







Languages
Pages
Legal


1
Introduction to Bioinformatics
Dr. rer. nat. Gong Jing
Cancer Research Center
Medicine School of Shandong University
2012.11.07
Introduction to Introduction to BioinformaticsBioinformatics

2
Chapter 3
Alignment
Introduction to Introduction to BioinformaticsBioinformatics

3
In the game of Mahjong Titans, you want to find the same symbol from a collection of symbols for a certain one. What you can do is to compare the symbol with every one, with your eyes.
Introduction to Introduction to BioinformaticsBioinformatics
Similarity Searches on Sequence Databases

4
For a protein or DNA sequence, similarity search means finding a similar one from a collection of sequences for a query sequence.
…… > 100,000
BLAST
Introduction to Introduction to BioinformaticsBioinformatics
Similarity Searches on Sequence Databases

5
Similar sequences often derive from a common ancestral sequence. They probably share similar structure and biological function. You can infer something you know about a particular DNA or protein sequence to all similar DNA or protein sequences.
Similar sequences
Similar structures Similar functions
Introduction to Introduction to BioinformaticsBioinformatics
The Importance of Similarity

6
Similar sequences often derive from a common ancestral sequence. They probably share similar structure and biological function. You can infer something you know about a particular DNA or protein sequence to all similar DNA or protein sequences.
Similar structure? Similar function? Brothers?
Introduction to Introduction to BioinformaticsBioinformatics
The Importance of Similarity

7
Residue: a letter; an amino acid in a protein; a base in a nucleotide.
Identity: If two sequences (protein or DNA) have the same length, the identity between them is defined as the percent of identical residues relative to their length.
Similarity: If two sequences (protein or DNA) have the same length, the similarity between them is defined as the percent of identical andsimilar residues relative to their length.
Similar or not: defined by a matrix, such as BLOSUM.
My name is Lampy.
Introduction to Introduction to BioinformaticsBioinformatics
Identity and Similarity

8
seq 1 : CLHKseq 2 : CIHL
Identity = 2/4 = 50%
Similarity = 3/4 = 75%
Introduction to Introduction to BioinformaticsBioinformatics
Identity and Similarity
seq 1 : C L H Kseq 2 : C I H L
seq 1 : C L H Kseq 2 : C I H L
Identical
similar
Residue: a letter; an amino acid in a protein; a base in a nucleotide.
Identity: If two sequences (protein or DNA) have the same length, the identity between them is defined as the percent of identical residues relative to their length.
Similarity: If two sequences (protein or DNA) have the same length, the similarity between them is defined as the percent of identical andsimilar residues relative to their length.
Similar or not: defined by a matrix, such as BLOSUM.

9
Residue: a letter; an amino acid in a protein; a base in a nucleotide.
Identity: If two sequences (protein or DNA) have the same length, the identity between them is defined as the percent of identical residues relative to their length.
Similarity: If two sequences (protein or DNA) have the same length, the similarity between them is defined as the percent of similar residues relative to their length. Who and who are similar, who and who not? They are defined by a matrix, such as BLOSUM.
What happens when two sequences have different lengths?
Identity? Similarity?
seq 1 : CLHKAseq 2 : CIHL
Introduction to Introduction to BioinformaticsBioinformatics
Identity and Similarity

10
Homologous: In general, if two protein sequences have an identity > 25%, or two DNA sequences have an identity > 70%, they can be regarded as homologous.
Nothing is sure about the meaning of observed similarity. Some protein sequences are less than 15% identical, but they have the same 3D structure, while some are 25% identical, but they have different structures.
Homology or non-homology is never granted. The 25% cutoff is mostly a common-sense indicator. In most cases, to make sure whether two sequences are true homologous, you need to consider many other things.
Homology is a binary relationship: yes or no; similarity or identity is a quantifiable property: 0%-100%.
Introduction to Introduction to BioinformaticsBioinformatics
Identity and Similarity

11
BLAST (Basic Local Alignment Search Tool) – A sequence comparison algorithm optimized for speed used to search sequence databases for optimal local alignments to a query.
Different kinds of BLAST (according to the query):
BLASTn: Search a nucleotide database using a nucleotide query.
BLASTp: Search protein database using a protein query.
BLASTx: Search protein database using a translated nucleotide query.
tBLASTn: Search translated nucleotide database using a protein query.
tBLASTx: Search translated nucleotide database using a translated nucleotide query.
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST

12
BLAST (Basic Local Alignment Search Tool) – A sequence comparison algorithm optimized for speed used to search sequence databases for optimal local alignments to a query.
Different kinds of BLAST (according to the algorithm):
standard BLAST
psi-BLAST
phi-BLAST
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST

13
The NCBI BLAST server http://www.ncbi.nlm.nih.gov/
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
13

14
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
14

15
blast.fasta
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
15

16
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
http://www.crc.sdu.edu.cn/bioinfo/2012
16

17
query only a part of your sequence
give a name to your job
blast.fasta
BLAST another sequence at the same time
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
17

18
select against which database you want to search
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
18

19
limit the search range to a certain species , e.g. human
select algorithm
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
19

20
Part 1 : a brief summary
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
20

21
sequence length and classification of the input protein.
Part 2 : graphic summary
an overview of similar sequences
……
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
21

22
Part 3 : descriptions
……
go to the alignment between your query sequence and the matching sequence
The NCBI BLAST server: BLASTp http://blast.ncbi.nlm.nih.gov
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
go to the corresponding database entry
22

23
Part 4 : Alignment
Introduction to Introduction to BioinformaticsBioinformatics
The Most Popular Search Tool: BLAST
23

24
Sometimes the standard BLAST is not enough. For instance, you want to catch all the members of a very large protein family, starting with one sequence that you have. When running BLAST, you catch only the most closely related sequences. The other distant members would not be found. In other words, you find your direct friends, but the friends of your friends are missing.
PSI (Position-Specific Iterated)-BLAST first looks for sequences that are closely related to yours; and then, gradually, it extends the circle of friends to include sequences that are distantly related.
- How does PSI-BLAST extend the circle of friends?
- A Position-Specific Weight Matrix and Iterations.
Introduction to BioinformaticsEnglish Courses for Graduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PSI-BLAST

25
Seq1: A B C DSeq2: B B C DSeq3: A C C DSeq4: A B D D
1 2 3 4A 75% 0 0 0B 25% 75% 0 0C 0 25% 75% 0D 0 0 25% 100%
A Position-Specific Weight Matrix describes the letter distribution of each position (column) for a family of sequences. The distributions can be presented as probabilities or other statistic values.
Introduction to BioinformaticsEnglish Courses for Graduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Position-Specific Weight Matrix

26
For the query sequence ABCD, the first round of search (first iteration) of PSI-BLAST is just like BLAST. All closely related sequences BBCD, ACCD and ABDD that have one different letter are found, but BCCD that has two different residues is missing.
Then, a Position-Specific Weight Matrix is made for ABCD, BBCD, ACCD and ABDD. This matrix is used in the second round of search (second iteration). Since BCCD matches the matrix, now it is found. And then, a second matrix is made for ABCD, BBCD, ACCD, ABDD and BCCD. And then new sequences will be found. …… Iterations ……
PSI-BLAST can detect distant evolutionary relationships, especially when the proteins returned by the first round of search are all hypothetical proteins, unknown proteins or predicted proteins.
BACD ……BBCD BBAD ……
BBCA BCADBCCD BCBD
ABCD ACCD ACBD BCDDACCB ……CBDD ……
ABDD ACDD ……ABDC ……
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PSI-BLAST

27
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PSI-BLASTThe NCBI BLAST server: PSI-BLAST http://blast.ncbi.nlm.nih.gov
27

28
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PSI-BLASTThe NCBI BLAST server: PSI-BLAST http://blast.ncbi.nlm.nih.gov

29
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PSI-BLASTThe NCBI BLAST server: PSI-BLAST http://blast.ncbi.nlm.nih.gov
29

30
PHI (Pattern-Hit Initiated)-BLAST: in every round of BLAST (iteration), you are required to give a sequence pattern to filter the results. Only the BLAST results that match the pattern are regarded as results.
Sequence pattern:
[LIVMF]-G-E-x-[GAS]-[LIVM]-x(3,7)
Yes: VGEAAMPRI
No: VGEAAYPRI
PHI-BLAST can find very exact “friends”.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PHI-BLAST

31
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PHI-BLASTThe NCBI BLAST server: PHI-BLAST http://blast.ncbi.nlm.nih.gov
31

32
BLAST
PSI-BLAST
PHI-BLAST
Query
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Upgraded BLAST: PHI-BLAST

33
http://blast.ddbj.nig.ac.jpDDBJJapanhttp://www.ebi.ac.uk/Tools/sssEBIEurope
http://www.ncbi.nlm.nih.gov/BLASTNCBIUSAhttp://web.expasy.org/blastExPASyEurope
URLServerLocationBLAST Servers around the World
WU-BLAST - WU stands for Washington University. More sensitive and more gifted at inserting gaps than NCBI-BLAST.Smith and Waterman (SSEARCH): It’s slower, but more accurate than BLAST.FASTA: It’s a bit slower than BLAST but more accurate when making DNA comparisons.BLAT: Use this for locating cDNA rapidly in a genome or finding close (mammalian vs. mammalian) proteins in a genome.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Similarity Searches for Free over the Internet

34
can help you to …Convince yourself that two sequences are in fact homologous;Find out that your sequences share a domain;Identify the exact location of common features, such as disulfide bridgesor catalytic active sites.
Domain: a structural and functional unit in a protein.
single-domain protein multiple-domain protein
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences

35
Methods: dot plot, global/local alignment
Dot plot is the simplest means of comparing two sequences. In fact, dot plot is the only type you can do with pencil and paper, without computer. Advantages: no biological hypothesis required; results can be analyzed with your eyes.
Seq1: THEFASTCAT
Seq2: THEFATCAT
T H E F A S T C A TT x x xH xE xF xA x xT x x xC xA x xT x x x
length(seq1) = 10length(seq2) = 910 x 9 = 90 comparisons
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Seq2
Seq 1
Comparing Two Sequences: Dot plot

36
Seq1: THEFASTCAT
Seq2: THEFATCAT
T H E F A S T C A TT x x xH xE xF xA x xT x x xC xA x xT x x x
The diagonals indicate the segments of similarity between the two sequences.
1. THEFA2. TCAT3. AT Seq 1
Seq2
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Dot plot

37
You can also do dot plot for one sequence to discover repeated subsequences hidden in it.
Seq1: THEFASTHE
T H E F A S T H ET x xH x xE x xF xA xS xT x xH x xE x x
Seq 1
Seq1
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Dot plot

38
http://arbl.cvmbs.colostate.edu/molkit/dnadotDnadot
http://sonnhammer.sbc.su.se/Dotter.htmlDotterhttp://emboss.sourceforge.netDottup
http://myhits.isb-sib.ch/cgi-bin/dotletDotletURLName
Dot plot servers
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Dot plot

39
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Comparing Two Sequences: Dot plot

40
dotlet.fasta
The Sequence Input Dialog
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Comparing Two Sequences: Dot plot

41
The dots window will display the diagonal plot.
Histogram window defines the grayscale
alignment window
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Substitution matrix, e.g. Blosum62 window size zoom
Comparing Two Sequences: Dot plot

42
Use Dot Plot to detect tandem repeats in a sequence.
Tandem repeat: two or more repeated units directly adjacent to each other.
Example: CCCABCABCABCDDD
They are often used by evolution to create new proteins or make them function more efficiently.
Short Tandem Repeat (STR) in DNA describes a pattern that helps determine an individual's inherited traits. A short tandem repeat polymorphism (STRP) occurs when homologous STR loci differ in the number of repeats between individuals. By identifying repeats of a specific sequence at specific locations in the genome, it is possible to create a genetic profile of an individual. There are currently over 10,000 published STR sequences in the human genome. STR analysis has become the prevalent analysis method for determining genetic profiles in forensic cases.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Comparing Two Sequences: Dot plot

43
Use Dot Plot to detect tandem repeats in a sequence.
Tandem repeat: two or more repeated units directly adjacent to each other.Example: CCCABCABCABCDDD
C C C A B C A B C A B C D D DC x C x C x A x x xB x x xC x x xA x x xB x x xC x x xA x x xB x x xC x x xD xD xD x
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Comparing Two Sequences: Dot plot

44
tandem.fasta
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Use Dot Plot to detect tandem repeats in a sequence.
Comparing Two Sequences: Dot plot

45
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Use Dot Plot to detect tandem repeats in a sequence.
Comparing Two Sequences: Dot plot

46
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Dot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
Use Dot Plot to detect tandem repeats in a sequence.
1. The number of repeats is equal to the number of diagonals including the main diagonal.
2. The distance between two adjacent diagonals represents the length of the repeat.
3. The shortest diagonal gives you a single repeat unit.
Comparing Two Sequences: Dot plot

47
Use Dot Plot to detect tandem repeats in a sequence.
Tandem repeat: two or more repeated units directly adjacent to each other.Example: CCCABCABCABCDDD
C C C A B C A B C A B C D D DC x C x C x A x x xB x x xC x x xA x x xB x x xC x x xA x x xB x x xC x x xD xD xD x
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Dot plotDot plot servers: Dotlet http://myhits.isb-sib.ch/cgi-bin/dotlet
1. The number of repeats is equal to the number of diagonals including the main diagonal.
2. The distance between two adjacent diagonals represents the length of the repeat.
3. The shortest diagonal gives you a single repeat unit.

48
An alignment is an arrangement of two protein or DNA sequences to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences.
Global alignment is most useful when the two sequences are similar and of roughly equal size.
Local alignment is more useful for dissimilar sequences that are suspected to contain segments of similarity.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Alignment

49
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
A substitution matrixBLOSUM62 gives a score for every pair of amino acids, defining “what is similar” and “how similar”.
Comparing Two Sequences: Alignment

50
Usages of global alignment:
Checking minor differences between two sequences.
Analyzing polymorphisms between closely related sequences.
Comparing two sequences that partly overlap.
Usages of local alignment:
Comparing two distantly related sequences that share only a few noncontiguous domains.
Analyzing repeated elements within a single sequence.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Alignment

51
How to generate a global alignment?
Input:
Seq1: PYMNVI
Seq2: PYELF
substitution matrix (BLOSUM62)
gap penalty (-1 by default ): The score of a residue vs. another residue is given by the substitution matrix; a gap penalty gives the score of a residue vs. a gap.
Output:
PYMNVI PYMNVI PYMNVI--PY-ELF or PYE-LF or PY---ELF or … ?
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Global Alignment

52
Seq1: PYMNVISeq2: PYELF
Step 1
IVNMYP-
F
L
E
Y
P
-
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Global Alignment

53
Step 2
IVNMYP-
F
L
E
Y
P
-
-5
-4
-3
-2
-1
-6-5-4-3-2-10
Seq1: PYMNVISeq2: PYELF
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Global Alignment

54
Step 3
IVNMYP-
F
L
E
Y
P
-
14131314113-5
14141415124-4
11121312135-3
10111213146-2
234567-1
-6-5-4-3-2-10
Seq1: PYMNVISeq2: PYELF
S(3, 3) = maxS(2, 2) + m(s13, s23) = 14+(-2) = 12S(3, 2) + gap = 13 + (-1) = 12S(2, 3) + gap = 13 + (-1) = 12
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Global Alignment
S(i, j) =maxS(i-1, j-1) + m(s1i, s2j)S(i, j-1) + gapS(i-1, j) + gap
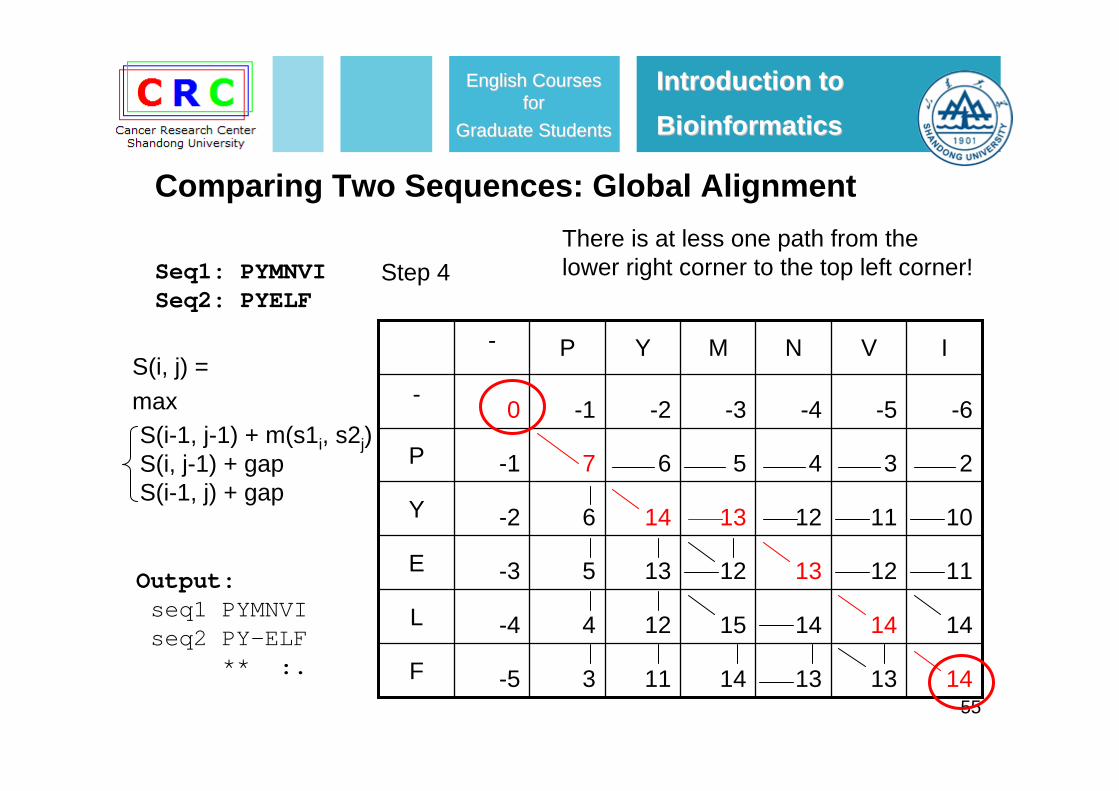
55
Step 4
IVNMYP-
F
L
E
Y
P
-
14131314113-5
14141415124-4
11121312135-3
10111213146-2
234567-1
-6-5-4-3-2-10
S(i, j) =max
Seq1: PYMNVISeq2: PYELF
S(i-1, j-1) + m(s1i, s2j)S(i, j-1) + gapS(i-1, j) + gap
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Output:seq1 PYMNVIseq2 PY-ELF
** :.
There is at less one path from the lower right corner to the top left corner!
Comparing Two Sequences: Global Alignment

56
Residue: a letter; an amino acid in a protein; a base in a DNA.
Identity: If two sequences (protein or DNA) have the same length, the identity between them is defined as the percent of identical residues relative to their length.
Similarity: If two sequences (protein or DNA) have the same length, the similarity between them is defined as the percent of similar residues (including identical residues) relative to their length. Who and who are similar, who and who not? They are defined by a matrix, such as BLOSUM.
What happens when two sequences have different lengths?
Identity? Similarity?
seq 1 : CVHKAseq 2 : CIHL
So far, we can define them for sequences with different lengths with the help of global alignment.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Identity and Similarity

57
Identity: The identity between two sequences is defined as the percent ofidentical residues in their global alignment.
Similarity: The similarity between two sequences is defined as the percent of similar residues (including identical residues) in their global alignment.
global alignment
PYMNVIPY-ELF** :.
Identity = 2 / 6 = 33.3%
Similarity = 3 / 6 = 50.0%
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Redefinition of Identity and Similarity

58
How to generate a local alignment?
Input:
Seq1: PYMNVI
Seq2: MN
substitution matrix (BLOSUM62)
gap penalty (-1 by default ): The score of an arbitrary residue vs. another arbitrary residue is given in the substitution matrix; a gap penalty gives the score of an arbitrary residue vs. a gap.
Output:
PYMNVI MN--MN-- or MN or … ?** **
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Comparing Two Sequences: Local Alignment

59
Seq1: PYMNVISeq2: MN
IVNMYP-
N
M
-
910114000
2345000
0000000
S(i, j) = max
0S(i-1, j-1) + m(s1i, s2j)S(i, j-1) + gapS(i-1, j) + gap
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Output:MNMN**
Comparing Two Sequences: Local Alignment

60
BLAST is an abbreviation of Basic Local Alignment Search Tool.
In a BLAST search, how does the most similar sequence found? Is the query sequence aligned to each sequence of the entire database?
–No. A BLAST search among 100,000 sequences needs ˂ 2 minutes, while calculation of 100,000 alignments needs > 10,000 minutes.
BLAST uses a heuristic algorithm:
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

61
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

62
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet
62

63
global.fastaglobal.fastaglobal.fastaglobal.fastaglobal.fasta
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

64
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

65
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the InternetEMBL Alignment Tool: http://www.ebi.ac.uk

66
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

67
small Gap Open +
large Gap Extend
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

68
small Gap Open +
large Gap Extend=
dispersive gaps in alignment
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

69
large Gap Open +
small Gap Extend=
concentrative gaps in alignment
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

70
adjust the gap openand gap extend
according to your expectation
Gap Open Gap Extend
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Global Alignment Over the Internet

71
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Local Alignment Over the Internet

72
local.fastalocal.fasta
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Local Alignment Over the Internet

73
>Seq1SEQUENCEMHHHHHHSSGVDLGTENLYFQSMKTTQEQLKRNVRFHAFISYSEHDSLWVKNELIPNLEKEDGSILICLYESYFDPGKSISENIVSFIEKSYKSIFVLSPNFVQNEWCHYEFYFAHHNLFHENSDHIILILLEPIPFYCIPTRYHKLKALLEKKAYLEWPKDRRKCGLFWANLRAAIN>Seq2GTENLYFQSMKTTQEQLKRNVRFHAFISYSEHDSLWVKNELIPNLEKEDGSILICLYESYFDPGKEWCHYEFYFAHHNLFHENSDHIILILLEPIPFYCIPTRAAAAAAAAAAA
EMBL Alignment Tool: http://www.ebi.ac.uk
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Making Local Alignment Over the Internet
73

74
Different between Global and Local Alignments
Global alignment
Length: 186Identity: 103/186 (55.4%)Similarity: 103/186 (55.4%)
Local alignment
Length: 130Identity: 103/130 (79.2%)Similarity: 103/130 (79.2%)
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

75
Free Pairwise Alignment over the Internet
http://www.bioinfo.mpg.de/AlignMe/AlignMe.html
Alignment of Membrane Proteins
AlignMe
http://pir.georgetown.edu/pirwww/search/pairwise.shtml
GlobalPIR
http://homepages.ed.ac.uk/eang33/mcalign/mcinstructions.html
alignment of non-coding DNA sequences
MCALIGN
http://lagan.stanford.edu/lagan_web/index.shtml
GlobalLAGAN
http://www.ebi.ac.uk/Tools/psaGlobal/LocalEMBL
http://www.ch.embnet.org/software/LALIGN_form.html
Global/LocalLalign
URLAlignment TypeNameOnline Pairwise Alignment Programs
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

76
Multiple Sequence AlignmentA multiple sequence alignment (MSA) is a global sequence alignment of three or more sequences.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

77
Multiple Sequence Alignment4 main criteria for building a multiple sequence alignment :
• Structural similarity - Amino acids that play the same role in each structure are expected in the same column. This is very difficult; only structure-superimposition programs can satisfy this criterion.
• Evolutionary similarity - Amino acids in the common ancestor of all the sequences are put in the same column. Indeed, no automatic program exactly uses this criterion, but they all try to respect it.
• Functional similarity - Amino acids with the same function are in the same column. Also, no automatic program exactly uses this criterion, but if the information is available, you can edit your alignmentmanually.
• Sequence similarity - Amino acids in the same column are those that yield an alignment with maximum similarity. Most programs take this, because it is the easiest criterion.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

78
Main applications of MSA:
1. Extrapolation: whether an uncharacterized sequence is really a member of a protein family.
2. Phylogenetic analysis: the phylogenetic tree of aligned sequences can be reconstructed.
3. Pattern identification: very conserved positions with a certain function can be sent to generate sequence pattern or sequence logo.
4. Domain identification: to turn an MSA into a profile (position-specific weight matrix) that describes a protein domain.
Multiple Sequence Alignment
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

79
Main applications of MSA:
5. DNA regulatory elements: to turn a DNA MSA of a binding site into a profile and scan other DNA sequences for potential binding sites.
6. Structure prediction: to predict protein/RNA secondary structures by similarity.
7. nsSNP analysis: MSA can help you predict whether a non-synonymous single-nucleotide polymorphism is likely to be harmful.
8. PCR analysis: a good multiple alignment can help you identify the less degenerated portions of a protein family, in order to fish out new members by PCR (polymerase chain reaction).
Multiple Sequence Alignment
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

80
Choosing the Right SequencesMSA is not for an arbitrary group of sequences. Instead, the sequences should be members of the same protein family, and they all share a common ancestor.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

81
Choosing the Right SequencesNaming sequences in the right way:
Never use white spaces in your sequence names. Use the underline (_) to replace spaces.
Do not use special symbols. (such as Chinese symbols, @, #, &, ^ etc.).
Never use names longer than 15 characters.
Never give the same name to two different sequences in your set.
If you don’t obey these naming rules, some MSA programs may automatically change the name of your sequences, without telling you.
e.g. This_is_my_favorite_sequence_about_mouse
e.g. 我的序列壹 [email protected]
e.g. My Seq 1 My_Seq_1
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

82
Choosing the Right SequencesChoosing the right number of sequences:
start with a relatively small number of sequence (10-15)
increase its size, after you get something interesting happening with this small set.
In any case, it’s hard to see any reason for generating a MSA with > 50 sequences.
If you start with hundreds of sequences, you immediately hit troubles:
Computing big alignments is difficult.
Building big alignments is difficult.
Displaying big alignments is difficult.
Using big alignments is difficult.
Making accurate big alignments is difficult.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

83
The most commonly used MSA packages.
Before you start making multiple sequence alignments, you must know that none of the methods available today is perfect. They all use approximations.
IVNMYP
F
L
E
Y
P
14131314113-5
14141415124-4
11121312135-3
10111213146-2
234567-1
-6-5-4-3-2-10
3 sequences = 3D
seq1
seq2 seq2seq1
seq3
2 sequences = 2D n sequences = nD
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

84
ClustalW - the most commonly usedMSA package.
Tcoffee - one of the latest MSA packages.
MUSCLE - one of the fastest alignment methods.
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

85
ClustalW is the latest of the Clustal software series. Clustal was the first multiple sequence alignment program. These days, with more than 35,000 citations, ClustalW is one of the most widely cited scientific publications in the history of biology.
ClustalW uses a progressive algorithm. This means that it adds sequences one by one, instead of aligning all the sequences at the same time.
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

86
http://clustalw.ddbj.nig.ac.jp/top-j.htmlJapanDDBJhttp://bips.u-strasbg.fr/fr/Documentation/ClustalW
EuropeStrasbourg
http://www.genome.jp/tools/clustalwJapanGenomeNet
http://pir.georgetown.edu/pirwww/search/multialn.shtml
USAPIR
http://searchlauncher.bcm.tmc.edu/multi-align/Options/clustalw.html
USABCM
http://www.ebi.ac.uk/Tools/msa/clustalw2EuropeEBI
http://www.ch.embnet.org/software/ClustalW.html
EuropeEMBnet
URLLocationNameA List of ClustalW Servers
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

87
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
87

88
msa.fasta
Human TLR1-10’s TIR domains
msa.fasta
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

89
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
90
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

91
The sequences in the alignment are sorted by the pairwise identity.
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

92
Red:hydrophobic
Blue:Acidic
Magenta:Basic
Green:Hydroxyl + Amine + Basic
Gray:Others
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

93
* Asterisk - an entirely conserved column.
: Double-dot - columns where all the residues have roughly the same size and the same hydropathy.
. Single-dot - columns where the size or the hydropathyhas been preserved in the course of evolution.
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

94
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

95
EMBL ClustalW http://www.ebi.ac.uk
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
The guide tree is NOT a phylogenetic tree!

96
Tcoffee is a recent method developed for conducting multiple sequence alignments. It uses a principle that’s a bit similar to ClustalW, but it yields more accurate alignments at the cost of a slightly longer running time. Tcoffeebuilds a progressive alignment like ClustalW, but it compares segments across the entire sequence set.
Home page : http://www.tcoffee.org
http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

97
http://www.es.embnet.org/Services/MolBio/t-coffeeEMBnethttp://cbsuapps.tc.cornell.edu/t_coffee.aspxCBSU
http://www.ebi.ac.uk/Tools/msa/tcoffeeEBI
http://toolkit.tuebingen.mpg.de/t_coffeeMax-Planck
http://tcoffee.vital-it.chSIB
http://www.igs.cnrs-mrs.fr/Tcoffee/tcoffee_cgi/ index.cgiCNRS
URLNameT-Coffee Mirror sites
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

98
Incorporate all the available structural information in your alignment. Will produce the best sequence alignments if the structures are available.
EXPRESSO
Evaluate the reliability of an existing multiple alignmentCORERun any requested Multiple sequence Alignment package and combine all the output into one final alignment.
MCOFFEE
Produce a multiple sequence alignment with Tcoffee.TCOFFEEDescriptionUsage
Available Tools on www.tcoffee.org
Aside from its accuracy, the main specificity of Tcoffee is its ability to align sequences and structures (EXPRESSO), the possibility of evaluating the accuracy of an alignment (CORE) and the possibility of combining many alternative multiple sequence alignments into one (Mcoffee).
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

99
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
99

100
Human TLR1-10’s TIR domains
msa.fasta
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
100

101
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
http://tcoffee.crg.cat
101

102
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
102

103
score_html file
clustalw_aln file
fasta_aln file
phylip file
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
103

104
When you choose to store your data in a specific format, you must ask yourself four questions:
Do most programs support this format?
Will my collaborators be able to use it?
Can I store all the information I need with this format?
Is it easy to manipulate?
If the program you’re using doesn’t produce alignments in the format you need, it is possible to use a third-party conversion tool to get the format you want.
fmtseq : http://www.bioinformatics.org/JaMBW/1/2 or
http://evol.mcmaster.ca/Pise/5.a/fmtseq.html
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

105
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

106
EXPRESSO is the latest development of Tcoffee, replacing what was known as 3D-Coffee. When you run Expresso, the program uses BLAST to search the PDB for structures whose sequences are similar to your sequences. It then uses theses structures to guide the alignment. Alignments based on structures are expected to be much more accurate than simple sequence alignments.
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
106

107
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
107

108
EXPRESSO T-Coffee
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

109
PDB ID
T-Coffee http://tcoffee.crg.cat
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

110
MUSCLE - is a newcomer in the MSA area but it is aremarkably efficient package for making fast, high-quality multiple sequence alignments. MUSCLE is ideal if you want to align several hundredssequences.
Home page : http://www.drive5.com/muscle
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

111
MUSCLE http://www.ebi.ac.uk/Tools/msa/muscle
The most commonly used MSA packages.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

112
Searching conserved patternsOne sentence summarizes what you really want from your multiple alignment:
You want to identify important positions!
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

113
Sequence Logos: WebLogo
Sequence logos - are a graphical representation of an amino acid or nucleic acid multiple sequence alignment developed by Tom Schneider and Mike Stephens. Each logo consists of stacks of symbols, one stack for each position in the sequence. The overall height of the stack indicates the sequence conservation at that position, while the height of symbols within the stack indicates the relative frequency of each amino or nucleic acid at that position. In general, a sequence logo provides a richer and precise description of, for example, a binding site.
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
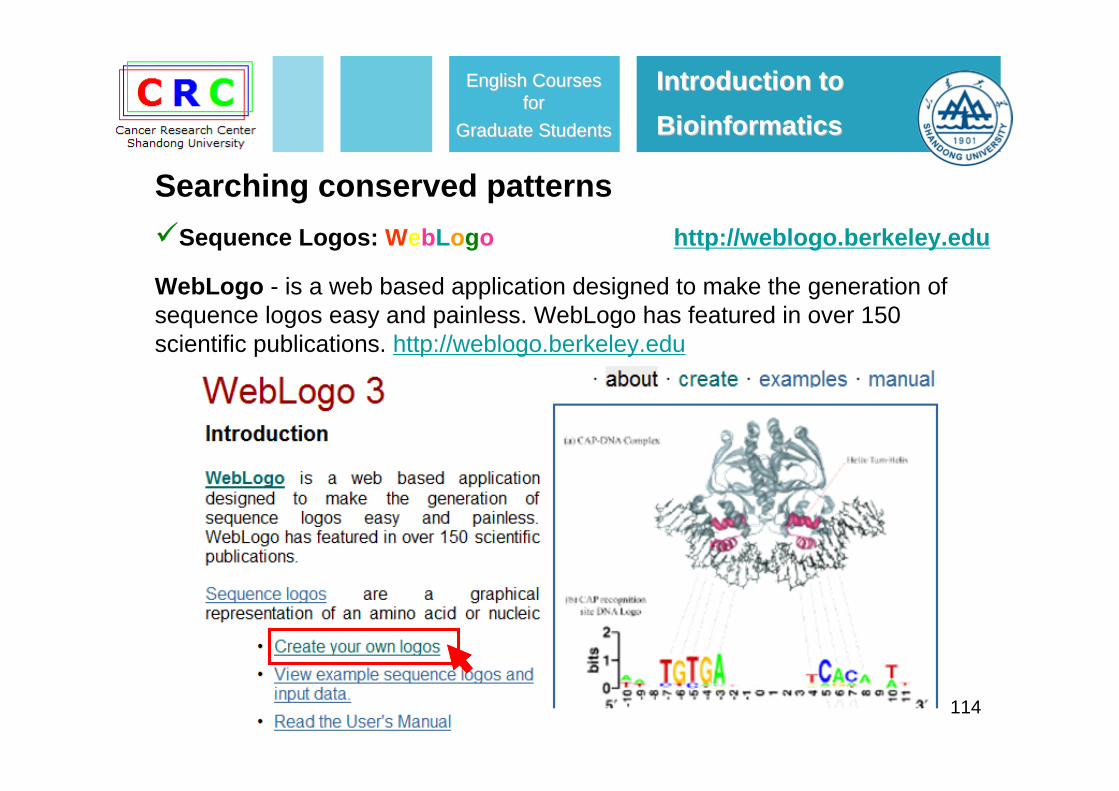
114
WebLogo - is a web based application designed to make the generation of sequence logos easy and painless. WebLogo has featured in over 150 scientific publications. http://weblogo.berkeley.edu
Sequence Logos: WebLogo http://weblogo.berkeley.edu
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

115
Sequence Logos: WebLogo http://weblogo.berkeley.edu
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
promoter.seqs
115

116
Sequence Logos: WebLogo http://weblogo.berkeley.edu
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

11720 30
Sequence Logos: WebLogo http://weblogo.berkeley.edu
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
117

118
In the promoter region of genes, we usually found a special fragment, called TATA box (also called Goldberg-Hogness box). The TATA box has the core DNA sequence 5'-TATAAT-3' or a variant. It is usually found as the binding site of RNA polymerase.
http://correlogo.abcc.ncifcrf.gov
Sequence Logos: WebLogo http://weblogo.berkeley.edu
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

119
Sequence Motif - a nucleotide or amino-acid sequence pattern that is widespread and has a biological significance.
Example: N-glycosylation site motif
Asn, followed by anything but Pro, followed by either Ser or Thr, followed by anything but Pro
This pattern can be written as N{P}[ST]{P}(Regular expression), where N=Asn, P=Pro, S=Ser, T=Thr; {X} means any amino acid except X; and [XY] means either X or Y. The notation [XY] does not give any indication of the probability of X or Yoccurring in the pattern. Observed probabilities can be graphically represented using sequence logos.
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html

120
The MEME Suite - Motif-based sequence analysis tools.
The MEME Suite allows you to:• discover motifs on groups of related DNA or protein sequences, • search sequence databases using motifs, • compare a motif to all motifs in a database of motifs. Home page : http://meme.sdsc.edu/meme/intro.html
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html

121
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html

122
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
meme.seqs

123
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html
123

124
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html
124

125
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html
125

126
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html
126

127
One sentence summarizes what you really want from your multiple alignment:
You want to identify important positions!
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Sequence Motifs: MEME http://meme.sdsc.edu/meme/intro.html
127

128
BB-Loop
BB-Loop - is important for the TIR domain dimerizationand interaction with downstream adaptors or inhibitors.
Human TLR 1-TIRHuman TLR 2-TIRHuman TLR 10-TIR
Searching conserved patterns
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
128

129
score_html file
clustalw_aln file
fasta_aln file
phylip file
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
129
Editing and Publishing Alignments

130
Editing and Publishing Alignments
For editing and publishing a multiple sequence alignment, bioinformaticanshave developed text editors that are specific for multiple sequence alignment. They make it easy for you to see exactly what’s going on.
Most of these editors require the installation of something on your computer. However, if you want to stick to your browser, you can use Jalview.
Jalview is a Java applet that you need only load into your Web browser for instant action. Home page : http://www.jalview.org
Do not load confidential sequences!Web interface is NOT secure.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
130

131
EMBL ClustalW http://www.ebi.ac.uk/Tools/msa/clustalw2
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

132
Jalview http://www.jalview.org/download.html
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

133
Jalview http://www.jalview.org/download.html
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

134
run
Jalview http://www.jalview.org/download.html
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

135
Close ALL the windows that appear within the Jalview Window, as they only contain sample data.
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
135

136
results.clustalwresults.clustalw
Jalview http://www.jalview.org/download.html
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
136

137
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
137

138
Jalview http://www.jalview.org/download.htmlhttp://www.jalview.org/help.html
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
138

139
Jalview http://www.jalview.org/download.html
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
139

140
Jalview http://www.jalview.org/download.htmlColour -> Clustalx
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
140

141
Jalview http://www.jalview.org/download.htmlColour -> Clustalx
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
http://www.jalview.org/help.html
141

142
When you edit an alignment, you usually want to do is collectively modify the alignment. To do this, you need to define them as a group, as follows:
Keep the Ctrl key pressed while you click names of sequences 1, 2, 3 and 4 to select them.
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
142

143
1. Keep the Ctrl key pressed.2. Put your mouse pointer right where you want to insert or remove the gap.3. Drag to the left or to the right to shift your sequences.
You can edit one sequence at a time by pressing the Shift key instead of Ctrl.
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
143

144
perform PairwiseAlignment for a pair of selected sequences
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
144

145
calculate tree for all selected sequences
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
145

146
predict secondary structure for a selected sequence.
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
146

147
JNet Secondary Structure Prediction result
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
147

148
save your alignment as a text/picture
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
148

149
Showtime has finally come: You have the multiple alignment you want, and you’re determined to show the world!
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
149

150
A multiple alignment editor written in Java
http://www.jalview.orgJalView
A very powerful shading and-coloring tool
http://espript.ibcp.fr/ESPript/ESPriptESPript
Shading in black and whitehttp://www.ch.embnet.org/software/BOX_form.html
Boxshade
Adding optional HTML markup to control coloring and web page layout
http://bio-mview.sourceforge.netMView
DescriptionURLNameMultiple Alignment Beautifying Tools
Editing and Publishing Alignments
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics

151
exercise.fasta
Can you make a MSA for these 5 protein sequences?Which two sequences are the most similar ones?How similar are they? (i.e. How about their sequence identity?)What kind of proteins are they?
Introduction to BioinformaticsEnglish Courses for Graduate Students
English Courses English Courses for for
Graduate StudentsGraduate Students
Introduction to Introduction to BioinformaticsBioinformatics
Top Related