
asq 13485.ppt
32
ISO-13485:2003 by: Aaron Lupo
-
Upload
darren-tan -
Category
Documents
-
view
357 -
download
3
description
ISO 13485
Transcript of asq 13485.ppt

ISO-13485:2003by: Aaron Lupo

Objectives Overview of ISO 13485:2003 Background / History of the new standard Structure Permissible exclusions from the standards Regulatory/Industry/International
acceptance

Why did they have to change it? Current ISO 13485:96 standard is based on
ISO 9001:94. As of December 15, 2003 ISO 9001:94 will be
obsolete Not recognized by oversight bodies
ISO 13485:96 would be modeled after an antiquated approach.
It was time. ISO standards are reviewed every 5 years

Why not adopt ISO 9001:2000 ? The Medical Device Industry did not agree with all
the changes to ISO 9001:2000 Harmonizing with the QSR (GMP) requirements ISO 9001:2000 places an emphasis on Continual
Improvement and Customer Satisfaction Harmonizing EN 4600X Standards with ISO 1348X Not user friendly Lacked Process orientation Could not tailor ISO to scope of registration
(service)

What they wanted to accomplish in the new revision
Useable by organizations of all sizes Specific to the Medical Device Community Easily Understood Compatible with “other” management systems
9001:2000, ISO 14001 Direct relationship with the activities involved
in running the business

ISO 13485 Series Standards - QMS
ISO 9000: Quality management system fundamentals and vocabulary Except as noted in section 3 of 13485
ISO 13485: Quality Management System Requirements combines ISO 13485 and 13488 limitations will be indicated on certificate
ISO/TS 14969: Quality Management Systems-Medical Devices-Guidance on the Application of ISO 13485

What has Changed?ISO 13485 now has a totally new structure,
‘20 elements’ is too linear, Adopted ‘process’ approach Emphasis is now on regulatory requirements for a quality
management system rather than a quality assurance system. Increased attention to production of a conforming product and
delivery of a conforming service is included in and is part of the quality management system.
There is now only one management system requirement standard, i.e. ISO 13485, where previously there were two
requirement standards: ISO 13485 and ISO 13488.

2003 Changes Continued Went from 20 elements to 8
Elements 4-8 contain all the requirements
Emphasis on Consistency through Documentation Does Allow for permissible exclusions
Exclusions need justification!
Organization replaces supplier to refer to the company implementing the standard
Supplier replaces subcontractor when referring to companies that your company purchases goods and services from

2003 Changes - Continued Top Management must be involved in
establishing and communicating the purpose and direction of the organization The management system begins and ends with
objectives established by Top management Top management – responsibility for profit or loss*
Top Management needs to create an atmosphere in which people are not afraid to become involved, and desire to help the company meet its goals and objectives

Why Can’t the FDA change Part 820 to harmonize it with 9000:2000
The FDA and medical device regulators take issue with following ISO 9001:2000 philosophies:
Customer Satisfaction- goes above and beyond the safety and efficacy of medical devices. Evaluating customer satisfaction beyond safety is not FDA’s purpose
Continuous Improvement- done simply for the sake of operating more efficiently is outside the scope of medical device regulatory agencies.
Documentation- documentation is needed to know what a company intends to do and what they actually did- ISO 9001:2000 requires less documentation than does 13485:2003
Process Approach- FDA does not feel its necessary for companies to change the structure of their documentation. FDA is assuming the purpose of the change in ISO is a change
in documentation and that companies do not already use the process approach.

FDA/QSR harmonization with the ISO Standards The FDA is planning on some level of harmonization
with ISO 13485 in late 2002 – early 2003. Provided that ISO 13485 will remain more true to the medical
device/regulatory viewpoint
Many other countries rely on ISO standards in regulating medical devices. Promotes International Consistency.
FDA and device regulatory agencies from other countries can more readily rely on one another's inspections and exchange inspection reports if the quality system requirements are similar. Very important for establishing MRAs and MOUs between
countries.

Why Would Medical Device Manufacturers want to maintain ISO 9001:2000 Certification as well as 13485:200X
Customers (Doctor’s, Hospitals, OEM’s) perceive a level of security in knowing they are buying from a manufacturer that has an ISO 9001 certified system.
Helps companies obtain product certification CE mark, Companies do not have to be ISO certified to get CE
Mark Depending on the product or service companies could opt
for ISO 17025

Permissible Exclusions - Allowable
(7.5.3) Identification and traceability - only partially applicable where there is no specific traceability requirement for the organization’s product
(7.5.2) Customer property - Nothing is provided from the customer to the supplier in the realization of its products or processes
(7.6) The organizations needs no measuring and test equipment to provide evidence of conformity

Differences:

Guidance:ISO 14969:200X will be the guidance document for implementation to 13485:2003
Section 3 of ISO 13485 has additional Terms and Definitions not included in ISO 9000:2000
ISO 9000:2000 and ISO 9004:2000 used as “consistent pair”
IAF Guidance documents www.iaf.nu

Differences: Section 4.1 13485: "Maintain the Effectiveness of
these processes" Records that you are meeting requirements
and stated objectives 9001:2000: “Continual Improvement of
these Processes“

Differences: Section 4.2.3 Organization must define the period for
retention of obsolete documents Duration must be for the life of the device or as specified by relevant regulatory
requirements (GMP/GLP) 9001:2000 Does not specify retention times

Differences: Section 4.2.4Additional Requirement in Section 4.2.4 13485:2003 that are not in 9001:2000 "Records shall be established and maintained to provide evidence of
conformity to requirements and of the effective operation of the quality management system. records shall remain legible, readily identifiable and retrievable. A documented procedure shall be established to define the controls needed for the identification, storage, protection, retrieval, retention time and disposition of records.
Records from suppliers which demonstrate conformance to specified requirements and the effective operation of the quality management system shall be maintained. The organization shall retain the records for a period of time at least equivalent to the lifetime of the medical device as defined by the organization, but not less than 2 years from the date of last shipment from the organization.
NOTE National or regional regulations may require a period longer than 2 years. The organization shall establish and maintain a record for each batch of medical devices that provides traceability
to the extent specified in 7.5.3 and identifies the quantity manufactured and quantity approved for distribution. The batch record shall be verified and approved.
NOTE A batch may be a single medical device."

Differences: Section 5 Section 5.1 of 13485:2003 does not require "Continually
Improving its effectiveness" ISO 9001:2000 does.
Section 5.2 of 13485:2003 focus is on meeting the customers needs 9001:2000 is focused on meeting and exceeding the customers
needs.
Section 5.3 of 13485:2003 emphasizes maintaining the system 9001:2000 talks about continual improvement

Differences: Section 5 Section 6.2.2 of 13485:2003 Requires a
procedure for training where as ISO 9001:2000 does not.
Section 6.4 Work Environment of 13485:2003 is more detailed (i.e. health records, cleanliness of clothing and personnel, environmental monitoring) than the same section in 9001:2000

Differences: Section 7 Section 7.3 Design and Development of 13485:2003
references ISO 14971 (Risk Analysis) 9001:2000 does not.
Section 7.5.1 of 13485:2003 has added sub-clause G, which talks about packaging and labeling.
Section 7.5.1.1 Cleanliness of Product and contamination control (Cleaning of product) is added for 13485:2003
Section 7.5.1.2 Installation (Installing the Device) is added for 13485:2003
Section 7.5.1.3 Servicing (After the Device is installed) is added for 13485:2003

Differences: Section 7 - cont’d Section 7.5.2 Validation of processes for production and service
provision- additional requirements for 13485: “The organization shall establish and maintain documented
procedures for the validation of the application of computer software (and changes to such software and/or its application) for production and service operations and measuring and monitoring operations (see 8.2) that affect the ability of the product to conform to specified requirements. Such software applications shall be validated prior to initial use. The results of validation shall be recorded (see 4.2.4).”
Particular requirement for sterile medical devices: “The organization shall subject the medical device to a validated
sterilization process and record all the process parameters of the sterilization process (see 4.2.4).”

Differences: Section 7 - cont’d Section 7.5.3 Identification and Traceability- additional requirements
for 13485: Particular requirements for active implantable medical devices and
implantable medical devices: a) “When defining the extent of traceability, the organization shall
include all components and materials used, and records of the environmental conditions when these could cause the medical device not to satisfy its specified requirements.”
b) “The organization shall require that its agents or distributors maintain records of the distribution of medical devices with regard to traceability and that such records are available for inspection.”
c) “The organization shall ensure that the name and address of the shipping package consignee is recorded (see 4.2.4).”

Differences: Section 7 - cont’dSection 7.5.5 Preservation of product- additional requirements for 13485: ”The organization shall establish and maintain documented procedures for the control of product with a limited shelf-life or requiring special storage conditions. Such special storage conditions shall be controlled and recorded.” (FIFO)

Differences: Section 8 Section 8.1 Measurement, analysis and
Improvement- additional Requirements for 13485:
If statistical techniques are used, organization shall establish and maintain documented procedures to implement and control their application.

Differences: Section 8 - cont’d Section 8.2.1 Customer Feedback-
Additional Requirements for 13485: “The organization shall establish and maintain a documented
feedback system to provide early warning of quality problems and for input into the corrective and preventive action system [see 7.2.3c)].”
“If regulations require the organization to gain experience from the post-production phase, the review of this experience shall form part of the feedback system (see 8.5.1).”

Differences: Section 8 - cont’dSection 8.2.4 Monitoring and measurement of Product- additional requirements for 13485:Particular requirement for active implantable devices and implantable devices:“The organization shall record (see 4.2.4) the identity of personnel performing any inspection or testing.”
Section 8.3 Control of nonconforming product- additional requirements for 13485:“If product needs to be reworked (one or more times), the organization shall document the rework in a work instruction that has undergone the same authorization and approval procedure as the original work instruction. Prior to authorization and approval, a determination of any adverse effect of the rework upon product shall be made and documented.”

Requirements for Documents – 92K ISO 9001:2000 requires a minimum of 6
procedures. Quality Manual 4.2.3 Control of Documents 4.2.4 Control of Records 8.2.2 Internal Audit 8.5.2 Corrective Action 8.5.3 Preventive Action
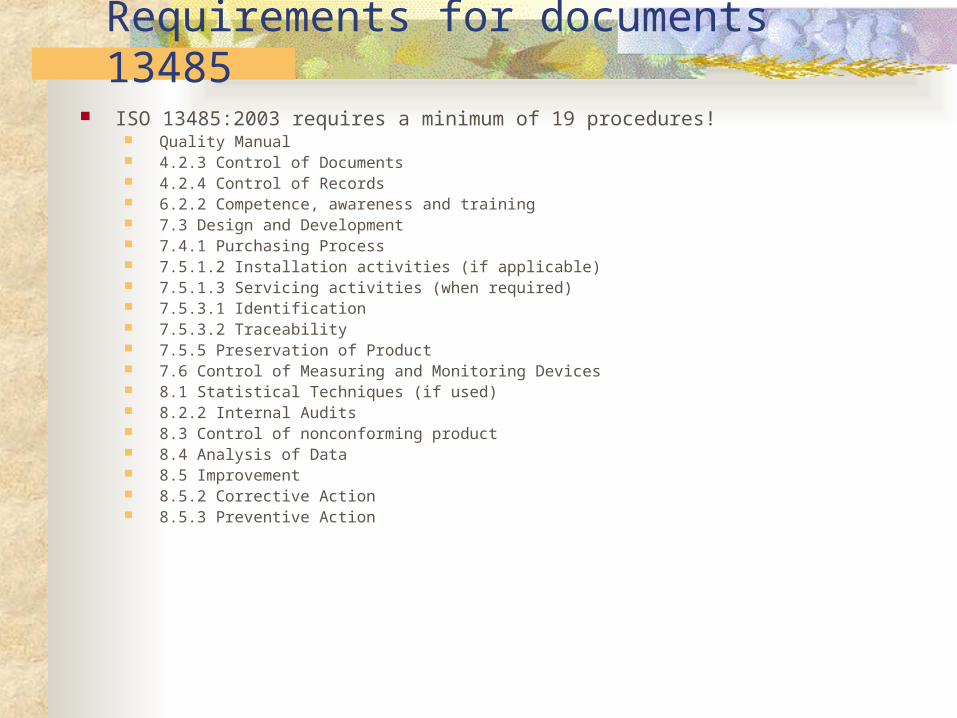
Requirements for documents 13485 ISO 13485:2003 requires a minimum of 19 procedures!
Quality Manual 4.2.3 Control of Documents 4.2.4 Control of Records 6.2.2 Competence, awareness and training 7.3 Design and Development 7.4.1 Purchasing Process 7.5.1.2 Installation activities (if applicable) 7.5.1.3 Servicing activities (when required) 7.5.3.1 Identification 7.5.3.2 Traceability 7.5.5 Preservation of Product 7.6 Control of Measuring and Monitoring Devices 8.1 Statistical Techniques (if used) 8.2.2 Internal Audits 8.3 Control of nonconforming product 8.4 Analysis of Data 8.5 Improvement 8.5.2 Corrective Action 8.5.3 Preventive Action

13485:2003 Certification = certification to ISO 9000:2000 ISO 13485 is a stand alone standard
It is based on ISO 9001:2000. Primary objective of ISO13485 is to facilitate
harmonized medical device regulatory requirements.
Includes some particular requirements for medical devices and excludes some of the ISO 9001:2000 requirements
These exclusions prevent users from claiming conformity with ISO9001:2000!

13485:2003 Registration Companies do not have to re-structure their
documentation to meet 13485:2003 format it would make for more efficient audits if a company did or at least
provided a cross-reference of documents from the old standard to the new
Certified companies will have 9 months from the release date of ISO 13485:2003 to update to the new revision Companies can be certified to ISO 13485:96 until 12/15/03.
Oversight bodies include SCC and UKAS ANSI/RAB developing accreditation scheme second quarter 2002
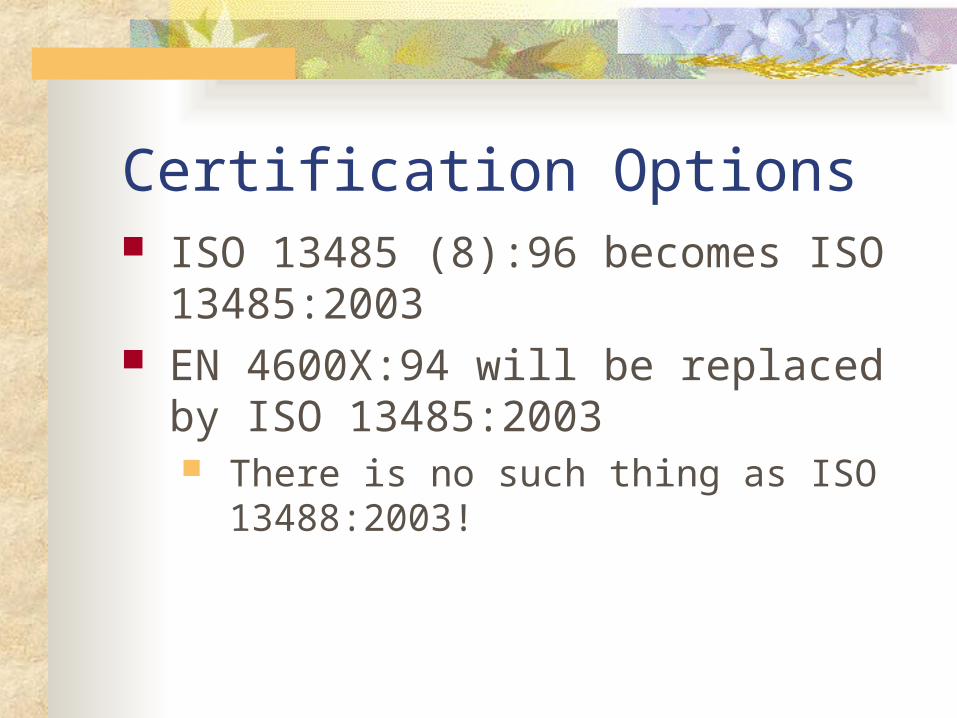
Certification Options ISO 13485 (8):96 becomes ISO
13485:2003 EN 4600X:94 will be replaced by ISO
13485:2003 There is no such thing as ISO 13488:2003!


















