
08 Hemoglobin 2013
74
Hemoglobin & Myoglobin Lippincott’s Chap. 3 (The Hemoglobinopathies) Genetics in Medicine, Chapter 11
description
hemoglobin
Transcript of 08 Hemoglobin 2013

Hemoglobin & Myoglobin
Lippincott’s Chap. 3
(The Hemoglobinopathies) Genetics in Medicine, Chapter 11

Objectives • Structure of hemoglobin (Hb) and myoglobin (Mb) • Heme binding site and the regulation of oxygen binding. State the
major structural differences between the oxygenated and deoxygenated forms of hemoglobin.
• O2 & CO2 transport and pH regulation. • Allosteric regulation O2, BPG, CO2, and H+ (the Bohr effect). • Cooperativity in O2 delivery. Fetal hemoglobin. • Right vs left shifts in the O2 binding curves - ie. a decrease or an
increase in affinity? • Oxygen dissociation curves, fractional saturation (Y) vs partial
pressure of oxygen (pO2). • Hemoglobinopathies

Hemeproteins
• Hemoglobins are globular proteins found in high concentration in red blood cells.
• They bind and transport O2 from lungs to capillary bed of tissue vascular system.
• Also transport metabolic CO2 and H+ from tissues to lungs.

Hemeproteins • Heme is a specialized prosthetic group (cofactor
or coenzyme) composed of protoporphyrin IX and ferrous iron (Fe2+)
• Heme synthesized in mitochondria (thus not in RBC) and cytosol
• Hemeprotein: apo vs holoprotein – cytochromes >> e- transfer (mito) – catalase >> H2O2 breakdown (peroxisome) – Hb & Mb >> O2 binding and transport


• Hemoglobin & myoglobin - two most abundant heme proteins in the human body.
• Fe held in place by 4 pyrrole N’s and 2 additional bonds ↑↓ plane of the ring.
• Free in solution this is typically H2O. • In Hb & Mb one of these bonds is due to the
proximal histidine (F8) imidazole on F helix, the other when O2 bound in OxyHb.
• The interaction with O2 stabilized by the distal histidine (E7) on E helix.

Mb 1st protein structure determined from a crystal (KendrewNature, 1958) globular proteins are comprised of α–helix with hydrophobic residues inside and polar and charged residues on the surface.

Myoglobin • Present in heart & skeletal muscle • O2 reservoir, carrier, ↑ rate of transport • Single chain (16 kD) 80% α-helical protein
similar (in sequence & fold) to Hb chains. • 8 helices (A to H) that each terminate with
proline, β-bends and loops • Nonpolar residues on the interior • Polar/charged residues mostly on the
surface for H-bonding with water.

Myoglobin • Heme binds in a crevice between F- and E-
helix that is lined with nonpolar residues. • 80 apolar interactions from 18 residues form
heme pocket, thus expulsion of solvated water & hydrophobic stabilization.
• Exception are two histidines, proximal and distal, that bind to Fe2+ and O2.
• Myoglobin creates a microenvironment for heme that favors reversible O2 binding & only rarely oxidation of ferrous iron (Fe2+) to ferric (Fe3+).

← Myoglobin
Hemoglobin → β-HbA1

Hb & Mb sequences similar • Mb 153 aa’s with 83 invariant in 23 species • α Hb 141 aa’s : β Hb 145 aa’s
– only 15/83 Mb sites invariant with α / β – eg. proximal & distal His
• most changes conservative (nonpolar:nonpolar) • specific changes reflect changes in function • Mb a monomer so surface - H2O interactions • Hb a tetramer so surface - quaternary interactions • changes in α, β, δ, γ chains reflect physiology • same tertiary fold from many different sequences

↓
↓
*
*
*
* * *
*
*

Hemoglobin • Hemoglobin found exclusively in RBC • Function to transport O2 from lungs to capillaries of
peripheral tissues • ~150 gm Hb/L plasma can carry 1.34 ml O2 /gm Hb,
or 150 x 1.34 = 201 ml O2 /L plasma~100x more than plasma (serum) alone (2.3 ml of O2/L).
• Also transports CO2 & H+ (& NO) while buffering the pH of plasma.
• A nitrite reductase, which generates nitric oxide (NO) for regulating hypoxic vasodilation under a variety of physiological and pathological ranges of temperature and pH.

Hemoglobin

Hemoglobin • HbA, major Hb in adults, comprised of 4
polypeptides, 2α & 2β chains (α2β2 ) held by noncovalent interactions.
• Each polypeptide highly α-helical (7 in α & 8 in β) with a heme binding pocket like Mb.
• O2 binding regulated by allosteric effectors (CO2 & H+ & BPG)
• Positive cooperativy binding due to changes in quaternary structure of α2β2 .

Stryer Fig 10.20

Hemoglobin • HbA tetramer α2β2 actually 2 αβ dimers
stabilized by hydrophobic, but also ionic and H-bonding interactions.
• In contrast the dimer:dimer interface is primarily ionic or polar interactions, & these weaker interactions allow the dimer to dimer interface to change with O2 binding.
• T (taut) form or low O2 affinity form – Favored by Deoxy Hb
• R (relaxed) form or high O2 affinity form – Favored by Oxy Hb

HEMOGLOBIN Cooperative O2 Binding Mechanism

Ligand (O2) binding
Heme
Proximal Histidine

O2 Binding • MbO2 ⇔ Mb + O2
• Keq = [Mb] [O2] / [MbO2]
• P50 = [Mb] pO2 / [MbO2]
• pO2 in units of torr or mm Hg
• [MbO2] = [Mb] pO2 / P50
Mb monomer has one heme & thus simple single site, noncooperative high affinity binding (P50 = 1 mm Hg) - hyperbolic when plotted vs O2 mm Hg

Stryer Fig 10.17

Hemoglobin • Hb tetramer α2β2 has 4 heme sites that bind
O2 cooperatively (P50 = 26 mm Hg) - sigmoidal when plotted vs O2 mm Hg
• Cooperativity reflects changes in the dimer:dimer interface upon going from Deoxy to Oxy Hb

D Fig 3.38a


Stryer Fig 10.22

Stryer Fig 10.21

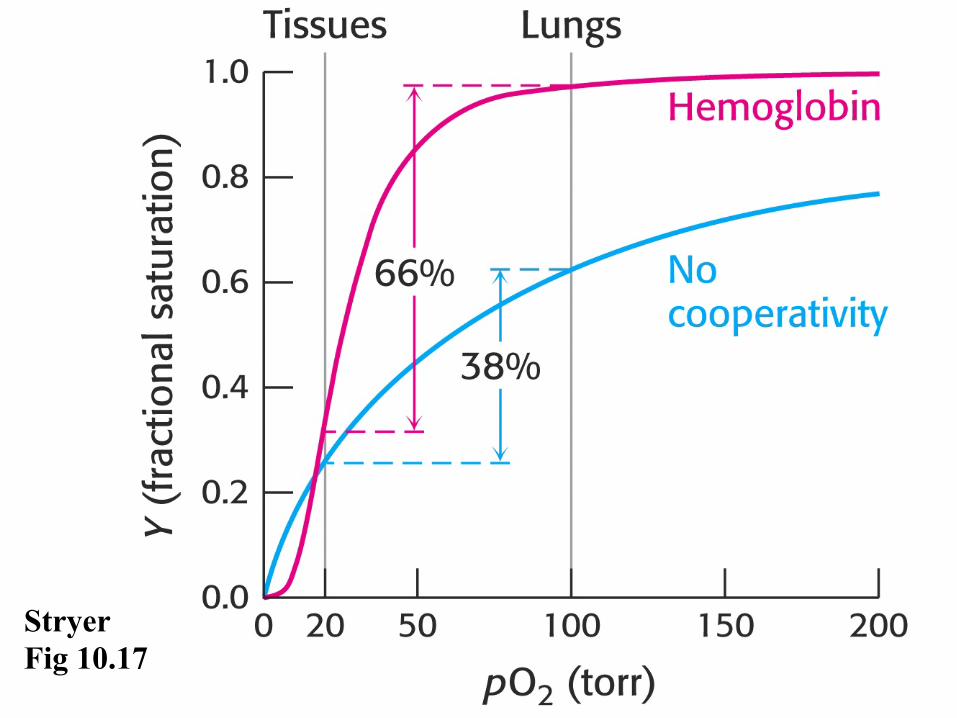
Stryer Fig 10.17

O2 Saturation Y • Y = [MbO2] / [Mb] + [MbO2]
• Y = {[Mb] pO2 / P50 / [Mb] + [Mb] pO2 / P50 } • cancel [Mb] & multiply & divide by {P50}
• Y = pO2 / P50 + pO2
• 1 - Y = P50 + pO2 - pO2 / P50 + pO2
• Y / 1 - Y = pO2 / P50

O2 Saturation Y • Y / 1 - Y = pO2 / P50
• taking log generates Hill equation
• log{Y / 1 - Y} = log pO2 - log P50
• for single site, or noncooperative sites a plot of • log{Y / 1 - Y} vs. log pO2
is linear with a slope of one • for cooperative sites a Hill plot is linear with a maximum slope of nH
• log{Y / 1 - Y} = nH log pO2 - constant

D Fig 3.36


Monod-Wyman-Changeux (MWC Model) All or None Model
Vs Sequential Model (Fig 10.16)

vs Hybrid Hemoglobin Models (Ackers) Sequential until you cross the dimer interface
with two O2’s binding & then MWC
distinguishes tertiary & quaternary conformational transitions

Hemoglobin Allosteria H+, CO2, Temperature, BPG

Bohr effect: inc. [H+] (lower pH) or inc. CO2 favors O2 dissociation (higher P50)

Stryer Fig 10.26
Bohr AAs

If the "Bohr Histidine" has a pK of 6.4, what fraction is protonated at pH 7.4?
A. B. C. D. E. F. G.
0% 0% 0% 0%0%0%0%
A. 1% B. 9.1% C. 33% D. 50% E. 66% F. 90.1% G. 99%

If the "Bohr Histidine" has a pK of 7.7, what fraction is protonated at pH 7.4? A. 1% B. 9.1% C. 33% D. 50% E. 66% F. 90.1% G. 99%
Do these problems

Molecular mechanism is quaternary transitions in Hb that link O2, CO2 and H+ binding.

D Fig 25.7

ADP ↓ ATP
The only way to make ATP in RBC’s, so diverting resources to release more O2 and enhance OxPhos in tissues, costs RBC ATP production.

Stored blood for transfusion loses BPG & with inc affinity becomes a trap for O2 rather than a transporter Why then does blood transfusion help bike racers?

D Fig 25.5
(DeOxy) (Oxy)
HbF binds BPG weakly (due to less positive charge in pocket) and thus has higher affinity for O2 than mother’s HbA (+ DPG).

Stryer Fig 10.23

chronic hypoxia chronic anemia obstructive pulmonary emphysema COPD.
? Consider BPG effect on

D Fig 25.6
Shift to the right, lower affinity for O2, occurs as HbF synthesis replaced by HbA1 synthesis, and thus BPG sensitivity restored.

Stryer Fig 10.24

Stryer Fig 10.25

Hb Classic Allosteric System
O2 - positive homotrophic effector CO2, BPG, H+ - negative heterotrophic effectors

Lung Tissue HbO2 + H+ <==> HHb + O2 CO2 + H2O <==> H+ + HCO3
- ------------------------------------------------------------ HbO2 + CO2 + H2O <==> HHb + HCO3
- + O2

Hb gene structure

Hb Diseases HEMOGLOBIN-OPATHIES Oxidation Low Hb concentrations High Hb concentrations Change in Amino Acids
Mutation ΔP50
Over 330 000 affected infants are born annually (83% sickle cell disorders, 17% thalassaemias). Hemoglobin disorders account for 3.4% of deaths in children less than 5 years of age.

Metmyoglobin & Methemoglobin • Oxidation of iron to ferric (Fe3+) by air,
H2O2, drugs, free radicals, mutation (HbM) • Met-hemoglobin Binds O2 with high
affinity Alters the O2 affinity of other hemes
• 6th coordination site contains H2O instead • reduced ability to release oxygen to tissues
HYPOXIA

Metmyoglobin & Methemoglobin • Blood a bluish or chocolate--‐brown color • HbM NADH methemoglobin reductase
deficient, or His mutations • Transfusion/bone marrow transplant • Ascorbic acid and glutathione enzymes • Supplemental oxygen and methylene blue
– 1% solution (10 mg/ml) 1 to 2 mg/kg administered intravenously
– artificial electron acceptor for NADPH – NADPH is generated via hexose
monophosphate shunt in Mitochondria

Methemoglobin • Methemoglobinemia can also arise in
patients with pyruvate kinase deficiency due to impaired production of NADH –normal tissue oxygen levels
• Congenital methemoglobinemia is seen in patients with abnormal hemoglobin variants such as hemoglobin M (HbM), or hemoglobin H (HbH) and mutations near Heme pocket.

HEMOGLOBIN species

Thalassemia α thalassemia patients have partial or complete defects in α globin production, leading to a relative abundance of β globin chains β globin chains aggregate to form HbH,
(B chains) Heinz bodies small round inclusions within the red cell body
Hb Barts (gamma chains)
Globally, 7% of pregnant women carry beta or alpha zero thalassaemia, or haemoglobin S, C, D Punjab or E, and over 1% of couples are at risk Beta thalassemia major, intermedia, minor (recessive inheritance, hbb)

A Syllabus of Human Hemoglobin Variants (1996) by Titus H.J. Huisman, Marianne F.H. Carver, and Georgi D. Efremov, published by The Sickle Cell Anemia Foundation, lists 693 mutants human hemoglobins affecting O2 binding affinity, cooperativity, Fe+2 oxidation, or stability, either dissociation or aggregation.

Hemoglobinopathies, Thalasemia & Sickle Cell. HbS induces aggregation of Hb, sickling of RBC’s, occlusion of microvascular channels & anemia – 1st molecular disease, predicted by Pauling - Sickle cell anemia is a genetically transmitted (homozygous recessive), chronic, hemolytic disease.

Hemoglobin S

Hemoglobin S

Hb C reduces the normal plasticity of host erythrocytes HETEROZYGOUS Hb AC
28–44% of total hemoglobin (Hb) is HbC, no anemia HbC does not polymerize as readily
Fewer sickle cells observable via peripheral smear Fewer acute vaso-occlusive events.
Individuals with HbS–HbC , HETEROZYGOUS (HbSC) Significant retinopathy, ischemic necrosis of bone, and priapism than those with pure SS disease
Hb C β6 Glu -> Lys

Mutant Hemoglobin (Human) O2 Binding
Turner, G. J. et al, (1992) Mutagenic Dissection of Hemoglobin Cooperativity…….Structure, Function, and Genetics 14:333-350

Hemoglobin Tetramer Cooperativity Map
Turner, G. J. et al, (1992) Mutagenic Dissection of Hemoglobin Cooperativity…….Structure, Function, and Genetics 14:333-350
Ribbon, Backbone Stick, Side chains

Hemoglobin Tetramer

Hemoglobin Tetramer
Dimers Disassociating

Hemoglobin Dimer Interface

Long term marker for diabetic sugar control
Carbamylation Hb A 1c


Questions

Which of the following statements concerning the hemoglobins is correct?
A. Fetal blood has a higher affinity for oxygen than does adult blood because HbF has a decreased affinity for 2,3-BPG.
B. Purified HbF (stripped of 2,3-BPG) has a higher affinity for oxygen than does purified HbA.
C. The chain composition of HbF is α2δ2. D. HbA1c differs from HbA by a single, genetically determined amino acid substitution.
E. HbA2 appears early in fetal life.

Which of the following statements concerning the binding of oxygen by hemoglobin is correct?
A. The Bohr effect results in a lower affinity for oxygen at higher pH values.
B. Carbon dioxide increases the oxygen affinity of hemoglobin by binding to the amino terminal groups of the polypeptide chains.
C. The oxygen affinity of hemoglobin increases as the percentage saturation increases.
D. The hemoglobin tetramer binds four molecules of 2,3-BPG
E. Oxyhemoglobin and deoxyhemoglobin have the same affinity for protons (H+).

• A woman with sickle cell trait and six months pregnant flies to visit her mother. What is the danger that the fetus will have a sickle cell crisis?


















