







Languages
Pages
Legal


DR SHIV DUBEYAIIMS Bhopal
11/19/17 1
A 11 months/F child came to emergency with severe right knee joint pain and swelling. There is h/o easy bruising and blue spots over body. She has just started to stand with support and fell.
History ?? D/D Investigations11/19/17 2

Differential Diagnosis Clinically impossible to differentiate from Hemophilia
B- FIX def- Christmas’ disease
Type 2N vWD, transmitted as an autosomal recessive trait, is characterized by mutations in VWF within the factor VIII binding domain. Affected patients present with low levels of factor VIII (usually 5 to 15 percent of normal), because of unimpeded proteolytic cleavage of factor VIII, along with a clinical pattern of bleeding similar to that seen in hemophilia A, rather than that associated with classical vWD Should be suspected in families in which an autosomal recessive (rather than X-linked) inheritance pattern is seen.
11/19/17 3

11/19/17 4

Definition Hemophilia- “love of bleeding” Hemophilia A: X linked recessive
hereditary disorder that is due to defective or deficient factor VIII
11/19/17 5

Incidence It is the second most common inherited
clotting factor abnormality (after von Willebrand disease)
1 in 5000-10000 live male births No difference between racial groups
11/19/17 6
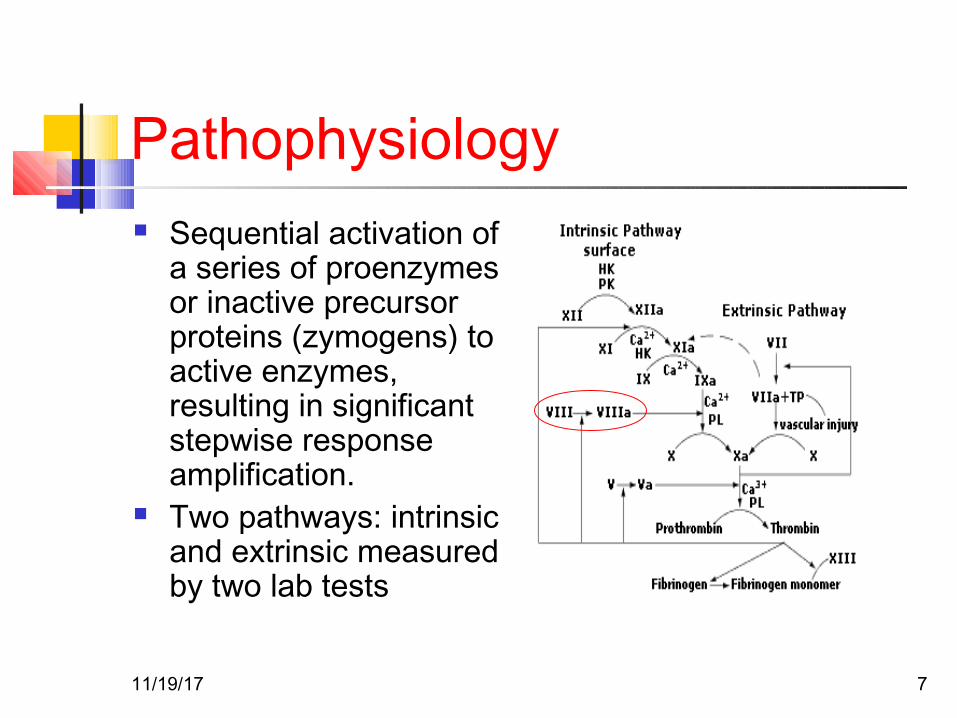
Pathophysiology Sequential activation of
a series of proenzymes or inactive precursor proteins (zymogens) to active enzymes, resulting in significant stepwise response amplification.
Two pathways: intrinsic and extrinsic measured by two lab tests
11/19/17 7

Pathophysiology
F VIII is a cofactor for intrinsic Xa
FvW is its carrier Activated by Xa and
thrombin Inactivated by activated
protein C in conjunction with protein S
11/19/17 8

Genetics
Transmitted by females, suffered by males The female carrier transmits the disorder to half their
sons and the carrier state to half her dtrs The affected male does not transmit the disease to his
sons (Y is nl) but all his dtrs are all carriers (transmission of defected X)
11/19/17 9

Genetics Hemophilia in females
If a carrier female mates with an affected male there’s the possibility that half their daughters are homozygous for the disease
Other possibility: Turner syndrome (45,X0) with a defective X
11/19/17 10
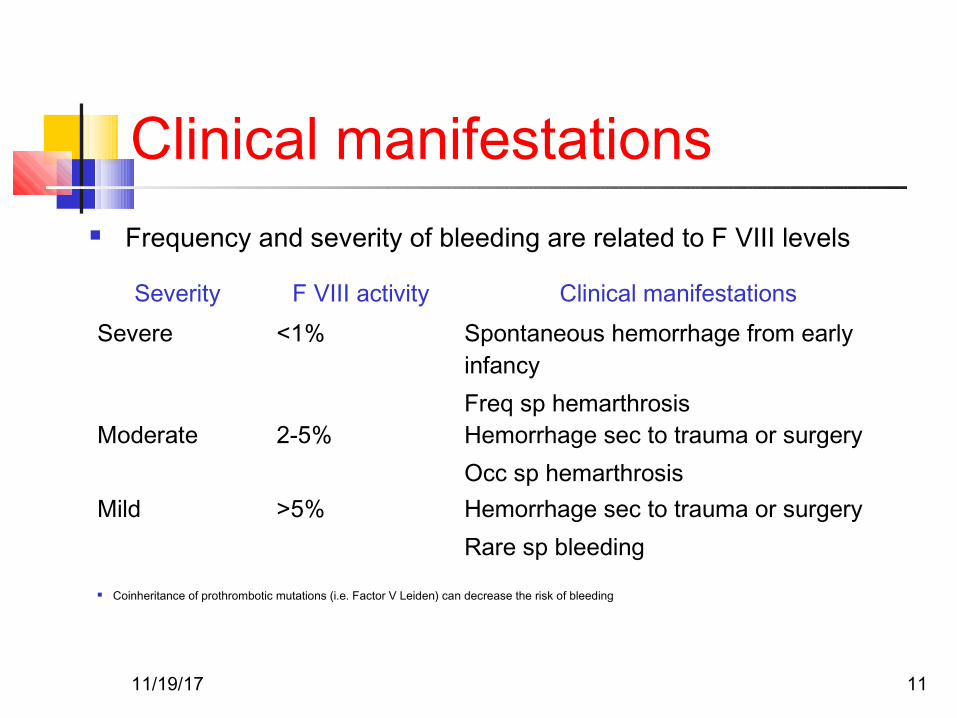
Clinical manifestations Frequency and severity of bleeding are related to F VIII levels
Severity F VIII activity Clinical manifestationsSevere <1% Spontaneous hemorrhage from early
infancyFreq sp hemarthrosis
Moderate 2-5% Hemorrhage sec to trauma or surgeryOcc sp hemarthrosis
Mild >5% Hemorrhage sec to trauma or surgeryRare sp bleeding
Coinheritance of prothrombotic mutations (i.e. Factor V Leiden) can decrease the risk of bleeding
11/19/17 11

Clinical Manifestations:Hemarthrosis The most common, painful and most
physically, economically and psychologically debilitating manifestation.
Clinically: Aura: tingling warm sensation Excruciating pain Generally affects one joint at the time Most commonly: knee; but there are others
as elbows, wrists and ankles. Edema, erythema, warmth and LOM If treated early it can subside in 6 to 8 hs
and disappear in 12 to 24 hs.
11/19/17 12

Clinical Manifestations:Hemarthrosis Pathophysiology: Bleeding probably starts from synovial vessels into
the synovial space. Reabsorption of this blood is often incomplete leading
to chronic proliferative synovitis, where the synovium is more thickened and vascular, creating a “target joint” with recurrence of bleeding.
There is destruction of surrounding structures as well-bone necrosis and cyst formations, osteophytes
Terminal stage: Chronic Hemophiliac arthropathy: fibrous or bony ankilosing of the joint.
11/19/17 13
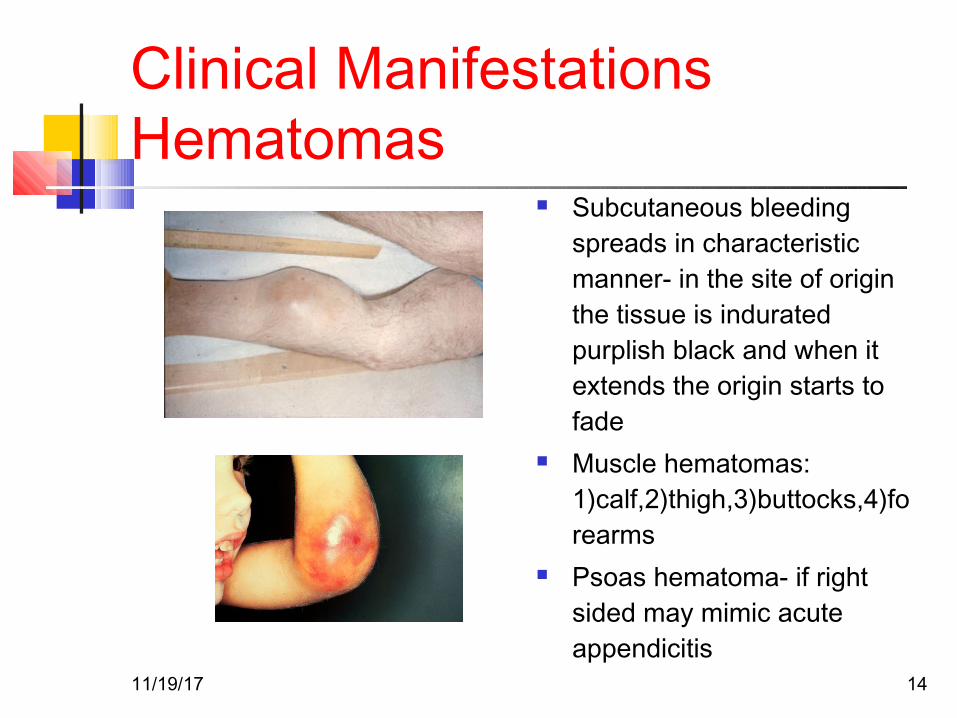
Clinical ManifestationsHematomas
Subcutaneous bleeding spreads in characteristic manner- in the site of origin the tissue is indurated purplish black and when it extends the origin starts to fade
Muscle hematomas: 1)calf,2)thigh,3)buttocks,4)forearms
Psoas hematoma- if right sided may mimic acute appendicitis
11/19/17 14

Clinical manifestationsIntracranial hemorrhage
Leading cause of death of hemophiliacs
Spontaneous or following trauma
Suspect always in hemophilic patient that presents with unusual headache
11/19/17 15

Clinical manifestationsOthers Gastrointestinal Bleeding:
Mucous Bleeding:Epistaxis, gum bleeding.
Genitourinary Bleeding:Frequently severe hemophiliac can experience
hematuria and a structural lesion should be ruled out.
11/19/17 16

Laboratory diagnosis
Deficit can be quantitative or qualitativeGeneral Lab: prolonged aPTT, nl PT and BTMixing studies: aPTT corrects with normal plasma –if there are no factor VIII antibodies presentClotting assays: F VIII activity, expressed in % of normal DecreasedQUANTITATIVEImmunoassays: “Cross Reactive Material” Positive- there is an antigen similar to the F VIII protein- QUALITATIVE
11/19/17 17

Carrier detection and Antenatal diagnosis Family history: if we follow the inheritance
pattern a female is a carrier if she: Has an hemophilic father Has two hemophilic sons Has one hemophilic son and has a family
history Has a son but no family history, there is a
67% chance that she is.
11/19/17 18

Carrier detection and Antenatal diagnosis
For prenatal diagnosis: DNA testing
on choronic villi samples obtained by
biopsy at week 12
11/19/17 19
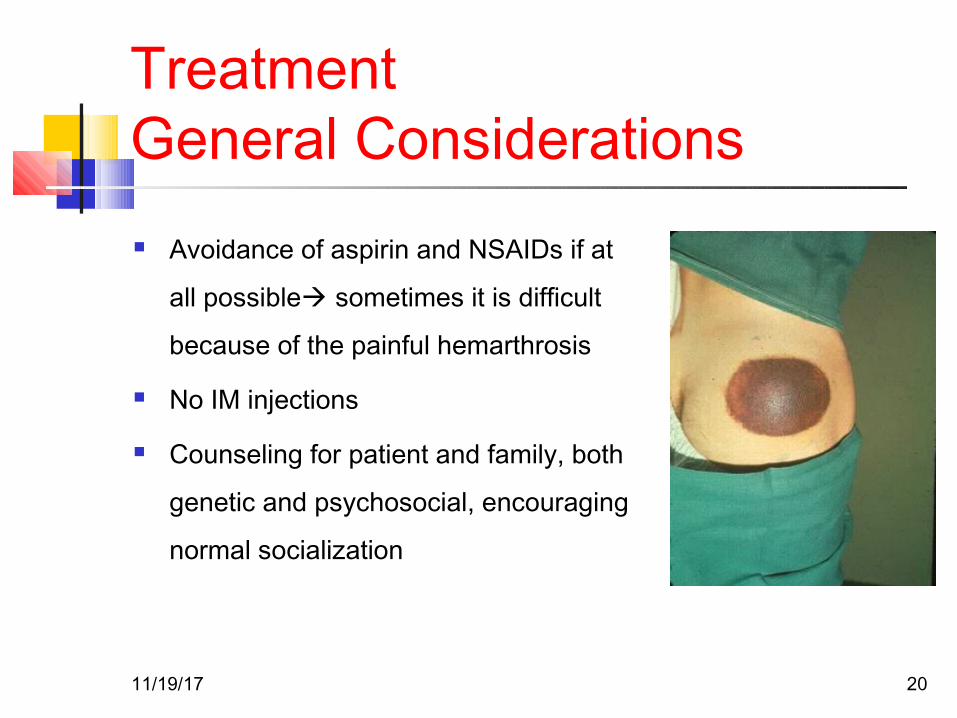
TreatmentGeneral Considerations Avoidance of aspirin and NSAIDs if at
all possible sometimes it is difficult
because of the painful hemarthrosis
No IM injections
Counseling for patient and family, both
genetic and psychosocial, encouraging
normal socialization
11/19/17 20

TreatmentFactor replacement Replacement of F VIII is the cardinal step to prevent
or reverse acute bleeding episodes Dosing: Replacement products can be given on the
basis of body weight or plasma volume ( aprox 5% of body weight)
1 U/ml = 100% factor activity Practically 1 unit of F VIII/kg increases F VIII about
0.02 U/ml In a severe hemophiliac, to raise F VIII to 100%
activity or 1 U/ml, we need 50 U/kg Redosing is based on half life: 8-12 hs Monitoring of Factor activity is crucial during therapy
11/19/17 21

TreatmentFactor replacement Choice of treatment: is based on Purity of the factor (how concentrated
or “purified” the factor is) Safety Cost Nowadays most used therapies are
believed to be effective and relatively safe
11/19/17 22

TreatmentFactor replacementSources of F VIII Plasma• FFP was used as the only replacement therapy until
1960s.• Not really much effective since it could only raise f
VIII to 20%, by giving the patient many liters• Usually patients experienced severe volume overload
(luckily furosemide was introduced around this time)• Patients used to have to spend most of their time in
the hospital
11/19/17 23

TreatmentFactor replacement Cryoprecipitate• By mid 1960s Pool et al demonstrated that cold
insoluble material obtained from plasma contained high levels of F VIII and fibrinogen, achieving a major advance in hemophilia treatment
• 1 unit of FFP prepared by cryoprecipitate contains 50-120 U of VIII
Plasma Derived f VIII prepared by monoclonal antibodies.
11/19/17 24

TreatmentFactor replacement Before 1985 all plasma derived products were
highly contaminated by blood borne virus such as HIV, HBV and HCV which is now incredibly reduced by the introduction of donor screening and viral inactivation techniques such as pasteurization, solvent detergent treatment and ultrafiltration.
However, there is still some theoretical concern about non lipid coated parvovirus, HAV and prion disease such as Creutzfeld-Jakob
11/19/17 25

TreatmentFactor replacement
Recombinant F VIII
• First generation: derived from hamster cell culture.
Contains human albumin for stabilization (possible
source of viral contamination)
• Second Generation: Mutated F VIII, lacking B domain
(no role in clotting) that can be stabilized by
sucrose “albumin free”
11/19/17 26

TreatmentFactor replacement Target level and duration of treatment: depend of
severity and site of bleeding
Site of hemorrhage Desired F VIII level
Duration of treatment(days)
Hemarthrosis 30-50 1-2
Superficial intramuscular hematoma 30-50 1-2
GI tract 50 7-10
Epistaxis 30-50 Until resolved
Oral Mucosa 30-50 Until resolved
Hematuria 30-100 Until resolved
CNS 50-100 At least 7-10 days
Retropharyngeal 50-100 At least 7-10 days
Retroperitoneal 50-100 At least 7-10 days
11/19/17 27

TreatmentOthers Fibrin Glue
Contains fibrinogen, thrombin and factor XIII It’s placed in the site of injury and stabilizes clot Used in dental procedures and after circumcision
Antifibrinolyitic Agents Epsilon aminocaproic acid Inhibit fibrinolysis by inhibiting plasminogen activator Adjuvant therapy for dental procedures Contraindicated in hematuria
Desmopressin Transient increase in F VIII levels in pts with mild hemophilia(2-4 times
above baseline) Mechanism: release from endothelial storage sites Has spared many hemophiliacs of blood borne products in the 1970s Repeated administration results in a diminished response-
tachyphylaxis Side effects: hyponatremia, facial flushing and headache
11/19/17 28

TreatmentGene Therapy
Hemophilia is an ideal disease to target for gene therapy since it is caused by mutations in a single identified gene.
A slight increase in factor activity can make a severe hemophilic in mild.
Tight regulation of gene expression is not essential. Many animal models trials have been studied, being
the main problems encountered: immunogenicity and short gene expression.
To date 3 hemophilia A trials in human (aprox 20 patients): transient increase of factor VIII activity and good safety profile.
Main issue remains: finding of a gene delivery system which is nonimmunogenic so as to allow for long term expression. 11/19/17 29

Course and prognosis Replacement therapy has its
complications and includes: Development of F VIII antibodies Liver disease resulting from hepatitis B
and C Infection with HIV
11/19/17 30

So…
WHY IS IT CALLEDTHE ROYAL DISEASE?!!?
11/19/17 31

HistoryWhy the Royal disease? This is because Queen Victoria, Queen of England from 1837 to
1901, was a carrier. Most likely a spontaneous mutation since the duke of Kent (her
father) was not affected and her mother did not have any affected children from the previous marriage.
Her eighth child, Leopold, had hemophilia and suffered from frequent hemorrhages. These were reported in the British Medical Journal in 1868.
Leopold died of a brain hemorrhage at the age of 31, but not before he had children. His daughter, Alice, was a carrier and her son, Viscount Trematon, also died of a brain hemorrhage in 1928.
The British family descends from Victoria’s first child Edward who was not affected. Hence this house is disease free.
11/19/17 32

HistoryWhy the Royal disease? Beatrice, Victoria’s youngest child had two
hemophilic sons and a daughter- Victoria Eugene that was a carrier
She introduced the hemophilia gene into the Spanish royal family by marrying king Alfonso XIII.
By this time, Queen Victoria’s blood was recognized as “defective” and the king may have been warned about Eugene’s carrier state. However, Royalty was more important than X chromosomes.
11/19/17 33

HistoryQueen Victoria’s pedigree
Spanish House
Russian House
British House
11/19/17 34

Case
11/19/17 35

A 3 year/F came in OPD with red spots/rash over lower limbs since morning. She is perfectly fine, no H/O Fever. There is H/O URI some 3 weeks back. Next ??
Examination – Normal Investigations – PC-20k TLC/Hb-N11/19/17 36

Idiopathic (Autoimmune) Thrombocytopenic Purpura(ITP)
The most common cause of acute onset of thrombocytopenia in an otherwise well child
A recent history of viral illness is described in 50-65% of cases of childhood ITP
11/19/17 37

ITP (cont.)
1 - 4 wk after exposure to a common viral infection
The peak age is 1-4 yr. ITP seems to occur more often in late winter
and spring after the peak season of viral respiratory illness.
11/19/17 38

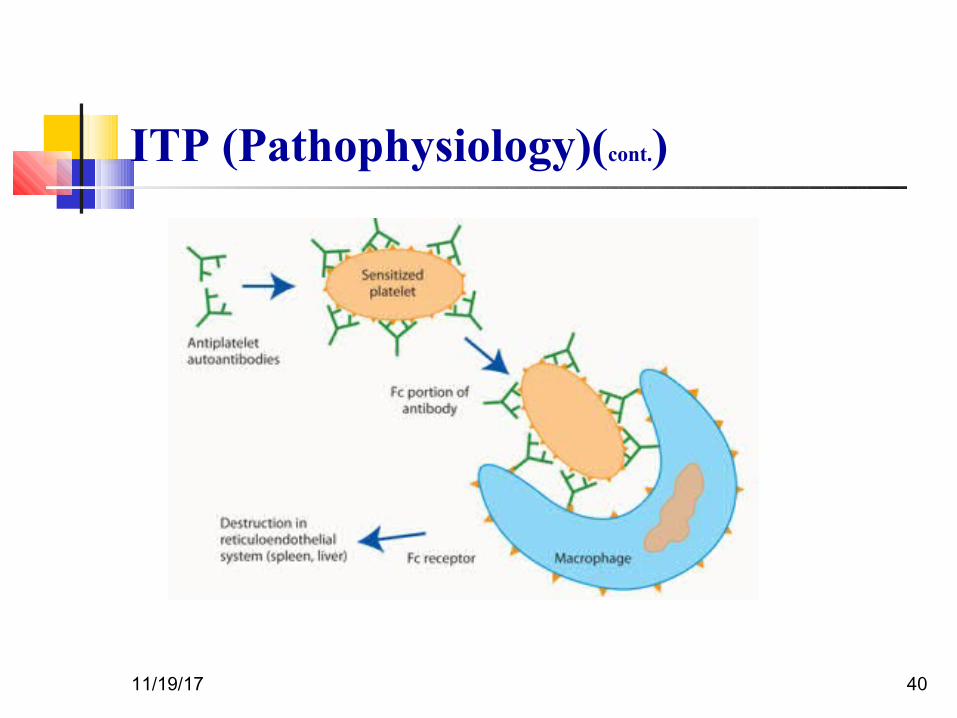
ITP (Pathophysiology)
An autoantibody directed against the platelet surface develops with resultant
sudden onset of thrombocytopenia After binding of the antibody to the platelet surface, circulating antibody-coated platelets
are recognized by the Fc receptor on splenic macrophages, ingested, and destroyed
11/19/17 39
ITP (Pathophysiology)(cont.)
11/19/17 40

ITP (Pathophysiology)(cont.)
Most common viruses have been described in association with ITP, including Epstein-Barr
virus In some patients ITP appears to arise in
children infected with Helicobacter pylori or rarely following the measles, mumps, rubella
vaccine
11/19/17 41

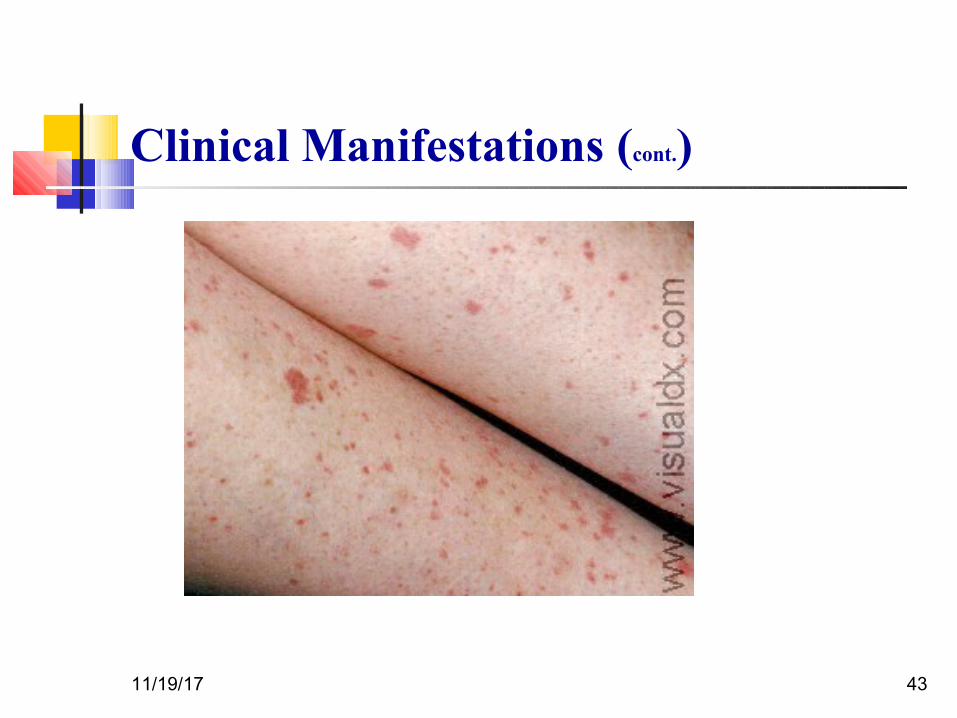
Clinical Manifestations (Cont.)
The classic presentation of ITP is a previously healthy 1-4 yr old child who has
sudden onset of generalized petechiae and purpura
Often there is bleeding from the gums and mucous membranes, particularly with
profound thrombocytopenia (platelet count <10 × 109/L).
11/19/17 42
Clinical Manifestations (cont.)
11/19/17 43

Clinical Manifestations (cont.)
11/19/17 44

Clinical Manifestations (Cont .)
The presence of abnormal findings such as hepatosplenomegaly, bone or joint pain, or
remarkable lymphadenopathy suggests other diagnoses
11/19/17 45

Prognosis
Severe bleeding is rare (<3% of cases) In 70-80% of children who present with acute
ITP, spontaneous resolution occurs within 6 mo
Fewer than 1% of patients develop an intracranial hemorrhage.
Approximately 20% of children who present with acute ITP go on to have chronic ITP
11/19/17 46

Prognosis(cont.) The outcome/prognosis may be related more to
age, as: ITP in younger children is more likely to
resolve The development of chronic ITP in
adolescents approaches 50%.
11/19/17 47

Laboratory Findings Severe thrombocytopenia (platelet count <20 ×
109/L) is common, and platelet size is normal or increased, reflective of increased platelet
turnover In acute ITP, the hemoglobin value, white
blood cell (WBC) count, and differential count should be normal.
11/19/17 48

Laboratory Findings(cont.)
Bone marrow examination shows normal granulocytic and erythrocytic series, with
characteristically normal or increased numbers of megakaryocytes
11/19/17 49

Diagnosis/Differential Diagnosis
Autoimmune thrombocytopenia may be an initial manifestation of :
1. SLE
2. HIV infection
3. Common variable immunodeficiency
4. Lymphoma(rarely)
11/19/17 50

Treatment
Platelet transfusion in ITP is usually contraindicated unless life-threatening
bleeding is present (Antiplatelet antibodies bind to transfused platelets as well as they do
to autologous platelets)
11/19/17 51

Treatment (cont.)
Initial approaches to the management of ITP include the following:
1. No therapy other than education and counseling of the family and patient for
patients with minimal, mild, and moderate symptoms, as defined earlier.
This approach is: Far less costly
Side effects are minimal
11/19/17 52

Treatment(cont.)
2.Intravenous immunoglobulin (IVIG). IVIG at a dose of 0.8- 1.0 g/kg/day for 1-2
days induces a rapid rise in platelet count (usually >20 × 109/L) in 95% of patients within 48 hr.
IVIG appears to induce a response by downregulating Fc-mediated phagocytosis of antibody-coated platelets.
11/19/17 53

Treatment(cont.)
2.Intravenous immunoglobulin (IVIG).(cont.)
IVIG therapy is :
Expensive Time-consuming to administer
After infusion, there is a high frequency of headaches and vomiting, suggestive of
IVIG-induced aseptic meningitis.
11/19/17 54

Treatment(cont.)
3.Intravenous anti-D therapy. For Rh positive patients: IV anti-D at a dose of 50-75 μg/kg causes a rise in platelet count to >20 × 109/L in 80-90% of patients within48-72 hr.
11/19/17 55

Treatment(cont.)
4. Prednisone. Doses of prednisone of 1-4 mg/kg/24 hr
Corticosteroid therapy is usually continued for 2-3 wk or until a rise in platelet count to >20 × 109/L has
been achieved, with a rapid taper long-term side effects of corticosteroid therapy:
1. Growth failure2. Diabetes mellitus
3. Osteoporosis
11/19/17 56

Treatment(Cont.)
The role of splenectomy in ITP should be reserved for 1 of 2 circumstances.
1. The older child (≥4 yr) with severe ITP that has lasted >1 yr (chronic ITP)
2. Whose symptoms are not easily controlled with therapy
3. Life-threatening hemorrhage (intracranial hemorrhage) complicates acute ITP
4. Platelet count cannot be corrected rapidly with transfusion of platelets and administration of IVIG and
corticosteroids
11/19/17 57

All Options Steroids IVIg / anti-D Rituximab (anti-CD20) Cyclophosphamide Danazol Accessory splenectomy H. pylori eradication
11/19/17 58

Hematopoietic Malignancies
Lymphoma is a general term used for proliferations that arise as discrete tissue masses.
Leukemia is used for neoplasms that present with widespread involvement of the bone marrow and the peripheral blood(usually).
59

What is LymphomaMalignant lymphoma is a term given to tumors
of the lymphoid system and specifically of lymphocytes and their precursor cells
i.e.Cancer of the lymphatic system.
Many lymphomas are known to be due to specific genetic mutations.
60

61

Associated with EBV infection mainly with mixed cellularity type.
High socio economic status. Prolonged use of of human growth
hormone.
11/19/17 62

Most common presentation is asymptomatic lymph node enlargement, typically in the neck.
Cervical lymphnodes are involved in 80% cases. Mediastinal involvement is seen in about 50% cases. They
produce symptoms like chest pain, cough and dyspnoea. Other less common symptoms are :Pruritis, alcohol induced pain over involved lymphnodes,
nephrotic syndrome, erythema nodosum, cerebellar degeneration, immune hemolytic anaemia, thrombocytopenia, hypercalcemia.
11/19/17 63

2008 WHO Classification of Hodgkin Lymphoma
64
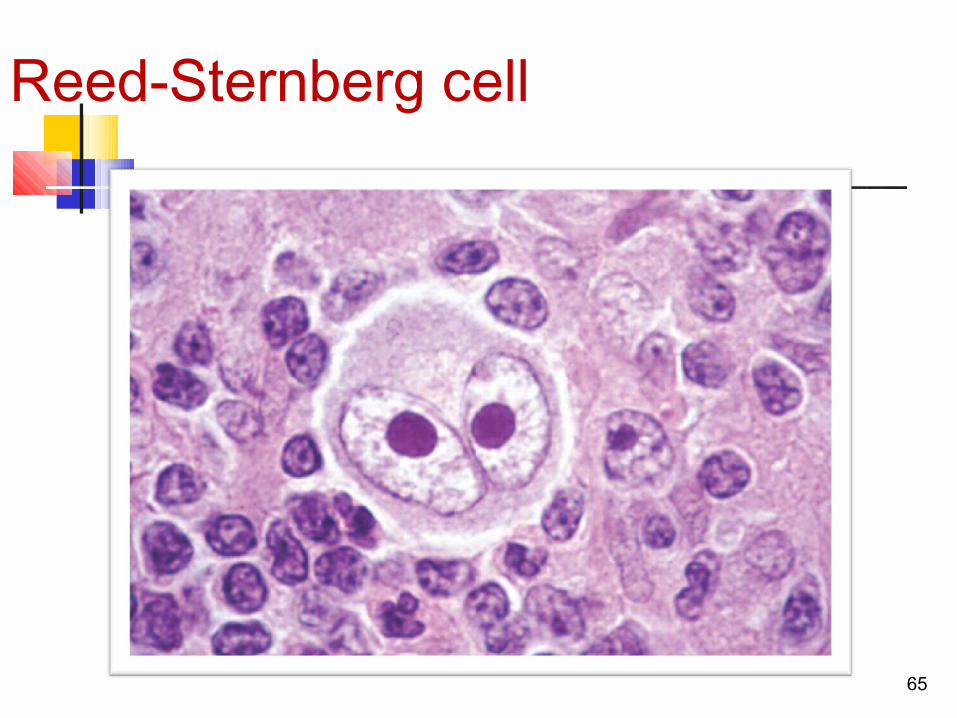
Reed-Sternberg cell
65

Ann Arbor Staging System Stage I: Single lymph node region (I) or single extralymphatic organ or
site (IE)
Stage II : > 2 lymph node regions on same side of diaphragm (II) or with limited, contiguous extra lymphatic tissue involvement (IIE)
Stage II I : both sides of diaphragm involved, may include spleen (IIIS) or local tissue involvement (IIIE)
Stage IV: multiple/disseminated foci involved with > 1 extralymphatic organs (i.e. bone marrow)
or (B) designates absence/presence of “B” symptoms*(E) Localized, solitary involvement of extralymphatic tissue, excluding liver
and bone marrow 66

Stage I Stage II Stage II I Stage IV
Staging of lymphoma
A: absence of B symptomsB: fever, night sweats, weight loss
67
68

Chemotherapy Regimen Medication Regimen Medication
1. ABVD(US)
•ADRIAMYCIN•BLEOMYCIN•VINBLASTINE•DACARBAZINE
2. STANFORD V(NEW)
•ADRIAMYCIN•BLEOMYCIN•VINBLASTINE•VINCRISTINE•PREDNISONE•MECHLORETHAMINEETOPOSIDE3. MOPP •Mechlorethamine
• Vincristine•Procarbazine• Prednisone
4. BEACOPP(EUROPE)
•BLEOMYCIN•ETOPOSIDE•ADRIAMYCIN•CYCLOPHOSPHAMIDE•ONCOVIN•PROCARBAZINE•PREDNISONE 69

THANK YOU!
11/19/17 70
Top Related