







Languages
Pages
Legal


Universidade de Lisboa
Faculdade de Ciencias da Universidade de Lisboa
Departamento de Biologia Vegetal
Determining the mechanism of inhibition ofTLR3 by the I329L ASFV protein
Pedro Luıs Vaz Belo Moura
Dissertacao
Mestrado em Biologia Molecular e Genetica
2015




Universidade de Lisboa
Faculdade de Ciencias da Universidade de Lisboa
Departamento de Biologia Vegetal
Determining the mechanism of inhibition ofTLR3 by the I329L ASFV protein
Pedro Luıs Vaz Belo Moura
Dissertacao orientada pela Doutora Sılvia Correia (IGC) e pela
Professora Doutora Maria Filomena Caeiro (DBV-FCUL)
Mestrado em Biologia Molecular e Genetica
2015


Contents
Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . i
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ii
Sumario . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . iii
1 Introduction 1
1.1 The Vertebrate Immune System . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.1.1 The Innate Response . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
1.1.2 The Adaptive Response . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2 Pathogen Associated Molecular Patterns (PAMPs) and Pattern Recognition Re-
ceptors (PRRs) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
1.2.1 Toll and Toll-like Receptors (TLRs) . . . . . . . . . . . . . . . . . . . . . 3
1.2.2 Toll-like Receptor 3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1.2.3 Viral inhibition of immune system components . . . . . . . . . . . . . . . 5
1.3 African Swine Fever Virus (ASFV) . . . . . . . . . . . . . . . . . . . . . . . . 6
1.3.1 The I329L protein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2 Objectives 8
3 Materials and methods 9
4 Results and Discussion 13
4.1 Computational prediction of I329L structure and function . . . . . . . . . . . . 13
4.2 Expression of I329L and its principal domains . . . . . . . . . . . . . . . . . . 14
4.3 Inhibition of TLR3 signaling by I329L . . . . . . . . . . . . . . . . . . . . . . 16
4.3.1 I329L, a dual-action TLR3 antagonist . . . . . . . . . . . . . . . . . . . . 16
4.3.2 Inhibition of TLR3 signaling by the intracellular domain of I329L . . . . . . . 16
4.3.3 Inhibition of TLR3 signaling by the extracellular-transmembrane domain of I329L 19
4.3.4 Impact of the full length I329L and ECDTM on TLR3-dependent activation of
c-Src . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4.4 I329L is a general inhibitor of TLR signaling . . . . . . . . . . . . . . . . . . 25
5 Conclusions 27
6 References 29
7 Appendix 1 - I329L is a type III membrane protein 33
8 Appendix 2 - I329L subconstructs used in this study 34


Acknowledgments
Para mim, este trabalho corresponde ao final do comeco de um longo percurso. Deixo
aqui um agradecimento especial a algumas das inumeras pessoas que, de uma forma ou
de outra, o trilharam comigo, tornando-o possıvel e/ou agradavel; e as mais sinceras des-
culpas aqueles que omitir, seja por falta de lembranca ou espaco (mas nunca de respeito).
To Michael, who gave me the wonderful opportunity to develop my thesis work at his
Infection and Immunity laboratory, and whose support and dedication to research proved
invaluable in the development of my capacity as an independent researcher;
A Sılvia, que coordenou directamente o desenvolvimento desta tese ensinando-me nao
so a tecnica mas tambem o rigor da Ciencia, e cuja amizade e paciencia incansaveis sao o
exemplo perfeito de como formar um estudante;
A Professora Filomena Caeiro, por coordenar o desenvolvimento desta tese ao nıvel
da FCUL, e pela sua sempre pronta disponibilidade e rapidez;
Aos restantes elementos do grupo de Infeccao e Imunidade, Diogo, Rute e Solange,
pela ajuda, apoio, amizade e companhia ao longo de todo este projecto;
To Krzysztof, who kindly and patiently taught me one of the most inherently chal-
lenging techniques in Molecular Biology, allowing me to successfully use it in this work;
Aos meus amigos de MBMG, Andreia, Ines, Tiago e Vania, pela sua amizade e car-
inho incondicionais, tanto nos melhores como nos piores momentos da minha vida ao
longo destes dois anos - serei sempre eternamente grato pela vossa amizade;
Aos meus amigos de LBCM, Alicia, Joao, Luıs, Miguel, Raquel, Sofia e Tiago - a prova
de que serao sempre parte de mim e ainda o serem agora;
A anTUNiA, em especial ao Diogo A., Goncalo F., Miguel V., Paulo F., Paulo G.,
Pedro G. e Pedro O. - nunca uma escola de musica ensinou tanto sobre vida, e vice-versa;
A Agne, Katia, Tania e Wiktoria, pelas chamas que acenderam e aticaram em mim;
e ao Hugo e Ze, que sempre estiveram ao meu lado quando as mesmas se apagaram;
E, finalmente, aos meus pais e avos, pelo amor incondicional durante toda a minha
vida, e sem cujo apoio certamente nao teria chegado ate aqui. Obrigado por tudo.
i

Abstract
The African Swine Fever Virus (ASFV) is a cytoplasmically replicating DNA virus
transmitted by ticks, which causes a highly contagious and frequently fatal disease in
domestic pigs. ASFV pathogenesis is typified by extensive haemorrhage and lymphoid
apoptosis, with an associated tropism for macrophages. Viral spread is inhibited through
the impact of interferon secreted from the early infected cells, which stimulates the ex-
pression of interferon stimulated genes (ISGs) that confer the development of an anti-viral
state to resist viral replication in both infected and nearby non-infected cells.
In order to survive in macrophages, the ASFV must have evolved multiple genes for
evasion and manipulation of interferon. Toll-like Receptors (TLRs) are particularly im-
portant in the induction of the innate immune response. This, and the fact that the virus
is adapted to survive in both vertebrate and invertebrate hosts, with only innate immu-
nity being common to both, stimulated us to search for and identify a TLR antagonist in
the ASFV genome, I329L.
The ASFV I329L ORF was interestingly predicted to be a type I transmembrane
protein containing two leucine-rich repeats (LRRs) in its extracellular domain and a barely
detectable homology with the TLR3 intracellular TIR domain, raising the possibility that
I329L might inhibit activation of IFN-β through an inhibitory interaction with the TIR
motif of the TLRs and the corresponding downstream signaling adaptor proteins.
This structural homology correlated with luciferase reporter assays which clearly demon-
strated that I329L inhibits TLR3-mediated activation of NF-κB and IRF3, therefore in-
hibiting production of IFN-β. Thus, this work attempted to clarify the mechanisms by
which I329L is able to inhibit the TLR3 signalling pathways. We have determined that
I329L has evolved two distinct strategies for inhibition of TLR3: the extracellular domain
interferes with dimerization of TLR3 and other TLRs, perhaps through the formation of
non-signaling heterodimers, while the intracellular domain directly interacts with TRIF
(a signaling protein downstream of TLR3), inhibiting signal transmission.
Keywords
Host evasion, innate immunity, Toll-like Receptors, African Swine Fever Virus, I329L
ii

Sumario
O vırus da Peste suına africana (VPSA) e um vırus pertencente ao grupo dos Vırus
nucleocitoplasmaticos de DNA com grandes dimensoes, com genoma de DNA de cadeia
dupla, capside com simetria icosaedrica e involucrado; e o unico vırus de DNA transmitido
por um vector artropode, sendo o agente etiologico da peste suına africana, uma doenca
extremamente contagiosa e de elevada mortalidade no porco domestico que e caracterizada
por extensas hemorragias e apoptose dos tecidos linfoides. Estes sintomas nao se verificam
nos hospedeiros naturais do VPSA (carraca, javali e porco selvagem africano), em que
causa apenas uma infeccao permanente e assintomatica.
A propagacao do vırus e inibida gracas ao efeito do interferao secretado por celulas
recem infectadas, que estimula por sua vez a expressao de genes estimulados por interferao;
estes genes levam a entrada da celula num estado anti-viral, que inibe a replicacao viral
de forma autocrina (por alteracao/paragem dos mecanismos basais celulares) e paracrina
(por secrecao de moleculas que induzem o mesmo efeito nas celulas vizinhas).
Os receptores do tipo Toll sao considerados como pertencentes a imunidade inata de-
vido a sua capacidade de reconhecer uma extensa variedade de moleculas associadas a
infeccao por patogenios, inexistentes na celula em condicoes regulares (p.ex., peptidogli-
cano da parede celular bacteriana, lipopolissacaridos da membrana externa de bacterias
Gram-negativas ou RNA de cadeia dupla). Apos reconhecimento deste tipo de moleculas,
estes receptores homodimerizam e induzem a producao de citocinas/quimiocinas e/ou in-
terferao, induzindo o estado anti-viral previamente mencionado. Os receptores do tipo
Toll existem apenas em vertebrados, mas sao semelhantes aos receptores Toll existentes
em invertebrados; assim, a resposta imune induzida pelos receptores do tipo Toll e pelos
receptores Toll nao e identica, mas tem a mesma origem evolutiva.
Em particular, o receptor de tipo Toll numero 3 (TLR3) dimeriza e induz sinalizacao
atraves dos factores de transcricao IRF-3 e NF-κB apos reconhecimento de RNA de cadeia
dupla. O RNA de cadeia dupla existe na celula pontualmente gracas aos mecanismos de
interferencia de RNA, sendo detectado e destruıdo de imediato; no entanto, RNA de cadeia
dupla de grandes dimensoes tende a nao existir na celula, e portanto e uma indicacao de
infeccao viral. Vırus de genoma de RNA de cadeia dupla ou RNA de sentido negativo
produzem RNA de cadeia dupla constitutivamente, mas os vırus de DNA (aos quais
pertence o VPSA) podem produzir moleculas de RNA de cadeia dupla por transcricao
convergente; assim, a infeccao por um destes tipos de vırus induz sinalizacao pelo TLR3
e a consequente inducao do estado anti-viral.
De forma a evitar este efeito, sobreviver e replicar-se com sucesso, existem inumeras
estrategias virais para evasao da resposta imune e/ou manipulacao da producao ou efeito
do interferao.
Esta dissertacao nao segue as normas do novo Acordo Ortografico.
iii

O facto de que o VPSA esta adaptado a sobrevivencia em hospedeiros vertebrados e
invertebrados (que partilham apenas alguns mecanismos conservados da imunidade inata)
levou-nos a procura de um antagonista dos receptores de tipo Toll no genoma do VPSA.
A proteına I329L do VPSA foi descrita como uma proteına transmembranar de tipo I,
com duas repeticoes ricas em leucina no domınio extracelular e uma homologia detectavel
com o domınio TIR (domınio homologo ao receptor Toll/interleucina-1) do TLR3. Estas
caracterısticas implicam a possibilidade de que a proteına viral I329L seja capaz de inibir
a inducao de IFN-β atraves de uma interaccao inibitoria com o motivo TIR dos receptores
de tipo Toll, assim como das suas proteınas adaptadores correspondentes (com a funcao
de propagar os sinais enviados pelos receptores de tipo Toll). O domınio TIR do TLR3
interage com a proteına adaptadora TRIF (“Adaptador indutivo do interferao-β com um
domınio TIR”), que transmite o sinal ao longo da via; em teoria, uma vez que o domınio
TIR da I329L e homologo ao domınio TIR do TLR3, a I329L podera interagir com o
TRIF, uma hipotese que foi estudada ao longo deste trabalho.
Esta homologia estrutural foi correlacionada com ensaios previos de actividade de lu-
ciferase expressa em genes reporter, que demonstram claramente uma inibicao da inducao
de IFN-β e activacao de NF-κB mediada pelo TLR3. Assim, este trabalho tem como
objectivo obter uma melhor compreensao dos mecanismos pelos quais a proteına I329L
inibe a activacao das vias de sinalizacao do TLR3. Gracas a informacao fornecida pelas
homologias previamente mencionadas, este objectivo tem, por sua vez, duas vertentes:
1) Explorar a possıvel formacao de heterodımeros sem capacidade de sinalizacao con-
stituıdos por I329L e TLR3, e as consequencias desta interaccao; e
2) Determinar o mecanismo de inibicao da sinalizacao do TLR3 pelo domınio intracelu-
lar da proteına I329L com regioes TIR, de forma a confirmar a interaccao do mesmo com
o seu alvo teorico, TRIF.
Para este efeito, segmentos do gene I329L correspondentes ao domınio extracelu-
lar/transmembranar e ao domınio intracelular foram clonados em plasmıdeos de ex-
pressao em mamıferos de forma a averiguar o impacto dos domınios constituintes da
proteına na sinalizacao da via do TLR3. Inicialmente, foi determinado que o domınio
extracelular da I329L interage apenas com o TLR3, inibindo a activacao da via apenas
quando esta e estimulada por um analogo sintetico do RNA de cadeia dupla, o acido poli-
inosınico/policitidılico (Poly (I:C)), mas nao quando esta e estimulada pela expressao
ectopica de TRIF; por outro lado, o domınio intracelular da I329L interage a jusante do
TLR3, visto inibir a via do TLR3 quando sujeito a ambos os tipos de activacao. Esta
interaccao por parte do domınio intracelular foi posteriormente caracterizada como sendo
uma interaccao directa com a proteına adaptadora TRIF atraves de ensaios de imunopre-
cipitacao.
iv

Assim, uma reconstrucao bioinformatica da estrutura da I329L foi feita atraves de
metodos de modelacao proteica com base em homologias evolutivas, com o objectivo de
delinear estrategias adequadas para a caracterizacao funcional da I329L.
A analise do domınio intracelular permitiu determinar a localizacao das tres regioes
descritas na literatura como sendo homologas do domınio TIR do TLR3; esta informacao
levou-nos a proceder com mutagenese dirigida da regiao central de forma a desregular
a possıvel interface de interaccao com TRIF. O resultado deste ensaio revelou-se in-
frutıfero, permitindo-nos inferir que a regiao central de homologia pode nao ser estri-
tamente necessaria para a interaccao com TRIF, ou que as duas outras regioes podem ter
um efeito de compensacao da actividade inibitoria apos desregulacao da regiao central.
Para averiguar estas hipoteses, serao feitos outros ensaios de mutagenese no futuro.
A analise estrutural do domınio extracelular permitiu-nos determinar a existencia na
I329L de domınios semelhantes ao domınio de ligacao ao RNA de cadeia dupla e domınio
de dimerizacao do TLR3. De forma a averiguar a hipotese de uma interacao directa com
o TLR3, um segmento do domınio extracelular sem o domınio de ligacao ao RNA foi
clonado no plasmıdeo de expressao adequado; este segmento manteve a sua actividade
inibitoria do TLR3, indicando que a interaccao da I329L com o TLR3 e independente da
ligacao ao RNA.
Foram desenvolvidos dois ensaios para deteccao de uma interaccao directa da I329L
com o TLR3: um ensaio de separacao electroforetica sem desnaturacao proteica (BN-
PAGE), cujos resultados foram por si so inconclusivos mas defendiam a possibilidade
desta interaccao; e um ensaio de ligacao cruzada de proteınas in vivo, cujos resulta-
dos demonstraram formacao de heterodımeros I329L-TLR3, assim como de homodımeros
I329L-I329L.
Uma vez que este tipo de ensaios nao permite afirmar sem qualquer duvida a existencia
de uma interaccao especıfica, averiguamos tambem o impacto da I329L e do domınio
extracelular ao nıvel da via de sinalizacao do TLR3 independente de TRIF; uma vez que
esta via e activada pelo domınio extracelular e inibida pela proteına completa, concluımos
que existe dimerizacao com o TLR3, sendo que o efeito diferencial se deve a disponibilidade
do local de interaccao com a proteına c-Src no domınio intracelular do TLR3.
Finalmente, com base nos resultados descritos previamente, decidimos averiguar a
possibilidade de a I329L ter um efeito inibitorio em outros TLR que nao o TLR3. Des-
crevemos a existencia de actividade inibitoria da I329L nas vias do TLR4, TLR5, TLR7
e TLR9, que reconhecem outros tipos de moleculas; este resultado cimenta a hipotese
da existencia de uma interaccao directa da I329L com TLRs atraves do seu domınio de
dimerizacao, sem necessidade de domınios de interaccao com ligandos.
v

Em resumo, concluımos que a proteına I329L do vırus da Peste suına africana e uma
proteına inibitoria da via de sinalizacao dos TLR com duas actividades distintas:
1) inibicao geral das vias de sinalizacao dos varios TLR atraves de interferencia com o
processo de dimerizacao, possivelmente pela formacao de heterodımeros sem capacidade
de sinalizacao;
2) inibicao das vias de sinalizacao do TLR3 e TLR4 atraves de interaccao directa com
a proteına adaptadora TRIF, inibindo a transmissao do sinal a jusante dos mesmos.
Palavras-chave
Evasao ao hospedeiro, imunidade inata, receptores de tipo Toll (TLRs), vırus da Peste
suına africana, I329L
vi

1 Introduction
1.1 The Vertebrate Immune System
The immune system is a highly complex system, composed of an interconnected net-
work of organs, cell types, humoral factors, cytokines and cell receptors that serves to
protect the host against an enormous variety of pathogens, ranging from submicroscopic
viruses to macroscopic worms.
The mechanisms for mobilizing an immune response to pathogens depend on the de-
tection of hallmark features that distinguish them as non-self, thus avoiding detrimental
damage to self and/or to beneficial commensal microbes.
Immunity is divided into two functional systems, characterized by the speed, specificity
and mechanisms of recognition: the early innate response and the late adaptive response.
1.1.1 The Innate Response
The innate immune system serves as the first line of defence during infection, being
decisive in recognizing invading pathogens and triggering a proinflammatory response1.
Essential components of the innate immune system are monocytes, dendritic cells (DCs),
macrophages, neutrophils, NK cells, cytokines, complement and acute phase proteins,
which provide an immediate defence response to pathogen invasion.
Some of the innate signaling pathways are highly conserved throughout evolution,
being found not only in vertebrate and invertebrate animals, but also in other organisms
such as plants and fungi2.
Innate immune responses are characterized by the recognition of specific and con-
served pathogen-associated molecular patterns (PAMPs) by host germ-line coded pattern
recognition receptors (PRRs)3. Recognition of PAMPs by PRRs signals the presence of
infection and rapidly triggers proinflammatory and antimicrobial responses that function
to contain and/or eliminate the infection. The proinflammatory response also controls
the direction of the adaptive response, activating the proliferation, differentiation and
migration of lymphocytes with specificity for the invading pathogen, thus ensuring an
appropriate adaptive immune response for the pathogen; for instance, cytotoxic T cells
for intracellular pathogens4.
Overall, the innate response is faster, less specific and less potent than the adaptive
response; it also lacks immunological memory – that is, unlike the adaptive response,
repeated exposure to the same pathogen results in a response with equal potency.
1

1.1.2 The Adaptive Response
In contrast to the innate response, the adaptive response is highly specific, recognizing
target antigens through antigen-specific receptors which are clonally expressed on the sur-
face of B and T lymphocytes and encoded by genes assembled by somatic rearrangement
of germ line genes5. This somatic recombination process, together with somatic mutation
of the variable region genes, potentially allows for the creation of millions of receptors
with different and unique antigen specificities from just a few hundred germ line-encoded
genes, thus generating the enormous repertoire and specificity of the adaptive response6.
Development of an efective adaptive immune response after pathogen entry is slow, as
only small numbers of cells with specificity for any given antigen are present in a normal
animal. They clonally proliferate after contact with the corresponding antigen, finally
developing into the effector cells of the adaptive response7.
Significantly, the adaptive response also produces memory cells, which rapidly undergo
clonal expansion after later reencountering the corresponding antigen; this allows for
greater potency of the immune response upon repeated exposure to the same pathogen8
and is the rational basis for vaccines. The proinflammatory response created by the innate
response and the exposure of lymphocytes to antigens by DCs and macrophages provides
a profile of secreted cytokines, which are critical to the functional direction of the adaptive
response. The adaptive response has a relatively recent evolutionary origin, having arisen
500 million years ago only in vertebrate animals9.
1.2 Pathogen Associated Molecular Patterns (PAMPs) and Pat-
tern Recognition Receptors (PRRs)
PAMPs are very highly conserved structures, which are present in large groups of
pathogenic microbes: They are defined as molecules that are unique to pathogens, existing
in the host only when there is infection by a pathogen; these tend to be essential for
the survival, pathogenicity or lifecycle of microorganisms, and are invariant in entire
classes of pathogens10. Examples of PAMPs are double-stranded RNA (dsRNA), which
is the genomic basis of some viruses (such as the family Reoviridae) and can be generated
during replication of positive-stranded RNA and DNA viruses11; lipopolysaccharide, which
is synthesized only by bacteria12; or unmethylated CpG sequences in DNA molecules,
common in bacterial DNA but very rare in vertebrate genomes13.
PRRs, being cellular receptors specific for given PAMPs, are thus as conserved and
restricted as their corresponding PAMPs. PRRs are typically expressed by cells of the
immune system14, but some specific types of PRR are also expressed on a larger variety
of cells15, providing an immediate response to pathogen invasion. Besides recognizing
PAMPs, some PRRs also recognize danger-associated molecular patterns (DAMPs), self
molecules in aberrant locations or abnormal conformations, signaling loss of homeostasis16.
2

After recognition of PAMPs, the corresponding PRRs trigger a proinflammatory re-
sponse by activating intracellular signaling pathways that result in the expression of cy-
tokines, chemokines, cell adhesion molecules and/or surface receptors, which together
limit the infection and control the direction of the adaptive immune response17. There
are three classes of PRRs, defined by their cellular location18: secreted, transmembrane
and cytosolic. Secreted PRRs, like ficolins and pentraxins, activate classical and lectin
pathways of the complement system by binding to microbial cell surfaces, and result in
opsonization of pathogens for phagocytosis; Transmembrane PRRs, mainly the Toll-like
receptor (TLR) family and the C-type lectin receptors (CLR), recognize extracellular
PAMPs if located on the cell surface and intracellular PAMPs if located in such intracel-
lular membranes as endosomes. The cytosolic PRRs include retinoic acid-inducible gene
I (RIG-I)-like receptors (RLRs) and nucleotide-binding domain and leucine-rich repeat-
containing receptors (NLRs); RLRs recognize viral pathogens, while NLRs detect both
bacterial and viral PAMPs and stress-associated DAMPs.
1.2.1 Toll and Toll-like Receptors (TLRs)
The Toll receptor and pathway were first described in Drosophila, as essential for
the establishment of the dorso-ventral pattern in developing embryos19. The signaling
pathway of Toll is similar to the mammalian IL-1 pathway, which activates NF-κB, a
transcription factor and a major player in the inflammatory immune response20. The
crucial experiment, linking the Toll signaling pathway with the innate response of the
immune system, was the demonstration that Toll-“knockout” flies were highly susceptible
to fungal infection21. Mammalian homologues of Toll with the capacity of recognizing
PAMPs were then identified and named as Toll-like Receptors, or TLRs22.
Currently, there are 13 recognized TLRs23, of which 10 are functional in humans and 12
in mice (TLRs 1-9 are homologous and functional in both mice and man; TLR 10 is only
expressed in humans, being nonfunctional in mice, and TLRs 11-13 are only expressed in
mice). Each TLR recognizes a different PAMP, but they can be divided into functional
subfamilies: TLR1, TLR2, TLR4 and TLR6 recognize lipids, whereas TLR3, TLR7,
TLR8 and TLR9 recognize nucleic acids24. Structurally, TLRs are glycosylated Type
I transmembrane proteins with leucine-rich repeats (LRRs) in the extracellular domain
(ECD) and a conserved intracellular domain (ICD), referred to as the Toll/IL-1 receptor
(TIR) domain, which is crucial for the activation of TLR signaling pathways25.
There are two main signaling pathways, which occur after ligand mediated dimeriza-
tion of TLR chains: the MyD88-dependent pathway26, requiring TIR domain-containing
adaptor MyD88 and used by every TLR except TLR3; and the TRIF-dependent path-
way27 (TRIF is also known as TIR-containing adaptor molecule-1, or TICAM-1), mainly
used by TLR3/4 and able to activate NF-κB, IRF-3 and AP-1, resulting in the production
of type I interferon (IFN) and proinflammatory cytokines/chemokines.
3

1.2.2 Toll-like Receptor 3
TLR3 recognizes viral dsRNA (and a synthetic analogue, polyriboinosinic : polyribo-
cytidylic acid [poly (I:C)]) and, using the TRIF-dependent signaling pathway28, induces
an antiviral state in the host cell. In immune cells, it is found in myeloid DCs, NK lym-
phocytes, macrophages and mast cells29; it can be present in other types of cells, such as
fibroblasts30, epithelial31 and neural cells32.
TLR3 mostly localizes to endosomes through UNC93B1-mediated transport33, through
its ECD, where it detects intracellular dsRNA. In a few cell types, such as macrophages34,
it can also localize to the cell surface and detect extracellular dsRNA.
Recognition of dsRNA by the ECD of TLR3 results in the receptor dimerization re-
quired for adaptor-mediated signal transduction35. The next obligatory step in TLR3
activation is phosphorylation of its tyrosines 759 and 858 by c-Src and EGFR, respec-
tively36, and binding of PI3-kinase37. After this phosphorylation, the ECD of TLR3 is
proteolytically cleaved38 and the phosphorylated ICD recruits TRIF through its newly
exposed TIR domain (Figure 1), thus mediating signaling through the recruitment of
tumour necrosis factor (TNF) receptor-associated factors (TRAF) 2, 3 and 6, and result-
ing in the activation of TANK-binding kinase 1 (TBK1/NAK) and IκB kinase-related
kinase-ε (IKK-ε), which activate IRF-3, NF-κB and AP-139. This induces transcription
of IFN-β, a type I IFN able to autocrinally and paracrinally stimulate infected cells to
develop an antiviral and antiproliferative state; as well as expression of proinflammatory
cytokines via NF-κB and AP-1 (the complete pathway is shown in Figure 2A).
Figure 1 – Activation of TLR3 signaling by dsRNA. (Adapted from Yamashita et al.36)
In addition to the gene expression-dependent process, the binding of c-Src to activated
TLR3 causes its own activation in a MyD88 and TRIF-independent form40, directly lead-
ing to a transient and gene expression-independent enhancement of cell migration, ad-
hesion and proliferation, functions thought to promote the early impact of the innate
immune response (Figure 2B).
4

Figure 2 – TLR3 signaling pathways. A) TRIF-dependent signaling pathway (Adapted from
Kimura et al.41). B) TRIF-independent signaling pathway (Adapted from Yamashita et al.40).
TLR3 signaling demonstrably contributes to morbidity and mortality in certain viral
infection models42, and drives pathogenic mechanisms in various immune-mediated and
autoimmune diseases, such as rheumatoid arthritis43 and asthma44. As a consequence,
down-regulation of TLR3 signaling may become a novel modulatory approach for treat-
ment of those diseases. Indeed, a considerable interest in agonists45 and antagonists46 of
TLRs for immune modulation currently exists.
1.2.3 Viral inhibition of immune system components
Since PAMP-PRR triggered responses are critical to the initiation and control of the
immune response, pathogens have unsurprisingly evolved counter-strategies to avoid the
anti-pathogen consequences of their recognition by TLRs.
Viruses, which have closely co-evolved with their hosts’ immune system, are paramount
in the search for examples of such counter-strategies: for instance, Enterovirus 71 3C
is able to inhibit the antiviral response mediated by TLR3, by cleaving TRIF47 and
thus suppressing interferon production and the induction of an antiviral state; Polyd-
navirus Ank proteins act as mimics of IκB48, binding to NF-κB and inhibiting many
of the proinflammatory signaling pathways. The Vaccinia virus protein A46 inhibits
TIR-domain-containing adaptors49, resulting in inhibition of both MyD88-dependent and
TRIF-dependent signaling through the disruption of receptor:adaptor interactions; the N2
protein of the same virus is able to inhibit production of IFN-β, by inhibiting activation
of IRF350. Finally, as an example of other possible pathways involved in the activation of
the IFN response, the V protein of Paramyxoviruses binds to a RLR, the dsRNA sensor
MDA5, preventing it from activating the interferon pathway51.
5

1.3 African Swine Fever Virus (ASFV)
The African Swine Fever Virus (ASFV) is a large, enveloped virus with icosahedral
morphology (Figure 3), a 170-190 Kb dsDNA genome and a mainly cytoplasmic repli-
cation cycle52. ASFV is the single member of its family, the Asfarviridae, and the only
known DNA arthropod-borne virus. Infection of domestic pigs (Sus scrofa domesticus)
causes a highly contagious and fatal disease, consisting of extensive haemorrhages and
lymphoid apoptosis. There is no vaccine, and control relies on restricting animal move-
ment and slaughtering infected pigs.
Figure 3 – Structural diagram of the African Swine Fever Virus (Adapted from Viralzone53)
Transmission between swine occurs through direct pig to pig contact and through soft
tick vectors, with the virus being able to replicate in ticks after ingestion of infected blood,
and subsequent transmission to healthy swine by the infected tick54.
In contrast to domestic pigs, only a persistent and inapparent infection is seen in the
natural wildlife hosts55, warthogs (Phacochoerus aethiopicus), bushpigs (Potamochoerus
Porcus) and soft ticks (Ornithodoros moubata). We may therefore assume that the ASFV
is a “newly emerged” pathogen, adapted to its wildlife host but, similarly to HIV-1,
pathogenic to its recent domestic pig host.
Although the virus principally replicates in macrophages, there is considerable collat-
eral apoptosis in T and B lymphocytes and endothelial cells during acute infections56.
Since ASFV is adapted to survive in both mammals and arthropods, some mechanism for
evasion of innate immunity must have evolved, as it is common to both vertebrate and
invertebrate hosts.
There are various known ASFV proteins involved in evading host immune systems,
such as A238L, which inhibits NF-κB activation and calcineurin phosphatase57; A276R,
which inhibits the induction of IFN-β58; and A528R, which inhibits the induction and
impact of type I and II IFNs58.
The ASFV may stimulate activation of TLR3 (and thus, production of IFN) due to
production of a dsRNA intermediate by convergent transcription59. Therefore, an evasion
mechanism against TLR3 is predicted to have evolved, and indeed one protein of the
ASFV was determined to function as a TLR3 antagonist: the I329L protein.
6

1.3.1 The I329L protein
The I329L protein is a highly glycosylated type I transmembrane protein60, composed
of a 237 aminoacid extracellular domain (ECD), a 23 aminoacid transmembrane domain
(TM) and a 69 aminoacid intracellular domain (ICD) (Figure 4). There are two LRRs in
its ECD, pointing to a degree of limited similarity with the ECDs of TLR family members.
Figure 4 – Structural diagram of the ASFV I329L protein. The ECD has 8 glycosylation sites
and 2 LRRs, plus 3 in-frame methionines. The ICD has 3 TIR-like regions (shown as a TIR-like
domain).
Although I329L is described in the literature as a 50 KDa protein, we have continuously
observed a heterogeneity of protein expression in HEK-293T cells, as shown in Figure
10, page 15.
The cause for this heterogeneity is currently under investigation in our laboratory,
but we hypothesize this phenomenon may be due to translation starting at the different
in-frame AUGs, since the estimated molecular weights of the different forms of I329L
correspond to the molecular weights of the segments starting from the different AUGs.
Thus, the three starting codons in the coding ORF of I329L are marked in the figure of
the I329L molecule. Further heterogeneity may arise from glycosylation, ubiquitination
and, as known to occur with TLR3, proteolytic processing38.
As part of an independent project to investigate alternative transcription/translation
from the three AUG codons, deletion ECDTM mutants (2MECDTM/3MECDTM) start-
ing at the 2nd and 3rd methionines and including the TM sequence were constructed.
Significantly, the 3MECDTM construct (shown in Figure 5) starts downstream of
the predicted dsRNA binding site, but upstream of the dimerization motif. Thus, after
structural prediction of the ECDTM interactions (Section 4.1) the 3MECDTM construct
was used to investigate the possibility of interaction of I329L with TLR3 without the need
for dsRNA binding (Section 4.3.3).
Figure 5 – Structural diagram of the 3MECDTM segment of I329L.
The 3MECDTM segment starts at the 3rd methionine of the full length I329L protein.
7

To pursue the similarity of the I329L ECD with TLRs, the ICD of I329L has a de-
tectable (although very low) homology with the TLR3 intracellular TIR domain, consist-
ing of three TIR-like regions with similar sequence and secondary structure65. Specifically,
as our previous work had shown that I329L is able to inhibit TLR3-mediated induction
of IFN-β and activation of NF-κB, we hypothesized that the ICD might interact with
the TIR motif in TLR3 and inhibit the corresponding signaling pathway of IFN-β activa-
tion. This hypothesis was supported by the observation that I329L-mediated inhibition
is observed both by stimulating TLR3 extracellularly with dsRNA and intracellularly by
ectopic expression of TRIF58. Direct evidence for the postulated I329L ICD-TRIF inter-
action now been demonstrated and is included in the experimental work of this thesis.
If I329L interferes with TRIF, it would inhibit activation of IRF3, IRF7 and NF-κB,
the transcription factors necessary for IFN-β and proinflammatory cytokine transcription;
Therefore, I329L would be considered a viral TLR3 antagonist which reduces the effec-
tiveness of the innate antiviral response. The use of bioinformatics was therefore deployed
(Section 4.2) to construct an adequate model for I329L and its possible interactions with
TLR3. Thus, the experimental work undertaken specifically addressed this question and
other clues related to the mechanism of inhibition of TLR3 signaling raised by the bioin-
formatic analysis, including the possible formation of TLR3-I329L heterodimers.
2 Objectives
The main objective of this project is to define the mechanism by which I329L is able
to inhibit TLR3 signaling, specifically by:
1) Determining the mechanism of inhibition of TLR3 signaling by the intracellular
TIR domain of I329L, by confirming the interaction of the I329L ICD with its putative
target, TRIF;
2) Determining the mechanism for I329L ECD-mediated inhibition of TLR3 through
functional analysis of I329L truncation mutants (by luciferase assays for the activation of
IFN-β and NF-κB) and exploration of the possible formation of non-signaling TLR3-I329L
heterodimers after stimulation with dsRNA, and
3) Determining an impact of I329L and its ECD truncation mutant on TLR3-activated
autophosphorylation of c-Src.
8

3 Materials and methods
Homology modeling of I329L: The sequence of I329L and its domains (ECDTM and
ICD), taken from the UniProt database (accession number A9JLD8), was investigated
for possible homologies using Phyre2, the Protein Homology/analogY Recognition En-
gine V2.0 Webserver61, which is able to predict protein structure by detection of sequence
homology with known structures. A preliminary set of models was obtained by use of the
I-TASSER server for protein prediction62 (data not shown), as a base and template for
further, more accurate modeling. I329L structures were homology modelled with Mod-
eller 9.1463, based on its alignment of I329L and the PDB templates obtained from Phyre2
and I-TASSER. Quality of the resulting models was assessed using PROCHECK v3.5.464.
The best quality models were used for determination of possible protein complexes by
superimposition with TLR3. Ray traced images were obtained with PyMOL v1.7.2 RC2.
PCR: The full length I329L gene (Fig. 4, Sec. 1.3.1) was a codon optimized sequence,
purchased from Invitrogen. PCR amplification of I329L truncated regions was performed
using primers for (1) the ECDTM (Forward, 1st ATG: 5’-CGAATTCATGCTGAGAG
TGTTC-3’; F, 3rd ATG: 5’-CGAATTCATGTGCGGCAAGCGGAAC-3’; Reverse: 5’-C
GATATCCAGCAGGCAGATGAACAGG-3’) and (2) the ICD (F: 5’-CGAATTCATG
CGCTCCATCTGCAAG-3’; R: 5’-CGATATCCAGCTTGCGGCGGCTGC-3’), with se-
quences for EcoRI and EcoRV restriction and using the high-fidelity enzyme Pfu DNA
polymerase. The following cycling parameters were used: 1) initial denaturation for 5
min at 94◦ C; 2) denaturation for 30 s at 94◦ C and annealing for 1 min at 44◦ C, followed
by extension for 2 min at 74◦ C, for 30 cycles; and 3) final extension at 74◦ C for 4 min.
Transformation/Cloning: The amplified products were identified with ethidium bro-
mide visualization on an agarose gel based on the expected size of the amplicons, and the
DNA was purified from the excised band using the GelPure kit (NZYTech). The frag-
ments were then cloned into plasmid pcDNA3-Myc-HA, which has an N-terminal Myc
peptide tag and a C-terminal influenza haemagglutinin (HA) peptide tag, with T4 DNA
Ligase (NEB). The resulting plasmids were used to transform Escherichia coli DH5α cells
by incubating cells with the DNA on ice during 30 min, followed by a 42◦ C heat shock of
45 sec and an incubation of 2 min on ice. SOC medium was added to the cells, followed by
an incubation of 1 h at 37◦ C, with shaker. DH5α cells were then plated onto LB plates
with ampicillin (100 mg/L). Single colonies were cultured on LB medium with ampicillin
overnight (O/N) at 37◦ C and plasmid DNA was isolated by use of a Miniprep kit. The
fidelity of the sequence of the cloned fragments was confirmed by automated sequencing.
9

Site-Directed Mutagenesis: With the purpose of obtaining a mutation in the sec-
ond TIR region of the I329L ICD, Phusion DNA polymerase (Fermentas) was used with
the following forward and reverse primers: 5’-CAGCTACACCTCCAGCCTGAAG-3’, 5’-
CTTCAGGCTGGAGGTGTAGCTG-3’. The following cycling parameters were used: 1)
30 s at 98◦ C; 2) 10 s at 98◦ C, 30 s at 57◦ C, and 3 min at 72◦ C; 3) 10 min at 72◦ C.
The resulting DNA was digested with DpnI for 1 hr at 37 ◦ C. Inactivation of the en-
zyme required 10 min of incubation at 80◦ C. DH5α competent cells were transformed as
previously described, plasmid DNA was extracted and the presence of the mutation was
confirmed by automated sequencing.
Cells: African green monkey Vero and human HEK-293T cell lines were cultured in 5%
CO2 at 37◦ C in Dulbecco’s modified Eagle medium (DMEM) with GlutaMAX and 1000
mg/l glucose, supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS) and
100 U/ml and 100 µg/ml penicillin and streptomycin, respectively (Invitrogen).
Antibodies: The antibodies used were as follows: rat monoclonal high-affinity antibody
(clone 3F10) against HA tag (AYPYDVPDYA) conjugated with horseradish peroxidase
(HRP) (Roche, 12013819001), rabbit polyclonal affinity-purified antibody against HA
(Sigma, H6908) and rabbit polyclonal affinity-purified antibody against TRIF (Cell Sig-
naling, 4596S) for immunoprecipitation assays, rabbit polyclonal affinity-purified antibody
against c-Myc tag (EQKLISEEDL) (Cell Signaling, 2272S), rabbit polyclonal affinity-
purified antibody against Phospho-Src (Tyr416) (Cell Signaling, 2101S), mouse mono-
clonal antibody (c. AC-15) against β-Actin conjugated with peroxidase (Sigma, A3854),
goat polyclonal antibody against rabbit IgG (H+L) conjugated with HRP (Thermo Fisher,
G-21234), rat monoclonal high-affinity antibody (c. 3F10) against HA for immunofluo-
rescence assays (Roche, 11867423001), goat polyclonal affinity-purified antibody against
mouse IgG (H+L) conjugated with Texas Red R©dye for immunofluorescence assays (Jack-
son ImmunoResearch, 115-075-003)
Immunoblotting: Vero and HEK-293T cells were cultured in 6-well plates at 37◦ C, un-
der 5% CO2 and at 2.5*105 cells/well; The cells were transfected with plasmid DNA (using
4 and 2 µg of DNA per well, respectively) by using the X-tremeGENE 9 and FuGENE 6
transfection reagents, respectively (Roche). In the case of polyinosinic:polycytidylic acid
(Poly (I:C)) stimulation, Poly (I:C) was added up to a final concentration of 100µg/mL
during 30 min pre-lysis. 48 h post-transfection, cells were lysed in cold lysis buffer (6 M
urea, 75 mM NaCl, 1 mM EDTA, 1% (v/v) NP40, 2% (v/v) glycerol, 1 mM PMSF, 1
µM DTT, 25 mM HEPES, pH 7.4). Extracts were boiled (5 min, 100◦ C) with sample
buffer (1.7% [w/v] SDS, 5% [v/v] glycerol, 0.1 M DTT, bromophenol blue [0.02 mg/ml],
58 mM Tris-HCl, pH 6.8).
10

Proteins were then separated by 12% SDS-PAGE, electroblotted onto polyvinylidene flu-
oride (PVDF, BioRad) membranes and blocked with 5% (w/v) non-fat dried milk in PBS
for 1 h at room temperature (RT). The membranes were probed with the following an-
tibodies: a rat antibody against HA conjugated with HRP, a rabbit polyclonal antibody
against c-Myc tag, a mouse monoclonal antibody against β-actin (Sigma) conjugated
with HRP to provide an internal control for protein loading, rabbit polyclonal antibod-
ies against TRIF and Phospho-Src and a goat polyclonal antibody against rabbit IgG
chains conjugated with HRP. Membranes were incubated with the indicated antibodies
overnight, at 4◦ C, and developed using ECL chemiluminescent reagents (PerBio Science)
or Luminata Forte (Merck), according to the manufacturer’s instructions.
Immunoprecipitation: HEK-293T cells seeded at 5*105 cells/60 mm dish were trans-
fected with 4 µg pcDNA3-HA empty vector or pcDNA3-Myc-ICD-HA and 2 µg TRIF
expression plasmid using Fugene 6, collected by scraping and lysed using cold lysis buffer.
Protein G dynabeads (Millipore) were incubated with 5 µg of polyclonal affinity-purified
anti-HA antibody or a 1:50 solution of polyclonal anti-TRIF antibody. The supernatant
of the protein extracts was collected and added to 30 µL of previously washed protein G
beads using commercially available lysis buffer (Cell Signaling). After rotating for 3 h at
4◦ C, the beads were washed three times with lysis buffer and heated at 100◦ C for 5 min
with sample buffer. The supernatants were then loaded onto SDS-PAGE gels and probed
through immunoblotting, with anti-HA and anti-TRIF antibodies.
Immunofluorescence: Vero cells were seeded onto coverslips in 6-well plates at 2*105
cells/well,, and incubated overnight at 37◦ C and 5% CO2. The cells were then transfected
with 4 µg of pcDNA3-HA empty vector or pcDNA3-Myc-HA expression constructs using
X-tremeGENE 9. Cells were fixed after 48 h with 4% (w/v) paraformaldehyde in PBS for
20 min at RT and then permeabilized by incubation in PBS containing 0.1% (v/v) Triton
X-100 (20 min, RT). Finally, the cells were blocked by incubation in PBS containing 0.05%
Tween-20 and 5% normal goat serum (30 min, RT). To detect recombinant proteins, a
primary rat monoclonal high-affinity antibody against HA diluted in blocking solution was
used (1 h, RT), followed by use of a secondary goat polyclonal anti-mouse IgG antibody
conjugated with Texas Red dye (1 h, RT). Cell nuclei were then stained with DAPI (Life
Technologies) by incubation with a 500 nM DAPI in PBS solution during 1 min, RT.
All washes after incubation with antibody were performed at RT with PBS containing
0.05% (v/v) Tween-20. Cells were mounted in Slowfade (Life Technologies) and observed
under a fluorescence microscope. HA expression is shown in red color for better visual
perception. Nuclei stained with DAPI are shown in blue. Fluorescent images were taken
with a Leica DMRA2 microscope equipped with a cooled CCD camera (Leica. Corp.,
Wetzlar, Germany).
11

Luciferase reporter assays: HEK-293T cells were cultured in 24-well plates at 6*104
cells/well, and 24 h later were transfected using Lipofectamine 2000 with reporter plas-
mids for IFN-β or NF-κB (100 ng) and the internal control β-galactosidase (25 ng),
TLR3 expression plasmid (50 ng) and pcDNA3-Myc-HA expression plasmids (300 ng)
or pcDNA3-HA empty vector (negative control), keeping the total amounts of DNA and
equimolar ratios constant in all assays by adding the appropriate amount of empty vector.
After 48 h, cells were either stimulated with 25 µg/ml of polyinosinic:polycytidylic acid
(poly (I:C)) (PeproTech) as indicated in the figures for 5 h, or left untreated. The cells
were lysed in 100 µl lysis solution (ABX210LM, Promega Systems) according to the man-
ufacturers instructions, and samples were assayed for both luciferase and β-galactosidase
activities. Luciferase activity was normalized to the β-galactosidase activity from the
co-transfected plasmid internal control and expressed as luciferase relative to galactosi-
dase activity. Values are expressed as mean relative stimulation ± SD (calculated from
triplicate determinations). A minimum of three independent assays were done for each
experiment reported. In an alternative protocol, IFN-β reporter assays were done using
HEK-293T cells ectopically expressing TRIF. Transfections were performed with equal
amounts of DNA mixtures comprising 25 ng of TRIF plasmid vector (TRIF) and the
above mentioned plasmids. After 48 h, the cells were harvested and lysed, following the
above protocol. Assays for other TLRs followed the first protocol, but using expression
plasmids for TLRs other than TLR3. Cells were stimulated as listed in Table 1, page 27.
Intracellular protein crosslinking: Disuccinimidyl suberate (DSS) (Thermo Scien-
tific) was stored at 4◦ C with desiccant. HEK-293T cells were cultured and transfected
as described above. 48 h post-transfection, the cells were washed twice with PBS and a
DSS solution was prepared at 20x final concentration in DMSO. The DSS solution was
added to 1x concentration (2 mM) directly to the wells, and incubated for 45 minutes
at RT. The reaction was quenched by removal of the solution and addition of TBS 1x.
After quenching, cells were lysed and protein extracts obtained as described above. After
sample buffer addition, the proteins were then separated on 4-12% SDS-PAGE gels and
probed according to the immunoblotting protocol.
Blue Native PAGE: HEK-293T cells were transfected (immunoblotting protocol), col-
lected by scraping from 6-well plates and lysed using cold lysis buffer. Protein super-
natants were obtained, to which BN-PAGE sample buffer was then added (5% Coomassie
Briliant Blue G-250 (Sigma), 100 mM BisTris-HCl (pH 7.0), 100 mM NaCl, 20% glycerol).
Proteins were then separated by PAGE using 4-16% Bis-Tris gels (Thermo Scientific) and
Bis-Tris-based running buffers (Cathode buffer: 50 mM Tricine, 15 mM Bis-Tris/HCl pH
7.0, 0.02% Coomassie Brilliant Blue G-250; Anode buffer: 50 mM Bis-Tris/HCl pH 7.0)
during a minimum of 5 h at 4◦ C, and probed through immunoblotting.
12

4 Results and Discussion
4.1 Computational prediction of I329L structure and function
Using homology modeling for protein structure simulation, and based on structural
assessments from a previous study65, it was decided to construct a model of I329L as a
platform for the subsequent strategies to determine structure-function correlations. Ini-
tially, the PHYRE 2.0 and I-TASSER servers were used to search for plausible templates.
Reflecting the clear two-domain structure of I329L, a distributed approach was used: the
I329L ECD was modelled using the Nogo receptor ECD (PDB ID: 1OZN), the decorin pro-
tein core (PDB ID: 1XKU), the Lingo-1 ECD (PDB ID: 2ID5), the TLR3 ECD (PDB ID:
3CIY) and the I-TASSER basic I329L model as templates; the I329L ICD was modelled
using the TIR domain of TLR10 (PDB ID: 2J67) as a single template. After modeling
each of the domains, the TM region was modelled as a single α-helix and a complete
model was constructed, as shown in Figure 6.
Figure 6 – Ribbon model of the full length I329L protein. α-helixes are colored in light blue,
β-sheets are colored as purple arrows.
Hallmarks of the protein, such as the putative AUG starting sites and LRRs present
in the ECD, or the TIR-like regions of homology in the ICD, are detailed in Figure 7.
Figure 7 – Ribbon model of I329L domains. N-terminal is shown in blue, C-terminal is shown in
black. A) ECD: the putative starting sites are labeled and shown in green, and the two LRRs are
shown in red; B) ICD: the three TIR-like regions are shown in red.
No structure is known for TRIF; therefore, we are limited in terms of computational
predictions for determination of functional domains for interaction.
Regarding the ECD, after analysis of the TLR3 ECD chain structure66, two regions of
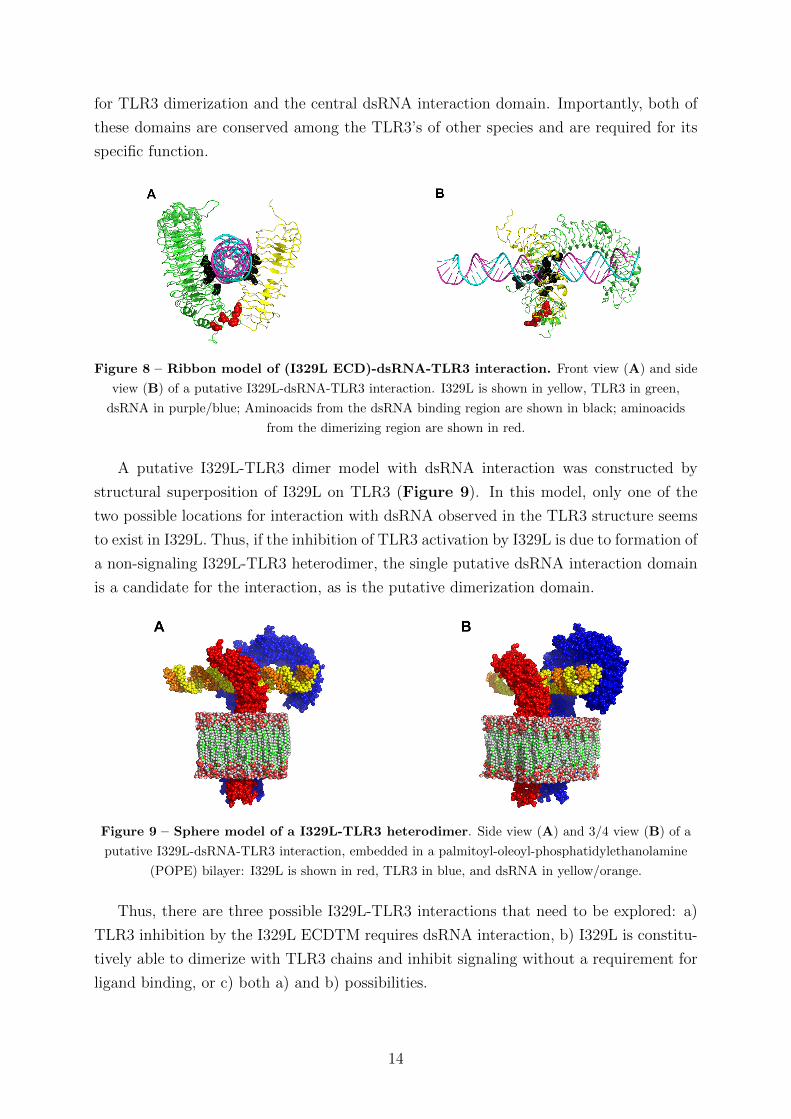
functional homology were identified (Figure 8), corresponding to the regions responsible
13
for TLR3 dimerization and the central dsRNA interaction domain. Importantly, both of
these domains are conserved among the TLR3’s of other species and are required for its
specific function.
Figure 8 – Ribbon model of (I329L ECD)-dsRNA-TLR3 interaction. Front view (A) and side
view (B) of a putative I329L-dsRNA-TLR3 interaction. I329L is shown in yellow, TLR3 in green,
dsRNA in purple/blue; Aminoacids from the dsRNA binding region are shown in black; aminoacids
from the dimerizing region are shown in red.
A putative I329L-TLR3 dimer model with dsRNA interaction was constructed by
structural superposition of I329L on TLR3 (Figure 9). In this model, only one of the
two possible locations for interaction with dsRNA observed in the TLR3 structure seems
to exist in I329L. Thus, if the inhibition of TLR3 activation by I329L is due to formation of
a non-signaling I329L-TLR3 heterodimer, the single putative dsRNA interaction domain
is a candidate for the interaction, as is the putative dimerization domain.
Figure 9 – Sphere model of a I329L-TLR3 heterodimer. Side view (A) and 3/4 view (B) of a
putative I329L-dsRNA-TLR3 interaction, embedded in a palmitoyl-oleoyl-phosphatidylethanolamine
(POPE) bilayer: I329L is shown in red, TLR3 in blue, and dsRNA in yellow/orange.
Thus, there are three possible I329L-TLR3 interactions that need to be explored: a)
TLR3 inhibition by the I329L ECDTM requires dsRNA interaction, b) I329L is constitu-
tively able to dimerize with TLR3 chains and inhibit signaling without a requirement for
ligand binding, or c) both a) and b) possibilities.
14
4.2 Expression of I329L and its principal domains
Based on the published results stating that I329L-mediated inhibition of the TLR3
signaling pathway is observed both by stimulating TLR3 with dsRNA and by ectopic
expression of TRIF, and considering the existence of homologies in both ECD and ICD
(Section 4.1), the following two constructs were cloned into the pcDNA3-Myc-HA ex-
pression plasmid: an N-terminal ECDTM construct, spanning aminoacids 1 to 260 and
containing two putative LRRs, eight putative glycosylation sites and a C-terminal hy-
drophobic signal-anchor sequence (Transmembrane, or TM); and a C-terminal ICD con-
struct, spanning aminoacids 261 to 329 and containing the TIR-like regions.
The pcDNA3-Myc-HA plasmid allows detection of expressed proteins using antibodies
either directed to the N-terminal tag (c-Myc) or C-terminal tag (HA). The full length
I329L and the two constructs (ECDTM and ICD) were expressed in Vero and 293T cells
and their expression was confirmed by immunoblotting and immunofluorescence (Figures
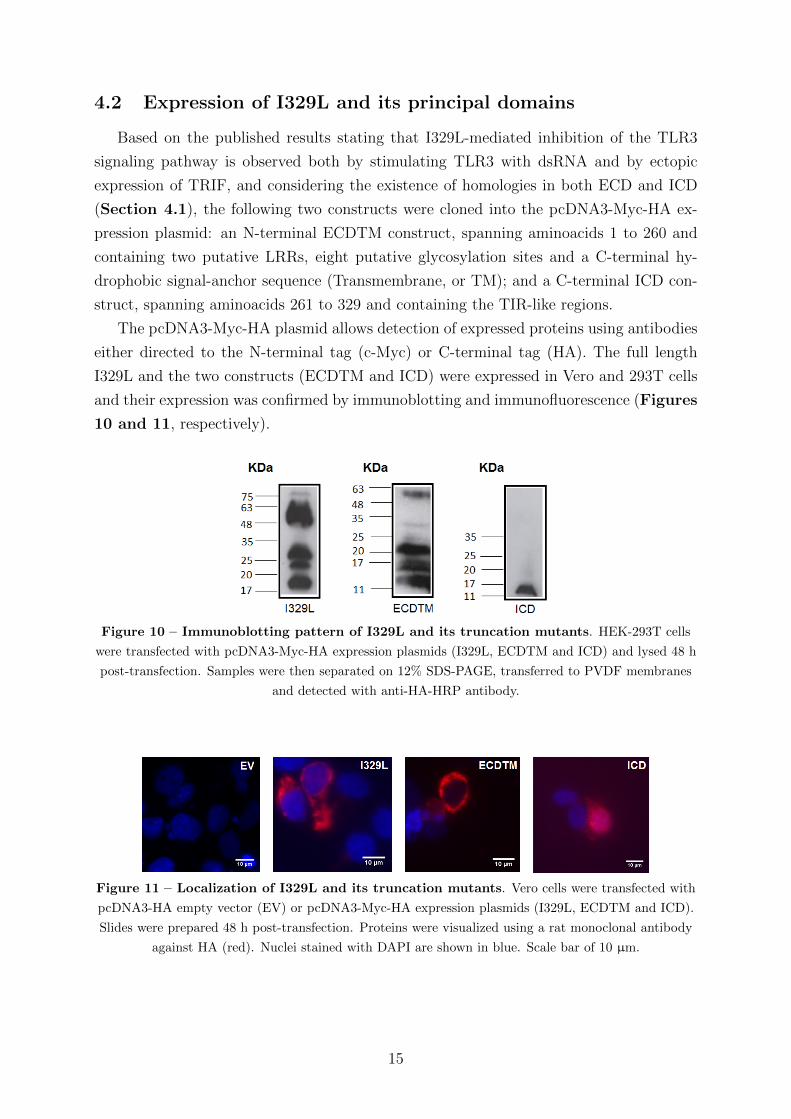
10 and 11, respectively).
Figure 10 – Immunoblotting pattern of I329L and its truncation mutants. HEK-293T cells
were transfected with pcDNA3-Myc-HA expression plasmids (I329L, ECDTM and ICD) and lysed 48 h
post-transfection. Samples were then separated on 12% SDS-PAGE, transferred to PVDF membranes
and detected with anti-HA-HRP antibody.
Figure 11 – Localization of I329L and its truncation mutants. Vero cells were transfected with
pcDNA3-HA empty vector (EV) or pcDNA3-Myc-HA expression plasmids (I329L, ECDTM and ICD).
Slides were prepared 48 h post-transfection. Proteins were visualized using a rat monoclonal antibody
against HA (red). Nuclei stained with DAPI are shown in blue. Scale bar of 10 µm.
15
The observed heterogeneity in the entire I329L and ECDTM proteins expressed after
transfection (Figure 10) is consistent and its origin is currently under investigation at the
laboratory; we hypothesize this phenomenon may be caused by the presence of different
transcripts (by alternative splicing, for instance) or translation starting at different AUGs
through special secondary structures of the I329L mRNA, as well as post-translational
modifications such as glycosylation, ubiquitination and/or proteolytic processing.
It is interesting to note the differential localization of the ICD after transfection, com-
pared to cells transfected with the entire I329L sequence or the I329L ECDTM (Figure
11). The ECDTM retains the same membrane localization as I329L, presumably due to
the anchoring sequence, while the ICD becomes distributed throughout the cytoplasm.
4.3 Inhibition of TLR3 signaling by I329L
4.3.1 I329L, a dual-action TLR3 antagonist
After confirmation of expression, the inhibitory activity of the constructs was as-
sayed through luciferase reporter assays of IRF3 transcription activation by both TLR3-
dependent signaling (Poly (I:C) stimulation, Figure 12A) and ectopic TRIF signaling
(transfection with TRIF expression plasmid, Figure 12B):
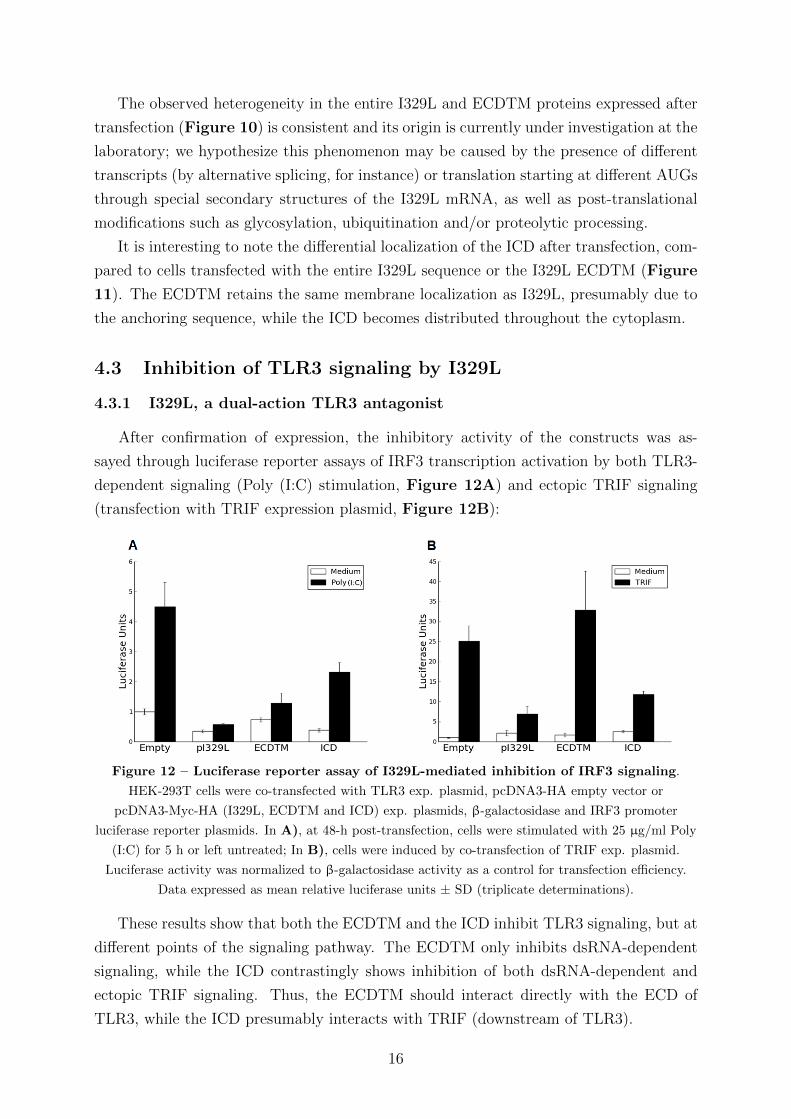
Figure 12 – Luciferase reporter assay of I329L-mediated inhibition of IRF3 signaling.
HEK-293T cells were co-transfected with TLR3 exp. plasmid, pcDNA3-HA empty vector or
pcDNA3-Myc-HA (I329L, ECDTM and ICD) exp. plasmids, β-galactosidase and IRF3 promoter
luciferase reporter plasmids. In A), at 48-h post-transfection, cells were stimulated with 25 µg/ml Poly
(I:C) for 5 h or left untreated; In B), cells were induced by co-transfection of TRIF exp. plasmid.
Luciferase activity was normalized to β-galactosidase activity as a control for transfection efficiency.
Data expressed as mean relative luciferase units ± SD (triplicate determinations).
These results show that both the ECDTM and the ICD inhibit TLR3 signaling, but at
different points of the signaling pathway. The ECDTM only inhibits dsRNA-dependent
signaling, while the ICD contrastingly shows inhibition of both dsRNA-dependent and
ectopic TRIF signaling. Thus, the ECDTM should interact directly with the ECD of
TLR3, while the ICD presumably interacts with TRIF (downstream of TLR3).
16

4.3.2 Inhibition of TLR3 signaling by the intracellular domain of I329L
After determining that the I329L ICD had an impact downstream of TLR3, we tested
for a direct TRIF-ICD interaction using the standard “pull down-Western blot” approach
using cells expressing TRIF with and without the full length I329L or the I329L ICD, and
using anti-TRIF or anti-HA (I329L or ICD) antibodies for precipitation of the protein
complexes. If an interaction exists, TRIF would be present in the sample after IP with
anti-HA antibodies, and I329L would be present after IP with anti-TRIF antibodies,
which would be shown by subsequent Western blot. The absence of TRIF after anti-HA
IP in the absence of I329L would serve as a negative control.
Indeed, an interaction between TRIF and the I329L ICD was observed, as shown in
Figure 13 by co-precipitation of the ICD with TRIF (Figure 13B and 13C) – thus
indicating that the ICD inhibits TLR3 signaling by means of a protein-protein interaction
with TRIF, which would interfere with signal transmission downstream of TLR3.
Figure 13 – The I329L ICD co-immunoprecipitates with TRIF. HEK-293T cells were
transfected with TRIF exp. plasmid and pcDNA3-HA empty vector or pcDNA3-Myc-HA exp. plasmids
(I329L and ICD), and lysed 48 h post-transfection. Samples were then separated on 12% SDS-PAGE,
transferred to PVDF membranes and detected with anti-HA or anti-TRIF antibodies, as indicated:
A) Expression in the samples was detected before immunoprecipitation through Western blot;
B) Immunoprecipitation with anti-TRIF followed by Western blot; C) Immunoprecipitation with
anti-HA followed by Western blot. IgGs were detected as a positive control for IP.
The I329L ICD contains two of its three TIR-like boxes as plausible candidates for
interaction with TRIF, and thus for mutation and loss-of-function studies. Interestingly,
the 2nd TIR-like box showed a conserved, centrally located structure and, after repeated
modeling attempts, seemed to mimic the structural basis of the TIR domain interaction.
To test whether this homology mediated the I329L ICD-TLR interaction, a mutational
approach was designed.
As the size of individual TIR-like boxes is relatively small (composed of 3 to 5
aminoacids) and no critical aminoacids for this particular interaction were known, a rel-
atively atypical approach was used. Rather than changing an aminoacid into an alanine
and determining the impact of that specific aminoacid on protein function, a mutation was
selected to have the most possible impact on the structure around the changed aminoacid
17
by inverting its properties, for instance, changing a charged aminoacid to an hydrophobic
aminoacid. Specifically, the 3rd aminoacid of the 2nd TIR-like box, a phenylalanine, was
mutated into a serine through point mutation (TTC >TCC). The predicted impact of
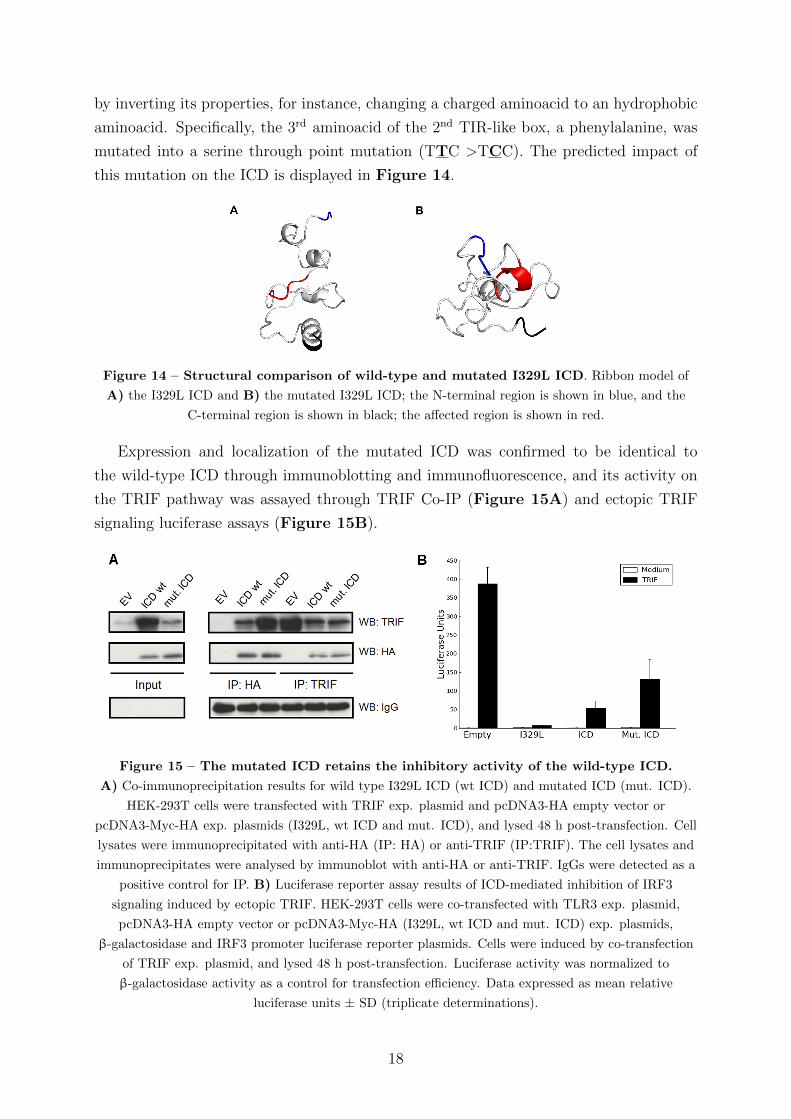
this mutation on the ICD is displayed in Figure 14.
Figure 14 – Structural comparison of wild-type and mutated I329L ICD. Ribbon model of
A) the I329L ICD and B) the mutated I329L ICD; the N-terminal region is shown in blue, and the
C-terminal region is shown in black; the affected region is shown in red.
Expression and localization of the mutated ICD was confirmed to be identical to
the wild-type ICD through immunoblotting and immunofluorescence, and its activity on
the TRIF pathway was assayed through TRIF Co-IP (Figure 15A) and ectopic TRIF
signaling luciferase assays (Figure 15B).
Figure 15 – The mutated ICD retains the inhibitory activity of the wild-type ICD.
A) Co-immunoprecipitation results for wild type I329L ICD (wt ICD) and mutated ICD (mut. ICD).
HEK-293T cells were transfected with TRIF exp. plasmid and pcDNA3-HA empty vector or
pcDNA3-Myc-HA exp. plasmids (I329L, wt ICD and mut. ICD), and lysed 48 h post-transfection. Cell
lysates were immunoprecipitated with anti-HA (IP: HA) or anti-TRIF (IP:TRIF). The cell lysates and
immunoprecipitates were analysed by immunoblot with anti-HA or anti-TRIF. IgGs were detected as a
positive control for IP. B) Luciferase reporter assay results of ICD-mediated inhibition of IRF3
signaling induced by ectopic TRIF. HEK-293T cells were co-transfected with TLR3 exp. plasmid,
pcDNA3-HA empty vector or pcDNA3-Myc-HA (I329L, wt ICD and mut. ICD) exp. plasmids,
β-galactosidase and IRF3 promoter luciferase reporter plasmids. Cells were induced by co-transfection
of TRIF exp. plasmid, and lysed 48 h post-transfection. Luciferase activity was normalized to
β-galactosidase activity as a control for transfection efficiency. Data expressed as mean relative
luciferase units ± SD (triplicate determinations).
18

Although the mutated ICD was repeatedly less inhibitory than the wild-type in the
luciferase assays (Figure 15B), its principal activity was clearly inhibitory and not sig-
nificantly different from the activity of the wild-type ICD. In addition, as the mutant
ICD still interacts with TRIF (as observed through the Co-IP assay, Figure 15A), this
mutation does not significantly abrogate I329L ICD-mediated inhibition of ectopic TRIF
activation. This suggests that the TIR-like boxes in the I329L ICD may have redundant
activity, with the 1st and 3rd TIR-like boxes perhaps compensating for the loss of the
2nd TIR-like box. Other mutations to test for this hypothesis are required in order to
determine the exact mechanism of interaction.
4.3.3 Inhibition of TLR3 signaling by the extracellular-transmembrane do-
main of I329L
In order to define the interaction of the I329L ECDTM with TLR3, a construct starting
at the 3rd ATG of I329L (and therefore, 3rd methionine, or 3M) and spanning aminoacids
159 to 260 was prepared (3MECDTM). The corresponding protein is predicted to lack the
dsRNA interaction region and contain one putative LRR, one putative glycosylation site
and the transmembrane anchor sequence. The structural prediction for this new construct
is shown in Figure 16.
Figure 16 – Sphere model of 3MI329L-dsRNA-TLR3 interaction. A) Side view and B) 3/4
view, embedded in a POPE bilayer: 3MI329L is shown in red, TLR3 in blue, and dsRNA in
yellow/orange.
Expression and localization of this construct was confirmed by immunoblotting and
immunofluorescence, and a luciferase reporter assay for IRF-3 induction was done to test
its activity on TLR3 inhibition (Figure 17). The surprising heterogeneity of the ex-
pressed molecule (Figure 17A) raises the possibility of ubiquitination of the 3MECDTM
(and therefore of I329L), as the different protein bands consist of a 8.5 KDa molecular
weight ladder (corresponding to the 8.5 KDa molecular weight of ubiquitin), but this
remains to be investigated. The membrane localization of 3MECDTM is identical to the
full length ECDTM (Figure 17B), suggesting that the signal peptide of I329L may not
be required for its correct localization (pursued in Appendix 1).
19
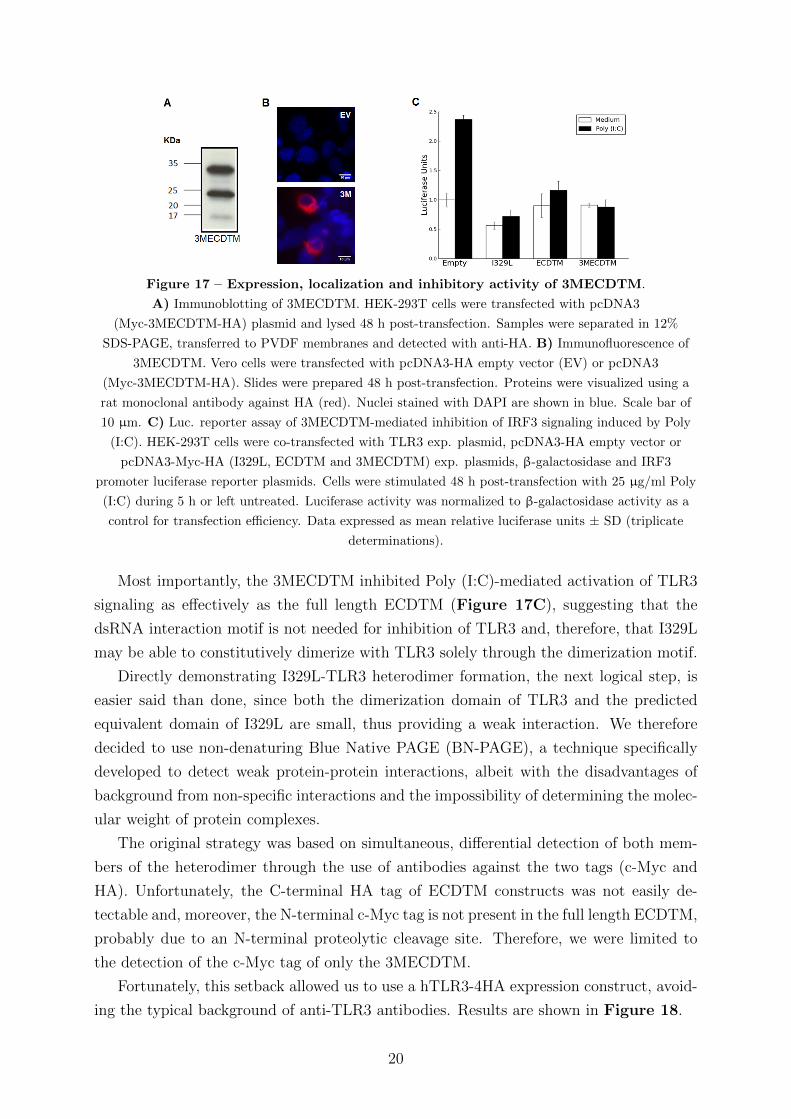
Figure 17 – Expression, localization and inhibitory activity of 3MECDTM.
A) Immunoblotting of 3MECDTM. HEK-293T cells were transfected with pcDNA3
(Myc-3MECDTM-HA) plasmid and lysed 48 h post-transfection. Samples were separated in 12%
SDS-PAGE, transferred to PVDF membranes and detected with anti-HA. B) Immunofluorescence of
3MECDTM. Vero cells were transfected with pcDNA3-HA empty vector (EV) or pcDNA3
(Myc-3MECDTM-HA). Slides were prepared 48 h post-transfection. Proteins were visualized using a
rat monoclonal antibody against HA (red). Nuclei stained with DAPI are shown in blue. Scale bar of
10 µm. C) Luc. reporter assay of 3MECDTM-mediated inhibition of IRF3 signaling induced by Poly
(I:C). HEK-293T cells were co-transfected with TLR3 exp. plasmid, pcDNA3-HA empty vector or
pcDNA3-Myc-HA (I329L, ECDTM and 3MECDTM) exp. plasmids, β-galactosidase and IRF3
promoter luciferase reporter plasmids. Cells were stimulated 48 h post-transfection with 25 µg/ml Poly
(I:C) during 5 h or left untreated. Luciferase activity was normalized to β-galactosidase activity as a
control for transfection efficiency. Data expressed as mean relative luciferase units ± SD (triplicate
determinations).
Most importantly, the 3MECDTM inhibited Poly (I:C)-mediated activation of TLR3
signaling as effectively as the full length ECDTM (Figure 17C), suggesting that the
dsRNA interaction motif is not needed for inhibition of TLR3 and, therefore, that I329L
may be able to constitutively dimerize with TLR3 solely through the dimerization motif.
Directly demonstrating I329L-TLR3 heterodimer formation, the next logical step, is
easier said than done, since both the dimerization domain of TLR3 and the predicted
equivalent domain of I329L are small, thus providing a weak interaction. We therefore
decided to use non-denaturing Blue Native PAGE (BN-PAGE), a technique specifically
developed to detect weak protein-protein interactions, albeit with the disadvantages of
background from non-specific interactions and the impossibility of determining the molec-
ular weight of protein complexes.
The original strategy was based on simultaneous, differential detection of both mem-
bers of the heterodimer through the use of antibodies against the two tags (c-Myc and
HA). Unfortunately, the C-terminal HA tag of ECDTM constructs was not easily de-
tectable and, moreover, the N-terminal c-Myc tag is not present in the full length ECDTM,
probably due to an N-terminal proteolytic cleavage site. Therefore, we were limited to
the detection of the c-Myc tag of only the 3MECDTM.
Fortunately, this setback allowed us to use a hTLR3-4HA expression construct, avoid-
ing the typical background of anti-TLR3 antibodies. Results are shown in Figure 18.
20

Figure 18 – The I329L ECDTM interacts with TLR3. HEK-293T cells were co-transfected with
TLR3-4HA exp. plasmid and pcDNA3-HA empty vector or pcDNA3-Myc-HA (ECDTM and
3MECDTM) exp. plasmids, and lysed 48 h post-transfection. A) Immunoblotting of TLR3-4HA.
Proteins were separated on 12% SDS-PAGE and detected with anti-HA. Processed (70 KDa) and
non-processed TLR3 (104 KDa) is detected; B) Immunoblotting of TLR3-4HA co-expressed with
ECDTM constructs. Proteins were separated on 4-16% BN-PAGE and detected with anti-HA. Bands
are indicated with arrows for clarity; C) Immunoblotting of TLR3-4HA co-expressed with 3MECDTM.
Proteins were separated on 4-16% BN-PAGE and detected with anti-c-Myc. The band corresponding to
a putative interaction with TLR3 is indicated with an arrow.
Differences between TLR3 and TLR3+I329L-TM expressing cells were observed, al-
beit with very low band resolution and without certainty that those interactions corre-
spond to TLR3 + I329LTM heterodimers. The appearance of new bands in the TLR3
+ 3MECDTM sample with anti-Myc antibody (Figure 18C) suggested that there may
be, indeed, an interaction between TLR3 and 3MECDTM, but this still requires a more
definitive proof.
Therefore, it was decided to use an in vivo protein crosslinking protocol using disuc-
cinimidyl suberate (DSS), a cell membrane-permeable crosslinking reagent with a reactive
N-Hydroxysuccinimide ester group at each end of an 8-carbon spacer arm. This reagent
crosslinks closely linked (<11.40A) nearest neighbour proteins by reacting with lysine
amino groups (Figure 19).
Figure 19 – Disuccinimidyl suberate (DSS). A) Structural formula; B) Crosslinking reaction67
As the reagent is membrane-permeable, it may be added to cell cultures before and
after Poly (I:C) stimulation, “freezing” any possible heterodimers in place. After this
protocol, cells are lysed and the protein extracts are analysed by SDS-PAGE.
In general, this protocol detects protein complexes by detection of a band in SDS-
PAGE gels which does not appear without the crosslinking reaction, and with a molecu-
lar weight (MW) corresponding to the sum of the interacting proteins’ MW. Results are
shown in Figure 20.
21

Figure 20 – Crosslinking reaction of ECDTM-TLR3. HEK-293T cells were co-transfected with
TLR3-4HA exp. plasmid and pcDNA3-HA empty vector or pcDNA3-Myc-HA (ECDTM and
3MECDTM) exp. plasmids, and lysed 48 h post-transfection. A) Immunoblotting of 3MECDTM
without crosslinking (Figure 17A); B) Immunoblotting of 3MECDTM with crosslinking. Cells were
crosslinked for 45 min at RT and then lysed. Samples were separated on 4-20% SDS-PAGE and detected
with anti-c-Myc. Novel bands corresponding to putative homodimers are indicated with arrows.
C) Immunoblotting of TLR3-4HA co-expressed with ECDTM constructs. Cells were crosslinked for 45
min at RT and then lysed. Samples were separated on 4-20% SDS-PAGE and detected with anti-HA.
Separation of the figure was done due to a need for overexposure to develop the lower half.
In control cells expressing the 3MECDTM, three expressed proteins were detected with
MWs 16, 24 and 33 KDa (Figure 20A). However, upon exposure to the crosslinking
reagent, two extra bands were observed with 45 and ∼70 KDa, perhaps dimers of the
24 and 33 KDa proteins (Figure 20B). In general, the immunoblotting revealed a large
amount of background with increased MW relative to the proteins to which the antibodies
are specific – this effect is created by crosslinking of nearby proteins of diverse sizes which
do not specifically interact with the proteins of interest. Interactions can be detected by
the appearance of defined bands that stand out from background ”noise”.
The possibility of 3MECDTM homodimerization is interesting, as it suggests the ca-
pacity of constitutive homodimerization of the full length protein, which might bind to
dsRNA and serve as a decoy without signaling capability. Only the 3MECDTM was used
from this point onwards, since it had a similar behaviour to the entire ECDTM and could
be detected with the anti-c-Myc antibody. In order to obtain a better resolution for the
high MW bands present in this experiment, the same samples were resolved on lower
concentration SDS-PAGE (6%) and then immunoblotted again with antibodies against
HA and c-Myc (Figure 21).
22

Figure 21 – Immunoblotting of TLR3 crosslinked with 3MECDTM. HEK-293T cells were
co-transfected with TLR3 exp. plasmid and pcDNA3-HA empty vector or pcDNA3-Myc-HA
(3MECDTM). 48 h post-transfection, cells were stimulated with 100 µg/ml Poly (I:C) for 30 min or left
untreated, crosslinked for 45 min at RT and then lysed. Samples were separated on 6% SDS-PAGE and
detected with A) anti-HA and B) anti-c-Myc. Novel bands corresponding to a putative TLR3
homodimer, 3MECDTM homodimer and TLR3-3MECDTM heterodimer are indicated with arrows
from left to right, respectively.
As can be seen with anti-HA, a band with apx. 150 KDa appeared (Figure 21A),
which is the expected MW of a processed TLR3 dimer; with anti-c-Myc, the previously ob-
served band below 63 KDa appeared once more when 3MECDTM is expressed alone, and
a new band with apx. 125-130 KDa (corresponding to the sum of MWs of non-processed
TLR3 and the central 3MECDTM band MW) appeared when expressed together with
TLR3 and stimulated with Poly (I:C) (Figure 21B). This band was later confirmed to
be present without need for Poly (I:C) stimulation.
These results support (but do not prove) the hypothesis that I329L directly interacts
and is constitutively able to form a heterodimer with TLR3.
4.3.4 Impact of the full length I329L and ECDTM on TLR3-dependent ac-
tivation of c-Src
Recognition of dsRNA by TLR3 and subsequent dimerization leads to availability of
a c-Src binding site on the TLR3 intracellular domain. The subsequently recruited c-Src
is then autophosphorylated, and stimulates a transient and gene expression-independent
increase in cell proliferation, functionally detected by a wound healing assay40.
Preliminary results of the wound healing assay (data not shown) indicated heightened
cell proliferation in TLR3-positive cells when co-expressing the I329L ECDTM. As a more
objective approach, we focused on detection of the autophosphorylated c-Src P-Tyr416
(P-Src), induced by TLR3 dimerization (Figure 22). Both Vero and HEK-293T cells
were used. Vero cells constitutively express TLR3, whereas the HEK-293T cells do not
23
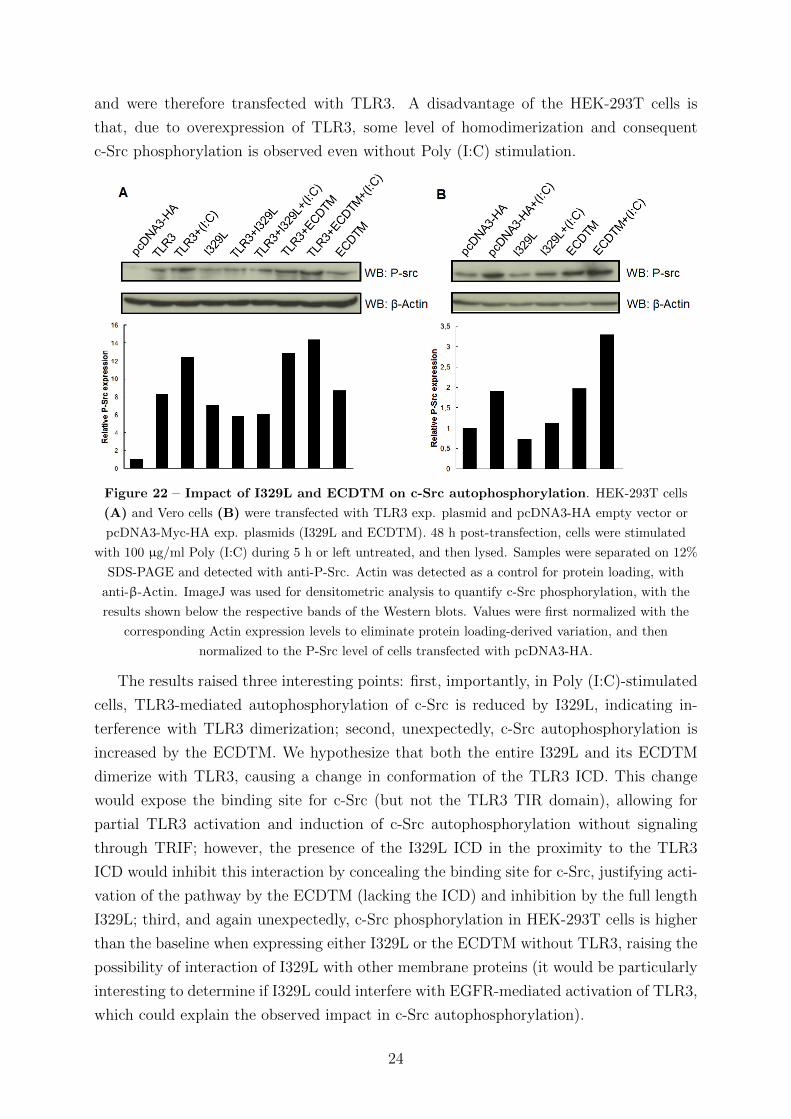
and were therefore transfected with TLR3. A disadvantage of the HEK-293T cells is
that, due to overexpression of TLR3, some level of homodimerization and consequent
c-Src phosphorylation is observed even without Poly (I:C) stimulation.
Figure 22 – Impact of I329L and ECDTM on c-Src autophosphorylation. HEK-293T cells
(A) and Vero cells (B) were transfected with TLR3 exp. plasmid and pcDNA3-HA empty vector or
pcDNA3-Myc-HA exp. plasmids (I329L and ECDTM). 48 h post-transfection, cells were stimulated
with 100 µg/ml Poly (I:C) during 5 h or left untreated, and then lysed. Samples were separated on 12%
SDS-PAGE and detected with anti-P-Src. Actin was detected as a control for protein loading, with
anti-β-Actin. ImageJ was used for densitometric analysis to quantify c-Src phosphorylation, with the
results shown below the respective bands of the Western blots. Values were first normalized with the
corresponding Actin expression levels to eliminate protein loading-derived variation, and then
normalized to the P-Src level of cells transfected with pcDNA3-HA.
The results raised three interesting points: first, importantly, in Poly (I:C)-stimulated
cells, TLR3-mediated autophosphorylation of c-Src is reduced by I329L, indicating in-
terference with TLR3 dimerization; second, unexpectedly, c-Src autophosphorylation is
increased by the ECDTM. We hypothesize that both the entire I329L and its ECDTM
dimerize with TLR3, causing a change in conformation of the TLR3 ICD. This change
would expose the binding site for c-Src (but not the TLR3 TIR domain), allowing for
partial TLR3 activation and induction of c-Src autophosphorylation without signaling
through TRIF; however, the presence of the I329L ICD in the proximity to the TLR3
ICD would inhibit this interaction by concealing the binding site for c-Src, justifying acti-
vation of the pathway by the ECDTM (lacking the ICD) and inhibition by the full length
I329L; third, and again unexpectedly, c-Src phosphorylation in HEK-293T cells is higher
than the baseline when expressing either I329L or the ECDTM without TLR3, raising the
possibility of interaction of I329L with other membrane proteins (it would be particularly
interesting to determine if I329L could interfere with EGFR-mediated activation of TLR3,
which could explain the observed impact in c-Src autophosphorylation).
24

4.4 I329L is a general inhibitor of TLR signaling
Inhibition of Poly (I:C)-stimulated activation of TLR3 by I329L was mediated by
the ECD of I329L (Section 4.3.1) and by the 3MECDTM truncation mutant (Section
4.3.3). The transcript of the latter encodes the putative dimerization sequence, but not
the dsRNA binding sequence. Although the specificity of signaling of the NF-κB pathway
by the different TLRs is determined by different ligands, a conserved dimerization domain
is also an important factor69. Thus, if the interaction of I329L with TLR3 depends on the
dimerization domain rather than the ligand binding structure, then it might also inhibit
signal transduction via TLRs other than TLR3. Therefore, it was decided to test if the
I329L 3MECDTM and full length I329L would also inhibit signaling by other TLRs.
The results from the luciferase assays are shown in Figure 23 and summarized in
Table 1, and clearly demonstrate that not only the full length I329L, but also the
3MECDTM, inhibited signaling from TLRs 3, 4, 5, 7 and 9.
Figure 23 – I329L is a general inhibitor of TLR signaling without need for a
ligand-binding domain. Luciferase reporter assay of inhibition of NF-κB by full length I329L and
3MECDTM through the signaling pathways of A) TLR4; B) TLR5; C) TLR7. Vero cells were
co-transfected with different TLR exp. plasmids and pcDNA3-HA empty vector or pcDNA3-Myc-HA
(I329L and 3MECDTM) exp. plasmids, and the β-galactosidase and PRD2 promoter luciferase reporter
plasmids (PRD2 responds to NF-κB). 24 h post-transfection, cells in the TLR7 assay were stimulated
with 1 µg/ml of R848 overnight, or left untreated; 48 h post-transfection, cells in the TLR4 assay were
stimulated with 100 ng/ml of E. coli lipopolysacharide, while cells in the TLR5 assay were stimulated
with 100 ng/ml of flagellin, both during 5 h (or left untreated). Luciferase activity was normalized to
β-galactosidase activity as a control for transfection efficiency. Data expressed as mean relative
luciferase units ± SD (triplicate determinations).
25
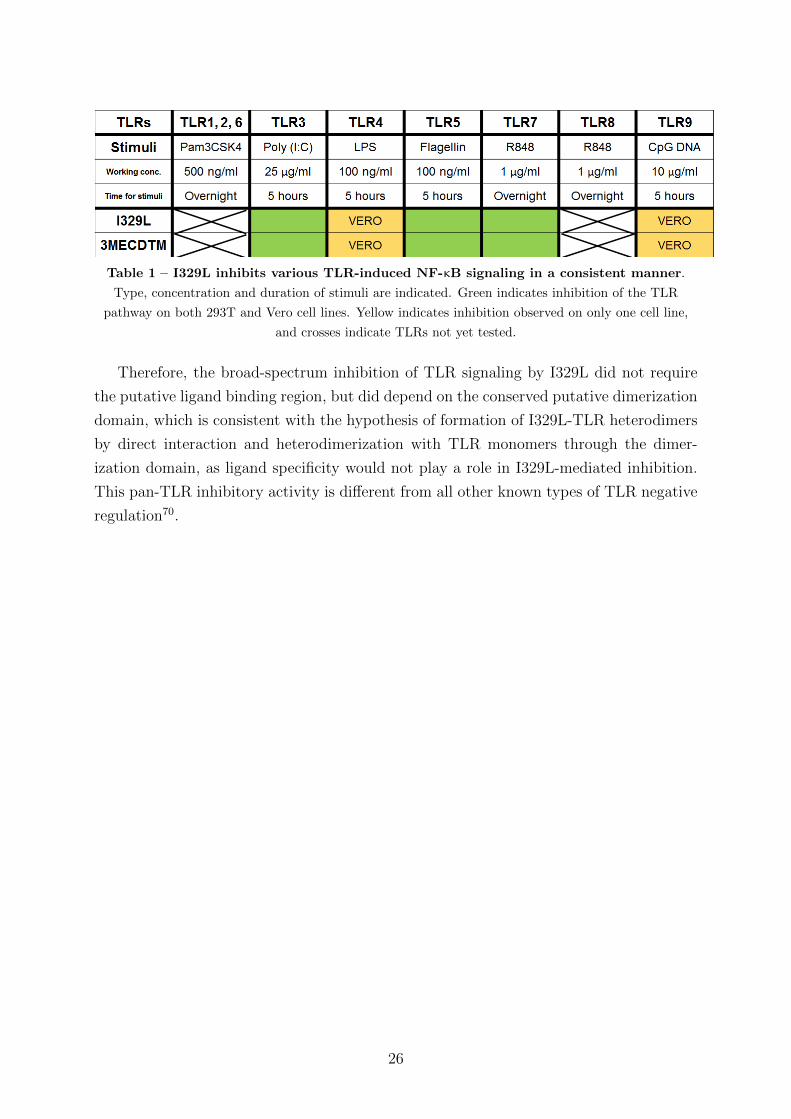
Table 1 – I329L inhibits various TLR-induced NF-κB signaling in a consistent manner.
Type, concentration and duration of stimuli are indicated. Green indicates inhibition of the TLR
pathway on both 293T and Vero cell lines. Yellow indicates inhibition observed on only one cell line,
and crosses indicate TLRs not yet tested.
Therefore, the broad-spectrum inhibition of TLR signaling by I329L did not require
the putative ligand binding region, but did depend on the conserved putative dimerization
domain, which is consistent with the hypothesis of formation of I329L-TLR heterodimers
by direct interaction and heterodimerization with TLR monomers through the dimer-
ization domain, as ligand specificity would not play a role in I329L-mediated inhibition.
This pan-TLR inhibitory activity is different from all other known types of TLR negative
regulation70.
26

5 Conclusions
Non-infectious disease, caused by a failure to control the inflammatory immune re-
sponse, is a growing problem as both the average age and associated disease susceptibility
are on the increase. The development of new treatments is logically focused on discovering
strategies to inhibit this inflammatory response.
Toll-like Receptors are an essential part of the innate inflammatory response, provid-
ing fast and effective triggering of a primary proinflammatory response to contain and/or
eliminate an infection and chronic disease of the host. However, an exacerbated and/or
prolonged inflammatory response also results in increased morbidity and chronic disease.
Viral strategies for down-regulation of signaling through TLRs thus provide a rational
route for controlling inflammatory responses.
The I329L protein of the African Swine Fever Virus was known to inhibit activation
of TLR3. Therefore, this work focused on further defining the mechanism of I329L inhi-
bition through bioinformatic, biochemical and biological investigations. Importantly, we
determined that the extracellular and intracellular domains of I329L both inhibit dsRNA-
stimulated activation of TLR3, but by different mechanisms.
Significantly, the extracellular domain (ECD) and, in addition, an ECD truncation
mutant containing the putative dimerization domain, but not the ligand binding do-
main (3MECDTM), were shown to inhibit TLR signaling not only of TLR3, but also
by TLRs 4, 5, 7 and 9. This observation is consistent with the hypothesis that the
inhibition of TLR signaling mediated by I329L occurs through interference with TLR
dimerization and possible formation of I329L-TLR non-signaling heterodimers. Further
mutation/truncation analysis of the 3MECDTM to formally demonstrate a structural
requirement for the dimerization domain (and a possible contributory role of the single
present LRR) is planned. It will also be interesting to determine the precise impact of
ECDTM on the signal transduction pathways stimulated by TLRs 3, 4, 5, 7 and 9.
Experiments to directly demonstrate the existence of an I329L-TLR3 heterodimer were
inconclusive. However, and as previously discussed (Section 4.3.4), the impact of I329L
and the I329L ECD on TLR3-stimulated c-Src autophosphorylation is consistent with
the formation of I329L-TLR3 heterodimers. Specifically, the induction of c-Src phospho-
rylation by the ECDTM truncation mutant lacking the intracellular domain (ICD) and
inhibition by the full-length I329L would imply an effect based on dimerization of I329L
with TLR3, that is then modulated by availability of the TLR3 binding site for c-Src. The
observation that the assumed interaction of the ECD of I329L with TLR3 leads to acti-
vation of c-Src, but inhibition of IRF-3 and NF-κB, is interesting, as it suggests that only
a partial change in conformation of the TLR3 ICD occurs upon I329L ECD interaction.
27

A combination of luciferase reporter gene assays and co-immunoprecipitation experi-
ments conclusively demonstrated that the ICD of I329L inhibits TLR3 signal transmission
through direct interaction with the adaptor signaling protein TRIF. Unfortunately, the
mutation of an ICD sequence predicted to be a candidate for interaction with TRIF did
not have a significant impact on ICD activity. Thus, further mutational analysis will be
required to define the structural basis for the I329L ICD-TRIF interaction.
Both the ECD and ICD may have potential as TLR antagonists. The pan-TLR in-
hibitory activity of the ECD is novel in terms of what is known about TLR negative
regulation, and harnessing this effect may be useful for inducing an anti-inflammatory
state to treat pathologic inflammation. The fact that this activity is also observed with
a relatively small segment of I329L (3MECDTM) is particularly useful for formulating
future applications of this protein. Therapeuthic use of this activity is, however, limited
by the need to deliver the protein to lipid membranes where TLRs are present.
The TRIF-ICD interaction could be harnessed to inhibit signal transmission in both
TLR3 and TLR4-driven pathologic inflammation, since TRIF is common to both signaling
pathways. The observed behavior of the ICD as a soluble protein would be useful, since its
delivery would be facilitated in regards to the highly glycosylated and membrane-located
ECD.
In conclusion, we have determined that the I329L protein of the African Swine Fever
Virus acts as an inhibitor of the TLR signaling pathway through two distinct activities:
1) pan-TLR inhibition by the I329L ECD, possibly through formation of non-signaling
heterodimers mediated by an I329L ECD domain that is homologous to the TLR ECD
dimerization domain;
2) inhibition of TLR3 signal transmission through interaction between the ICD of
I329L and the intracellular downstream signaling intermediate, TRIF.
28

6 References
1. Akira, S. et al. Pathogen recognition and innate immunity. Cell 124, 783–801. issn: 0092-
8674 (Feb. 2006).
2. Litman, G. W. et al. Reconstructing immune phylogeny: new perspectives. Nature Reviews.
Immunology 5, 866–879. issn: 1474-1733 (Nov. 2005).
3. Takeuchi, O. et al. Pattern recognition receptors and inflammation. Cell 140, 805–820.
issn: 1097-4172 (Mar. 2010).
4. Iwasaki, A. et al. Toll-like receptor control of the adaptive immune responses. Nature
Immunology 5, 987–995. issn: 1529-2908 (Oct. 2004).
5. Dempsey, P. W. et al. The art of war: Innate and adaptive immune responses. Cellular
and molecular life sciences: CMLS 60, 2604–2621. issn: 1420-682X (Dec. 2003).
6. Arstila, T. P. et al. A direct estimate of the human alphabeta T cell receptor diversity.
Science (New York, N.Y.) 286, 958–961. issn: 0036-8075 (Oct. 1999).
7. Sablitzky, F. et al. Somatic mutation and clonal expansion of B cells in an antigen-driven
immune response. The EMBO Journal 4, 345–350. issn: 0261-4189 (Feb. 1985).
8. Sprent, J. Immunological memory. Current Opinion in Immunology 9, 371–379. issn: 0952-
7915 (June 1997).
9. Pancer, Z. et al. The evolution of adaptive immunity. Annual Review of Immunology 24,
497–518. issn: 0732-0582 (2006).
10. Janeway, C. A. The immune system evolved to discriminate infectious nonself from non-
infectious self. Immunology Today 13, 11–16. issn: 0167-5699 (Jan. 1992).
11. Weber, F. et al. Double-stranded RNA is produced by positive-strand RNA viruses and
DNA viruses but not in detectable amounts by negative-strand RNA viruses. Journal of
Virology 80, 5059–5064. issn: 0022-538X (May 2006).
12. Nicaise, V. et al. Recent Advances in PAMP-Triggered Immunity against Bacteria: Pattern
Recognition Receptors Watch over and Raise the Alarm. Plant Physiology 150, 1638–1647.
issn: 0032-0889, 1532-2548 (Jan. 2009).
13. Vollmer, J. et al. Characterization of three CpG oligodeoxynucleotide classes with distinct
immunostimulatory activities. European Journal of Immunology 34, 251–262. issn: 0014-
2980 (Jan. 2004).
14. Mogensen, T. H. Pathogen Recognition and Inflammatory Signaling in Innate Immune
Defenses. Clinical Microbiology Reviews 22, 240–273. issn: 0893-8512 (Apr. 2009).
15. Elphick, D. A. et al. Paneth cells: their role in innate immunity and inflammatory disease.
Gut 54, 1802–1809. issn: 0017-5749 (Dec. 2005).
16. Lotze, M. T. et al. The grateful dead: damage-associated molecular pattern molecules and
reduction/oxidation regulate immunity. Immunological Reviews 220, 60–81. issn: 0105-
2896 (Dec. 2007).
29

17. Lee, M. S. et al. Signaling pathways downstream of pattern-recognition receptors and their
cross talk. Annual Review of Biochemistry 76, 447–480. issn: 0066-4154 (2007).
18. Lee, M. S. et al. Pattern-recognition receptor signaling initiated from extracellular, mem-
brane, and cytoplasmic space. Molecules and Cells 23, 1–10. issn: 1016-8478 (Feb. 2007).
19. Hashimoto, C. et al. The Toll gene of Drosophila, required for dorsal-ventral embryonic
polarity, appears to encode a transmembrane protein. Cell 52, 269–279. issn: 0092-8674
(Jan. 1988).
20. Gay, N. J. et al. Drosophila Toll and IL-1 receptor. Nature 351, 355–356. issn: 0028-0836
(May 1991).
21. Lemaitre, B. et al. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls
the potent antifungal response in Drosophila adults. Cell 86, 973–983. issn: 0092-8674
(Sept. 1996).
22. Medzhitov, R. et al. A human homologue of the Drosophila Toll protein signals activation
of adaptive immunity. Nature 388, 394–397. issn: 0028-0836 (July 1997).
23. Piras, V. et al. Beyond MyD88 and TRIF Pathways in Toll-Like Receptor Signaling. Fron-
tiers in Immunology 5. issn: 1664-3224. doi:10.3389/fimmu.2014.00070. <http://www.
ncbi.nlm.nih.gov/pmc/articles/PMC3932418/> (visited on 07/25/2014) (Feb. 2014).
24. Roach, J. C. et al. The evolution of vertebrate Toll-like receptors. Proceedings of the Na-
tional Academy of Sciences of the United States of America 102, 9577–9582. issn: 0027-
8424 (July 2005).
25. Takeda, K. et al. Toll-like receptors in innate immunity. International Immunology 17,
1–14. issn: 0953-8178, 1460-2377 (Jan. 2005).
26. Barrio, L. et al. TLR4 signaling shapes B cell dynamics via MyD88-dependent pathways
and Rac GTPases. Journal of Immunology (Baltimore, Md.: 1950) 191, 3867–3875. issn:
1550-6606 (Oct. 2013).
27. Takeda, K. et al. TLR signaling pathways. Seminars in Immunology 16, 3–9. issn: 1044-
5323 (Feb. 2004).
28. Alexopoulou, L. et al. Recognition of double-stranded RNA and activation of NF-kappaB
by Toll-like receptor 3. Nature 413, 732–738. issn: 0028-0836 (Oct. 2001).
29. Vercammen, E. et al. Sensing of viral infection and activation of innate immunity by toll-
like receptor 3. Clinical Microbiology Reviews 21, 13–25. issn: 1098-6618 (Jan. 2008).
30. Karpus, O. N. et al. Triggering of the dsRNA Sensors TLR3, MDA5, and RIG-I Induces
CD55 Expression in Synovial Fibroblasts. PLoS ONE 7, e35606 (May 2012).
31. Slater, L. et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to
rhinovirus in bronchial epithelium. PLoS pathogens 6, e1001178. issn: 1553-7374 (2010).
32. Liu, T. et al. TLR3 deficiency impairs spinal cord synaptic transmission, central sensiti-
zation, and pruritus in mice. The Journal of Clinical Investigation 122, 2195–2207. issn:
1558-8238 (June 2012).
30

33. Lee, B. L. et al. UNC93B1 mediates differential trafficking of endosomal TLRs. eLife 2,
e00291. issn: 2050-084X (Feb. 2013).
34. Matsumoto, M. et al. Subcellular localization of Toll-like receptor 3 in human dendritic
cells. Journal of Immunology (Baltimore, Md.: 1950) 171, 3154–3162. issn: 0022-1767
(Sept. 2003).
35. Leonard, J. N. et al. The TLR3 signaling complex forms by cooperative receptor dimer-
ization. Proceedings of the National Academy of Sciences of the United States of America
105, 258–263. issn: 1091-6490 (Jan. 2008).
36. Yamashita, M. et al. Epidermal growth factor receptor is essential for Toll-like receptor 3
signaling. Science Signaling 5, ra50. issn: 1937-9145 (July 2012).
37. Sarkar, S. N. et al. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-
stranded RNA signaling. Nature Structural & Molecular Biology 11, 1060–1067. issn: 1545-
9993 (Nov. 2004).
38. Qi, R. et al. Proteolytic processing regulates Toll-like receptor 3 stability and endosomal lo-
calization. The Journal of Biological Chemistry 287, 32617–32629. issn: 1083-351X (Sept.
2012).
39. Yamamoto, M. et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor
signaling pathway. Science (New York, N.Y.) 301, 640–643. issn: 1095-9203 (Aug. 2003).
40. Yamashita, M. et al. A TRIF-independent branch of TLR3 signaling. Journal of Immunol-
ogy (Baltimore, Md.: 1950) 188, 2825–2833. issn: 1550-6606 (Mar. 2012).
41. Kimura, H. et al. Cytokine production and signaling pathways in respiratory virus infec-
tion. Virology 4, 276 (2013).
42. Gowen, B. B. et al. TLR3 Deletion Limits Mortality and Disease Severity due to Phle-
bovirus Infection. The Journal of Immunology 177, 6301–6307. issn: 0022-1767, 1550-6606
(Jan. 2006).
43. Brentano, F. et al. The role of Toll-like receptor signalling in the pathogenesis of arthritis.
Cellular Immunology 233, 90–96. issn: 0008-8749 (Feb. 2005).
44. Stowell, N. C. et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and
impairs lung function in mice. Respiratory Research 10, 43. issn: 1465-993X (2009).
45. Kaczanowska, S. et al. TLR agonists: our best frenemy in cancer immunotherapy. Journal
of Leukocyte Biology 93, 847–863. issn: 0741-5400 (June 2013).
46. Lin, E. et al. Innate immunity and toll-like receptor antagonists: a potential role in the
treatment of cardiovascular diseases. Cardiovascular Therapeutics 27, 117–123. issn: 1755-
5914 (2009).
47. Lei, X. et al. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral
responses mediated by Toll-like receptor 3. Journal of Virology 85, 8811–8818. issn: 1098-
5514 (Sept. 2011).
31

48. Bitra, K. et al. Polydnavirus Ank Proteins Bind NF-kB Homodimers and Inhibit Processing
of Relish. PLoS Pathog 8, e1002722 (May 2012).
49. Kim, Y. et al. Structure of vaccinia virus A46, an inhibitor of TLR4 signaling pathway,
shows the conformation of VIPER motif. Protein Science: A Publication of the Protein
Society 23, 906–914. issn: 1469-896X (July 2014).
50. Ferguson, B. J. et al. Vaccinia virus protein N2 is a nuclear IRF3 inhibitor that promotes
virulence. The Journal of General Virology 94, 2070–2081. issn: 0022-1317 (Sept. 2013).
51. Wu, B. et al. Viral counterattack against the host innate immune system. Cell Research
23, 735–736. issn: 1748-7838 (June 2013).
52. Salas, L. et al. African swine fever virus morphogenesis. Virus Research 173, 29–41. issn:
1872-7492 (Apr. 2013).
53. Hulo, C. et al. ViralZone: a knowledge resource to understand virus diversity. Nucleic
Acids Research 39, D576–582. issn: 1362-4962 (Jan. 2011).
54. Costard, S. et al. Epidemiology of African swine fever virus. Virus Research 173, 191–197.
issn: 1872-7492 (Apr. 2013).
55. Gomez del Moral, e. a. African swine fever virus infection induces tumor necrosis factor
alpha production: implications in pathogenesis. Journal of Virology 73, 2173–2180. issn:
0022-538X (Mar. 1999).
56. Wardley, R. C. et al. African swine fever virus replication in porcine lymphocytes. The
Journal of General Virology 37, 425–427. issn: 0022-1317 (Nov. 1977).
57. Silk, R. N. et al. African swine fever virus A238L inhibitor of NF-kappaB and of calcineurin
phosphatase is imported actively into the nucleus and exported by a CRM1-mediated
pathway. The Journal of General Virology 88, 411–419. issn: 0022-1317 (Feb. 2007).
58. Correia, S. et al. Identification and utility of innate immune system evasion mechanisms
of ASFV. Virus Research 173, 87–100. issn: 1872-7492 (Apr. 2013).
59. Bowie, A. G. et al. Viral evasion and subversion of pattern-recognition receptor signalling.
Nat Rev Immunol 8, 911–922. issn: 1474-1733 (Dec. 2008).
60. De Oliveira, V. L. et al. A novel TLR3 inhibitor encoded by African swine fever virus
(ASFV). Archives of Virology 156, 597–609. issn: 1432-8798 (Apr. 2011).
61. Kelley, L. A. et al. Protein structure prediction on the Web: a case study using the Phyre
server. Nature Protocols 4, 363–371. issn: 1750-2799 (2009).
62. Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC bioinformatics 9,
40. issn: 1471-2105 (2008).
63. Eswar, N. et al. Comparative protein structure modeling using MODELLER. Current
Protocols in Protein Science / Editorial Board, John E. Coligan ... [et Al.] Chapter 2,
Unit 2.9. issn: 1934-3663 (Nov. 2007).
32

64. Laskowski, R. A. et al. PROCHECK: a program to check the stereochemical quality of
protein structures. Journal of Applied Crystallography 26, 283–291 (Apr. 1993).
65. Henriques, E. S. et al. Modeling of the Toll-like receptor 3 and a putative Toll-like receptor
3 antagonist encoded by the African swine fever virus. Protein Science : A Publication of
the Protein Society 20, 247–255. issn: 0961-8368 (Feb. 2011).
66. Liu, L. et al. Structural basis of Toll-Like Receptor 3 Signaling with double-stranded RNA.
Science (New York, N.Y.) 320, 379–381. issn: 0036-8075 (Apr. 2008).
67. Hermanson, G. T. in Bioconjugate Techniques (Third edition) (ed Hermanson, G. T.) Third
edition, 275–298 (Academic Press, Boston, 2013). isbn: 978-0-12-382239-0.
68. Theofilopoulos, A. N. et al. Sensors of the innate immune system: their link to rheumatic
diseases. Nature Reviews Rheumatology 6, 146–156. issn: 1759-4790 (Mar. 2010).
69. Zhang, H. et al. Integrin-nucleated Toll-like receptor (TLR) dimerization reveals subcellular
targeting of TLRs and distinct mechanisms of TLR4 activation and signaling. FEBS Letters
532, 171–176. issn: 0014-5793 (Dec. 2002).
70. Liew, F. Y. et al. Negative regulation of Toll-like receptor-mediated immune responses.
Nature Reviews Immunology 5, 446–458. issn: 1474-1733, 1474-1741 (June 2005).
33
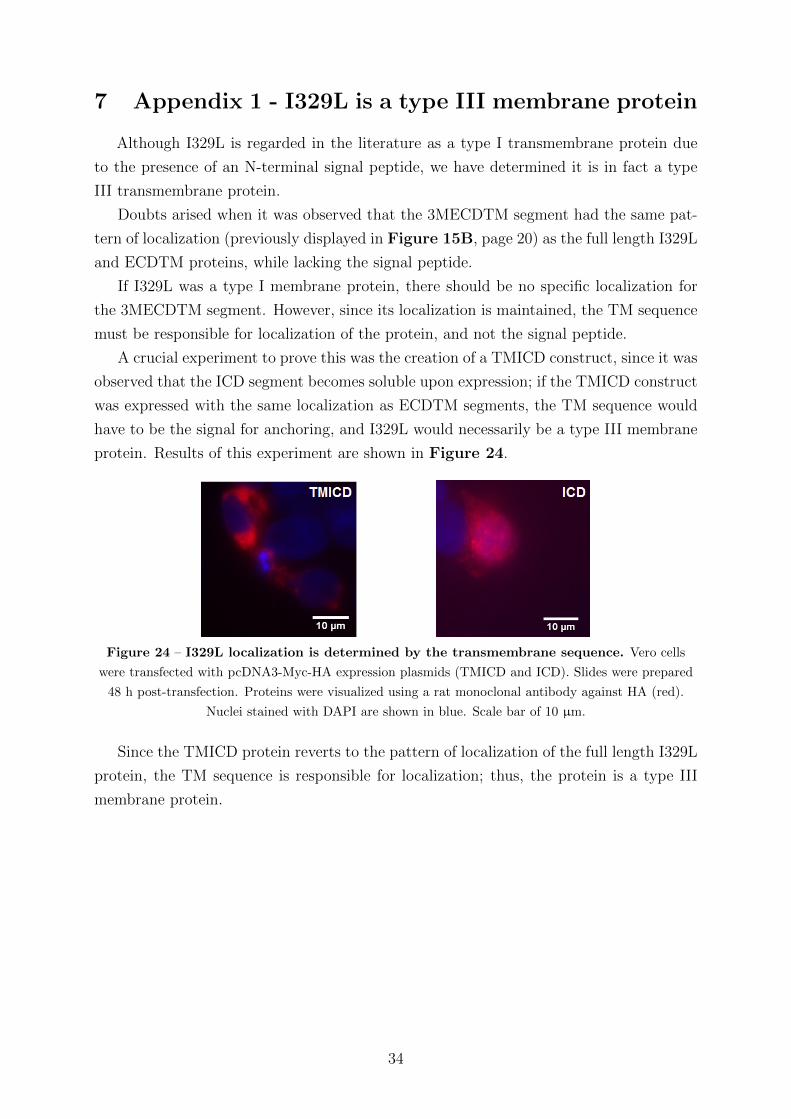
7 Appendix 1 - I329L is a type III membrane protein
Although I329L is regarded in the literature as a type I transmembrane protein due
to the presence of an N-terminal signal peptide, we have determined it is in fact a type
III transmembrane protein.
Doubts arised when it was observed that the 3MECDTM segment had the same pat-
tern of localization (previously displayed in Figure 15B, page 20) as the full length I329L
and ECDTM proteins, while lacking the signal peptide.
If I329L was a type I membrane protein, there should be no specific localization for
the 3MECDTM segment. However, since its localization is maintained, the TM sequence
must be responsible for localization of the protein, and not the signal peptide.
A crucial experiment to prove this was the creation of a TMICD construct, since it was
observed that the ICD segment becomes soluble upon expression; if the TMICD construct
was expressed with the same localization as ECDTM segments, the TM sequence would
have to be the signal for anchoring, and I329L would necessarily be a type III membrane
protein. Results of this experiment are shown in Figure 24.
Figure 24 – I329L localization is determined by the transmembrane sequence. Vero cells
were transfected with pcDNA3-Myc-HA expression plasmids (TMICD and ICD). Slides were prepared
48 h post-transfection. Proteins were visualized using a rat monoclonal antibody against HA (red).
Nuclei stained with DAPI are shown in blue. Scale bar of 10 µm.
Since the TMICD protein reverts to the pattern of localization of the full length I329L
protein, the TM sequence is responsible for localization; thus, the protein is a type III
membrane protein.
34
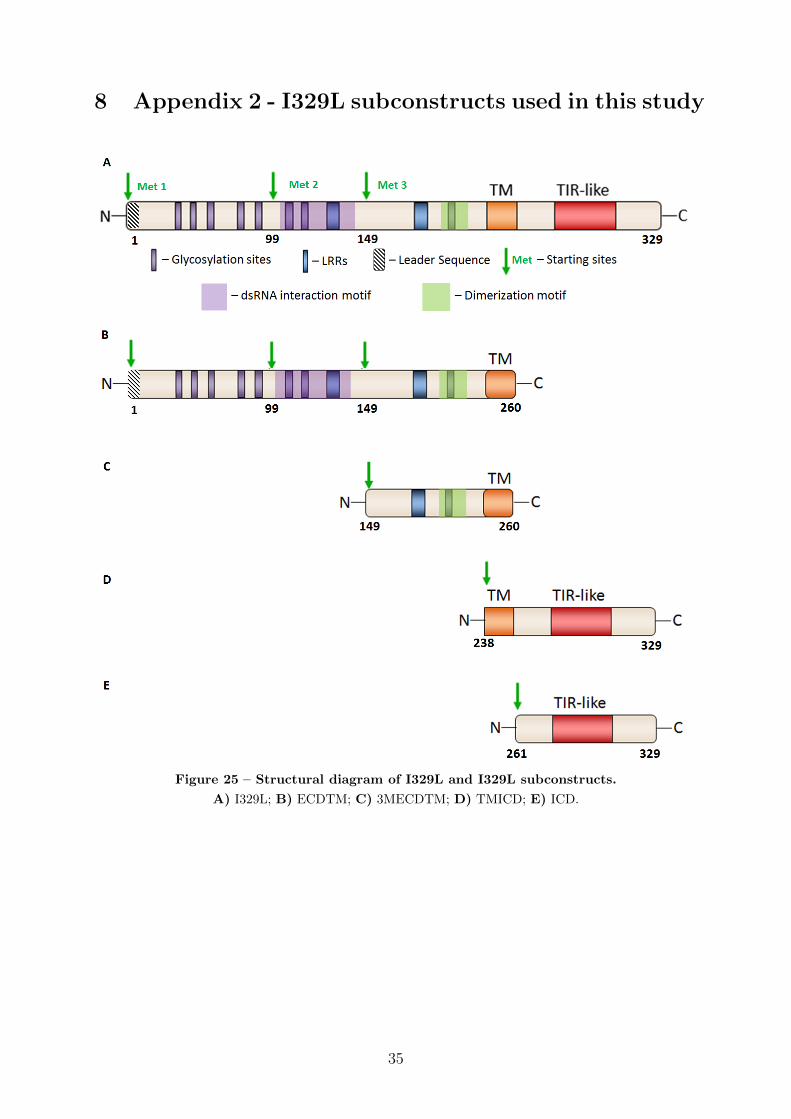
8 Appendix 2 - I329L subconstructs used in this study
Figure 25 – Structural diagram of I329L and I329L subconstructs.
A) I329L; B) ECDTM; C) 3MECDTM; D) TMICD; E) ICD.
35
Top Related