
71st ICREA Colloquium - The unbearable lightness of being (a protein) by Xavier Barril
28
e unbearable lightness of being (a protein) Xavier Barril ICREA Research Professor, Barcelona University 71 st ICREA Colloquium Barcelona, 14 th June 2016
-
Upload
mayi-suarez -
Category
Science
-
view
46 -
download
0
Transcript of 71st ICREA Colloquium - The unbearable lightness of being (a protein) by Xavier Barril

The unbearable lightness of being (a protein)
Xavier Barril ICREA Research Professor, Barcelona University
71st ICREA Colloquium Barcelona, 14th June 2016


Protein?

Proteins come in all shapes and forms…
…because form follows function


Structure Determination: X-ray Crystallography
PDBtodayTotal:119480X-ray:106777(89.3%)
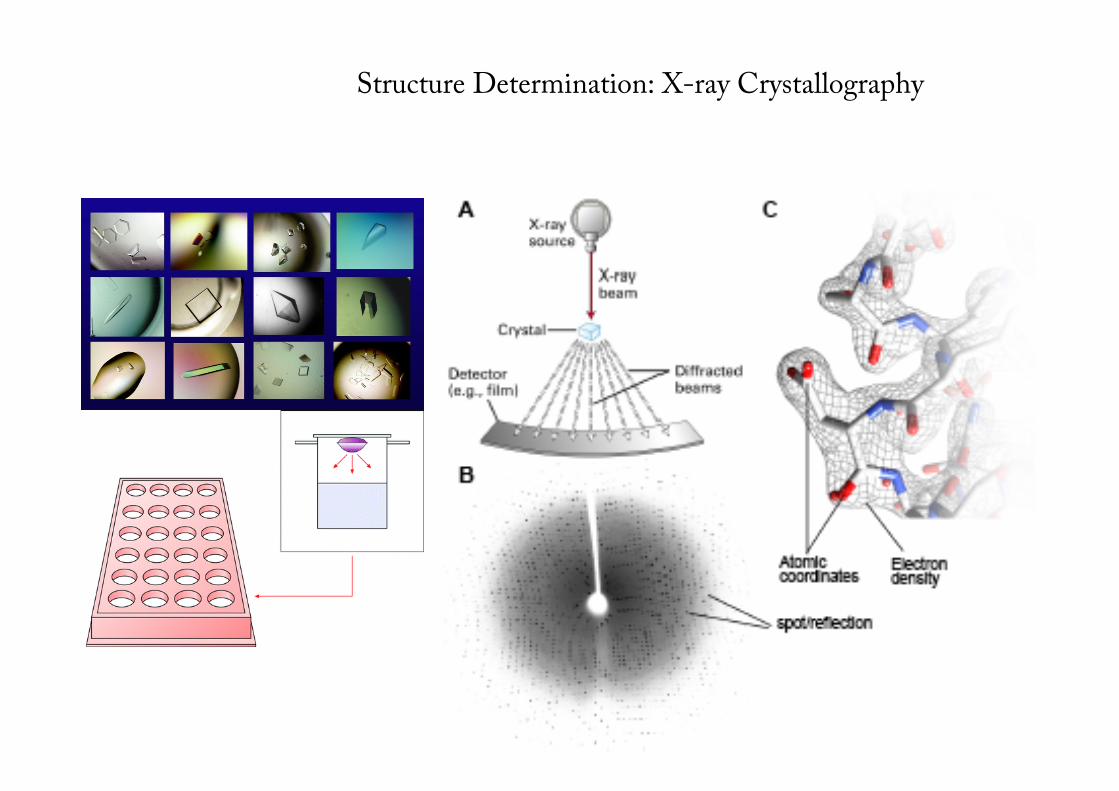
Structure Determination: X-ray Crystallography

Structure-Based Drug Design

A New Hope
Progressinsciencedependsonnewtechniques,newdiscoveriesandnewideas,probablyinthatorder.—SydneyBrenner






Score
Score
Score
A
B
C
Score
Score
Score
A B C
!Ei=-kBT·ln(Pi)Ei=-kBT·ln(Pi)
NATURECHEMISTRY|VOL6|JULY2014
SBDD: More is less

Finding Hot Spots: MDmix

High-Throughput Docking
http://rdock.sourceforge.net
ü Fast (~10k cpds x CPUday) ü Free & Open Source ü Advanced features:
• Limited Protein flexibility • Interfacial water molecules
ü User control: • Pharmacophoric restraints • Tethered scaffold docking

Dynamic Undocking
• Different number of dynamic undocking runswere tested (2, 8 or 22). As shown in the picturefor CDK2, increasing the number of runsimproved the results. However, the differencebetween 8 and 22 runs is really small.
Workflow & Details
On one hand, SMD has already been used todifferentiate between true binders and inactivebinders. Colizzi and colleagues [4] pulled out inhibitorsfrom the protein binding pocket and observed that thedifferent energy profiles obtained allowed them toidentify active compounds.On the other hand, we studied in the past the effect ofthe local environment and water molecules onshielded hydrogen bonds as structural determinants inbinding kinetics, where an early unbinding event caninfluence the whole dissociation process [5].Combining this two ideas, we decided to study if thepotency of a ligand could be related to the breaking ofa key hydrogen bond. Thus, we use model receptorsthat comprise the residues around the key interactionand pull the two atoms involved in the hydrogen bondfrom 2.5 to 5 A ("dynamic undocking"), the workinvolved in this breaking will then used to calculatethe free energy of the "quasi-bound" state [6].The main benefits of this approach are the reduction ofcomputational time (small chunks are 10 times smaller
than full proteins) and the simplification of thereaction coordinate.However, as we are focusing in a particular interactioninstead of a full dissociation, there must be a keyanchoring point that explains most of the activity.
AbstractIt is known that the productivity of the pharmaceutical industry is decreasingyear after year, as the investments are growing and the number of new chemicalentities remains constant [1,2]. Hence, alternative methods that improve andincrease the effectiveness of rational drug design are needed.We present a novel approach combining molecular dynamics (MD) and steeredmolecular dynamics (SMD) simulations with free energy (FE) methods. Toovercome main limitations of such low throughput methods our approach isfocused on the breaking of a key interaction point and a reduction of the systemsize, which simplifies the choice of the reaction coordinate and speeds up thecalculations.As the main point is the usage of a reduced portion of the system instead of thewhole protein, it is not possible to recover the full dissociation profile. However,most of the work needed to break a complex is used in the initial stages wherenative hydrogen bonds are broken. We show that the named "quasi-bound" state,defined as the conformation of a ligand that has just broken the key interactionsand obtained by our "dynamic undocking" approach, is good to identify thoseligands with higher resistance to dissociate.A validation of the method has been carried out with cyclin-dependent kinase 2(CDK2) which, in combination with docking [3], yielded very good enrichmentfactors.
Dynamic undocking of protein chunks: applications fordrug discoverySergio Ruiz-Carmona1,2,*, Peter Schmidtke4, Xavier Barril1,2,31Physical Chemistry Dept., Pharmacy Faculty, University of Barcelona. Av. Joan XXIII s/n, 08028 Barcelona, Spain2Biomedicine Institute of the University of Barcelona (IBUB), Barcelona, Spain3Catalan Institution for Research and Advanced Studies (ICREA), Barcelona, Spain4Discngine, Paris, France
*E-mail: [email protected]
Conclusions• SMD had been proposed as a tool for ligand screening, but was impractical
due to computational cost and difficulty in defining the RC.• Approximations introduced: initial step of dissociation explains most of
the activity, small part of the system is sufficient, which increasesefficiency and simplifies choice of RC.
• Potential to become the first automatic VS method based on Free Energycalculations as results are robust to the choice of parameters (system size,pulling velocity, spring constant).
• In a retrospective validation with kinase CDK2 and in combination withmolecular docking, we obtained very good enrichment factors.
• Prospective application in system HSP90: novel ligands were identified andSPR, NMR and X-ray confirmed positive results.
References[1] J. P. Overington et al. Nat Rev Drug Discov, 2006.[2] A. L. Hopkins and C. R. Groom. Nat Rev Drug Discov, 2002.[3] Ruiz-Carmona, S. et al. PloS Comput Biol, 2014.[4] Colizzi, F. et al. J Am Chem Soc, 2010.[5] Schmidtke, P. et al. J Am Chem Soc, 2011.[6] Jarzynski, C. Phys Rev E, 1997.
Acknowledgements•We want to thank Vernalis fortheir collaboration.
• All input ligands are preparedwith Schrödinger tool ligprep.• rDock is the docking tool andhas been guided using restraints,forcing all ligands to fulfill thedefined key interaction point.• For each ligand, the resultingbinding modes with best scorewill act as input for next stages.
• A set of active fragments was exported from PDB structures and a set of decoys wasgenerated using the DUDe decoy generator.• rDock was used to generate all the starting binding modes, forcing all ligands to fulfill thedefined key hydrogen bond with the hinge region of CDK2.• About 1100 molecules were run with Amber for 10 ns of free MD simulation and 12replicas of steered MD simulations.
Application: CDK2
• Active ligands are shown in redand decoys in black or gray (forbest 25% docking poses).The enrichment of each sector isalso shown, highlighting thesector with the best enrichmentfactor.• The lines correspond to thedensity functions and are colouredaccording to the set of the mainplot they represent.
• MD simulations: 1000 steps of minimization followedby 1 ns of equilibration and steps of 1 ns of MDsimulations• SMD simulations: starting from each ns of MD, 2SMD are run at 300K and 325K, respectively. Thespring constant, pulling velocity, displacement andreaction coordinate have already been defined in MOEsetup stage.• In all MD and SMD runs, the chunk is restrained witha force of 1 kcal/mol·A2 to avoid denaturation.• The GPU time required for each ligand and stage is20 min for minimisation and equilibration, 15 min foreach ns of MD simulation and 10 min for each SMDrun.
• The system is reduced to get aprotein chunk around a keyinteraction point. All residues at adistance threshold of 6A are keptand a manual inspection adds anyimportant residues in the cavityfurther than this threshold.• The size of the protein chunk is 10times smaller than the full protein.
• The protein chunk is prepared with MOE.The atom which acts as the key anchoringpoint is selected and the reactioncoordinate is defined.• MOE svl scripts automate the process:MD and SMD parameters can be changedin this stage: MD queueing template, SMDlength (500 ps), SMD displacement (2.5 A)or SMD spring constant (50 kcal/mol·A2).• Input ligands are stored in a MOEdatabase and they are automaticallyparameterized using forcefield PFROSSTwith tleap.
• Jarzynski's equality isused to calculate freeenergy differences in anon-equilibrium processbetween two differentequilibrium states.• Due to ourapproximation, wecalculate the minimumvalue of each replicamaximum value, instead ofaveraging all the replicas(orange line in plot).
SettingsSampling
• As the number of runs increased, the dissociation energyprofiles increased their standard deviation, finding morefavourable dissociation pathways.• Concept of minimum of the maximum values of allreplicas.
• The predicted DG for the CDK2 decoys decreased as the numberof runs increased, whereas for the active the change was smaller.
Pulling velocity
Background
• Average predicted DG in kcal/mol for each set and number of runs.
• We also tested if softening the pulling velocity in SMDsimulations improved the results: the correlation wasalmost 1 between 5 A/ns and 1.25 A/ns.

CO-0001
Conc. (µM)
ΔTm
0 500 1000 15000
2
4
6
Virtual
Library
VirtualHits

Nucleus
ER
Correct Folding
Ribosome
Protein Function
Normal gene
Target Organelle
RareDiseases
HEALTHY STATE

Nucleus
ER
Ribosome
Protein Deficiency
Mutated gene
Target Organelle
Proteasomal degradation
Incorrect Folding
RareDiseases
DISEASE STATE

Nucleus
ER
Ribosome
Protein Inhibited
Mutated gene
Target Organelle
PharmacologicalChaperones
Correct Folding
Proteasomal degradation
Substrate-compeLLvepharmacologicalchaperone
PC THERAPY
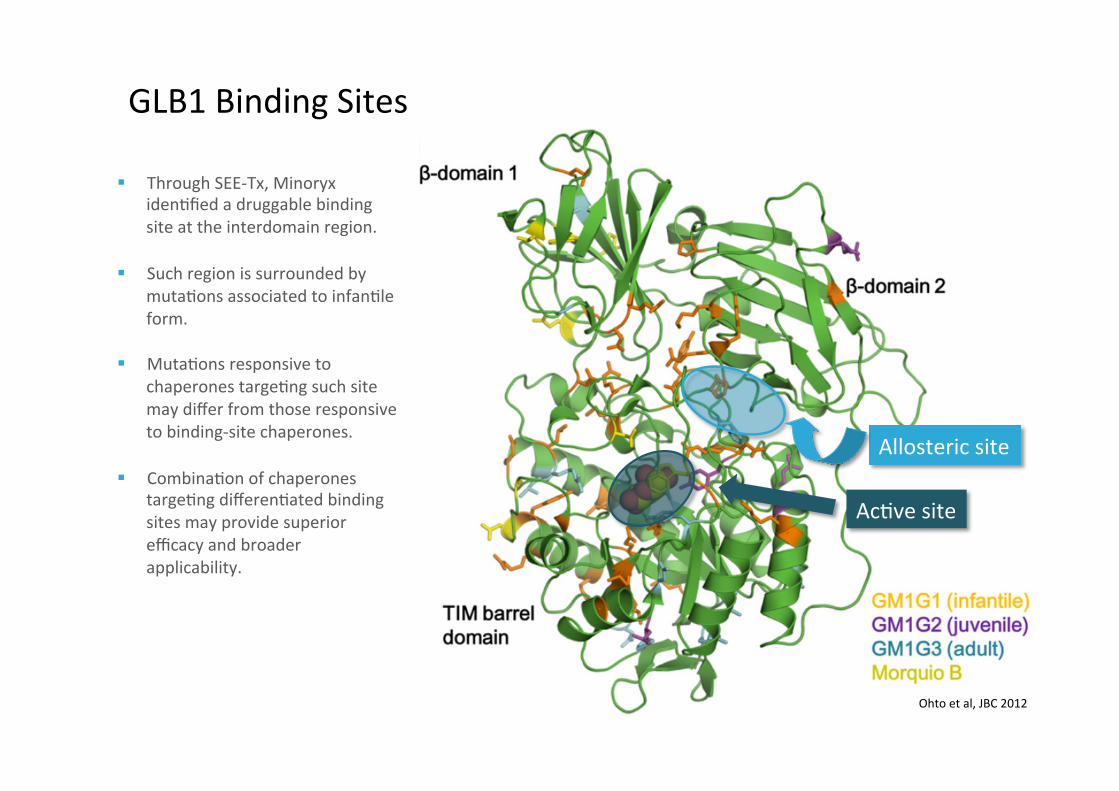
GLB1BindingSites
§ ThroughSEE-Tx,MinoryxidenLfiedadruggablebindingsiteattheinterdomainregion.
§ SuchregionissurroundedbymutaLonsassociatedtoinfanLleform.
§ MutaLonsresponsivetochaperonestargeLngsuchsitemaydifferfromthoseresponsivetobinding-sitechaperones.
§ CombinaLonofchaperonestargeLngdifferenLatedbindingsitesmayprovidesuperiorefficacyandbroaderapplicability.
Ohtoetal,JBC2012
AcLvesite
Allostericsite

Virtual
Library
~1.5M cpds
AllostericSite
VirtualHits 140tested
CatalyLcSite
VirtualHits 50tested
DSF
1AcLveSeries
2AcLveSeries
HitIdenLficaLon&PrimaryScreening
Mel,ngcurve
ΔTm
CO-0001
Conc. (µM)
ΔTm
0 500 1000 15000
2
4
6
DGJKD=242±40µM
KD~240µMKD~2µM
DSF
KD~15µM
DGJΔTm@100µM=2.3ºC
Compound@30µM
§ Hitseries:Verysmallmolecules(av.Mw=250Da)withexcellentligandefficiency(potencyinmidmicroMrange).
§ TargeLngofnon-exploitedbindingsite.
§ >150patentablenon-sugarlikeNCE’salreadysynthesized.
§ IPfiledonmainseries.
§ Back-upscaffoldsidenLfied.

SeriesOpLmizaLon
Minoryx’scpds.
§ Enzymeenhancement>300%(at10µM)
§ Highlyefficient(MW=270Da)
§ Drug-likeprofile
§ Highin-vivoBBBpenetraLon(Brain/plasma>>1)§ Morethat150compoundshavebeen
synthesized.
§ ThisallowedidenLfyingseveralacLveseriesandsubseries.
§ MostpromisingseriesareseriesA1(whichincludescpd#3frompreviousslide)andseriesC1(cpd#8).
§ SeriesC1,showincreasedcell-basedpotencyaswellopLmizedproperLesforBBBpenetraLon.
EnzymeenhancementupontreatmentofCOScellstransientlytransfectedwithmutanthGLB1(DGJ:substratecompeLLvePCTwithmicroMaffinity;NN-DGJ:SubstratecompeLLvewithnMaffinity;#3,4and10:Minoryx’snon-compeLLvePCTs;SeriesA1;SeriesC1)
T420KGM1type3

Non-compeLLveHitseries:InhibiLonofthelysosomalenzymeinlysatesfromnormalfibroblasts
ü Control1andControl2exhibiteddose-responseinhibitoryacLvityontheenzymewithlysatesfromnormalhumanfibroblasts,aspreviouslydescribed.
ü Incontrast,Minoryx�scompoundsdonotexhibitanyinhibitoryacLvityontheenzyme.
Allostericchaperonesdonotinhibit
Control1
Control2
IC50=17µM
IC50=0.1µM
Non-compeLLvechaperones


Acknowledgements
Group Members • Sergio Ruiz – rDock; DUck • Carles Galdeano – Fbw7, BRD4 • Serena Piticchio – Fragments • Guillermo Gutiérrez – Automaton, Flex • Míriam Martínez – BRD4, Fbw7
Former Group Members • Daniel Álvarez – MDmix • Peter Schmidtke – DUck Collaborators • F. Javier Luque • Minoryx • Vernalis


















