







Languages
Pages
Legal


Update Pain Management
Presenter : Dr Norlida Binti SuhaimiModerator : Dr Wan Rohaidah

Intravenous Paracetamol
PCA Oxynorm
Bupenorphine patch
Perioperative lignocaine infusion
Outline presentation…

Intravenous Paracetamol

“A Fatal Accident Inquiry in Scotland in 2011 concluded that a young adult died from liver failure due to an overdoseof paracetamol. The Sheriff found ‘there was, at the time of the death, a prevailing culture of assumed familiarity withthe administration of IV paracetamol, a familiarity derived from the common use of oral paracetamol’. The patient,who weighed 35kg, died nine days after receiving paracetamol 1g IV on a sustained and regular basis.”

IV paracetamol was licensed in the UK in 2004 and
is used routinely in anaesthetic practice.
Paracetamol is frequently used for perioperative
analgesia, alone or in combination with an opioid.
Clinical studies have shown that paracetamol has
potent analgesics and reduce opioid consumption.
Introduction…

for the short-term treatment of moderate pain -
especially following surgery
for the short-term treatment of fever
when administration by IV route is clinically justified by
an urgent need to treat pain or hyperthermia and/or
when other routes of administration are not possible.
IV Paracetamol - therapeutic indications…

in patients with hypersensitivity to
paracetamol
in cases of severe hepatocellular
insufficiency.
IV Paracetamol – contraindications…

Precautions for use:
IV Paracetamol should be used with caution in cases of:
hepatocellular insufficiency
severe renal insufficiency (creatinine clearance ≤ 30
mL/min)
chronic alcoholism
chronic malnutrition (low reserves of hepatic
gluthatione)

Paracetamol has essentially no effect on cyclo-
oxygenase in vitro – but it has been classified as a
NSAID because of its moderate analgesic and
antipyretic properties.
The drug is not associated with the increased
incidence of platelet dysfunction, gastritis, and renal
toxicity that are sometimes associated with NSAIDs
IV Paracetamol – clinical pharmacology…

MOA
Unclear
it is thought to exert its analgesic activity by
inhibiting the synthesis of prostaglandins in the CNS
(central acting) and peripherally blocking pain
impulse generation.
In addition, it has been proposed that
acetaminophen has a serotonergic (5-HT)
mechanism and a cannabinoid agonism mechanism,
which may contribute to its analgesic effect.

It has been proposed that its antipyretic actions
are due to :
- inhibition of the hypothalamic heat-
regulating center,
- inhibition of prostaglandin synthesis within
the central nervous system , by inhibition
of COX-3 (a COX – 1 ) variant.
- cannabinoid agonism.

The pharmacokinetics of intravenous
acetaminophen have been described in several
studies, and the serum therapeutic level required
to produce an analgesic effect is 16 mcg/mL in
adults and 10 mcg/mL in children.
Pharmacokinetics and Pharmacodynamics.

IV PCM has a faster onset and results in more predictable pharmacokinetic than oral or rectal PCM formulations
provides onset of pain relief within five to 10 minutes after administration.
Intravenous
1 hour 2 hours
Bloodconcentration
Intramuscular
Suppository
Effervescent tabletEffective
concentration
Tablet

mean IV C max (maximum plasma concentration of drug) was
nearly twice that observed with oral administration and
nearly four times that observed with rectal administration.
The lag time after oral administration is 20-30 min
The lag time after rectal administration often exceeds 1
hour

A major benefit is that IV PCM may be
administered before or during surgery, permitting
the initiation of effective analgesic therapy in the
early phase of the postoperative period.
When patients are able to tolerate oral intake,
they may be switched from IV to oral PCM to
maintain the predictable analgesia established
by the IV route.

Distribution:
The volume of distribution of paracetamol is
approximately 1 L/kg.
Oral bioavailability 80%
Paracetamol is not extensively bound to plasma
proteins (10%)
Following infusion of 1 g paracetamol, significant
concentrations of paracetamol (about 1.5 μg/mL)
were observed in the Cerebro Spinal Fluid as and
from the 20th minute following infusion.

Paracetamol metabolized by the liver mainly to
glucuronide conjugates but also sulphate and cysteine
conjugates
These are actively excreted in the urine, only small
fraction being excreted unchanged.
N- acetyl-p-amino-benzoquinoneimine is a highly toxic
metabolite of paracetamol that produced in small amount
in therapeutic doses.
It is rapidly conjugated with hepatic glutathione to render
it harmless

Following toxic dose – gluthatione is exhausted, NAPQI
accumulates – then free to form covalent bonds with
sulphydryl groups on hepatocytes resulting cell death and
centrilobular hepatic necrosis
Treatment:
- with oral methionine(enhances gluthatione
synthesis) and/or oral or IV acetylcysteine which is
hydrolysed to a precursor of glutathione
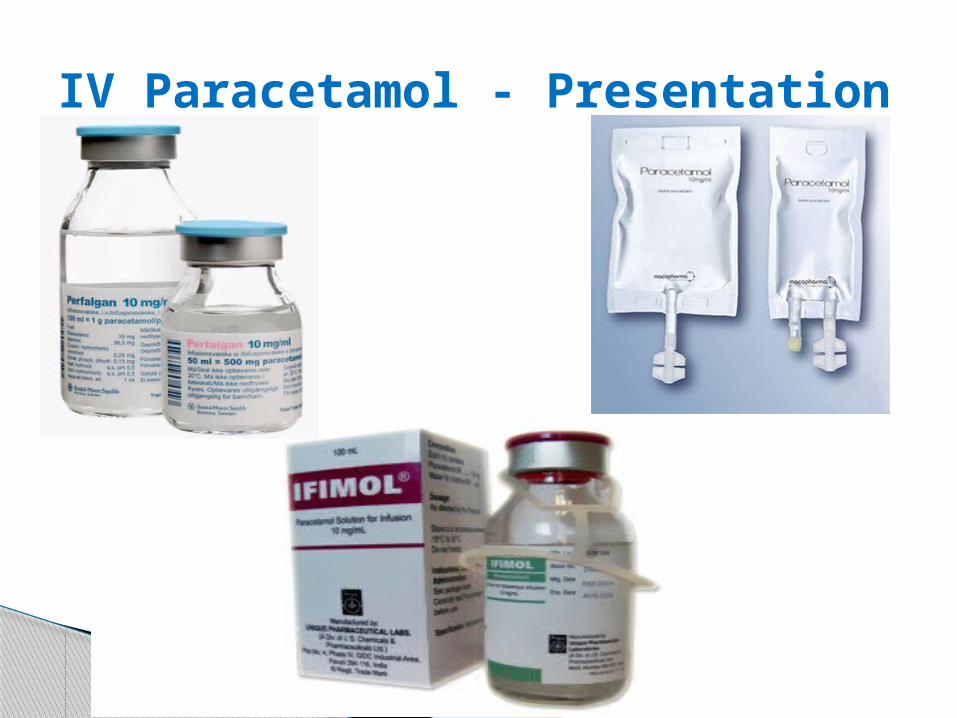
IV Paracetamol - Presentation

Composition:
One ml contains 10mg paracetamol
One 50 ml vial contains 500mg paracetamol
One 100 ml vial contains 1000mg
paracetamol

The 100 ml vial or 100 ml bag is restricted to
adults, adolescents and children weighing more
than 33 kg.
The 50 ml vial is adapted to infants, toddlers and
children weighing less than 33 kg.

Recommended dose of IV paracetamol…

IV Paracetamol should be given by infusion over
15 minutes
minimum dose interval should not be less than
four hours (six hours in patients with renal
impairment).
IV Paracetamol : administration…

Once the vacuum seal of the glass vial has been
penetrated, the dose of IV must be administered
within 6 hours.
IV Paracetamol is a single-use vial, and the
unused portion must be discarded.
To prevent the possibility of an air embolism, it is
important to observe the end of the infusion

can also be diluted in a 0.9% sodium chloride
solution or 5% glucose solution
Use the diluted solution within the hour following
its preparation (infusion time included).

NON–WEIGHT-BASED DOSING:
For 1000 mg (10 mg/mL) doses, deliver from the bottle
Use aseptic technique to prepare the vial and IV line
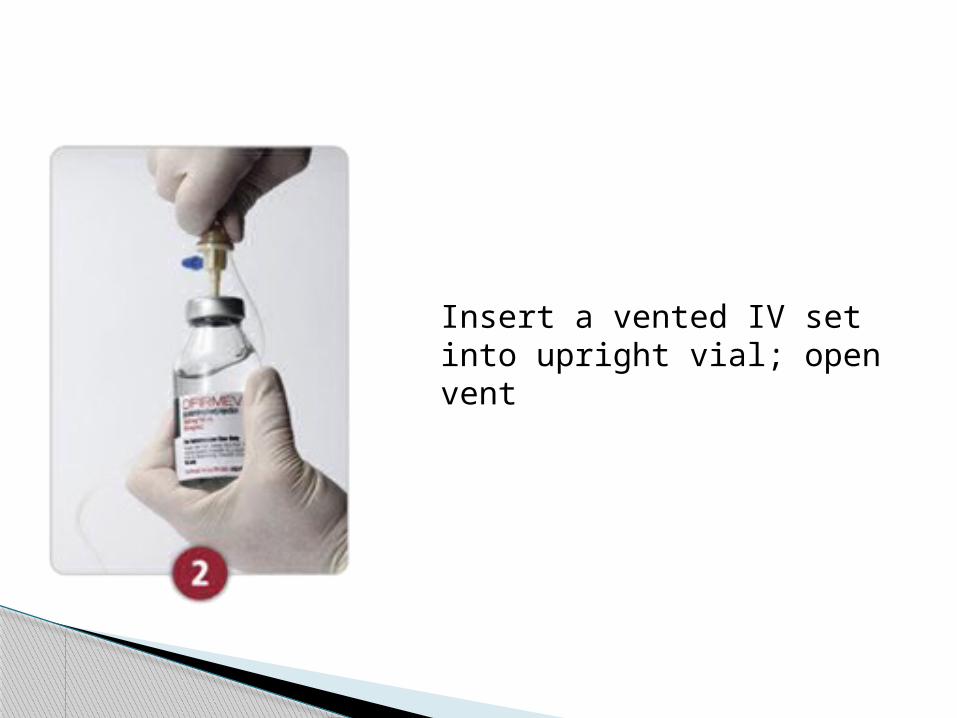
Insert a vented IV set into upright vial; open vent
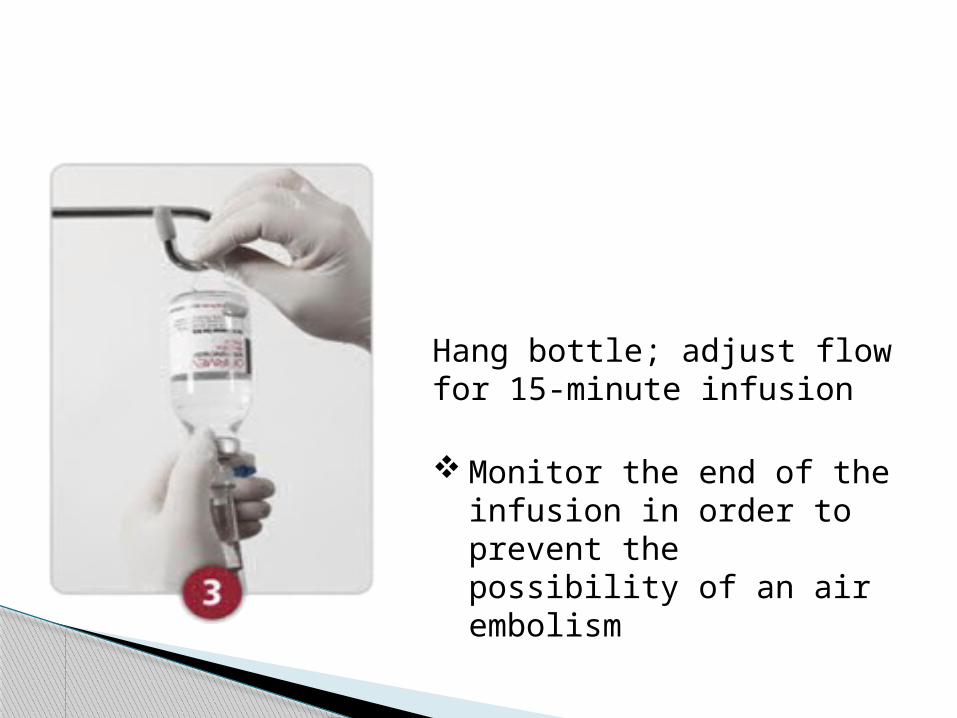
Hang bottle; adjust flow for 15-minute infusion
Monitor the end of the infusion in order to prevent the possibility of an air embolism
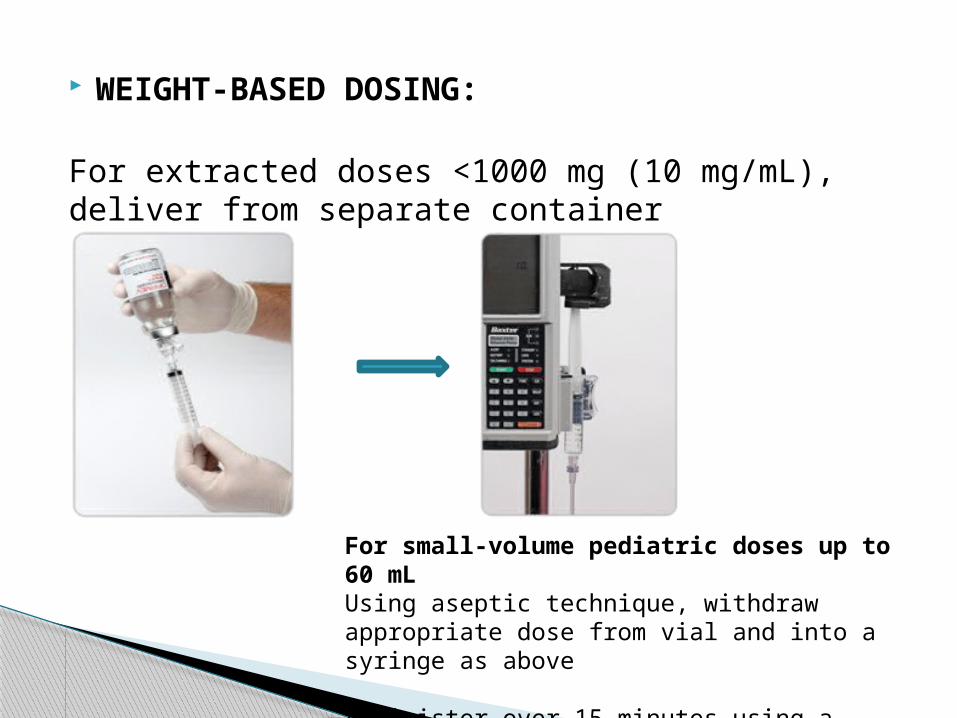
WEIGHT-BASED DOSING:
For extracted doses <1000 mg (10 mg/mL), deliver from separate container
For small-volume pediatric doses up to 60 mL Using aseptic technique, withdraw appropriate dose from vial and into a syringe as above
Administer over 15 minutes using a syringe pump

Since introduction, there have been concerns
about accidental overdose of IV paracetamol due
to errors in drug prescription and administration,
particularly in children, small adults, the elderly,
alcoholics and those with pre-existing
hepatocellular insufficiency.

Reported errors include:
- incorrect dose in adults with high or low body mass index;
- accidental overdose in children associated with use of
100ml- vials;
- 10-fold drug calculation errors;
- confusion between dose volume in millilitres and dose of
drug in milligrams;
- errors when setting up infusion pumps;
- and duplication of doses between the ward and
the operating theatre or recovery.

IV paracetamol should be prescribed carefully,
according to the weight, age and co-morbidities of the
patient. The upper dose limit for each single dose and
in each 24-hour period should not be exceeded.
50ml vials of IV paracetamol should be used for
patients less than 33kg. In infants and small children,
doses should be measured accurately using a syringe.
SALG recommendations…

Enquiry about recent paracetamol ingestion
should form part of routine pre-operative
assessment.
All doses of paracetamol administered in the
operating theatre should be recorded on the drug
administration chart and in the anaesthetic
record.

Advice should be sought from the local poisons information service in all cases of overdose of intravenous paracetamol.
Treatment with acetylcysteine is suggested following a single dose greater than 60mg/kg.
IV paracetamol remains under intensive monitoring by the MHRA.
All suspected adverse reactions to IV paracetamol should be reported to the Yellow Card Scheme and discussed with the local poisons information service.
PCA OxyNorm

Oxycodone is a semi-synthetic derivatives, full
opioid agonist with no antagonist properties.
It has an affinity for kappa, mu and delta opiate
receptors in the brain and spinal cord.
Oxycodone is similar to morphine in its action.
- The therapeutic effect is mainly
analgesic, anxiolytic and sedative.

Oxynorm…
belongs to a group of medicines called strong
analgesics
Indication:
* commonly used as an analgesic in
moderate to severe acute pain
* also used in moderate to severe cancer pain,
and sometimes in chronic non-cancer pain

Reseptor type Location Action when stimulated
µ receptor *brain especially - analgesia
areas involved - physical with sensory & dependence
motor perception - resp depression
and integration - reduced peristalsis
- Abundant in - euphoria
preaqueductal grey. - meiosis
*Spinal cord
δ receptor * brain - Analgesia - anti depressant - physical dependence
Opiod receptor classification…

Ƙ receptor * Brain - Spinal analgesia * Spinal cord - Sedation - Meiosis
NOP receptor * Brain - Anxiety ( nociceptin * spinal cord - DepressionOrphanin FQ - Affect learningPeptide receptor and memory- Most recently - involved in identified tolerance
Reseptor type Location Action whenstimulated

Other pharmacological effects of oxycodone : - respiratory depression, antitussive,
bronchospasm- smooth muscle (constipation, reduction in gastric, biliary and pancreatic secretions, spasm of sphincter of Oddi and transient elevations in serum amylase) - nausea and vomiting : CTZ stimulation via
5- HT3 and dopamine receptors- cardiovascular system (release of histamine and/or peripheral vasodilatation, possibly causing pruritus, flushing, sweating and/or orthostatic hypotension).

Opioids may influence the hypothalamic-pituitary-
adrenal or –gonadal axes.
- Some changes that can be seen include an
increase in serum prolactin, and decreases
in plasma cortisol and testosterone.
Clinical symptoms may be manifest from
these hormonal changes.

Absorption
- 40% to 85% bioavailability after oral
administration
- presence of methoxy group at C3
position of the phenanthrene structure
protects drug against glucuronide
conjugation, and hence first pass effect
-100% bioavailability after iv
administration
Pharmacokinetics…

Distribution:
Distributed to skeletal muscle, liver, intestinal tract,
lungs, spleen and brain.
- tissues with mu, kappa and delta opiod
receptors
Pka 8.5. at pH 7.4, 7.4% unionized
Vd 2-6 L/kg
45% protein binding

Metabolism & Elimination:
Oxycodone has an elimination half-life of approximately 3 hours
metabolised principally in the liver to noroxycodone and
oxymorphone.
Oxymorphone has some analgesic activity but present in plasma
in low concentrations and is not considered to contribute to
oxycodone’s pharmacological effect.
CYP3A4 and CYP2D6 are involved in the formation of
noroxycodone and oxymorphone, respectively.
Metabolites mainly excreted in urine and sweat, accumulates in
patients with renal impairement.

Concentration: Oxycodone hydrochloride 10 mg/ml
Dilute to 1 mg/ml in 0.9% saline, 5% dextrose or
water for injections.
Setting:
* Bolus doses : 0.03 mg/kg (e.g. 1-2mg per 70 kg)
* lock-out time : minimum 5 minutes.
* Background infusion : nil
* Four hour dose limit : 30 mg
PCA oxynorm : administration…

Discontinuation of PCA...
PCA should be discontinued when minimal use is
required and the patient is able to tolerate oral
analgesia
Full explanation and reassurance must be given to the
patient.
Ensure that adequate analgesia is prescribed.
Continue regular pain assessment after the pain
control system has been discontinued and act
accordingly.

Transferring patients between oral and parenteral
oxycodone…
The dose should be based on the following ratio: 2 mg
of oral oxycodone is equivalent to 1 mg of parenteral
oxycodone.
It must be emphasized that this is a guide to the dose
required.
Inter-patient variability requires that each patient is
carefully titrated to the appropriate dose.
Buprenorphine patch

is a semisynthetic, highly lipophilic derivative of the opium
alkaloid thebain
approximately 25-50 times as potent as morphine, usual dose ~
0.3-0.6 mg
Buprenorphine is a partial agonist .has high affinity for, but low
intrinsic activity at, mu receptors.
however, its maximal opioid effects are less than that of full
agonists, and reach a ceiling where higher doses do not result in
increasing effect.
produces analgesia and other effects similar to morphine,
including CVS
BUPRENORPHINE…

peak blood concentration appear at 5 min/i.m., and at 2
hrs/s.l. or oral
plasma protein binding is ~ 96%
plasma half-life is ~ 3 hrs, however, the duration of
action is longer, sometimes up to 6 hrs,
probably due to tissue binding
both N-dealkylation and conjugation occur in the liver,
however most of the drug is excreted unchanged in the
faeces

Transdermal buprenorphine may be used in chronic severe pain
when lower doses of strong opioids are indicated; especially in
patients who need continuous, around-the-clock narcotic pain
relief for an extended period of time.
However, the place of transdermal buprenorphine in pain
management is not well established.
The 7-day patch formulation may have a particular role for
patients who are vomiting or have swallowing difficulties.
Place in therapy…

Transdermal buprenorphine is not suitable for the
management of acute pain because it has a slow
onset and extended duration of action.

transdermal buprenorphine
5 mg (releasing buprenorphine 5
micrograms per hour) every 7 days
10 mg (10 micrograms per hour) every 7
days
20 mg (20 micrograms per hour)every 7 days
Presentation..

Transdermal buprenorphine patches deliver
buprenorphine at a constant rate over 7 days.
The dose equivalence of transdermal
buprenorphine and oral morphine is not
established.
The manufacturer suggests that the dose range
covered by the three patch strengths may be
equivalent to oral morphine up to 90 mg/day.

Other literature and the dose relativities
suggested that the buprenorphine 20-microgram-
per-hour patch might be equivalent to oral
morphine up to 36 mg/day or 53 mg/day

Buprenorphine: a partial agonist
Buprenorphine is a partial agonist so there is a ceiling dose
to its analgesic effect — that is, above a certain dose there
is no further analgesic effect.
The dose at which this occurs in humans is not established
but it is unlikely at the doses in the transdermal patches.
Buprenorphine may trigger opioid withdrawal symptoms in
people who have developed physical dependence on other
opioids.

Buprenorphine has high affinity for mu opioid
receptors and is not easily displaced by opioid
antagonists.
Consequently, the effects of buprenorphine in
overdose are only partially reversed by naloxone

Buprenorphine produces typical opioid adverse
effects (such as constipation, headache, nausea,
vomiting, dizziness).
Local irritation may occur at the application site.
Buprenorphine has a long half-life, so plasma
concentrations fall slowly after the patch is
removed.
Another opioid should not be started within 24 hours of
removing a patch
Safety issues…

Dependence and abuse potential
Physical dependence may develop with chronic use of
buprenorphine.
If a withdrawal syndrome does occur when buprenorphine is
discontinued, it is usually of mild to moderate intensity,
occurs within 2 days and resolves
within 2 weeks.
Avoid prescribing buprenorphine to people who
may be dependent on other opioids because it can
precipitate withdrawal symptoms, including pain.

Transdermal buprenorphine may have lower
abuse
potential than other buprenorphine dosage forms
because of the relatively low plasma
concentrations
achieved, the slow onset of effect and because it
is
likely to be difficult to extract the drug from the
matrix design.

Misuse could take the form of using excessive
amounts of the intact patch or applying it to
sites that would enhance systemic absorption.
It should be used with caution in people with a
past history of dependence on alcohol or other
drugs.

In overdose the effects of buprenorphine are only
partially reversed by naloxone.
The manufacturer states that the dose of naloxone
should start in the usual range but that naloxone 5–12
mg intravenously may be required.
Repeated naloxone doses may be needed because
naloxone has a shorter duration of action than
buprenorphine.
Management of overdose should focus on maintaining
adequate ventilation.
Overdose: effects only partially reversedby naloxone

the effect of buprenorphine on respiratory
depression reaches a ceiling, with higher doses
not increasing respiratory depression to a
significant degree.
However, if buprenorphine is used in combination
with other central nervous system depressants,
such as benzodiazepines, the combined effect on
respiration can be life threatening.

Opioid-naïve patients should start at the lowest
strength.
Supplemental analgesics should be continued as
needed during titration because buprenorphine
concentrations rise slowly.
Patients converting from other opioids (up to the
equivalent of oral morphine 90 mg/day) can also
begin on a low strength of buprenorphine and should
continue with their previous regimen during titration.
Dosing issues…

The dose should be titrated to effect and should not be increased at intervals of less than 3 days.
To increase the dose, remove the current patch and apply a higher strength patch or a combination of 2 patches.
No more than two 20-microgram-per-hour patches should be used at once.

Apply Buprenorphine patch to a hairless or nearly
hairless skin site at the upper outer arm, upper
chest, upper back or the side of the chest.
These 4 sites (each present on both sides of the
body) provide 8 possible application sites.
Rotate Buprenorphine patch among the 8
described skin sites.
Application…


New patches should always be applied to a
different site from the previous one.
Any site should not be re-used for 3–4 weeks to
minimize the risk of local skin irritation
immediately re-using a site can increase the rate
of absorption of buprenorphine.

Discuss the potential adverse effects of
buprenorphine.
Most adverse effects reduce with time.
Constipation may persist; advise patients to drink
adequate amounts of water, increase their fibre
intake and remain as mobile as possible.
Regular laxatives such as lactulose should be
started when buprenorphine is initiated and
continued for long as buprenorphine is used.

PERIOPERATIVE LIGNOCAINE INFUSION


Intravenous infusion of lidocaine decreases
postoperative pain and speeds the return of
bowel function.
tested the hypothesis that perioperative lidocaine
infusion facilitates acute rehabilitation protocol in
patients undergoing laparoscopic colectomy.
Background…

the concept of fast-track surgery has been developed to
reduce postoperative morbidity and duration of
hospitalization, and to accelerate postoperative recovery
and convalescence.
Acute rehabilitation programs combine preoperative
optimization of patients’ physical and psychological status,
attenuation of surgical stress, dynamic pain relief, enforced
mobilization, and early oral (enteral) nutrition, as well as
changes in surgical care de-emphasizing tubes and drains.
Introduction…

Effective postoperative analgesia is key to acute rehabilitation.
An alternative approach to accelerate postoperative recovery
after colon surgery is administration of intravenous
lidocaine, which has analgesic, antihyperalgesic,
and antiinflammatory properties and has been reported
to speed the return of bowel function after surgery.
In a case series, acute rehabilitation after laparoscopic
colectomy
using intravenous lidocaine yielded outcomes similar
to those reported using epidural.

Furthermore,nontoxic plasma lidocaine
concentrations reduce requirements for various
volatile anesthetics in several animal species
although the benefits in humans remain unclear.
intravenous lidocaine is inexpensive, easy to
administer, and relatively safe.

enrolled 45 ASA status I–III patients scheduled to undergo elective
laparoscopic colectomy for nonmalignant disease at
the Centre Hospitalier Universitaire de Lie`ge. (Lie`ge, Belgium)
Patients were enrolled from January 2003 until December 2004.
Exclusion criteria :
- age greater than 70 yr,
- history of gastroduodenal peptic ulcer or renal failure -
hepatic insufficiency,
- psychiatric disorder
- steroid treatment
- chronic treatment with opioid.
Materials and Methods…

Protocol
Patients fasted at least 6 h and were orally
premedicated with 50 mg hydroxyzine and 0.5
mg alprazolam 2 h before surgery.
Lactated Ringer’s solution (8 ml/kg/h) was infused
throughout surgery.

Anesthesia
Patients were randomly allocated to two groups
double blinded study.
Just before induction of anesthesia, patients
assigned to receive lidocaine (n " 22) :
- given an intravenous bolus injection of 1.5 mg/kg
lidocaine followed by a continuous infusion of
2 mg/kg/h.
- The lidocaine infusion was continued at a rate of 1.33
mg/kg/h for 24 h postoperatively.
Patients assigned to the control group (n " 23) were given
equal volumes of saline

General anesthesia was induced with 0.15 ug/kg
sufentanil and 2 mg/kg propofol.
Orotracheal intubation was facilitated with
cis-atracurium,
cis-atracurium was also used for intraoperative
muscle relaxation full muscle relaxation during
surgery

Anesthesia was maintained with sevoflurane in a
mixture of oxygen and air with 2 l/min fresh gas flow.
Sevoflurane concentration was adjusted to maintain
mean arterial pressure within 15% of the preinduction
value.
The use of opioid was restricted during surgery:
Sufentanil, 5 ug, was injected only if mean arterial
pressure increased more than 15% or if heart rate was
greater than 100 beats/min despite the administration
of sevoflurane to an end-tidal concentration of 3.5%.

BIS was monitored
allowed increases in inspired sevoflurane concentration
if BIS exceeded 50.
Core temperature was kept above 36.0°C using a
forced-air warming system.
All patients were given 0.625 mg droperidol and 2 mg
tropisetron, a 5-hydroxytryptamine type 3 antagonist, as
prophylaxis against postoperative nausea and vomiting
1h before the end of surgery.

Surgical Procedure
Two experienced laparoscopic surgeons (B.J.D.,
S.R.L.) performed procedures using a standard four- or five-
trocar technique.
For right colectomy, after intracorporeal dissection of the
ascending colon and the Bauhin valve, the specimen was
exteriorized through a 5- to 6-cm minilaparotomy in the right
lower abdomen.
After resection of the pathologic colon, the anastomosis was
hand-sewn and returned to the abdominal cavity.
The minilaparotomy was then closed.

In laparoscopic sigmoid colectomy, the sigmoid
colon
was first mobilized intracorporeally up to the
rectosigmoid junction.
The rectosigmoid junction was cut using a stapler.
The sigmoid colon was retrieved through a 5- to 6-
cm minilaparotomy in the left lower abdomen and
then resected.
The surgeons were unaware of the patient’s group
assignment.

Postoperative Analgesia
Postoperative analgesia was provided in both groups by
the combination of the paracetamol (acetaminophen)
precursor propacetamol - 2 g propacetamol= 1 g
paracetamol),
2 g intravenously 30 min before the end of surgery and
then every 6 h,
and ketorolac, 30 mg intravenously every 8 h.
Patient-controlled analgesia with piritramide
was used as rescue medication (bolus=1
mg, lockout interval =5 min, no basal infusion).

Twenty-four hours after the end of surgery, the
intravenous infusion of lidocaine or placebo was
stopped
analgesia was provided with oral paracetamol, 1
g every 6 h;
- diclofenac 75 mg twice daily;
- and 100 mg tramadol, if necessary.

An abdominal drain was left in contact with the anastomosis for 24 h.
The bladder catheter was removed on the first postoperative morning. An intravenous infusion of 5% glucose was started after surgery at a rate of 80 ml/h. Patients were allowed to drink water 6 h after surgery. If patients did not report nausea or vomiting, they were given 200
ml of nutritive supplement without residue (Clinutren® 1.5 kcal/ml; Nestle 1 h later.
On the first postoperative day, patients had a light breakfast and lunch. If this food was tolerated, the intravenous infusion was stopped
and a normal diet was resumed. Patients were asked to drink three 200-ml cartons of nutritive
supplement each day.
Acute Rehabilitation…

Active mobilization was started in bed 4 h after
surgery.
Assisted ambulation was enforced on the
subsequent days: 20 m in the morning and 50 m
in the afternoon on postoperative day 1, then 100
m in the morning and the afternoon on day 2.
Defecation and tolerance of normal diet were
required before discharge.

Arterial pressure, heart rate, and end-tidal sevoflurane
concentrations were measured on a Datex-Ohmeda S/5
monitor every 15 min during anesthesia.
BIS scores were also recorded at 15-min intervals.
After surgery, piritramide consumption was recorded every 4
h.
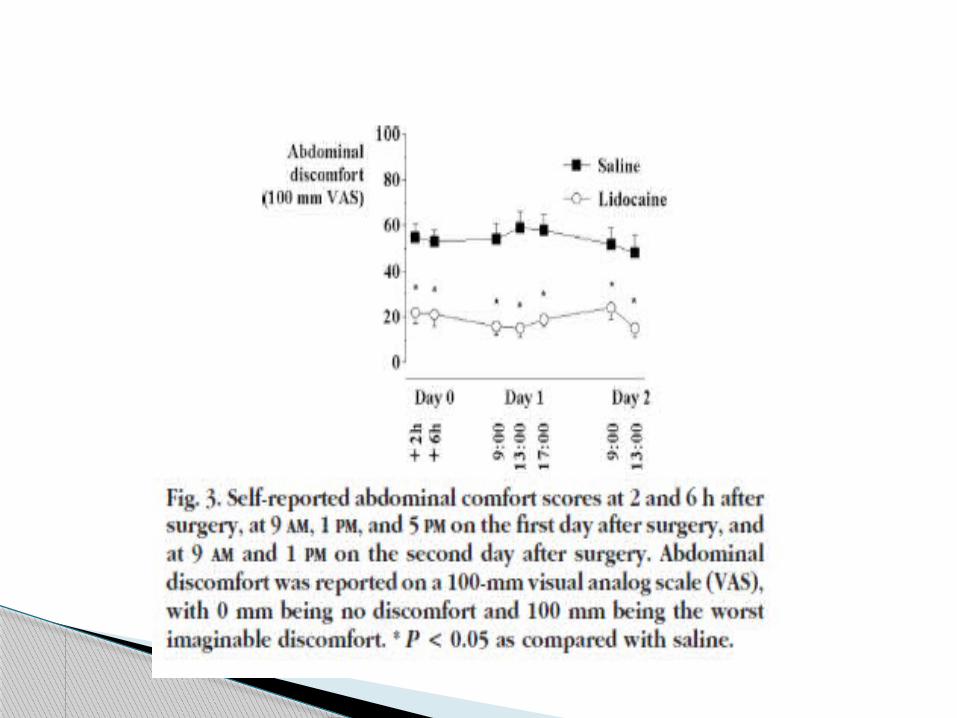
Pain scores were obtained on a visual analog scale at rest,
during mobilization from the supine to the sitting position,
and during coughing at 2 and 6 h postoperatively and at 9:00
AM, 1:00 PM, and 5:00 PM on postoperative days 1 and 2.
Measurements…

Postoperative fatigue scores and gastrointestinal
discomfort (colic, abdominal fullness, internal
discomfort) were also assessed on a visual analog
scale at the same times.
Times to first flatus, defecation, and hospital
discharge were recorded. Episodes of postoperative nausea and vomiting
were noted.

Immediately after induction of anesthesia, the bladder
was catheterized and emptied.
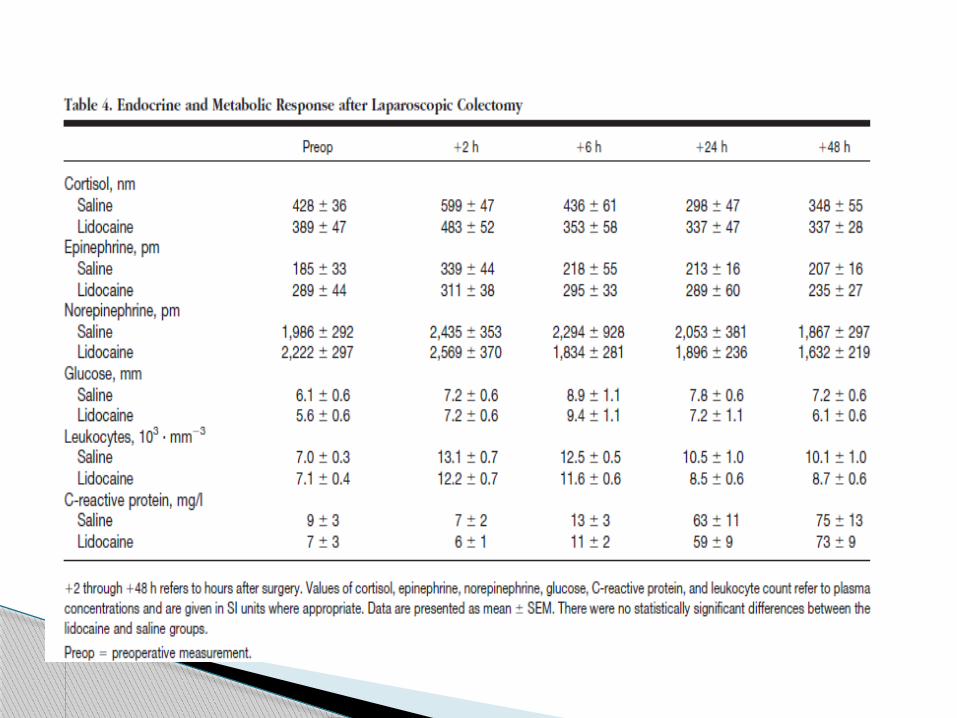
In the first 30 patients (n " 15 in each group), urine was
then collected to measure urinary secretion of cortisol,
epinephrine, and norepinephrine to assess the stress
response during anesthesia and surgery.
Blood samples were drawn in the same patients before
surgery and after surgery at 2, 6, 24, and 48 h.
Plasma concentrations of glucose, C-reactive
protein, cortisol, catecholamines, and leukocyte counts
were measured.

Blood samples were drawn at 5, 15, and 60 min
after anesthetic induction, at the end of surgery,
and 24 h after the end of surgery to measure
plasma lidocaine concentrations

A previous study at same institution using a similar protocol indicated that18 patients per group allowed detecting a 12-h difference
in the recovery of bowel function between the groups, at an alpha level of 0.05 and with 80% power.
therefore enrolled patients until 40 patients (n " 20 in each group) completed the study.
Statistical Analysis

Continuous variables are presented as mean ± SD; they were
compared using analysis of variance for repeated measures for
two criteria (time and treatment)
followed by the Scheffe´ test for multiple comparisons or
the Student t test, as appropriate.
If the Kolmogorov-Smirnov normality test did not demonstrate
gaussian distributions, the Mann–Whitney test was used;
data are then presented as median [25–75% interquartile range].
Categorical data were analyzed with chi-square tests.
P ≤0.05 was considered significant.

Results…
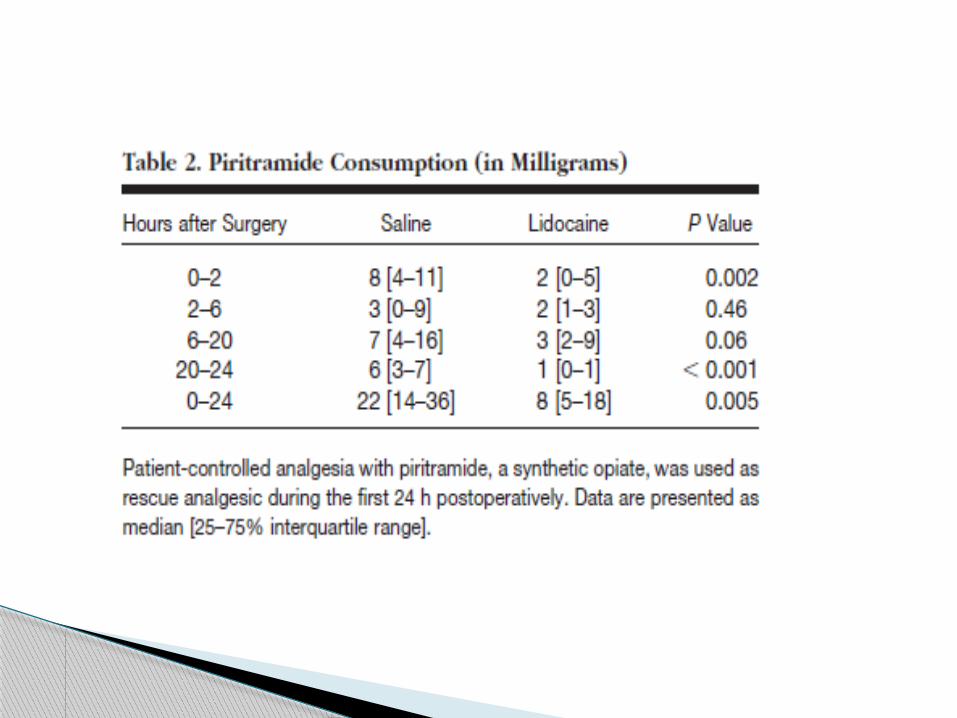

Three patients in the control group, but none in
the lidocaine group, requested tramadol after the
interruption of piritramide patient-controlled
analgesia between the 24th and 48th
postoperative hours:
These patients were given 100, 200, and 400 mg.


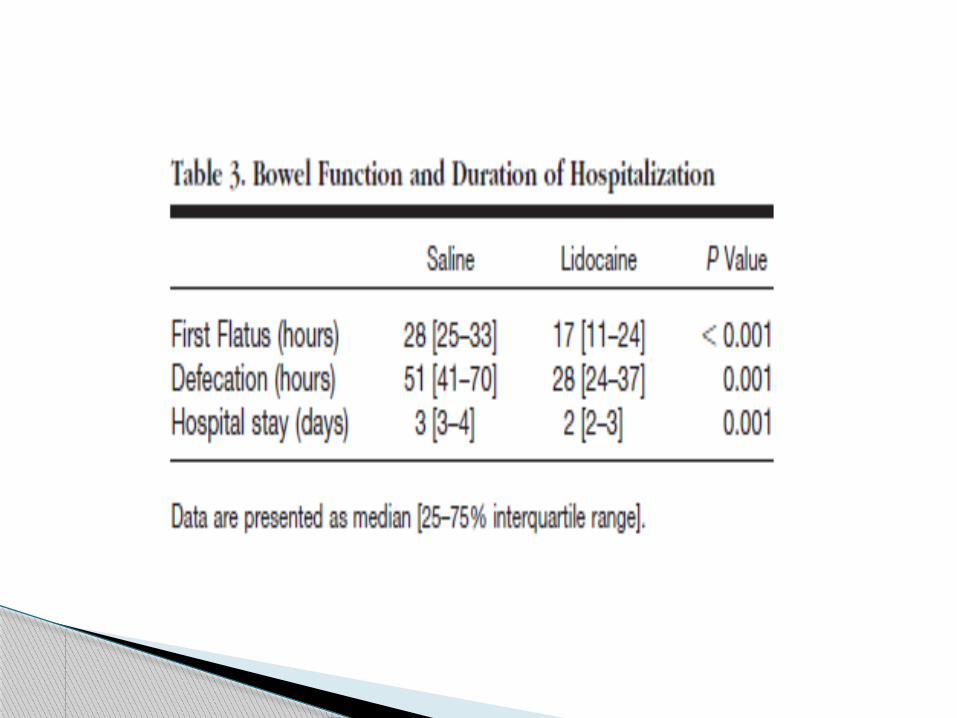

patients in the lidocaine group tolerated a normal diet the day
after surgery and had their intravenous infusion interrupted 24
h after surgery,
whereas three patients in the control group required
prolongation of postoperative fasting and intravenous infusion
(31, 54, and 72 h) (P " 0.22).
Four patients in the saline group but only one in the lidocaine
group experienced nausea (P =0.17).
Vomiting occurred in two patients in the saline group and none
in the lidocaine group (P =0.23)

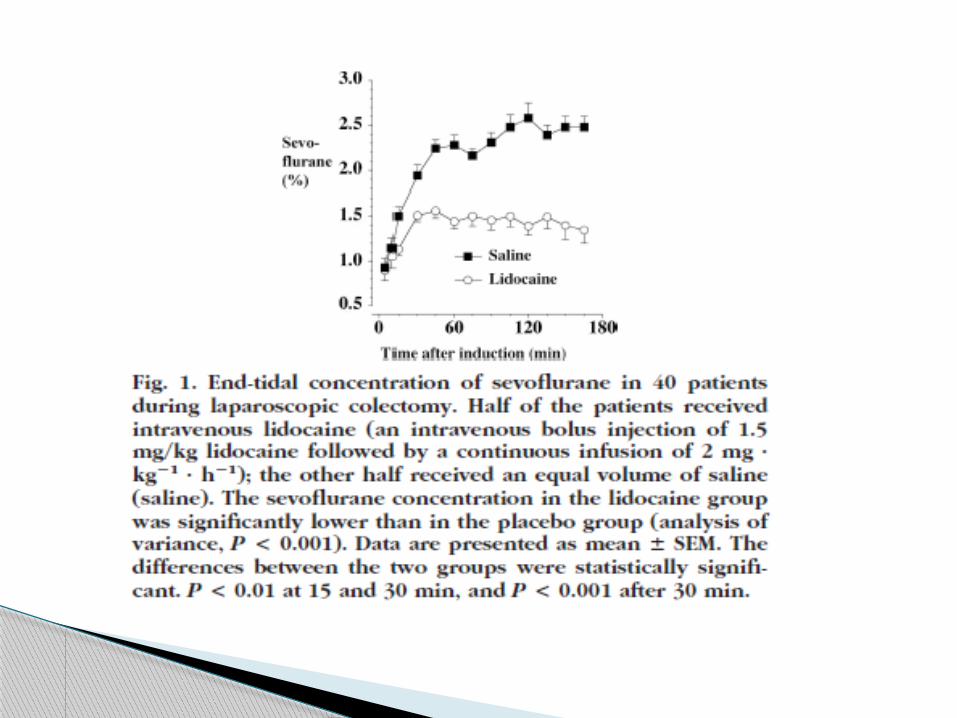
Lidocaine plasma concentrations were measured in 15
patients and were 1.6 ±0.9 ug/ml at 5 min, 1.3 ± 0.4 ug/ml at
15 min, and 1.8 ±0.5 ug/ml at 60 min after the bolus injection
of lidocaine;
2.4 ±0.6 ug/ml at the end of surgery
2.7 ±1.1 ug/ml at the end of the 24-h infusion.
The highest plasma concentrations of lidocaine measured at
each of these time points were 3.5, 2.1, 2.6, 4.0, and 4.6
ug/ml, respectively.

This study demonstrated that perioperative intravenous
infusion of nontoxic doses of lidocaine improved:
- postoperative analgesia,
- reduced postoperative opioid requirements,
- accelerated postoperative recovery of bowel
function,
- attenuated postoperative fatigue,
- reduced the duration of hospitalization,
- facilitated acute rehabilitation
in patients undergoing laparoscopic abdominal surgery.
results further indicated that moderate plasma lidocaine
concentrations reduced sevoflurane requirements necessary for
maintaining intraoperative hemodynamic stability and anesthetic
depth.

Intravenous lidocaine is analgesic,
antihyperalgesic, and antiinflammatory.
These properties are mediated by a variety of
mechanisms, including sodium channel blockade,
as well as inhibition of G protein–coupled
receptors and N-methyl-D-aspartate receptors.
In this study, intravenous lidocaine reduced
postoperative opioid consumption, as well as pain
scores during activity.

the analgesic effect persisted after the lidocaine
infusion was discontinued, which suggests a prevention
of spinal or peripheral hypersensitivity or both.
Inhibition of N-methyl-D-aspartate receptors which play a
major role in postoperative hyperalgesia
Abdominal discomfort was significantly reduced by
lidocaine, which is consistent with the ability of lidocaine
to alleviate visceral pain in animal models.

Systemic lidocaine also improved postoperative bowel
function.
Defecation occurred almost 1 day earlier in the lidocaine
group.
The reduction in ileus duration by intravenous lidocaine
may be mediated by the reduction of postoperative opioid
consumption, the antiinflammatory properties of lidocaine,
and/or a direct inhibition of the sympathetic myenteric
plexus.

In summary, this study demonstrated that perioperative
administration of low doses of intravenous lidocaine
reduces intraoperative anesthetic requirements and has
a clinically relevant beneficial effect on postoperative
recovery after colectomy.
These data suggest that intravenous local anesthetics
can contribute to postoperative acute rehabilitation
programs.

Thank you
Top Related