







Languages
Pages
Legal


POLYMERASE CHAIN REACTION
Tapeshwar Yadav(Lecturer)BMLT, DNHE, M.Sc. Medical Biochemistry
Tapeshwar Yadav(Lecturer)BMLT, DNHE, M.Sc. Medical Biochemistry


• The Polymerase Chain Reaction (PCR) was not a discovery, but rather an invention
• In vitro technique for generating large quantities of a specified DNA.
• Karry Mullis, the inventor of PCR in 1989, was awarded the 1993 Nobel Prize in Chemistry


• PCR work was first published (1985)using Klenow polymerase – unstable with heat
• New enzyme had to be added manually at each step• Maximum length 400bp• Great idea – not very practical
• First reports using DNA polymerase
from Thermus aquaticus (1988)• Taq-polymerase (Saiki et al, 1988) from
Yellow stone National Park hot springs• Developed automatic “thermocycler” programmable
heat block
Development….

Polymerase Chain Reaction (PCR)
• PCR is a technique which is used to amplify the number of copies of a specific region of DNA, in order to produce enough DNA to be adequately tested.
• Cell-free amplification for synthesizing multiple identical copies (billions) of any DNA of interest.
• Basic tool for the molecular biologist.• The purpose of a PCR is to make a huge number of
copies of a gene. As a result, it now becomes possible to analyze and characterize DNA fragments found in minute quantities in places like a drop of blood at a crime scene or a cell from an extinct dinosaur.
• Like Xerox machine for gene copying.
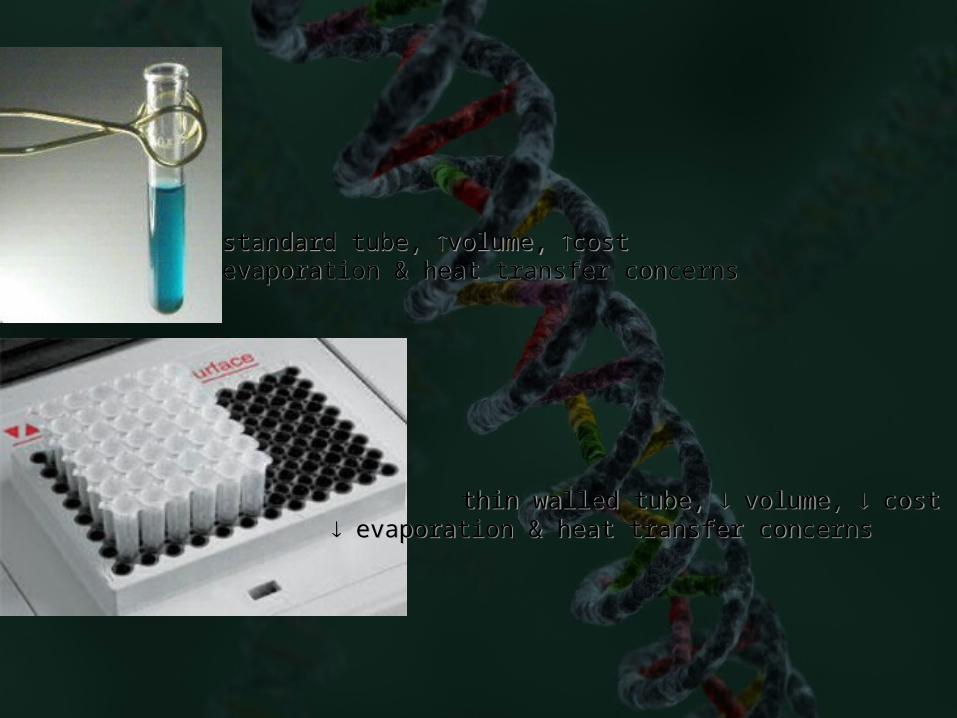
standard tube, volume, costevaporation & heat transfer concerns
thin walled tube, volume, cost evaporation & heat transfer concerns
standard tube, volume, costevaporation & heat transfer concerns
thin walled tube, volume, cost evaporation & heat transfer concerns

PCR Thermocycler

heated lidsadjustable ramping timessingle/multiple blocksgradient thermocycler blocks
heated lidsadjustable ramping timessingle/multiple blocksgradient thermocycler blocks
ThermocyclersThermocyclers

What all PCR Can Do ?
• Starting with one original copy an almost infinite number of copies can be made using PCR
• “Amplified” fragments of DNA can be sequenced, cloned, probed or sized using electrophoresis
• Defective genes can be amplified to diagnose any number of illnesses
• Genes from pathogens can be amplified to identify them (i.e., HIV, Vibrio sp., Salmonella sp. etc.)
• Amplified fragments can act as genetic fingerprints

PROCEDURE …..

PCR Reagents• 1X Buffer
– 10mM Tris-HCl, 50mM KCl• MgCl2
– 1mM - 4mM (1.5mM)• dNTPs
– 200μM• Primers
– 100nM-1μM, 200nm (or less) for real time analysis• DNA polymerase
– Taq DNA polymerase is thermostable– 1-4 Units (1 unit)
• DNA– 10pg-1μg (20ng)

MgCl2 (mM) 1.5 2 3 4 5
Magnesium Chloride (MgCl2 - usually 0.5-5.0mM)
Magnesium ions have a variety of effectsMg2+ acts as cofactor for Taq polymerase
Required for Taq to function
Mg2+ binds DNA - affects primer/template interactions
Mg2+ influences the ability of Taq pol to interact with primer/template sequences
More magnesium leads to less stringency in binding

Different types of buffers

Directional SynthesisDirectional Synthesis

Steps:1.Denaturation (Separation):- by heating at 95’C for 15 sec to 2 min.
2. Annealing (Priming):- primera are annealed by cooling to 50’C for 0.5 to 2 min.
3. Amplification (Polymerisation):-DNA strands are synthesized by Taq polymerase 72’C for 30 sec. in presence of dNTPS.
Steps:1.Denaturation (Separation):- by heating at 95’C for 15 sec to 2 min.
2. Annealing (Priming):- primera are annealed by cooling to 50’C for 0.5 to 2 min.
3. Amplification (Polymerisation):-DNA strands are synthesized by Taq polymerase 72’C for 30 sec. in presence of dNTPS.

Sources of DNA Polymerase: In the original technique of PCR, Klenow fragments of E.coli DNA polymerase was used.
This enzymes gets denatured at higher temp. therefore fresh enzyme had to be added each cycle.
Therefore introduction of Taq DNA Polymerase (Lawyer 1989) from thermophilic bacterium, Thermus aquaticus.
Taq DNA Polymerase is heat resistant, hence it is not necessary to freshly add this enzyme for each cycle to PCR.

“Xeroxing” DNA
1 copy
Cyc
le 1
PLU
S d
NTP
s, b
uffe
r,
salts
,Taq
pol
,
prim
ers
Cycle 35
n36 = 68,719,476,736 copies in ~ 2 hrs
2 copies
Cycle 2
4 copies
Cycle 3
8 copies

A simple thermocycling protocolA simple thermocycling protocol
annealingannealing
94ºC94ºC 94ºC94ºC
55ºC55ºC
72ºC72ºC
4ºC4ºC
3 min3 min 1 min1 min
45 sec45 sec
1 min1 min
∞ hold∞ hold
Initial denaturation of DNAInitial denaturation of DNA
1X1X 35X35X 1X1X
extensionextension
denaturationdenaturation
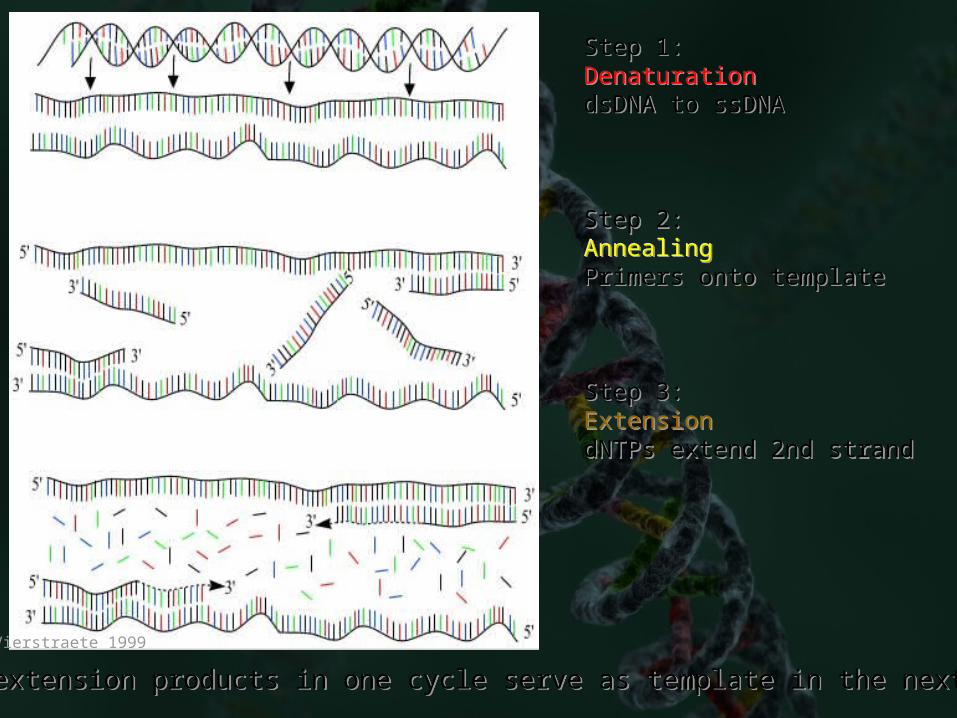
Step 1:DenaturationdsDNA to ssDNA
Step 2:AnnealingPrimers onto template
Step 3:ExtensiondNTPs extend 2nd strand
Step 1:DenaturationdsDNA to ssDNA
Step 2:AnnealingPrimers onto template
Step 3:ExtensiondNTPs extend 2nd strand
Vierstraete 1999
extension products in one cycle serve as template in the nextextension products in one cycle serve as template in the next

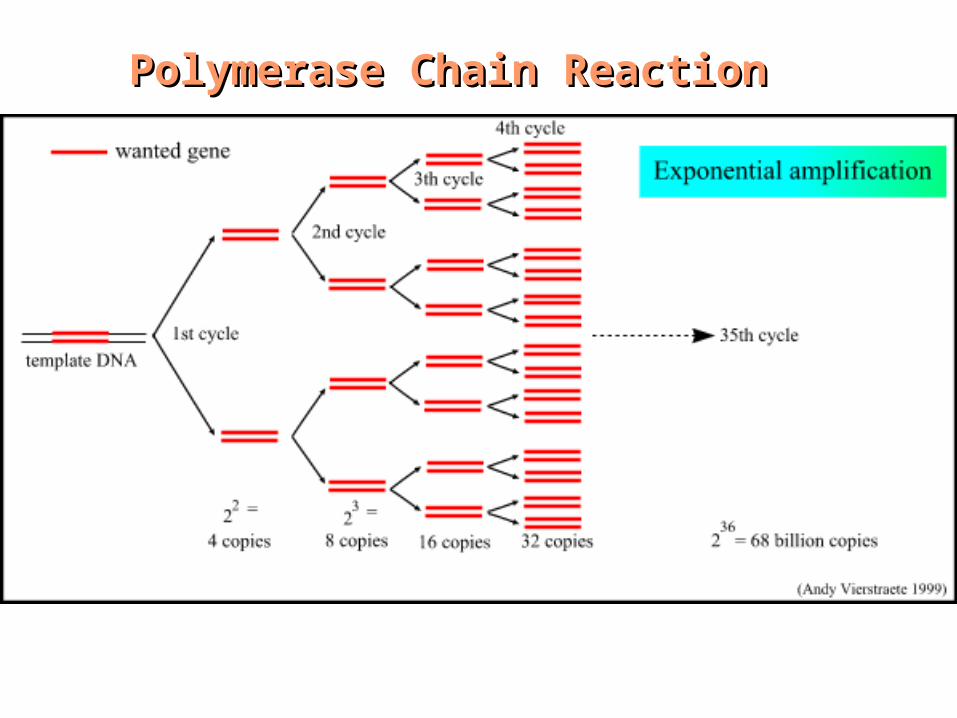
Polymerase Chain ReactionPolymerase Chain Reaction
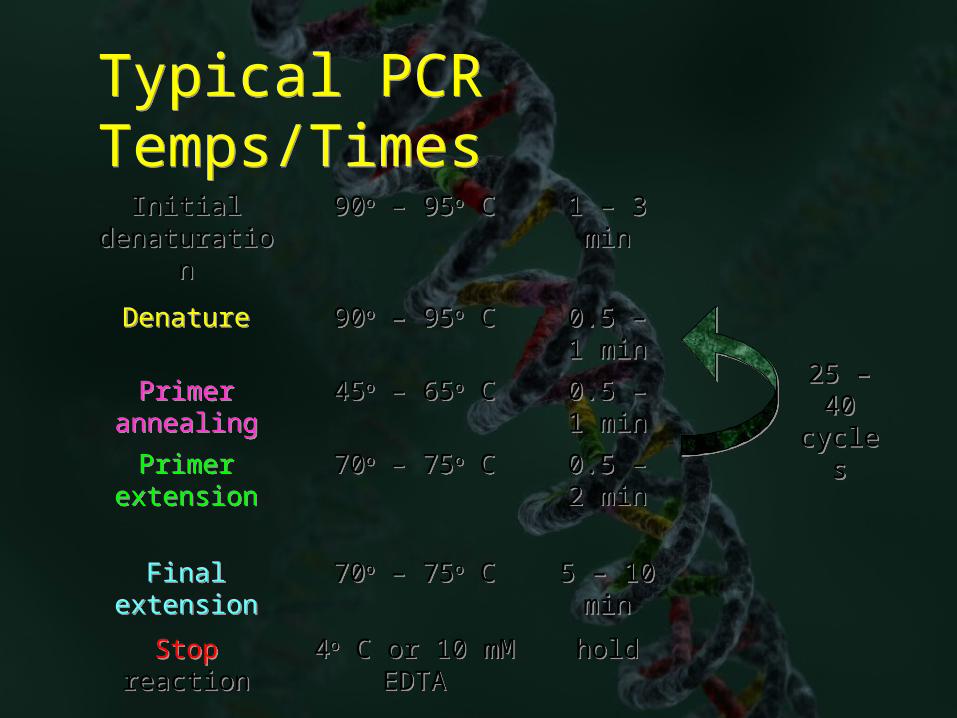
Typical PCR Temps/TimesTypical PCR Temps/Times
holdhold4o C or 10 mM EDTA
4o C or 10 mM EDTA
Stop reactionStop reaction
5 – 10 min
5 – 10 min
70o – 75o C70o – 75o CFinal extension
Final extension
0.5 – 2 min
0.5 – 2 min
70o – 75o C70o – 75o CPrimer extension
Primer extension
0.5 – 1 min
0.5 – 1 min
45o – 65o C45o – 65o CPrimer annealing
Primer annealing
0.5 – 1 min
0.5 – 1 min
90o – 95o C90o – 95o CDenatureDenature
1 – 3 min
1 – 3 min
90o – 95o C90o – 95o CInitial denaturation
Initial denaturation
25 – 40 cycles
25 – 40 cycles

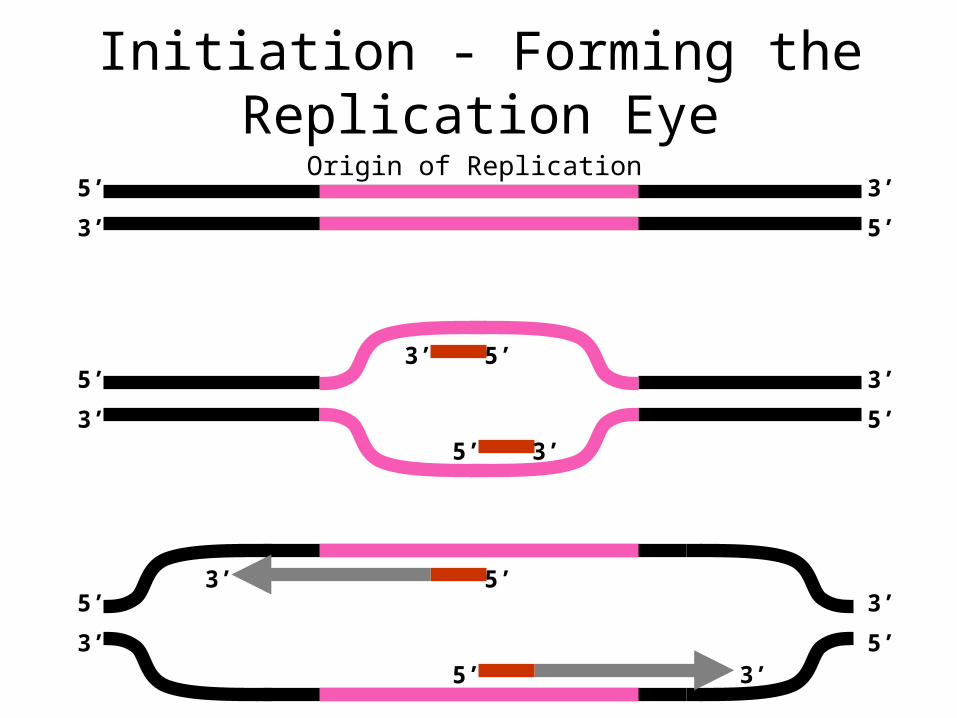
Initiation - Forming the Replication Eye
3’ 5’
3’5’5’
5’
3’
3’
Origin of Replication
5’
3’
3’
5’
5’3’
5’
5’
5’
3’
3’3’

Leading StrandLeading Strand
Laging StrandLaging Strand
3’
5’3’
5’
Extension - The Replication Fork5’
5’5’3’
3’
5’3’3’
5’
Single strand binding proteins
DNA Polymerase
Okazaki fragment
RNA Primers
Primase
5’3’
5’
Helicase

How are the functions of replication achieved during PCR ???
. N/A as fragments are short
Joining nicks
. . Taq Polymerase Polymerizing DNA
. . Primers added to the reaction mix
Providing primer
PCRFunction
. . Heat Melting DNA
ENZYMES
• Helicase•SSB proteins•Topoisomerase
•DNA pol
•Primase
•Ligase

PCRMelting
94 oC
Tem
pera
ture
100
0
50
T i m e
5’3’
3’5’

PCRMelting
94 oC
Tem
pera
ture
100
0
50
T i m e
3’5’
5’3’
Heat

PCRMelting
94 oCAnnealing
Primers50 oC
Extension72 oC
Tem
pera
ture
100
0
50
T i m e
3’5’
5’3’5’
5’
Melting94 oC
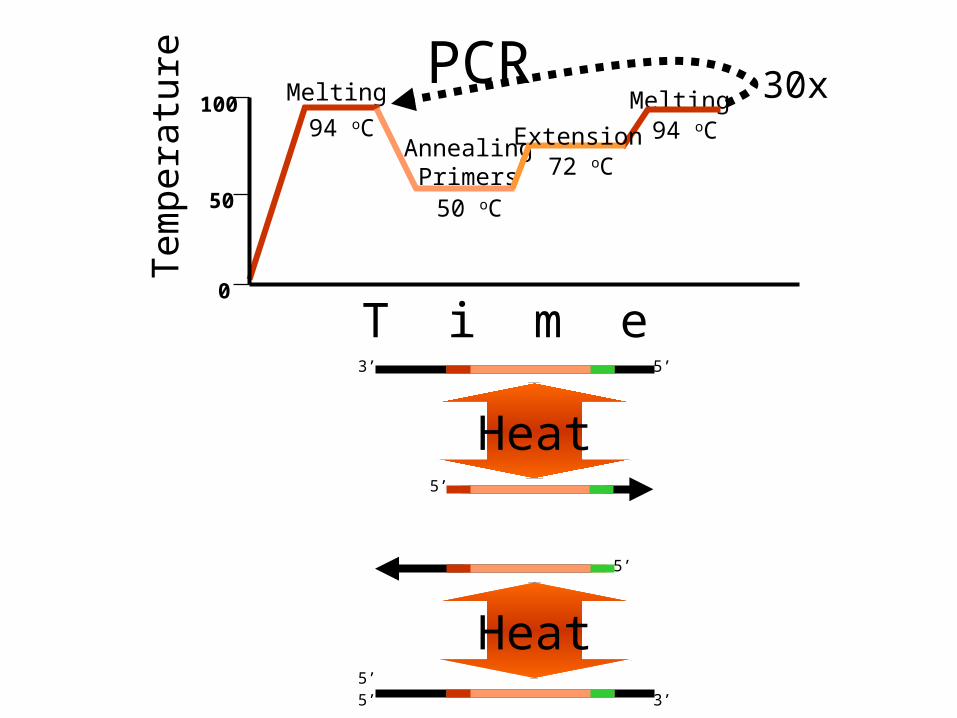
PCRMelting
94 oCMelting
94 oCAnnealing
Primers50 oC
Extension72 oC
Tem
pera
ture
100
0
50
T i m e
30x
3’5’
5’3’
Heat
Heat
5’
5’
5’

PCRMelting
94 oCMelting
94 oCAnnealing
Primers50 oC
Extension72 oC
Tem
pera
ture
100
0
50
T i m e
30x
3’5’
5’3’5’
5’
5’
5’
5’
5’

PCRMelting
94 oCMelting
94 oCAnnealing
Primers50 oC
Extension72 oC
Tem
pera
ture
100
0
50
T i m e
30x
3’5’
5’3’ 5’
5’5’
5’
5’
5’
Heat
Heat

PCRMelting
94 oCMelting
94 oCAnnealing
Primers50 oC
Extension72 oC
Tem
pera
ture
100
0
50
T i m e
30x
3’5’
5’3’ 5’
5’5’
5’
5’
5’
5’
5’
5’
5’

Fragments of defined length
PCRMelting
94 oCMelting
94 oCAnnealing
Primers50 oC
Extension72 oC
Tem
pera
ture
100
0
50
T i m e
30x
3’5’
5’3’ 5’
5’5’
5’
5’
5’
5’
5’
5’
5’
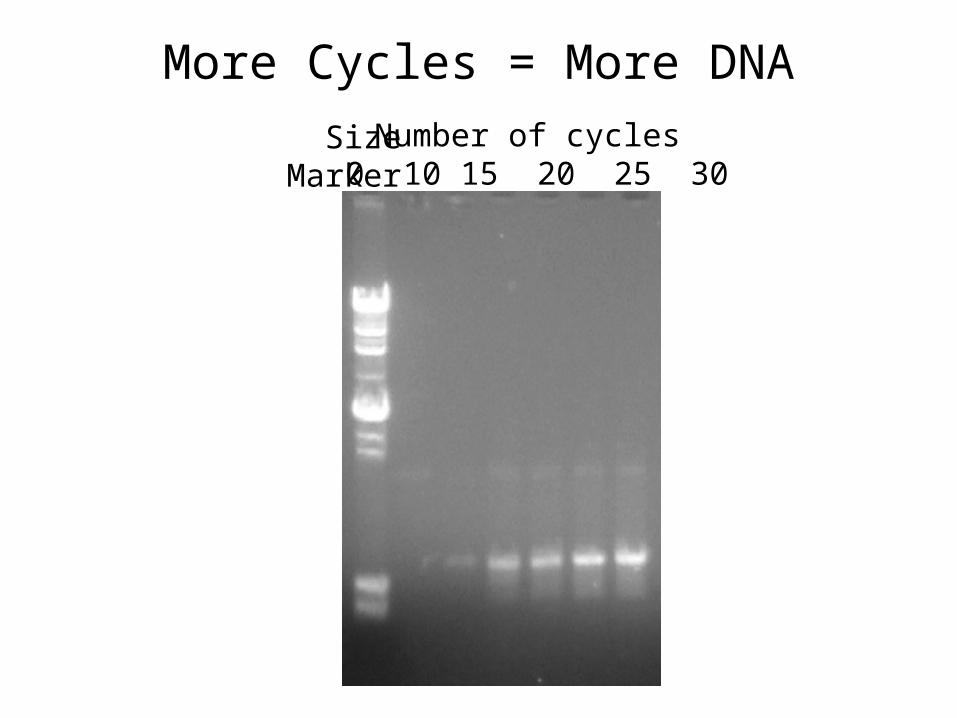
More Cycles = More DNA
Number of cycles 0 10 15 20 25 30
SizeMarker

PCR Optimisation 1: Buffers• Most buffers have only KCl (50mM) and Tris
(10mM)– Concentrations of these can be altered– KCl facilitates primer binding but
concentrations higher than 50mM inhibit Taq
• DMSO, BSA, gelatin, glycerol, Tween-20, Nonidet P-40, Triton X-100 can be added to aid in the PCR reaction– Enhance specificity, but also can be inhibitory
• Pre-mixed buffers are available

PCR Optimisation 2: MgCl2• MgCl2: required for primer binding
– MgCl2 affects primer binding, Tm of template DNA,
product- and primer-template associations, product
specificity, enzyme activity and fidelity
– dNTPs, primers and template chelate and sequester the
Mg ion, therefore concentration should be higher than
dNTPs (as these are the most concentrated)
– Excess magnesium gives non-specific binding
– Too little magnesium gives reduced yield

PCR Optimisation 3: Primer Design• Specific to sequence of interest
– Length 18-30 nucleotides• Annealing temperature 50oC-70oC
– Ideally 58oC-63oC• 3’ end critical (new strand extends from here)
• 3’ complementarity:– <3-4 bases similar to other primer regions

PCR Optimisation 4: Cycling Conditions
• Denaturation: – Some Taq polymerases require initial denaturation (hot
start)
• Annealing temperature: – ~ 5oC less than Tm of primers– Tm = 4(G + C) + 2(A + T)oC (or use of primer software)– Decrease in annealing temperature result in non-
specific binding– Increase in annealing temperature result in reduced
yield

PCR Optimisation 5: Cycle Number• 25-40 cycles• Half-life of Taq is
30 minutes at 95oC
• Therefore if you use more than 30 cycles at denaturation times of 1 minute, the Taq will not be very efficient at this point
Theoretical yield = 2n
ie. cycle 1 = 2, cycle 2 = 4, cycle 3 = 8, etc
eg. if you start with 100 copies after 30 cycles you will have 107, 374, 182, 400 copies

PRIME
• The PRIME is a good tool for the design of primers for PCR and sequencing– For PCR primer pair selection, you can choose a target range of
the template sequence to be amplified
• In selecting appropriate primers, PRIME allows you to specify a variety of constraints on the primer and amplified product sequences. – upper and lower limits for primer and product melting temperatures – a range of acceptable primer sizes – a range of acceptable product sizes. – required bases at the 3' end of the primer (3' clamp) – maximum difference in melting temperatures between a pair of PCR primers

PC Software• There are a number of (expensive) dedicated PCR
primers design programs for personal computers that have “special features” such as nested and multiplex PCR :– Oligo (Molecular Biology Insights, Inc.)– Primer Premier (Premier Biosoft)
• Many of the comprehensive MolBio. programs also have PCR features
– Mac Vector– OMIGA– Vector NTI– Gene Tool

Primer Problems
• primers should flank the sequence of interest
• primer sequences should be unique
• primers that match multiple sequences will give multiple products
• repeated sequences can be amplified - but only if unique flanking regions can be found where primers can bind

Variations of the PCR• Colony PCR• Nested PCR• Multiplex PCR• AFLP PCR• Hot Start PCR• In Situ PCR• Inverse PCR• Asymmetric PCR• Long PCR• Long Accurate PCR• Reverse Transcriptase PCR• Allele specific PCR• Real time PCR

Types of PCR
Long PCR: Used to amplify DNA over the entire length up to 25kb of genomic DNA
segments cloned.
Nested PCR: Involves two consecutive PCR reactions of 25 cycles. The first PCR uses primers external to the sequence of interest. The second PCR uses the product of the first PCR in conjunction with one or more nested primers to amplify the sequence within the region flanked by the initial set of primers.
Inverse PCR: Used to amplify DNA of unknown sequence that is adjacent to known DNA sequence.
Quantitative PCR: Product amplification w r t time, which is compared with a standard DNA.
Hot start PCR: Used to optimize the yield of the desired amplified product in PCR and simultaneously to suppress nonspecific amplification.

Colony PCRColony PCR- the screening of bacterial (E.Coli) or yeast clones for
correct ligation or plasmid products.
Pick a bacterial colony with an autoclaved toothpick, swirl it into 25 μl of TE autoclaved dH2O in an microfuge tube.
Heat the mix in a boiling water bath (90-100C) for 2 minutes
Spin sample for 2 minutes high speed in centrifuge.
Transfer 20 μl of the supernatant into a new microfuge tube
Take 1-2 μl of the supernatant as template in a 25 μl PCR standard PCR reaction.

Hot Start PCR• This is a technique that reduces non-specific amplification
during the initial set up stages of the PCR
• The technique may be performed manually by heating the reaction components to the melting temperature (e.g., 95°C) before adding the polymerase
• DNA Polymerase- Eubacterial type I DNA polymerase, Pfu
• These thermophilic DNA polymerases show a very small polymerase activity at room temperature.

Nested PCR
• Two pairs (instead of one pair) of PCR primers are used to
amplify a fragment.
• First pair -amplify a fragment similar to a standard PCR.
Second pair of primers-nested primers (as they lie / are
nested within the first fragment) bind inside the first PCR
product fragment to allow amplification of a second PCR
product which is shorter than the first one.
• Advantage- Very low probability of nonspecific amplification


Multiplex PCR
• Multiplex PCR is a variant of PCR which enabling simultaneous amplification of many targets of interest in one reaction by using more than one pair of primers.

Inverse PCR• Inverse PCR (Ochman et al., 1988) uses standard PCR
(polymerase chain reaction)- primers oriented in the reverse direction of the usual orientation.
• The template for the reverse primers is a restriction fragment that has been selfligated
• Inverse PCR functions to clone sequences flanking a known sequence. Flanking DNA sequences are digested and then ligated to generate circular DNA.
Application• Amplification and identification of flanking sequences such
as transposable elements, and the identification of genomic inserts.

Long PCR
• Extended or longer than standard PCR, meaning over 5 kilobases (frequently over 10 kb).
• Long PCR is useful only if it is accurate. Thus, special mixtures of proficient polymerases along with accurate polymerases such as Pfu are often mixed together.
• Application- to clone large genes

Reverse Transcriptase PCR
• Based on the process of reverse transcription, which reverse transcribes RNA into DNA and was initially isolated from retroviruses.
• First step of RT-PCR - "first strand reaction“-Synthesis of cDNA using oligo dT primers (37°C) 1 hr.
• “Second strand reaction“-Digestion of cDNA:RNA hybrid (RNaseH)-Standard PCR with DNA oligo primers.
• Allows the detection of even rare or low copy mRNA sequences by amplifying its complementary DNA.

Why real time PCR ?
• QUANTITATION OF mRNA
– northern blotting– ribonuclease protection assay– in situ hybridization– RT-PCR
• most sensitive
• can discriminate closely related mRNAs
• technically simple
• but difficult to get truly quantitative results using conventional PCR

Real-Time PCRReal-Time PCR
Real-time PCR monitors the fluorescence emitted during the reaction as an indicator of amplicon production at each PCR cycle (in real time) as opposed to the endpoint detection

• Traditional PCR has advanced from detection at
the end-point of the reaction to detection while the
reaction is occurring (Real-Time).
• Real-time PCR uses a fluorescent reporter signal
to measure the amount of amplicon as it is
generated. This kinetic PCR allows for data
collection after each cycle of PCR instead of only
at the end of the 20 to 40 cycles.

Real-time PCR advantagesReal-time PCR advantages
* amplification can be monitored real-time
* no post-PCR processing of products (high throughput, low contamination risk)
* ultra-rapid cycling (30 minutes to 2 hours)
* wider dynamic range of up to 1010-fold
* requirement of 1000-fold less RNA than conventional assays
(6 picogram = one diploid genome equivalent)
* detection is capable down to a two-fold change
* confirmation of specific amplification by melting curve analysis
* most specific, sensitive and reproducible
* not much more expensive than conventional PCR(except equipment cost)

Real-time PCR disadvantagesReal-time PCR disadvantages
* Not ideal for multiplexing
* setting up requires high technical skill and support
* high equipment cost
* intra- and inter-assay variation
* RNA liability
* DNA contamination (in mRNA analysis)

Applications of PCRApplications of PCR
• Classification of organisms
• Genotyping• Molecular archaeology
• Mutagenesis• Mutation detection
• Sequencing• Cancer research
• Detection of pathogens
• DNA fingerprinting
• Drug discovery• Genetic matching• Genetic engineering
• Pre-natal diagnosis

1.PCR in clinical diagnosis:
specificity & sensitivity of PCR is highly useful for the diagnosis of various diseases in humans.
Eg- Inherited disorders (genetic diseases), Viral diseases, Bacterial diseases etc
i) Prenatal diagnosis of inherited diseases
ii) Diagnosis of retroviral infections
iii) Diagnosis of bacterial infections
iv) Diagnosis of cancers
v) Sex determination of embryos

2. PCR in DNA sequencing
3. PCR in forensic medicine
4. PCR in comparative studies of genomes.

PCR Virtues
• High sensitivity• Can detect and quantify specific events• Higher stability of DNA permits analysis of food
samples.• Quantitative and qualitative

Thank you
THANK YOU
Top Related