







Languages
Pages
Legal


1
FHOD1 regulates stress fiber organization by controlling transversal arc 1
and dorsal fiber dynamics 2
3 4
Nina [Schulze]1, Melanie [Graessl]1, Alexandra [Blancke Soares]2, Matthias [Geyer]3, Leif 5
[Dehmelt]4, Perihan [Nalbant]1,* 6
7
8 1Department of Molecular Cell Biology, Center for Medical Biotechnology, University of 9
Duisburg-Essen, 45141 Essen, Germany 10 2Bernhard Nocht Institute for Tropical Medicine, 20359 Hamburg, Germany 11 3Research Center Caesar, 53175 Bonn, Germany 12 4Department of Systemic Cell Biology, Max-Planck-Institute of Molecular Physiology, and 13
Dortmund University of Technology, Fakultät Chemie, Chemische Biologie, 44227 14
Dortmund, Germany 15
16
17
18
19
20
21
*Corresponding author: 22
Perihan Nalbant, Ph. D. 23
Molecular Cell Biology 24
University of Duisburg-Essen 25
Universitätsstrasse 2 26
45141 Essen, Germany 27
Phone: +49 201 183 3206 28
Fax: +49 201 183 7339 29
Email: [email protected] 30
31
Running title: FHOD1 in stress fiber organization 32
This is an Open Access article distributed under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium providedthat the original work is properly attributed.
© 2014. Published by The Company of Biologists Ltd.Jo
urna
l of C
ell S
cien
ceA
ccep
ted
man
uscr
ipt
JCS Advance Online Article. Posted on 30 January 2014

2
Summary 33
34
The formin FHOD1 can act as a capping and bundling protein in vitro. In cells, active FHOD1 35
stimulates the formation of ventral stress fibers. However, the cellular mechanism by which 36
this phenotype is produced and the physiological relevance of FHOD1 function were not 37
understood so far. Here, we first show that FHOD1 differentially controls the formation of 38
two distinct stress fiber precursors. On the one hand, it inhibits dorsal fiber growth, which 39
requires polymerization of parallel, long actin filament bundles. On the other hand, it 40
stimulates transversal arcs that are formed by fusion of short antiparallel actin filaments. This 41
combined action is critical for stress fiber maturation and their spatio-temporal organization 42
and lack of FHOD1 function perturbs dynamic cell behavior during cell migration. 43
Furthermore, we show that the GBD-FH3 domains are responsible for stress fiber association 44
and co-localization with Myosin. Surprisingly, FHOD1 that lacks those domains nevertheless 45
retains its full capacity to stimulate arc and ventral stress fiber formation. Based on our 46
findings we propose a mechanism, in which FHOD1 promotes the formation of short actin 47
filaments and transiently associates with transversal arcs to tightly control their formation and 48
turn-over into mature ventral stress fibers in space and time during dynamic cell behavior. 49
50
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

3
Introduction 51
52
Diaphanous related formins (DRFs) form an evolutionarily conserved family of actin 53
regulators, which are involved in numerous cellular processes such as morphogenesis, cell 54
division and cell polarity. In general, formins are well known for their ability to act as 55
processive actin nucleators, or “leaky cappers” for actin filaments: they catalyze the 56
nucleation of new actin filaments and stay attached to the growing filament barbed end 57
(Kovar and Pollard, 2004; Higashida et al., 2004). However, the efficiency of actin nucleation 58
and subsequent, processive monomer addition varies greatly among individual family 59
members (Paul and Pollard, 2009). In addition to their nucleation activity, several formins 60
have been described to have other modulatory roles such as actin filament bundling or actin 61
severing/de-polymerization (Harris et al., 2006; Harris et al., 2010), suggesting that they 62
might also play additional roles in cells (Harris et al., 2004; Chhabra and Higgs, 2006; 63
Chesarone et al., 2010). 64
65
For multiple family members, the highly conserved FH2 domain was shown to be sufficient to 66
catalyze actin nucleation in vitro. The FH1 domain is thought to promote processive filament 67
elongation by recruiting profilin bound monomeric actin (Sagot et al., 2002; Paul and Pollard, 68
2008; Romero et al., 2004). Two additional modules shared among DRFs, the C-terminal 69
DAD (diaphanous autoregulatory domain) and the N-terminal DID (diaphanous inhibitory 70
domain) region, interact with each other, keeping the protein in the inactive conformation 71
(Alberts, 2001; Faix and Grosse, 2006; Schönichen and Geyer, 2010). In mDia1, this auto-72
inhibitory, intramolecular interaction was shown to be released by binding of the Rho GTPase 73
RhoA to the N-terminal GTPase binding domain (GBD) stimulating the actin nucleating 74
activity of this formin and promoting actin polymerization into stress fibers from focal 75
adhesions (Watanabe et al., 1997; Hotulainen and Lappalainen, 2006). ROCK, another 76
prominent effector of RhoA, phosphorylates Myosin II and increases its motor activity to 77
promote bundling and contractility of existing actin filaments (Ishizaki et al., 1997). Thus, 78
RhoA controls stress fiber formation via the concerted activation of at least two downstream 79
effectors: Rho kinase (ROCK) and mDia1 (Watanabe et al., 1999). 80
81
The formin FHOD1 is also thought to play a role in stress fiber formation (Koka et al., 2003). 82
FHOD1 shares a similar domain organization with other DRFs, and is also activated by a 83
mechanism involving release of auto-inhibition (Schönichen et al., 2006). Truncation of the 84
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

4
C-terminal amino acids 1012-1164 including the DAD region and a short sequence with 85
unknown function (called X) leads to constitutive activation of this formin and formation of 86
prominent stress fibers throughout the cell (Koka et al., 2003; Gasteier et al., 2003). The 87
RhoA effector ROCK phosphorylates FHOD1 at three residues within this C-terminal region 88
(S1131, S1137, T1141) ablating the inhibitory interaction between the N-terminal FH3 89
domain (DID in mDia1) and the DAD-region (Takeya et al., 2008). Similar to the C-terminal 90
truncation mutant, overexpression of the corresponding triple phosphomimetic mutant induces 91
the formation of thick linear stress fibers, suggesting that ROCK mediated phosphorylation is 92
involved in the activation of FHOD1 (Takeya et al., 2008). In addition, the Rho GTPase Rac1 93
was suggested to release the auto-inhibition of FHOD1 by binding to the GBD region similar 94
to the interaction between RhoA and mDia1 (Westendorf, 2001). However, there is little 95
experimental evidence to support this claim and it is currently unclear, how Rac1 activity 96
affects FHOD1 in cells. Active FHOD1 has been shown to localize to stress fibers and its 97
overexpression in cells induces the formation of thick rigid actin fibers (Koka et al., 2003; 98
Gasteier et al., 2005). Truncation of a region encompassing the GBD-, FH3 and a part of the 99
coiled-coil domain (1-573) was shown to ablate FHOD1 stress fiber binding (Takeya et al., 100
2003; Schönichen et al., 2013), however, the functional role of stress fiber association was not 101
explored in cells. 102
103
Based on a recent study by Krainer et al. FHOD1 expression was among the highest of all 15 104
human formins tested in a large variety of cell lines and tissues suggesting its global 105
physiological relevance in cytoskeletal organization (Krainer et al., 2013). Yet, it is currently 106
not known how FHOD1 is involved in the regulation of the complex actin meshwork and its 107
coordinated reorganization during cellular morphogenesis or directional cell migration. In 108
adherent cells, several distinct types of stress fibers exist, each of which could be regulated 109
differentially by endogenous FHOD1. Contractile transversal arcs are curved structures 110
formed by the anti-parallel assembly of short actin filaments (Zhang et al., 2003). After their 111
initial formation in the lamellipodium, they undergo retrograde flow towards the cell center 112
(Heath, 1983; Small et al., 1998; Hotulainen and Lappalainen, 2006). Myosin II motors and 113
the filament bundling protein α-actinin are incorporated into arcs in a periodically alternating 114
pattern (Hotulainen and Lappalainen, 2006). During their retrograde flow arcs are not linked 115
to focal adhesions but associate with dorsal stress fibers which are also generated in the 116
leading edge of motile cells. These fibers are associated to focal adhesions with their distal 117
ends whereas their proximal end is linked to the retrograde flow of the transversal arc 118
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

5
meshwork (Small et al., 1998; Hotulainen and Lappalainen, 2006). In contrast to arcs, dorsal 119
stress fibers are formed by polymerization of long parallel actin filaments at focal adhesions. 120
Lastly, ventral stress fibers are coupled with both ends to focal adhesions and thought to be 121
formed by fusion of transversal arcs with two flanking dorsal stress fibers or by end-to-end-122
fusion of two dorsal stress fibers with each other (Small et al., 1998; Hotulainen and 123
Lappalainen, 2006). Stress fibers induced by active FHOD1 mutants might be related to such 124
ventral stress fibers. However, the precise formation mechanism and spatial organization of 125
these thick actin fibers are still poorly understood. Based on its homology to other DRFs, 126
FHOD1 would be expected to play a role in those processes by nucleating and stimulating 127
actin polymerization in stress fibers. However, a recent study by Schönichen et al. showed 128
that FHOD1 does not act as an actin filament nucleator, but instead can cap and bundle actin 129
filaments in vitro (Schönichen et al., 2013). It is currently unclear, what role those molecular 130
functions of FHOD1 play in cells, and if they are involved in the enhanced formation of 131
ventral stress fibers by the activated formin. 132
133
Here we used a combination of live cell microscopy, expression of truncation mutants and 134
acute pharmacological perturbation to investigate FHOD1 function in cells. Our studies 135
revealed that this formin distinctly regulates the formation and growth of different stress fiber 136
types. RNAi-mediated silencing of FHOD1 expression resulted in the collapse of the cellular 137
stress fiber meshwork and defects in cell migration and cell spreading. Together, our work 138
supports a model, in which FHOD1 plays a central role in the spatial and temporal 139
coordination of cellular stress fiber dynamics. 140
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

6
Results 141
142
FHOD1 depletion leads to the collapse of the stress fiber meshwork in U2OS cells 143
144
To study the cellular mechanism how FHOD1 induces stress fibers in cells, we first used 145
RNAi to deplete the endogenous formin. Efficient knock-down of the protein was confirmed 146
by western blot analysis (Fig. S1A). First, transversal arcs and dorsal stress fibers were 147
quantified in RNAi treated cells. We distinguished between those distinct stress fiber types 148
based on their association pattern to focal adhesions (Hotulainen and Lappalainen, 2006): 149
actin arcs are not associated to focal adhesions but instead linked to dorsal stress fibers, and 150
dorsal stress fibers are associated with their distal end to focal adhesions and with their 151
proximal end to actin arcs. After FHOD1 depletion, both well-organized actin arcs and dorsal 152
stress fibers were observed less frequently (Fig. 1A,B). Instead, cells preferentially formed 153
thick and less dynamic peripheral actin bundles (Fig. 1A, yellow arrow, B). These findings 154
were confirmed with four individual siRNA oligonucleotides to exclude off-target effects 155
(Fig. S1B). As reported earlier (Small et al., 1998; Hotulainen and Lappalainen, 2006), 156
transversal arcs were localized on the dorsal cell surface (Fig. 1C). We next analyzed, if 157
FHOD1 plays a role in the spatial organization of arcs within individual cells. We restricted 158
our morphometric measurements to those cells that were still able to form arcs, and found that 159
FHOD1 depletion decreased the arc-covered cell area (Fig. 1D, see Fig. S1C for definition of 160
area quantification). Furthermore, the average actin fluorescence intensity was reduced in this 161
area, suggesting that the density of arcs is reduced as well (Fig. 1E). Interestingly, we 162
observed a substantial increase of cells that contained prominent stellate accumulations of 163
actin fibers (Fig. 1F-H). The exclusive presence of the focal adhesion marker paxillin at the 164
distal ends of the individual fibers and the strong Myosin II staining in the center suggests that 165
those unusual structures might be generated by collapse of the transversal arc and dorsal stress 166
fiber meshwork (Fig. 1G,H). Live-cell imaging revealed that these structures are only formed 167
transiently (Fig. 1I) to leave behind the less dynamic peripheral actin bundles described above 168
(Fig. 1A, yellow arrow). In addition to the clear long-term effects of FHOD1 depletion on 169
actin network organization, Cytochalasin D washout experiments revealed impaired stress 170
fiber recovery in cells lacking FHOD1, suggesting that this formin is not only critical for 171
long-term network organization, but also for the dynamic de novo formation of stress fibers 172
(Fig. S2). These striking defects in actin stress fiber organization were accompanied by a 173
significant decrease in migration efficiency (Fig. S3A-D). 174
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

7
175
The observed disruption in actin network organization was also paralleled by alterations in the 176
size of focal adhesions. We quantified this effect by measuring the average number and focal 177
adhesion size in confocal micrographs of mKate-Paxillin expressing cells (Fig. 2A). We 178
found that the average size of focal adhesions in FHOD1 depleted cells was significantly 179
decreased (Fig. 2B) while the number of focal adhesions per cell was significantly increased 180
(Fig. 2C). In contrast, average cell area was not altered after formin depletion (ntsiRNA: 1740 181
µm2 ± 123.8; FHOD1 siRNA: 1679 µm2 ± 97.05 (mean ± s.e.m.)). This apparent change in 182
focal adhesion organization was accompanied by only moderately perturbed cell spreading 183
(Fig. 2D,E; Fig. S3E). We found that the early phase of U2OS cell spreading on collagen type 184
I was significantly impaired as reflected by a smaller average adhesion area compared to 185
control cells (Fig. 2D,E, 30 min). Interestingly, this difference was ablated at a later time-186
point (Fig. 2D,E, 60 min) suggesting compensation by other, possibly Rac1 mediated 187
mechanisms (Price et al., 1998; del Pozo et al., 2000). 188
189
190
FHOD1 promotes transversal arc formation and their turnover into ventral stress fibers 191
192
In order to study the cellular mechanism, by which FHOD1 affects the cellular stress fiber 193
organization, we combined acute Cytochalasin D washout experiments with the expression of 194
FHOD1 mutants. Compared to control EGFP-transfected cells, overexpression of wild-type 195
FHOD1 did not significantly alter stress fiber recovery dynamics (Fig. 3A,B). To study the 196
effect of activated FHOD1 we used a point-mutant (FHOD1 V228E) which induces an open 197
conformation and strongly stimulates stress fiber formation in cells (Schulte et al., 2008). In 198
contrast to C-terminally truncated constitutively active FHOD1 (FHOD1 1-1011), this point-199
mutant retains an intact C-terminus. After Cytochalasin D washout, FHOD1 V228E induced 200
enhanced generation of dorsal transversal arcs as compared to EGFP control and wild-type 201
FHOD1 (Fig. 3A,B). This observation is further supported by an analysis of confocal F-actin 202
image stacks, which revealed enhanced F-actin staining on the dorsal cell surface induced by 203
FHOD1 V228E (Fig. 3C). 204
205
In addition, live-cell imaging during acute Cytochalasin D washout revealed that the enhanced 206
arc formation was paralleled by an accelerated transformation of arcs into straight stress fibers 207
(Fig. 3C-E and Movie S1). This can explain the known long-term effect of enhanced stress 208
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

8
fiber formation after overexpression of constitutively active FHOD1 mutants (Koka et al., 209
2003; Gasteier et al., 2003; Takeya and Sumimoto, 2003; Schulte et al., 2008). Our data now 210
show that FHOD1 stimulates the generation of transversal arcs, which then more rapidly 211
mature into linear stress fiber bundles (Fig. 3D-F). 212
213
We next examined the effect of FHOD1 activation or inactivation on dorsal stress fibers. In 214
cells lacking this formin, we found that the average growth rate of dorsal stress fibers and the 215
average maximal fiber-length were significantly increased (Fig. 4A,B). Furthermore, in 216
FHOD1-depleted cells, the majority of mature dorsal stress fibers bent or buckled instead of 217
growing straight and perpendicular to the cell edge as in control cells (Fig. 4C,D and Movie 218
S2). This suggests that the formation of dorsal stress fibers is stimulated in the absence of 219
FHOD1 and that their regulation is perturbed leading to aberrant turnover dynamics. 220
Conversely, dorsal stress fibers were observed less frequently and their average length was 221
decreased after Cytochalasin D washout if activated FHOD1 was expressed (Fig. 4E-G). In 222
contrast to other formins, such as mDia1 which was proposed to stimulate actin 223
polymerization by processive actin filament elongation (Higashida et al., 2004), this 224
inhibitory effect of FHOD1 might be due to an alternative mechanism involving filament end 225
capping. 226
227
228
FHOD1 localizes to contractile anti-parallel actin arrays at sites of Myosin association 229
230
To gain more insight into the mechanism of FHOD1 mediated actin arc formation we next 231
studied its intracellular localization via confocal scanning and TIRF microscopy at low 232
expression levels. Wild-type FHOD1 is largely cytosolic, however, due to the increased 233
signal-to-background ratio in TIRF microscopy, we found that wild-type FHOD1 was 234
distributed in an irregular, punctate pattern, with some preference for ventral stress fibers. 235
Those structures are known to form anti-parallel actin filament arrays (i.e. arrays of mixed 236
actin filament polarity (Naumanen et al., 2008)) (Fig. 5A, yellow arrow). Actin arcs, which 237
also contain anti-parallel actin arrays undergo rapid retrograde flow along the dorsal side of 238
the cell and are therefore hardly detected via TIRF microscopy. The constitutively active 239
mutant FHOD1 1-1011 was strongly associated with both ventral stress fibers and arcs and 240
therefore easily detected in the complete cell volume via confocal scanning microscopy (Fig. 241
5B, yellow and green arrows, respectively). 242
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

9
243
Interestingly, active FHOD1 did not bind to the majority of dorsal stress fiber structures, 244
which are thought to be composed of parallel actin arrays (i.e. unidirectional actin filament 245
arrays) (Fig. 5B, orange arrows). Thus, the preferential binding of FHOD1 to ventral stress 246
fibers and actin arcs compared to dorsal fibers suggests that FHOD1 selectively binds to anti-247
parallel actin arrays. However, FHOD1 did bind to the distal ends of dorsal fibers that 248
overlapped with focal adhesions (Fig. 5B, red arrows). Interestingly, similar to FHOD1, 249
Myosin was also strongly localized to the distal ends of dorsal stress fibers that substantially 250
overlap with focal adhesions (Fig. 5C, red arrows, S4A), suggesting coordinated 251
functionalities of the two proteins in these areas. 252
253
Arcs are formed in the lamellipodium by bundling and fusion of short α-actinin bound actin 254
filaments that undergo rapid retrograde flow (Hotulainen and Lappalainen, 2006; Burnette et 255
al., 2011; Tojkander et al., 2011). Myosin II is incorporated into these actin filament bundles 256
generating an alternating pattern with α-actinin. As FHOD1 associates with stress fibers, it 257
might play a role in coordinating those processes. Previously, it was unclear, if FHOD1 co-258
operates with α-actinin mediated filament assembly or Myosin mediated contractility. Here, 259
we found that activated FHOD1 (EGFP-FHOD1 V228E) substantially co-localized with 260
Myosin and was excluded from α-actinin enriched regions (Figs 5D, S4B). Thus, our results 261
suggest that FHOD1 might co-operate with Myosin to regulate the formation of contractile 262
stress fibers. 263
264
265
Recruitment of FHOD1 to stress fibers and Myosin co-localization requires an N-terminal 266
targeting region 267
268
Stress fiber localization of FHOD1 is mediated by sequences within its N-terminal region 1-269
573 that consists of multiple parts including the GBD, FH3-, linker and helical domain 270
(Takeya and Sumimoto, 2003; Schönichen et al., 2013). The FH3 domain adjacent to the 271
GBD bears the capacity of FHOD1 to interact with the C-terminal DAD to mediate auto-272
inhibition (Takeya and Sumimoto, 2003; Schönichen et al., 2006; Schulte et al., 2008). The 273
helical domain was suggested to be responsible for FHOD1 side-binding to actin filaments in 274
vitro and might therefore mediate filament bundling (Schönichen et al., 2013). However, the 275
precise N-terminal region that is responsible for stress fiber localization was not mapped so 276
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

10
far. To address this question, we generated various FHOD1 truncation mutants and analyzed 277
their subcellular localization (Figs 6A,B, S5B). 278
279
As expected, full-length, activated FHOD1 V228E robustly associated with stress fibers 280
similar to the previously described constitutive active mutant FHOD1 1-1011 (Schulte et al., 281
2008; Koka et al., 2003; Gasteier et al., 2003; Takeya and Sumimoto, 2003) (Figs 6A,B). In 282
agreement with previous studies, the entire N-terminus (1-573) was found along stress fibers 283
(Fig. 6B). Here, we show that GBD alone (1-115) was predominantly cytosolic, whereas 284
GBD-FH3 (1-339) yielded distinct association with stress fibers (Fig. 6B) and co-localization 285
with Myosin II (Fig. 6C). In contrast, a construct lacking these domains (340-1164) was again 286
mostly cytosolic as judged by confocal microscopy (data not shown) and did not localize 287
along stress fibers. However, using TIRF microscopy, we found a weak but reproducible 288
accumulation of FHOD1 340-1164 along the entire length of focal adhesions (Figs 6B, S5C). 289
Interestingly, when stress fiber formation was stimulated acutely by Cytochalasin D washout, 290
this N-terminal truncation mutant strongly accumulated in the lamellipodium (Fig. 6D). 291
Lamellipodia were rarely observed after long-term expression of FHOD1 340-1164, however, 292
if they were present we also observed enrichment of this mutant in this cell region in steady 293
state conditions (data not shown). Together, these observations show that the GBD-FH3 294
domains can mediate FHOD1 stress fiber association and co-localization with Myosin II 295
independent of the two domains that were previously proposed to mediate F-actin interaction: 296
the FH2 domain (617-1011) and the helical domain (396-573) (Pruyne et al., 2002; 297
Schönichen and Geyer, 2010; Schönichen et al., 2013). In agreement with this, a construct 298
containing only the linker region and the helical domain (340-573) localized mostly diffusely 299
in the cells (Fig. S5B). 300
301
As shown recently, truncation of the entire N-terminus (574-1164) effectively ablated stress 302
fiber association (Fig. 6B) (Schönichen et al., 2013). Here, the majority of this truncated 303
FHOD1 was mostly cytosolic, but also found in dot-like accumulations throughout the cell 304
body with a preference for focal adhesions suggesting that this construct has a tendency to 305
form aggregates but nevertheless retains some subcellular targeting functionality (Fig. 6B). 306
307
We next studied the functional role of the FHOD1 N-terminal stress fiber localization domain 308
and the helical domain. Surprisingly, in long-term expression as well as acute Cytochalasin D 309
washout experiments FHOD1 340-1164 which lacks the stress fiber targeting GBD-FH3 310
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

11
domains (Figs 6B), nevertheless stimulated the formation of thick linear stress fibers to a 311
similar extent as the constitutively active FHOD1 V228E (Fig. 7A-C). FHOD1 340-1164 still 312
encompasses the helical domain (396-573) which was suggested to bind actin filaments in 313
vitro via side-binding and therefore might mediate filament bundling (Schönichen et al., 314
2013). Thus, the helical domain might play a functional role in stress fiber formation. Indeed, 315
the truncated mutant that lacks the helical domain (FHOD1 574-1164) failed to stimulate 316
stress fiber formation (Fig. 7A,B). 317
318
Thus, FHOD1 can induce stress fibers independently of the N-terminal GBD-FH3 domains 319
(1-339), which are responsible for stress fiber targeting. Our results therefore suggest that the 320
helical domain 396-573 plays a key role in the enhanced stress fiber formation by the FHOD1 321
340-1164 mutant, potentially via its accumulation in the lamellipodium (Fig. 6D), which 322
might lead to the formation of short actin filament bundles via its proposed role in actin 323
bundling (Schönichen et al., 2013). 324
325
We next characterized the spatial organization of the thick actin stress fiber bundles that are 326
induced by activated FHOD1 V228E in more detail using confocal F-actin image stacks. We 327
found that the majority of those bundles were localized on the ventral side of cells, where they 328
associated with focal adhesions on both ends. Thus, those bundles were mainly composed of 329
ventral stress fibers (Fig. 7D, left). Interestingly, we also observed a minor fraction of thick 330
actin bundles that formed an arch-like structure, being localized dorsal only in their central 331
region (Fig. 7D, right, white arrow) and ventral in the flanking fiber ends (Fig. 7D, right, 332
white arrow tip), which were associated with focal adhesions. Thus, while sharing partial 333
dorsal localization with actin arcs, those arch-like structures also shared properties of ventral 334
stress fibers. 335
336
337
The C-terminal FHOD1 domain is required for efficient formation of ventral stress fibers 338
339
The C-terminal DAD region of FHOD1 mediates its auto-inhibitory conformation by 340
interacting with the N-terminal FH3 domain (Takeya and Sumimoto, 2003; Schönichen et al., 341
2006). The C-terminally truncated FHOD1 construct (FHOD1 1-1011) was therefore used in 342
previous studies as a model for constitutively active FHOD1 with open conformation. To test 343
if the FHOD1 C-terminus might play additional functional roles, we compared stress fiber 344
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

12
formation of FHOD1 1-1011 with the FHOD1 point mutant (V228E), which contains the 345
entire C-terminus. Similar to FHOD1 1-1011, FHOD1 V228E is in the active conformation, 346
as this mutant blocks the intramolecular auto-inhibitory interaction (Schulte et al., 2008). We 347
analyzed the effect of these FHOD1 constructs on actin organization by quantifying the 348
number of cells that contain an increased number of mature ventral stress fibers (Fig. 8A). As 349
expected, wild-type FHOD1 only weakly enhanced formation of ventral stress fibers, whereas 350
the FHOD1 1-1011 mutant significantly stimulated their formation almost 3 fold as compared 351
to EGFP (Fig. 8A). Interestingly, the FHOD1 V228E mutant had an even more pronounced 352
effect on ventral stress fiber formation. This was surprising, as confocal imaging and cross 353
correlation analysis of FHOD1 fluorescence with the corresponding actin signals revealed that 354
the FHOD1 V228E mutant was only weakly associated with stress fibers as compared to 355
FHOD1 1-1011 (Fig. 8B,C). 356
357
While FHOD1 1-1011 lacks the three C-terminal residues phosphorylated by the upstream 358
regulator ROCK, these sites are still present in FHOD1 V228E. To study the effects of ROCK 359
inhibition on localization of those mutants, we combined acute pharmacological inhibition of 360
the kinase with live-cell TIRF microscopy. We found that wild-type FHOD1 rapidly 361
dissociated from actin fibers after addition of the ROCK inhibitor Y27632 (50 µM, 5 min). 362
This suggests that FHOD1 phosphorylation by ROCK antagonizes constitutive de-363
phosphorylation, thereby allowing dynamic control of FHOD1 stress fiber association (Fig. 364
8D,E). As expected, FHOD1 1-1011 remained mostly at stress fibers after ROCK inhibition. 365
However, FHOD1 V228E dissociated substantially from stress fibers after Y-27632 treatment 366
similar to wild-type FHOD1 (Fig. 8D, arrows, E). This finding suggests that even in the 367
FHOD1 V228E mutant, phosphorylation of the C-terminus still controls the actin binding 368
function mediated by the N-terminal GBD-FH3 domains. 369
370
Together, our observations suggest that the C-terminus plays not only an important role in 371
FHOD1 auto-inhibition, but also a more direct functional role in the stimulatory effect of 372
FHOD1 on stress fiber formation. 373
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

13
Discussion 374
375
Here, we show that FHOD1 controls the coordinated maturation of stress fibers by its distinct 376
effects on two dynamic precursor stress fiber types: stimulation of transversal arcs and slower 377
growth of dorsal fibers. This combined action of FHOD1 leads to efficient formation of less 378
dynamic ventral stress fibers. Our detailed analysis of this process offers novel mechanistic 379
insight into the role of FHOD1 in the dynamic processes that build contractile actin structures 380
in cells. 381
382
Interestingly, FHOD1 was preferentially associated with actin structures that have anti-383
parallel orientation, such as ventral stress fibers and transversal arcs. Conversely, FHOD1 was 384
largely absent from dorsal fibers, which mainly consist of parallel actin-bundles (Cramer et 385
al., 1997; Svitkina et al., 1997; Pellegrin and Mellor, 2007). This preference of FHOD1 for 386
anti-parallel actin fibers is shared by Myosin II (Verkhovsky and Borisy, 1993), which has to 387
bind such anti-parallel fibers to generate contractile forces (Clark et al., 2007). The only 388
region close to dorsal stress fibers, which showed FHOD1 and Myosin II association, 389
overlapped substantially with focal adhesions suggesting that a subpopulation of anti-parallel 390
oriented actin filaments albeit with yet unknown function might be present in that region. It is 391
currently unclear, how FHOD1 might be able to selectively associate with anti-parallel actin 392
bundles. One possibility would be binding to Myosin II either directly or indirectly via 393
adapter proteins. Our observation, that FHOD1 is co-localized with Myosin II rich stripes and 394
not with α-actinin within stress fibers, supports this idea. The N-terminal GBD-FH3 region is 395
required for FHOD1 co-localization with Myosin II and might thus mediate such coordinated 396
recruitment. 397
398
Both Myosin II and FHOD1 are activated by phosphorylation via the serine/threonine kinase 399
ROCK (Amano et al., 1996a; Kimura et al., 1996; Hannemann et al., 2008; Takeya et al., 400
2008). The upstream activator of ROCK, the small GTPase RhoA, is known to form spatial 401
activity gradients that correlate with cell motility, cell shape and polarity (Pertz et al., 2006; 402
Nalbant et al., 2009). Thus, the RhoA/ROCK pathway is a likely candidate to coordinate the 403
spatio-temporal organization of stress fiber formation via FHOD1 and their contractility via 404
Myosin II in those cellular processes. 405
406
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

14
Although regulation of FHOD1 by ROCK phosphorylation was proposed earlier (Hannemann 407
et al., 2008; Takeya et al., 2008), the functional role of this modification was not fully 408
explored. On the one hand, the effect of ROCK on subcellular FHOD1 localization and the 409
time-scale at which ROCK mediates FHOD1 regulation were unknown. On the other hand, 410
experiments that implemented C-terminal truncation mutants or phosphomimic mutations are 411
difficult to interpret as they might affect other functions of the C-terminus that are not directly 412
related to auto-inhibition. Indeed, the C-terminal DAD region of the formin mDia1 was 413
recently suggested to play an active role in filament nucleation (Gould et al., 2011). Here, we 414
show that in comparison to the C-terminal truncation (FHOD1 1-1011) the point mutation 415
V228E is an even stronger activator of stress fiber formation, although it is significantly less 416
co-localized with stress fibers. This suggests that the C-terminus of FHOD1 indeed plays 417
additional roles in FHOD1 function, potentially by augmenting actin monomer recruitment 418
similar as in the formins mDia1 and FMNL2 (Gould et al., 2011; Block et al., 2012). 419
Alternatively, the sensitivity of FHOD1 stress fiber association to ROCK inhibition suggests 420
that dynamic binding and unbinding of FHOD1 to stress fibers by 421
phosphorylation/dephosphorylation cycles might be necessary to fully activate FHOD1 422
function. 423
424
One of the most intriguing observations in this study was that FHOD1 has at least two distinct 425
cellular functions: a) stimulating stress fibers that contain anti-parallel filaments (arcs and 426
ventral stress fibers) and b) inhibiting the growth of stress fibers that contain long parallel 427
filaments (dorsal stress fibers). Our studies give critical novel insight into the molecular basis, 428
how FHOD1 could perform those functions: 1) We were able to narrow down the region of 429
FHOD1 that mediates its association with stress fibers and co-localization with Myosin II to 430
amino-acids 1-339. 2) We found that a truncation mutant lacking the GBD/FH3 domains 431
accumulated in the lamellipodium and along the entire length of focal adhesions. 3) We found 432
that persistent stress fiber association is not necessary for stimulation of stress fiber formation. 433
4) Further truncation of a helical domain (396-573), which was recently suggested to mediate 434
actin filament bundling in vitro (Schönichen et al., 2013), ablated FHOD1 mediated stress 435
fibers stimulation. Together, these observations suggest that the helical, FH1-, FH2- and C-436
terminal domains are critical for both the stimulatory function of FHOD1 for arcs and their 437
turn-over into ventral stress fibers. Previous studies proposed that the short bundled actin 438
filaments that form transversal arcs originate from the lamellipodium (Hotulainen and 439
Lappalainen, 2006; Burnette et al., 2011; Tojkander et al., 2011). Therefore, the 440
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

15
lamellipodium localization of the highly active truncation mutant 340-1164 during acute arc 441
stimulation suggests that FHOD1 might mediate bundling of preformed short actin filaments 442
via the helical domain at the leading edge of the cell, which are then efficiently assembled 443
into transversal arcs at the interface to the lamellum. 444
445
Classically, formins are thought to act as processive actin capping proteins that can stimulate 446
actin polymerization (Zigmond et al., 2003; Shemesh et al., 2005b; Otomo et al., 2005). 447
However, recent biochemical studies show that purified active FHOD1 inhibits actin 448
polymerization in addition to filament bundling (Schönichen et al., 2013). In cells, dorsal 449
stress fibers require rapid filament elongation at the associated focal adhesion by the efficient 450
processive capping protein mDia1 (Hotulainen and Lappalainen, 2006; Oakes et al., 2012). At 451
those focal adhesions, FHOD1 might act as a capping protein, which would inhibit growth of 452
associated dorsal stress fibers by competing with mDia1. Our observation that a FHOD1 453
mutant lacking the stress fiber targeting GBD-FH3 domains associates more prominently with 454
focal adhesions in steady state conditions further supports this idea. In the lamellipodium, 455
linear filaments are generated by mDia2 or FMNL2 (Yang et al., 2007; Tojkander et al., 2011; 456
Block et al., 2012). Here, FHOD1 might enrich the pool of short actin filaments either by 457
permanent or slow processive capping. Concomitantly, FHOD1 might promote transversal arc 458
assembly by bundling those preformed, short filaments. Thus, it is intriguing that FHOD1 by 459
the same molecular activity might control opposing processes at dorsal stress fibers and arcs. 460
Finally, while the central and C-terminal domains of FHOD1 are responsible for robust 461
filament formation, the N-terminal stress fiber targeting region (1-339) might facilitate precise 462
spatial FHOD1 targeting to fine tune its cellular function. These processes in the generation of 463
mature contractile stress fibers are summarized in a working model which integrates the two 464
proposed molecular functions of FHOD1 – filament capping and bundling – into a dynamic 465
process tightly controlled in space and time (Fig. S6). 466
467
In summary, our study establishes a key role for the formin FHOD1 in the spatio-temporal 468
control of actin filament meshwork dynamics in adherent cells. FHOD1 promotes the efficient 469
formation of transversal actin arcs in the leading edge and, by restricting the length of dorsal 470
stress fibers, their coordinated turn-over into mature contractile stress fibers. 471
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

16
Materials and Methods 472
473
Cell culture and reagents 474
Human U2OS osteosarcoma cells (ATCC HTB-96) were maintained in DMEM-GlutaMAX 475
(Life Technologies) supplemented with 10% fetal bovine serum (PAN Biotech), 50 U/ml 476
penicillin, and 50 µg/ml streptomycin (Life Technologies) at 37°C and 5% CO2. Prior to cell 477
seeding, coverslips (#1.5 glass, Thermo Scientific) and dishes (#1.5 glass bottom, MatTEK 478
Corporation) were coated with 10 µg/ml collagen type I (C8919, Sigma-Aldrich) for 1 h at 479
37°C and washed with PBS. 480
Cells were treated with the mycotoxin Cytochalasin D (2 µM, 90 minutes) (C8273, Sigma 481
Aldrich) to allow complete depolymerization of F-actin. Treatment started 72 h after 482
transfection of siRNAs or 16 h after transfection of EGFP or EGFP-FHOD1 constructs. Stress 483
fiber recovery was stimulated by five repeated washing steps with growth medium. The 484
selective ROCK inhibitor Y-27632 (Y0503) was purchased from Sigma-Aldrich. 485
486
Constructs and siRNAs 487
EGFP-Actin was provided by Melissa Rolls (Pennsylvania State University, USA) and 488
mKate-Paxillin was a gift from Eli Zamir (MPI Dortmund, Germany). EGFP-NMHCIIA 489
(Addgene Plasmid 11347, Wei und Adelstein, 2000) and mCherry-NMHCIIA (Addgene 490
Plasmid 35687, Dulyaninova et al., 2007) plasmids were obtained from Addgene. mCherry-α-491
actinin 1 and mCherry-Actin constructs were generated by replacing the EGFP from pEGFP-492
α-actinin 1 (Addgene plasmid 11908, Edlund et al., 2001) and pEGFP-Actin with mCherry 493
from pmCherry-C1 (Clontech) using AgeI/BsrGI restriction sites. For FHOD1 localization 494
studies low expression EGFP-FHOD1-contructs (delCMV-EGFP-FHOD1) were used as 495
described recently (Schönichen et al., 2013). Low cellular expression using a truncated, less 496
efficient CMV promoter (delCMV) has been introduced by a previous study by Watanabe and 497
Mitchison (Watanabe and Mitchison, 2002). FHOD1 V228E mutant was generated by site-498
directed mutagenesis (QuikChange Mutagenesis Kit, Agilent Technologies). Truncated 499
FHOD1 mutants were generated by PCR from wild-type delCMV-EGFP-FHOD1 or FHOD1 500
V228E, respectively. Plasmid DNA constructs were transfected using Lipofectamine™2000 501
(Life Technologies). 502
All siRNAs were purchased from Qiagen. The FlexiTube GeneSolution set (GS29109) 503
recommended by Qiagen to specifically target human FHOD1 contained four different 504
siRNAs (termed #2, #5, #6 and #7 by the supplier), which were used either as a mix (referred 505
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

17
to as siFHOD1) or as individual oligonucleotides: siFHOD1#2 (5’-506
CAGCGAGAGGAGCATCTACAA-3’), siFHOD1#5 (5’- 507
AGGGTCAACGCTATCTTGGAA-3’), siFHOD1#6 (5’-CCCGCCGTGTTGCCATGCTAA-508
3’) and siFHOD#7 (5’-CCCGTGCACCCAGGCTCTCTA-3’). AllStars negative control 509
siRNA (1027280) was used as control (ntsiRNA). With the exception of wound healing 510
assays, cells were transfected with 5 nM of the indicated siRNAs directly after plating (fast 511
forward transfection) using HiPerFect (Qiagen) and replated 24 h after transfection. 512
Experiments were carried out 72 h after siRNA transfection. Knockdown efficiency was 513
determined by Western blotting. 514
515
Immunofluorescence 516
Cells were fixed with 4% formaldehyde in PBS (20 minutes, 37°C), washed with PBS and 517
permeabilized using 0.2% Triton X-100 in PBS (10 minutes, room temperature (RT)). 518
Samples were washed and incubated in blocking solution (2% BSA in PBS, 1 h, RT) followed 519
by incubation with anti-Paxillin antibody (1:500 in blocking solution, 1h; clone 349, BD 520
Transduction Laboratory) or anti-NMHCIIa antibody (1:200 in blocking solution, 1h; 521
ab24762, Abcam), respectively. Coverslips were washed and incubated with AlexaFluor488- 522
or AlexaFluor633-conjugated goat-anti-mouse (1:500; Life Technologies) or AlexaFluor488-523
conjugated goat-anti-rabbit (1:1000; Life Technologies) secondary antibody together with 524
Rhodamine-Phalloidin to visualize filamentous actin (1:1,000; Life Technologies) in blocking 525
solution (1 h, RT). Samples were washed and, if required, stained with 4',6-diamidino-2-526
phenylindole (DAPI) (1:2,000, 10 minutes, RT; Sigma-Aldrich). Coverslips were mounted 527
using ProLong Gold (Life Technologies). 528
529
Microscopy 530
Epifluorescence and phase contrast imaging was carried out with an Eclipse Ti inverted 531
microscope (Nikon) equipped with a motorized stage, a build-in Perfect Focus System and a 532
CoolSNAP HQ2 camera (Photometrics). Images were acquired using a 20x/0.45 NA air or a 533
60x/1.40 NA oil immersion objective. Acquisition was controlled by NIS-Elements Imaging 534
Software (Nikon). Random positions were chosen using the “multi-position tool”. 535
Confocal laser scanning microscopy was performed on a TCS SP5 AOBS system (Leica 536
Microsystems) supported by LASAF software (Leica Microsystems) and equipped with an 537
HCX PL APO 63x/1.4 NA oil immersion objective. Laser lines used for excitation were 538
488 nm (EGFP, Alexa Fluor 488), 561 nm (RFP, Rhodamine, mCherry) and 633 nm (Alexa 539
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

18
Fluor 633). Confocal Z-Stacks were acquired with 210 nm spacing and maximum projections 540
were generated with ImageJ software (http://rsbweb.nih.gov/ij/). Color-coded maximum 541
projections of confocal Z-Stacks were generated using the “Time Series Color Coder” Macro 542
(http://cmci.embl.de/downloads/timeseriescolorcoder) with a modified LUT. 543
544
Time-lapse TIRF microscopy was performed on an Eclipse Ti-E (Nikon) inverted microscope 545
with a motorized TIRF Illuminator Unit (Nikon), an Andor AOTF Laser Combiner (Andor 546
Technology), and a Clara Interline CCD camera (Andor Technology). Laser lines used for 547
excitation of EGFP and mCherry were 488 nm and 561 nm, respectively. Images were 548
acquired using an Apo TIRF 100×1.49 NA oil immersion objective (Nikon). Acquisition was 549
controlled by Andor IQ Software (Andor Technology). 550
All microscopes were equipped with temperature-controlled incubation chambers. Time-lapse 551
microscopy experiments were carried out at 37°C in CO2-independent medium (HBSS buffer, 552
10% FBS, 2 mM L-Glutamine, 10 mM HEPES, 1 mM MgCl2, 1 mM CaCl2, supplemented 553
with 10 µl/ml Oxyrase (Oxyrase, Inc.)) with indicated frame rates. 554
555
Wound healing assay 556
IBIDI culture inserts (IBIDI) were used to study wound healing efficiency. Cells (8x103) were 557
seeded in each compartment of the culture inserts and directly transfected with 10 nM of 558
siRNAs using HiPerFect (Qiagen). Inserts were removed 72 h after siRNA transfection and 559
growth medium was replaced by growth medium supplemented with 10 mM HEPES. Cell 560
migration was monitored by time-lapse phase contrast microscopy (frame rate: 30 seconds). 561
For each condition, ten different positions along the wound were recorded in each experiment. 562
Migration efficiency was quantified by measuring the migrated distance (ImageJ). Lines were 563
drawn along the wound edge at start and end points of migration assay and the average 564
movement was calculated from all positions. 565
566
Cell adhesion and spreading 567
For adhesion and spreading assays, cells were detached from culture dishes using 568
Trypsin/EDTA and plated onto collagen type I coated glass coverslips in growth medium 72 h 569
after siRNA transfection. After 15, 30 or 60 minutes, coverslips were rinsed once with PBS, 570
fixed with 4% formaldehyde and stained with DAPI (nuclei) and Rhodamine-Phalloidin (F-571
actin). Epifluorescence images were acquired using a 20x/0.45 NA air objective. Adhered 572
cells were quantified from 25 random regions for each condition and time point. To quantify 573
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

19
cell spreading, 20 random positions were acquired using a 60x/1.40 NA oil immersion 574
objective for each condition and cell area was measured using ImageJ software. Focal 575
adhesion size and number per cell were quantified from confocal images of cells expressing 576
mKate-Paxillin using ImageJ. Thresholds were set manually to isolate focal adhesions 577
properly. Average area and number of focal adhesion per cell were quantified from binary 578
images using the analyze particle tool in ImageJ. Particles smaller than 0.25 µm2 and larger 579
than 10 µm2 were excluded to avoid unspecific background signal and not properly separated 580
neighboring adhesions. 581
582
RNA isolation, reverse transcription and quantitative real-time PCR analysis 583
Total RNA was extracted using the RNeasy Mini kit (Qiagen). Reverse transcription was 584
performed with Superscript®II Reverse Transcriptase (Life Technologies) according to the 585
manufacturer’s protocols using 500 ng total RNA. Quantitative real-time PCR was performed 586
with a StepOnePlus™ Real-Time PCR system (Life Technologies) using the Fast SYBR® 587
Green Master Mix (Life Technologies) and human FHOD1 specific primer pairs (forward: 5´-588
TACACGGTCACCCTCATCAA-3´, reverse: 5´-AGTGCATCCGTCACATCGTA-3´). 589
GAPDH, ACTB and RRN18S were used as housekeeping genes (QuantiTect Primer Assays, 590
Qiagen). Relative quantification was performed using the efficiency corrected relative 591
quantification method (Pfaffl, 2001). Primer efficiencies were calculated for each primer pair 592
by performing dilution series experiments. 593
594
Western blot analysis 595
Cells were washed once with ice-cold PBS and lysed in ice-cold radioimmunoprecipitation 596
assay buffer (50 mM Tris, pH7.5, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 597
1 mM EDTA, 1x protease inhibitor cocktail and 1x PhosSTOP phosphatase inhibitor cocktail 598
(Roche)). Insoluble cell debris were removed by centrifugation at 13,000 g for 10 minutes at 599
4°C. Protein concentration in the supernatants was determined by the Bradford method 600
(BioRad). Equal amounts of total protein were mixed with 5x Laemmli sample buffer, boiled 601
at 95°C for 10 minutes and separated by SDS-PAGE. After electrophoresis, proteins were 602
transferred on a PVDF membrane (Thermo Scientific) using a Biometra fastblot B34 blotting 603
device (Biometra). Blots were blocked for 1 h at RT with 5% nonfat dry milk in TBS-T 604
(20 mM Tris, pH 7.6, 137 mM NaCl and 0.1% Tween-20) and incubated overnight at 4°C 605
with the primary antibodies anti-FHOD1 (1:200; FM3521; ECM Bioscience) or anti-α-606
Tubulin (1:20,000; clone B-5-1-2, Sigma-Aldrich). Membranes were washed three times with 607
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

20
TBS-T and incubated with HRP-conjugated anti-mouse secondary antibody (1:20,000; Santa 608
Cruz Biotechnology, Inc.) for 1 h at RT. After additional washing steps with TBS-T and TBS 609
(20 mM Tris, pH 7.6, 137 mM NaCl), protein bands were visualized using ECL Western 610
blotting substrate (Pierce). Images were captured with a Fusion Fx7 system and quantified 611
with Bio-1D software (Peqlab). 612
613
Statistical analysis 614
Independent two-tailed student´s t-tests were used if not stated otherwise. In case of unequal 615
variances Welch´s t-test was used. The resulting p-values are indicated in the figures and 616
legends. 617
618
619
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

21
Acknowledgments 620
621
The authors thank Michael Ehrmann, Hemmo Meyer and Andrea Vortkamp for helpful 622
discussions. This work was supported by the Deutsche Forschungsgemeinschaft Priority 623
Programme funding (SPP 1464) to PN (NA 413/3-1) and by a grant from the Deutsche 624
Forschungsgemeinschaft to MG (GE 976/4). 625
626
627
Author contributions 628
629
NS and PN conceived or designed the experiments. NS performed the majority of 630
experiments with contributions from MGraessl and ABS. MGraessl generated TIRF imaging 631
data on FHOD1 localization and regulation. ABS contributed data on FHOD1 localization and 632
stress fiber stimulation. NS performed most of the data analysis with contributions from 633
MGraessl, ABS and PN. NS generated the figures. MG and LD contributed reagents. MG 634
commented on the manuscript. LD and PN wrote the paper. 635
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

22
References 636
637
Alberts, A. S. (2001). Identification of a carboxyl-terminal diaphanous-related formin homology 638 protein autoregulatory domain. J. Biol. Chem. 276, 2824–2830. 639
Amano, M., Ito, M., Kimura, K., Fukata, Y., Chihara, K., Nakano, T., Matsuura, Y. and 640 Kaibuchi, K. (1996). Phosphorylation and activation of myosin by Rho-associated kinase (Rho-641 kinase). J. Biol. Chem. 271, 20246–20249. 642
Block, J., Breitsprecher, D., Kühn, S., Winterhoff, M., Kage, F., Geffers, R., Duwe, P., Rohn, J. 643 L., Baum, B., Brakebusch, C. et al. (2012). FMNL2 drives actin-based protrusion and migration 644 downstream of Cdc42. Curr. Biol. 22, 1005–1012. 645
Burnette, D. T., Manley, S., Sengupta, P., Sougrat, R., Davidson, M. W., Kachar, B. and 646 Lippincott-Schwartz, J. (2011). A role for actin arcs in the leading-edge advance of migrating 647 cells. Nat. Cell Biol. 13, 371–381. 648
Chesarone, M. A., DuPage, A. G. and Goode, B. L. (2010). Unleashing formins to remodel the actin 649 and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 11, 62–74. 650
Chhabra, E. S. and Higgs, H. N. (2006). INF2 Is a WASP homology 2 motif-containing formin that 651 severs actin filaments and accelerates both polymerization and depolymerization. J. Biol. Chem. 652 281, 26754–26767. 653
Clark, K., Langeslag, M., Figdor, C. G. and van Leeuwen, F. N. (2007). Myosin II and 654 mechanotransduction: a balancing act. Trends Cell Biol. 17, 178–186. 655
Cramer, L. P., Siebert, M. and Mitchison, T. J. (1997). Identification of novel graded polarity actin 656 filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J. 657 Cell Biol. 136, 1287–1305. 658
del Pozo, M. A., Price, L. S., Alderson, N. B., Ren, X. D. and Schwartz, M. A. (2000). Adhesion to 659 the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. 660 EMBO J. 19, 2008–2014. 661
Dulyaninova, N. G., House, R. P., Betapudi, V. and Bresnick, A. R. (2007). Myosin-IIA heavy-662 chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol. Biol. Cell 18, 663 3144–3155. 664
Edlund, M., Lotano, M. A. and Otey, C. A. (2001). Dynamics of alpha-actinin in focal adhesions 665 and stress fibers visualized with alpha-actinin-green fluorescent protein. Cell Motil. Cytoskeleton 666 48, 190–200. 667
Faix, J. and Grosse, R. (2006). Staying in shape with formins. Dev. Cell 10, 693–706. 668
Gasteier, J. E., Madrid, R., Krautkrämer, E., Schröder, S., Muranyi, W., Benichou, S. and 669 Fackler, O. T. (2003). Activation of the Rac-binding partner FHOD1 induces actin stress fibers 670 via a ROCK-dependent mechanism. J. Biol. Chem. 278, 38902–38912. 671
672
Gould, C. J., Maiti, S., Michelot, A., Graziano, B. R., Blanchoin, L. and Goode, B. L. (2011). The 673 formin DAD domain plays dual roles in autoinhibition and actin nucleation. Curr. Biol. 21, 384–674 390. 675
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

23
Hannemann, S., Madrid, R., Stastna, J., Kitzing, T., Gasteier, J., Schönichen, A., Bouchet, J., 676 Jimenez, A., Geyer, M., Grosse, R. et al. (2008). The Diaphanous-related Formin FHOD1 677 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J. Biol. Chem. 678 283, 27891–27903. 679
Harris, E. S., Gauvin, T. J., Heimsath, E. G. and Higgs, H. N. (2010). Assembly of filopodia by the 680 formin FRL2 (FMNL3). Cytoskeleton (Hoboken) 67, 755–772. 681
Harris, E. S., Li, F. and Higgs, H. N. (2004). The mouse formin, FRLalpha, slows actin filament 682 barbed end elongation, competes with capping protein, accelerates polymerization from 683 monomers, and severs filaments. J. Biol. Chem. 279, 20076–20087. 684
Harris, E. S., Rouiller, I., Hanein, D. and Higgs, H. N. (2006). Mechanistic differences in actin 685 bundling activity of two mammalian formins, FRL1 and mDia2. J. Biol. Chem. 281, 14383–14392. 686
Heath, J. P. (1983). Behaviour and structure of the leading lamella in moving fibroblasts. I. 687 Occurrence and centripetal movement of arc-shaped microfilament bundles beneath the dorsal cell 688 surface. J. Cell. Sci. 60, 331–354. 689
Higashida, C., Miyoshi, T., Fujita, A., Oceguera-Yanez, F., Monypenny, J., Andou, Y., 690 Narumiya, S. and Watanabe, N. (2004). Actin polymerization-driven molecular movement of 691 mDia1 in living cells. Science 303, 2007–2010. 692
Hotulainen, P. and Lappalainen, P. (2006). Stress fibers are generated by two distinct actin 693 assembly mechanisms in motile cells. J. Cell Biol. 173, 383–394. 694
Ishizaki, T., Naito, M., Fujisawa, K., Maekawa, M., Watanabe, N., Saito, Y. and Narumiya, S. 695 (1997). p160ROCK, a Rho-associated coiled-coil forming protein kinase, works downstream of 696 Rho and induces focal adhesions. FEBS Lett. 404, 118–124. 697
Katoh, K., Kano, Y. and Noda, Y. (2011). Rho-associated kinase-dependent contraction of stress 698 fibres and the organization of focal adhesions. J R Soc Interface 8, 305–311. 699
Kimura, K., Ito, M., Amano, M., Chihara, K., Fukata, Y., Nakafuku, M., Yamamori, B., Feng, 700 J., Nakano, T., Okawa, K. et al. (1996). Regulation of myosin phosphatase by Rho and Rho-701 associated kinase (Rho-kinase). Science 273, 245–248. 702
Koka, S., Neudauer, C. L., Li, X., Lewis, R. E., McCarthy, J. B. and Westendorf, J. J. (2003). 703 The formin-homology-domain-containing protein FHOD1 enhances cell migration. J. Cell. Sci. 704 116, 1745–1755. 705
Kovar, D. R. and Pollard, T. D. (2004). Insertional assembly of actin filament barbed ends in 706 association with formins produces piconewton forces. Proc. Natl. Acad. Sci. U.S.A. 101, 14725–707 14730. 708
Krainer, E. C., Ouderkirk. J. L., Miller, E. W., Miller, M. R., Mersich, A. T. and Blystone S. D. 709 (2013). The multiplicity of human formins: Expression patterns in cells and tissues. Cytoskeleton 710 Apr 29. doi: 10.1002/cm.21113. [Epub ahead of print] 711
Nalbant, P., Chang, Y.-C., Birkenfeld, J., Chang, Z.-F. and Bokoch, G. M. (2009). Guanine 712 nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the 713 leading edge. Mol. Biol. Cell 20, 4070–4082. 714
Naumanen, P., Lappalainen, P. and Hotulainen, P. (2008). Mechanisms of actin stress fibre 715 assembly. J Microsc 231, 446–454. 716
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

24
Oakes, P. W., Beckham, Y., Stricker, J. and Gardel, M. L. (2012). Tension is required but not 717 sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 196, 363–374. 718
Otomo, T., Tomchick, D. R., Otomo, C., Panchal, S. C., Machius, M. and Rosen, M. K. (2005). 719 Structural basis of actin filament nucleation and processive capping by a formin homology 2 720 domain. Nature 433, 488–494. 721
Paul, A. S., Paul, A., Pollard, T. D. and Pollard, T. (2008). The role of the FH1 domain and profilin 722 in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9–19. 723
Paul, A. S. and Pollard, T. D. (2009). Review of the mechanism of processive actin filament 724 elongation by formins. Cell Motil. Cytoskeleton 66, 606–617. 725
Pellegrin, S. and Mellor, H. (2007). Actin stress fibres. J. Cell. Sci. 120, 3491–3499. 726
Pertz, O., Hodgson, L., Klemke, R. L. and Hahn, K. M. (2006). Spatiotemporal dynamics of RhoA 727 activity in migrating cells. Nature 440, 1069–1072. 728
Pfaffl, M.W. (2006). A new mathematical model for relative quantification in real-time RT-PCR. 729 Nucleic Acids Res. 29, e45. 730
Price, L. S., Leng, J., Schwartz, M. A. and Bokoch, G. M. (1998). Activation of Rac and Cdc42 by 731 integrins mediates cell spreading. Mol. Biol. Cell 9, 1863–1871. 732
Pruyne, D., Evangelista, M., Yang, C., Bi, E., Zigmond, S., Bretscher, A. and Boone, C. (2002). 733 Role of formins in actin assembly: nucleation and barbed-end association. Science 297, 612–615. 734
Ridley, A. J. (2011). Life at the leading edge. Cell 145, 1012–1022. 735
Romero, S., Le Clainche, C., Didry, D., Egile, C., Pantaloni, D. and Carlier, M.-F. (2004). Formin 736 is a processive motor that requires profilin to accelerate actin assembly and associated ATP 737 hydrolysis. Cell 119, 419–429. 738
Rose, R., Weyand, M., Lammers, M., Ishizaki, T., Ahmadian, M. R. and Wittinghofer, A. (2005). 739 Structural and mechanistic insights into the interaction between Rho and mammalian Dia. Nature 740 435, 513–518. 741
Sagot, I., Rodal, A. A., Moseley, J., Goode, B. L. and Pellman, D. (2002). An actin nucleation 742 mechanism mediated by Bni1 and profilin. Nat. Cell Biol. 4, 626–631. 743
Schönichen, A., Alexander, M., Gasteier, J. E., Cuesta, F. E., Fackler, O. T. and Geyer, M. 744 (2006). Biochemical characterization of the diaphanous autoregulatory interaction in the formin 745 homology protein FHOD1. J. Biol. Chem. 281, 5084–5093. 746
Schönichen, A. and Geyer, M. (2010). Fifteen formins for an actin filament: a molecular view on the 747 regulation of human formins. Biochim. Biophys. Acta 1803, 152–163. 748
Schönichen, A., Mannherz, H. G., Behrmann, E., Mazur, A. J., Kühn, S., Silván, U., 749 Schoenenberger, C.-A., Fackler, O. T., Raunser, S., Dehmelt, L. et al. (2013). FHOD1 is a 750 combined actin filament capping and bundling factor that selectively associates with actin arcs and 751 stress fibers. J. Cell. Sci. 752
Schulte, A., Stolp, B., Schönichen, A., Pylypenko, O., Rak, A., Fackler, O. T. and Geyer, M. 753 (2008). The human formin FHOD1 contains a bipartite structure of FH3 and GTPase-binding 754 domains required for activation. Structure 16, 1313–1323. 755
756
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

25
Shemesh, T., Otomo, T., Rosen, M. K., Bershadsky, A. D. and Kozlov, M. M. (2005b). A novel 757 mechanism of actin filament processive capping by formin: solution of the rotation paradox. J. 758 Cell Biol. 170, 889–893. 759
Small, J. V., Rottner, K., Kaverina, I. and Anderson, K. I. (1998). Assembling an actin 760 cytoskeleton for cell attachment and movement. Biochim. Biophys. Acta 1404, 271–281. 761
Svitkina, T. M., Verkhovsky, A. B., McQuade, K. M. and Borisy, G. G. (1997). Analysis of the 762 actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell 763 Biol. 139, 397–415. 764
Takeya, R. and Sumimoto, H. (2003). Fhos, a mammalian formin, directly binds to F-actin via a 765 region N-terminal to the FH1 domain and forms a homotypic complex via the FH2 domain to 766 promote actin fiber formation. J. Cell. Sci. 116, 4567–4575. 767
Takeya, R., Taniguchi, K., Narumiya, S. and Sumimoto, H. (2008). The mammalian formin 768 FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress 769 fibre formation in endothelial cells. EMBO J. 27, 618–628. 770
Tojkander, S., Gateva, G. and Lappalainen, P. (2012). Actin stress fibers--assembly, dynamics and 771 biological roles. J. Cell. Sci. 125, 1855–1864. 772
Verkhovsky, A. B. and Borisy, G. G. (1993). Non-sarcomeric mode of myosin II organization in the 773 fibroblast lamellum. J. Cell Biol. 123, 637–652. 774
Watanabe, N., Kato, T., Fujita, A., Ishizaki, T. and Narumiya, S. (1999). Cooperation between 775 mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1, 136–143. 776
Watanabe, N., Madaule, P., Reid, T., Ishizaki, T., Watanabe, G., Kakizuka, A., Saito, Y., Nakao, 777 K., Jockusch, B. M. and Narumiya, S. (1997). p140mDia, a mammalian homolog of Drosophila 778 diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16, 779 3044–3056. 780
Watanabe, N. and Mitchison, T. J. (2002). Single-molecule speckle analysis of actin filament 781 turnover in lamellipodia. Science 295, 1083–1086. 782
Wei, Q. and Adelstein, R. S. (2000). Conditional expression of a truncated fragment of nonmuscle 783 myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol. Biol. Cell 11, 3617–3627. 784
Westendorf, J. J. (2001). The formin/diaphanous-related protein, FHOS, interacts with Rac1 and 785 activates transcription from the serum response element. J. Biol. Chem. 276, 46453–46459. 786
Yang, C., Czech, L., Gerboth, S., Kojima, S.-i., Scita, G. and Svitkina, T. (2007). Novel roles of 787 formin mDia2 in lamellipodia and filopodia formation in motile cells. PLoS Biol. 5, e317. 788
789
Zhang, X.-F., Schaefer, A. W., Burnette, D. T., Schoonderwoert, V. T. and Forscher, P. (2003). 790 Rho-dependent contractile responses in the neuronal growth cone are independent of classical 791 peripheral retrograde actin flow. Neuron 40, 931–944. 792
Zigmond, S. H., Evangelista, M., Boone, C., Yang, C., Dar, A. C., Sicheri, F., Forkey, J. and 793 Pring, M. (2003). Formin leaky cap allows elongation in the presence of tight capping proteins. 794 Curr. Biol. 13, 1820–1823 795
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

26
Figure Legends 796
Figure 1: Stress fiber organization is perturbed upon depletion of FHOD1. (A) Representative 797
confocal images of the actin cytoskeleton in fixed siRNA treated U2OS cells. Stress fiber types were 798
identified by F-actin (rhodamine-phalloidin) and Paxillin staining (focal adhesion marker; not shown). 799
Green and red arrows indicate dorsal stress fibers and transversal arcs, respectively. Yellow arrow 800
points to thick peripheral stress fiber bundles. Scale bar, 20 µm. (B) Percentage of cells with dorsal 801
stress fibers (dSF), transversal arcs (TA), both stress fiber types (dSF + TA) or thick peripheral actin 802
bundles only (peripheral bundles; yellow arrow in A). Data are mean ± s.d. from 3 experiments (n > 803
360 cells for each condition). (C) 3D organization of transversal arcs in U2OS cells. Depicted is the 804
maximum projection of a confocal Z-Stack (F-actin), in which the Z coordinates are indicated by the 805
color bar. Scale bar, 10 m. (D) Arc-covered lamellum regions were measured based on confocal Z-806
stacks (F-actin) and normalized to the whole cell adhesion area (ImageJ, free-hand line tool) (See Fig. 807
S1C for illustration of the analysis process). Only cells that contain arcs were included in the analysis. 808
Data are mean ± s.e.m. of n = 90 cells for each condition from 3 experiments. (E) Mean actin intensity 809
in arc-covered lamellum regions from (D). Red lines represent mean values. (F) Quantification of cells 810
with stellate stress fiber aggregates. Data are mean ± s.d. from 3 experiments (n > 360 cells for each 811
condition). (G) Representative image of a stellate stress fiber aggregate in a FHOD1 depleted cell. 812
Maximum projection of a confocal Z-Stack showing F-actin (rhodamine-phalloidin, red) and focal 813
adhesions (anti-paxillin antibody, green). Scale bar, 10 µm. (H) Actin aggregates contain Myosin IIA. 814
Representative maximum projection of a confocal Z-Stack of a FHOD1 depleted cell expressing 815
EGFP-NMHC IIA (green). F-Actin was visualized by rhodamine-phalloidin staining (red). Myosin 816
IIA is excluded from most of the straight actin fibers (white arrows) whereas strong localization is 817
detected within the aggregate. Scale bar, 10 µm. (I) Representative time-lapse confocal images 818
depicting the disassembly of an actin aggregate in an EGFP-Actin expressing FHOD1 depleted cell. 819
Scale bar, 10 µm. n = 20 movies. *** = p<0.001; ** =p<0.01; * = p<0.05. 820
821
Figure 2: FHOD1 depletion leads to decreased focal adhesion size and moderate spreading 822
defects. (A-C) FA size and number per cell were quantified 72 h after siRNA transfection. (A) 823
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

27
Representative confocal images of mKate-Paxillin and EGFP-Actin expressing control (ntsiRNA) and 824
FHOD1 depleted (siFHOD1) cells. Quantification by threshold based image analysis. Lower panel: 825
Binary images and outlines of focal adhesions. Scale bar, 10 µm. (B) Focal adhesion size and (C) 826
number were quantified using the analysis as shown in (A). Data are mean ± s.e.m. of n = 20 cells for 827
each condition from 5 experiments. (D-E) Cell spreading of ntsiRNA and FHOD1 depleted 828
(siFHOD1) cells. F-actin (rhodamine phalloidin; grey) and nuclear (DAPI; blue) staining at the 829
indicated time-points after replating. (D) Representative images during spreading. Scale bar, 20 µm. 830
(E) Quantification of cell area after replating. Data are mean ± s.e.m. of n > 100 cells (15 min) or 831
> 200 cells (30 and 60 min) from 3 experiments. ** = p<0.001** = p<0.01; * = p<0.05. 832
833
Figure 3: Active FHOD1 promotes transversal arc formation and enhanced maturation into 834
linear stress fibers. Acute effects of FHOD1 on stress fiber formation. Cells expressing EGFP, 835
EGFP-FHOD1 wild-type (FHOD1 WT), and active EGFP-FHOD1 V228E (FHOD1 V228E) were 836
treated with Cytochalasin D (2 µM, 90 min) to disrupt actin filaments. Stress fiber recovery was 837
stimulated by drug washout. Cells were either fixed and stained with rhodamine-phalloidin (F-actin) 838
(A -C) or subjected to confocal time-lapse imaging of the actin cytoskeleton (D-F). (A) Upper panel: 839
3D organization of stress fibers. Depicted are representative maximum projections of confocal Z-840
Stacks (F-actin) 30 min after washout, in which the Z-coordinates are indicated by the color bar. 841
Lower panel: Enlarged areas from boxed regions of upper panels. Scale bar, 10 µm. (B) Arc-covered 842
lamellum regions (in percent of total cell adhesion area) in fixed cells 30 minutes after drug washout 843
(see Fig. S1C for illustration of the analysis process). Data are mean ± s.e.m. of 100 cells for each 844
condition from 3 experiments. (C) FHOD1 V228E enhances stress fiber formation on the dorsal cell 845
side. Z-sections (middle) were generated from confocal Z-Stacks (F-actin) along lines indicated by red 846
arrows in the maximum projections (ImageJ, MultipleKymograph tool) (left). Scale bar, 10 µm. Mean 847
actin intensity was measured on the dorsal (red dotted line) or ventral (green dotted line) as indicated 848
in the schematic (middle) and plotted as “ratio dorsal/ventral” (right). n = 67 and 85 cells for the two 849
conditions, respectively (3 independent experiments; red lines indicate mean). (D) Representative 850
time-lapse confocal images of stress fiber recovery after Cytochalasin D washout in cells expressing 851
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

28
mCherry-Actin and EGFP-FHOD1 constructs (not shown). Acquisitions started immediately after 852
washout. Scale bar, 10 µm. (E) Time-point of first linear stress fiber appearance in time-lapse images. 853
n = 40, 33, and 44 cells for the three conditions, respectively (3 experiments; red lines indicate mean). 854
(F) Data from D were plotted as Histogram. ns = not significant; *** = p<0.001; ** = p<0.01. 855
856
Figure 4: FHOD1 inhibits growth of dorsal stress fibers. (A-D) Time-lapse confocal imaging of 857
control (ntsiRNA) and FHOD1 depleted (siFHOD1) cells expressing EGFP-Actin and mKate-Paxillin. 858
(A) Representative confocal time-lapse images depicting dorsal stress fiber growth. Boxed regions 859
show enlarged dorsal stress fibers. Arrowheads indicate distal (red) and proximal (green) end of dorsal 860
fibers. (B) Length of nascent dorsal stress fibers at each frame after their initiation (duration 30 861
minutes; frame rate = 30 seconds). Data are mean ± s.e.m. for each time point of 25 control and 27 862
FHOD1 depleted cells from 4 experiments. (C) Representative confocal time-lapse images showing 863
aberrant dorsal stress fiber bending in a FHOD1 depleted cell. (D) Quantification of cells with dorsal 864
stress fiber bending over time. Data are mean ± s.d. from 5 experiments (n > 40 cells for each 865
condition). (E-G) Constitutively active FHOD1 inhibits growth of dorsal stress fibers. Dorsal stress 866
fibers were quantified 30 min after Cytochalasin D washout by rhodamine-phalloidin (F-actin, green) 867
and Paxillin staining (focal adhesion marker, red). (E) Representative maximum projections of 868
confocal Z-Stacks showing stress fiber organization (F-actin, green) together with focal adhesions 869
(Paxillin, red) of FHOD1 WT or FHOD1 V228E expressing cells. Boxed regions are shown enlarged. 870
Red arrows point to typical dorsal stress fiber appearance for each condition. Scale bar, 10 µm. (F) 871
Quantification of dorsal stress fibers in fixed cells expressing EGFP-FHOD1 constructs 30 minutes 872
after Cytochalasin D washout. Data are mean ± s.d. from 3 experiments (n > 100 cells for each 873
condition). (G) Quantification of dorsal stress fiber length at 30 minutes after Cytochalasin D washout. 874
All detectable dorsal stress fibers were measured. Data are mean ± s.e.m. of 34-44 cells for each 875
condition from 3 experiments. Welch´s t-test was performed in (G). *** = p<0.001; ** = p<0.01; 876
ns = not significant. 877
878
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

29
Figure 5: FHOD1 localizes to contractile stress fibers with anti-parallel actin arrays. (A) FHOD1 879
localizes to ventral stress fibers (yellow arrow). TIRF images of EGFP-FHOD1 (green) and RFP-880
Actin (red). Ventral stress fibers are attached to the cell cortex with both ends and therefore entirely 881
visible in the TIRF field. Scale bar, 20 µm. (B) Constitutively active FHOD1 1-1011 localizes to distal 882
ends of dorsal stress fibers. Maximum projections of confocal Z-stacks are shown. Overlay: F-actin 883
(red, phalloidin staining), EGFP-FHOD1 (1-1011) (green), Paxillin staining (blue). Yellow arrow: A 884
ventral stress fiber associated with both ends to the substrate. Orange arrow: main portion of a dorsal 885
stress fiber. Red arrow: distal end of a dorsal stress fiber. Scale bar, 10 µm. (C) Myosin II localization 886
at distal ends of dorsal stress fibers at the interface to focal adhesions (red arrows). Confocal single 887
plane images of F-actin (Rhodamine Phalloidin: red), anti-NMHC IIA (green) and anti-Paxillin (blue) 888
staining. (D) Active FHOD1 V228E co-localizes with Myosin on contractile stress fibers. Confocal 889
images of EGFP-FHOD1 V228E (green) and Myosin (mCherry-NMHC IIA; red) co-localization on 890
transversal arcs. Yellow arrow: Punctuate pattern with FHOD1 and Myosin co-localization. 891
Representative images of n = 60 cells from 4 experiments. Scale bars, 10 µm. 892
893
Figure 6: Localization of N-Terminally truncated FHOD1 constructs. (A) Domain architecture of 894
FHOD1 mutant constructs. Red star marks the V228E mutation to activate FHOD1. (B) TIRF images 895
of EGFP-FHOD1 mutants (green) and mCherry-Actin (red). The N-terminally truncated construct 896
340-1164 is not associated along stress fibers. Yellow arrows point to regions of FHOD1 897
accumulation at focal adhesions (n >60 cells from 4 experiments for each construct). (C) FHOD1 1-898
339 co-localizes with Myosin II. Single plane confocal images of EGFP-FHOD1 1-339 (green) and 899
Myosin (mCherry-NMHCIIA; red) (n = 36 cells from 3 experiments). (D) N-terminally truncated 900
FHOD1 localizes to the lamellipodium during acute stress fiber stimulation by Cytochalasin D 901
washout. Left: Representative confocal images of FHOD1 wild-type and FHOD1 340-1164 (EGFP: 902
green) with F-actin (Rhodamine Phalloidin: red). Right: Number of cells with accumulated EGFP-903
FHOD1 signal in the lamellipodium at the leading edge 30 minutes after washout of Cytochalasin D. 904
Data are mean ± s.d. of >50 cells for each condition from 3 experiments. ns = not significant; 905
* = p<0.05. Scale bars, 10 µm. 906
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

30
907
Figure 7: Stress fiber binding is dispensable for robust stress fiber formation. (A) Representative 908
maximum projections of confocal Z-stacks (F-actin) of cells expressing EGFP-FHOD1 constructs. 909
Transfected cells are indicated by asterisks. (B) Cells entirely decorated with thick linear stress fibers 910
(red arrows in A) were quantified. Data are mean ± s.d. (n = 220-300 cells per condition from 3 911
experiments). (C) Acute stress fiber formation upon Cytochalasin D washout. Cells expressing EGFP 912
alone, EGFP-FHOD1 wild-type (FHOD1 WT) and active EGFP-FHOD1 mutants were treated with 913
Cytochalasin D (2 µM, 90 min) followed by extensive washout to stimulate stress fiber recovery. Cells 914
were fixed and stained with rhodamine-phalloidin (F-actin) 30 min after washout. Relative area of 915
cells covered with stress fibers was quantified. Data are mean ± s.e.m. of n = 56-74 cells for each 916
condition from 3 experiments. Welch´s t-test was performed in (C). ns = not significant; 917
*** = p<0.001. (D) 3D organization of stress fiber bundles stimulated by FHOD1 V228E. Depicted is 918
a single confocal plane on the ventral cell side (left) and maximum projections of confocal Z-Stacks 919
(F-actin, red; Paxillin, green). Z coordinates are indicated by the color bar (right). Scale bar, 10 m. 920
921
Figure 8: The C-terminal regulatory domain is required for FHOD1 effector function to 922
stimulate efficient stress fiber maturation. (A) FHOD1 V228E mutant is a more potent inducer of 923
ventral stress fibers as compared to the C-terminal truncated FHOD1 1-1011. Maximum projections of 924
confocal Z-stacks (F-actin) of cells transfected with EGFP-FHOD1 constructs and EGFP control were 925
generated.. Cells covered entirely with linear stress fibers (linear stress fiber phenotype) were 926
quantified. Data are means ± s.d. from 3 experiments (n = 46-62 cells). (B) Stress fiber association of 927
constitutively active FHOD1 mutants. Upper panel: Representative maximum projections of confocal 928
Z-stacks showing EGFP-FHOD1 and F-actin. Lower panel: Line scans were used to analyze the 929
correlation of FHOD1 (green) and F-actin (red) distribution (ImageJ, plot profile function). Lines were 930
drawn perpendicular to stress fibers from the leading edge to the center. Normalized intensity profiles 931
are shown. Scale bars, 10 µm. (C) Pearson´s correlation coefficient (GraphPad Prism) to quantify the 932
correlation of FHOD1 and F-actin intensities obtained from line scans (n = 26 for FHOD1 1-1011 and 933
24 FHOD1 V228E cells from 3 experiments). Red lines indicate mean. (D) TIRF images of a 934
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

31
representative cell expressing EGFP-FHOD1 wild-type and mCherry-Actin prior and 5 min post 935
ROCK inhibition via Y-27632 (50 µM). Scale bar, 20 µm. (E) Quantification of stress fiber 936
dissociation of EGFP-FHOD1 wild-type and the two constitutively activated constructs EGFP-FHOD1 937
V228E, EGFP-FHOD1 1-1011. Average fluorescence intensity decrease along individual stress fibers 938
in each EGFP-FHOD1 TIRF image was measured using ImageJ software and normalized to the 939
change of the corresponding mCherry-Actin signal (n = 14–16 cells from 3-4 experiments for each 940
construct; red line marks mean). *** = p < 0.001, ** = p < 0.01, * = p < 0.05, ns = not significant. 941
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
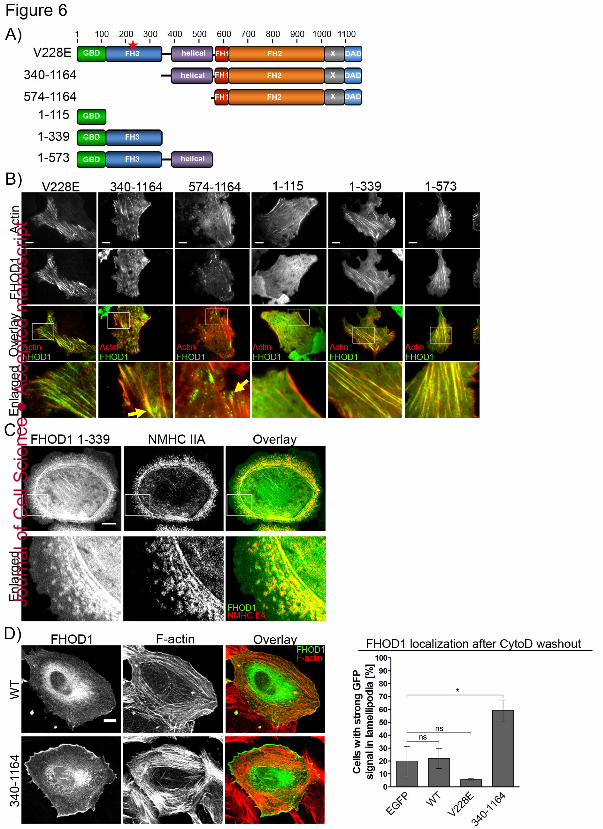
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
Top Related