
BONDING AND ELECTRONIC STRUCTURE OF MINERALS · BONDING AND ELECTRONIC STRUCTURE OF MINERALS ......
64
BONDING AND ELECTRONIC STRUCTURE OF MINERALS RONALD E. COHEN Carnegie Institution of Washington Geophysical Laboratory and Center for High Pressure Research 5251 Broad Branch Rd., N.W. Washington, D.C. 20015 USA Abstract. Minerals are crystalline solids, and their properties are gov- erned by quantum mechanics. Density functional theory in the local den- sity approximation or the generalized gradient approximation gives accu- rate predictions for energetic properties of closed shell systems, as well as ionic/covalent crystals, and open-shelled transition metals and transition metals oxides. The electronic structure and phase transitions in transition metal oxides are forefront problems in solids state physics and high pressure physics, and much progress is being made. 1. Introduction–Minerals are Crystalline Solids The first thing that one must realize in order to understand minerals, is that minerals are solids, and the same physical laws that govern mineral behavior are those for all other solids. It is quantum mechanics that governs the behavior of electrons and nuclei in solids, and it is quantum mechanics that rules mineral behavior. The atomic positions in minerals have space- group symmetry, and the behavior of the electrons and nuclei, and thus the properties of minerals depend on symmetry. Thus one must study solid state physics and crystallography to understand mineral behavior. No at- tempt will be made here to duplicate the manifold excellent texts on solid state physics and crystallography. The student is pointed to texts such as Ashcroft and Mermin [1], Kittel [2], and Harrison [3] for the general background needed to understand minerals. It used to be that important minerals were generally more complex than the important materials of solid state physics. Mineralogists were interested in feldspars, and physicists were
Transcript of BONDING AND ELECTRONIC STRUCTURE OF MINERALS · BONDING AND ELECTRONIC STRUCTURE OF MINERALS ......

BONDING AND ELECTRONIC STRUCTURE OF MINERALS
RONALD E. COHEN
Carnegie Institution of WashingtonGeophysical Laboratory and Center for High Pressure Research5251 Broad Branch Rd., N.W.Washington, D.C. 20015USA
Abstract. Minerals are crystalline solids, and their properties are gov-erned by quantum mechanics. Density functional theory in the local den-sity approximation or the generalized gradient approximation gives accu-rate predictions for energetic properties of closed shell systems, as well asionic/covalent crystals, and open-shelled transition metals and transitionmetals oxides. The electronic structure and phase transitions in transitionmetal oxides are forefront problems in solids state physics and high pressurephysics, and much progress is being made.
1. Introduction–Minerals are Crystalline Solids
The first thing that one must realize in order to understand minerals, isthat minerals are solids, and the same physical laws that govern mineralbehavior are those for all other solids. It is quantum mechanics that governsthe behavior of electrons and nuclei in solids, and it is quantum mechanicsthat rules mineral behavior. The atomic positions in minerals have space-group symmetry, and the behavior of the electrons and nuclei, and thusthe properties of minerals depend on symmetry. Thus one must study solidstate physics and crystallography to understand mineral behavior. No at-tempt will be made here to duplicate the manifold excellent texts on solidstate physics and crystallography. The student is pointed to texts suchas Ashcroft and Mermin [1], Kittel [2], and Harrison [3] for the generalbackground needed to understand minerals. It used to be that importantminerals were generally more complex than the important materials of solidstate physics. Mineralogists were interested in feldspars, and physicists were

2
interested in NaCl or Cu. This is not generally true anymore, with the greatinterest in cuprate superconductors, complex ferroelectrics and multicom-ponent and complicated alloys in physics, and the greater interest in simplematerials such as MgO in mineralogy. Many of the problems are also thesame, predicting and understanding phase transitions, equations of state,electrical and chemical transport, etc. The goals remain somewhat differ-ent however. The goal of mineralogy is to understand natural materials inorder to interpret better the way the world works, without regard to utilityor applications. In this brief review the underlying physics of minerals willbe touched on, and bonding in minerals will be discussed. No attempt ismade here to thoroughly review different theoretical methods, to study anysystems in detail, or to provide a practical tutorial on computation. Also,this is not meant to be a balanced review of all methods and work doneon theory of materials; rather it is a biased picture and represents just onepoint of view.
2. Minerals are made of atoms, or electrons and nuclei
No attempt will be made here to review solid state theory in any significantway. Instead the flavor of our current understanding of the fundamentalphysics of minerals will be presented, and some key points and milestonesin our understanding of the theory of minerals will be discussed. First onemust understand that minerals are made out of atoms, and that all mineralproperties are governed by the atoms that make up a mineral, and theinteractions between the atoms. As a graduate student I remember tellingmy advisor, J.B. Thompson, Jr. that I didn’t want to have to deal withelectrons, but he said that “you may have to worry about the electrons!”And indeed, if you are interested in the fundamentals of mineral behavior,you will have to worry about the electrons, because it is the interactionsbetween electrons in atoms in a material that are responsible for everythingabout the mineral. Quantum theory tells us that the time-independentground state of a system is given by a complex antisymmetric many bodywave function, whose square gives the probability of finding a particle ineach point in space
ρ(r) =
∫dr2dr3dr4...Ψ
∗(r, r2, r3, r4, r5...)Ψ(r, r2, r3, r4, r5...) (1)
In principle, the wave function can be represented as a sum of determi-nants of orthogonal single particle states, φi, of the system. In practice thisapproach, known as Configuration Interaction (CI) is intractable for solids,and even for molecules and many-electron atoms converges very slowly. Inan atom or molecule, the eigenvalues are the well known energy levels of

3
the electronic system. In a crystal, these states are modified by the otheratoms in the crystal. The core states, or deep levels for each atom, remainsharp delta function-like states, which may be raised or lowered in energyrelative to their positions in isolated atoms. These core-level shifts are duelargely to the screened Coulomb potential from the rest of the atoms inthe crystal. The occupied valence and empty conduction states no longerlook like atomic states, but are broadened into energy bands (fig. 1). Thereare often intermediate states between the core levels and the valence statescalled semi-core states which are slightly broadened at low pressures, butwhich become broader and more different from atomic states with increas-ing pressure. Atoms in crystals can also donate or accept electrons from
1s
2s2p
3s3p
1s
2s
2p
Si Ocrystal
1s
2s2p
3s3p
1s
2s
2p
Si Omolecule
Figure 1. Schematics of electronic energy states in (a) an Si-O molecule and (b) aSiO2 crystal. The atom level hybridize, and are broadened into energy bands rather thandiscrete levels in the crystal. The three hatched bands in the crystal indicate bonding,non-bonding, and conduction (anti-bonding) bands from bottom to top.
other atoms, or in other words the states can be filled differently than inatoms, leading to the formation of ions. This ionicity is very important inthe bonding of minerals, and is driven by the increased stability of an ionwhen it has a filled shell of electrons. For example, oxygen has a nuclearcharge Z=8, which means that it would have 2 1s, 2 2s, and 4 2p electrons,but a filled p shell has 6 electrons. Thus the O atom is highly reactive, and

4
in the gas combines to form O2 or other molecules with other atoms. In anoxide or silicate crystal, the O grabs two more electrons to form an O2−
anion from another atom, which becomes a positively charged cation. Thisleads to a strong attractive electrostatic, or Madelung, interaction betweenthe ions which greatly enhances the crystal stability.
The band states in a crystal can be characterized by a continuous quan-tum number k, so that the eigenvalues are ε(k), and the eigenfunctions canrepresented as Bloch states
φi(k, r) = u(r) exp−ik·r (2)
where u(r) is a periodic function of position r. The eigenvalues as a functionof k are known as the band structure. The density of states gives the densityof eigenvalues around each energy E. Each eigenfunction also has a certainamount of weight around each atom, and a partial density of weights canbe found that indicates the amount each atom contributes to each energyrange. By studying the band structure and densities of states (figs. 2,3,4),and the charge densities, one can understand the nature of bonding andhow it changes with chemistry, distortions, and pressure.
(a) (b)
Figure 2. Computed band structures for MgO. (a) MgO B1 V = 18.1A3, -0.05 GPa. (b)MgO B1 V = 10A3, 290 GPa. States from the bottom are Mg 2p, O 2s, O 2p, Mg 3s, O3s at zero pressure. The band gap is above the O 2p states. Note that the O 2s band hassignificant width even at zero pressure, and a large width at high pressures, indicatingit is not really an atomic-like state. Such a state is called a “semi-core” state. Note thatthe gap widens and changes from direct at Γ to indirect Γ to X with increasing pressure.From Ref. [4].
5
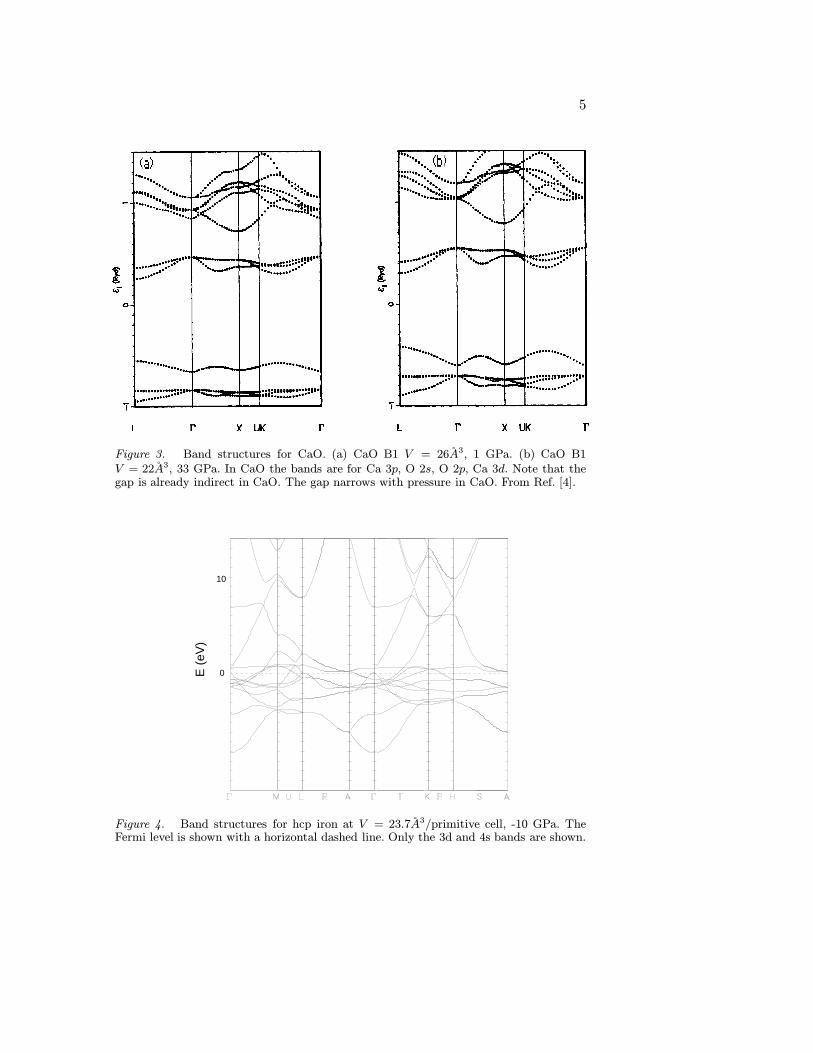
Figure 3. Band structures for CaO. (a) CaO B1 V = 26A3, 1 GPa. (b) CaO B1V = 22A3, 33 GPa. In CaO the bands are for Ca 3p, O 2s, O 2p, Ca 3d. Note that thegap is already indirect in CaO. The gap narrows with pressure in CaO. From Ref. [4].
E (
eV)
0
10
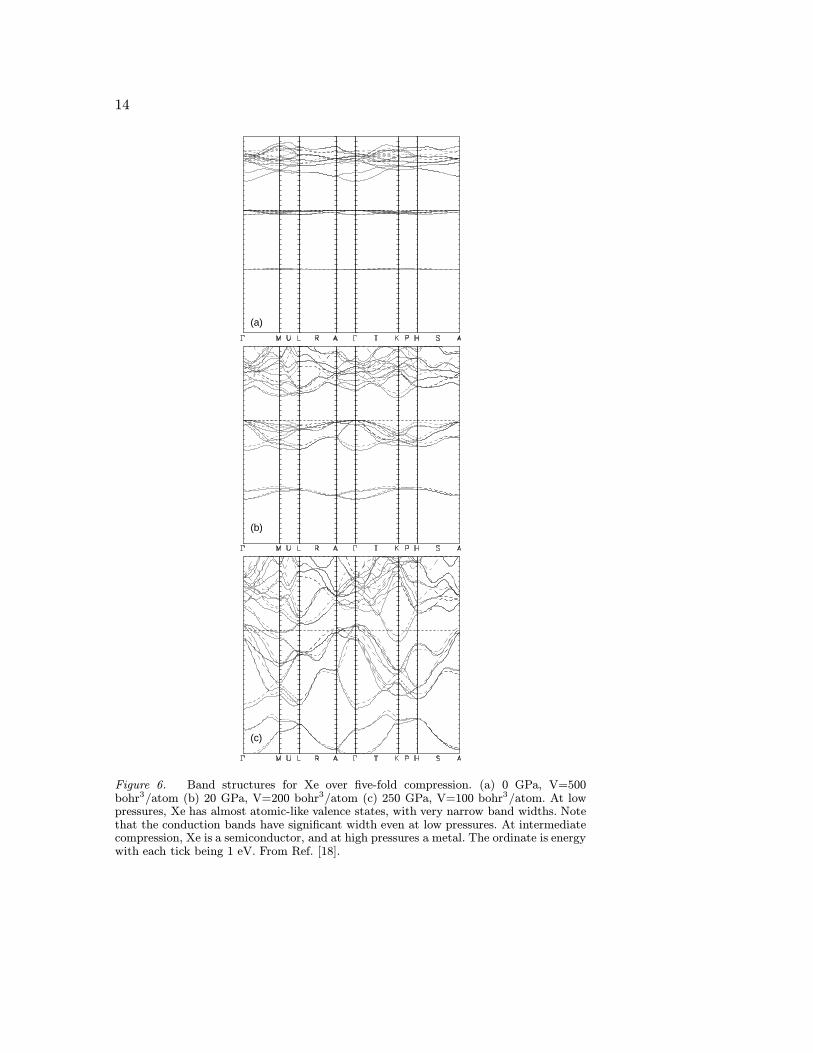
Figure 4. Band structures for hcp iron at V = 23.7A3/primitive cell, -10 GPa. TheFermi level is shown with a horizontal dashed line. Only the 3d and 4s bands are shown.

6
Figure 5 shows the density of states for MgO compared with XPS spec-troscopy (injecting x-ray photons and measuring the energies of electronsthat are ejected) [5]. The close agreement between LDA band energies andexperimental quasiparticle energies is evident. Matrix elements are not in-cluded in the density of states calculation, and the experiment is surfacesensitive and instrumentally broadened, contributing to the differences inintensities shown.
Figure 5. MgO density of states computed with the LDA compared with XPS measure-ments. From Ref. [5].
3. Bonding in crystals
Crystals must have a lower energy, or be bound, relative to separated atomsor molecules, or the atoms would just fly off into space. The binding forcesthat hold the atoms together are generally electrostatic in origin. Secondly,there must be some force that keeps the atoms from collapsing into eachother. This force is largely due to the increased kinetic energy as atomsget closer, the Pauli exclusion principle that keeps electrons apart, theelectrostatic repulsion between electrons, and ultimately as the atoms arebrought closer and closer together, the electrostatic repulsion of the nuclei.Crystals can be characterized by the primary source of their binding energy.

7
In ionic crystals, the primary source of binding is from the electro-static attraction among ions. Examples are NaCl (Na+Cl−) and MgO(Mg2+O2−). Covalent materials primarily bind through hybridization(sometimes called “sharing”) of valence electrons, which is a way to lowerthe energy of the electrons, good examples being diamond and the semicon-ductor Si. The potential energy is lowered since the electrons can see theattractive positive potential of two nuclei or atom cores rather than justone. The electron distribution is modified, and charge builds up betweenthe atoms. The lowering of the potential energy more than overcomes theincrease in kinetic energy due to the increased electron density in the regionbetween the atoms. In metals, the primary binding comes from embeddingatom cores in a sea of itinerant electrons, for example in Na metal. Most ma-terials have combinations of these interactions. Thus silicates are about halfionic and half covalent. Transition metals such as Fe are both metallic andcovalent. Another source of bonding is from dispersion, or van der Waalsforces, fluctuating dipoles on separated atoms or molecules. Although theseforces are important at long range, where atoms do not overlap, these forcesare quenched as the atoms or molecules overlap. So called “van der Waals”solids are probably not held together by dispersion forces, but rather bylocal many-body exchange and correlation interactions among electrons.
Usually we are not asking whether a crystal will be bound at all, butrather questions like what crystal structure will be stable over another, andwhat are the physical properties, such as elasticity and equation of state,of a given mineral. Many of these properties, including all thermodynamicproperties (at ordinary temperatures, i.e. most all geophysically relevantconditions) can be found by computing ground state properties alone, forexample the energy as a function of moving an atom or straining the lattice.Excited state properties are more difficult to obtain accurately, and involvethe energetics of exciting electrons out of their ground state configurations.Examples of such properties include optical spectra.
4. Band theory
The eigenstates of a crystal can be characterized by a quantum number k(eq. 2). The energy bands are dispersive as functions of k, and the energyversus k is known as the band structure. Much can be learned from studyingband structures and how they change as the crystal structure or chemistrychanges. Much of solid state electronics is based on relative simple ideasbased on band structures, occupancy of different bands with doping, andhow bands line up across interfaces. Bands can also be studied experimen-tally using photoemission, for example the study of the relative energiesof emitted electrons as functions of input photon energy and wavevector,

8
or from x-ray spectroscopy, or combinations of x-ray and electron spec-troscopy. Crystals with band gaps between occupied and unoccupied statesshould be insulators, and those with partially filled bands should be metals.(If the gap is small or if the material is useful in electronic applications bychemical doping to inject electrons in the conduction bands, or holes in thevalence bands, a crystal is called a semiconductor.) In a non-magnetic sys-tem, each band holds two electrons, and thus a crystal with an odd numberof electrons in the unit cell should be a metal since it will have at least onepartially filled band. This is not always true, as will be discussed below inthe section on Mott insulators.
A most important concept to understand band theory of crystals is theidea of a quasiparticle. The real energy states in a crystal are not singleparticle eigenstates at each value of k; rather there is an energy spectrumwhich has more or less strong peaks at the quasiparticle energies. If there arestrong peaks in the spectrum, the energies coincide with objects that behavelike independent particles that are dressed by all of the interactions withother real particles in the system. These objects are known as quasiparticles,and these are what are generally observed in an experiment.
There is often some confusion by what one means by a band structure,especially when comparing experimental and theoretical results. Usuallyone calls the eigenvalue spectrum of whatever theory one is using the bandstructure. However, in Density Functional Theory (DFT, see below) theeigenvalue spectrum has no fundamental relationship to what one wouldobserve in an experiment, or to true quasiparticle energies. Nevertheless,DFT band structures are often in good agreement with experiment (fig. 5)and are widely used to give insight into bonding in crystals. The experimen-tal picture is also not straightforward, and band structures obtained usingdifferent methods may have different meanings. In angle resolved photoe-mission, for example, the observed band structure is the energy spectrumfor removing electrons from the surface of the crystal, which is sometimes acomplex phenomenon, and may be broadened or shifted from the intrinsicenergy levels in the interior of the crystal.
5. Density Functional Theory
By far the most computations for solids have been based on the densityfunction theory of Hohenberg and Kohn [6] and Kohn and Sham [7]. TheHohenberg-Kohn theorem states that all ground state properties of a sys-tem can be obtained from the ground state charge density, and Kohn andSham showed a practical way to do this, as described below. An alterna-tive approach is based on Hartree-Fock theory [8]. Traditionally quantumchemistry has been based on Hartree-Fock as a first approximation, followed

9
by further approximations to include correlation effects. Hartree-Fock in-cludes exchange exactly for a single Slater determinantal wave function,but neglects a large part of the energy which is called “correlation” fromelectron-electron interactions, culminating eventually in full configurationinteraction (CI) in which the wave function is a sum over all possible de-terminants formed from the basis. The correlated methods are extremelycomputationally intensive, and cannot be applied completely to crystals, inthe sense that one can never exhaust all of the possible correlated statessince there are an infinite number of many-body states one can form evenfrom a limited basis by coupling different k-vectors. Instead of relying onthe traditional Hartree-Fock to CI path, many quantum chemists are alsomoving towards using density functional theory (DFT), which has been sosuccessful for crystals, for molecules.
6. Total energy
Following the Kohn-Sham (KS) procedure, the total energy is given by
E = T0(ρ) + En−n +Ee−n +Eh + Exc, (3)
where T0 is the kinetic energy of a non-interaction gas of Fermions of thesame density ρ as the real system, En−n is the nuclear-nuclear repulsion,Ee−n electrostatic electron-nuclear interaction, Eh is the Hartree energy,that is the electrostatic energy for the electron charge density, and Exc isthe exchange-correlation energy. Exc is defined such that equation 3 is exact;thus it contains the correction from the non-interacting kinetic energy aswell as the exchange and correlation interactions between the electrons.
One solves the single-particle Schrodinger-like equation:
(−h2
2m∇2 + VKS)Ψi = εiΨi (4)
where the Kohn-Sham potential VKS is the functional derivative of the totalenergy with respect to the density,
VKS =δE(ρ)
δρ, (5)
and
Vxc =δExc(ρ)
δρ. (6)
The charge density ρ is given by:
ρ =∑
Ψ∗iΨif(εi −EF ). (7)

10
where f is 1 for εi − EF < 0 and zero otherwise (or the Fermi function atfinite T). This set of equations is solved self-consistently (i.e. iteratively).One starts with an initial guess for ρ, computes VKS, solves equation 4for Ψi, computes a new ρ from equation 7, and iterates until the inputand output densities ρ agree. Equation 4 is solved by using a basis φ forΨi =
∑j cijφij, and the secular equation∑
j
HijΨj (k) =∑j
εi (k)OijΨj , (8)
must be diagonalized, where the Hamiltonian matrix H is given by
Hij (k) = 〈Ψi (k) |H |Ψj (k)〉 , (9)
the overlap matrix O is given by
Oij (k) = 〈Ψi (k) |Ψj (k)〉 , (10)
and the Hamiltonian operator is
H (r) = −∇2 + Vn + Vh (n (r)) + Vxc (n (r)) , (11)
where the first term is the kinetic energy operator, the second is the nuclearpotential, the third is the classical electrostatic Hartree potential, and thelast term is the quantum mechanical exchange correlation potential whichaccounts for the many-body electron-electron interactions. The total energyis given by
Etot (n) = Ebs −Eh + ∆Exc + EEwald, (12)
where the band structure energy is given by
Ebs =∑i
εif(εi − EF ). (13)
The term Eh corrects for double counting of the Hartree energy in Ebs,∆Exc =
∫dr [εxc −Vxc] ρ (r) is the difference in exchange-correlation en-
ergy functional and the potential, and EEwald is the nuclear-nuclear (orcore-core) electrostatic energy. If the functional Exc(ρ) were known, theKohn-Sham procedure would be exact, and the computed energy and den-sity would be exact. As discussed below, even in the exact theory, the KSeigenvalues of equations 4 or 8 are not the actual quasiparticle energies,and the band structure constructed from these eigenvalues is not the bandstructure that would be measured experimentally. Empirically, it is oftenfound that that the KS band structure is a good approximation to the realband structure.

11
The exact Exc(ρ) (or Vxc(ρ)) is not known, although excellent approx-imations have been developed that work well for most materials. In theLocal Density Approximation (LDA), Vxc(ρ) is the exchange-correlationpotential for the homogeneous electron gas of density ρ(r), for each pointr). This assumption is similar to the idea of “local equilibrium” in meta-morphic petrology. It works surprisingly well, and has been the backbone ofcomputation solid state theory for the past thirty years. Several different pa-rameterizations of Vxc(ρ) for the homogeneous electron gas exist, with mostbeing very similar, and based on quantum Monte Carlo simulations for theelectron gas. The LDA systematically produces volumes that are too smallcompared with experiment, and thus at zero pressure it tends to give elasticconstants such as the bulk modulus accordingly too high. The Wigner ap-proximation to the LDA exchange-correlation potential gives volumes thatare slightly too high, and is significantly different from other LDA’s. It wasderived in a different way from other LDA’s, as an interpolation formulabetween known high density and low density behavior.
The Generalized Gradient Approximation (GGA) includes gradients ofthe charge density from the linear response of the electron gas [9, 10, 11].Although the exact density functional is unknown, many sum rules andconstraints on the exact functional are known. The GGA was developedsuch that sum rules that are obeyed in the LDA continue to be obeyed(thus the term “generalized”); a simple addition of gradient terms in aTaylor expansion to Vxc actually results in a worse approximation than theLDA, due to the loss of these sum rules. The GGA generally results inbetter agreement with experiment than the LDA, though in many cases itovercorrects, and in some cases the volume is larger than experiment by thesame amount that LDA underestimates the volume. There are a number ofcases, such as the stability of quartz versus stishovite, or magnetic bcc ironversus non-magnetic hcp, where the GGA gives accurate results and LDAfails.
One way of understanding the exchange-correlation potential is throughthe exchange-correlation hole. The exchange correlation hole is the regioncarved out around an electron due to the Pauli principle and electrostat-ics, and integrates to one-electron. Most work assumes that the exchange-correlation hole is local (i.e. short-ranged), but it can be demonstrated tohave long-range behavior that is not properly included in LDA or GGA.One general failure of the LDA and GGA is their behavior when an electronis removed from an atom or a crystal surface. The exchange-correlation holeshould stay behind where the rest of the electrons are, but in the LDA andGGA the xc-hole follows the electron that is being removed. This is alsorelated to “self-interaction” errors. An electron cannot interact with itself,yet in self-consistent field methods, a Hartree interaction is computed for

12
the change density, so that a spurious interaction is included between anelectron and itself. If the exchange were computed exactly, the spuriousself-interaction would cancel out. This is very clear in a one-electron sys-tem. There should be no Hartree-potential in a one-electron system, or itshould be completed canceled in the exchange potential. This cancellationis not complete in the LDA and GGA. There should also be no correla-tion energy in a one-electron system, but the LDA and GGA include afalse self-correlation. Self-interaction corrections (SIC) attempt to correctfor this, and though they work very well in atoms and show promise forcrystals [12, 13, 14], they are not generally used due to some arbitrarinessin how they are applied. There are some other improved methods whichshow great promise. The Weighted Density Approximation (WDA) evalu-ates the exchange-correlation energy for a local system with density of thatin the local exchange-correlation hole[15, 16]. The WDA has shown greatpromise with very accurate predictions of the volume and other cohesiveproperties. Unfortunately, a spin-polarized WDA has not yet been devel-oped so magnetic materials cannot be treated. Other improved methodsinvolve using exact (i.e. Hartree-Fock) exchange with density functionalsfor the correlation energy [11]. There are not many applications of WDAor exact exchange plus correlation corrections methods to minerals so theywill not be considered further here, but the reader should expect to seesuch work in the future. A hybrid method that is discussed below is theLDA+U method, where LDA (or GGA) is supplemented with a local orbitaldependent potential to correct problems in transition metal materials.
Different types of bonding in crystals are now discussed: van der Waals,ionic, covalent, covalent/ionic, and metallic. Finally Mott insulators will bediscussed, which are metallic according to band theory, but are actuallyionic insulators.
7. Rare gas and “van der Waals” solids
Crystals made of rare gas atoms and/or weakly bound molecules are of-ten called “van der Waals” solids, and are considered as if the atoms ormolecules were bound by dispersion forces that arise from fluctuations in thecharge densities (often called fluctuating dipoles) of the atoms or molecules.Separated, non-overlapping charge densities do indeed have such an attrac-tive force between them, and it varies as C6/r
6 at large distances. Theconstant C6 is known as a van der Waals coefficient, and it can be obtainedfrom studying the scattering of atoms or molecules in the gas phase. TheLDA or GGA do not give 1/r6 behavior at long distances, but rather anexponential decay. Exact DFT, however, should give proper van der Waalsbehavior since the energy is a ground state property, and recently Kohn et

13
al. [17] presented a formalism, and obtained very accurate results for simplepairs of atoms. Some potential models include 1/r6 terms, assuming thatvan der Waals continues to be important in crystals. Such forces have alsobeen called on to explain the binding of neutral planar units in some min-erals, such as graphite. However, the importance of van der Waals forces insolids is unclear, especially at elevated pressures, and in general these forcesare probably quenched. Unlike the gas phase, there is appreciable overlapof charge densities in a crystal, especially at high pressures, but this is alsotrue at low pressure. This is reflected in the band structures, which do showsome dispersion even at low pressures (fig. 6a). Thus the effects attributedto van der Waals, such as the binding energy of rare gas crystals, is moreproperly ascribed to correlation forces, such as those present in LDA orGGA. In fact, such computations do quite well for the crystalline proper-ties of rare gas solids [18] and molecules. For the Ne2 dimer, for example,the PBE GGA [11] gives a bond length of 5.83 bohr and anharmonic fre-quency of 11 cm−1 compared with 5.84 and 14 from experiment [19]. Ifone were to add unquenched, long-range 1/r6 terms in the interactions incrystals, agreement could only degrade. On the other hand, it is probablytrue that LDA and GGA leave out some of the long-range correlation, butgenerally speaking these effects must be small or not very dependent onatomic configurations, because all insulating materials could have such in-teractions in principle. It must be that the charge density fluctuations arestrongly screened, even in insulators, and thus die off rapidly than 1/r6 incrystals, leaving the more local correlation effects dominant [20].
One approach has been to take accurate pair potentials from atomicscattering experiments, and apply these potentials to the crystalline phase[21]. The resulting crystalline properties and structures are not accurateunless one includes many-body forces [22].
At high pressures, the attractive terms in molecular solids become lessand less important, and one could rather think of the atoms as soft spheres.Much of the chemistry and phase diagrams of such materials can be un-derstood as the packing of spheres of various sizes. However, as pressureis increased, the electronic structure also changes. This happens first inthe crystals of heavier atoms, such as Xe. Figure 6 shows the computedband structure of Xe at three pressures, where it is an atomic-like “vander Waals” solid, a semiconductor and a metal. The metallization of Xehas been studied experimentally [23, 24] and using various electronic struc-ture methods, including the GW approach which is an approximation tocompute quasiparticle energies or excitation spectra, and thus gives a moreaccurate gap than the KS eigenvalues [25]. A complex stacking phase transi-tion appears in Xe in the range of the metallic transition [26]. It is typical tofind changes in the crystal structure when the bonding type changes, such
14
(a)
(b)
(c)
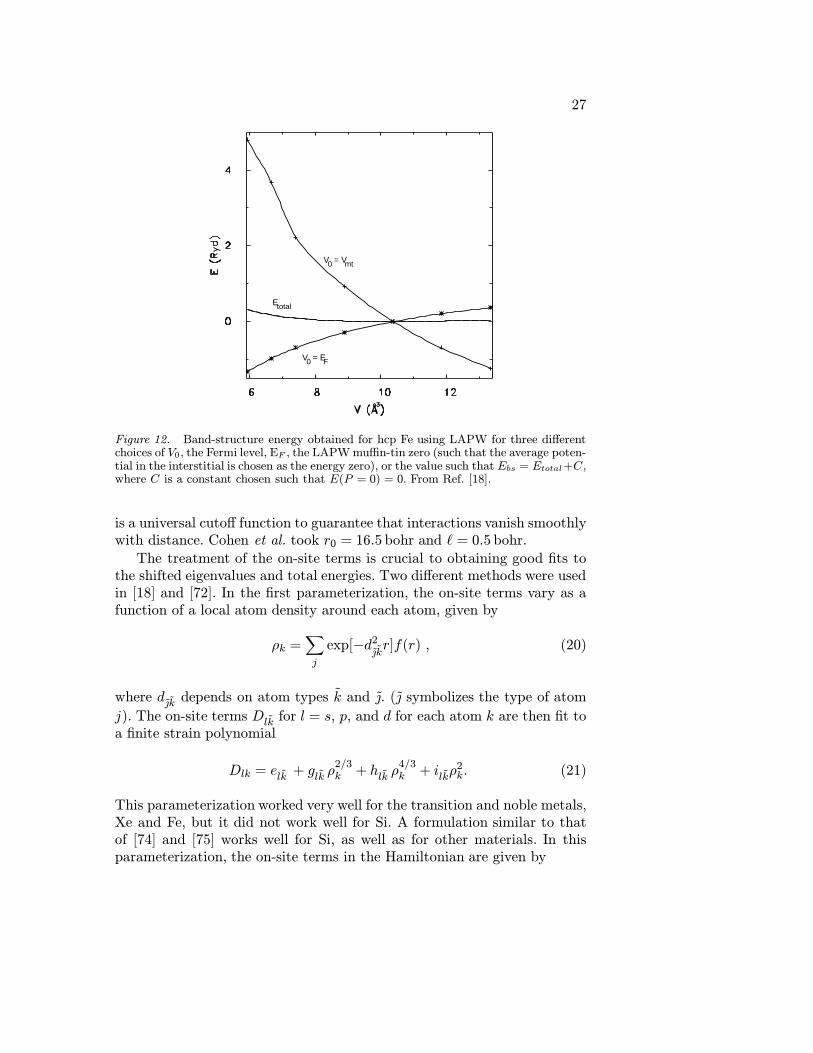
Figure 6. Band structures for Xe over five-fold compression. (a) 0 GPa, V=500bohr3/atom (b) 20 GPa, V=200 bohr3/atom (c) 250 GPa, V=100 bohr3/atom. At lowpressures, Xe has almost atomic-like valence states, with very narrow band widths. Notethat the conduction bands have significant width even at low pressures. At intermediatecompression, Xe is a semiconductor, and at high pressures a metal. The ordinate is energywith each tick being 1 eV. From Ref. [18].

15
as at a metal-insulator transition, because it is the nature of the bondingthat gives rise to the stability of one crystal structure over another.
NeAr Kr Xe
GKLDA GGA
GK
GK
GK LDA
fcc
hcp
Figure 7. Equations of state of rare gas solids. Gordon-Kim (GK) results use quasi-harmonic lattice dynamics at 300 K. The LAPW LDA and GGA-PBE results are forstatic lattice. Points are from experiment. Both fcc and hcp are shown for Xe, and theother results are for fcc. Agreement is quite good with the self-consistent results. Notethat there is a small but significant error in the Gordon-Kim results, due to the pairapproximation, the Thomas-Fermi overlap kinetic energy, and, with increasing pressure,covalency and metallization. Van der Waals interactions are not included in any of thecomputations, and are apparently small effects for the crystal. However, Ne in GGA-PBEappears to be slightly unbound without van der Waals.
Figure 7 shows the equations of state of Ne, Ar, Kr and Xe computedusing the LAPW method and the LDA (for Xenon) and PBE-GGA (forNeon), compared with experiments. Agreement is good, and there is noevidence that some large piece of the interactions is missing. On the otherhand, it is difficult to reach definitive conclusions around zero pressure,since the crystals are so soft, zero point and thermal vibrational correctionsare very important, and the binding energies are very small. As mentionedabove, indications for molecules are that the GGA is quite accurate [19].It remains to be seen if this is true for crystals. Preliminary computationsusing GGA for Ne show anomalous behavior at zero pressure, with a netrepulsive potential between the atoms at large volume leading to expansionof the crystal without bound, but it remains to be seen if this is a numericalproblem due to the tiny energy differences in the low pressure regime, or areal failure of the PBE-GGA. LDA does not show this, but rather shows ashallow minimum in energy.

16
One theme in research on rare gas solids has been an interest in theimportance of many-body forces between three or more atoms [29]. Theseforces arise in three main ways. Firstly, when a crystal is compressed, re-gions of density where three atoms overlap significantly introduce three-body terms, since the exchange-correlation functional is not linear in den-sity. These contributions appear to be small, on the order of a few percentor less of the energy [30], at least at pressures that are not extreme. Asecond contribution comes from multiple atom dispersion forces, the lowestorder being the attractive Axelrod-Teller forces which are from fluctuatingdipoles on three atom neighbors [31]. The expansion in terms of higher or-der multipoles and dispersion interactions is only slowly convergent. It wasthought for some time that many-body forces would stabilize the fcc phaseover the hcp structure. Pair potentials that accurately describe molecularbeam scattering fail to give the fcc phase as the ground state structure,but Ne, Ar, and Kr are fcc at zero pressure, and transform to hcp withincreasing pressure. However, many-body interactions do not fix this ap-parent problem. Rather it is zero point and thermal energy that stabilizesthe fcc phase.
Quantum anharmonicity has been another area of much interest in raregas solids, and the most extreme cases are 3He and 4He, the former aFermion system, and the latter a Bose system. Quantum Monte Carlo sim-ulations have been used to study the solid and liquid phase diagram, andthe properties of the solid and the liquid [32]. The liquid state exhibits su-perfluidity, which is a quantum state in which the viscosity vanishes. 4He isalso unusual in that it remains liquid down to 0 K. Only pressure stabilizesthe crystalline phase, zero point motions being so large. The bcc structureof the low pressure crystalline phase in 4He is also stabilized by zero pointenergy. This is not a review of methods, but if it were, quantum MonteCarlo would have to be described in some detail, since in the future suchcomputations are expected to be much more common.
Gordon and Kim [27] developed a model for rare-gas solids, that turnedout to be more generally useful and has lead to significant advances inour understanding of thermal properties of solids. They developed a modelthat is really the prototypical example of LDA, and shows the power ofthe LDA even when not used self-consistently. In Gordon-Kim models, thetotal charge density is modeled by overlapping atomic or ionic charge densi-ties, and then the total energy is computed for that charge density using theLDA. The method is less accurate than the self-consistent KS method, evenif the model density is good, because the local density form for the kineticenergy, T0 = ρ5/3 is not accurate enough in many cases (for example it doesnot give the proper shell structure for atoms). The KS approach does notmake this approximation for the kinetic energy even in the LDA and the ki-

17
netic energy derives from the occupied orbitals. The electrostatic energiesare computed exactly, and the LDA is used for the exchange-correlationenergy. Gordon and Kim also modified the kinetic and correlation interac-tions (leading to the term modified electron gas, or MEG) to give betterresults for atoms. Gordon-Kim models will be discussed more generally inthe next section on ionic models, but the prototypical application was raregas solids. The MEG model generally gave equations of state that were toostiff for the rare gas solids, but was quite successful considering the simplic-ity of the model. The main problem with the simple Gordon-Kim model isresolved by modifying the rare gas density in response to the embeddingcrystal potential, so that the rare gas atom is compressed with increasingpressure [28]. These studies have also shown that the dispersion forces aredamped or screened as the charge densities overlap [20].
The Gordon-Kim model is the prototypical ab initio model. An ab initiomodel is a way to compute ground state properties without performingfull self-consistent computations, but without fitting experimental data.Ab initio models are generally based on the DFT. The total energy is afunctional of the charge density, so given the crystal charge density the totalenergy is in principle constrained, and the total energy is variational withrespect to the changes in the density (i.e. the total energy is a minimum withrespect to variations in the density). In Gordon-Kim models, the chargedensity is approximated as overlapping ions or atoms. The overlapping ionmodel appears to be an excellent model for the charge density comparedwith self-consistent computations for system of closed shell ions.
8. Ionic solids
Attractive forces in ionic solids are dominated by the electrostatic, orMadelung forces between charged ions. The most easily understood ioniccrystals are those made of closed-shell ions, such as Mg2+, O2−, Na+, Cl−
etc. Thus the alkali halides and alkaline earth oxides are prototypical ionicsolids. One can study these systems using the full machinery of DFT, andresults have been obtained for equations of state [33, 34, 35, 36], elasticity[37], and a range of defect [38, 39, 40], surface [41, 42], electronic [43, 44],and optical [45] properties of these materials using self-consistent methods.
Since the physics of closed-shell systems is straightforward, more effi-cient models have been developed, based on first-principles, which allow thecomputation of time-dependent properties such as transport properties andthe performance of long molecular dynamics simulations not possible usingself-consistent methods. Most of these methods are based on the Gordon-Kim model discussed above. In ionic crystals, one must add the Madelungenergy to the overlap energies. A further important complication arises in

18
oxides, since the O2− ion is unstable in the free state. Rather it is stabilizedby the crystal field in an oxide. An O2− charge density can be obtained byincluding a stabilizing potential in the atomic computation, and most stud-ies have used a “Watson sphere” [46], which is a charged sphere, usually ofopposite charge of the ion. Thus for O2− a sphere of charge 2 is includedas well as the nuclear charge Z = 8 [47]. Then when an electron moves farfrom the atom it sees an object of positive charge +1 behind it, the electronis bound and the configuration remains stable. The remaining question ishow to choose the radius of the sphere. Muhlhausen and Gordon [47] stabi-lized the ion with a sphere whose radius was chosen so that the electrostaticpotential in the sphere equals the Madelung potential at the site at a givenvolume, a potential appropriate to that charge density was found, and thenrigid-ion calculations were performed with that potential.
Figure 8. Difference in charge density of MgO generated with overlapping ions usingthe PIB model and computed self-consistently using the LAPW method. The contourinterval is 0.005 e−/bohr3. From Ref. [4].
In the Potential Induced Breathing (PIB) model, the Watson-sphereradius is given by the Madelung potential, Vmad, as atoms are displaced orthe lattice strained, Rwat = Zwat/Vmad, giving a non-rigid ion, many-body,potential, where Zwat is the charge on the sphere (2+ for O2−). Figure 8

19
shows the difference in charge density of MgO computed with overlappingPIB ions and computed self-consistently using the LAPW method. Theagreement is excellent. Furthermore, the bands computed from the crystalpotential generated from the PIB charge density is in excellent agreementwith the self-consistent band structure (figs. 9, 10, and 11). Thus the PIBcharge density is a good approximation to the self-consistent charge densityfor ionic materials such as MgO. Presumably similar agreement would befound with Gordon-Kim rare gas solids, though such a comparison has notbeen made. In the PIB model, the total energy consists of the Madelungenergy, the overlap energy (which contains the Thomas-Fermi kinetic en-ergy approximation, the LDA exchange and correlation energies, and thelocal electrostatic energies among electrons and between electrons and nu-clei), and the self-energy. The self-energy is the energy of each ion densityas generated with the Watson sphere, with the Watson-sphere interactionenergy removed. The self-energy for each ion thus depends on the Watsonsphere radius. The dependence of the energy on the Watson sphere radiigives rise to effective many-body non-pair-wise interactions between theatoms that give improved elastic properties [48]. In the rocksalt structure(or any structure with inversion symmetry at each site), if all atoms inter-act through central pairwise forces, the elastic constant C44 = C12 at zeropressure (the Cauchy relation) [49], and yet the experimental elastic con-stants deviate from this relation in the alkaline earth oxides (the Cauchyviolation). Mehl et al. [48] found reasonable values of the Cauchy violationfor the alkaline earth oxides, and the correct trend from MgO to BaO. Thispromising result lead to the development of the lattice dynamics of the PIBmodel [50] and reasonable dispersion curves were obtained for the alkalineearth oxides using the PIB model.
All was not well, though. In the PIB model, the Watson sphere radiiare given by the Madelung potential, but the Madelung potential is notwell behaved in the long-wave limit. A longitudinal wave exp[−q · r] givesrise to a potential wave, and as q → 0 the Madelung potentials on Osites approach a linear slope from +∞ to −∞ across the infinite crystal.Since the self-energy depends on the value of the Madelung potential, itdiverges as q → 0, and the longitudinal optic (LO) mode diverges [51]. Itis possible to remove this divergent part and recover reasonable results,but the resulting model is still not completely satisfactory since it predictsspherical breathing of atoms in a linear field. Nevertheless, many usefulresults were obtained with the PIB model. It is fast, and often nearly asaccurate as self-consistent calculations for ionic materials. The key effectof PIB is that the anion changes size with crystal environment, shrinkingwith increasing pressure or local compression.
A better procedure, though several times slower, is to optimize the total

20
Figure 9. MgO band structure at V=18.1 A(zero pressure) computed using the LAPWmethod (lines) compared with that from the potential generated by the overlapping ionPIB charge density. From Ref. [4].
energy with respect to Watson sphere radii rather than to chose the radiususing the Madelung potential [52]. This gives a Watson sphere radius closeto that of PIB at zero pressure. However, it changes more rapidly withcompression than PIB due to the compression of the atom by short-rangeforces, in addition to the electrostatic crystal field (fig. 11). This model isknown as the VIB, or variationally induced breathing, model. The anoma-lous behavior shown by the PIB model is absent in the VIB model. In theVIB model, the LO-TO splitting is the same as given by a rigid ion model,since all atomic deformations are spherical. There is no dipolar charge re-laxation. In spite of the absence of atomic polarizability, the VIB model isvery accurate and gives results that compare quite well with self-consistentresults and experiment for non-polar materials and for properties for whichdielectric behavior is not crucial. It is clear that the spherical breathingincluded in PIB and VIB is more important than dipolar polarizability,and one should not neglect spherical breathing in models for oxides andminerals containing other unstable ions such as S2−. Even in alkali halides,the spherical breathing effect is important [53, 54].
The advantage of fast methods such as PIB or VIB is that one can

21
Figure 10. Differences in eigenvalues from the LAPW band structure and those derivedfrom PIB as functions of the Watson sphere potential (Vwat =Zwat/Rwat) (a) at zeropressure (18.1 A3) and at (b) 290 GPa (10 A3). The Madelung potential is shown byan arrow. At low pressures PIB gives an excellent approximation for the band structure(almost as good as self-consistent). At high pressures, the best fit Watson sphere potentialwould be larger (i.e. a smaller Watson sphere radius, and a smaller O2− ion) due toshort-range forces that compress the oxygen ion.
do lattice dynamics and long molecular dynamics simulations on reason-able sized systems, and study thermodynamic properties, phase transitions,transport properties, etc. For example, Isaak et al. performed lattice dy-namics on MgO as a function of lattice strain, going beyond the normalquasiharmonic approximation, and studied the high effects of temperatureand pressure on elasticity and the equation of state [55]. In that study,the dynamical matrix was found throughout the Brillouin zone in order toobtain the free energy, and this was repeated for different lattice strains.In spite of increased computational power in the last eight years, no suchstudy has yet been done self-consistently. The Isaak et al. study gave whatare still one of the few estimates of cross derivatives of pressure and tem-perature on elasticity that are available. The results were then used to helpunderstand the increase in seismic parameter d lnVs/d lnVp with depth inthe Earth [56], a quantity that has been difficult to constrain experimen-tally. Going beyond lattice dynamics, Inbar and Cohen [57] determined thethermal equation of state of MgO using molecular dynamics and the PIBmodel. Such studies are just becoming possible using self-consistent meth-ods, and still have not been performed. Using molecular dynamics and the

22
Figure 11. Behavior of best fit, VIB, and PIB Watson sphere potentials as functions ofcompression and composition in alkaline earth oxides. (a) The Watson sphere potentialsVwat that give band structures that best fit self-consistent LAPW band structures versusMadelung potential for different alkaline earth oxides versus pressure. Pressure increasesto the right for each curve. PIB (represented by the 1:1 dashed line) works well at lowpressures, but less well with increasing pressure. (b) Watson sphere potential obtainedusing VIB (minimizing the total energy with respect to the Watson sphere radii) versusMadelung potential. VIB gives a better approximation to the slope seen in (a). (c) VIBpotential versus best-fit potential. PIB is represented again as the 1:1 heavy dashed line.The VIB results parallel the best fit results, giving the correct compressive behavior.There is a small offset to lower potentials for VIB, which may compensate for the kineticenergy approximation in VIB.
VIB model, it is also possible to study complex phenomena, such as ther-mal conductivity [58] and diffusion [59, 60]. The diffusivity of O in MgO,for example, is obtained in agreement with measurements within experi-mental error. Ab initio models can also be used to study the stability of avariety of structures to search for possible phase transitions. This was donesuccessfully for Al2O3, where the PIB model showed a high pressure elasticinstability [61], detailed PIB computations for different structures showeda phase transition at high pressures to the Rh2O3 II structure [62], andthe transition was confirmed and pressure computed accurately using the

23
LAPW method with the PIB structural parameters [63]. These computa-tions predicted a phase transition at 90 GPa, in excellent agreement withlater experiments [64].
Ab initio models have undergone further development by including thecrystal potential in the atomic calculation, and performing a self-consistentcycle between the atomic densities and the crystal potential [28, 65]. Inthe Self-Consistent Charge Deformation model (SCAD) [66, 67, 68] atomicdensities are computed in the crystal potential, and states are occupied inorder of energy, allowing charge flow between the atoms. The inclusion ofnon-spherical charge deformations has increased the accuracy of the models,but at the cost of much increased complexity. Perhaps fast implementationsof these new self-consistent model methods will be developed that will al-low molecular dynamics and other types of simulations for crystals wherenon-spherical charge distortions are important. Whether covalency can beproperly modeled by these methods remains to be seen.
Ab initio models also give insights into bonding and electronic structurethat are not obvious from self-consistent computations alone. One exampleis the relationship among and meaning of ionicity, covalency, and bandwidth. One might think for example that a purely ionic model would haveatomic-like energy levels, and that band width arises from hybridization orcovalency. However, band structures computed from the potential generatedfrom overlapping ionic charge densities (fig. 9) not only have width, but arein excellent agreement with self-consistent computations for ionic crystalssuch as MgO. On the other hand, if one were to ask the origin of the bandwidth in a tight-binding representation, one would find that the O 2p bandwidth in MgO, for example, comes primarily from O-O ppσ interactions[69]. Thus we see that even a purely ionic charge density, generated byoverlapping spherical ions, has a charge density that generates a potential,that when used in the KS equations implies a band width consistent withhybrid electronic states. Thus there is a sort of duality in the description ofionic materials, and they can be described from a charge density or tight-binding (or LCAO) perspective. In either case, there must be long-rangeMadelung terms in the total energy, that gives rise to LO-TO splitting inthe lattice dynamics. Therefore, it would not be correct to say that MgOcould be treated as an ionic or as a covalent crystal. It is an ideal ioniccrystal (as indicated by the LO-TO splitting with effective charges nearthe nominal values [68, 70]), but this ionic charge density implies a bandwidth in the KS eigenvalues from its potential. Note that the SCAD modeldoes indeed have atomic like eigenvalues in its spectrum, but presumablyif one generated the band structure from the SCAD charge density onewould find reasonable agreement with the self-consistent band structure.This would be a useful test for SCAD and other improved ab initio models.

24
Not all ionic solids are formed from closed shell ions. For example, FeO(wustite) and solid solutions between MgO and FeO (magnesiowustite) be-have like ionic solids, yet Fe2+ is a d6 ion and is not closed shell. FeO is alsoa Mott insulator, and is discussed below. Such materials are very difficult totreat, and in spite of the importance of Fe in minerals, there is not yet a goodmethod for obtaining first-principles results that are completely correct. Abinitio models such as PIB fail to give accurate predictions for the equationof state and other properties for these materials as well, even if one spher-icalizes the Fe, and treats it as ionic. Nevertheless, much can be learnedabout these materials from self-consistent computations as described below.Perhaps simple and accurate ab initio models can be developed for thesematerials, but this has not yet been done.
The hydrogen bond is really another type of ionic bond, but usuallyclassified separately. A proton has of course a 1+ charge, and in an oxideor silicate, hydrogen almost always occurs as the (OH)−1 ion or bound asH2O. In either case there is a residual positive charge around the proton,even though it is enveloped by the polarized oxygen charge density, andthis residual charge can bind with an excess negative charge on anotheratom or molecular unit. This binding strongly influences the H vibrationalfrequencies and may also influence the crystalline configuration if there arefloppy structural units attached to either the positive or negative side ofthe bond. Whether hydrogen bonds are favorable at a site or not may alsostrongly influence the position of H in the crystal.
9. Covalent solids
In covalent solids the dominant bonding interaction is caused by hybridiza-tion among the states on different atoms. Such interactions can be verystrong, and are responsible for the strong bonds between C’s in diamondand less so in Si. In a covalent bond, charge concentrates in the bondingregion, increasing the potential and kinetic energy of interaction betweenelectrons, but reducing the energy through the electron-nuclear interactions(since each electron is now on average close to both nuclei or atomic cores,which have a net positive charge to the electrons). Atoms that form strongcovalent bonds have decreased repulsion relative to atoms that do not. ThePauli exclusion principle says that two electrons cannot be in the same state,so as electron density increases, electrons must be in higher energy states.In strong covalent solids, orbitals are not fully occupied on the constituentatoms. Hybrid states can form without occupying higher energy levels, andthus the total energy decreases due to the favorable electron-core interac-tions. For example, in H2, each H atom separately has one 1s electron, andwhen brought close, there is no problem forming one molecular bonding

25
orbital out of the two 1s atomic orbitals, which is then doubly occupied.In contrast, in He, the 1s states are filled, and as two He atoms approach,electrons must be boosted into higher energy states. Thus H forms strongcovalent bonds, and He does not.
Covalent bonds tend to be very directional since they are formed fromlinear combinations of directional orbitals on the two atoms. Self-consistentcomputations can of course be done for these materials, and many suchstudies of all kinds of properties of electronic and optical semiconductorshave been performed. It is also straightforward to develop tight-bindingmodels for such covalent materials, and such models have ranged frommodels with empirical parameters to those with parameters obtained fromfirst-principles. The advantage of tight-binding models is that they are veryfast compared with self-consistent methods, and one can perform molecu-lar dynamics and other simulations on large systems for relatively longtimes. Tight-binding models also give insight into the bonding interactions,especially in covalent solids. Tight-binding models give a complementarypicture to ab initio models, the former being based on matrix elements andband structures, and the latter focused entirely on the charge density. Sincethe formalism is instructive, and may give some further understanding ofbonding and energetics of crystals it will be described in detail.
In tight-binding total energy models (TBTE) the total energy E as afunction of the atomic or nuclear positions r is represented as a sum of aband structure term, Ebs,
Ebs =∑i
εif (εi (k)−EF , T ) , (14)
where εi are the band eigenvalues and f is the Fermi function, with EFchosen to give the correct number of electrons, and additional structuredependent term F , which is often represented as a pair potential, so that:
E = Ebs + F . (15)
In tight-binding methods [71], the radial parts of the integrals (eqs. 9-10) are parameterized, and there is no self-consistency loop. In the two-center approximation, only integrals between pairs of atoms are included;integrals that involve a potential on one atom and orbitals on two othersare neglected. Thus the Hamiltonian and overlap matrices can be writtenas:
Hijαβ =∑l
exp [ik· (Rjl −Ril)]hijαβ
∫dΩφiαφjb (16)
Oijαβ =∑l
exp [ik· (Rjl −Ril)] oijαβ
∫dΩφiαφjb (17)

26
where the integrals are the angular integration for orbital α on atom iwith orbital β on atom j, and the sums are over lattice vectors l. Angularmomentum is quantized along z, the vector from atom jl to atom i, so thatthe indices αβ are contracted to a total of ten bonding states up to d-states,ssσ, spσ, ppσ, ppπ, sdσ, pdσ, pdπ, ddσ, ddπ, and ddδ. The parameters h ando are fit to results of first-principles band structures or are fit empirically forindividual interactions in a given crystal structure. The on-site parametershiiαα are fit or set to atomic energies, and a set of parameters hssσ, etc. areobtained, either by coordination shell (i.e. first-neighbor, second-neighbor,etc.) or as functions of distance.
If O is assumed to be the identity matrix the method is “orthogonaltight-binding,” otherwise it is “non-orthogonal.” A non-orthogonal fit givesa more accurate band structure at the expense of large correlations amongparameters, but generally a non-orthogonal model is required to fit a largerange of structures and/or volumes. Non-orthogonal models sometimes areproblematic when used outside the range of parametrization, since the over-lap matrix may become non-positive definite, which makes solution of thesecular equations impossible (or unphysical).
In standard tight-binding models there is a large source of ambiguitythat arises from separating and fitting separately Ebs from the function F(eq. 15). The problem is that for a periodic crystal the zero of energy, V0,for a band structure is arbitrary, so that the band structure energy (eq. 13)contains an arbitrary and unknown structure dependent term. This termarises from the G=0 term in Fourier expansion of the Coulomb potential,and is canceled by an identical contribution in F in self-consistent calcula-tions. This cancellation is not guaranteed in tight-binding methods wherethe band structures and F are fit separately. Figure 12 illustrates the sever-ity of the problem. This problem is solved by shifting the eigenvalues sothat the band structure energy is the total energy, and F = 0 [18, 72]. Analternative approach is to fit eigenvalue differences, rather than absoluteeigenvalue energies, let the zero of energy float, and then simultaneously fittotal energy differences [73].
The problem is then reduced to finding a parametrization that accu-rately reproduces not only the band structure, represented by the shiftedeigenvalues, but also the total energies. Such a parametrization was foundand tested for several transition and noble metals [72]. Cohen et al. [18, 72]parameterized the Hamiltonian h and overlap o parameters as a functionsof distance:
Pi = (ai + bir) exp[−c2i r]f(r) , (18)
where
f(r) = (exp[(r− r0)/`] + 1)−1 (19)
27
V = V
V = E
0 mt
0 F
Etotal
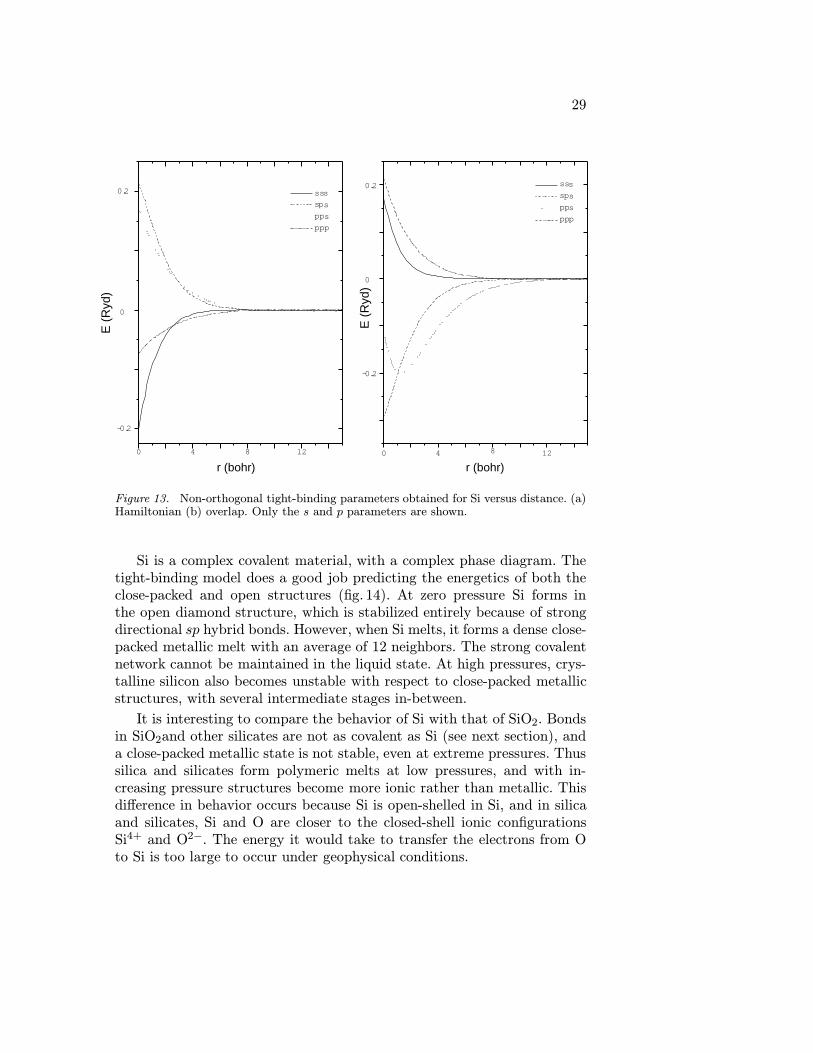
Figure 12. Band-structure energy obtained for hcp Fe using LAPW for three differentchoices of V0, the Fermi level, EF , the LAPW muffin-tin zero (such that the average poten-tial in the interstitial is chosen as the energy zero), or the value such that Ebs = Etotal+C,where C is a constant chosen such that E(P = 0) = 0. From Ref. [18].
is a universal cutoff function to guarantee that interactions vanish smoothlywith distance. Cohen et al. took r0 = 16.5 bohr and ` = 0.5 bohr.
The treatment of the on-site terms is crucial to obtaining good fits tothe shifted eigenvalues and total energies. Two different methods were usedin [18] and [72]. In the first parameterization, the on-site terms vary as afunction of a local atom density around each atom, given by
ρk =∑j
exp[−d2kr]f(r) , (20)
where dk depends on atom types k and . ( symbolizes the type of atom
j). The on-site terms Dlk for l = s, p, and d for each atom k are then fit toa finite strain polynomial
Dlk = elk + glk ρ2/3k + hlk ρ
4/3k + ilkρ
2k. (21)
This parameterization worked very well for the transition and noble metals,Xe and Fe, but it did not work well for Si. A formulation similar to thatof [74] and [75] works well for Si, as well as for other materials. In thisparameterization, the on-site terms in the Hamiltonian are given by

28
Dlml′m′k = qll′k
+∑j
(slmk
+ tlmk
rjk)
exp[u2
lmkrjk]Ylm (rjk)Yl′m′ (rjk)
(22)This form arises from the onsite integrals 〈Ψilm|Vj|Ψil′m′〉 for interactions ofan orbital on atom i with the same or another orbital on i with a sphericalpotential on atom j. This can be reduced to similar angular representationsas the hopping and overlap terms to ssσ, spσ, ppσ, ppπ, sdσ, pdσ, pdπ,ddσ, ddπ, and ddδ if off-diagonal m 6= m′are excluded. For pp onsite terms,equation 22 reduces to [74, 75]
Dpzk = qpp +∑j
l2jk (rjk) Ippσ (rjk) + (1− l2jk (rjk))Ippπ (rjk) (23)
where l2jk (rjk) = z2/r2, for example, and Ippσ (rjk) are the distance depen-
dent terms, represented in our model by(sσk
+ tσkrjk)
exp[−u2
σkrjk].
Excellent fits were found for Si using this formalism, for which we in-cluded s, p,and d interactions using only the diagonal terms correspondingto ssσ, ppσ, and ddσ, giving twelve onsite parameters:
Ds = qs + Issσ (24)
Dpx = qp +x2
r2Ippσ
Dpy = qp +y2
r2Ippσ
Dpz = qp +z2
r2Ippσ
Ddxz = qd +x2z2
r4Iddσ
Ddyz = qd +y2z2
r4Iddσ
Ddxy = qd +x2y2
r4Iddσ
Dd(x2−y2) = qd +
(x2 − y2
)r4
Iddσ
Dd(3z2−r2) = qd +3z2 − r2
r4Iddσ
Figure 13 shows the fitted parameters versus distance. This gives a graphicdepiction of how the bonding interactions vary in Si with distance.
29
0 4 8 12
-0.2
0
0.2
sss sps pps ppp
r (bohr)
E (
Ryd
)
0 4 8 12
-0.2
0
0.2
sss sps pps ppp
r (bohr)
E (
Ryd
)
Figure 13. Non-orthogonal tight-binding parameters obtained for Si versus distance. (a)Hamiltonian (b) overlap. Only the s and p parameters are shown.
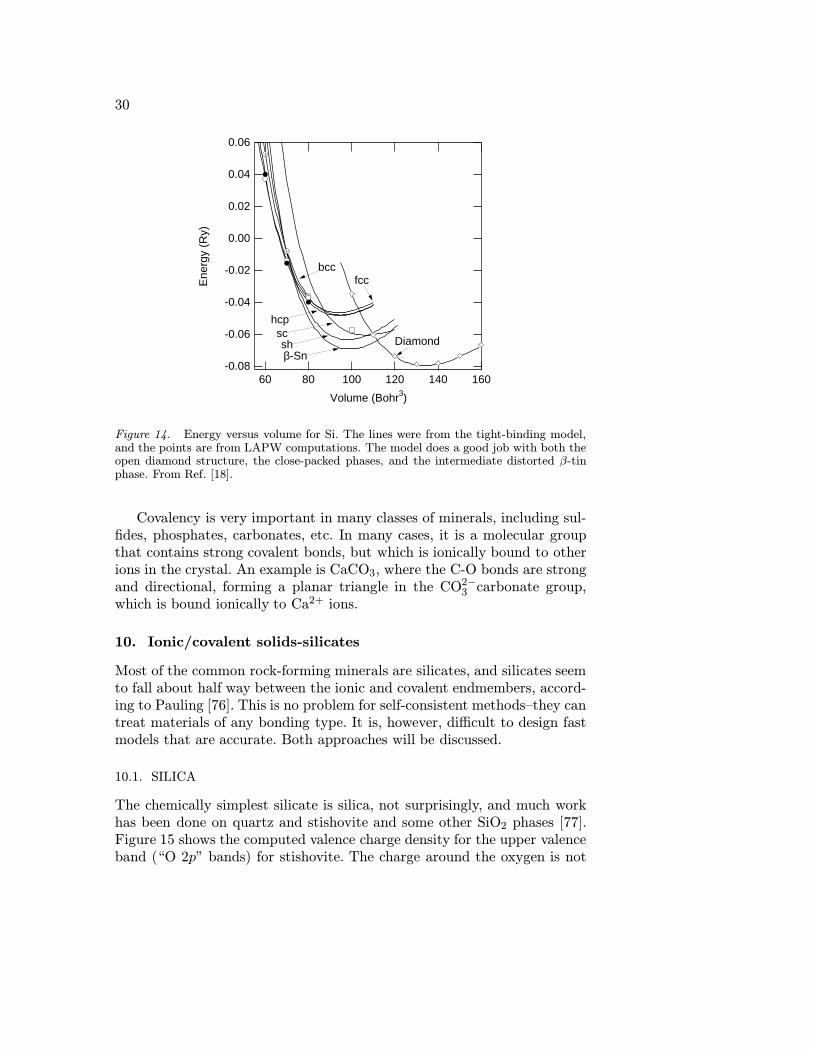
Si is a complex covalent material, with a complex phase diagram. Thetight-binding model does a good job predicting the energetics of both theclose-packed and open structures (fig. 14). At zero pressure Si forms inthe open diamond structure, which is stabilized entirely because of strongdirectional sp hybrid bonds. However, when Si melts, it forms a dense close-packed metallic melt with an average of 12 neighbors. The strong covalentnetwork cannot be maintained in the liquid state. At high pressures, crys-talline silicon also becomes unstable with respect to close-packed metallicstructures, with several intermediate stages in-between.
It is interesting to compare the behavior of Si with that of SiO2. Bondsin SiO2and other silicates are not as covalent as Si (see next section), anda close-packed metallic state is not stable, even at extreme pressures. Thussilica and silicates form polymeric melts at low pressures, and with in-creasing pressure structures become more ionic rather than metallic. Thisdifference in behavior occurs because Si is open-shelled in Si, and in silicaand silicates, Si and O are closer to the closed-shell ionic configurationsSi4+ and O2−. The energy it would take to transfer the electrons from Oto Si is too large to occur under geophysical conditions.
30
-0.08
-0.06
-0.04
-0.02
0.00
0.02
0.04
0.06
Ene
rgy
(Ry)
1601401201008060
Volume (Bohr3)
Diamondβ-Snsh
sc
fcc
hcp
bcc
Figure 14. Energy versus volume for Si. The lines were from the tight-binding model,and the points are from LAPW computations. The model does a good job with both theopen diamond structure, the close-packed phases, and the intermediate distorted β-tinphase. From Ref. [18].
Covalency is very important in many classes of minerals, including sul-fides, phosphates, carbonates, etc. In many cases, it is a molecular groupthat contains strong covalent bonds, but which is ionically bound to otherions in the crystal. An example is CaCO3, where the C-O bonds are strongand directional, forming a planar triangle in the CO2−
3 carbonate group,which is bound ionically to Ca2+ ions.
10. Ionic/covalent solids-silicates
Most of the common rock-forming minerals are silicates, and silicates seemto fall about half way between the ionic and covalent endmembers, accord-ing to Pauling [76]. This is no problem for self-consistent methods–they cantreat materials of any bonding type. It is, however, difficult to design fastmodels that are accurate. Both approaches will be discussed.
10.1. SILICA
The chemically simplest silicate is silica, not surprisingly, and much workhas been done on quartz and stishovite and some other SiO2 phases [77].Figure 15 shows the computed valence charge density for the upper valenceband (“O 2p” bands) for stishovite. The charge around the oxygen is not

31
spherical, but rather is polarized towards the Si atoms. Since there arethree planar Si’s around each oxygen, the O’s have a triangular-prismaticshape. They are polarized strongly towards the oxygens. Most of the chargeis around the oxygen. All of it would be around the oxygen if it were O2−
and the Si were Si4+.
There is no unique way to extract a static ionicity from the charge den-sity (though the dynamic effective are well defined, as described below forCaSiO3.) One way to extract an effective ionicity is to compare the self-consistent charge density with that of a model system, such as overlappingions. Model charge densities made of ions with different valence can becompared with the self-consistent density (for example by comparing theamount of charge in a sphere around each atom with that of the modeloverlapping ion system). Alternatively, one can compute the band struc-ture from the potential generated by model charge densities with differentionicities, and compare this with the self-consistent band structure. Bothprocedures were performed for stishovite and gave similar results, with abest fit of between O1.4− and Si2.8+ and O1.2− and Si2.4+ [78], which sup-ports the idea that the Si-O bond is a mix of ionic and covalent bonding.There was no evidence for changes in these values with pressure. The sameexercise has not been performed for quartz but it would be interesting tosee if there is any conspicuous difference in the ionicity of tetrahedral andoctahedrally coordinated Si using this measure.
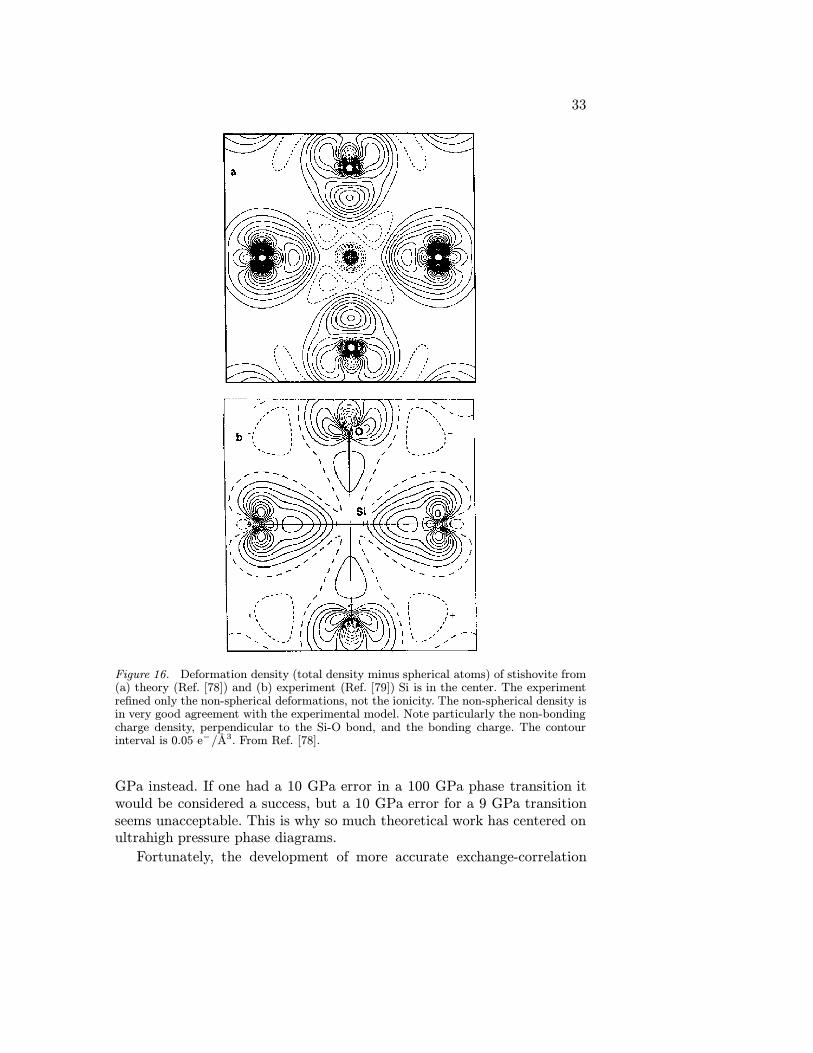
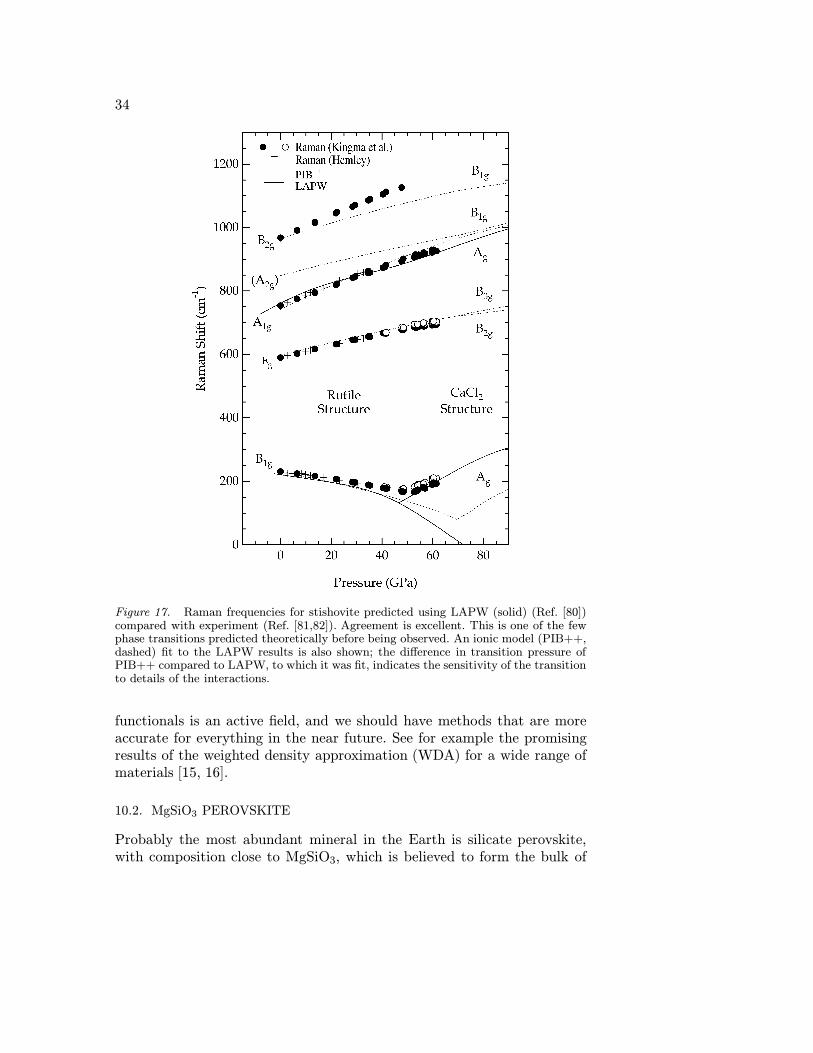
The LDA did a very good job predicting the charge density of stishovite(rutile-structured SiO2) compared with experiment [79](fig. 16), and theequation of state, structure parameters, and Raman frequencies computedwere also in excellent agreement with experiment [78]. Further computa-tions [80] showed an elastic instability in c11-c12 and a phase transition at45 GPa to the CaCl2 structure, and suggested that the transition couldbe detected by following the B1g Raman mode, which would decrease infrequency with pressure until the phase transition, and then increase. Suchexperiments were done, and were in excellent agreement with the theoreticalpredictions [81]. This illustrates the great power of accurate computationswithin the LDA for making reliable predictions.
LDA computations for quartz also showed good agreement with exper-iment [83, 84, 85, 86, 87], suggesting that LDA should be predictive for avariety of SiO2 phases. Pseudopotential computations showed energies ofstishovite lower than quartz, making stishovite the ground state, but thiswas attributed to inaccuracies in the pseudopotentials [85]. It was quite sur-prising, therefore, when more accurate computations showed that indeedLDA predicts stishovite to be the ground state structure for SiO2, contraryto experiment [88]. The energy error is not huge (0.6 eV), but clearly meth-ods with smaller errors are needed to predict phase diagrams. Fortunately,

32
Figure 15. Isosurfaces of the valence density of stishovite. The ionic and covalent char-acter are evident. The isosurface densities are at 0.06 and 0.08 electrons/bohr3 .
the GGA is a great improvement, and gives quartz as the correct groundstate phase [88]. Interestingly, the Wigner LDA gives the correct orderingof quartz and stishovite, but the energy difference is much too small (0.01eV). Analysis of the charge density differences between GGA and LDA forquartz and stishovite show that the main differences are in quartz, andare greater in the atomic rather than bonding regions (fig. 18). In otherwords, the “sizes” of the atoms in GGA are slightly different than in LDA.The bonding doesn’t change at all between GGA and LDA. This is evidentby comparing the densities of states computed using the LDA and GGA,which are identical (fig. 19). Unfortunately these rather subtle changes leadto significant energy differences when comparing different phases. Note thatwithin a given phase, these errors can cancel out, for example, in comput-ing an equation of state. In fact the LDA equation of state for stishoviteis somewhat better than the GGA. Another way of looking at the stabil-ity problem is to realize that the accuracy of predicting phase transitionsbetween considerably differently structured phases may exceed 10 GPa inLDA (and perhaps GGA), yet the quartz-stishovite transition is at 9 GPa,and the LDA errors are such that the transition is predicted at about 0
33
Figure 16. Deformation density (total density minus spherical atoms) of stishovite from(a) theory (Ref. [78]) and (b) experiment (Ref. [79]) Si is in the center. The experimentrefined only the non-spherical deformations, not the ionicity. The non-spherical density isin very good agreement with the experimental model. Note particularly the non-bondingcharge density, perpendicular to the Si-O bond, and the bonding charge. The contourinterval is 0.05 e−/A3. From Ref. [78].
GPa instead. If one had a 10 GPa error in a 100 GPa phase transition itwould be considered a success, but a 10 GPa error for a 9 GPa transitionseems unacceptable. This is why so much theoretical work has centered onultrahigh pressure phase diagrams.
Fortunately, the development of more accurate exchange-correlation
34
Figure 17. Raman frequencies for stishovite predicted using LAPW (solid) (Ref. [80])compared with experiment (Ref. [81,82]). Agreement is excellent. This is one of the fewphase transitions predicted theoretically before being observed. An ionic model (PIB++,dashed) fit to the LAPW results is also shown; the difference in transition pressure ofPIB++ compared to LAPW, to which it was fit, indicates the sensitivity of the transitionto details of the interactions.
functionals is an active field, and we should have methods that are moreaccurate for everything in the near future. See for example the promisingresults of the weighted density approximation (WDA) for a wide range ofmaterials [15, 16].
10.2. MgSiO3 PEROVSKITE
Probably the most abundant mineral in the Earth is silicate perovskite,with composition close to MgSiO3, which is believed to form the bulk of

35
Figure 18. Isosurface of the difference in charge density of quartz computed using GGAand LDA. Isosurface level is 0.004 electrons/bohr3 . The lumpy objects are oxygen ions,and the spheres are silicons.
the Earth’s lower mantle. Extensive computations have been performed forMgSiO3, although it is difficult to study because there are 20 atoms inthe primitive unit cell. MgSiO3 appears to be more ionic than stishovite[89, 90], but the fully charged ionic model is not as successful as it is forsimple minerals such as MgO. Nevertheless, the predictions of single crys-tal elastic constants for MgSiO3 [91] were in reasonable agreement withlater experiments [92, 93], and high pressure elastic constants are still notavailable experimentally. More recently, large scale computations of elas-tic constants have been performed for MgSiO3 using a plane wave basiswith pseudopotentials [94]. Agreement with available experiments is excel-lent, and these computations give elasticity data for perovskite for pressuresthroughout the mantle. These computations will be benchmarks for modelsthat allow computation of thermoelasticity, to obtain properties at mantletemperatures.

36
(c)
(b)
(a)
Figure 19. Density of state of quartz in the (a) Hedin-Lunqvist LDA, (b) GGA, and(c) Wigner LDA. The valence band densities of states are essentially identical.
An accurate and fast model for MgSiO3 would still be very useful. Re-cent results show that if one reduces the amount of charge transfer, usingfixed ion charges Mg2+, Si3.4+, and O1.8−, consistent with the LAPW chargedensity, excellent agreement with LAPW computations can be obtained (Ita

37
and Cohen, unpublished) (fig. 20). Preliminary results also show excellentagreement with the experimental thermal equation of state. These resultssuggest that the primary effect of covalency in silicate perovskite may bethe reduction of the effective ionic charges. This implies that ionic chargesare different for oxygen, say, in perovskite and magnesiowustite. If this istrue, then energetics and electrical properties of interfaces between silicateperovskite may be more interesting than expected.
Figure 20. Energy versus M-point and R-point rotations in perovskite. The points arefrom LAPW computations, and the lines are from VIB with reduced ionic charges asdescribed in the text.
10.3. LINEAR RESPONSE COMPUTATIONS
Computation of phonon frequencies allows investigation of crystal stability,and derivation of the thermal free energy in the quasiharmonic approx-imation. Computations are quite fast for ab initio models, once the ex-pressions for the dynamical matrix are derived, and one can compute thephonon frequencies for any wavevector q throughout the Brillouin zone. Inself-consistent methods the computational burdens are much higher. Thestraightforward procedure is to do “frozen phonon” calculations, in whichthe energy is computed as a function of displacement of the atoms alongsymmetry or normal mode directions. This method becomes less tractablefor wavevectors other than Γ (q = 0) since supercells must be used, and thecomputational burden goes as N 3 using conventional diagonalization tech-

38
niques, where N is the number of atoms in the supercell. Linear responsetheory [95] avoids this by expanding the total energy expression and Kohn-Sham equations for second-order changes in external potential, V (q), whichallows direct determination of phonon frequencies, the dielectric constant,and transverse effective charges with a computational burden that is con-stant for arbitrary wavevector. The dynamical matrix elements are derivedfrom the perturbing potential being displacements of atoms (i.e. nuclei oratom cores). For the high frequency (electronic) dielectric constant theperturbing potential is a potential (electric field) wave. For the transverseeffective charges the perturbing potential is an atomic displacement and anelectric field wave. With linear response, the computational complexity in-volves only N atoms, where N is the number of atoms in the primitive cell,regardless of the wavevector q. It is still a large job to derive the phonondispersion curves with self-consistent linear response theory, since in gen-eral there are 3N phonon modes at each k-point, so one must perform 3Nseparate linear response computations at a general symmetry k-point foran atom at the general position (i.e. with no symmetry). Each computa-tion still scales at N 3 using conventional diagonalization techniques, so thisprocedure rapidly becomes intractable for large low symmetry structures.However, for high symmetry phases and high symmetry k-points only afew computations are necessary to find the dynamical matrix. Typically,one does a small number of such computations, and interpolates the fre-quencies by finding short-range force constants from the dynamical matrixat a few high symmetry points in conjunction with the effective chargesand dielectric constant, also obtained from linear response computations.Thus the entire set of phonon dispersion curves throughout the Brillouinzone can be obtained.
The transverse, or Born, effective charges are important for understand-ing the degree of ionicity and type of bonding in crystals. The Born effectivecharges Qiαβ = ∂2F/∂uiα∂Eβ = ∂Pβ/∂uiαwhere F is the free energy, E isthe electric field and uiα is the displacement of atom i in the α direction,and P is the polarization (dipole moment per volume). As the notation in-dicates, the effective charge is a tensor. It indicates how much charge movesas an atom is displaced. In an ideal rigid ion crystal, the effective chargeswould be independent of direction (i.e. scalars), and would equal the nom-inal ionic charges. Deviations from this behavior indicate covalency, andgive information on the directionality and nature of the bonding.
10.4. CaSiO3 PEROVSKITE
Stixrude et al. [96] performed linear response computations with the LAPWmethod on CaSiO3 perovskite in the cubic perovskite structure. Unlike

39
MgSiO3, which is orthorhombic, experimental studies have found CaSiO3 tobe cubic. Unexpectedly, however, the linear response computations showedcubic CaSiO3 to be unstable at the M- and R-points. Frozen phonon cal-culations confirmed this result. The results suggest strongly that CaSiO3 isnot cubic but tetragonal or lower symmetry. The small strain predicted isconsistent with the x-ray studies that were interpreted as showing a cubicstructure for CaSiO3.
Perhaps more important, from the point of view of this survey of “bond-ing in minerals,” are the effective charges (Table 1). The effective charges inCaSiO3 are close to the nominal ionic charges, and are only slightly affectedby pressure, indicating that the degree of covalency does not vary over thepressure range of the Earth’s mantle. This makes clear that the much re-duced numbers that come from Mulliken populations, for example, are notindicative of the amount of charge that moves when a nucleus is displaced,nor are they good measurements of the amount of static charge on eachatom since in reality the charge densities strongly overlap, so that atomicbasis functions represent states that extend from one atom, for example,O2− onto another.
TABLE 1. Born effective charges for CaSiO3 (at a volume of 310 bohr3, -8 GPa) (Ref. [97])and PbTiO3 (Ref. [98,99]) at approximately zero pressure. Note that the effective chargescan differ greatly from their nominal values, reflecting covalency, and that they can bedirectional, and strongly dependent on structural distortions. For tetragonal PbTiO3, O1
is the O is closest to Ti, along [001], and O2 is the O on the xz face, farther from the Ti.
CaSiO3 Ca Si O
2.60 4.12 -1.83(x)/-3.06(z)
PbTiO3 Pb Ti O1 O2
(cubic) 3.92 6.71 -2.56(x,y)/-5.51(z)
(tetragonal) 3.74 6.20(x)/5.18(z) -2.15(x)/-4.38(z) -2.61(x)/-5.18(y)/-2.16(z)
Effective charges have only been computed for a few materials, andvery few minerals. Although PbTiO3 is not a mineral, the effective chargescomputed for it are instructive as compared with CaSiO3. PbTiO3 is muchmore covalent than CaSiO3, and this is evident in the effective charges.Though some values are close to the nominal values, such as for O motionstransverse to the Ti-O bond, the effective charges parallel to the Ti-Obond are greatly enhanced, as are the Ti effective charges. This is a generalfeature of effective charges in more covalent, but still ionic materials. Whena nucleus or atom core is displaced, much more charge redistributes thanthe nominal value due to the bonding states formed from hybridizationbetween the atoms. The sensitivity of the effective charges to structural

40
distortions in PbTiO3 suggests a large amount of covalency, consistent withother diagnostics such as the partial density of states. The large effectivecharges in PbTiO3 contribute to its ferroelectric properties and piezoelectricresponse. Detailed study of how effect charges change with distortion inminerals is liable to shed more quantitative details on bonding and how itchanges with structural charges, but little such work has been done.
11. Metals
Metals are distinguished by the fact that they easily conduct electricity,which is due to the fact that they have partially occupied states at the Fermilevel, the highest occupied energy level in a crystal. One can understandthe insulating behavior of materials with fully filled bands by consideringwhat happens when one applies an electric field. An electric field raises thepotential at one part of the sample relative to another, and one would thinkthat electrons would then flow down that potential gradient. In fact boththe electrons move and the nuclei are displaced in response to the appliedfield; the material becomes polarized. The electrons do not continue toflow however. The material becomes polarized by mixing in part of theexcited state, or conduction band, eigenvectors into the valence band. Onlyso much can mix in, however, due to the finite gap, and since the bandsare filled in an insulator, there can be no continual flow under a small fieldsince electrons would have to be excited above the gap, which takes a finiteenergy. Thus the materials has a finite susceptibility and the material isinsulating. In a metal, the partially filled states at the Fermi level meansthat current will flow for any applied field, since there is no energy gapbetween occupied and unoccupied states, and the susceptibility is infinite.
Metals are not often considered as in mainstream of mineralogy or min-eral physics, though there are natural metals, and much of the mass ofthe Earth, that is the Earth’s core, is metallic iron. Recent progress in un-derstanding metals such as iron at high pressures is thus reviewed brieflyhere. In self-consistent computations, metals are treated in the same wayas insulators or semiconductors. The same methods work regardless of thedifferent type of bonding. In metals, however results can be more sensitiveto convergence parameters such as the type of basis and basis set size, andthe number of k-points used to integrate over the Brillouin zone. Many k-points are needed since different electronic states are occupied at differentk-points, due to the fact that bands cross the Fermi level. Also, the bondingin metals is not localized; rather one can think of atoms or ions embedded inan inhomogeneous sea of electrons. Since there is significant charge densityeverywhere, localized basis sets such as Gaussians are not optimal. Thisbecomes even more important when considering metallic surfaces, where

41
the charge density fades off gradually into the vacuum. The LAPW basisis ideal for metals, as for other materials, in that it uses a plane basis inthe region between atoms, which can represent any density. Plane wavebases with pseudopotentials are also ideal, as long as the pseudopotentialis accurate, which is sometimes difficult for 3d metals.
The LDA works quite well for metals in general, but the GGA is amarked improvement for magnetic 3d metals such as Fe. Iron is magneticand forms in the bcc structure at low pressures. The LDA however, in-correctly, gives a non-magnetic hcp structure as the ground state. It wasthought that the GGA overstabilized magnetic phases, but LAPW compu-tations showed that GGA gives not only the proper magnetic bcc groundstate, but also an accurate transition pressure to non-magnetic hcp at 11GPa, compared with an experimental bracket of 10-15 GPa, indicatingthat the magnetic stabilization is accurately predicted in GGA[99]. Theoryshows that it is magnetism that stabilizes the bcc structure, and that oth-erwise bcc iron would not form. There had been much discussion of bcc asthe possible structure for iron in the Earth’s inner core, but calculationsshow that bcc iron is mechanically unstable at high pressures due to theloss of magnetism with pressure [100].
Models have also been developed for metals that work quite well. Em-bedded atoms models are similar to Gordon-Kim models for closed shellsystems, except that the main term is the embedding energy for puttingan atom into an electron gas [101, 102]. Embedded atom models have notyet been much applied to high pressure or mineralogical problems. Tight-binding models work well for metals, and both non-empirical and empiricalmodels have been developed. Very accurate models have been developed foriron, for example, that are fit to self-consistent calculations, and that workwell over a factor of two of compression [18]. Hybrid models have beendeveloped that appear to be quite accurate for transition metal systems[103, 104, 105, 106].
High temperature properties are difficult to obtain self-consistently, soWasserman et al. applied the particle-in-a-cell model to compute thermody-namic properties of iron at high pressures and temperatures in conjunctionwith a new fast and accurate tight-binding model which allowed computa-tions for large unit cells [107]. Excellent agreement with shock compressiondata was obtained, up to ultrahigh pressures and temperatures.
Metals have contributions to thermodynamic properties from thermalelectronic excitations in addition to phonons at finite temperatures. Figure21 shows the computed thermal pressure for iron, and shows the phonon andelectronic contributions. Metallization of iron bearing oxides, if it occursin the mantle, will significantly change the density as well as transportproperties due to the electrons at the Fermi surface.
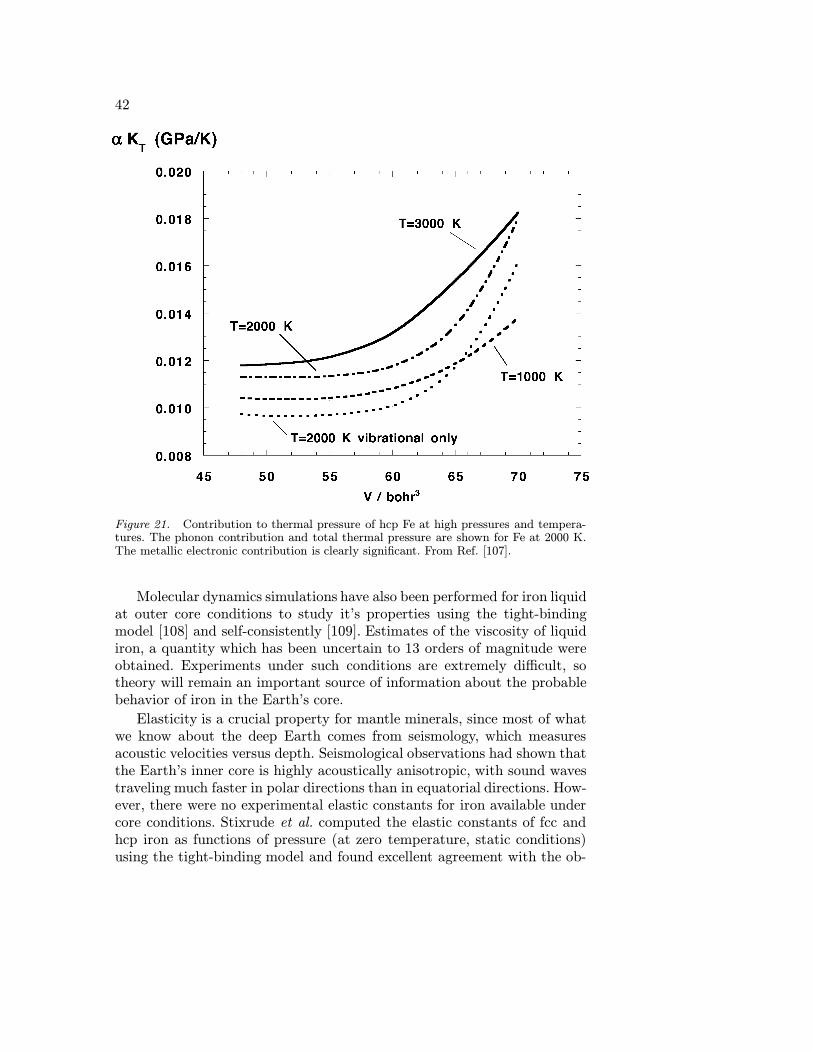
42
Figure 21. Contribution to thermal pressure of hcp Fe at high pressures and tempera-tures. The phonon contribution and total thermal pressure are shown for Fe at 2000 K.The metallic electronic contribution is clearly significant. From Ref. [107].
Molecular dynamics simulations have also been performed for iron liquidat outer core conditions to study it’s properties using the tight-bindingmodel [108] and self-consistently [109]. Estimates of the viscosity of liquidiron, a quantity which has been uncertain to 13 orders of magnitude wereobtained. Experiments under such conditions are extremely difficult, sotheory will remain an important source of information about the probablebehavior of iron in the Earth’s core.
Elasticity is a crucial property for mantle minerals, since most of whatwe know about the deep Earth comes from seismology, which measuresacoustic velocities versus depth. Seismological observations had shown thatthe Earth’s inner core is highly acoustically anisotropic, with sound wavestraveling much faster in polar directions than in equatorial directions. How-ever, there were no experimental elastic constants for iron available undercore conditions. Stixrude et al. computed the elastic constants of fcc andhcp iron as functions of pressure (at zero temperature, static conditions)using the tight-binding model and found excellent agreement with the ob-

43
TABLE 2. Elastic constants for non-magnetic hcp Fe at V=60bohr3 . From Ref. [110].
C11 C33 C12 C13 C44 C66
(GPa) (GPa) (GPa) (GPa) (GPa) (GPa)
Iron GGA
930 1010 320 295 260 305
Iron LDA
860 950 280 260 235 290
Iron Tight-Binding [18]
845 900 350 340 235 245
Iron FP-LMTO GGA[111]
870 810 255 320 235 310
Diamond Anvil Cell Strain Exp.[112]
640(55) 650(85) 300(55) 255(40) 420(25) 170(55)
served anisotropy if the inner core is a large hcp single crystal, or orientedsmaller crystals [113]. Self-consistent computations using the full-potentialLMTO method gave similar elastic constants [111]. The situation has be-come more complicated, in that recent diamond anvil experiments find verydifferent anisotropy from what has been predicted [112], and seismologistsfind much more complicated patterns of anisotropy in the inner core whichare difficult to reconcile with experimental or theoretical elastic constants.It may take some time until this is sorted out, because experiments are ex-tremely difficult, and the interpretation of the seismic data is also fraughtwith problems.
In order to better understand the accuracy of predictions of high pres-sure elasticity in hcp metals, we have performed detailed self-consistentLAPW computations for hcp Co and Re in addition to Fe, and find verygood agreement with experimental data for Co and Re (Table 3) [110].
12. Magnetism
Perhaps this is a good place to mention some general ideas about mag-netism. Magnetism is due to the presence of unpaired electrons. Electronshave magnetic moments, but if they are paired up in states as “up” and“down” pairs, there will be no net magnetism in the absence of a mag-netic field. However, in open shelled atoms there may be a net moment.For example, ferrous iron, Fe2+, has 6 d-electrons, and two endmember sit-uations can be considered. In low-spin ferrous iron, the 6 d-electrons arepaired up to fill 3 t2g states, and there is no net-magnetic moment. In high-spin ferrous iron, five “up” d-states are split in energy from five “down”

44
TABLE 3. Elastic constants for hcp Co and Re. After Ref. [111].
Volume C11 C33 C12 C13 C44 C66
(Bohr3) (GPa) (GPa) (GPa) (GPa) (GPa) (GPa)
Cobalt Experiment [114]
74.9 306 357 165 102 75 71
Cobalt GGA
75.0 325 365 165 105 90 80
Cobalt LDA
75.0 295 340 135 85 95 80
Rhenium Experiment[115]
99.3 616 683 273 206 161 172
Rhenium GGA
100.0 640 695 280 220 170 180
Rhenium LDA
100.0 605 650 235 195 175 185
d-states by the exchange energy, and 5 d-electrons go into the 3 t2g and 2eg lower-energy, majority spin states, and one electron into a high-energyt2g state, giving a net magnetic moment component of 4 µB (Bohr mag-netons) (fig. 22). According to Hund’s rules, atoms (or isolated ions) willmaximize their net magnetic moment, which lowers their total energy dueto a decrease in electrostatic energy. In a crystal, interactions with otheratoms, and formation of energy bands (hybrid crystalline electronic states)may lead to intermediate or low spin magnetic structures.
In the Stoner model, the effect of magnetism on the total energy is givenby:
∆E =−M2I
2+
M2
2N(0), (25)
where M is the magnetic moment or magnetization, the Stoner IntegralI is an atomic property, and N(0) is the density of states at the Fermilevel (or top of the valence band). The first-term is the magnetic energyand the second is the change in the band energy with magnetic moment.Minimization of ∆E gives the Stoner criterion:
IN(0) > 1 (26)
for a magnetic state (M > 0) to be stable. In the more sophisticated ex-tended Stoner model, the average density of states is introduced,
N(m) = M/∆ε , (27)

45
Figure 22. Schematic of occupancies of states in (a) high-spin and (b) low-spin ferrousiron.
where ∆ε is the spin (exchange) splitting,
N(0) = N(0) = ∂M/∂∆ε , (28)
and the instability criterion is IN(M) > 1. The Stoner model works quitewell in predicting magnetic states. It will be discussed further below. Aspressure is increased, band widths increase due to increasing hybridization,so that in the absence of new bands crossing the Fermi level or changes inband topology at the Fermi level or top of the valence band, the effectivedensity of states will decrease, and magnetism will decrease and disappearwith increasing pressure.
13. Transition metal ions and Mott insulators
One might not infer that there was anything special about transition metalions such as ferrous iron (Fe2+) and ferric iron (Fe3+) from examining manyphysical properties of minerals. There is complete solid solution between Mgand Fe endmembers for most minerals, and in general thermodynamic mix-ing properties and thermoelastic properties are smooth and even close toideal. This evident simplicity hides a problem that is at the frontier of solidstate physics. Whereas Mg2+ is a simple closed-shell ion, and it forms simpleionic bonds in oxides and silicates, Fe2+ is an opened-shell, magnetic, andvery non-spherical ion that forms complex ionic/covalent bonds. Althoughthe transition metal oxides such as FeO, MnO, NiO, and CoO form simplerocksalt (or distorted rocksalt) structures, and seem like ordinary ionic insu-lators in some ways, they are actually very complex and poorly understood

46
materials, experimentally as well as theoretically. Wustite, Fe1−xO, hasadditional complications since it is non-stoichiometric, and contains someferric iron even in equilibrium with iron metal. The vacancies in wustiteform complex defect clusters[116]. With increasing pressure, more stoichio-metric wustite can be stabilized. Theoretical work on wustite has mostlyconcentrated on the difficult enough problem of pure FeO, and that is thefocus here.
13.1. ELECTRONIC STRUCTURE
The band structures computed for these materials using conventional bandtheory (LDA or standard GGA methods) give a metallic ground state atzero pressure for FeO and CoO, and a small band gap for MnO and an-tiferromagnetic NiO, whereas these materials are believed to be wide gapinsulators. Magnetic crystals that are insulators by virtue of local magneticmoments are known as Mott insulators [117]. The failure of conventionalband theory to predict proper band gaps is not a reason by itself to be-lieve that band theory is completely wrong, since there are other “normal”materials for which LDA has problems. For example, Ge is found to bemetallic in LDA, rather than a semiconductor [118], and LDA always givesband gaps that are too small by a large factor compared with experiments.However, this failure to predict correct gaps in ordinary materials is wellunderstood, and is really not a failure of the theory, but a failure of afundamentally unjustified interpretation of the Kohn-Sham eigenvalues asquasiparticle energies, which they are not. We are used to the Kohn-Shameigenvalues being good approximations for the band structure, but thereis no fundamental justification for this correspondence, except that theKohn-Sham equations look similar to the quasiparticle equations.
Actually, the root cause of the “failure” of band theory for the transitionmetal oxides may be similar to the failure to predict accurate band gapsin other materials, but it is also believed that there is a more specificfailure of LDA-like theories for the transition metal oxides, which is thatLDA does consider energetic differences for a electron at a given point inspace depending on what orbital it is in. The Hohenberg-Kohn theoremsays that such orbital dependent potentials should not be necessary to findthe energy and ground state charge density of a system, but the exactfunctional that would give this behavior is unknown, and would likely beextremely complex in order to give the proper charge density of transitionmetal oxide compounds, especially those that involve orbital ordering [119,120]. The main problem with LDA-like theories is believed to be the mean-field treatment of the local Coulomb repulsion U , which is a measure of theincrease in energy when an electron is added to an atom. For example, Fe2+

47
has 6 d-electrons per atom, and a conducting electron for d-states at theFermi level, as predicted by LDA, must hop from Fe to Fe. However, thereis actually a large increase in energy when an electron hops from one Fe toanother. The simplest model that contains local interactions of this sort iscalled the Hubbard model. Even this simple model, though, has not beensolved, even in two-dimensions (it has been solved in infinite dimensions,in the dynamical mean field model [121, 122], but no one yet knows ifthis model is accurate for smaller numbers of dimensions). Even numericalsimulations of the Hubbard model have proven to be very difficult. RecentMonte Carlo results for the Hubbard model [123] are shown in fig. 23 andwill be discussed further below. The critical parameter that indicates theimportance of U is U/W , where W is the effective single-particle bandwidth (i.e. not including magnetism or hybridization with O 2p states.)
Figure 23. Monte Carlo results for the Hubbard model. Pressure increases to the lefton the absicca. The transition at the right (at low pressures) is TN , the Neel transition,(antiferromagnetic ordering). The high pressure transition is the high-spin low-spin tran-sition. Between is a metal insulator transition which is discontinuous at low temperatures,and is continuous out at high temperatures, above TN . From Ref. [123].
Hartree-Fock calculations include an unscreened orbital dependent U ,

48
and thus give large band gaps for these materials. they also give reasonableenergetics [124, 125]. However, they give band gaps that are way too large,and values of U that are much too high. Hartree-Fock seems to be a goodway to study crystals at low pressures in the high U limit, and it givesproper orbital ordering in cases where LDA fails completely. On the otherhand, Hartree-Fock cannot be used for metals, where it gives pathologicalresults, and the huge overestimation of band gaps makes it unsuitable tostudy metal-insulator transitions, or materials at high pressure which maybe properly described as metallic by LDA.
A promising method for realistic computations for Mott insulators, butwhich needs further investigation is the LDA+U model [126, 127, 128].In this method an orbital dependent potential is added to simulate U .U can be computed by using constrained LDA computations, where oneestimates U by computing the change in energy with orbital occupancy.LDA+U appears to be an excellent approximation at zero pressure in thehigh U/W limit, but it is unclear whether it will give reasonable results athigh pressures, where U/W is much smaller.
The parameter U/W decreases with pressure since U is relatively insen-sitive to pressure, but the band width W increases rapidly with pressuredue to increased hybridization.
13.2. ENERGETICS
In spite of the problems with the electronic structure of transition metal ox-ides, energetic properties can be predicted with accuracy similar to simplematerials using the LDA and GGA, with the exception of distortions in or-bitally ordered structures. The energy versus magnetic moment must alsobe reasonable, because magnetic moments are well predicted. The GGAequation of state is superior to the LDA, in that the LDA predicts signifi-cantly too small volumes. Nevertheless, even the LDA gives many proper-ties correctly, such as the qualitative change in rhombohedral angle withpressure in FeO [129]. This result indicates the power of theory, becausewe were able to study why the rhombohedral angle changes with pressure,which would be very difficult to understand from experiments alone. Visual-ization of the charge density as a function of rhombohedral angle [129, 130]indicated that Fe-Fe bonding causes the rhombohedral strain, and the in-crease in Fe-Fe bonding with pressure is responsible for the increased strainwith pressure. The rhombohedral strain brings Fe ions closer together, andthus is favored with increasing Fe-Fe interactions. It is dangerous, however,to extrapolate such conclusions to other materials. NiO has the oppositesense rhombohedral strain, and Isaak et al. suggested, without doing com-putations for NiO, that the sign of that strain would change with pressure

49
due to increased Ni-Ni bonding with pressure, and the strain would changein a similar way to FeO. Actually, the strain in NiO does not change signwith pressure, and increases in the sense that Ni ions move apart due tothe strain [131]. Evidently the Ni-Ni interactions are antibonding in nature,compared with the interactions in FeO.
13.3. MAGNETISM AND MAGNETIC COLLAPSE
Isaak et al. found that LDA predicted that the magnetic moment in FeO col-lapsed at pressures above 100 GPa [129]. They suggested that the magneticcollapse may lead to a, possibly simultaneous, metal-insulator transition.The predicted magnetic collapse is a type of high-spin low-spin transition,which has been observed in other materials such as NiI2[132, 133] and MnS[134, 135]. High-spin low-spin transitions can be either continuous higher-order phase transitions, or first-order phase transitions.
A more comprehensive study of the transition metals oxides MnO, FeO,CoO, and NiO was then performed using the GGA and the LMTO-ASAmethod [136]. The GGA LMTO equation of state results for FeO were bet-ter than LDA at low pressures, and the magnetic moments versus volumewere quite similar to the earlier LAPW results, except that it appearedthat LMTO could access metastable high-spin states in the low-spin sta-bility field, and low-spin states in the high spin stability field. The LMTOcomputations gave a 200 GPa high-spin low-spin transition for FeO, andat the time we believed that difference from the 100 GPa transition pre-dicted earlier was the difference between GGA and LDA, and that theLMTO computations gave good agreement with the earlier LAPW LDAcomputations of Isaak et al. Publication of the new results was followed byexperiments that showed a 100 GPa transition in FeO [137], which lead usto question whether the agreement with the earlier LDA calculations wasfortuitous, whether LDA could be more accurate than GGA, or whetherthe observed transition was different from that predicted. Detailed LAPWGGA calculations [108] have now shown that the LMTO results were inerror, and they give a transition pressure of 100 GPa in agreement with thePasternak et al. experiments.
Figure 24 shows the predicted magnetic moments versus volume forFeO, and figure 25 shows the equation of state. A discontinuous transitionis predicted for AFM FeO, and a continuous transition for ferromagneticFeO. There is a large ∆V for the high-spin low-spin transition.
Published diffraction data for FeO of Yagi et al. showed no discontinuityin the equation of state [138], so either the sample examined by Yagi et al.did not transform for some reason, or the transition was spread out overa large pressure range so that no discontinuity was seen, perhaps due to
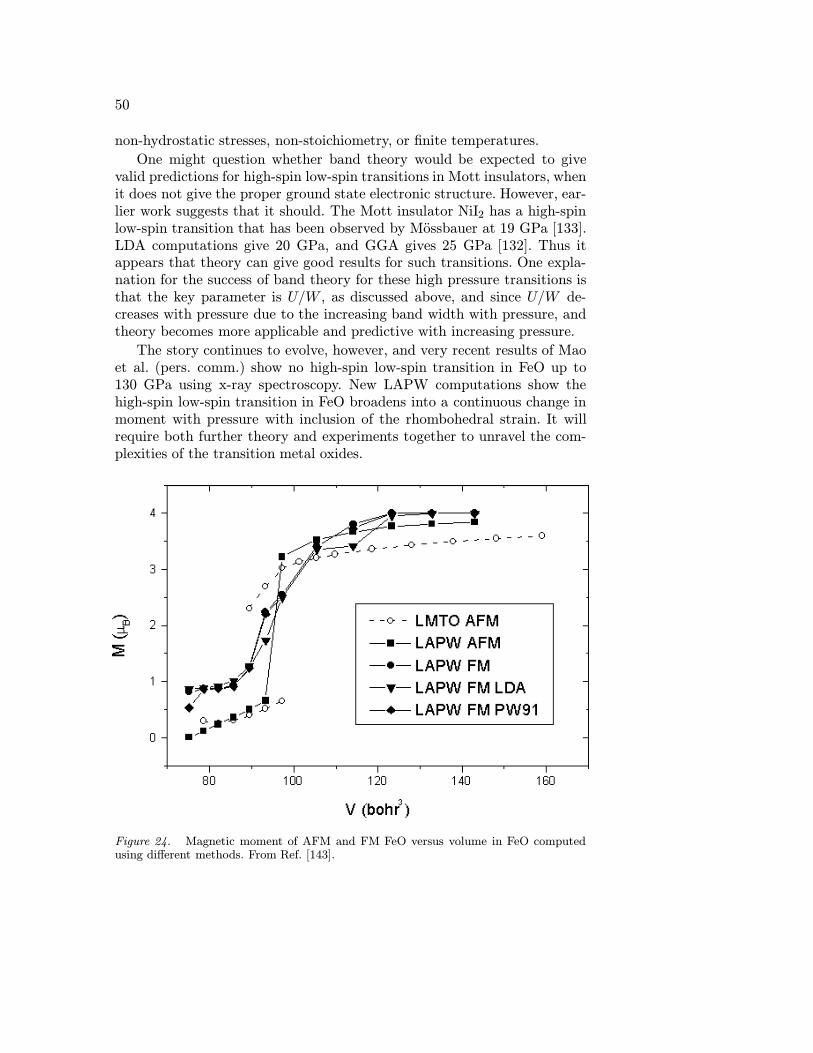
50
non-hydrostatic stresses, non-stoichiometry, or finite temperatures.
One might question whether band theory would be expected to givevalid predictions for high-spin low-spin transitions in Mott insulators, whenit does not give the proper ground state electronic structure. However, ear-lier work suggests that it should. The Mott insulator NiI2 has a high-spinlow-spin transition that has been observed by Mossbauer at 19 GPa [133].LDA computations give 20 GPa, and GGA gives 25 GPa [132]. Thus itappears that theory can give good results for such transitions. One expla-nation for the success of band theory for these high pressure transitions isthat the key parameter is U/W , as discussed above, and since U/W de-creases with pressure due to the increasing band width with pressure, andtheory becomes more applicable and predictive with increasing pressure.
The story continues to evolve, however, and very recent results of Maoet al. (pers. comm.) show no high-spin low-spin transition in FeO up to130 GPa using x-ray spectroscopy. New LAPW computations show thehigh-spin low-spin transition in FeO broadens into a continuous change inmoment with pressure with inclusion of the rhombohedral strain. It willrequire both further theory and experiments together to unravel the com-plexities of the transition metal oxides.
Figure 24. Magnetic moment of AFM and FM FeO versus volume in FeO computedusing different methods. From Ref. [143].

51
The study of Mott insulators such as FeO is fascinating since no oneknows what to expect;even the generic behavior of these systems is un-known. Even the simplest pertinent model, the Hubbard model, has neverbeen solved. Quantum Monte Carlo simulations for the Hubbard model[123] have shown behavior similar to what has been observed in FeO andother systems studied so far. Figure 23 shows a proposed phase diagramfrom Hubbard model simulations designed to study f-electron systems, suchas Ce, which also show strong volume collapse transitions. There has beensome controversy as to the nature of these transitions, but one view is thatthese transitions are essentially high-spin low-spin transitions. Figure 23shows ordered and disordered “high-spin” magnetic phases, a non-magnetic“low-spin” phase, and a metal insulator transition. The metal-insulatortransition is particularly interesting, in that it occurs not at the high-spinlow-spin transition boundary, but before this transition. Furthermore themetal-insulator transition is sharp at low temperatures, but becomes con-tinuous at high temperatures, with a funny triple-peaked density of states.Conductivity may still onset discontinuously at a Mott-Anderson transi-tion when there are sufficient densities of states at the Fermi-level to havebulk conduction, the so-called mobility edge [117]. Secondly, note that themagnetic collapse boundary at high pressures may be the same as the Neeltransition boundary at low pressures, and one may be able to go contin-uously from a phase with disordered local moments, to a phase with nolocal moments, since these phases have the same symmetry. Thus the Neeltemperature initially increases, and then decreases with increasing pressuredue to the decreasing local moments.
Now it is possible to better understand the results of Pasternak et al.[137] The Mossbauer experiments showed a transition with increasing pres-sure to a low spin phase, and also a transition to a non-magnetic phasewith increasing temperature. This would be difficult to understand if themoments were ordered in all cases, that is if the temperature was well belowTN as in their proposed phase diagram, because the high-spin phase shouldbe the high entropy phase; thus increasing temperature should promote thehigh-spin magnetic phase, rather than the low-spin non-magnetic phase.However, if one understands that the transition with increasing tempera-ture is a disordering of the local moments, rather than a loss of local mo-ments, all is well, since temperature should promote disordering. Mossbauerdoes not distinguish between loss of local moments, and disordering of mo-ments. So it appears that the experiments on FeO are consistent with whatis known about the behavior of the Hubbard model except that the locationof the metal insulator transitions in FeO and other transition metal oxidesare unknown. At about 100 GPa the B1 phase appears to be insulating,not metallic, which suggests that the insulator metal transition should be
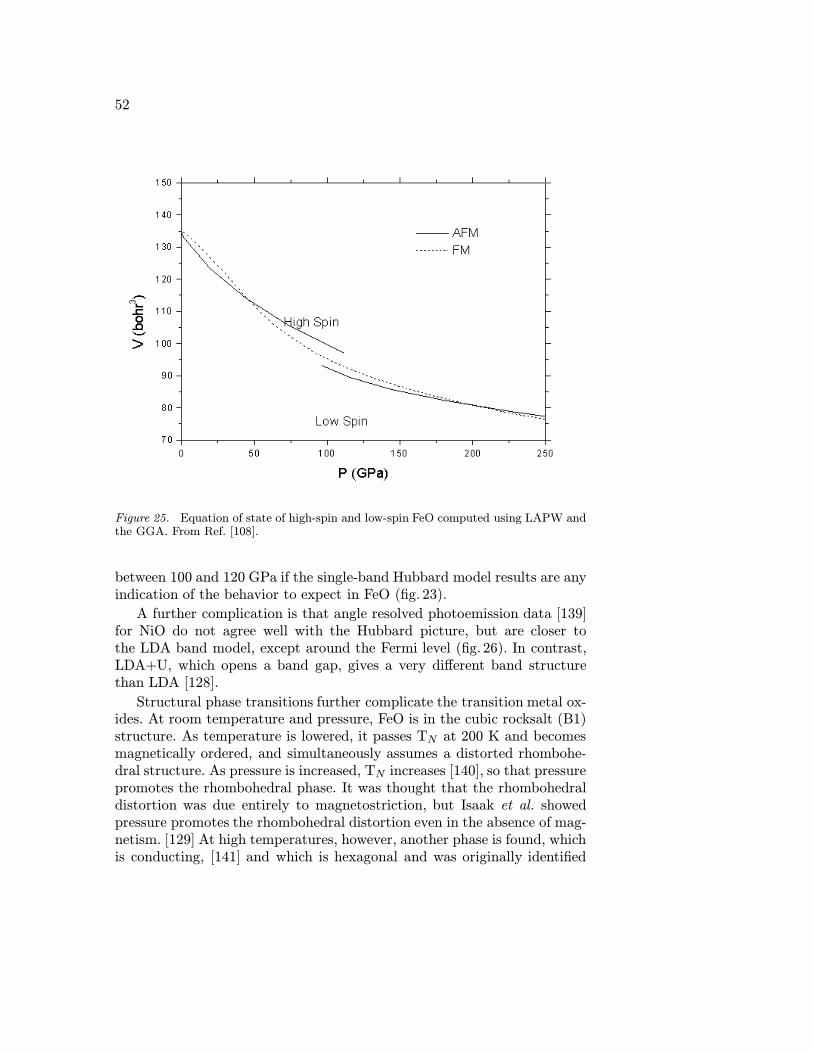
52
Figure 25. Equation of state of high-spin and low-spin FeO computed using LAPW andthe GGA. From Ref. [108].
between 100 and 120 GPa if the single-band Hubbard model results are anyindication of the behavior to expect in FeO (fig. 23).
A further complication is that angle resolved photoemission data [139]for NiO do not agree well with the Hubbard picture, but are closer tothe LDA band model, except around the Fermi level (fig. 26). In contrast,LDA+U, which opens a band gap, gives a very different band structurethan LDA [128].
Structural phase transitions further complicate the transition metal ox-ides. At room temperature and pressure, FeO is in the cubic rocksalt (B1)structure. As temperature is lowered, it passes TN at 200 K and becomesmagnetically ordered, and simultaneously assumes a distorted rhombohe-dral structure. As pressure is increased, TN increases [140], so that pressurepromotes the rhombohedral phase. It was thought that the rhombohedraldistortion was due entirely to magnetostriction, but Isaak et al. showedpressure promotes the rhombohedral distortion even in the absence of mag-netism. [129] At high temperatures, however, another phase is found, whichis conducting, [141] and which is hexagonal and was originally identified

53
Figure 26. Angle resolved photoemission data for NiO compared with the LDA bandstructure. (a) Photoemission data. The marked peaks are interpreted as quasiparticlestates. The breadth of peaks is due to instrumental resolution, surface effects, multi-ple bands, and lifetime broadening. (b) Comparison with the antiferromagnetic bandstructure. Agreement is quite good except for the band crossing the Fermi level. FromRef. [139].
from in situ diffraction studies to be the NiAs (B8) structure [142].This phase has not yet been seen at room temperatures, although the
phase diagram seems to suggest that it should occur at low temperatures aswell; perhaps the transition is kinetically inhibited. Although the positionsof the diffraction peaks agreed with the B8 structure, the intensities did not.By reanalyzing the diffraction data, Mazin et al. [143] concluded that themost likely interpretation of the data was that of a polytype or superlatticebetween B8 and anti-B8, with Fe in the As-site and O in the Ni-site. The B8and anti-B8 structures can be joined together smoothly, and the boundarybetween them is the rhombohedrally-distorted B1 structure. Thus one canjoin together all three phases into a single coherent structure (fig. 27). Thisphase could form either due to lack of equilibrium, or could even form aunique continuous structure transition between the different phases. Theinverse-B8 structure is predicted to be an insulator (the only known LDA

54
Figure 27. Normal and inverse NiAs superlattice proposed for FeO. From Ref. [108].
band insulator in FeO) whereas normal B8 is predicted to be a metal. Thisis due to the fact that the Fe-Fe distances are very short in normal B8,and much farther apart in inverse-B8. GGA band theory predicts similarenergies for normal and inverse B8 in the non-magnetic or ferromagneticcases, but when allowed to be anti-ferromagnetic, inverse B8 is stabilized somuch that it is actually predicted to be the ground state phase, i.e. lower inenergy than B1. This is very unlikely to be correct, although it isn’t entirelyimpossible. B1 could be found due to a combination of non-stoichiometry,which would stabilize B1, and kinetic constraints, since inverse-B8 is onlymore stable in the low-temperature antiferromagnetically ordered phase,which would likely not form spontaneously at low temperatures from B1.The fact that the high pressure and temperature phase has not been suc-cessfully quenched to room conditions argues against its stability relativeto B1, though.
Finally, we consider the origin of high-spin low-spin magnetic transi-tions, and the likely magnetic structure of transition metal ions in solidsolution in other phases. The conventional, local atomic, picture of high-spin low-spin transitions is that the crystal field splitting between eg andt2g states gets larger than the exchange splitting between spin-up and spin-down states, thus leading to a transition to a low spin electronic structure

55
(figs. 28 and 29). Furthermore, the conventional view is that the eg and t2g
splitting is due to the Coulomb potential from the O2− coordination poly-hedron, and that the splitting increases as ρ5, where ρ is the crystallinedensity [135, 144, 145]. Neither part of this conventional picture appears tobe correct, in spite of the fact that the observed crystal field splitting variesapproximately as ρ5 in many cases. Firstly, if one considers the eg and t2g
splitting itself, it is not due to the electrostatic interaction of surrounding2− charges. In fact, the on-site contributions to the crystal field splittingeven have the wrong sign, and would give eg lower than t2g by a smallamount, rather than the observed t2g being lower. This has been knownsince the seminal work of Mattheiss [146, 147, 148], (and we have verifiedthis fact), but it appears not to have been appreciated by the mineralogicaland high pressure communities. The main contribution to the eg-t2g split-ting is due to d−d hybridization between transition metal ions, which alsovaries as ρ5 [3]. Now the eg-t2g symmetry is only appropriate at the Γ-point,and Mattheiss showed that only the d − d interactions operate at Γ, butthe band width at other points in the Brillouin zone is due to hybridizationwith O 2p states. Optical spectroscopy measurements do not measure onlythe states at Γ, but rather are sensitive to vertical transitions throughoutthe zone, and thus are sensitive to the entire bandwidth. The d−p interac-tions lead to a band width that behaves approximately as ρ7[3]. Thus theband width and apparent crystal field splitting should vary between ρ5 andρ7 with increasing pressure, and this is due to increased hybridization withincreasing pressure. The distinction between the present picture and theconventional view can be seen most easily in terms of the behavior of theHamiltonian. In the convention view, it is the on-site, diagonal, elementsthat vary rapidly with pressure, giving rise to increased splitting, whereasband theory says that it is the off-diagonal, covalent, contributions whichchange the band widths with pressure. The transition does not requirethe band widths to get larger than the exchange splitting, but rather theeffective density of states must go below a critical value, as discussed abovefor the extended Stoner model.
Figure 30 shows the Stoner diagram for CoO. With increasing pressurethe effective density of states drops, and the stable magnetic structurebecomes low-spin due to this effect. We have studied high-spin low-spintransitions in a number of structures, and it appears that the effective bandwidth is dominated by the local coordination (e.g. d−p hybridization). Thusa transition metal ion in a smaller site will transform at lower pressures thanone in a larger site. Thus ferric iron in solid solution substituting for Mg2+,which is a smaller ion than Fe2+, is likely to transform at lower pressuresthan in the pure ferrous iron compound. This suggests that magnesiowustitewill be low spin at even lower pressures than 100 GPa in the Earth. On the
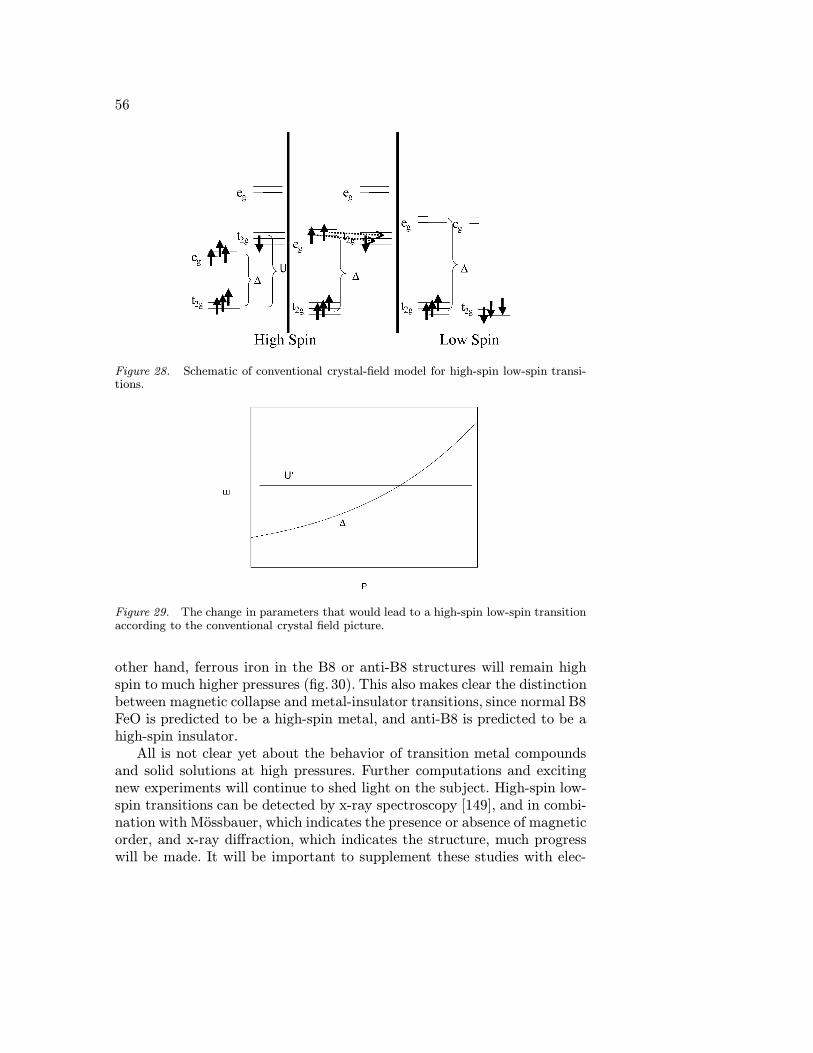
56
Figure 28. Schematic of conventional crystal-field model for high-spin low-spin transi-tions.
Figure 29. The change in parameters that would lead to a high-spin low-spin transitionaccording to the conventional crystal field picture.
other hand, ferrous iron in the B8 or anti-B8 structures will remain highspin to much higher pressures (fig. 30). This also makes clear the distinctionbetween magnetic collapse and metal-insulator transitions, since normal B8FeO is predicted to be a high-spin metal, and anti-B8 is predicted to be ahigh-spin insulator.
All is not clear yet about the behavior of transition metal compoundsand solid solutions at high pressures. Further computations and excitingnew experiments will continue to shed light on the subject. High-spin low-spin transitions can be detected by x-ray spectroscopy [149], and in combi-nation with Mossbauer, which indicates the presence or absence of magneticorder, and x-ray diffraction, which indicates the structure, much progresswill be made. It will be important to supplement these studies with elec-

57
Figure 30. Stoner diagram for CoO. The effective density of states is plotted againstmoment for several different volumes. The inverse Stoner parameter, 1/I is also shown,and places where the effective density of states crosses 1/I with a negative slope arestable solutions. At high volumes (low pressures) there is only a high-spin (large moment)solution. At intermediate pressures both high-spin and low-spin solutions exist. (Whethersuch coexistence is possible, i.e. whether the transition is first-order or second-order,depends on the shape of the effective density of states.) At high pressures, there is only alow-spin solution. The Stoner model behaves very differently from the localized, crystalfield, picture which would always predict a discontinuous transition and would not allowintermediate moments. From Ref. [136].
trical conductivity measurements and/or susceptibility measurements andto vary the temperature to find all of the boundaries of the expected com-plex phase diagrams for Mott insulating systems. High pressure studies areclearly necessary to reach a full understanding of these complex crystals.
14. Conclusions
Progress in the field of mineralogy now requires a deep understanding ofsolid state physics and chemistry, as well as geology and geophysics. Miner-als are simply natural materials, and given the fact that most of the basicstructural and descriptive chemical questions about minerals have beenstudied thoroughly, the next phase of research in the field is necessary tounderstand the fundamental origins of mineral structure, chemistry, andbehavior. There is much work left for mineralogy in the next century andbeyond, but only if the study of minerals is integrated with the science ofother materials. On the other hand, the study of minerals is as fundamentalas any other branch of Earth science, and a rich frontier of fundamentalquestions remain to be explored using newly developed theoretical methodsand modern experimentation.

58
15. Acknowledgments
I would like to thank all of my collaborators. Much of the work presentedwas done in collaboration with I. Inbar, D. Isaak, J. Ita, H. Krakauer,F. Marton, I. Mazin, G. Saghi-Szabo, D. Singh, G. Steinle, L. Stixrude,and E. Wasserman. Thanks also to L. Finger, S. Gramsch, R. Hemley, D.Mao, A. McMahan, W. Pickett, C. Prewitt, and R. Scalettar for helpfuldiscussions. Research was supported by NSF EAR-9305060, EAR-9870328,EAR-9614363 and the Carnegie Institution of Washington. Computationswere performed on the Cray J90/16-4096 at the Geophysical Laboratorysupported by NSF EAR-9512627, and the Keck Foundation.
References
1. Ashcroft, N.W. and Mermin, N.D. (1976) Solid State Physics, Holt Rinehart andWinston, New York.
2. Kittel, C. (1996) Introduction to Solid State Physics, Wiley, New York.3. Harrison, W.A. (1980) Electronic Structure and the Properties of Solids: The
Physics of the Chemical Bond, W.H. Freeman and Company, San Francisco.4. Mehl, M.J., Cohen, R.E., and Krakauer, H. (1988) Linearized Augmented Plane
Wave electronic structure calculations for MgO and CaO, J. Geophys. Res. 93,8009-8022; Correction: 94, 1977, 1989.
5. Taurian, O.E., Springborg, M. and Christensen, N.E. (1985) Self-consistent elec-tronic structures of MgO and SrO, Solid State Comm., 55, 351-355.
6. Hohenberg, P. and Kohn, W. (1964) Inhomogeneous electron gas, Phys. Rev. 136,864-871.
7. Kohn, W. and Sham, L.J. (1965) Self-consistent equations including exchange andcorrelation effects, Phys. Rev. A, 140,1133-1140.
8. Pisani, C., Dovesi, R. and Roetti, C. (1988) Hartree-Fock ab Initio Treatment ofCrystalline Systems, Springer, New York.
9. Perdew, J.P. and Wang, Y. (1992) Accurate and simple analytic representation ofthe electron-gas correlation energy, Phys. Rev. B 45, 13244-13249.
10. Perdew, J.P., Chevary, J.A., Vosko, S.H., Jackson, K.A., Pederson, M.R., Singh,D.J. and Fiolhais, C. (1992) Atoms, molecules, solids, and surfaces: Applicationsof the generalized gradient approximation for exchange and correlation, Phys. Rev.B 46, 6671-6687.
11. Perdew, J.P., Burke, K. and Ernzerhof, M. (1996) Generalized gradient approxi-mation made simple, Phys. Rev. Lett. 77, 3865-3868.
12. Zunger, A., Perdew, J.P. and Oliver, G.L. (1980) A self-interaction corrected ap-proach to many-electron systems: beyond the local spin density approximation,Solid State Comm. 34, 933-936.
13. Perdew, J.P. and Zunger, A. (1981) Self-interaction correction to density-functionalapproximations for many-electron systems, Phys. Rev. B 23, 5048-5079.
14. Szotek, Z. and Temmerman, W.M. (1993) Application of the self-interaction cor-rection to transition-metal oxides, Phys. Rev. B 7, 4029-4032.
15. Mazin, I.I. and Singh, D.J. (1998) Nonlocal density functionals and the linearresponse of the homogeneous electron gas, Phys. Rev. B 57, 6879-83.
16. Mazin, I.I. and Singh, D.J. (1998) Weighted density functionals for ferroelectricmaterials, in R. E. Cohen (ed.), First-Principles Calculations for Ferroelectrics,AIP, 251-264.
17. Kohn, W., Meir, Y. and Makarov, D.E. (1998) van der Waals energies in density

59
functional theory, Phys. Rev. Lett. 80, 4153-4156.18. Cohen, R.E., Stixrude, L. and Wasserman, E. (1997) Tight-binding computations
of elastic anisotropy of Fe, Xe, and Si under compression, Phys. Rev. B 56, 8575-8589.
19. Patton, D.C. and Pederson, M.R. (1997) Application of the generalized gradientapproximation to rare-gas dimers, Phys. Rev. A 56, R2495.
20. LeSar, R. (1984) Electron-gas plus damped-dispersion model for intermolecularforces. The rare-gas and H2-He, H2-Ne, and H2-Ar potentials, J. Phys. Chem. 88,4272-4278.
21. Dham, A.K., Meath, W.J., Allnatt, A.R., Aziz, R.A. and Slaman, M.J. (1990) XCand HFD-B potential energy curves for Xe-Xe and related physical properties, J.Chem. Phys. 142, 173-189.
22. Ross, M., Mao, H.K., Bell, P.M. and Xu, J. (1986) The equation of state of denseargon: A comparison of shock and static studies, J. Chem. Phys. 85, 1028-1033.
23. Reichlin, R., Brister, K., McMahan, A.K., Ross, M., Martin, S., Vohra, Y.K. andRuoff, A.L. (1989) Evidence for the insulator-metal transition in xenon from opti-cal, x-ray, and band-struture studies to 170 GPa Phys. Rev. Lett. 62, 669-672.
24. Goettel, K.A., Eggert, J.H., Silvera, I.F. and Moss, W.C. (1989) Optical evidencefor the metallization of Xenon at 132(5) GPa, Phys. Rev. B 62, 665-668.
25. Chacham, H., Zhu, X. and Louie, S.G.(1992) Pressure induced insulator-metaltransitions in solid xenon and hydrogen: A first-principles quasiparticle study,Phys. Rev. B 46, 6688-6699.
26. Jephcoat, A.P., Mao, H.-K., Finger, L.W., Cox, D.E., Hemley, R.J. and Zha, C.-S. (1987) Pressure-induced structural phase transition in solid xenon, Phys. Rev.Lett. 59, 2670-2673.
27. Gordon, R.G. and Kim, Y.S. (1972) Theory for the forces between closed-shellatoms and molecules, J. Chem. Phys. 56, 3122-3133.
28. LeSar, R. (1983) Ground- and excited-state properties of solid argon under pres-sure, Phys. Rev. B 28, 6812-6820.
29. Barker, J.A. (1986) Many-body interactions in rare gases: krypton and xenon,Phys. Rev. Lett. 57, 230-233.
30. Kim, S.K. (1975) Nonadditive three-body interactions of rare-gas atoms. I. Smalland intermediate distances, Phys. Rev. A 11, 796-803.
31. Barker, J.A. and Klein, M.L. (1973) Monte Carlo calculations for solid and liquidargon Phys. Rev. B, 7, 4707-4712.
32. Ceperley, D.M. (1995) Path integrals in the theory of condensed helium, Rev. Mod.Phys. 67, 279-355.
33. Bukowinski, M.S.T. (1980) Pressure effects on bonding in MgO, J. Geophys. Res.85, 285-292.
34. Bukowinski, M.S.T. (1985) First-principles equations of state of MgO and CaO,Geophys. Res. Lett., 12, 536-539.
35. Bukowinski, M.S.T. and Wolf, G.H. (1986) Equation of state and stability offluorite-structured SiO2, J. Geophys. Res. 91, 4704-4710.
36. Feldman, J.L., Klein, B.M., Mehl, M.J. and Krakauer, H. (1990) Metallizationpressure for NaCl in the B2 structure, Phys. Rev. B 42, 2752-2760.
37. Karki, B.B., Stixrude, L., Clark, S.J., Warren, M.C., Ackland, G.J. and Crain, J.(1997) Structure and elasticity of MgO at high pressure, Amer. Mineral. 82, 51-60.
38. De Vita, A., Gillan, M.J., Lin, J.S., Payne, M.C., Stich, I. and Clarke, L.J. (1992)Defect energetics in oxide materials from first principles, Phys. Rev. Lett. 68, 3319-3322.
39. De Vita, A., Gillan, M.J., Lin, J.S., Payne, M.C., Stich, I. and Clarke, L.J. (1992)Defect energetics in MgO treated by first-principles methods, Phys. Rev. B 46,12964-12973.
40. Gibson, A., Haydock, R. and LeFemina, J.P. (1994) Stability of vacancy defects inMgO: the role of charge neutrality, Phys. Rev. B 50, 2582-2592.

60
41. LaFemina, J.P. (1994) Total energy computations of oxide surface reconstructions,Critical Reviews in Surface Chemistry 3, 297-386.
42. Refson, K., Wogelius, R.A., Fraser, D.G., Payne, M.C., Lee, M.H. and Milman, V.(1995) Water chemisorption and reconstruction of the MgO surface, Phys. Rev. B52, 10823-10826.
43. Zhang, H., Daniels, W. and Cohen, R.E. (1994) The exciton energy and its pressuredependence in alkali halides, Phys. Rev. B 50, 70-74.
44. Akkerman, A., Boutboul, T., Breskin, A., Chechik, R., Gibrekhterman, A. andLifshitz, Y. (1996) Inelastic electron interactions in the energy range 50 eV to 10keV in insulators: alkali halides and metal oxides, Physica Status Solidi B 198,769-784.
45. Shirley, E.L. (1996) Simulating x-ray fluorescence in graphite and boron nitride,in D. L. Ederer and J. H. McGuire (eds.) , Raman Emission by X-ray Scattering,World Scientific, 50-60.
46. Watson, R.E. (1958) Analytic Hartree-Fock solutions for O=, Phys. Rev. 111,1108-1110.
47. Muhlhausen, C. and Gordon, R.G. (1981) Electron-gas theory of ionic crystals,including many-body effects, Phys. Rev. B 23, 900-923.
48. Mehl, M.J., Hemley, R.J. and Boyer, L.L. (1986) Potential induced breathing modelfor the elastic moduli and high-pressure behavior of the cubic alkaline-earth oxides,Phys. Rev. B, 33, 8685-8696.
49. Born, M. and Huang, K. (1954) Dynamical Theory of Crystal Lattices, ClarendonPress, Oxford.
50. Cohen, R.E., Boyer, L.L. and Mehl, M.J. (1987) Lattice dynamics of the PotentialInduced Breathing model: First principles phonon dispersion in the alkaline earthoxides, Phys. Rev. B 35, 5749-5760.
51. Cohen, R.E., Boyer, L.L. and Mehl, M.J. (1987) Theoretical studies of chargerelaxation effects on the statics and dynamics of oxides, Phys. Chem. Minerals 14,294-302.
52. Wolf, G.H. and Bukowinski, M.S.T. (1988) Variational stabilization of the ioniccharge densities in the electron-gas theory of crystals: Applications to MgO andCaO, Phys. Chem. Minerals, 15, 209-220.
53. Feldman, J.L., Mehl, M.J., Boyer, L.L. and Chen, N.C. (1988) Ab initio calculationsof static lattice properties for NaCl and a test of the Decker equation of state, Phys.Rev. B 37, 4784-4787.
54. Hemley, R.J. and Gordon, R.G. (1985) Theoretical study of solid NaF and NaClat high pressures and temperatures, J. Geophys. Res. B90, 7803-7813.
55. Isaak, D.G., Cohen, R.E. and Mehl, M.J. (1990) Calculated Elastic and ThermalProperties of MgO at High Pressure and Temperatures, J. Geophys. Res. 95, 7055-7067.
56. Isaak, D.G., Anderson, O.L. and Cohen, R.E. (1992) The relationship betweenshear and compressional velocities at high pressures: reconciliation of seismic to-mography and mineral physics, Geophys. Res. Lett. 19, 741-744.
57. Inbar, I. and Cohen, R.E. (1995) High pressure effects on thermal properties ofMgO, Geophys. Res. Lett. 22, 1533-1536.
58. Cohen, R.E. (1998) Thermal conductivity of MgO at high pressures, Rev. HighPressure Sci. Technology 7, 160-162.
59. Ita, J. and Cohen, R.E. (1997) Effects of pressure on diffusion and vacancy for-mation in MgO from non-empirical free-energy integrations, Phys. Rev. Lett. 79,3198-3201.
60. Ita, J. and Cohen, R.E. (1998) Diffusion in MgO at high pressure: Implications forlower mantle rheology, Geophys. Res. Lett. 25, 1095-1098.
61. Cohen, R.E. (1987) Calculation of elasticity and high pressure instabilities in corun-dum and stishovite with the potential induced breathing model, Geophys. Res. Lett.14, 37-40.

61
62. Cynn, H., Isaak, D.G., Cohen, R.E., Nicol, M.F. and Anderson, O.L. (1990) Ahigh pressure phase transition of corundum predicted by the Potential InducedBreathing model, Amer. Mineral., 75, 439-442.
63. Marton, F.C. and Cohen, R.E. (1994) Prediction of a high pressure phase transitionin Al2O3, Amer. Mineral., 79, 789-792.
64. Funamori, N. and Jeanloz, R. (1997) High pressure transformation of Al2O3, Sci-ence 278, 1109-1111.
65. Edwardson, P.J. (1989) Corridors-between-adjacent-sites model for the four phasesof KNbO3, Phys. Rev. Lett. 63, 55-59.
66. Boyer, L.L., Stokes, H.T. and Mehl, M.J. (1995) Self-consistent potential inducedbreathing model calculations for longitudinal modes in MgO, Ferroelectrics 164,177-181.
67. Stokes, H.T., Boyer, L.L. and Mehl, M.J. (1996) Spherical self-consistent atomicdeformation model for first-principles energy calculations in ionic crystalline solids,Phys. Rev. B 54, 7729-7736.
68. Boyer, L.L., Stokes, H.T. and Mehl, M.J.(1997) Application of a Kohn-Sham-likeformulation of the self-consistent atomic deformation model, Ferroelectrics 194,173-186.
69. Kohan, A.F. and Ceder, G. (1996) Tight-binding calculation of formation energiesin multicomponent oxides: application to the MgO-CaO phase diagram, Phys. Rev.B 54, 805-811.
70. Bilz, H. and Kress, W. (1979) Phonon Dispersion Curves in Insulators, Springer-Verlag.
71. Slater, J.C. and Koster, G.F. (1954) Simplified LCAO method for the periodicpotential problem, Phys. Rev. 94, 1498-1524.
72. Cohen, R.E., Mehl, M.J. and Papaconstantopoulos, D.A. (1994) Tight-bindingtotal energy method for transition and noble metals, Phys. Rev. B 50, 14694-14697.
73. Schelling, P.K., Yu, N. and Halley, J.W. (1998) Self-consistent tight-bindingatomic-relaxation model of titanium dioxide, Phys. Rev. B 58, 1279-1293.
74. Chadi, D.J. (1989) Transferability of tight-binding matrix elements, in V. Vitek andD. J. Srolovitz (eds.), Atomistic Simulation of Materials: Beyond Pair Potentials,Plenum Press, 309-315.
75. Mercer, J.L., Jr. and Chou, M.Y. (1994) Tight-binding model with intra-atomicmatrix elements, Phys. Rev. B 49, 8506-8509.
76. Pauling, L. (1960) The Nature of the Chemical Bond, Cornell University Press,Ithaca, New York.
77. Cohen, R.E. (1994) First-principles theory of crystalline SiO2, in P.J. Heaney,C.T. Prewitt, and G.V. Gibbs (eds.), Silica: Physical Behavior, Geochemistry, andMaterials Applications, Reviews in Mineralogy, Vol. 29, Mineralogical Society ofAmerica, Washington, D.C., 369-402.
78. Cohen, R.E. (1991) Bonding and elasticity of stishovite at high pressure: Linearizedaugmented plane wave calculations, Amer. Mineral. 76, 733-742.
79. Spackman, M.A., Hill, R.J. and Gibbs, G.V. (1987) Exploration of structure andbonding in stishovite with Fourier and pseudoatom refinement methods using singlecrystal and powder X-ray diffraction data, Phys. Chem. Minerals 14, 139-150.
80. Cohen, R.E. (1992) First-principles predictions of elasticity and phase transitionsin high pressure SiO2 and geophysical implications, in M. H. Manghnani and Y.Syono (eds.), High Pressure Research in Mineral Physics: Application to Earth andPlanetary Science, AGU, 425-432.
81. Kingma, K.J., Cohen, R.E., Hemley, R.J. and Mao, H.-K. (1995) Transformationof silica to a denser phase at lower-mantle pressures, Nature 374, 243-245.
82. Hemley, R.J. (1987) Pressure dependence of Raman spectra of SiO2 polymorphs:α-quartz, coesite, and stishovite, in M. H. Manghnani and Y. Syono (eds.), High-Pressure Research in Mineral Physics, Terra Scientific, 347-359.

62
83. Tse, J.S. and Klug, D.D. (1991) Mechanical instability of α-quartz: A moleculardynamics study, Phys. Rev. Lett., 67, 3559-3562.
84. Tsuneyuki, S., Aoki, H., Tsudaka, M. and Matsui, Y. (1990) Molecular-dynamicsstudy of the α to β structural phase transition of quartz, Phys. Rev. Lett. 64,776-779.
85. Keskar, N.R. and Chelikowsky, J.R. (1992) Structural properties of nine silicapolymorphs, Phys. Rev. B 46, 1-13.
86. Liu, F., Garofalini, S.H., King-Smith, D. and Vanderbilt, D. (1994) First-principlesstudy of crystalline silica, Phys. Rev. B 49, 12528-12534.
87. Xu, Y.-N. and Ching, W.Y. (1991) Electronic and optical properties of all poly-morphic forms of silicon dioxide, Phys. Rev. B 44, 11048-11059.
88. Hamann, D.R. (1996) Generalized gradient theory for silica phase transitions, Phys.Rev. Lett. 76, 660-663.
89. Hemley, R.J. and Cohen, R.E. (1992) Silicate perovskite, Ann. Rev. Earth Planet.Sci. 20, 553-600.
90. Stixrude, L. and Cohen, R.E. (1993) Stability of orthorhombic MgSiO3 perovskitein the Earth’s lower mantle from first principles, Nature 364, 613-615.
91. Cohen, R.E. (1987) Elasticity and equation of state of MgSiO3 perovskite, Geophys.Res. Lett. 14, 1053-1056.
92. Yeganeh-Haeri, A., Weidner, D.J. and Ito, E. (1989) Elasticity of MgSiO3 in theperovskite structure, Science 243, 787-789.
93. Yeganeh-Haeri, A. (1994) Synthesis and re-investigation of the elastic propertiesof single-crystal magnesium silicate perovskite, Phys. Earth Planet. Inter. 87, 111-121.
94. Karki, B.B., Stixrude, L., Clark, S.J., Warren, M.C., Ackland, G.J. and Crain,J. (1997) Elastic properties of orthorhombic MgSiO3 perovskite at lower mantlepressures, Amer. Mineral., 82, 635-638.
95. Yu, R. and Krakauer, H. (1994) Linear-response calculations within the linearizedaugmented plane-wave method, Phys. Rev. B 49, 4467-4477.
96. Stixrude, L., Cohen, R.E., Yu, R. and Krakauer, H. (1996) Prediction of phasetransition in CaSiO3 perovskite and implications for lower mantle structure, Amer.Mineral., 81, 1293-1296.
97. Zhong, W., Kingsmith, R.D. and Vanderbilt, D. (1994) Giant LO-TO splitting inperovskite ferroelectrics, Phys. Rev. Lett. 72, 3618-3621.
98. Saghi-Szabo, G. and Cohen, R.E. (1998) First-principles study of piezoelectrcityin PbTiO3, Phys. Rev. Lett. 80, 4321-4324.
99. Stixrude, L., Cohen, R.E. and Singh, D. (1994) Iron at high pressure: Linearizedaugmented plane wave computations in the generalized gradient approximation,Phys. Rev. B 50, 6442-6445.
100. Stixrude, L. and Cohen, R.E. (1995) Constraints on the crystalline structure ofthe inner core: Mechanical instability of BCC iron at high pressure, Geophys. Res.Lett. 22, 125-128.
101. Daw, M.S., Foiles, S.M. and Baskes, M.I.(1993) The embedded-atom method: areview of theory and applications, Mater. Sci. Rep., 9, 251-310.
102. Chantasiriwan, S. and Milstein, F.(1998) Embedded-atom models of 12 cubic met-als incorporating second-and third-order elastic-moduli data, Phys. Rev. B 58,5996-6005.
103. Moriarty, J.A. and Widom, M. (1997) First-principles interatomic potentials fortransition-metal aluminides: theory and trends across the 3d series, Phys. Rev. B56, 7905-7917.
104. Moriarty, J.A. (1994) Angular forces and melting-in BCC transition metals: a casestudy of molybdenum, Phys. Rev. B 49, 12431-12445.
105. Moriarty, J.A. and Phillips, R.(1991) First-principles interatomic potentials fortransition-metal surfaces, Phys. Rev. Lett. 66, 3036-3039.
106. Moriarty, J.A. (1990) Analytic representation of multi-ion interatomic potentials

63
in transition metals, Phys. Rev. B 42, 1609-1628.107. Wasserman, E., Stixrude, L. and Cohen, R.E. (1996) Thermal properties of iron
at high pressures and temperatures, Phys. Rev. B 53, 8296-8309.108. Cohen, R.E., Fei, Y., Mazin, I.I. and Isaak, D.G.(1998) Magnetic collapse and the
behavior of transition metal oxides: FeO at high pressures, in R. Wentzcovitch, R. J.Hemley, W. J. Nellis, and P. Yu (eds.), High-Pressure Materials Research, MaterialsReseach Society Proceedings, Vol. 499, Materials Research Society, Pittsburgh, PA,in press.
109. de Wijs, G.A., Kresse, G. and Price, G.D. (1998) The viscosity of liquid iron atthe physical conditions of the Earth’s core, Nature 392, 805-807.
110. Steinle-Neumann, G., Stixrude, L. and Cohen, R.E. (1998) First-Principles Elas-tic Constants for the hcp Transition Metals Fe, Co, and Re at High Pressure,submitted for publication.
111. Soderlind, P., Moriarty, J.A. and Wills, J.M. (1996) First-principles theory of ironup to earth-core pressures–structural, vibrational, and elastic properties, Phys.Rev. B, 53, 14063-14072.
112. Singh, A.K., Mao, H.-K., Shu, J. and Hemley, R.J. (1998) Estimation of single-crystal elastic moduli from polycrystalline X-ray diffraction at high pressure: Ap-plication to FeO and iron, Phys. Rev. Lett. 80, 2157-2160.
113. Stixrude, L. and Cohen, R.E. (1995) High pressure elasticity of iron and anisotropyof Earth’s inner core, Science 267, 1972-1975.
114. Schober, H.R. and Dederichs, H. (1981) Landolt Bornstedt, New Series III,Springer.
115. Manghnani, M.H., Katahara, K. and Fisher, E.S. (1974) Ultrasonic equation ofstate of rhenium, Phys. Rev. B 9, 1421-1431.
116. Hazen, R.M. and Jeanloz, R. (1984) Wuestite (Fe1−xO): A review of its defectstructure and physical properties., Rev. Geophys. Space Phys. 22, 37-46.
117. Mott, N.F. (1990) Metal-Insulator Transitions, Taylor Francis, New York.118. Fillippi, C., Singh, D.J. and Umrigar, C.J. (1994) All-electron local-density and
generalized-gradient calculations of the strutural properties of semiconductors,Phys. Rev. B 50, 14947-14951.
119. Sawada, H., Hamada, N., Terakura, K. and Asada, T. (1996) Orbital and spinorderings in YVO3 and LaVO3 in the generalized gradient approximation, Phys.Rev. B 53, 12742-12749.
120. Anisimov, V.I., Elfimov, J.S., Korotin, M.A. and Terakura, K. (1997) Orbital andcharge ordering in Pr1−xCaxMnO3 (x = 0 and 0.5) from the ab initio calculations,Phys. Rev. B 55, 15494-15499.
121. Fleck, M., Liechtenstein, A.I., Oles, A.M., Hedin, L. and Anisimov, V.I. (1998)Dynamical mean-field theory for doped antiferromagnets, Phys. Rev. Lett. 80,2393-2396.
122. Lichtenstein, A.I. and Katsnelson, M.I. (1998) Ab initio calculations of quasipar-ticle band structure in correlated systems: LDA++ approach, Phys. Rev. B 57,6884-6895.
123. McMahan, A.K., Huscroft, C., Scalettar, R.T. and Pollock, E.L. (1998) Volumecollapse in the rare earth metals, Journal of Computer Aided Materials Design, inpress.
124. Towler, M.D., Hines, R.I., Allan, N.L., Braithwaite, M. and Mackrodt, W.C. (1994)Equations of state for polar solids at high pressures and elevated temperatures, AIPConf. Proc. 309, 129-132.
125. Mackrodt, W.C., Harrison, N.M., Saunders, V.R., Allan, N.L., Towler, M.D., Apra,E. and Dovesi, R. (1993) Ab initio Hartree-Fock calculations of CaO, VO, MnOand NiO, Phil. Mag. A 68, 653-666.
126. Anisimov, V.I., Zaanen, J. and Andersen, O.K. (1991) Band theory and Mottinsulators: Hubbard U instead of Stoner I, Phys. Rev. B 44, 943-954.
127. Anisimov, V.I., Solovyev, I.V., Korotin, M.A., Czyzyk, M.T. and Sawatzky, G.A.

64
(1993) Density functional theory and NiO photoemission spectra, Phys. Rev. B48, 16929-16934.
128. Mazin, I.I. and Anisimov, V.I. (1997) Insulating gap in FeO: Correlations andcovalency, Phys. Rev. B 55, 12822-12825.
129. Isaak, D.G., Cohen, R.E., Mehl, M.J. and Singh, D.J. (1993) Phase stability ofwustite at high pressure from first-principles LAPW calculations, Phys. Rev. B47, 7720-7731.
130. Hemley, R.J. and Cohen, R.E. (1996) Structure and bonding in the deep mantleand core, Phil. Trans. R. Soc. London A 354, 1461-1479.
131. Sasaki, T. (1996) Lattice distortion of NiO under high pressure, Phys. Rev. B 54,R9581-R9584.
132. Dufek, P., Blaha, P. and Schwarz, K. (1995) Theoretical investigation of thepressure-induced metallization and the collapse of the antiferromagnetic state ofNiI2, Phys. Rev. B 51, 4122-4127.
133. Pasternak, M.P., Taylor, R.D., Chen, A., Meade, C., Falicov, L.M., Giesekus, A.,Jeanloz, R. and Yu, P.Y. (1990) Pressure induced metallization and the collapseof the magnetic state in the antiferromagnetic insulator NiI2, Phys. Rev. Lett. 65,790-793.
134. Bargeron, C.B., Avinor, M. and Drickamer, H.G. (1971) The effect of pressureon the spin state of iron (II) in manganese (IV) sulfide, Inorganic Chemistry 10,1338-1339.
135. Drickamer, H.G. and Frank, C.W.(1973) Electronic Transition and the High Pres-sure Chemistry and Physics of Solids, Chapman and Hall, London.
136. Cohen, R.E., Mazin, I.I. and Isaak, D.G. (1997) Magnetic collapse in transitionmetal oxides at high pressure: Implications for the Earth, Science 275, 654-657.
137. Pasternak, M.P., Taylor, R.D., Jeanloz, R., Li, X., Nguyen, J.H. and McCammon,C. (1997) High pressure collapse of magnetism in Fe0.94O: Mossbauer spectroscopybeyond 100 GPa, Phys. Rev. Lett. 79, 5046-5049.
138. Yagi, T., Suzuki, K. and Akimoto, S. (1985) Static compression of wustite(Fe0.98O) to 120 GPa, J. Geophys. Res. 90, 8784-8788.
139. Shen, Z.X., List, R.S., Dessau, D.S., Wells, B.O., Jepsen, O., Arko, A.J., Barttlet,R., Shih, C.K., Parmigiani, C.K., Ghuang, J.C., Lindberg, P.A.P. (1991) Electronicstructure of NiO: Correlation and band effects, Phys. Rev. B 44, 3604-3626.
140. Zou, G., Mao, H.-K., Bell, P.M. and Virgo, D. (1980) High pressure experimentson the iron oxide wustite (Fe1−xO), Carnegie Inst. Wash. YB 79, 374-376.
141. Knittle, E. and Jeanloz, R. (1986) High-pressure metallization of FeO and impli-cations for the earth’s core, Geophys. Res. Lett., 13, 1541-1544.
142. Fei, Y. and Mao, H.-K. (1994) In situ determination of the NiAs phase of FeO athigh pressure and temperature, Science 266, 1668-1680.
143. Mazin, I.I., Fei, Y., Downs, J.W. and Cohen, R.E. (1998) Possible polytypism inFeO at high pressures, Amer. Mineral. 83, 451-457.
144. Watanabe, H. (1966) Operator Methods in Ligand Field Theory, Prentice-Hall,Englewood Cliffs, New Jersey.
145. Burns, R.G. (1993) Mineralogical Applications of Crystal Field Theory, CambridgeUniversity Press, Cambridge.
146. Mattheiss, L.F. (1972) Electronic structure of the 3d transition-metal monoxides.I. Energy-band results, Phys. Rev. B 5, 290-306.
147. Mattheiss, L.F. (1970) Crystal-field effects in the tight-binding approximation:ReO3 and perovskite structures, Phys. Rev. B 2, 3918-3935.
148. Mattheiss, L.F. (1972) Electronic structure of the 3d transition-metal monoxides.II. Interpretation, Phys. Rev. B, 5, 306-315.
149. Hamalainen, K., Kao, C.-C., Hastings, J.B., Siddons, D.P., Berman, L.E., Sto-janoff, V. and Cramer, S.P. (1992) Spin-dependent x-ray absorption of MnO andMnF2, Phys. Rev. B 46, 14274-14277.


















