Asthma 105
causes of airway obstruction which produce similar symptoms that need to be excluded. Inadults, such conditions include mechanical obstruction of the airways, laryngealdysfunction, chronic bronchitis, pulmonary emphysema, congestive cardiac failure,pulmonary embolism, pulmonary infiltration with eosinophilia, and cough secondary todrugs. Of particular interest is the confusion with chronic bronchitis more so in elderlysmokers. Presence of crepitations; absence of eosinophils in the nasal secretion, sputum,and blood eosinophilia; lack of good reversibility after bronchodilators; and an abnormaldiffusion capacity favours chronic bronchitis with emphysema. Occasionally, it is notpossible to differentiate the two conditions.
Of all the battery of tests utilised to diagnose asthma (methacholine challenge testing, peakexpiratory flow variability over a 2-week period, the FEV1/FVC ratio, the reversibility testing,and the differential count of eosinophils in blood and sputum), methacholine airwayresponsiveness and the sputum differential eosinophil count seems to be the most usefulobjective tests in patients with mild asthma. The sensitivity of these two tests are 91 and 72%respectively, and the specificity is 90 and 80% respectively.44
Increase bronchial responsiveness demonstrated by methacholine or histamine challengeis associated with symptomatic asthma, but is also common in the general population and inpatients with COPD. However, failure to demonstrate hyperresponsiveness in an untreatedperson with suspected asthma should prompt reconsideration of the diagnosis.
Other Tests
Lung function tests may show changes suggestive of an alternative lung disease. For example,COPD may be suspected in the presence of obstructive spirometry, reduced diffusing capacity(CO uptake) and pressure dependent airway collapse on flow volume curves, but these changesare not diagnostic and do not exclude asthma, which may anyway coexist with otherconditions. Failure to respond to asthma treatment should prompt a search for an alternative,or additional, diagnosis. Chest X-rays in all patients with atypical symptoms should be done.
The differential diagnosis of bronchial asthma includes: COPD, cardiac diseases, laryngealtumours, tracheal tumours, bronchogenic carcinoma, bronchiectasis, foreign body, interstitiallung disease, pulmonary embolism, aspirations, vocal cord dysfunction, pulmonaryinfiltrations with eosinophilia, cough due to drugs (beta blockers, ACE inhibitors) andhyperventilation. A detailed clinical history as well as investigations as outlined will behelpful in differentiating these conditions.
In spite of a cautious and careful approach, there may be situations when one has to referthe case to a specialist for opinion and further investigations. These situations include:• Diagnosis unclear or in doubt• Unexpected clinical findings (like crepitations, collapse, effusion, cardiac murmur, clubbing,
heart failure, cyanosis, etc.)• Spirometry or PEFR does not fit the diagnosis (like restrictive defect)• Suspected occupational asthma• Persistent shortness of breath (non-episodic, or without wheeze)• Unilateral or fixed wheeze• Stridor• Persistent chest pain or atypical features

106 Bronchial Asthma
• Weight loss• Persistent cough or sputum production• Non-resolving pneumonia
A suggested algorithm for the diagnostic work up in younger subjects with suspectedasthma is shown in Figure 6.1.
Cough, Wheezing, Dyspnoea
Spirometry with bronchodilators (Reversibility testing)
Positive Negative
Skin testing
Positive Negative
Exercise/Methacholine Consider other diagnosis
Positive Negative
Bronchial asthma
Fig. 6.1: Diagnostic work-up for bronchial asthma
COPD and Bronchial Asthma
Most often there is a confusion whether the patient is having bronchial asthma or COPD asboth the conditions has similar symptoms like cough, wheezing and breathlessness. Thereare some similarities also between the two conditions. Tissue eosinophilia, sputumeosinophilia, increased bronchial hyperreactivity, inflammatory cells, cytokines, etc. canbe similar in COPD, but the types of cells and degree of involvement differ. Because theoverall prognosis and course of the disease are entirely different in both the conditions.Hence, the differentiation should always be made. It must, however, be possible that bothconditions may coexist.
The important differentiating points between the two are shown in Table 6.1.45
Diagnosis of Occupational Asthma
Careful history and temporal relationship of symptoms with work place will clinch thediagnosis. However, it is important to establish objectively a relationship between workand asthma symptoms. Specific challenge tests of occupational exposure tests are oftenconsidered a reference standard for the diagnosis of occupational asthma. The various testsused are:
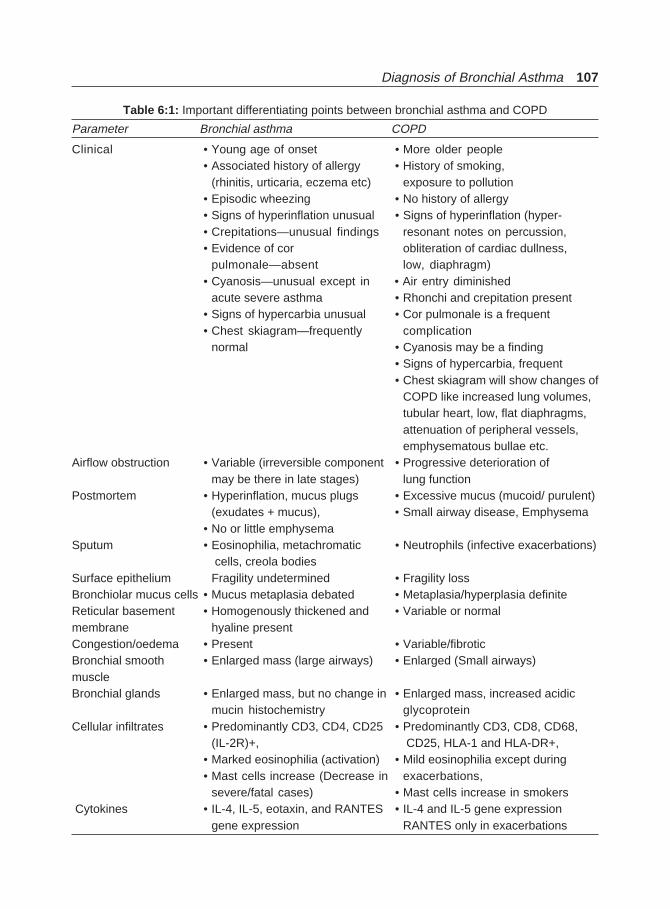
Diagnosis of Bronchial Asthma 107
Table 6:1: Important differentiating points between bronchial asthma and COPD
Parameter Bronchial asthma COPD
Clinical • Young age of onset • More older people• Associated history of allergy • History of smoking,
(rhinitis, urticaria, eczema etc) exposure to pollution• Episodic wheezing • No history of allergy• Signs of hyperinflation unusual • Signs of hyperinflation (hyper-• Crepitations—unusual findings resonant notes on percussion,• Evidence of cor obliteration of cardiac dullness,
pulmonale—absent low, diaphragm)• Cyanosis—unusual except in • Air entry diminished
acute severe asthma • Rhonchi and crepitation present• Signs of hypercarbia unusual • Cor pulmonale is a frequent• Chest skiagram—frequently complication
normal • Cyanosis may be a finding• Signs of hypercarbia, frequent• Chest skiagram will show changes of
COPD like increased lung volumes,tubular heart, low, flat diaphragms,attenuation of peripheral vessels,emphysematous bullae etc.
Airflow obstruction • Variable (irreversible component • Progressive deterioration ofmay be there in late stages) lung function
Postmortem • Hyperinflation, mucus plugs • Excessive mucus (mucoid/ purulent)(exudates + mucus), • Small airway disease, Emphysema
• No or little emphysemaSputum • Eosinophilia, metachromatic • Neutrophils (infective exacerbations)
cells, creola bodiesSurface epithelium Fragility undetermined • Fragility lossBronchiolar mucus cells • Mucus metaplasia debated • Metaplasia/hyperplasia definiteReticular basement • Homogenously thickened and • Variable or normalmembrane hyaline presentCongestion/oedema • Present • Variable/fibroticBronchial smooth • Enlarged mass (large airways) • Enlarged (Small airways)muscleBronchial glands • Enlarged mass, but no change in • Enlarged mass, increased acidic
mucin histochemistry glycoproteinCellular infiltrates • Predominantly CD3, CD4, CD25 • Predominantly CD3, CD8, CD68,
(IL-2R)+, CD25, HLA-1 and HLA-DR+,• Marked eosinophilia (activation) • Mild eosinophilia except during• Mast cells increase (Decrease in exacerbations,
severe/fatal cases) • Mast cells increase in smokers Cytokines • IL-4, IL-5, eotaxin, and RANTES • IL-4 and IL-5 gene expression
gene expression RANTES only in exacerbations

108 Bronchial Asthma
i. Measurement of lung function before and after a work shift. This is not very helpful inestablishing a causal relationship between symptoms and work exposure.
ii. Measurements of lung function (FVC and FEV1) when the patient has been away from thework environment for a period of time and again when he returns to work. An improvementin symptoms and lung functions away from work and recurrence of symptoms anddeterioration in lung function after returning to work, confirms that the symptoms arerelated to the work environment.
iii. Prolonged recording of PEFR by the patient at work and at home is a good method ofestablishing the causal relationship. The patient is asked to measure and record thePEFR every 2 hours from waking to sleep for at least 2 to 3 weeks at work, followed byat least 10 days off work. Different patterns of PEFR are described. The method has thedisadvantage of falsification of data and inaccurate readings.
iv. Serial measurements of nonspecific airway responsiveness in conjunction withprolonged recording of PEFR has been proposed as an additional test to provide objectiveevidence of sensitisation. Significant increases in airway responsiveness when awayfrom work, associated with appropriate changes in PEFR, suggest an occupationalrelationship. Specific challenge tests are required to identify the substances in the workplace causing the symptoms. However, this is time consuming and not devoid of danger.They should be performed by experienced personnel in hospital settings whereresuscitation facilities are available and frequent observations can be made.
Allergy skin tests with high molecular weight compounds may be useful in identifyingthe responsible agent. Animal products, flour, coffee, and castor bean produce immediatepositive reactions on skin testing in sensitised subjects.
Specific IgE antibodies to various occupational allergens may be demonstrated by RASTor by ELISA. Such specific antibodies against low molecular weight compounds conjugatedto a protein like trimellite anhydride and isocyanate have been demonstrated in someexposed subjects. However, positive skin tests and the presence of IgE antibodies indicatesensitisation and may occur in exposed workers without asthma.
The clinical investigation of occupational asthma is shown in Figure 6.2.46
Classification of Asthma
Bronchial asthma can be defined as mild, moderate, and severe on severity of disease.47
This enables the clinician to categorize the overall assessment of a patient’s asthma andselect appropriate therapy. The characteristics are shown in Table 6.2 and are recommendedby the Expert Panel of the National Asthma Education Program by the National Heart,Lung, and Blood Institute, USA.47 The characteristics are general, and because asthma ishighly variable, these characteristics may overlap. Furthermore, an individual may switchinto different categories over time.
Thus, severity of bronchial asthma, as defined by the National Asthma EducationProgramme (NAEP) Expert Panel of 1991, can be summarised as:
Mild: It is characterised by intermittent daytime symptoms up to two times in a week, briefwheezing, cough, or breathlessness with activity, and infrequent nocturnal cough orwheezing less than two times in a month. The FEV1 or PEFR is expected to be greater than80% when asymptomatic and to vary 20% with symptoms.
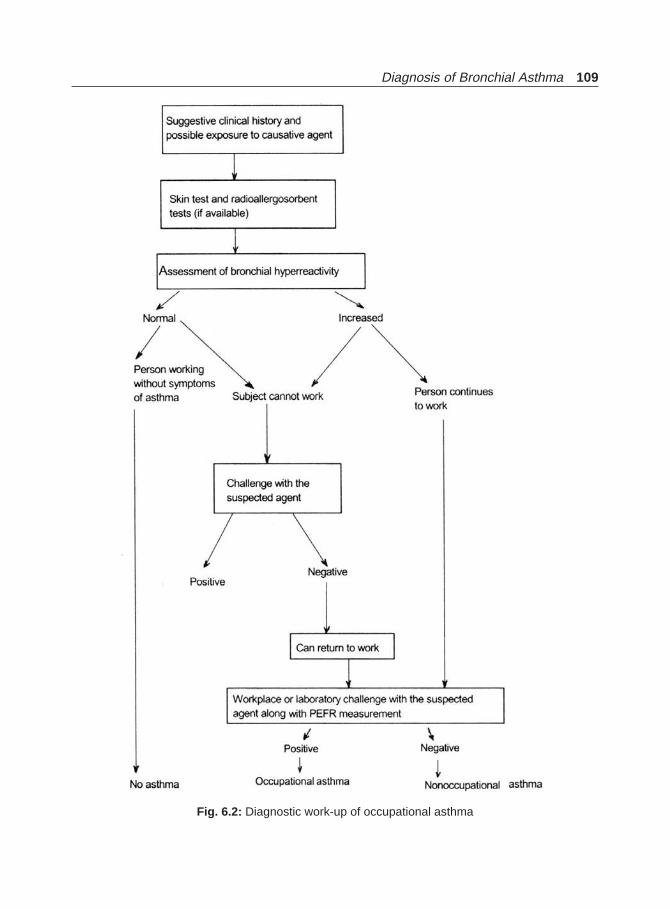
Diagnosis of Bronchial Asthma 109
Fig. 6.2: Diagnostic work-up of occupational asthma
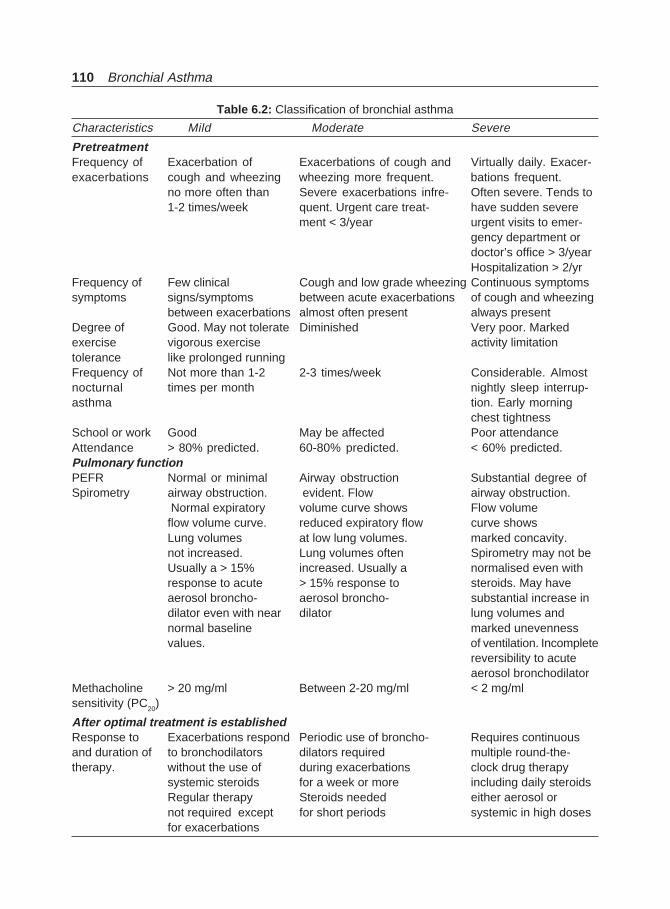
110 Bronchial Asthma
Table 6.2: Classification of bronchial asthma
Characteristics Mild Moderate Severe
PretreatmentFrequency of Exacerbation of Exacerbations of cough and Virtually daily. Exacer-exacerbations cough and wheezing wheezing more frequent. bations frequent.
no more often than Severe exacerbations infre- Often severe. Tends to1-2 times/week quent. Urgent care treat- have sudden severe
ment < 3/year urgent visits to emer-gency department ordoctor’s office > 3/yearHospitalization > 2/yr
Frequency of Few clinical Cough and low grade wheezing Continuous symptomssymptoms signs/symptoms between acute exacerbations of cough and wheezing
between exacerbations almost often present always presentDegree of Good. May not tolerate Diminished Very poor. Markedexercise vigorous exercise activity limitationtolerance like prolonged runningFrequency of Not more than 1-2 2-3 times/week Considerable. Almostnocturnal times per month nightly sleep interrup-asthma tion. Early morning
chest tightnessSchool or work Good May be affected Poor attendanceAttendance > 80% predicted. 60-80% predicted. < 60% predicted.Pulmonary functionPEFR Normal or minimal Airway obstruction Substantial degree ofSpirometry airway obstruction. evident. Flow airway obstruction.
Normal expiratory volume curve shows Flow volumeflow volume curve. reduced expiratory flow curve showsLung volumes at low lung volumes. marked concavity.not increased. Lung volumes often Spirometry may not beUsually a > 15% increased. Usually a normalised even withresponse to acute > 15% response to steroids. May haveaerosol broncho- aerosol broncho- substantial increase indilator even with near dilator lung volumes andnormal baseline marked unevennessvalues. of ventilation. Incomplete
reversibility to acuteaerosol bronchodilator
Methacholine > 20 mg/ml Between 2-20 mg/ml < 2 mg/mlsensitivity (PC
20)
After optimal treatment is establishedResponse to Exacerbations respond Periodic use of broncho- Requires continuousand duration of to bronchodilators dilators required multiple round-the-therapy. without the use of during exacerbations clock drug therapy
systemic steroids for a week or more including daily steroidsRegular therapy Steroids needed either aerosol ornot required except for short periods systemic in high dosesfor exacerbations

Diagnosis of Bronchial Asthma 111
Moderate: Moderate asthma is characterised by symptoms more than 1-2 times weekly affectingsleep and activity levels, exacerbations lasting several days, and occasional emergency care.The FEV1 or PEFR is expected to be 60-80% at baseline and vary between 20-30% withsymptoms.
Severe: This is characterised by continuous symptoms including nocturnal symptoms,limited activity levels, frequent exacerbations, and occasional hospitalisation, and emergencytreatment. The FEV1 or PEFR is less than 60% at baseline and highly variable.
The British Guidelines are parallel to the NAEP guidelines. However, the Global Strategyfor Asthma Management and Prevention Workshop, a joint effort of the National Heart,Lung, and Blood Institute and the WHO, 1995 (NIH Publication No. 96-3659A) classifiesseverity of asthma into different (discussed subsequently).
REFERENCES
1. Empey DW, Laitinen LA, Jacobs L, Gold WM, Nadel A. Mechanisms of bronchial hyperreactivityin normal subjects after upper respiratory tract infection. Am Rev Respir Dis 1976; 113:131.
2. Behera D. Normal values of Pulmonary Function Tests. In: Pulmonary functions tests in Healthand Disease (Ed). PS Shankar. Indian College of Physicians, 1998; 150-59.
3. Wagner EM, Liu MC, Weinnman GG, Permutt S, Bleecker ER. Peripheral lung resistance innormal and asthmatic subjects. Am Rev Respir Dis 1990;141:584.
4. Cibella F, Cuttitta G, Bella V et al. Lung function decline in bronchial asthma. Chest 2002;122:1944-48.
5. Gelb AF, Licuanan J, Shinar CM, Zamel N. Unsuspected loss of lung elastic recoil in chronicpersistent asthma. Hest 2002;121:715-21.
6. Wassermann K. Is asthma another interstitial lung disease? Chest 2002;121:673-74.7. Gupta ML, Behera D: Pattern of airflow obstruction in Bronchial Asthma—An observation on
Home-Monitoring of Peak Expiratory Flow Rate. J Ass Phy India, 1997;45:94-96.8. Clark TJH, Hetzel MR. Diurnal variation of asthma. Br J Dis Chest 1977;71:87-92.9. Jamison JP, McKinley RK. Validity of peak expiratory flow rate variability for the diagnosis of
asthma. Clin Sci 1993;85:367-71.10. Hunter CJ, Brightling CE, Woltmann G, Wardlaw AJ, Pavord ID. A comparison of the validity
of different diagnostic tests in adults with asthma. Chest 2002;121:1051-57.11. Allergy skin testing. Board of Directors,; American Academy of Allergy and Immunology
J Allergy Clin Immunol 1993;92:636-37.12. Corne J, Smith S, Schreiber J et al. Prevalence of atopy in asthma. Lancet 1994;344:344-45.13. Holt PG, Macaubas C, Stumbles PA et al. The role of allergy in the development of asthma.
Nature 1999;402:B12-B17,14. Holgate ST. The epidemic of allergy and asthma. Nature 1999;402:B2-B4.15. Busse WW, Lemanske RF. Advances in immunology: Asthma. N Engl J Med 2001;344:350-62.16. Graif Y, Yigla M, Tov N, Kramer MR. Value of a negative aeroallergen skin-prick test result in
the diagnosis of asthma in young adults. Co-relative study with methacholine challenge testing.Chest 2002;122:821-25.
17. Higgins BG, Britton JR, Chinn S et al. The distribution of peak flow variability in a populationsample. Am Rev Respir Dis 1989;140:1368-72.
18. Kesten S, Rebuck AS. Is the short-term response to inhaled beta-adrenergic agonist sensitive orspecific for distinguishing between asthma and COPD! Chest 1994;105:1042-1045.
19. Thiadens HA, De Bock GH, Dekker FW et al. Value of measuring diurnal peak flow variabilityin the recognition of asthma: a study in general practice. Eur Respir J 1998;12:842-47.

112 Bronchial Asthma
20. Kunzli N, Stutz EZ, Perruchaoud AP et al. Peak flow variability in the SAPALDIA study and itsvalidity in screening for asthma-related conditions. The SAPALDIA Team. Am J Respir CritCare Med 1999;160:427-34.
21. Siersted HC, Mostgaard G, Hyldebrandt N et al. Interrelationships between diagnosed asthma,asthma like symptoms, and abnormal airway behaviour in adolescence. The Odense SchoolchildStudy. Thorax 1996;51:503-09.
22. Quackenboss JL, Libowitz MD, Krzyzanoski M. The normal range of diurnal changes in peakexpiratory flow rates. Relationship to symptoms, and respiratory disease. Am Rev Respir Dis1991;143:323-30.
23. Reddel HK, Salome CM, Peat JK et al. Which index of peak expiratory flow is most useful in themanagement of stable asthma? Am J Respir Crit Care Med 1995;151:1320-25.
24. Tweeddale PM,Alexander F, McHardy GJ. Short-term variability in FEV1 and bronchodilatorresponsiveness in patients with obstructive ventilatory defects. Thorax 1987;42:487-90.
25. Newton MF, O’Donnell E, Forkert L. Response of lung volumes to inhaled salbutamol in a largepopulation of patients with severe hyperinflation. Chest 2002;121:1042-50.
26. Anderson SD. Nonisotonic aerosol challenge in the evaluation of bronchial hyper-responsiveness.Allergy Proc 1991;12:143.
27. Boulet LP, Legris C, Thibault L, Turcotte H. Comparative bronchial response to hyperosmolarsaline and methacholine in asthma. thorax 1987;42:953-58.
28. Boushey HA, Holtzman MJ, Sheller JR, Nadel JA. Bronchial hyper-reactivity. Am Rev RespirDis 1980;121:389-414.
29. Hopp RJ, Townley RG, Biven RE, Bewtra AK, Nair NM. The presence of airway reactivity beforethe development of asthma. Am Rev Respir Dis 1990;141;2-8.
30. Jones A. Asymptomatic bronchial hyper-reactivity and the development of asthma and otherrespiratory tract illnesses. Thorax 1994;49;757-61.
31. Chatham M, Bleecker ER, Smith PL, Rosenthal RR, Mason P, Norman PS. A comparison ofhistamine, methacholine, and exercise airway reactivity in normal and asthmatic subjects. AmRev Respir Dis 1982;126:235-40.
32. American Thoracic Society Guidelines for methacholine and exercise challenge testing, 1999.Am J Respir Crit Care Med 2000;161:309-329.
33. American Thoracic Society Guidelines for bronchial inhalation challenges with pharmacologicand antigenic agents. ATS News 1980 (Spring).
34. Parker AL, McCool FD. Pulmonary function characteristics in patients with different patterns ofmethacholine airway hyper-responsiveness. Chest 2002;121:1818-23.
35. Laprise C, Boulet LP. Asymptomatic airway hyper-responsiveness: A three year follow-up. AmJ Respir Crit Care Med 1997;156:403-409.
36. Pattemore PK, Asher MH, Harrison AC et al. The interrelationship among bronchial hyper-responsiveness, the diagnosis of asthma, and asthma symptoms. Am Rev Respir Dis 1990;142:549-554.
37. Burney PGJ, Luczynska G, Chinn S et al. The European Community Respiratory Health Survey.Eur Respir J 1994;7:954-60.
38. Schwartz J, Schindler C, Zemp E et al. Predictors of methacholine responsiveness in a generalpopulation. Chest 2002;122:812-20.
39. Perkins PJ, Morris MJ. Vocal cord dysfunction induced by methacholine challenge testing. Chest2002;122:1988-93.
40. Hargreave FE, Ryan G, Thomson NC et al. Bronchial responsiveness to histamine or methacholinein asthma: Measurement and clinical significance. J Allergy Clin Immunol 1981;68:347-55.
41. Chatham M, Bleecker ER, Norman P, Smith PL, Mason P. A Screening test for airways reactivity.Chest 1982;82:15-18.

Diagnosis of Bronchial Asthma 113
42. Ryan G, Latimer KM, Dolovich J, Hargreave FE. Bronchial responsiveness to histamine:relationship to diurnal variation of peak flow rate, improvement after bronchodilator, airwaycaliber. Thorax 1982;37:423-29.
43. Galvez RA, McLaughlin FJ, Levison H. The role of the methacholine challenge in children withchronic cough. J Allergy Clin Immunol 1987;79:331-35.
44. Hunter CJ, Brightling CE, Voltman G et al. A comparison of the validity of different diagnostictests in adults with asthma. Chest 2002;122:1051-57.
45. Jeffery P. Immunopathology: Comparison of COPD and asthma. In: Hansel TT, Barnes PJ (Eds):New Drugs for Asthma, Allergy, and COPD. Prog Respir Res. Basel, Karger, 2001; 31:24-29.
46. Chan-Yeung M, Malo JL. Occupational asthma. New Engl J Med 1995;333:107-12.47. National Asthma Education Programme. Expert Panel Report. Guidelines for the diagnosis and
management of asthma. National Heart, Lung, and Blood Institute, National Institute of Health,Bethesda, Maryland, USA, Publication No. 91-3042A, June 1991.

114 Bronchial Asthma
Prognosis of Bronchial Asthma
7
FACTORS FOR ASTHMA MORTALITY
Although the possibility of asthma-related death exists for all patients with asthma, severalstudies have revealed factors associated with an increased risk of such deaths.1-6 Severalstudies from many countries of the world including Britain, New Zealand, United States,France, Germany, and Canada have shown increases over the last two decades in theincidence of deaths from asthma. The cause of such increase in deaths remains a puzzle.There are many hypotheses to explain this, but little emphasis has been placed on thepossibility that confidence in better drug treatment may modify patient’s behaviour so asto place him at greater risk of illness. Excessive confidence in bronchodilator inhalers andnebulisers can make patients stay away from hospitals too long during acute attacks. It isalso very likely that prevention of symptoms by use of antiasthma drugs could allow patientsto spend more time in environments containing antigens or other agents that provokeasthma, resulting in more serious and long-lasting bronchial inflammation and reactivity.
Some of these recognised factors that increases the susceptibility to death from asthma areas follows.
Age and Ethnicity
Asthma-related death rates are higher among older patients than in any other age group.Although the death rate is relatively low in younger patients, an increased trend in asthmadeaths among these individuals between the age group of 5 to 34 years have been notedduring the last 10 years. People in their late teens and early twenties, particularly membersof minority groups, are over represented in asthma mortality statistics groups. African-Americans have asthma related mortality rates that are higher than those of Caucasians,especially in relatively young age groups, and the mortality rate in this group has increasedsignificantly during the past decade. In 1979, African-Americans of both sexes were abouttwice as likely to die of asthma as Caucasians.
Previous Life-threatening Acute Asthma Exacerbations
Individuals who have had acute exacerbations of asthma that resulted in respiratory failureand required intubation are at increased risk for subsequent fatal exacerbations. Those whohave experienced respiratory acidosis without requiring intubation are also high-riskpatients.

Prognosis of Bronchial Asthma 115
Hospital Admission for Asthma within the Last Year
Those patients hospitalised for asthma within the last year have a greatly increased risk ofdying from asthma when compared to severity-matched asthma patients in the communitythat have not been hospitalised. Those with more than two hospitalisations for statusasthmaticus in spite of long-term oral steroid therapy are at the highest risk of dying fromasthma.
In some patients, deterioration during an acute exacerbation occurs very rapidly.Underestimation of the severity of such exacerbations may lead to a life-threatening delayin starting medical treatment or seeking medical care.
Some patients may fail to appreciate a poor response to treatment during an acuteexacerbation and may rely on frequent repetitive use of inhaled β2-agonist far in excess ofrecommended doses for therapy at home. This treatment may temporarily blunt symptomsbut mask increasing inflammation and airway hyperresponsiveness, which may in turn,lead to abrupt and severe deterioration of lung function. Without the documented objectivemeasures of pulmonary function or realisation by the patient and/or the physician of theseverity of the disease, risk of death is increased.
Psychological and Psychosocial Problems
Depression leads to increased death particularly in children. Other psychological problemsthat have been documented as associated with those at increased risk include alcohol abuse,documented depression, recent family loss and disruption, recent unemployment, andschizophrenia. Patients who have experienced a life-threatening asthma exacerbation havebeen reported, on the whole, to deny that they are at risk of death. Following a near fatalexacerbation, they tend to either develop decompensating psychiatric disease and symptomsof extreme anxiety or develop even higher levels of denial. Some tend to minimise theirsymptoms and avoid access to health care. Other associations include life crises, familyconflict, and social isolation.
Regardless of the possible physiologic and psychological interactions that link anxiety,depression, and asthma fatality, it is evident that patients who have these psychologicaldisruptions are at increased risk for death.7-15
Lack of Access to Medical Care
Lack of access to medical care is another risk factor associated with asthma-related death.Patients of lower socioeconomic class are unable to obtain routine preventive asthma care.As a result, these patients seek help only when their asthma symptoms are severe andreport to the emergency room for initial care.16 In rural areas, lack of access to adequateemergency care can result in life-threatening delay in medical treatment duringexacerbations. Even in some urban centers, adequate facilities like ventilatory support arenot available.
Medication Use
Medications, particularly steroids, are underused at the time of death. The controversy ofthe asthma mortality because of β-agonist use is still on.17-19

116 Bronchial Asthma
REFERENCES
1. Wissow LS, Gittelsohn AM, Szklo M, Starfield B, Mussman M. Poverty, race, and hospitalisationfor childhood asthma. Am J Public Health 1988;78:777.
2. Miller BD. Depression and asthma: A potentially lethal mixture. J Allergy Clin Immunol1987;80:481.
3. Strunk RC. Identification of the fatally-prone subject with asthma. J Allergy Clin Immunol1989;83:477.
4. Rea HH, Scragg R, Jackson R et al. A case-controlled study of deaths from asthma. Thorax1986;41:833.
5. Benatar SR. Fatal asthma. N Engl J Med 1986;314:423.6. Barriot P, Riou B. Prevention of fatal asthma. Chest 1987;92:460.7. Campbell DA, McLennan G, Coates JR et al. A comparison of asthma deaths and near fatal
asthma attacks in South Australia. Eur Respir J 1994;7:490-97.8. Strunk RC, Mrazek DA, Fuhrmann GSW, LeBreque JF. Death from asthma in childhood. Can
they be predicted? JAMA 1985;254:1193-98.9. Wareham NJ, Harrison BDW, Jenkins PF, Nicholls J, Stableforth DE. A district confidential enquiry
into death due to asthma. Thorax 1993;48;1117-20.10. Campbell DA, Yellowlees PM, McLennan G, et al. Psychiatric and medical features of near fatal
asthma. Thorax 1995;50;254-59.11. Creer TL. Psychological factors and deaths from asthma; Creation and critique of a myth. J Asthma
1986;23;261-69.12. Boseley CM, Fosbury JA, Cochrane GM. The psychological factors associated with poor
compliance with treatment in asthma. Eur Respir J 1995;8;899-904.13. Fitzgerald JM. Psychological barriers to asthma education. Chest 1994;1069(Suppl4):2S-3S.14. Gibson GJ. Perception, personality, and respiratory control in life-threatening asthma. N Engl J
Med 1994;330:1329-34.15. Weiss KB, Wagener DK. Geographical variations in US asthma mortality: Small area analysis of
exercise mortality, 1981-85. Am J Epidemiol 1990;132:s107.16. Barger LW, Vollmer WM, Felt RW, Buist AS. Further investigations into the recent increase in
asthma death rates; a review of 41 asthma deaths in Oregon in 1982. Ann Allergy 1988;60:31-39.17. Crane J, Flatt A, Jackson R et al. Prescribed fenoterol and death from asthma in New Zealand,
1981-83: Case control study. Lancet 1989;1:917-27.18. Crane J, Pearce N, Burgess C, Beasley R. Asthma and the beta agonist debate. Thorax
1995;50(Suppl 1):S5-S10.19. Suissa S, Ernst P, Boivin JF et al. A cohort analysis of excess mortality in asthma and the use of
inhaled beta agonists. Am J Respir Crit Care Med 1994;149:604-10.

Complications of Bronchial Asthma 117
Complications ofBronchial Asthma
8
Infections, pneumothorax, pneumomediastinum, and atelectasis due to mucus pluggingare the complications of acute bronchial asthma. Allergic broncho-pulmonary mycosis(ABPM) is an important complication of asthma.1 The most common fungus involved isAspergillus fumigatus. Sensitisation to aspergillus antigens may occur in asthmatics withoutfull-blown picture of ABPA. The prevalence of such sensitisation reportedly occurs in20-50% of cases of bronchial asthma and the incidence of full-blown pictures of ABPA occursin about 65 of cases, although higher figures have been reported.2-11 Other organisms thatcan cause such bronchopulmonary reactions include other species of Aspergillus, Candidaalbicans, Pseudoallescheria boydii, Stemphylium sp, Helminthosporium sp, Pseudomonasaeruginosa, Curvularia lunata, Drechslera hawaiiensis, Torulopsis glabrata, Rhizopus, Penicillium,Bipolaris, and Fusarium vasinifectum.12 Cor pulmonale secondary to bronchial asthma isextremely uncommon and in fact, the presence of this complication should be an indicationthat the underlying problem is not asthma. Respiratory failure is common during acutesevere asthma.
ALLERGIC BRONCHOPULMONARY ASPERGILLOSIS (ABPA)
Allergic bronchopulmonary aspergillosis is a complex hypersensitivity reaction toAspergillus antigens because of the presence of the fungus in the bronchial tree and thedisorder characterised by bronchospasm, pulmonary infiltrates, eosinophilia, andimmunologic evidence of allergy to the antigens of Aspergillus species. Aspergillus fumigatusis the one responsible for the condition although other species may also be responsible.
The first three cases were diagnosed in 1952 in England by Hinson et al.13 Subsequentlythe entity has been reported more frequently from that country as well as from other regionsof the world like Australia,14 North America15 and parts of Asia.16 It was first reported fromIndia in 197117 and a few case series have subsequently been documented.18-27 The disease istypically seen in patients with long-standing asthma or cystic fibrosis. The incidence of thecondition in asthmatics is reported to vary from 3 to 20% of corticosteroid dependent asthmapatients28 and 6% of patients with cystic fibrosis meet the diagnostic criteria of ABPA.29
Pathophysiology
Patients with ABPA are usually atopic and have a history of bronchial asthma. The basicunderlying pathophysiologic process in ABPA is a hypersensitivity reaction to the presence
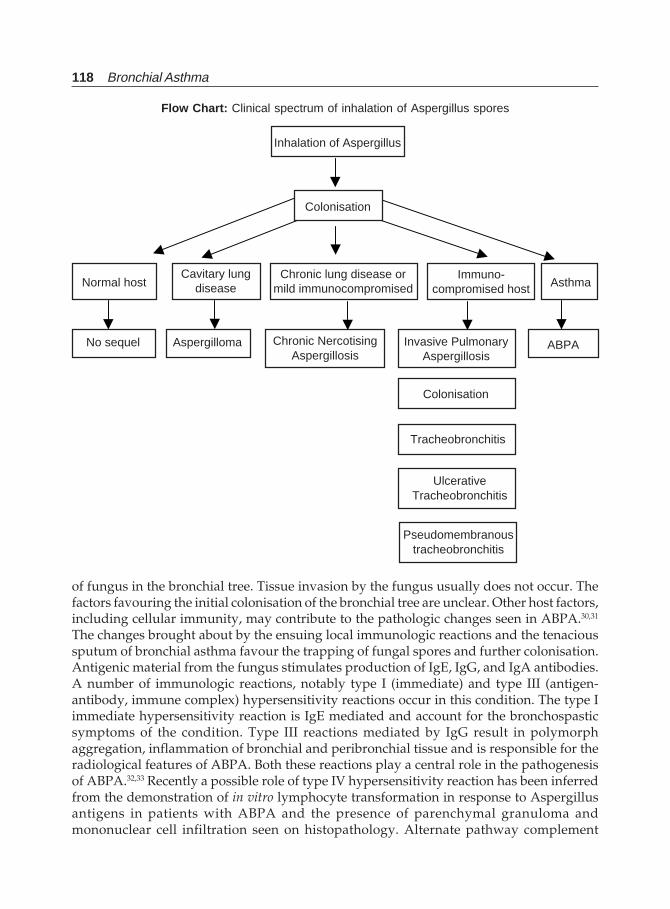
118 Bronchial Asthma
of fungus in the bronchial tree. Tissue invasion by the fungus usually does not occur. Thefactors favouring the initial colonisation of the bronchial tree are unclear. Other host factors,including cellular immunity, may contribute to the pathologic changes seen in ABPA.30,31
The changes brought about by the ensuing local immunologic reactions and the tenacioussputum of bronchial asthma favour the trapping of fungal spores and further colonisation.Antigenic material from the fungus stimulates production of IgE, IgG, and IgA antibodies.A number of immunologic reactions, notably type I (immediate) and type III (antigen-antibody, immune complex) hypersensitivity reactions occur in this condition. The type Iimmediate hypersensitivity reaction is IgE mediated and account for the bronchospasticsymptoms of the condition. Type III reactions mediated by IgG result in polymorphaggregation, inflammation of bronchial and peribronchial tissue and is responsible for theradiological features of ABPA. Both these reactions play a central role in the pathogenesisof ABPA.32,33 Recently a possible role of type IV hypersensitivity reaction has been inferredfrom the demonstration of in vitro lymphocyte transformation in response to Aspergillusantigens in patients with ABPA and the presence of parenchymal granuloma andmononuclear cell infiltration seen on histopathology. Alternate pathway complement
Inhalation of Aspergillus
Colonisation
Normal hostCavitary lung
diseaseChronic lung disease or
mild immunocompromisedImmuno-
compromised host Asthma
No sequel Aspergilloma Chronic NercotisingAspergillosis
Invasive PulmonaryAspergillosis
ABPA
Colonisation
Tracheobronchitis
UlcerativeTracheobronchitis
Flow Chart: Clinical spectrum of inhalation of Aspergillus spores
Pseudomembranoustracheobronchitis

Complications of Bronchial Asthma 119
activation may also take part in the inflammatory response of ABPA. Long-standinginvolvement of the bronchial tree leads on to bronchiectasis, fibrosis, lung contraction, andlobar shrinkage.
Lung biopsy in ABPA (done rarely as diagnosis is mainly clinical and laboratory findings)demonstrates different stages of chronic inflammatory process involving bronchial wallsand peribronchial tissues. There is no tissue invasion by the fungus and granulomas maybe seen. The most significant findings involve bronchi and bronchioles34 with bronchocentricgranulomas and mucoid impaction. Other findings include granulomatous inflammation.The cellular infiltration consists of eosinophils, monocytes, plasma cells and multinucleatedgiant cells. The bronchi are dilated and are filled with tenacious exudates containingeosinophilic material and mycelia. In long-standing cases variable degrees of interstitialand alveolar fibrosis are seen. Presence of immune complexes has been demonstrated insome cases with immunofluorescent studies. Vasculitis is very rare.
Bronchi contain tenacious mucus, fibrin, Curschmann’s spirals, Charcot-Leyden crystals,eosinophils, and mononuclear cells. Fungal hyphae may be seen in the bronchial lumenwithout tissue invasion.34
Clinical Features of ABPA
The patient is usually an atopic individual with established bronchial asthma of many years.There is no clear relationship between exposure to antigens and the onset of symptoms.The onset is insidious with nonspecific complaints like anorexia, progressive fatigue,headache, generalised aches and pains, low grade fever, and loss of weight. The underlyingasthma usually increases in frequency and severity with less degree of control with theusual anti-asthmatic medications. The increased frequency of wheezing is associated withintermittent or continuous sputum production. Rubbery golden-brown plugs of sputumproduction are characteristic of this condition and have been reported in 5 to 54% of cases.Expectoration of such plugs is associated with a dramatic improvement in symptomsparticularly wheezing.35 These plugs consist of fungal hyphae with eosinophils and mucus.Cough is universal and dyspnoea may be present in a substantial number of cases.Haemoptysis has been reported in 34 to 85% of cases. Pleuritic chest pain may be present inabout half of the patients and is usually localised to the side involved on chest X-ray. Chroniccases may present with symptoms compatible with bronchiectasis. Patients may exhibitminimal symptoms, yet demonstrate extensive pulmonary consolidation on chest radio-graphy. Wheezing and diffuse crepitations are the common findings on chest examination.
Five stages have been identified in patients with ABPA,36,37 which help to guide themanagement of the disease. It is not necessary for a patient to progress through all thesestages. The stages are:
• Stage I (Acute stage);• Stage II (Remission stage);• Stage III (Exacerbation stage);• Stage IV (Corticosteroid-dependent asthma stage); and• Stage V (Fibrotic stage).
Stage I The classic signs, symptoms, and laboratory findings present at diagnosischaracterize the acute stage. Bronchial asthma, a markedly elevated IgE levels, peripheraleosinophilia, pulmonary infiltrates, and the presence of IgE and IgG antibodies to A.fumigatuscharacterize this stage. In practice, patients are rarely identified at this stage.

120 Bronchial Asthma
Stage II The remission stage is characterised by radiological clearing, a decline in totalserum IgE levels, but not to the normal levels, eosinophilia is absent, control of respiratorysymptoms, and a discontinuation of corticosteroid therapy over a six month period withoutrecurrence of ABPA. Serum IgG antibodies to Aspergillus may be slightly elevated.Prolonged and permanent remissions may occur after treatment of the acute stage withsteroids, and maintenance therapy is not required in these patients. In some, asthma maybecome refractory to aminophylline, β-agonists and Cromolyn and inhaled steroids maybe necessary.
Stage III The exacerbation stage is the one when the patent is a known case of ABPA anddemonstrates all characteristics of the acute stage or when there is a two-fold rise in the totalserum IgE levels in association with radiological finding in the absence of other causes ofinfiltrates like bacterial or viral pneumonias. Remission after an exacerbation is induced inthese patients with corticosteroids and prolonged therapy is not necessary.
Stage IV The corticosteroid-asthma stage is present when patients require oral steroid therapyto control asthma (steroid-dependent asthma) or to prevent recurrent exacerbations. The doseof steroids needed to control asthma usually is not sufficient for preventing the exacerbationsof ABPA or the occurrence of both. Attempt to taper steroid therapy will result in worseningof symptoms and the development of pulmonary infiltrates.
Stage V The fibrotic lung disease stage is present when there are extensive fibrotic changeson chest X-ray (end-stage lung disease) with irreversible obstructive lung disease onpulmonary testing. Steroid therapy is not able to reverse these changes completely.Dyspnoea, cyanosis, crepitations, clubbing, cor pulmonale, respiratory failure and deathmay occur in some patients. The serum IgE level and eosinophil count may be low or high.A minority of patients progress to this stage.
ABPA may precede the clinical recognition of the disease for many years. Usually thereare two sets of ABPA patients based on the onset of asthma before the age of 30 who havegreater skin reactivity to other common allergens, and who show additional features ofallergic disease such as eczema and allergic rhinitis. In the other subset of patients whohave the onset of their asthma after the age of 30, generally have less cutaneous skin reactivityto common allergens and no other clinical symptom suggests allergic disease.
Radiology
The roentgenography changes in ABPA may be normal in early stages of the disease orthey may be transient or permanent.38 (Figs 8.1 to 8.5 plate 1 and 2) During acute exacerbations,the typical changes are fleeting pulmonary infiltrates that tend to be in the upper lobe andcentral in location. Transient changes, which may clear with or without steroid therapy isdue to parenchymal infiltrates, mucoid impactions or secretions in damaged bronchi. Thesetransient findings include:
i. Perihilar infiltrates simulating adenopathy;ii. Air-fluid levels from dilated central bronchi filled with fluid and debris;
iii. Massive homogenous consolidation which may be unilateral or bilateral;iv. Radiographic infiltrates;v. “Tooth-paste shadows” due to impaction of mucus in the damaged bronchi;vi. “Gloved-finger” shadows due to distally occluded bronchi filled with secretions;
and,

Complications of Bronchial Asthma 121
vii. Tram-line shadows, which are two parallel hairline shadows extending out fromthe hilum. Permanent changes include:
i. Proximal bronchiectasis;ii. Parallel line shadows which are tram-line shadows resulting from bronchi-
ectasis; andiii. Ring shadows which are dilated bronchi. Other rare findings may be cavitation,
local emphysema, contracted upper lobes, honeycomb fibrosis, total lungcollapse due to mucus impaction, and spontaneous pneumothorax. Normalchest X-ray does not exclude the diagnosis of ABPA. The chest CT may bemore sensitive in demonstrating the above changes and has replaced thenecessity of bronchography.
Laboratory Findings
Peripheral eosinophilia is common, and sputum contains eosinophils in most of the patients.Leucocytosis and raised ESR are found during acute episodes.
The serological abnormalities include a marked increase in total serum IgE and specificIgE and IgG antibodies against A.fumigatus. The levels of both total and specific serum IgElevels are high during the development of pulmonary infiltrations; the levels decline afterremission. Serial determination of total serum IgE may thus be helpful in detecting patientswith ABPA or following the course of ABPA and determining the onset of an acuteexacerbation.39 Occasionally, the serum IgE may be low.
Skin testing with potent A.fumigatus extracts demonstrates an immediate wheal and flarereaction in most cases. This reaction is frequently followed by a late onset of erythema andedema occurring at the injection site over the next 4 to 6 hours. The reaction reaches its peakby 8 hours and subsides by 24 hours. These late reactions are due to deposits of IgG, IgM,IgA, and complement components. Serologic tests using double gel diffusion method revealprecipitating antibodies in most patients of ABPA. Radio immunoassay or ELISA techniquesdetects antibodies specific for Aspergillus belonging to several immunoglobulin classes. Ithas been demonstrated that up to 25% of patients of asthma have immediate skin reactivityto A.fumigatus and 10% demonstrate positive precipitating antibodies against this. Thus,neither of these parameters is specific for ABPA.
Aspergillus can be cultured from sputum of nearly two-thirds of patients during acuteepisodes of ABPA. Repeated cultures are necessary to demonstrate the fungi.
Bronchial challenges with A.fumigatus characteristically show a dual response in patientswith ABPA. β-2 agonists can prevent the immediate reaction and the late reaction may beprevented by corticosteroids. Cromolyn sodium may prevent both types of reactions.However, bronchial challenge test is not required to confirm ABPA and may be risky.
Abnormalities of pulmonary function tests in ABPA depend upon the stage at whichthey are performed. During the earlier stages of pure bronchospasm there will be anobstructive physiologic profile, whereas during the irreversible stages of the disease withbronchiectasis and fibrosis, the tests will reflect a restrictive physiologic profile. The degreeof reversibility is much less compared to that in classic extrinsic-asthma. The diffusioncapacity is reduced in most patients with a good correlation with the duration of thedisease.

122 Bronchial Asthma
Diagnosis
There are no universally accepted criteria for the definite diagnosis of ABPA. Rosenberget al35 have suggested the following which is accepted by most investigators. Greenbergerand Patterson recently modified the diagnostic criteria for ABPA.39 They are listed in Tables8.1 and 8.2.
Table 8.1: Rosenberg criteria for diagnosis of ABPA
Primary1. Episodic bronchial obstruction2. Peripheral blood eosinophilia3. Immediate skin reactivity to Aspergillus antigens4. Precipitating antibodies against Aspergillus antigens5. Elevated serum IgE6. History of infiltrates in the chest X-ray7. Central bronchiectasis
Secondary1. Aspergillus in sputum2. History of mucus plug expectoration3. Late skin (Arthus) reactivity to Aspergillus antigen
The diagnosis of ABPA is considered likely if the first six primary criteria are present; certain if allseven are present.
Table 8.2: Modified diagnostic criteria of ABPA
1. Bronchial asthma2. Immediate skin reactivity to Aspergillus3. Serum precipitin to A.fumigatus4. Increased serum IgE and IgG to A.fumigatus5. Total serum IgE > 1000 ng/ml6. Current or previous pulmonary infiltrates7. Central bronchiectasis8. Peripheral eosinophilia (1,000 cells/μL)
Not all of these criteria need to be present to diagnose ABPA. Withholding therapy untilthe development of all clinical symptoms and evidence of bronchiectasis may lead to amissed diagnosis in a significant number of patients and to delayed treatment resulting inirreversible pulmonary damage. Therefore, ABPA may be subdivided into the followinggroups of patients with or without central bronchiectasis.40
A. Essential criteria for the diagnosis of ABPA with central bronchiectasis :Asthma,Immediate skin reactivity to Aspergillus antigenSerum IgE > 1000 ng/mlCentral bronchiectasis
B. Minimal criteria for the diagnosis of ABPA without central bronchiectasis: (labelledABPA-seropositive)
Asthma,Immediate skin reactivity to Aspergillus antigenSerum IgE > 1000 ng/mlHistory of pulmonary infiltratesElevated levels of serum IgE and IgG antibodies to A.fumigatus

Complications of Bronchial Asthma 123
From a North Indian hospital (PGIMER, Chandigarh) a total of 651 patients with clinicalsuspicion of ABPA27 were reported during a period of 8 years (January 1991 to December1998). Overall, 338 cases (52%) were positive either by sputum microscopy/culture (66 of203 patients), by skin reactivity (150 of 309 cases), or by precipitating antibodies (122 of 338cases) against Aspergillus species. However, in 89 patients, diagnosis was confirmed onthe basis of Rosenberg’s criteria. Clinical profile and laboratory findings showed that thedisease was more common among males. Poor control of asthma, constitutional symptoms,mucopurulent expectoration, increased dyspnoea or wheezing and rhonchi were the mainpresenting symptoms. Skin reactivity against aspergillin was seen in 73 (82%), precipitatingantibodies against aspergillus species were positive in 64 (72%) and sputum microscopy/culture was positive in 56 (63%) of these 89 patients. Central bronchiectasis and fleetingshadows were the most common radiological findings.
Differential Diagnosis
A number of disorders may be confused with ABPA. Tuberculosis, because of its similar upperlobe involvement on chest X-ray, may be the initial diagnosis. It is not uncommon to findpatients receiving antitubercular therapy. A high degree of suspicion is necessary to avoidthis confusion.19 History of asthma with such chest X-ray should arouse the suspicion.Repeated sputum examination will be negative for acid-fast bacilli. In that situation adiagnostic work-up for ABPA is warranted. Cystic fibrosis patients also may be confusedwith ABPA. In fact, these patients have a number of features in common with ABPAincluding isolation of the fungus from the sputum, bronchospasm, skin test reactivity andelevated serum IgE levels. However, the age of onset of cystic fibrosis, sweat chloride test,and other associated nonpulmonary features will help to distinguish the two conditions.Carcinoma of the lung, particularly, bronchoalveolar cell carcinoma, may some times beconfused with ABPA particularly in elderly individuals. The other etiologies of eosinophilicpneumonias can usually be differentiated on clinical and immunological grounds. Althoughclassically ABPA is caused by Aspergillus fumigatus, some cases can also be due to otherspecies of Aspergillus. In recent years, allergic bronchopulmonary reactions have also beenobserved due to moulds or bacteria. Stemphylum species, Helminthosporium species,Pseudomonas aeruginosa, Curvularia lunata, Candida albicans, Dreschslera hawaiiensis, andTorulopsis globata are examples which have been shown to cause such reactions similar toABPA in the lungs.
Treatment
Therapeutic approach to treat ABPA may be directed to achieve two goals: (i) to removethe source of antigenic stimulation by eliminating the fungus from the bronchial tree; and(ii) suppressing the bronchial hypersensitivity reactions and their associated localparenchymal changes. The first one was thought to be achieved by employing inhalation ofanti-fungal agents such as amphotericin B, nystatin, natamycin, and cotrimazole. Howeverthis approach has now largely been abandoned because of frequent recurrences and becauseof the need for repetitive treatments more often.
Oral corticosteroid therapy is the treatment of choice in ABPA. They act by suppressingthe allergic inflammatory reaction by suppressing the immunologic response to aspergillusantigen and decrease sputum production. Because of the later effect the bronchus becomes

124 Bronchial Asthma
less favourable for further fungal colonisation. Resolution of radiographic infiltrates andimprovements in symptoms have been observed in most patients. Prednisone therapymaintains clinical improvement in over 80% of patients by relief of bronchospasm, clearingof pulmonary infiltrates, and decreasing serum IgE level and peripheral eosinophilia.41,42
The current treatment of exacerbation of ABPA consists of daily administration ofprednisone in a dose of 0.5 mg/kg, given as a single morning dose for a period of twoweeks and then gradually decreasing the dose.43 This dose is usually sufficient to improvepulmonary lesions in two weeks, at which time the same dosage is changed to a singlealternate-day regimen. This dosage is maintained for a minimum of three months. Mostpatients, however, require more prolonged therapy to control their symptoms and minimizerelapse.43,44 If the chest X-ray shows improvement and there is a substantial reduction intotal serum IgE levels, slow reduction of prednisone, at a rate of 5 mg/day may be attempted.Treatment must be individualised depending upon the stage of ABPA, frequency ofexacerbations, and severity of asthma. Monthly serum IgE levels are to be obtained, andwhen a twofold increase is present, a chest X-ray should be obtained to rule out exacerbation.Usually there is an exacerbation of symptoms during particularly seasons due to an increasein the fungal spores in the atmosphere. This varies with geographic locations and accordinglythe steroid therapy should be reduced with caution during these months. The frequency ofchest X-ray to be taken in following a patient of ABPA is not known. It is perhaps best toobtain the X-ray every three to six months during the first year of follow-up and on a yearlybasis thereafter to avoid missing intercurrent pulmonary damage. Serum IgE levels shouldalso be monitored regularly. Pulmonary function tests should be obtained yearly.
Inhaled therapy may be beneficial, but its use is limited by the degree of obstruction andmucus plugging. Inhaled steroids are not helpful in preventing the progression of lungdamage associated with ABPA.45,46
Since there are side effects associated with long-term use of corticosteroid therapy,including an increased risk of invasive aspergillosis,47 attempts were made to use alternativedrugs. The role of itraconazole, an anti-fungal agent has been evaluated.48 When the drug isused in a dose of 200 mg twice daily for 4 months, 46% of the patients showed a significantresponse (a 50% reduction in corticosteroid dose, a decrease of at least 25% in the serum IgElevel, and a 25% improvement in exercise tolerance or pulmonary function test results, orthe resolution or absence of pulmonary infiltrates). The study concluded that patients withABPA generally benefit from concurrent itraconazole therapy without much side effectand suggested that a lower dose of 200 mg daily is equally beneficial and may be used as amaintenance therapy to sustain remission.
The disease has been seen throughout the world and has been a subject of extensivereview from across the globe.49-53
REFERENCES
1. Bredin CP, Donnely S. Period prevalence of allergic bronchopulmonary mycosis in an outpatientpopulation is over 1 percent. Eur Respir J 1991;4(Suppl 14):1715.
2. Aggarwal AK, Behera D, Malik SK, Kumar L, Talwar P. Skin hypersensitivity and precipitatingantibodies against A.fumigatus in bronchial asthma. Lung India 1989;7:67-69.
3. Behera D, Guleria R, Jindal SK, Chakrabarti A, Panigrahi D. Allergy BronchopulmonaryAspergillosis: A Retrospective study of 35 cases. Indian J Chest Dis All Sci 1994;36:173-79.
4. Malik SK, Talwar P. Allergic bronchopulmonary aspergillosis. Bull PGI 1980;14:95-98.

Complications of Bronchial Asthma 125
5. Subramanium S, Viswanathan R. Allergic bronchopulmonary aspergillosis. Ind J Chest Dis1972;14:72-77.
6. Shah JR. Allergic bronchopulmonary aspergillosis. J Ass Phys India 1971;19: 835-841.7. Sandhu RS, Mishra SK, Randhawa HS, Prakash D. Allergic bronchopulmonary aspergillosis. in
India. Scand J Respir Dis 1972;503:289-301.8. Pamra SP, Khan ZU, Sandhu RS, Ilyas M. Allergic bronchopulmonary aspergillosis. Ind J Tubercl
1972;19:61-67.9. Khan ZU, Sandhu RS, Randhwa HS et al. Allergic bronchopulmonary aspergillosis. A study of
46 cases with special reference to laboratory aspects. Scand J Respir Dis 1976;57:73-87.10. Shivpuri DN, Aggarwal MK. Studies on the allergic fungal spores of Delhi., India, metropolitan
area. J allergy 1969;44:204-13.11. Chetty A, Bhargava S, Jain RK. Allergic bronchopulmonary aspergillosis in Indian children with
bronchial asthma. Ann Allergy 1985;54:46-49.12. Backman KS, Roberts M, Patterson R. Allergic bronchopulmonary mycosis caused by Fusarium
vasinfectum. Am J Respir Crit Care Med 1995;152:1379-81.13. Hinson KFW, Moon AJ, Plummer NS. Bronchopulmonary aspergillosis. A review and a report
of eight cases. Thorax 1952;7:317-33.14. Elder JL, Smith JT. Allergic bronchopulmonary aspergillosis. Med J Australia 1967;1:231-33.15. Patterson R, Golbert F. Hypersensitivity disease of the Lung. University Michigan Med Centre
J 1968;34:8-11.16. Subramianiam S, Viswanathan R. Allergic aspergillosis. Ind J Chest Dis 1972;14:72-77.17. Shah JR. Allergic bronchopulmonary aspergillosis. J Ass Phys Ind 1971;19:835-41.18. Khan ZU, Sandhu RS, Randhawa HS, Menon MPS, Dusaj IS. Allergic bronchopulmonary
aspergillosis: A study of 46 cases with special reference to laboratory aspects. Scand J Respir Dis1976;57:73-87.
19. Behera D, Guleria R, Jindal SK, Chakrabarti A, Panigrahi D. Allergic bronchopulmonaryaspergillosis: a retrospective study of 35 cases. Ind J Chest Dis All Sci 1994;36:173-79.
20. Bedi RS. Allergic bronchopulmonary aspergillosis: Review of 20 cases. Ind J Chest Dis All Sci1994;36:181-86.
21. Shah A, Khan ZU, Chaturvedi S, Bazaz Malik G, Randhawa HS. Concomittant allergic Aspergillussinusitis and allergic bronchopulmonary aspergillosis with familial occurrence of allergicbronchopulmonary aspergillosis. Ann allergy 1990;64:507-12.
22. Shah A, Khan ZU, Sircar M, Chaturvedi S, Bazaz Malik G, Randhawa HS. Allergic aspergillussinusitis; an Indian report. Respir Med 1990;84:249-51.
23. Aggarwal AK, Behera D, Malik SK, Kumar L, Talwar P. Skin hypersensitivity and precipitatingantibodies against A.Fumigatus in bronchial asthma. Lung India 1989;7:67-69.
24. Chetty A, Bhargava S, Jain RK. Allergic bronchopulmonary aspergillosis in Indian children withbronchial asthma. Ann Allergy 1985;54:46-49.
25. Sandhu RS, Mishra SK, Randhawa HS, Prakash D. Allergic bronchopulmonary aspergillosis inIndia. Scand J Respir Dis 1972;53:289-301.
26. Chakrabarti A, Sharma SC, Chander J. Epidemiology and pathogenesis of Para nasal sinusmycoses. Otolaryngol Head Neck Surg 1992;107:745-50.
27. Chakrabarti A, Sethi S, Raman DSV, Behera D. Eight-year study of allergic bronchopulmonaryaspergillosis in an Indian teaching hospital. Mycoses 2002;45:295-99.
28. Basich JE, Graves TS, Baz MN et al. Allergic bronchopulmonary aspergillosis in corticosteroiddependent asthmatics. J Allergy Clin Immunol 1981;68:98-102.
29. Mrouch S, Spok A. Allergic bronchopulmonary aspergillosis in patients with cystic fibrosis.Chest 1994;105:32-36.
30. Knutsen AP, Slavin RG. Invitro T-cell response in patients with cystic fibrosis and allergicbronchopulmonary aspergillosis. J Lab Clin Med 1989;113:428-35.

126 Bronchial Asthma
31. Chauhan B, Santiago I, Kirschmann DA et al. The association of HLA-DR alleles and T-cellactivation with allergic bronchopulmonary aspergillosis. J Immunol 1997;159:4072-76.
32. Wang JL, Patterson R, Rosenberg M et al. Serum IgE and IgG antibody activity against Aspergillusfumigatus as a diagnostic aid in allergic bronchopulmonary aspergillosis. Am Rev Respir Dis1978;117:917-27.
33. Cockrill BA, Hales CA. Allergic bronchopulmonary aspergillosis. Ann Rev Med 1999;50:303-16.34. Bosken CH, Myers JL, Greenberger PA et al. Pathologic features of allergic bronchopulmonary
aspergillosis. Am J Surg Pathol 1988;12:216-22.35. Rosenberg M, Patterson R, Mintzer R et al. Clinical and immunological criteria for the diagnosis
of allergic bronchopulmonary aspergillosis. Ann Intern Med 1977;86:405-14.36. Patterson R, Greenberger PA, Radin RC et al. Allergic bronchopulmonary aspergillosis: staging
as an aid to management. Ann Intern Med 1982;96:286-91.37. Patterson R, Greenberger PA, Hawig JM et al. Allergic bronchopulmonary aspergillosis; natural
history and classification of early disease by serologic and roentgenographic studies. Arch InternMed 1986;146:916-18.
38. Mintzer RA, Rogers LF, Kruglik GD et al. The spectrum of radiologic findings in allergicbronchopulmonary aspergillosis. Radiology 1978;127:301-07.
39. Greenberger PA, Patterson R. Diagnosis and management of allergic bronchopulmonaryaspergillosis. Ann allergy 1986;56:444-48.
40. Greenberger PA. Immunologic aspects of lung diseases and cystic fibrosis. JAMA 1997;278:1924-30.
41. Rosenberg M, Patterson R, Robert M et al. The assessment of immunologic and clinical changesoccurring during corticosteroid therapy for allergic bronchopulmonary aspergillosis. Am J Med1978;64:599-606.
42. Wang JL, Patterson R, Roberts M et al. The management of allergic bronchopulmonaryaspergillosis. Am Rev Respir Dis 1979;120:87-92.
43. Capewell S, Chapman BJ, Alexander F et al. Corticosteroid treatment and prognosis in pulmonaryeosinophilia. Thorax 1989;44:925-29.
44. Safirstein BH, D’Souza MF, Simon g et al. Five-year follow-up of allergic bronchopulmonaryaspergillosis. Am Rev Respir Dis 1973;108:450-59.
45. British Thoracic Association. Inhaled beclamethasone dipropionate in allergic bronchopulmonaryaspergillosis: Report to the Research Committee of the British thoracic Association. Br J DisChest 1979;79:349-56.
46. Soubani AO, Chandrasekar PH. The clinical spectrum of pulmonary aspergillosis. Chest2002;121:1988-99.
47. Ganassinni A, Cazzadori A. Invasive pulmonary aspergillosis complicating allergic broncho-pulmonary aspergillosis. Respir Med 1995;89:143-45.
48. Stevens DA, Schwartz HJ, Lee JY et al. A randomised trial of itraconazole in allergicbronchopulmonary aspergillosis. N Engl J Med 2000;342:756-62.
49. Davis SF, Sarosi GA. Role of serodiagnostic tests and skin tests in the diagnosis of fungal disease.Clin Chest Med 1987;8:135.
50. Pennington JE. Aspergillus lung disease. Med Clin North Am 1980;64:475.51. Glimp RA, Bayer AS. Fungal pneumonias. Part 3. Allergic bronchopulmonary aspergillosis.
Chest 1981;80:8552. Ricketti AJ, Greenberger PA, Mintzer RA, Patterson R. Allergic bronchopulmonary aspergillosis.
Chest 1984;86:773.53. Fink JN. Allergic bronchopulmonary aspergillosis. Chest 1987;87(Suppl):81S.

Management of Bronchial Asthma 127
Management ofBronchial Asthma
9
A number of guidelines on the management of bronchial asthma, both in children andadults are developed in recent years.1-11 They include those of the British Thoracic Society,NHLB, USA, and the Global Initiative for Asthma, etc. The recommendations are based onthe same principle and basically the same. The goals of management of bronchial asthmaas recommended by these agencies are as follows:
i. To recognise asthmaii. To maintain a normal activity level including exercise.
iii. To maintain a normal or near normal (best) pulmonary function rates.iv. To prevent chronic and troublesome symptoms like coughing or breathlessness in the
night, early in the morning, or after exertion.v. To prevent recurrent exacerbations.
vi. To minimise absence from work or schoolvii. To enable normal growth to occur in children, and
viii. To use the least minimum drugs to avoid adverse reactions from medications usedfor asthma.
Since bronchial asthma is a chronic condition with acute exacerbations, treatment requiresa continuous care approach to control symptoms, to prevent exacerbations, to treatadequately such exacerbations, and to reduce chronic airway inflammation. Prevention ofexacerbation is an important principle of therapy. This includes avoidance of triggers andallergens. Round-the-clock medication may be beneficial to many patients. Children andadults, who have poor exercise tolerance, recurrent symptoms, and frequent nocturnalattacks and patients with moderate asthma will often benefit from the regular administrationand more aggressive use of antiasthma medication, particularly anti-inflammatory drugs.In contrast, patients with mild intermittent asthma with uninterrupted sleep at night, andgood exercise tolerance may need only occasional treatment for the relief of symptoms.Periodic assessment of these patients is essential to assure that their therapy is appropriate.
The treatment of asthma should also be based on the understanding of the underlyingpathophysiologic mechanisms and the objective assessment of severity of the disease. It isnow appreciated that asthma is an inflammatory disease and therapy should include anti-inflammatory agents to reduce inflammation and to relieve or prevent symptomatic airwaynarrowing. Anticipatory or early interventions in treating acute exacerbations of asthmareduce the likelihood of developing severe airway narrowing.

128 Bronchial Asthma
Thus, the integral components of asthma therapy include patient education, environ-mental control, and medication with the use of objective measures to monitor the severityof disease and the efficacy of therapy. The interrelationship of all these approaches is shownin Figure 9.1.
Basically the treatment of asthma consists of both;i. Nonpharmacologic therapy and
ii. Pharmacologic therapy.The optimal nonpharmacological treatment consists of
i. Patient and family education;ii. Avoidance of agents that induce or trigger asthma like allergens, irritants like
smoke, and reasonable attempts at reducing exposure to respiratory viruses; andiii. Immunotherapy.
The pharmacologic therapy is used to treat reversible airflow obstruction andairway hyper-responsiveness. Medications include bronchodilators and anti-inflammatory agents with some acting as both.
NONPHARMACOLOGIC MANAGEMENT
Patient and Family Education
Patient education by the treating physician is a powerful tool for helping patients to gainself-confidence to control their asthma.12,13 Since much of the day-to-day responsibility for
Fig. 9.1: General principles of management of asthma

Management of Bronchial Asthma 129
managing asthma falls on the patient and the patient’s family, encouraging activeparticipation in a partnership with the clinician can improve patient adherence to thetreatment plan and stimulate family effort to improve control of asthma.14,15 In fact a patientis his best physician since he alone can recognise well about his illness, its progression,regression, response to treatment, and imminent acute attack. It should start at the time ofdiagnosis and should be continued throughout as an integral component during continuedcare. Family participation is an essential component of this programme. Establishment of apartnership with the patient, encouraging adherence to the treatment plan, teaching aboutthe triggers (exercise, viral respiratory tract infections, allergens and irritants) and how toavoid, eliminate, or control them, explaining the patient regarding medications bothpreventive and rescue therapy, their adverse effects and educating about the adverse drugreactions are important components of this plan. Moreover teaching the patient how torecognise the severity of asthma and the appropriate time to seek medical advice duringacute exacerbations are important. Giving information alone does not alter behaviour.Written and audiovisual reinforcement of spoken language further helps patient confidence.Giving these informations along with written self management plans will help the patientwho may adjust treatment to keep themselves symptom free that reduces morbidity andhealth costs.16,17 Although now there is definite evidence of benefit from patient educationand issuing of self management plans, certain areas like who need them, and what formthey should take (number of action levels, thresholds for intervention) are poorly defined.Proper use of inhalers is very essential.18,19 Patient should demonstrate use of the metered-dose inhaler to the physician, and the technique should be reviewed at every visit. Sincehome-monitoring of PEFR is an essential component of asthma management, the patientneeds to be taught how to use a peak flow meter correctly and how to interpret it.20,21
Psychosocial issues as outlined above which increases asthma morbidity and mortalityneed to be taken care of.
Management of Allergy
Since allergy has a very significant role in the pathophysiology of asthma, interventions tocontrol this are important. There can be two ways to approach this problem: (i) environmentalcontrols; and (ii) immunotherapy.
Environmental Control
Outdoor allergens like pollens and mould are best avoided by staying indoors particularlyduring the midday and afternoons. An air conditioned environment is the best way. Variousnasal filters are available, which may be helpful to prevent penetration of allergens. However,this has not been proved to be very effective.
Indoor allergen elimination is possible by paying special attention to the following. Toavoid exposure to animal danders, the animal should be removed from the house. Removalof pets may not afford immediate relief even when followed by vigorous cleaning, sinceallergens continue to stay in the home for many months. Application of 3% tannic acid willdenature and render such substances nonallergic. If the pet cannot be kept out of the house,there should be least contact with the patient and the animal should not be allowed at all tothe bed room. Washing and bathing the pet frequently may reduce the amount of danderand dried saliva to be deposited on carpets and furnitures.22

130 Bronchial Asthma
Reducing exposure to dust mites can be achieved by the following four plans ofattack:23-25
a. By placing barriers between the patient and reservoirs of dust mite Elimination of mite exposureis possible by encasing the mattress in an airtight cover and encasing the pillow,particularly plastic mattress covers. These are not only inexpensive, but they effectivelyreduce dust mite exposure and clinical symptoms of asthma. Microporous covers arealso available which allow passage of water vapour for patient comfort while excludingmites and their allergens.
b. To kill and remove mites Regular washing of bedding and pillows by washing it at leastonce weekly. The bedding should be washed in hot water (>58°C) frequently. This killsmites and removes mites from an important exposure source. Ascaricides, tannic acid,dry heating, and liquid nitrogen have been used to kill mites, but they need furtherstudy particularly in terms of side effects to the patient and they need professionalapplication.26 It is also important to remove the dead mites once they are killed, byvacuuming otherwise they continue to be the source antigen. HEPA filtration removesair-borne mites but leaves undisturbed the major reservoir antigen in carpets, beddings,and upholstery.
c. Making the environment less hospitable for mites The patient should avoid sleeping or lyingon upholstered furnitures. The carpets and other dust collectors that are laid on concreteare to be removed. Reduction of indoor humidity to less than 50% by air conditioning ormechanical ventilation are less favourable to the growth of mites. Although not so effectivein removing live mites, regular vacuuming removes their food and shelter.
d. To remove the patient to dust-free environment Although practically inconvenient andexpensive, this is a very effective measure, and can be adopted whenever feasible whiledust busting is completed at home.To prevent growth of moulds, special attention should be paid to areas with increased
humidity. Such areas like bathrooms, kitchens, and basements require adequate ventilationand frequent cleaning using chlorine bleach. Sweat on foam pillows encourage mouldgrowth. They should be encased or changed frequently. While cleaning, the patient shouldwear a dust mask. Climate control by air conditioning is beneficial, because it allowswindows and doors to be closed and by reducing indoor humidity, discourages mould andmite growth. Humidifiers are potentially hazardous. If not cleaned regularly and properly,they facilitate the growth and aerosolise mould spores. A number of other devices areavailable for cleaning allergens from the indoor air. Two such major devices are mechanicalfilters and electrical filters.
Other indoor irritants like tobacco smoke,27 wood smoke, strong odours or sprays(perfumes, talcum powders), household cleaning substances, and fresh paints irritate theairway and trigger asthma symptoms. Therefore, these should be avoided. Exposure toozone and sulphur dioxide worsen asthma by interacting with allergens or other triggersand should be avoided as far as possible. Since occupational exposure is an important causeof bronchial asthma in adults, avoidance to such exposure is important. However, patientswith suspected occupational asthma should not be advised to cease work until the diagnosisis proven and until all methods for reducing exposure at the work place have been explored.Specialist respiratory physician, occupational physicians, and employers will all need to beinvolved in this process.

Management of Bronchial Asthma 131
Immunotherapy
Allergenic extract immunotherapy is in use since the early 1900’s in an attempt to protectagainst grass pollen. Allergy immunotherapy has been shown to reduce the symptoms ofasthma in a number of double-blind studies with a wide variety of allergens, includinghouse-dust , grass pollen , cat dander and cladosporium and alternaria.28-31 Such therapyreduces the late reaction to allergens in the lung, reduces asthma symptoms followinginjections. Long-term use also reduces bronchial hyperresponsiveness. These suggest thatallergen immunotherapy can be employed to prevent the development of allergicinflammation and perhaps the resulting bronchial hyperresponsiveness.32-37 However, theBritish Thoracic Society guidelines recommends that hyposensitisation (immunotherapy)is not indicated in the management of bronchial asthma.9
This therapy is employed only after performing a careful diagnostic study of history andskin tests to identify possible offending inhalant allergens. The history of symptoms mustcorrelate accurately with allergen exposure with confirmed IgE-mediated reactivity to oneor more suspected allergens, usually by wheal and flare skin reactivity or by serology suchas RAST.
The decision regarding immunotherapy depends upon three important considerations.(i) It must be established that there is a clinically important allergic component to asthma.(ii) In patients with a significant allergic components who are not obtaining full clinicalimprovement with standard environmental control and medication, and (iii) Failure ofmaximal environmental control measures.
Currently the methods and frequency of administration of allergenic extractimmunotherapy vary considerably. The dosage and frequency vary considerably. Theallergens used are often poorly standardised and characterised, and the methodology is ill-defined. With most forms of allergenic extracts, the initial frequency of injections is usuallyonce weekly, with doubling of the dosage at regular intervals and progression to a series ofmonthly maintenance injections, depending upon the antigen preparation employed andthe individual patient requirements. The therapy is dose-dependent and specific for theallergen employed, the higher the dose, the greater the clinical improvement. Allergic signsand symptoms may develop subsequent to injections, manifested either as local or systemicanaphylactic reactions (rare). There are no well-defined guidelines regarding the durationof therapy.
Most physicians attempt to discontinue therapy after three or four years of a successfulregimen, The National Blood, Heart, and Lung Institute, USA1 recommends that once patientachieves maintenance levels of immunotherapy, the interval between injections should beextended, with a goal of monthly injections. If the patient’s symptoms improve, treatmentis usually continued for 3-5 years, although under some circumstances more prolongedtherapy at monthly intervals may be warranted. If there is no evidence of response followingtwo allergy seasons after reaching the maintenance or highest level tolerated by the patient,immunotherapy should be discontinued. Allergy immunotherapy should be administeredonly under the direct supervision of a physician who is adequately trained.
The mechanisms for clinical improvement are unknown, but one or more immunologicalchanges may be responsible for such improvement. Among these changes are:
a. A rise in serum IgG blocking antibodies;b. Suppression of the usual seasonal rise in IgE antibodies, which follows environmental
exposure,

132 Bronchial Asthma
c. An increase in blocking IgG and IgA antibodies in respiratory secretions,d. A reduced basophil reactivity to allergens,e. Reduced lymphocyte responsiveness to allergen, andf. An increase in specific T-suppressor cell generation.
However, the problem of immunotherapy is the recognisation of the allergen. Most ofthe times, identification is not feasible and as mentioned, immunotherapy is only allergen-specific. The duration of treatment is often prolonged and costly. Moreover relapse occursin most patients after discontinuation of therapy. Therefore, immunotherapy is not widelyused as an important component in the management of bronchial asthma.
REFERENCES
1. National Asthma Education Programme. Expert Panel Report. Guidelines for the diagnosis andmanagement of asthma. National Heart, Lung, and Blood Institute, National Institute of Health,Bethesda, Maryland, USA, Publication No. 91-3042A, June 1991.
2. Guidelines for the management of asthma in adults. 1-Chronic persistent asthma. Statement bythe British Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:651-53.
3. Guidelines for the management of asthma in adults. 2-Acute severe asthma. Statement by theBritish Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:797-800.
4. Warner JO, Gotz M, Landau LI et al. Management of asthma: A consensus statement. Arch DisChild 1989;64;1065-79.
5. International Paediatric asthma Consensus Group. Asthma, a follow-up statement. Arch DisChild 1992;67:240-48.
6. International Consensus report on the diagnosis and management of asthma. Clin Exp Allergy1992;22(Suppl):1-72.
7. British thoracic Society and others. Guidelines for the management of asthma: A summary. BMJ1993;9:287-92.
8. The British Guidelines on Asthma Management. 1995 Review and Position Statement. Thorax1997;52(Suppl 1): S2-S8.
9. British thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, national asthma Campaign et al. Guidelines on the management ofasthma. Thorax 1993;48:S1-S24.
10. British thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, national asthma Campaign et al. Summary charts. BMJ1993;306:776-82.
11. Global Initiative for Asthma. A practical guide for public health officials and health careprofessionals. US Department of Health and human services. NIH Publication No. 96-3659A,December 1995.
12. Brewis RAL. Patient education, self-management plans and peak flow measurements. RespirMed 1991;85:457.
13. Feldman CH, Clark NM, Evans D. The role of health education in medical management in asthma.Clin Rev Allergy 1987;5:195-205.
14. Mellians RB. Patient education is key to successful management of asthma. J Rev Respir Dis1989;Suppl:S47-S52.
15. Clark NC. Asthma self-management education: Research and implications for clinical practice.Chest 1989;95:1110-13.
16. D’Souza W, Crane J, Burgess C, et al. Community-based asthma care; trial of a “credit card”asthma self-management plan. Eur Respir J 1994;7:1260-65.

Management of Bronchial Asthma 133
17. Iganacio-Gracia JM, Gonzalez-Santos P. Asthma self management education programme byhome monitoring of peak expiratory flow. Am J Respir Crit Care Med 1995;151:353-59.
18. Shim C, Williams MH. The adequacy of inhalation of aerosol from canister nebullisers. Am JMed 1980;69:891-94.
19. Newman SP, Pavia D, Clarke SW. Simple instructions for using pressurised aerosol broncho-dilators. J R Soc Med 1980;73:776-79.
20. Vathenen AS, Cooke NJ. Home peak flow meters. Br Med J 1991;302:738.21. Mendoza GR. Peak flow monitoring. J Asthma 1991;28:161.22. Glinert R, Wilson P, Wedner HJ. Fel; D1 is markedly reduced following sequential washing of
cats. J Allergy Clin Immunol 1990;85:225.23. Wallshaw MJ, Evans CC. allergen avoidance in house dust mite sensitive adult asthma. Q J Med
1986;58:199-215.24. Ehnert B, Lau-Schadendorf S, Weber A, Buettner P, Sehou C, Wahn U. Reducing domestic
exposure to dust mite allergen reduces bronchial hyper-reactivity in sensitive children withasthma. J Allergy Clin Immunol 1993;90:135-38.
25. Murray AB, Fergusson AC. Dust free bedroom in the treatment of asthmatic children with housedust mite allergy: A controlled trial. Paediatrics 1983;91:418-22.
26. Colloft MJ, Ayres J, Carswell F, et al. The control of dust mites and domestic pets: A positionpaper. Clin Exp Allergy 1992;22(Suppl 2):1-28.
27. Andrae S, Axelson O, Bjorksten B, Fredriksson M, Kiellman NM. Symptoms of bronchial hyperreactivity and asthma in relation to environmental factors. Arch Dis Child 1988;63:474-78.
28. Reid MJ, Moss RB, Hsu YP. Seasonal asthma in Northern California; allergy causes and efficacyof immunotherapy. J Allergy Clin Immunol 1986;78:590-600.
29. Aas K. Controlled trial of hyposensitisation to house dust. Acta Paediatrc Scand 1971;60:264-68.30. Ohman JL Jr, Findlay SR, Leiterman KM. Immunotherapy in cat-induced asthma: Double-blind
trial with evaluation of in vivo and invitro responses. J Allergy Clin Immunol 1984;74:230-39.31. Horst M, Hejjaoui A, Horst V, Michel FB, Bousquet J. Double-blind, placebo-controlled rush
immunotherapy with a standardised alternaria extract. J Allergy Clin Immunol 1990;85:460-72.32. Lilja G, Sundin B, Graff-Lonnevig v, et al. Immunotherapy wit cat and dog dander extracts IV.
Effect of 2 years treatment. J Allergy Clin Immunol 1989;83:37-44.33. Bousquet J, Maasch HJ, Hejjaoui A et al. Double-blind, Placebo-controlled immunotherapy with
mixed grass-pollen allergoids. III. Efficacy and safety of unfractionated and high-molecular-weight preparations in rhinoconjunctivitis and asthma. J Allergy Clin Immunol 1989;84:546-56.
34. Boulet LP, Cartier A, Thomson NC et al. Asthma and increases in nonallergic bronchialresponsiveness from seasonal exposure. J Allergy Clin Immunol 1983;71:399-406.
35. Van Bever HP, Stevens WJ. Suppression of late asthmatic reaction by hyposensitisation inasthmatic children allergic to house dust mites (Dermatophagoides pteronyssiunus). Clin ExptAllergy 1989;19:399.
36. Chapman MD. Use of nonstimulatory peptides: A new strategy for immunotherapy? J AllergyClin Immunol 1991;88:300.
37. Hoshino K, Hawasaki A, Mizushima Y, Yano S. Effect of antiallergic agents and bronchialhypersensitivity in short-term bronchial asthma. Chest 1991;100:57.

134 Bronchial Asthma
PharmacologicManagement of Asthma
10
The drugs used for the treatment of bronchial asthma are classified as:1. Bronchodilators
• β-adrenergic agonists• Anticholinergics• Methylxanthines (now can be classified as anti-inflammatory)
2. Anti-inflammatory agents• Corticosteroids• Cromolyn sodium or cromolyn-like compounds• Methylxanthines• Leukotriene antagonists• Miscellaneous compounds including antihistamines.
METHYLXANTHINES
Theophylline, the principal methylxanthine used in asthma therapy over the past six decadesand the most widely prescribed anti-asthma treatment worldwide, is a dimethylxanthinesimilar in structure to the common dietary xanthines, caffeine and theobromine.1-3 Othersubstituted xanthines have also bronchodilator property and include: Dyphylline(dihydroxypropyl theophylline), Etophylline (β-hydroxyethyl theophylline), Proxyphylline(β-hydroxypropyl theophylline), and Enprophylline (3-propylxanthine).
Many “salts” of theophylline preparations are commonly marketed and have been inuse over many years. Aminophylline, the ethylenediamine salt is perhaps the commonestcompound used in many countries. Other commonly salts include formulations with calciumsalicylate, sodium glycinate, and choline (oxtryphylline).
Mechanism of action of theophylline remains unclear despite the long history andwidespread use of the drug.4 Various mechanisms proposed for the molecular mechanismof action has been proposed and are shown in Table 10.1.
Phosphodiesterase Inhibition
Earlier it was believed that theophylline acts as an anti-asthma drug as it relaxes bronchialsmooth muscle. Although the exact mechanism of such relaxation was not known, in vitro,theophylline inhibits phosphodiesterase (PDE) which breaks down cyclic nucleotides inthe cell, that results in delayed degradation of cAMP and cGMP. Several families of PDE

Pharmacologic Management of Asthma 135
are now recognised,5 of which PDE III is predominant in airway smooth muscle relaxationand PDE IV is important in inflammatory cells.5-8 Theophylline is a nonselective PDEinhibitor. Such inhibition occurs at concentrations ten-fold higher than those usually attainedclinically. Total PDE activity in human lung extracts is inhibited by only 5-20% at therapeuticconcentrations of theophylline.9,10 However, this modest inhibition may be sufficient to causea substantial increase in intracellular cyclic nucleotide levels in the presence of endogenousactivators of adenylyl cyclase.11 Inhibition of PDE could also lead to synergistic interactionwith β-agonists. Since there is some evidence that PDE levels may be higher in asthmaticsthan normal individuals, theophylline may have a greater than expected inhibitory effectson PDE in asthmatic airways than in normal airways.12 Bronchodilating effects oftheophylline appear to closely parallel the serum concentrations. Although a steady-stateserum concentration between 10-20 μg/ml gives optimal effect, a more conservativeapproach would be to aim for levels between 5-15 mg/ml. Since there is a linear relationshipbetween log serum concentration and bronchodilator effect within this range, the dose shouldbe increased if symptoms persist and the patient is at the lower end of the serumconcentration range.
Adenosine Receptor Antagonism
Adenosine causes bronchoconstriction in bronchial asthma both in vitro and clinically whengiven by inhalation.13,14 This involves the release of histamine and leukotrienes from airwaymast cells. This bronchoconstricting effect of adenosine is prevented by theophylline.15 Thisshows that theophylline is capable of antagonising the effects of adenosine at therapeuticconcentrations. Theophylline is a potent inhibitor of adenosine receptors (both A1 and A2receptors) at therapeutic concentrations and this may be the basis of its bronchodilatoreffect.16 Since the potent bronchodilators enprophylline doxofylline, do not have action againstadenosine receptors, adenosine antagonism may not be the exact cause of bronchodilatation.However, inhibition of different adenosine receptor types and subtypes may be importantfor this differential action.
Increased Catecholamine Release
Intravenous theophylline increases the secretion of adrenaline from the adrenal medulla.17,18
Although the increase is small, it may be important.
Anti-inflammatory Effect
Recent evidence shows that theophylline may also possess some anti-inflammatory activity.19
Theophylline reduces both bronchial hyper-reactivity20,21 and the inflammatory response.The anti-inflammatory effect has been shown both in vitro and in vivo studies. The effects
Table 10.1: Mechanism of action of theophylline
Phosphodiesterase inhibitionAdenosine receptor antagonistIncrease in circulating adrenalineMediator antagonism (anti-inflammatory effect)Inhibition of calcium ion fluxEffect on respiratory muscles

136 Bronchial Asthma
include decreased mediator release from mast cells, decreased release of reactive oxygenspecies from macrophages, decreased cytokine release from monocytes, decreased basicprotein release by eosinophils, decreased proliferation of T-lymphocytes, decreased releaseof ROS (reactive oxygen species), and inhibition of late response to allergens, increasedCD8
+ cells in peripheral blood and decreased T-lymphocytes in airways in asthmatic patients.
Theophylline inhibits plasma exudation in guinea pigs.20 It also demonstrates immuno-modulatory effects in vivo because of the inhibitory effects on T-lymphocytes. The anti-inflammatory effect is seen in much lower concentrations than its bronchodilatoryconcentrations.
Effect on Respiratory Muscles
In addition to bronchodilatation, it improves respiratory function by increasing the strengthand reducing the fatigue of respiratory muscles particularly diaphragm.21,22 A number ofstudies suggest that during various contractile maneuvers theophylline increases Pdi/Edi,where Pdi denotes intrathoracic pressure swings across the diaphragm which reflects muscleforce and Edi is the electromyographic recordings taken at the skin surface opposite thediaphragm insertion to measure the nervous input to the muscle. The ratio represents force/unit of input.
Inhibition of Calcium Ion Flux and Other Extrapulmonary Effects
Some evidence suggest that theophylline may interfere with calcium mobilisation in airwaysmooth muscle. Although it has no effect on entry of calcium ions through voltage-dependentchannels, it may influence calcium entry via receptor-operated channels. Other possibleeffects may be release of calcium from intracellular stores or may have some effect ionphosphatidylinositol turnover which is linked to release of calcium ion from intracellularstores. Theophylline may increase calcium uptake into the intracellular stores also.23,24
The drugs also increase mucociliary clearance. Other pharmacological effects oftheophylline include a transient diuretic effect, stimulation of the central nervous system,cerebral vascular constriction, gastric acid secretion, and inhibition of uterine contractions.These effects are of little clinical importance when appropriate doses are used for thetreatment of asthma (or apnea of prematurity). Theophylline also exert activity on cardiacventricular contractility.
Theophylline is rapidly and completely absorbed from the gastrointestinal tract when itis administered in the form of solutions and tablets. Once absorbed, it is distributed rapidlythrough extracellular body fluid, and to some extent into intracellular space. Theophyllineis then eliminated through multiple parallel pathways that include demethylation andoxidation. Approximately 90% of orally administered theophylline is metabolised in liver.The drug’s elimination is reduced by such factors as liver disease, congestive heart failure,sustained high fever, and with drugs like cimetidine, troleandomycin, and erythromycin.Therefore, the dose of the drug should be reduced in these circumstances. Cigarette andmarijuana smoking, phenobarbital, phenytoin, and intravenous isoproterenol increases theelimination of the drug. Major changes in diet also have a potential effect with 25% increasesin clearance associated with a low carbohydrate, high protein diet and about a 25% meandecrease in clearance associated with a high carbohydrate low protein diet. The drug is alsoeliminated rapidly from the body by some individuals, especially children. In obese individuals,

Pharmacologic Management of Asthma 137
with greater than 120% ideal body weight, initial theophylline should be calculated on thebasis of ideal rather than actual body weight to avoid overdosing.
Theophylline has long been marketed in a wide variety of formulations. The traditionalpreparation for oral and parenteral use has been theophylline with ethylenediamine knownas aminophylline. Suppository and rectal solutions are also available. Fixed dose combina-tions of theophylline with ephedrine that were the most frequently used formulationspreviously were associated with synergistic toxicity while providing a small additive effect.They are now not been used. During the past decade, newer formulations have beendeveloped with slower controlled release preparations because of unacceptable fluctuationsduring the use of plain tablets. Both twice-dosing and once-a day dosing are now available.Although once-a-day dosing may be satisfactory in adults who eliminate the drug slowly,substantial peak-to-trough differences in serum concentrations are found in individualswho eliminate the drug rapidly. Furthermore, intestinal transit time in some patients maybe so rapid that sustained-release preparations designed to release drugs especially slowlywith long absorption half-lives, will pass out of the gut before absorption is complete. Theselonger acting preparations may also be affected by the presence of food in the gut or by thefat content. In some cases, the rate of drug release is greatly accelerated, and in other casesdrug absorption is impaired. Other products are relatively unaffected by food administration.One should be familiar with these properties of the product selected.
Theophylline is used for the treatment of both acute and chronic asthma. In chronicasthma, the usual starting dose is 10 mg/kg/day up to 800 mg maximum dose. In childrenthe starting dose is 10 mg/kg/day; with usual maximum is as;
1 year or more 16 mg/kg/day< 1 year 0.2(age in weeks) + 5 = mg/kg/day
For the management of acute asthma, the drug may have an additive effect on othermedications. Intravenous aminophylline has been used in the management of acute severeasthma for over 50 years. However, its has been questioned recently in view of the risk ofadverse effects compared with nebulised β-agonists. Intravenous aminophylline is lesseffective than nebulised β-agonists.25 Thus some authors recommend that the drug shouldbe reserved for those patients who fail to respond to β-agonists. On the other hand, thereare evidences to suggest that use of aminophylline in the emergency room reducessubsequent admissions to hospitals.26 There is no added advantage if aminophylline is usedin addition to β-agonists. Use of intravenous aminophylline may increase the death rates.27
However, this is a drug which is cheap and still used as an important drug in many hospitalsin the management of acute severe asthma. Whenever a decision is taken to useaminophylline intravenously, it should be given as a slow intravenous infusion with carefulmonitoring and a plasma theophylline concentration should be monitored, if possible, priorto infusion. The loading dose is aimed for a target serum concentration no higher than themid point of the 10 to 20 μg/ml that is determined by multiplying the desired change inserum concentration by an average volume distribution of about 0.5 L/kg. In other words,each μg/ml increase in serum concentration requires 0.5 mg/kg of a loading dose. A repeatserum concentration 30 minutes after the loading infusion determines the need for anadditional loading dose and provides a baseline for monitoring change during a subsequentmaintenance infusion. A conservative maintenance infusion based on mean clearance andtargeting a steady state serum concentration of 10 μg/ml is maintained with as follows:

138 Bronchial Asthma
Infants under age 1 0.008 × age in weeks + 0.22 mg/kg/hrChildren (1-9 years) 0.8 mg/kg/hrChildren (9-16 years) 0.6 mg/kg/hrAdults 0.4 mg/kg/hr
The adult dose should be decreased by one half for those with heart failure or liver disease.Subsequent infusion is adjusted according to the serum concentration.
The other important use of theophylline is its use as maintenance therapy for chronicasthma. To attend an optimal dosage one should proceed with patience. Rapid attainmentof therapeutic concentrations is associated with a high degree of minor complaints whichmay discourage the patient from continuing therapy. Therefore, the aim should be to attendsuch optimum concentration over a period of 1-2 weeks. Because of the variability in therates of elimination, the final doses requirements are highly variable. While average dosesare higher on a weight-adjusted basis for children than adults, considerable variability isobserved at all ages. According to these principles the initiation of therapy should be atdoses of 400 mg/day or 16 mg/kg/day, whichever is less. Since this dosage is low, adequatecontrol of symptoms is not expected and for that period, another drug should be used forcontrol of symptoms. The dose is then to be increased every three days to 600 mg/day forthose more than 45 mg/kg or if the patient weighs less than 45 mg/kg, either 600 mg/kg or16-20 mg/kg, whichever is less. After the next three days, the dose is to be increased to800 mg/kg for those more than 45 kg in weight and if less than 45 kg in weight, the doseshould be 800 mg/day or 18-24 mg/kg/day, whichever is less. The dose is then adjustedaccording to the serum concentration which should be measured about 4 hours after a dosewhen none have been missed or added for three days.
Theophylline has little or no effect on bronchomotor tone in normal airways, but it reversesbronchoconstriction in asthmatics. The routine of theophylline in chronic stable asthma hasrecently been questioned.28-30 In various guidelines of management of bronchial asthma(discussed subsequently), theophylline is used as an additional bronchodilator if asthmaremains difficult to control after moderate to high dose inhaled steroids. The recent use ofsalmeterol and formoterol may still threaten the position of theophylline. Nonetheless, thedrug is cheap and is in use for several decades in many developing countries as a main stayof treatment.
Monitoring serum concentrations is an important part of acute or chronic care of asthma.The frequency of measurements depend upon the specific clinical situation. Monitoring isrequired in those who fail to exhibit the expected clinical effect while receiving an appropriatetherapeutic regimen and in patients who develop an adverse effect to an usual dose. It isuseful to monitor serum theophylline concentrations when a patient begins his therapyand then at 6-12 months, as long as no adverse effects are observed. The therapeutic rangeof theophylline was based on measurements of acute bronchodilatation in response to theacute administration of theophylline.31 However, it is possible that the nonbronchodilatoreffects of theophylline may be exerted at lower plasma concentrations. Since side effectsare also related to plasma concentration, these may be markedly reduced by aiming forplasma concentrations of 5-15 mg/l (28-55μM), rather than the previously recommendeddoses of 10-20 mg/l (55-110μM). This level should be in the steady state (at least 48 hours inthe same dose). Improvements in slow-release preparations, including that of once-a-dayproducts, have further improved the problem of fluctuations in plasma concentrations.

Pharmacologic Management of Asthma 139
Side Effects
The signs and symptoms of theophylline intoxication involve many organ systems. Thecommonest toxicity are caffeine-like side effects including minor degrees of central nervousstimulation, headache, restlessness and nausea and vomiting or a queasiness of the stomachoccur frequently after a loading dose and have no direct relationship to the serumconcentration. Most patients rapidly acquire tolerance of these side effects when therapy ismaintained and avoid them when the dose is gradually built up. As serum concentrationsexceed 20 μg/ml, there is an associated progressively increasing risk of more serious sideeffects including seizures and death, most commonly when the level exceeds 40 μg/ml.The seizures may not be preceded by other central nervous system symptoms.Cardiopulmonary effects include tachycardia, and arrhythmias even at serum concentrationsconsidered to be therapeutic. Multifocal atrial tachycardia may herald sudden cardiacdeath.32 Other adverse effects include stimulation of respiratory center causing tachypnoea,diuresis, relaxation of the detrusor muscle causing difficulty in urination in older men withprostatism, and important metabolic effects such as hyperglycaemia and hypokalaemia.The effect of theophylline on behaviour and learning of children have received attentionrecently. Because the drug stimulates the central nervous system, it may produce behaviourdisturbances in children. Of more serious consequence are the reports that its use is associatedwith impairment of learning,33-35 although a carefully designed study could not confirmthis.36
Some of the side effects of theophylline like central stimulation, gastric secretion, diuresis,and arrhythmias may be due to adenosin receptor antagonism and may, therefore, beavoided by drugs such as enprofylline, which has no significant adenosine antagonism atbronchodilator doses.37 However, the commonest side effects of theophylline like nausea,vomiting and headache are also seen with enprofylline.38
Prevention of toxicity is important by monitoring the serum concentrations and by aimingfor lower plasma concentrations as indicated earlier to some extent, side effects may bereduced by gradually increasing the dose until therapeutic concentrations are achieved .39-41
Acute accidental or suicidal overdoses of theophylline are better tolerated than sustainedhigh levels encountered due to uncontrolled therapy. Since theophylline-induced seizuresare more dangerous including brain damage and death, an aggressive approach to thetreatment of an overdose is necessary. Initial therapy with ipecac or other measures toinduce vomiting removes remaining aminophylline in the stomach. Activated charcoal stopsfurther absorption, and simultaneous administration of a cathartic such as sodium sulphateincreases the transit time of charcoal and any remaining undisclosed drug. Repeated dosesof activated charcoal increases the rate of elimination of theophylline already absorbed bytwo folds, possibly due to the result of a gastrointestinal dialysis. Extracorporeal charcoalhaemoperfusion allows more rapid clearance. There are many factors which affect serumtheophylline concentrations. These factors and actions to be taken are shown in Table 10.2.
βββββ-ADRENERGIC AGONISTS
Normal βββββ-adrenergic Receptor Physiology
The autonomic nervous system is responsible for regulating the airway tone through therelease of neurotransmitters that activate specific autonomic receptors. The autonomic
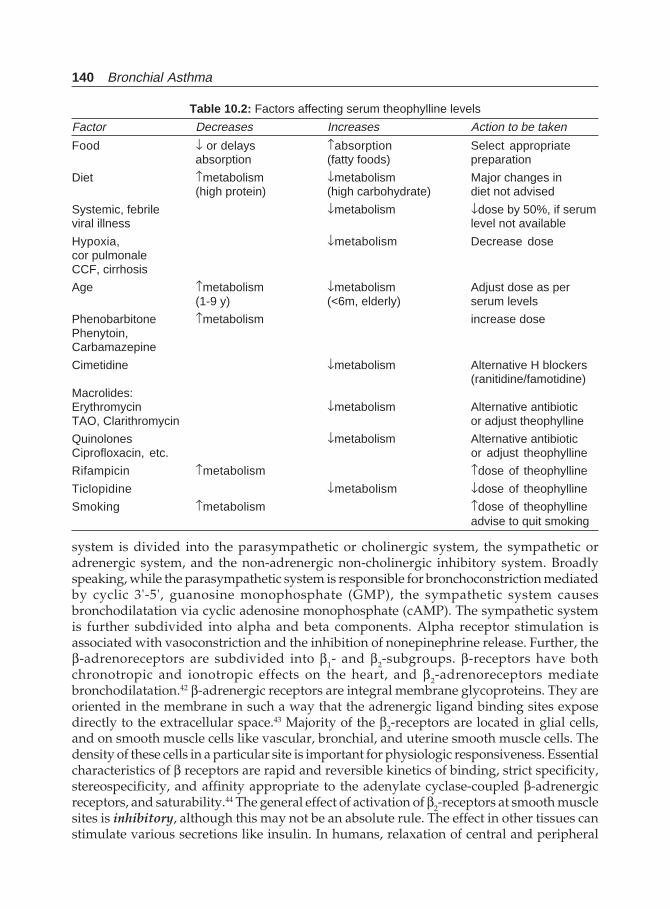
140 Bronchial Asthma
system is divided into the parasympathetic or cholinergic system, the sympathetic oradrenergic system, and the non-adrenergic non-cholinergic inhibitory system. Broadlyspeaking, while the parasympathetic system is responsible for bronchoconstriction mediatedby cyclic 3'-5', guanosine monophosphate (GMP), the sympathetic system causesbronchodilatation via cyclic adenosine monophosphate (cAMP). The sympathetic systemis further subdivided into alpha and beta components. Alpha receptor stimulation isassociated with vasoconstriction and the inhibition of nonepinephrine release. Further, theβ-adrenoreceptors are subdivided into β1- and β2-subgroups. β-receptors have bothchronotropic and ionotropic effects on the heart, and β2-adrenoreceptors mediatebronchodilatation.42 β-adrenergic receptors are integral membrane glycoproteins. They areoriented in the membrane in such a way that the adrenergic ligand binding sites exposedirectly to the extracellular space.43 Majority of the β2-receptors are located in glial cells,and on smooth muscle cells like vascular, bronchial, and uterine smooth muscle cells. Thedensity of these cells in a particular site is important for physiologic responsiveness. Essentialcharacteristics of β receptors are rapid and reversible kinetics of binding, strict specificity,stereospecificity, and affinity appropriate to the adenylate cyclase-coupled β-adrenergicreceptors, and saturability.44 The general effect of activation of β2-receptors at smooth musclesites is inhibitory, although this may not be an absolute rule. The effect in other tissues canstimulate various secretions like insulin. In humans, relaxation of central and peripheral
Table 10.2: Factors affecting serum theophylline levels
Factor Decreases Increases Action to be taken
Food ↓ or delays ↑absorption Select appropriateabsorption (fatty foods) preparation
Diet ↑metabolism ↓metabolism Major changes in(high protein) (high carbohydrate) diet not advised
Systemic, febrile ↓metabolism ↓dose by 50%, if serumviral illness level not available
Hypoxia, ↓metabolism Decrease dosecor pulmonaleCCF, cirrhosis
Age ↑metabolism ↓metabolism Adjust dose as per(1-9 y) (<6m, elderly) serum levels
Phenobarbitone ↑metabolism increase dosePhenytoin,Carbamazepine
Cimetidine ↓metabolism Alternative H blockers(ranitidine/famotidine)
Macrolides:Erythromycin ↓metabolism Alternative antibioticTAO, Clarithromycin or adjust theophylline
Quinolones ↓metabolism Alternative antibioticCiprofloxacin, etc. or adjust theophylline
Rifampicin ↑metabolism ↑dose of theophylline
Ticlopidine ↓metabolism ↓dose of theophylline
Smoking ↑metabolism ↑dose of theophyllineadvise to quit smoking

Pharmacologic Management of Asthma 141
airways is mediated entirely by β2 receptors. Increased bronchial reactivity could resultfrom a decreased β-adrenergic or nonadrenergic inhibitory activity. Most probably it iscaused by decreased responsiveness of β-adrenergic receptors.45
β-receptors mainly work through the enzyme adenylate cyclase activation and cyclicAMP formation. The enzyme adenylate cyclase is stimulated by catecholamines in virtuallyall tissues in which β-receptors can be found. The principal type of receptor coupling toadenylate cyclase by β2-adrenergic receptors is referred to as a “stoichiometric coupling”,in which the biological response elicited is directly proportional to the percentage of receptoroccupied (occupancy theory). A reduction in receptor number will alter the sensitivity ofthe tissue to catecholamines. The tissue will require more drug to provide the same degreeof receptor occupation as the receptor concentration is lowered. There is a general mechanismof hormone to receptor to adenylate cyclase interaction. Mammalian cells controlled byβ-adrenergic hormones contain plasma membrane-bound adenylate cyclase and specifichormone receptors. These systems have a protein(s) which couples their receptors to theadenylate cyclase catalytic protein. This coupling protein contains a guanine nucleotidebinding site, and is labelled as Ns. The interaction of the hormone, receptor, and guaninenucleotide binding protein with the adenylate cyclase catalytic unit and in the presence ofmagnesium ions, results in the formation of cAMP from adenosine triphosphate (ATP).46,47
Further, cAMP functions as a second messenger of catecholamine or hormonal action bymodifying enzyme activities and permeability barriers. This is possible by the activation ofprotein kinases. These enzymes transfer terminal phosphate groups from ATP to aminoacid residues of certain proteins.44 In bronchial smooth muscle, the kinases cause a reductionof calcium dependent coupling of actin and myosine and this results in smooth musclerelaxation. Thus, β2 agonists increase intracellular cAMP concentrations which are essentialin the relaxation response.
Recently, it has been become clear that β2 agonists may cause bronchodilatation, at least inpart, via maxi-K channels in airway smooth muscle cells which are directly linked torelaxation.48-51 Maxi-K channels are opened by low concentrations of β2 agonists which arelikely to be therapeutically relevant. There is now evidence that β receptors may be coupleddirectly to maxi-K channels via the alpha-subunit of Gs,
48 and may therefore, inducerelaxation without any increase in cAMP.
The sympathomimetic bronchodilators are the keystone of therapy of bronchialasthma.52-62 Β-2 agonists are often the first and most commonly used drugs for the treatmentof bronchial asthma. Modern bronchodilator therapy started in 1900 when the use of adrenalextract to treat asthma was described.42 In 1924, ephedrine was introduced into westernmedicine, although its parent plant, ma huang, was known to the Chinese for more than5000 years. These drugs had both alpha and beta-adrenoceptor activity which were describedby Ahlquist in 1948.42 The older sympathomimetic agents ephedrine, epinephrine, andisoproterenol have been generally replaced by the newer, longer acting, more β-2 specificbronchodilators. Nonetheless, they are still important antiasthma medications. Recognitionof the pharmacologic differences between β1, β2, and α receptors has led to the developmentof adrenergic agonists that can preferably act on β2 receptors of bronchial smooth musclewith little direct stimulation of the β1 receptors of the myocardium. At present, β2 agonistsor sympathomimetics, are the preferred and most effective bronchodilators available forthe treatment of bronchial asthma and are often the first and most important drug to beused worldover.

142 Bronchial Asthma
Biochemistry
The β-adrenoceptor agonists are sympathomimetic amines whose parent compound isβ-phenylethylamine. They consist of a benzene ring attached to an amine group by twocarbon atoms. The distinctive features of different β-agonists depend on the basic structure,and on the substitution on the amine group in particular. Increasing the size of the terminalamino group substituent protects the drug against degradation by monoamine oxidase,and further increases the duration of bronchodilatation.43 Modification of thephenylethylamine nucleus has helped to increase β2-specificity and duration of action.
Catecholamines refer generically to all compounds containing a catechol nucleus (benzenewith two adjacent hydroxyl groups) and an amine group. They have a relatively short half-life, because they are subject to removal by active uptake mechanisms and to rapidmetabolism by catechol-o-methyltransferase (COMT) and monoamine oxidase (MAO). Theyare orally inactive because of their inactivation by gastrointestinal sulphatases. Bymodification of the 3,4-hydroxyl groups on the benzene ring, which are the sites of actionof COMT, prolonged bronchodilating action and oral administration is possible. The variousβ2-agonists are shown in Table 10.3.
Mechanisms of Action
Sympathomimetic amines have six general types of action: peripheral excitatory, peripheralinhibitory, cardiac excitatory, metabolic, endocrine, and central nervous system actions.β2-agonists cause a direct relaxation of the pre-constricted or spontaneously contracting humanbronchial smooth muscle. Their bronchodilator action is evident in normal persons, in patientswith chronic obstructive pulmonary disease, and asthma. They cause a marked reduction innonspecific bronchial reactivity to stimuli such as histamine, methacholine, or exercise. Themode of bronchodilatation seems to be due to a decrease in catecholamine-stimulated adenylate
Table 10.3: Adrenergic bronchodilators
Classification Receptor activity Availability Duration of action(hours)
Ephedrine α, β1, β2. Oral 2-3Catecholamines
Epinephrine α, β1, β2 Injection, inhaler 1-2Isoproterenol β1, β2 Oral, inhaler, Injection 1-2Isoetharine (β1), β2 Inhaler 3
ResorcinolsMetaproterenol β2 Oral, inhaler 3-5Terbutaline β2 Oral, inhaler, Injection 4-6Fenoterol β2 Inhaler 4-6
SaligeninSalbutamol β2 Oral, inhaler, Injection 4-6
MiscellaneousBitolterol β2 Inhaler 6-8Pirbuterol β2 Inhaler 4-6Procaterol β2 Oral, inhaler 6-8
Long acting drugsFormoterol β2 Inhaler > 12Salmeterol β2 Inhaler > 12

Pharmacologic Management of Asthma 143
cyclase activity. The final effect is an increase in cellular cyclic adenosine monophosphate.This effect derives from and is mediated through a plasma membrane-associated β-adrenergicreceptor: the guanine nucleotide regulatory protein, which in turn activates adenylate cyclaseand leads to generation of cAMP. β2-adrenoceptor agonists vary in their selectivity for β2-adrenoreceptors, but none is β2-specific. They all stimulate β- receptors to a lesser but dose-dependent extent. The duration of action is dose dependent, but to a limited extent.
Since the human airway smooth muscle cells express β2-receptors from the trachea to theterminal bronchioles,63,64 these drugs as functional antagonists can prevent and reverse theeffects of all substances,65 including leukotrienes, acetylcholine, bradykinin, prostaglandins,histamine and endothelins. Because of the widespread presence of β-receptors, the β2- agonistsmay affect many cells like stabilisation of mast cells,66 which may be the cause of effectivenessof these agents in blocking the bronchoconstricting effects of allergens, exercise, and fog.Further, β2-agonists inhibit cholinergic neurotransmission in the human airway, which canresult in reduced cholinergic-reflex bronchoconstriction. The other mechanisms of action ofβ-agonists, although not proved conclusively, include, inhibition of mediator release,modulation of neural pathways, reduction of microvascular leak, and increased mucociliaryclearance.67
Long acting β2-agonists, salmeterol xinafoate and formoterol fumarate, are currentlyavailable in many countries.68-70 They are available in inhaled forms. While salmeterol actslonger but is a partial agonist, formoterol is a nearly full agonist.71 Both provide effectivebronchodilatation over a 12-hours period and thus, they are more useful for patients whohave nocturnal asthma.72,73 Because these drugs have no anti-inflammatory effect, they shouldalways be used with an inhaled glucocorticoid. Both drugs also protect against airwayschallenge with methacholine for a period of 12 hours.72,74 International guidelines haverecommended both drugs to be added in the treatment of bronchial asthma. Several studieshave demonstrated the superiority of salmeterol and formoterol to regular treatment witheither salbutamol or placebo.75-77 Both these drugs differ pharmacologically, but there is nodifference in the efficacy between the two drugs in any severity of bronchialasthma,78-80 although formoterol is more potent than salmeterol in vitro, with a faster onset buta shorter duration of action,81 but with similar bronchodilator action at 12h. Relative potencyestimates show that 50 mg salmeterol corresponds to 9 mg formoterol.82
Optimal Pharmacological Profile of βββββ-adrenoceptor Agonists
They exhibit a range of physico-chemical properties, which arise from differences in molecularstructure, and determine their pharmacological profiles with respect to affinity, efficacy, andduration of action at subtypes of β-adrenoceptors in a number of target cells. All β-agonistsare racemic mixtures of optical isomers, there being two isomers, R and S in salmeterol andfour isomers (RR, RS, SR, and SS) in fenoterol and formoterol. β-agonist activity residespredominantly in the R-form, which ranges from 40-fold to 1000-fold more potent than theS-isomer. At β2-adrenoceptors, salmeterol, formoterol, and fenoterol have a higher affinitythan isoprenaline and salbutamol, the associated rank order of potency being: formoterol >salmeterol > fenoterol = isoprenaline > salbutamol. Fenoterol and formoterol are full agonists,and salmeterol and salbutamol are partial agonists, compared with isoprenaline. Salbutamol,and particularly salmeterol are weak and have low efficacy at β1 and β2-adrenoceptors,whereas formoterol and fenoterol are potent, full agonists. The functional β2-adrenoceptor

144 Bronchial Asthma
selectivity is lowest for fenoterol and highest for salmeterol. β2-agonists such as salbutamoland fenoterol are hydrophilic and interact with the β-receptor directly, whereas formoterol ismoderately lipophilic, and salmeterol is highly lipophilic, gaining access to the active site ofthe β2-adrenoceptor through the cell membrane. The rates of onset of action of salbutamol,fenoterol, and formoterol are more rapid than those of salmeterol. The duration of action isconcentration-dependent for all β-agonists, with the exception of salmeterol, which appearsto be intrinsically long-acting (salmeterol >> formoterol>fenoterol>salbutamol) due toadditional exo-site binding in the β2-receptor protein.
Aerosol or oral inhaled therapy is comparable or better than oral therapy in producingbronchodilatation and cause fewer systemic side effects such as cardiovascular stimulation,anxiety, and tremor. Inhaled therapy has a more rapid onset of action when compared withoral formulations and a similar duration of action, even when administered in substantiallylower dosages. Furthermore, inhaled therapy appears superior to oral therapy because thelatter causes more adverse effects and require higher doses to achieve similar effects. β2-agonists are the medications of first choice for treatment of acute exacerbations and for theprevention of exercise-induced asthma. They can be used either intermittently to controlepisodic airway narrowing or chronically to aid in the control of persistent airway narrowing.Salbutamol inhalation reduces hyperinflation of the lungs. Measurements of lung volumesbefore and after bronchodilators add sensitivity when examining for bronchodilatorresponsiveness.83
Recently there is a trend to use more of inhaled form of these drugs rather than oralpreparations because of adverse effects and slow onset of action. The advantage of slow-release oral agents has been taken over by the introduction of long-acting inhaled β2- agonists,which are more effective in preventing induced bronchoconstriction than equivalent dosesof oral β2-agonists.84 Further, inhaled drugs may reach superficial cells in the airways, suchas mast cells and epithelial cells, that are less easily reached by oral drugs. Thus, nebulisedβ2-agonists are the first choice for acute severe asthma and may be life saving.85 Since theonset of action is rapid, and the therapeutic ratio of bronchodilatation to side effects isgreatly improved, inhaled administration is preferred. Since there is a rapid action, this canbe attributable to the direct effect of the drug on the smooth muscle β-adrenoceptor. Whengiven by inhalation, all currently available β-agonists achieve a measurable effect within5 minutes and by 10 minutes, 80-90% of the maximal response has actually been achieved.42
Another advantage of giving bronchodilators by inhalation is that they are not distributedto the rest of the body in large concentrations and therefore may be given in much smallerdoses.
The doses of some of these drugs are given in Table 10.4.
Side Effects
The predictable side effects of β-agonists include tachycardia, palpitation, dysrhythmia,hypokalemia, tremor, restlessness and rarely hypoxemia. Tremor, due to stimulation of β2-adrenoceptors in skeletal muscles is a common side effect of these class of drugs. Tremor isinseparable from bronchodilator action, but, incidence usually declines with continuedadministration.86 Since the frequency of adverse effects are directly proportional to theplasma concentration, administration via inhalation results in less drug absorption andtherefore fewer adverse effects than either oral or inject able routes. Although, the adrenergicaerosols are currently among the safest drugs available for asthma therapy, there are some
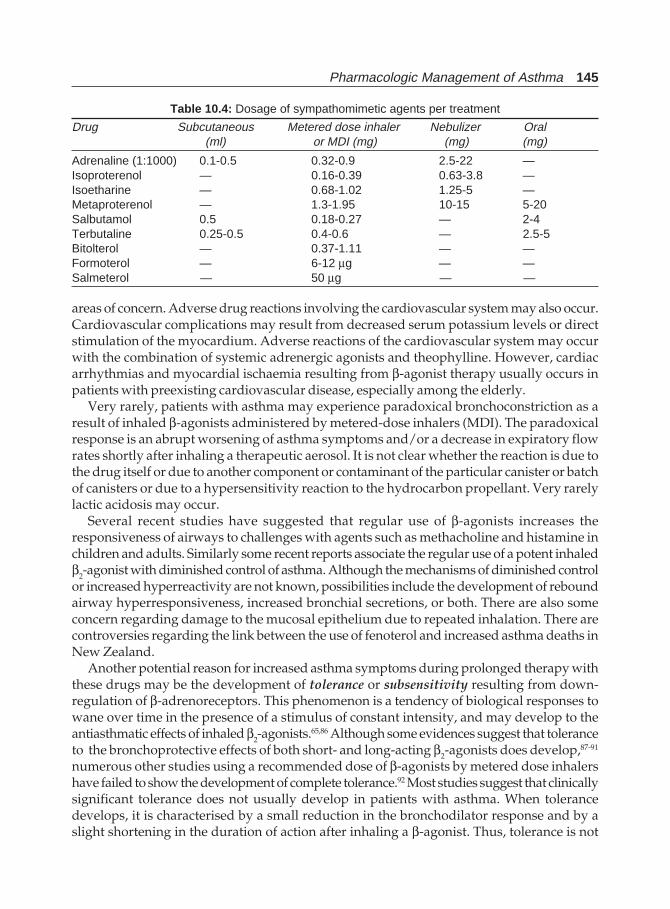
Pharmacologic Management of Asthma 145
areas of concern. Adverse drug reactions involving the cardiovascular system may also occur.Cardiovascular complications may result from decreased serum potassium levels or directstimulation of the myocardium. Adverse reactions of the cardiovascular system may occurwith the combination of systemic adrenergic agonists and theophylline. However, cardiacarrhythmias and myocardial ischaemia resulting from β-agonist therapy usually occurs inpatients with preexisting cardiovascular disease, especially among the elderly.
Very rarely, patients with asthma may experience paradoxical bronchoconstriction as aresult of inhaled β-agonists administered by metered-dose inhalers (MDI). The paradoxicalresponse is an abrupt worsening of asthma symptoms and/or a decrease in expiratory flowrates shortly after inhaling a therapeutic aerosol. It is not clear whether the reaction is due tothe drug itself or due to another component or contaminant of the particular canister or batchof canisters or due to a hypersensitivity reaction to the hydrocarbon propellant. Very rarelylactic acidosis may occur.
Several recent studies have suggested that regular use of β-agonists increases theresponsiveness of airways to challenges with agents such as methacholine and histamine inchildren and adults. Similarly some recent reports associate the regular use of a potent inhaledβ2-agonist with diminished control of asthma. Although the mechanisms of diminished controlor increased hyperreactivity are not known, possibilities include the development of reboundairway hyperresponsiveness, increased bronchial secretions, or both. There are also someconcern regarding damage to the mucosal epithelium due to repeated inhalation. There arecontroversies regarding the link between the use of fenoterol and increased asthma deaths inNew Zealand.
Another potential reason for increased asthma symptoms during prolonged therapy withthese drugs may be the development of tolerance or subsensitivity resulting from down-regulation of β-adrenoreceptors. This phenomenon is a tendency of biological responses towane over time in the presence of a stimulus of constant intensity, and may develop to theantiasthmatic effects of inhaled β2-agonists.65,86 Although some evidences suggest that toleranceto the bronchoprotective effects of both short- and long-acting β2-agonists does develop,87-91
numerous other studies using a recommended dose of β-agonists by metered dose inhalershave failed to show the development of complete tolerance.92 Most studies suggest that clinicallysignificant tolerance does not usually develop in patients with asthma. When tolerancedevelops, it is characterised by a small reduction in the bronchodilator response and by aslight shortening in the duration of action after inhaling a β-agonist. Thus, tolerance is not
Table 10.4: Dosage of sympathomimetic agents per treatment
Drug Subcutaneous Metered dose inhaler Nebulizer Oral(ml) or MDI (mg) (mg) (mg)
Adrenaline (1:1000) 0.1-0.5 0.32-0.9 2.5-22 —Isoproterenol — 0.16-0.39 0.63-3.8 —Isoetharine — 0.68-1.02 1.25-5 —Metaproterenol — 1.3-1.95 10-15 5-20Salbutamol 0.5 0.18-0.27 — 2-4Terbutaline 0.25-0.5 0.4-0.6 — 2.5-5Bitolterol — 0.37-1.11 — —Formoterol — 6-12 μg — —Salmeterol — 50 μg — —

146 Bronchial Asthma
usually of major clinical significance and does not diminish the overall usefulness of inhaledβ2-agonists in asthma therapy. It is possible, however, that receptor down-regulation couldaccount for some of the diminished control of asthma and increased airway hyper-reactivityreported during chronic regular use of these drugs.
Subsensitisation occurs because of the receptors in the tissue are exposed to persistentstimulation by agonists. The problem can occur at one or several different points in theformation of cAMP. It could occur at the level of the receptor, stimulatory or inhibitory,and/or involve down regulation mechanisms. These will involve an uncoupling of thehormone-receptor complex from the guanine nucleotide binding protein. Further, repeatedexposure to catecholamines may reduce the number of β-receptors in the airways that arefree to interact with catecholamine bronchodilators.43 Thus repeated administration ofβ-agonists makes the airways even less responsive. Tolerance is seen most commonly withtriggers that operates through mast cell activation, such as adenosine, allergens, and exercise.Whether steroids protect against development tolerance is not known. The problem maybe avoided by taking long acting β2-agonists only at night. Recent studies of thepolymorphism of human β2-receptors suggest that some forms of the receptors may bemore likely to be down regulated.93 Patients having Arg-16 → Gly form of the receptor, whichis more likely to be down regulated have more frequent asthma in the night.94 In contrast, theGln→Glu form, resist down regulation and is having less airway hyperreactivity.95
There is some concern recently regarding the use of β2-agonists and excess asthma mortality.The two epidemics of asthma death recorded in the literature, one in several countries in the1960’s and the other in New Zealand in the late 1970s, were associated with a rapid increasein the use of a β-agonist formulation delivering a high dose by metered-dose inhalers,isoprenaline in the 1960s and fenoterol in the late 1970s. Although, reports are conflicting, itseems likely that those epidemics were due to high-dose β2-agonist use. There is noepidemiological evidence to suggest that β-agonists have an appreciable effect on mortalityoutside these epidemics.96-101
Some Controversial Facts About βββββ2-Agonists
Despite the worldwide use and the significant contributions of inhaled synthetic sympatho-mimetic agents in the therapeutic management of bronchial asthma, the risk/benefit ratioof these agents have evoked controversy throughout the last half of the 20th century.Concerns about possible deleterious effects of the first reported from the United Kingdom,Australia and New Zealand in the mid-1960s, when a sudden increase in asthma mortalitywas attributed to overuse of a short-acting, dose-fortified formulation of isoproterenol.102 Asimilar phenomenon occurring a decade later in New Zealand appeared to be associatedspecifically with regular use of inhaled fenoterol, a more selective, relatively short-actingβ2-agonist (SABA),.97 A Canadian retrospective case-control analysis of pressurised SABAin patients with asthma suggested that increased asthma mortality was not necessarily dueto fenoterol alone but also occurred after overuse of any pressurised SABA of the sameclass.96 A subsequent meta-analysis of six similar surveys not only failed to confirm thisconclusion but found that mortality was increased to a slight extent only in patients whoused SABA on a regular basis.103 Even if this controversy keeps on appearing off and on, mostclinicians believe that the mortality attributable to SABA is most likely based on over dosage and/orabuse by poorly controlled patients.104

Pharmacologic Management of Asthma 147
The above controversies led to more intensive exploration of the nonbronchodilatorproperties of SABAs and also long-acting β2-agonists (LABAs). The interactive effects ofthese agents as well as individual agents have been studied extensively.105,106 Suchinvestigations have revealed a complex and contradictory array of biological activities thatencompass both proinflammatory and anti-inflammatory effects. As examples of anti-inflammatory effects, β2-agonists are known to attenuate release of mediators from mastcells, suppress airway smooth muscle growth, and inhibit the function of immunocompetentlymphocytes. By contrast, the proinflammatory effects include suppression of interleukin-12 production in antigen-presenting cells, intensification of the T-helper type 2 immuneresponse, augmentation of eosinophil survival and enhancement of the late allergic response.SABAs may also favor the synthesis of receptors associated with neurogenic inflammationthat could play a role in the phenomenon of increased airway hyperresponsiveness that hasbeen noted after long-term use of these agents.
Similar concerns were expressed about the LABAs. The lipophilic nature of these agentswould enable them to partition into the outer phospholipid layer of cell membranes, wherethey have better access to receptors and downstream signalling cascades. Fortunately, down-regulation of β2-agonist receptors on smooth muscle is not clinically relevant, presumablybecause of their overabundant distribution and relative refractoriness to tachyphylaxis inthis tissue site. A number of studies in the mid 1980s and early 1990s demonstrated thatregular use of SABAs increased airway hyperresponsiveness and actually worsen asthmacontrol, and many asthma management guidelines recommended against their regular useover prolonged periods. Similar concerns were also expressed when LABAs were available,and in fact early clinical trials reported that both short-term and long-term use of LABAsdampened the β2-agonist protective effect against methacholine-induced bronchospasmwithout evidence of smooth muscle tachyphylaxis.90,107 However, more recent studiesdemonstrated that the LABA-induced protective effect against airway hyperresponsivenesswas unimpaired after relatively long-term, continuous use of LABAs without evidence of arebound effect after cessation of therapy.108,109 These contradictory results have been ascribedto patient-specific differences in sensitivity to the deleterious effects of bronchodilators,variability of allergic status among patient groups, or a masking activity of β2-agonists .110
The later effect might occur because these agents inhibit only the early allergic responseand might exacerbate the ongoing inflammation associated with the late allergic response.A recent controlled study111 performed over a 6-week period using placebo or salmeterolthat utilised a well-defined allergic phenotype of mild asthma, (pollen sensitive asthmatics),and a well-defined exposure period (a grass pollen season), measured both direct and indirectairway hyperresponsiveness using methacholine and adenosine monophosphate and exhaledNO was measured as an index of airway inflammation. Airway caliber (FEV1), airwayhyperresponsiveness indices and exhaled NO were measured before the administration ofsalmeterol or placebo and at mid season. Patients receiving salmeterol experienced significantprotection against a fall in FEV1 during the height of the allergy season. The increase inairway hyperresponsiveness showed only a small insignificant increase in the treated groupcompared to the placebo group. The result emphasised the difference between natural exposureand a single experimental allergen challenge studies reported earlier. There was a failure todetect a significant difference in adenosine monophosphate-induced airway responsivenessbetween salmeterol-treated and placebo-treated patients when they were challenged with theagent during the height of the pollen season. Since the adenosine monophosphate indirect

148 Bronchial Asthma
challenge reflects bronchoconstriction caused by mast cell mediators, long-term salmeteroldid not attenuate the chronic effects of mediators during the season and therefore did notfunction as an anti-inflammatory agent. The exhaled NO levels were increased both treatmentarms during the height of the pollen season, but there was neither an augmentative norinhibitory effect in the salmeterol group. These results strengthened the safety profile ofsalmeterol and indicated that long-term use of a LABA alone will not provide a clinicallyeffective anti-inflammatory effect.
Ultimately perhaps, the balance between the salutary and adverse effect of both SABAsand LABAs are tilted towards a more clinical benefit to the patient in the management ofbronchial asthma.
Anticholinergics
Anticholinergics are the oldest forms of bronchodilator therapy for asthma and arerecommended as early as the 17th century.112 The recreational and medicinal properties ofatropine have been well-known to many cultures for many centuries. Atropine, in the formof the leaves and roots of Datura stramonium, was very well known to Indians for use inrespiratory disorders, and it was introduced to Western medicine by the British militaryofficers in the early 1800s, who in turn learnt its usefulness from Indians. At that time,stramonium, belladonna, and their alkaloid extract, atropine, had their place in mostpharmacopoeias. Atropine was used for many years for the management of bronchialasthma. With the availability of potent β-adrenergic agonists in the 1920s, its use declined.In recent years there has been an increased interest in inhaled atropine sulphate, especiallyin patients with chronic bronchitis. Atropine is usually given as a powder nebuliser with aβ-adrenergic agent. Its side effects include tachycardia, dryness of the oral mucosa, blurredvision, urinary retention, and constipation. The drug has a delayed onset of action. Atropineshould not be used in patients with narrow angle glaucoma and prostatic hypertrophy.With the advent of newer more selective drugs without these unpleasant side effects ofatropine, the later is almost no more used.112,113 The newer anticholinergic agents are water-soluble, quaternary ammonium compounds that are poorly absorbed, and when they aregiven by inhalation, they cause fewer systemic side effects.114-118 A better understanding ofthe cholinergic mechanisms that control airway caliber in health and disease and thedevelopment of newer synthetic analogs of atropine that are poorly absorbed, but retainthe anticholinergic properties of the atropine, have revitalised the interest in anticholinergictherapy. Several anticholinergic agents that are in use worldwide include:
• Atropine• Ipratropium bromide• Thiazinamum• Oxytropium bromide• Glycopyrrolate• Tiotropium bromide
Rationale for the Use of Anticholinergics
To understand the rationale of use of these agents it is important to understand themechanisms of bronchoconstriction and bronchodilatation that are mediated by theautonomic nervous system. The majority of the autonomic nerves in human airways are

Pharmacologic Management of Asthma 149
branches of the vagus nerve, the efferent paraganglionic fibres of which enter the lungs at thehilum and travel along the airways into the lungs.119 The efferent innervations is derived fromthe postganglionic fibres that end in the epithelium, submucosal glands, and smooth muscleof the airways as well as in the vascular structures. Thus, the release of acetylcholine at thesesites results in smooth muscle contraction and the release of secretions from submucosalglands stimulated by their muscuranic receptors. Cholinergic pathways are important toregulate the acute bronchomotor responses, and many stimuli can provoke bronchoconstrictionvia vagal pathways. Anticholinergic medications antagonise transmission at the muscarinicreceptors. They will only block reflex cholinergic bronchoconstriction and will have no effecton bronchoconstriction resulting from the action of, for example, histamine on the airways.
Cholinergic-induced bronchoconstriction appears to involve primarily the larger airways,whereas β-agonist medications relax both large and small airway contraction equally. Inhumans, there are at least three pharmacologically distinct subtypes of muscarinic receptorswithin the airways, which are known as M1, M2, and M3 receptors.120 Recently, the typesdescribed are up to 5, M1 to M5. The M1 receptors are present within the parasympatheticganglion and mediate increased cholinergic transmission. They may facilitate nicotineictransmission or be responsible for maintaining cholinergic tone. Inhibition would reducecholinergic tone and thus would reduce bronchoconstriction. M1 receptors are also found onalveolar walls, although their function is unknown. Prejunctional M2 receptors on thepostganglionic nerves act as negative feedback loop in neuronal transmission. They areactivated by the release of acetylcholine and promote its reuptake, thereby limiting the degreeof bronchoconstriction produced. These receptors are thought to be dysfunctional in asthma,resulting in exaggerated cholinergic reflexes. The loss of M2 receptor function has beendemonstrated after viral infections. Similar changes can be seen after ozone exposure orantigen challenge.121 When the M2 receptors are dysfunctional, the resulting excessiveconcentrations of acetylcholine at the motor endplate can promote significantbronchoconstriction. Finally, M3 receptors are located on the airway smooth muscle. Thereceptor activation leads to a release of calcium ions from intracellular stores and a decreasein intracellular adenosine 3’,5’-cyclic monophosphate levels, resulting in the contraction ofairway smooth muscle. M3 receptors also are located on submucosal glands, where they arelikely to be involved in mucus secretion.
Ipratropium bromide and oxytropium bromide are quaternary ammonium derivatives ofatropine and are bronchoselective when delivered by inhalation.122,123 Ipratropium bromide isa muscarinic cholinergic antagonist that inhibits smooth muscle contraction by competingwith the neurotransmitter acetylcholine at the muscarinic receptor.124 These drugs are thusless effective than inhaled β2-agonists because they counteract only cholinergic neuralbronchoconstriction, which may be a relatively minor part of the broncho-constrictormechanism in asthma. As discussed earlier, recently it is recognised that there are at least fivesubtypes of muscarinic receptors expressed in the airways.120, 125 The M3 receptors play themajor role in causing bronchoconstriction, whereas the M2receptors mediate the feedbackinhibition of acetylcholine release from airway sensory nerves.126 Atropine, ipratropiumbromide and oxytropium bromide are nonselective antagonists and produce their beneficialeffect by blocking M3 receptors. However by blocking prejunctional M2 receptors, they increasethe release of acetylcholine and thus may have relatively deleterious effects.126 This mayweaken the effect of the postjunctional M3 muscarinic receptor blockade on airway smooth

150 Bronchial Asthma
muscle and submucosal glands. This suggests that antagonists that bind selectively to M1
and M3 receptors may be more effective in inhibiting cholinergic effects on the airways. Thedrugs also inhibit hypersecretion of mucus in the airways.127 The anticholinergic drugs act byreducing intrinsic vagal tone to the airways. They also block reflex bronchoconstriction causedby inhaled irritants. The agents also block postganglionic efferent vagal pathways. They arerelatively free of systemic side effects because they are minimally absorbed into the systemiccirculation and do not cross blood-brain barrier.
The natural antichiolinergic, atropine, is rarely used in patients at the present time,however, this drug was used extensively as a nebulised solution by intensivists andemergency department specialists for years.128 It is readily absorbed across the oral andrespiratory mucosa and when higher doses are used to maximize bronchodilator effect, theincidence of dry mouth, blurred vision, urinary retention, nausea and tachycardia maylimit the usefulness of atropine. The principal anticholinergic agent is ipratropium bromide,a nonselective muscarinic antagonist.129,130 The drug is topically active, and the compoundis poorly lipophilic and not significantly absorbed from the respiratory or GI tract. It has noor very little systemic effect. The drug has been found to be an effective bronchodilators inpatients with COPD and selective patients with asthma both alone and when used incombination β2-agonists and theophylline. When used via MDI aerosol, the recommendeddose of ipratropium bromide is 2 puffs (40 μg) 4 times daily. The drug has been shown to beeffective during status asthmaticus when used in nebulised form in combination withβ-adrenergics.131-133 It does not appear to affect mucus secretion and ciliary movement.Another significant advantage of ipratropium bromide in the critically ill asthma patientsis the lack of tachycardia, which does occur with β2-agonist use.134 The only remarkableside effect is the inhibition of salivary secretions at high doses. It has no effect on urinaryflow, or intraocular tension, and possible effects on the eye (glaucoma) can be prevented byusing a mouth piece during nebulisation. The onset of action is 3 to 30 minutes with up to50% of the response occurring in 3 minutes and 80% in 30 minutes, with a peakbronchodilator effect observed within 1 to 2 hours, and the duration of action is up toapproximately 6 hours. These properties are ideal for acute asthma treatment. Oxytropiumbromide is a quaternary ammonium anticholinergic compound that is based on scopolamineinstead of atropine. It is also a nonselective muscarinic antagonist. The drug is used in adose of 200-400 μg per day and is perhaps less effective in chronic asthma.134 It has a longerduration of action, up to 8 hours than ipratropium bromide, but has a slower onset of effect.135
The peak onset of action is 1-2 hours. In children ipratropium has bronchodilator action inacute exacerbations of asthma. However, the benefits of its use in day-to-day managementof asthma in children and adults have not been established, although its use appears to bemost effective in patients with COPD with partially reversible airflow obstruction. Tiotropiumbromide is a recently developed, long acting, selective, anti-muscarinic medication. Thisagent is selective for both M1 and M3 receptors. In human bronchi, the drug has a similarinhibitory effect with a slow onset of action with the peak bronchodilator effect observedafter 1.5 to 2 hours and a very prolonged effect compared to ipratropium bromide. Theeffect lasts for 10-15 hours.136,137 The drug has a prolonged inhibitory effect acetylcholinereleased from postganglionic nerve endings in the airways, probably via an inhibitory effecton M1 receptors. The drug is available as a lactose based powder formulation containing18 mg of active substance and is used once daily.

Pharmacologic Management of Asthma 151
In certain clinical situations these drugs may be useful bronchodilators for the treatmentof bronchial asthma.138 They are recommended for patients who cannot tolerate β-adrenergicagonists because of severe tremor or underlying cardiac disease and for patients withbronchospasm precipitated by β-adrenergic antagonists139 or acetylcholinesterase inhibitors.They can be used in combination with β-agonists.
Corticosteroids
Glucocorticosteroids are the most potent anti-inflammatory drugs useful in the treatmentof bronchial asthma. With the realisation of the role of inflammation as an essential andimportant component of asthma, their frequent use is justified. Inhaled glucocorticosteroidshave revolutionised the treatment of asthma and are highly effective in controlling asthmain all patients.140
Glucocorticosteroids are active against bronchial asthma, mainly through their anti-inflammatory effects.141,142 The anti-inflammatory action of corticosteroids is as follows. Thehormone penetrates freely into the cell and binds to the receptor forming an inactive complex,which is further activated or transformed to an active complex having an enhanced affinityfor DNA forming the nuclear-bound complex. Then it is translocated to the nucleus whereit binds to specific sequences (glucocorticoid-responsive element) on the upstream regulatorypart of steroid-responsive gene.143 This complex then by binding to regulatory elementsassociated with certain genes, can activate or inhibit transcription of these genes. Thehormone thereby increases or decreases the levels of mRNA and usually of the proteinsthat the genes encode. These proteins may be enzymes, secretory products, and regulatorsof various functions including transcription of other genes, which are the primary effectorsof hormone actions. The particular genes and proteins regulated by corticosteroids dependon the type of cells. This may increases the production of a substance called lipocortin-1,which inhibits the enzyme phospholipase A2, an enzyme essential for activation ofarachidonic acid metabolism. The complex may cause reduced transcription with inhibitionof protein synthesis like cytokines. An important effect of steroids in asthma may be theinhibition of synthesis of key cytokines like IL-3, Il-5, and GM-CSF, which play significantrole in perpetuating the inflammatory response.144
It is also likely that steroids act on many different cells of the airways. Although they donot reduce the release of mediators from mast cells themselves,145 they lead to a significantreduction in mast cell numbers, possibly due to inhibition of IL-3, which is necessary formast cell survival in the airways.146 Steroids inhibit release of mediators by macrophages,147
but eosinophils are less responsive.148 But, eosinophil survival is markedly reduced due toblockage of the effect of cytokines like IL-3, Il-5, and GM-CSF.149 Inhaled steroids also reducemarkedly the proportion of circulating low-density eosinophils in asthmatic patients throughinhibition of IL-5 secretion.150 The other most important effect of steroids is on T lymphocyteswhere the synthesis of cytokines is reduced. Additional effects directly related to anti-inflammatory action include reduced plasma exudation from postcapillary venules in theairways,151 and inhibition of mucus glycoprotein secretion.152 Further, inhaled steroid therapycauses a reduction in bronchial hyperresponsiveness to histamine and the underlyingT-cell-dominated inflammation in the bronchial wall.153
Although the molecular mechanisms of the anti-inflammatory action of steroids are betterunderstood,154 the key cellular targets in asthma have not yet been conclusively established.It appears that airway epithelial cells are important target cells and besides the above

152 Bronchial Asthma
mentioned mechanisms including the inhibition of expression of cytokines like IL-1, IL-8,regulated on activation normal T-expressed and secreted (RANTES) and GM-CSF, they alsoinhibit lipid mediators,155 nitric oxide,156 and adhesion molecules.157 They also may inhibit theexpression of inducible genes in airway epithelial cells by blocking key transcription factorssuch as nuclear factor-kappa B and activator protein-1.154
Thus, the important mechanisms of anti-inflammatory action of corticosteroids can besummarised as follows:
i. Interference with arachidonic acid metabolism through alteration of lipocortinsynthesis and that of the synthesis of leukotrienes, cytokines and prostaglandins. Theyinhibit the production of IL-1, collagenase, elastase, and plasminogen activator.
ii. Prevention of the direct migration and activation of inflammatory cells. Dampeningof the recruitment and activation of eosinophils results from their direct effect onthese cells as well as upon T-lymphocytes, endothelial cells, and macrophages. Localactivation of a variety of cell types including neutrophils, basophils, macrophagesand possibly eosinophils by γ-interferon may be blocked by inhibition of this substancefrom T-lymphocytes by glucocorticoids.
iii. Inhibition of cytokine gene transcription and translation leading to inhibition ofcytokine secretion and increased intranuclear breakdown of these mediators.
iv. Inhibition of cellular response to cytokines, such as increased release of mast cellmediators, expression of adhesion molecules, and prolonged survival of inflammatorycells.
v. An acute anti-inflammatory action mediated via inhibition of microvascular leakage.
Direct evidence for the anti-inflammatory effect of inhaled steroids is provided by biopsystudies in asthmatic patients. After regularly inhaling steroids over one to three months,bronchial biopsy shows many fewer eosinophils, mast cells, and lymphocytes,146, 153, 158 andin patients with mild inflammation of the airways, there is complete resolution. In biopsiesof patients after ten years of inhaled steroids, inflammatory cells disappear completely,although basement membrane thickening may persist.159
Steroids facilitate the action of adrenergic bronchodilators, apparently by altering theratio of α to β- adrenergic receptors on cell surface.160,161 Oral prednisolone therapy preventsthe development of down regulation and subsensitivity of lymphocyte β2-adrenoceptors insubjects given long-term treatment with oral β2-agonists.162 Effects of corticosteroids inasthma patients are considerable.163-168 Treatment with inhaled corticosteroids improves FEV1,peak expiratory flow, and symptoms within weeks. Improvements in airways hyper-responsiveness are slower in onset, and gradual amelioration usually continues up to atleast 1 year.164 Exacerbation rates are markedly reduced by treatment with inhaledcorticosteroids in asthma.164,167,168 Some studies have even indicated that delayed introductionof inhaled corticosteroids results in an impaired response.169,170 Recent studies on the long-term effect in patients who are treated with terebutaline and beclomethasone dipropionateindicate that the initial improvement in lung function are well preserved over 5 years.171
Inhaled steroids prevent the accelerated decline of FEV1.172
The wide-ranging clinical benefits associated with corticosteroids are shown in Table 10.5.Corticosteroids can be administered parenterally, orally, or as aerosols. Because of the
availability of inhaled steroids, there has been less fear now to treat patients with steroids

Pharmacologic Management of Asthma 153
either with a short course therapy or for longer times. It is now clear that the duration andseverity of an acute asthma attack can be substantially reduced by therapy with corticoste-roids.
Early treatment of severe acute exacerbations of asthma with oral corticosteroids preventsprogression of the exacerbation, decreases the need for emergency visits and hospitalisation,and reduces the morbidity of the illness. When oral steroids are used to treat acute severeasthma, the onset of action is gradual, occurring approximately 3 hours after administrationwith peak effectiveness occurring about 6-12 hours after administration.
Acute short-term therapy is begun usually with a relatively high dose of 40-80 mg ofprednisone daily and can be maintained up to 5-10 days or tapered over the same interval.Therapy with oral steroids should be maintained until peak expiratory flow rates are stablenear the best predictable value. The major adverse effects associated with high-dose short-term systemic therapy are: reversible abnormalities in glucose metabolism, increased appetite,fluid retention, weight gain, rounding of face, mood alteration, hypertension, peptic ulcer,and aseptic necrosis of the femur.
In all patients requiring chronic maintenance therapy with steroids, a trial of inhaledsteroids, which have minimal systemic side effects, should be attempted to see if oralcorticosteroids could be reduced or eliminated. Oral therapy can be continued only if thatshows to reduce chronic symptoms substantially or reduce the frequency of severe episodes.
Table 10.5: Clinical benefits of glucocorticosteroids
* Improved pulmonary function
Diurnal variability in pulmonary function
Protection against antigen-induced bronchoconstriction
Asthma exacerbation rate
Hospital admission rate
Asthma mortality rate
* Prevention of long-term lung damage and therefore irreversible airflow obstruction
The anti-inflammatory effects of glucocorticosteroids are shown in Figure 10.1:
Glucocorticosteroids
EosinophilsMast cells CYTOKINEST-LymphocytesMucus secretionPlasma exudationMediator formation
β-Adrenoceptors
INFLAMMATION
Fig.10.1: Anti-inflammatory effects of glucocorticosteroids

154 Bronchial Asthma
Oral steroids should not be used alone without maximising other forms of therapy. Long-termoral steroid therapy is associated with significant side effects such as osteoporosis,hypertension, Cushing’s syndrome, cataracts, myopathy, hypothalamo-pituitary-adrenal axissuppression, and in rare instances, impaired immune mechanisms. Therefore, prolonged useof oral steroids should be reserved for patients with severe asthma despite use of high-doseinhaled corticosteroids. The lowest possible drug dose should be employed including attemptsof alternate-day therapy. The drug should be given as a single-morning dose and pulmonaryfunction tests should be used to objectively assess efficacy.
Inhaled steroids are safe and effective for the treatment of asthma. They are very effectivein controlling the symptoms of asthma and usually achieve rapid control. As a companiondrug to β2-agonists, inhaled steroids reduce symptoms, reduce the need for rescuebronchodilators, and improved lung function compared to regular treatment with β2-agonistalone.173 Inhaled steroids inhibit the late response reflecting inflammation to allergen andprevent the increase in airway hyperresponsiveness that follows allergen exposure.174 Theyalso reduce AHR when given regularly, although the reduction takes place slowly overtwo months or more as the chronically inflamed airway heals slowly.175 When discontinued,symptoms and AHR revert to pretreatment levels.176 In patients with mild asthma treatedwith inhaled steroids for a long time, there may be long symptom free periods beforerecurrence.177 In patients with atopic asthma, changes in the bronchial eosinophils and lungfunction during steroid therapy occur , but independently.178
Some basic principles regarding inhaled corticosteroids include:179,180
• Both efficacy and side effects of aerosol glucocorticoids are dose dependent, andpatients vary in their dose requirement. Patients with chronic asthma severe enoughto need large oral maintenance doses are unlikely to respond adequately to inhaledtreatment alone.
• Aerosol treatment is not effective in acute severe asthma.• A part of the inhaled drug is absorbed resembling parenteral injection bypassing liver
with reduced hepatic degradation of the active compound and able to produce systemiceffects.
• Aerosol treatment is more effective if divided into several doses throughout the day.The introduction of beclomethasone dipropionate to asthma therapy in the early 1970’s
represented a major advance in asthma management. Various guidelines describedsubsequently advocate use of inhaled corticosteroids for longer periods of time thanpreviously recommended in patients with mild asthma and at higher doses than previouslyconsidered feasible in patients with severe asthma. Inhaled corticosteroids are now gainingwidespread acceptance as safe and effective agents for the management of childhood asthma.They are unique among anti-asthma medicines that no other anti-asthma drug currentlyavailable share such a wide ranging profile of clinical benefits.
An important unresolved question is whether inhaled steroids exert a therapeutic effecton the airways through a systemic action. Since they reduce the number of circulating low-density eosinophils, it is suggested that inhaled steroids have an effect in the circulation orin the bone marrow.150 However, this phenomenon can be as a result of local airway effectthrough inhibition of synthesis of the eosinophil-stimulating cytokine IL-5 and RANTES.Studies in dogs have suggested that inhaled steroids affect the production of leucocyteprogenitors in the bone marrow, but it is not clear whether this results from affecting thesynthesis of some stimulatory factor in the airways or from the action of the systemically

Pharmacologic Management of Asthma 155
absorbed fraction of the inhaled steroid on the bone marrow.181 It is also uncertain whethersteroids deposited in the proximal airways can be distributed via the airway circulation to themore distal airways.
The inflammation of asthma affects the whole of the bronchial tree, from the large centralairways down to the small peripheral airways.182,183 Steroid receptors, the site of action ofinhaled corticosteroid therapy, are likewise located through out the bronchial tree.184 Variousinhaled steroids available for clinical use include Beclomethasone dipropionate,betamethasone valerate, Budesonide, Flunisolide, Triamcinolone acetonide, Fluticasonepropionate, Mometasone furoate, and Ciclesonide.185-189 Beclomethasone is the first inhalersteroid available nearly for the past 30 years and is used widely. The dose varies from < 400μg per day to as high as 1600 μg depending upon the severity of bronchial asthma.Budesonide is a glucocorticoid aerosol with high ratio between topical and systemiccorticosteroid effects.190,191 The drug is usually administered in a dose of 200-400 μg twicedaily. Fluticasone propionate introduced in the 1990s, is one of the most potent inhaledsteroids currently available, and is developed from the androstane 17 β-carboxylic acidand is a highly potent, selective anti-inflammatory steroid which binds with a high affinityto the glucocorticoid receptor of the human lung (18 times that of dexamethasone and 3 timesthat of budesonide). It has greater airway selectivity, rapid fast-pass metabolism (so lesssystemic side effects, and increased uptake and retention in the lungs as a result of its highlipophilicity. It is approximately 2-fold more potent than beclomethasone dipropionate and4-fold more potent than budesonide.192 500 μg b.d. Fluticasone propionate is at least aseffective as beclomethasone dipropionate 1000 μg b.d.193 Estimated clinical comparabilityof doses for inhaled corticosteroids are shown in Table 10.6.
It is estimated that beclomethasone and budesonide achieve comparable effects at similarmicrogram doses by MDI. Beclomethasone has similar effects to twice the dose oftriamcenoline acetonide on a microgram basis. However, fluticasone has effects similar totwice the dose of budesonide and beclomethasone when given via MDI in a microgrambasis. Budesonide given by a Turbuhaler has effects similar to twice the dose delivered byMDI, implying greater bronchial delivery by the delivery device. These observations aremade on the basis of clinical trials comparing effects in reducing symptoms and improvingPEFR.
The potency of a glucocorticosteroid is described by its receptor affinity and intrinsicactivity. For all therapeutically used corticosteroids in asthma, the intrinsic activity directlycorresponds to the receptor affinity, which is a compound-specific property. If the receptoractivity of a corticosteroid is determined under standardised conditions (usually withdexamethasone as reference), the relative receptor affinity can be calculated and comparedwith other corticosteroids. The same is shown in Table 10.7.194
Table 10.6: Comparison of potency of inhaled corticosteroids
Drug Topical potency Corticosteroid receptor Receptor(skin blanching) binding half-life (hrs) binding affinity
Beclomethasone 600 7.5 13.5Budesonide 980 5.1 9.4Flunisolide 330 3.5 1.8Fluticasone 1,200 10.5 18.0Triamcinolone 330 3.9 3.6
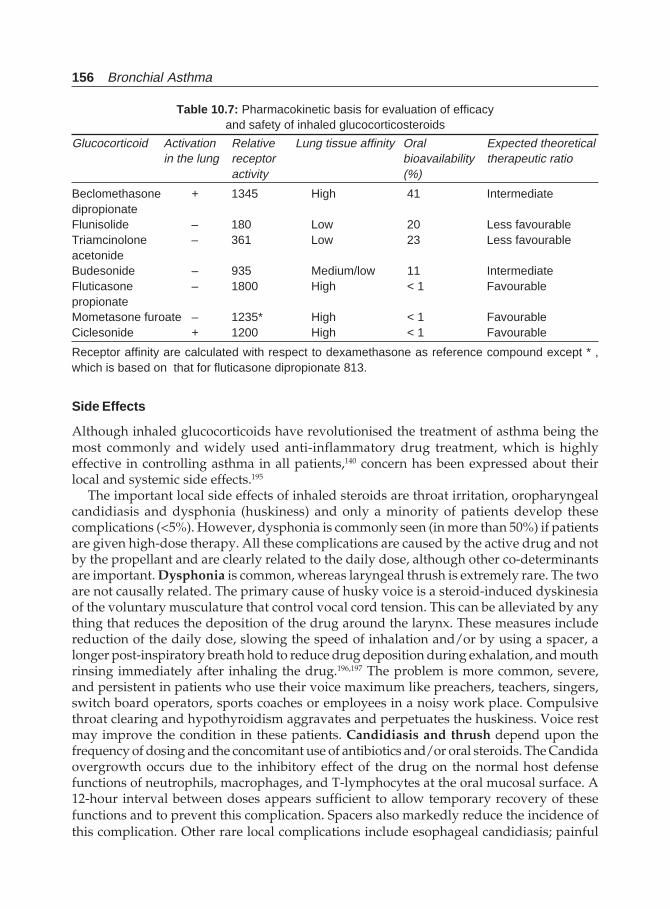
156 Bronchial Asthma
Side Effects
Although inhaled glucocorticoids have revolutionised the treatment of asthma being themost commonly and widely used anti-inflammatory drug treatment, which is highlyeffective in controlling asthma in all patients,140 concern has been expressed about theirlocal and systemic side effects.195
The important local side effects of inhaled steroids are throat irritation, oropharyngealcandidiasis and dysphonia (huskiness) and only a minority of patients develop thesecomplications (<5%). However, dysphonia is commonly seen (in more than 50%) if patientsare given high-dose therapy. All these complications are caused by the active drug and notby the propellant and are clearly related to the daily dose, although other co-determinantsare important. Dysphonia is common, whereas laryngeal thrush is extremely rare. The twoare not causally related. The primary cause of husky voice is a steroid-induced dyskinesiaof the voluntary musculature that control vocal cord tension. This can be alleviated by anything that reduces the deposition of the drug around the larynx. These measures includereduction of the daily dose, slowing the speed of inhalation and/or by using a spacer, alonger post-inspiratory breath hold to reduce drug deposition during exhalation, and mouthrinsing immediately after inhaling the drug.196,197 The problem is more common, severe,and persistent in patients who use their voice maximum like preachers, teachers, singers,switch board operators, sports coaches or employees in a noisy work place. Compulsivethroat clearing and hypothyroidism aggravates and perpetuates the huskiness. Voice restmay improve the condition in these patients. Candidiasis and thrush depend upon thefrequency of dosing and the concomitant use of antibiotics and/or oral steroids. The Candidaovergrowth occurs due to the inhibitory effect of the drug on the normal host defensefunctions of neutrophils, macrophages, and T-lymphocytes at the oral mucosal surface. A12-hour interval between doses appears sufficient to allow temporary recovery of thesefunctions and to prevent this complication. Spacers also markedly reduce the incidence ofthis complication. Other rare local complications include esophageal candidiasis; painful
Table 10.7: Pharmacokinetic basis for evaluation of efficacyand safety of inhaled glucocorticosteroids
Glucocorticoid Activation Relative Lung tissue affinity Oral Expected theoreticalin the lung receptor bioavailability therapeutic ratio
activity (%)
Beclomethasone + 1345 High 41 IntermediatedipropionateFlunisolide – 180 Low 20 Less favourableTriamcinolone – 361 Low 23 Less favourableacetonideBudesonide – 935 Medium/low 11 IntermediateFluticasone – 1800 High < 1 FavourablepropionateMometasone furoate – 1235* High < 1 FavourableCiclesonide + 1200 High < 1 Favourable
Receptor affinity are calculated with respect to dexamethasone as reference compound except * ,which is based on that for fluticasone dipropionate 813.

Pharmacologic Management of Asthma 157
and protracted atrophic glossitis; chronic oesophagitis resulting from combined Candida-herpes simplex infection; reflex cough and bronchospasm; and nonspecific symptomsincluding nausea, headache, dry throat, gas, pruritus, rash, impaired taste or smell,abdominal pain, diarrhoea, constipation, and heartburn.
Systemic bioavailability varies with the preparation selected. The systemic activity ofany particular dose in different patients and patient groups depends largely on the fractionof the emitted dose that reaches the important absorptive surface in the lung periphery.This fraction is determined by the interaction of numerous factors, including variations innormal lung anatomy, the degree of pulmonary function impairment and presence orabsence of associated chronic bronchitis, each of which reduces peripheral delivery of theinhaled drug, and in particular, by the inspiratory techniques used. There is some fear ofsystemic effects because of oral, gastrointestinal, and pulmonary absorption of the drugs.140,198
However, they are infrequent. Approximately 80% of an inhaled corticosteroid dose willbe deposited in the mouth and subsequently swallowed, thus giving the potential forsystemic adverse events during long-term therapy. This can be reduced by using a large-volume spacer and mouth rinsing or other steroid sparing agents like cromolyn sodium.Side effects can also be reduced by choosing a steroid such as budesonide or fluticasonepropionate that undergoes extensive first-pass hepatic metabolism, allowing little of thedrug to enter the systemic circulation. Then, the only source of systemic absorption will befrom the fraction absorbed from lung deposits. The side effects may include bone and skinthinning, easy bruising, cataract formation, inhibition of longitudinal bone growth in childrenand suppression of adrenocortical function. Clinically significant adrenal suppression andaltered bone metabolism are rare below 800 μg/day,199-201 but a minimum daily dose shouldbe sought once clinical response has been achieved. Very large doses of inhaled drugs maycross the placental barrier as shown in experimental animals.
Because of the importance of airway inflammation in the pathogenesis of asthma, inhaledcorticosteroids are being used more frequently as primary therapy for moderate and severeasthma. This approach not only provides symptomatic benefit but also reduces airwayhyperresponsiveness.
CROMONES (CROMOLYN SODIUM AND NEDOCROMIL SODIUM)
Cromolyn Sodium
Cromolyn sodium is the best nonsteroidal anti-inflammatory drug for asthma availablecurrently.202,203 This drug has been available for 35 years. When administered prophyl-actically, Cromolyn sodium inhibits early- and late phase allergen-induced airwaynarrowing and acute airway narrowing after exercise (less than inhaled adrenergic agents),exposure to cold dry air, and sulphur dioxide. They are also effective in controlling symptomsin patients with mild asthma.204 There is no way to predict reliably whether a patient willrespond to Cromolyn sodium. A 4-6 week trial may be required to determine efficacy inindividual patients. The drug is available in a capsule form (5 mg) taken as an inhaler aswell as metered dose inhaler and even as a nebuliser solution. It controls the symptoms ofbronchoial asthma and bronchial hyperresponsiveness and reduces the number of acuteexacerbations with an acceptable safety profile.205-207 Cromolyn sodium produces only minimalside effects, such as occasional coughing upon inhalation of the powder formulation.

158 Bronchial Asthma
Nedocromil Sodium
This is a pyranoquinoline derivative and is shown to inhibit mediator release prophylacti-cally in a variety of in vitro systems.208 It also inhibits allergen-induced acute and late-phaseasthmatic reactions and modulates allergen-induced increases in bronchial hyper-responsiveness. It also reduces the acute airway narrowing response to exercise,hyperventilation, mist, and sulphur dioxide. Various clinical trials have proved that long-term therapy reduces nonspecific airway reactivity in atopic and nonatopic asthmatics. Inclinical trials the drug has been used in a dose of 4 mg four times a day with most beneficialtherapeutic effects. Therapy with nedocromil is not associated with any significant adverseeffects. The drug, however, is not yet used extensively in clinical practice. Although the exactmechanism of action of cromones as anti-inflammatory drugs is not clear, it is believed thatthe drugs stabilise and prevent mediator release from mast cells.209-213 The drugs are also likelyto affect several other inflammatory cells including sensory nerves.204,208 However, anotherstudy has found no evidence of a decrease of inflammatory cells after treatment withcromones.214 Other studies suggest that cromones may block swelling-dependent chloridechannels.215 Additional chloride channels in mast cells, sensory nerves, and epithelial cellsmay be important.
Advantages of cromones are that they control symptoms of asthma and effectively blockbronchoconstriction induced by a number of agents and factors. Both drugs are safe and haveno significant side effects.216 One study has found that nedocromil has steroid sparing effectsbut other studies have not confirmed this. The disadvantage of cromones are that they areweak anti-inflammatory drugs compared to inhaled steroids, and more costly. They appear towork best in patients with mild asthma, but not always. It is difficult to predict which patientswill respond. Recent studies have shown that cromones may be most beneficial for patientswhose predominant symptom is coughing. They may be considered as the first line therapy inchildren and as a prophylactic agent against allergen-induced asthma. The other disadvantageof these drugs is their short action, and therefore they are to be used four times a day which isan inconvenient regimen for long-term prophylaxis.
Ketotifen
Ketotifen is an orally active, prophylactic drug used in many countries in the management ofasthma.217,218 Originally it was thought that the drug is a mast cell stabiliser that has theadditional property of being a potent H1-receptor antagonist. Large double-blind, placebo-controlled trials have proved the efficacy of the drug in the prophylaxis of asthma particularlyin children. Recent data suggests that the ability of the drug to act as a prophylactic agent isnot related to its mast cell stabilising effect nor the H1-receptor antagonistic properties. Thedrug like many other prophylactic antiasthma drugs, inhibits PAF-induced eosinophilinfiltration and bronchial hyperresponsiveness. The drug is most effective in mild asthmaand require at least 4-12 weeks to show any clinically significant effect. It is given in a dose of2-4 mg twice daily and this dose is roughly equivalent to 4 puffs of Cromolyn sodium. Themajor advantage of ketotifen over other prophylactic drugs is that it can be used orally. Themajor side effect of the drug is sedation.

Pharmacologic Management of Asthma 159
Antihistamines
With the development of new classes of nonsedating antihistamines, there has been renewedinterest in their use.219,220 The rationale for their use was that subjects with asthma demonstratehyperresponsiveness airways to histamine and require only small quantities of this mediatorto demonstrate changes in their pulmonary functions. These agents block the acutebronchoconstricting effect produced by inhaled histamine, but not that produced bymethacholine. They have also bronchodilating action. The newer antihistamines also inhibitmediator release from in vitro cell systems. Clinical trials have shown their superiority overplacebo in grass and pollen induced asthma. Most of these drugs (terfenadine, astemizole,azelastine and cetirizine) moderately inhibit the early asthmatic response. Only terfenadineinhibits exercise induced asthma. The drug also has some calcium channel blocking properties.Although some studies with H1 antihistamines in asthma have demonstrated some therapeuticbenefits, their role and usefulness in treatment of asthma require additional studies and theyare not recommended as anti-asthma drugs.
Leukotriene Antagonists and Synthesis Inhibitors
Cysteinyl leukotrienes (LT) play a significant part in the pathogenesis of bronchial asthmaas discussed earlier.221-238 These leukotrienes are produced and released from proinflammatorycells, including eosinophils and mast cells, and are at least 1000 times more potent broncho-constrictors than histamine or methacholine in normal subjects and patients with bronchialasthma. They mediate many of the pathophysiologic processes associated with asthmaincluding microvascular leakage, bronchoconstriction and eosinophil recruitment into theairways. Since the structure of leukotrienes was described in 1979 ,239 attempts were made tomodulate their pharmacological actions so that they can be of some clinical use by the way ofblocking the leukotriene receptors or inhibition of their synthesis. The therapeutic strategieswere
i. Dietary provision of alternative fatty acid substrates within membrane phospholipidswhich will products with less proinflammatory activity,240 but the attempt wasunsuccessful;241
ii. Pharmacological inhibition of specific enzymes, particularly 5-lipoxyenase; andiii. Modulation of end organ effects with selective cysteinyl leukotriene receptor
antagonists.221,238
Generally four classes of drugs are currently under development and some of them areavailable for clinical use as anti-asthma or anti-inflammatory therapy, which interferes withLT synthesis or activity. They are depicted in Table 10.8 and Figure 10.2.242
Although a number of the above compounds were tried initially, only a few could beused clinically in human beings because of safety factors.243 Zafirlukast is active both orallyand when administered by inhalation and is the most potent oral cysteinyl LT antagonist.244
Pranlukast, another orally active drug is marked in Japan in the mid-1995.245, 246 Thesedrugs are at least 200 times more than the early LT antagonists and they cause a shift up to100-fold in the bronchoconstrictor dose-response curve. They are very active in preventingbronchoconstriction induced by agonists both in healthy and asthmatic individuals. Theyalso reduce bronchoconstriction induced by several natural triggers of asthma includingexercise, cold air, allergen and aspirin. A single 20 mg oral dose of zafirlukast producesmarked protection against exercise-induced bronchoconstriction with the maximum effect
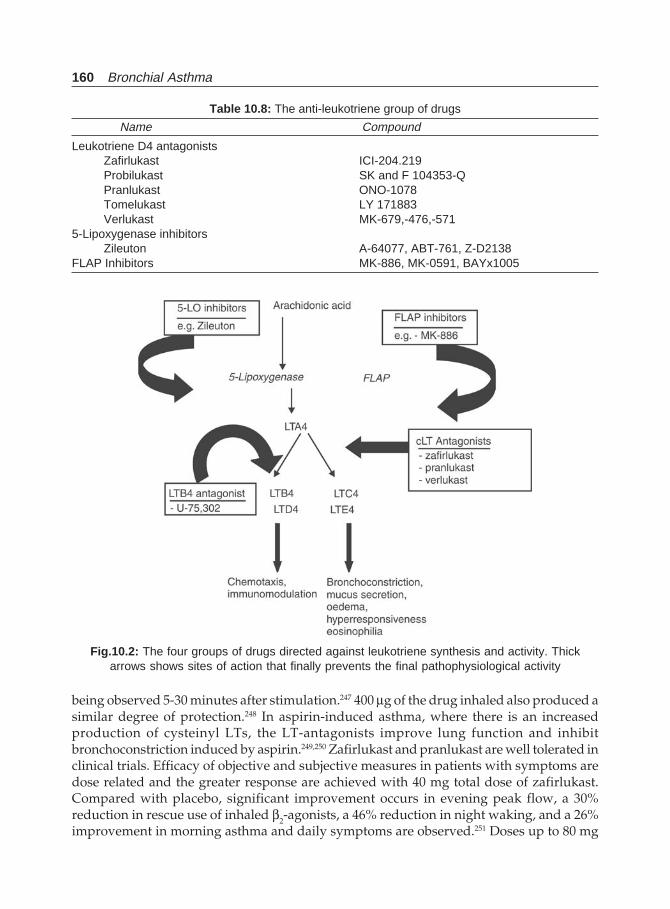
160 Bronchial Asthma
being observed 5-30 minutes after stimulation.247 400 μg of the drug inhaled also produced asimilar degree of protection.248 In aspirin-induced asthma, where there is an increasedproduction of cysteinyl LTs, the LT-antagonists improve lung function and inhibitbronchoconstriction induced by aspirin.249,250 Zafirlukast and pranlukast are well tolerated inclinical trials. Efficacy of objective and subjective measures in patients with symptoms aredose related and the greater response are achieved with 40 mg total dose of zafirlukast.Compared with placebo, significant improvement occurs in evening peak flow, a 30%reduction in rescue use of inhaled β2-agonists, a 46% reduction in night waking, and a 26%improvement in morning asthma and daily symptoms are observed.251 Doses up to 80 mg
Table 10.8: The anti-leukotriene group of drugs
Name Compound
Leukotriene D4 antagonistsZafirlukast ICI-204.219Probilukast SK and F 104353-QPranlukast ONO-1078Tomelukast LY 171883Verlukast MK-679,-476,-571
5-Lipoxygenase inhibitorsZileuton A-64077, ABT-761, Z-D2138
FLAP Inhibitors MK-886, MK-0591, BAYx1005
Fig.10.2: The four groups of drugs directed against leukotriene synthesis and activity. Thickarrows shows sites of action that finally prevents the final pathophysiological activity

Pharmacologic Management of Asthma 161
twice daily have been given to patients with beneficial effects increasing still further at thehigher doses.252 Besides being effective in preventing bronchoconstriction due to varioustriggers, these drugs also affect eosinophil reflux, microvascular permeability, proliferation ofairway smooth muscle cells in chronic severe asthma, mucus secretion, mucociliary transport,and interaction with nerves. The reported side effects of these drugs are: headache, dry mouth,and somnolence.251,253-255
The 5-lipoxygenase inhibitor zileuton is the best studied drug.256 Oral zyleuton in a doseof 800 mg inhibits the early response to allergen challenge and reduced LT synthesis. It alsoprevents the response to cold air challenge and to aspirin in aspirin-induced asthma. It alsoincreases the FEV1. The drug is as effective as theophylline in moderate asthma. It helps inreducing the symptoms of asthma, β-agonist use, and inhaled steroid doses can also bereduced. The drug also has a steroid-sparing effect in severe asthma. The only disadvantageof zileuton is its relatively low potency and short half-life (2.5 hours). Therefore, dosing isto be made four times a day. Even if the drug appears to be safe in clinical trials, this is anantioxidant and potentially can interfere with redox reactions in other metalloenzymes.
Of the above mentioned drugs, four oral antileukotriene drugs are now available for thetreatment of asthma: monteleukast, zafirleukast, and pranleukast, and the 5-lipoxygenaseinhibitor, zyluton. Clinical studies have shown improvement in FEV1, improvement indaytime and nocturnal asthma symptom scores, and reduction in reliever β2-agonist use inpatients with patients treated with leukotriene antagonists.257,258 For this reason, recentasthma guidelines have recommended that antileukotrienes have a role in the managementof bronchial asthma (see below). However, not all patients will show a significant clinicalimprovement. No factor has been identified to predict such a response except that cysteinylleukotrienes release from leukocytes is correlated with leukotriene receptor antagonistresponse.259
In summary, while cysteinyl leukotrienes are important pro-inflammatory and broncho-constrictor mediators in the pathogenesis of asthma, leukotriene receptor antagonistsdemonstrate hybrid anti-inflammatory and bronchodilatory properties.260 A meta-analysisfound that these agents reduced exacerbations by 50% and reduced the requirement ofadditional asthma therapy.261 Another meta-analysis from 13 trials showed weightedestimated protection of leukotriene receptor antagonists amounted to a 0.85 doubling doseshift, thus the estimated protection amounted to almost one doubling dose reinforcing therole of these agents as anti-inflammatory therapy in asthma.262 Current InternationalGuidelines recommend using an leukotriene receptor antagonist as first-line therapy inpatients with mild, persistent asthma, or as second-line therapy in conjunction with inhaledcorticosteroids, as an alternative to increasing the dose of inhaled corticosteroid. Further,leukotriene-receptor antagonists confers significant additive pro-inflammatory effects totherapy with a low-dose inhaled corticosteroid.263
Alternative Treatment for Oral Steroid Dependence
Approximately 10% of patients with asthma have severe disease and require high doses ofinhaled or oral glucocorticoids. Some of these patients may have more severe disease becauseof relative resistance to the effect of glucocorticoids.264 Although this group may constitutea very small proportion of the total cases, they consume more than 50% of the resources. Theyrequire more frequent medical attention, need more expensive drugs, more often hospitalised,

162 Bronchial Asthma
and miss more time for work or school than patients with milder form of the disease. Treatmentof patients with severe, persistent asthma who require high doses of systemic steroids presentsa therapeutic challenge. Such high doses of steroids for longer periods of time have multiplesystemic side effects. Several modalities of therapeutic regimens have been advocated/triedto help reduce oral steroid dependence in severe asthma. Some of these approaches are stillexperimental and should be used only in specialised centers.
Before labelling someone having steroid resistance preventable factors need to be considered.These include poor compliance, occupational factors, gastro-oesophageal reflux, specificantigens, and dietary factors. Anxiety about asthma deaths might lead to overuse of steroids.Superimposed psychosocial factors and hyperventilation syndrome may cause furtherproblem. Some patients are truly corticosteroid resistant, i.e. shows response to bronchodilatorsbut none to corticosteroids.265,266 These patients should not be prescribed corticosteroids. Somepatients having severe asthma and who show some response to steroids should not be labelledas steroid resistant cases, but as severe asthma only. Before attempting any of thefollowing experimental drugs, a trial of high doses of inhaled corticosteroids(2 to 4 times the usual doses) is essential. This approach has the lowest incidence of adverseeffects and has a high likelihood of clinical efficacy. In the steroid dependent asthma, apatient, treatment with high-dose inhaled corticosteroids should be maintained over a periodof several weeks to months, and the dose of oral steroids should be reduced slowly whilemonitoring pulmonary function. This approach is often helpful.
Various drugs have been proposed as alternatives to systemic steroids, includingtroleandomycin, gold, azathioprine, methotrexate, and cyclosporine, intravenousimmunoglobulins, hydroxychloroquine, dapsone, inhaled frusemide, and intravenousmagnesium sulphate.267-270 Since asthmatic inflammation may be regulated by Th2lymphocytes, some of these drugs which are immunosuppressive, act due to their action ofinhibition of T-lymphocytes.
Methotrexate is an antimetabolite which antagonizes folic acid by inhibiting dihydro-folate reductase. This interferes with thymidine synthesis and thus blocks DNA synthesisand cell division. At higher doses, it is an antineoplastic agent and in low doses (5-25 mg/week) it acts as an anti-inflammatory and immuno-suppressive agent. The mechanism ofaction as an anti-inflammatory drug include inhibition of histamine release from basophils,inhibition of cytokine release (IL-1) from mononuclear cells, and reduction of neutrophilchemotaxis.271-273 Although some clinical trials have shown benefit from low dose methotrexate,(15 mg/wk) others do not support this.273-279 The drug is also found useful in children.280,281
There is significant reduction in steroid doses, when methotrexate is added in addition tosubjective improvement. The common side effects are nausea, vomiting, hepatic dysfunction,alopecia, oral ulcers, and neutropenia. In spite of the controversy and confusion, the followingprovisional conclusion may be made from available literature:
i. Methotrexate may be a steroid agent in some steroid-dependent patients;ii. No predictive factor could be found about the responders;
iii. No consistent effect on airflow or bronchial responsiveness is expected;iv. To have an appreciable effect, the treatment may be continued for long periods (> 3
months) and unlikely to occur after 1 year;v. Steroid-sparing effect disappears on discontinuation of methotrexate;
vi. Doses more than 15 mg/wk may have unacceptable side effects;

Pharmacologic Management of Asthma 163
vii. Steroid weaning should be done before methotrexate trial; andviii. The drug should be viewed as a risky preposition in comparison to long-term oral
steroid therapy.282
Cyclosporin A is a fungal cyclic polypetide used mostly for transplant patients. It inhibitsthe activation of T lymphocytes and the synthesis and release of lymphokines like IL-2,IL-3, Il-4, IL-5, and TNF (tumor necrosis factor). It also inhibits histamine and LTC4 releasefrom mast cells and basophils and inhibits neutrophil chemotaxis, monocytes-macrophages.283
Use of cyclosporin has shown some result.284,285 Hypertrichosis, hypertension, parasthesia,tremour, headache, and flue-like symptoms are some of the side effects of cyclosporin treatment.Cyclosporin appears to be a promising drug in the treatment of steroid-dependent bronchialasthma.
Gold salts (auranofin) has anti-inflammatory properties and are commonly used for thetreatment of rheumatoid arthritis. The drug has been shown to inhibit IgE-mediated release ofhistamine and LTC4 from basophils and mast cells.286 It also inhibits tracheal smooth musclecontraction in response to histamine and specific antigens in guinea pigs.287 Oral auranofin 3mg twice daily has been found useful.288,289 Mucocutaneous reactions are the common sideeffects of gold therapy.
Other agents like azathioprine,290 intravenous immunoglobulin,291 troleandomycin,292
colchicine,293,294 and hydroxychloroquine295 have been suggested to be alternatives in steroid-dependent asthma. But the experience in clinical practice is not much, and they are notrecommended for the routine use in these patients.
REFERENCES
1. Weinberger M. The pharmacology and therapeutic use of theophylline. J Allergy Clin Immunol1984;73:525.
2. Arkinstall WW. The role of theophyllines in a preventive approach for subjects with both mildand severe asthma. Postgrad Med J 1991;67(Suppl 4):S25.
3. Isles A, MacLeod SM, Levison H. Theophylline. New thoughts about an old drug. Chest1982;82(Suppl):49S-54S.
4. Barnes PJ, Pauwels RA. Theophylline in the management of asthma: Time for reappraisal? EurRespir J 1994;7:579-91.
5. Beavo JA, Reifsynder DH. Primary sequence of cyclic nucleotide phosphodiesterase isoenzymesand the design of selective inhibitors. Trends Pharmacol Sci 1990;11:150-55.
6. Nicholson CD, Challiss RAJ, Shahid M. Differential modulation of tissue function and therapeuticpotential of selective inhibitors of cyclic nucleotide phosphodiesterase isoenzymes. TrendsPharmacol Sci 1991;12:19-27.
7. Trophy TJ, Undern RJ. Phosphodiesterase inhibitors; new opportunities for the treatment ofasthma. Thorax 1991;46:499-503.
8. Giembycz MA. Could selective cyclic nucleotide phosphodiestrase inhibitors renderbronchodilator therapy redundant in the treatment of bronchial asthma? Biochem Pharmacol1992;43:2041-51.
9. Bergstrand H. Phosphodiesterase inhibition and theophylline. Eur J Respir Dis 1980;61(Suppl109):37-44.
10. Polson JB, Kazanowski JJ, Goldman AL, Szentivanyl A. inhibition of human pulmonaryphosphodiesterase activity by therapeutic levels of theophylline. Clin Exp Pharmacol Physiol1978;5:535-39.

164 Bronchial Asthma
11. Kuehl FA, Zanetti ME, Soderman DD, Miller DK, Ham EA. Cyclic AMP-dependent regulation oflipid mediators in white cells. A unifying concept of explaining the efficacy of theophylline inasthma. Am Rev Respir Dis 1987;136:210-13.
12. Estenne M, Yernault J, de Troyer A. Effect of parenteral aminophylline on lung mechanics innormal humans. Am Rev Respir Dis 1980;121:967-71.
13. Cushley MJ, Tattersfield AE, Holgate ST. Adenoine-induced bronchoconstriction in asthma;antagonism by inhaled theophylline. Am Rev Respir Dis 1984;129:380-84.
14. Bjork T, Gustafsson LE, Dahlen SE. Isolated bronchi from asthmatics are hyperresponsive toadenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am RevRespir Dis 1992;145:1087-91.
15. Mann JS, Holgate ST. Specific antagonism of adenosine induced bronchoconstriction in asthmaby oral theophylline. Br J Clin Pharmacol 1985;19:85-92.
16. Fredholm BB, Persson CGA. Xanthine derivatives as adenosine receptor antagonists. Eur JPharmacol 1982;81:673-76.
17. Higbee MD, Kumar M, Galant SP. Stimulation of endogenous catecholamine release bytheophylline, a proposed additional mechanism of theophylline effects. J Allergy Clin Immunol1982;70:377-82.
18. Ishizaki T, Minegishi A, Morishita A et al. Plasma catecholamine concentration during a 72-houraminophylline infusion in children with acute asthma. J Allergy Clin Immunol 1988;92:146-54.
19. Jenne JW. Two roles for theophylline in the asthmatics? J Asthma 1995;32:89-95.20. Cresioli S, Spinazzi A, Plebani M et al. Theophylline inhibits early and late asthmatic reaction
induced by allergens in asthmatic subjects. Ann Allergy 1991;66:245-51.21. Cresioli S, de Marzo N, Boschetto P et al. Theophylline inhibits late asthmatic reactions induced
by toluene diisocyanate in sensitised subjects. Eur J Pharmacol Environ Toxicol 1992;228:45-50.22. Erjefalt I, Persson CGA. Pharmacologic control of plasma exudation into tracheobronchial airways.
Am Rev Respir Dis 1991;143:1008-14.23. Decramer M, Deschepper K, Jiang T, Derom E. Effects of aminophylline on respiratory muscle
interaction. Am Rev Respir Dis 1991;144:797.24. Kolbeck RC, Spier WA, Carrier GO, Bransome ED. Apparent irrelevance of cyclic nucleotides to
the relaxation of tracheal smooth muscle induced by theophylline. Lung 1979;156:173-83.25. Bowler SD, Mitchell CA, Armstrong JG. Nebulised fenoterol and IV aminophylline in acute
severe asthma. Eur J Respir Dis 1987;70:280-83.26. Wrenn K, Slovis CM, Murphy F, Greenberg RS. Aminophylline therapy for acute bronchspastic
disease in the emergency room. Ann Intern Med 1991;115:241-47.27. Eason J, Makowe HLJ. Aminophylline toxicity: How many hospital deaths does it cause? Resp
Med 1989;83:219-26.28. Lam A, Newhouse MT. Management of asthma and chronic airflow limitation. Are methyl-
xanthines obsolete? Chest 1990;98:44-52.29. Trigg CJ, Davies RJ. Use of slow-release theophylline in asthma: It is justified? Respir Med
1990;84:1-3.30. Marks MB. Theophylline: Primary or tertiary drug? A brief review. Ann Allergy 1987;59:85-89.31. Mitenko PA, Ogilvie RI. Rational intravenous doses of theophylline. N Engl J Med 1973;289:600-
03.32. Fittar G, Friedman HS. The arrhythmogenicity of theophylline. A multivariate analysis of clinical
determinants. Chest 1991;99:1415.33. Furukawa CT, Sapiro CG, Duhamel T et al. Learning and behavioural problems associated with
theophylline therapy. Lancet 1985;1:621.34. Furukawa CT, Duhamel T, Weimer L et al. Cognitive and behavioural findings in children taking
theophylline. J Allergy Clin Immunol 1988;81:83-85.

Pharmacologic Management of Asthma 165
35. Rachelefsky WOJ, Adelson J, Mickey MR et al. Behavioural abnormalities and poor schoolperformance due to oral theophylline use. Paediatrics 1986;78:1113-38.
36. Lindgren S, Loksin B, Stromquist A et al. Does asthma or treatment with theophylline limitchildren’s academic performance. N Engl J Med 1992;327:926-30.
37. Persson CGA. Development of safer xanthine drugs for the treatment of obstructive airwaydisease. J Allergy Clin Immunol 1986;78:817-24.
38. Chapman KR, Bryant D, Marlin GE et al. A placebo-controlled dose-response study of enprofyllinein the maintenance therapy of asthma. Am Rev Respir Dis 1989;139:688-93.
39. Weinberger M. The pharmacology and therapeutic use of theophylline. J Allergy Clin Immunol1984;73:525-40.
40. Weinberger M., Hendeles l. Sloe-release theophylline: Rationale and basis for product selection.N Engl J Med 1983;308:760-764.
41. Hendeles L, Weinberger M. Theophylline: A state of the art review. Pharmacotherapy 1983;3:2-44.
42. Shenfield GM, Brogaden RN, Ward A. Pharmacology of bronchodilators. In: Bronchodilatortherapy: The basis of asthma and chronic obstructive airways management., Cochrane GM Ed:ADIS Press Limited, Auckland, NZ, 1984.
43. Fraser CM, Potter PC, Venter JC, Nelson HS, Middleton E: Adrenergic agents. In: Middleton E etal, (Eds). Allergy: Principles and Preactice, Vol I, CV Mosby Company, St.louis,1988.
44. Szentivanyi A, Krzanowski JJ, Polson JB: The autonomic nervous system. In: Middleton E et al,(Eds). Allergy: Principles and Practice, Vol I, CV Mosby Company, St.louis,1988.
45. Holtzmzn MJ: Pathophysiology in asthma: An overview of mechanisms of bronchialhyperreactivity. In: Morley J (Eds). Bronchial hyperreactivity (Perspectives in asthma;1), AcademicPress, London,1982.
46. Rang HP. Drug receptors. University Park Press, Baltimore, 1973.47. Cooke BA: β−adrenoceptor-adenylate cyclase: Basic mechanisms of control. In: Morley J (Eds).
Β-adrenoceptors in asthma (Perspectives in asthma;2), Academic Press London,1982.48. Kume H, Graziano MP, Kotokoff MI. Stimulatory and inhibitory regulation of calcium-activated
potassium channels by guanine nucleotide binding proteins. Proc Natl Acad Sci USA1992;89:11051-55.
49. Jones TR, Charette L, Gracia ML, Kaczorowski GJ. Selective inhibition of relaxation of guinea-pigtrachea by charybodotoxin, a potent Ca++-activated K+ channel inhibitor. J Pharmacol Exp Ther1990;225:697-706.
50. Miura M, Belvisi MG, Stretton CD, Yacoub MH, Barnes PJ. Role of potassium channels inbronchodilator response in human airways. Am Rev Respir Dis 1992;146:132-36.
51. Jones TR, Charette L, Gracia ML, Kaczorowski GJ. Interaction of iberiotoxin with β−adrenoceptoragonists and sodium nitroprusside on guinea-pig trachea. J Appl Physio 1993;74:1879-84.
52. Rees J. β−2 agonists and asthma. Still the main stay of symptomatic treatment. Br Med J1991;302:1166.
53. Meltzer DL, Kemp JP. β2-agonists: Pharmacology and recent developments. J Asthma 1991;28:179-86.
54. Erjefalt I, Persson GA. Long duration and high potency of anti-exudative effects of fermoterol inGuinea-pig tracheobronchial airways. Am Rev Respir Dis 1991;144:788.
55. Kesten S, Chapman KR, Broder I et al. A three-month comparison of twice daily inhaled formoterolversus four times daily inhaled albuterol in the management of stable asthma. Am Rev RespirDis 1991;144:622.
56. Gongora HC, Wisniewski AFZ, Tattersfield AE. A single dose comparison of inhaled albuteroland two formulations of salmeterol on airway reactivity in asthmatic subjects. Am Rev RespirDis 1991;144:626.
57. Janson C. Plasma levels and effects of salbutamol after inhaled or IV administration in stableasthma. Eur Respir J 1991;4:544.

166 Bronchial Asthma
58. Tinkelman DG, Berkowitz RB, Cole WQ III. Aerosols in the treatment of asthma. J Asthma1991;28:243.
59. Woolcock AJ. Inhaled drugs in the prevention of asthma. Am Rev Respir Dis 1977;115:191.60. Maesen FP, Costongs R, Smeetys JJ, Brobacher BJ, Zweers PGMA. The effect of maximal doses
of fermoterol from a metered dose inhaler on pulse rates, ECG, and serum potassiumconcentrations. Chest 1991;99:1367.
61. Haahtela T, Jarvinen M, Kava T et al. Comparison of a β2-agonist, terbutaline, with an inhaledcorticosteroid, budesonide, in newly detected asthma. N Engl J Med 1991;325-88.
62. Nelson HS, Szefler S, Martin RJ. Regular inhaled β−adrenergic agonists in the treatment ofbronchial asthma: Beneficial or detrimental? Am Rev Respir Dis 1991;144-249.
63. Carstairs JR, Nimmo AJ, Barnes PJ. Autoradiographic visualisation of β-adrenoceptor subtypesin human lung. Am Rev Respir Dis 1985;132:541-47.
64. Hamid QA, Mak JC, Sheppard MN et al. Localisation of β2-adrenoceptor messenger RNA inhuman and rat lung using in situ hybridisation: Correlation with receptor autoradiography. EurJ Pharmacol 1991;206:133-38.
65. Barnes PJ. β−adrenergic receptors and their regulation. Am J Respir Crit Care Med 1995;152:838-60.
66. Butchers PR, Skidmore IF, Vardey CJ et al. Characterisation of the receptor mediatingantianaphylactic effects of β-adrenoceptor agonists in human lung tissues in vitro. Br J Pharmacol1980;71:663-67.
67. Devalia JL, Sapsford RJ, Rusznak C et al. The effects of salmeterol and salbutamol on cilliary beatfrequency of cultured human bronchial epithelial cells in vitro. Pulm Pharmacol 1992;5:257-63.
68. Boulet LP. Long versus short-acting β2-agonists. Drug 1994;47:207-22.69. Rossi A, Kristufek P, Levine BE et al. Comparison of the efficacy, tolerability, and safety of
formoterol dry powder and oral, slow-release theophylline in the treatment of COPD. Chest2002;121:1058-69.
70. Moore RH, Khan A, Dickey BF. Long-acting inhaled β2-agonists in asthma therapy. Chest1998;113:1095-1108.
71. Jeppsson AB, Kallstrom BL, Waldeck B. Studies on the interaction between formoterol andsalmeterol in guinea pig trachea in vitro. Pharmacol Toxicol 1992;71:272-77.
72. Derom EY, Pauwels RA, Van der Straeten ME. The effect of inhaled salmeterol on methacholineresponsiveness in subjects with asthma up to 12 hours. J Allergy Clin Immunol 1992;89:811-15.
73. Derom EY, Pauwels RA. Time course of bronchodilating effect of inhaled formoterol, a potentand long acting sympathomimetic. Thorax 1992;47:30-33.
74. Ramsdale EH, Otis J, Kline PA et al. Prolonged protection against methacholine inducedbronchoconstriction by the inhaled β2-agonist formoterol. Am Rev Respir Dis 1991;143:998-1001.
75. Dahl R, Earnshaw JS, Palmer JBD. Salmeterol: A four week study of a long acting β-adrenoceptoragonist for the treatment of reversible airway disease. Eur Respir J 1991;4:1178-84.
76. Midgren B, Melander B, Persson G. Formoterol, a new long acting β2-agonist, inhaled twicedaily, in stable asthmatic subjects. Chest 1992;101:1019-22.
77. Ulman A, Hedner J, Svedmyr N. Inhaled salmeterol and salbutamol in asthmatic patients: Anevaluation of asthma symptoms and the possible development of tachyphylaxis. Am Rev RespirDis 1990;142:571-75.
78. Campbell LM, Anderson TJ, Paraschak MR et al. A comparison of the efficacy of long-acting β2-agonists: Eformoterol via Turbohaler and salmeterol via pressurised metered dose inhaler orAccuhaler, in mild to moderate asthmatics. Respir Med 1999;93:236-44.
79. Vervoet D, Ekstrom T, Pela R et al. A six-month comparison between formoterol and salmeterolin patients with reversible obstructive airways disease. Respir Med 1998 1998;92:836-42.
80. Nightingale J, Rogers DF, Barnes PJ. Comparison of the effects of salmeterol and formoterol inpatients with severe asthma. Chest 2002;121:1401-06.

Pharmacologic Management of Asthma 167
81. Naline E, Zhang Y, Qian Y et al. Relaxant effects and duration of action of formoterol andsalmeterol on the isolated human bronchus. Eur Respir J 1994;7:914-20.
82. Palmqvist M, Persson G, Lazer L et al. Inhaled dry powder formoterol and salmeterol in asthmaticpatients: onset of action, duration of effect, and potency. Eur Respir J 1997;10:2484-89.
83. Newton MF, O’Donnell DE, Forkert L. Response of lung volumes to inhaled salbutamol in alarge population of patients with severe hyperinflation. Chest 2002;121:1042-50.
84. Anderson SD, Seale JP, Rozea P et al. Inhaled and oral salbutamol in exercise-induced asthma.Am Rev Respir Dis 1976;114:493-500.
85. McFadden ER Jr, Hejal R. Emergency medicine: Asthma. Lancet 1995;345:1215-20.86. Grove A, Lipworth BJ. Tolerance with β2-adrenoceptor agonists: Time for reappraisal. Br J Clin
Pharmacol 1995;39:109-18.87. Yates DH, Worsdell M, Sussman H et al. Regular formoterol treatment in mild asthma: Effect on
bronchial reactivity during and after treatment (abstract). Am J Respir Crit CareMed 1995;151:A272.
88. O’Connor BJ, Aikman SL, Barnes PJ. Tolerance to the non-bronchodilator effects of inhaled β2-agonists. N Engl J Med 1992;327:1204-08.
89. Cockroft D, McParland CP, Britton SA et al. Regular inhaled salbutamol and airway responsive-ness to allergen. Lancet 1993;342:833-37.
90. Cheung D, Timmers MC, Zwinderman AH et al. Long-term effect of a long-acting β2-adrenoceptoragonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. New Engl JMed 1992;327:1198-1203.
91. Ramage L, Lipworth BJ, Ingran CG et al. Reduced protection against exercise induced broncho-constriction after chronic dosing with salmeterol. Respir Med 1994;88:363-68.
92. Tattersfied AE, Britton JR. β-adrenoceptor agonists. In: Barnes PJ (Eds). Asthma—Basicmechanisms and clinical management. Academic Press, London, 1988.
93. Green SA, Turki J, Innis M et al. Amino-terminal polymorphisms of the human β2-adrenergicreceptor impart distinct agonist-promoted regulatory properties. Biochemistry 1994;33:9414-19.
94. Turki J, Pak J, Green SA et al. Genetic polymorphism of the β2-adrenergic receptor in nocturnaland non-nocturnal asthma: Evidence that Gly16 correlates with thenocturnal phenotype. J ClinInvest 1995;95:1635-41.
95. Hall IP, Wheatley A, Wilding P et al. Association of Glu 27 β2-Adrenoceptor polymorphism withlower airway reactivity in asthmatic subjects. Lancet 1995;345:1213-14.
96. Speitzer WO, Suissa S, Ernst P et al. The use of β-agonists and the risk of death and near deathfrom asthma. N Engl J Med 1992;326:501-06.
97. Crane J, Pearce N, Flatt A. et al. Prescribed fenoterol and death from asthma in New Zealand,1981-83: A case control study. Lancet 1989;1:917-22.
98. Grainger J, Woodman K, Pearce N et al. Prescribed fenoterol and death from asthma in NewZealand, 1981-7: A further case control study. Thorax 1991;46:105-11.
99. Suissa S, Ernst P, Boivin J et al. A cohort analysis of excess mortality in asthma due to use ofinhaled β-agonists. Am J Respir Crit Care Med 1994;149:604-10.
100. Barrett TE, Strom BL. Inhaled β-adrenergic receptor agonists in asthma: More harm than good?Am J Respir Crit Care Med 1995;151:574-77.
101. Crane J, Pearce N, Burgess C, Beasley R. Asthma and the β-agonist debate. Thorax 1995;50(Suppl):S5-S10.
102. Speizer FE, Doll R, Heaf P. Observations on recent increase in mortality from asthma. BMJ1968;1:335-39.
103. Muller MI, Mullen B, Carey M. The association between β2-agonist use and death from asthma.JAMA 1993;270:1842-45.
104. Bernstein IL. β2-agonists: déjà vu all over again. The second generation controversy. Chest2002;122:763-65.

168 Bronchial Asthma
105. Greeen SA, Turki J, Bejarano P et al. Influence of β2-adrenergic receptor genotype on signaltransduction in human airway smooth muscle cells. Am J Respir Cell Mol Biol 1995;13:25-33.
106. Barnes PJ. Scientific rationale for inhaled combination therapy with long-acting β2-3agonists andcorticosteroids. Eur Respir J 2002;19:182-91.
107. Wong CS, Wahedna I, Pavord ID et al. Effects of budesonide and terbutaline on bronchialreactivity to allergy in subjects with mild atopic asthma. Thorax 1992;47:231P(Abstract).
108. Booth H, Fishwick K, Harkawat R et al. Changes in methacholine induced bronchoconstrictionwith the long acting β2-agonist salmeterol in mild to moderate asthmatic patients. Thorax1993;48:1121-24.
109. Rosenthal RR, Busse WW, Kemp JP et al. Effect of long-term salmetrol therapy compared withas-needed albuterol use on airway hyperresponsiveness. Chest 1999;116:595-602.
110. Van Schayck CP, Cloosterman SGM, Hofland ID et al. How detrimental is chronic use ofbronchodilators in asthma and chronic obstructive lung disease? Am J Respir Crit Care Med1995;151:1317-19.
111. Prieto L, Gutierrez V, Torres V, et al. Effect of salmeterol on seasonal changes in airwayresponsiveness and exhaled nitric oxide in pollen-sensitive asthmatic subjects. Chest 2002;122:798-805.
112. Gandevia B. Historical review of the use of parasympatholytic agents in the treatment ofrespiratory disorders. Postgrad Med J 1975;51(Suppl 7):13-20.
113. Gross NJ, Skorodin MS. Anticholinergic antimuscarinic bronchodilators. Am Rev Respir Dis1984;129:856-70.
114. Mann JS, George CF. Anticholinergic drugs in the treatment of airway disease. Brit J Dis Chest1985;79:209.
115. Ziment I, Au JP. Anticholinergic agents. Clin Chest Med 1986;7:454-58.116. Weber R. Role of anticholinergics in asthma. Ann Allergy 1990;65:348-50.117. Beakes DE. The use of anticholinergics in asthma. J Asthma 1997;34:357-68.118. Ward MJ, Fentem PH, Roderick-Smith WH et al. Ipratropium bromide in acute asthma. BMJ
1981;282:598-600.119. Richardson JB. Nerve supply to the lungs. Am Rev Respir Dis 1979;119:785-802.120. Barnes PJ, Minette P, Maclagan J. Muscarinic subtypes in airways. Trends Pharmacol Sci 1988;9:
412-16.121. Fryer AD, Jacoby DB. Effect of inflammatory cell mediators on M2 muscarinic receptors in the
lungs. Life Sci 1993;52:529-36.122. Baigelman W, Chodosh S. Bronchodilator action of the anticholinergic drug, ipratropium bromide,
as an aerosol in chronic bronchitis and asthma. Chest 1977;71:324.123. Frith PA, Jenner B, Dangerfield R, Atkinson J, Drennan C.. Oxytropium bromide. Dose-response
and time-response study of a new anticholinergic bronchodilator drug. Chest 1986;89:249-53.124. Storms WW, Bodman SF, Nathan RA et al. Use of ipratropium bromide in asthma. Am J Med
1986;81(Suppl 5A):61-66.125. Barnes PJ. Muscarinic receptor subtypes in airways. Life Sci 1993;52:521-27.126. Patel HJ, Barnes PJ, Takahasi T et al. Evidence for prejunctional muscarinic autoreceptors in
human and guinea pig trachea. Am J Respir Crit Care Med 1995;152:872-78.127. Tamaoki J, Chiyotani A, Tagaya E et al. Effect of long-term treatment with oxytropium bromide
in airway secretion in chronic bronchitis and diffuse panbronchiolitis. Thorax 1994;49:545-48.128. Seifkin AD,. Optimal pharmacologic treatment of the critically ill patients with obstructive airway
disease. Am J Med 1996;100(Suppl):545-615.129. Schlueter DP. Ipratropium bromide in asthma: A review of the literature. Am J Med 1986;
81(Suppl):55-59.130. Chapman KR, Smith DL, Rebuck AS et al. Haemodynamic effects of inhaled ipratropium bromide
alone and combined with an inhaled β-agonist. Am Rev Respir Dis 1985;132:845-47.

Pharmacologic Management of Asthma 169
131. O’Driscoll BR, Taylor RJ, Horsley MG et al. Nebulised salbutamol with and without ipratropiumbromide in acute airflow obstruction. Lancet 1989;1:1418-20.
132. Rebuck AS, Chapman KR, Abboud R et al. Nebulised anticholinergic and sympathomimetictreatment of asthma and chronic obstructive airways disease in the emergency room. Am J Med1987;82:59-64.
133. Cugell DW,. Clinical pharmacology and toxicology of ipratropium bromide. Am J Med 1986;81:18-22.
134. Gross NJ. Ipratropium bromide. N Engl J Med 1988;319;486-94.135. Peel ET, Cheong AB, Broderick N. A comparison of oxytropium bromide and ipratropium
bromide in asthma. Eur Respir J 1984;65:106-08.136. O’Connor BJ, Towse LJ, Barnes PJ. Prolonged effect of tiotropium bromide on methacholine
induced bronchoconstriction in asthma. Am J Respir Crit Care Med 1996;154:876-80.137. Disse B, Witek TJ Jr. Anticholinergics: Tiotropium. In: Hansel TT, Barnes PJ (Eds): New drugs for
Asthma, Allergy and COPD. Prog Respir Res, Basel, Karger, 2001; 31,72-76.138. Bethel RA, Irvin CG. Anticholinergic drugs and asthma. Semin Respir Med 1987;8:366-71.139. Ind PW, Dixon CMS, Fuller RW, Barnes PJ. Anticholinergic blockade of β-blocker-induced
bronchoconstriction. Am Rev Respir Dis 1989;139:1390-94.140. Barnes PJ. Inhaled glucocorticosteroids for asthma. N Engl J Med 1995;332:868-70.141. Corticosteroids: their biological mechanisms and application to the treatment of asthma. Am
Rev Respir Dis 1990;141(Suppl):S2.142. Kaliner M. Mechanisms of glucocorticoid action in bronchial asthma. J Allergy Clin Immunol
1985;76:321.143. Beato M. Gene regulation by steroid hormones. Cell 1989;56:335-344.144. Guyre PM, Girard MT, Morganelli P, Managaniello PD. Glucocorticoid effects on the production
and action on immune cytokines. J Steroid Biochem 1988;30:88-93.145. Schleimer RP. Effects of glucocorticoids on inflammatory cells relevant to their therapeutic
applications in asthma. Am Rev Respir Dis 1990;141:559-69.146. Laitinen LA, Laitinen A, Haahtela T et al. A comparative study of the effects of an inhaled
corticosteroid, budesonide, and a β2-agonist , terbuline, on airway inflammation in newlydiagnosed asthma. J Allergy Clin Immunol 1992;90:32-42.
147. Lee TH, Lane SJ. The role of macrophages in the mechanisms of airways inflammation inasthma. Am Rev Respir Dis 1992;145:527-30.
148. Kita II, Abu-Ghazaleh R, Sanderson CJ, Gleich GJ. Effect of steroids on immunoglobulin inducedeosinophil degranulation. J Allergy Clin Immunol 1991;87:70-77.
149. Her E, Frazer J, Austen KF, Owen WF Jr. Eosinophil haemopoitins antagonise the programmedcell death of eosinophils. Cytokine and glucocorticoid effects on eosinophils maintained byendothelial cell-conditioned medium. J Clini Invest 1991;88:1982-87.
150. Evans PM, O’Connor B, Fuller RW, Barnes PJ, Chung KF. Effect of inhaled corticosteroids onperipheral eosinophil counts and density profiles in asthma. J Allergy Clin Immunol 1993; 91:643-49.
151. Boschetto P, Rogewrs DF, Faibbri LM, Barnes PJ. Corticosteroid inhibition of airway micro-vascular leakage. Am Rev Respir Dis 1991;141:1044-1049.
152. Shimura S, Sasaki HT. Ikeda K, Yammauchi K, Sasaki H, Takishim T. Direct inhibitory action ofglucocorticoid on glycoconjugate secretion from airway submucosal glands. Am Rev Respir Dis1990;141:1041-9.
153. Burke C, Power CK, Norris A, Condez A, Schmekel B, Poulter LW. Lung function and immuno-pathological changes after inhaled corticosteroid therapy in asthma. Eur Respir J 1992;5:73-79.
154. Barnes PJ, Adcock IM. Anti-inflammatory action of steroids: Molecular mechanisms. TrendsPharmacol Sci 1993;14:436-41.

170 Bronchial Asthma
155. Mitchell JA, Belvisi MG, Akarasereenont P et al. Induction of cyclooxygenase-2 by cytokines inhuman pulmonary epithelial cells: Regulation by dexamethasone. Br J Pharmacol 1994;113:1008-14.
156. Robbins RA, Barnes PJ, Springall DR et al. Expression of inducible nitric oxide synthase in humanbronchial epithelial cells. Biochem Biophs Res Commun 1994;203:209-18.
157. Van de Stolpe A, Caldenhoven E, Raaijmakers JAM et al. Glucocorticoid-mediated repression ofintercellular adhesion molecule-1 expression in human monocytic and bronchial epithelial celllines. Am J Respir Cell Mol Biol 1993;8:340-47.
158. Djukanovie R, Wilson JW, Britten KM, et al. Effect of an inhaled corticosteroid on airwaysinflammation and symptoms in asthma. Am Rev Respir Dis 1992;145:669-74.
159. Lundgren R, Soderberg M, Horstedt P, Stenling R. Morphological studies of bronchial mucosalbiopsies from asthmatics before and after ten years treatment with inhaled steroids. Eur RespirJ 1988;1:883-89.
160. Barnes PJ, Dolery Ct, MacDernot J. Increased pulmonary alpha-adrenergic and reduced β2-adrenergic receptors in experimental asthma 1980;285:569-71.
161. Mano K, Akharzadeh A, Kosesnadi K et al. The effect of hydrocortisone on β-adrenergic receptorsin lung tissue. J Allergy Clin Immunol 1979;63:147.
162. Brodde OE, Brinkman M, Schermuth R et al. Terbutaline induced desensitisation of humanlymphocyte β2-adrenoceptors: accelerated restoration of β-adrenergic responses by prednisoloneand ketotifen. J Clin Invest 1985;76:1096-1101.
163. Kraan J, Koeter GH, van der Mark THW, et al. Changes in bronchial hyperreactivity induced by4 weeks of treatment with antiasthma drugs in patients with allergic asthma: A comparisonbetween budesonide and terbutaline. J Allergy Clin Immunol 1985;76:628-36.
164. Kerstjens HAM, Brand PLP, Hughes MD et al, and the Dutch chronic non-specific (CNSLD)study group. A comparison of the bronchodilator therapy with or without inhaled corticosteroidtherapy for obstructive airways disease. N Engl J Med 1992;327:1413-19.
165. Kerrebijn KF, van Essen-Zandvliet EEM, Neijens HJ. Effect of long-term treatment with inhaledcorticosteroids and beta-agonists on the bronchial responsiveness in children with asthma.J Allergy Clin Immunol 1987;79:653-659.
166. Juniper EF, Kline PA, Vanzieleghem MA et al. Long-term effect of budesonide on airwayresponsiveness and clinical asthma severity in inhaled steroid-dependent asthmatics. Eur RespirJ 1990;3:1122-1127.
167. Juniper EF, Kline PA, Vanzieleghem MA et al. Effect of long-term treatment with an inhaledcorticosteroid (budesonide) on airway hyperresponsiveness and clinical asthma in nonsteroid-dependant asthmatics. Am Rev Respir Dis 1990;142:832-36.
168. Haahtela T, Jarvinen M, Kava T et al. Comparison of a agonist, terbutaline, with an inhaledcorticosteroid, budesonide, in newly detected asthma. N Engl J Med 1991;325:388-92.
169. Overbeek SE, Kerstjens HAM, Bogaard JM et al. Is delayed introduction of inhaled corticosteroidsharmful in patients with obstructive airways disease (asthma and COPD)? The Dutch chronicnon-specific lung disease (CNSLD) study group. Chest 1996;110:35-41.
170. Haahtela T, Jarvinen M, Kava T et al. Effects of reducing or discontinuing inhaled budesonide inpatients with mild asthma. N Engl J Med 1994;331:700-05.
171. Rob Douma W, Kerstjens HAM, Gooijer AD et al. Initial improvement in lung function andbronchial hyperresponsiveness are maintained during a 5 years of treatment with inhaledbeclomethasione dipropionate and terbutaline. Chest 2002;121:151-57.
172. Dompeling E, van Schayck CP, van Grunsven PM et al. Showing the deterioration of asthma andchronic obstructive pulmonary disease observed during bronchodilator therapy in adding inhaledcorticosteroids: A 4-year prospective study. Ann Intern Med 1993;118:770-78.
173. Haahtela T, Jarvinen M, Kava T et al. Comparison of a β2-agonist terbutaline with an inhaledcorticosteroid, budesonide, in newly detected asthma. N Engl J Med 1991;325:388-92.

Pharmacologic Management of Asthma 171
174. Cockcroft DW, Murdock KY. Comparative effects of salbutamol, sodium cromoglycate andbeclomethasone dipropionate on allergen-induced early asthmatic response, late asthmaticresponse, and increased bronchial responsiveness to histamine. J Allergy Clin Immunol1987;79:734-40.
175. Barnes PJ. Effect of corticosteroids on airway hyperresponsiveness. Am Rev Respir Dis1990;141:S70-S76.
176. Vathenen AS, Knox AJ, Wisniewiski A, Tatterfield AE. Time course of change in bronchialreactivity with an inhaled corticosteroid in asthma. Am Rev Respir Dis 1991;143:1317-21.
177. Juniper EF, Kline PA, Vanzieleghem MA et al. Airway responsiveness and clinical asthma severityin steroid-dependent asthmatics. Eur Respir J 1990;3:1122-27.
178. Faul JL, Demers EA, Burke CM, Poulter LW. Alterations in airway inflammation and lung functionduring corticosteroid therapy for atopic asthma. Chest 2002;121:1414-20.
179. Clark TJH. Inhaled corticosteroid therapy: A substitute for theophylline as well as prednisolone?J Allergy Clin Immunol 1985;76:330.148.Reed CE. Aerosol steroids as primary treatment of mildasthma. N Engl J Med 1991;325:425.
180. Reed CE. Aerosol steroids as primary treatment of mild asthma. N Engl J Med 1991;325:425.181. Wooley MJ, Denberg JA, Ellis R et al. Allergen-induced changes in bone marrow progenitors
and airway responsiveness in dogs and the effect of inhaled budesonide on these parameters.Am J Respir Cell Mol Biol 1994;11:600-06.
182. Howarth P. The relevance of and site of airway inflammation in asthma and targeted aerosoldelivery. Int J Clin Pract Suppl 1999;106:3-10.
183. Hamid Q, Song Y, Kotsimbos TC et al. Inflammation of small airways in asthma. J Allergy ClinImmunol 1997;100:44-51.
184. Adcock IM, Gilbey T, Gelder CM et al. Glucocorticoid receptor localisation in normal andasthmatic lung. Am J Respir Crit Care Med 1996;154:771-82.
185. Laursen LC, Taudorf E, Weeke B. High dose inhaled budesonide in treatment of severe steroid-dependent asthma. Eur J Respir Dis 1986;68:19-28.
186. Nikolaizik WH, Marchant JL, Preece MA, Warner JO. Nocturnal cortisol secretion in healthyadults before and after inhalation of budesonide. Am J Respir Crit Care Med 1996;153:97-101.
187. Connett GJ, Warde C, Wooler E, Lenny W. Use of budesonide in severe asthmatics aged 1-3years. Arch Dis Child 1993;69:351-55.
188. Vaz de azevedo M, Coelho M, Pinto Mendes JA, Bugalho de Almeida A. Can a low or moderatedose of inhaled budesonide replace oral non-steroidal anti-asthma treatment? J InternationalMed Res 1991;19:280-88.
189. Tan KS, Grove A, Cargill RI, McFarlane LC, Lipworth BJ. Effects of inhaled fluticasone propionateand oral prednisolone on lymphocyte β2-adrenoceptor function in asthmatic patients. Chest1996;109:343-47.
190. Clissold SP, Hell RC. Budesonide: A preliminary review of its pharmacodynamic propertiesand therapeutic efficacy in asthma and rhinitis. Drugs 1984;28:485-518.
191. Field HV, Jenkinson PMA, Frame MH, Warner JO. Asthma treatment with a new corticosteroidinhaler, budesonide, administered twice daily by spacer inhaler. Arch Dis Child 1982;57:864-66.
192. Johnson M. Development of fluticasone propionate and comparison with other inhaledcorticosteroids. J Allergy Clin Immunol 1998;101:S434-S439.
193. Bootsman GP, Koenderman L, Dekhuijzen PNR et al. Effects of fluticasone propionate andbeclomethasone dipropionate on parameters of inflammation in peripheral blood of patientswith asthma. Allergy 1998;53:653-61.
194. Hogger P. Dose response and therapeutic index of inhaled corticosteroids in asthma. Curr OpinPulm Med 2003;9:1-8.
195. Barnes PJ, Pedersen S. Efficacy and safety of inhaled steroids in asthma. Am Rev Respir Dis1993;148(4 pt2):S1-S26.

172 Bronchial Asthma
196. Brown PH, Greening AP, Compton GK. Large volume spacer devices and the influence of highdose beclomethasone dipropionate on hypothalamo-pituitary-adrenal axis function. Thorax1993;48:233-38.
197. Selroos O, Halme M. Effect of a volumetric spacer and mouth rinsing on systemic absorption ofinhaled corticosteroids from a metered-dose inhaler and dry powder inhaler. Thorax 1991;46:891-94.
198. Lipworth BJ. New perspectives on drug delivery and systemic bioactivity. Thorax 1995;50:105-10.
199. Geddes DM. Inhaled corticosteroids: Benefits and risks. Thorax 1992;47:404-07.200. Brown PH, Blundell G, Greening AP, Crompton GK. Screening for hypothalamo-pituitary-
adrenal axis suppression in asthmatics taking high dose inhaled corticosteroids. Respir Med1991;85:511.
201. Brown PH, Blundell G, Greening AP, Crompton GK. Hypothalamo-pituitary-adrenal axissuppression in asthmatics inhaling high dose corticosteroids. Respir Med 1991;85:501.
202. Shapiro GG, Coneg P. Cromolyn sodium: A review. Pharmacotherapy 1985;5:156-70.203. Bernstein IL. Cromolyn sodium. Chest 1985:87:68S.204. Hoag JE, McFadden ER Jr. Long-term effect of cromolyn sodium on nonspecific bronchial hype
responsiveness: A review. Ann allergy 1991;66:53-63.205. Schwartz HJ, Blumenthal M, Brady R et al. A comparative study of the clinical efficacy of
nedocromil sodium and placebo: How does cromolyn sodium compare as an active controltreatment? Chest 1996;109:945-52.
206. Furukawa CT, Shapiro GG, Bierman CW et al. A double blind study comparing the effectivenessof cromolyn sodium and sustained release theophylline in childhood asthma. Paediatrcs1984;74:453-59.
207. Petty TL, Rollins DR, Christopher K et al. Cromolyn sodium is effective in adult chronic asthma.Am Rev Respir Dis 1989;139:694-701.
208. Thomson NC. Nedocromil sodium: an overview. Respir Med. 1989;83:269-76.209. Petty TL, Rollins DR, Christopher K et al. Cromolyn sodium is effective in adult chronic asthmatics.
Am Rev Respir Dis. 1989;139:694-701.210. Eigen H, Reid JJ, Dahl R et al. Evaluation of the addition of cromolyn sodium to bronchodilator
maintenance therapy in the long-term management of asthma. J Allergy Clin Immunol1987;80:612-21.
211. Bel EH, Timmers MC, Hermmans J, Dijkman JH, Sterk PJ. The long-term effects of nedocromilsodium and beclomethasone dipropionate on bronchial responsiveness to methacholine innonatopic asthmatic subjects. Am Rev Respir Dis 1990;141:21.
212. North American Tilade study group: A double blind multicentre group comparative study ofthe efficacy and safety of nedocromil sodium in the management of asthma. Chest 1990;97:1299.
213. Bone MF, Kubik MM, Keaney NP et al. Nedocromil sodium in adults with asthma dependent oninhaled corticosteroids: A double-blind, placebo controlled study. Thorax 1989;44:654-59.
214. Manolitsas ND, Wang J, Devalia JL et al. Regular albuterol, nedocromil sodium, and bronchialinflammation in asthma. Am J Respir Crit Care Med 1995;2152:1925-30.
215. Heinke S, Szues G, Norris A et al. Inhibition of volume activated chloride currents in endothelialcells by cromones. Br J Pharmacol 1995;115:1392-98.
216. Barnes PJ, Holgate ST, Lattinen LA et al. Asthma mechanisms, determinants of severity andtreatment,: The role of nedocromil sodium. Clin Expt Allergy 1995;25:771-87.
217. Tinkelman DG, Moss BA, Bukantz SC et al. A multicentre trial of the prophylactic effect ofketotifen, theophylline and placebo in atopic asthma. J Allergy Clin Immunol 1985;76:487.
218. Magnussen H.The inhibitor effect of azelastine and ketotifen on histamine induced broncho-constriction in asthmatic patients. Chest 1987;91:855.
219. Phillips G, Rafferty P, Beasley R, Holgate ST. Effect of oral terfenadine on the bronchoconstrictorresponse to inhaled histamine and adenosine-5'-monophosphate in nonatopic asthma. Thorax1987;42:939.

Pharmacologic Management of Asthma 173
220. Bruttmann G, Pedrali P, Arendt C, Rihoux JP. Protective effect of cetrizine in patients sufferingfrom pollen asthma. Ann Allergy 1990;64:224-28.
221. Taylor IK. Cysteinyl leukotrienes in asthma: Current state of therapeutic evaluation. Thorax1995;50:1005-10.
222. Samuelsson B. Leukotrienes: Mediators of immediate hypersensitive reactions and inflammation.Science 1983; 220:568-75.
223. Lewis RA, Austen KF. The biologically active leukotrienes. J Clin Invest 1984;73:889-97.224. Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of lipoxygenase pathway.
New Engl J Med 1990;323:645-55.225. Barnes PJ, Chung KF, Page CP. Inflammatory mediators and asthma. Pharmacol Rev 1988;40:
49-84.226. Piacentini GL, Kaliner MA. The potential roles of leukotrienes in bronchial asthma. Am Rev
Respir Dis 1991;143:S96-S99.227. Arm JP, Lee TH. Sulphidopeptide leukotrienes in asthma. Clin Sci 1993;84:501-510.228. Dahlen SE, Hansson G, Hedquist P et al. Allergen challenge of lung tissue from asthmatics elicits
bronchial contraction that correlated with the release of leukotrienes C4, D4, and E4. Proc NatlAcad Sci, USA 1983;80:1712-16.
229. MacGlashan DW, Schleimer RP, Peters SP et al. Generation of leukotrienes by purified humanlung mast cells. J Clin Invest 1982;70:747-51.
230. Weller PF, Lee CW, Foster DW et al. Generation and metabolism of 5-lipoxygenase pathwayleukotrienes by human eosinophils: Predominant production of leukotriene C4. Proc Natl acadSci USA. 1983;80:7626-30.
231. Kern R, Smith LJ, Patterson R et al. Characterisation of the airway response to inhaled leukotrieneD4 in normal subjects. Am rev Respir Dis 1986;133:1127-32.
232. Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4, LTD4,LTE4 and LTB4) in human skin. J Invest Dermatol 1983;80:115-19.
233. Coles SJ, Neill KH, Leid LM et al. Effects of leukotrienes C4 and D4 on glycoprotein and lysozymesecretion by human bronchial mucosa. Prostaglandins 1983;25:155-70.
234. O’Byrne PM, Leikauf GD, Aizwa H et al. Leukotriene B4 induces airway hyper-responsiveness indogs. J Appl Physiol 1985;59:1941-46.
235. Black PN, Fuller RW, Taylor GW et al. Bronchial reactivity is not increased after inhalation ofleukotriene B4 and prostaglandin D2. Br J Clin Pharmacol 1988;25:667P.
236. Bigby T. Inflammatory mediators and asthma. Pulm Perspectives 1992;9(4):6-9.237. Holgate ST, Bradding P, Sampson AP. Leukotriene antagonists and synthesis inhibitors: New
directions in asthma therapy. J Allergy Clin Immunol 1996;98:1-13.238. Fischer AR, McFadden CA, Frantz R, Awni WM, Cohn J, Drazen JM, Israel E. Effect of chronic
5-lipoxygenase inhibition on airway hyper-responsiveness in asthmatic subjects. Am J RespirCrit Care Med 1995;152:1203-07.
239. Murphy RC, Hammarstorm S, Samuelsson B et al. Leukotriene C: A slow-reacting substancefrom murine mastocytoma cells. Proc Natl Acad Sci USA 1979;76:4275-79.
240. Lee TH. pharmacological modulation of leukotriene and platelet-activating factor biosynthesisand activities by alternative dietary fatty acids. Clin Exp Allergy 1989;19:15-23.
241. Arm JP, Horton CE, Mencia-Huerta JM et al. Effects of dietary supplementation with fish oillipids on mild asthma. Thorax 1988;43:84-92.
242. Miller BD. Depression and asthma: A potentially lethal mixture. J Allergy Clin Immunol1987;80:481-86.
243. Hay DWP. Pharmacology of leukotriene receptor antagonists. More than inhibitors ofbronchoconstriction. Chest 1997;111:35S-45S.
244. Krell RD, Aharony D, Buckner CK et al. The preclinical pharmacology of ICI 204,219, a peptideleukotriene antagonist. Am Rev Respir Dis 1990;141:978-87.
245. Miyamoto T. The clinical evaluation of a leukotriene antagonist, ONO-1078. Presented at theEuropean Congress of Allergology and Clinical Immunology, May 1992; Paris, France.

174 Bronchial Asthma
246. Fujimura M, Sakahato S, Kamis Y, Matsuda T. The effect of a leukotriene antagonist ONO-1078on bronchial hyperresponsiveness in patients with asthma. Respir Med 1993;87:133-38.
247. Finnerty JP, Wood-Baker R, Thomson H, Holgate ST. Role of leukotriene in exercise-inducedasthma: Inhibitory effect of iCI 204219, a potent leukotriene D4 receptor antagonist. Am RevRespir Dis 1992;145:746-49.
248. Makkar HK, Lau LC, Thomson HW, Binks SM, Holgate ST. The protective effect of inhaledleukotriene D4 antagonist ICI 204,219 against exercise-induced asthma. Am Rev Respir Dis1993;147:1413-18.
249. Arm JP, Lee TH. Sulphidopeptide leukotrienes in asthma. Clin Sci 1993;84:501-10.250. Dahlen B, Margolskee DJ, Zetterstrom O, Dahlen SE. Effect of the leukotriene antagonist MK-0679
on baseline pulmonary function in aspirin-sensitive asthmatic subjects. Thorax 1993;48:1205-10.251. Spector SL, Smith LJ, Glass M. Accolate Asthma Trialist Group. Effects of 6 weeks of therapy with
oral doses of iCI 204, 219, a leukotriene D4 receptor antagonist, in subjects with bronchialasthma. Am J Respir Crit Care Med 1994;150:618-23.
252. Spector S, Miller CJ, Glass M. Thirteen-week dose response study with Accolate (zafirlukast) inpatients with mild to moderate asthma (abstract). Am J Respir Crit Care Med 1995;151:A379.
253. Specter SL, Glass M, Minkwitz MC. The effect of six-week therapy with oral doses of ICI 204,219in asthmatics (abstract). Am Rev Respir Dis 1992;145:A16.
254. Specter SL, Glass M, Minkwitz MC. The effect of six-week therapy with oral doses of ICI 204,219in asthmatics (abstract). Am Rev Respir Dis 1992;145:A16.
255. Barnes NC, Pujet JC. First clinical experience of the oral leukotriene antagonist, pranlukast(SB205312/ONO 1078) in North European patients with mild to moderate asthma (Abstract).Am J Respir Crit Care med 1995;151:A378.
256. Cohn J. Zileuton (A-64077): A 5-lipoxygenase inhibitor. In: Lewis A, Furst DE (Eds). Nonsteroidalanti-inflammatory drugs: Mechanisms and Clinical uses. 1994;367-90.
257. Barnes NC, Pujet JC. Pranlukast, a novel leukotriene receptor antagonist: Results of the firstEuropean, placebo controlled, multicenter clinical study in asthma. Thorax 1997;52:523-27.
258. Reiss TF, Chervinsky P, Dockhorn RJ et al. Monteleukast, a once daily leukotriene receptorantagonist, in the treatment of chronic asthma. Ann Intern Med 1998;158:1213-20.
259. Terasima T, Amakawa K, Matsumaru A, Yamaguchi K. Correlation between cysteinyl leukotrienerelease from leukocytes and clinical response to a leukotriene inhibitor. Chest 2002;122:1566-70.
260. Lipworth BJ. Leukotriene-receptor antagonists. Lancet 1999;353:57-62.261. Barnes NC, Miller CJ. Effect of leukotriene-receptor antagonist therapy on the risk of asthma
exacerbations in patients with mild to moderate asthma: An integrated analysis of zafirlukasttrials. Thorax 2000;55:478-83.
262. Currie GP, Lipworth BJ. Bronchoprotective effects of leukotriene-receptor antagonists in asthma:A meta-analysis. Chest 2002;122:146-50.
263. Dempsey OJ, Fowler SJ, Wilson A et al. Effects of adding either a leukotriene-receptor antagonistor low-dose theophylline to a low or medium dose of inhaled corticosteroid in patients withpersistent asthma. Chest 2002;122:151-59.
264. Barnes PJ, Adcock IM. Steroid-resistance in asthma. Q J Med 1995;88:455-68.265. Schwartz HJ, Lowell FC, Melby JC. Steroid resistance in asthma. Ann Intern Med 1968;69:
493-99.266. Carmichael J, Paterson IC, Diaz P et al. Corticosteroid resistance in chronic asthma. Br Med J
1981;282:1419-22.267. Hill JM, Tattersfield E. Corticosteroid sparing agents in asthma. Thorax 1995;50:577-82.268. Moss RB. Alternative pharmacotherapies for steroid-dependent asthma. Chest 1995;107:817-25.269. Shiner RJ, Nunn AJ, Chung KF et al. Randomised, double-blind, placebo-controlled trial of
methotrexate in steroid dependent asthma. Lancet 1990;336:137-40.270. Alexander AG, Barnes NC, Kay AB. Trial of cyclosporin in corticosteroid-dependent chronic
severe asthma. Lancet 1992;339:324-28.

Pharmacologic Management of Asthma 175
271. Hu S, Mitcho YL, Oronsky AL, Kerwar SS. Studies on the effect of methotrexate on macrophagefunction. J Rheumatol 1988;15:206-09.
272. Nolte H, Skov PS. Inhibition of basophil histamine release by methotrexate. Agents Actions1988;23:173-76.
273. Mullarkey ME, Blumenstein BA, Andrade WP et al. Methotrexate in the treatment ofcorticosteroid-dependent asthma. N Engl J Med 1988;318:603-07.
274. Shiner RJ, Nunn AJ, Kan Chung K, Geddes DM. Randomised, double-blind, placebo controlledtrial of methotrexate in steroid dependent asthma. Lancet 1990;336:137-40.
275. Dyer PD, Vaughan TR, Weber RW. Methotrexate in the treatment of steroid dependent asthma.J Allergy Clin Immunol 1991;88:208-12.
276. Stewart GE, Diaz JD, Locky RF, et al. Comparison of oral pulse methotrexate with placebo in thetreatment of severe glucocorticoid-dependent asthma. J Allergy Clin Immunol 1994;94:482-89.
277. Erzurum SC, Leff JA, Cochran JE et al. Lack of benefit of methotrexate in severe steroid-dependentasthma. Ann Intern Med 1991;114:353-60.
278. Trigg CJ, Davies RJ. Comparison of methotrexate 30 mg per week with placebo in chronicsevere asthma: A 12-week double-blind, cross-over study. Respir Med 1993;87:211-16.
279. Coffey MJ, Sanders G, Eschenbacher WL et al. The role of methotrexate in the management ofsteroid-dependent asthma. Chest 1994;105:649-50.
280. Stemple DA, Lammert J, Mullarke MF. Use of methotrexate in the treatment of steroid-dependentadolescent asthmatics. Ann allergy 1991;67:346-48.
281. Guss S, Portnoy J. Methotrexate treatment for severe asthma in children. Pediatrics 1992;89:635-39.
282. Moss RB. Alternative pharmacotherapies for steroid-dependent asthma. Chest 1995;107:817-25.283. Calderon E, Lockey RF, Bukantz Sc et al. Is there role for cyclosporin in asthma? J Allergy Clin
Immunol 1992;89:629-36.284. Alexander AG, Barnes NC, Kay AB. Trial of cyclosporin in corticosteroid-dependent severe
asthma. Lancet 1992;339:324-28.285. Szczeklik A, Nizankowaska F, Dworosky R et al. Cyclosporin for steroid dependent asthma.
Allergy 1991;46:312-15.286. Bernstein DI, Bernstein IL, Bodenheimer SS et al. An open study of auranofin in the treatment of
steroid-dependent asthma. J Allergy Clin Immunol 1988;81:6-16.287. Szczeklik A, Nizankowaska F, Dworosky R et al. Cyclosporin for steroid dependent asthma.
Allergy 1991;46:312-315.288. Muranaka MM, Miyamoto T, Shida T et al. Gold salt in the treatment of bronchial asthma—A
double blind study. Ann allergy 1978;40:132-37.289. Nierop G, Gijzel WP, Bel EH et al. Auranofin in the treatment of steroid dependent asthma:
A double blind study. Thorax 1992;47:349-54.290. Hodges NG, Brewis RAL, Howell JBL. An evaluation of azathioprine in severe chronic asthma.
Thorax 1971;26:734-39.291. Mazor BD, Giclas PC, Gelfand EW. Immunomodulatory effects of intravenous immunoglobulin
in severe steroid dependent asthma. Clin Immunol Immunopathol 1989;53:S156-S163.292. Spector SL, Katz FH, Farr RS. Troleandomycin: Effectiveness in steroid dependent asthma and
bronchitis. J Allergy Clin Immunol 1974;54:367-79.293. Ilfield D, Kivity S, Feirman E, Topilsky M, Rojkind M. Effect of invitro colchicine and oral
theophylline on suppressor cell function of asthmatic patients. Clin Exp Immunol 1985;61:360-67.
294. Schwartz YA, Kivity S, Ilfield DN, et al. A clinical and immunological study of colchicine inasthma. J Allergy Clin Immunol 1990;85:578-82.
295. Charous BL. Open study of hydroxychloroquine in the treatment of severe symptomatic orcorticosteroid-dependent asthma. Ann Allergy 1990;65:53-58.

176 Bronchial Asthma
Inhalation Therapy
11
Current therapeutics emphasises the importance of effective delivery of a drug to its site ofaction—the Targeted Drug Delivery.1-6 The advantage of this approach is avoidance ofunnecessary and undesirable exposure of tissues/organs not involved in the disease processto the drug. It is sufficient and advantageous to have the drug delivered only to the sitewhere the drug is needed. Targeted delivery of the drug to the desired site of action meansthat smaller doses are enough to produce the desired effect and generalised systemic sideeffects can be eliminated or minimised and a rapid action of the drug can be obtained. Thevalue of inhalation as a route of drug administration has been recognised for thousands ofyears by the ancient civilisation in India, China, the Middle East and as well as by Hippocratesand Galen. The Ayurvedic system of medicine advocated the use of datura smoked in apipe for a variety of ailments and atropa belladonna was given by smoking as a standardremedy for asthma. Asthma cigarettes made from Datura leaves are also being used byherbalists.
Bronchodilator aerosols have been in use since 1935. In the past adrenergic broncho-dilators have been given by hand-held squeeze-bulb nebulisers. This was cumbersome,and modern pressurised aerosols were introduced in 1956 and constituted a breakthroughin inhalation treatment. In recent times, inhalation therapy for asthma has been developedto a high level of sophistication although they are simple to use.
The key to inhalation therapy is the aerosol particle. An aerosol is a suspension of fineliquid or solid particles in air. The efficacy of an inhaled drug depends largely on howmuch of the drug is deposited in the peripheral airways. On the other hand, the depositionof inhaled particles is determined both by the physical characteristics of the air-borneparticles and physiological parameters like airflow to the lungs. Particle size is an importantdeterminant of aerosol deposition in the lungs. Most devices (discussed below) generateparticles in the size range of 1-10 micron. Only particles in the size range of 2-5 micron canbe inspired deep into the lungs; particles 5 micron or more in diameter are impacted in thethroat or in larger airways. Particles less than 1 micron behave like a gas and are exhaled inthe expired air. Most aerosols contain a wide range of particle sizes and are known asheterodisperse aerosols. The mass median aerodynamic diameter (MMAD) is the mediandiameter of the aerosol multiplied by the square root of particle density. MMAD is importantas regards aerosol deposition rather than the particle sizes. The propellant surrounding thedrug particles evaporates on emerging from the canister and the particle steadily decreasesas the aerosol moves away from the canister. Other factors that count for aerosol depositioninclude velocity, inertial impaction, and gravitational sedimentation. However measure-ments using radioactive teflon particles labelled with technetium-99m with a gamma camera

Inhalation Therapy 177
have shown that about 10% of the drug released from an MDI is deposited in the lung.About 80% is deposited in the oropharynx and about 10% is trapped on the walls of theinhaler device.
For the purpose of inhalation therapy an aerosol of the drug can be generated in threeways:
i. Pressurised aerosol systems;ii. Dry powder system;
iii. Nebulisers.
Pressurised Aerosol Systems (Metered Dose Inhalers—MDIs)
Most medications prescribed for the treatment of bronchial asthma for maintenance or rescue,are administered via a metered dose inhaler (MDI). In Pressurised aerosol systems or metereddose inhalers micronised finely powdered drug is dissolved or suspended in a liquidpropellant mixture, and packed in a sealed container (Fig. 11.1). The liquid propellants arehighly volatile chlorofluorocarbons, CFC, (Freons) with a high vapor pressure of about 400 kPa.These freons are gases at room temperature, have a low boiling point, are inert, non-inflammable, and odourless. Some surfactant is added so that they are not clumped together.On actuation, propellants emerge and break up into aerosol particles each consisting of adrug particle surrounded by the propellant. The valve is metered so that each actuationreleases a fixed amount of the drug-propellant mixture. Therefore, it is named as the metereddose inhaler (MDI).
During recent years, CFCs have been criticised for their harmful effects on the environ-ment, especially the depletion of the stratospheric ozone layer. The production and use ofCFCs were banned by international treaty (the Montreal protocol) in 1987, although theiruse in medications is not as the amount for this use is very small. Such exemptions to theNational and International bans were made for MDIs to allow time for comparative clinicaltrials of alternative propellants, as required by worldwide regulatory agencies. Althoughno deadline has been set for the United States, Canada aims to achieve total transition by2005, and the European Commission predicts that there will be no need for CFC-basedMDIs in the European Community by the year 2003. So far, the most promising alternativepropellants for MDIs are derivatives of hydrofluoroalkane (HFA). The HFA agents lackchlorine, and thus have zero ozone depletion potential.7-9 Preclinical studies have
Fig. 11.1: Components of meter dose inhalers

178 Bronchial Asthma
demonstrated the acceptability of these propellants in terms of pharmacology, toxicology,and safety.10 Clinical studies also have shown that these propellant are as equivalent oreven better than those use CFCs as propellants.11-18 Clinical trials have demonstrated thatthe level of asthma control achieved with CFC-beclomethasone dipropionate may beobtained with approximately half the total daily dose of HFA- beclomethasone dipropionate.This is probably due to improved lung deposition with the extra fine aerosol of HFA-beclomethasone dipropionate compared with the suspension of CFC based aerosol lungdeposition is also greater with HFA- beclomethasone dipropionate compared with CFC-beclomethasone dipropionate and CFC-fluticasone propionate. Deposition values are relatedto the particle size distribution of each inhaler, with the smaller particles of HFA-beclomethasone dipropionate providing the greatest lung deposition and least oropharyn-geal deposition.19
Evaluation of adherence to treatment is one important step in asthma management.Patients tend to overestimate the usage of MDIs presumably secondary to recall bias or asan effort to avoid criticism Prescription refill histories as from the issuing authorities orverification of the medical bill of the patient and canister weighing are more objectivemeasures, but they do not reflect pattern of usage. Electronic monitors are recently availablefor accuracy of MDI use.20
Dry Powder Inhalers
In an attempt to overcome the coordination problem that is required for the successful useof the pressurised MDIs, a number of dry powder inhalers have been developed. In the drypowder system, micronised drug is mixed with a carrier substance (lactose) and the mixtureis filled into a gelatin capsule. The capsule is loaded into the inhaler device and is cut openin the device before inhalation (Fig. 11.2). After piercing or fracturing the gelatin capsule,the patient only needs to do is to inhale through the device to draw the powder out of thecapsule. The aerosol is generated by means of the energy contained in the inspired air. Theair stream passes the powder in such a way that a turbulent flow is formed which breaksup the particles into a dust or aerosol. The higher the inspiratory flow rate (>60L/min),higher the number of respirable particles. Although it is easier to use than a MDI, it is lessconvenient because of the need to load the capsule before use. Because of a high flow rate,many patients, particularly children, cannot generate a sufficient inspiratory flow requiredto break up the aggregates during an acute attack. Thus, too large particles are unable topenetrate into lung periphery. Further, in the panic and distress of an acute situation, thepatient may have difficulty in inserting the capsule into the device. Inhalation of the dry
Fig. 11.2: Dry powder system

Inhalation Therapy 179
powder may cause some irritation and cough in some patients. The gelatin capsules aresubject to environmental influences of moisture and temperature during storage. This willmake the capsule soggy and could not be broken by the system efficiently. Thus, they maybe reserved for those who cannot master the technique of MDI. Recently, a multi-dose,ready to use, additive free dry powder inhaler effective at low inspiratory flow rates areavailable which can overcome the above difficulties.
Different types of DPI such as Turbohalers, Diskhalers, and Accuhalers are availablenow. These devices have the advantage of being breath activated, and delivery of an accuratedose is less dependent on patient technique. Recently, new generation multi-dose drypowder inhaler (MDPI) is available, which has a triple inhalation control system, so thatthe patient has acoustic (click), visual (dose counter), and sensory (oropharyngeal sensationconfirmation of dosing. Other mandatory features of the DPI are an accurate meteringsystem, a dose counter, and a robust compact design. A unique feature in terms of cost andflexibility is that the inhaler utilises replaceable cartridges that contain up to 200 doses,with the future potential for a wide range of therapies.21
Nebulisers
In nebulisation, small droplets are generated suitable for inhalation from a nebulisingsolution containing the drug. Two types of nebulisers are used for this purpose:a. the Jet nebuliser (Fig. 11.3) which is powered by compressed air or oxygen from a
compressor or a cylinder; andb. the ultrasonic nebuliser which derives the energy required to make an aerosol from high
frequency sound waves (Fig. 11.4). Nebulisers need a power source and use of nebulisersis time consuming. However, they are useful in very young children or adult patientswho cannot manage the use of inhalers and for delivery of large doses of bronchodilatorsas in acute severe asthma. In addition they are used for delivery of drugs that cannot beformulated in a MDI because of technical reasons since very high doses cannot be packedand for bronchial challenge tests and lung ventilation scanning. The main advantage ofnebulisers is the ease of use by patients. It can be inhaled with normal tidal breathingthrough a mouthpiece or a face mask. Since they can be driven by oxygen, this becomesan extra advantage in acute asthma. Moisture obtained from wet aerosol may be helpfulin loosening the mucus. Nebulised bronchodilators can also be administered throughpressure-cycled ventilators. Jet nebulisers driven by oxygen/compressed air needs aflow rate of at least 6-8 litres/min generate aerosol particles in the respirable range. Thesolution used for nebulisation needs to be diluted with isotonic and preservative-freesolutions to reduce drug loss due to impacting of aerosols in the dead space of theapparatus. A minimum volume fill of 4 ml (drug + normal saline) with a flow of 6 litres/min. is recommended to ensure a high aerosol output, small particle size, And shorttreatment time. In infants, the small minute volume of 3-3.5 litres compared to thenebuliser output of 6 litres/min limits the amount of the drug to be inhaled. Hence, theywill need a higher dose. Other precaution to be taken is that the interior of the containerbe cleaned thoroughly after use to avoid bacterial (Pseudomonas aeruginosa) contaminationand the air intake grill and filters to avoid Aspergillus contamination. The choice of theparticular nebuliser is important as not all of them produce desired aerosols. As withMDI, only about 10-12% of the drug can reach the lungs, most of it being retained as
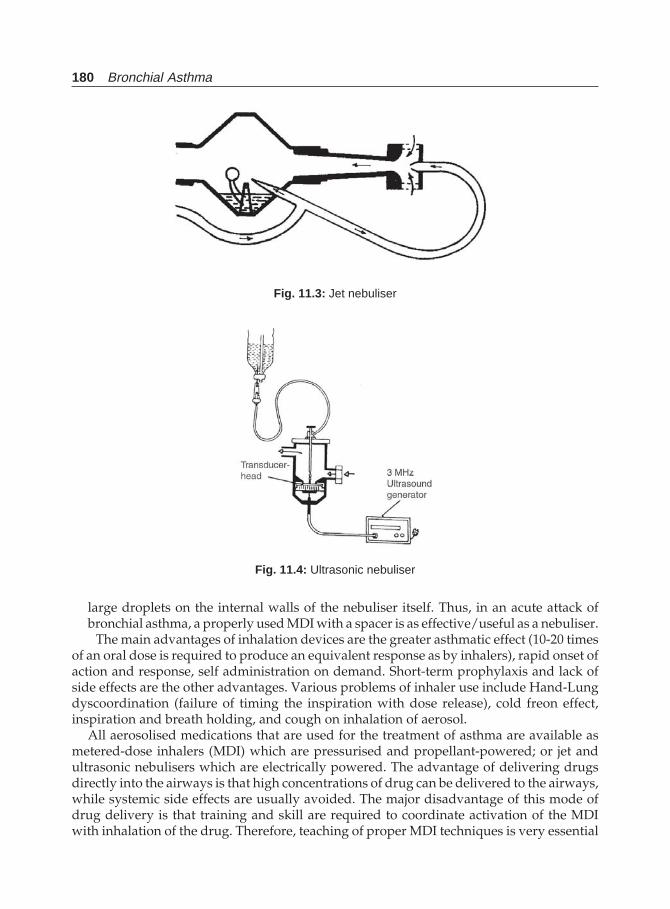
180 Bronchial Asthma
large droplets on the internal walls of the nebuliser itself. Thus, in an acute attack ofbronchial asthma, a properly used MDI with a spacer is as effective/useful as a nebuliser.
The main advantages of inhalation devices are the greater asthmatic effect (10-20 timesof an oral dose is required to produce an equivalent response as by inhalers), rapid onset ofaction and response, self administration on demand. Short-term prophylaxis and lack ofside effects are the other advantages. Various problems of inhaler use include Hand-Lungdyscoordination (failure of timing the inspiration with dose release), cold freon effect,inspiration and breath holding, and cough on inhalation of aerosol.
All aerosolised medications that are used for the treatment of asthma are available asmetered-dose inhalers (MDI) which are pressurised and propellant-powered; or jet andultrasonic nebulisers which are electrically powered. The advantage of delivering drugsdirectly into the airways is that high concentrations of drug can be delivered to the airways,while systemic side effects are usually avoided. The major disadvantage of this mode ofdrug delivery is that training and skill are required to coordinate activation of the MDIwith inhalation of the drug. Therefore, teaching of proper MDI techniques is very essential
Fig. 11.3: Jet nebuliser
Fig. 11.4: Ultrasonic nebuliser

Inhalation Therapy 181
since only about 10% of the inhaled dose penetrates the lower airways, even with optimaltechniques. It is generally agreed that maximum delivery of aerosol into the airways isobtained by inhaling an aerosol bolus from functional residual capacity. A flow rate of lessthan 1 L/sec, with an inhalation time of over 5 second and a breath holding time of 10 secondsis believed to be the optimal technique. There are two different general approaches toinhalant techniques with MDI: the open mouth and closed mouth techniques. In the openmouth technique the inhaler is held approximately 4 cm in front of an open mouth. Otherdelivery devices which have been developed in an attempt to overcome the disadvantagesof pressurised MDI include the Roto-Haler, Gentle-Haler, Autohaler, and Turbohalers. Themost important device is the “spacer” which may be a cone spacer or a tube spacer.
Spacer Devices
Spacer device is an extension chamber interposed between the mouth piece of the MDI andthe mouth of the patient (Fig. 11.5). This device allows time and distance for the aerosol totravel in space before it is inhaled. The particle size is reduced because of evaporation ofthe liquid propellant. The aerosol velocity is also reduced because of resistance offered byair in the space. Smaller particle size helps better deposition in the peripheral airways andreduces deposition in the oropharynx. The slowed down particles held in the chamber canbe inhaled few seconds later after the release and there is no need of synchronisation ofinspiration and actuation. This simplifies inhaler use and quite helpful in those who havecoordination problems. Spacer devices are usually of two types: small volume spacers (tubespacers) and large volume spacers or valved spacers of the volume of about 750 ml. Theaerosol cloud emerging from the MDI expands into a conical shape as it moves away. Theconical shape of the spacer accommodates the enlarging cloud. The spacer is such thatimpaction loss on the walls is minimal. The one way valve opens during inspiration andcloses during expiration. This helps retaining the modified aerosol in the chamber until it isemptied by inhalation. The expired air leaves the mouth piece through a side port.
It is calculated by laser holography studies that one cubic millimeter of air within aspacer is about 5500 at the end of 5 seconds and is still 3300 after 30 seconds of actuation.The mass median diameter of the aerosol from an MDI is reduced from 8.4 to 4.9 micronsmaking possible most of the particles to be in the respirable range. Lung deposition isincreased by 133% and reduces throat deposition by 90%.
Fig. 11.5: Spacer

182 Bronchial Asthma
REFERENCES
1. Hillman B. Aerosol deposition and delivery of therapeutic aerosols. J Asthma 1991;28:239.2. Newman SP, Clarke SW. Therapeutic aerosols 1- Physical and practical considerations. Thorax
1983;38:881-86.3. Newman SP. Aerosol deposition consideration in inhalation therapy. Chest 1985;88(Suppl):S152-
60S.4. Sackner MA, Kim CS. Auxillary MDI delivery systems. Chest 1985;88(Suppl):S161-S69.5. Newhouse MT, Dolovich MB. Control of asthma by aerosols. N Engl J Med 1986;315:870-74.6. Summer W, Elston R, Tharpe L, Nelson S, Haponik EF. Aerosol bronchodilator delivery methods.
Arch Intern Med 1989;149:618-23.7. Noakes TJ. CFCs, their replacements and the ozone layer. J Aerosol Med 1995;8(Suppl):S3-S7.8. Smith IJ. The challenge of reformulation. J Aerosol Med 1995;8(Suppl):S19-S27.9. Fischer DA, Hales CS, Wang WC et al. Model calculations of the relative effects of CFCs and
their replacements on global warming. Nature 1990;344:513-16.10. CPMP on possible alternatives to CFCs. Scrip 1994;1943:26.11. Furukawa C, Atkinson D, Forster TJ et al. Controlled trial of two formulations of cromolyn
sodium in the treatment of asthmatic patients > 12 years of age. Chest 1999;116:65-72.12. Busse WW, Brazinsky S, Jackobson K et al. Efficacy response of inhaled beclomethasone
dipropionatein asthma is proportional to dose and is improved by formulation with a newpropellant. J Allergy Clin Immunol 1999;104:1215-22.
13. Davies RJ, Stampone P, O’Conner BJ. Hydrofuoroalkane 134a beclomethasone dipropionateextrafine aerosol provides equivalent asthma control to chlorofluorocarbon beclomethasonedipropionate to approximately half the total daily dose. Respir Med 1998;92(Suppl A):23-31.
14. Gross G, Thompson PJ, Chervinsky P et al. Hydrofuoroalkane 134a beclomethasone dipropionate,400 μg is as effective as chlorofluorocarbon beclomethasone dipropionate 800 μg for treatmentof moderate asthma. Chest 1999;115:343-51.
15. Leach CL. Improved delivery of inhaled steroids to the large and small airways. Respir Med1998;92(Suppl A):3-8.
16. Juniper EF, Price DB, Stampone PA et al. Clinically important improvements in asthma-specificquality of life, but no difference in conventional clinical indexes in patients changed from conven-tional beclomethasone dipropionate to approximately half the dose of extra fine beclomethasonedipropionate. Chest 2002;121:1824-32.
17. Langley SJ, Holden J, Derham A et al. Fluticasone propionate via the disk haler or hydrofluo-roalkane-134a metered-dose inhaler on methacholine-induced airway hyper-responsiveness.Chest 2002;122:806-11.
18. Nayak A, Lanier R, Weinstein S et al. Efficacy and safety of beclomethasone dipropionate extrafine aerosol in childhood asthma. A 12-week, randomised, double-blind, placebo-controlledstudy. Chest 2002;122:1956-65.
19. Leach CL, Davidson PJ, Hasselquist BE, Boudreau RJ. Lung deposition of Hydrofluoroalkane134a beclomethasone is greater than that of chlorofluorocarbon fluticasone and chlorofluoro-carbon beclomethasone. Chest 2002;122:510-16.
20. Julus S, Sherman JM, Hendeles L. Accuracy of three electronic monitors for metered dose inhalers.Chest 2002;121:871-76.
21. Hansel TT, Kunkel G. New horizons in asthma therapy: The Novoliser dry powder inhaler.Curr Opinion Pulm Med 2001;7(Suppl 1):S1-S2.

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 183
Therapeutic Approach inPatients with Asthma
I. Chronic Bronchial Asthma
12
Asthma is a chronic condition with acute exacerbations having variable course. The courseof disease is not uniform with periods of exacerbations and remissions which varies fromdays to weeks to months to years. Management therefore, requires a continuous careapproach to control symptoms, prevent exacerbations, and reduce chronic airwayinflammation. The course of asthma varies among patients. The degree of an individual’sasthma severity may change from one season or year to the next. Therefore, specific asthmatherapy must be selected to fit the need of individual patients. The therapy must be adaptableto change as the disease changes in the individual.
Over the years a number of guidelines have been developed. Notable amongst them arethose of the National Heart, Lung and Blood Institute of the NIH, USA, British ThoracicSociety, Research Unit of the Royal College of Physicians of London, the King’s Fund Center,the National Asthma Campaign, Global Initiative for Asthma and WHO.1-11 The basicprinciples are the same in all these guidelines. Management of bronchial asthma can bedivided into that for chronic asthma and acute severe asthma.
CHRONIC BRONCHIAL ASTHMA—AIMS OF THERAPY
The basic goals or aims of management of chronic asthma are:i. To recognise asthma and its severity
ii. To abolish symptoms particularly those of the chronic troublesome ones like nocturnalcough and dyspnoea, and early morning symptoms
iii. To maintain normal activity levels including exerciseiv. To maintain a normal or near normal or the best possible long-term pulmonary
functionsv. To prevent recurrent exacerbations of asthma and the risk of severe attacks
vi. To minimise absence from school or workvii. To enable normal growth to occur in children
viii. To minimise the need for as needed (quick-relief) β2-agonist therapyix. To avoid adverse effects from asthma medicationsx. To meet patients’ and families’ expectations of and satisfaction with asthma care.

184 Bronchial Asthma
PRINCIPLES OF MANAGEMENT
Certain basic principles of bronchial asthma needs to be considered before administeringany specific therapeutic modalities.
i. Since there are many conditions which mimic bronchial asthma, the diagnosis shouldbe established.
ii. It should also be realised that asthma is a chronic condition with acute exacerbationswith varying periods of remissions. There is no cure of bronchial asthma but if thepatient follows certain guidelines including medications, the disease can be controlledand the patient can lead life like a normal individual. Treatment requires a continuouscare approach to control symptoms, to prevent exacerbations, and to reduce airwaysinflammation.
iii. Asthma is an inflammatory disease and inflammation may continue even duringperiods of clinical remission and even in patients with mild asthma. Therefore anti-inflammatory treatment is an essential component of management of bronchial asthma.
iv. The severity of asthma must be evaluated by assessing the activity limitation, byevaluation of night time symptoms and by assessing pulmonary function.
v. The therapy selected should not have adverse effects that are perceived by the patientto be worse than the underlying disease. The therapy is usually dictated by the severityof disease, medication tolerance, and sensitivity to environmental allergens. All thesefactors need to be incorporated in the formulation of therapy.
vi. It is essential to deal with common asthma triggers. Environmental control measuresmust be under taken to avoid allergens. All types of smoking should be stopped andexposure to passive smoke should be eliminated. Inhaled β2−agonist or Cromolynsodium or both taken prior to an anticipated encounter with a known trigger canprevent or diminish an asthmatic response. This is particularly true for exercise-induced asthma. The same principle can also be applied to other situations, includingexposure to antigens like animal danders, cold air, or other irritants. Both adults andchildren who has upper respiratory tract viral infections, and start to have acute asthmasymptoms may need to add or increase anti-inflammatory asthma medications inorder to control the asthma symptoms. Bacterial otitis and sinusitis should be treatedwith antibiotic therapy. Sometimes aggressive antiasthma therapy fails because anupper respiratory infection has been overlooked. Allergic and nonallergic rhinitisshould be treated with antihistamines, Cromolyn sodium nasal spray, or topical nasalsteroids. The patient must be taught to avoid:a. Beta blockers (tablets and eyedrops) are contraindicated.b. If aspirin or NSAIDs are known to induce asthma, they should be avoided.c. Allergens as outlined above (e.g.; house-dust mite, domestic pets, and pollens.
should be avoided where relevant.d. Occupational causes must be considered and appropriate steps be taken.e. Active smoking should be avoided.f. Passive smoking should also be avoided.g. Prophylactic treatment for exercise or before exposure to triggers.
vii. Anticipatory or early interventions in treating acute exacerbations of asthma reducethe likelihood of developing severe airway narrowing.
viii. Asthma therapy has the following integral components:

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 185
a. Patient education and family participationb. Avoidance of identified causes where possiblec. Use of the lowest effective dose with a target of controlling asthma but with the
minimum of short and long term side effects.The above three approaches are interrelated in the management and pathogenesis of
bronchial asthma are shown in Figure 12.1.
PATIENT EDUCATION
Health education by the physician is a powerful tool for helping patients gain the motivationand skill to control their asthma.12-15 There is definite evidence of benefit from patienteducation and the issuing of self management plans. Management of asthma requires apartnership between the patient and family and the health care provider. It should be madeclear from the very beginning that treatment and supervision are likely to be required overa prolonged period of time. Education should be the basis of sharing of information and theacquisition by the patient and family of understanding and skills. This will bring aboutappropriate change in behaviour only if patients and family are given adequate opportunityto express any fears or concerns, and time to discuss their expectations of both the diseaseand its treatment. Patients and parents require both verbal and written advice and manywill require guided self management plans, so that the patient can keep well and adjusttreatment according to a plan developed with the physician. Giving information alone doesnot alter behaviour, but written and audiovisual reinforcement of spoken message aidspatient confidence. All patients should be given information about features which indicatewhen their asthma is worsening, and what to do under those circumstances. Giving thosewith asthma written self management plans so that they may adjust treatment to keepthemselves well reduces morbidity and health costs.16,17
Fig. 12.1: Approach to therapy of bronchial asthmadepending upon the aetiopathogenesis

186 Bronchial Asthma
Various components of patient education plan includes:i. Establishing a partnership which improves the patient adherence to the treatment
plan and stimulate family effort to improve control of patient’s asthma18,19
ii. Encouraging adherence to the treatment plan is the next step which can be achievedby clarifying patient’s expectations for treatment and answering questions,20 involvingthe patient and family in the development of a treatment plan, simplifying the treatmentplan, providing the patient with diaries to record antecedents of asthma exacerbations,symptoms, actions taken, and peak expiratory flow rates. Further, an importantquestion to be kept in mind whether that patient can afford to buy the medicationsprescribed, and if not, alternative therapies must be considered. Ignoring this fact is avery important cause of non-adherence to treatment prescribed. Evaluation of theresult of treatment from time to time helps positive reinforcement plans. If the patientis not adhering to the treatment plan, then the cause for the same to be identified byasking the patient the likely problems and a possible solution for the same should beprovided.
iii. Various essentials of patient education includes the content of teaching both writtenand audiovisual, explaining to the patient about asthma like what is it, what are thekey points about the symptoms and signs of asthma, the role of inflammation and therole of various medications, asthma triggers and how to avoid them, the need fortreatment, how the medicines work, adverse effects of drugs and their prevention,preventive treatment, and early treatment during exacerbations, alleviating patientfears concerning medications, providing written guidelines, and steps to manageasthma episodes at home.
iv. The correct use of inhalers should be demonstrated to the patient.21,22 Similarly, thepatient should demonstrate the use of the MDI to the clinician. The patient’s MDItechnique should be reviewed during each visit. When several inhalers are prescribed,labelling them is essential and also explaining when to use which one and whichinhaler to be used first.
v. The patient should be able to recognise the early warning symptoms or signs of airflowobstruction which will enable him to begin treatment immediately. Early warningsigns include a peak flow level below 20% predicted or personal best level; cough orwheeze, particularly during daily activity; an individual pattern of early signs suchas chest tightness, shortness of breath, or dark circles under the eyes in children.Indications of immediate report to emergency include cyanosis, difficulty in breathing,talking or walking, retraction of the chest, neck or ribs and nasal flaring, failure ofmedications to control worsening of symptoms, and a steady decline of PEFR.
vi. Knowledge of the optimal use of home peak expiratory flow meter—both its recordingand interpretation are now one of the essential components of asthma management.
vii. Materials and guidelines for individuals and group education and support networkare very helpful. The education must continue in a long-term basis. discussions,demonstration, group classes, and dramas help patients learn guided self-managementskills. The most effective is for the health care professional to give information verbally,demonstrate techniques, and then provide reinforcement by several routes. The specificmethods should be selected on the basis of patient and cultural preferences. Somepatients benefit from joining asthma patient support groups or clubs. These groupsvary from area to area, but most provide informative materials, group education, and

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 187
mutual support. Group members exchange personal tips on managing asthma, makingchanges at home, and coping with the stress of a chronic disorder in the family.
Currently asthma education worldwide websites are available. However, asthmaeducation material contains many accessibility barriers, is highly variable in quality, andcontent, and takes little innovative use of technology. These informations currently availablein the web fails to meet the information needs of the patient.23
The patient must be made to understand that there are new ways to manage their diseaseso that they can prevent problems, be free of symptoms both day and night, and liveproductive, active lives. They can learn to control their asthma, handle mild attacks promptlyat home, and prevent serious attacks. Emergency visits should no longer be needed. Regularmedical visits provide periodic opportunities to address concerns, solve problems, andreach agreement on long-term treatment. This is achieved by having the patient becomeactively involved as a partner in his or her care through guided self-management. Guidedself-management means a patient can take medications correctly; understand the differencebetween quick-relief and long-term preventive medications, avoid triggers; monitor personalstatus using symptoms and if possible, PEFR indicators; recognise signs that asthma isworsening and take action; follow personalised action steps and stop attacks; and seekmedical help at the appropriate time to stop serious attacks.
Long-term asthma control requires a written management plan that describes what todo to prevent symptoms and attacks and what to do in case an attack occurs. An asthmamanagement zone system is effective for guided self-management and should be included inthe management plan. This system classifies levels of asthma control as different zonesbased on the frequency and severity of symptoms and peak expiratory flow measurements.The system then indicates the appropriate therapy for each zone. The zone system helpspatients understand the chronic and variable nature of asthma, monitor their condition,identifies the earliest possible signs that day to day control of asthma is deteriorating, andact quickly to regain control. When PEFR readings are available, the patient’s current readingmust be compared to his or her personal best—the highest PEFR value achieved when thepatient’s asthma is under control—is his benchmark for asthma control. The patient thenfollows the prearranged action steps appropriate to each of the following three zones.
Green zone It indicates all clear. Asthma is under control with no symptoms or interruptionof activities or sleep. PEFR are usually 80-100 percent of personal best. The variability isless than 20%. If the patient has stayed for at least 3 months in this zone, a careful step-down of the therapy can be considered as outlined below.
Yellow zone This signals caution. Some mild asthma symptoms are present. PEFR readingsare 60-80% of personal best. There is 20-30% variability. Readings in this zone indicatesthat an acute attack may be present for which a temporary increase in medication is needed,essentially β2−agonist inhalers for quick relief. The patient should develop a treatment planwith the physician. Also it is possible that an overall deterioration of asthma might haveoccurred that require further treatment. A short burst of corticosteroids will be required tillthe PEFR comes back to the green zone. In case the patient is taking inhaled steroids, thatshould be doubled for 12 weeks or until PEFR improves. Frequent fluctuations into theyellow zone may indicate poor control of asthma and the green zone therapy has to beincreased.
Red zone This indicates medical alert. Asthma symptoms are present even when the patientis at rest or interfere with activity. PEFR readings are below 60% of the personal best. The

188 Bronchial Asthma
patient should follow medication plan. An inhaled short-acting β-agonist should be takenimmediately. If the PEFR improves after initial bronchodilator therapy, the yellow zone actionsshould be continued. After the attack is controlled, the green zone therapy and patientadherence to the management plan should be reviewed and adjusted accordingly.
IMMUNOLOGICAL MANAGEMENT
Although the benefits of immunotherapy remains unproven, this form of therapy is stillwidely practiced by many physicians. Since allergy has a significant role in thepathophysiology of bronchial asthma, environmental control measures to avoid allergens isan important step in the control of bronchial asthma. The main method of identifying allergyis by clinical history. Skin prick tests and in vitro specific IgE measurements are rarely helpfulin diagnosis and management and should be interpreted by a physician familiar with suchtests. In general both active and passive smoking should be avoided.
Outdoor Allergens
Exposure to outdoor allergens is best reduced by remaining indoors, preferably in an airconditioned environment,24,25 particularly during the midday and afternoon when pollenand some mould spore counts are highest. Since there is a geographical and seasonal variationof pollens and other aeroallergens, knowledge of the same is helpful. Use of nasal filters ormasks have been tried with little success. Pollen particles greater than 10 microns are usuallycleared in the nose and mouth and do not generally penetrate the lower airway.26 However,some plants produce allergen containing particles that are in the respirable range like ragweed,and congress grass pollination, which are clearly associated with asthma. Mould spores aregenerally smaller than pollen grains and are more likely to penetrate the lower airway. Mouldspores exist primarily out of doors and tend to be seasonal. Some fungi sporulate on worm,dry summer days; others in the rainy seasons. Keeping windows closed during seasons ofhigh mould production will reduce exposure.
Indoor Allergens
Environmental control to reduce exposure to indoor allergens is a critical component of asthmamanagement. House dust is an important source of indoor allergens. Although house dust perse is not an allergen, there are allergic components in house dust. The most important includemites, cockroach allergen, and animal danders.
Animal Allergens
The best way to avoid animal allergens are just to remove the animal from the house (dog, cat,rodents, birds, etc). Removal of the animal may not afford immediate relief even when followedby vigorous cleaning as allergen has been shown to remain in the home for many months.27
Residual allergen can be denatured and rendered nonallergenic by application of 3% tannicacid solution. If the pet cannot be removed from the house, it should at least be kept out of theallergic person’s bedroom at all times. If the animal is in the bedroom at all, the dander andsaliva will remain for a long time even after the pet leaves. Weekly washing of the pet mayreduce the amount of dander and dried saliva deposited on carpets and furnishings. The

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 189
most reliable reduction of exposure to allergens derived from furred pets is done by completelyexcluding the pet from the home and avoiding exposure to pets elsewhere. It is of course notpossible always. It is important to make patients aware of the choices they are making likemore medications, decrease in health related quality of life and to reexamine these choices ona regular basis. Patients are often interested in alternatives to excluding the pet from the home.In such a situation, there is hardly anything other than complete avoidance can benefitasthmatic patients with a documented pet allergy.28 Since there is a dose-response relationshipbetween parameters of asthma control, and exposure to allergens, it seems reasonable to keepthe cat out of the bedroom, living room, and play- room. Washing of pets as mentioned abovehas given variable results and must be repeated so frequently as to be impractical in mostsituations. Settled allergens will be removed by methods of removal of house dust (see on page191). Cat and dog allergens, as opposed to dust mite allergen, are often found on smallparticles that are easily airborne. This renders the use of air filtration devices attractive. Dueto small particle size of airborne allergens, HEPA cleaners are likely required. Significanthealth benefits are not documented despite the reduction in the amount of airborne particles,29
although an improvement in bronchial hyperresponsiveness has been found in cat allergicchildren.30 In a recent meta-analysis, it is reported that use of air filtration systems in patientswith asthma or allergy showed a reduction in symptoms, but not of medication use, and noimprovement in measures of peak flow.31
House-dust Mite
House-dust mites are important causes of allergic asthma. They occur in environments withsufficient humidity since they are quite dependent for survival on moisture from atmosphere.Mite antigen is found throughout the home, wherever human dander, the food for the mite, isfound. High levels are obtained in dust obtained from mattresses, pillows, carpets, upholsteredfurnitures, bedcovers, clothes, and soft toys.32 The principal allergen is found in the mitefaeces. A gram of dust may contain 1,000 mites and 250,000 faecal pellets. These faecal pelletsare quite large varying in size from 10-40 microns, and therefore do not easily penetrate thelower respiratory tract. Mite antigen is easily demonstrated in the air during housecleaningactivities, but it is present in only very small amounts in undisturbed air.33 Some mite allergenis associated with smaller size particles that may be in the respirable range for the lowerairways.34 House-dust mite control measures include encasing the mattress in an airtightcover; the pillows are also to be encased and be washed weekly; the bedding should bewashed in water at 130° F or 55° C weekly and dry thoroughly in a hot dryer or in the sun;curtains and children’s soft toys are to be washed regularly; the patient should avoid sleepingor lying on upholstered furnitures; and carpets laid on concrete are to be removed, if possiblethe indoor humidity is to be reduced to less than 50%; carpets may be removed from thebedroom; and ascaricides may be useful in killing the mites.35-38 If carpet removal is not possible,vacuuming should be carried out with a high-efficiency particulate air (HEPA) vacuum cleanerwith a double reservoir bag, preferably when the asthmatic patient is not present. Centralvacuum cleaners exhausted to the outside are also likely to be effective.39 Such a multifacetedand concerted approach to reducing levels of dust mite allergen among asthmatic subjectswith positive allergy skin test results to mite allergens, if successful, is clearly associated withimprovement in the parameters of asthma control like symptoms, need for medications,measures of airflow, and measures of bronchial hyperresponsiveness.40

190 Bronchial Asthma
Cockroach Allergen
It is important in warmer climates.41 The infested homes should be cleaned thoroughly andregularly. Pesticides and pesticide sprays are used to eliminate cockroaches. If sprays areused the patient should not be present at that time.
Indoor Moulds
These are found in environments with increased humidity. Bathrooms, kitchens and base-ments require adequate ventilation and frequent cleaning using chlorine bleach if necessary.Dehumidifiers for damp basement areas should be considered, with humidity levels forless than 50% but above 25%. The unit should be cleaned regularly. Perspiration on foampillows may encourage mould growth. Pillows should be encased or changed regularly.
Other Precautions
Since vacuum cleaners are prone to mobilise fine respirable allergen particles, allergicpatients should not vacuum or if they do so, they should use a dust mask, or use a vacuumcleaner with a high efficiency particulate air filter. Air conditioning is helpful since thewindows and doors need to be closed and it reduces indoor humidity discouraging mouldand mite growth. Humidifiers are potentially harmful. The patient should avoid tobaccosmoke, both active and passive. Wood smoke and other smoke from domestic cookingshould be avoided as they are known to increase respiratory symptoms. To avoid this, allfurnaces and stoves are to be vented outside and the room is to be kept well-ventilated.Strong odours or sprays produced by cosmetics like perfumes, talcum powder, roomdeodorisers, frying, household cleaning products, and fresh paints irritate some patient’sairways and trigger asthma symptoms. Those affected by such odours should avoid them.Exposure to air pollutants like oxidants and sulphur oxides and ozone has been associatedwith worsening pulmonary function and increased airway hyperresponsiveness in personswith asthma. These environmental exposures may interact with allergens and other triggersin the causation of bronchial asthma.
Other precipitating factors like gastroesophageal reflux, sulphite sensitivity, and medica-tion sensitivities need to addressed in all patients.
All patients with asthma deserve an allergy evaluation to identify sensitisation to commoninhaled allergens. Avoidance of allergens to which a patient with asthma is sensitised is anintegral and effective part of asthma management. Indoor allergens are of particularimportance because of the most part of the time spent indoors. The indoor allergens mostlikely to be relevant are dust mites, cockroaches, and furred pets. Avoidance measures fordust mites,42 and cockroaches,42 are probably effective at improving asthma control if themeasures are strictly adhered to. Air filtration devices are unlikely to be important or effectiveover and above the more usual measures, given the characteristic distribution of theseallergens at home. Air filtration devices are effective at reducing levels of pet allergen inhome and may improve asthma control when combined with exclusion of the pet from thebedroom. This is likely to be less effective than ridding off the pet from the home completely.43
IMMUNOTHERAPY
The role of specific immunotherapy in asthma management is controversial and is undercontinual investigation. In fact, the British Thoracic guidelines clearly mentions that the

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 191
hyposensitisation or immunotherapy is not indicated in the management of chronic bronchialasthma in adults.9 Currently available medications and avoidance strategies usually providegood control of asthma. Allergen avoidance is always the first recommendation for managingasthma symptoms. However, when avoidance is not possible and appropriate medicationsfail to control symptoms of allergic asthma, referral for allergy immunotherapy may beconsidered.1 Allergy immunotherapy has been shown to reduce symptoms of asthma with avariety of allergens including house dust,44 cat dander,45 grass pollen,46 and alternaria .47
They also reduce the threshold for skin or lungs to the allergen employed and the late reactionto allergens in the lungs are also reduced.48 However, their efficacy in the overall clinicalmanagement remains controversial. Response to allergy immunotherapy decreases with ageand with lower baseline levels of pulmonary function.49 If immunotherapy is administered, itis recommended that once patient achieves maintenance levels of immunotherapy, the intervalbetween injections should be extended, with a goal of monthly injections. If the patient’ssymptoms improve, treatment is usually continued for 3-5 years, although under somesituations more prolonged therapy at monthly intervals may be needed. If there is no evidenceof response following two allergy seasons after reaching the maintenance or the highest leveltolerated by the patient, immunotherapy should be discontinued. Allergy immunotherapyshould only be administered in a physician’s office who is well versant with the therapy andwhere facilities and trained personnel are available to treat any life-threatening reaction thatcan occur, which is a very rare situation.
PHARMACOLOGICAL THERAPY
Pharmacological therapy for bronchial asthma is often described as “step care”, in which thenumber of medications and frequency of administration are increased or decreased asnecessary. The possibility of toxicity is also increased with this approach. The patient musthave medication, an inhaled β2−agonist, available for acute relief of symptoms. If symptomsoccur frequently (more than two times a week), preventive therapy is necessary in addition torescue treatment. Rescue treatment itself has a step-care pattern, adding medications asnecessary to control symptoms. Increasing use of rescue treatment by the patient is an indicationto review the medication plan and possibly to increase preventive therapy. As asthma is achronic inflammatory condition, anti-inflammatory treatment should be given to most patients.Treatment is to be considered in a step-wise manner as shown in Figure 12.2. In addition tothose depicted in the figure it is essential that:
a. The patient should avoid provoking factors whenever possibleb. Patient involvement and education should be an integral partc. The best inhaler device should be selectedd. Treatment may be stepped up as necessary to achieve good responsee. Treatment may be stepped down if control of asthma is good andf. A peak expiratory flow meter may be prescribed to monitor response to treatment.The patient should start treatment at the step most appropriate to the initial severity. A
reuse course of prednisolone tablets will be necessary at any time and at any stepA short “rescue” course of corticosteroid tablets may be needed at any time and at any step
to gain control of asthma. The indications of rescue courses of steroids are as follows:• Symptoms and PEFR get progressively worse day-by-day• PEFR falls below 60% of the patient’s best
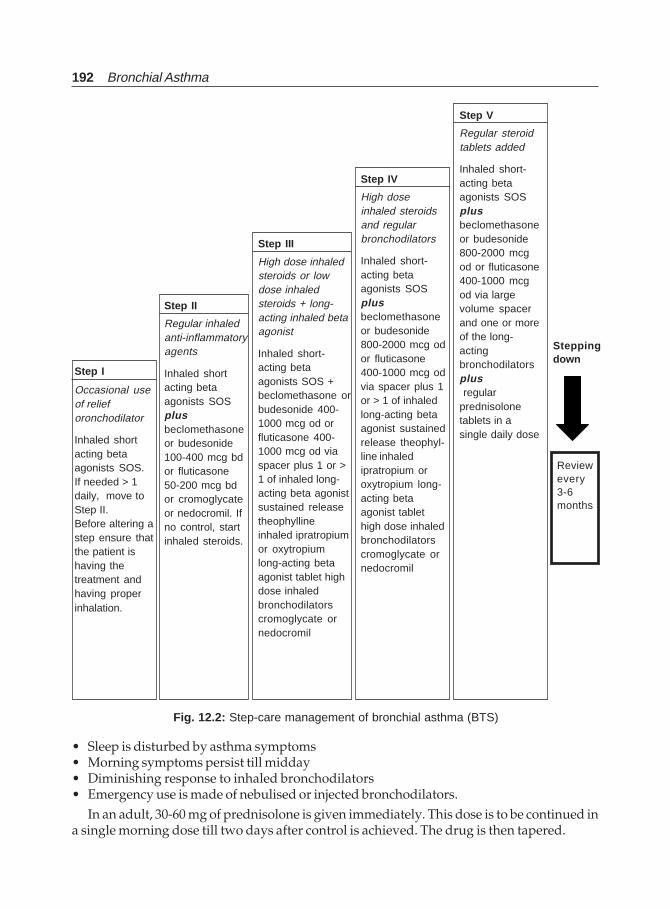
192 Bronchial Asthma
• Sleep is disturbed by asthma symptoms• Morning symptoms persist till midday• Diminishing response to inhaled bronchodilators• Emergency use is made of nebulised or injected bronchodilators.
In an adult, 30-60 mg of prednisolone is given immediately. This dose is to be continued ina single morning dose till two days after control is achieved. The drug is then tapered.
Step II
Regular inhaledanti-inflammatoryagents
Inhaled shortacting betaagonists SOSplusbeclomethasoneor budesonide100-400 mcg bdor fluticasone50-200 mcg bdor cromoglycateor nedocromil. Ifno control, startinhaled steroids.
Step III
High dose inhaledsteroids or lowdose inhaledsteroids + long-acting inhaled betaagonist
Inhaled short-acting betaagonists SOS +beclomethasone orbudesonide 400-1000 mcg od orfluticasone 400-1000 mcg od viaspacer plus 1 or >1 of inhaled long-acting beta agonistsustained releasetheophyllineinhaled ipratropiumor oxytropiumlong-acting betaagonist tablet highdose inhaledbronchodilatorscromoglycate ornedocromil
Step IV
High doseinhaled steroidsand regularbronchodilators
Inhaled short-acting betaagonists SOSplusbeclomethasoneor budesonide800-2000 mcg odor fluticasone400-1000 mcg odvia spacer plus 1or > 1 of inhaledlong-acting betaagonist sustainedrelease theophyl-line inhaledipratropium oroxytropium long-acting betaagonist tablethigh dose inhaledbronchodilatorscromoglycate ornedocromil
Step V
Regular steroidtablets added
Inhaled short-acting betaagonists SOSplusbeclomethasoneor budesonide800-2000 mcgod or fluticasone400-1000 mcgod via largevolume spacerand one or moreof the long-actingbronchodilatorsplus regularprednisolonetablets in asingle daily dose
Fig. 12.2: Step-care management of bronchial asthma (BTS)
Step I
Occasional useof relieforonchodilator
Inhaled shortacting betaagonists SOS.If needed > 1daily, move toStep II.Before altering astep ensure thatthe patient ishaving thetreatment andhaving properinhalation.
Steppingdown
Reviewevery3-6months

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 193
The appropriate treatment with different drugs in various steps are summarised below:
Step 5 Addition of Regular Steroid Tablets Inhaled short-acting beta agonists as requiredwith inhaled beclomethasone or budesonide 800-2000 μg daily or fluticasone 400-1000 μgdaily by a large volume spacer and one or more of the long-acting bronchodilators withregular prednisolone in a single daily dose.Step 4 High dose inhaled steroids and regular bronchodilators Inhaled short-actingβ-agonists as required with inhaled beclomethasone or budesonide 800-2000 μg daily orfluticasone 400-1000 μg daily through a large volume spacer plus a sequential therapeutictrial of one or more of inhaled long-acting beta agonist or sustained release theophyllineor inhaled ipratropium or oxytropium or long-acting beta agonist tablets high doseinhaled bronchodilators or cromoglycate or nedocromil.Step 3 High dose inhaled steroids or low dose inhaled steroids plus long-acting inhaled betaagonist bronchodilator Inhaled short-acting beta agonists as required plus eitherbeclomethasone or budesonide increased to 800-2000 μg daily or fluticasone 400-1000 μgdaily through a large volume spacer or beclomethasone or budesonide 100-400 μg twicedaily or fluticasone 50-200 μg twice daily. In a very small number of patients who experienceside effects with high doses of inhaled steroids, either the long-acting inhaled beta agonistor a sustained release theophylline may be added to step 2 medications. Cromoglycate ornedocromil may also be tried.Step 2 Regular inhaled anti-inflammatory agents Inhaled short-acting beta agonists asrequired plus beclomethasone or budesonide 100-400 μg twice daily or fluticasone50-200 μg twice daily. Alternatively, use cromoglycate or nedocromil sodium. If controlis not achieved start inhaled steroids.Step 1 Occasional use of relief bronchodilators Inhaled short-acting beta-agonists as requiredfor symptom relief are acceptable. If they are needed more than once daily move tostep 2.
Before altering a treatment step, it must be ensured that the patient is having the treatmentand has a good inhaler technique. Any fear the patient might be having must be addressed.
The possible outcome with various steps of treatment are shown below.
Outcome of steps 1-3: Control of asthma Outcome of step 4-5: Best possible result• Minimal or no chronic symptoms • Least possible symptoms• No nocturnal symptoms • Least need for relief bronchodilators• Infrequent exacerbations • Least possible activity limitations• Minimal need of relief bronchodilators • Least possible variation in PEFR• No activity or exercise limitations • Best PEFR• PEFR variation < 20%, PEFR 80%/more • Least adverse reactions from drugs• Minimum side effects from drugs
The expected line of therapy and possible outcome in different grades of asthma aredepicted in Figure 12.3.
For patients who have established control of their asthma, regular follow up visits atapproximately 1 to 3 month intervals are necessary to review the treatment plan, the patientsmanagement techniques including use of medicines, peak flow measurements, etc. For many

194 Bronchial Asthma
patients with moderate to severe asthma, control of the disease as reflected in normalisationof pulmonary function and activity levels without symptoms, can be maintained with onlycontinuous preventive therapy. The aim of therapy should be to use the minimum medica-tion needed to maintain control with minimum risk for adverse effects. Reduction of therapycan be carefully considered if PEFR variability is less than 10% and there are no asthmasymptoms for a reasonable period (2-3 days for the exacerbation in mild asthma, severalweeks for moderate or severe asthma). If PEFR variability is greater than 10-20%, evaluationof the patient’s technique in using the medication, find out environmental aggravators andthe patient’s efforts to control them will be necessary. The possibility of concomitant upperrespiratory tract disease, and the possibility of increasing the dosage or change of medicationsmay also need to be considered.
Recently the Expert Panel Report 2 of the National Institute of Health has published guide-lines for the diagnosis and management of bronchial asthma in adults and children .50
Although the basic principles of management are the same, some more facts have beenhighlighted. The basic components of management include:A. Measures of assessment and monitoring. The new guidelines includes additional goal
of therapy to meet the expectations of the patient and its family with satisfaction. Periodicassessment of six domains of patient health is highlighted. These include signs andsymptoms, pulmonary function, quality of life, history of exacerbations, pharmaco-therapy, patient-provider communication and patient satisfaction. The panel alsorecommends home monitoring of PEFR from twice daily to once in the morning only. Ifthe morning reading is less than 80% of the personal best PEFR, more frequent monitoringmay be required. The use of the individual patient’s personal best PEFR is emphasised.It is emphasised that patients of all severity levels are to monitor symptoms to recogniseearly signs of deterioration. Sample questions to use in periodic assessments are alsoadded.
B. Control of factors contributing to asthma severity is important. Skin testing or in vitrotesting is now specifically recommended for at least those patients with persistent asthma
Fig.12.3: Step-care management of asthma and expected outcome

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 195
exposed to perennial indoor allergens. Adult patients with severe persistent asthma, nasalpolyps, or a history of sensitivity to aspirin or non-steroidal anti-inflammatory drugs areto be counselled regarding the risk of severe and even fatal exacerbations from using thesedrugs. Routine use of chemicals to kill house-dust mite and denature the antigen is nolonger recommended as a control measure. Patients should be treated for rhinitis, sinusitis,and gastro-oesophageal reflux. Annual influenza vaccinations are specificallyrecommended for patients with persistent asthma. Pneumococcal vaccination is considerednot important.
C. The importance of pharmacological therapy is very much there and is an importantcomponent of asthma management. However, medications are now categorised into twogeneral classes; (a) long-term control medications; and (b) quick-relief medications. Whilethe former are used to achieve and maintain control of persistent asthma, the later are usedto treat acute symptoms and exacerbations. The most effective medications for long-termcontrol are those having anti-inflammatory effects and include drugs like corticosteroids,long-acting β2-agonists, and leukotriene antagonists. The stepwise approach to asthmatherapy emphasises initiating a higher level of therapy at the onset to establish promptcontrol and then stepping down. Corticosteroids are the most important anti-inflammatorydrugs and include various inhaled forms as discussed earlier and systemic drugs likemethylprednisolone, prednisolone, and prednisone.
D. Education for a partnership in asthma care includes providing patients both a writtentreatment plan for daily self-management and a written action plan for management ofexacerbations. However they do not replace, though supplement, the education by thephysician. Patient education by the principal clinician as well as other members of thehealth care team is important. To enhance the delivery of education, detailed questions toelicit information and educational messages for each visit are to be provided. Key messagesare to be reinforced during each visit. Further, it is necessary to evaluate the outcome interms of patient perceptions of improvement, specially quality of life and the ability toengage in desired activities. Patient education for CFC-free inhalers is gaining importancein view of the ban on the use of CFC (chlorofluorocarbons).
The Expert Panel50 has changed the classification of the severity of asthma from mild,moderate, severe to mild intermittent, mild persistent, moderate persistent, and severe persistentasthma. The clinical features before treatment are shown in Table 12.1.
Stepwise management of bronchial asthma in adults and children over 5 years of agedepending upon the above severity criteria as recommended by the NIH Expert Panel isshown in Table 12.2.
GENERAL PRINCIPLES OF APPROACH TO TREATMENT OFCHRONIC PERSISTENT ASTHMA
i. Treatment of chronic persistent asthma should be considered in a stepwise manner asdescribed above with the patients starting treatment at the step most appropriate forthe initial severity of their conditions.
ii. The stepwise approach presents general guidelines to assist clinical decision making;it is not intended to be a specific prescription.
iii. Since asthma is highly variable; clinicians should tailor specific medication plans tothe needs and circumstances of individual patients.
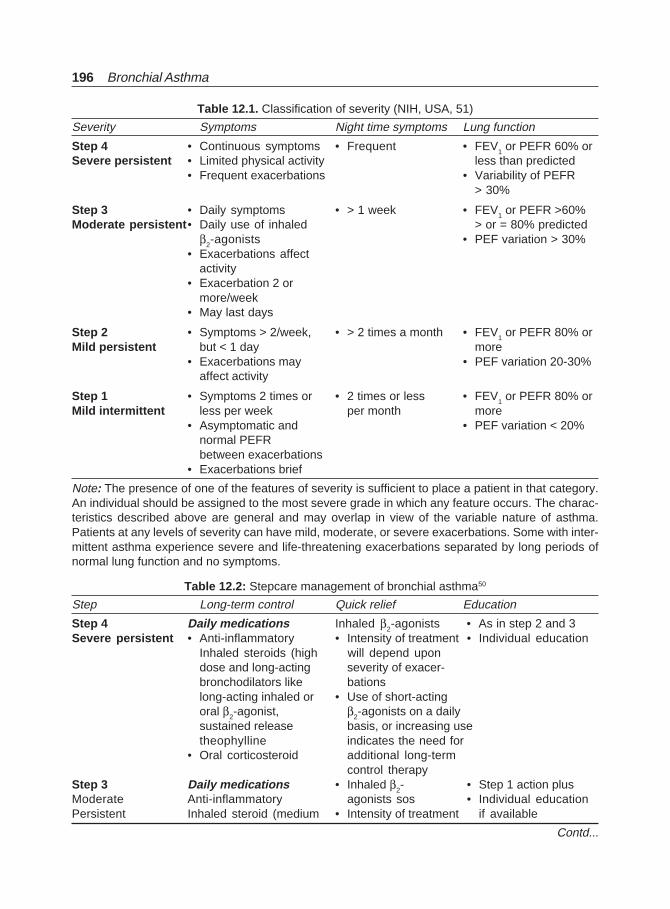
196 Bronchial Asthma
Table 12.2: Stepcare management of bronchial asthma50
Step Long-term control Quick relief Education
Step 4 Daily medications Inhaled β2-agonists • As in step 2 and 3Severe persistent • Anti-inflammatory • Intensity of treatment • Individual education
Inhaled steroids (high will depend upondose and long-acting severity of exacer-bronchodilators like bationslong-acting inhaled or • Use of short-actingoral β2-agonist, β2-agonists on a dailysustained release basis, or increasing usetheophylline indicates the need for
• Oral corticosteroid additional long-termcontrol therapy
Step 3 Daily medications • Inhaled β2- • Step 1 action plusModerate Anti-inflammatory agonists sos • Individual educationPersistent Inhaled steroid (medium • Intensity of treatment if available
Table 12.1. Classification of severity (NIH, USA, 51)
Severity Symptoms Night time symptoms Lung function
Step 4 • Continuous symptoms • Frequent • FEV1 or PEFR 60% orSevere persistent • Limited physical activity less than predicted
• Frequent exacerbations • Variability of PEFR> 30%
Step 3 • Daily symptoms • > 1 week • FEV1 or PEFR >60%Moderate persistent• Daily use of inhaled > or = 80% predicted
β2-agonists • PEF variation > 30%• Exacerbations affect
activity• Exacerbation 2 or
more/week• May last days
Step 2 • Symptoms > 2/week, • > 2 times a month • FEV1 or PEFR 80% orMild persistent but < 1 day more
• Exacerbations may • PEF variation 20-30%affect activity
Step 1 • Symptoms 2 times or • 2 times or less • FEV1 or PEFR 80% orMild intermittent less per week per month more
• Asymptomatic and • PEF variation < 20%normal PEFRbetween exacerbations
• Exacerbations brief
Note: The presence of one of the features of severity is sufficient to place a patient in that category.An individual should be assigned to the most severe grade in which any feature occurs. The charac-teristics described above are general and may overlap in view of the variable nature of asthma.Patients at any levels of severity can have mild, moderate, or severe exacerbations. Some with inter-mittent asthma experience severe and life-threatening exacerbations separated by long periods ofnormal lung function and no symptoms.
Contd...

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 197
dose) or low-medium will depend upon • Review and updatedose and add a long- severity of exacer- self management planacting bronchodilator, bationsespecially for night- • Use of short-actingtime symptoms: either β2-agonists onlong-acting inhaled daily basis indicateβ2-agonist, sustained need for term-controlrelease theophylline therapyor long-actingβ2-agonist tablets
If needed:Anti-inflammatory:Inhaled corticosteroids(medium-high dose) andlong-acting broncho-dilators, especially fornight-time symptoms;either long-actinginhaled β2-agonist,sustained releasetheophylline, or long-acting β2-agonist tablets
Step 2 Daily medications: Short-acting • Step 1 action +Mild persistent Anti-inflammatory bronchodilator: teach self monitoring
Either inhaled steroid • Inhaled β2- • Group education.(low doses) or cromolyn agonist as needed. • Review andor nedocromil • Intensity of treatment update self manageAlternatively, sustained will depend on exa- ment plan.release theophylline. cerbation severity.Leukotriene antagonists • Use of short actingmay also be tried for the inhaled β2-agonistspatients of 12 yrs or >, daily or increasingalthough their role in use indicates needtherapy is not fully for additional long-established. term control therapy.
Step 1 Daily medication not Short-acting broncho- • Teach basic facts ofMild intermittent needed dilator: Inhaled β2- bronchial asthma
agonist sos • Teach inhaler spacer/• Intensity of treatment holding chamber
will depend on techniqueseverity of exacer- • Discuss role of medi-bation cation management
• Use of above more planthan 2 times a week • Develop self manage-may need to start ment plan
Step Long-term control Quick relief Education
Contd...
Contd...

198 Bronchial Asthma
iv. The control should be gained as soon as possible and then the treatment is to bedecreased to the least medications necessary to maintain control. Gaining control maybe accomplished by either starting treatment at the step most appropriate to the initialseverity of their condition or by starting a high level of therapy like a short course oforal corticosteroids may be needed at any time and at any step to control their asthmaas mentioned above. Alternatively, a higher dose of inhaled corticosteroid may beused.
v. Patients should avoid all known factors precipitating their asthma. They may bechemicals, certain drugs like aspirin and non-steroidal anti-inflammatory drugs.β−blockers are contraindicated in asthma.
vi. A rescue course of systemic corticosteroids may be required at any step at any time.vii. Some patients with intermittent asthma experience severe and life-threatening
exacerbations separated by long periods of normal lung function and no symptoms.This may be especially common with exacerbations provoked by respiratoryinfections. A short course of systemic corticosteroids is recommended in that situation.
viii. At each step the patient should control his environment to avoid or control factorsthat make their asthma worse like allergens, irritants, etc. This requires specificdiagnosis and education.
ix. Referral to an asthma specialist for consultation or co-management is recommendedif there are difficulties in achieving or maintaining control of asthma or if the patientrequires step 4 care. Referral may be considered if the patient requires step 3 care.
Quick-relief Medications
Bronchodilators. Short-acting β2-agonists are the therapy of choice for relief of acute symptomsand prevention of exercise-induced asthma. They are the most effective medications forrelieving acute bronchospasm. A β2-agonist such as salbutamol 100-200 μg or terbutaline250-500 μg should be used as required rather than regularly. Patients should be encouragedto use the minimum dose to control their symptoms. They can be used 2 puffs three to fourtimes a day. Salbutamol is also available as rotahalers with 100-200 mcg/capsule which can
Step Long-term control Quick relief Education
Contd...
long-term control • Develop action plan totherapy when and how to take
rescue actions• Discuss appropriate
environment controlmeasures
Step down Step upReview treatment every 1-6 months If control is not maintained, considera gradual stepwise reduction in treatment step up. First, review patient medicationmay be possible technique adherence, and environment
control

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 199
be used 1-2 capsules every 4-6 hours as needed and prior to exercise. Increasing use of thesedrugs or roughly use of more than one canister in one month indicates inadequate control ofasthma and the need for initiating or intensifying anti-inflammatory therapy. Regularlyscheduled or daily use of these drugs are generally not recommended. Solutions for use innebulisers are also available (salbutamol 5 mg/ml or terbutaline 5 mg/ml) for use duringacute exacerbations.
Anticholinergic drugs like ipratropium bromide may provide additive benefit to inhaledβ2-agonists in severe exacerbations. It may be an alternative bronchodilator for patients whodo not tolerate inhaled β2-agonists. They may be used as regular maintenance therapy in step4 (British Thoracic Society) patients who already require high dose inhaled steroids. The drugis also available in solution forms for nebulisation. The MDI dose is 20 mcg for each puff and2-3 puffs are used every 6 hours.
Although systemic corticosteroids are strictly not bronchodilators, they are used for moderateto severe exacerbations to speed recovery and prevention of exacerbations along with othermedications.
Long-term Control Medications
Inhaled anti-inflammatory agents. Patients who need to inhale a bronchodilator more than oncedaily or who have night time symptoms require regular inhaled anti-inflammatory drugs.Various drugs that can be used include corticosteroids, sodium cromoglycate (5-20 mg fourtimes a day), and nedocromil sodium (4 mg four times daily). Inhaled steroids are the drugs ofchoice since they are the most potent and effective anti-inflammatory drugs available currentlyfor mild, moderate and severe persistent asthma. The small but potential risk of adversereactions from the use of inhaled corticosteroids is well balanced by their efficacy. Inhaledform is used for the long-term control of asthma. Systemic corticosteroids are used in long-term therapy to gain prompt control of the disease and also to manage severe persistentasthma. Beclomethasone dipropionate or budesonide should be started in doses of 100-400μg twice daily. As the severity increases, the doses are to be increased. Patients with nocturnalsymptoms and more severe and persistent form of disease may need more frequent and higherdoses. Once symptoms and PEFR are controlled, the dose should be reduced to be maintainedwith the minimum.
High doses of inhaled steroids. If control is not achieved, spacers and higher doses are neededafter checking the compliance and proper use of inhalers. When the dose exceeds 800 μg, alarge volume spacer is recommended to reduce systemic and local effects. The delivery systemis an important determinant of the systemic effects of inhaled corticosteroids. The Turbohalerdelivers approximately twice as much inhaled steroid to the lung,52 and, therefore the dosemay be halved when this device is used. The patient should also be advised to rinse mouth(rinse and spit) following inhalation. The lowest possible dose of inhaler should be used tomaintain control. Current guidelines recommend that patients should double the dose ofinhaled steroids temporarily if their asthma deteriorates or at the first sign of an upperrespiratory tract infection. To maintain control asthma, particularly nocturnal symptoms, along-acting inhaled β2-agonist is to be added to a low-to medium dose of inhaled corticosteroidrather than using a high dose. For children growth monitoring is essential.
Inhaled corticosteroids are typically associated with a flat dose-response curve whentraditional efficacy values are examined by measurement of FEV1. Thus, by increasing the
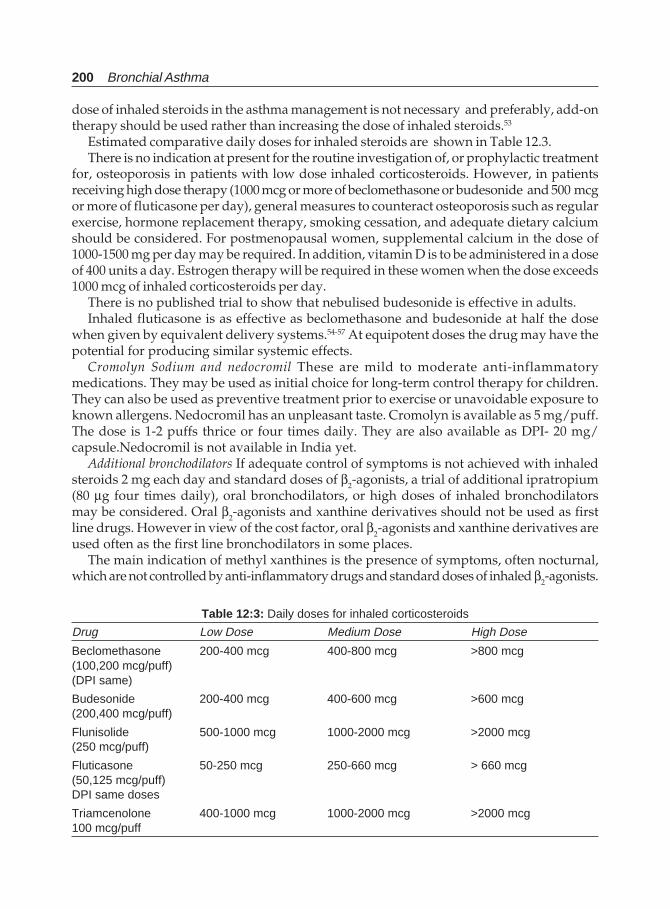
200 Bronchial Asthma
Table 12:3: Daily doses for inhaled corticosteroids
Drug Low Dose Medium Dose High Dose
Beclomethasone 200-400 mcg 400-800 mcg >800 mcg(100,200 mcg/puff)(DPI same)
Budesonide 200-400 mcg 400-600 mcg >600 mcg(200,400 mcg/puff)
Flunisolide 500-1000 mcg 1000-2000 mcg >2000 mcg(250 mcg/puff)
Fluticasone 50-250 mcg 250-660 mcg > 660 mcg(50,125 mcg/puff)DPI same doses
Triamcenolone 400-1000 mcg 1000-2000 mcg >2000 mcg100 mcg/puff
dose of inhaled steroids in the asthma management is not necessary and preferably, add-ontherapy should be used rather than increasing the dose of inhaled steroids.53
Estimated comparative daily doses for inhaled steroids are shown in Table 12.3.There is no indication at present for the routine investigation of, or prophylactic treatment
for, osteoporosis in patients with low dose inhaled corticosteroids. However, in patientsreceiving high dose therapy (1000 mcg or more of beclomethasone or budesonide and 500 mcgor more of fluticasone per day), general measures to counteract osteoporosis such as regularexercise, hormone replacement therapy, smoking cessation, and adequate dietary calciumshould be considered. For postmenopausal women, supplemental calcium in the dose of1000-1500 mg per day may be required. In addition, vitamin D is to be administered in a doseof 400 units a day. Estrogen therapy will be required in these women when the dose exceeds1000 mcg of inhaled corticosteroids per day.
There is no published trial to show that nebulised budesonide is effective in adults.Inhaled fluticasone is as effective as beclomethasone and budesonide at half the dose
when given by equivalent delivery systems.54-57 At equipotent doses the drug may have thepotential for producing similar systemic effects.
Cromolyn Sodium and nedocromil These are mild to moderate anti-inflammatorymedications. They may be used as initial choice for long-term control therapy for children.They can also be used as preventive treatment prior to exercise or unavoidable exposure toknown allergens. Nedocromil has an unpleasant taste. Cromolyn is available as 5 mg/puff.The dose is 1-2 puffs thrice or four times daily. They are also available as DPI- 20 mg/capsule.Nedocromil is not available in India yet.
Additional bronchodilators If adequate control of symptoms is not achieved with inhaledsteroids 2 mg each day and standard doses of β2-agonists, a trial of additional ipratropium(80 μg four times daily), oral bronchodilators, or high doses of inhaled bronchodilatorsmay be considered. Oral β2-agonists and xanthine derivatives should not be used as firstline drugs. However in view of the cost factor, oral β2-agonists and xanthine derivatives areused often as the first line bronchodilators in some places.
The main indication of methyl xanthines is the presence of symptoms, often nocturnal,which are not controlled by anti-inflammatory drugs and standard doses of inhaled β2-agonists.

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 201
The addition of long-acting xanthine derivatives may be good enough to control symptoms.Sustained release theophylline is a mild to moderate bronchodilator used principally asadjuvant to inhaled corticosteroids for prevention of nocturnal symptoms. They may havemild anti-inflammatory effect also. Usual starting dose is 10 mg/kg of body weight up to amaximum of 800 mgm per day.
Long-acting β2-agonists are to be used concomitantly with anti-inflammatory drugs (low-to-medium dose corticosteroid inhalers) for long-term control of symptoms, particularlynocturnal symptoms. They also prevent exercise-induced bronchospasm. Long-acting β2-agonists are not to be used for the treatment of acute symptoms or exacerbations. High doses ofinhaled bronchodilators should be considered only if the patient does not respond to standarddoses. Daily use of long-acting β2-agonists should generally not exceed 80-100 mcg. Salmeterolis currently available in India and Formoterol is being tried for clinical use in this country.Oral sustained release salbutamol 8 mgs tablets are available which can be used twice daily.
Leukotriene Modifiers These drugs may be considered as alternative therapy to low doses ofinhaled corticosteroids or cromolyn or nedocromil in mild persistent asthma in patients 12years or older. These drugs are not yet marketed in India. Further study and clinical experienceare needed to help establish their role in asthma therapy. The leukotriene receptor antagonistis available in USA as 20 mg tablets and the adult dose is 40 mg daily (1 tablet twice daily).The 5-lipoxygenase inhibitor Zileuton is available as 300 and 600 mg tablets and the dailydose is 2400 mgs in four divided doses.
Oral steroids Systemic corticosteroids are used in long-term therapy to gain prompt controlof the disease (Rescue therapy) and also to manage severe persistent asthma. High doses ofinhaled steroids should always be continued in patients receiving oral steroids. These patientsneed to be referred to a specialty clinic where additional measures can be considered. Forlong-term treatment of severe persistent asthma, single dose is to be administered in a.m.either daily or on alternate days which may produce less adrenal suppression. Short coursesor “bursts” are effective for establishing control when initiating therapy or during a period ofgradual deterioration. The burst should be continued till the patient achieves 80% of personalbest PEFR or the symptoms resolve. This usually requires 3-10 days, but may require longer.The most commonly available oral corticosteroid is prednisolone, although methylprednisoloneor prednisone can also be used. Prednisolone tablets are available in 5 mg, and 20 mg, tablets.For control of asthma symptoms, a dose of 10-60 mgms daily single doses may be required. Forshort-course “burst” 40-60 mgm per day as single or 2 divided doses may be given for 3-10days.
Alternative and Complementary Therapies
Alternative and complementary therapies are sought most frequently for many chronicdisabling conditions as a desperate (sometimes with true belief) measure as there is nodefinite “cure” for these conditions. These include back pain, anxiety, depression, arthritis,headaches, HIV infection, and chronic allergic disorders like allergic rhinitis, and asthma.58,59
These forms of treatment include acupuncture, homeopathy, fish therapy, other herbal therapyincluding ayurvedic drugs, ionisers, and antihistamines including ketotifen, chiropractice,megavitamins, and spiritual healing, which are tried by many but have not stood the test ofcontrolled clinical trials, although some patients claim benefit. Results of a 1998 surveyindicates that 42% of adults in the United States consult alternative medical practitioners and

202 Bronchial Asthma
spend an estimated $27 billion annually on alternative medical therapies.58 Many physicianseither practice for complimentary/alternative medicine themselves or refer patients for suchtreatment.60
Acupuncture is one of the oldest and most widespread complimentary techniques, althoughhomeopathy and Aurvedic forms of therapy are other forms of therapy in many countries. Thefirst documented history of acupuncture is ascribed to the legendary Yelloe Emperor (HuangDi) in China (circa 2000 BC). The first paragraph of the second part of his classic book HuangDi Nei Jing describes the desire of the benevolent emperor to relieve the suffering of his subjectsaffected with disease, instead of the use of poisons of medicine in favour of fine needles toharmonise the blood and Qi energy.61 British Medical Association has recently approvedacupuncture for practice,62 and many medical schools in the USA offer elective courses incomplimentary/alternative medicine.63 Traditional Chinese medicine has claimed the abilityto favourably influence the course and symptoms of bronchial asthma. However, the publisheddata on this subject are controversial. Real acupuncture has been shown to have an immediateeffect,64-66 but not a lasting effect on asthma.67-69 Methacholine challenges and exercise havebeen shown to be affected. However, most studies performed to date are not controlled orcross-over in design.70,71 Immunomodulatory changes in lymphocyte subsets and cytokineshave been shown to be affected by acupuncture. In a recent randomised cross over study, itwas shown that a short course of acupuncture treatment in patients with moderate asthmaresulted in no change in lung function, bronchial hyperreactivity, or patient symptoms.72
Thus, acupuncture may not be recommended for bronchial asthma, although still thousandsof patients continue to seek for this form of therapy. If these modalities of complimentary/alternative medicine are tried, then the conventional treatment should also be continued.
Hyposensitisation or immunotherapy is not indicated in the management of asthma.Ascaricides may be effective in controlling number of mites but have not produce clinically
relevant benefit.
Referral to a Specialist
Referral to a specialist should be considered for:i. Patients in whom there is a doubt about the diagnosis for example in elderly and smokers
with wheeze; those with persistent unexplained persistent cough; and those withsystemic symptoms like fever, rash, weight loss, or proteinuria that might suggestassociated disorders such as systemic eosinophilia or vasculitis.
ii. Patients with possible occupational asthma.iii. Patients with difficult asthma and management problems. Such patients include
• those who are recently discharged from a hospital• those with catastrophic severe (brittle) asthma• those with continuing symptoms despite high doses of inhaled steroids• those being considered for long-term treatment with nebulised bronchodilators• pregnant women with worsening asthma, and• patients in whom asthma is interfering with their lifestyles despite changes in
treatment.iv. Patients suspected to have developed complication like bronchopulmonary mycosis
(i.e.; ABPA).

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 203
These patients most often misdiagnosed as pulmonary tuberculosis. This complicationmay be suspected if the patient has uncontrolled symptoms, systemic symptoms, haemoptysis,weight loss, profuse expectoration, and infiltrates in chest skiagram.
Assessment of Asthma Control
Once a treatment plan is established for a patient of bronchial asthma, it is essential to assessthe asthma control. Most asthma guidelines recommend assessing asthma control accordingto a series of criteria based on symptoms and pulmonary function.11,73 As the aim of asthmatreatment is to minimise symptoms, rescue bronchodilator need, and exacerbations, whileoptimizing pulmonary function. Recently, methods for assessment of airway inflammationnoninvasively have been developed, but they are not currently integrated into the assessmentof asthma control.74,75 Studies or surveys on asthma generally use an all or none approach ora strictly qualitative evaluation of asthma control, without specific quantification of itsmagnitude or degree compared with optimal goals. Other means of assessing these parametersinclude evaluating or scoring each separate component of asthma control and comparing theeffects of treatment or intervention on these specific parameters. This global assessmentapproach may overestimate the adequacy of asthma control by the physician and even moreso, by the patient, and may consequentially contribute to the poor asthma control whencurrent guideline criteria are used.76-78 Quantification of control with tools such as the validatedquestionnaire developed by Juniper et al79 are the common ones, but they are too exhaustive tobe used by the busy clinician. A new, simple method of global assessment and quantificationof asthma control has been developed.80 This easy-to-use asthma control scoring system isbased on a percentage of optimal control. The percentage symptom score but not the globalcontrol score of this new method correlates with patient’s global assessment of asthma control.This method provides a percentage control for symptoms, baseline expiratory flows, and anoptional parameter for airway inflammation assessed from induced sputum eosinophil count.
REFERENCES
1. National Asthma Education Programme. Expert Panel Report. Guidelines for the diagnosis andmanagement of asthma. National Heart, Lung, and Blood Institute, National Institute of Health,Bethesda, Maryland, USA, Publication No. 91-3042A, June 1991.
2. Guidelines for the management of asthma in adults. 1-Chronic persistent asthma. Statement bythe British Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:651-53.
3. Guidelines for the management of asthma in adults. 2-Acute severe asthma. Statement by theBritish Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:797-800.
4. Warner JO, Gotz M, Landau LI et al. Management of asthma: A consensus statement. Arch DisChild 1989;64;1065-79.
5. International Paediatric Asthma Consensus Group. Asthma, a follow-up statement. Arch DisChild 1992;67:240-48.
6. International Consensus report on the diagnosis and management of asthma. Clin Exp Allergy1992;22(Suppl):1-72.
7. British thoracic Society and others. Guidelines for the management of asthma: A summary. BMJ1993;9:287-92.

204 Bronchial Asthma
8. The British Guidelines on Asthma Management. 1995 Review and Position Statement. Thorax1997;52(Suppl 1): S2-S8.
9. British thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, national asthma Campaign et al. Guidelines on the management ofasthma. Thorax 1993;48:S1-S24.
10. British thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, national asthma Campaign et al. Summary charts. B M J 1 9 9 3 ; 3 0 6 :776-82.
11. Global Initiative for Asthma. A practical guide for public health officials and health careprofessionals. US Department of Health and human services. NIH Publication No. 96-3659A,December 1995.
12. Brewis RAL. Patient education, self-management plans and peak flow measurements. RespirMed 1991;85:457.
13. Feldman CH, Clark NM, Evans D. The role of health education in medical management inasthma. Clin Rev Allergy 1987;5:195-205.
14. Mellians RB. Patient education is key to successful management of asthma. J Rev Respir Dis1989;Suppl:S47-S52.
15. Clark NC. Asthma self-management education: Research and implications for clinical practice.Chest 1989;95:1110-13.
16. D’Souza W, Crane J, Burgess G et al. Community based asthma care: Trial of a “credit card”asthma self-management plan. Eur Respir J 1994;7:1260-65.
17. Ignacia-Garcia JM, Gonzalez-Santos P. Asthma self management education programme byhome monitoring of peak expiratory flow. Am J Respir Crit Care Med 1995;151:353-59.
18. Schulman BA. Active patient orientation and outcomes in hypertensive treatment. Med Care1979;17:267-80.
19. Clark NC. Asthma self-management education: Research and implications for clinical practice.Chest 1989;95:1110-13.
20. Korsch BM, Gozzi EK, Francis V. Gaps in doctor-patient communication. I. Doctor-patientinteraction and patient satisfaction. Pediatr 1958;42:855-71.
21. Shim C, Williams MH. The adequacy of inhalation of aerosol from canister nebulisers. Am J Med1980;69:891-94.
22. Newman SP, Pavia D, Clarke SW. Simple instructions for using pressurised aerosol broncho-dilators. Jr Soc Med 1980;73:776-79.
23. Croft DR, Peterson MW. An evaluation of the quality and contents of asthma education on theWorld Wide Web. Chest 2002;121:1301-07.
24. Solomon WR, Burge HA, Bloise JR. Exclusion of particulate allergens by window air conditioners.J Allergy Clin Immunol 1980;65:305-08.
25. Koostra JB, Pasch R, reed CE. The effects of air cleaners on hay-fever symptoms in air-conditionedhomes. J allergy Clin Immunol 1978;61:315-19.
26. Wilson AF, Novy HS, Berke RH, Sufprenant EC. Deposition of inhaled pollen and pollen extractin human airways. N Engl J Med 1973;288:1056-58.
27. Woods RA, Chapman MD, Adkinson FN Jr, Eggleston PA. The effect of cat removal on allergencontent in household-dust samples. J Allergy Clin Immunol 1989;83:730-34.
28. Chapman MD, Wood RA. The role and remediation of animal allergens in allergic diseases.J Allergy Clin Immunol 2-001;107(3 Suppl):S414-S421.
29. Wood RA, Johnson EF, van Natta ML et al. A placebo controlled trial of a HEPA air cleaner in thetreatment of cat allergy. Am J Respir Crit Care Med1998;158:115-20.
30. Van Der Heise S, van Aalderen WM, Kauffman HF et al. Clinical effects of air cleaners at homesof asthmatic children sensitised to pet allergens. J Allergy Clin Immunol 1999;104(2 pt 1):447-51.

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 205
31. McDonald E, Cook D, Newman T et al. Effects of air filtration systems on asthma. A systematicreview of randomised trials. Chest 2002;122:1535-42.
32. Tovey ER, Chapman MD, Platts-Mills TAE. Mite faeces are a major source of house-dust allergens.Nature 1981;289:592-93.
33. Platts-Mills TAE, de Weck AL. Dust-mite allergens and asthma: Worldwide problem. J AllergyClin Immunol 1989;83:416-26.
34. Swanson MC, Campbell AR, Klauck MJ, Reed CE. Correlation between levels of mite and catallergens in settled and airborne dust. J Allergy Clin Immunol 1989;83:776-83.
35. Walshaw MJ, Evans CC. Allergen avoidance in house dust mite sensitive adult asthma. Q J Med1986;58:199-215.
36. Ehnert B, Lau-Sehadendorf S, Weber A, Buettner P, Sehou C, Wahn U. Reducing domesticexposure to dust mite allergen reduces bronchial hyperreactivity in sensitive children withasthma. J Allergy Clin Immunol 1993;90:135-38.
37. Murray AB, Fergusson AC. Dust free bed room in the treatment of asthmatic children withhouse dust mite allergy: A controlled trial. Pediatrics 1983;91:418-22.
38. Colloff MJ, Ayeres J, Carswell F et al. The control of dust mites and domestic pets: A positionpaper. Clin Exp Allergy 1992;22(Suppl 2):1-28.
39. Arlian LG, Platts-Mills TA. The biology of dust mites and the remediation of mite allergens inallergic diseases. J Allergy Clin Immunol 20-01;107(3 Suppl):S406-S13.
40. Custovic A, Simpson A, Chapman MD et al. Allergen avoidance in the treatment of asthma andatopic disorders. Thorax 1998;53:63-72.
41. Lan JL, Lee DT, Wu CH et al. Cockroach hypersensitivity: Preliminary study of allergic cockroachasthma in Taiwan. J Allergy Clin Immunol 1988;82:736-40.
42. Eggleston PA, Arruda LK. Ecology and elimination of cockroaches allergens in the home.J Allergy Clin Immunol 2001;107(3 Suppl):S422-S29.
43. Ernst P. Environmental measures and asthma. Chest 2002;122:1509-10.44. Aas K. Controlled trial of hyposensitisation to house dust. Acta Pediat Scand 1971;60:264-68.45. Ohman JL Jr, Findlay SR, Leiterman KM. Immunotherapy in cat-induced asthma: Double-blind
trial with evaluation of in vivo and in vitro responses. J Allergy Clin Immunol 1984;74:230-39.46. Reid MJ, Moss RB, Hsu YP. Seasonal asthma in northern California: Allergy causes and efficacy
of immunotherapy. J Allergy Clin Immunol 1986;78:590-600.47. Horst M, Hejjaoui A, Horst V, Michel FB, Bouquest J. Double-blind, placebo-controlled rush
immunotherapy with a standardised alternaria extract. J Allergy Clin Immunol 1990;85:460-72.48. Van Bever HP, Stevens WJ. Suppression of the late asthmatic reaction by hyposensitisation in
asthmatic children allergic to house dust mites (dermatophagoides pteronyssiunus). Clin ExpAllergy 1989;19:399-404.
49. Bousquet J, Maasch HJ, Hejjaoui A et al. Double-blind, placebo-controlled immunotherapy withmixed grass-pollen allergoids. III. Efficacy and safety of unfractionated and high-molecular-weight preparations in rhino conjunctivitis and asthma. J Allergy Clin Immunol 1989;84:546-56.
50. Expert Panel Report 2: Guidelines for the Diagnosis and Management of Asthma. AmericanCollege of Allergy, Asthma and Immunology. National Heart, Lung and Blood Institute, NationalInstitute of Health. NIH Publication No. 97-4051A, May 1997.
51. Glinert R, Wilson P, Wedner HJ. Fel; D1 is markedly reduced following sequential washing ofcats. J Allergy Clin Immunol 1990;85:225.
52. Thorsson L, Edsbacker S, Conardson TB. Lung deposition of budesonide from turbohaler istwice that from a pressurised metered dose inhaler p-MDI. Eur Respir J 1994;7:1839-44.
53. O’Sullivan S, Cormican L, Murphy M et al. Effects of varying doses of fluticasone propionate onthe physiology and bronchial wall immunopathology in mild-to-moderate asthma. Chest2002;122:1966-72.

206 Bronchial Asthma
54. Lundback B, Alexander M, Day J et al. Evaluation of fluticasone propionate (500 mcg per day)administered either as dry powder via diskhaler or pressurised inhaler and compared withbeclomethasone dipropionate (1000 mcg/day) administered by pressurised inhaler. RespirMed 1993;87:609-20.
55. Gustaffson P, Tanakas J, Gold M, Primhak R, Radford M, Gillies E. Comparison of the efficacyand safety of inhaled fluticasone pripionate 200 mcg per day with beclomethasone dipropionate2000 mcg per day in mild and moderate asthma. Arch Dis Child 1993;19:206-11.
56. Leblane P, Minks S, Keistinen T, Saaelainen PA, Ringdal N, Payne SL. A comparison of fluticasonepropionate 200 mcg/day with beclomethasone dipropionate 400 mcg/day in adult asthma.Allergy 1994;49:380-85.
57. Conolly A. A comparison of fluticasone propionate 100 mcg twice daily with budesonide 200 mcgtwice daily via their respective powder devices in the treatment of asthma. Eur J Clin Res1996;7:15-29.
58. Eisenberg DM, Davis RB, Ettner SL et al. Trends in alternative medicine use in the United States.1990-1997: Results of a follow-up national survey. JAMA 1998;280:1569-75.
59. Fairfield KM, Eisenberg DM, Davis RB et al. Patterns of use, expenditures, and perceived efficacyof complimentary and alternative therapies in HIV-infected patients. Arch Intern Med 1998;158:2257-64.
60. Austin JA, Marie A, Pelletier KR et al. A review of the incorporation of complementary andalternative medicine by mainstream physicians. Arch Intern Med 1998;158:2303-10.
61. Wu JN. A short history of acupuncture. J Altern Complement Med 1996;2:19-21.62. Silvert M. Acupuncture wins BMA approval. BMJ 2000;321:11.63. Wetzel MS, Eisenberg DM, Kaptchuk TJ. Courses involving complementary and alternative
medicine in US medical schools. JAMA 1998;280;784-87.64. Yu DY, Lee SP. Effects of acupuncture on bronchial asthma. Clin Sci Mol Med 1976;51:503-09.65. Virsik K, Kristufek D, Bangha O, et al. The effect of acupuncture on pulmonary function in
bronchial asthma. Progr Respir Res 1980;14:271-75.66. Takishima T, Mue S, Tamura G et al. The bronchodilating effect of acupuncture in patients with
acute asthma. Ann Allergy 1982;48:44-49.67. Christensen PA, Laursen LC, Tauderf E et al. Acupuncture and bronchial asthma. Allergy
1984;39:379-85.68. Jobst K, Chen JH, McPherson K et al. Controlled trial of acupuncture for disabling breathlessness.
Lancet 1986;2:416-19.69. Biernacki W, Peake MD. Acupuncture in treatment of stable asthma. Respir Med 1998;92:
1143-45.70. Lewth GT, Watkins AD. Unconventional therapies in asthma: An overview. Allergy 1996;51:
761-69.71. Varon J, Fromm RE Jr, Marik PE. Acupuncture for asthma. Fact or fiction? Chest 2002;121:
1387-88.72. Shapira MY, Berkman N, Ben-David G et al. Short-term acupuncture therapy is of no benefit in
patients with moderate persistent asthma. Chest 2002;121:1396-1400.73. Boulet LP, Becker A, Berube D et al. Canadian Asthma Consensus Report; 1999. Can Med Assoc
J 1999;61(Suppl 11):S1-S62.74. Jayaram L, Paramswaran K, sears MR et al. Induced sputum cell counts: Their usefulness in
clinical practice. Eur Respir J 2000;16:150-58.75. Kips JC, Pauwels RA. Use of induced sputum in the diagnosis and follow up of asthma and
chronic obstructive pulmonary disease. Monaldi Arch Chest Dis 2000;55:93-95.76. Boulet LP, Phillips R, O’Byrne P et al. Evaluation of asthma control by physicians and patients:
Comparison with current guidelines. Can Respir J 2003.

Therapeutic Approach in Patients with Asthma (Chronic Bronchial Asthma) 207
77. Rabe KF, Vermiere PA, Soriano JB et al. Clinical management of asthma in 1999: The asthmaInsights and Reality in Europe (AIRE) study. Eur Respir J 2000;16:802-807.
78. Kips JC, Pauwels RA. Asthma control. Where do we fall? Eur Respir J 2000;16:797-98.79. Juniper EF, O’Byrne PM, Guyatt GH et al. Development and validation of a questionnaire to
measure asthma control. Eur Respir J 1999;14:902-07.80. Boulet LP, Boulet V, Milot J. How should we quantify asthma control? A proposal. Chest
2002;122:2217-23.

208 Bronchial Asthma
Therapeutic Approach inPatients with Asthma
II. Acute Severe Asthma (SA)
13
Definition
The term status asthmaticus was previously used to describe a severe attack of asthma, whichhad continued for more than 24 hours. Although most severe attacks of asthma developover days or weeks prior to presentation to medical care, all recent studies of asthma deathshave described patients who have died within hours or even minutes of the onset ofsymptoms. It is, therefore, not appropriate to include the duration of the attack in a definitionof acute severe asthma. The most important aspect of such an attack is its severity. Anothersuggested definition of an acute asthmatic attack is severe airflow obstruction that hadbecome unresponsive to the patient’s normal bronchodilator treatment. All patients withbronchial asthma are at risk of developing a severe asthma attack that places them at risk ofdeveloping respiratory failure—the disorder referred to as status asthmaticus.1-19 The attackcan occur at any time and at any speed. In most cases of severe life-threatening asthmadevelops against a background of poorly controlled disease. However, in 10-20% of casesof fatal or near fatal asthma the onset appears to be sudden and unexpected, with deathoccurring at times within a couple of hours. Such episodes are called “Sudden asphyxicasthma”.20 This form is quite different from the more slowly progressive forms of airflowobstruction. These are accompanied pathologically by only mild inflammatory changesand little mucus plugging of the airways.21 This sudden and unexpected increased airflowobstruction results primarily from bronchial smooth muscle-mediated bronchospasm. Animportant feature of this near fatal asthma is that attacks are often recurrent and a previouslife-threatening episode represents one of the most important factors predicting asthmadeaths.22 Acute severe asthma said to “run to type”, meaning thereby, if hypercapniadevelops during one severe attack, it is likely to recur in a subsequent episode.23 Both fataland near fatal asthmatic attacks have similar features.24
Patients dying of sudden exacerbations of asthma have diminished eosinophils andincreased neutrophils in the airway submucosa20 and less intraluminal mucus.25 This is incontrast with the relatively slower onset disease. Patients who develop progressivesymptoms over days before finally presenting to the emergency room do so with respiratorydistress. In these patients, inflammation of the airway wall and edema play a significant

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 209
role. Yet many patients fail to perceive the severity to treat effectively their worsening airwayinflammation.26 They fail to appreciate the severity of the final episode because of poorperception. Reduced chemosensitivity to hypoxia and blunted perception of dyspnoeaperhaps predispose patients to fatal asthma.27
About 1.1 to 7.0% of patients with bronchial asthma die from an acute attack.28-30 It isdifficult to predict which asthma patients will have a fatal or near-fatal asthma attack. Inboth these conditions (fatal and near-fatal asthma), there is a female predominance,31,32
history of frequent hospital admissions and emergency department visits,33 non-compliance,34 psychosocial abnormalities,35 and socioeconomic factors linked to poverty.36
In addition, a significantly decreased response to inspiration against resistance and tohypoxic hypercapnia27 and a low perception of dyspnoea are other risk factors.37 In a casecontrol study, the following eight variables were found to be associated with near-fatalasthma: history of seizures, conflict with parents and hospital staff, inappropriate self-care,decrement in prednisolone dose by 50%, use of inhaled beclomethasone, increased asthmasymptoms during the week prior to discharge, depressive symptoms, and disregard ofasthma symptoms.38 The variables associated with subsequent deaths include older age,female gender, smoking, labile asthma, poor compliance, and psychiatric treatment foranxiety or depression.39 In a recent unmatched, case-control study, the near fatal asthmawas most often associated with the use of bronchodilators or corticosteroids during the last12 months and these patients had nocturnal symptoms in the previous two weeks, moresevere form of the asthma, and more likely to have had a previous intubation.40 Further,patients with near fatal asthma have more food allergies and onset of their episodes followsa visit to the bar, party or restaurant.41 This may be related to hypersensitivity to foods,such as nuts, or exposure to smoking exposure or substance abuse in this group.42 Anotherimportant contributory factor may be an inadequate access to health care facilities.43
Pathophysiology
Abnormalities of gas exchange occur due to airways obstruction as a result of inflammationand bronchial smooth muscle contraction. Mucus plugging of both large and small airwaysis found at autopsy. Dead space increases as result of hypoperfusion of hyperinflated lungregions. Mechanical abnormalities of the lung include marked elevation of airwaysresistance, inspiratory transpulmonary pressure during quiet tidal breathing increases toas high as 50 cm H2O and expiration becomes active. Despite increased work of breathing,FEV1 is reduced to almost 10-20 percent of normal and PEFR is less than 100 L/min insevere cases. Expiratory time is prolonged and alveolar emptying is not complete at theend of expiration. Intrinsic or occult positive-end expiratory pressure (PEEPi) is theconsequence of alveolar pressure not reaching atmospheric pressure under the conditionof prolonged expiratory time.
Abnormal circulatory effects of severe airways obstruction result mostly from pleuralpressure excursions associated with breathing. During expiration, increases in intrathoracicpressure diminish blood return to the right heart. During inspiration, right ventricularvolume may increase sufficiently to shift the interventricular septum towards the leftventricle compromising the volume of this chamber and resulting in incomplete filling.These cyclical events result in pulsus paradoxus. Additionally, large negative pleural pressuremay directly impair left ventricular emptying by increasing left ventricular after load.44,45
Further, lung hyperinflation may represent a further after load on the right ventricle by

210 Bronchial Asthma
increasing pulmonary artery pressure.46 During quiet breathing without airways obstruction,the pulsus measured as the maximal drop in systolic blood pressure during inspiration, isless than 10 mmHg. During severe asthma this may be greater than 15 mm Hg and is usedas a measure of severity of asthma,47 although presently this is not included as a guidelinein assessing the severity of asthma. Pulsus paradoxus may be faulty if the patient ceasesmaking sufficient effort to cause large intrathoracic pressure swing, which is possible in afatigued asthmatic unable to generate significant changes in the pleural pressure. Thus, theabsence of a wide pulsus paradoxus does not always mean a mild attack.48,49
Further progression to ventilatory failure in status asthmaticus may result fromrespiratory muscle fatigue, increased work of breathing due to increased airways resistance,diaphragm failure as a force generator because of dynamic hyperinflation, and intrinsicPEEP associated with dynamic gas trapping. All these events are responsible for respiratorymuscle fatigue. Lactate excess is thought to be due to increased muscle production, theaction of catecholamines used during treatment, or diminished clearance related to hypo-perfusion. Regardless of etiology, lactic acidosis is a metabolic marker which may be usedto predict an increase risk of progression to ventilatory failure.
Clinical Features
The clinical presentation of a patient with status asthmaticus includes increased breathless-ness, cough, wheezing, and chest tightness. The patient is typically anxious, breathless,fatigued, sitting upright in bed, and is preoccupied with the task of breathing. He is inapparent respiratory distress. Clinical signs include tachypnoea, tachycardia, hyperinflatedlungs, wheeze, use of accessory muscles, pulsus paradoxus and diaphoresis. The absenceof wheezing does not exclude a diagnosis of bronchial asthma and the classical sign ofwheezing correlates poorly with the degree of airflow obstruction.50 Rather, a silent chest ina case of status asthmaticus indicates severe airway obstruction with little movement of airto produce respiratory sound. In the period immediately preceding respiratory arrest thechest may be completely silent. Presence of localised wheeze and crepitations may indicatemucus plugging, atelectasis, pneumonitis or some other cause. Adults with statusasthmaticus who assume the upright position usually has a significantly higher pulse rate,respiratory rate, and pulsus paradoxus and a significantly low arterial oxygen tension, andlow PEFR than patients who are able to lie supine. If upright patients are also diaphoretic,the PEFR is even lower.51 Use of sternocleidomastoid muscle indicates severe airwaysobstruction. Inability to lie supine, diaphoresis, impaired sensorium, inability to speak anduse of accessory muscles are all signs severe disease. However, even patients with severeobstruction and exhaustion can lie down supine giving a false impression of less severedisease.
The possible complications of bronchial asthma are shown in Table 13.1.While examining a patient of status asthmaticus clue for the possible complications should
be looked for.Electrocardiogram (ECG) in such a patient will show tachycardia. Successful treatment
of airflow obstruction is usually associated with a decrease in heart rate, although someimproving patients may remain tachycardic because of the use of drugs. Successful therapywill reduce heart rate.52 The rhythm is usually sinus tachycardia. Rhythm abnormalitiesthat are possible include supraventricular arrhythmias, and atrial, ventricular or combinedarrhythmias.53 Patients with arrhythmias are usually older compared to patients without

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 211
them. Status asthmaticus can cause right ventricular strain, which is usually transient andwhich resolves within hours of therapy.52,54 However, presence of right heart failure shouldsuggest some other disease. The possibility of coronary ischaemia should be considered inolder patients with coronary artery disease. Such patients with status asthmaticus are inincreased risk of myocardial oxygen supply/demand imbalance when large decreases inintrathoracic pressure increases left ventricular after load and possibly decreased coronaryblood flow.55 High dose beta agonists, theophylline and hypoxia can further tilt this balance.
Although effort-dependent, objective physiologic measurements of airflow obstructioncan be made by bedside determination of FEV1 and PEFR. In most patients PEFR can bemeasured more easily even if the maneuver is difficult in severely dyspnoic patients. Inthese cases, this may be deferred because deep inhalation may worsen bronchospasm56 andin rare cases, precipitate respiratory arrest.57 However, the measurements are safe in mostpatients.
Measurement of arterial blood gas is essential in patients with status asthmaticus. Themeasurement is generally recommended when the FEV1 is less than 1 litre or PEFR is lessthan 120 L/min. Patients in the early stages of status asthmaticus will exhibit mild hypoxemiaand respiratory alkalosis with low carbon dioxide tension. If respiratory alkalosis persistsfor hours to days, compensatory renal bicarbonate wasting may take place manifesting asa non-anion-gap metabolic acidosis. As the severity of obstruction increases, PaCO2 increases.This is because of patient exhaustion, inadequate alveolar ventilation, and/or an increasein physiologic dead space. Hypercapnia has a good correlation with FEV1 and usually doesnot occur unless the FEV1 is less than 25% of the predicted.58 Presence of hypercapnia, evennormal PaCO2, indicates a severe degree of airflow obstruction and the possible need formechanical ventilation. However, in certain situations, hypercapnia alone is not an indicationfor intubation. Such patients may respond to aggressive drug therapy.54,59 On the otherhand the absence of hypercapnia does not exclude the possibility of severe airflowobstruction and impending respiratory arrest.3 Metabolic acidosis can occur in as high as28% of patients with status asthmaticus.60 This may occur more likely in men and in patientswith more severe degrees of airflow obstruction and hypoxemia. The possible cause isbecause of the elevated anion gap. Although the pathogenesis of lactic acidosis in this settingis not clearly understood, the possible mechanisms are:61
Table 13.1: Complications of acute bronchial asthma
PneumothoraxPneumomediastinumSubcutaneous emphysemaPneumopericardiumMyocardial infarctionMucus pluggingAtelectasisDrug toxicity (theophylline)Electrolyte imbalances (hypokalemia, hypophosphatemia, hypomagnesemia)DehydrationMyopathyLactic acidosisHypoxic brain injury

212 Bronchial Asthma
• Increased work of breathing resulting in anaerobic metabolism, tissue hypoxia,intracellular alkalosis, decreased lactate clearance by the liver because of passivecongestion in the setting of high intrathoracic pressure, and use of parenteral beta-adrenergic agonists.62 Repeated blood gas sampling are not necessary to determinewhether the patient is improving or deteriorating. In most cases important clinicalsigns described above will be sufficient to judge whether the patient is to be intubatedor not. Patients who are deteriorating on these grounds to the point of respiratoryarrest should be intubated whether or not the PaCO2 is rising. On the contrary, patientswho are more comfortable on pharmacologic drug therapy are to continue with thesame treatment despite an elevated PaCO2. Patients on mechanical ventilation willhowever, need serial measurement of arterial blood gas.
Chest radiography has little role in the management of mild to moderate asthma likearterial blood gas. The benefits of routine chest skiagram are minimal and they contributeonly 1-5% of studies where the treatment is influenced.63 Such changes include normal,hyperinflation, a minimal increase in interstitial markings. Other changes include focal/major atelectasis, pneumonitis or one of the above described complications. However, chestskiagram is definitely indicated in patients with fever, purulent sputum, signs or symptomsof barotrauma, (chest pain, mediastinal crunch, subcutaneous emphysema, cardiovascularinstability, or asymmetric breath sounds), suggestion of pneumonia, other localising chestsigns, or when it is not clear that asthma is the cause of respiratory distress.
Differential Diagnosis
The differential diagnosis of severe dyspnoea with wheezing includes status asthmaticus,upper airways obstruction, foreign body aspiration, left ventricular failure or ischaemia,acute exacerbations of COPD, asthma complicated by pulmonary embolism, pneumonia orbarotrauma. History and physical findings will differentiate many of these conditions.
Assessment of Severity
Assessment of severity of an acute asthma is very important since one has to decide whetherthe patient can be managed at home, or needs to be hospitalised or he is to be admitted toan intensive care unit with or without ventilatory support. It is necessary to manage apatient with acutely severe asthma with the same sense of emergency as for a 50-year oldperson with crushing substernal chest pain suspected of having myocardial ischaemia. Theinitial assessment should consist of a brief history pertinent to the exacerbation which includethe time of onset and cause of present exacerbation; severity of symptoms including exerciselimitation and disturbance of sleep, all current medication with the time of last administeredmedication and any recent use of systemic corticosteroids, prior hospitalisation, priorepisodes of respiratory insufficiency, and significant prior cardiopulmonary disease. Itgenerally requires an analysis of several factors, including the medical history, physicalexamination, bedside monitoring of airflow obstruction, response to initial therapy, arterialblood gas measurements, and radiographic studies. This multifactorial analysis is necessarybecause no single clinical measurements have been found to predict outcome reliably.64
A brief cardiopulmonary examination should be performed, with emphasis on findingsrelevant to assessing the severity or identifying complications (pneumonia, atelectasis,pneumothorax, and pneumomediastinum). The overall assessment of the patient should

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 213
include alertness, colour, respiratory distress, and fluid status. Although wheezing is aprominent finding on chest auscultation, extremely severe obstruction may be accompaniedby “silent chest”. Routine laboratory studies and sputum culture are not necessary for initialmanagement of patients. A chest X-ray may be needed afterwards to identify anycomplication. Measurement of PEFR and FEV1 are very essential. Arterial blood gas shouldbe obtained in all cases as far as possible, particularly if the patient is not able to performpulmonary function tests or for whom intubation and mechanical ventilation are beingconsidered.
Various indices of acute severe asthma are shown in Table 13.2.Various clinical parameters helpful for the overall assessment of the patient are shown
in Table 13.3.
Table 13:2. Sherwood Jones’ index of severity of asthma
Grade 1a Able to carry out house work or job with moderate difficulty.Sleep occasionally disturbed.
Grade 1b Able to carry out house work or job with great difficulty.Sleep frequently disturbed.
Grade 2a Confined to a chair or bed but able to get up with moderate difficulty.Sleep disturbed. Little or no relief from inhalers.
Grade 2b Confined to a chair or bed but able to get up with great difficulty. Unable to sleep.Pulse rate > 120/min.
Grade 3 Totally confined to a chair or bed. No asleep. No relief from inhaler.Pulse rate > 120/min.
Grade 4 Moribund.
Table 13.3: Indices of acutely severe asthma
Symptoms/historySevere breathlessness, cough, chest tightness, and wheezingDifficulty in walking for 100 feet or moreSpeech fragmented by rapid breathingSyncope or near syncope
Physical examinationPulsus paradoxus of > 12 mmHgUse of accessory muscles of respirationDiaphoresis, unable to lie supineHeart rate > 120/minRespiratory rate > 30/ minSilent chest
Pulmonary functionsFEV
1 or PEFR , 30-50% baselineFailure of PEFR to improve at least 10% after initial treatment
Arterial blood gasPO
2 < 60 mmHg or O2 saturation of less than 90%PCO2 > 40 mmHg
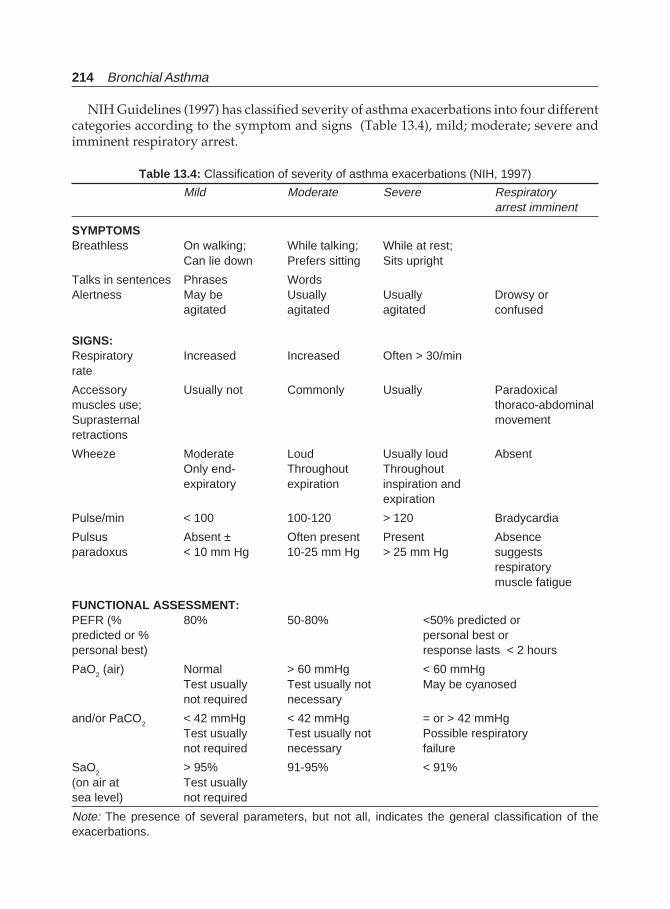
214 Bronchial Asthma
NIH Guidelines (1997) has classified severity of asthma exacerbations into four differentcategories according to the symptom and signs (Table 13.4), mild; moderate; severe andimminent respiratory arrest.
Table 13.4: Classification of severity of asthma exacerbations (NIH, 1997)
Mild Moderate Severe Respiratoryarrest imminent
SYMPTOMSBreathless On walking; While talking; While at rest;
Can lie down Prefers sitting Sits upright
Talks in sentences Phrases WordsAlertness May be Usually Usually Drowsy or
agitated agitated agitated confused
SIGNS:Respiratory Increased Increased Often > 30/minrate
Accessory Usually not Commonly Usually Paradoxicalmuscles use; thoraco-abdominalSuprasternal movementretractions
Wheeze Moderate Loud Usually loud AbsentOnly end- Throughout Throughoutexpiratory expiration inspiration and
expiration
Pulse/min < 100 100-120 > 120 Bradycardia
Pulsus Absent ± Often present Present Absenceparadoxus < 10 mm Hg 10-25 mm Hg > 25 mm Hg suggests
respiratorymuscle fatigue
FUNCTIONAL ASSESSMENT:PEFR (% 80% 50-80% <50% predicted orpredicted or % personal best orpersonal best) response lasts < 2 hours
PaO2 (air) Normal > 60 mmHg < 60 mmHg
Test usually Test usually not May be cyanosednot required necessary
and/or PaCO2 < 42 mmHg < 42 mmHg = or > 42 mmHgTest usually Test usually not Possible respiratorynot required necessary failure
SaO2 > 95% 91-95% < 91%(on air at Test usuallysea level) not required
Note: The presence of several parameters, but not all, indicates the general classification of theexacerbations.

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 215
Recent data also showed that peak expiratory flow measurements must be interpretedin the light of other features of severity and the patient’s past history, particularly previousadmissions to hospital, attendance at the emergency departments and current treatment,especially corticosteroids. Life-threatening asthma is one where the PEFR is less than 33%of the personal best. There is also a difference between a patient with PEFR of 50% who hasbeen on prednisolone for a week and a patient who has a short history and has not yetstarted oral steroids. Similarly, pulsus paradoxus need not be measured as it adds nothingto the assessment and its interpretation is subject to many factors (BTS, 1995). Facilities forthe monitoring of oxygen saturation should be available in all clinical areas that treat patientswith acute asthma. Interpretation of saturation in patients who are on or who have recentlybeen on oxygen, is difficult, but if the value is 92% or more and there is no feature of animminently life-threatening attack, arterial puncture may be deferred (BTS, 1995).
Therapeutic Management
The best strategy for management of asthma exacerbations is early treatment to preventdeterioration and abort the exacerbation. Therefore early recognition of worsening lungfunction, prompt communication between the patient and the physician, appropriateintensification of antiasthma medication, and removal of the allergen or irritant are importantcomponents of management. Some patients are at increased risk for exacerbations and thecategory of high risk for asthma-related death includes patients who have history of :
• Prior intubation for asthma,• Two or more hospitalisation for asthma in the past year,• Three or more emergency care visits for asthma in the past year,• Hospitalisation or emergency care visit within the past month’• Current use of systemic steroids or recent withdrawal from systemic steroids,• Past history of syncope/hypoxic seizure due to asthma,• Prior admission to hospital based intensive care unit (ICU), and• Serious psychiatric disease or psychosocial problems.
The basic principle of care of acute asthma exacerbations is the rapid reversal of airflowobstruction with relief of accompanying respiratory distress. This can be achieved byrepetitive administration of inhaled β2-antagonists, early addition of systemic steroids, andcorrection of hypoxaemia.
Some acute exacerbations can be managed at home, as an emergency outpatient basis, orthe patient needs hospitalisation with or without admission into the intensive care unit(ICU). The primary goal of home management of acute exacerbations of asthma is to avoiddelays in initiating antiasthma therapy by having the patient begin treatment at home. It isequally important that the patient does not delay seeking professional medical help if theasthma exacerbation is severe or if the response to therapy is not prompt and sustained.
The initial treatment should consist of subcutaneous adrenaline (1:1000) in a dose of0.5 ml slowly. This can be repeated every 20 minutes for three times. The drug should beavoided in patients with hypertension, and elderly individuals particularly with underlyingheart disease. Although these drugs were commonly used as the first line therapy earlier,because of the availability of more β2-antagonist selective inhaled drugs recently, their usehas declined dramatically. Subcutaneous adrenaline, terbutaline, or salbutamol has no

216 Bronchial Asthma
advantage over inhaled β-agonists. However, there are situations when these drugs are veryuseful. These situations are:
i. when the patient is unable to cooperate to inhale;ii. patients with impaired sensorium; and
iii. patients with cardiopulmonary arrest.65-67
These drugs can also be tried in intubated patients not responding adequately to inhaledtherapy. If parenteral β-agonists are used, potassium monitoring is essential to avoidhypokalemia, lactic acidosis, and cardiac arrhythmias. In very emergency cases, epinephrinemay be delivered effectively through the endotracheal tube. Other injectable preparationsthat can be used include terbutaline and salbutamol.68 Some patients will not need furtherparenteral therapy and can be stabilised with oral or inhaled bronchodilators and steroids.There is no great advantage of more β2-specific drugs over subcutaneous epinephrine. Ratherthey cause more cardiovascular side effects like tachycardia for the same degree ofbronchodilatation.69 Adrenaline is contraindicated during pregnancy because it is associatedwith congenital malformations and decreases uterine blood flow.70 Terbutaline is thepreferred drug in this setting. However, terbutaline or salbutamol may inhibit uterinecontractility at term. Routine use of infused β-agonists are not necessary and they have noextra advantage. On an individual basis however, intravenously administered β-agonistsmay be considered in patients below the age of 40 years who do not respond to inhaled orsubcutaneous therapy, and in whom respiratory arrest is imminent or in whom persistentsevere airflow obstruction is associated with alarming levels of lung hyperinflation duringmechanical ventilation. In a recent meta-analysis, it is shown that the clinical benefit ofintravenous β-agonists appears questionable, while the potential clinical risks are obvious.The only recommendations of intravenous β2-agonist use should be in those patients in whominhaled therapy is not feasible.71,72
Inhalation of selective β2-agonist bronchodilators by nebulisation is favoured for bothchildren and adults as the immediate and first-line therapy of status asthmaticus of allseverity. The onset of action is very rapid and their side effects are usually tolerated.Salbutamol is the most commonly used drug. This has a slightly longer duration of actionand it is more β2-selective. Long-acting β-agonists like salmeterol or formoterol are not indicatedin acute asthma since their onset of action is very slow. Terbutaline (2.5-5 mg), or salbutamol2.5-5 mg, or metaproterenol 15 mg or isoetharine 5 mg can be given 4-6 hourly diluted withnormal saline. This dose can be used every twenty minutes for 1 hour (three doses) followedby administration every hourly during the first several hours of therapy. A single dose of 7.5mg nebulised salbutamol and sequential doses of 2.5 mg nebulised salbutamol are clinicallyequivalent in the treatment of patients with moderate-to-severe acute asthma and result insimilar disposition from the emergency room.73 Fewer doses can be given in patients with lesssevere airflow obstruction who demonstrate a good response. On the other hand, inhaledtreatment can be given continuously to severely obstructed patients until an adequate clinicalresponse is achieved or adverse effects limit further use. Prior use of inhaled drugs at homedoes not prevent further use in hospital. Lower doses are preferred initially but they can berepeated or increased if necessary. Larger and more frequent doses are necessary in acuteasthma because the dose-response curve and duration of action of activity of these drugs areaffected adversely by the degree of bronchoconstriction.74 Airway narrowing and thecooperation and breathing pattern of the patient may further reduce the dose of the drugdelivered by inhalation.

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 217
Recent studies have shown that in nonintubated patients, MDIs combined with a spacerare as useful/effective as that used by a nebulizer.75-78 Further, MDIs are more quicker actionand cheaper still, many prefer the use of nebulizers as they need fewer instructions, lesscoordination is required and less supervision is necessary. Moreover, both the patient andphysician feel satisfied psychologically. Both MDIs and nebulizers can also be used inpatients on ventilators,79 although there are controversies regarding the optimal delivery ofinhaled drugs in the intubated and/or mechanically ventilated patient, optimal ventilatorysettings to be used during ventilation for drug delivery, ideal site on the ventilator circuitfor connection of the nebulizer, and maximal acceptable drug dosage.80 Higher dosages arerequired to achieve physiologic effects than in nonintubated patients since pulmonaryaerosol deposition is poor in these patients, in the range of 1.2-2.9%.81, 82 In recently extubatedpatients, MDIs can be used successfully.
Inhaled β2-agonists are the drugs of choice with which to treat patients with acute severeasthma. In comparison to systemic approach, inhalation is associated with a more rapidonset of action, and fewer systemic side effects. There is a consensus that frequent intermittentnebulisation, every 20 minutes within the first hour, are appropriate, although continuousnebulisation also has been proposed. Since the late 1980s, there has been considerable clinicaland academic interest in the use of continuous aerosolised bronchodilators for the treatmentof acute asthma.83 A recent meta-analysis supports the equivalence of continuous andintermittent salbutamol nebulisation in the treatment of acute adult asthma.84 This methodof therapy has potential advantage in terms of time, cost, and medication delivery. Thisfeature may allow deeper penetration into the airways and greater reduction of broncho-constriction. Furthermore, this may result in fewer side effects. In children, continuoussalbutamol nebulisation is considered to be better than intermittent therapy.85-87
Since airway inflammation is an important component of airflow obstruction of bronchialasthma, treatment with corticosteroids is the most important and an integral part ofmanaging a case of acute severe asthma. Ideally, an intensifying treatment of worsening ofone’s asthma should start with aggressive use of corticosteroids, which both the patientand many physicians do not appreciate. If not intervened early, airway inflammationproceeds unchecked. Whether patients are using corticosteroids or not when they arrive inthe emergency with severe attacks of asthma, the dose is inadequate. There is evidence tosuggest that failure to use or under use of steroids contribute to asthma deaths.88 Benefits ofsteroids are well established in recently confirmed meta-analysis of large number of studies.89
The facts that emerge are that steroids given in the emergency room significantly reducethe rates of admission and the number of future relapses in the first 7-10 days. It did notmatter whether steroids are given intravenously or orally even if intravenous therapy ispreferred in patients at risk of intubation, as long as a minimum of 30 mg of prednisoloneor its equivalent is given every 6 hourly. Lower doses are less effective and there will be noobvious benefit by giving higher doses.90-92 It is recommended that 150-225 mg/day ofprednisolone or its equivalent is required to reach maximal therapeutic benefit. Either 40 mgmsof intravenously administered methyl prednisolone every 6 hours or prednisolone 60 mgorally every 6-8 hours for 36-48 hours depending upon the condition of the patient will bemost ideal. Corticosteroids both intravenous and oral should be started simultaneously.However, these drugs have a slow onset of action because of their intracellular mode of actionwhatever their route of administration. Intravenous administration speeds up their actionmarginally by about an hour. Hydrocortisone should be administered 200 mg intravenously

218 Bronchial Asthma
initially followed by 100 mg every 4 hourly in place of methyl prednisolone because the lateris costly. This can also be administered as an infusion in a dose of 0.5 mg/kg/hr. Prednisonein a dose of 40-60 mg should be administered from the first day for several days followed bytapering of the dosage according to the response.
A meta-analysis has suggested that the administration of corticosteroids in addition toinhaled β2-agonists in patients with acute asthma on their arrival at the emergencydepartment does not improve airflow obstruction nor reduces the need for hospitalisation.93
The failure of steroids to influence the early course of patients with acute asthma is due tothe fact that it may take up to 24 hours for the effect of corticosteroids to become evident.However, other randomised, placebo-controlled trials94 have shown that high doses ofinhaled glucocorticosteroids together with salbutamol in patients with acute asthma whoare treated in the emergency department significantly improve pulmonary function whencompared to the use of salbutamol alone. The difference becomes evident by 90 minutes. Itis due to the fact that locally acting inhaled corticosteroids cause local vasoconstriction andthereby decrease edema formation and plasma exudation.95 However, glucocorticosteroids,either in inhaled form or oral form or parenteral form are the important mode of therapyfor managing a case of acute bronchial asthma. Some studies have shown that after 48 hoursof intravenous treatment with corticosteroids, the use of high-dose inhaled flunisolide(250 μg per activation, eight puffs twice daily is as effective as systemic corticosteroids, inadults hospitalised for a severe asthma exacerbation.96 A meta-analysis indicates that thereis some evidence that therapy with high-dose inhaled corticosteroids (beclomethasonedipropionate, > 2,000 mg or equivalent per day) may replace therapy with oral corticosteroidsfollowing the emergency department discharge of patients who have been treated for anacute asthma exacerbation.97 However, this meta-analysis has not shown any concreteevidence to change the practice of administering oral prednisolone for a short while for7-10 days at a dose of 40 mg per day, which is cheap, effective, and safe.98 Addition ofinhaled corticosteroid (budesonide 1600 μg/day to oral corticosteroid reduces the numberof relapses.95
If the patient continues to deteriorate despite treatment, with beta-agonists and steroids,the alternatives are to add nebulised ipratropium bromide 500 μg 6 hourly. The drugaugments the bronchodilating effect of β2-agonists in acute asthma.99-101 The role for anti-cholinergic medications is not well-defined. Thus, the use of therapy with anticholinergicsand β2-agonists, either simultaneously or in sequence, has produced positive as well asnegative results in different trials. Most recent relevant reviews had proved that the use ofmultiple doses of ipratropium bromide are indicated in emergency department treatmentof children and adults with severe acute asthma. The studies reported a substantial reductionin hospital admission (30-60%, number needed to treat 5-11) and significant difference inlung function favouring the combined treatment. No apparent increase in the occurrence ofside effects was observed. The use of single-dose protocols of ipratropium bromide withβ2-agonist treatment produced, particularly in children with more severe acute asthma, amoderate improvement in pulmonary function without reduction in hospital admission.;in adults, the evidence showed a similar increase in pulmonary function with an approxi-mately 35% reduction in hospital admission rate. In patients with mild-to-moderate acuteasthma, there is no apparent benefit adding a single dose of an anticholinergic medication.102
Theophylline: Aminophylline is commonly used as an important drug in many developingcountries both as a maintenance drug in chronic asthma as well for treatment during an

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 219
acute attack of bronchial asthma in the form of a continuous drip in many countries becauseof its lower cost and if given in proper doses, the drug is not all that unsafe. Moreover,other modalities may not be available at all places. However, the ready availability of betaagonists and their safety and quick onset of action has replaced theophylline therapy inacute asthma. The usual loading dose and maintenance dose is shown in the table below. Inmost situations roughly 250 mg, 400 mg, and 500 mg 8 hourly will be required for a small,medium or large person respectively for maintenance. Serum theophylline level should beobtained whenever possible. Use of theophylline confers additional benefit on beta agonistsand parenteral steroid therapy.103 Further, administration of theophylline results in fewerhospitalisations.104 A recent analysis of the use of theophylline had concluded that there isinadequate evidence to support or reject the use of theophylline in the emergency setting.105
However, theophylline continues to be an important adjunct in the management of acuteasthma particularly in the setting of poor or incomplete response to treatment with betaagonists and steroids and where economy is a consideration. Theophylline can be, and hasbeen used, safely if attention is paid to the possible side effects, serum drug levels, and tofactors that increase serum levels. Serum levels should be checked within 6 hours ofintravenous loading to avoid toxic levels.
High concentrations of oxygen (35%) will increase arterial PO2 and will not lead to carbondioxide retention unless there is some other associated problem. Obstruction of peripheralairways will result in V/Q mismatch and hypoxemia. However, true shunt in acute asthmais only 1.5% of the pulmonary blood flow.106 Therefore, correction of hypoxemia requiresonly modest flow of oxygen, 1-3 L/min through a nasal cannula. Only a small proportionof patients below the age of 45 years develop hypoxaemia. Although there is a goodcorrelation of FEV1 or PEFR with that of arterial oxygen tension, there is no cut-off value foreither measurement to accurately predict hypoxaemia. The routine administration of lowflow oxygen is an entirely safe practice that is recommended if routine pulse oxymetry isnot available and if there is co-morbid condition such as coronary artery disease. Oxygentherapy improves oxygen delivery to the peripheral tissues including respiratory muscles,reverses hypoxic pulmonary vasoconstriction, airway bronchodilatation, and protectionagainst the modest fall in PaO2 often seen after β2-agonist administration resulting frompulmonary vasodilatation and increased blood flow to low V/Q units.
Since the attack is frightening reassurance to the patient is essential.Routine use of antibiotics is not necessary unless there is evidence of bacterial infection
in form of fever, purulence of sputum or radiological evidence of consolidation. However,sputum that looks purulent, may be due to abundant eosinophils and not polymorpho-nuclear leukocytes.
Adequate hydration should be maintained.Sedatives of all kinds should be avoided. Periodic assessment of progress of disease or
the effect of therapy is very essential.There are reports supporting the usefulness of magnesium sulphate107-111 or heliox in the
treatment of refractory bronchial asthma. This benefit has been described in patients withnormal magnesium levels, although hypomagnesemia has been reported in 50% of patientswith acute asthma.112 Magnesium was first reported as a treatment for acute bronchial asthmain 1936.113 It has since been shown to be a bronchodilator.114-116 Many case reports showed clini-cal benefits in patients with respiratory failure complicating bronchial asthma.117,118 Severaltrials in paediatric patients demonstrated a benefit from IV magnesium.119,120 Many

220 Bronchial Asthma
randomised trials of IV magnesium in acute asthma in adults have shown mixedresults.121-123 A recent multicentric trial has shown that administration of 2g of IV magnesiumsulphate improves pulmonary function when used as an adjunct to standard therapy inpatients with very severe, acute asthma.124 The mechanism of action of magnesium is notknown. One hypothesis is that magnesium inhibits calcium channels of airway smooth muscleand thus, interferes in calcium-mediated smooth muscle contraction. Magnesium decreasesacetylcholine release at the neuromuscular junction, which may interfere with broncho-constriction. Magnesium also reduces histamine-induced and methacholine-inducedbronchoconstriction in asthmatics and affects respiratory muscle force generation. Although1-2 gm of the drug is safe, care should be taken to monitor renal function. Further, hypotension,and loss of deep tendon reflexes can result from magnesium intoxication. Mild complicationsinclude flushing and mild sedation. The drug is not recommended for routine use.
Heliox is a mixture of helium and oxygen (80:20,70:30, or 60:40) with a gas density lessthan that of air. It can be delivered through a tight fitting nonrebreathing face-mask innonintubated patients and through the inspiratory limb of the ventilator circuit inmechanically ventilated patients. Because the mixture is lighter than air, airways resistanceis decreased in bronchi with turbulent flow. This decreases the work of breathing and delaysthe inspiratory muscle fatigue until definite bronchodilator and anti-inflammatory therapybecomes effective.125,126
Drugs used in the treatment of status asthmaticus are shown in Table 13.5.
Indications for Admission in the Intensive Care Unit
Each patient should be assessed for response to treatment instituted initially in the emergencyroom. Patients who can be discharged include:127
• Significant improvement in shortness of breath• Improved air entry on clinical examination• PEFR or FEV1 greater than or equal to 70% predicted.
Observation for a minimum period of 30 minutes after the last dose of β-agonist isadministered is necessary to ensure stability before discharge. An incomplete response totreatment may be defined as the persistence of wheezing or shortness of breath and a PEFRor FEV1 between 40-70% of predicted. These groups of patients require ongoing treatmenteither in the emergency room or in the medical ward where facilities are available. Beforedischarge, these patients should be provided with a detailed follow-up plan which includeswritten medical instructions and a written plan of action to be followed if there is worseningof symptoms. Follow-up schedule should be made before the patient is sent home.
However patients having the following problems should be admitted and observed fortreatment in the intensive care unit. These are:
• Patients with severe airflow obstruction;• Use of accessory muscles of respiration,• Pulsus paradoxus of > 12 mmHg• Diaphoresis• Inability to recline• Hypercapnia• PEFR < 40% predicted

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 221
Table 13.5: Drugs used in status asthmaticus
Drug Dosage/Mode of use
Adrenaline 0.3 ml of 1:1000 solution subcutaneous every 20 minutes 3 times. Terbutaline isfavoured in pregnancy. The drug is to be avoided in patients with hypertension,older patients, and those with coronary artery disease.
Salbutamol 0.5 ml of 0.5% solution (2.5/5 mg) in 3 ml normal saline by nebulisation or 4 puffs byMDI with spacer every 20 min. for three doses; intubated patients can also giventhis drug.
Corticosteroids Methyl prednisolone 60-125 mg given intravenously every 6 hours or prednisolone30-40 mg orally every 6 hours. Hydrocortisone can also be given 200 mg start,followed by 100 mg every 6-8 hourly. The intravenous drugs are to be continued forat least for a period of 24-48 hours. Oral prednisolone is to be substituted as soonas possible. In fact, oral drugs are to be started simultaneously so that when theinjectable steroids are withdrawn, they can take-over.
Anticholinergics Ipratropium bromide 0.5 mg by nebulisation hourly or 4-10 puffs by MDI with Spacerevery 20 minutes for three doses.
Theophylline
- Loading dose: No previous drug -6mg/kg of aminophylline (lean body weight) infused over30 minutes or can be given slowly diluted with 25% of glucose over 20 minutesAlready on theophylline: Reduce according to serum level as follows: If serum levelis Cs mg/L, thenVol. of distribution (Vd) = 0.5L/kg × Wt (kg)Loading dose = Vd × desired change in serum level (mg/L)
= Vd × (Cs desired -Cs known)For a patient of 50 kg Wt; with Cs of 8 mg/L and desired Cs of 18mg/L;Loading dose = 0.5 × 50 × (18 mg/L -8 mg/L) = 250 mg.
Maintenance dose: Infusion Rate (mg/kg/hr)Adults 0.5Smokers 0.8Liver disease 0.4Severe COPD 0.4CCF 0.2Viral illness 0.4Children 0.9-1.2drug interactions As discussed in section under theophylline
Oxygen 1-3 l/min by nasal canula. Titration by pulse oxymetry
Unproved alternativesMagnesium 1 gm intravenously over 20 minutes (Total dose 2 gm). If hypomagnesemic, dosesulfate adjusted to normalize serum levels.
Heliox 80:20, 70:30, or 60:40 helium:oxygen mixture by tight-fitting, nonrebreathing facemask. Higher helium concentrations are needed for maximal effect.
• Poor response to initial therapy• Less than 10% improvement in PEFR
• Those deteriorate despite therapy• Respiratory arrest

222 Bronchial Asthma
• Altered mental status• Cardiac toxicity
• Tachyrrhythmias, Angina, Myocardial infarction
Further, patients with imminently life-threatening features such as unconsciousness,confusion, drowsiness, hypoxia (PaO2 < 60 mmHg despite 60% oxygen) or patients with araised PaCO2 should be admitted into the ICU. Similarly, patients whose condition isdeteriorating and PaCO2 is rising need to be monitored in an intensive care unit.
Mechanical Ventilation
Many, but not all, of these patients will need ventilatory support. Patients receivingpharmacological therapy needs to be watched closely for the need for intubation in patientsshowing clinical deterioration. Indications for intubation will depend upon:
a. Changes in posture, alertness, speech, use of accessory muscles, and respiratory rate,all of which indicates worsening respiratory failure. These signs do not need anyarterial blood gas measurement or peak flow documentation;
b. Fatigued patient despite PaCO2 levelsc. Altered mental status despite PaCO2 levelsd. PaCO2 is not an important predictor for intubation since patients who are more
comfortable, better able to speak, and less respiratory distress can continue medicaltherapy despite a rise in PaCO2;
Immediate intubation:e. Patients presenting with cardiopulmonary arrestf. Near cardiopulmonary arrest (patients unable to speak and/or gasping for air)g. Comah. Significant obtundation
A. Non-invasive Ventilation
Non-invasive ventilation through face mask mechanical ventilation is an option used bysome clinicians as a short-term ventilatory support in patients with hypercapnic ventilatoryfailure who are not
i. Responding adequately to drug therapy; andii. Where immediate intubation and mechanical ventilation is not indicated/required.
Patients with encephalopathy or with a need of airway protection are not suitable forthis form of therapy. Various advantages of noninvasive ventilation are; decreasedneed of anaesthesia, sedation, and paralysis, decreased incidence of nosocomialinfections, decreased incidence of sinusitis and otitis, and better patient comfort.128-131
Usually a nasal CPAP of 5-7.5 cm H2O will be necessary. Potential disadvantages ofthis mode of ventilation include aspiration of gastric contents due to gastric insufflation,facial pressure necrosis, and less control of the patient’s ventilatory status.
B. Intubation
Once it is decided to ventilate the patient intubation should be done quickly by an experi-enced person as manipulation of the upper airways may precipitate laryngospasm. Oralintubation allows insertion of larger bore endotracheal tubes (8 mm or more), which has

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 223
advantage of better suction and a decrease in airway resistance beneficial at high airflow.Nasal intubation is safe in most patients particularly if he is awake, breathless and in obesepatients with short necks. Fibre optic guided intubation may be helpful in difficult cases.The disadvantage with the nasal route is that a smaller endotracheal tube can only be usedand there is a high incidence of nasal polyps and sinusitis in them.
C. Sedation
Sedation will invariably be required in awake patients to prepare for intubation and toallow for effective mechanical ventilation (to avoid fighting the ventilator). Further, sedationimproves patient comfort, facilitates various procedures, and decreases oxygen demandand consumption and decreased carbon dioxide production besides decreasing the risk ofbarotrauma. Various sedatives that can be used in status asthmaticus are shown inTable 13.6.
D. Paralysis
Muscle paralysis is required in patients fighting with the ventilator despite sedation and inthose who continue to have asynchronous breathing which is a grave risk factor forgeneration of high airway pressure and loss of airway access. The decision, however, willbe based on the clinician’s own judgment whether to use neuroparalytic agents or not toachieve a therapeutic strategy to maintain stable respiratory parameters. Paralysis furtherhelps to reduce oxygen consumption, carbon dioxide production, and lactic aciodosis inaddition to decreasing the risk of barotrauma with an overall augmentation of sedativesalready used. Because the expiratory effort is eliminated, there is less airway collapse.
The paralytic drugs of choice are vecuronium and atracurium. These are nondepolarisingagents and are free of cardiovascular side effects, although larger doses may causehypotension. The drugs are either given intermittently by bolus injection or by continuous
Table 13.6: Sedatives that can be used in status asthmaticus
Drug Dose Comments
Before intubationMidazolam 1 mg IV push slowly, can be Hypotension, respiratory depression
repeated every 2-3 min. as needed.
Ketamine 1-2 mg/kg IV at a rate of Sympathomimetic effects,0.5 mg/kg/min. respiratory depression,
mood change, delirium
Propofol 60-80 mg/min IV Respiratory depressionup to 2 mg/kg followed byan infusion of 5-10 mg/kg/hras needed
After intubation for prolonged ventilation:
Lorazepam 1-5 mg/hr IV continuous Drug accumulationinfusion or bolus
Morphine sulphate 1-5 mg/h IV continuous infusion Ileus
Ketamine 0.1-0.5 mg/kg IV As described abovePropofol 1-4.5 mg/kg/h IV Seizures, hypertriglyceridemia

224 Bronchial Asthma
intravenous infusions. The disadvantages of neuroparalytic agents in acute asthma are:difficulties in assessing the mental status, greater danger of developing deep vein thrombosis,disuse muscle atrophy and myopathy.132 The last complication is more pronounced in thepresence of concomitant use of high dose steroids.
E. Mechanical Ventilation
Hypotension and hypoperfusion often follow intubation and positive pressure ventilation.Slowly bagging (4-6 breathes/min) with 100% oxygen for about 1 min will allow forprolonged expiratory time to decrease PEEPi which will result in a rise of blood pressure. Ifthis occurs a fluid bolus of 0.5-1L of normal saline should be given every 10-20 minutesuntil adequate circulation is restored.
The goals of mechanical ventilation in status asthmaticus are:• To achieve adequate alveolar ventilation;• Low levels of PEEPi;• Minimal circulatory compromise;• Low risk of barotrauma.
The patient should initially be fully sedated and adequate muscle-relaxation be achievedto minimize airway pressures. Dynamic hyperinflation is often seen in these patients if anattempt is made to make them eucapnic. This can be prevented by using small tidal volumesand high inspiratory flow. Mechanical ventilation should be initiated with a tidal volumeof 8 ml/kg, rate of 12-15/min, and inspiratory flow rate of 60L/min. Measurement of PEEPiin the sedated patient who has also received muscle relaxant, the expiratory limb of theventilator is occluded at end expiration while omitting the subsequent breath. The proximalairway pressure rises to a plateau (PEEPi).
133 If peak airway pressures exceed 55 cmH2Oand PEEPi is greater than 15 cmH2O despite full sedation and muscle relaxation, tidal volumecan be varied in 100-ml increments with corresponding changes in rate and flow in anattempt to optimize peak airway pressure (Ppk), PEEPi, and PCO2. In some patients, a highlevel of PCO2 in the range of 70-90 mmHg is the only way to bring Ppk below 55 cm H2Oand PEEPi below 15 Cm H2O. This is known as “controlled hypoventilation”. This ispreferable to the risk of barotrauma during an attempt to bring the PCO2 to normal, attemptto achieve this should never be tried. If associated acute respiratory acidosis is severe (pH< 7.2), bicarbonate infusion can be given to achieve a serum pH of approximately 7.25.External PEEP should not be used during ventilation of a patient of status asthmaticus as itcan result in dangerous increases in lung volumes and pressures.
Weaning from mechanical ventilation of the patient of status asthmaticus requires goodplanning. The paralytic agents should be discontinued briefly every 4 hours and re-administered only if evidence of muscle activity is seen. While some patients with verylabile asthma may respond to therapy within hours, more typically the patient will require24-48 hours of aggressive bronchodilator therapy until airway pressures and PEEPi fall.Once this begins, improvement is usually rapid, with resolution of all dynamic hyperinflationby 12 hours. As airway pressures fall, sedatives, muscle relaxants, and bicarbonate infusionscan be reduced to prepare the patient for a brief period of spontaneous ventilation and thenextubation. Assessment of respiratory muscle strength should be made by determinationof negative inspiratory pressure. If the patient has adequate muscle strength and no sign ofrespiratory failure emerge during the brief period of spontaneous breathing, extubationshould be performed since the endotracheal tube itself can perpetuate bronchospasm. A

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 225
quick return to spontaneous breathing can be achieved through a T-piece or by decreasingthe respiratory rate on SIMV. The use of 5-8 cm H2O of pressure support helps overcomeendotracheal tube resistance. Once the patient tolerates a trial of spontaneous breathing,quick extubation is done. Vigorous bronchodilator and chest physiotherapy should becontinued in the ICU until the next day. During this period and following discharge fromthe ICU, a careful programme for education should be implemented to help the patientidentify signs of worsening asthma and optimize the drug regimen to reduce/obviate futureepisodes of life-threatening asthma.
F. Inhalant Anaesthetics
Rarely, the above strategy fails to allow adequate ventilation at a safe level of lung inflation.In that situation inhalation of general anaesthetic is—halothane and enflurane—can beconsidered. These have bronchodilatation activity that can acutely reduce Ppk, andPaCO2.
134,135 But the effect lasts as long as the drugs are in use. The bronchodilating effectare lost once the drugs are stopped. Inhalation of these drugs are. Inhalation of these drugshas significant cardiovascular side effects including myocardial depression, arterialvasodilatation, and arrhythmias.
Recently, nitric oxide has been used to induce bronchodilatation, however, the experienceis limited and at a dose of 80 ppm it exerts a weak bronchodilator effect in asthmatics.136
G. Bronchoalveolar Lavage
Since mucus plugging is one of the notable features of status asthmaticus, all attemptsshould be made to remove the same from the respiratory tract to improve airflow. Strategiesof mucus removal short of bronchodilators or steroids, like chest physiotherapy or mucolyticsare not efficacious.137,138 Bronchoalveolar lavage using saline or acetylcystine, is being triedto remove mucus plug.139-143 Although there is improved airflow in intubated patients, theprocedure is not without risk in intubated patients which further compromises the airwaylumen. This will increase the expiratory airway resistance and may lead to high levels oflung hyperinflation. At present, BAL is not recommended as part of a routine managementof patients with status asthmaticus.
Hospital Discharge and Future Plan of Action
After the patient has come out of the ICU, in the general ward itself the question of preventionand treatment of subsequent asthma attacks should be addressed. This process starts withextensive patient education, how to recognise the worsening of asthma and what to do athome in such an eventuality, and when to contact a physician or to visit the emergencydepartment. Patients should be provided with written medication instructions as well as awritten plan of action to be followed in the event of worsening of symptoms. The properagents like inhaled medications with or without spacer as per the need. Their doses,frequency, etc. should be clearly explained. Similarly, any oral medication required is to betold to the patient in writing. It is important to explain also the purpose of such medicationplans and the techniques are to be taught, particularly for inhalers. The importance of airwayinflammation and use of anti-inflammatory drugs needs special explanation to the patient.When inhaled corticosteroids are prescribed, patients must be told not to expect immediate

226 Bronchial Asthma
relief of respiratory symptoms as they are not bronchodilators. Possible side effects of variousdrugs are to be told. The purpose and importance of peak flow measurements is to beexplained. The technique of the manoeuvre, frequency of measurement maintaining a diary,timings of measurements are to be told. The patient should be told that it is to be measuredat am and pm and the best of three readings each time is to be noted as the representativePEFR. Most important is to tell the patient what is the best for him and at what level ofreading (severity) he should report to the physician. Appointments are to be made for follow-up care with primary clinician or asthma specialist. The patient should be given the date,time and location of appointment within seven days of hospital discharge. Before or atdischarge the action plan has to be decided. The patient or its caregiver should be instructedon simple plan of action to be taken for symptoms, signs, and PEFR values suggestingrecurrent airflow obstruction.
Assessment of severity:• PEFR measurement— < 50% personal best or predicted suggests severe exacerbation.• Signs and symptoms correlate poorly with severity• Accessory muscle use and suprasternal retractions suggest severe exacerbations.
Initial treatment:• Inhaled short-acting β2-agonist; up to three treatments of 2-4 puffs by MDI at 20-minutes
intervals or single nebuliser treatment.
Good response Incomplete response Poor response
Mild episode Moderate episode severe episode
PEFR > 80% predicted or 50-80% predicted or < 50% predicted orpersonal best personal best personal best
No wheezing or shortness Persistent wheezing and Marked wheezingof breath shortness of breath and breathlessness
Response to β2-agonist Add oral corticosteroid Add oral steroidsustained for 4 hours
May continue above every Continue β2-agonist Repeat β2-agonist3-4 hours for 24-48 hrs immediately
For patients on inhaled • If distress is severecorticosteroids, double and non-responsive, consultdose for 7-10 days physician and report to
emergency.
Follow up with physician Contact physician Visit emergencyurgently the same day department
Fig. 13.1: Home treatment of asthma exacerbations
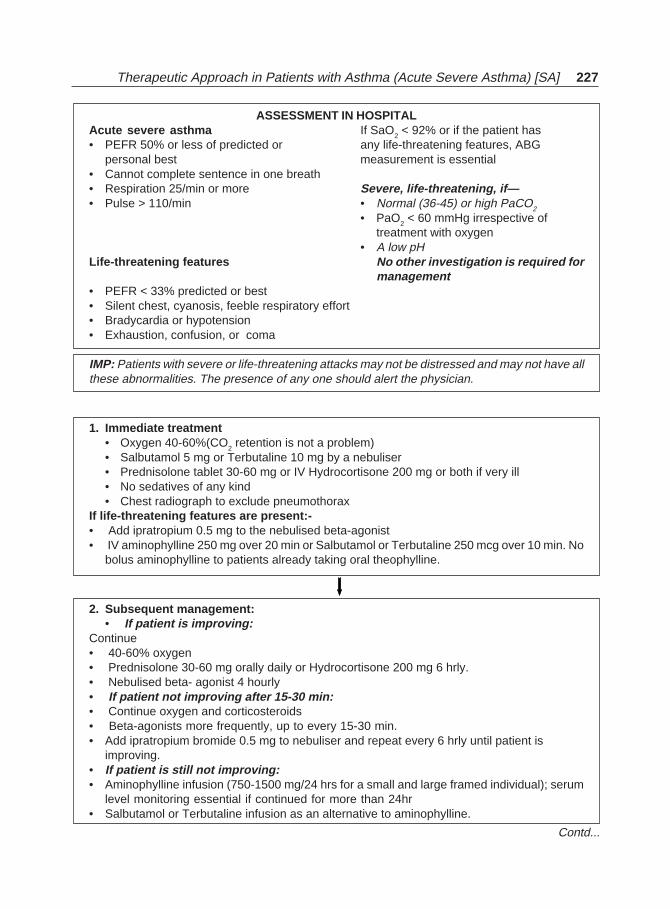
Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 227
ASSESSMENT IN HOSPITALAcute severe asthma If SaO2 < 92% or if the patient has• PEFR 50% or less of predicted or any life-threatening features, ABG
personal best measurement is essential• Cannot complete sentence in one breath• Respiration 25/min or more Severe, life-threatening, if—• Pulse > 110/min • Normal (36-45) or high PaCO2
• PaO2 < 60 mmHg irrespective oftreatment with oxygen
• A low pHLife-threatening features No other investigation is required for
management• PEFR < 33% predicted or best• Silent chest, cyanosis, feeble respiratory effort• Bradycardia or hypotension• Exhaustion, confusion, or coma
IMP: Patients with severe or life-threatening attacks may not be distressed and may not have allthese abnormalities. The presence of any one should alert the physician.
1. Immediate treatment• Oxygen 40-60%(CO2 retention is not a problem)• Salbutamol 5 mg or Terbutaline 10 mg by a nebuliser• Prednisolone tablet 30-60 mg or IV Hydrocortisone 200 mg or both if very ill• No sedatives of any kind• Chest radiograph to exclude pneumothorax
If life-threatening features are present:-• Add ipratropium 0.5 mg to the nebulised beta-agonist• IV aminophylline 250 mg over 20 min or Salbutamol or Terbutaline 250 mcg over 10 min. No
bolus aminophylline to patients already taking oral theophylline.
2. Subsequent management:• If patient is improving:
Continue• 40-60% oxygen• Prednisolone 30-60 mg orally daily or Hydrocortisone 200 mg 6 hrly.• Nebulised beta- agonist 4 hourly• If patient not improving after 15-30 min:• Continue oxygen and corticosteroids• Beta-agonists more frequently, up to every 15-30 min.• Add ipratropium bromide 0.5 mg to nebuliser and repeat every 6 hrly until patient is
improving.• If patient is still not improving:• Aminophylline infusion (750-1500 mg/24 hrs for a small and large framed individual); serum
level monitoring essential if continued for more than 24hr• Salbutamol or Terbutaline infusion as an alternative to aminophylline.
Contd...
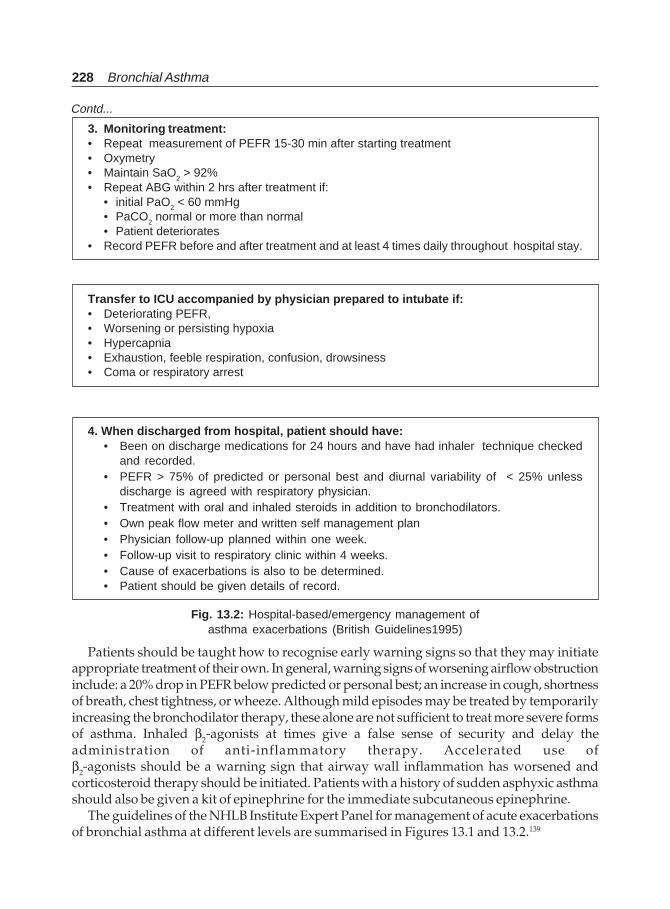
228 Bronchial Asthma
3. Monitoring treatment:• Repeat measurement of PEFR 15-30 min after starting treatment• Oxymetry• Maintain SaO2 > 92%• Repeat ABG within 2 hrs after treatment if:
• initial PaO2 < 60 mmHg• PaCO2 normal or more than normal• Patient deteriorates
• Record PEFR before and after treatment and at least 4 times daily throughout hospital stay.
Transfer to ICU accompanied by physician prepared to intubate if:• Deteriorating PEFR,• Worsening or persisting hypoxia• Hypercapnia• Exhaustion, feeble respiration, confusion, drowsiness• Coma or respiratory arrest
4. When discharged from hospital, patient should have:• Been on discharge medications for 24 hours and have had inhaler technique checked
and recorded.• PEFR > 75% of predicted or personal best and diurnal variability of < 25% unless
discharge is agreed with respiratory physician.• Treatment with oral and inhaled steroids in addition to bronchodilators.• Own peak flow meter and written self management plan• Physician follow-up planned within one week.• Follow-up visit to respiratory clinic within 4 weeks.• Cause of exacerbations is also to be determined.• Patient should be given details of record.
Fig. 13.2: Hospital-based/emergency management ofasthma exacerbations (British Guidelines1995)
Contd...
Patients should be taught how to recognise early warning signs so that they may initiateappropriate treatment of their own. In general, warning signs of worsening airflow obstructioninclude: a 20% drop in PEFR below predicted or personal best; an increase in cough, shortnessof breath, chest tightness, or wheeze. Although mild episodes may be treated by temporarilyincreasing the bronchodilator therapy, these alone are not sufficient to treat more severe formsof asthma. Inhaled β2-agonists at times give a false sense of security and delay theadministration of anti-inflammatory therapy. Accelerated use ofβ2-agonists should be a warning sign that airway wall inflammation has worsened andcorticosteroid therapy should be initiated. Patients with a history of sudden asphyxic asthmashould also be given a kit of epinephrine for the immediate subcutaneous epinephrine.
The guidelines of the NHLB Institute Expert Panel for management of acute exacerbationsof bronchial asthma at different levels are summarised in Figures 13.1 and 13.2.139

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 229
REFERENCES
1. Rebuck AS, Read J. Assessment and management of severe asthma. Am J Med 1971;51:788.2. McFadden ER, Kiser R, deGroot WJ. Acute bronchial asthma: Relation between clinical and
physiological manifestations. N Engl J Med 1973;288:221.3. McFadden ER, Lyons HA. Arterial blood gas tension in asthma. N Engl J Med 1968;278:1027-32.4. Nowak RM, Pensler MI, Parker DD. Comparison of peak expiratory flow and FEV1: admission
criteria for acute bronchial asthma. Ann Emerg Med 1982;11:64.5. Bucknall CE. Who needs referral to the hospital asthma specialist? Respir Med 1991;85:453.6. Beasley R, Burgess C, Crane J, Bremner P, Pearce N. Respiratory arrest in near fatal asthma.
N Engl J Med 1991;325:205.7. Barnes PJ. A new approach to the treatment of asthma. N Engl J Med 1989;321:1517.8. Reed CE. New therapeutic approaches in asthma. J Allergy Clin Immunol 1986;11:537.9. Hargreave FE, DolorichJ, Newhouse MT. The assessment and treatment of asthma: A conference
report. J Allergy Clin Immunol 1990;85:1098.10. Whitelaw WA. Asthma deaths. Chest 1991;99:1507.11. Robin ED. Risk-benefit analysis in chest medicine: Death from bronchial asthma. Chest
1988;93:614.12. Shim CS, Williams MS. Evaluation of severity of asthma patient versus physician. Am J Med
1980;68:11.13. McFadden ER Jr. Therapy of acute asthma. J Allergy Clin Immunol 19898;84:151.14. White CS, Cole RP, Lubetsky HW, Austin JH. Acute asthma: Admission chest radiography in
hospitalised adult patients. Chest 1991;100:14.15. Daley J, Kopelman RI, Comeau E, Ginns LC, Rossing TH. Practice patterns in the treatment of
acutely ill hospitalised asthmatic patients at three teaching hospitals. Variability in resourceutilisation. Chest 1991;100:51.
16. Tattersfield AE. Current management of acute severe asthma. Br J Hosp Med 1991;46:38.17. Jones Es. The intensive therapy of asthma. Proc R Soc Med 1971;64:1151.18. Neville E, Gribbin H, Harrison BD. Acute severe asthma. Respir Med 1991;85:463.19. Corbridge TC, Hall JB. The assessment and management of adults with status asthmaticus. Am
J Respir Crit Care Med 1995;151:1296-16.20. Wasserfallen JB, Schaller MD, Feihl F, Perret C. Sudden asphyxic asthma: A distinct entity? Am
Rev Respir Med 1990;142:108-11.21. Sur S, Crotty TB, Cephart GM et al. Sudden-onset fatal asthma: A distinct entity with few
eosinophils and relatively more neutrophils in the airway submucosa? Am Rev Respir Dis1993;148:713-19.
22. Rea HH, Scragg R, Jackson R et al. A case control study of death from asthma. Thorax 1986;41:833-39.
23. Mountain RD, Sahn SA. Clinical features and outcome in patients with acute asthma presentingwith hypercapnia. Am Rev Respir Dis 1988;138:535-39.
24. Campbell DA, McLennan G, Coates JR et al. A comparison of asthma deaths and near-fatalasthma attacks in South Australia. Eur Respir J 1994;7:490-97.
25. Reid LM. The presence or absence of bronchial mucus in fatal asthma. J Allergy Clin Immunol1987; 80:415-16.
26. Gibson GJ. Perception, personality, and respiratory control in life-threatening asthma. Thorax1995;50(Suppl 1):S2-S4,
27. Kikuchi Y, Okabe S, Tamura G et al. Chemosensitivity and perception of dyspnoea in patientswith a history of near-fatal asthma. N Engl J Med 1994;330:1329-34.
28. Rackeman FM, Edwards MC. Asthma in children: A follow up study of 688 patients after aninterval of 20 years. N Engl J Med 1952;246; 815-23 and 858-63.

230 Bronchial Asthma
29. Blair H. Natural history of childhood asthma: Twenty year follow-up. Arch Dis Child 1977;52:613-19.
30. Ogilvie AG. Asthma: A study in prognosis of 1000 patients. Thorax 1962;17:183-189.31. Ruffin RE, Latimer KM, Schembri DA. Longitudinal study of near fatal asthma. Chest 1991;99:
77-83.32. Boulet LP, Deschenses F, Torcotte H et al. Near fatal asthma; clinical and physiological features,
perception of bronchoconstriction, and psychological profile. J Allergy Clin Immunol 1991;88:838-46.
33. Campbell DA, McLennan G, Coates JR et al. A comparison of asthma deaths and near fatalasthma attacks in south Australia. Eur Respir J 1994;7:490-497.
34. Sears M, Rea HH. Patients at risk for dying of asthma: The New Zealand experience. J AllergyClin Immunol 1987;80:477-481.
35. Rea HH, Scragg R, Jackson R et al. A case control study of deaths from asthma. Thorax 1986;41:833-39.
36. McDonald JB, Mcdonald ET, Seaton A et al. Asthma deaths in Cardiff 1963-74: 53 deaths inhospital. BMJ 1976;ii:721-23.
37. Magadle R, Berar-Yanay N, Weiner P. The risk of hospitalisation and near fatal and fatal asthmain relation to the perception of dyspnoea. Chest 2002;121:329-33.
38. Strunk R, Mrazek D, Furmann G et al. Physiologic and psychological characteristics associatedwith deaths due to asthma in childhood: A case-controlled study. JAMA 1985;254:1193-98.
39. Strunk R, Nicklas R, Milgrom H et al. Risk factors for fatal asthma: In: Sheffer A, (Ed). FatalAsthma (vol 115). New York, NY Marcel Dekker, 1997; 115:31-44.
40. Mitchell I, Tough SC, Semple LK et al. Near-fatal asthma. A population based study of riskfactors. Chest 2002;121:1407-13.
41. Sampson H, Mendelson L, Rosen J. Fatal and near fatal anaphylactic reaction to food in childrenand adolescents. N Engl J Med 1992;327:380-84.
42. Greenberger P, Miller T, Lifschultz B. Circumstances surrounding deaths from asthma in CookCounty (Chicago), Illinois. Allergy Proc 1993;14:321-26.
43. Castro M. Near fatal asthma. What have we learned? Chest 2002;121:1394-95.44. Scharf S, Brown R, Saunders N, Green L. Effects of normal and loaded spontaneous inspiration
on cardiovascular function. J Appl Physiol 1979;47:582-90.45. Scharf S, Brown R, tow D, Parisi A. Cardiac effects of increased lung volume and decreased
pleural pressure. J Appl Physiol 1979;47:257-62.46. Permutt S, Wise RA. Mechanical interaction of respiration and circulation. In: Fishman A (Ed).
Handbook of Physiology; American Physiological Society. Williams and Wilkins., Baltimore.1986; 3:647-56.
47. Knowles G, Clark TJH. Pulsus paradoxus as a valuable sign indicating severity of asthma. Lancet1973;2:1356-59.
48. Kelson SG, Kelson DP, Fleegier BF, Jones RC, Rodman T. Emergency room assessment andtreatment of patients with acute asthma. Am J Med 1978;64:622-28.
49. Fitzgerald JM, Hargreave FE. The assessment and management of acute life-threatening asthma.Chest 1989;95:888-94.
50. Shim CS, Williams MH. Relationship of wheezing to the severity of obstruction in asthma. ArchIntern Med 1983;143:890-92.
51. Brenner BE, Abraham E, Simon RR. Position and diaphoresis in acute asthma. Am J Med1983;74:1005-09.
52. Grossman J. The occurrence of arrhythmias in hospitalised asthma patients. J Allergy ClinImmunol 1976;57:310-17.
53. Josephson GW, Kennedy HL, MacKenzie EJ, Gibson G. Cardiac dysrrhythmias during thetreatment of acute asthma: A comparison of two treatment regimens by a double blind protocol.Chest 1980;78:429-35.

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 231
54. Rebuck AS, read J. Assessment and management of severe asthma. Am J Med 1971;51:788-98.55. Scharf S. Mechanical cardiopulmonary interaction with asthma. Clin Rev allergy 1985;4:
487-500.56. Lim TK, Ang SM, Rossing TH, Ingenito EP, Ingram RH Jr. The effect of deep inhalation on
maximal expiratory flow during intensive treatment of spontaneous asthmatic episodes. AmRev Respir Dis 1989;140:340-43.
57. Lemarchand P, Labrune S, Herer B, Huchon GJ. Cardiorespiratory arrest following peakexpiratory flow measurement during attacks of asthma. Chest 1991;100:1168-69.
58. Nowak RM, Tomalanovich MC, Sarkar DD, Kvale PA, Anderson JA. Arterial blood gases andpulmonary function testing in acute bronchial asthma: Predicting patient outcomes. JAMA1983;249:2043-46.
59. Mountain RD, Sahn S. Clinical features and outcomes in patients with acute asthma presentingwith hypercapnia. Am Rev Respir Dis 1988;138:535-39.
60. Mountain RD, Heffner JE, Brackett NC, Sahn SA. Acid-base disturbances in acute asthma. Chest1990;98:651-655.
61. Appel D, Rubenstein R, Schrager K, Williams MH. Lactic acidosis in severe asthma Am J Med1983;75:580-84.
62. O’Connel MB, Iber C. Continuous intravenous terbutaline infusions for adult patients with statusasthmaticus. Ann Allergy 1990;64:213-18.
63. Findley LJ, Sahn SA. The value of chest roentgenograms in acute asthma in adults. Chest1980;5:535-36.
64. Rodrigo G, Rodrigo C. Assessment of the patient with acute asthma in the emergency department;a factor analytic study. Chest 1993;104:1325-28.
65. Fanta CH, Rossing TH, McFadden ER. Treatment of acute asthma: Is combination therapy withsympathomimetics and methylxanthines indicated. Am J Med 1986;80:5-10.
66. Uden DL, Goetz DR, Kohen DP, Fifield GC. Comparison of nebulised terbutaline and subcuta-neous epinephrine in the treatment of acute asthma. Ann Emerg Med 1985;14:229-32.
67. Becker AB, Nelson NA, Simons FER. Inhaled salbutamol (albuterol) vs injected epinephrine inthe treatment of acute asthma in children. J Pediatr 1983;102:465-469.
68. Behera D, Malik SK and Jindal SK: Subcutaneous salbutamol in acute bronchial asthma. Ind JChest Dis All Sci 1982; 24:290-92.
69. Amory DW, Burnham Sc, Chenery FW Jr. Comparison of the cardiopulmonary effects ofsubcutaneously administered epinephrine and terbutaline in patients with reversible a i r w a yobstruction. chest 1975;67:279-86.
70. Rosenfeld CR, Barton MD, Meschia G. Effects of epinephrine on distribution of blood flow inthe pregnant woman. Am J Obstet Gynecol 1972;124:156-63.
71. Tracers AH, Rowe B, Barker S et al. The effectiveness of IV β-agonists in treating patients withacute asthma in the emergency department. A meta-analysis. Chest 2002;122:1200-07.
72. Varon J, Fromm RE Jr, Marik PE. IV 2β or not 2β, that is the question! Chest 2002;122:1116-17.73. Cydulka RK, McFadden ER, Sarver JH, Emerman CL. Comparison of single 7.5 mg dose treatment
vs sequential multidose 2.5 mg treatments with nebulised albuterol in the treatment of acuteasthma. Chest 2002;122:1982-87.
74. Kelly HW. New β-2 agonist aerosols. Clin Pharmacol Ther 1985;4:393-403.75. Idris AH, Mcdermott MF, Raucci JC, Morrabel A, McGorray S, Hendeles L. Emergency
department treatment of severe asthma: metered dose inhaler plus holding-chamber is equivalentin effectiveness to nebulizer. Chest 1993;103:665-72.
76. Jasper AC, Mohsenifar Z, Kahan S, Goldberg HS, Koerner SK. Cost-benefit comparison of aeroslbronchodilator delivery methods in hospitalised patients. Chest 1987;91:614-18.
77. Newhouse MT. Emergency department management of life-threatening asthma: Are nebulizersobsolete? Chest 1993;103:661-63.

232 Bronchial Asthma
78. Newman KB, Milne S, Hamilton C, Hall K. A comparison of albuterol administered by metered-dose inhaler and spacer with albuterol by nebulizer in adults presenting to an urban emergencydepartment with acute asthma. Chest 2002;121:1036-41.
79. Harvey CJ, O’Doherty MJ, Page CJ, Thomas SHL, Nunan TO, Treacher DF. Effect of a spacer onpulmonary aerosol deposition from a jet nebulizer during mechanical ventilation. Thorax1995;50:50-53.
80. Hess D. How should bronchodilators be administered to patients on ventilators. Respir Care1991;36:399-404.
81. Thomas SHL, O’Doherry MJ, Fidler HM, Page CJ, Treacher DF, Nunan TO. Pulmonary depositionof a nebulised aerosol during mechanical ventilation. Thorax 1993;48:154-59.
82. Fuller HD, Dolovich MB, Posmituck G, Wong Pack W, Newhouse MT. Pressurised aerosol versusjet aerosol delivered to mechanically ventilated patients. Comparison of dose to the lung. Amrev Respir Dis 1990;141:440-44.
83. Olshaker J, Jerrand D, Barish RA et al. The efficacy and safety of a continuous albuterol protocolfor the treatment of acute adult asthma attack. Am J Emerg Med 1993;11:131-33.
84. Rodrigo GJ, Rodrigo C. Continuous vs intermittent β-agonists in the treatment of acute adultasthma. A systemic review with meta-analysis. Chest 2002;122:160-165.
85. Khine H, Fuchs SM, Saville AL. Continuous vs intermittent nebulised albuterol for emergencymanagement of asthma. Acad Emerg Med 1996;3:1019-24.
86. Papo M, frank J, Thompson AE. A prospective, randomised study of continuous versusintermittent nebulised albuterol for severe status asthmaticus in children. Crit Care Med1993;21:1489-86.
87. Portnoy J, Aggarwal J. Continuous terbutaline nebulisation for the treatment of severeexacerbations of asthma in children. Ann Allergy 1988;60:368-71.
88. Benatar SR. Fatal asthma. N Engl J Med 1986;314:423-29.89. Rowe BH, Keller JL, Oxman AD. Effectiveness of steroid therapy in acute exacerbations of asthma:
a meta analysis. Am J Emerg Med 1992;10:301-10.90. McFadden ER Jr. Dosages of corticosteroids in asthma. Am Rev Respir Dis 1993;147:1306-1310.91. Haskel RJ, Wong EM, Hansen JE. A double-blind, randomised clinical trial of methyl prednisolone
in status asthmaticus. Arch Intern Med 1983;143:1324-27.92. Littenberg B, Gluck EH. A controlled trial of methyl prednisolone in the emergency treatment of
acute asthma. N Engl J Med 1986;314:150-52.93. Rodrigo G, Rodrigo C. Corticosteroids in the emergency department therapy of acute adult
asthma: An evidence based evaluation. Chest 1999;116:285-95.94. Rodrigo C, Rodrigo G. Inhaled flunisolide for acute severe asthma. Am J Respir Crit Care Med
1998;157:698-703.95. McFadden ER. Inhaled glucocorticosteroids and acute asthma: Therapeutic breakthrough or
nonspecific effect? Am J Respir Crit Care Med 1998;157:677-78.96. Lee-Wong M, Dayrit FM, Kohli AR et al. Comparison of high-dose inhaled flunisolide to systemic
corticosteroids in severe adult asthma. Chest 2002;122:1208-13.97. Edmonds ML, Camargo CA Jr, Brenner BE, Rowe BH. Replacement of oral corticosteroids with
inhaled corticosteroids in the treatment of acute asthma following emergency departmentdischarge. Chest 2002;121:1798-1805.
98. Mark PE, Varon J. Oral vs inhaled corticosteroids following emergency department dischargeof patients with acute asthma. Chest 2002;121:1735-36.
99. Rebuck AS, Chapman KR, Abboud R et al. Nebulised and sympathomimetic treatment of asthmaand chronic obstructive airways disease in the emergency room. Am J Med 1987;82:59-64.
100. Bryant DH, Rogers P. Effects of ipratropium bromide solution nebulisation with and withoutpreservatives in the treatment of acute and stable asthma. Chest 1992;102:742-47.

Therapeutic Approach in Patients with Asthma (Acute Severe Asthma) [SA] 233
101. Kelly HW, Murphy S. Should anticholinergics be used in acute severe asthma? Ann Pharma-cothera 1990;24:409-14.
102. Rodrigo GJ, Rodrigo C. The role of anticholinergics in acute asthma treatment: An evidencebased evaluation. Chest 2002;121:1977-1987.
103. Lalla S, Saleh A, Faroog J et al. Intravenous aminophylline in acute, severe bronchial asthma.Chest 1991;(Suppl):60S.
104. Wrenn K, Slovis CM, Murphy F, Greenberg RS. Aminophylline therapy for acute bronchospasticdisease in the emergency room. Ann Intern Med 1991;115:241-47.
105. Littenberg B. Aminophylline in severe, acute asthma: A meta-analysis. JAMA 1988;259:1678-1684.
106. Rodriguez-Roisin R, Ballester E, Roca J, Torres A, Wagner PD. Mechanisms of hypoxia in patientswith status asthmaticus requiring ventilation. Am Rev Respir Dis 1989;139:732-39.
107. Skobeloff EM, Spivey WH, McNamara RM, Greenspon L. Intravenous magnesium for thetreatment of acute asthma in the emergency department. JAMA 1989;262:1210-13.
108. Noppen M, Vanmaele L, Impens N, Schandevyl W. Bronchodilating effect of intravenousmagnesium sulfate in acute severe asthma. Chest 1990;97:373-76.
109. Rolla G, Bucca C, Caria E et al. Acute effects of intravenous magnesium sulfate on airwayobstruction of asthmatic patients. Ann allergy 1986;61:388-91.
110. Okayama H, Aikawa T, Okayama M, Sasaki H, Mue S, Takishima T. Treatment of statusasthmaticus with intravenous magnesium sulfate, J Asthma 1991;28:11-17.
111. Bloch H, Silverman R, Mancherje N, et al. Magnesium sulfate is a useful adjunct to standardtherapy for acute severe asthma. Chest 1992;102(Suppl):83S.
112. Haury VG. Blood serum magnesium in bronchial asthma and its treatment by the administrationof magnesium sulfate. J Lab Clin Med 1940;25:340-344.
113. Boselo SJ, Pla JC. Sulfato de magnesio en la crisis de asthma. Prensa Med Argent 1936;23:1677-80.
114. Noppen M, Vanmaele I, Impens N et al. Bronchodilating effect of intravenous magnesiumsulfate in acute severe bronchial asthma. Chest 1990;97:373-76.
115. Rolla G, Bucca C, Caria E et al. Acute effect of intravenous magnesium sulfate on airwayobstruction of asthmatic patients. Ann allergy 1988;61:388-91.
116. Okayama H.Aikawa T, Okayama M et al. Bronchodilating effects of intravenous magnesiumsulfate in bronchial asthma. JAMA 1987;257:1076-78.
117. McNamara RM, Spivey WH, Skobeloff E et al. Intravenous magnesium in the management ofacute respiratory failure complicating asthma. Ann Emerg Med 1989;18:197-99.
118. Kuitert LM, Kletchko SL. Intravenous magnesium sulfate in life-threatening asthma. Ann EmergMed 1991;20:1243-45.
119. Devi PR, Kumar L, Singhi S et al. Intravenous magnesium sulfate in acute severe asthma notresponding to conventional therapy. Indian Paediatr 1997;34:389-97.
120. Clarallo L, Brousseau D, Reinert S. Higher-dose intravenous magnesium therapy for childrenwith moderate to severe acute asthma. Arch Pediatr Adolesc Med 2000;154:979-83.
121. Blosch H, Silverman R, Mancherje N et al. Intravenous magnesium sulfate as an adjunct in thetreatment of acute asthma. Chest 1995;107:1576-81.
122. Green SM, Rothrock SG. Intravenous magnesium for acute asthma: Failure decrease emergencytreatment duration or need for hospitalisation. Ann Emerg Med 1992;21:260-65.
123. Tiffany BR, Berk WA, Todd IK et al. Magnesium bolus or infusion fails to improve expiratoryflow in acute asthma exacerbations. Chest 1993;104:831-34.
124. Silverman RA, Osborn H, Runge J et al. IV magnesium sulfate in the treatment of acute severeasthma; a multicenter randomised controlled trial. Chest 2002;122:489-97.
125. Gluck EH, Onorato DG, Castriotta R. Helium-oxygen mixtures in intubated patients with statusasthmaticus and respiratory acidosis. Chest 1990;98:693-98.

234 Bronchial Asthma
126. Manthouse CA, Hall JB, Melmed A et al. Heliox improves pulsus paradoxus and peak expiratoryflow in nonintubated patients with severe asthma. Am J Respir Crit Care Med 1995;151:310-14.
127. National Asthma Education Programme. Expert panel report. Guidelines for the diagnosis andmanagement of asthma. Bethesda, US Department of Health and Human Services. PublicationNo.91-3042.
128. Meduri GU, Abou-Shala N, Fox RC, Jones CB, Leeper KV, Wunderink RG. Non-invasive facemask mechanical ventilation in patients with acute hypercapnic respiratory failure. Chest1991;100:445-54.
129. Shivram U, Miro AM, Cash ME, Finch PJP, Heurich AE, Kamolhz SL. Cardiopulmonary responsesto continuous positive airway pressure in acute asthma. J Crit Care 1993;8:87-92.
130. Martin JG, Shore S, Engel LA. Effect of continuous positive airway pressure on respiratorymechanics and pattern of breathing in induced asthma. Am Rev Respir Dis 1982;126:812-17.
131. Mansel JK, Stogner SW, Norman JR. Face-mask CPAP and sodium bicarbonate infusion in acutesevere asthma and metabolic acidosis. Chest 1989;96:943-44.
132. Pollard BJ. Which drug-steroid or benzylisoquinolium? Intensive Care Med 1993;19:46-60.133. Pepe PE, Marini JJ, Occult end expiratory pressure ion mechanically ventilated patients with
airflow obstruction. Am Rev Respir Dis 1982;126:166-70.134. Saunier FF, Durocher AV, Deturck RA, Lefebvre MC, Wattell FE. Respiratory and haemodynamic
effects of halothane in status asthmaticus. Intensive Care Med 1990;16:104-07.135. Echeverria M, Gelb AW, Wexier HR, Ahmad D, Kenefick P. Enflurane and halothane in status
asthmaticus. Chest 1986;89:153-54.136. Hogman M, Frostell CG, Hedenstrom G. Inhalation of nitric oxide modulates adult human
bronchial tone. Am Rev Respir Dis 1993;148:1474-78.137. Falliers CJ, McCann WP, Chai H, Ellis EF, Yazdi N. Controlled therapy of iodotherapy for
childhood asthma. J Allergy 1966;38:183-92.138. Wanner A, Rao A. Clinical indications of and effects of bland, mucolytic, and antimicrobial
aerosols. Am Rev Respir Dis 1980;122:79-87.139. Helm WH, Barran KM, Mukerjeee SC. Bronchial lavage in asthma and bronchitis. Ann Allergy
1972;30:518-23.140. Lang DM, Simon RA, Mathison DA, Tims RM, Stevensopn DD. Safety and possible efficacy of
Fibre optic bronchoscopy with lavage in the management of refractory asthma with mucusimpaction. Ann Allergy 1991;67:324-30.
141. Rogers RM, Shuman JF, Zubrow AB. Bronchoalveolar lavage in asthma. Chest 1973;63(Suppl):62S-64S.
142. Smith DL, Desnazo RD. Bronchoalveolar lavage in bronchial asthma. Am Rev Respir Dis1993;148:523-32.
143. Millman M, Millman FM, Goldstein IM, Mercandetti AJ. Use of acetylcystein in bronchial asthma:another look. Ann Allergy 1985;54:294-96.

Management of Asthma with Special Problems 235
Management of Asthmawith Special Problems
14
EXERCISE-INDUCED ASTHMA (EIA)
The term “exercise-induced asthma” (EIA) is often used to describe the asthma of personsin whom exercise is the predominant or at times the only identified trigger to airflowlimitations. The goal of EIA is to enable patients to participate in any activity they choosewithout experiencing asthma symptoms and to enable the patient to achieve a normalexercise capacity. For some asthmatics, this goal means an ability to walk short distancesand to work regularly without limitation due to breathlessness. While for other, like childrenand young adults, it implies the freedom to run about and to compete in sports withoutrespiratory disability. For highly trained athletes with asthma, it means to compete atextremely high levels of physical activity and ventilatory performance. Therefore, the goalsof managing asthma in relation to exercise are: (i) to maximise lung function prior to exercise,(ii) to protect bronchoconstriction during exercise.
The presence or absence of bronchospasm induced by exercise can be established byspirometry before and after the exercise task in question. Some difficulty is encounteredwhen the exercise cannot readily be performed in the laboratory. In that situation, thecondition must be mimicked as closely as possible in the laboratory by having the patientexercise with a stationary bicycle or treadmill to the levels of ventilation and with inspiredair temperature and humidity that simulates the actual exercising condition. Testing world-class athletes in an otherwise well equipped pulmonary laboratory becomes still difficultbecause of difficulty in providing a sufficiently strenuous exercise work load. In this setting,eucapnic voluntary hyperventilation can be substituted for physical exertion, and the targetlevel of ventilation to be sustained can be set without limit. Formal pulmonary evaluationbefore and after exercise can also be used effectively to evaluate the impact of preventivetherapies. In athletes failing to respond to conventional prophylactic treatment, identicalexercise challenges can be performed following administration of various medications invaried doses and combinations, thereby objectively assessing their success.
Bronchoconstriction induced by exercise are generally rapidly eliminated by adminis-tration of inhaled β2-agonists. However, the bigger problem is preventing the developmentof significant airflow obstruction during and following exercise so as to minimise the impactof asthma on athletic participation and performance. Optimised control of asthma in generalas outlined is the first step towards prevention. The less is the underlying bronchial hyper-responsiveness and higher the pre-exercise level of expiratory airflow, the less likely it is

236 Bronchial Asthma
that a particular exercise task will provoke asymptomatic airflow obstruction. In someasthmatics, regular use of inhaled corticosteroids will be needed to achieve these goals.Inhaled corticosteroids are currently approved by the International Sports authorities forthis purpose.
Most patients with EIA are otherwise asymptomatic. In these patients, inhaled β2-agonists,used prior to exercise, will prevent EIA in more than 80% of subjects. These may be takenfrom less than 10 to 20 minutes prior to exercise in standard doses (1-2 inhalations) and arehelpful for up to several hours. They provide excellent protection for 2 hours. Because theydo not enhance exercise performance in any way other than prevention of air-flowobstruction, they are permitted and approved for use in world-class competitions. Fenoterolis an exception as its metabolite parahydroxyamphetamine, which is not allowed for athletes.The newer, longer-acting β-agonists, salmeterol and formoterol, offer effective preventionof exercise-induced symptoms for as long as 8 hours following a single, pre-exercisedose.1,2
Cromolyn sodium (2 puffs) before exercise is another acceptable pre-treatment,particularly in those who cannot tolerate beta-agonists. This drug is virtually devoid of anyside effect. The peculiarity of the drug is that when used as an anti-inflammatory agent forchronic asthma, it takes several weeks for its effect. But as a prophylactic in EIA, a singledose prior to exercise is effective. Regular use of the drug does not enhance the protectiveaction.3 The small percentage of patients who still encounter difficulty are helped by anincrease in the dosage of β2-agonist or use of both this and Cromolyn.
Many other drugs have been shown to inhibit the bronchoconstrictive effect of exercise.They are ipratropium bromide, theophylline, calcium channel blocking agents, nedocromil,terfenadine, leukotriene D4 antagonists and lipoxygenase inhibitors, inhaled frusemide4,and inhaled heparin5 are some of the conventional and newer therapies active in modifyingthe response to exercise. However, use of many of these are rarely required. Their use canbe considered when conventional therapy with β-agonists and cromolyn fails.
Patients who experience a refractory period during continuous exercise may benefitfrom a warm-up period before exercise utilising submaximal exercise and may not needrepeated medications during periods of continuous exercise. They should avoid a suddenchange to a warm, moist environment immediately after exercise which will help inmodifying the post-exercise response. Specially designed low-resistance “heat and moistureexchange” face mask has been shown to help asthmatics in cold environments.6
Medications that are approved by the US Olympic Committee for use in competitioninclude β2-agonists (aerosol or inhalant forms of salbutamol, bitolterol, and terbutaline),Cromolyn sodium, theophylline and inhaled steroids.
PREGNANCY AND ASTHMA
The natural history of asthma during pregnancy is variable. Several physiological changesoccur during pregnancy that could worsen or improve asthma. However, it is not clearwhich are the factors that will decide the ultimate course. It is reported that symptoms ofasthma worsens during pregnancy in 43% of patients and improves in 14%.7 In a perspectivecohort study of 366 pregnancies in 330 women with asthma, the disease worsened duringpregnancy in 35%.8 Other studies suggest that 11-18% of pregnant women with asthmawill have at least one emergency department visit for acute asthma and of these, 62% willrequire hospitalisation.9,10 Exacerbations of asthma symptoms usually occurs during the

Management of Asthma with Special Problems 237
last trimester. In a large cohort study the most severe symptoms were experienced by patientsbetween the 24th and 36th week of pregnancy. Thereafter symptoms decreased significantlyin the last four weeks and 90% had no asthma symptoms during labor or delivery. Only twopatients required additional medications beyond bronchodilators.8 Another study has alsoconfirmed that during the last months of pregnancy asthma is least likely to exacerbate.11
Some other reports suggest that there is no change in the course of asthma during pregnancy.12
Women tend to follow the same pattern during all pregnancies with respect to the course oftheir asthma.13, 8 Women with extrinsic (atopic) asthma tend to have fewer symptoms duringpregnancy than patients with intrinsic asthma.14 A cohort study comparing 198 pregnantwomen with asthma with 198 women without asthma observed that non-atopic patientswith asthma tend to have more severe asthma. Pre-eclampsia was also more common in thisgroup. However, with proper surveillance and treatment, pregnancy and delivery relatedcomplications could be avoided. A systematic review has shown that baseline asthma severitydoes determine what happens to asthma in pregnancy and asthma may affect the risk ofadverse outcome.15 Severe asthma is likely to worsen during pregnancy than mild asthma.8
However, some patients with severe asthma may experience improvement, whilst symptomsmay deteriorate in some with mild asthma. A meta-analysis of 14 studies concluded andagreed with the commonly quoted generalisation that during pregnancy about one-third ofasthma patients experience an improvement in their asthma, one-third experience a worseningof symptoms, and one-third remains the same.16 During pregnancy 40% of the patients managedwith the same asthma medications as before pregnancy; 18% needed less; and in 42% theneed was more.17
Bronchial asthma in the mother has been associated with increased perinatal morbidityand mortality,18,19 and also with increased morbidity during infancy.18 Obstetric complicationshave been observed more often in asthmatics than in control subjects.18 Pre-eclampsia occurredmore often in asthmatics than controls, especially in patients with severe asthma. Hypoglycemiaoccurred more often in infants of mothers with severe asthma than in infants of mothers withless severe disease. Uncontrolled asthma is associated with many maternal and fetalcomplications, including hyperemesis, hypertension, pre-eclampsia, vaginal hemorrhage,complicated labor, intrauterine growth retardation, pre-term birth, increased perinatalmortality, and neonatal hypoxia.20-23 A large Swedish population based study using recordlinkage data demonstrated increased risks for preterm birth, low birth weight, perinatalmortality, and pre-eclampsia in women with asthma. The risk of prematurity and low birthweight were higher in women with severe asthma necessitating admission.24 In contrast, ifasthma is well controlled throughout pregnancy there is little or no increased risk of adversematernal or fetal complications.9,10 Pregnancy, therefore, should be an indication foroptimisation of therapy and maximising lung function in order to reduce the risk of acuteexacerbations.
Theophylline at term did not influence labor or delivery. Thus, severe asthma or systemiccorticosteroid treatment or both during pregnancy seems to increase the incidence of mildpre-eclampsia in the mother and hypoglycaemia in the newborn.19 Poorly controlled asthmahas adverse effect on the fetus, resulting in increased perinatal mortality, increased prematurity,and low birth weight. For this reason, the use of drugs to obtain optimal control of asthma isjustified even when their safety in pregnancy has not been unequivocally proven. For mostantiasthma drugs, clear documentation of teratogenic effects is lacking. On the other hand,these drugs have not yet been proved to be safe. Thus all drugs can be used without increased

238 Bronchial Asthma
risk to the fetus25,26 except alpha-adrenergic compounds. Reports on the adverse effects ofcorticosteroids during pregnancy are contradictory. Apgar reported an increased incidenceof cleft palate in two series of pregnancies in women treated with systemic corticosteroids inearly pregnancy.27 The incidence of stillbirths was eight times higher in women havingcontinuous steroid treatment than in a corresponding group of untreated women with thesame diseases.28 However, the alarming findings in these studies may be a part of theunderlying severe disease rather than the treatment.29 However, two other studies of pregnantasthmatic women, some having short-term and some long-term systemic corticosteroids,showed no complications attributable to steroid treatment.30,31 Thus, although the direct effectsof systemic steroids on the fetus appears to be small, their indirect effects (hypertension, pre-eclampsia, hypoglycemia) may still form a risk for the infant. However, as an acute exacerbationof asthma carries even greater risks for the mother and child, systemic steroids should not bewithheld when the need arises. Higher theopylline concentrations in the mother might beassociated with complications of labor and delivery due to uterine atony. On the other hand,inhaled steroids may be quite safe in recommended doses. Immunotherapy should not bestarted during pregnancy, if that is considered. It is important to avoid fetal hypoxia duringacute exacerbations. No special treatment is required for asthma during labor except for thosewho have received daily parenteral steroids for a week or three separate courses in thepreceding year, in whom hydrocortisone supple-mentation (100 mg every 8 hours) should begiven for the stress of delivery unless there is documentation of normal adrenal responsiveness.Terbutaline and salbutamol can cause delayed/poor uterine contraction during labor. Duringacute attacks, drug therapy should be the same as for the non-pregnant patient. Oxygenshould be delivered to maintain saturation above 95%. Continuous fetal monitoring isrecommended for severe asthma. In women with poor control of asthma, there should be closeliaison between the respiratory physician and obstetrician.
In general, the medicines used to treat patients of asthma are quite safe in pregnancy.32 Therisk of harm to the fetus from severe or chronically under treated asthma outweighs any smallrisk from the medications used to control asthma. β2-agonists, corticosteroid inhalers are to beused as normal during pregnancy.33,34 No significant association has been demonstratedbetween major congenital malformations or adverse outcome and exposure tomethylxanthines.32,35 Although there were some concerns of use of oral steroids during earlypregnancy, they were unfounded and steroid tablets are to be used as normal when indicatedduring pregnancy for severe asthma and they should not be withheld because of pregnancy.Data regarding the safety of leukotriene antagonists are limited. Animal studies and post-marketing surveillance for zafirlukast and monteleukast are reassuring. There are concernsabout animal data on zyleuton.36 Leukotriene antagonists should not be started duringpregnancy. They may be continued in women who have demonstrated significant improvementin asthma control with these agents prior to pregnancy not achievable with other medications.
Acute attacks of asthma are very rare in labor due to endogenous steroid production. Inwomen receiving steroid tablets there is a theoretical risk of maternal hypothalamic-pituitary-adrenal axis suppression. Women with asthma may safely use all forms of pain relief inlabor. In some studies there is an association between asthma and an increased caesareansection rate., which may not be conjectural. Risk of postpartum exacerbation of asthma isincreased in women undergoing caesarean section. This may be related to the severity ofasthma rather than caesarean section per se. The women should continue their usual asthmamedications in labor. In the absence of acute severe asthma, caesarean section should be

Management of Asthma with Special Problems 239
reserved for usual obstetrics indications. If anaesthesia is required, regional blockade ispreferable to general anaesthesia in women with asthma. Women receiving steroid tablets ata dose exceeding prednisolone 7.5 mg per day for more than two weeks prior to deliveryshould receive parenteral hydrocortisone 100 mg 6-8 hourly during labor. Prostaglandin F2αshould be used with extreme caution in these women with asthma because of the risk ofinduction of bronchoconstriction.
Antiasthma drugs may pass into the breast milk, and earlier, mothers have, therefore, beenadvised not to breastfeed. However, no evidence has shown that inhaled drugs or moderatedoses of theophylline or systemic steroids taken by mouth by the lactating mother would beharmful to the infant.37 Breastfeeding, thus, is to be encouraged.19 All medications used to treatasthma including steroid tablets have been shown in early studies to be safe to use in nursingmothers.15 There is less data with newer agents. Less than 1% of the maternal dose oftheophylline is excreted into the breast milk. Prednisolone is secreted in breast milk, but milkconcentrations of prednisolone are only 5-25% of those in serum.16. The proportion of an oralor intravenous dose of prednisolone recovered in breast milk is less than 0.01%. For maternaldoses of at least 20 mg once or twice daily, the nursing infant is exposed to minimal amountsof steroid with no clinically significant risk. Thus, asthma drugs should be used as normalduring lactation, in line with manufacturer’s recommendations.
SURGERY AND ASTHMA
Possible intraoperative and postoperative complications can occur in patients of bronchialasthma because of bronchial hyper-reactivity, airflow obstruction, and mucus hyper-secretion.38-40 Possible complications are many and may include the following. Acute attacksmay be triggered during intubation which stimulates sensory receptors in the upper airwaythat can lead to reflex efferent neurotransmission via the vagus nerve, resulting in bronchialsmooth muscle contraction. Increased airway obstruction may result due to suppressed coughand mucus plugging following surgery. This will result in ventilation-perfusion mismatchingand may contribute to impaired gas exchange resulting in hypoxaemia and possiblyhypercapnia during and following surgery. Severe airflow obstruction along withpostoperative pain, can impair the effective cough. Retained airway secretion can furtherimpair airflow and gas exchange. Further, mucus impaction can cause atelectasis andassociated diminished respiratory excursion will result in respiratory infection and furtherimpairment of gas exchange. The likelihood of these complications will depend upon theseverity of the patient’s airway hyperresponsiveness, the degree of airways obstruction, andthe amount of mucus plugging at the time of surgery. These variables can be assessed byhistory, physical examination, and spirometry. Other factors that influence the rate ofpostoperative complication include the type of surgery (thoracic and upper abdominal surgeryhave the greatest risk) and type of anaesthesia (general anaesthesia with endotrachealintubation carries the greatest risk).
All patients with active asthma should undergo preoperative respiratory evaluation. Evenasymptomatic patients with bronchial asthma should undergo evaluation as they may havesignificant airflow obstruction and bronchial hyper-responsiveness. Patients with moderateto severe disease, the evaluation should begin several days prior to surgery. When the asthmais uncontrolled, the patient should be hospitalised for a day or more prior to surgery foroptimisation of lung function. Patients having frequent nocturnal awakening, frequent or

240 Bronchial Asthma
continuous use of steroids, prior perioperative complications related to asthma, large volumeof sputum production, and co-morbid cardiovascular disease are associated with a high riskfor perioperative complications. Spirometry is the best way to assess the degree of airflowobstruction and all attempts should be made to achieve a normal or near normal lung functionprior to surgery.
Asthma patients experiencing wheezing, productive cough, chest tightness, or dyspnoeashould receive intensified treatment of their asthma prior to elective therapy, even if thisnecessitates delay of surgery. An attempt should be made to improve lung function in patientswith an FEV1 or PEFR < 80% of predicted or < 80% of their recent best value. Frequently a briefcourse of corticosteroids will be required to achieve this goal. Modification of anaestheticapproach may be possible in some at increased perioperative risk. Spinal, epidural, or localanaesthesia may in some cases be substituted for general anaesthesia and postoperative paincontrol may be achieved with epidural analgesia rather than parenteral narcotics. Even in theasymptomatic or minimally symptomatic patient, it is useful to administer an inhaled β2-agonist bronchodilator immediately prior to surgery. Patients receiving daily medications forasthma should generally be maintained on these drugs. Intravenous aminophylline can beused to maintain therapeutic levels who are taking this drug, but are not permitted to takeanything by mouth. The usual maintenance dose of theophylline is intravenous aminophyllineof 0.6 mg/kg/hour by continuous infusion. Inhaled bronchodilators can be maintained duringsurgery even patients receiving general anaesthesia and mechanical ventilation. To preventdepressed adrenal-pituitary response to stress, intraoperative and postoperative steroidsupplementation should be given to patients who have received systemic steroids for morethan 2 weeks within the last 6 months or more than two courses of systemic steroids withinthe last 12 months. Patients who have been taking high-dose inhaled corticosteroids morethan the conventional recommended doses, should also be considered at risk of relativeadrenal-pituitary suppression and should be given perioperative steroid replacement therapy.The usual dose of replacement corticosteroid therapy during stress is 300 mg of hydrocortisoneper day. Hydrocortisone of 100 mg each should be given intravenously on the day of surgeryin the morning, intraoperatively, and postoperatively. The dose is then tapered over the nextfew days. Clearance of airways is an important aspect of postoperative care.
OLDER PATIENTS WITH ASTHMA
Although asthma affects all age groups, morbidity and mortality is particularly high in theelderly. In New York City, asthma-attributable mortality rates in adults > 65 years of age aresix times higher than those in adults < 40 years of age.4 Whereas many factors contribute tothe urban asthma problem, sensitisation to indoor allergens may play a particularly significantrole. The presence of cockroach-specific serum IgE in a population of elderly urban patientswith asthma, is associated with more severe asthma, as reflected by an increase in airwayobstruction and hyperinflation.42 Asthma is frequently under diagnosed in the elderly becauseof a number of differential diagnosis, difficulty in measurement of lung function, and under-reporting of symptoms.43 The later may occur because of reduced expectations or because ofan age related reduction in perception of breathlessness.44 A similar age related differencein the physical signs associated with severe asthma may lead to underestimation of severityand under treatment.45 Simple tests of mental functioning may be necessary to ensure thatelderly people with asthma are capable of acquiring the necessary skills for treating andmonitoring their condition.46 Increased asthma mortality is more common in elderly patients

Management of Asthma with Special Problems 241
above the age of 55 years. This may be due to difficulty in diagnosis (COPD and congestivecardiac failure). The precise cause of severe airflow obstruction is difficult to diagnose attimes. Some cases asthma diagnosed in older individuals may actually be a combination ofasthma and COPD or of asthma and congestive heart failure. Further, coexistence of otherdiseases (myocardial ischaemia), can cause additional problems. For example, ischaemicheart disease in a case of bronchial asthma may be more dangerous because of associatedhypoxaemia which could result in decreased myocardial oxygenation followed by myocardialinfarction or rhythm disturbances. Frequent intake of drugs by people of this age are knownto aggravate asthma (beta blockers for hypertension and eye drops, aspirin and othernonsteroidal anti-inflammatory drugs). Since arthritis is a known disease of this age, if thepatient takes aspirin and other non-steroidal anti-inflammatory drugs may cause suddenand severe asthma exacerbations. Further, epinephrine and theophylline have the potentialof precipitating the underlying heart disease. Although treatment of chronic and acute asthmaexacerbations should be the same as per the recommended guidelines, certain specialconsiderations are necessary in elderly individuals. All patients of asthma above the age of55 years old should be evaluated for coexisting disease conditions. Particular attention shouldbe given to the monitoring of hypoxemia if he has concomitant heart disease. Theophyllinemay increase the risk of urinary retention in older men with prostatism apart from its potentialcardiac side effects. These patients should carefully be monitored for steroid side effects.Monitoring of hematocrit and blood sugar periodically is essential to rule out hyperglycemia,hypokalemia, and gastrointestinal bleeding. Eye examination is to be conducted annually torule out cataract and glaucoma. Evaluation of possible alterations in calcium homeostasis isessential particularly in whom there is a greater concern of bone loss as in postmenopausalwomen. Anticholinergic bronchodilator therapy may have a greater role in this age groupthan in the younger patients.47 Oxygen should also be used with caution if there is associatedCOPD to avoid precipitation of carbon dioxide retention. Depression and other associatedserious psychiatric illness needs detail assessment. Older individuals are more prone fordepression, which in turn is identified as a risk factor for fatally-prone asthma. Family lossand disruption, difficult adjustments to retirement, and other psychosocial problems aremore common in this age group. Certain other impairments more common in older individualsmay interfere worth treatment. These include arthritis, which may require special devicessuch as a spacer to assist actuation. Nebulisers might be more useful. Patients with visualimpairment may be unable to read the numbers on the peak flow meter in which colour codesmight help. Further, there may be difficulty in reading instructions either on the drug orprescriptions given by the physician. Some other family member may be helpful in this situation.Patients with memory difficulties might forget to adhere to medical regimens that requireseveral drugs and frequent schedules. Patients with hearing difficulties may not tell thehealth provider that they have not heard or understood the instructions. Asking the patientsto state the information and/or instructions in their own words will help ensure understanding.
OCCUPATIONAL ASTHMA
The diagnosis should be suspected in all adults with airflow obstruction and with a positivehistory of high-risk occupations or exposures. Patients with pre-existing asthma aggravatednon-specifically by dust and fumes at work (work aggravated asthma) should be distinguishedfrom those with pre-existing asthma who become additionally sensitized to an occupationalagent. The subject is usually better on days away from work and better on holidays. Although

242 Bronchial Asthma
they are not specific for occupational asthma, but are important clues for investigation for anoccupational agent. These symptoms are also present in asthmatics due to sensitizing agentsat home and in those who do much less physical exertion. An accurate history taking isimportant which should include exposures to chemicals, organic dusts and other possibleagents both at the current time as well as in the past.
The diagnosis and management of occupational asthma are difficult and can be dividedinto three parts:• Confirming the diagnosis of asthma;• Confirmation of the relationship between asthma and work exposures; and• Finding the specific cause
The first step is to confirm that the patient is having asthma by using standard measureslike peak expiratory flow measurements, lung function tests, and reversibility testing. COPDand non-respiratory causes of breathlessness should be excluded. The next step is theestablishment of a relationship between asthma and work exposure. These include serialmeasurements of PEFR at home, and at work, measurements of non-specific airway hyperreactivity after days at and away from work, measurements of specific IgE to an occupationalagent, and specific bronchial provocation testing. The decision of making a case ofoccupational-induced asthma remains a matter of clinical judgment. Measurements of PEFRshould be made every two hours from working to sleeping for four weeks keeping treatmentconstant and documenting times at work.48 Minimum standards for diagnostic sensitivityof > 70% and specificity of > 85% are (a) at least three days in each consecutive work period;(b) At least three series of consecutive days at work with three periods away from work(usually about three weeks); and (c) at least four evenly spaced readings per day. Non-specific responsiveness measurements with methacholine or histamine can be undertakenafter a period at end away from work exposure. A more than 3.2-fold changes in PC20indicates a significant change outside the 95% confidence intervals for repeat measurements.The diagnostic sensitivity, however, is only 40%, which is substantially worse than serialPEFR measurements.49 The third step is identification of the cause of occupational asthma,which is often difficult. There should be information about the sources of exposure (riskassessment). IgE measurements are possible for most biological agents and a few lowmolecular weight chemicals. These include latex in health care workers, flour and enzymesin bakers, rodent urine extracts, and animal epithelia in laboratory animal workers andveterinary surgeons, and acid anhydrides in exposed workers. Carefully controlled exposureto workplace agents and suitable controls is the gold standard for diagnosis.50 Tests aredifficult to do and are not widely available, and are not always possible for some types ofworkplace exposures.
The ideal treatment for patients with occupational asthma with a latency period is removalfrom exposure. Early diagnosis and removal from exposure is associated with a favourableprognosis. Eliminating exposure should be tried. In some instances, reducing exposure byimproving ventilation or providing a respirator may allow a person to return to the samejob. However, once sensitisation occurs, bronchoconstriction will often be triggered bysubsequent minimal exposure. Once well established, occupational asthma may not becompletely reversible. Recovery, if occurs may take months to years after removal fromexposure. A worker might be transferred to a job without exposure in the same company. Mostpeople with occupational asthma have to be retained for a job with another employer in adifferent field. Before advising the worker to leave the job, attempt should be made to see if

Management of Asthma with Special Problems 243
changing the job process or activities can be changed to reduce exposure or if protectiveequipment is useful. When the employees have irritant-induced asthma, or work-aggravatedasthma, employer should make every effort towards reasonable accommodation byimproving the workplace as required. The physician should advise the patient regardingcompensation as per the law of the particular country.
If the patient returns to the same job, should have close medical follow-up. Worsening ofasthma should lead to immediate removal from exposure. Pharmacological treatment ofoccupational asthma is similar to the treatment of patients with other forms of asthma.However, continuing monitoring is important. Although removal from the source ofexposure lead to improvement, patients may continue to require medication and have airflowlimitation or bronchial hyperresponsiveness for many months or years.
Patients should be referred to compensation boards or similar other agencies. Patientsshould be evaluated for temporary impairment and disability when their asthma is undergood control. Evaluation for potential permanent impairment and disability should takeplace after two years, when improvement in asthma has plateaued.
Irritant-induced asthma (Reactive airway dysfunction syndrome—RADS) is another form ofasthma associated with the workplace, in which a wheezing illness starts within 24 hours,and usually less than that, of a single large exposure to an irritant. The condition isinflammatory and does not involve immunological recognition of the irritant, so thatcontinued low levels of exposure to the causative agent can be tolerated without problems.RADS is diagnosed by the presence of non-specific responsiveness and a compatible history.The prognosis varies, but there is a good likelihood of improvement.
DRUG-INDUCED ASTHMA
Even an initial reaction to aspirin or other nonsteroidal anti-inflammatory drug (NSAID)may be severe and an adverse reaction can occur at any time, typically following years ofemploying these drugs without difficulty. Therefore, all patients of asthma should avoidthis group of drugs. Usually safe and alternative drugs are acetaminophen, sodium salicylate,or disalcid. Reaction to aspirin or an NSAID produce a refractory state lasting 2-7 days anddo not occur if patients ingest the drugs on a daily basis.51 If the patient is avoiding thesedrugs, the initial dose in the form of a rapid graded challenge should be given in the presenceof a physician. If the patient has severe asthma requiring steroids or has severe asthmawith compromised pulmonary function, or if the patient reports a previous broncho-constrictive reaction to these drugs, a more conservative treatment approach is indicatedand should be undertaken by a physician familiar with the technique.51 Aspirin use may bea special problem in patients with nasal polyps, chronic rhino sinusitis, and steroiddependency. If there is a concern regarding the use of aspirin in these patients, a sensitivitychallenge should be conducted.
GASTRO-OESOPHAGEAL REFLUX AND ASTHMA
The relationship of asthma to gastro-oesophageal reflux is a matter great debate, althoughmost people now believe that this is an important precipitating factor for asthma.52,53 In somestudies, medical and surgical treatment of gastro-oesophageal reflux has resulted inimprovement in symptoms of oesophagitis and also a decrease in asthma symptoms,

244 Bronchial Asthma
particularly those occurring in the night. On the other hand, other studies have failed todocument similar benefits.
Medical management of the reflux include elevation of the head of the bed 6-8 inches,eating smaller but frequent meals, avoiding food or drink between dinner and bed time,inhibition of gastric acid production using H2-antagonists and maintenance of loweroesophageal sphincter pressure by avoiding fatty meals, spices, ethanol, theophylline, caffeine,and avoiding drugs like metoclopromide that increase lower oesophageal sphincter pressure.Surgery is indicated for severely symptomatic oesophagitis that is not responsive to medicaltherapy, for complications like stricture, and for established pulmonary complications ofnocturnal reflux. Since surgery is extensive and is not successful for everyone, emphasisshould be on medical management.
REFERENCES
1. Anderson SD, Rodwell LT, Toit JD, Young IH. Duration of protection by inhaled salmeterol inexercise-induced asthma. Chest 1991;100:1254-60.
2. Henriksen JM, Agertoff L, Pederson S. Protective effect and duration of action of inhaled fenoteroland salbutamol in exercise-induced asthma in children. J Allergy Clin Immunol 1992;89:1176-82.
3. Rohr AS, Siegel SC, Katz RM et al. A comparison of inhaled albuterol and cromolyn in theprophylaxis of exercise induced bronchospasm. Ann Allergy 1987;59:107-09.
4. Bianco S, Robuschi M, Vaghi A, Pasorgikliom. Prevention of exercise induced bronchial asthmaby inhaled frusemide. Lancet 1988;2:252-55.
5. Ahmed T, Garnigo J, Danta I. Preventing bronchoconstriction in exercise induced asthma withinhaled heparin. N Engl J Med 1993;329:90-95.
6. Nisar M, Spence DPS, West D, et al. A mask to modify inspired air temperature and humidityand its effects on exercise induced asthma. Thorax 1992;47:446-50.
7. Gluck JC, Gluck PA. The effects of pregnancy on asthma: A prospective study. Ann Allergy1976;37:164-68.
8. Schatz M, Harden K, Forsythe A et al. The course of asthma during pregnancy, postpartum andwith successive pregnancies: A prospective analysis. J allergy Clin Immunol 1988;81:509-17.
9. Schatz M, Zeiger RS, Hoffman CP et al. Potential outcome in the pregnancies of asthmaticwomen: A prospective controlled analysis. Am J Respir Crit Care Med 1995;151:1170-74.
10. Wandel OJ, Ramin SM, Barnett-Hamm C et al. Asthma treatment in pregnancy: A randomizedcontrolled study. Am J Obstet Gynecol 1996;175:150-54.
11. Stenius-Armiala B, Hedman J, Terano KA. Acute asthma during pregnancy. Thorax 1996;51:411-14.
12. Sims CD, Chamberlain GVP, de Swiet M. Lung function tests in bronchial asthma during andafter pregnancy. Br J Obstet Gynaec 1976;83:434-37.
13. Schatz M, Harden K, Forsythe A et al. Course of asthma post-partum (PP) and during successivepregnancies; A prospective analysis (Abstract). J Allergy Clin Immunol 1986;77(Suppl):161.
14. Hiddlestone HJ. Bronchial asthma and pregnancy. N Z Med J 1964;63:521-23.15. Schtz M. Interrelationship between asthma and pregnancy; a literature review. J Allergy Clin
Immunol 1999;103:5330-35.16. Juniper EF, Newhouse MT. Effect of pregnancy on asthma: a systematic review and meta-
analysis. In Schatz m, Zeiger RS, Claman HC (Eds). Asthma and immunology of diseases inpregnancy and early infancy. New York, Marcel Dekker, 1993;401-27.
17. Stenius-Aarniala B, Piirila P, Teramo K. Asthma and pregnancy; A prospective study of 198pregnancies. Thorax 1988;43:12-18.

Management of Asthma with Special Problems 245
18. Gordon M, Niswander KR, Berendes H, Kantor AG. Fetal morbidity following potentiallyanoxigenic obstetric conditions. VII. Bronchial asthma. Am J Obstet Gynaecol 1970;106:421-29.
19. Bahna SL, Bjerkedahl T. The course and outcome of pregnancy in women with bronchial asthma.Acta Allergol 1972;27:397-406.
20. Fitzsimons R, Greenberger PA, Patterson R. Outcome of pregnancy in women requiringcorticosteroids for severe asthma. J Allergy Clin Immunol 1986;78:349-53.
21. Perlow JH, Montogomery D, Morgan MA et al. Severity of asthma and perinatal outcome. AmJ Obstet Gynecol 1992;167:964-67.
22. Schatz M, Zeiger RS, Hoffman CP. Intrauterine growth is related to gestational pulmonaryfunction in pregnant asthmatic women. Kaiser-Permanents Asthma and Pregnancy Study Group.Chest 1990;98:389-92.
23. Demissie K, Breckbridge MB, Rhods GG. Infant and maternal outcomes in the pregnancies ofasthmatic women. Am J Respir Crit Care Med 1998;158:1091-95.
24. Kallen B, Rydhstroem H, Aberg A. Asthma during pregnancy: A population based study. Eur JEpidemiol 2000;16:167-71.
25. Spector SL. The treatment of the asthmatic during pregnancy and lactation. Ann allergy1983;51:173-77.
26. Greenberger PA, Patterson R. Beclomethasone dipropionate for severe asthma during pregnancy.Ann Intern Med 1983;98:478-80.
27. Apgar V. The drug problem during pregnancy. Clin Obstet Gynec 1966;9:623-30.28. Warrell DW, Taylor R. Outcome for the fetus of mothers receiving prednisolone during pregnancy.
Lancet 1968;i:117-18.29. Walsh SD, Clark FR. Pregnancy in patients on long-term corticosteroid therapy. Scot Med J
1967;12:302-06.30. Schatz M, Patterson R, Zitz S, O’Rourke J, Melam H. Corticosteroid therapy for the pregnant
asthmatic patient. JAMA 1975;233:804-07.31. Fitzsimons R, Greenberger PA, Patterson R. Outcome of pregnancy in women requiring
corticosteroids for severe asthma. J Allergy Clin Immunol 1986;78:349-53.32. Schatz M, Zieger RS, Harden K et al. The safety of asthma and allergy medications during
pregnancy. J Allergy Clin Immunol 1997;100:301-06.33. Rayburn WF, Atkinson BD, Gilbert K et al. Short term effects of inhaled albuterol on maternal
and fetal circulation. Am J Obstet Gynecol 1994;171:770-73.34. Dombrowski M, Thom E, McNellis D. Maternal Fetal Medicine Units (MFMU) studies of inhaled
corticosteroids during pregnancy. J Allergy Clin Immunol 1999;103:5356-59.35. Stenius-Armiala B, Rikonen S, Terano K. Slow-release theophylline in pregnant asthmatics.
Chest 1995;107:642-47.36. The use of newer asthma and allergy medications during pregnancy. The American College of
Obstetricians and Gynaecologists (ACOG) and the American College of Allergy, Asthma andImmunology (ACAAI). Ann Allergy Asthma Immunol 2000;84:475-80.
37. Chung KF, Barnes PJ. Prescribing in pregnancy. Treatment of asthma. Br Med J 1987;294:103-05.38. Banatar SR. Anaesthesia for the asthmatic. S Afr Med J 1981;59:409.39. Kingston HG, Hirshman CA. Perioperative management of the patient with asthma. Anaesth
Analg 1984;63:844.40. Oh SH, Patterson R. Surgery in corticosteroid-dependent asthmatics. J Allergy Clin Inmmunol
1974;53:345.41. New York City department of Health. Asthma facts. New York, NY: New York City Department
of Health, 2000.42. Rogers L, Cassino C, Berger KL et al.Asthma in the elderly. Cockroach sensitization and severity
of airway obstruction in elderly nonsmokers. Chest 2002;122:1580-86.43. Dow L, Coggon D, Campbell MJ, Ormond C, Holgate ST. The interaction between immuno-
globulin E and smoking in airflow obstruction in the elderly. Am Rev Respir Dis 992;146:402-07.

246 Bronchial Asthma
44. Connoly MJ, Crowley JJ, Chatran NB, Nielson CP, Vestel RE. Reduced subjective awareness ofbronchoconstriction provoked by methacholine in elderly asthmatic and normal subjects asmeasure in a simple awareness scale. Thorax 1992;47:410-13.
45. Petheram IS, Jones DA, Collins JV. Assessment and management of acute asthma in the elderly:a comparison with the younger asthmatic. Postgrad Med J 1982;58:149-52.
46. Allien AC, Prior A. What determines whether an elderly patient can use a metered dose inhalercorrectly? Br J Dis Chest 1986;80:45-59.
47. Ullah MI, Newman GB, Saunders KB. Influence of age on response to ipratropium bromide andsalbutamol in asthma. Thorax 1981;36:523-29.
48. Burge PS, Pantin CF, Newton DT et al. Development of an expert system for the interpretationof serial peak expiratory flow measurements in the diagnosis of occupational asthma. MidlandsThoracic Society Research Group. Occup Med 1999;56:758-64.
49. Perrin B, Malo JL, l’Archeveque J et al. Comparison of monitoring of peak expiratory flow ratesand bronchial responsiveness with specific inhalation challenges in occupational asthma. AmRev Respir Dis 1990;141:A79.
50. Cartier A, Bernstein IL, Burge PS et al. Guidelines for bronchoprovocation on the investigationof occupational asthma. Report of the Subcommittee on Bronchoprovocation for OccupationalAsthma. J Allergy Clin Immunol 1989;84:823-29.
51. Stevenson DD, Simon RO: Aspirin sensitivity: Respiratory and cutaneous manifestations, In:Middleton E, Reed C, Ellis EF et al (Eds): Allergy Principles and Practice, (3rd Ed). St. Louise,CV Mosby, 1988.
52. Larrin A, Carrasco E, Galleguillos F, Sepulveda R, Pope CE. Medical and surgical treatment ofnonallergic asthma associated with gastroesophageal reflux. Chest 1991;99:1330.
53. Nelson HS. Worsening asthma: is reflux esophagitis to blame? J Rev Respir Dis 1990;11:827-44.

New Treatment Modalities/Newer Drugs for Bronchial Asthma 247
New Treatment Modalities/NewerDrugs for Bronchial Asthma
15
A number of advances had taken place in the management of bronchial asthma and a numberof national and international guidelines have been developed on this regard.1-10 The mostrecent one is the British Thoracic Society Guidelines-(2003).11 The main therapy consists ofinhaled β2-agonists, and corticosteroids along with other supplementary drugs. All theseguidelines emphasize that ‘add-on’ or adjunctive therapies to inhaled corticosteroids,including long acting β2-agonists and leukotriene antagonists, are clearly a better option forasthmatic persons receiving lower doses of inhaled corticosteroids than merely increasingthe dose. A number of studies have demonstrated that in patients on moderate doses ofinhaled corticosteroids, addition of a long acting β2-agonist to the regimen is a better optionthan merely increasing the dose of inhaled corticosteroids.12 The addition of a long acting β2-agonist is also effective in reducing the number and severity of exacerbations.13 Combinationtherapy of inhaled corticosteroid and long-acting β2-agonist, salmeterol or formoterol deliveredsimultaneously by a single device may be beneficial.14
Although, current asthma therapy is effective and well-tolerated, there are certainlimitations. There are still concerns about side effects of corticosteroids, particularly in childrenand in patients requiring very high doses of the drug. Many patients are also reluctant to takesteroids because of the fear of side effects. In general there is a corticophobia in the mind ofmany. There are also concerns about the osteoporosis and fracture which is in direct correlationwith the overall drug intake. Inhaled β2-agonists, although effective bronchodilators, thereare concerns about side effects particularly tremor, tachycardia, and tachyphylaxis.Theophylline, similarly has potentially serious side effects. About ~5% of asthma patients aresteroid-resistant. They do not respond to high doses of corticosteroid therapy. This group ofpatients will need some other forms of therapy. Thus, there is a need for the development ofnewer drugs for the treatment of asthma.
It is now firmly established about the cellular and mediator basis of the inflammatoryprocesses in bronchial asthma. This knowledge has been harvested through advancement inbiotechnology, by which new treatment options are being provided.15 Currently, a number ofdifferent therapies are under investigation, and in some stages of clinical trial, which include:
• Anti-immunoglobulin E (IgE) antibodies• Soluble interleukin-4 receptors (sIL-4Rs), and• Anti-interleukin-5 antibodies.The basis of these therapeutic strategies are depicted in Figure 15.1.

248 Bronchial Asthma
Anti-immunoglobulin E (IgE) antibodies-(E25)
Anti-IgE is a specific monoclonal antibody that inhibits IgE activity by binding both tocirculating IgE and to IgE on the surface of B cells.16 However, it does not bind to IgE on theIgE receptors (both low-affinity and high-affinity types, Fcε receptors). They block thebinding of IgE to its receptors on effector cells, like mast cells and basophils, but do nottrigger the activation of these cells. The molecule is nonanaphylactogenic. It forms anti-IgE-IgE complexes, which are central to the action of the drug. The maximum demonstrablesize of these complexes is as hexamers, which consist of three molecules of soluble IgE andthree molecules of anti-IgE. Soluble IgE has a half-life of only a few days. The half-life of thehexamer, on the other hand, is considerably longer, and it acts as a sump for the allergen,and also has a role in antigen presentation. They lower serum free IgE in rodents and blockpassive sensitisation of lung fragments by serum from sensitised individuals.
Humanised version of murine monoclonal anti-IgE antibodies - rhuMab-E25 and CGP51910- are developed by grafting the variable immunoglobulin region of murine originonto a backbone of the constant region of human IgG1.17 This reduced the immunogenicityof the monoclonal antibody and thus, enabled examination of the role of IgE in humandisease. The safety and efficacy profile of the anti-IgE antibody – E25, was studied byrepeated injections of the antibody to allergic human subjects which did not provokeanaphylaxis and lowered the serum IgE levels by > 99%.18 The treatment also reducedbasophil receptor density and histamine release by > 96% and 90% respectively. The effectswere then examined to see the preventive effect on the allergen provokingbronchoconstriction. The drug had protective effects on both the early and late responsesto allergen challenges.19,20 E25 treatment also reduced the number of circulating eosinophilsand the increases in bronchial reactivity and in sputum eosinophilia provoked by allergenchallenge. The drug also reduced the eosinophilic-inflammation of the airways provokedby antigen challenge in sensitised mice.21 The treatment also inhibited production of IL-4and IL-5.
Antigen
(Late asthma reaction)
Fig. 15.1: Therapeutic basis of anti-IgE antibody, IL-4 and IL-5
B-cell
IL-4
IgE
Acute symptoms(early asthmareaction)
Mast cell
Th2 IL-5 Eosinophil

New Treatment Modalities/Newer Drugs for Bronchial Asthma 249
The first large scale clinical trial of the efficacy of the humanised, monoclonal anti-IgEantibody, E25, examined the effects of repeated dosing on the severity of allergic rhinitis,22
and showed beneficial results. When given as regular treatment to patients with moderateto severe asthma requiring regular treatment with corticosteroids, inhaled or oral, E25showed safety and efficacy.23 The double blind, prospective study of 317 patients comparedthe effects of placebo with that of a low dose E25 (2.5 μg /kg/ngIgE/ml) or a high dose E25(5.8 μg /kg/ngIgE/ml) as adjuvant therapy every two weeks for 20 weeks. In both activeIgE treatment groups, serum free IgE dropped rapidly, and remained low throughout thestudy period, significantly improved the morning PEFR, quality of life scores, the need forrescue treatments, and the severity of asthma symptoms. There was a significant reductionin the reduction in dose for corticosteroid treatment. Apart from a slight increase in urticaria,adverse events were not more than those in the placebo group. Another large study alsohas shown E25 to be effective in adults and children with ragweed allergic rhinitis.24 Otherstudies of E25 in adults and asthma with moderate severity confirm the safety and efficacyin terms of a significant reduction in the frequency of exacerbations as an adjunct or duringtapering off of steroids.25 The above clinical studies, thus shows that the monoclonal anti-IgE antibody, E25 treatment, significantly improves asthma, but does not cure asthma.Development of more selective, apparently safe anti-IgE monoclonal antibody holds greatpromise not only as a research tool for defining the role of IgE in health and disease, butalso as a novel therapeutic tool in the treatment of bronchial asthma.
Soluble Interleukin-4 Receptors (sIL-4Rs)
Interleukin-4 plays a number of important roles in the allergic process and is criticallyimportant for the development of allergic inflammation. Important functions includesecretion of IgE by B lymphocytes, induction of VCAM-1 expression on vascular endotheliumin promoting cellular inflammation by which it directs the migration of T lymphocytes,monocytes, basophils, and especially eosinophils to the site of inflammation. Othermechanisms of eosinophilic inflammation are increasing eotaxin expression, inhibition ofeosinophil apoptosis, and mucus gene expression and hypersecretion. The most importantof all these is the biological activity of its ability to drive the differentiation of Th-0lymphocytes into Th-2 cells. It shares a number of activities with IL-13 and because of itsability to prevent apoptosis of T lymphocytes, it is important in allergic immune responses.The IL-4 levels are increased in the BAL fluid of allergic individuals, and peripheral bloodmononuclear cells produce IL-4 in response to dust mite antigen. Aerosolised IL-4significantly increases airway hyper-responsiveness in patients with mild asthma. Further,there is altered regulation of IL-4 in atopic individuals as well atopic individuals havehigher number of T cells. In view of a wide variety of important contribution of IL-4 inasthma pathogenesis,26-41 anti- IL-antibody is an important target for asthma therapy.
A soluble IL-4 receptor is currently under investigation for the treatment of bronchialasthma.40,41 Preclinical studies in mice has shown prevention of the development of allergen-specific IgE and reduction of eosinophilic inflammation. The drug is proved to be safe andeffective in patients of bronchial asthma. In 25 patients of mild to moderate persistentasthma on inhaled corticosteroid therapy, were randomised to placebo or IL-4R at doses of0.5 or 1.5 mg once by nebuliser42 and their steroid therapy was discontinued. There was noside effect of the drug, neither there was development of any antibody. Treatment with

250 Bronchial Asthma
1.5 mg IL-4R resulted in significantly better FEV1 at 2 hrs after treatment, and theimprovement persisted till 2 weeks. Asthma symptom score also improved significantly.Bronchial reactivity to methacholine was reduced in 6 of the 8 patients tested. Its anti-inflammatory effect was obvious by a reduction of exhaled NO scores. In further phaseI/II randomised, double blind, placebo-controlled study in 62 patients with moderatelysevere persistent asthma, treatment was given with 0.75, 1.5, or 3.0 mg of IL-4R twice weeklyby nebulisation or placebo administration.43 The steroid inhalation was discontinued. Il-4Rwas safe and well tolerated. Antibodies to IL-4R developed in <3% without any symptom.The results were similar to that of the earlier study.
Thus, it is apparent that IL-4 receptor is potentially a safe and effective treatment forasthma without the use of corticosteroids. Once-weekly dosing targeting the lungs willimprove patient compliance. Long-term disease progression can be prevented by IL-4 as itinhibits the central inflammatory process.41
Anti-interleukin-5 Antibodies
Eosinophils are important in the inflammatory pathophysiology of bronchial asthma.Interleukin-5 is thought to be associated with the late stages of maturation and release ofeosinophils that occur within the bone marrow in different allergic diseases. Two differentmonoclonal antibodies against IL-5 in clinical trials have shown that single intravenousinfusion with 2.5 or 10 mg/kg reduced the eosinophil levels to low normal values in theblood and sputum from patients with asthma, which is maintained up to 4 months. No effect,however was noticed on bronchial hyperresponsiveness.44 Another study using a singledose of anti-IL-5 provided only a small increase in FEV1 in noneosinophilic bronchialasthma.45
Other Cytokine-directed Therapy
a. TNF Antagonism. Based on findings from experimental animals and the presence of elevatedTNF levels in patients with bronchial asthma, there is a rationale for TNF antagonism to bedeployed in asthmatics. TNF seems to play an important role in determining the severity ofasthma. TNF antagonism may be useful in treating cases of severe persistent asthma aswell as acute severe asthma.46
b. Interleukin-13 antagonism. IL-13 plays an important role in the induction of airway hyper-reactivity, mucus formation, and airway remodelling in animal models of pulmonarydisease. There is an increased production of IL-13 in atopic and non-atopic asthma, atopicdermatitis, allergic rhinitis, and chronic sinusitis. Recent human genetic data also showassociation of genes of the IL-13 signalling pathway to allergic disease and bronchialasthma. These findings suggest the possible role of IL-13 antagonists in the treatment ofbronchial asthma.47
c. IL-10 therapy. IL-10 deficiency is associated with several inflammatory diseases likebronchial asthma and cystic fibrosis. Animal studies have shown that endogenous IL-10suppresses excessive inflammation in the lung. Clinical studies have shown thatconstitutive IL-10 protein concentrations are reduced in BAL fluid from asthmaticcompared to normal subjects and the IL-10 production by monocytes is reduced. Theeffect of IL-10 may thus, provide a physiological form of anti-inflammatory therapy.48

New Treatment Modalities/Newer Drugs for Bronchial Asthma 251
d. Antihistamine drugs like H1- antihistamines (cetirizine, terfenadine, loratadine, astemizole,azelastine, fexofenadine, mizolastine etc.) are useful drugs for associated conditions likeallergic rhinitis. They do not have any direct effect on bronchial asthma.49 Similarly,H3–antihistamines, particularly in combination with H1-antihistamines, are useful forthe treatment of allergic rhinitis. This combination provides a better treatment approachfor allergic nasal congestion without the hypertensive liability of current alpha-adrenergicagonist decongestive therapy.50
e. Other future targeted therapeutic approaches. Because of the role of a large number ofcytokines and other mediators in the pathophysiology of bronchial asthma, theoreticallythere is a wide scope of manipulating these products, which can be used as anti-asthmatherapy. These are
i. Mediator inhibitors and agonists like kinin receptor antagonists, endothelinantagonists, tachykinin antagonists, selective iNOS inhibitors, mucus regulation,and P2Y receptor antagonists;
ii. Allergen-and IgE-directed therapy;iii. T-cell immunomodulation like GATa-3, mycobacterial immunisation, macrocyclic
immunosuppressants;iv. Chemokine receptor inhibition;v. Adhesion molecule inhibitors;
vi. Inhibition of cell signalling;vii. Therapies acting on transcription andviii. Genetic therapy, which includes antisense therapy, ribozyme therapy and gene
therapy.f. Vaccines for bronchial asthma. Asthma is not curable by the currently available drugs.
However, some investigators believe that the disease is potentially curable throughstrategies that prevent or reverse the immunological abnormalities in atopy. There areseveral approaches to reduce the preponderance of Th2 cells in atopy by switching thebalance in favour of Th1 cells. This can be achieved in animals by exposure to bacterialproducts such as BCG, Mycobacterium vaccae, or unmethylated cytosine-guanosinedinucleotidecontaining oligonucleotides (CpG ODN).51-53 This suggests that vaccinationwith allergens, immunomodulators and adjuvants may be a future strategy for theprevention or cure of asthma.54
PDE4 Inhibitors
Phosphodiesterase 4 (PDE4) is present in a number of inflammatory cells responsible forthe development of asthma. One of the mechanisms of action of theophylline forbronchodilatation is inhibition of PDE4. However, the potency of the drug is relativelypoor at therapeutic concentrations. A number of highly selective PDE4 inhibitors have beendeveloped to overcome this problem. Phosphodiesterase inhibition increases the intracellularlevels of cyclic AMP, which is important for regulation of cell function.55,56 PDE4 inhibitorsreduce eosinophil survival, inhibit eosinophil chemotaxis, degranulation, adhesion moleculeexpression, and leukotriene synthesis. Other functions of PDE4 inhibitors include attenuationof proliferation of mononuclear cells in atopic individuals and Th1 and Th2 cells. They alsoinhibit cytokine generation. Thus, PDE4 inhibition suppresses various characteristic featuresof inflammation including recruitment of inflammatory cells to the lungs, airway hyper-reactivity, and airway edema.

252 Bronchial Asthma
The drugs of this class include:• Rolipram,• Cilomilast , and• RoflumilastThese drugs suppress various aspects of allergic inflammation. The orally active PDE4
selective inhibitor CDP840 when administered orally for 9.5 days, attenuates the developmentof the late asthmatic response in mild asthmatics while showing effect on the acute responseand with no significant side effects.57 The inhibition of the late asthma response is due to theiranti-inflammatory property and not due to bronchodilator action per se. Single oraladministration of the drug is without any significant bronchodilator activity. Hence, it issuggested that a mixed PDE3/4 inhibitor would seem to be a better option provided that theyhave minimal side effects. Another PDE4 inhibitor RP73401 has also no acute effect.58
Roflumilast is more potent than CDP840 acts against late asthma response, suppresses thedevelopment of bronchial hyper-responsiveness following antigen challenge in asthma, andimproves various indices of lung function, and effective in allergic rhinitis.59-61 Cilomilast,another orally active PDE4 inhibitor attenuates bronchoconstriction following exercise, andalso various features of allergic rhinitis62 and improves various pulmonary function parameters.The drug is well tolerated up to a dose of 15 mg bid.
Apart from the potential anti-inflammatory action, they perhaps act by suppression ofneural reflexes by suppressing neuropeptide release from sensory C-fibres and inhibition ofbronchoconstriction by vagal nerves. The major side effect of this class of drugs isgastrointestinal disturbances.63
REFERENCES
1. Guidelines for the management of asthma in adults. 1-Chronic persistent asthma. Statement bythe British Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:651-53.
2. Guidelines for the management of asthma in adults. 2-Acute severe asthma. Statement by theBritish Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:797-800.
3. Warner JO, Gotz M, Landau LI et al. Management of asthma: A consensus statement. Arch DisChild 1989;64;1065-79.
4. International Paediatric asthma Consensus Group. Asthma, a follow-up statement. Arch DisChild 1992;67:240-48.
5. International Consensus report on the diagnosis and management of asthma. Clin Exp Allergy1992;22(Suppl):1-72.
6. British thoracic Society and others. Guidelines for the management of asthma: A summary. BMJ1993;9:287-92.
7. The British Guidelines on Asthma Management. 1995 Review and Position Statement. Thorax1997;52(Suppl 1): S2-S8.
8. British thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, national asthma Campaign et al. Guidelines on the management ofasthma. Thorax 1993;48:S1-S24.
9. British thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, National Asthma Campaign et al. Summary charts. BMJ 1993;306:776-82.
10. Global Initiative for Asthma. A practical guide for public health officials and health care

New Treatment Modalities/Newer Drugs for Bronchial Asthma 253
professionals. US Department of Health and human services. NIH Publication No. 96-3659A,December 1995.
11. New British Guidelines on management of Asthma –Thorax 1993;58(Suppl 1):1-94.12. Shrewsbury S, Pyke S, Britton M. Meta-analysis of increased doses of inhaled steroid or addition
of salmeterol in symptomatic asthma. (MIASMA). BMJ 2000;320:1368-73.13. Pauwels RA, Lofdahl CG, Postma DS et al. Effect of inhaled formoterol and budesonide on
exacerbation of asthma. New Engl J Med 1997;337:1405-11.14. Kavuru M, Melamed J, Gross G, et al. Salmeterol and fruticasone propionate combined in a new
powder inhalation device for the treatment of asthma: A randomised, double-blind, placebo-controlled trial. J Allergy Clin Immunol 2000;105:1108-16.
15. Hansel TT, Barnes PJ (Editors); New drugs for asthma, allergy and COPD, In: Progress inRespiratory Research, 31, Basel: Karger;2001.
16. Chang TW. The pharmacological basis of anti-IgE therapy. Nature Biotechnol 2000;18:157-62.17. Presta LG, Lahr SJ, shields RL, et al. Humanisation of an antibody directed against IgE. J Immunol
1993;151:2623-32.18. Mac-Glashan DW, Bochner BS, Adel man DC, et al. Down regulation of FceR1 expression on
human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol1997;158:1438-45.
19. Boulet LP, Chapman KR, Cote J, et al. Inhibitory effects of an anti-IgE antibody E25 on allergen-induced early asthmatic response. Am J Respir Crit Care Med 1997;155:1835-40.
20. Fahy JV, Fleming HE, Wong HH, et al. The effect of an anti-IgE monoclonal antibody on theearly- and late-phase responses to allergen inhalation in asthmatic subjects. Am J Respir CritCare Med 1997;155:1828-34.
21. Coyle AJ, Wagner K, Bertrand C, et al. Central role of immunoglobulin (Ig) E in the induction oflung eosinophil infiltration and T helper 2 cell cytokine production: Inhibition by a nonanaphy-lactogenic anti-IgE antibody. J Exp Med 1996;183:1303-10.
22. Casale TB, Benstein IL, Busse WW et al. Use of an anti-IgE humanised monoclonal antibody inragweed-induced allergic rhinitis. J Allergy Clin Immunol 1997;100:110-21.
23. Milgrom H, Fick RB, Su JQ et al. Treatment of allergic asthma with monoclonal anti-IgE antibody.New Engl J Med 1999;341:1966-73.
24. Casale T, Condemi J, Miller SD et al. rhu-MAB-E25 in the treatment of seasonal allergic rhinitis(SAR). Ann Allergy Asthma Immunol 1999;82:75.
25. Boushey HA, Fick R, Fahy JV. Anti-IgE antibody. In. Hansel TT, Barnes PJ (Eds). New drugs forAsthma, Allergy and COPD, In: Progress in Respiratory Research, 31, Basel: Karger;2001,31:201-05.
26. Coffman RL, Ohara J, Bond MW et al. B cell stimulatory factor-1 enhances the IgE response oflipopolysaccharide–activated B cell. J Immunol 1986;136:4538-41.
27. Pawankar R, Okuda M, Yssel H, et al. Nasal mast cells in perennial allergic rhinitis exhibitincreased expression of the Fc epsilon R1, CD40L, IL-4, and IL-13, and can induce IgE synthesisin B cells. J Clin Invest 1997;99:1492-99.
28. Moser R, Fehr J, Bruijnzeel P. IL-4 controls the selective endothelium-driven transmigration ofeosinophils from allergic individuals. J Immunol 1992;149:1432-38.
29. Dabbagh K, Takeyama K, Lee HM, et al. IL-4 induces mucin gene expression and goblet cellmetaplasia in vitro and in vivo. J Immunol 1999;162:6233-37.
30. Hsieh CS, Hiemberger AB, Gold JS, et al. Differential regulation of T helper phenotypedevelopment by interleukins 4 and 10 in an alpha beta T-cell receptor transgenic system. ProcNatl Acad Sci USA 1992;89:6065-69.
31. Sedar RA, Paul WE, Davis MM, et al. The presence of interleukin 4 during in vitro primingdetermines the lymphokine-producing potential of CD4+T cells from T cell receptor transgenicmice. J Exp Med 1992;176:1091-98.

254 Bronchial Asthma
32. Kopf M, Le Gros G, Bachmann M et al. Disruption of the mucin IL-4 gene blocks Th2 cytokinesresponses. Nature 1993;362:245-48.
33. Vella A, Teague TK, Ihle J et al. Interleukin 4 (IL-4) or Il-7 prevents the death of resting T cells: stat6 is probably not required for the effect of IL-4. J Exp Med 1997;186:325-30.
34. Xie H, Seward RJ, Huber BT. Cytokine rescue from glucocorticoid induced apoptosis in T cells ismediated through inhibition of I kappa-Balpha. Mol Immunol 1997;34:987-94.
35. Haher S, Santos IM, Sole D, et al. Interleukin-4 and soluble CD23 serum levels in asthmaticatopic children. J Invest Allergol Clin Immunol 1995;5:251-54.
36. Walker C, Bauer W, Braun RK, et al. Activated T cells and cytokines in bronchoalveolar lavagesfrom patients with various lung diseases associated with eosinophilia. Am J Respir Crit CareMed 1994;150:1038-48.
37. Leonard C, Tormey V, Burke C, et al. Allergen-induced cytokine production in atopic diseaseand its relationship to disease severity. Am J Respir Cell Mol Biol 1997;17:368-75.
38. Shi HZ, Deng JM, Xu H, et al. Effect of inhaled interleukin–4 on airway hyper-reactivity inasthmatics. Am J Respir Crit Care Med 1998;157:1818-21.
39. Parronchi P, De Carli M, Manetti R, et al. Aberrant interleukin(IL)-4 and IL-5 production in vitroby CD4+ helper T cells from atopic subjects. Eur J Immunol 1992;22:1615-20.
40. Chan SC, Brown MA, Wilcox TM et al. Abnormal IL-4 gene expression by atopic dermatitis Tlymphocytes is reflected in altered nuclear protein interactions with IL-4 transcriptional regulatoryelement. J Invest Dermatol 1996;106:1131-1136.
41. Borish L, Agosti JM. Interleukin-4 antagonism. In: Hansel TT, Barnes PJ (Eds). New drugs forAsthma, Allergy and COPD, In: Progress in Respiratory Research;31, Basel: Karger;2001;31:256-59.
42. Borish LC, Nelson H, Lanz MJ, et al. Interleukin-4 receptor in moderate atopic asthma: A phaseI/II randomised, placebo-controlled trial. Am J Respir Crit Care Med 1999;160:1816-23.
43. Borish LC, Steinke JW, 2. Cytokines and chemokines. J Allergy Clin Immunol. 2003;111(2 Suppl):S460-75.
44. Lecki MJ, ten Brinke A, Khan J, et al. Effects of an interleukin-5 blocking monoclonal antibodyon eosinophils, airway hyper-responsiveness and the late asthmatic response. Lancet2000;356:2144-48.
45. Kipa JC, O’Conner BJ, Langley SJ et al. Results of a phase I trial with SCH55700, a humanisedanti-IL-5 antibody in severe persistent asthma (Abstract). Am J Respir Crit Care Med2000;161:A505.
46. McDonnell N, Abbott NN, Mohler KM et al. TNF antagonism. In: Hansel TT, Barnes PJ (Eds).New drugs for Asthma, Allergy and COPD, In: Progress in Respiratory Research; Basel:Karger;2001; 31:247-50.
47. Donaldson DD, Elias JA, Wills-Karp M. Interleukin-13 antagonism. In: Hansel TT, Barnes PJ(Eds). New drugs for Asthma, Allergy and COPD, In: Progress in Respiratory Research; Basel:Karger;2001,31:260-64.
48. Narula S, Cuss F. Interleukin-10. In. Hansel TT, Barnes PJ (Eds). New drugs for Asthma, Allergyand COPD, In: Progress in Respiratory Research, Basel: Karger;2001; 31:269-73.
49. De Vos C, Rihoux JP H-antihistamines. In: Hansel TT, Barnes PJ (Eds). New drugs for Asthma,Allergy and COPD, In: Progress in Respiratory Research, volume 31, Basel: Karger;2001;31:28-132.
50. McLeod RL, Egan RW, Cuss FM et al. Histamine H3 antagonists. In: Hansel TT, Barnes PJ (Eds).New drugs for Asthma, Allergy and COPD, In: Progress in Respiratory Research, Basel:Karger;2001; 31:133-36.
51. Hertz U, Gerhold K, Gruber C et al. BCG infection suppresses allergic sensitisation anddevelopment of increased airway reactivity in an animal model. J Allergy Clin Immunol1998;102:867-74.

New Treatment Modalities/Newer Drugs for Bronchial Asthma 255
52. Wang CC, Rook GA,. Inhibition of an established allergic response to ovalbumin in BALB/cmice by killed Mycobacterium vaccae. Immunology 1998;93:307-13.
53. Sur S, Wild JS, Chaudhury BK, et al. Long term prevention of allergic lung inflammation in amouse model of asthma by CpG oligodeoxynecleotides. J Immunol 1999;162:6284-93.
54. Holt PG. A potential vaccine strategy for asthma and allied atopic diseases during infancy.Lancet 1994;344:456-58.
55. Torphy TJ. Phosphodiesterase isoenzymes: Molecular targets for novel anti-asthma agents. AmJ Respir Crit Care Med 1998;157:325-32.
56. Essayan DM. Cyclic nucleotide phosphodiesterases. J Allergy Clin Immunol 2001;108:671-80.57. Harbinson PL, McLeod D, Hawksworth R, et al. The effect of a novel orally active selective
PDE4 isoenzyme inhibitor (CDP840) on allergen-induced response in asthmatic subjects. EurRespir J 1997;10:1008-14.
58. Jonker GJ, Tijhuis GJ, de Monchery JGR. RP73401 (a phosphodiesterase IV inhibitor) single dosedoes not prevent allergen induced bronchoconstriction during the early phase reaction inasthmatics. Eur Respir J 1996;9:82S.
59. Nell H, Louw C, Leichtl S, et al. Acute anti-inflammatory effect of the novel phosphodiesterase4 inhibitor roflumilast on allergen challenge in asthmatics after a single dose (abstract) Am JRespir Crit Care Med 2000;161:A200.
60. Leichtl S, Schmid-Wirlitsch C, Bredenbroker D, et al. Dose-related efficacy of once-dailyroflumilast, a new, orally active, selective phosphodiesterase 4 inhibitor, in asthma (abstract)Am J Respir Crit Care Med 2002;165:A85.
61. Schmidt BM, Kusma M, Feuring M, et al. The phosphodiesterase 4 inhibitor roflumilast is effectivein the treatment of allergic rhinitis. J Allergy Clin Immunol 2001;108:530-36.
62. Torphy TJ, Barmette MS, Underwood DC, et al. Airflow (SB 207499): A second generationphosphodiesterase 4 inhibitor for the treatment of asthma and COPD: From concept to clinic.Pulm Pharmacol Ther 1999;12:131-35.
63. Spina D. Theophylline and PD4 inhibitors in asthma. Curr Opin Pulm Med 2003;9:57-64

256 Bronchial Asthma
New Guidelines forAsthma Management
(Non-pharmacological Management)
16
The British Thoracic Society has recommended the new guidelines for the management ofbronchial asthma in 2003.1 The first British Guidelines on Asthma Management in adultswere first published in 1990 after a joint initiative between the Thoracic Society, the RoyalCollege of Physicians of London, the King’s Fund Center, and the National AsthmaCampaign. These were updated in 1993 when the addition of childhood asthma and furtherupdated in 1995. Simultaneously Guidelines were also developed by the AmericanPhysicians, by the Global Initiative for Asthma (GINA), the International Consensus Reportand the WHO. These guidelines are already discussed earlier.2-12 The Scottish IntercollegiateGuidelines Network (SIGN) published its first asthma guidelines in 199613 and hassubsequently published on primary care management of asthma in 199814 and managementof acute asthma in 1999.15 Further, both the British Thoracic Society and the SIGN haverecognised the need to update their asthma guidelines, using evidence-based methodologyto cover all aspects of asthma care. The two organisations jointly produced thecomprehensive new guideline, which was strengthened by collaboration with the NationalAsthma Campaign, the Royal College of Physicians of London, the Royal College ofPaediatrics and Child Health, General Practice Airways Group, and the British Associationof Accident and Emergency Medicine. The outcome is the new British Guideline on theManagement of Asthma.1
One important and new feature in this new guideline is the levels of evidence and gradesof recommendations used in these guideline. The panel of experts have taken into accountthe evidence-based medicine, a more scientific way of expressing a particular conclusion .16
However, it is emphasised that the grade of recommendation relates to the strength of theevidence and not necessarily the clinical importance of the recommendation in patientmanagement. Where there are only low grade recommendations in important clinical areas,this should be seen as a stimulus for further research. The recommendations levels aregraded into 8 subtypes (1++, 1+, 1-, 2++, 2+, 2-, 3, and 4) depending on whether the inferencefrom high-level meta-analyses, systemic review of randomised controlled-trials (RCTs),RCTs with a very low-risk of bias, well-conducted meta-analysis, systemic reviews, RCTswith low-risk bias, meta-analyses, systemic reviews, or RCTs with high-risk of bias, Highquality systemic reviews of case-control or cohort studies, High quality case control or

New Guidelines for Asthma Management (Non-pharmacological Management) 257
cohort studies with a very low-risk of confounding or bias or a high probability that therelationship is causal, well conducted case control or cohort studies with low-risk ofconfounding or bias and a moderate probability that the relationship is causal, case controlor cohort studies with a high-risk of confounding or bias and a significant risk that therelationship is not causal, non-analytical studies like case reports, case series, and expertopinion. The grades of recommendations are from A-D depending on the levels of evidence.Good practice points are also given based on clinical experience of the guideline developmentgroup.
NON-PHARMACOLOGICAL MANAGEMENT
There is increasing interest in factors which, if avoided might facilitate the management ofasthma, reducing the requirement for pharmacotherapy, and which may have the potentialto modify fundamental causes of asthma. However, evidence has been difficult to obtainfor many approaches and more studies are required.
Primary Prophylaxis
Primary prophylaxis is employed before there is any evidence of disease in an attempt toprevent its onset. A number of potential strategies are suggested.
Allergen Avoidance
There is a strong correlation between allergic sensitisation to common aeroallergens andthe subsequent development of asthma. There is also a strong association between allergenexposure in early life and sensitisation to these allergens, although it has not been possibleto demonstrate an association between allergen exposure and the development of asthma.Majority of allergen avoidance studies focus on dietary manipulation to prevent atopiceczema and have paid little attention to aeroallergen avoidance. Two trials in progress areinvestigating the consequences of introducing house dust mite reduction in early pregnancy,and are following up the children born to the participating mothers. Although accurateasthma phenotyping is not possible in infancy, outcomes at one year of age indicate a modestbut significant reduction in wheezing illnesses.
Allergen avoidance after birth has been studied in a number of controlled (but no doubleblind) trials. There appears to be a transient reduction in the prevalence of atopic eczemain the first two years of life but no evidence of sustained benefit in relation to asthma. Anumber of epidemiological studies suggest that close contact with a cat or dog in very earlyinfancy reduced subsequent prevalence of allergy and asthma. This may be a consequenceof high allergen exposure inducing tolerance.
However, no recommendations on prenatal or postnatal allergen avoidance can be madein relation to primary prevention of asthma.
Breastfeeding
A systematic review and meta-analysis involving 8183 subjects followed for a mean of fouryears revealed a significant protective effect of breastfeeding against the development ofasthma. The effect was greatest in children with a family history of atopy. In contrast, amore recent study in 1246 patients found that breastfeeding was associated with a reduced

258 Bronchial Asthma
risk of infant wheeze, but also with an increased risk of asthma at six years. Thus, breast-feeding should be encouraged and its benefits include a protective effect in relation to earlylife wheezing.
Modified Infant Milk Formulae
Trials of modified milk formulae using partial and extensive hydrolysates of whey or caseinor soy formulae compared with conventional formulae have not shown any consistentsignificant long-term benefits in relation to asthma. Variation in study design, interventionused, co-interventions and outcome definition make meta-analysis problematical.
Other Dietary Modifications
Limited epidemiological evidence suggests that fish oil consumption may protect againstasthma in childhood. Trials of lipid supplementation during pregnancy and postnatally toprevent atopic disease are in progress.
Microbial Exposure
The “hygiene hypothesis” suggests that early exposure to microbial products will switchoff allergic responses preventing allergic diseases such as asthma. Epidemiological studiescomparing large populations who have or have not had such exposures support thehypothesis. A double blind placebo trial of the probiotic, lactobacillus CG, reported a reducedincidence of atopic eczema but no effect on IgE antibody sensitisation. Small sample sizeand early outcome age limit the interpretation of this study. In the absence of good qualityintervention studies, no recommendation can be made at present.
Immunotherapy and Primary Prevention
Three observational studies, in over 8000 patients, have shown that immunotherapy inindividuals with a single allergy reduces the numbers subsequently developing new allergiesover a three to four years follow-up compared with contemporaneous untreated controls.No double blind placebo controlled trials of immunotherapy as primary prevention havebeen conduced, and at present immunotherapy cannot be recommended for primaryprevention. Preliminary results from an ongoing parallel group study using contempo-raneous untreated children as the control group for pollen immunotherapy in childrenwith allergic rhinitis suggest a lower rate of onset of asthma in the treated group.
Avoiding Pollutants
No evidence was found to support a link between exposure to environmental tobacco smokeand other air pollutants and the induction of atopic asthma. An early meta-analysis suggestedan association between gas cooking and respiratory illness, but this has not been broughtout in larger studies. Increased risk of infant wheeze is associated with smoking duringpregnancy and maternal postnatal smoking. Pregnancy smoking affects an infant’s airwayfunction, increasing susceptibility to wheeze. There are many other adverse effects on theyoung child of such exposures.
Pharmacotherapy
There are some pharmacological trials of treatments designed to prevent onset of the disease.Children given ketotifen (206 infants, in two trials) showed significantly less asthma at one

New Guidelines for Asthma Management (Non-pharmacological Management) 259
and three years follow-up compared with those receiving placebo. In the third study, usingcetirizien, 18 months’ treatment had no effect in the intention to treat population butsignificantly reduced asthma in children with atopic dermatitis sensitised to either grasspollen or house dust mite. Cetirizien had additional benefits for atopic dermatitis alone andreduced the frequency of urticaria.
Secondary Prophylaxis
Allergen Avoidance
Allergen avoidance measures may be helpful in reducing the severity of existing disease.Increasing allergen exposure in sensitised individuals is associated with an increase in asthmasymptoms, bronchial reactivity and deterioration in lung function. Treatment requirements,hospital attendance and respiratory arrest are associated with increased exposure to highconcentrations of indoor allergens.
Threshold concentrations of allergens that can be regarded as risk factors for acute attacksinclude:
• 10 μg/g dust of group l mite allergen• 8 μg/g dust of Fel d l, the major cat allergen• 10 μg/g dust of Can f l, the major dog allergen• 8 μg/g dust of cockroach allergen.Evidence that reducing allergen exposure can reduce morbidity and mortality is tenuous.
In uncontrolled studies, children and adults have both shown benefit from exposure to a verylow allergen environment. However, the benefits in such circumstances cannot be necessarilyattributed to the allergen avoidance.
House dust mite control measures There have been two Cochrane reviews on house dustmite control measures and the management of asthma. The first concluded that currentchemical and physical methods were ineffective and could not be recommended asprophylactic treatment for asthma patients with sensitivity to house dust mites. An amendmentconcluded that physical reduction methods may reduce asthma symptoms. The reviewedstudies used various chemical, physical or combinations of methods to reduce mite exposure.The combined meta-analysis showed no difference in improvement in asthma between patientsin experimental groups compared with controls. There was heterogeneity between studieswith regard to intervention, and in some studies intervention allocation was not adequatelyconcealed. Larger and more carefully controlled studies are required to demonstrate any clearbenefit from house dust mite avoidance. At present, this does not appear to be a cost-effectivemethod of achieving benefit.
In committed families with evidence of house dust mite allergy and who wish to try miteavoidance, the following are recommended.
• Complete barrier bed covering systems• Removal of carpets• Removal of soft toys from bed• High temperature washing of bed linen• Acaricides to soft furnishings• Dehumidification

260 Bronchial Asthma
Other allergens Animal allergens, particularly cat and dog, are a potent cause of asthmasymptoms. Observational studies have not found that removing a pet from a home improvesasthma control. In a study in adults with cat sensitivity, randomisation to either bedroom aircleaner and covers for bedding or no active intervention with restriction of cats away from thebedroom, resulted in no differences between groups with regard to symptoms, peak flow,lung function of bronchial reactivity. Alternatively, there is a suggestion that maintaining ahigh exposure to cat allergen in the domestic environment might actually induce some degreeof tolerance. Many experts still feel that removal of pets from the home of individuals withasthma who also have an allergy to that pet should be recommended. Cockroach allergy is nota common problem in some countries like UK, but important in some other areas. There is noconclusive evidence regarding the impact of cockroach allergen reduction on asthmasymptoms. Although fungal exposure has been strongly associated with hospitalisation andincreased mortality in asthma, to date no controlled trials have addressed fungal exposurereduction and asthma.
Environmental Factors
Smoking The association between passive smoking and respiratory health has beenextensively reviewed. There is a direct causal relationship between parental smoking andlower respiratory illness in children up to three years of age. Infants whose mothers smokeare four times more likely to develop wheezing illnesses in the first year of life. Theindependent contributions of prenatal and postnatal maternal smoking to the developmentof asthma in children are difficult to distinguish. Maternal pregnancy smoking has beenshown to have an adverse influence on lung development. There is little evidence thatmaternal pregnancy smoking has an effect on allergic sensitisation. Exposure to tobaccosmoke in the home contributes to the severity of childhood asthma. A US Institute ofMedicine review identified a causal relationship between environmental tobacco smoke(ETS) exposure and exacerbations of asthma in pre-school children. Average exposure isassociated with a 30% increased risk of symptoms. One small study suggests that by stoppingsmoking, parents decrease the severity of asthma in their children. Parents who smokeshould be advised about the dangers for themselves and their children and offeredappropriate support to stop smoking.
Starting smoking as a teenager increases the risk of persisting asthma. Only one studywas identified that examined the incidence of asthma related to taking up smoking. Thisshowed a relative risk of 2.1 for the development of asthma over six years in 14 years oldchildren who have started to smoke. No studies were identified that directly related to thequestion of whether smoking affects asthma severity. One controlled cohort study suggestedthat exposure to passive smoke at home delayed recovery from an acute asthma attack.Studies of interventions designed to reduce environment tobacco smoke exposure in thehome have been largely ineffective in reducing the degree of exposure and none weredesigned with primarily clinical (as opposed to smoking outcomes. In one observationalstudy giving up smoking in adults was associated with improved severity of asthma scores.Smoking cessation should be encouraged as it is good for general health and may decreaseasthma severity.
Air pollution There is evidence that changing from a high particulate sulphur dioxide(coal burning) environment to a low sulphur dioxide/high diesel particulate environment

New Guidelines for Asthma Management (Non-pharmacological Management) 261
increases the incidence of asthma and atopy. In the UK, asthma is more prevalent in 12-14years olds in non-metropolitan rather than metropolitan areas. However, many differencesbetween environments might explain the variation in asthma and allergy risk. There issome laboratory evidence that various pollutants can enhance the response of patients withasthma to allergens, but there is no firm epidemiological evidence that this has occurred inthe UK or elsewhere.
Time series studies suggest that air pollution may provoke acute asthma attacks oraggravate existing chronic asthma, although the effects are minimal in comparison withfactors such as infection. The short-term fluctuations in levels of air pollution currentlyencountered in the UK may be responsible for small changes in numbers of hospitaladmissions and emergency attendances for asthma.
No evidence was identified regarding asthma and indoor air pollutants, such as volatileorganic compounds, formaldehyde or nitrogen oxides. Further research in this area isrequired.
Complementary and Alternative Medicine
Herbal and Traditional Chinese Medicine
Currently available evidence does not allow any firm judgment to be made on herbalremedies in general or individual preparations in particular. Seventeen trials were identifiedbut the combined results are inconclusive. Nine of the 17 trials reported some improvementin lung function, but it is not clear that the results reported would be generalised.
Acupuncture
A Cochrane review of 21 trials raised many methodological concerns. Only seven trials(174 patients) achieved randomisation to active (i.e. recognised in traditional Chinesemedicine to be of benefit in asthma) or sham acupuncture (i.e. points with no recognisedactivity) for the treatment of persistent or chronic asthma. Binding was a common problem,and only achieved for those making the observations. The difficulty in making shamacupuncture convincing and part of the holistic approach of traditional Chinese medicinewas emphasised. There was wide inconsistency in methodology. Acute trials show thatacupuncture has a beneficial effect, but this is less in magnitude than that achieved byinhaled bronchodilators or cromones. Demonstrating that this effect can be transferred topersistent asthma using regular treatment was achieved in one RCT reported in the Cochranereview. The Cochrane review found no evidence for a clinically valuable benefit fromacupuncture, with no statistically significant improvement in lung function being demons-trated. More rigorous research methodology and attention to outcomes other than lungfunction are required.
Air Ionisers
Ionisers are widely advertised and marketed as being of benefit to patients with asthma,however, there is no evidence that they are of value in ameliorating the symptoms of asthmaor improving lung function. They do reduced mite allergen levels in the room in whichthey are used, and could be incorporated into a coordinated allergen avoidance programme,but this has not been formally tested. One study has raised concerns that ionisation may

262 Bronchial Asthma
produce an increase in nocturnal cough. The use of ionisers cannot be encouraged, as thereis no evidence of benefit and a suggestion of adverse effect.
Homeopathy
A Cochrane review identified only three methodologically sound randomised controlledtrials. In the first trials (24 patients), homeopathy improved symptom scored and forced vitalcapacity (FVC) but had no effect on FEV1 or bronchial reactivity. The second studydemonstrated improvements in both active and placebo groups. The third, poorly reported,trial demonstrated an increase in lung function in patients receiving the active preparation.There is insufficient information regarding the value of homeopathy in the treatment ofasthma. Large well designed trials using defined remedies and a spectrum of patients arewarranted.
Hypnosis
Studies of hypnosis in patients with asthma are generally poorly controlled and patientcharacteristics and outcome measured vary enormously. The conclusions from a criticalreview were that hypnosis may be effective for asthma with the biggest effect in susceptiblesubjects, but more randomised and appropriately controlled studies are required.
Manual Therapy including Massive and Spinal Manipulation
A Cochrane review identified four relevant randomised controlled trials. The two trials ofchiropractice suggest that there is no place for this modality of treatment in the managementof asthma. No conclusions can be drawn on massage therapy.
Physical Exercise Training
A Cochrane review has shown no effect of physical training on PEF, FEV1, FVC or VEmax.However, oxygen consumption, maximum heart rate, and work capacity all increasedsignificantly. Most studies discussed the potential problems of exercise-induced asthma,nut none made any observations on this phenomenon. As physical training improves indicesof cardiopulmonary efficiency, it should be seen as part of a general approach to improvinglifestyle and rehabilitation in asthma, with appropriate precautions advised about exercise-induced asthma.
Breathing Exercise Including Yoga and Buteyko
The underlying principle of Yoga and Buteyko is to reduce hyperventilation by loweringrespiratory frequency. A Cochrane review found no change in routine measures of lungfunction. Two studies reported a reduction in use of medication, and two a reducedfrequency of attacks. At present it is not possible to make an evidence-based recommendationabout breathing exercises for asthma.
Family Therapy
A Cochrane review identified two trials, in 55 children showing that family therapy may bea useful adjunct to medication in children with asthma. Small study size limits therecommendations.

New Guidelines for Asthma Management (Non-pharmacological Management) 263
Dietary Manipulation
Minerals
Low magnesium intakes have been associated with higher prevalence of asthma. Anintervention study of magnesium supplementation has suggested a reduced rate of bronchialhyperresponsiveness and wheeze. Studies of sodium and antioxidant supplements such asselenium and vitamin C have produced little or no evidence of benefit amongst patients withasthma.
Fish Oils and Fatty Acids
In vitro studies suggest that supplementing diet with the omega n-3 fatty acids foundpredominantly in fish oils might reduce the inflammation associated with asthma. Controlledclinical studies in small numbers have on the whole been negative, with a Cochrane reviewconcluding that there was little evidence to recommend fish oil supplements in asthma.
Weight Reduction in Obese Patients with Asthma
A small randomised parallel group study has shown improved asthma control followingweight reduction in obese patients with asthma.
REFERENCES
1. New British Guidelines on Management of Asthma. Thorax 1993;58(Suppl 1):1-94.2. Guidelines for the Management of Asthma in Adults. 1-Chronic persistent asthma. Statement
by the British Thoracic Society, Research Unit of the Royal College of Physicians of London,King’s Fund Center, National Asthma Campaign. BMJ 1990;301:651-53.
3. Guidelines for the Management of Asthma in Adults. 2-Acute severe asthma. Statement by theBritish Thoracic Society, Research Unit of the Royal College of Physicians of London, King’sFund Center, National Asthma Campaign. BMJ 1990;301:797-800.
4. Warner JO, Gotz M, Landau LI, et al. Management of Asthma: A consensus statement. Arch DisChild 1989;64;1065-79.
5. International Paediatric Asthma Consensus Group. Asthma, a follow-up statement. Arch DisChild 1992;67:240-48.
6. International consensus report on the diagnosis and management of asthma. Clin Exp Allergy1992;22(Suppl):1-72.
7. British Thoracic Society and others. Guidelines for the management of asthma: A summary.BMJ 1993;9:287-92.
8. The British Guidelines on Asthma Management. 1995 Review and Position Statement. Thorax1997;52(Suppl 1): S2-S8.
9. British Thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, National Asthma Campaign, et al. Guidelines on the management ofasthma. Thorax 1993;48:S1-S24.
10. British Thoracic Society, British Paediatric Association, Royal College of Physicians of London,The King’s Fund Center, National Asthma Campaign, et al. Summary charts. BMJ 1993;306:776-82.
11. Global Initiative for Asthma. A practical guide for public health officials and health careprofessionals. US Department of Health and human services. NIH Publication No.1995;96:3659A,.
12. National Asthma Education and Prevention Program. Expert Panel Report II: Guidelines for thediagnosis and management of asthma. Bethesda, MD: National Institute of Health. 1997.

264 Bronchial Asthma
13. Scottish Intercollegiate Guidelines Network (SIGN). Hospital inpatient management of acuteasthma attack. SIGN Publication No. 6, Edinburgh, SIGN, 1996.
14. Scottish Intercollegiate Guidelines Network (SIGN). Primary care management of asthma. SIGNPublication No. 33, Edinburgh, SIGN, 1998.
15. Scottish Intercollegiate Guidelines Network (SIGN). Emergency management of acute asthma.SIGN Publication No. 38, Edinburgh, SIGN, 1999.
16. Harbour R, Miller J. A new system for grading recommendations in evidence based guidelines.BMJ 2001;323:334-36.

New Guidelines for Asthma Management (Pharmacological Management) 265
New Guidelines forAsthma Management
(Pharmacological Management)
17
The aims of pharmacological management of asthma are:• The control of symptoms, including nocturnal symptoms and exercise-induced asthma• Prevention of exacerbations• Achievement of best possible pulmonary function• With minimal side effects.
It is not appropriate to define a fixed level of lung function or symptom control whichmust be achieved, as individual patients will have different goals and may also wish tobalance these aims against the potential side effects or inconvenience of taking the medicationnecessary to achieve “perfect” control. In general terms, control of asthma is assessed againstthese standards:• Minimal symptoms during day and night• Minimal need for reliever medication• No exacerbations• No limitation of physical activity• Normal lung function (in practical terms FEV1 and/or PEF>80% predicted or best)
A stepwise approach aims to abolish symptoms as soon as possible and to optimise peakflow by starting treatment at the level most likely to achieve this. Patients should starttreatment at the step most appropriate to the initial severity of their asthma. The aim is toachieve early control and to maintain control by stepping up treatment as necessary andstepping down when control is good. Before initiating a new drug therapy practitionersshould check compliance with existing therapies, inhaler technique and eliminate triggerfactors.
All doses of inhaled steroids in this section refer to beclomethasone (BDP) given via ametered dose inhaler (pMDI). Adjustment may be necessary for fluticasone and/or otherdevices.
STEP 1: MILD INTERMITTENT ASTHMA
The following medicines act as short acting bronchodilators:• Inhaled short acting β2-agonists• Inhaled ipratropium bromide

266 Bronchial Asthma
• β2-agonist tablets or syrup• Theophyllines
Short acting inhaled β2-agonists work more quickly and/or with fewer side effects than thealternative. An inhaled short acting β2-agonist should be prescribed as short-term reliever therapy forall patients with symptomatic asthma.
Frequency of Dosing of Inhaled Short Acting βββββ2-Agonists
There is no consistent evidence of any benefit or harm from regular (four times daily) use ofshort acting β2-agonists compared with “as required” (sos) use. Unless individual patientsare shown to benefit from regular use of inhaled short acting β2-agonists, then “as required”use is recommended.
Using two or more canisters of β2-agonists per month or > 10-12 puffs per day is a markerof poorly controlled asthma. Patients with high usage of inhaled short acting β2-agonistsshould have their asthma management reviewed.
STEP 2: INTRODUCTION OF REGULAR PREVENTER THERAPY
For steps 2, 3 and 4, treatments have been judged on their ability to improve symptoms,improve lung function, and prevent exacerbations, with an acceptable safety profileimprovement of quality of life while important is the subject of two few studies to be usedto make recommendations at present.
Inhaled Steroids
Inhaled steroids are the most effective preventer drug for adults and children for achievingoverall treatment goals. They are the recommended preventer drug for adults and childrenfor achieving overall treatment goals. The threshold for introduction of inhaled steroidshas never been firmly established. There is strong evidence that patients requiring shortacting β2-agonists more than two to three times a day should be treated with inhaled steroids,but patients with lower inhaler requirements may also benefit. Inhaled steroids should bestarted for patients with;• Recent exacerbations,• Nocturnal asthma,• Impaired lung function, or• Use of inhaled β2-agonists more than once a day.
Starting Dose of Inhaled Steroids
In mild to moderate asthma, starting at very high doses of inhaled steroids and steppingdown confers no benefit. Start patients at a dose of inhaled steroids appropriate to theseverity of disease. In adults, a reasonable starting dose will usually be 400 μg per day andin children 200 μg per day. In children under 5 years of age, higher doses may be requiredif there are problems in obtaining consistent drug delivery. The dose is to be titrated to thelowest dose at which effective control of asthma is maintained.
Frequency of Dosing of Inhaled Steroids
Current inhaled steroids are slightly more effective when taken twice rather than oncedaily. There is little evidence of benefit for dosage frequency more than twice daily. Initially,

New Guidelines for Asthma Management (Pharmacological Management) 267
the inhaled steroids are to be given twice daily. Once a day inhaled steroids at the same totaldaily dose can be considered if good control is established.
Safety of Inhaled Steroids
The safety of inhaled steroids is of crucial importance and a balance between benefits andrisks for each individual needs to be assessed. Account should be taken of other topicalsteroid therapy when assessing systemic risk. There is little evidence that doses below 800 μgday cause any short-term detrimental effects apart from the local side effects of dysphoniaand oral candidiasis. However, the possibility of long-term effects on bone has been raised.Cross-sectional studies have shown possible dose related reduction in bone density. Otherstudies have shown effects on adrenocortical function of uncertain significance.
In children, inhaled stereoids of 400 μg day of beclomethasone dipropionate or equivalentmay be associated with systemic side effects like growth retardation, and adrenal suppression.The later may manifest as hypoglycaemic episodes. The smallest dose of inhaled steroidsrequired to maintain adequate asthma control must be used. At higher doses, add-on therapylike long acting β2-agonists should be considered. The height of the children should bemonitored on a regular basis.
Comparison of Inhaled Steroids
Many studies comparing different inhaled steroids are of inadequate design and have beenomitted from further assessment. In view of the clear differences between normal volunteersand asthma patients in the absorption of inhaled steroids, data from normal volunteers havenot been taken into account. Only studies in which more than one dose of at least one of theinhaled steroids or both safety and efficacy has been studied together in the same trial wereevaluated. Non-blinded studies also has to be considered because of the problems of obtainingcompetitors’ delivery devices. All comparison used BDP-CFC (chlorofluoro-carbons) as thereference.
Beclomethasone dipropionate (BDP) and budesonide are approximately equivalent inclinical practice although there may be variations with different delivery devices. There islimited evidence from two open studies of less than ideal design that budesonide via theturbohaler is more clinically effective. However, at present a 1:1 ratio should be assumedwhen changing between BDP and budesonide. Fluticasone provides equal clinical activity toBDP and budesonide at half the dosage. The evidence that it causes fewer side effects at doseswith equal clinical effect is limited.
Other Preventive Therapies
Inhaled steroids are the first choice preventive drugs. Alternative, less effective preventivetherapies in patients taking short acting β2-agonists alone are:• Cromones (have an inconvenient dosing frequency).• Sodium cromoglycate is ineffective in children.• Nedocromil sodium is of benefit in 5-12 years old.• Leukotriene receptor antagonists have some beneficial effect (side effects are common and
monitoring of plasma levels is required).• Long acting inhaled β2-agonists have some beneficial effects but they are not recommended
as first line preventive therapy.• Antihistamines and ketotifen are ineffective.

268 Bronchial Asthma
STEP 3: ADD-ON THERAPY
Before initiating a new drug therapy one should recheck compliance, inhaler technique andeliminate trigger factors. The duration of a trial of add-on therapy will depend on the desiredoutcome. For instance, preventing nocturnal awakening may require a relatively short trial oftreatment (days or weeks), whereas preventing exacerbations of asthma or decreasing steroidtablet use may require a longer trial of therapy (weeks or months). If there is no response totreatment the drug should be discontinued.
CRITERIA FOR INTRODUCTION OF ADD-ON THERAPY
No exact dose of inhaled steroid can be determined, the correct dose at which to add anothertherapy. The addition of other treatment options to inhaled steroids has been investigatedat doses from 200-1000 μg in adults and up to 400 μg in children. Many patients will benefitmore from add-on therapy than from increasing inhaled steroids above doses as low as200 μg/day. Furthermore, at doses of inhaled steroid above 800 μg/day side effects becomemore frequent. An absolute threshold for introduction of add-on therapy in all patientscannot be defined. Thus, one should carry out a trial of other treatment before increasingthe inhaled steroid dose above 800 μg/day in adults and 400 μg/day in children.
Add-On Therapy
Option for add-on therapy are summarised in Figure 17.1. In adult patients taking inhaledsteroids at doses of 200-800 μg/day and in children taking inhaled steroids at a dose of400 μg/day the following interventions are of value.
First choice would be the addition of an inhaled long acting β2-agonists (LABA), whichimproves lung function and symptoms, and decreases exacerbation. The first choice asadd-on therapy to inhaled steroids in adults and children (5-12 years) is an inhaled longacting β2-agonists. If, as may happen occasionally, there is no response to inhaled longacting β2-agonist, the LABA should be stopped and the dose of inhaled steroid to be increasedto 800 μg/day (adults) or 400 μg/day (children) if not already on this dose. If there is aresponse to LABA, but control remains poor, one should continue with the LABA andincrease the dose of inhaled steroid to 800 μg/day (adults) or 400 μg/day (children 5-12years).• Leukotriene receptor antagonists provide improvement in lung function, a decrease in
exacerbations, and an improvement in symptoms.• Theophylline improves lung function and symptoms, but side effects occur more
commonly.• Slow release β2-agonists tablets also improve lung function and symptoms, but side effects
occur more commonly.If control is still inadequate after a trial of LABA and after increasing the dose of inhaled
steroid, one may consider a sequential trial of add-on therapy, i.e. leukotriene receptorantagonists, theophyllines, slow release β2-agonist tablets in adults.
Addition of anticholinergics is generally of no value. Addition of cromones is of marginalbenefit. In patients on inhaled steroids whose asthma is stable, no intervention has beenconsistently shown to decrease inhaled steroid requirement in a clinically significant mannercompared to placebo.

New Guidelines for Asthma Management (Pharmacological Management) 269
Combination Inhalers
There is no difference in efficacy in giving inhaled steroid and long acting β2-agonist incombination or in separate inhalers.
STEP 4: POOR CONTROL ON MODERATE DOSE OF INHALED STEROID +ADD-ON THERAPY: ADDITION OF FOURTH DRUG
In a small proportion of patients asthma is not adequately controlled on a combination ofas required short acting β2-agonist, inhaled steroid (800 μg/day), and an additional drug,usually a long acting β2-agonist. There are very few clinical trials in this specific patientgroup to guide management. The following recommendations are based on extrapolationfrom trials of add-on therapy to inhaled steroids and on previous guidelines.
If control remains inadequate on 800 μg daily in adults and 400 μg/day in children, of aninhaled steroid plus a long acting agonist, β2-agonist, the following interventions are to beconsidered:
• Increase the inhaled steroids to 2000 μg/day in adults or 800 μg/day in children of5-12 years of age.
• Leukotriene receptor antagonists• Theophyllines• Slow release β2-agonist tablets, (caution needs to be taken in patients on long acting
β2-agonist)
Inadequate control on low-dose inhaled steroids
Add inhaled long-acting β2-agonist (LABA)
Assess control of asthma
Good response to LABA and Benefit from LABA, but control still No response to LABA:good control: inadequate: • Stop LABA• Continue LABA • Continue LABA and • Increase inhaled steroid
• Increase inhaled steroid dose to dose to 800 µg / day800 µg/day (adults) and 400 µg/ (adults) and 400 µg/dayday (children, 5-12 years) (children 5-12 years)
If control still inadequate, go Control still inadequate: Trial ofto step 4 other add-on therapy, e.g. leuko-
triene receptor antagonist ortheophylline
If control still inadequatego to step 4
Fig. 17.1: Summary of step 3: add-on therapy

270 Bronchial Asthma
There is no control trial indicating which of these is the best option. If a trial of an add-ontreatment is ineffective, the drug is to be stopped, or in case of increased dose of inhaledsteroid, the dose is to be reduced to the original dose). Before proceeding to step 5, the physicianshould consider referring the patient with inadequately controlled asthma, especially children,to specialist care.
STEP 5: CONTINUOUS OR FREQUENT USE OF ORAL STEROIDS
Prevention and Treatment of Steroid Tablet-induced Side Effects
Patients on long-term steroid tablets (e.g. longer than three months) or requiring frequentcourses of steroid tablets (e.g. three to four per year) will be at risk of systemic side effects• Blood pressure should be monitored• Diabetes mellitus may occur• Osteoporosis commonly occurs and should be monitored and treated.• Growth should be monitored in children• Cataracts should be screened for in children
Steroid Tablet—Starting Medication
The aim of treatment is to control the asthma using the lowest possible dose, or if possible, tostop long-term steroid tablets completely. Inhaled steroids are the most effective drug fordecreasing requirement for long-term steroid tablets. There is limited evidence for the abilityof long acting β2-agonists, theophyllines, or leukotriene receptor antagonists to decrease therequirements for steroid tablets, but they may improve symptoms and pulmonary function.
In adults, the recommended method of eliminating or reducing the dose of steroid tablets isinhaled steroids, at doses of up to 2000 μg/day if required. In children of aged 5-12 years,doses above 1000 μg/day should be added cautiously. There is a role for a trial of treatment inadults with long acting β2-agonists, leukotriene receptor antagonists, and theophyllines forabout six weeks. They should be stopped if no improvement in steroid dose, symptoms orlung function is detected.
Immunosuppressants (methotrexate, cyclosporin and oral gold) decrease long-term steroidtablet requirements but all have significant side effects. There is no evidence of persistingbeneficial effect after stopping them; and there is marked variability in response.Immunosuppressants (methotrexate, cyclosporin and oral gold) may be given as a three monthtrial, once other drug treatments have proved unsuccessful. Their risks and benefits should bediscussed with the patient and their side effects carefully monitored. Treatment should be ina center with experience of using these medicine. Colchicine and intravenous immunoglobulinhave not been shown to have any beneficial effect in adults.
Continuous subcutaneous terbutaline infusion has been reported to be beneficial in severeasthma but efficacy and safety have not been assessed in randomised controlled trials.
Steroid Formulations
Prednisolone is the most widely used steroid for maintenance therapy in chronic asthma.There is no evidence that other formulations offer any advantage. Although popular in

New Guidelines for Asthma Management (Pharmacological Management) 271
paediatric practice, there are no studies to show whether alternate day steroids produce fewerside effects than daily steroids.
βββββ-Blockers
β-blockers, including eye drops, are contraindicated in patients with asthma.
Stepping Down
Stepping down treatment once asthma is controlled is recommended, but often notimplemented leaving some patients over treated. There is little evidence regarding the mostappropriate way to step down treatment. Regular review of patients as treatment is steppeddown is important. When deciding which drug to step down first and at what rate, theseverity of asthma, the side effects of the treatment, the beneficial effect achieved, and thepatient’s preference should all be taken into account. Patients should be maintained at thelowest possible dose of inhaled steroid. Reduction in inhaled steroid dose should be slow aspatients deteriorate at different rates. Reductions should be considered every three months,decreasing the dose by approximately 25-50% each time.
SPECIFIC MANAGEMENT PROBLEMS
Onset of Exacerbation Asthma
Although, recommended for both adults and children in previous guidelines and as part ofasthma action plants, doubling the dose at the time of an exacerbation is of unproven value.In adult patients on a low dose (220 μg) of inhaled steroids, a five-fold increase in dose at thetime of exacerbation leads to a decrease in the severity of exacerbations. This five-fold increaseshould not be extrapolated to higher doses of inhaled steroids.
Exercise-induced Asthma
For most patients exercise-induced asthma is an expression of poorly controlled asthma andregular treatment including inhaled steroids should be reviewed.
The following medicines give protection against exercise-induced asthma:• Inhaled steroids• Short acting β2-agonists• Long acting β2-agonists• Theophyllines• Leukotriene receptor antagonists• Cromones• β2-agonist tablets
The following medicines do not give protection against exercise-induced asthma at normaldoses:• Anticholinergics• Ketotifen• Antihistamines
Long acting β2-agonists and leukotriene antagonists provide more prolonged protection

272 Bronchial Asthma
than short acting β2-agonists, but a degree of tolerance develops with LABA particularly withrespect to duration of action. No tolerance has been demonstrated with leukotriene receptorantagonists.
If exercise is a specific problem in patients taking inhaled steroids who are otherwise wellcontrolled, the following therapies are to be considered:• Leukotriene receptor antagonists• Long acting β2-agonists• Cromones `• Oral β2-agonists• Theophyllines
Immediately before exercise, inhaled short acting β2-agonists are the drug of choice.
Rhinitis
Patients with asthma often have rhinitis. The most effective therapy is intranasal steroids.Treatment of allergic rhinitis has not been shown to improve asthma control.
Allergic Bronchopulmonary Aspergillosis
In adult patients with allergic bronchopulmonary aspergillosis (ABPA), itraconazole maydecrease steroid tablet dose and improve asthma control. In adult patients with ABPA, afour-month trial of itraconazole should be considered. Careful monitoring of side effects,particularly hepatic dysfunction, is recommended.
Aspirin Intolerant Asthma
There are theoretical reasons to suggest that leukotriene receptor antagonists might be ofparticular value in the treatment of aspirin intolerant asthma. However, there is littleevidence to justify managing patients with aspirin intolerant asthma in a different mannerto patients tolerant of aspirin, apart from the rigorous avoidance of non-steroidal anti-inflammatory medications.
NOVEL THERAPIES
Anti-IgE Monoclonal Antibody
In highly selected patients an anti-IgE monoclonal antibody has some beneficial effect, butits role in the stepwise treatment of asthma is unclear. At present this drug does not have alicense in many countries.
Mometasone
Mometasone is a new inhaled steroid and the relatively limited number of studies suggestsit is equivalent to twice the dose of BDP-CFC. The relative safety of mometasone is not fullyestablished.
Tiotropium Bromide
Tiotropium bromide is a once daily long acting anticholinergic agent. Its value in the treatmentof asthma has not been evaluated.
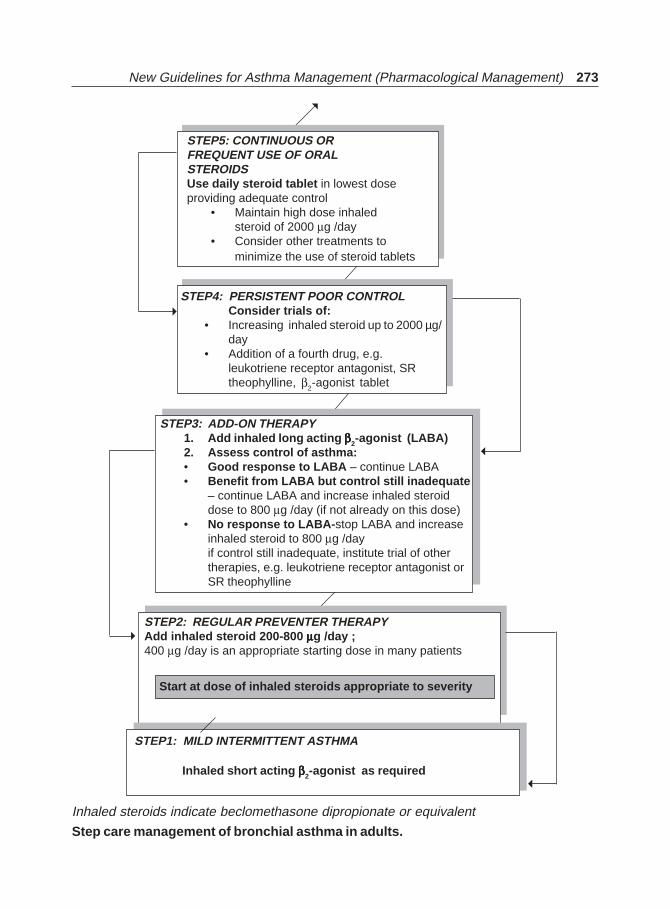
New Guidelines for Asthma Management (Pharmacological Management) 273
STEP5: CONTINUOUS ORFREQUENT USE OF ORALSTEROIDSUse daily steroid tablet in lowest doseproviding adequate control
• Maintain high dose inhaledsteroid of 2000 μg /day
• Consider other treatments tominimize the use of steroid tablets
STEP4: PERSISTENT POOR CONTROLConsider trials of:
• Increasing inhaled steroid up to 2000 µg/day
• Addition of a fourth drug, e.g.leukotriene receptor antagonist, SRtheophylline, β2-agonist tablet
STEP3: ADD-ON THERAPY1. Add inhaled long acting βββββ2-agonist (LABA)2. Assess control of asthma:• Good response to LABA – continue LABA• Benefit from LABA but control still inadequate
– continue LABA and increase inhaled steroiddose to 800 μg /day (if not already on this dose)
• No response to LABA-stop LABA and increaseinhaled steroid to 800 μg /dayif control still inadequate, institute trial of othertherapies, e.g. leukotriene receptor antagonist orSR theophylline
STEP2: REGULAR PREVENTER THERAPYAdd inhaled steroid 200-800 μμμμμg /day ;400 μg /day is an appropriate starting dose in many patients
Start at dose of inhaled steroids appropriate to severity
STEP1: MILD INTERMITTENT ASTHMA
Inhaled short acting βββββ2-agonist as required
Inhaled steroids indicate beclomethasone dipropionate or equivalent
Step care management of bronchial asthma in adults.
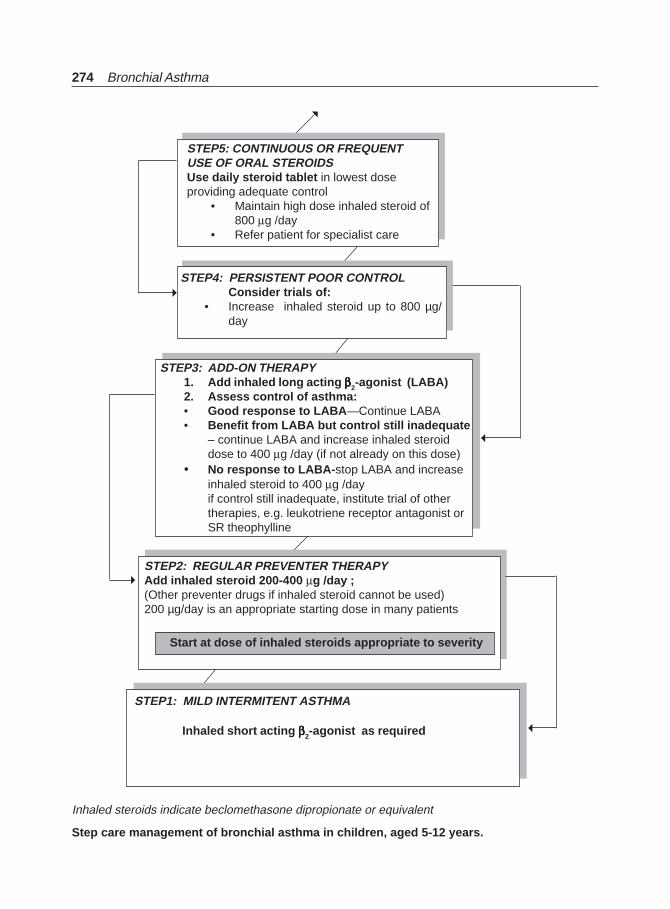
274 Bronchial Asthma
Inhaled steroids indicate beclomethasone dipropionate or equivalent
Step care management of bronchial asthma in children, aged 5-12 years.
STEP5: CONTINUOUS OR FREQUENTUSE OF ORAL STEROIDSUse daily steroid tablet in lowest doseproviding adequate control
• Maintain high dose inhaled steroid of800 μg /day
• Refer patient for specialist care
STEP4: PERSISTENT POOR CONTROLConsider trials of:
• Increase inhaled steroid up to 800 µg/day
STEP3: ADD-ON THERAPY1. Add inhaled long acting βββββ2-agonist (LABA)2. Assess control of asthma:• Good response to LABA—Continue LABA• Benefit from LABA but control still inadequate
– continue LABA and increase inhaled steroiddose to 400 μg /day (if not already on this dose)
• No response to LABA-stop LABA and increaseinhaled steroid to 400 μg /dayif control still inadequate, institute trial of othertherapies, e.g. leukotriene receptor antagonist orSR theophylline
STEP2: REGULAR PREVENTER THERAPYAdd inhaled steroid 200-400 μg /day ;(Other preventer drugs if inhaled steroid cannot be used)200 µg/day is an appropriate starting dose in many patients
Start at dose of inhaled steroids appropriate to severity
STEP1: MILD INTERMITENT ASTHMA
Inhaled short acting βββββ2-agonist as required

New Guidelines for Asthma Management (Pharmacological Management) 275
Inhaled steroids indicate beclomethasone dipropionate or equivalent. Higher nominal doses maybe required if drug delivery is difficult.
Step care management of bronchial asthma in children less than 5 years.
STEP4: PERSISTENT POOR CONTROL• Refer to Paediatrician with
respiratory specialization
STEP 3: ADD-ON THERAPYIn children aged 2-5 years consider trial of leukotrienereceptor antagonist
In children under 2 years consider proceeding to Step 4.
STEP 2: REGULAR PREVENTER THERAPYAdd inhaled steroid 200-400 µg/day ; orLeukotriene receptor antagonist if inhaled steroid cannot be used
Start at dose of inhaled steroids appropriate to severity
STEP 1: MILD INTERMITTENT ASTHMA
Inhaled short acting β2-agonist as required

276 Bronchial Asthma
New Guidelines forAsthma Management
(Acute Asthma)
18
Confidential enquires into over 200 asthma deaths in the UK have concluded that threeimportant factors associated with the disease—the medical management and the patient’sbehaviour or psychosocial status-contributed to the death. Most deaths occurred beforeadmission to hospital.
Disease Factors
Most patients who died of asthma had chronically severe asthma. In a minority the fatalattack occurred suddenly in a patient with only mild or moderately severe backgrounddisease.
Medical Management
Many of the deaths occurred in patients who had received inadequate treatment with inhaledsteroid or steroid tablets and/or inadequate objective monitoring of their asthma. Followup was inadequate in some and others should have been referred earlier for specialist advice.There was widespread underuse of written management plans. Heavy or increasing use ofβ2-agonist therapy was associated with asthma death.
Deaths have continued to be reported following inappropriate prescription of β-blockertherapy or heavy sedation. A small proportion of patients with asthma were sensitive tononsteroidal anti-inflammatory agents; all asthma patients should be asked about pastreactions to these agents.
Adverse Psychosocial and Behavioural Factors
Behavioural and adverse psychosocial factors were recorded in the majority of patientswho died of asthma. The most important are:I. A combination of severe asthma:
• Previous near fatal asthma, e.g. previous ventilation or respiratory acidosis• Previous admission for asthma especially if in the last year• Requiring three or more classes of asthma medication

New Guidelines for Asthma Management (Acute Asthma) 277
• Heavy use of β2-agonist• Repeated attendances at emergency for asthma care especially in the last year• Brittle asthma.
II Adverse behavioural or psychological features:• Non-compliance with treatment or monitoring• Failure to attend appointments• Self-discharge from hospital• Psychosis, depression, other psychiatric illness or deliberate self-harm• Current or recent major tranquilliser use• Denial• Alcohol or drug abuse• Obesity• Learning difficulties• Employment problems• Income problems• Social isolation• Childhood abuse• Severe domestic, marital or legal stress
Case control studies support most of these observations. Compared with control patientsadmitted to hospital with asthma, those who died were significantly more likely to havelearning difficulties; psychosis or prescribed antipsychotic drugs; financial or employmentproblems; repeatedly failed to attend appointments or discharged themselves from hospital;drug or alcohol abuse; obesity; or a previous near fatal attack.
Compared with control patients with asthma in the community, patients who died hadmore severe disease; more likelihood or a hospital admission or visit to emergencydepartment for their asthma in the previous year; more likelihood or a previous near fatalattack; poor medical management; failure to measure pulmonary function; and non-compliance. Health care professionals must be aware that patients with severe asthma andone or more adverse psychosocial factors are at risk of death.
Studies comparing near fatal asthma with deaths from asthma have concluded thatpatients with near fatal asthma have identical adverse factors to those described above,and that these contribute to the near fatal asthma attack. Compared with patients who die,those with near fatal asthma are significantly younger, are significantly more likely to havehad a previous near fatal asthma attack, are less likely to have concurrent medical conditions,are less likely to experience delay in receiving medical care, and more likely to have readyaccess to acute medical care.
Not all patients with near fatal asthma require intermittent positive pressure ventilation.For those with near fatal asthma, adults as well as children, it is always wise to involve aclose relative when discussing future management. Patients with brittle asthma shouldalso be identified. Patients who have had near fatal asthma or brittle asthma should be keptunder specialise supervision indefinitely.
In the UK there is a peak of asthma deaths in young people (aged up to 44 years) in Julyand August and, in December and January in older people, and similar seasonal deathsmight also be true for other countries.

278 Bronchial Asthma
PREDICTION AND PREVENTION OF A SEVERE ASTHMA ATTACK
Most (88-92%) attacks of asthma severe enough to require hospital admission developrelatively slowly over a period of six hours or more. In one study, over 80% developed overmore than 48 hours. There should therefore be time for effective action and the potential toreduce the number of attacks requiring hospitalisation. There are many similarities betweenpatients who die from asthma, patients with near fatal asthma and asthmatic controls whoare admitted to hospital. A respiratory specialist should follow up patients admitted withsevere asthma for at least one year after the admission.
I. ACUTE ASTHMA IN ADULTS
Recognition of Acute Asthma
Definitions of increasing levels of severity of acute asthma exacerbations are as follows:Near fatal asthma Raised PaCO2 and/or requiring mechanical ventilation with raised
inflation pressuresLife threatening asthma Any one of the following in a patients with severe asthma:
• PEF<33% best or predicted • Bradycardia• SpO2<92% • Dysrhythmia• PaO2<60 mmHg • Hypotension• Normal PaCO2 (4.6-6.0 kPa) • Exhaustion• Silent chest • Confusion• Cyanosis • Coma• Feeble respiratory effort
Acute severe asthma Any one of• PEF 33-50% best or predicted• Respiratory rate > 25/min• Heart rate > 100/min• Inability to complete sentences in one breath
Moderate asthma exacerbation• Increasing symptoms• PEF >50-75% best or predicted• No features of acute severe asthma
Brittle asthma• Type 1: Wide PEF variability (> 40% diurnal variation for 50%
of the time over a period > 150 days) despite intense therapy• Type 2: Sudden severe attacks on a background of apparently
well controlled asthma.Predicted PEF values should be used only if the recent best PEF (within two years) is
unknown.
Self Treatment by Patients Developing Acute or Uncontrolled Asthma
Many patients with asthma and all patients with severe asthma should have an agreedwritten action plan and their own peak flow meter, with regular checks of inhaler techniqueand compliance. They should know when and how to increase their medication and when

New Guidelines for Asthma Management (Acute Asthma) 279
to seek medical assistance. Asthma action plans have been shown to decrease hospitalisationfor and deaths from asthma.
Initial Assessment
All possible initial contact personnel, should be aware that asthma patients complaining ofrespiratory symptoms may be at risk and should have immediate access to a doctor ortrained asthma nurse. The assessments required to determine whether the patients issuffering from an acute attack of asthma, the severity of the attack and the nature of treatmentrequired are detailed above as well as follows: It may also be helpful to use a systematicrecording process. Proformas have proved useful in the emergency settings.
Clinical features Clinical features, symptoms and respiratory and cardiovascular signsare helpful in recognising some patients with severe asthma, e.g.severe breathlessness (including too breathless to complete sentencesin one breath), tachypnea, tachycardia, silent chest, cyanosis orcollapse.
None of these singly or together is specific and their absence does notexclude a severe attack
PEF or FEV1 Measurements of airway caliber improve recognition of the degreeOr severity, the appropriateness or intensity of therapy, and decisionsabout management in hospital or at home.
PEF or FEV1 are both useful and valid measures of airway caliberPEF is more convenient and cheaper.
PEF expressed as a percentage of the patient’s previous best value ismore useful clinically. PEF as a percentage of predicted gives a roughguide in the absence of a known previous best value. Different peakflow meters give different readings. Where possible the same orsimilar type of peak flow meter should be used.
Pulse oximetry Measurement of oxygen saturation (SpO2) with a pulse oxymeter isnecessary in acute severe asthma to determine the adequacy of oxygentherapy and the need for arterial blood gas (ABG) measurement. Theaim of oxygen therapy is to maintain SpO2 > 92%.
Blood gases (ABG) Patients with SpO2<92% or other features of the threatening asthmarequire ABG measurement.
Chest X-ray Chest X-ray is not routinely recommended in patients in the absenceof:• Suspected pneumomediastinum or pneumothorax• Suspected consolidation• Life threatening asthma• Failure to respond to treatment satisfactorily• Requirement for ventilation.
Systolic paradox Systolic paradox (pulsus paradoxus) has been abandoned as anindicator of the severity of an attack for pragmatic reasons.

280 Bronchial Asthma
Prevention of Acute Deterioration
A register of patients at risk may help primary care health professionals to identify patientswho are more likely to die from their asthma. A system should be in place to ensure thatthese patients are contacted if they fail to attend for follow up.
Criteria for Admission
One should refer to a hospital if one comes across any patients with features of acute severeor life threatening asthma. Other factors, such as failure to respond to treatment, socialcircumstances or concomitant disease, may warrant hospital referral.
Criteria for Admission
Patients with any feature of a life threatening or near fatal attack should be admitted. Alsopatients with any feature of a severe attack persisting after initial treatment need to beadmitted. Patients whose peak flow is greater than 75% best or predicted one hour afterinitial treatment may be discharged from emergency, unless they meet any of the followingcriteria, when admission may be appropriate:
• Still have significant symptoms• Concerns about compliance• Living alone/socially isolated• Psychological problems• Physical disability or learning difficulties• Previous near fatal or brittle asthma• Exacerbation despite adequate dose steroid tablets pre-presentation• Presentation at night• Pregnancy
Criteria for admission in adults are summarised subsequently.
Treatment of Acute Asthma in Adults
Oxygen
Patients with acute severe asthma are hypoxaemic. This should be corrected urgently usinghigh concentrations of inspired oxygen (usually 40-60%) and a high flow mask such as aHudson mask. Unlike patients with COPD there is little danger of precipitating hypercapniawith high flow oxygen. Hypercapnia indicates the development of near fatal asthma andthe need for emergency specialist/anaesthetic intervention. Oxygen saturations of at least92% must be achieved. In view of the theoretical risk of oxygen desaturaion while using airdriven compressors to nebulise β2-agonist bronchodilators, oxygen-driven nebulisers arethe preferred method of delivery in hospitals, ambulances and primary care. (in order togenerate the flow rate of 61/min required to drive most nebulisers, a high flow regulatormust be fitted to the oxygen cylinder). The absence of supplemental oxygen should notprevent nebulised therapy from being administered where appropriate. In hospital,ambulance and primary care, nebulised β2-agonist bronchodilators should be driven byoxygen. Outside hospital, high dose β2-agonist bronchodilators may be delivered via large

New Guidelines for Asthma Management (Acute Asthma) 281
volume spacers or nebulisers. Whilst supplemental oxygen is recommended, its absenceshould not prevent nebulised therapy being given if indicated.
β2-Agonist Bronchodilators
In most cases of acute asthma, inhaled β2-agonist given in high doses act quickly to relievebronchospasm with few side effects. There is no evidence for any difference in efficacybetween salbutamol and terbutaline, although rarely patients may express a preference.
In acute asthma without life threatening features, β2-agonist can be administered byrepeated activations of a pMDI via an appropriate large volume spacer or by wet nebulisationdriven by oxygen, if available. Inhaled β2-agonist are at least as efficacious and preferableto intravenous β2-agonist (meta-analysis has excluded subcutaneous trials) in adult acuteasthma in the majority of cases. High-dose inhaled β2-agonist are to be used as first lineagents in acute asthma and should be administered as early as possible. Intravenousβ2-agonist should be reserved for those patients in whom inhaled therapy cannot be usedreliably.
In acute asthma with life threatening features the nebulised route (oxygen-driven) isrecommended. Parenteral β2-agonists, in addition to inhaled β2-agonist may have a role inventilated patients or those patient in extremes in whom nebulised therapy may fail; howeverthere is limited evidence to support this.
Continuous nebulisation of β2-agonist is at least as efficacious as bolus nebulisation in relievingacute asthma. Most cases of acute asthma will respond adequately to bolus nebulisation ofβ2-agonist. In severe asthma (PEF or FEV1 <50% best or predicted) and asthma that is poorlyresponsive to an initial bolus dose of β2-agonist, continuous nebulisation may be considered.Repeated doses of β2-agonist should be given at 15-30 minute intervals or continuousnebulisation of salbutamol at 5-10mg/hour (requires appropriate nebuliser) used if there isan inadequate response to initial treatment. Higher bolus doses, e.g. 10 mg of salbutamol,are unlikely to be more effective.
Steroid Therapy
Steroid tablets reduce mortality, relapses, subsequent hospital admission and requirementfor β2-agonist therapy. The earlier they are given in the acute attack the better the outcome.Steroid tablets are to be given in adequate doses in all cases of acute asthma. Steroid tabletsare as effective as injected steroids, provided tablets can be swallowed and retained. Dosesof prednisolone of 40-50 mg daily or parenteral hydrocortisone 400 mg daily (100 mg six-hourly) are as effective as higher doses. For convenience, steroid tablets may be given as2 × 25 mg tablets daily rather than 8-12 × 5 mg tablets. The duration of prednisolone40-50 mg daily is for at least five days or until recovery.
Following recovery from the acute exacerbation steroid tablets can be stopped abruptlyand doses do not need tapering provided the patient receives inhaled steroids (apart frompatients on maintenance steroid treatment or rare instances where steroids are required forthree or more weeks). There is no evidence to suggest that inhaled steroids should besubstituted for steroid tablets in the treatment of patients with acute severe, or life threateningasthma. Further randomised controlled trials to determine the role of inhaled steroids inthese patients are required. Inhaled steroids do not provide benefit in addition to the initial

282 Bronchial Asthma
treatment, but should be continued (or started as soon as possible) to form the start of thechronic asthma management plan.
Ipratropium Bromide
Combining nebulised ipratropium bromide with a nebulised β2-agonist has been shown toproduce significantly greater bronchodilation that a β2-agonist alone, leading to a fasterrecovery and shorter duration of admission. Anticholinergic treatment is not necessaryand may not be beneficial in milder exacerbations of asthma or after stabilisation.
Nebulised ipratropium bromide (0.5 mg 4-6 hourly) should be added to β2-agonisttreatment for patients with acute severe or life threatening asthma or those with a poorinitial response to β2-agonist therapy.
Intravenous Magnesium Sulphate
A single dose of IV magnesium sulphate has been shown to be safe and effective in acutesevere asthma. The safety and efficacy of repeated doses have not been assessed in patientswith asthma. Repeated doses could give rise to hypermagnesaemia with muscle weaknessand respiratory failure.
Indications of giving a single dose of IV magnesium sulphate for patients are:• Acute severe asthma who have not had a good initial response to inhaled
bronchodilator therapy• Life threatening or near fatal asthma
IV magnesium sulphate (1.2- 2g IV infusion over 20 minutes) should only be usedfollowing consultation with senior medical staff. More studies are needed to determine theoptimal frequency and dose of IV magnesium sulphate therapy.
Intravenous Aminophylline
In acute asthma, the use of intravenous aminophylline is not likely to result in any additionalbronchodilation compared to standard care with inhaled bronchodilators and steroid tablets.Side effects such as palpitations, arrhythmias and vomiting are increased if IV aminophylineis used. Intravenous aminophylline is to be used only after consultation with senior medicalstaff.
Some individual patients with near fatal asthma or life threatening asthma with a poorresponse to initial therapy may gain additional benefit from IV aminophylline (5mg/kgloading dose over 20 minutes unless on maintenance oral therapy, then infusion of 0.5-0.7mg/kg/h). Such patients are probably rare and could not be identified in a meta-analysisof trials involving 739 subjects. If IV aminophylline is given to patients, on oral aminophy-lline or theophylline, blood levels should be checked on admission. Levels should be checkeddaily for all patients on aminophylline infusions.
Leukotriene Receptor Antagonists
There is no published study of the use of leukotriene receptor antagonists in the managementof acute asthma.

New Guidelines for Asthma Management (Acute Asthma) 283
Antibiotics
When an infection precipitates an exacerbation of asthma, it is likely to be viral in type. Therole of bacterial infection has been overestimated. Routine prescription of antibiotics is notindicated for acute asthma.
Heliox
The use of heliox (Helium/oxygen mixture in a ratio of 80:20 or 70:30) in acute adult asthmacannot be recommended on the basis of present evidence.
Intravenous Fluids
There are no controlled trials or even observational or cohort studies of differing fluid regimesin acute asthma. Some patients with acute asthma require rehydration and correction ofelectrolyte imbalance. Hypokalaemia can be caused or exacerbated by β2-agonist and/orsteroid treatment must be corrected.
Referral to Intensive Care
Indications for admission to intensive care facilities or a high dependency unit includepatients requiring ventilatory support and those with severe acute or life threatening asthmawho are failing to respond to therapy, as evidenced by:
• Deteriorating PEF• Persisting or worsening hypoxia• Hypercapnea• Arterial blood gas analysis showing fail in pH or rising H+ concentration• Exhaustion, feeble respiration• Drowsiness, confusion• Coma or respiratory arrest
Not all patients admitted to the Intensive Care Unit (ICU) need ventilation, but thosewith worsening hypoxia or hypercapnea, drowsiness or unconsciousness and those whohave had a respiratory arrest require intermittent positive pressure ventilation. Intubationin such patients is very difficult and should ideally be performed by an anaesthetist or ICUconsultant. All patients transferred to intensive care units should be accompanied by adoctor suitably equipped and skilled to intubate if necessary.
Non-invasive Ventilation
Non-invasive ventilation (NIV) is now well established in the management of ventilatoryfailure caused by extrapulmonary restrictive conditions and exacerbations of COPD.Hypercapneic respiratory failure developing during the evolution of an acute asthmaticepisode is regarded as an indication for urgent admission to the ICU. It is unlikely the NIVwould ever replace intubation in these very unstable patients but it has been suggested thatthis treatment can be used safely and effectively. Future studies might usefully examine itsrole in the gradually training patient, but at present this treatment cannot be recommendedoutside randomised controlled trials.

284 Bronchial Asthma
Further Investigation and Monitoring
Measurement and recording of PEF 15-30 minutes after starting treatment, and thereafteraccording to the response is necessary. Measurement and recording of PEF before and afternebulised or inhaled β2-agonist bronchodilator (at least four times daily) throughout thehospital stay and until controlled after discharge is quite helpful.
Recording of oxygen saturation by oximetry and maintaining arterial SaO2 >92% is veryhelpful. Repeat measurements of blood gas tensions within two hours of starting treatmentis indicated if:
• The initial PaO2 is < 8 kPa unless SaO2 is >92%; or• The initial PaCO2 is normal or raised; or• The patient’s condition deterioratesOne should ensure them again if the patient’s condition has not improved by 4-6 hours.• Measure and record the heart rate.• Measure serum potassium and blood concentrations.• Measure the serum theophylline concentration if aminophylline is continued for more
than 24 hours (aim at a concentration of 55-110 μmol/l.
Asthma Management Protocols and Proformas
The use of structured proformas has been shown to facilitate improvements in the processof case in emergency departments and hospital wards and to improve patient outcomes. Theuse of this type of documentation can assist data collection aimed at determining quality ofcare and outcomes.
Hospital Discharge and Follow-Up
Timing of Dsicharge
There is no single physiological parameter that defines absolutely the timing of dischargeform an admission with acute asthma. Patients should have clinical signs compatible withhome management, be on medical therapy they can continue safely at home. Althoughdiurnal variability of PEF is not always present during an exacerbation, evidence suggeststhat patients discharged with PEF<75% best or predicted and with diurnal variability >25%are at greater risk of early relapse and readmission.
Patient Education
Following discharge from hospital or emergency departments, a proportion of patients re-attend emergency departments, with more than 15% re-attending within two weeks. Somerepeat attendees need emergency care, but many delay seeking help, and are under-treatedand/or under-monitored. Prior to discharge, trained staff should give asthma education.This should include education on inhaler technique and PEF record keeping , with a writtenPEF and symptom based action plan being provided allowing the patient to adjust theirtherapy within recommendations. These measures have been shown to reduce morbidityafter the exacerbation and reduce relapse rates. There is some experience of a discretepopulation of patients who inappropriately use emergency departments rather than theprimary care services for their asthma care. For these groups there is a role for a trainedasthma liaison nurse based in, or associated with the emergency department.

New Guidelines for Asthma Management (Acute Asthma) 285
Follow-Up
A careful history should elicit the reasons for the exacerbation and explore possible actions,the patient should take to prevent future emergency presentations. Medication should bealtered depending upon the assessment, and the patient provided with an asthma actionplan aimed at preventing relapse, optimising treatment and preventing delay in seekingassistance in the future. Follow-up should be arranged prior to discharge with the patient’sgeneral practitioner or asthma nurse within two working days; and with a hospital specialistasthma nurse or respiratory physician at about one month after admission. It is essentialthat the patient’s primary care practice is informed within 24 hours of discharge fromemergency or hospital following an asthma exacerbation treated in hospital. Ideally thiscommunication should be directly with a named individual responsible for asthma carewithin the practice, by means or fax or e-mail.
ACUTE ASTHMA IN CHILDREN AGED OVER 2 YEARS
Initial Assessment
The details of criteria for assessment of severity of acute asthma attacks in children are:
Acute severe Life threateningCan’t complete sentences in one breathOr too breathless to talk or feed
Silent ChestPulse >120 in children aged > 5 years Cyanosis
>130 in children aged 2-5 years Poor respiratory effortHypotension
Respiration >30 breaths/min aged>5 yrs Exhaustion>50 breaths/min aged 2-5 yrs Confusion
ComaBefore children can receive appropriate treatment for acute asthma in any setting, it is
essential to assess accurately the severity of their symptoms. The following clinical signsshould be recorded:
• Pulse rate (increasing tachycardia generally denotes worsening asthma; a fall in heartrate in life threatening asthma is a pre-terminal event).
• Respiratory rate and degree of breathlessness (i.e. too breathless to complete sentencesin one breath or to feed).
• Use of accessory muscles of respiration (best noted by palpation of neck muscles)• Amount of wheesing (which might become biphasic or less apparent with increasing
airways obstruction).• Degree of agitation and conscious level (always give calm reassurance).Clinical signs correlate poorly with the severity of airways obstruction. Some children
with acute severe asthma do not appear distressed. Objective measurements of PEF andSpO2 are essential. Suitable equipment should be available for use by all health professionalsassessing acute asthma in both primary and secondary care settings. Low oxygen saturationsafter initial bronchodilator treatment selects a more severe group of patients. Intensiveinpatient treatment for children with SpO2 <92% on air after initial bronchodilator treatmentshould be considered.

286 Bronchial Asthma
Decisions about admission should be made by trained physicians after repeatedassessment of the response to further bronchodilator treatment. A measurement of <50%predicted PEF or FEV1 with poor improvement after initial bronchodilator treatment ispredictive of more prolonged asthma attack. An attempt has to be made to measure PEF orFEV1 in all children aged >5 years, taking the best of three measurements; ideally expressedas percentage of personal best for PEF (as detailed in a written action plan) or alternativelyas percentage of predicted for PEF or FEV1. Chest X-rays and ABG measurements rarelyprovide additional useful information and are not routinely indicated.
TREATMENT OF ACUTE ASTHMA IN CHILDREN AGED OVER 2 YEARS
Emergency units attending to children with acute asthma should have a registered sickchildren’s nurse available on duty at all times and staff familiar with the specific needs ofchildren. The use of proformas can increase the accuracy of severity assessment. Anassessment driven algorithm has been shown to reduce treatment costs and hospital stay.The use of structured care protocols detailing bronchodilator usage, clinical assessment,and specific criteria for safe discharge is recommended.
Oxygen
Children with life threatening asthma or SpO2 < 92% should receive high flow oxygen viaa tight fitting face mask or nasal cannula at sufficient flow rates to achieve normal saturations.
β2-Agonist Bronchodilators
Inhaled β2-agonist are the first line treatment for acute asthma. pMDI + spacer is an effectivealternative to nebulisers for bronchodilator inhalation to treat mild to moderate asthma.Children receiving β2-agonist via pMDI+ spacer are less likely to have tachycardia andhypoxia than when the same drug is given via a nebuliser. pMDI+ spacer are the preferredoption in mild to moderate asthma. Information about implementing evidence-basedguidelines using such devices has been published. Children aged < 3 years are likely torequire a face mask connected to the mouthpiece of a spacer for successful drug delivery.Inhalers should be actuated into the spacer in individuals puffs and inhaled immediatelyby tidal breathing. Frequent doses of β2-agonist are safe for the treatment of acute asthma,although children with mild symptoms benefit from lower doses. Drug dosing is to beindividualised according to severity and adjust according to the patient’s response.
Two to four puffs repeated every 20-30 minutes according to clinical response might besufficient for mild attacks although up to 10 puffs might be needed for more severe asthma.Children with acute asthma in primary care show have not improved after receiving up to10 puffs of β2-agonist should be referred to hospital. Further doses of bronchodilator shouldbe given as necessary, whilst awaiting transfer. Treatment of children should be given beforethey are transported to hospital by ambulance with oxygen and nebulised β2-agonist duringthe journey. Children with severe or life threatening asthma should be transferred urgentlyto hospital to receive frequent doses of nebulised β2-agonist (2.5-5 mg albuterol or 5-10 mgterbutaline). Doses can be repeated every 20-30, omits. Continuous nebulised β2-agonistare of no greater benefit than the use of frequent intermittent doses in the same total hourlydosage.

New Guidelines for Asthma Management (Acute Asthma) 287
IV Salbutamol
The role of intravenous β2-agonist in addition to nebulised treatment remains unclear. Onestudy has shown that an IV bolus of salbutamol given in addition to near maximal doses ofnebulised salbutamol results in clinically significant benefits. The early addition of a bolusdose of intravenous salbutamol (15 μg/kg) can be an effective adjunct to treatment in severecases. Continuous intravenous infusion should be considered when there is uncertaintyabout reliable inhalation or for severe refractory asthma. Doses above 1-2 μg/kg/min(200 μg/ml solution) should be given in a Paediatric intensive Care Unit (PICU) setting (upto 5 μg/kg/min) with regular monitoring of electrolytes.
Steroid Therapy
Steroid Tablets
The early use of steroids for acute asthma can reduce the need for hospital admission andprevent a relapse in symptoms after initial presentation. Benefits can be apparent withinthree to four hours. Prednisolone is to be given early in the treatment of acute asthmaattacks. A soluble preparation dissolved in a spoonful of water is preferable in those unableto swallow tablets. The dose is 20 mg for children 2-5 years old and 30-40 mg for children> 5 years. Oral and intravenous steroids are of similar efficacy. Intravenous hydrocortisone(4 mg/kg repeated four hourly) should be reserved for severely affected children who areunable to retain oral medication. Larger doses do not appear to offer a therapeutic advantagefor the majority of children. There is no need to taper the dose of steroid tablets at the endof treatment. A dose of 20 mg prednisolone for children aged 2-5 years and a dose of30-40 mg for children > 5 years is appropriate. Those already receiving maintenance steroidtablets should receive 2 mg/kg prednisolone up to a maximum dose of 60 mg. The dose ofprednisolone in children who vomit may be repeated and intravenous steroids in thosewho are unable to retain orally, ingested medication should be considered. Treatment forup to three days is usually sufficient but the length of course should be tailored to thenumber of days necessary to bring about recovery.
Inhaled Steroids
There is insufficient evidence to support the use of inhaled steroids as alternative oradditional treatment to steroid tablets for acute asthma. One need not initiate inhaled steroidsin preference to steroid tablets to treat acute childhood asthma. Children with chronic asthmanot receiving regular preventive treatment will benefit from initiating inhaled steroids aspart of their long-term management. There is no evidence that increasing the dose of inhaledsteroids is effective in treating acute symptoms, but it is good practice for children alreadyreceiving inhaled steroids to continue with their usual maintenance doses.
Ipratropium Bromide
There is good evidence for the safety and efficacy of frequent doses of ipratropium bromideused in addition to β2-agonist for the first two hours of a severe asthma attack. Benefits aremore apparent in the most severe patients. If symptoms are refractory to initial β2-agonisttreatment, add ipratropium bromide (250 mg/dose mixed with the nebulised β2-agonist

288 Bronchial Asthma
solution). Frequent doses up to every 20-30 minutes (250 μg/dose mixed with the β2-agonistsolution in the same nebuliser) should be used early. The dose frequency should be reducedas clinical improvement occurs. Repeated doses of ipratropium bromide should be givenearly to treat children poorly responsive to β2-agonist. Children with continuing severeasthma despite frequent nebulised β2-agonist and ipratropium bromide and those withlife-threatening features need urgent review by a specialist with a view to transfer to aHigh Dependency Unit or PICU.
IV Amniophylline
There is no evidence that aminophylline is of benefit for mild to moderate asthma and sideeffects are common and troublesome. However, one well conducted study has shownevidence for benefit in severe acute asthma unresponsive to multiple doses of β2-agonistand steroids. Aminophylline is not recommended in children with mild to moderate acuteasthma. However, one may consider aminophylline in a High Dependency Unit or PICUsetting for children with severe or life-threatening bronchospasm unresponsive to maximaldoses of bronchodilators and steroid tablets. A 5 mg/kg loading dose should be given over20 minutes with ECG monitoring (omit in those receiving maintenance oral theophyllines)followed by a continuous infusion at 1 mg/kg/hour. Estimation of serum theophyllinelevels in patients already receiving oral treatment and in those receiving prolonged treatmentwill be necessary.
Other Therapies
There is no evidence to support the use of heliox or leukotriene receptor antagonists for thetreatment of acute asthma in childhood. There is insufficient evidence to support or refutethe role of antibiotics in acute asthma, but the majority of acute asthma attacks are triggeredby viral infection.
Antibiotics are not to be given routinely in the management of acute childhood asthma.
Intravenous Fluids
Children with prolonged severe asthma not tolerating, oral fluids will require intravenoushydration. Two-third of the child’s maintenance requirement should be given because ofthe possibility of inappropriate antidiuretic hormone secretion. Serum electrolytes shouldbe measured and hypokalamia corrected, if detected. ECG monitoring is mandatory for allintravenous treatments.
IV Magnesium Sulphate
Intravenous magnesium sulphate is a safe treatment for acute asthma although its place inmanagement is not yet established. Doses of up to 40 mg/kg/day (maximum 2g) by slowinfusion has been used. Studies of efficacy for severe childhood asthma unresponsive tomore conventional therapies have been inconsistent in providing evidence of benefit.
Further Investigation and Monitoring
Children can be discharged when stable on 3-4 hourly inhaled bronchodilators that can becontinued at home. PEF and/or FEV1 should be >75% of best or predicted and SpO2>94%.

New Guidelines for Asthma Management (Acute Asthma) 289
Adult studies show that “optimal care” comprising self-monitoring, regular review anda written asthma action plan can improve outcomes. Acute asthma attacks should beconsidered a failure of preventive therapy and thought should be given about how to helpfamilies avoid further severe episodes. Discharge plans should address the following:
• Check inhaler technique• Consider the need for regular inhaled steroids• Provide a written asthma action plan for subsequent asthma with clear instructions
about the use of bronchodilators, seeking urgent medical attention in the event ofworsening symptoms and, if appropriate, starting a course of oral steroids
• Arrange follow-up by a General Practitioner within one week• Arrange follow-up in a paediatric asthma clinic within one to two months.
ASSESSMENT OF ACUTE ASTHMA IN CHILDREN AGED LESS THAN 2 YEARS
The assessment of acute asthma in early childhood can be difficult. Intermittent wheesingattacks are usually due to viral infection and the response to asthma medication isinconsistent. Prematurity and low birth weight are risk factors for recurrent wheesing. Thedifferential diagnosis of symptoms includes aspiration pneumonitis, pneumonia,bronchiolitis, tracheomalacia, and complications of underlying conditions such as congenitalanomalies and cystic fibrosis. These guidelines are intended for those who are thought tohave asthma causing acute wheeze. They should not be used as a guide for treating acutebronchiolitis.
TREATMENT OF ACUTE ASTHMA IN CHILDREN AGED < 2 YEARS
βββββ2-Agonist Bronchodilators
A trial of bronchodilator therapy should be considered when symptoms are of concern. Ifinhalers have been successfully administered but there is no response, review the diagnosisand consider the use of other treatment options. Oral β2-agonists have not been shown toaffect symptom score or length of hospital stay for acute asthma in infancy when comparedto placebo. Oral β2-agonists are not recommended for acute asthma in infants. Inhaledβ2-agonists are the treatment of choice for the initial treatment of acute asthma. Close fittingface masks are essential for optimal drug delivery. The dose received is increased if thechild is breathing appropriately and not taking large gasps because of distress and screaming.There is good evidence that pMD+ spacer is as effective as, if not better than, nebulisers fortreating mild to moderate asthma in children aged <2 years. For mild to moderate acuteasthma, a pMDI+ spacer is the optimal drug delivery device. Whilst β2-agonists offermarginal benefits to children aged < 2 years with acute wheeze, there is little evidence foran impact on the need for hospital admission or length of hospital stay.
Steroid Therapy
Steroid tablets in conjunction with β2-agonists have been shown to reduce hospital admissionrates when used in the emergency department. Steroid tablets have also been shown toreduce the length of hospital stay. Steroid tablets are to be considered in infants early in themanagement of moderate to severe episodes of acute asthma in the hospital setting. One

290 Bronchial Asthma
study has shown similar benefits when comparing oral and nebulised steroids for acuteasthma. Steroid tablet therapy (10 mg of soluble prednisolone for up to three days) is thepreferred steroid preparation for use in this age group.
Ipratropium Bromide
The addition of ipratropium bromide to β2-agonists for acute severe asthma may lead tosome improvement in clinical symptoms and reduce the need for more intensive treatment.It does not reduce the length of hospital stay either in combination with β2-agonists or incomparison with placebo. Inhaled ipratropium bromide in combination with an inhaledβ2-agonist may be considered for more severe symptoms.
Further Investigation and Monitoring
Many children with recurrent episodes of viral-induced wheesing in infancy do not go onto have chronic atopic asthma. The majority do not require treatment with regular inhaledsteroids. Parents should be advised about the relationship between cigarette smoke exposureand wheezy illnesses. Referral to suitable agencies should be offered to those who wish togive up smoking. Parents of wheezy infants should receive appropriate discharge plansalong similar lines to those given for older children. Management of acute severe asthmain adults and children is shown in the following diagrams.
Management of acute severe asthma in adults in general practice
Many deaths from asthma are preventable, but Assess and record:delay can be fatal. • Peak expiratory flow (PEF)Factors leading to poor outcome include: • Symptoms and response to• Doctors failing to assess severity by objective self treatment
measurement • Heart and respiratory rates• Patients or relatives failing to appreciate • Oxygen saturation by pulse oxymetry, if
severity available• Under use of corticosteroids Patients with severe or life threatening
Regard each emergency asthma consultation attacks may not be distressed and mayas for acute severe asthma until it is shown to be not have all the abnormalities listed below.otherwise. The presence of any should alert the doctor.
Moderate asthma Acute Severe Asthma Life Threatening Asthma ↓ ↓ ↓
Initial Assessment
PEF > 50% best or predicted PEF 33-50% best or predicted PEF < 33% best or predicted
Further Assessment
• Speech normal • Can’t complete sentences • SpO2 < 92%• Respiration , 25/min • Respiration > 25/min • Silent chest, cyanosis, or• Pulse < 110/min • Pulse > 110/min feeble respiratory effort
• Bradycardia, dysrrhythmia,or hypotension
• Exhaustion, confusion or coma
↓ ↓ ↓Contd...

New Guidelines for Asthma Management (Acute Asthma) 291
Management
Treat at home Consider admission Arrange immediate ADMISSIONAssess response to treatment
↓ ↓ ↓
Treatment
• High-dose β2-bronchodilator: • Oxygen 40-60% if available • Oxygen 40-60%• ideally via oxygen-driven • High-dose β2-bronchodilator: • Prednisolone 40-50 mg
nebulizer (salbutamol 5 mg • ideally via oxygen-driven or IV hydrocortisoneor terbutaline 10mg) nebulizer (salbutamol 5 mg 100 mg immediately
• Or via spacer or air-driven or terbutaline 10mg) • High-dose β2-broncho-nebulizer (1 puff 10-20 times) • Or via spacer (1 puff β2-agonist dilator and ipratropium:
If PEF > 50-70% predicted: via a large volume spacer and • Ideally via oxygen-• Give prednisolone 40-50 mg repeat 10-20 times) or air- driven nebulizer (sal-• Continue or step up usual driven nebulizer butamol 5 mg or
treatment • Prednisolone 40-50 mg or IV terbutaline 10 mg andIf good response to first nebulised hydrocortisone 100 mg ipratropium 0.5 mg)treatment (symptoms improved, • If no response in acute • Or via spacer (1 puffrespiration and pulse setting, and severe asthma, Admit β2-agonist via a largePEF > 50%) continue or step up volume spacer andusual treatment and continue repeat 10-20 times)prednisolone or air-driven nebulizer
↓ ↓ ↓Admit to hospital if any: If admitted patient to hospital: Follow up after treatment• Life threatening features • Stay with patient till ambulance or discharge from hospital:• Features of acute severe arrives • General Practitioner
asthma present after initial • Send written assessment and review within 48 hrtreatment referral details to hospital • Monitor symptoms and
• Previous near-fatal asthma • Give high dose β2-broncho- PEFLower threshold for admission if: dilator via oxygen-driven • Check inhaler technique• Afternoon or evening attack, nebulizer in ambulance • Written asthma action• Recent nocturnal symptoms plan
or hospital admission • Modify treatment accord-• Previous severe attacks ing to guidelines for• Patient unable to assess own chronic persistent asthma
condition • Address potentially• Concern over social circumstances preventable contributors
to admission
Contd...
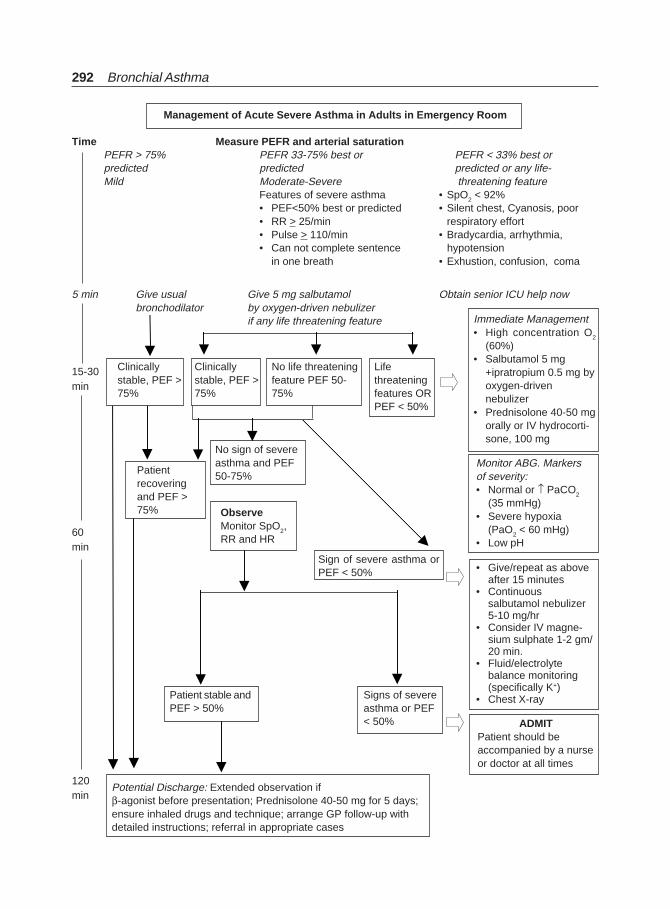
292 Bronchial Asthma
Management of Acute Severe Asthma in Adults in Emergency Room
Time Measure PEFR and arterial saturationPEFR > 75% PEFR 33-75% best or PEFR < 33% best orpredicted predicted predicted or any life-Mild Moderate-Severe threatening feature
Features of severe asthma • SpO2 < 92%• PEF<50% best or predicted • Silent chest, Cyanosis, poor• RR > 25/min respiratory effort• Pulse > 110/min • Bradycardia, arrhythmia,• Can not complete sentence hypotension
in one breath • Exhustion, confusion, coma
5 min Give usual Give 5 mg salbutamol Obtain senior ICU help nowbronchodilator by oxygen-driven nebulizer
if any life threatening feature
Clinicallystable, PEF >75%
Clinicallystable, PEF >75%
No life threateningfeature PEF 50-75%
Lifethreateningfeatures ORPEF < 50%
Immediate Management• High concentration O2
(60%)• Salbutamol 5 mg
+ipratropium 0.5 mg byoxygen-drivennebulizer
• Prednisolone 40-50 mgorally or IV hydrocorti-sone, 100 mg
Monitor ABG. Markersof severity:• Normal or ↑ PaCO2
(35 mmHg)• Severe hypoxia
(PaO2 < 60 mHg)• Low pH
• Give/repeat as aboveafter 15 minutes
• Continuoussalbutamol nebulizer5-10 mg/hr
• Consider IV magne-sium sulphate 1-2 gm/20 min.
• Fluid/electrolytebalance monitoring(specifically K+)
• Chest X-ray
ADMITPatient should beaccompanied by a nurseor doctor at all times
15-30min
60min
120min
Patientrecoveringand PEF >75%
No sign of severeasthma and PEF50-75%
Sign of severe asthma orPEF < 50%
ObserveMonitor SpO2,RR and HR
Patient stable andPEF > 50%
Signs of severeasthma or PEF< 50%
Potential Discharge: Extended observation ifβ-agonist before presentation; Prednisolone 40-50 mg for 5 days;ensure inhaled drugs and technique; arrange GP follow-up withdetailed instructions; referral in appropriate cases

Alternate Treatments in Asthma 293
AlternateTreatments in Asthma
19
Standard asthma therapy, as defined by various management guidelines includes oral andinhaled corticosteroids, leukotriene antagonists, short-acting and long-acting β-agonists,cromolyn, theophyllines, and nedocromil. Although these agents are generally successfulin controlling asthma symptoms, a small but significant number of patients will continuewith persistent symptoms, frequent exacerbations, and no improvement in objectivepulmonary function parameters despite maximum standard therapy. The long-term use oforal and high-dose inhaled corticosteroids is often associated with significant side effects.Thus there is need for alternate agents that are effective in the treatment of asthma.
Alternate agents those have been evaluated in prospective randomized trials or havenovel mechanisms of action are shown in Table 19.1.1-4
Table 19.1: Alternate agents for bronchial asthma
MethotrexateAzathioprineGoldHydroxychloroquineTroleandomycinCyclosporineIVIGInhaled heparinInhaled furosemideDapsoneAnti-IgE and soluble interleukin (IL)-4 receptor therapy
Methotrexate
Mechanism of Action
Methotrexate is a folate antagonist. It has anti-inflammatory properties at low doses.Therefore, it is widely used in a variety of autoimmune and inflammatory diseases, includingsevere steroid-dependent asthma. A number of potential reasons for its effectiveness havebeen proposed, the mechanism of action of methotrexate in asthma remains unclear. Thedrug inhibits leukotriene B4-mediated and leukotriene C5a-mediated neutrophil chemotaxisin vitro5 although inflammatory cell numbers in vivo appear to be unaltered duringtreatment.6 It affects function of many cytokines. The drug inhibits the expression of Ia, a

294 Bronchial Asthma
marker of macrophage activation and monocyte IL-1 production, IL-6, IL-8, and histaminerelease, and platelet-activating factor-induced eosinophil chemotaxis.6-8 The inhibition ofpurine metabolism by methotrexate diminishes lymphocyte proliferation and antibodyformation6 and increases in CD8+ T-suppressor cells and suppression of B-cell differentiationhave been observed. No significant interference with steroid metabolism has been noted.9
Methotrexate may enhance the sensitivity of peripheral blood monocytes to glucocorticoidsin cases of steroid-refractory asthma.10
A number of randomized trials11-21 have been made using methotrexate in bronchialasthma and three meta-analyses have been performed since then with mixed results.22-24 Ofthe 11 prospective randomized trials, 10 evaluated the use of oral or IM low-dosemethotrexate vs placebo in steroid-dependent asthma patients using either parallel orcrossover design. A three-arm study compared methotrexate, 15 mg weekly orally, with asingle dose of triamcinolone, 360 mg IM, or placebo.20 Most trials required that patients hadreceived a minimum of 12 months of long-term corticosteroid therapy to be eligible forenrolment (range, 5 months to 28 years). The majority received oral methotrexate, 15 mgweekly, and many used a variety of run-in periods to maximize asthma therapy prior tostarting treatment. The duration of methotrexate therapy ranged from 12 to 24 weeks, withno further reported long-term follow-up. Most studies reported significant reductions inoral steroid use in their placebo groups, which was attributed to the close-interval follow-up and the education that patients received during enrolment. Others reported a statisticallysignificant reduction in corticosteroid dose using methotrexate, some demonstratingreduction of mean oral corticosteroid use up to 50%.12 Other trials, however, could notdemonstrate a significant difference in corticosteroid reduction between the methotrexateand placebo groups. Small patient numbers further limits the interpretation. Many trialshad significant dropout rates. No statistically significant changes in peak flow, spirometry,or mean values for the dose of methacholine provoking a 20% fall in FEV1 were observedin patients taking methotrexate neither reduction in steroid-related side effects werereported.12-21
Although responses to methotrexate have been reported after 3 months in patients withimmunologic diseases such as rheumatoid arthritis, treatment for 12 weeks may beinsufficient to demonstrate an adequate therapeutic response in patients with asthma. Somestudies reported statistically significant reductions in the baseline prednisone dose, withover half were weaned off all steroid therapy.25, 26
Common side effects of methotrexate include liver function test abnormalities, GIsymptoms (including abdominal pain, nausea, and diarrhea), oral ulcers and stomatitis,constitutional symptoms (including fatigue and decreased concentration), headache, rash,and less common ones like opportunistic infections (Pneumocystis carinii pneumonia),increased incidence of bacterial pneumonia, disseminated varicella zoster. However, sideeffects were transient and reversible and only minimal from the administration of low-dosemethotrexate. No bone marrow toxicity was observed at the dosages used, and follow-upwas too short to address issues of potential hepatic fibrosis. Methotrexate competes withthe hepatic metabolism of theophylline, with an average decrease in theophylline clearanceof 19% in one case series.
Methotrexate is well-known to cause a variety of pulmonary manifestations, includingdrug-induced hypersensitivity reaction, chronic pneumonitis and fibrosis, bronchiolitis

Alternate Treatments in Asthma 295
obliterans with organizing pneumonia, noncardiogenic pulmonary edema, and broncho-spasm.27
Troleandomycin
Troleandomycin (TAO) is a macrolide antibiotic and was first described as a treatment forsteroid-dependent asthma in 197428 The drug is believed to have a synergistic effect whenadministered with oral corticosteroids. In vitro data using its parent compound,oleandomycin, at a concentration of 5 µg/mL demonstrated a 44% reduction in theconcentration of methylprednisolone that was required to inhibit human lymphocyte blasttransformation by 50% (p < 0.005).29
It is believed that Troleandomycin alters corticosteroid bioavailability by decreasinghepatic metabolism and excretion.30-32 Subjective patient improvement has not beencorrelated with evidence of infection using sputum culture, suggesting that a directantimicrobial effect is less likely.28,32
Clinical efficacy data on troleandomycin showed improvement in clinical symptomsand/or a reduction in corticosteroid dosage in when used as 14 mg/kg/d TAO (maximumdose, 1 g.33-35 These benefits, however, were less convincingly demonstrated in another trialof TAO efficacy.36 The results were limited by significant patient The authors concludedthat patients who had been randomized to TAO experienced no advantage and appearedto develop greater steroid-related side effects than did placebo subjects.
Steroid-related side effects are common with the use of TAO, especially in earlier trialswhen patients were given doses of 1 g daily.33,34 Cushingoid features, weight-gain, fluidretention, and glucose intolerance were the most common findings. GI distress andhepatotoxicity, ranging from transient liver enzyme abnormalities37 to prolonged cholestasis38
and jaundice39 have been reported, predominantly at higher doses. The doses of theophyllineand other medications with hepatic metabolism must be adjusted to avoid toxicity. DecreasedIgG levels and one case of varicella zoster has been reported with its use.
Gold
Gold is an immunomodulatory agent that has been used commonly for the treatment of avariety of inflammatory and autoimmune conditions. Although the complete mechanismof anti-inflammatory activity is unknown, gold has been demonstrated to decrease neutrophiland macrophage phagocytosis, and lymphocyte reactivity to antigenic stimulation.40 It alsoinhibits antibody production and lysosomal enzyme release from phagocytic leukocytes.41
Gold inactivates C1 (complement), decreases prostaglandin and leukotriene production invitro, and inhibits IgE-mediated release of histamine from isolated basophils and lung mastcells.40-44 The enhancement of eosinophil survival with IL-5 is inhibited by the presence ofgold.45
The beneficial effects of gold in the treatment of asthma were reported as early as 1932.Improvements have been reported in bronchial reactivity and oral corticosteroidrequirements have been decreased46-48 oral or parenteral gold treatment was used for 12 to22 weeks. There have been three prospective randomized studies examining the efficacy ofgold. Other studies have reported benefit. The Auranofin Multicenter Drug Trial49 has beenthe largest clinical trial to examine the efficacy of oral gold. After a 4-week observationperiod 275 patients with daily oral prednisone requirements of ≥ 10 mg mg were randomized

296 Bronchial Asthma
to auranofin, 3 mg twice daily, or placebo for a period of 6 months. The results were limitedby a significant patient dropout rate (auranofin group, 40%; placebo group, 46%) due toadverse effects, protocol violations, and voluntary withdrawals, and no intention-to-treatanalysis was performed. No significant differences were found in symptoms or objectivemeasurements of pulmonary function, although more patients treated with gold were ableto reduce their daily oral corticosteroid dose by ≥ 50% compared to those receiving placebo(60% vs 32%, respectively; p < 0.001). Statistically significant reductions in serum IgE level(reduction, 44.63 IU/mL; p = 0.003) were also observed in the auranofin group.
Gold has been associated with a variety of side effects, including GI upset and diarrhea,pruritic rash, cytopenias, oral ulcerations, proteinuria, and frank nephrotic syndrome. Nearly40% of patients in the prospective randomized trials that were detailed earlierexperienced.49-51 All side effects were self-limited with discontinuation or reduction of therapy.Some experts have argued that the relative lack of severe side effects with gold therapy,compared to methotrexate therapy, make it a preferable agent for the treatment of severe,glucocorticoid-dependent asthma, but no clear consensus exists on the issue.
Cyclosporine
Cyclosporine is an immunomodulatory and anti-inflammatory drug and is a fungal metabolitethat is commonly used in organ transplantation. Cyclosporine binds to cyclophilin, inhibitingcytokine messenger RNA transcription and CD4+ T-cell activation.52 The drug also reducesthe synthesis and release of inflammatory mediators from mast cells and basophils, and itdecreases B-cell IgE synthesis and release.53 Cyclosporine has been demonstrated to reducethe macrophage synthesis of IL-1, tumor necrosis factor, superoxide, and hydrogen peroxide,and has been shown to decrease neutrophil chemotaxis and serum soluble IL-2 receptorconcentrations.40,54 The production of granulocyte macrophage colony-stimulating factor(GMCSF) and IL-5 from stimulated monocytes is also reduced with drug therapy, inhibitingeosinophil proliferation and survival activity.55,56
Cyclosporine has been shown to block the late asthmatic reaction and to inhibit theproduction of eosinophil-related cytokines after allergen challenge.52,57 Statistically significantimprovement in airway hyperreactivity has been observed in steroid-dependent asthmapatients after 12 weeks of therapy with cyclosporine. Three prospective randomized trialsthat have examined the effect of cyclosporine in asthma patients, which have shown a 12%increase in morning peak expiratory flow rates (PEFRs) (p < 0.004), a 17.6% increase inFEV1 (p < 0.001), and a 48% reduction in exacerbations requiring increased steroid dosing(p < 0.02) compared to those receiving placebo.58 In another study treatment withcyclosporine (initial dose, 5 mg/kg/d) for 36 weeks in resulted in a statistically significantreduction in the median daily prednisolone dosage (62% vs 25%, respectively; p = 0.043)along with improvements in PEFR.59 However, another study with a longer follow-up perioddemonstrated no statistically significant effects of cyclosporine using the objective markersof pulmonary function and steroid-sparing effects.60
The side effects of cyclosporine are dose-dependent nephrotoxicity, tremor, hirsutism,hypertension, gum hyperplasia, and infectious complications. The majority of these sideeffects are not observed in the low doses that were used in the trial listed above. Severalpatients experience hypertrichosis and a worsening of pre-existing hypertension that mayresult in the discontinuation of therapy. Treatment-limited neuropathy also has beenobserved.

Alternate Treatments in Asthma 297
IVIG
IVIG has been shown to reduce immediate skin test reactivity to allergens, to decrease totalserum IgE levels, to inhibit lymphocyte activation and the production of IL-2 and IL-4 invivo, and to suppress cytokine-dependent lymphocyte proliferation in vitro.61-63 IVIG alsohas been shown to increase lymphocyte sensitivity to the suppressive effects ofdexamethasone, even in patients with prior documented steroid resistance.64
Monthly administration of high-dose IVIG resulted in a three-fold reduction in the oralglucocorticoid dose, a reduction in symptoms, and in improved PEFRs with decreases inserum total IgE levels and skin test reactivity to allergens.61 These steroid-sparing effectsare observed both in adult and pediatric asthma patients.65,66 Other investigators haveobserved after 3 months of therapy a reduction of oral corticosteroid doses67, others failedto identify a significant difference in steroid dose reductions, pulmonary function testingresults, or the number of clinical exacerbations among patients in the IVIG groups and theplacebo group.68
The minor adverse effects of therapy include headache and nausea, which generally occurwith the infusion and are self-limiting. Although current commercial preparations shouldhave no risk of transmission of viral hepatitis, there remains the remote possibility of IVIGtransmission of a yet-undefined viral illness. More serious reactions can be associated withpatients who have IgA deficiency, and IVIG administration should be avoided in thispopulation. It also has been rarely associated with interstitial nephritis and aseptic meningitis,as was mentioned above.68
Heparin
Heparin is an endogenous glycosaminoglycan and is extensively used as an anticoagulant.Its flexible structure and high anionic charge allow heparin to interact with a variety ofmolecules in vivo and it is involved in airway inflammation. Elevated levels of heparin-likeanticoagulants have been demonstrated in atopic asthma patient and have been induced insome patients after antigen inhalational challenge leading to further interest in investigatingthe role of heparin in this disease.69-71
Heparin binds and inhibits a variety of cytotoxic and inflammatory mediators, includingeosinophilic cation protein and peroxidase.72 It also increases the association rate of secretoryleukocyte protease inhibitor with human neutrophil elastase and cathepsin G, reducingtheir activity.73 Heparin has been associated with the inhibition of lymphocyte activation74,neutrophil chemotaxis75, smooth muscle growth, and vascular tone.76 It also reducescomplement activation.77 It has been suggested78 that the sulfate groups on the heparinmolecule may attenuate antigen-induced bronchoconstriction via the inhibition of inositol1,4,5 triphosphate-dependent, IgE-mediated mast cell histamine release. Perhaps heparinis bound to cell surface proteins in the airway epithelium may modulate smooth muscletone either by inhibiting inositol 1,4,5 triphosphate-mediated calcium release or bypreventing C-fiber stimulation, decreasing bronchial responsiveness, and reducing airwayhyperreactivity.69
Subjective improvements in asthma symptoms have been reported with the use of IVheparin.79-81 Inhaled heparin therapy administered at a maximum dose of 80,000 U preservedspecific airway conductance (sGaw) better than did 20 mg inhaled cromolyn or placebofollowing exercise82 but not following histamine challengen.83-85 Conflicting data exist

298 Bronchial Asthma
regarding the effects of heparin pretreatment on the early asthmatic response to inhaledallergen challenge (dust mite extract), with mild but statistically significant protective effectsin FEV1 seen 7 to 8 h postchallenge (p < 0.05). Trials examining the effects of inhaled heparinon bronchoprovocation using methacholine also have yielded mixed results, with variableeffects on the provocative concentration of methacholine causing a 20% fall in FEV1, butwith no significant effects on FEV, airway resistance, or sGaw postchallenge. Two cases ofcorticosteroid-resistant asthma patients who responded to 100,000 U inhaled heparin duringasthma exacerbations have been reported.
No adverse effects associated with the use of inhaled heparin at the doses describedabove have been reported. Heparin inhalation alone has not been demonstrated to affectbaseline FEV1 despite the frequent use of isotonic saline solution as its carrier. No bleedingcomplications have been reported, and no significant changes in serum partial prothrombintime or anti-factor Xa activity have been observed with unfractionated and low-molecular-weight heparin, respectively.
Furosemide and Other Diuretics
Changes in water concentration and surface osmolarity of the airway epithelium areimportant contributing factors to exercise-induced bronchospasm that prompted the firstuse of inhaled frusemide (ie, furosemide) as a potential treatment for asthma.86 Furosemideis a loop diuretic that acts in the kidney by inhibiting the Na+/K+/2 Cl- cotransporter in theascending limb of the loop of Henle. Despite early speculations about the effects of furosemideon airway water concentration, its mechanism of action does not appear to be related to thediuretic effects of the drug. Furosemide is not effective against asthma when administeredorally at the usual diuretic doses and must be inhaled at relatively high doses (i.e. 20 to 40 mg)for significant antiasthma effects.86
Furosemide attenuates bronchoconstriction by reducing apical chloride channel activityand by decreasing the potential difference and short-circuit current in airway epithelialcells.87,88 The drug’s inhibition of chloride transport also appears to inhibit the release ofeosinophil mediators89 and may be related to the modulatory effects observed on presynapticneuropeptide release from noncholinergic, nonadrenergic sensory nerves and cholinergicneural responses in animal models.90 Furosemide acts by inhibiting the release of histamineand leukotrienes from passively sensitized human lung.91 Conflicting data exist regardingthe effects of furosemide on airway prostaglandins. Furosemide is well-known to enhancerenal synthesis of prostaglandin E2
92 and the stimulation of inhibitory prostaglandins fromthe airway epithelium may be the cause of its protective role in some challenges.93 Further,it is suggested that the drug inhibits the production of bronchoconstricting prostaglandins.94
Effects of cyclooxygenase inhibitors on the activity of furosemide have been mixed95,96
reinforcing the lack of clarity in this area. Other postulated mechanisms of action based onanimal data include the reduction of airway temperature variation through local airwayvasodilation following dry air challenge97 and the enhancement of paracellular watermovement in response to an osmotic stimulus.98 However, other data have shown thatfurosemide has little99 or no effect100 on mucociliary clearance, an indirect measure of therate of recovery of periciliary fluid volume after isocapnic hyperventilation.
Furosemide appears to attenuate the effects of indirect bronchoconstrictor mechanisms,including early and late responses to allergen101 and the effects of exercise86 distilled water102
adenosine 5'-monophosphate103 sodium metabisulfite104,105, aspirin105 and propranolol.106

Alternate Treatments in Asthma 299
Bronchoconstrictors that work directly on airway smooth muscle like histamine103
methacholine104,107 and prostaglandin F2α108 are not affected by furosemide. The similarities
between the protective spectrum of furosemide and cromolyn have led to speculation abouta common mechanism of action, although cromolyn has been shown to have a statisticallygreater protective effect on airway reactivity when equal doses of the two inhaled drugswere compared.109
Inhaled furosemide has been shown to inhibit the cough response induced by theinhalation of low-chloride-content solutions110 in healthy volunteers, but not in asthmaticpatients.111 The presumed effect of the drug is due to changing the local concentration ofchloride ions in the vicinity of myelinated afferent nerve fibers that are acting as coughreceptors at the airway surface.112
A significant steroid-sparing effect using a combination of lysine acetylsalicylate (LASA)and furosemide is observedin severe steroid-dependent asthma for 10 to 28 weeks.113-115
Effect of inhaled furosemide in acute asthma exacerbations showed mixed results.115 SerialFEV1 measurements showed a mean increase of 14.9 ± 10.5% in FEV1 with furosemide therapyalone (difference not significant) compared to an increase of 42.9 ± 15.2% with metaproterenoltherapy (p = 0.003) and no additive benefit with combination therapy.116 Others have reportedsimilar results.117 Others reported statistically significant improvements in PEFRs (p < 0.05)in acute asthma exacerbations, FEV1 had increased by > 2 SDs compared to those receivingplacebo, although no differences between the good responders and the poor responderscould be identified.118 A follow-up case series119 reported clinical improvement in 9 of 11patients with severe asthma exacerbations that were refractory to conventional medicaltherapy with the addition of inhaled furosemide.
Furosemide can cause allergic reactions due to its incorporated sulfa moiety and hasbeen reported to cause ototoxicity with high-dose rapid IV infusion. None of the clinicaltrials using inhaled furosemide have reported significant side effects, and no diuretic effecthas been reported.
CONCLUSION
Standard anti-asthma therapy is highly successful in most patients. Therefore, the use ofalternate agents for treating asthma should be reserved for the steroid-resistant asthmapatient or for the steroid-dependent asthma patient in whom a thorough evaluation toexclude other diagnoses and exacerbating factors has been performed.
Of all the agents that have been examined in prospective randomized trials, methotrexateand gold appear to be the most important in terms of steroid-sparing and side effect.Methotrexate has been shown to reduce oral corticosteroid requirements modestly in steroid-dependent asthma patients in some short-term, randomized, clinical trials, although itsmechanism of action remains unclear and the data examining this issue remain limited andconflicting. Two case series have suggested that long-term therapy with methotrexate maybe required to demonstrate objective benefit. Gold also has significant steroid-sparing effectsin patients with high daily corticosteroid requirements, but this conclusion must be madewith caution due to the confounding effects of the high dropout rate in the AuranofinMulticenter Drug Trial. Side effects with gold therapy are common but generally are minorand self-limited with dose reduction or the cessation of therapy. Until further data fromcontrolled clinical trials are available, however, it is unclear whether methotrexate or gold

300 Bronchial Asthma
offers a significant risk/benefit ratio compared to close follow-up, intensive standardtherapy, and patient education alone.
Cyclosporine offers an attractive mechanism of action and reasonable efficacy data intwo of three small prospective randomized trials, but it also carries with it a significant sideeffect profile. Because of the risk of permanent renal damage and the need for intensivemonitoring, further studies with larger prospective randomized trials should be performedbefore cyclosporine is considered as an appropriate alternate agent for asthma therapy.
Although troleandomycin (TAO) appears to be an effective methylprednisolone dose-reducing agent, the drug has not been shown to significantly improve asthma control or toreduce steroid-related side effects, and it was associated with an increased rate ofosteoporosis and hypercholesterolemia in one clinical trial. Data demonstrating the beneficialeffects of IVIG also are limited, while cost, convenience, and a possible risk of asepticmeningitis are all potential detractors to this therapy. At this time, the use of TAO appearsto offer no advantage over conventional asthma therapy and patient education, and therapywith IVIG should be limited to clinical trials.
Although its postulated mechanism of action and effects on exercise-inducedbronchospasm are intriguing, the majority of clinical data currently available on inhaledheparin therapy is limited to single-blind trials involving < 20 patients. The enoxaparindata suggest that the accepted dosing regimens may be too low to demonstrate a fulltherapeutic effect, and larger prospective placebo-controlled trials are needed to determinethe efficacy, dose, and patient population that may benefit from this therapy. Furosemideand other loop diuretics appear to attenuate a variety of indirect bronchoconstrictormechanisms, although their exact mechanism of action remains unknown. The fact thatloop diuretics seem to have little direct effect on bronchial smooth muscle is a likelyexplanation for their lack of effect as a single agent or in combination with β-agonists alone inpatients with asthma exacerbations. The current data and the lack of significant side effectsmake them potential steroid-sparing agents in the long-term treatment of mild persistent-to-severe asthma. The current clinical data are limited, however, and larger randomizedtrials are necessary to confirm the efficacy of loop diuretics and their role in the treatment ofasthma.
REFERENCES
1. Niven AS, Argyros G. Alternate treatment in asthma. Chest 2003;123:1254-65.2. Boushey HA. Experiences with monoclonal antibody therapy for allergic asthma. J Allergy Clin
Immunol 2001;108:S77-S83.3. Oettgen HC, Geha RS. IgE regulation and roles in asthma pathogenesis. J Allergy Clin Immunol
2001;107:429-40.4. Mohapatra SS, San Juan HS. Novel immunotherapeutic approaches for the treatment of allergic
diseases. Immunol Allergy Clin North Am 2000;20:625-42.5. Suarez CR, Pickett WC, Bell DH, et al. Effect of low dose methotrexate on neutrophil chemotaxis
induced by leukotriene B4 and complement C5a. J Rheumatol 1987;14:9-11.6. Cronstein, BN Molecular mechanism of methotrexate action in inflammation. Inflammation
1992;16:411-423.7. Lynch JP, McCune, WJ Immunosuppressive and cytotoxic pharmacotherapy for pulmonary
disorders. Am J Respir Crit Care Med 1997;155:395-420.8. Tsai JJ, Wang TJ, Wang SR. The inhibitory effect of methotrexate on PAF-induced neutrophil
and eosinophil locomotion in asthmatic patients. Asian Pac J Allergy Immunol 1994;12:65-71

Alternate Treatments in Asthma 301
9. Glynn-Barmhart, AM Erzurum, SC Leff, JA, et al. Pharmacokinetics of low-dose methotrexatein adult asthmatics. Pharmacotherapy 1992;12:383-390.
10. Vrugt B, Wilson S, Bron A, et al. Low-dose methotrexate treatment in severe glucocorticoid-dependent asthma: effect on mucosal inflammation and in vitro sensitivity to glucocorticoids ofmitogen-induced T-cell proliferation. Eur Respir J 2000;15:478-85.
11. Mullarkey MF, Blumenstein BA, Andrade WP, et al. Methotrexate in the treatment ofcorticosteroid dependent asthma. N Engl J Med 1988;318,603-07.
12. Shiner RJ, Nunn AJ, Chung KF, et al. Randomized, double-blind, placebo controlled trial ofmethotrexate in steroid dependent asthma. Lancet 1990;336:137-40.
13. Erzurum SC, Leff JA, Cochran JE, et al. Lack of benefit of methotrexate in severe, steroid-dependent asthma. Ann Intern Med 1991;114:353-60.
14. Dyer PD, Vaughan TR, Weber RW. Methotrexate in the treatment of steroid-dependent asthma.J Allergy Clin Immunol 1991;88:208-12.
15. Trigg, CJ, Davies, RJ Comparison of methotrexate 30 mg per week with placebo in chronic steroid-dependent asthma: A 12-week double-blind, cross-over study. Respir Med 1993;87,211-16.
16. Taylor DR, Flannery EM, Herbison GP, et al. Methotrexate in the management of severe steroid-dependent asthma. N Z Med J 1993;106:409-11.
17. Stewart GE, Diaz JD, Lockey RF, et al Comparison of oral pulse methotrexate with placebo inthe treatment of severe glucocorticoid-dependent asthma. J Allergy Clin Immunol 1994;94:482-89.
18. Coffey MJ, Sanders G, Eschenbacher WL, et al. The role of methotrexate in the management ofsteroid-dependent asthma. Chest 1994;105:117-21.
19. Kanzow G, Nowak D, Magnussen, H Short-term effects of methotrexate in severe steroid-dependent asthma. Lung 1995;173:223-31.
20. Ogirala, RG, Sturm, TM, Aldrich, TK, et al. Single, high-dose intramuscular triamcinoloneacetonide vs weekly oral methotrexate in life-threatening asthma: A double-blind study. Am JRespir Crit Care Med 1995;152,1461-66.
21. Hedman J, Seideman P, Albertioni F, et al. Controlled trial of methotrexate in patients withsevere chronic asthma. Eur J Clin Pharmacol 1996;49:347-49.
22. Marin MG. Low-dose methotrexate spares steroid usage in steroid-dependent asthmatic patients:A meta-analysis. Chest 1997;112:29-33.
23. Aaron SD, Dales RE, Pham B. Management of steroid-dependent asthma with methotrexate: Ameta-analysis of randomized clinical trials. Respir Med 1998;92:1059-65.
24. Davies H, Olson L, Gibson P. Methotrexate as a steroid sparing agent for asthma in adults(Cochrane Review). The Cochrane Library, Issue 4 2000 Update Software. Oxford: UK.
25. Mullarkey MF, Lammert JK, Blumenstein BA. Long-term methotrexate treatment in cortico-steroid-dependent asthma. Ann Intern Med 1990;112:577-81.
26. Shiner RJ, Katz I, Shulimzon T, et al. Methotrexate in steroid-dependent asthma: Long-termresults. Allergy 1994;49:565-68
27. Jones G, Mierins E, Karsh J. Methotrexate-induced asthma. Am Rev Respir Dis 1991;143:179-81.28. Spector SL, Katz FH, Farr RS. Troleandomycin: Effectiveness in steroid-dependent asthma and
bronchitis. J Allergy Clin Immunol 1974;54:367-79.29. Ong KS, Grieco MH, Rosner W. Enhancement by oleandomycin of the inhibitory effect of
methylprednisolone on phytohemagglutinin-stimulated lymphocytes. J Allergy Clin Immunol1978;62:115-18.
30. Szefler SJ, Rose JQ, Ellis EF, et al. The effect of troleandomycin on methylprednisolone elimination.J Allergy Clin Immunol 1980;66:447-51.
31. Townley RG, Selenke WM. Metabolic effects of macrolide antibiotics on bronchial asthma,experimental anaphylaxis and corticosteroid metabolism: Ninth International Congress of Allergy(vol 144) 1967;90 Excerpta Medica. Hillsborough, NJ.

302 Bronchial Asthma
32. Itkin IH, Menzel ML. The use of macrolide antibiotic substances in the treatment of asthma.J Allergy 1970;45:146-62.
33. Siracusa A, Brugnami G, Fiordi T, et al. Troleandomycin in the treatment of difficult asthma.J Allergy Clin Immunol 1993;92:677-82.
34. Zeiger RS, Schatz M, Sperling W, et al. Efficacy of troleandomycin in outpatients with severe,corticosteroid-dependent asthma. J Allergy Clin Immunol 1980;66:438-46.
35. Wald JA, Friedman BF, Farr RS. An improved protocol for the use of troleandomycin (TAO) inthe treatment of steroid-requiring asthma. J Allergy Clin Immunol 1986;78:36-43.
36. Nelson HS, Hamilos DL, Corsello PR, et al. A double-blind study of troleandomycin andmethylprednisolone in asthmatic subjects who require daily corticosteroids. Am Rev Respir Dis1993;147:398-404.
37. Dasgupta A, Marcoux JP Hepatic abnormalities associated with long-term use of troleandomycinin asthma: A case report. Ann Allergy 1978;41:297-98.
38. Larrey D, Amouyal G, Danan G, et al. Prolonged cholestasis after troleandomycin-induced acutehepatitis. J Hepatol 1987;4:327-29.
39. Uzzan B, Vassy R, Nicholas P, et al. Troleandomycin hepatotoxicity: A case report of overtjaundice and a placebo-controlled trial. Therapie 1993;48:61-62.
40. Ledford DK. Treatment of steroid-resistant asthma. Immunol Allergy Clin North Am 1996;16:777-796.
41. Walz DT, DeMartino MJ, Griswold DE, et al. Biologic actions and pharmacodynamic studies ofauranofin. Am J Med 1983;75,90-108.
42. Marone G, Columbo M, Galeone D, et al. Modulation of the release of histamine and aracidonicacid metabolites from human basophils and mast cells by auranofin. Agents Actions 1986;18:100-02.
43. Parente J, Wong K, David P, et al. Effects of gold compounds on leukotriene B4, leukotriene C4and prostaglandin E2 production by polymorphonuclear leukocytes. J Rheumatol 1986;3:47-51.
44. Bernstein DI, Berstein IL, Bodenheimer SS, et al. An open study of auranofin in the treatment ofsteroid-dependent asthma. J Allergy Clin Immunol 1988;81:6-16
45. Suzuki S, Okubo M, Kaise S, et al. Gold sodium thiomalate selectively inhibits interleukin-5-mediated eosinophil survival. J Allergy Clin Immunol 1995;96:251-56.
46. Alvarez J, Szefler SJ. Alternative therapy in severe asthma. J Asthma 1992;29:3-11.47. Honoma M, Tamura G, Shirato K, et al. Effect of an oral gold compound, auranofin, on non-
specific bronchial hyperresponsiveness in mild asthma. Thorax 1994;49:649-651.48. Klaustermeyer WB, Noritake DT, Kwong FK, et al. Chrysotherapy in the treatment of steroid-
dependent asthma. J Allergy Clin Immunol 1987;79:720-25.49. Bernstein IL, Bernstein DI, Dubb JW, et al. A placebo-controlled multicenter study of auranofin
in the treatment of patients with corticosteroid-dependent asthma: Auranofin Multicenter DrugTrial. J Allergy Clin Immunol 1996;98:317-24.
50. Muranaka MM, Miyamoto T, Shida T, et al. Gold salt in the treatment of bronchial asthma: Adouble blind study. Ann Allergy 1978;40:132-37.
51. Nierop G, Gijzel WP, Bel EH, et al. Auranofin in the treatment of steroid dependent asthma: Adouble blind study. Thorax 1992;47:349-54.
52. Sihra BS, Kon OM, Durham SR, et al. Effect of cyclosporin A on the allergen-induced late asthmaticreaction. Thorax 1997;52:447-52.
53. Frew AJ, Plummeridge MJ. Alternative agents in asthma. J Allergy Clin Immunol 2001;108:3-10.54. Alexander AG, Barnes NC, Kay AB, et al. Clinical response to cyclosporin in chronic severe
asthma is associated with reduction in serum soluble interleukin-2 receptor concentrations. EurRespir J 1995;8:574-78.
55. Sano T, Nakamura Y, Matsunaga Y, et al. FK506, and cyclosporin A inhibit granulocyte/macrophage colony-stimulating factor production by mononuclear cells in asthma. Eur Respir J1995;8:1473-79.

Alternate Treatments in Asthma 303
56. Mori A, Suko M, Nishizaki Y, et al. IL-5 production by CD4+ T cells of asthmatic patients issuppressed by glucocorticoids and the immunosuppressants FK506 and cyclosporine A. IntImmunol 1995;7:449-57.
57. Khan LN, Kon OM, MacFarlane A. Attenuation of the allergen-induced late asthmatic reactionby cyclosporin A is associated with inhibition of bronchial eosinophils, interleukin-5, granulocytemacrophage colony-stimulating factor and eotaxin. Am J Respir Crit Care Med 2000;162:1377-82.
58. Alexander AG, Barnes NC, Kay AB. Trial of cyclosporin in corticosteroid-dependent chronicsevere asthma. Lancet 1992;339:324-28.
59. Lock SH, Kay AB, Barnes NC. Double-blind, placebo-controlled study of cyclosporin A as acorticosteroid-sparing agent in corticosteroid-dependent asthma. Am J Respir Crit Care Med1996;153:509-14.
60. Nizankowska E, Soja J, Pinis G, et al. Treatment of steroid-dependent bronchial asthma withcyclosporin. Eur Respir J 1995;8:1091-99.
61. Mazer BD, Gelfand EW. An open-label study of high dose intravenous immunoglobulin in severechildhood asthma. J Allergy Clin Immunol 1991;87:976-83.
62. Amran D, Renz H, Lack G, et al. Suppression of cytokine-dependent human T-cell proliferationby intravenous immunoglobulin. Clin Immunol Immunopathol 1994;73:180-86.
63. Leung DY, Burns J, Newburger J, et al. Reversal of immunoregulatory abnormalities in Kawasakisyndrome by intravenous gammaglobulin. J Clin Invest 1987;79:468-72.
64. Spahn JD, Leung DY, Chan MT, et al. Mechanisms of glucocorticoid reductions in asthmaticpatients treated with intravenous immunoglobulin. J Allergy Clin Immunol 1999;103:421-26.
65. Jakobsson T, Croner S, Kjellman N, et al. Slight steroid-sparing effects of intravenousimmunoglobulin in children and adolescents with moderately severe bronchial asthma. Allergy1994;49:413-20.
66. Landwehr LP, Jeppson JD, Katlan MG. Benefits of high-dose IV immunoglobulin in patientswith severe steroid-dependent asthma. Chest 1998;114:1349-56.
67. Salmun LM, Barlan I, Wolf HM, et al. Effect of intravenous immunoglobulin on steroidconsumption in patients with severe asthma: A double-blind, placebo-controlled, randomizedtrial. J Allergy Clin Immunol 1999;103:810-15.
68. Kishiyama JL, Valacer D, Cunningham-Rundles C, et al. A multi-center, randomized, double-blind, placebo-controlled trial of high-dose intravenous immunoglobulin for oral corticosteroid-dependent asthma. Clin Immunol 1999;91:126-33.
69. Ragazzi E, Chinellato A. Heparin: pharmacological potentials from atherosclerosis to asthma.Gen Pharmacol 1995;26:697-701.
70. Lasser EC, Lang JH, Curd JG, et al. The plasma contact system in atopic asthma. J Allergy ClinImmunol 1983;72:83-88.
71. Lasser EC, Simon RA, Lyon SG, et al. Heparin-like anticoagulants in asthma. Allergy 1987;42:619-25.
72. Motojima S, Frigas E, Wegering DA, et al. Toxicity of eosinophil cationic protein from guinea-pig tracheal epithelium in vitro. Am Rev Respir Dis 1989;139:801-05.
73. Fath MA, Wu X, Hileman RE, et al. Interaction of secretory leukocyte protease inhibitor withheparin inhibits proteases involved in asthma. J Biol Chem 1998;273:13563-569.
74. Lider O, Mekori YA, Miller T, et al. Inhibition of T lymphocyte heparanase by heparin preventsT cell migration and T cell-mediated immunity. Eur J Immunol 1990;20:493-99.
75. Matzner Y, Marx G, Drexler R, et al. The inhibitory effect of heparin and related glyco-saminoglycans on neutrophil chemotaxis. Thromb Haemost 1984;52:134-37.
76. Karnovsky MJ, Edelman ER. Heparin/heparin sulphate regulation of vascular smooth musclebehavior. Black, J Page, CP (Eds). Airways and vascular remodeling in asthma and cardiovasculardisease: Implications for therapeutic intervention. 1994,45-70 Academic Press. London, UK.

304 Bronchial Asthma
77. Ekre HPT, Fjellner B, Hagermark O. Inhibition of complement dependent experimentalinflammation in human skin by different heparin fractions. Int J Immunopharmacol 1986;8:277-86.
78. Ahmed T, Syriste T, Mendelssohn R, et al. Heparin prevents antigen-induced airway hyper-responsiveness: Interference with IP3-mediated mast cell degranulation. J Appl Physiol1994;76:893-901.
79. Dolowitz DA, Dougherty TF. The use of heparin as an anti-inflammatory agent. Laryngoscope1960;70:873-84.
80. Boyle JP, Smart RH, Shirey JK. Heparin in the treatment of chronic obstructive broncho-pulmonary disease. Am J Cardiol 1964;14:25-28.
81. Fine NL, Shim C, Williams MH. Objective evaluation of heparin in the treatment of asthma. AmRev Respir Dis 1968;98:886-87.
82. Ahmed T, Abraham WM, D’Brot, J. Effects of inhaled heparin on immunologic and nonimmuno-logic bronchoconstrictor responses in sheep. Am Rev Respir Dis 1992;145:566-70.
83. Ahmed T, Garrigo J, Danta I. Preventing bronchoconstriction in exercise-induced asthma withinhaled heparin. N Engl J Med 1993;329:90-95.
84. Garrigo J, Danta I, Ahmed T. Time course of the protective effect of inhaled heparin on exercise-induced asthma. Am J Respir Crit Care Med 1996;153:1702-07.
85. Ahmed T, Gonzalez BJ, Danta I. Prevention of exercise-induced bronchoconstriction by inhaledlow-molecular-weight heparin. Am J Respir Crit Care Med 1999;160:576-81.
86. Bianco S, Vaghi A, Robuschi M, et al. Prevention of exercise-induced bronchoconstriction byinhaled frusemide. Lancet 1988;2:252-55.
87. Welsh MJ. Inhibition of chloride secretion by furosemide in canine tracheal epithelium. J MembrBiol 1993;71:218-26.
88. Alton EW, Kingsleigh-Smith DJ, Munkonge FM, et al. Asthma prophylaxis agents alter thefunction of an airway epithelial chloride channel. Am J Respir Cell Mol Biol 1996;14:380-87.
89. Perkins R, Dent G, Chung KF, et al. Effect of anion transport inhibitors and extracellular Cl-concentrations on eosinophil respiratory burst activity. Biochem Pharmacol 1992;107;481-88.
90. Elwood W, Lotvall JO, Barnes PJ, et al. Loop diuretics inhibit cholinergic and non-cholinergicerves in guinea pig airways. Am Rev Respir Dis 1991;143:1340-44.
91. Anderson SD, Wei HE, Temple DM. Inhibition by furosemide of inflammatory mediators fromlung fragments [letter]. N Engl J Med 1991;324:131.
92. Miyanoshita A, Terada M, Endou H. Furosemide directly stimulates prostaglandin E2 productionin the thick ascending limb of Henle’s loop. J Pharmacol Exp Ther 1989;251:1155-59.
93. Barnes PJ. Diuretics and asthma. Thorax 1993;48:195-96.94. Levasseur-Acker GM, Molimard M, Regnard J, et al. Effect of furosemide on prostaglandin
synthesis by human nasal and bronchial epithelial cells in culture. Am J Respir Cell Mol Biol1994;10:378-83.
95. Pavord ID, Wisniewski A, Tattersfield AE. Inhaled furosemide and exercise induced asthma:Evidence of a role for inhibitory prostanoids. Thorax 1992;47:797-800.
96. O’Connor BJ, Barnes PJ, Chung KF. Inhibition of sodium metabisulphite induced broncho-constriction by frusemide in asthma: Role of cyclooxygenase products. Thorax 1994;49:307-11.
97. Gilbert IA, Lenner KA, Nelson JA, et al. Inhaled furosemide attenuates hyperpnea-inducedobstruction and intra-airway thermal gradients. J Appl Physiol 1994;76:409-15.
98. Freed AN, Taskar V, Schofield B, et al. Effect of furosemide on hyperpnea-induced airwayobstruction, injury and microvascular leakage. J Appl Physiol 1996;81:2461-67.
99. Daviskas E, Anderson SD, Gonda I, et al. Mucociliatory clearance during and after isocapnichyperventilation with dry air in the presence of frusemide. Eur Respir J 1996;9:716-24.
100. Hasani A, Pavia D, Spiteri MA, et al. Inhaled frusemide does not affect lung mucociliary clearancein health and asthmatic subjects. Eur Respir J 1994;7:1497-1500.

Alternate Treatments in Asthma 305
101. Bianco S, Pieroni MG, Refini RM, et al. Protective effect of inhaled furosemide on allergen-induced early and late asthmatic reactions. N Engl J Med 1989;321,1069-73.
102. Moscato G, Dellabianca A, Falagiani P, et al. Inhaled furosemide prevents both the broncho-constriction and the increase in neutrophil chemotactic activity induced by ultrasonic “fog” ofdistilled water in asthmatics. Am Rev Respir Dis 1991;143:561-66.
103. O’Conner BJ, Chung KF, Chen-Worsdell, YM, et al. Effect of inhaled furosemide and bumetanideon adenosine 5'-monophosphate- and sodium metabisulfate-induced bronchoconstriction inasthmatic subjects. Am Rev Respir Dis 1991;143:1329-33.
104. Nichol GM, Alton EW, Nix A, et al. Effect of inhaled furosemide on metabisulfite- and methacho-line-induced bronchoconstriction and nasal potential difference in asthmatic subjects. Am RevRespir Dis 1990;142:576-80.
105. Vargas FS, Croce M, Teizeira LR, et al. Effect of inhaled furosemide on the bronchial response tolysine-aspirin inhalation in asthmatic subjects. Chest 1992;102:408-11.
106. Myers JD, Higham MA, Shakur BH, et al. Attenuation of propranolol-induced broncho-constriction by furosemide. Thorax 1997;52:861-65.
107. Vaghi A, Robuschi M, Chilaris M, et al. Inhaled furosemide does not alter the bronchial responseto methacholine in asthmatics. Eur Respir J 1988;1:85.
108. Stone RA, Yeo TC, Barnes PJ, et al. Frusemide inhibits cough but not bronchoconstriction toprostaglandin F2α in patients with asthma. Am Rev Respir Dis 1991;143:A548.
109. Stone RA, Barnes PJ, Chung KF. Effect of frusemide on cough responses to chloride-deficientsolution in normal and mild asthmatic subjects. Eur Respir J 1993;6:862-67.
110. Siffredi M, Mastropasqua B, Pelucchi A, et al. Effect of inhaled furosemide and cromolyn onbronchoconstriction induced by ultrasonically nebulized distilled water in asthmatic subjects.Ann Allergy Asthma Immunol 1997;78:238-43.
111. Ventresca PG, Nichol GM, Barnes PJ, et al. Inhaled frusemide inhibits cough induced by low-chloride solutions but not by capsaicin. Am Rev Respir Dis 1990;142,143-46.
112. Stone RA, Barnes PJ, Chung KF. Effect of frusemide on cough response to low-chloride solutionin subjects with mild asthma. Thorax 1991;46:752P.
113. Chung KF Furosemide and other diuretics in asthma. J Asthma 1994;31:85-92.114. Bianco S, Robuschi M, Vaghi A, et al. Steroid sparing effect of inhaled lysine-aspirin and
furosemide in steroid-dependent asthma. Melillo, G O’Byrne, PH Marone, G (Eds). RespiratoryAllergy 1993,261-69 Elsevier. Amsterdam, the Netherlands.
115. Bianco S, Vaghi A, Robuschi M, et al. Steroid-sparing effect of inhaled lysine acetylsalicylateand furosemide in high-dose beclomethasone-dependent asthma. J Allergy Clin Immunol1995;95,937-43.
116. Karpel JP, Dworkin F, Hager D, et al. Inhaled furosemide is not effective in acute asthma. Chest1994;106:1396-1400.
117. Pendino JC, Nannini LJ, Chapman KR. Effect of inhaled furosemide in acute asthma. J Asthma1998;35,89-93.
118. Ono Y, Kondo T, Tanigaki T, et al. Furosemide given by inhalation ameliorates acute exacerbationsof asthma. J Asthma 1997;34:283-89.
119. Tanigaki T, Kondo T, Hayashi Y, et al. Rapid response to inhaled frusemide in severe acuteasthma with hypercapnia. Respiration 1997;64:108-10.

306 Bronchial Asthma
INTRODUCTION
Severe or fatal asthma or refractory asthma constitutes about <5% of all the asthmatics. Theentity is poorly understood clinically, physiologically, and pathologically. Severe forms ofthe disease often remain refractory to the best current medical care. Although some patientswith severe asthma have had severe disease for most of their lives, a second group developssevere disease in adulthood. It is not clear which genetic and environmental elements maybe the most important in the development of severe disease. Physiologically, these patientsoften have air-trapping and may have loss of elastic recoil, as well. The pathologydemonstrates a wide variety of findings, those include continued eosinophilic inflammation,structural changes, distal disease, and, in at least one third of patients, a different pathology.Treatment remains problematic. These patients respond poorly to the usual treatment andare very difficult to manage. Accordingly the cost of treatment is very high with pooroutcomes. The introduction of high-potency inhaled corticosteroids (CS) had a markedimpact on the numbers of patients who were dependent on therapy with oral CS. However,beyond those medications, little further progress has been made in understanding the diseaseand improving its treatment.
Definition
Severe or “refractory” asthma was defined by the workshop sponsored by the AmericanThoracic Society.1 This definition includes the following:
Major Criteria
• Continuous high-dose inhaled corticosteroids or• Oral corticosteroids for > 50% of the previous year
Minor Criteria
• Aspects of lung function,• Exacerbations,• Disease stability,• Amount of additional medications
For a diagnosis of fatal asthma at least one major and additional two of the seven minorcriteria are to be fulfilled. Patients also must have had compliance and exacerbating factors
Severe Asthma(Fatal Asthma, Refractory Asthma)
20

Severe Asthma (Fatal Asthma, Refractory Asthma) 307
should be fully addressed. These definitions are a guide, but the list of criteria still may not bedefinitive and may have many pitfalls. Some suggest that expanding the minor criteriarequirements to three would likely improve the capture of those who fulfill the “spirit” of thedefinition, rather than the “letter” of the definition.
Epidemiology
Very little is known about the development of severe asthma. It is not clear whether mostpatients with severe asthma have a life-altering event in childhood that irreversibly alterstheir lungs, from which they will never recover, or whether they slowly but steadily declineover the years. It is also not certain whether those patients with a history of adult-onsetdisease actually have some level of asthma as children those were ignored, or if at all theyhave a more rapid decline in function once the asthma begins. No satisfactory answer tothese questions has been found although some information has come from the large cohortof asthma patients studied in Melbourne, Australia followed for 35 years.2 Those datasuggest that reduced lung function in childhood leads to reduced lung function in adulthood,although there is little “progressive decline” of the mean data. Two studies3,4 from Europehave suggested that late-onset asthma is associated with a more rapid decline in lungfunction. In the database of > 100 patients with severe asthma who were seen at NationalJewish Medical and Research Center (Denver, CO), approximately two-thirds of patientshad onset in childhood, and the remaining one third experienced onset after the age of20 years.5 Existence of any distinct phenotypic differences in adult-onset and childhoodonset asthma or severe asthma is not known.
Aetiology
Various risk factors for development of severe asthma are shown in Table 20.1.
Table 20.1: Various risk factors for development of severe asthma
GeneticMutations in both the interleukin-4 gene or the interleukin-4 receptorNon-T helper (Th) type 2 factors
Transforming growth factor (TGF)-β1
Monocyte chemotactic protein-1
Environmental factorsAllergens (house dust mite; cockroach; alternaria exposure)SmokingPet allergy
InfectionsRespiratory syncytial virus infections in childhoodMycoplasma and Chlamydia infections in adults
Lung-externa factorsObesity (Increased body mass index)Gastroesophageal reflux diseaseChronic sinusitis
Compliance/adherence to medicationsInadequate response to therapy

308 Bronchial Asthma
As is the case for many diseases, risk factors can be divided into genetic and environ-mental. Unfortunately, asthma itself is a disease involving multiple genes. Severe asthma isnot likely to be different and is less well-studied. There are reports6,7 of relevant mutations inboth the interleukin-4 gene or the interleukin-4 receptor, some of which have been linked toloss of lung function, and others to near-fatal events. Interestingly, two non-T helper (Th) type2 factors also have been associated with severity of asthma, transforming growth factor (TGF)-β1
and monocyte chemotactic protein-1, both of which can promote fibrotic reactions.8,9 Whethermutations of the receptors for the primary treatments for asthma (β2
and glucocorticoid receptors[GRs]) decrease responsiveness to medications and influence outcomes is not yet clear.
Environmental factors include both allergen and tobacco exposure, with the strongest datafor house dust mite, cockroach, and Alternaria exposures.10-12 Additionally, many patientswill continue to smoke or own a cat despite being aware of the negative effects.13 Infection alsomay contribute to severe disease, with respiratory syncytial virus infections implicated inchildhood, while pathogens like Mycoplasma and Chlamydia may play a role in adults.4
Although not precisely “environmental,” additional “lung-external” factors may includeobesity, gastroesophageal reflux disease, and chronic sinusitis. Epidemiologic study of patientswith severe/difficult-to-treat asthma suggested that body mass index increases with increasingseverity of disease and that 76% of the cohort of patients with severe disease were eitheroverweight or obese.14 However, similar to gastroesophageal reflux disease and chronicsinusitis, the relationship of effective treatment of obesity to severity of disease is not clear.
Another external factor related to severity of disease is compliance/adherence tomedications. Studies15 have suggested that in children and adolescents, instability of diseaseis related to adherence to therapy with corticosteroids. Adherence to medication may beinfluenced by lack of responsiveness to medication. If the patient is receiving therapy withoral corticosteroids, the early-morning measurement of cortisol level can be helpful indetermining compliance. If this is not helpful, then treatment trials with injectable long-acting steroids, such as depomethylprednisolone or triamcinolone, can be informative.16
Physiology
Progressive increase in airflow limitation, which is often irreversible leads to a morerapid decline in the FEV1, although there is poor correlation between FEV1 and diseasesymptoms .17 This may be true in some patients. Others may have severe airflow limitationat presentation, while others, particularly adults, may develop a more rapid decline in lungfunction over a 10-year-period of time.4 These changes are not completely irreversible. Theremay be irreversibility to current aggressive medical management. However, it does notnecessarily mean the lungs are in a fixed fibrotic state.
Airway hyperreactivity also plays a role in the severity of asthma. The provocativeconcentration causing a 20% fall in FEV1 with disease severity is often present but is poorindicators.18 This instability may be an important aspect to the symptomatology of a subgroupof patients with severe asthma, in whom continuous airflow limitation may play a role.19
FEV1 and airway reactivity changes do not adequately explain disease severity. It ispossible that other physiologic factors, such as changes in elastic recoil and/or small airwayphysiology, are important. The elastic recoil properties of the lung in asthma patients arenot normal.20,21 Compliance is increased in patients with moderate persistent asthma,although the precise pathologic mechanism behind the change is not clear. It is suggestedthat the airways and the parenchyma are more collapsible than are the airways in healthy

Severe Asthma (Fatal Asthma, Refractory Asthma) 309
individuals.22 The FVC1 slow vital capacity ratio is decreased in a group of patients withsevere asthma who had persistent eosinophilia.23 These patients are at a higher risk of near-fatal events than those with a more normal (1:1) ratio.
There is air-trapping in patients with severe asthma, without associated hyperinflation.Residual volumes are routinely > 200% of predicted in severe asthma, with only modestlyincreased thoracic gas volumes.23 Whether this increase in residual volume may be reflectiveof small airway disease. There is no correlation of physiologic measures with inflammatoryor structural changes.
Pathology
Up to two-thirds of patients with severe asthma have persistent tissue eosinophils, despitecontinued therapy with high-dose systemic steroids. There are associated increases in Tlymphocytes and markers for activation of a Th-2 pathway.23 This pattern of inflammationrepresents steroid resistance, whereas a Th-2 pattern of inflammation persists despite thepresence of high-dose steroid therapy. This lack of effect is due to a number of factors thoseinclude high levels of proinflammatory mediators sequestering the glucocorticoid receptors(GR), diminished binding of the GR to the genome, or increased levels of an alternativelyspliced GR (i.e. GR-β), which has lessened inhibitory capabilities.24-26 Other, non-Th-2,proeosinophilic factors also play a role in the process. These changes lead to poor/modifieddrug response in patients with more unstable asthma.
The apparent progressive loss of lung function in more severe forms of asthma is due tostructural or remodeling changes in the airways and perhaps the parenchyma as wellalthough tthe precise changes are unclear. Numerous structures have been implicated,including the sub-basement membrane (SBM), epithelium, smooth muscle, nerves, and bloodvessels. Although the SBM is thickened in asthma patients, the relationship to disease severityis unclear. Patients with severe asthma with persistent eosinophil levels had the thickestSBM when compared to those of healthy control subjects, patients with milder cases ofasthma. This thickened SBM was seen in association with high numbers of TGF-β-positivecells in the submucosa.23,27 However, the absolute increase in thickness is small and cannotexplain the increase in airflow limitation. It may be used as a marker for abnormalities incomposition, distribution, or quantity of extracellular matrix elements in other regions ofthe airway or parenchyma.
The epithelium is abnormal in asthma patients. There is an increase in the ratio of gobletcells to ciliated epithelial cells. Mucus plugging of the small and medium airways contributesfurther to airflow limitation and air trapping in patients with severe asthma. Other studies28,29
suggested alterations in epithelial growth factor receptor and TGF-β1 and/or TGF-β2 inasthma patients, which may contribute to inappropriate and inadequate repair process,augmenting goblet cell metaplasia and mucus production.
The amount (and perhaps phenotypes) of smooth muscle in the airways of patients withsevere asthma is also increased. Patients dying of status asthmaticus have increased smoothmuscle mass in the airways from the largest airways to nearly the smallest.30 Relationshipof increased airway smooth muscle to severity of disease is possible although relationshipof any of these structural changes to functional changes is not very clear. In addition toairflow limitation, airtrapping, hyperresponsiveness, and loss of elastic recoil/collapsibilityare important. Alterations in the alveolar attachments to the airways and the airwaysthemselves play a role in collapsibility, but the cause of changes in elastic recoil is not clear.

310 Bronchial Asthma
Elastin levels have been shown to be abnormal (i.e. decreased or disordered) in patients whohave died of asthma. The numbers of proteolytic enzymes that alter elastin composition areincreased in several instances in asthma.31,32 It is possible that changes in elastin composition,secondary to chronic inflammatory elements, contribute to the unique structural/functionalrelationships of patients with severe asthma.
Physiologic and pathologic data suggest that inflammatory changes exist in the lungperiphery. Autopsy studies33,34 have suggested that both increased inflammation and wallthickness may exist in patients who have died of asthma, as opposed to those with milderasthma and healthy control subjects. Studies35,36 of living asthma patients also have suggestedthat distal lung inflammation may be more important than proximal lung inflammation.These observations have implications for current drug therapy, as most inhaled medicationsare unlikely to reach the lung periphery in high amounts.37 These structural and inflammatorychanges in the small airway and parenchyma may interact to a greater degree in the smallairways than the large airways due to the smaller general mass of the airway structure.
Classically bronchial asthma has continued eosinophilic inflammation but, patients withsevere asthma have neutrophil predominance or very little inflammation23,38, 39 The patientswithout eosinophils also do not appear to have the same degree of collapsibility and haveless severe asthma attacks, also supporting a different presentation for this disease subtype.Recently CT scan findings confuse the issue whether other, less well-defined obstructivediseases like bronchiolitis obliterans also could masquerade as severe asthma.40
Management
The treatment of severe asthma remains difficult. Corticosteroids remain the drug of choicebecause of their broad and nonspecific effects, and there are few alternatives in existence.Leukotriene modifiers may be helpful in some cases, especially as a large percentage ofpatients with severe asthma may be aspirin-sensitive.41 Anti-IgE also appears to be efficaciousin patients with more severe forms of asthma and may be of benefit in some of these patients.42
Other forms of therapy, such as cyclosporine and methotrexate have limited value.43
However, use of alternate agents in treating asthma patients, whose disease remains poorlycontrolled while receiving standard therapy, may be considered. It is important to recognizethat the reasons for lack of response to treatment are numerous, and the clinical approach tothe patient with poorly controlled asthma must be systematic and individualised. Theobjective confirmation of asthma and the exclusion of other pulmonary conditions withscreening blood tests, chest radiograph, spirometry, bronchoprovocation challenge, andcardiopulmonary exercise testing is vital in any patient who does not respond to asthmatherapy. It is of importance of maximising standard asthma therapy with close outpatientfollow-up, patient education, and compliance monitoring.
The treatment of concomitant gastroesophageal reflux44 and chronic sinusitis45,46 and theremoval of environmental triggers of asthma have been shown to improve asthma control.Glucocorticoid absorption and metabolism can be affected by thyroid disease and a varietyof drugs, including antacids, rifampin, cholestyramine, and numerous antiepileptic agents.47
The increased detection of Mycoplasma pneumoniae and Chlamydia pneumoniae by polymerasechain reaction in the airways of patients with chronic asthma has led to questions regardingtheir role in pathogenesis48 and several small case series49,50 have demonstrated statisticallysignificant improvements in lung function and reductions in bronchial reactivity to histamineafter treatment with macrolides. Due to the significant side effects of many alternate asthma

Severe Asthma (Fatal Asthma, Refractory Asthma) 311
therapies, it is essential to thoroughly address these issues before going for a novel treatmentstrategy.
It is also important to distinguish the “difficult-to-manage” asthma patient from the patientwho is steroid-resistant. This asthma subgroup, which was first described in 196851 ischaracterised by patients with larger than usual daily oral corticosteroid requirements andpoor symptom control, a blunted eosinopenic response to cortisol-21-succinate, and increasedclearance of cortisol. Other clinical characteristics that are associated with steroid resistanceinclude African-American race, symptoms requiring oral glucocorticoid agents at an earlyage, and < 15% improvement in FEV1 following 7 to 14 days of treatment with high-dose(i.e. > 40 mg daily) oral glucocorticoids.52 The recognition and early identification of thesepatients may isolate a subgroup of patients who could benefit from early intervention withalternate asthma therapies with better long-term asthma control and reduction in cortico-steroid side effects.
REFERENCES
1. Wenzel SE, Fahy JV, Irvin CG, et al. Proceedings of the ATS Workshop on Refractory Asthma:Current understanding, recommendations and unanswered questions. Am J Respir Crit CareMed 2000;162:2341-51.
2. Oswald H, Phelan PD, Lanigan A, et al. Childhood asthma and lung function in mid-adult life.Pediatr Pulmonol 1997;23:14-20.
3. Ulrik CS, Lange P. Decline of lung function in adults with bronchial asthma. Am J Respir CritCare Med 1994;150:629-34.
4. Ten Brinke A, van Dissel JT, Sterk PJ, et al. Persistent airflow limitation in adult-onset nonatopicasthma is associated with serologic evidence of Chlamydia pneumoniae infection. J Allergy ClinImmunol 2001;107:449-54.
5. Gibbs R, Miranda C, Wenzel S. Initial demographic information from an extensive data base ofsevere, steroid dependent asthmatics studied at National Jewish. Am J Respir Crit Care Med2002;165,A119.
6. Sandford AJ, Chagani T, Zhu S, et al. Polymorphisms in the IL4, IL4RA, and FCERIB genes andasthma severity. J Allergy Clin Immunol 2000;106:135-40.
7. Burchard EG, Silverman EK, Rosenwasser LJ, et al. Association between a sequence variant inthe IL-4 gene promoter and FEV1 in asthma. Am J Respir Crit Care Med 1999;160:919-22.
8. Pulleyn LJ, Newton R, Adcock IM, et al. TGF-β-1 allele association with asthma severity. HumGenet 2001;109:623-27.
9. Szalai C, Kozma GT, Nagy A, et al. Polymorphism in the gene regulatory region of MCP-1 isassociated with asthma susceptibility and severity. J Allergy Clin Immunol 2001;108:375-81.
10. Squillace SP, Sporik RB, Rakes G, et al. Sensitisation to dust mites as a dominant risk factor forasthma among adolescents living in central Virginia: Multiple regression analysis of a population-based study. Am J Respir Crit Care Med 1997;156:1760-64.
11. Halonen M, Stern DA, Wright AL, et al. Alternaria as a major allergen for asthma in childrenraised in a desert environment. Am J Respir Crit Care Med 1997;155:1356-61.
12. Rosenstreich DL, Eggleston P, Kattan M, et al. The role of cockroach allergy and exposure tocockroach allergen in causing morbidity among inner-city children with asthma. N Engl J Med1997;336:1356-63.
13. Siroux V, Pin I, Oryszczyn MP, et al. Relationships of active smoking to asthma and asthmaseverity in the EGEA study: Epidemiological study on the genetics and environment of asthma.Eur Respir J 2000;15:470-77.
14. Weiss ST, Tager IB, Speizer FE, et al. Persistent wheeze: Its relation to respiratory illness, cigarette

312 Bronchial Asthma
smoking and level of pulmonary function in a population sample of children. Am Rev Respir Dis1980;122:697-707.
15. Milgrom H, Bender B, Ackerson L, et al. Noncompliance and treatment failure in children withasthma. J Allergy Clin Immunol 1996;98:1051-57.
16. Ogirala RG, Sturm TM, Aldrich TK, et al. Single, high-dose intramuscular triamcinolone acetonideversus weekly oral methotrexate in life-threatening asthma: A double-blind study. Am J RespirCrit Care Med 1995;152:1461-66.
17. Teeter JG, Bleecker ER Relationship between airway obstruction and respiratory symptoms inadult asthmatics. Chest 1998;113:272-77.
18. Weiss ST, Van Natta ML, Zeiger RS. Relationship between increased airway responsiveness andasthma severity in the childhood asthma management program. Am J Respir Crit Care Med2000;162:50-56.
19. Chan MT, Leung DY, Szefler SJ, et al. Difficult-to-control asthma: Clinical characteristics ofsteroid-insensitive asthma. J Allergy Clin Immunol 1998;101:594-601.
20. Woolcock AJ, Rebuck AS, Cade JF, et al. Lung volume changes in asthma measured concurrentlyby two methods. Am Rev Respir Dis 1971;104:703-09.
21. Woolcock AJ, Read J. The static elastic properties in the lungs in asthma. Am Rev Respir Dis1968;98:788-94.
22. Gelb AF, Zamel N. Unsuspected pseudophysiologic emphysema in chronic persistent asthma.Am J Respir Crit Care Med 2000;162:1778-82.
23. Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be dividedpathologically into two inflammatory subtypes with distinct physiologic and clinical characteri-stics. Am J Respir Crit Care Med 1999;160:1001-08.
24. Kam JC, Szefler SJ, Surs W, et al. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J Immunol 1993;151:3460-66.
25. Lane SJ, Adcock IM, Richards D, et al. Corticosteroid-resistant bronchial asthma is associatedwith increased c-fos expression in monocytes and T lymphocytes. J Clin Invest 1998;102:2156-64.
26. Leung DY, Hamid Q, Vottero A, et al. Association of glucocorticoid insensitivity with increasedexpression of glucocorticoid receptor beta. J Exp Med 1997;186:1567-74.
27. Minshall EM, Hogg JC, Hamid QA. Cytokine mRNA expression in asthma is not restricted to thelarge airways. J Allergy Clin Immunol 1998;101:386-90.
28. Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factor receptors to gobletcell production in human bronchi. Am J Respir Crit Care Med 2001;163:511-16.
29. Howat WJ, Holgate ST, Lackie PM. TGF-β isoform release and activation during in vitro bronchialepithelial wound repair. Am J Physiol 2002;282:L115-L23.
30. James AL, Pare PD, Hogg JC. The mechanics of airway narrowing in asthma. Am Rev Respir Dis1989;139:242-46.
31. Vignola AM, Riccobono L, Mirabella A, et al. Sputum metalloproteinase-9/tissue inhibitor ofmetalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis.Am J Respir Crit Care Med 1998;158:1945-50.
32. Lemjabbar H, Gosset P, Lamblin C, et al. Contribution of 92 kDa gelatinase/type IV collagenasein bronchial inflammation during status asthmaticus. Am J Respir Crit Care Med 1999;159:1298-1307.
33. Carroll NG, Elliot J, Morton AR, et al. The structure of large and small airways in nonfatal andfatal asthma. Am Rev Respir Dis 1993;147:405-10.
34. Carroll NG, Mutavdzic S, James AL. Distribution and degranulation of airway mast cells innormal and asthmatic subjects. Eur Respir J 2002;19:879-85.
35. Kraft M, Djukanovic R, Wilson S, et al. Alveolar tissue inflammation in asthma. Am J Respir CritCare Med 1996;154:1505-10.

Severe Asthma (Fatal Asthma, Refractory Asthma) 313
36. Balzar S, Wenzel SE, Chu HW. Transbronchial biopsy as a tool to evaluate small airways inasthma. Eur Respir J 2002;20:254-59.
37. Leach CL, Davidson PJ, Boudreau RJ. Improved airway targeting with the CFC-free HFA-beclomethasone metered-dose inhaler compared with CFC-beclomethasone. Eur Respir J1998;12:1346-53.
38. Wenzel SE, Szefler SJ, Leung DYM, et al. Bronchoscopic evaluation of severe asthma: Persistentinflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med 1997;156:737-43.
39. Louis R, Lau LCK, Bron AO, et al. The relationship between airways inflammation and asthmaseverity. Am J Respir Crit Care Med 2000;161:9-16.
40. Jensen SP, Lynch DA, Brown KK, et al. High-resolution CT features of severe asthma andbronchiolitis obliterans. Radiology 2000;217(suppl):595.
41. Virchow JC, Jr. Prasse A, Naya I, et al. Zafirlukast improves asthma control in patients receivinghigh-dose inhaled corticosteroids. Am J Respir Crit Care Med 2000;162:578-85.
42. Holgate S, Bousquet J, Wenzel S, et al. Efficacy of omalizumab, an anti-immunoglobulin Eantibody, in patients with allergic asthma at high risk of serious asthma-related morbidity andmortality. Curr Med Res Opin 2001;17:233-40.
43. Wenzel S. Severe/Fatal Asthma. Chest 2003;123:405S-10S.44. Irwin RS, Curley FJ, French CL. Difficult-to-control asthma: Contributing factors and outcome
of a systematic management protocol. Chest 1993;103:1662-69.45. Rachelefsky GS, Goldberg M, Katz RM, et al. Sinus disease in children with respiratory disease.
J Allergy Clin Immunol 1978;61:310-14.46. Rachelefsky GS, Katz RM, Siegel SC Chronic sinus disease with associated reactive airway disease
in children. Pediatrics 1984;73:526-29.47. Spahn JD, Covar R. Steroid-resistant asthma. Immunol Allergy Clin North Am 2001;21:569-87.48. Kraft M, Cassell GH, Henson JE, et al. Detection of Mycoplasma pneumoniae in the airways of
adults with chronic asthma. Am J Respir Crit Care Med 1998;158:998-1001.49. Ekici A, Ekici M, Erdemoglu AK. Effect of azithromycin on the severity of bronchial hyper-
responsiveness in patients with mild asthma. J Asthma 2002;39:181-85.50. Kraft M, Cassell GH, Pak J, et al. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma:
Effect of clarithromycin. Chest 2002;121:1782-88.51. Schwartz HJ, Lowell FC, Melby JC. Steroid resistance in bronchial asthma. Ann Intern Med
1968;69:493-99.52. Chan MTS, Leung DYM, Szefler SJ, et al. Difficult-to-control asthma: Clinical characteristics of
steroid insensitive asthma. J Clin Immunol 1998;101:594-601.

314 Bronchial Asthma
Asthma in Children
21
The previous chapters have dealt with bronchial asthma in general, which is applicable, bothin cases of adult as well as childhood asthma. However, this chapter will highlight certainimportant points about childhood asthma.
PREVALENCE
International Scene
A worldwide rise in the prevalence of asthma is being reported with increase in wheeze at analarming rate of 5% per year. From 1983 onwards an increase in asthma mortality andmorbidity has been noticed worldwide.1 Data on prevalence of bronchial asthma on childrenare few from most countries but many from countries like Australia and UK.2 Table 21.1shows the prevalence of current asthma, diagnosed asthma, wheeze ever, airwayhyperresponsiveness, and atopy in children.
There are large differences in the prevalence among the rich, partly rich, and poorpopulations, with the highest prevalence found in Australia. It is possible that the differencesmay be as a consequence of responses to different allergens, to different allergen loads, or toother factors in the environment in the affluent and not-so-affluent populations. There aresome suggestions that patients with high levels of parasitic infections are less atopic, althoughthere is no convincing experimental confirmation. This protection of parasitic infectionsagainst asthma may be a cause of less prevalence of the later in many developing countries.Diet may also be a factor. Exposure to allergens may be important although the most commonallergen, the house dust mite has been found everywhere it has been looked for. However,these mites are mainly found in bedding and it is possible that steeping on a bed rather thanon a floor, which many poor children do, increases exposure to them.
There was considerable concern that the prevalence of asthma and allergic diseases isincreasing in Western and developing countries. However, the etiology of these conditionsremains poorly understood, despite a large volume of clinical and epidemiological researchwithin populations that has been directed at explaining why some individuals and notothers develop asthma and allergies. Little is known about such worldwide variations in theprevalence of asthma and allergic diseases. More authentic data was available from theInternational Study of Asthma and Allergies in Childhood (ISAAC) designed in late 90’s.3
The study allowed comparisons between populations in different countries. ISAAC PhaseOne used standardized simple surveys conducted among representative samples of schoolchildren from centres in most regions of the world. Two age groups (13-14 years and
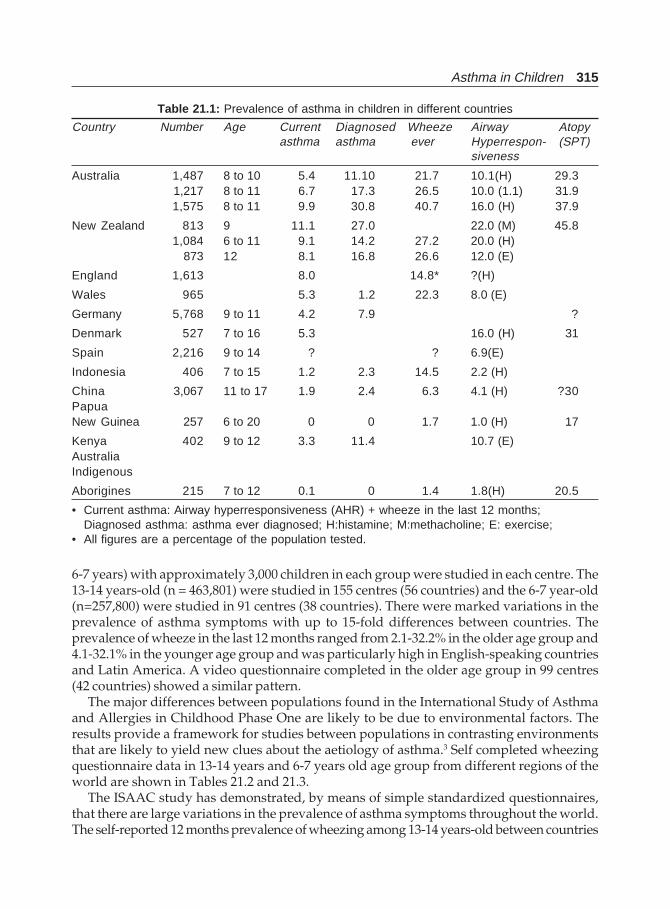
Asthma in Children 315
6-7 years) with approximately 3,000 children in each group were studied in each centre. The13-14 years-old (n = 463,801) were studied in 155 centres (56 countries) and the 6-7 year-old(n=257,800) were studied in 91 centres (38 countries). There were marked variations in theprevalence of asthma symptoms with up to 15-fold differences between countries. Theprevalence of wheeze in the last 12 months ranged from 2.1-32.2% in the older age group and4.1-32.1% in the younger age group and was particularly high in English-speaking countriesand Latin America. A video questionnaire completed in the older age group in 99 centres(42 countries) showed a similar pattern.
The major differences between populations found in the International Study of Asthmaand Allergies in Childhood Phase One are likely to be due to environmental factors. Theresults provide a framework for studies between populations in contrasting environmentsthat are likely to yield new clues about the aetiology of asthma.3 Self completed wheezingquestionnaire data in 13-14 years and 6-7 years old age group from different regions of theworld are shown in Tables 21.2 and 21.3.
The ISAAC study has demonstrated, by means of simple standardized questionnaires,that there are large variations in the prevalence of asthma symptoms throughout the world.The self-reported 12 months prevalence of wheezing among 13-14 years-old between countries
Table 21.1: Prevalence of asthma in children in different countries
Country Number Age Current Diagnosed Wheeze Airway Atopyasthma asthma ever Hyperrespon- (SPT)
siveness
Australia 1,487 8 to 10 5.4 11.10 21.7 10.1(H) 29.3 1,217 8 to 11 6.7 17.3 26.5 10.0 (1.1) 31.91,575 8 to 11 9.9 30.8 40.7 16.0 (H) 37.9
New Zealand 813 9 11.1 27.0 22.0 (M) 45.81,084 6 to 11 9.1 14.2 27.2 20.0 (H)
873 12 8.1 16.8 26.6 12.0 (E)
England 1,613 8.0 14.8* ?(H)
Wales 965 5.3 1.2 22.3 8.0 (E)
Germany 5,768 9 to 11 4.2 7.9 ?
Denmark 527 7 to 16 5.3 16.0 (H) 31
Spain 2,216 9 to 14 ? ? 6.9(E)
Indonesia 406 7 to 15 1.2 2.3 14.5 2.2 (H)
China 3,067 11 to 17 1.9 2.4 6.3 4.1 (H) ?30PapuaNew Guinea 257 6 to 20 0 0 1.7 1.0 (H) 17
Kenya 402 9 to 12 3.3 11.4 10.7 (E)AustraliaIndigenous
Aborigines 215 7 to 12 0.1 0 1.4 1.8(H) 20.5
• Current asthma: Airway hyperresponsiveness (AHR) + wheeze in the last 12 months;Diagnosed asthma: asthma ever diagnosed; H:histamine; M:methacholine; E: exercise;
• All figures are a percentage of the population tested.
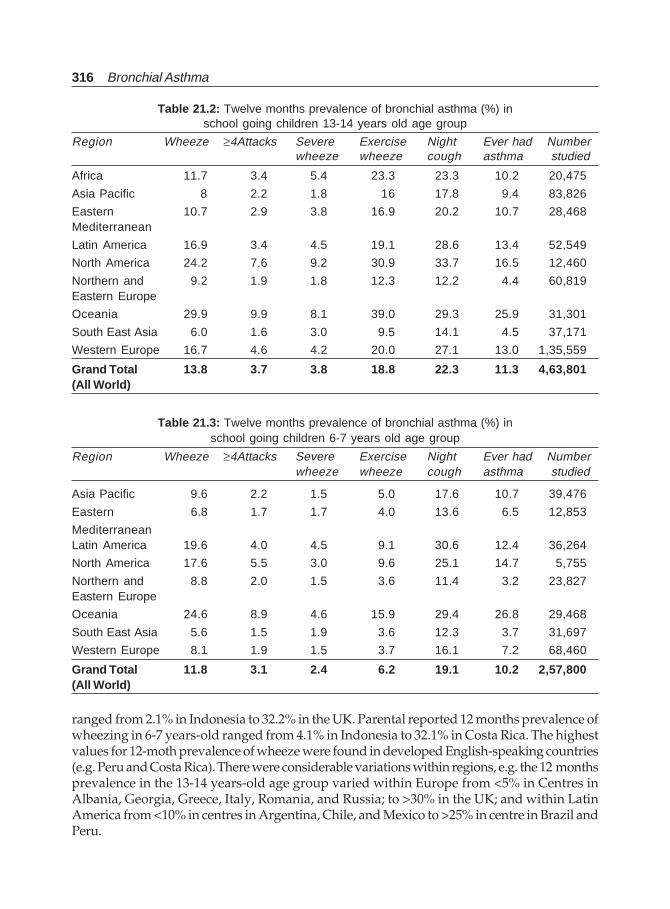
316 Bronchial Asthma
Table 21.2: Twelve months prevalence of bronchial asthma (%) inschool going children 13-14 years old age group
Region Wheeze ≥4Attacks Severe Exercise Night Ever had Numberwheeze wheeze cough asthma studied
Africa 11.7 3.4 5.4 23.3 23.3 10.2 20,475
Asia Pacific 8 2.2 1.8 16 17.8 9.4 83,826
Eastern 10.7 2.9 3.8 16.9 20.2 10.7 28,468Mediterranean
Latin America 16.9 3.4 4.5 19.1 28.6 13.4 52,549
North America 24.2 7.6 9.2 30.9 33.7 16.5 12,460
Northern and 9.2 1.9 1.8 12.3 12.2 4.4 60,819Eastern Europe
Oceania 29.9 9.9 8.1 39.0 29.3 25.9 31,301
South East Asia 6.0 1.6 3.0 9.5 14.1 4.5 37,171
Western Europe 16.7 4.6 4.2 20.0 27.1 13.0 1,35,559
Grand Total 13.8 3.7 3.8 18.8 22.3 11.3 4,63,801(All World)
Table 21.3: Twelve months prevalence of bronchial asthma (%) inschool going children 6-7 years old age group
Region Wheeze ≥4Attacks Severe Exercise Night Ever had Numberwheeze wheeze cough asthma studied
Asia Pacific 9.6 2.2 1.5 5.0 17.6 10.7 39,476
Eastern 6.8 1.7 1.7 4.0 13.6 6.5 12,853
MediterraneanLatin America 19.6 4.0 4.5 9.1 30.6 12.4 36,264
North America 17.6 5.5 3.0 9.6 25.1 14.7 5,755
Northern and 8.8 2.0 1.5 3.6 11.4 3.2 23,827Eastern Europe
Oceania 24.6 8.9 4.6 15.9 29.4 26.8 29,468
South East Asia 5.6 1.5 1.9 3.6 12.3 3.7 31,697
Western Europe 8.1 1.9 1.5 3.7 16.1 7.2 68,460
Grand Total 11.8 3.1 2.4 6.2 19.1 10.2 2,57,800(All World)
ranged from 2.1% in Indonesia to 32.2% in the UK. Parental reported 12 months prevalence ofwheezing in 6-7 years-old ranged from 4.1% in Indonesia to 32.1% in Costa Rica. The highestvalues for 12-moth prevalence of wheeze were found in developed English-speaking countries(e.g. Peru and Costa Rica). There were considerable variations within regions, e.g. the 12 monthsprevalence in the 13-14 years-old age group varied within Europe from <5% in Centres inAlbania, Georgia, Greece, Italy, Romania, and Russia; to >30% in the UK; and within LatinAmerica from <10% in centres in Argentina, Chile, and Mexico to >25% in centre in Brazil andPeru.

Asthma in Children 317
The analysis shows that there is consistently more variation between countries than withincountries. Three countries with a very large number of centres were represented across therange of prevalence, India with 14 centres representing the low prevalence group, Italy with14 centres representing the middle prevalence group and the UK with 15 centres representingthe high prevalence group. However, it must be remembered that the countries, and centreswithin countries were self-selected, and it is possible that countries with larger within-countryvariation did not participate.
The only other comparable international survey of asthma is the European CommunityRespiratory Health Survey (ECRHS),4,5 which studied males and females aged 20-44 years.,mainly from European centres. Among the 13 centres 10 countries that were reported in bothstudies, the ranking of prevalence of wheeze in the last 12 months was similar, with theEnglish-speaking countries (Australia, New Zealand, Republic of Ireland, and the UK) havingthe highest and Italy and Greece the lowest rates. Subsequent other studies from differentparts of the world also show similar trends.6-14
Indian Scene
The ISAAC data from 12 different parts of the country shows wide variability in the history ofwheeze over a 12 months period in children between 13-14 years-old age group ( Table 21.4).In Akola the prevalence was 1.6% whereas the highest figures was reported from Kottayam(17.8%) in the South. The children from this town also had history of “Ever had asthma” of12.4%. The prevalence was also the highest 24.6% from Kottayam in the 6-7 years-old agegroup (Table 21.5). There is a difference in the prevalence of asthma in children from Northernand Southern part of the country. From the Northern part of the country the figure variedbetween 5.4 to 6.9% in the 6-7 years-old age group. The figures from the Western part were lesscompared to those from the Northern and Southern regions.5 Another hospital based studyfrom South India, Bangalore on 20,000 children under the age of 18 years from 1979, 1984,1989, 1994 and 1999 in the city of Bangalore showed a prevalence of 9%, 10.5%, 18.5%, 24.5%and 29.5% respectively. The increased prevalence correlated well with demographic changesof the city. Further to the hospital study, a school survey in 12 schools on 6,550 children in theage group of 6 to 15 years was undertaken for prevalence of asthma and children werecategorized into three group-depending upon the geographical situation of the school inrelation to vehicular traffic and the socioeconomic group of children. Group I—children fromschools of heavy traffic area showed prevalence of 19.34%, group II—children from heavytraffic region and low socioeconomic population had 31.14%, and group III—children fromlow traffic area school had 11.15% respectively. A continuation of study in rural areas showed5.7% in children of 6-15 areas. The persistent asthma also showed an increase from 20% to27.5% and persistent severe asthma 4% to 6.5% between 1994-99.15 Another study from Delhiin 1999 revealed the prevalence of current asthma was 11.9% while past asthma was reportedby 3.4% of children. Exclusive exercise-induced asthma was reported by 2.1% while thatassociated with colds by 2.4% of children. Boys had significantly higher prevalence of currentasthma as compared with girls (12.8% and 10.7%, respectively). Multiple logistic regressionanalysis showed that male sex, a positive family history of atopic disorders, and the presenceof smokers in the family were significant factors influencing the development of asthma whileeconomic class, air pollution (total suspended particulates), and type of domestic kitchen fuelwere not. The prevalence of current asthma in children in Delhi is 11.9%. Significant riskfactors for its development are male sex, a positive family history of atopic disorders, and the
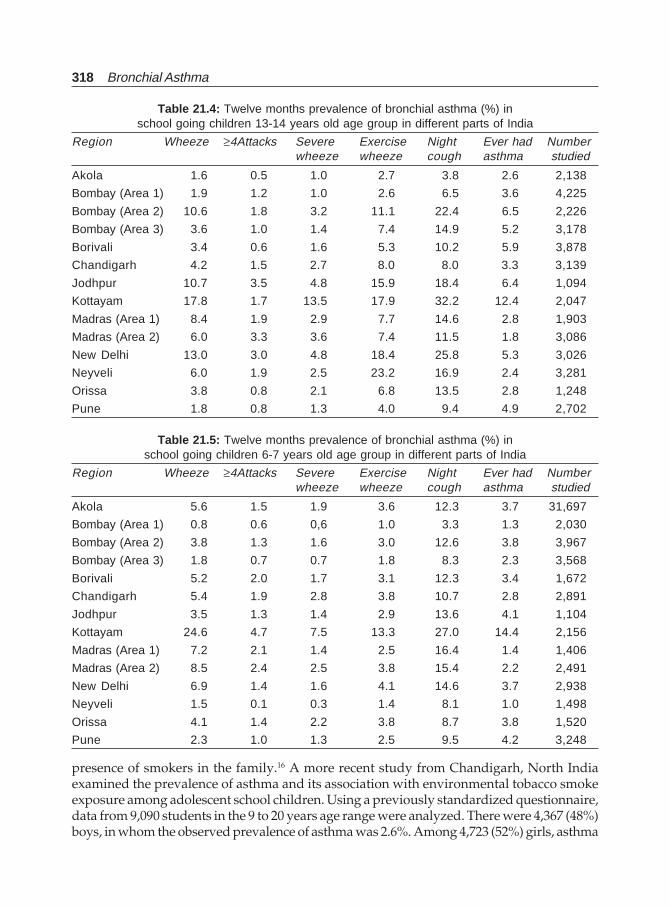
318 Bronchial Asthma
Table 21.4: Twelve months prevalence of bronchial asthma (%) inschool going children 13-14 years old age group in different parts of India
Region Wheeze ≥4Attacks Severe Exercise Night Ever had Numberwheeze wheeze cough asthma studied
Akola 1.6 0.5 1.0 2.7 3.8 2.6 2,138
Bombay (Area 1) 1.9 1.2 1.0 2.6 6.5 3.6 4,225
Bombay (Area 2) 10.6 1.8 3.2 11.1 22.4 6.5 2,226
Bombay (Area 3) 3.6 1.0 1.4 7.4 14.9 5.2 3,178
Borivali 3.4 0.6 1.6 5.3 10.2 5.9 3,878
Chandigarh 4.2 1.5 2.7 8.0 8.0 3.3 3,139
Jodhpur 10.7 3.5 4.8 15.9 18.4 6.4 1,094
Kottayam 17.8 1.7 13.5 17.9 32.2 12.4 2,047
Madras (Area 1) 8.4 1.9 2.9 7.7 14.6 2.8 1,903
Madras (Area 2) 6.0 3.3 3.6 7.4 11.5 1.8 3,086
New Delhi 13.0 3.0 4.8 18.4 25.8 5.3 3,026
Neyveli 6.0 1.9 2.5 23.2 16.9 2.4 3,281
Orissa 3.8 0.8 2.1 6.8 13.5 2.8 1,248
Pune 1.8 0.8 1.3 4.0 9.4 4.9 2,702
Table 21.5: Twelve months prevalence of bronchial asthma (%) inschool going children 6-7 years old age group in different parts of India
Region Wheeze ≥4Attacks Severe Exercise Night Ever had Numberwheeze wheeze cough asthma studied
Akola 5.6 1.5 1.9 3.6 12.3 3.7 31,697
Bombay (Area 1) 0.8 0.6 0,6 1.0 3.3 1.3 2,030
Bombay (Area 2) 3.8 1.3 1.6 3.0 12.6 3.8 3,967
Bombay (Area 3) 1.8 0.7 0.7 1.8 8.3 2.3 3,568
Borivali 5.2 2.0 1.7 3.1 12.3 3.4 1,672
Chandigarh 5.4 1.9 2.8 3.8 10.7 2.8 2,891
Jodhpur 3.5 1.3 1.4 2.9 13.6 4.1 1,104
Kottayam 24.6 4.7 7.5 13.3 27.0 14.4 2,156
Madras (Area 1) 7.2 2.1 1.4 2.5 16.4 1.4 1,406
Madras (Area 2) 8.5 2.4 2.5 3.8 15.4 2.2 2,491
New Delhi 6.9 1.4 1.6 4.1 14.6 3.7 2,938
Neyveli 1.5 0.1 0.3 1.4 8.1 1.0 1,498
Orissa 4.1 1.4 2.2 3.8 8.7 3.8 1,520
Pune 2.3 1.0 1.3 2.5 9.5 4.2 3,248
presence of smokers in the family.16 A more recent study from Chandigarh, North Indiaexamined the prevalence of asthma and its association with environmental tobacco smokeexposure among adolescent school children. Using a previously standardized questionnaire,data from 9,090 students in the 9 to 20 years age range were analyzed. There were 4,367 (48%)boys, in whom the observed prevalence of asthma was 2.6%. Among 4,723 (52%) girls, asthma

Asthma in Children 319
was present in 90 (1.9%) students. 31% students reported presence of one or more respiratorysymptoms. More students with asthma had either parents or other family members smokingat home as compared to nonasthmatics (41% vs. 28%, p<0.0001). The odds ratio for beingasthmatic for patients exposed to ETS compared to those not exposed to ETS was 1.78 (95%confidence interval 1.33-2.31). ETS was also positively associated with prevalence of all therespiratory symptoms, with odds ratios varying between 1.6 and 2.25.17
Risk Factors
A number of risk factors have been identified for the causation of bronchial asthma in children.They include male sex a positive family history of atopic disorders, and the presence ofsmokers in the family,16 urbanization, air-pollution, environmental tobacco smoke,15,17 andother socio environmental factors.18
Intrauterine Exposure
Some studies have suggested a link to maternal, not paternal, allergy in the development ofallergy and asthma. While it has been suggested that preferential acquisition of the mother’sgenes (genomic imprinting) may account for this phenomenon, such an occurrence isexcessively uncommon and the intrauterine environment is a more likely cause. A positiverelationship has been found between greater head circumference at birth and the laterdevelopment of allergy and high serum IgE levels. At first, such an association may soundstrange, but placental and nutritional factors that increase brain growth in the last trimesterof pregnancy may well influence the maturation of the thymus gland, the origin of the immunesystem. It has been suggested that after 26 weeks gestation, the fetus adopts a T’hl immune-phenotype, to prevent maternal rejection and that, in the last trimester, with increased IFN-γproduction, this phenotype converts to a more T’hl picture. IL-4 is produced by the humanamnion epithelium throughout pregnancy, and IL-10, a cytokine that inhibitors have beenfound in human placenta. If, on account of factors, the Th2 mode is maintained rather thanconverting to a T’hl mode, an allergic diathesis might be expected to occur. Such a mechanismmight also be invoked as a factor in sudden infant death syndrome in which mast cell tryptaseand eosinophils are encountered in the lung and circulation.
It is also possible that allergens crossing the placenta may be involved in subsequentdevelopment of allergy and asthma since mothers exposed to high concentrations of allergens,such as birch pollen, during the last trimester of pregnancy are more likely to have childrenwho develop allergy and asthma. It is not known, however, how minute amounts of allergentaken in by the mother can cross the placenta to sensitive offspring. However, it has beenshown, that children who subsequently develop allergy or asthma, have impaired cord bloodT-lymphocyte production of IFN-γ at birth in response to exposure to specific allergens. Thisimpaired response suggests an impaired inhibitory mechanism for shutting down a Th2response rather than one that primarily enhances it. Other environmental factors that maydirect the placental-fetal relationship towards a Th2 response include young maternal ageand smoking during pregnancy.
Several studies have reported an increased prevalence of respiratory symptoms like cough,wheeze and reduction in lung function in children or adolescents who were born as prematureinfants or who had a low birth weight.19,20

320 Bronchial Asthma
Viral Infection
It has long been recognized that viral infections, especially the common cold viruses, can leadto deterioration of asthma lasting several weeks. In infants, respiratory syncytial virus (RSV)is responsible for most wheezing illnesses. These observations have suggested that viralinfections may be intimately involved in the development of asthma and allergy. It has beenshown that over 80% of acute asthma exacerbations in school children and about 60% inadults result from viral infections (mostly common cold viruses). One explanation of thesusceptibility of the asthmatic airway to viral inflammation is that persistent allergic mastcell and eosinophil-driven inflammation stimulates the release of cytokines such as tumournecrosis factor-alpha, which cause an increase in the expression of receptors for humanrespiratory viruses on the airway lining epithelium. In the case of most rhinoviruses, thereceptor is an adhesion molecule, intracellular adhesion molecule-1. Once the virus enters theepithelial cells, it replicates and is able to generate wide variety of proinflammatory cytokines,which further enhance eosinophil and mast cell inflammation.
Protective Infections
Curiously, an important additional socioeconomic factor may be a reduction in early childhoodinfections (viral, bacterial, or parasitic) associated with improved living conditions. Whileviral infections can undoubtedly cause deterioration of established asthma, there is evidencethat viral or bacterial infection during the first 3 years of life may serve a protective functionagainst the development of allergic diseases.
One of the most consistent risk factors for allergy relates to family size. The prevalence ofmucosal allergy and positive skin tests in children declines markedly in the last-born childwith increasing numbers of siblings. A working hypothesis is that over the past 30 years,opportunities for acquiring infections from siblings or playmates in early childhood havedeclined with reduction in average family size, vaccination programs, and higher standardsof personal hygiene. Most viruses and some bacteria are able to evoke a Thl like protectiveresponse with the generation of IFN-α and IL-12. Thus, if multiple infections occur during thefirst few years of life, high concentrations of these Th1, cytokines could inhibit the release ofTh2 cytokines, thereby biasing the mucosal immune response away ‘for this hypothesis isseen in an African study of, from allergen sensitization. Support adolescents infected withmeasles during the first year of life compared to those vaccinated later. Those infected earlyhad a 63% lesser chance of developing positive skin tests to common aeroallergens. RepeatedBacille Calmette-Guerin (BCG) vaccination in young Japanese children also exerts a protectiveeffect against the development of allergy. Both measles and BCG are potent stimulators—ofthe Th1 cytokine response’.
It has also been suggested that the increase in asthma and allergy with movements tourban centres may be related to the decrease in early exposure to parasitic infections commonin some rural areas. One study tested the effect of anti-helminthic treatment on the allergicreactivity of children in a slum area of Caracas, Venezuela. One group was treated for 22 monthswhile a second group who declined treatment was used as a control. Active treatmenteliminated worms in children (from 68 to 5%) and resulted in a decrease in total serum IgElevels (from 2,543 to 1,124 IU/ml) but was accompanied by an increase in skin test reactivityto house dust mite (from 17 to 68%). In contrast, in the untreated group, parasite colonizationcontinued to increase (43 to 70%), IgE levels increased (1,649 to 3,697 IU/ml), but dust mite

Asthma in Children 321
sensitization fell (26 to 16%). Further testing showed that polyclonal stimulation of IgEsynthesis by the parasites resulted in mast cell receptor saturation and suppression of specificIgE antibody synthesis. From a public health stand point; high levels of nonspecific IgE mayprotect rural dwellers exposed to parasites from allergy and asthma. It follows that eradicationof parasites or reduced opportunities for infection could, in part, explain the rural to urbandifferences in the prevalence of allergic diseases.
Some investigators believe that early childhood respiratory symptoms are a risk factor forasthma.21 This inference however, is weakened by the possibility of recall bias. Perhaps,respiratory symptoms reported by parents very early in life are not significantly associatedwith future asthma, but those symptoms that begin at or persist through age 3 to 4 years arelikely to be associated with asthma.22
Diet
As societies become affluent, the dietary habits change and such changes are linked withincreased prevalence of asthma observed in recent years.23-25 Prospective studies have shownthat breastfeeding has a transient beneficial effect on the incidence of eczema, food allergy,atopic sensitization, and wheezing illness in the first three years of life.26,27 However, there islittle evidence for a persistent protective effect of breastfeeding on the ‘incidence of childhoodasthma.28-30 In the UK, the amount of salt eaten with food seems to be correlated with bronchialhyperreactivity and asthma mortality.31 The severity of asthma—not its inception—has beenlinked to increased salt intake, but only in males.32 Recent studies have shown lower prevalenceof asthma and bronchial hyperresponsiveness in children with a high intake of fresh oilyfish,33 a source of’ polyunsaturated oils. Other studies also have shown an association of a,high fish consumption and improved baseline FEV1.34 Children who eat fish regularlyconsume more omega-3 fatty acids, which may protect them from bronchial hyperresponsive-ness.
Air-Pollution
Air-pollution has been cited as—a causal factor in the development of asthma. The USEnvironmental Protection Agency concludes that35 passive exposure to tobacco smoke iscausally related to:
i. An increased risk of lower respiratory tract infections, such as bronchitis and pneumoniain infants and young children,
ii. A small but significant dose-dependent reduction in pulmonary function, andiii. Additional episodes and increased severity of asthma symptoms in asthmatic children.
Exposure to tobacco smoke is also considered to be a risk factor for the development of newcases of asthma in children.36 Trucson Children’s Respiratory Study has shown thatmaternal smoking is related to both transient early wheezing and persistent wheezing.37 Therole of sulphur dioxide and particulate matters in the causation of asthma is not wellestablished.38-41 Traffic pollutions42 and effects of ozone may also be of consequence forchildhood asthma.43
Evolution of Asthma
Asthma may develop during the first few months of life, but it is often difficult to make adefinite diagnosis until the child is older. In infants, the most common cause of wheezing is

322 Bronchial Asthma
respiratory viral infections. However, there is a correlation of early wheeze with reduced lungfunction before the onset of symptoms, which suggests that small lungs may be responsiblefor some infant wheeze that resolves with the child’s growth. Those children with asthmacontinue to wheeze in later childhood. Recurring exacerbations of asthma may be associatedwith exposure to allergens. In the susceptible infant, atopy may predispose the airways tosensitization by environmental allergens or irritants and the child experiences recurrentepisodes of wheezing. In particular, early exposure to Alternaria, housedust mite, and animalallergens in high quantities appears to be important as discussed above.
During early childhood, wheezing and cough may occur at infrequent intervals. In someinfants wheezing becomes more frequent and asthma is well established at an early age. It isreported that the majority of 7-years-old children with airway hyperresponsiveness sufferedfrom atopy during their infancy.44 Asthma also affects development of the lung. Asthma ininfancy can result in a decrease ‘in lung function by approximately 20% in adulthood,45
although subsequent studies did not confirm the same.46
The predominant feature associated with asthma in children is allergy, and house dustmite represent major allergens worldwide in, both affluent and partly affluent countries.47
The role of viral infection in the causation of asthma in older children is less clear, althoughin atopic children viral infection is clearly important triggers of asthma exacerbations. By theage of 8 years, a proportion of children develop airway hyperresponsiveness and the associatedsymptoms of moderate to severe persistent asthma, while others continue to have mildintermittent asthma.48 Lung growth is unaffected in most children with asthma, but it can bereduced throughout childhood and adolescence in those with severe and persistent symptoms.A longitudinal study in New Zealand concluded that improved spirometric function wasimpaired in children with airway hyperresponsiveness and/or allergy to house dust mite orcat allergen.49
Although childhood asthma has long been considered as a single, easily recognizabledisease characterized by reversible airflow limitations,50 recent findings have challenged thisconcept. Martinez et al21 studied the natural history of children (0-6 years) and found thatapproximately half of them experienced wheezing at some time during the study period. Theyrecognized three patterns of wheezing:1. Transient early wheezing. Wheezing occurred in life but resolves by the age of three years2. Late onset wheezing. Some experience wheezing between the ages of three and six years3. Persistent wheezing. Wheezing illness throughout the entire study period.
The outcomes of these patterns are associated with different risk factors. Children withtransient early wheezing had reduced pulmonary function as measured by functional residualcapacity shortly after—birth and before any lower respiratory tract illness had occurred. Therisk also increases in children—of mothers who smoked during pregnancy, had lower lungfunction values compared to those whose mothers did not smoke. Thus, the authors concludedthat congenitally smaller airways might predispose children to wheeze illness later in life.Persistent and late onset wheezing is more likely associated with atopy with their mothersbeing asthmatics. Lung function in persistent wheezers is also less.
The long-term prognosis of childhood asthma is a matter of controversy and of majorconcern. It has often been believed that the child grows out of its asthma when he or shereaches adulthood (asthma disappears). However, epidemiological studies are lessconvincing.46,51,52 Although there are methodological difficulties it is estimated that asthmadisappears in 30 to 50% of children at puberty, but often reappears in adult life. Up to

Asthma in Children 323
two-third of children with asthma continue to suffer from the disorder through puberty andadulthood. Even when asthma symptoms disappear, the lung function frequently remainsaltered or airway hyperresponsiveness or cough persists. The prognosis of asthma becomesworse when the child has eczema or there is a family history of eczema. Wheezing in the firstyear of life is not a prognostic indicator for asthma or for more severe asthma or for moresevere asthma later in childhood. About 5 to10 % of children with asthma that is consideredtrivial will have severe-asthma in later life. Therefore, childhood asthma should never beneglected with the hope that the child will grow out of it. Children with mild asthma are likelyto have a good prognosis, but those with a moderate to severe asthma probably continue tohave some degree of airway hyperresponsiveness and will be at risk of the long-term effects ofasthma throughout life.53 Some, clinical studies have reported that up to 80% of asthmaticsbecome asymptomatic during puberty.54,55 In a cohort study of Australian school children56
tested initially at the age of 8 to 10 years and then again at 12-14 years of age, the persistenceif bronchial hyperresponsiveness at 12 to 14 years of age was found to be related to theseverity of disease at 8 to 10 years of age, the atopic status of the child, and the presence ofasthma in the parents. Most of the children who had a slight or mild degree of bronchialhyperresponsiveness at 8 to 10 years of age lost their increased response by the age of 12-14years. However, only 15.4% of children with severe or moderate bronchial hyperresponsivenessat initial assessment had normal levels of bronchial responsiveness at the later assessment.There are several factors why asthma often goes unrecognized and tends to be under treatedin teenagers because usually this is a period of turmoil, awkwardness, rebelliousness, andintolerability.53-59
Notwithstanding the factors described above as the factors responsible for the induction ofasthma in childhood, occurrence of asthma within families is the strongest risk factor for thedevelopment of asthma in children.60-63
DIAGNOSIS
Various symptoms and signs of bronchial asthma are not different than those in adults asdiscussed earlier. However, in children there is more chance of under diagnosis in this agegroup. This is a frequent problem and occurs most often when young children who wheezeonly when they have respiratory infections and are dismissed as having wheezy bronchitis,asthmatic bronchitis, bronchitis, bronchiolitis, or pneumonia, despite evidence that the signsand symptoms are most compatible with a diagnosis of bronchial asthma.
Although, recurrent episodes of cough and wheezing are almost always due to asthma inboth children and adults, it is to be remembered that all that wheezes is not asthma always.There are other causes of airways obstruction leading to wheezing. The differential diagnosiswill be as follows.
Infants and Children
Obstruction in the Large Airways
1. Foreign body in trachea, bronchus2. Vascular rings3. Laryngotracheomalacia

324 Bronchial Asthma
4. Enlarged lymph nodes or tumors5. Laryngeal webs6. Tracheal stenosis7. Bronchial stenosis
Obstruction Involving both Large and Small Airways
1. Bronchial asthma2. Viral bronchiolitis3. Cystic fibrosis4. Chlamydia trachomatis infection5. Obliterative bronchiolitis6. Bronchopulmonary dysplasia7. Aspiration8. Vascular engorgements9. Pulmonary oedema
Miscellaneous
1. Primary ciliary dyskinesia syndrome2. Primary immune deficiency3. Congenital heart disease4. Congenital malformations causing narrowing of intrathoracic airways.
Asthma in childhood can present a particularly difficult problem largely because episodicwheezing and cough are among the most common symptoms encountered in childhoodillnesses, particularly in the under-3-years-old. Although health care professionals areincreasingly encouraged to make a positive diagnosis of asthma whenever recurrent wheezing,breathlessness, and cough occur (particularly if associated with nocturnal and early morningsymptoms), the underlying nature of the disorder’s process may differ in infants from that inolder children and adults. The use of the label “asthma” to describe such children has importantclinical consequences. It implies a syndrome in which there is airway inflammation and forwhich there is a specific protocol of management. The younger the child, particularly belowages 5, the greater the possibility of an alternative diagnosis for recurrent wheeze as describedabove. Chest radiography is important as a diagnostic test to exclude alternative causes.Features such as a neonatal onset of symptoms, associated failure to thrive, vomiting-associated symptoms, and localized lung or cardiovascular signs all suggest an alternativediagnosis and indicate the need for investigations, such as a sweat test to exclude cysticfibrosis, measurements of immune function, and reflux studies.
Among those with no alternative diagnosis, there is the possibility that the problem doesnot have a uniform underlying pathogenesis. Nonetheless, there are two general patterns ofwheezing in infancy. Some infants who have recurrent episodes of wheeze associated withacute viral respiratory infections, often with a first episode in association with respiratorysyncytial virus (RSV) bronchiolitis, come from nonatopic families and have no evidence ofatopy themselves. These infants usually outgrow their symptoms in the preschool years andhave no evidence of subsequent asthma, though they may have minor defects of lung function

Asthma in Children 325
and airway hyperresponsiveness. This syndrome may have more to do with airway geometrythan airway inflammation, and thus may differ mechanistically from the more establishedchronic inflammatory condition that underlies asthma in older children and adults.
Other infants with asthma have an atopic background often associated with eczema anddevelop symptoms later in infancy that persists through childhood and into adult life. Inthese children, characteristic features of airway inflammation can be found even in infancy.However, there are no practical clinical tests that can be done to establish the presence ofairway inflammation. Only associated atopic problems can be used as a guide to prognosis.Early age (less than 2 years) of onset of wheeze is a poor predictor of continuing problem inlater childhood.
It is likely that the issue of asthma associated with recurrent virus-related episodes and thelater development of persistent asthma requires further study. Apart from the confusion overaetiological mechanisms of asthma in childhood, there is also considerable reluctance inestablishing a diagnosis and, as a consequence, initiating appropriate therapy. Because lowerrespiratory tract symptoms similar to symptoms of asthma are so common in childhood (andfrequently occur in association with upper respiratory tract symptoms), either a correctdiagnosis is not made or an inappropriate diagnosis is given, thereby, depriving the child ofantiasthma medication.
Although in these young children there is the possibility of over treatment, the episodes ofwheezing may be foreshortened and reduced in intensity by the effective use of anti-inflammatory drugs and bronchodilators rather than antibiotics, and it is for this reason thathealth care professionals are encouraged to use the word “asthma” rather than otherterminology to describe this syndrome.
Asthma in all age groups may present only as repeated coughing especially at night, withexercise, and with viral illness, but these are particularly common forms of presentation ofasthma in childhood. The presence of recurrent nocturnal cough in an otherwise healthychild should raise awareness of asthma as a probable diagnosis. Although repeated infectionsof the sinuses, tonsils, and adenoids may explain nocturnal coughing, the occurrence of thissymptom awaking the child in the early hours of the morning is almost always diagnostic ofasthma.
Under the age of 5 years, the diagnosis of asthma has to rely largely on clinical judgmentbased on a combination of symptoms and physical findings. Because the measurement ofairflow limitation and airway hyperresponsiveness infants and small children requirescomplex equipment and is difficult, it can therefore only be recommended as a research tool.A trial of treatment is probably the most confident-way in which a diagnosis of asthma can besecured in children (and in many adults as well). Prognostic features include a family historyof asthma or eczema and presence of eczema in a young child with respiratory symptoms.Children aged 4 to 5 can be taught to use a peak expiratory flow (PEF) meter and obtainreliable readings. However, unless there is careful parental supervision over when and howthe measurements are made, PEF recording in childhood can be unreliable.
Some children with asthma only present with exercise-induced symptoms. In this group,or when there is doubt over the existence of low-grade asthma in childhood, exercise testingis helpful. A 6-minute running protocol is easily performed in clinical practice, and whenused in-conjunction with measurements of airflow limitation (FEV, or PEF), it can be most

326 Bronchial Asthma
helpful in establishing a firm diagnosis, especially if the cough produced by the exercise issimilar to that occurring spontaneously at night.
MANAGEMENT OF ASTHMA IN CHILDREN
Several guidelines have been published since 1990 with the aim of improving management ofasthma both in children and adults. However, a systematic analysis of guidelines till 1995had brought out several controversial-issues as well as gaps in knowledge. In May 1997, anexpert committee of the National Heart Lung and Blood Institutes of USA published guidelinesabout management of asthma where they have tried to overcome many of the previous lapses.These guidelines along with the recent British thoracic society guideline have been discussedin previous chapters.
The need for similar guidelines has always been felt amongst the physicians managingchildren in India, but no uniform guidelines are available for the disease as seen in India. It.was felt that the guidelines originating in India would have much more relevance to theground situation and the status of health services. To setup the process of achieving consensus,towards suitable guidelines, a Consensus Conference was held on April 17 and 18, 1998 atthe Advanced Paediatric Centre of the Post Graduate Institute of Medical Education andResearch, Chandigarh in which, 15 experts who manage asthma patients and have publishedpapers in this field, participated. Recent evidence was accessed using searches on Medline,Embase, Index Medicus, and Excerpta Medica. Some of the contentious issues were resolvedwith the help of the Cochrane Library.64 Since the consumer of health care in India is notsufficiently literate, physicians have been assigned a lot of responsibility in decision makingfor the patients. The guidelines are required to be updated periodically and provide flexibilityto individualize patients. Since in a large area in our country, all the recommended modalitiesmay not be available then suitable improvisations must be made.
The objectives of the conference were:i. To reach at a uniform treatment approach towards children with asthma keeping in
mind the limitations of resources in the Indian context and to develop guidelines basedon available evidence for the pediatricians, and
ii. To prepare a consensus document for management of children with asthma whichwould provide guidelines to a general pediatricians managing asthma in India.
Various components discussed included pathogenesis, definition, classification of severity,measure of assessment and monitoring, referral, control of factors contributing to asthma inseventy, pharmacological therapy and education of patient, family and health professionalsregarding asthma care.
The Expert Group I recommended that for the diagnosis of asthma in children a detailedmedical history, careful physical examination and peak-expiratory flow rate (PEFR)measurement to demonstrate obstruction with reversibility of variable airflow obstruction areneeded. To establish the diagnosis of asthma the clinician must determine that:
i. Episodic symptoms of airflow obstruction, more than 3 episodes are presentii. Airflow-obstruction is at least partially reversible
iii. Alternative diagnoses are excluded.

Asthma in Children 327
Physicians who care for children with asthma should be well versed in PEFR monitoring.They should perform spirometery wherever possible.
Measures of Assessment and Monitoring
A child with asthma is to be monitored for clinical signs and symptoms of asthma with thehelp of asthma diary given to the patients/parents and record of PEFR with a standardizedpeak flow meter. PEFR must be monitored at the physician’s office, asthma clinics (wherespirometry should be available) and in the emergency room, and patients must be encouragedand trained to monitor their PEFR at home once a day routinely and twice a day if the morningreading is abnormal to determine their PEFR variability. Patient’s personal best should beassessed and used subsequently. Spirometry has been kept optional, and emphasis must begiven to patient’s quality of life. Emphasis must be on self-management but physician’ssupervision must still be the prima mode. Patients must be given a written crisis managementplan where literate. Otherwise verbal communication at each contact must continue.
Classification of Asthma Severity
Asthma severity classification was accepted as changed to be mild intermittent, mild persistent,moderate persistent and severe persistent asthma (Table 21.6). Since spirometry is not routinelyavailable to pediatricians in this country it was felt that more emphasis be placed on PEFRmeasurement, especially at the physician’s office. Patient’s personal best be used as thestandard but in its absence expected PEFR according to norms published on children in Indiamust be used.65 It was also mentioned that a severe form of asthma requiring daily oralsteroids or stronger treatments like immunosuppressants is extremely uncommon in Indianchildren, and most of the children get controlled with inhaled medications.
Goals of Asthma Therapy
The goals of asthma therapy are:• Prevent chronic and troublesome symptoms• Maintain near normal (PEFR)• Maintain normal activity levels (including exercise and physical activity)• Prevent recurrent exacerbations of asthma and minimize the need for emergency room
visits and hospitalization• Provide optimal pharmacotherapy with minimal side effects• Meet patient’s and family’s expectations of satisfaction with asthma care.
Pharmacological Therapy
Pharmacological therapy is the cornerstone of management. It must be instituted with properenvironmental control measures. Medications are classified into two broad categories:
i. Long-term control medications or the preventatives, andii. Medications or rescue medications. Long-term control medications, are, inflammatory
compounds. Early intervention with inhaled steroids can improve control and normalizelung function, and preliminary studies show that it might prevent irreversible airwayinjury. These are to be administered with the help of a metered dose inhaler (MDI) and
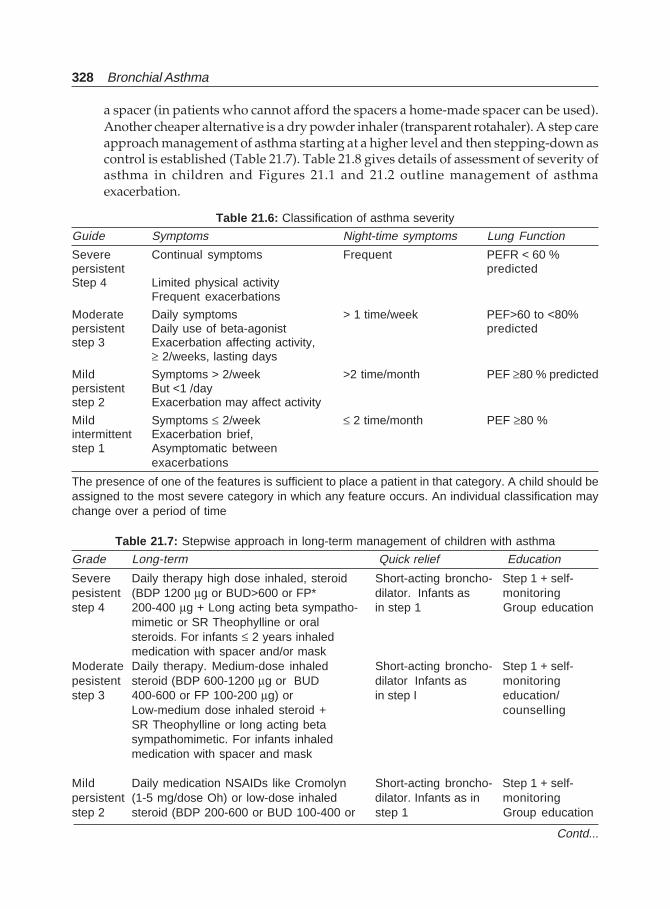
328 Bronchial Asthma
Table 21.6: Classification of asthma severity
Guide Symptoms Night-time symptoms Lung Function
Severe Continual symptoms Frequent PEFR < 60 %persistent predictedStep 4 Limited physical activity
Frequent exacerbations
Moderate Daily symptoms > 1 time/week PEF>60 to <80%persistent Daily use of beta-agonist predictedstep 3 Exacerbation affecting activity,
≥ 2/weeks, lasting days
Mild Symptoms > 2/week >2 time/month PEF ≥80 % predictedpersistent But <1 /daystep 2 Exacerbation may affect activity
Mild Symptoms ≤ 2/week ≤ 2 time/month PEF ≥80 %intermittent Exacerbation brief,step 1 Asymptomatic between
exacerbations
The presence of one of the features is sufficient to place a patient in that category. A child should beassigned to the most severe category in which any feature occurs. An individual classification maychange over a period of time
Table 21.7: Stepwise approach in long-term management of children with asthma
Grade Long-term Quick relief Education
Severe Daily therapy high dose inhaled, steroid Short-acting broncho- Step 1 + self-pesistent (BDP 1200 μg or BUD>600 or FP* dilator. Infants as monitoringstep 4 200-400 μg + Long acting beta sympatho- in step 1 Group education
mimetic or SR Theophylline or oralsteroids. For infants ≤ 2 years inhaledmedication with spacer and/or mask
Moderate Daily therapy. Medium-dose inhaled Short-acting broncho- Step 1 + self-pesistent steroid (BDP 600-1200 μg or BUD dilator Infants as monitoringstep 3 400-600 or FP 100-200 μg) or in step I education/
Low-medium dose inhaled steroid + counsellingSR Theophylline or long acting betasympathomimetic. For infants inhaledmedication with spacer and mask
Mild Daily medication NSAIDs like Cromolyn Short-acting broncho- Step 1 + self-persistent (1-5 mg/dose Oh) or low-dose inhaled dilator. Infants as in monitoringstep 2 steroid (BDP 200-600 or BUD 100-400 or step 1 Group education
Contd...
a spacer (in patients who cannot afford the spacers a home-made spacer can be used).Another cheaper alternative is a dry powder inhaler (transparent rotahaler). A step careapproach management of asthma starting at a higher level and then stepping-down ascontrol is established (Table 21.7). Table 21.8 gives details of assessment of severity ofasthma in children and Figures 21.1 and 21.2 outline management of asthmaexacerbation.
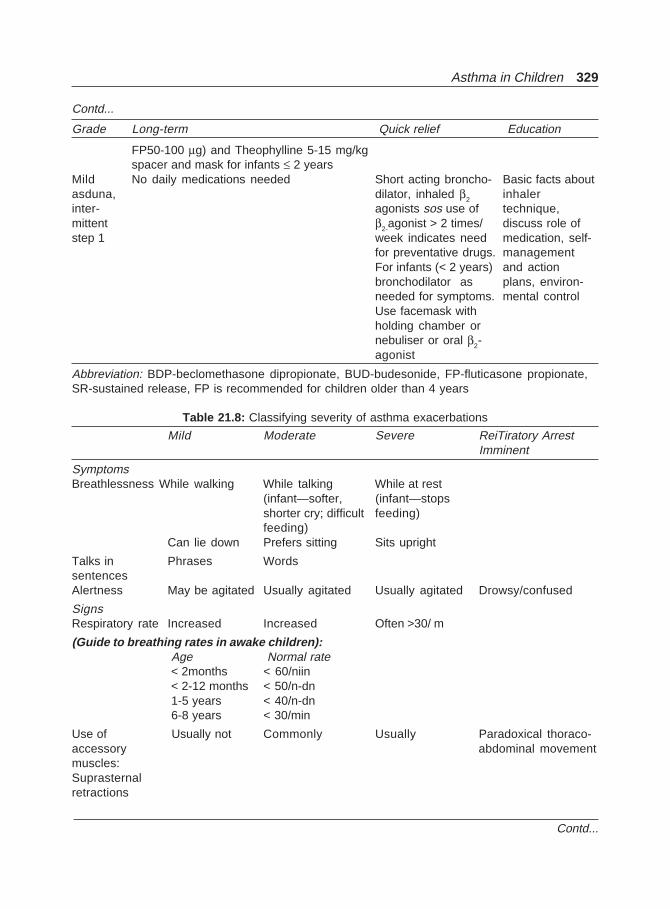
Asthma in Children 329
FP50-100 μg) and Theophylline 5-15 mg/kgspacer and mask for infants ≤ 2 years
Mild No daily medications needed Short acting broncho- Basic facts aboutasduna, dilator, inhaled β2 inhalerinter- agonists sos use of technique,mittent β2-agonist > 2 times/ discuss role ofstep 1 week indicates need medication, self-
for preventative drugs. managementFor infants (< 2 years) and actionbronchodilator as plans, environ-needed for symptoms. mental controlUse facemask withholding chamber ornebuliser or oral β2-agonist
Abbreviation: BDP-beclomethasone dipropionate, BUD-budesonide, FP-fluticasone propionate,SR-sustained release, FP is recommended for children older than 4 years
Grade Long-term Quick relief Education
Contd...
Table 21.8: Classifying severity of asthma exacerbations
Mild Moderate Severe ReiTiratory ArrestImminent
SymptomsBreathlessness While walking While talking While at rest
(infant—softer, (infant—stopsshorter cry; difficult feeding)feeding)
Can lie down Prefers sitting Sits upright
Talks in Phrases WordssentencesAlertness May be agitated Usually agitated Usually agitated Drowsy/confused
SignsRespiratory rate Increased Increased Often >30/ m
(Guide to breathing rates in awake children): Age Normal rate < 2months < 60/niin < 2-12 months < 50/n-dn 1-5 years < 40/n-dn 6-8 years < 30/min
Use of Usually not Commonly Usually Paradoxical thoraco-accessory abdominal movementmuscles:Suprasternalretractions
Contd...
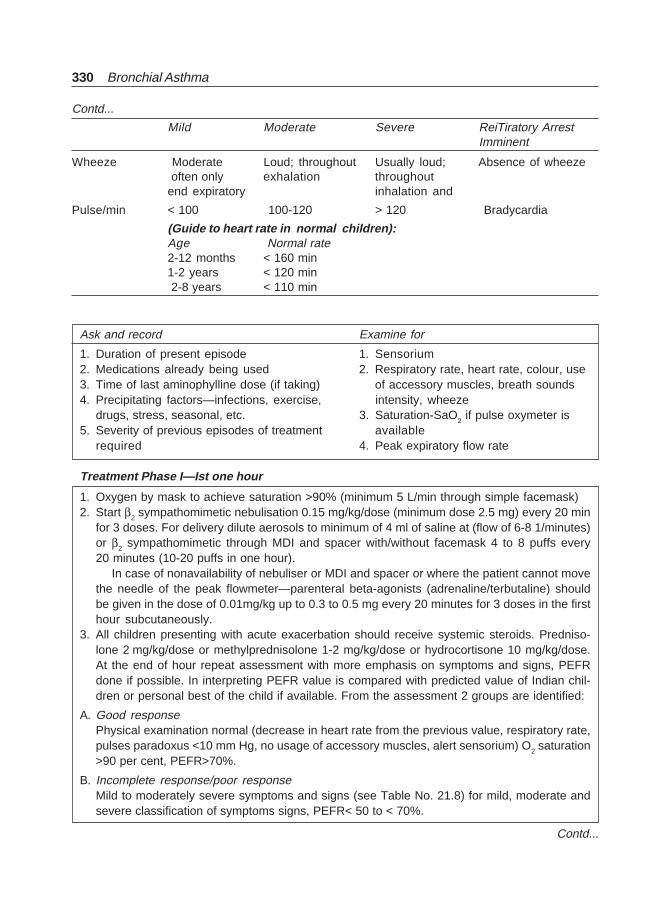
330 Bronchial Asthma
Mild Moderate Severe ReiTiratory ArrestImminent
Contd...
Wheeze Moderate Loud; throughout Usually loud; Absence of wheeze often only exhalation throughoutend expiratory inhalation and
Pulse/min < 100 100-120 > 120 Bradycardia
(Guide to heart rate in normal children):Age Normal rate2-12 months < 160 min1-2 years < 120 min 2-8 years < 110 min
Ask and record Examine for
1. Duration of present episode 1. Sensorium2. Medications already being used 2. Respiratory rate, heart rate, colour, use3. Time of last aminophylline dose (if taking) of accessory muscles, breath sounds4. Precipitating factors—infections, exercise, intensity, wheeze
drugs, stress, seasonal, etc. 3. Saturation-SaO2 if pulse oxymeter is5. Severity of previous episodes of treatment available
required 4. Peak expiratory flow rate
Treatment Phase I—Ist one hour
1. Oxygen by mask to achieve saturation >90% (minimum 5 L/min through simple facemask)2. Start β2 sympathomimetic nebulisation 0.15 mg/kg/dose (minimum dose 2.5 mg) every 20 min
for 3 doses. For delivery dilute aerosols to minimum of 4 ml of saline at (flow of 6-8 1/minutes)or β2 sympathomimetic through MDI and spacer with/without facemask 4 to 8 puffs every20 minutes (10-20 puffs in one hour).
In case of nonavailability of nebuliser or MDI and spacer or where the patient cannot movethe needle of the peak flowmeter—parenteral beta-agonists (adrenaline/terbutaline) shouldbe given in the dose of 0.01mg/kg up to 0.3 to 0.5 mg every 20 minutes for 3 doses in the firsthour subcutaneously.
3. All children presenting with acute exacerbation should receive systemic steroids. Predniso-lone 2 mg/kg/dose or methylprednisolone 1-2 mg/kg/dose or hydrocortisone 10 mg/kg/dose.At the end of hour repeat assessment with more emphasis on symptoms and signs, PEFRdone if possible. In interpreting PEFR value is compared with predicted value of Indian chil-dren or personal best of the child if available. From the assessment 2 groups are identified:
A. Good responsePhysical examination normal (decrease in heart rate from the previous value, respiratory rate,pulses paradoxus <10 mm Hg, no usage of accessory muscles, alert sensorium) O2 saturation>90 per cent, PEFR>70%.
B. Incomplete response/poor responseMild to moderately severe symptoms and signs (see Table No. 21.8) for mild, moderate andsevere classification of symptoms signs, PEFR< 50 to < 70%.
Contd...
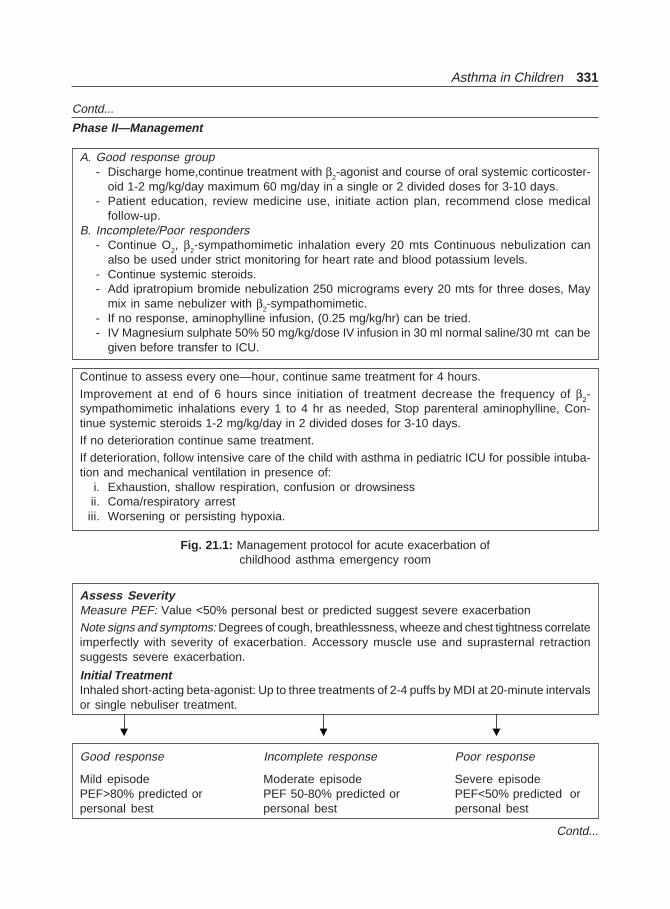
Asthma in Children 331
Contd...
Phase II—Management
A. Good response group- Discharge home,continue treatment with β2-agonist and course of oral systemic corticoster-
oid 1-2 mg/kg/day maximum 60 mg/day in a single or 2 divided doses for 3-10 days.- Patient education, review medicine use, initiate action plan, recommend close medical
follow-up.B. Incomplete/Poor responders
- Continue O2, β2-sympathomimetic inhalation every 20 mts Continuous nebulization canalso be used under strict monitoring for heart rate and blood potassium levels.
- Continue systemic steroids.- Add ipratropium bromide nebulization 250 micrograms every 20 mts for three doses, May
mix in same nebulizer with β2-sympathomimetic.- If no response, aminophylline infusion, (0.25 mg/kg/hr) can be tried.- IV Magnesium sulphate 50% 50 mg/kg/dose IV infusion in 30 ml normal saline/30 mt can be
given before transfer to ICU.
Continue to assess every one—hour, continue same treatment for 4 hours.
Improvement at end of 6 hours since initiation of treatment decrease the frequency of β2-sympathomimetic inhalations every 1 to 4 hr as needed, Stop parenteral aminophylline, Con-tinue systemic steroids 1-2 mg/kg/day in 2 divided doses for 3-10 days.If no deterioration continue same treatment.If deterioration, follow intensive care of the child with asthma in pediatric ICU for possible intuba-tion and mechanical ventilation in presence of:
i. Exhaustion, shallow respiration, confusion or drowsinessii. Coma/respiratory arrestiii. Worsening or persisting hypoxia.
Fig. 21.1: Management protocol for acute exacerbation ofchildhood asthma emergency room
Assess SeverityMeasure PEF: Value <50% personal best or predicted suggest severe exacerbationNote signs and symptoms: Degrees of cough, breathlessness, wheeze and chest tightness correlateimperfectly with severity of exacerbation. Accessory muscle use and suprasternal retractionsuggests severe exacerbation.
Initial TreatmentInhaled short-acting beta-agonist: Up to three treatments of 2-4 puffs by MDI at 20-minute intervalsor single nebuliser treatment.
Good response Incomplete response Poor response
Mild episode Moderate episode Severe episodePEF>80% predicted or PEF 50-80% predicted or PEF<50% predicted orpersonal best personal best personal best
Contd...
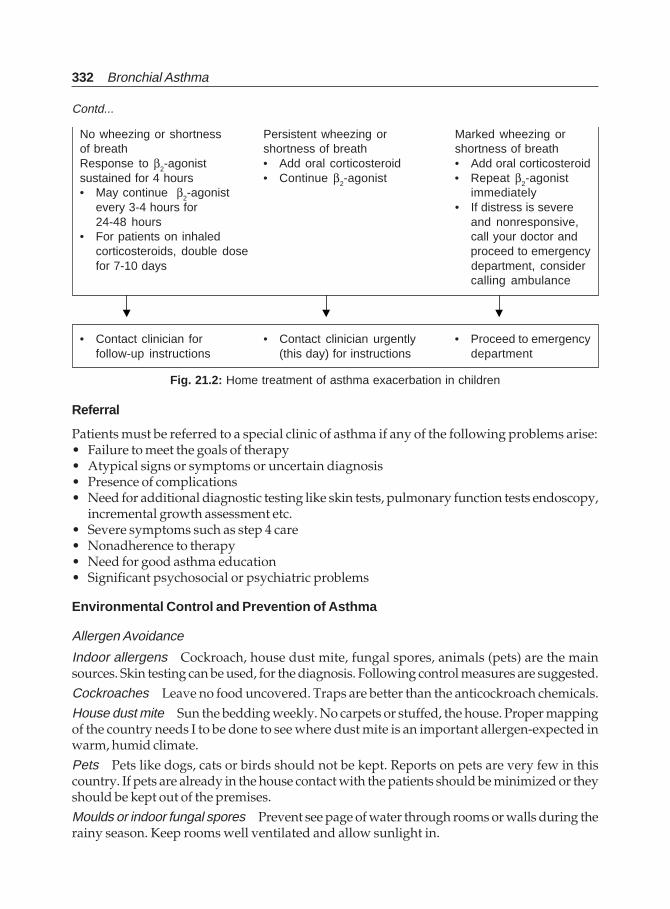
332 Bronchial Asthma
No wheezing or shortness Persistent wheezing or Marked wheezing orof breath shortness of breath shortness of breathResponse to β2-agonist • Add oral corticosteroid • Add oral corticosteroidsustained for 4 hours • Continue β2-agonist • Repeat β2-agonist• May continue β2-agonist immediately
every 3-4 hours for • If distress is severe24-48 hours and nonresponsive,
• For patients on inhaled call your doctor andcorticosteroids, double dose proceed to emergencyfor 7-10 days department, consider
calling ambulance
• Contact clinician for • Contact clinician urgently • Proceed to emergencyfollow-up instructions (this day) for instructions department
Fig. 21.2: Home treatment of asthma exacerbation in children
Contd...
Referral
Patients must be referred to a special clinic of asthma if any of the following problems arise:• Failure to meet the goals of therapy• Atypical signs or symptoms or uncertain diagnosis• Presence of complications• Need for additional diagnostic testing like skin tests, pulmonary function tests endoscopy,
incremental growth assessment etc.• Severe symptoms such as step 4 care• Nonadherence to therapy• Need for good asthma education• Significant psychosocial or psychiatric problems
Environmental Control and Prevention of Asthma
Allergen Avoidance
Indoor allergens Cockroach, house dust mite, fungal spores, animals (pets) are the mainsources. Skin testing can be used, for the diagnosis. Following control measures are suggested.Cockroaches Leave no food uncovered. Traps are better than the anticockroach chemicals.House dust mite Sun the bedding weekly. No carpets or stuffed, the house. Proper mappingof the country needs I to be done to see where dust mite is an important allergen-expected inwarm, humid climate.Pets Pets like dogs, cats or birds should not be kept. Reports on pets are very few in thiscountry. If pets are already in the house contact with the patients should be minimized or theyshould be kept out of the premises.Moulds or indoor fungal spores Prevent see page of water through rooms or walls during therainy season. Keep rooms well ventilated and allow sunlight in.

Asthma in Children 333
Seasonal exposure to pollens and fungi can be reduced by keeping the doors and windowsclosed from every morning till evening. Wherever affordable, air-conditioning can be used. Incase an allergen is found to contribute significantly to patient problem, he or she should bereferred to a specialist for skin testing and if required, for immunotherapy. In children lessthan 5 to 6 years of age immunotherapy is avoided.
Irritants or Chemicals
Avoid tobacco smoke, strong odours, fumes from various kinds of stoves/chullah, usingkerosene, wood, cowdung.
In high risk families (atopy on both sides or even one side), exclusive breastfeed to continuefor 4 to 6 months and mother to avoid well-known allergenic food in diet while baby isexclusively breastfeed.
Psychosocial Aspects of Asthma Management in Children
Children with chronic illnesses are at an increased risk for developing psychologicaldisturbances.
Children with severe asthma have been found to be three times more likely to developemotional/behavioural problems as compared to healthy children. It was decided at themeeting that the primary physician to the patient be able to deliver the necessary preventiveservices like explaining the basic facts about the disease and try to improve the quality of lifeby optimum care. Mental health workers can provide important services to asthmatic children,who have obvious psychological or behavioural problems, experience school difficulties andare noncompliant with treatments.
Family therapy aimed at modifying family interaction problems and parent-childrelationships can help in improved management of asthma and also improve the overallquality of life. Hence, family therapy is considered an adjunct to the conventional treatment inasthma in children with severe disease. It is important that psychologists be part of themultidisciplinary treating team in order to provide comprehensive services to children withasthma.
Health Education
The experts stressed the need for health education not only in asthma clinic or hospital butalso on TV, radio and other communication media. The attitudes and practices concerningthis disease demonstrate a high degree of ignorance and misinformation. Written materialcontaining information regarding basic facts of asthma should be made available to thepatient and the parent at the time of transmission of information regarding the diagnosis.Special measures were recommended to be taken to educate the people about the harms ofpassive smoking.
Future Directions for Research
The data presented indicates gross inadequacy of information regarding basic facts of asthmato patients and their parents. Intervention in the form of written material significantly improvesthe knowledge of these individuals. More studies need to be done to assess the knowledge,attitudes and practices of these patients and specific materials developed to improve thebaseline information and change attitudes towards inhalational therapy. All the participantsfelt that there was a local social stigma attached to the disease, and parents of the patients

334 Bronchial Asthma
were specially concerned about the inhalatinal therapy having potential for producing drugdependence. Why incidence of asthma is relatively less in India and the disese is less severeas compared to some of the Western countries, information regarding it is not available. Moredata need to be generated towards epidemiology of asthma in this country. Research intousefulness of yogic breathing exercises and role of Ayurveda needs to be evaluated, althoughat present they have no proven scientific value.
REFERENCES
1. Global Strategy for Asthma Management and Prevention. NHLBI/WHO Workshop report.Global Initiative for Asthma. NIH Publication No. 95-3659; January 1995, reprinted 1996;Epidemiology; Chapter 2; 1-24.
2. Gergen Pj, Mullay DY, Evans R M. National Survey of prevalence of asthma among children inthe United States, 1976 to 1980. Pediatrics 1988;81:1-7.
3. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee.Worldwide variations in the prevalence of asthma symptoms: The International study ofAsthma: The International Study of Asthma and Allergies in Childhood (ISAAC). Eur Respir J1998;12:315-335.
4. Burney PGJ. Luczynska C. Chinn S, Jarvis D. The European Community Respiratory HealthSurvey. Eur Respir J 1994; 7:954-60.
5. European Community Respiratory Health Survey (ECRHS). Variations in the prevalence ofrespiratory symptoms, self-reported asthma attacks, and use of asthma medication in theEuropean Community Respiratory Health Survey (ECRHS). Eur Respir J 1996; 9:687-695.
6. Habbick BF, Pzzichini MM, Taylor B, Rennie D, Senthilselvan A, Sears MR. Prevalence of asthma,rhinitis and eczema among children in 2 Canadian cities: in International Study of Asthma andAllergies in Childhood. CMAJ. 1999; 160:1824-28.
7. Shamssain MH, Shamsian N. Prevalence and severity of asthma, rhinitis, and atopic eczema in13 to 14 years-old schoolchildren from the northeast of England. An Allergy Asthma Immunol.2001; 86:428-32.
8. EI-Sharif NS, Nemery B, Barghuthy F, Mortaja S, Qasrawi R, Abdeen Z. Geographical variationsof asthma and asthma symptoms among schoolchildren aged 5 to 8 years and 12 to 15 years inPalestine: The International Study of Asthma and Allergies in Childhood (ISAAC). Ann AllergyAsthma Immunol. 2003; 90:63-71.
9. AI-Dawood K. Epidemiology of bronchial asthma among schoolboys in AI-Khobar city, SaudiArabia: Cross-sectional study. Croat Med J 2000;41:437-41.
10. AI-Riyami BM, AI-Rawas OA, AI-Riyami AA, Jasim LG, Mohammed AJ. A relatively highprevalence and severity of asthma, allergic rhinitis and atopic eczema in schoolchildren in theSultanate of Oman. Respirology 2003;8:69-76.
11. Trakultivakorn M. Prevalence of asthma, rhinitis, and eczema in Northern Thai children fromChiang Mai (International Study of aAsthma and Allergies in Childhood, ISAAC). Asian Pac JAllergy Immunol 1999; 17:243-48.
12. Shohat T, Golan G, Tamir R, Green MS, Livne I, Davidson Y, Harari G, Garty BZ. Prevalence ofasthma in 13-14 years-old schoolchildren across Israel. Eur Respir J 2000;15:725-29.
13. Crane J, Mallol J, Beasley R, Stewart A, Asher MI. International study of Asthma and Allergies inChildhood Phase I study group. Agreement between written and video questions for comparingasthma symptoms in ISAAC. Eur Respir J 2003; 21:455-61.
14. Beasley R, Ellwood P, Asher I, International patterns of the prevalence of pediatric asthma inISAAC program. Pediatr Clin North Am. 2003; 50:539-53.
15. Paramesh H. Epidemiology of asthma in India. Indian J Pediatr 2002;69:309-12.

Asthma in Children 335
16. Chhabra SK, Gupta CK, Chhabra P, Rajpal S. Risk factors for development of bronchial asthmain children in Delhi. Ann Allergy Asthma Immunol. 1999;83:385-90.
17. Gupta D, Aggarwal AN, Kumar R, Jindal SK. Prevalence of Bronchial Asthma and Associationwith Environmental Tobacco Smoke Exposure in Adolescent School Children in Chandigarh,North India. Journal of Asthma, 2001; 38:501-07.
18. Palmer LJ, Valinsky IJ, Pikora T, Zubrick SR, Landau LI. Environmental factors and asthma andallergy in schoolchildren from Western Australia. Eur Respir J. 1999;14:1351-57.
19. Chan KN, Elliman A, Bryan et al. Respiratory symptoms in children of low birth weight Arch DisChild 1989; 64:1294-1304.
20. Chan KN, Noble-jamieson Cm, Elliman A et al. Lung function of children of low birth weight-Arch Dis Child 1989;1284-1393.
21. Burrows B, Taussig ML. “As the twig is bent, the tree inclines” (perhaps). Am Rev Respir Dis19K-122:813-16.
22. Dodge R, Martinez FD, Chm MG et al. Early childhood respiratory symptoms and the subsequentdiagnosis of asthma. J Allergy Clin Immunol 1996,98:48-54.
23. Roberson CF, Heycock E, Bishop J et al. Prevalence of asthma in Melbourne schoolchildren-changes over 26 years. BMI 1991;302:1116-18.
24. Ninan T, Ryssekk G. Respiratory symptoms and atopy in Aberdeen school children -evidencefrom two surveys 25 years apart- BMI 1992;304:873-75.
25. Peat JK-van den Berg RK Green WF et al. Chanding prevalence of Asthma in Australian children.BMJ 1994;308:1591-96.
26. Chandra RK. Prospective studies of the effect of breast feeding on incidence of infection andallergy. Acta Paediatr Scand 1979;68:691-94.
27. Fergusson DK Horwood JL, Shannon FT et al. Breast feeding gastrointestinal and lowerrespiratory illness in the first two years. Aust Paediatr J 1981;17:191-95.
28. Burr ML, Limb ES, Maguire MJ et al. Infant feeding wheezing, and allergy—A prospective.study.Arch Dis Child 19930:724-28.
29. Poysa L, Korppi M, Remes K et al. Atopy in childhood and diet in infancy a nine years follow-upstudy-I-clinical manifestations. Allergy Proc 1991;12:107-11.
30. Rust GS, Thompson CJ, Minor P, Davis-Mitchell W, Holloway K, Murray V. Does breastfeedingprotect children from asthma? Analysis of NHANES III survey data. J Natl Med Assoc. 2001;93:139-49.
31. Burney P. A diet rich in sodium may potentiate asthma—epidemiology evidence for a newhypothesis. Chest l987;91(Suppl):143S-148S.
32. Carey OJ, Locke C, Cooksoh JB. Effect of alterations of dietary sodium on the severity of asthmaIn men. Thorax 1993;48:714-18.
33. Peat JK, Hodge T, Salome CM et al. Dietary fish intake and asthma in children. Am J Respir CritCare Med :1995;151(Suppl):A469.
34. Schwartz J, Weiss ST. The relationship of dietary fish intake to level of pulmonary function in thefirst National Health and Nutrition Survey (NHNS). Eur Respir 1 1994;7:1821-24.
35. National Research Council, Environmental tobacco smke’- nwzsunng -exposures and assessingeffects. Washington: National Academy Press, 1986.
36. Cunningham J, O’Connor GT, Dockery DW et at. Environmental tobacco smoke, wheezing andasthma children in 24 communities. Am I Respir Crit Care Med 1996;153:218-24.
37. Martinez FD, Wright AL, Taussig LM et al. Asthma and wheezing in the first six years of Iife:N 1995;332:133-38.
38. Von Mutius E, Sherrill DL, fritzsch C et al. Air-pollution and upper respiratory-symptoms inchildren from East Germany. Eur Respir j 1995;8:23-28.
39. Braback L, Breborowicz A, Knutpson A et al. Atopic sensitisation and respiratory symptoms,and Swedish school children. Clin Exp Allergy 1994;24:826-35.

336 Bronchial Asthma
40. Behera D, Sood P, Singhi S. Passive smoking, domestic fuels, and lung function in North IndiaChildren. Ind J Chest Dis All Sci 1998; 40:89-98.
41. Behera D, Sood P, Singhi S. Respiratory, symptoms in Indian children exposed to differentcooking fuels. J Assoc Phys India 1998;46:182-18
42. Wjst M, Reitmeir P, Dold S et al. Road traffic and adverse effects on respiratory health in children.BMJ 1993;307:596-600.
43. Weinmann.GG, Bowes SK Gerbase MW et al. Response to acute ozone exposure in healthy men.Results of a screening procedure. Am I.Respir Crit Care Med 1995;151:33-40.
44. Clough JB, WAHom JD, Holgate ST. Effect of atopy on the natural history of flow, and bronchial,hyperresponsiveness in 7 and 8-years-old children with 4 Respir Dis 1991;143:755-60.
45. Martin AJ, Landau LI, Phelan PD. Lung functions in young adults who had asthma in childhood.Am Respir Dis 1980;122:609-16.
46. Gerritsen J. Prognosis of asthma from childhood to adulthood. Am Rev Respir Dis 1989;140:1325-30.
47. Peat JK Wookock AJ. Sensitivity to common allergens; relation to respiratory symptoms andbronchial hyperesponsiveness in children from different climatic areas of Australia. Clin ExptAllergy 1991;21:573-81.
48. van Asperen PP, Kemp AS, Mukhi A. Atopy in infancy predicts the severity of bronchialhyperresponsiveness in later childhood. J Allergy Cline Immune 1990; 85:790-95.
49. Sherril D. The effect of airway hyperresponsiveness, wheezing and atopy on longitudinalpulmonary function in children—A six ear follow-up study. Pediatr Pulmonol 1992;13:78-85.
50. Murphy S. Asthma: An inflammatory disease: In Hillman BC (Ed): Paediatric Respiratory Disease:Diagnosis and Management. Philadelphia: WB Saunders, 1993;621-26.
51. von Mutius E. Progression of allergy and asthma through childhood to adolescence. Thorax19%;51(Suppl 1):S3-S6.
52. Kelly WJ. Childhood asthma and adult lung function. Am Rev Respir Dis 1988;138:26-30.53. Martin AJ, Landau LI, Phelan PD. Asthma from childhood at age 21—the patient and his (or her)
disease. Br Med J 1982;284:380-8254. Williams H, McNicol KN. Prevalence, natural history, and relationship of wheez bronchitis and
asthma in children. An epiden-dologic study. Br Med J 1969;4:321-25.55. Park ES, Golding J, Carswell F et al. Preschool wheezing and prognosis at 10. Arch Dis Child
1986;61:642-46.56. Balfour-Lynn. Childhood asthma and puberty. Arch Dis Child 1985;60:231-35.57. Peat JK, Salome CM, Segwick CS et al. A prospective study of bronchial hyperresponsiveness
and respiratory symptoms in a population of Australian school children. Clin Expt Allergy1989;19:299-306.
58. Roorda RJ. Prognostic factors for the outcome of childhood asthma in adolescence, Thorax1996;51(Suppl 1):S7-Sl2.
59. Price JF. Issues in adolescent asthma: What are the needs? Thorax 1996;51(Suppl 1):Sl3-Sl7.60. Ownby DR. Enviroronmental factors versus genetic determinants of childhood inhalant allergies.
J Allergy Clin Immunol 1990;86:279-87.61. Frischer T, Kuehr J, Meinert R et al. Risk factors for childhood asthma and recurrent wheezy
bronchitis. Eur J Pediatr 1993;152:771-75.62. Sherman CB, Tosteson TD, Tager IB et al. Early childhood predictors of asthma. Am I Epidemiol
1990;132:83-95.63. Sibbald B, Horn MEC, Gregg 1. A family study of the genetic basis of asthma and wheezy
bronchitis. Arch Dis Child 1980;55:354-57.64. The Cochranc Library: Update Soft Ware, Oxford, UK.65. Parmar V, Kumar L, Malik SK. Normal values of peak expiratory flow rate in healthy north
Indian school children 6-16 years of age. Ind Pediatr 1977;14:591-94.

Index 337
Index
A
Acupuncture 261Acute severe asthma 208, 276
anti-immunoglobulin E 248,272
assessment 212,226in children 285clinical features 210complications 211definition 208differential diagnosis 212indices 213management 215, 290pathophysiology 209therapeutic approach 227
Add-on therapy 268Adhesion molecules 48Adrenergic bronchodilators 142Allergens 16, 20, 188Allergic bronchopulmonary
aspergillosis 117, 272Allergy 14, 129
management 129Alternative and complementary
therapies 201Alternative treatment
oral steroid dependence 161Animal allergens 19Anticholinergics 148
comparison 155side effects 156
Antihistamines 159Arachidonic acid 43Aspirin 22, 67, 272Assessment of asthma control
202Asthma
definition 1sign 99symptoms 98
Asthma remission 89Atopy 14
B
β2-agonists 281Basophils 48Beta-adrenergic receptors 62,
139, 143Bradykinin 52Brittle asthma 96Bronchial asthma
aetiology 14complication 117diagnosis 98epidemiology 1management 127pathology 86pathophysiology 40pharmacologic manage-
ment 134prognosis 114
Bronchial asthma, management127, 235acute severe asthma 208diet modification 258nonpharmacologic 128, 256,
257pharmacologic 134, 191step-care 192, 194, 196
Bronchial hyperreactivity 40, 61Bronchoprovocation test 103
C
Childhood asthma 7Chronic bronchial asthma 183Chronic eosinophilic bronchitis
44Complementary and alternative
medicine 261Corticosteroids 151Cough variant asthma 92Cromones 157Cyclosporin 163, 296Cytokines 49, 53, 55, 250
D
Dietary manipulation 263Diuretics 298Drug-induced asthma 243Drugs 22Dry powder inhalers (DPI) 178
E
Early asthmatic reaction 40Endocrinal factors 30Environmental factors 32, 129,
260Environmental tobacco smoke
30Eosinophils 44Epithelial-mesenchymal trophic
unit 89Exercise-induced asthma 23, 69,
235, 271Extrinsic asthma 93
F
Fatal asthma 306Food allergen 20
G
Gastro-oesophageal reflux(GER) 27, 243
Genes 32Genetics 31Gold salts 163, 295Gut hormones 59
H
Heparin 297Histamine 52Hospital discharge 284House-dust mite 18, 189, 259Hygiene hypothesis 258

338 Bronchial Asthma
I
Immunotherapy 131,190Infection 20,21Inflammation 55, 64, 65Inflammatory mediators 49Inhalation therapy 176Interleukin 249Intrinsic asthma 93
K
Ketotifen 158
L
Laboratory findings 100Late asthmatic reaction 40Late onset asthma 93Leukotriene 43, 49
antagonists 159Lymphocytes 45
M
Macrophages 47Mast cells 42Mechanical ventilation 224Mediators 58Metered dose inhalers 177Methotrexate 162, 293Methylxanthines 134Monocytes 47Morning dippers 96Mortality 4,6,114Mould 16
N
Natural history 6Nebulisers 179Nedocromil sodium 158Neural control 59Neurogenic inflammation 60Neuropeptides 59Neutrophils 48Neutrophins 58New guidelines for asthma
management 256, 265, 276Newer drugs 247Nitric oxide 58, 64Nocturnal asthma 72, 95
O
Objective tests 102Occupational asthma 24, 70, 94,
106, 109, 241Oxygen radicals 57
P
Patient education 128, 185,284
Phosphodiesterase inhibition134, 251
Platelet 52Pollen 16Pollution 29, 260Pregnancy 236Prostaglandins 52Provocateurs 21
R
Refractory asthma 306Remodelling 57, 88Rhinitis 27, 272
S
Secondary prophylaxis 259Severe asthma 306Sherwood-Jones index 213Sinusitis 27Smoking 260Spacer devices 181Status asthmatics 208
drugs 221Step-care management 273,
274, 275Sulphite sensitivity 26Surgery 239Sympathomimetic agents 145
T
Tartrazine 26Theophylline 140Tiotropium 272Tokyo-Yokohama asthma 29Troleandomycin 295
V
Ventilator 222noninvasive 283
Virus-induced asthma 69
Y
Yin-Yang hypothesis 60