







Languages
Pages
Legal


Parathyroid and Adrenal Glands,
Endocrine Pancreas

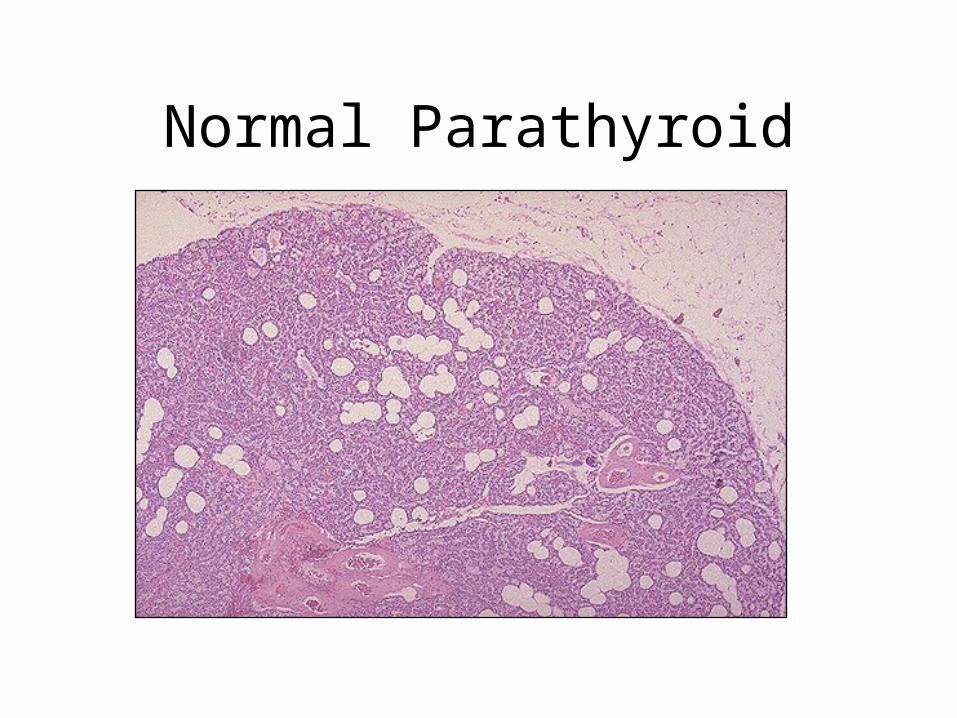
Normal Parathyroid Gland
• Parenchyma consists of chief cells that secrete parathyroid hormone (parathormone, PTH) under the influence of decreasing serum calcium
• There are also variable numbers of oxyphil cells in small nodules which have pink cytoplasm

Parathyroid Glands
• Normal number 4 (but can be 2 or 6)
• Normal combined weight 120 mg
• Normal maximum dimension 6mm
• Derived from epithelium and 3rd and 4th branchial clefts
Normal Parathyroid

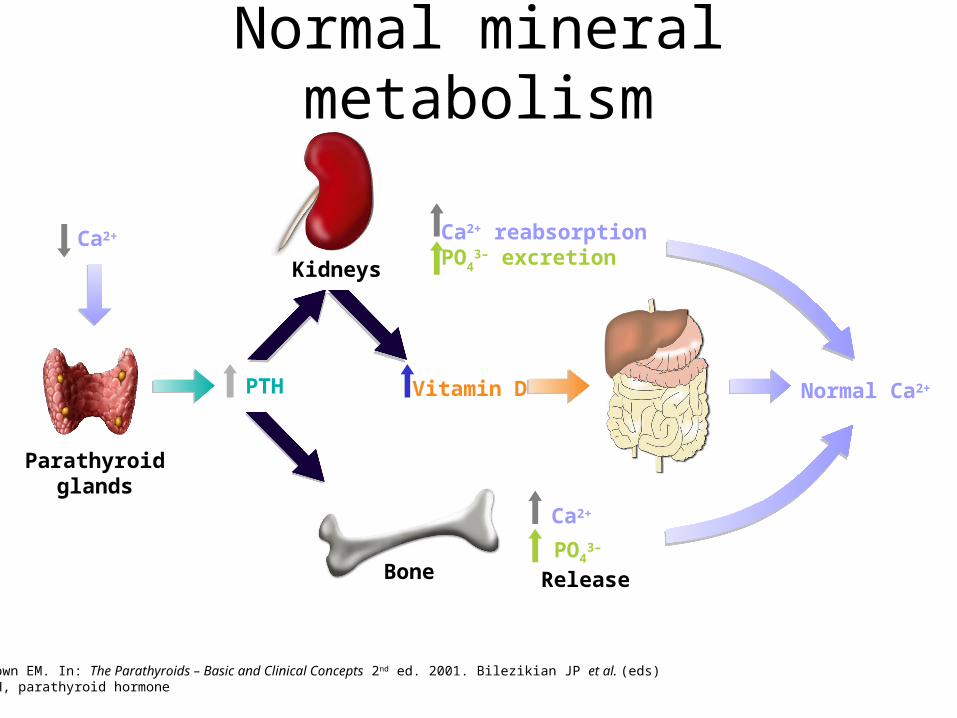
Actions of Parathormone PTH
• Kidney: – a.increased Ca resorbtion by tubule – b.decreased phosphate resorption– c. stimulate 1,25-OH2D3 synthesis by the kidney, thus promoting Ca
absorption from the gut
• Bone: increased calcium and phosphate resorption by osteoclasts• Bowel: increased calcium and phosphate absorbtion by
enterocytes
Net effect: raises serum calcium, lowers serum phosphate
Normal Ca2+
Ca2+
PO43–
ReleaseBone
Kidneys
Ca2+ reabsorptionPO4
3– excretion
PTH
Normal mineral metabolism
Brown EM. In: The Parathyroids – Basic and Clinical Concepts 2nd ed. 2001. Bilezikian JP et al. (eds)PTH, parathyroid hormone
Ca2+
Parathyroidglands
Vitamin D

Causes and Types of Hyperparathyroidism
• Primary: found in 1:1000 adults. Usually female, 30+. Adenoma 70%, hyperplasia 30%
• Secondary: less common. Chronic renal disease, Vit D deficiency, malabsorbtion, ectopic hormone production
• Tertiary: rare. Autonomous adenoma developing in secondary hyperplasia
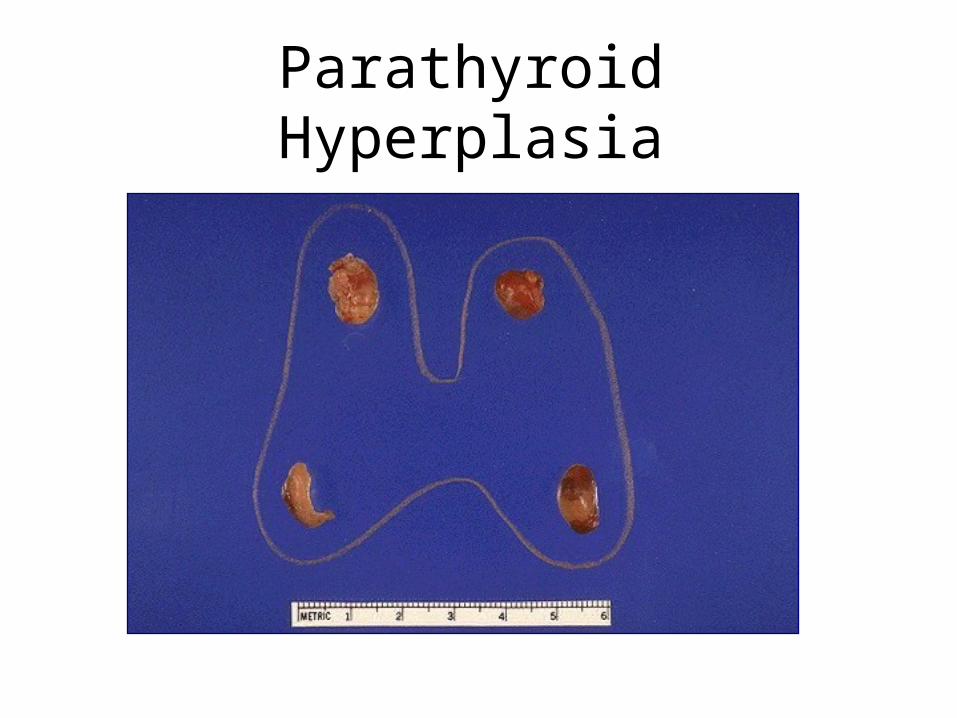
Parathyroid Hyperplasia

Parathyroid Hyperplasia
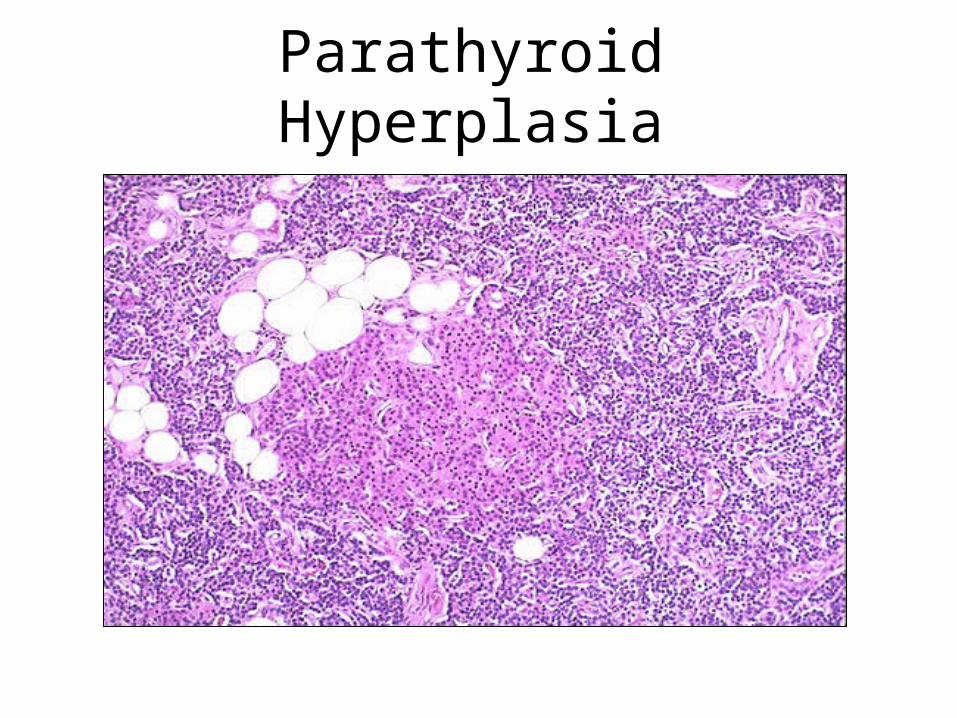
Parathyroid Hyperplasia

Clear cell Hyperplasia

Parathyroid Adenoma
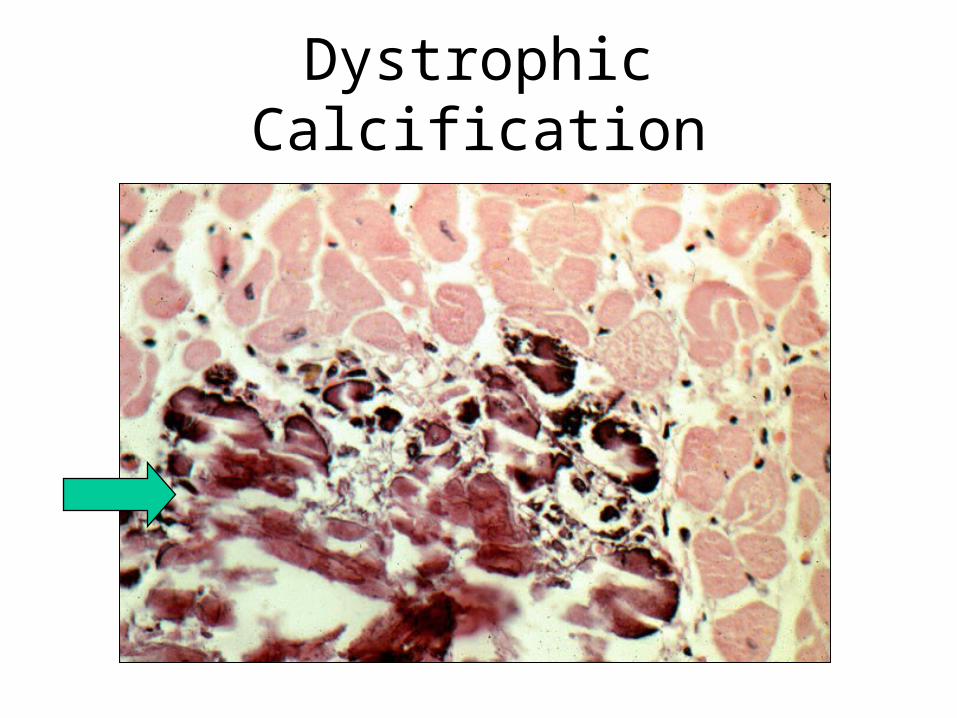
Dystrophic Calcification

Parathyroid Carcinoma

Features of Hyperparathyroidism
• Malaise, constipation, muscle weakness, neuropsychiatric disorders
• renal colic due to stones (60%)• bone pain due to generalised Ca loss• peptic ulcer (10%)• acute pancreatitis• nephrocalcinosis• raised serum calcium and PTH• raised urinary PO4 and serum alk phos• raised urinary hydroxyproline

Osteitis Fibrosa Cystica
• Classic localised bone lesion of hyperparathyroidism. Bone is lysed by osteoclasts driven by elevated PTH. Marrow replace by highly vascularised fibrous tissue. Stress on weakened bone causes haemorrhage and cyst formation.
• Old term for this lesion was “brown tumour”. Colour due to massive haemosiderin deposition
• Typically found in jaw and long bones and may cause pathological fractures
• Can be distinguished from other giant cell tumours of bone by estimation of serum Ca.

Causes and features of Hypoparathyroidism
• Injury or removal: surgery, birth trauma, autoimmune destruction
• Receptor defect: X-linked dominant receptor deficiency- so-called pseudohypoparathyroidism
• Clinical features: tetany, low Ca, high PO4, low urine PO4

Pathogenesis of diseases of the Adrenal Glands and
Endocrine Pancreas
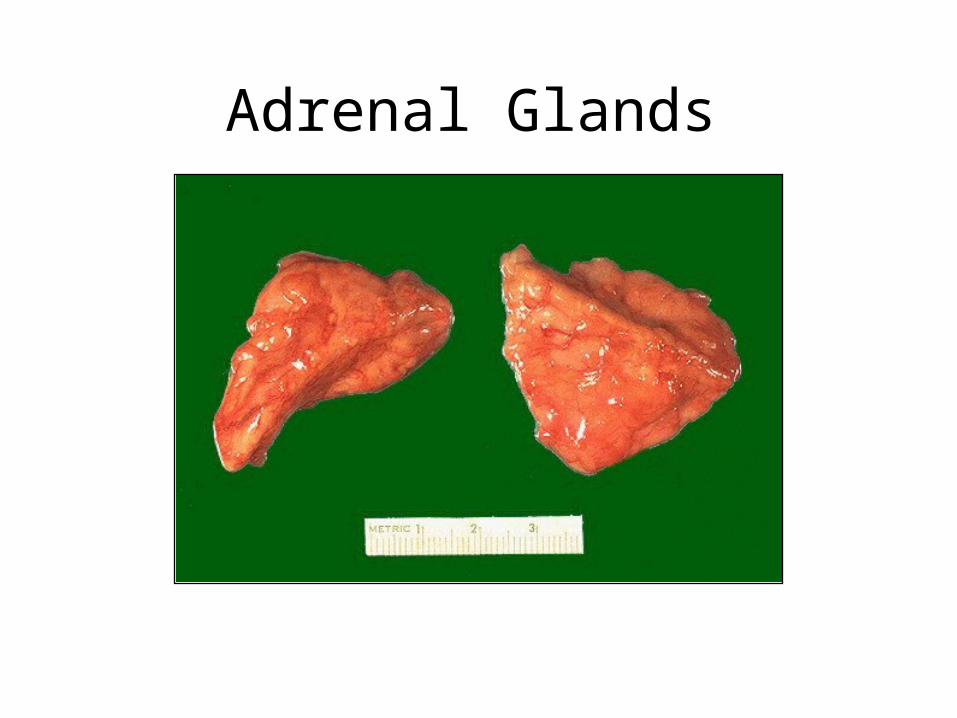
Adrenal Glands

Adrenal Glands
• Cortex: zona glomerulosa, zona fasicularis, zona reticulosa– Aldosterone: re-absorbtion of NA, loss of K– Glucocorticoids: gluconeogenesis, anti-
inflammatory– Androgens: anabolic, virilizing
• Medulla: sympathetic nerve cells, produce adrenaline and noradrenaline

Adrenal Gland : normal

Causes of Cushing Syndrome (hypersecretion of glucocorticoids)
• Iatrogenic: corticosteroid therapy• Pituitary origin: basophilic adenoma or
hypothalamic dysfunction• Adrenal origin: cortical adenoma,
carcinoma, hyperplasia• Ectopic ACTH production: small cell
carcinoma of lung, islet cell tumours, other neoplasms

Manifestations of Cushing Syndrome
• Psychosis• Polycythaemia• Moon face• Buffalo hump• Striae• Truncal obesity• Muscle Wasting• Acne, hirsutism, hypertension,
hyperglycaemia, osteoporosis• Amenorrhoea , impotence

Hypocorticoadrenalism (acute)
• Increased demand for steroids in the face of chronic insufficiency (e.g. Addison’s)
• Bilateral adrenal necrosis due to viremia (e.g.herpes), Gram- septicemia, and/or DIC. Necrosis often haemorrhagic- typified by the Waterhouse-Friederichsen syndrome
• Features of acute insufficiency: profound hypotension (shock), vomiting, dehydration, low serum Na, high K

Hypocorticoadrenalism (chronic)
• Usually referred to as Addison’s disease, and caused by either:
• Autoimmune destruction of adrenal cortex in which there is a strong association with other autoimmune diseases e.g. PA, Type 1 diabetes, Hashimoto thyroiditis or
• Metastatic tuberculosis of the adrenals

Addison’s Disease
• Clinical manifestations:Clinical manifestations:
extreme muscle weakness,hypotension, hypoglycaemia, pigmentation buccal mucosa
• Biochemical manifestationsBiochemical manifestations:
low serum sodium, glucose and cortisol
high serum potassium and ACTH
high urinary sodium
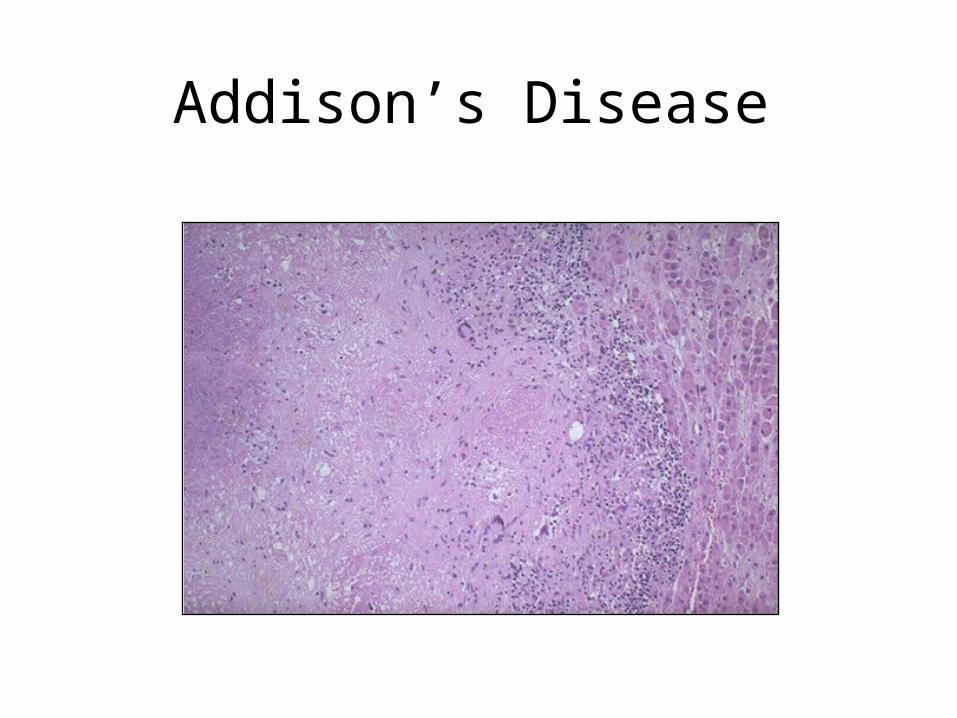
Addison’s Disease

Adrenal Haemorrhage

Tuberculosis Adrenal

Amyloid Adrenal

Tumours of Adrenal Cortex
• Adenoma: common, benign, and mostly non-functional. Found at autopsy in 2% of population. More frequent in hypertensives (20%), women > 80 (30%), obese diabetics (30%)
• Carcinoma: rare, functional in 50%. May cause Cushing’s syndrome (50%), virilization (30%), feminisation (12%), Conn’s syndrome (4%)
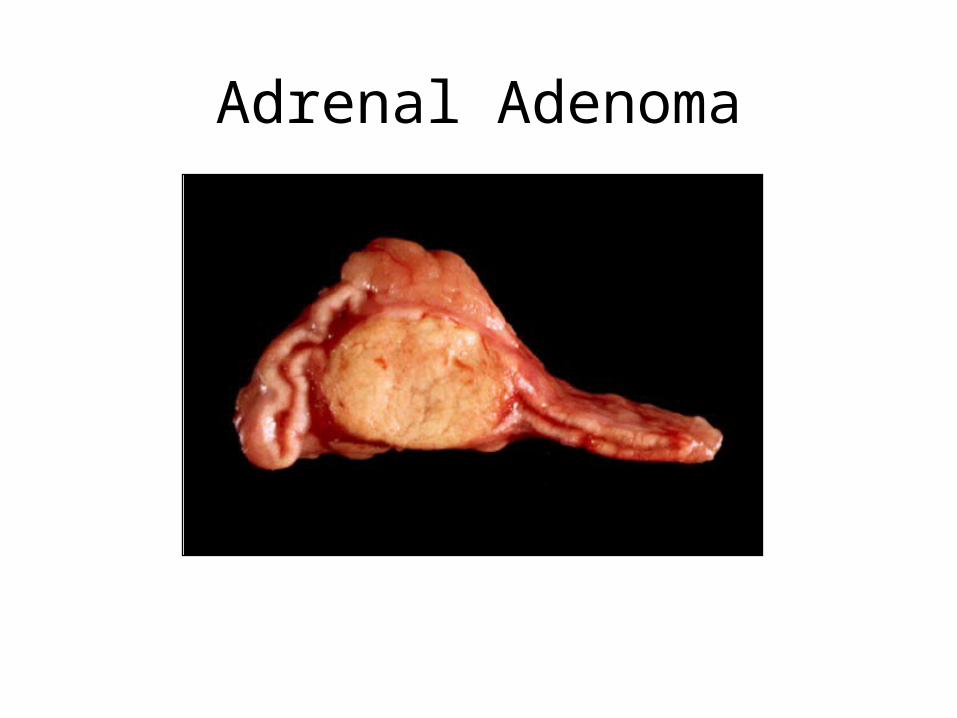
Adrenal Adenoma

Cortical Adenoma

Adrenocortical Carcinoma

Adrenocortical Carcinoma
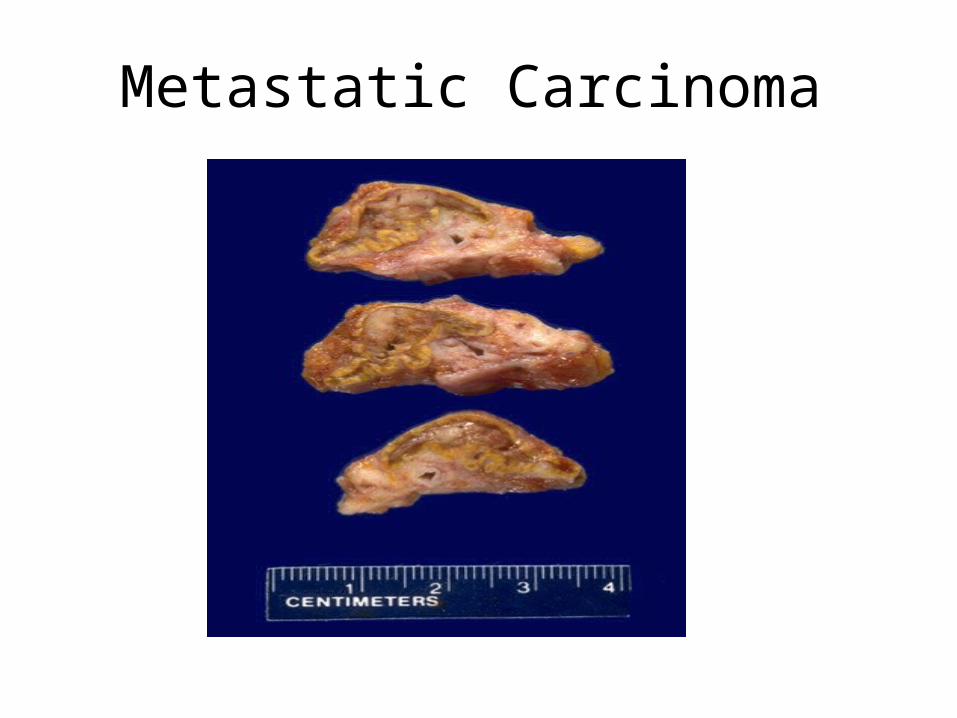
Metastatic Carcinoma

Conn’s Syndrome (hyper-mineraloadrenocorticism)
• Clinical Features:
hypertension due to sodium retention
muscle paralysis due to hypokalemia
polyuria and polydipsia
susceptibility to pyelonephritis
low plasma rennin

Conn’s Syndrome: pathogenesis
• Cortical adenoma (aldosteronoma) 90%
• Nodular cortical hyperplasia 10%
• Adrenocortical carcinoma- very rare

Tumours of Adrenal Medulla: neuroblastoma
• < 3 years, M=F• highly malignant PNET that commonly
presents with mets e.g. skull (Hutchinson) or liver (Pepper)
• sporadic and familial forms• N-myc amplified in up to 80% of cases have
deletion of distal short arm of Ch1• Early lesions with ganglion cells have much
better prognosis (94% 2 yr survival)
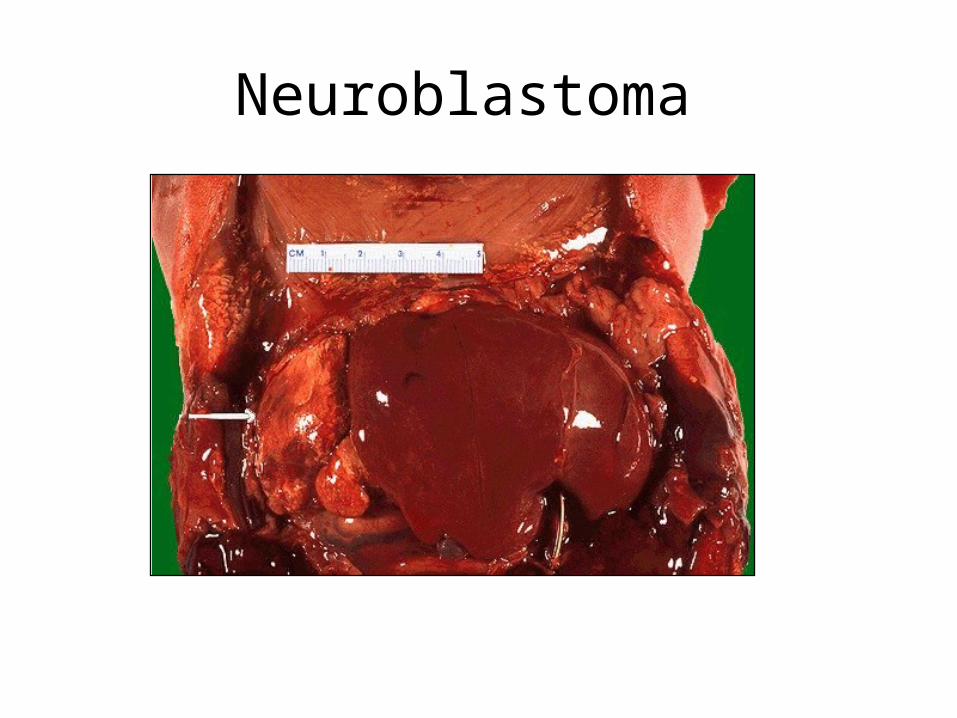
Neuroblastoma

Neuroblastoma

Tumours of Adrenal Medulla: phaeochromocytoma
• a paraganglioma, 30-50 yrs, M=F
• 10% bilateral, 10% malignant, 10% extra-adrenal
• secretes noradrenaline producing hypertension, tachycardia, glycosuria, pallor and sweating
• elevated urinary VMA

Phaeochromocytoma
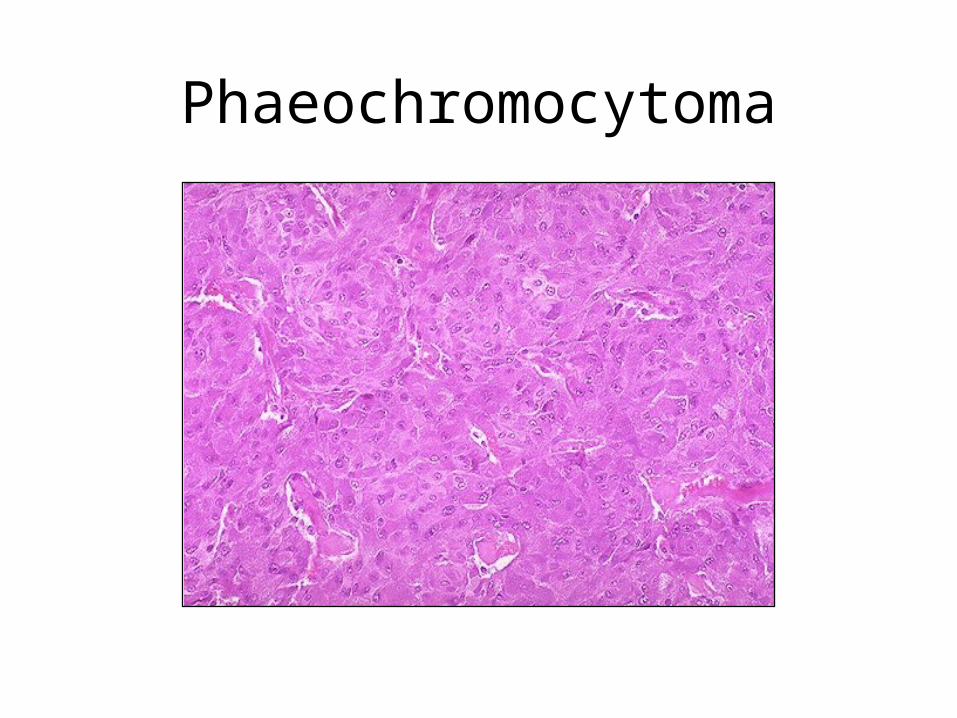
Phaeochromocytoma

Diseases of the Endocrine Pancreas
[slides 45-103- for personal revision]

Diabetes Mellitus
• Definition
A chronic systemic disease characterised by absolute or relative lack of insulin and abnormalities of structure and function of blood vessels

Diabetes Mellitus
• Disorder of Metabolism- carbohydrate, protein and fat
• Characterised by hyperglycaemia
• Clinically; polyuria, polydipsia, polyphagia

Diabetes Mellitus
• Diabetes mellitus (sweet urine)• 3% world population, 100 million people• Incidence increasing alarmingly (40% in the
past decade, more in future, 259m by 2025)• Most common non-communicable disease• High morbidity and mortality• Shortens life span by 15 years• Leading cause of blindness and kidney
disease

Normal Pancreatic Islet:
ß cells ß cells (Insulin) αα cells cells (Glucagon)
δδ cells cells (Somatostatin) pp Cells pp Cells (polypep)
ßß αα
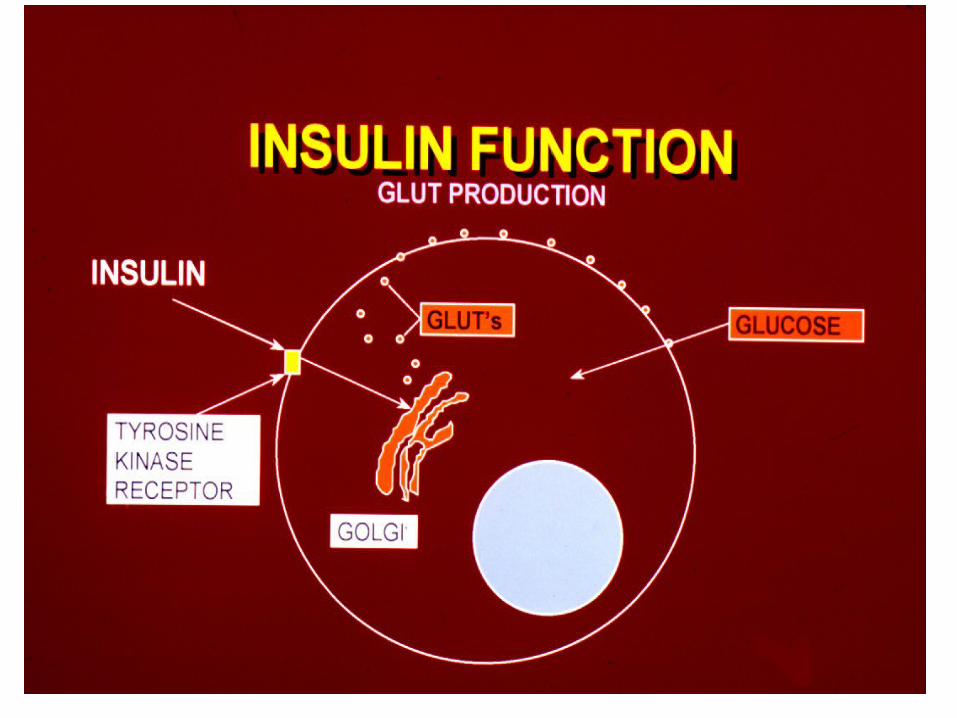

Cellular Glucose Uptake
Insulin Requiring• Striated Muscle• Cardiac Muscle• Fibroblasts• FAT
Non-Insulin Requiring• Blood Vessels• Nerves• Kidney• Eye Lens

Blood glucose and hormones
• Insulin lowers blood glucose
• Glucocorticoids, Glucagon, Growth hormone and adrenaline all raise blood glucose

Insulin - Anabolic Steroid
• Transmembrane transport of glucose
• Liver, muscle & fat blood glucose
• Liver & skeletal muscle - glycogen
• Converts glucose to triglycerides
• Nucleic acid & Protein synthesis
• Diabetes Diabetes Increased catabolism. Increased catabolism.
• Hyperglycemia, protein synthesis, Lipolysis, wasting, weight loss.

Pathology in Diabetes
• Low glucose inside cell : decreased cell metabolism (muscle and liver)
• High glucose outside cell: glycosylation damage and polyol products damage

Pathophysiology of Insulin Deficiency
Normal
• Transmembrane transport of glucose (GLUTS) and amino acids. Maintains normoglycaemia.
• Glycogen formation in liver and muscle
• Conversion of glucose to triglycerides in fat
• Protein synthesis from amino acids
Diabetes• Failure of transmembrane
transport gives rise to hyperglycaemia
• Glycogen not formed and existing stocks broken down by unopposed Glucagon activity
• Breakdown of fat leads to increased free fatty acids which are oxidised in liver to acetone, beta-OH-butyric and acetoacetic acid
• Breakdown of existing protein to produce amino acids, converted to glucose in liver

Classification
Primary DM (primary- no other disease)• Type 1- IDDM/Juvenile 10%• Type 2- NIDDM/Adult onset 80%• MODY- 5% maturity onset- genetic beta cell
dysfunction• Gestational diabetesSecondary DM (secondary to other disease)• Pancreatitis/tumours/haemochromatosis• Infectious- congenital rubella, CMV• Endocrinopathy• Drugs- corticosteroids

Diabetes Mellitus
• Type 1 or “juvenile” IDDM- almost all HLA-DR3 or HLA-DR4 yet
- monozygous twin concordance only 40-45%
- this suggests that factors other than, or as well as, genetic predisposition are responsible
• Type II or “adult” NIDDM- no HLA relationship but strong genetic influence
indicated by
- monozygous twin concordance of 50-90%

Type 1 Diabetes Mellitus: current hypothesis 1
• Evidence for organ-specific autoimmunity- antibodies to islet cells in acute cases: these
are directed against glutamic acid decarboxylase (GAD) and other cytoplasmic proteins but may be a secondary phenomenon
- associated familial autoimmunity- lymphocytic insulitis: T cells, mainly CD8+ and
variable numbers of CD4+. Transfer of the CD4+ cells can induce DM in animals

Type 1 Diabetes Mellitus: current hypothesis 2
• Evidence for viral damage to islets- High titres of coxackie antibodies in acute
cases in the first year
- Seasonal variation in incidence; onset higher in autumn/winter than spring/summer
- Lymphocytic insulitis

Type 1 Diabetes Mellitus: current hypothesis 3
• Many viruses are beta-cell tropic but direct viral injury alone is rarely severe enough to cause DM. During viral insulitis cytokines, such as gamma-interferon, are secreted locally by T-cells and cause beta-cells to express Class II MHC (HLA) antigens. These antigenically altered cells provide a target for cytotoxic CD8+ T-cells.

Pathogenesis of Type I DM
Genetic Genetic
HLA-DR3/4HLA-DR3/4 EnvironmentEnvironment
Viral infe..?Viral infe..?
Insulin deficiencyInsulin deficiencyType I / IDDMType I / IDDM
Autoimmune InsulitisAutoimmune Insulitis
Ab to ß cells/insulin Ab to ß cells/insulin
ß cell ß cell DestructionDestruction
• Glomerulonephritis• Graves, Hashimoto thyroiditis.• Rheumatic heart disease• SLE, Collagen vascular disease• Rheumatoid arthritis.
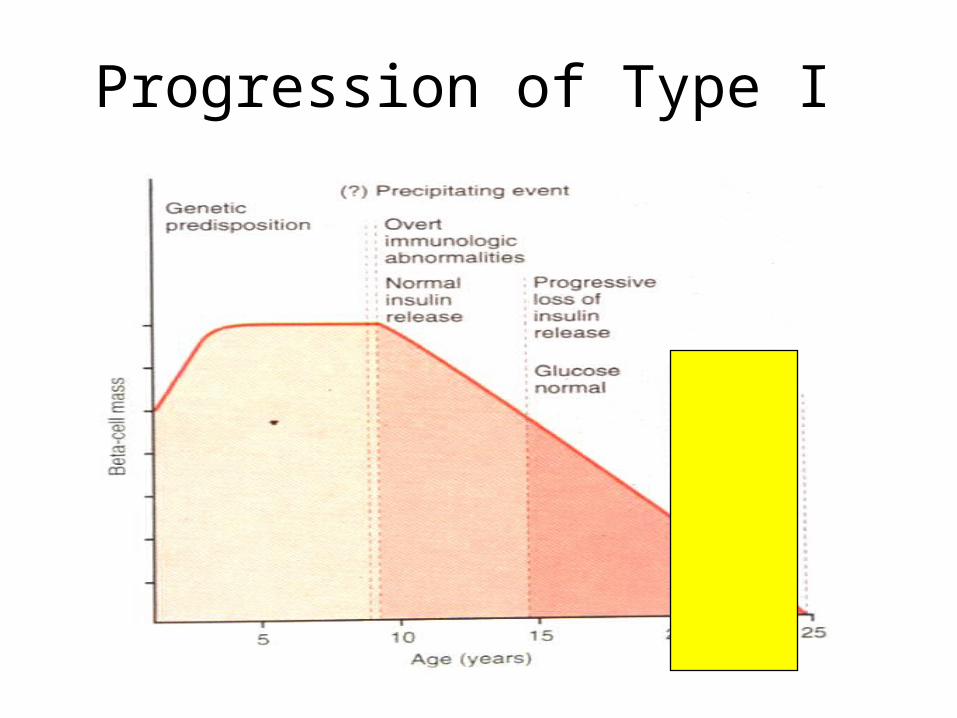
Progression of Type I

Pathogenesis of Type 2 DM
• Genetic susceptibility very important• Obesity (80%)/Lifestyle/Age• No HLA linkage• No autoimmune mechanisms• Metabolic defects are:
– Derangement of beta cell secretion of insulin– Decreased response of peripheral tissue in
responding to insulin (receptor downregulation/dysfunction; insulin antagonism)

IDDMIDDM
Genetic /Genetic /
ß cell defectß cell defect
Pathogenesis of Type II DM
Obesity /Obesity /
Life style ?Life style ?
ß cell ß cell
exhaustionexhaustion
Type II NIDDMType II NIDDM
Abnor. SecretionAbnor. Secretion
Insulin ResistanceInsulin Resistance
Relative Relative
Insulin Def.Insulin Def.


Islet Pathology in Diabetes
• Pancreas in long-standing Type 1 diabetes shows absence of beta cells in islets
• In Type 2 diabetes the islets may appear normal or may show replacement by amyloid, due to deposition of amylin, a protein which is co-secreted with insulin by the beta cells.

Insulitis – Type I
InsulinitisInsulinitis
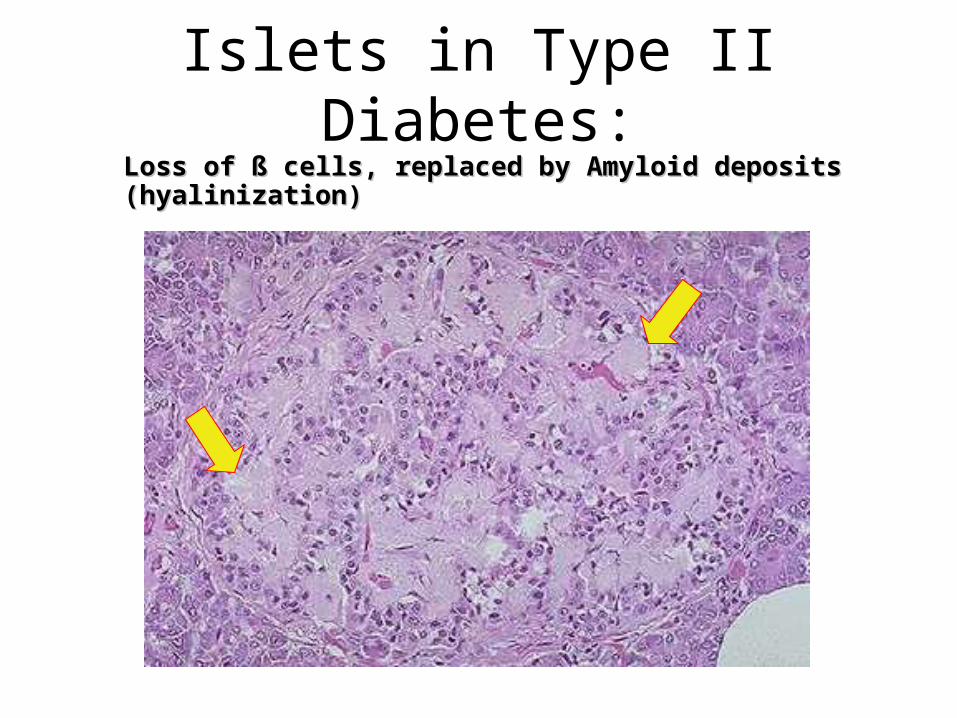
Islets in Type II Diabetes:Loss of ß cells, replaced by Amyloid deposits (hyalinization)Loss of ß cells, replaced by Amyloid deposits (hyalinization)

Type 1 Type 2
• Less common• Children < 25 yrs• Insulin dependant• Duration: weeks• Acute metabolic
complications• Autoantibody- yes• Family history- no• Insulin levels low• Islets – insulitis• 50% twin concordance
• More common• Adults > 25 yrs• Insulin independent• Months to years• Chronic vascular
complications• No autoantibodies• Family history- yes• Insulin levels N or high• Normal/exhaustion• 60-80% in twins

The acute complications of diabetes
• Hyperglycemia
• Diabetic ketoacidosis
• Non-ketotic hyperosmolar diabetic coma
• Lactic acidosis

Acute Complications of DM

The Chronic Complications of Diabetes
• Retinopathy
• Neuropathy
• Nephropathy

Complications of Diabetes: Basement Membrane Thickening
Non-enzymatic glycosylation of proteins leads to irreversible cross-linkage to BM collagen IV in vessels
This results in trapping of filtered protein and loss of proteoglycans from a thickened and leaky basement membrane, and is the cause of diabetic nephropathy and retinopathy.

Advanced Glycosylation end-products
• AGE’s cause accelerated atheroma by damage to basement membrane of vessels allowing low-density lipoprotein accumulation in the intima
• AGE’s derived from glycosylated plasma proteins also bind to receptors on lymphocytes and macrophages and block effective cell-cell signalling. This is probably the cause of relative immune incompetance and increased susceptibility to infection in diabetes

Complications of Diabetes: intracellular hyperglycaemia
• Neural and vascular cells do not require insulin for glucose importation
• High intracellular glucose levels due to hyperglycaemia lead to sorbitol and fructose accumulation
• This results in osmotic damage to Schwan cells and pericytes
• Neuropathy and Microaneurysms develop

Conditions affecting diabetics atypically or more frequently than normal
• ATHEROMA• INFECTION • CATARACT• GALL-STONES• FOETAL DEATH/DEFECT
The latter is probably a result of varying maternal glucose levels during gestation. Neonatal hypoglycaemia may be due to increased foetal beta-cell mass induced by persistently high maternal glucose levels.

Microangiopathy Pathogenesis:
Hyperglycemia chronic. Glycosylation of basement membrane
proteins Leaky blood vessels. Excess deposition of proteins –
glycosylation cycle. Thick and Leaky blood vessels. Narrow lumen Ischemic Organ damage...
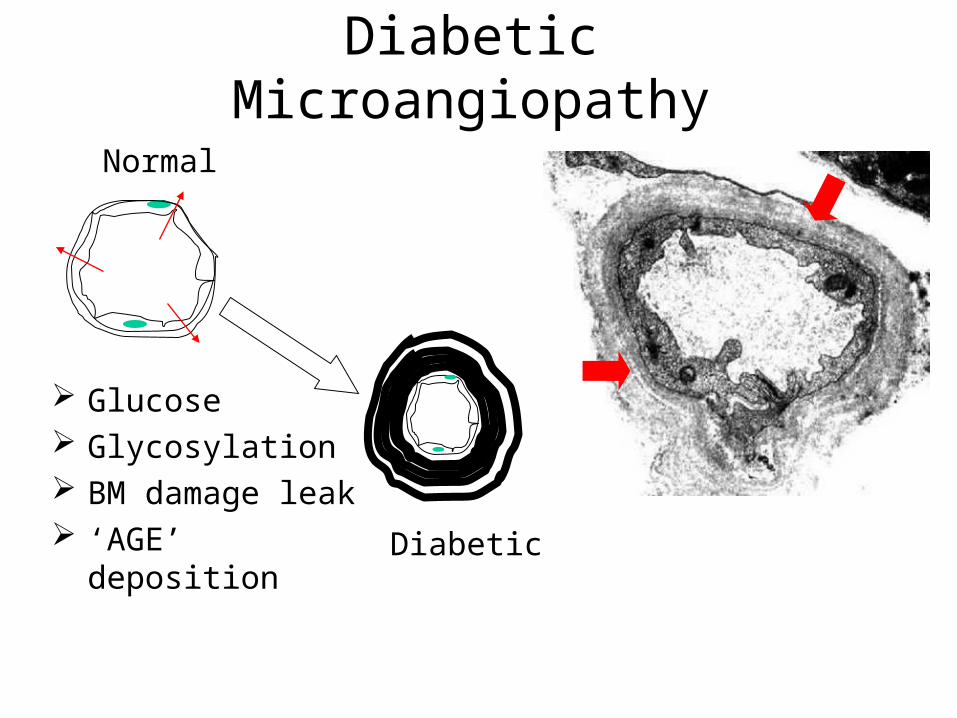
Diabetic Microangiopathy
Normal
Diabetic
Glucose Glycosylation BM damage leak ‘AGE’ deposition

Diabetic Nephropathy
• Nodular and/or diffuse glomerulosclerosis
• Arteriolar nephrosclerosis
• Pyelonephritis
• Renal papillary necrosis
Combination of microangiopathy, atherosclerosis and infections

Diabetic Glomerulosclerosis
• Diffuse basement membrane thickening, capsular fibrin drops and Kimmelstein-Wilson nodule (AGE products)
• On EM diffuse basement membrane thickening plus massive increase in mesangial matrix

Nodular Glomerulosclerosis – KW lesion

Diabetic Neuropathy
• Sensory, motor (focal demyelination)• Peripheral neuropathy: bilateral, symmetric,
progressive, irreversible. Parasthesia, pain, muscle atrophy
• Visceral neuropathy: GIT, constipation and diarrhoea. CVS- orthostatic hypotension
• Neuropathic ulcers- peripheral sensory neuropathy, trauma and deformity, ischemia
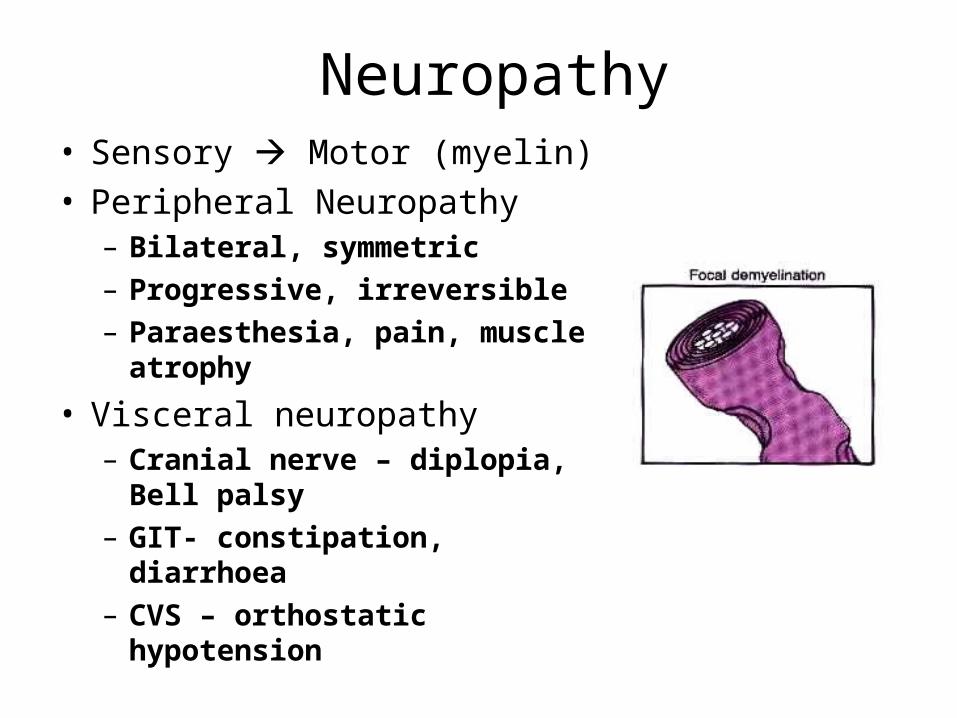
Neuropathy• Sensory Motor (myelin)• Peripheral Neuropathy
– Bilateral, symmetric– Progressive, irreversible– Paraesthesia, pain, muscle
atrophy
• Visceral neuropathy– Cranial nerve – diplopia, Bell
palsy– GIT- constipation, diarrhoea– CVS – orthostatic hypotension

Neuropathy
Myelin loss in nerve
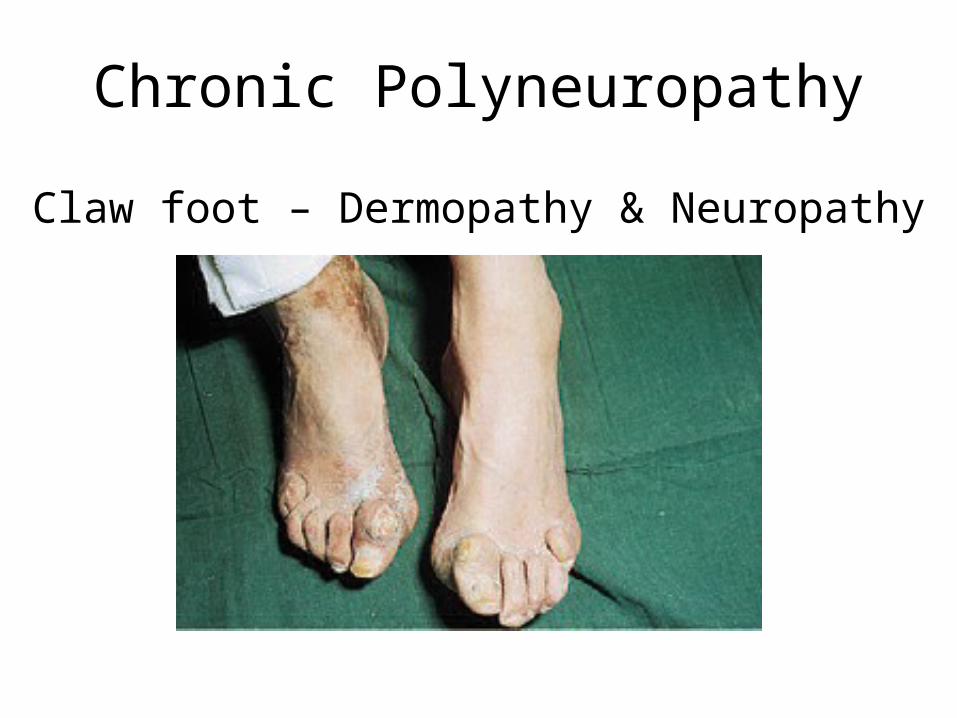
Chronic Polyneuropathy
Claw foot – Dermopathy & Neuropathy
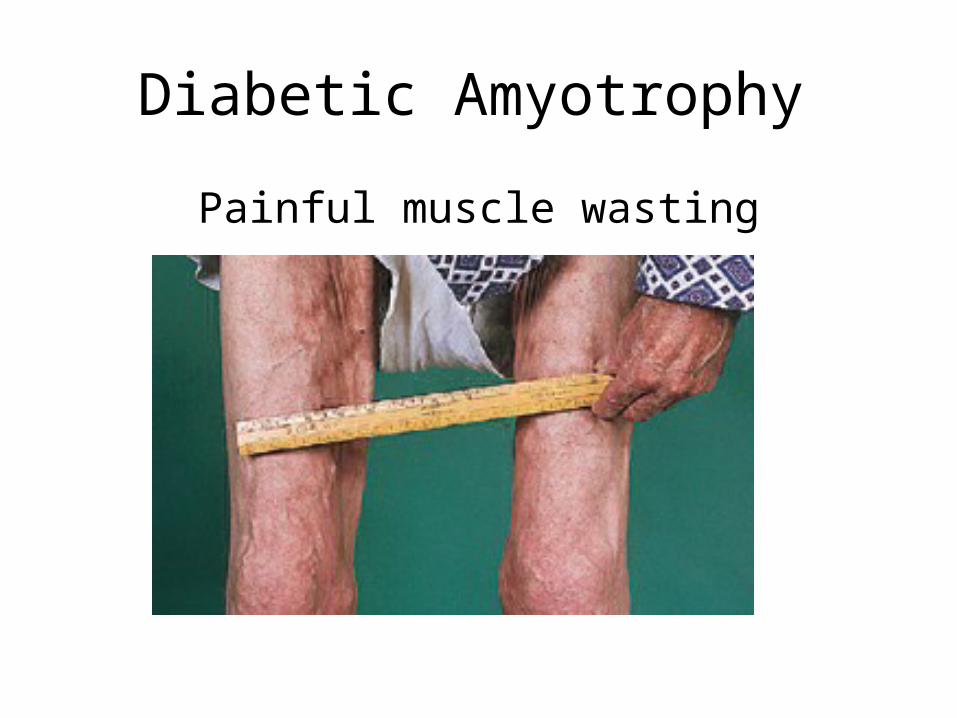
Diabetic Amyotrophy
Painful muscle wasting

Neuropathic ulcer
Etiology: peripheral sensory
neuropathy, Trauma & deformity.
Factors: Ischemia, callus formation,
and edema.

Diabetic Retinopathy
• Saccular micro-aneurysms with apparent thickening of walls due to leakage of plasma.
• Ischemic areas develop in association with collapsed capillaries

Diabetic Retinopathy
• Non-proliferative:
Microaneurysms, dot blot haemorrhages,
Exudates, infarcts
• Proliferative:
Neovascularisation, large haemorrhages,
retinal detachment

Proliferative Retinopathy• Neo-vascularization - which grows into the vitreous cavity. • In advanced disease, neo-vascular membranes can
occur, resulting in a traction retinal detachment. • Vitreous hemorrhages may result.• sudden severe loss of vision can occur when there is
intra-vitreal hemorrhage. • Poor visual prognosis if severe retinal ischaemia,
extensive neo-vascularization, or extensive fibrous tissue formation.
• Pan-retinal photocoagulation may diminish or eliminate proliferative retinopathy

Non Proliferative Retinopathy
• Venous dilation and small red dots posterior retinal pole - capillary micro-aneurysms.
• Dot and blot retinal hemorrhages and deep-lying edema and lipid exudates impair macular function.
• Late generalized diminution of vision due to ischaemia and macular edema - common cause of visual defect (best detected by fluorescein angiography)
• Cotton-wool spots (soft exudates) – micro-infarcts due to ischaemia. They are white and obscure underlying vessels. Hard exudates are caused by chronic edema. They are yellow and generally deep to retinal vessels.

Retinopathy
• Non Proliferative– Microaneurysms, – Dot blot hemorrhages– Hard and soft exudates– Cotton wool – infarcts– Macular edema.
• Proliferative.– Neovascularization – Large hemorrhages– Retinal detachment.
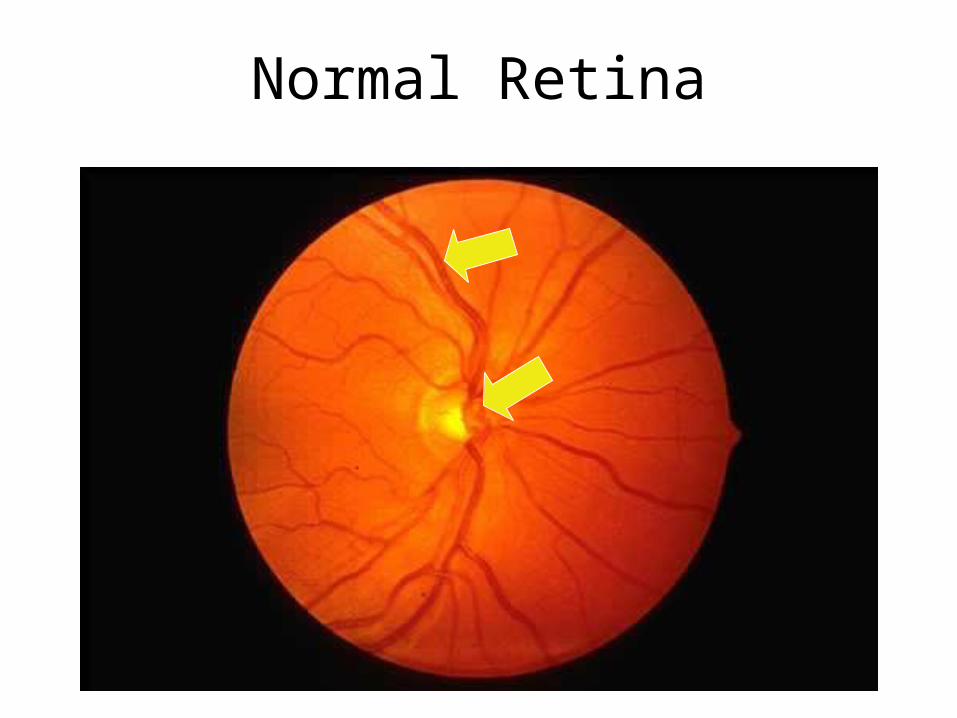
Normal Retina
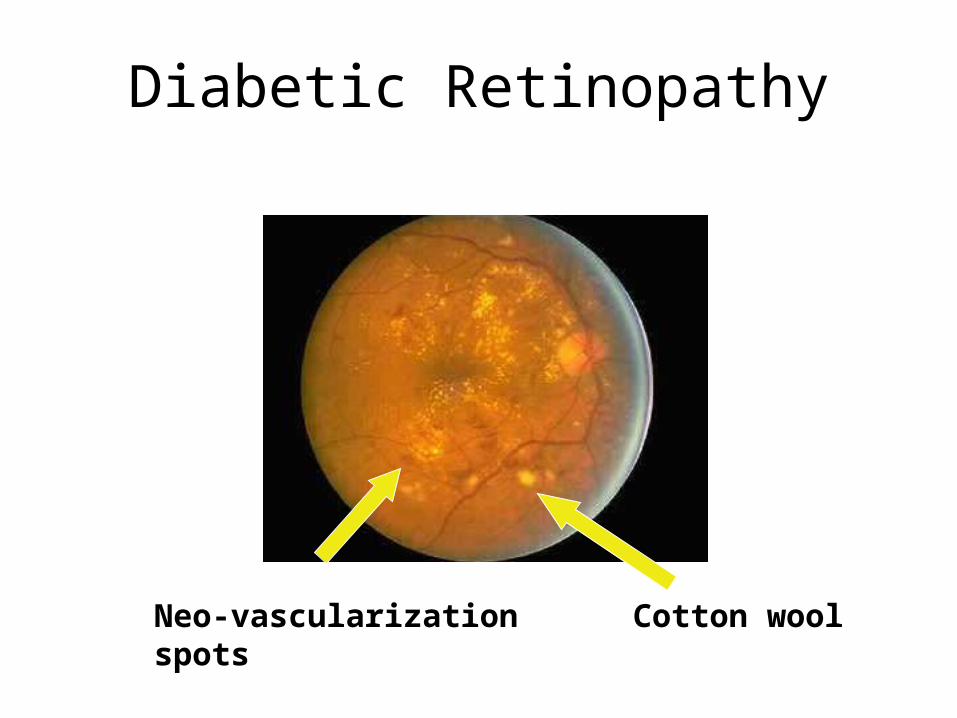
Diabetic Retinopathy
Neo-vascularization Cotton wool spots

Diabetic Retinopathy
Pre retinal Hemorrhage - detachment

Diabetic Retinopathy
Advanced fibrous plaques
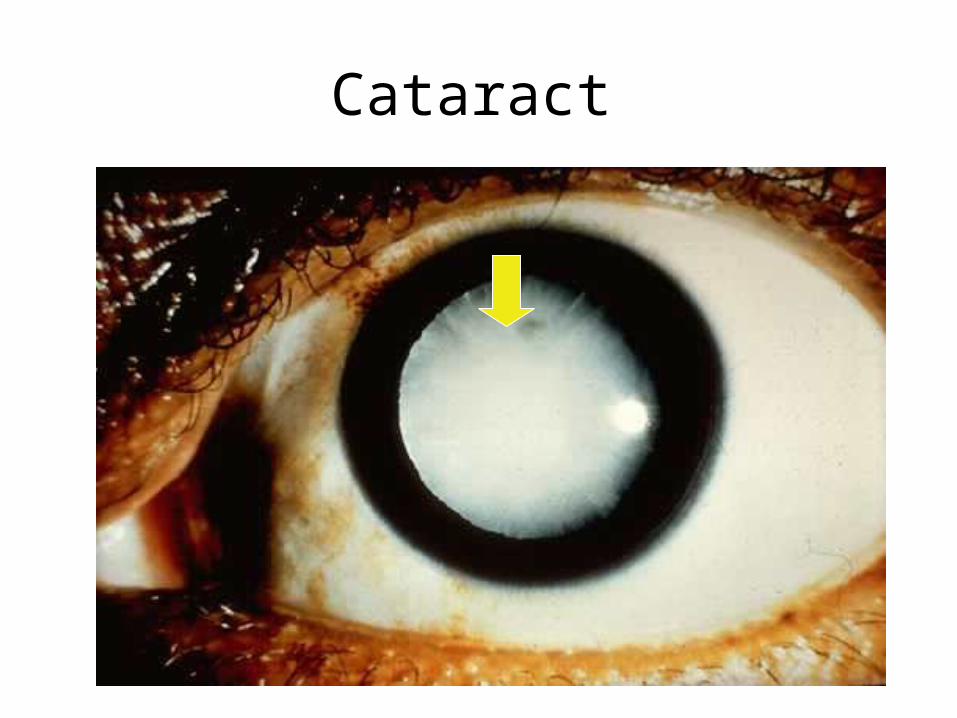
Cataract

Macroangiopathy Atherosclerosis
• Dyslipidemia
HDL
• Non-Enzymatic Glycosylation
Platelet Adhesiveness
Thromboxane A2
Prostacyclin
• Endothelial damage Atherosclerosis
• MI, CVA, Gangrene of Leg (PVD), Renal Insufficiency
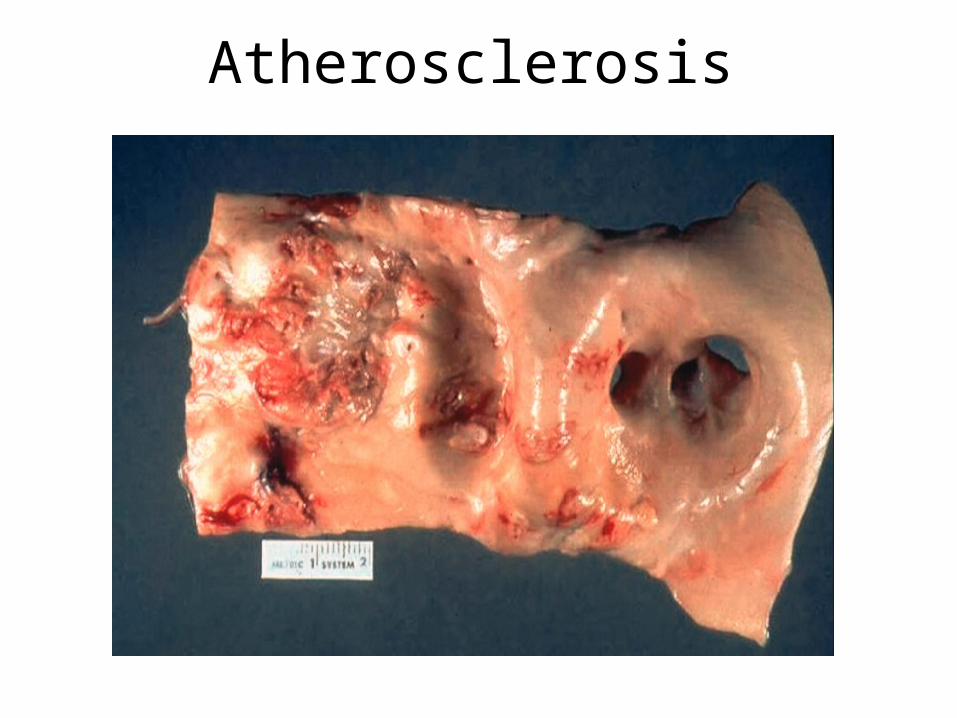
Atherosclerosis

Infections in DM
• Decreased metabolism- low immunity• Decreased function of lymphocytes and
neutrophils- glycosylation• Glycosylation of immune mediators• Capillary thickening; impaired inflammation• Ischemia and infarcts• Increased blood glucose alone is not the
cause
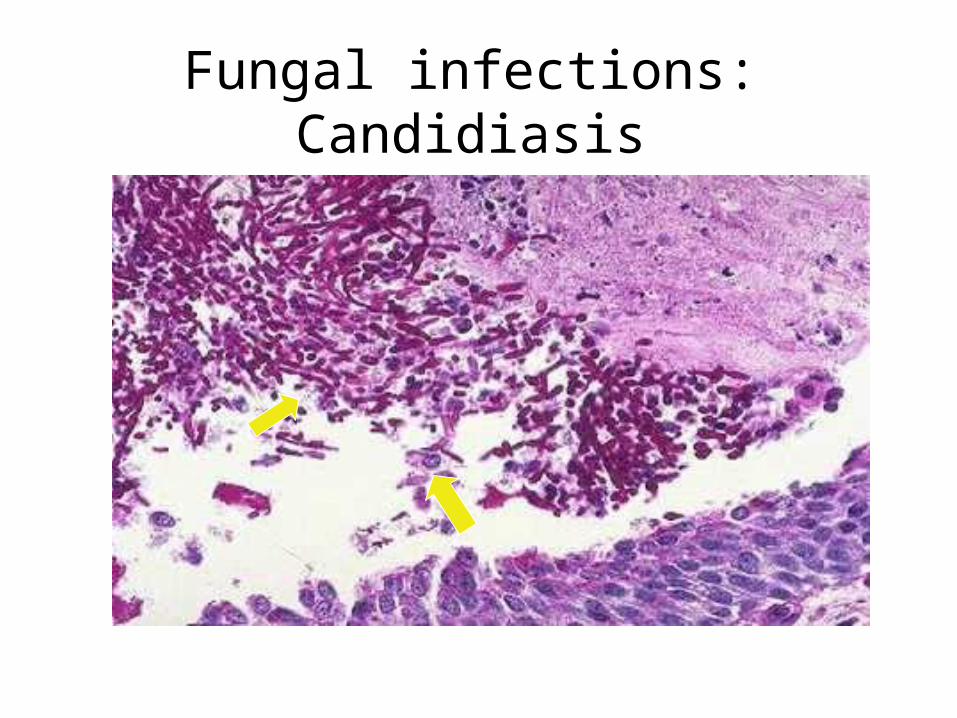
Fungal infections: Candidiasis

Laboratory Diagnosis
• Urine glucose- dipstick- screening
• Random or fasting blood glucose (N is < 11)
• Fasting > 7mmol, random > 11 then OGGT
• HbA1c- for follow up, not diagnosis

Points to remember
• Disorder of metabolism- insulin• Type I- children, acute, metabolic
complications• Type 2- adults, chronic, vascular complications• Increased infections• Hypoglycemia more dangerous than
hyperglycaemia acutely• Glucose control is critical to prevent
complications

Pancreatic Endocrine Neoplasms

Pancreatic Endocrine Neoplasms
• 2% of all pancreatic neoplasms• Benign/Malignant• Single/Multiple• Functioning/non-functioning• β-cell tumours (insulinomas)• Gastrinomas• α-cell tumours (glucagonomas)• δ-cell tumours (somatostatinomas)

Insulinomas
1. Attacks of hypoglycemia with blood glucose below 50mg/dL
2. Attacks manifest by CNS symptoms such as confusion, stupor, LOC
3. Attacks precipitated by exercise and relieved by food/IV glucose
4. 90% benign5. Located anywhere in the gland

Zollinger-Ellison Syndrome (gastrinomas)
• Hypersecretion of gastrin
• Arise in duodenum, peri-pancreatic soft tissue, pancreas
• 50% locally invasive/metastatic at presentation
• Hypersecretion of gastric acid and peptic ulceration; diarrhoea

Glucagonomas
• Raised serum glucagon- can be extremely high
• Mild diabetes, skin rash (necrolytic migratory erythema), anaemia
• Peri-menopausal and post-menopausal women

Somatostatinomas
• Diabetes,cholelithiasis, steatorrhoea, hypochlorhydria
• High somatostatin levels

Multiple Endocrine Neoplasia Syndromes
• MEN 1 (Wermer syndrome):
• 3 “P”’s: parathyroid, pancreas, pituitary– Primary hyperparathyroidism– Endocrine Tumours of pancreas (and
duodenum)– Pituitary adenoma (prolactinoma)– Germline mutation in MEN1 gene at 11q13

Multiple Endocrine Neoplasia Type 2
• MEN-2A (Sipple syndrome): phaeochromocytoma, medullary carcinoma, parathyroid hyperplasia– Germline mutation in RET gene on10q11.2
• MEN-2B: medullary carcinoma, phaeochromocytoma, skin and mucosal neuromas– Different RET mutation to 2A
Top Related