







Languages
Pages
Legal


PAPER www.rsc.org/materials | Journal of Materials Chemistry
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online / Journal Homepage / Table of Contents for this issue
Charge ordering, symmetry and electronic structure issues and Wignercrystal structure of the quarter-filled band Mott insulators and highpressure metals d-(EDT-TTF-CONMe2)2X, X ¼ Br and AsF6†
Leokadiya Zorina,ab Sergey Simonov,ab C�ecile M�ezi�ere,a Enric Canadell,c Steve Suh,d Stuart E. Brown,d
Pascale Foury-Leylekian,e Pierre Fertey,f Jean-Paul Pougete and Patrick Batail*a
Received 15th April 2009, Accepted 6th July 2009
First published as an Advance Article on the web 4th August 2009
DOI: 10.1039/b906287d
We report on the synthesis and application of an internal chemical pressure to effectively control,
and reduce, the Mott gap in the system d-(EDT-TTF-CONMe2)2X, X ¼ Br, AsF6; the detailed
accounts of its Pmna, averaged room temperature structure and reversible phase transition at ca. 190 K
towards a low temperature P21/a structure; the synthesis of (13C-EDT-TTF-CONMe2)2Br, where one
carbon atom of the inner double bond is 100% 13C-enriched and single crystal 13C solid state NMR
spectroscopy and relaxation revealing that charge ordering occurs at room temperature and ambient
pressure; the discovery of weak superstructure Bragg reflections in d-(EDT-TTF-CONMe2)2Br and
subsequent analysis of the superstructure symmetry and refinement of an exhaustive synchrotron
radiation data set; suggesting an alternation at room temperature of neutral and oxidized molecules
along both the stacking a and transverse b directions in orthorhombic, non-centrosymmetric space
group P2nn, a CO pattern compatible with ferroelectricity. The charge disproportionation and long
range order crystallization of the electron gas onto every other molecular site within a three-
dimensional Wigner lattice is coupled to a concerted activation-deactivation of large collections of
transverse Csp2–H/O hydrogen bonds and an anti-phase, static modulation of the bromide anions
displacements along b. Despite the occurrence of charge ordering, the stacks remain essentially
uniform, in agreement with the rich low temperature Mott physics of the system.
Introduction
The ability to control what makes electrons (or holes) travel long
distances, as fast as one thousandth of the speed of light
(the Fermi velocity, 105 m s�1), over molecular sites along
a metallic wire or, instead, become prevented to do so and stay
put, localized onto the molecular backbone, is critical for the
development of molecular circuitry and electronic nanodevices,
and all comes down to playing with, and mastering, the interplay
of electron correlations and Coulomb interactions. This interplay
is recognized to be at the heart of the mechanism of metal-to-
insulator transitions (MIT), one of the most fascinating key
aLaboratoire de Chimie et Ing�enierie Mol�eculaire d’Angers,CNRS-Universit�e d’Angers, 49045 Angers, France. E-mail: [email protected] of Solid State Physics, Russian Academy of Sciences, 142432Chernogolovka, MD, RussiacInstitut de Ci�encia de Materials de Barcelona (ICMAB-CSIC), Campusde la UAB, E-08193 Bellaterra, SpaindDepartment of Physics and Astronomy, University of California, LosAngeles, USAeLaboratoire de Physique des Solides, CNRS-Universit�e Paris-Sud, 91405Orsay, FrancefSynchrotron SOLEIL, L’Orme des Merisiers - Saint Aubin, B.P. 48,91192 Gif-sur-Yvette, France
† Electronic supplementary information (ESI) available: Schemes S1–S3,Table S1. CCDC reference numbers 727931–727934. For ESI andcrystallographic data in CIF or other electronic format see DOI:10.1039/b906287d
6980 | J. Mater. Chem., 2009, 19, 6980–6994
phenomena in condensed matter materials chemistry and physics
whose approach was initiated by pioneering considerations by
Wigner, Mott and Hubbard more than 50 years ago,1–3 one
which pervades over, and keep inspiring, much current inter-
disciplinary research.4 For example, systems with an odd number
of electrons per site (sites here may be either discrete atomic
entities or covalent molecular units instead) can undergo a Mott-
Hubbard electron localization for large U/t ratios, where U is the
on-site Coulomb repulsion and t is the transfer integral.2,3 Elec-
tron localization can also occur for a non-integer number of
electrons per site in the presence of long range Coulomb repul-
sions, of which the nearest neighbour’s interaction V is the most
prominent,5 leading to a so-called Wigner crystallization. While
the study of such transitions in materials with open d and f
electron shells has a long history,6 MIT of similar nature were
recently observed in low dimensional molecular conductors.7
These materials, with their soft anisotropic lattices directed by
a subtle balance of a series of weak intermolecular interactions
such as hydrogen bonds,8 are currently receiving much attention
because the MIT can be tuned not primarily by doping but rather
by physical9,10 or chemical pressure, providing an avenue for
bandwidth control of the Mott-Hubbard gap.2,3,11
The importance of long range Coulomb interactions in organic
conductors was established very early by the discovery of the so-
called 4kF instability in the one dimensional (1D) charge transfer
salt TTF-TCNQ,12 the signature, as seen from the reciprocal
space, of a 1D Wigner lattice of localized charges in direct space
This journal is ª The Royal Society of Chemistry 2009

Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
accompanied by a concomitant lattice deformation.13 The 4kF
instability was found to be enhanced in radical cation or anion
salts, with a 2:1 or 1:2 stoichiometry, respectively, where the net
charge of conducting mixed valence molecular stacks is balanced
by neighboring strings of closed shell anions or cations which
here do not provide for mobile charge to screen the Coulomb
repulsions.14 In these salts, whose (electron or hole like) band is
quarter filled, the insulating ground state is achieved for
reasonable U/t and V/t ratios.15–17 With one carrier every two
sites, the 4kF insulating ground state is achieved by a doubling of
the chain repeat periodicity, a consequence of the localization of
the carriers either on one intermolecular bond out of two between
successive molecules (hence, here all molecules are in a mixed
valence state), or rather right onto one molecular site only out of
two successive molecular sites. The first case corresponds to the
formation of a mixed valence Mott dimer associated with a stack
dimerization, as observed for example at the 335 K MIT in
MEM(TCNQ)2.18 The second case, where charge rich molecules
alternate with charge poor molecules along the stack, corre-
sponds to a charge ordering (CO) or charge dis-
proportionation.6d,e This charge disproportionation is expected
to yield a doubling of the number of NMR lines at the CO
transition, as indeed observed in (DI-DCNQI)2Ag19 as well as for
the (TMTTF)2X series with X ¼ PF6, SbF6, etc.20 For
(DI-DCNQI)2Ag it has been found that the 210 K transition
corresponds to the divergence of a 1D 4kF structural instability,21
yet the 50 K Wigner structure consists of different stacks where
the charges are localized either on the sites, or on the bonds
(dimers) or in an intermediate position.19c A single type of stack is
identified in the CO state of (TMTTF)2X, but as this stack was
not uniform to start with at room temperature but already
slightly yet significantly dimerized at higher temperature, the low
temperature structure is postulated to consist of a mixture of 4kF
bond and site orders, in agreement with the observation of
ferroelectricity due to the removal of all inversion centers at
CO.22 Although the Extended Hubbard Hamiltonian produces
the CO ground state,3,16 the role of coupling to the lattice is
necessary to describe the complete temperature/pressure phase
diagram of materials such as (TMTTF)2X.23,24 The counterion
sublattice is likely also important,22,25–27 but there are no sup-
porting structural data. In contrast, the present report will
conclude there is a strong involvement of the lattice in producing
the CO state at ambient pressure.
In this context, the report in 2003 of the quasi-one-dimen-
sional, 3⁄4 -filled band Mott insulator d-(EDT-TTF-CON-
Me2)2AsF6 (3⁄4 -filled with electrons or ¼-filled with holes),28
where there is no centre of symmetry between the molecular units
along the stack whose strict uniformity is imposed by glide plane
symmetry, has triggered an in-depth exploration of its rich phase
diagram.29 The Mott gap in d-(EDT-TTF-CONMe2)2AsF6, an
insulator at ambient pressure and room temperature, decreases
upon hydrostatic pressure up to 20 kbar where it is suppressed
and the system becomes metallic. Here, we report on d-(EDT-
TTF-CONMe2)2Br and demonstrate that substitution of AsF6�
for Br�, whose volume is 76 A3 smaller, builds up an internal
chemical pressure which results in a shrinking of the stacking axis
and, conversely, an increase of the transfer integral along the
stack, as discussed. This crystal engineering approach effectively
allows for a control and a sizeable decrease of the Mott gap, as
This journal is ª The Royal Society of Chemistry 2009
the localization in d-(EDT-TTF-CONMe2)2Br is suppressed at
a smaller hydrostatic pressure with a net gain of as much as
7 kbar, as reported independently.29
Then, for the reasons described previously, it is likely that in
the insulating state the charge density will no longer remain
uniform along the stacks, that is, charge disproportion is
expected to occur. Therefore, in order to search for CO, we set
out to synthesize (13C-EDT-TTF-CONMe2)2Br, where one
carbon atom of the inner double bond is 100% 13C-enriched, in
order to perform high resolution 13C solid state NMR experi-
ments, reported here, and IR reflectivity and Raman experi-
ments, reported independently,30 which both reveal that CO and
Wigner crystallization occur already at room temperature and
ambient pressure in these salts. This prompted the present
complementary investigation of the crystal structure of d-(EDT-
TTF-CONMe2)2Br using a combination of X-ray diffuse scat-
tering experiments and synchrotron radiation leading to the
discovery of superstructure Bragg reflections which in turn
imposes two independent molecules to co-exist in a double unit
cell. An assessment of the symmetry base and the successful
refinement of the ambient pressure, room temperature synchro-
tron data are further discussed herein, allowing for a complete
description of the three-dimensional pattern of CO, here in the
non-centrosymmetric space group P2nn, which proved to
confirm earlier models.
Experimental
Synthesis of 13C-enriched 4,5-ethylenedithio-40-(N,N-dimethylcarbamoyl)
tetrathiafulvalene
13C-enriched 4,5-ethylenedithio-40-(N,N-dimethylcarbamoyl)-
tetrathiafulvalene is prepared with one important modification
of the procedure reported earlier.31 Since the synthesis of the13C-enriched 4,5-ethylenedithio-2-thioxo-1,3-dithiole necessary
to obtain the key molecule 13C-EDT-TTF-(CO2Me)2 is not
practical because it requires large amounts of CS2, we chose to
prepare its oxo equivalent instead and then adjust the coupling
conditions to obtain a correct yield. The enriched precursor
4,5-ethylenedithio-2-oxo-1,3-dithiole is prepared as described via
the Larsen-Lenoir route (ESI,† Scheme S1).32
13C-enriched potassium O-(isopropyl)dithiocarbonate
3.64 g (0.065 mol) of potassium hydroxide are dissolved in 35 mL
of 2-propanol distilled on CaO with heating. The hot solution is
filtered into a 100 mL round bottom flask and cooled to 0 �C with
an ice bath. 13C-enriched CS2 from Sigma-Aldrich Company
(5 g, 0.065 mol) is added dropwise by a syringe with stirring.
After 30 minutes the potassium xanthate is collected by filtration
and washed with four 10 mL portions of diethyl ether and dried
under pressure. Recrystallization from ethanol affords 6.88 g of
the desired compound (Mp 265 �C).
Coupling procedure
The coupling of the 13C-enriched 4,5-ethylenedithio-2-oxo-1,3-
dithiole moiety with the 4,5-dimethyloxycarbonyl-2-oxo-1,3-
dithiole has been optimized and the best yields are obtained using
five equivalents of the diester moiety (ESI,† Schemes S2 and S3).
J. Mater. Chem., 2009, 19, 6980–6994 | 6981

Fig. 1 Atom labelling and molecular structure for the averaged, room
temperature orthorhombic structure of d-(EDT-TTF-CONMe2)2Br
(DIAMOND diagram with 50% thermal ellipsoids). Atoms S5, C7, C8
and C11 are disordered about the mirror plane in Pmna.
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
13C-EDT-TTF-CONMe2
13C-EDT-TTF-COCl (800 mg, 2.23 mmol) dissolved in dry THF
(32 mL) was added dropwise to a 2M THF solution of dime-
thylamine (5.6 mL, 11.2 mmol) diluted in dry THF (32 mL).
After stirring for one hour the solution is filtered and evaporated.
The orange solid is recrystallized in CH3CN to afford the
compound as orange platelets (620 mg, 76%), mp 166 �C; anal-
ysis found: C, 36.28; H, 2.77; N, 3.91. 12C1013C1H11ONS6
requires: C, 36.31; H, 3.02; N, 3.82%; NMR (500 MHz, DMSO-
d6) dH 7.28 (s, 1H, C]CH), 3.38 (s, 4H, CH2–CH2), 3.00 (s, 6H,
CH3); dC 160.36 (C]O), 130.23 (C–CO), 124.58 (]CH), 112.82–
116.91 (C]C), 103.97 (13C]C), 35.90 (CH3) and 29.58 (CH2–
CH2); m/z (EI) 366 (M+, 100%), 338 (M+ � CH2 � CH2, 50%).
Electrocrystallization experiments. (EDT-TTF-CONMe2)2Br
Black, elongated crystalline platelets were grown at 25 �C upon
constant current (1.5 mA) oxidation at a Pt wire electrode (length
2 cm, diameter 1 mm) of a dichloromethane/acetonitrile 1/1
solution of EDT-TTF-CONMe2 (5 mg) containing PPh4Br
(35 mg) as electrolyte. The crystals were harvested on the anode
after one week.
(13C-EDT-TTF-CONMe2)2AsF6
Black, elongated crystalline platelets were grown at 30 �C upon
constant current (0.5 mA) oxidation at a Pt wire electrode (length
2 cm, diameter 1 mm) of a 1,1,2-trichloroethane solution (24 mL)
of 13C-EDT-TTF-CONMe2 (10 mg) containing TBAAsF6
(100 mg) as electrolyte. The crystals were harvested on the anode
after two weeks.
(13C-EDT-TTF-CONMe2)2Br
Black, elongated crystalline platelets were grown at 20 �C upon
constant current (3 mA) oxidation at a Pt wire electrode (length 2
cm, diameter 1 mm) of a dichloromethane/acetonitrile 1/1 solu-
tion (12 mL) of 13C-EDT-TTF-CONMe2 (10 mg) containing
PPh4Br (70 mg) as electrolyte. The crystals were harvested on the
anode after one week.
Standard X-ray diffraction experiments
Experimental X-ray structural data were collected on a d-(EDT-
TTF-CONMe2)2Br crystal mounted on a glass fiber using
a Bruker Nonius KappaCCD diffractometer with mono-
chromatized MoKa radiation (l ¼ 0.71073A, graphite mono-
chromator) equipped with an Oxford Cryosystems cryostream
cooler. Preliminary measurements of unit cell parameters were
performed in the temperature range of 300–100 K with 10 degree
steps. The as-grown crystal has an orthorhombic unit cell (space
group Pmna) characterized by sharp discrete Bragg reflections at
room temperature. Significant changes in the diffraction patterns
occur upon lowering the temperature. The former sharp reflec-
tions broaden upon reaching 200 K to the point where two
different systems of reflections are identified at 180 K and below
(down to 100 K). The latter define two equivalent monoclinic
lattices, as revealed by the DIRAX program,33 both in space
group P21/a. This qualifies an orthorhombic-to-monoclinic
phase transition accompanied by twinning.
6982 | J. Mater. Chem., 2009, 19, 6980–6994
In order to clarify the nature of the reversible phase transition,
data were collected at room temperature and 150 K for the
orthorhombic and monoclinic phases, respectively, by
a combined 4- and u-scan method. Diffraction intensities from
both twin domains obtained at 150 K were integrated using the
twinning option in the EVALCCD package of programs34 and
combined into SHELX HKLF5 file with additional indexes 1 or
2 marking the reflections from one or another twin cell, respec-
tively. Analytical absorption correction of experimental intensi-
ties taking into account the crystal shape and size (prism
0.05 � 0.23 � 0.04 mm) was applied for both phases using the
WINGX package of programs.35
The structures were solved by a direct method followed by
Fourier syntheses and refined by a full-matrix least-squares
method in an anisotropic approximation for all non-hydrogen
atoms using the SHELX-97 programs.36 The twin relationship
was taken into account in the HKLF5 refinement of the low-
temperature data with SHELXL-97.37 The twin fraction was
refined to a value of 0.4723(4).
Orientational disorder affects one amide methyl group and the
outer ethylene group at room temperature (Fig. 1). A slight
disorder of the ethylene group (10% of the alternative position)
remains in the monoclinic phase at 150 K. H atoms in both
phases were introduced in calculated positions with isotropic
displacement parameters fixed at 120% (or 150% for –CH3
groups) of the corresponding parameters of the attached
C atoms. Torsion angles of idealized hydrogen positions in the
–CH3 groups were refined from electron density (by HFIX 137
instruction). Details of unit cell data, data collection and
refinement are summarized in Table 1.†
A similar temperature-dependent study performed for one
single crystal of the isostructural salt, d-(EDT-TTF-CON-
Me2)2AsF6, concludes that the very same twinning occurs in the
low-temperature monoclinic phase. The complete experimental
data set was collected at 100 K. Twin data integration combining
intensities of both twin domains into the HKLF5 file as well as
structure solution and refinement were conducted with proce-
dures identical to those used for d-(EDT-TTF-CONMe2)2Br at
150 K.
X-Ray diffuse scattering experiments
The diffuse scattering study was performed using copper radia-
tion (l ¼ 0.1542 nm) issued from a rotating anode equipped with
a confocal multilayer monochromator. The investigation was
first performed with the so-called fixed film-fixed crystal method
in order to detect any weak X-ray diffuse scattering. Then,
This journal is ª The Royal Society of Chemistry 2009

Table 1 Crystal data, data collection and refinement details for d-(EDT-TTF-CONMe2)2X (X ¼ Br, AsF6)
X ¼ Br X ¼ AsF6
l/A 0.71073 0.71073 0.92072 0.71073Temperature/K 295(2) 150(2) 293(2) 100(2)Chemical formula C22H22BrN2O2S12 C22H22BrN2O2S12 C22H22BrN2O2S12 C22H22AsF6N2O2S12
Molecular weight 811 811 811 920Crystal system Orthorhombic Monoclinic Orthorhombic Monoclinica/A 7.1097(4) 6.9762(9) 7.1126(5) 7.0157(8)b/A 6.5049(9) 6.480(1) 13.0250(7) 6.422(1)c/A 32.691(3) 32.671(9) 32.7590(13) 35.465(5)a/� 90 90 90 90b/� 90 90 90 90g/� 90 92.45(2) 90 92.26(1)V/A3 1511.9(3) 1475.6(5) 3034.8(3) 1596.6(3)Space group, Z Pmna, 2 P21/a, 2 P2nn, 4 P21/a, 2F(000) 822 822 1644 926rcalc./g$cm�3 1.782 1.825 1.775 1.914m/cm�1 22.16 22.71 22.61 19.15Tmin, Tmax 0.732, 0.922 0.735, 0.920 0.467, 0.918 0.748, 0.9152qmax/� 60.0 60.4 95.4 64.1Reflections collected 18647 37386 42335 46120Independent reflections 2333 — 12895 —Rint 0.067 — 0.077 —No. of parameters 134 189 356 208GooF on F2 1.002 1.002 1.006 1.012R1 [I > 2s(I)] 0.0331 0.0571 0.0521 0.0784wR2[I > 2s(I)] 0.0599 0.1274 0.1418 0.1832
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
accurate measurements of the superstructure reflection intensity
and of the possible forbidden reflections were performed using
a four-circle diffractometer. The set-up was equipped with
a closed-cycle helium cryostat operating from 300 K down to
15 K. For the fixed film-fixed crystal technique, it was possible to
investigate the temperature range 300–1.8 K using a home-made
cryocooler equipped with a Joule-Thomson He closed-cycle third
stage. Three single crystals (�1 mm � 0.1 mm2) were used in the
experiments.
Synchrotron experiments at SOLEIL
X-Ray synchrotron data were collected at the Cristal beamline
using a Newport 4-circle diffractometer equipped with a 2D
CCD Ruby detector from Oxford Diffraction. A 13.466 keV
energy was chosen (0.92072 A) just below the Br absorption edge
in order to reinforce the anomalous signal of the Br atom
(f0 ¼ �7.74289 e�). 4- and u-scans were collected using the
oscillation method (1�/frame) at four different detector positions
(2qD¼�26� and 2qD¼�55�) with 1 s/� and 3 s/� exposure times,
respectively. During these data collections, the incident beam
was attenuated to limit the detector saturation. However, an
extra data set was collected without attenuation of the incident
beam to improve the signal of weak reflections. All the frames
were then processed using the CrysAlis software suite (integra-
tion of the intensities, incident beam monitor corrections, frame
scaling, empirical absorption corrections and reciprocal space
layer reconstructions).38
The structure was solved and refined in different space groups
with SHELX-9736 using special dispersion and absorption coef-
ficients for all chemical elements for a synchrotron wavelength of
0.92072 A (the DISP instruction was applied). A direct method
followed by Fourier syntheses and a full-matrix least-squares
method in an anisotropic approximation for all non-hydrogen
This journal is ª The Royal Society of Chemistry 2009
atoms were used for the structure solution and refinement,
respectively. Results in space group P2nn were considered as the
best description of the data, as discussed below. Large thermal
parameters are observed for one C atom only within the ethylene
outer group of each EDT-TTF-CONMe2 molecule, pointing to
two essentially equiprobable positions with anisotropic
displacement parameters restrained to be equal. Orientational
disorder affects some other atoms (S, Me groups), yet with lesser
thermal anisotropy and two atom sites could not be properly
distinguished. Hydrogen atoms were introduced as described for
the standard X-ray diffraction data. Details of unit cell data, data
collection and refinement are summarized in Table 1.†
Single crystal 13C NMR
The 13C spin-labelled samples with Br and AsF6 counterions were
studied over a range of temperatures (4–300K) and with a field
strength of 10 T oriented in the bc plane. The range of temper-
atures is accessed using a standard variable temperature cryostat
inserted into a cold-bore superconducting magnet. The rf tuning
coil was wound specifically for the small single crystals used in
this study so as to optimize signal/noise, then mounted onto
a rotatable platform. Evidence for two inequivalent environ-
ments, consistent with charge disproportionation, was obtained
from spectroscopy and spin–lattice relaxation measurements.
Electronic structure
The tight-binding band structure calculations used an extended
H€uckel type Hamiltonian.39 The off-diagonal matrix elements of
the Hamiltonian were calculated according to the modified
Wolfsberg-Helmholz formula.40 A basis set composed of double-
z Slater type orbitals for all atoms was used. The exponents and
ionization potentials (eV) employed were 1.817 and �20.0 for S
J. Mater. Chem., 2009, 19, 6980–6994 | 6983

Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
3s, 1.817 and�13.3 for S 3p, 1.626 and�21.4 for C 2s, 1.625 and
�11.4 for C 2p, 1.95 and �26.0 for N 2s, 1.95 and �13.4 for N
2p, 2.275 and �32.3 for O 2s, 2.275 and�14.8 for O 2p, 1.30 and
�13.6 for H 1s.
Results and discussion
The synthesis of 13C-EDT-TTF-CONMe2, described in Schemes
S1–S3 (ESI†), was adapted from earlier procedures31 for EDT-
TTF-CONMe2. Electrocrystallization of the p-donor molecule
on a platinum wire electrode provides elongated, ruler-like
crystals appropriate for X-ray, transport, ESR, IR and Raman
and solid state NMR experiments.
The high and low temperature, averaged Pmna and P21/a
structures, both have uniform stacks imposed by glide plane
symmetry
Standard X-ray diffraction single crystal determinations revealed
the room and low temperature structures of d-(EDT-TTF-
CONMe2)2Br where discrete anions are interspersed between
successive radical cation slabs along c, as exemplified in the two
projections of the 150 K monoclinic structure shown in Fig. 2.
The unit cell contains one bromine anion on an inversion centre
and one independent donor molecule located in a general posi-
tion in the low-temperature monoclinic phase or on the mirror
plane in the room temperature orthorhombic form, some of the
Fig. 2 Projections of the averaged, P21/a structure of the low-temper-
ature monoclinic form of d-(EDT-TTF-CONMe2)2Br at 150 K along the
a-direction (a) and b-direction (b). Note that the locations in the lattice of
atoms C1, which will eventually be 13C-enriched (see text), have been
single out by coloring the atom in black.
6984 | J. Mater. Chem., 2009, 19, 6980–6994
atoms (methyl and ethylene groups) being disordered about the
m-plane, as shown in Fig. 1. These are denoted as the averaged
Pmna and P21/a structures, on account that, in each one of them,
the single independent molecule bears the formal charge 0.5+.
It is important to note that a defining, characteristic feature of
the average structures of this 2:1 mixed-valence system is the
presence of exactly uniform stacks of half-charged p-conjugated
molecules generated by the repetition of any donor molecule by
the crystallographic glide plane a. Adjacent molecules in the
stack adopt a criss-cross mode of overlap and are 3.488(1) and
3.555(1) A apart in the monoclinic and orthorhombic phases,
respectively, i.e., a/2. Within the stack, CEt–H/O hydrogen
bonds (Fig. 2b, Table 2) are eventually strong enough to provide
additional, slightly shortened van der Waals S/C as well as
C/C intrastack contacts, while all S/S intrastack distances
exceed 3.7 A. Coplanar molecules from neighboring stacks are
connected by cell translation along b, creating a d-type layer, and
interact by one Csp2–H/O contact and three S/S contacts, two
of which are very short (ESI,† Table S1).
Likewise, the ethylene and methyl groups are hydrogen
bonded to bromide atoms (Fig. 2), the CEt–H/Br hydrogen
bonds being significantly shorter than CMe–H/Br contacts
(Table 2). Such dependence of hydrogen bond strength on the
nature of the carbon atom is a common tendency for C–H/X
contacts.8 One of the two independent methyl C-atoms in EDT-
TTF-CONMe2 (C11 in Fig. 1) does not form any hydrogen
bonds at 150 K despite a C/O separation as short as 3.254(2) A
from the carbonyl oxygen atom. Hence, C11 is more loosely
packed in the crystal and exhibits the largest thermal parameter
at low temperature in comparison with all other atoms.
Structural phase transition accompanied by twinning
The lattice distortion identified in d-(EDT-TTF-CONMe2)2Br
below 190 K proved to be associated with an orthorhombic-to-
monoclinic phase transition. The occurrence of twinning
complicates the analysis of this transition. Two sets of Bragg
reflections are distinguished in the diffraction patterns of the
twinned monoclinic crystals at 180 K and below. This structural
phase transition is found to be entirely reversible upon warming
the crystal back to room temperature. Extensive unit cell
parameter determinations performed over a wide temperature
range demonstrate that there is no evidence for any cell volume
discontinuity or temperature hysteresis. Therefore, this is
a second order phase transition.
The temperature dependence of the monoclinic g angle, shown
in Fig. 3, qualifies g as an order parameter characteristic of the
transition. Note that the lattice distortion is also revealed by
a change of slope of the linear temperature dependence of the
ESR linewidth and the narrowing of the latter at 190 K, both for
the AsF6� and Br� salts,41 as well as by a deviation at this
temperature of the spin susceptibility from its high temperature
Bonner-Fisher dependence.41 The separation in terms of the g
angle between the two twin parts becomes more appreciable
upon further cooling, as illustrated in Fig. 3.
The nature of the twinning is an important question. One of
two ways is possible: either the crystals are twinned already at
room temperature or twin micro-domains form during the phase
transition. Note that, as exemplified in Fig. 4a, several crystals are
This journal is ª The Royal Society of Chemistry 2009

Table 2 C–H/O donor/donor and C–H/Br donor/anion hydrogen contacts in d-(EDT-TTF-CONMe2)2Br at room temperature and 150 K
Contact type
Geometry of D–H/A contactsa
150 K 295 K
293 K (double lattice)
H-bond donor I0 H-bond donor II�+
CEt–H/O (intrastack) 2.32 [3.26A, 163�] 2.39 [3.29A, 154�] 2.45 [3.38A, 162�] 2.39 [3.33A, 163�]2.48 [3.37A, 153�] 2.54 [3.46A, 157�] 2.56 [3.30A, 134�] 2.67 [3.54A, 150�]
Csp2–H/O (interstack) 2.89 [3.80A, 165�] 2.93 [3.84A, 165�] 2.96 [3.86A, 165�] 2.91 [3.82A, 167�]
CEt–H/Br 2.73 [3.70A, 175�] 2.76 [3.72A, 173�] 2.84 [3.72A, 152�] 2.78 [3.75A, 175�]2.95 [3.64A, 129�] 2.98 [3.68A, 130�] 3.19 [3.86A, 127�] 3.05 [3.71A, 127�]
CMe–H/Br 3.21 [4.10A, 155�] 3.38 [4.21A, 145�] 3.32 [4.15A, 145�] 3.36 [4.19A, 146�]3.28 [4.19A, 159�] 3.40 [4.09A, 130�] 3.37 [4.22A, 148�] 3.42 [4.27A, 148�]
a Values for each donor–H/acceptor contact are given in following order: H/A distances, A [D/A distances, D–H–A angles].
Fig. 3 Change in the g lattice angle across the structural phase transi-
tion. Blue rhombuses and red circles below the transition are data points
for the two different twin domains in the monoclinic phase.
Fig. 4 Different shapes of the crystals of d-(EDT-TTF-CONMe2)2Br.
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
composed of two identical fragments separated by a long thin
crystal plate in between, as though the inner thin plate is actually
the twin boundary. However, X-ray experiments on those crystals
as well as on crystal cut-offs along the assumed twin boundary, or
alternatively on crystals of strictly rectangular shape (Fig. 4b),
always yielded the orthorhombic symmetry at room temperature
and the same twin diffraction pattern at low temperature (the full,
final X-ray data set was obtained from a rectangular crystal).
The observation of positional disorder in the orthorhombic
form of the crystal also suggests possible twinning at room
temperature. As mentioned earlier, in the orthorhombic phase
This journal is ª The Royal Society of Chemistry 2009
the asymmetric donor molecule lies flat on a mirror plane, hence
the methyl and ethylene groups are disordered over two equi-
probable sites about this plane, as exemplified in Fig. 1 and 5a. In
other words, the donor disorder in the structure of the high-
temperature phase is imposed by the existence of the mirror
plane; yet the choice of another, non-centrosymmetric space
group (with possible twinning) is ruled out by taking into
account the nature of the observed phase transition. In agree-
ment with a second order transition, the symmetry group of the
orthorhombic phase should necessarily be a super-group of that
of the monoclinic phase.42 Analysis of systematic absences makes
for the unambiguous assignment of P21/a as the space group for
the low temperature monoclinic form. The twin structure
refinement based on this space group delivers a high quality
structure without any indication of the disorder caused by
symmetry elements 21 or a. Thus, the centrosymmetric space
group Pmna, a supergroup of P21/a, was chosen for the high-
temperature orthorhombic average structure.
Note finally that the twinning operation is deciphered as the
result of a two-fold rotation about a in the direct lattice (Fig. 5b),
and that this two-fold axis is an integral part of the complete set
of symmetry elements of P2/m2/n21/a. Hence, no evidence for the
presence of a twinning similar to that found below the phase
transition is expected at room temperature. Across the phase
transition, although the 2x symmetry is lost at the macroscopic,
full length-scale of the crystal, it is kept locally as a twin opera-
tion. Temperature and pressure-induced structural transitions
are susceptible to this kind of twinning and even more so for
molecular compounds whose structure is directed by non-cova-
lent intermolecular interactions such as hydrogen bonds and van
der Waals interactions and where alternative orientations of
molecules in the single crystal and across the twin boundary can
be energetically similar.43 As all the crystals tested (more than
ten, from different syntheses) have repeatedly shown the same
twinning across the phase transition, d-(EDT-TTF-CON-
Me2)2Br appears to be another typical example of concomitant
twinning and structural transition.
As shown in Fig. 5b, the orientational disorder affecting the
methyl groups in the room temperature orthorhombic phase has
vanished in the monoclinic structure at 150 K, as any of the two
twin domains now selectively contains one or the other methyl
group orientations. Note that a similar situation applies for the
ethylene-end groups albeit each of the two twin domains still
J. Mater. Chem., 2009, 19, 6980–6994 | 6985

Fig. 5 (a) d-Layer of the orthorhombic Pmna phase viewed along c.
(b) The twinned, monoclinic structure of the low-temperature P21/
a phase finds its origin in a stacking fault at the domain ac-interface in
keeping with the local two-fold axial symmetry about a. Two twin
domains are shown in different colours. Note here that the methyl and
ethylene groups are ordered in the low temperature, monoclinic structure
with a neat partition inside individual twin domains. That is, the outer,
ordered ethylene C–C bonds are parallel inside each twin domain while
the spatial orientations of these bonds differ from one domain to the
other. The same applies to all ordered C–Me bonds.
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
remains affected by some small amounts (like 10%) of disorder.
Most likely, some amounts of disorder remain because the
transition appears not to be completed at 150 K, yet the data in
Fig. 3 suggest that a fully ordered structure is expected at lower
temperatures. The two twin domains are almost equiprobable at
150 K as the refined partition is 53% and 47%.
A similar twinning also affects the low temperature monoclinic
phase of d-(EDT-TTF-CONMe2)2AsF6. Here, analysis of single
crystal X-ray diffraction data collected at 100 K reveals that the
Table 3 Chemical pressure effect in the series d-(EDT-TTF-CONMe2)2X (X
X ¼ Br�
Temperature/K 150 295a/A 6.9762(9) 7.1097(4)b/A 6.480(1) 6.5049(9)c/A 32.671(9) 32.691(3)a/� 90 90b/� 90 90g/� 92.45(2) 90V/A3 1475.6(5) 1511.9(3)
6986 | J. Mater. Chem., 2009, 19, 6980–6994
molecules are fully ordered and that, within the two twin
domains, the same difference in spatial orientations of the methyl
and ethylene groups prevails. The twin domain ratio amounts
to 62:38.
Chemical pressure
Several monoclinic phases with a d-type, criss-cross pattern of
overlap within the stack and similar lattice parameters have been
identified amongst BEDT-TTF salts and analyzed using
CSD.44,45 Note that they all have other symmetry groups with
their monoclinic axis always parallel to the d-slab while it is here
normal to the layers. Hence, the sole isostructural salt known to
date is d-(EDT-TTF-CONMe2)2AsF6.28
The structures of the Br� and AsF6� salts and their trans-
formation at low temperatures are quite similar yet the donor
stacks in d-(EDT-TTF-CONMe2)2Br are both more compact
than those in d-(EDT-TTF-CONMe2)2AsF6 and exhibit more
and shorter S/C and C/C intra-stack contacts as a result of the
chemical pressure which sets in upon installing the bromide
anion, whose effective volume is 76 A3 smaller than that of AsF6�
(Table 3). Note that, conversely, the cell dimension in the
direction b transverse to the stacks increases in the Br� salt so
that the effective area of the donor layer in the ab plane barely
changes by 1%. Accordingly, side-by-side S/S and H/O
contacts are slightly shorter in the AsF6� salt. The largest
compression of the lattice upon changing the anion from AsF6�
to Br� occurs along the direction across the layers. Remarkably
here, AsF6� is found to be strongly disordered upon cooling the
crystal from room temperature down to 100 K while Br� is
ordered in the whole temperature range, hence providing for
a better fit to the donor packing.
Uniform stacks and chemical pressure effect both nicely
exemplified in the compared electronic structures of Br� and AsF6�
salts
Because of the symmetry of the crystal, there are only two
different types of donor/donor interactions in the donor layers
of these salts: one along the stack (//) and one along the
perpendicular direction (t). Although the S/S contacts along
the stacks are all longer than 3.7 A whereas there are some quite
short contacts along the interstack direction (around 3.3 A or
shorter; see ESI† Table S1), the HOMO/HOMO interaction
along the stacks is considerably stronger. For instance, the
calculated transfer integrals for d-(EDT-TTF-CONMe2)2Br at
150 K are t// ¼ 87 meV and tt ¼ �32 meV. With the previous
¼ Br�, AsF6�)
X ¼ AsF6�
100 150 2957.0157(8) 7.0618(9) 7.2419(4)6.422(1) 6.4459(8) 6.4622(3)
35.465(5) 35.562(4) 35.557(2)90 90 9090 90 9092.26(1) 91.73(1) 90
1596.6(3) 1618.0(3) 1664.0(2)
This journal is ª The Royal Society of Chemistry 2009

Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
values of the S/S contacts in mind, the latter value can seem
surprisingly small. However, a closer look shows that the above-
mentioned short contacts are associated with one (or two)
S atoms of the six-member rings, which have small contributions
to the HOMO (i.e. less than one-third that of the inner core
S atoms). In addition, the associated orbital interactions are of
the intrinsically weak p-type. Thus, the relatively weak lateral
interaction is not that surprising. In contrast, the interactions
along the stacks, although associated with longer S/S contacts,
are of the stronger s-type. This leads to the pseudo-one dimen-
sional (pseudo-1D) band structure of Fig. 6a. The calculated
Fermi surface assuming a metallic filling of the bands
(see Fig. 6b) is a pair of warped but perfectly nested open lines.
The pressure effect discussed above is clearly seen in the
calculated band structures. We report in Table 4 the main
characteristics of the calculated band structures for the mono-
clinic structure of d-(EDT-TTF-CONMe2)2Br at 150 K and
d-(EDT-TTF-CONMe2)2AsF6 at 100 K as well as the ortho-
rhombic room temperature structure of d-(EDT-TTF-CON-
Me2)2Br. It is clear from the comparison of the low-temperature
values for the Br and AsF6 salts that there is a pressure effect
affecting both the intra and interstack interactions, which is
consistent with the fact that the Br salt becomes metallic for
a pressure which is 7 kbar lower than for the AsF6 salt.29 Thermal
contraction also noticeably affects the electronic structure since
there is an increase of approximately 15% in the intrastack
interaction for the Br salt when going from room temperature to
150 K. However thermal contraction is also associated with
a decrease of the interstack interactions (compare the values for
Fig. 6 Calculated band structure (a) and Fermi surface assuming
a metallic filling of the bands (b) for the donor layers of the low-
temperature, P21/a monoclinic form of d-(EDT-TTF-CONMe2)2Br at
150 K. G ¼ (0, 0), X ¼ (a*/2, 0), Y ¼ (0, b*/2).
Table 4 Main parameters of the calculated band structure for d-(EDT-TTF-CONMe2)2Br at room temperature (RT) and 150 K, and d-(EDT-TTF-CONMe2)2AsF6 at 100 K
Wc, meV Wa, meV Wa0, meV
d-(EDT-TTF-CONMe2)2Br at RT 371 84 194d-(EDT-TTF-CONMe2)2Br at 150
K423 66 149
d-(EDT-TTF-CONMe2)2AsF6 at100 K
309 42 103
This journal is ª The Royal Society of Chemistry 2009
Wa and Wa0 in Table 4) despite the fact that the S/S contacts
decrease somewhat (see ESI† Table S1), a reminder of the
importance of the orbital overlap effects which do not always
follow purely metric considerations. Thus, pressure and
thermal contraction seem to have slightly different effects on the
electronic structure although both reinforce the intrastack
interactions.
Let us note the similarity of the electronic band structure of
these salts with that of (TMTTF)2Br,46 even if one must bear in
mind that the stacks in the two salts are slightly different
because of the peculiar criss-cross intrastack overlap of the d-
type salts.47 This leads to the fact that the band dispersion
along the interstack direction goes down slightly in
(TMTTF)2Br but up slightly in the present d-type salts. Indeed,
when comparing the room temperature structures of the two Br
salts, the total dispersion along the stack direction (Wc)
amounts to 0.371 eV in d-(EDT-TTF-CONMe2)2Br, i. e., only
4.8% larger than in (TMTTF)2Br (0.354 eV). The interstack
dispersions are somewhat stronger in d-[EDT-TTF-CON-
Me2]2Br (0.084, 0.194 eV for the upper and lower bands along
G / Z, to be compared with 0.030, 0.082 eV in (TMTTF)2Br).
Thus, the two bromine salts are electronically quite similar but
(TMTTF)2Br has an enhanced 1D character. An important
difference comes from the fact that the present d-type salts do
not exhibit a dimerization gap (D in Fig. 6a) because of the
existence of the glide planes, whereas there is a small but non-
zero band dimerization (0.027 eV) in (TMTTF)2Br. Thus the
present d-type salts can be described as genuine quarter-empty
pseudo-1D systems.
Single crystal 13C NMR spectroscopy and relaxation in (13C-
enriched EDT-TTF-CONMe2)2X (X ¼ AsF6� and Br�)
NMR measurements were carried out at UCLA on samples 13C
spin labeled on the C1 site (Fig. 1–2 and ESI† Scheme S2) for
both AsF6 and Br salts. The objective for the experiments was to
determine whether the materials are in a charge-ordered phase
for T < 300K. Owing to the relatively larger crystals available for
the former, a more detailed set of experiments were made on the
AsF6 compound. However, over the range of temperatures
measured, both materials exhibited spectra and spin–lattice
relaxation consistent with unusually large CO amplitude.
Specifically, the charge ratio for the two sites of the AsF6 salt is
estimated at rB:rA � 9:1, and essentially independent of
temperature over the range 10K < T < 200K. The Br salts are
also in a CO phase over the range 16K < T < 150K, most likely
with a similarly large charge imbalance.
The results reported here were obtained from crystals with
approximate dimensions 1.5 � 0.2 � 0.05 mm3 and 1.5 � 0.1 �0.05 mm3 for the AsF6 and Br salts, respectively. They were
oriented so that the stacking (a) axis is orthogonal to the applied
field. Then, the 13C sites are magnetically equivalent in the
orthorhombic structure when the magnetic field is directed along
b or c; for other orientations, two independent sites should be
observed when all molecular environments are equivalent. Below
we present the results of spectroscopy and relaxation measure-
ments taken on the AsF6 crystal and conclude with a brief
discussion of spectroscopy on the Br crystal.
J. Mater. Chem., 2009, 19, 6980–6994 | 6987

Fig. 8 Shift vs. angle for the four spectral lines. The rotation is about the
a-axis. The results in Fig. 7 were recorded at 4 ¼ �36�.
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
A spectrum recorded at T ¼ 200 K and 4 ¼ �36� relative to
the c-axis is shown in Fig. 7, in which four absorption lines are
resolved and labelled A1, A2, B1, B2. The spectrum provides the
evidence that the AsF6 salt is in the CO phase: the doubling of
the number of resolved peaks indicates that for each of the
two molecular orientations, there are two distinct magnetic
environments.
A rotation pattern, also obtained at T ¼ 200 K, is shown in
Fig. 8, and in which the four distinct absorption features are
labelled as above. The shifts along a were not measured, so the
full shift tensor is not known. However, the variation Kii(4) in
the bc plane is rotated away from the crystal axes, and
approximately determined by the molecular orientations.
For the two distinct molecular environments, we obtain
Kxx(A,B) ¼ 150, �260 ppm and Kyy(A,B) ¼ 50,�400 ppm at
T ¼ 200K, with principal axes rotated relative to the crystal
axes by 4A1,2 ¼ �14(5)�, 4B1,2 ¼ �68(10)�. The results are
consistent with rotation of the principal axes in the bc plane by
approximately 90� for sites A,B. Any additional shifts produced
by the relatively small distortion below the orthorhombic/
monoclinic transition at TOM ¼ 190 K are not resolved in these
experiments.
The variation with temperature of the spectra in environ-
ments A,B as well as the spin–lattice relaxation rate T1�1 vs. T
for sites A,B are discussed separately in a physics paper
reporting on the (P,T) phase diagram.29 The spin–lattice
relaxation rates are shown to differ by about 2 orders of
magnitude (AT1�1 � O(100 BT1
�1) over the entire range of
measured temperatures. The observation of very different rates
is qualitatively similar to that observed in other charge transfer
salts with CO symmetry breaking, such as (TMTTF)2X,20b
Ag(DI-DCNQI)2,19 or a-(BEDT-TTF)2I3.48 Following the
discussions of, for example, the TMTTF materials, we consider
the charge imbalance ratio as
T�1
1 ðBÞT�1
1 ðAÞ¼�
QB
QA
�2
and consequently the charge ratios for sites A,B is roughly 9:1.
Finally, we note that there is no distinction between the two
compounds in the physical properties as measured by NMR at
ambient pressure. For example, consider the spectra for the Br
salt recorded for a sequence of temperatures and shown in
Fig. 9. The distinction between the A, B sites is again clear,
Fig. 7 13C NMR spectrum recorded at T ¼ 200 K and applied field
B ¼ 10.007 T. For arbitrary orientations, there are four non-equivalent
sites (labelled A1, A2, B1, B2; see text).
6988 | J. Mater. Chem., 2009, 19, 6980–6994
even at T ¼ 150 K. Upon cooling, one of the two absorption
lines broadens significantly. In this case, the orientation of the
crystal relative to the magnetic field is sufficiently close to the
crystalline axes that no splitting of either the A or B site is
observed.
In summarizing the results from the NMR, it is useful to
contrast with other compounds exhibiting the CO state, such as
the TMTTF family.20 In this case, the charge imbalance is
significantly larger. Presumably, this is a consequence of the
combination of a large V/t, and strong coupling to the counterion
sublattice. Such a coupling is necessary to explain the details of
the CO order parameter revealed by the X-ray diffraction
measurements described below.
Fig. 9 Temperature evolution of 13C spectra for the Br salt. The CO is
evident already at T ¼ 150 K, and significant line broadening occurs as
the sample is cooled.
This journal is ª The Royal Society of Chemistry 2009

Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
Back to X-ray experiments: charge ordering consistent with
a doubling of b and change of symmetry
13C NMR, IR and Raman30 all provide evidence for charge
ordering already above the orthorhombic-to-monoclinic transi-
tion for both the AsF6 and Br salts. Therefore, a re-investigation
of their crystal structures is in order to try and identify the
Wigner crystal structure. As the structures described above
contain one independent molecule only, allowing for charge
ordering necessitates two non-equivalent molecules, which
implies either a change of symmetry or a doubling of one lattice
parameter. Therefore, an exploration of the reciprocal space of
(EDT-TTF-CONMe2)2Br was conducted using highly sensitive
experimental set ups at Orsay and readily revealed additional
weak Bragg reflections satisfying both of these criteria. A
doubling of the unit cell occurs along b, the direction transverse
to the stacking axis. Fig. 10 shows one example of a scan across
one superstructure reflection using an X-ray diffractometer with
a point detector. Furthermore, a number of additional Bragg
reflections are found to violate the extinction conditions for
several symmetry elements of Pmna, namely n t y, a t z and
21 k z, yet their intensity does not exceed 1& of the intensity of
the main Bragg reflections. Accordingly, an extensive explora-
tion of the reciprocal space using a standard CCD detector in
Angers, with very long X-ray data collection times, only deliv-
ered a very few of these additional reflections which ultimately
proved to be useless for structure refinements in the double
lattice.
In contrast, single crystal synchrotron experiments for the
detection of the superstructure proved very successful. Crisp
superstructure patterns are obtained upon using the SOLEIL
synchrotron source. Fig. 11a represents the (0kl) reciprocal plane
drawn out of the experimental diffraction data. Rows of super-
structure Bragg reflections are located in between the lines of
main strong reflections. Of particular note in Fig. 11b is the (h7l)
plane entirely composed of superstructure weak reflections.
Notice the sharpness of the reflections, with evidence of reflec-
tions at high diffraction angles indicating long range ordering
with low positional shift. These features are seen to be primarily
associated with the ordering of the heavy bromide atoms since
the experiment was done at the bromine absorption edge.
Fig. 10 Scan across (2, �1.5, 3) at room temperature. Note that the net
intensity of this reflection is 3 to 4 orders of magnitude weaker than that
of the neighbouring Bragg reflections qualifying the averaged Pmna
structure.
This journal is ª The Royal Society of Chemistry 2009
In contrast with the former clear case for a doubling of the
lattice parameter along b, the pattern of systematic absences for
the new lattice is rather a complex one, as exemplified in Table 5
where the comprehensive set of synchrotron data intensities is
systematically averaged for all possible symmetry elements.
Space group determination proved to be challenging since there
are quite a few possibilities to choose from on the basis of the
inventory of symmetry elements satisfying the observed system-
atic absence conditions.
First, recall that EDT-TTF-CONMe2 has no symmetry
elements. In the double lattice of the orthorhombic unit cell two
independent molecules, bearing different charges, can exist either
Fig. 11 (a) (0kl) and (b) (h7l) reciprocal planes drawn out of the room
temperature synchrotron data.
J. Mater. Chem., 2009, 19, 6980–6994 | 6989

Table 5 Number of systematic absence exceptionsa in the orthorhombic double unit cell of d-(EDT-TTF-CONMe2)2Br at room temperature
a ¼ 7.1126(5) A b ¼ 13.0250(7) A (double axis) c ¼ 32.759(1) A
b t x 21 k x n t y 21 k y a t z b t z n t z 21 k z
Nb 6678 31 2549 79 1017 1097 828 217N, I > 3s(I) 231 4 60 0 17 24 17 6<I/s(I)> 0.7 1.2 0.6 0.3 0.5 0.5 0.4 0.6Absence
conditions0kl:k ¼ 2n + 1
h00:h ¼ 2n + 1
h0l:h + l ¼ 2n + 1
0k0:k ¼ 2n + 1
hk0:h ¼ 2n + 1
hk0:k ¼ 2n + 1
hk0:h + k ¼ 2n + 1
00l:l ¼ 2n + 1
a Only possible symmetry elements (i.e. with low <I/s(I)> ratio) are listed in the table. b N is the number of reflections satisfying the absence conditions.
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
in a non-centrosymmetric space group where they occupy general
positions or in an m-containing centrosymmetric group if the two
molecules lie on a mirror plane. In order to allow for the possi-
bility of charge ordering within the stacks (and also, if any, of an
intrastack dimerization), one excludes glide plane a t z. Then,
the symmetry elements 21 k x and b t z are not allowed as they
would move the molecules over into improper positions in the
structure. b t x cannot exist since rows of weak superstructural
peaks with odd k indices are found in the 0kl reciprocal plane.
One can also exclude 2 k y since it connects molecules inside the
stack and therefore is not compatible with a charge ordered
stack. Thus, according to the structure pattern and those
systematic absences remaining after deleting the former dis-
carded symmetry elements, one is left with the following
symmetry elements: 2 k x, m t x, 21 k y, n t y, 21 k z, n t z.
These elements are consistent with three possible orthorhombic
space groups that would all ultimately allow for non-uniform
molecular stacks with two independent molecules, that is, Pmnn,
P2nn, P22121.
Fig. 12 Three dimensional pattern of alternating neutral and charged
molecules based on refinements of the room temperature synchrotron
data taking into account weak superstructural reflections in space group
P2nn: (a) along the stacking axis direction a; and (b) for one single layer
viewed along b, the direction transverse to the stacking axis. The geom-
etry of hydrogen contacts is given in Table 2. Anti-phase displacements of
several bromide anions belonging to this layer are symbolized by red
arrows.
Refinement of room temperature synchrotron data favors non-
centrosymmetric group P2nn and reveals the three-dimensional
pattern of charge ordering
Refinements in all of the former three groups, Pmnn, P2nn and
P22121, each consistent with a double lattice with two indepen-
dent molecules of different charge, yield approximately the same
picture for the three-dimensional charge disproportionation
pattern, the essence of the Wigner crystal structure. Yet, refine-
ment in the acentric P2nn group is seen as the best outcome of the
analysis and refinement of the room temperature synchrotron
data, as discussed below.
The primary outcome of the synchrotron data refinements in
the double cell of P2nn symmetry is that the two independent
molecules have different charges (Fig. 12) as demonstrated by the
striking difference between the bond lengths of the inner C]C
double bonds within the TTF cores. Indeed, the calculated
HOMO energy of donor I is 0.100 eV lower than that of donor II,
suggesting a smaller charge for I. Hence, as exemplified in
Fig. 12, molecule I0 with a short inner C]C of 1.354(2) A is
formally neutral. Conversely, it is likely that most of the charge is
localized on radical cation IIc+ whose inner C]C bond is elon-
gated up to 1.392(2) A in agreement with the respective bonding
character of the adjacent inner carbon p orbitals contribution to
the HOMO, and entirely consistent with the former analysis
of the temperature dependence of the single crystal 13C NMR
6990 | J. Mater. Chem., 2009, 19, 6980–6994
spin–lattice relaxation rate T1�1 for the two discrete molecular
sites which suggested a record high charge ratio of 9:1. Therefore,
radical cations IIc+ and neutral molecules I0 alternate along the
stacks, as shown in Fig. 12a.
The Wigner structure keeps uniform stacks
Although P2nn symmetry does allow for any amount of
dimerization, there is only a minute, if any, dimerization along
the stacking direction since the separations between successive
molecular planes, amounting to 3.550(9) and 3.563(8) A, are
This journal is ª The Royal Society of Chemistry 2009
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
identical within experimental accuracy. Nevertheless, the
difference in the CEt–H/O contact distances is significant.
Each pair of molecules within the stack is connected by two
hydrogen contacts, and both of them are shorter in one pair
(interaction A) in comparison with another (interaction B,
Fig. 12a and Table 2).
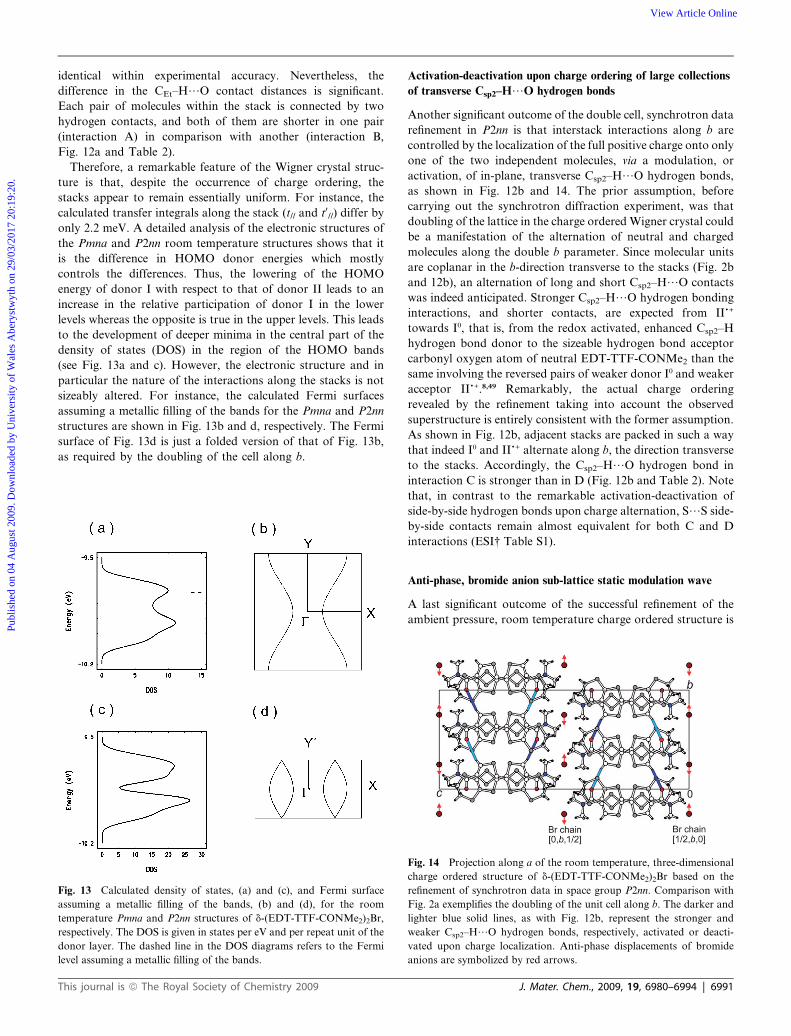
Therefore, a remarkable feature of the Wigner crystal struc-
ture is that, despite the occurrence of charge ordering, the
stacks appear to remain essentially uniform. For instance, the
calculated transfer integrals along the stack (t// and t0//) differ by
only 2.2 meV. A detailed analysis of the electronic structures of
the Pmna and P2nn room temperature structures shows that it
is the difference in HOMO donor energies which mostly
controls the differences. Thus, the lowering of the HOMO
energy of donor I with respect to that of donor II leads to an
increase in the relative participation of donor I in the lower
levels whereas the opposite is true in the upper levels. This leads
to the development of deeper minima in the central part of the
density of states (DOS) in the region of the HOMO bands
(see Fig. 13a and c). However, the electronic structure and in
particular the nature of the interactions along the stacks is not
sizeably altered. For instance, the calculated Fermi surfaces
assuming a metallic filling of the bands for the Pmna and P2nn
structures are shown in Fig. 13b and d, respectively. The Fermi
surface of Fig. 13d is just a folded version of that of Fig. 13b,
as required by the doubling of the cell along b.
Fig. 13 Calculated density of states, (a) and (c), and Fermi surface
assuming a metallic filling of the bands, (b) and (d), for the room
temperature Pmna and P2nn structures of d-(EDT-TTF-CONMe2)2Br,
respectively. The DOS is given in states per eV and per repeat unit of the
donor layer. The dashed line in the DOS diagrams refers to the Fermi
level assuming a metallic filling of the bands.
This journal is ª The Royal Society of Chemistry 2009
Activation-deactivation upon charge ordering of large collections
of transverse Csp2–H/O hydrogen bonds
Another significant outcome of the double cell, synchrotron data
refinement in P2nn is that interstack interactions along b are
controlled by the localization of the full positive charge onto only
one of the two independent molecules, via a modulation, or
activation, of in-plane, transverse Csp2–H/O hydrogen bonds,
as shown in Fig. 12b and 14. The prior assumption, before
carrying out the synchrotron diffraction experiment, was that
doubling of the lattice in the charge ordered Wigner crystal could
be a manifestation of the alternation of neutral and charged
molecules along the double b parameter. Since molecular units
are coplanar in the b-direction transverse to the stacks (Fig. 2b
and 12b), an alternation of long and short Csp2–H/O contacts
was indeed anticipated. Stronger Csp2–H/O hydrogen bonding
interactions, and shorter contacts, are expected from IIc+
towards I0, that is, from the redox activated, enhanced Csp2–H
hydrogen bond donor to the sizeable hydrogen bond acceptor
carbonyl oxygen atom of neutral EDT-TTF-CONMe2 than the
same involving the reversed pairs of weaker donor I0 and weaker
acceptor IIc+.8,49 Remarkably, the actual charge ordering
revealed by the refinement taking into account the observed
superstructure is entirely consistent with the former assumption.
As shown in Fig. 12b, adjacent stacks are packed in such a way
that indeed I0 and IIc+ alternate along b, the direction transverse
to the stacks. Accordingly, the Csp2–H/O hydrogen bond in
interaction C is stronger than in D (Fig. 12b and Table 2). Note
that, in contrast to the remarkable activation-deactivation of
side-by-side hydrogen bonds upon charge alternation, S/S side-
by-side contacts remain almost equivalent for both C and D
interactions (ESI† Table S1).
Anti-phase, bromide anion sub-lattice static modulation wave
A last significant outcome of the successful refinement of the
ambient pressure, room temperature charge ordered structure is
Fig. 14 Projection along a of the room temperature, three-dimensional
charge ordered structure of d-(EDT-TTF-CONMe2)2Br based on the
refinement of synchrotron data in space group P2nn. Comparison with
Fig. 2a exemplifies the doubling of the unit cell along b. The darker and
lighter blue solid lines, as with Fig. 12b, represent the stronger and
weaker Csp2–H/O hydrogen bonds, respectively, activated or deacti-
vated upon charge localization. Anti-phase displacements of bromide
anions are symbolized by red arrows.
J. Mater. Chem., 2009, 19, 6980–6994 | 6991

Scheme 1 Schematics of the full set of four possible arrangements of
neutral (open ovals) and charged (solid black ovals) EDT-TTF-
CONMe2 molecules, and Br anions (pink circles), in the bc plane of
a double lattice, perpendicular to the stacking a-direction. Closed and
dashed symbols refer to positions at a ¼ 0 or 1/2, respectively. The
charge ordered state is associated with an activation of hydrogen bond
interactions between Br� and [EDT-TTF-CONMe2]+c, shown by black
dotted lines, resulting in anti-phase shifts of the Br anions towards the
radical cations as indicated by red arrows. (a) and (b) correspond
to structures where stacks of [EDT-TTF-CONMe2]+c and stacks of
[EDT-TTF-CONMe2]0 would be segregated. In (a), charge-rich and
charge-poor stacks alternate along c, and there is no Br shift due to
symmetrical hydrogen interactions. In (b), with no alternation of charge
along c, the modulation waves are in-phase for all Br� strings. (c) and
(d) correspond to mixed stacks, that is where charges order along both
the stacking a and transverse b directions. There is no bromide atoms
displacement in (c), while in (d) displacement waves for adjacent Br�
Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
that the Br/Br separations along b, which are equivalent in the
averaged Pmna and P21/a structures, are found to differ in P2nn,
amounting to 6.4985(6) and 6.5265(6) A. Strings of Br� anions
are threading along in the bc-planes across pockets bordered by
ethylene groups of differently charged donors. Electrostatic
interactions between I0 and IIc+ and Br ions are not equivalent,
pointing to a modulation of the anion surroundings upon charge
ordering, which leads the bromide anions to shift away from
neutral organic molecules towards the positively charged donors
(Fig. 12b and Table 2 for CEt–H/Br contacts). In agreement
with the pattern of charge alternation in the bc-plane, adjacent
anions along the Br ion strings experience counter displacement
primarily along b in such a way that pairs of charged molecules
become bracketed in between pairs of closer bromide anions. It
should be emphasized here that the concerted anti-phase move-
ment of heavy Br atoms within any such string accounts for most
of the intensity of the 2b superstructure reflections measured with
the synchrotron radiation (Fig. 11). As a result, the anion string
becomes slightly dimerized. Moreover, as exemplified in Fig. 14,
the static longitudinal modulation wave of the bromine anions
string running along [0,b,1/2] is in anti-phase with the similar
wave along [1/2,b,0], in tune with the pattern of charge ordering
within neighboring donor stacks, hence the characteristic 3D
pattern of dimerization of the anion sub-lattice (Fig. 12).
It is of interest to note that the demonstration here of
a synchronized displacement of the cation and anion sites in the
crystal upon charge ordering provides convincing support for
earlier similar models of displacements postulated for the
(TMTTF)2X series which could not be tested by such full
structure determination.27
strings along c are necessarily in anti-phase, as observed in the present
P2nn structure.
Final considerations on symmetry and possible patterns of chargeordering
In order to reach a comprehensive view of the symmetry of the
CO state, we now consider all possible patterns of charge
ordering in the radical cation sub-lattice and assess their
compatibility with relevant atomic displacements in the anion
sub-lattice. Let us remember that, right at the onset of the
choice of space group, it was assumed that the structure in the
double lattice contains charge ordered stacks. Yet, the charge
ordering pattern could be principally different if one allows for
segregated stacks, that is, either charge-rich or charge-poor
(neutral) stacks to be repeated along b. Scheme 1 gives an
overview of the full set of four possible CO patterns with either
segregated stacks (a and b) or mixed stacks (c and d) where
neutral and charged molecules are shown by white and black
ovals, respectively. Quite remarkably, each of the diverse, four
CO topologies (a-d) appears to be associated with divergent
types of transformations of the anion sub-lattices. In fact, in
each case the movement of the anions is seen as being directed
by the activated hydrogen bond interactions between Br� and
charge-rich radical cations represented by dotted lines in
Scheme 1. Accordingly, for segregated stacks, the bromide
anion sub-lattice would eventually be uniform (Scheme 1a), or
one single, anti-phase longitudinal modulation wave for the
bromide strings could prevail and it should be in-phase in all
chains (Scheme 1b). For mixed stacks, there is either no
bromide atoms displacement (Scheme 1c) or anti-phase
6992 | J. Mater. Chem., 2009, 19, 6980–6994
modulation waves in adjacent anion chains (Scheme 1d). In this
X-ray experiment carried out at the Br absorption edge, the Br
atom sub-lattice is better defined than the organic sub-lattice
and thereby captures most of the structural features of the
concerted radical cation-anion ordering. Indeed, refinement of
the structure in an asymmetric P1 double lattice allowing for
unconstrained displacements of the heavy Br ions demonstrates
that only pattern d, which associates mixed stacks and anti-
phase Br modulation, ultimately holds.
Note that pattern d is compatible with two space groups
only, P2nn and P22121, both of which are non-centrosym-
metrical. However, refinement in P22121 indicates that the
differences in bond lengths and intermolecular contact distances
between I0 and IIc+ are quite subtle and, besides, this space
group merely accounts for the full pattern of systematic
absences, given in Table 5, in comparison with P2nn which
describes it all. Note also that centrosymmetric Pmnn, the last
of three previously selected space groups, corresponds to c
where the Br modulation wave cannot be observed. Thus, we
conclude that the acentric space group P2nn, compatible with
a ferroelectric charge ordered state, is the only one to provide
for a full description of the orthorhombic room temperature
charge ordered structure.
Finally, we note in closing that the present report meets with
the challenge outlined in the introductory comments by
This journal is ª The Royal Society of Chemistry 2009

Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
delivering a full structure determination of a Wigner crystal
structure which hereby demonstrates a rather simple 3D pattern
where the charges are found to order on every other molecules
both along the stacking direction a as well as from one stack to
another along b in an orthorhombic unit cell and non-centro-
symmetric space group. This appears to be in sharp contrast with
the complex pattern proposed recently for (DI-DCNQI)2Ag19c
where the charge is localized both on molecular sites and onto
dimeric molecular units, with a complex helico€ıdal pattern in
between, and also for the (TMTTF)2X series whose CO pattern
has been postulated to consist of a mixture of 4kF bond (mixed
valence dimers) and (single molecular) site orders.20,22,25,27
Conclusions
The whole of the foregoing results, which include the synthesis
and successful application of an internal chemical pressure to
effectively control, and reduce, the Mott gap in d-(EDT-TTF-
CONMe2)2Br; the detailed accounts of its Pmna, averaged
room temperature structure as well as reversible phase transi-
tion at ca. 190 K, accompanied by twinning; the full description
of its averaged low temperature P21/a structure; the synthesis of
(13C-EDT-TTF-CONMe2)2[Br and AsF6], where one carbon
atom of the inner double bond is 100% 13C-enriched, allowing
high resolution 13C solid state NMR experiments to be con-
ducted, as well as IR reflectivity and Raman experiments
reported independently, which both reveal that charge ordering
occurs already at room temperature and ambient pressure; the
discovery of weak superstructure Bragg reflections in d-(EDT-
TTF-CONMe2)2Br and subsequent analysis and refinement of
an exhaustive synchrotron radiation data set; suggests an
alternation at room temperature of neutral and oxidized
molecules along both the stacking a and transverse b directions
in orthorhombic, non-centrosymmetric space group P2nn, and
demonstrates a rather simple 3D CO pattern compatible with
a ferroelectric charge ordered state. The charge disproportion-
ation, and subsequent long range order crystallization of the
electron gas within the three-dimensional Wigner lattice is
found to be coupled to a concerted activation-deactivation of
large collections of transverse Csp2–H/O hydrogen bonds
coupled to an anti-phase, static modulation of the bromide
anions displacements along b, thereby demonstrating a cooper-
ative participation of the anion and cation sub-lattices to the
stabilization of the CO state. In addition to (DI-DCNQI)2Ag
discussed earlier, where superstructure Bragg reflections
condensate from diffuse scattering at the CO transition, only
a few CO structures of molecular systems have been investi-
gated using synchrotron radiation. The analysis of powder
synchrotron data for (EDO-TTF)2PF6 suggested a doubling of
the unit cell upon CO.50 Single crystal synchrotron data were
used for the investigation of the P1 CO structure51 of a-(BEDT-
TTF)2I3 where no superstructure reflections are identified, in
contrast to q-(BEDT-TTF)2RbZn(SCN)4 where a doubling of
the unit cell was found52 upon CO transition at high pressure.
Note in closing that a remarkable feature of the Wigner crystal
structure is that the stacks appear to remain essentially uniform
in the charge ordered state in agreement with the rich low
temperature Mott physics of the system.29
This journal is ª The Royal Society of Chemistry 2009
Acknowledgements
This work was supported by the French National Research
Agency, ANR Project CHIRASYM 2005-08 (NT05-2 42710),
the Interdisciplinary ANR project 3⁄4 -Filled 2009-2011 (ANR-
08-BLAN-0140-01); the CNRS; the INTAS Grant 04-03-4001;
the Spanish Ministerio de Educaci�on y Ciencia (Project FIS2006-
12117-C04-01) and the Generalitat de Catalunya (2005 SGR
683); and NSF grants DMR-0520552 and DMR-0804625. We
thank Jean-Charles Ricquier for expert assistance in preparing
the graphical abstract illustration. We thank the CNRS for an
associated researcher position (LZ) and the R�egion des Pays de
Loire for a post-doctoral fellowship (SS).
References
1 N. F. Mott, Metal-insulator transitions, Taylor and Francis, London/Philadelphia, 1990.
2 T. Giamarchi, Chem. Rev., 2004, 104, 5037.3 H. Seo, C. Hotta and H. Fukuyama, Chem. Rev., 2004, 104, 5005.4 P. P. Edwards, H. B. Gray, M. T. J. Lodge and R. J. P. Williams,
Angew. Chem. Int. Ed., 2008, 47, 6758.5 See Fig. 9 of reference 2.6 (a) P. Fulde, Electron Correlations in Molecules and Solids, 3rd
edition, Springer-Verlag; Berlin, 2002; (b) N. Grewe, F. Steglich, inHandbook on the Physics and Chemistry of Rare Earths, Volume 14,K. A. Gscheidner Jr. and L. Eyring, Eds., Elsevier; Amsterdam -New York, 1991, p. 343; (c) M. Imada, A. Fujimori and Y. Takura,Rev. Mod. Phys., 1998, 63, 1; (d) J. P. Attfield, Solid State Sci.,2006, 8, 861–867; (e) J. P. Wright, J. P. Attfield and P. G. Radaelli,Phys. Rev. Lett., 2001, 87, 266401.
7 (a) P. Batail, Special Issue on Molecular Conductors, Chem. Rev.,2004, 11, p104; (b) Special Topic: Organic conductors, J. Phys. Soc.Jpn, 2006, 5, 75.
8 M. Fourmigu�e and P. Batail, Chem. Rev., 2004, 104, 5379.9 (a) D. J�erome, Chem. Rev., 2004, 104, 5565; (b) L. Degiorgi and
D. J�erome, J. Phys. Soc. Jpn, 2006, 75, 051004.10 S. Kagoshima and R. Kondo, Chem. Rev., 2004, 104, 5593.11 R. Rousseau, M. Gener and E. Canadell, Adv. Funct. Mater., 2004,
14, 201.12 J. P. Pouget, S. K. Khanna, F. Denoyer, R. Com�es, A. F. Garito and
A. J. Heeger, Phys. Rev. Lett., 1976, 37, 437.13 In reciprocal space, 4kF corresponds to the first Fourier component of
a 1D Wigner lattice of localized charges in direct space (in reciprocalunits, 4kF amounts to the inverse average charge separation), see:(a) J. Kondo and K. Yamaji, J. Phys. Soc. Jpn, 1977, 43, 424;(b) J. Hubbard, Phys. Rev. B, 1978, 17, 494.
14 A. J. Epstein, J. S. Miller, J. P. Pouget and R. Com�es, Phys. Rev. Lett.,1981, 47, 741.
15 F. Mila and X. Zotos, Europhys. Lett., 1993, 24, 133.16 H. Seo, J. Merino, H. Yoshioka and M. Ogata, J. Phys. Soc. Jpn,
2006, 75, 051009.17 M. Dressel and N. Drichko, Chem. Rev., 2004, 104, 5689.18 S. Huizinga, J. Kommandeur, A. Sawatzky, B. T. Thole, K. Kopinga,
W. J. M. de Jonge and J. Joos, Phys. Rev. B, 1979, 19, 4723.19 (a) K. Hiraki and K. Kanoda, Phys. Rev. Lett., 1998, 80, 4737;
(b) K. Miyagawa, K. Kanoda and A. Kawamoto, Chem. Rev.,2004, 104, 5635; (c) T. Kakiuchi, Y. Wakabayashi, H. Sawa,T. Itou and K. Kanoda, Phys. Rev. Lett., 2007, 98, 066402.
20 (a) D. S. Chow, E. Zamborszky, B. Alavi, D. J. Tantillo, A. Baur,C. A. Merlic and S. E. Brown, Phys. Rev. Lett., 2000, 85, 1698;(b) F. Zamborzky, W. Yu, W. Rass, S. E. Brown, B. Alavi,C. A. Merlic and A. Baur, Phys. Rev. B, 2002, 66, 081103.
21 Y. Nogami, R. Moret, J. P. Pouget, Y. Yamamoto, K. Oshima,K. Hiraki and K. Kanoda, Synth. Met., 1999, 102, 1778.
22 P. Monceau, F. Ya Nad and S. Brazovskii, Phys. Rev. Lett., 2001, 86,4080.
23 R. T. Clay, S. Mazumdar and D. K. Campbell, Phys. Rev. B, 2003, 67,115121.
24 M. Kuwabara, H. Seo and M. Ogata, J. Phys. Soc. Jpn., 2003, 72, 225.25 J. Riera and D. Poilblanc, Phys. Rev. B, 2001, 63, 241102(R).
J. Mater. Chem., 2009, 19, 6980–6994 | 6993

Publ
ishe
d on
04
Aug
ust 2
009.
Dow
nloa
ded
by U
nive
rsity
of
Wal
es A
bery
stw
yth
on 2
9/03
/201
7 20
:19:
20.
View Article Online
26 W. Yu, F. Zhang, F. Zamborszky, B. Alavi, A. Baur, C. A. Merlic andS. E. Brown, Phys. Rev. B, 2004, 70, 121101.
27 M. de Souza, P. Foury-Leylekian, A. Moradpour, J. P. Pouget andM. Lang, Phys. Rev. Lett., 2008, 101, 216403.
28 K. Heuz�e, M. Fourmigu�e, P. Batail, C. Coulon, R. Cl�erac,E. Canadell, P. Auban-Senzier, S. Ravy and D. J�erome, Adv.Mater., 2003, 15, 1251.
29 P. Auban-Senzier, C. Pasquier, D. J�erome, S. Suh, S. E. Brown,C. M�ezi�ere and P. Batail, Phys. Rev. Lett., 2009, 102, 257001.
30 T. Yamamoto, C. M�ezi�ere, P. Batail, unpublished work.31 K. Heuz�e, M. Fourmigu�e and P. Batail, J. Mater. Chem., 1999, 9, 2373.32 J. Larsen and C. Lenoir, Synthesis, 1989, 2, 134.33 A. J. M. Duisenberg, J. Appl. Cryst., 1992, 25, 92.34 A. J. M. Duisenberg, L. Kroon-Batenburg and A. M. M. Schreurs,
J. Appl. Cryst., 2003, 36, 220.35 L. J. Farrugia, J. Appl. Cryst., 1999, 32, 837.36 G. M. Sheldrick, SHELXS-97 and SHELXL-97, University of
G€ottingen, Germany, 1997.37 R. Herbst-Irmer and G. M. Sheldrick, Acta Cryst., 1998, B54, 443.38 Oxford Diffraction (2007). Oxford Diffraction Ltd., Xcalibur CCD
system, CrysAlisPro Software system, Version 1.171.32.39 M.-H. Whangbo and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 6093.40 J. Ammeter, H.-B. B€urgi, J. Thibeault and R. Hoffmann, J. Am.
Chem. Soc., 1978, 100, 3686.41 B. N�afr�adi, R. G�aal, A. Olariu, L. Forr�o, C. M�ezi�ere, P. Batail,
A. J�anossy, unpublished work.
6994 | J. Mater. Chem., 2009, 19, 6980–6994
42 B. K. Vainstein, V. M. Fridkin, V. L. Indenbom, ModernCrystallography, 2nd ed., Springer-Verlag; Berlin – Heidelberg –New York, 1995, V. 2.
43 S. Parsons, Acta Cryst., 2003, D59, 1995.44 Cambridge Structural Database. Version 5.26. University of
Cambridge, UK.45 I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae,
P. McCabe, J. Pearson and R. Taylor, Acta Cryst., 2002, B58, 389.46 (a) L. Balicas, K. Behnia, W. Kang, E. Canadell, P. Auban-Senzier,
D. J�erome, M. Ribault and J. M. Fabre, J. Phys. I, 1994, 4, 1539; (b)for an earlier band structure, although not directly comparable tothe present results because of the different computational details, see:L. Ducasse, A. Abderraba and B. Gallois, J. Phys. C, 1986, 19, 3805.
47 T. Mori, Bull. Chem. Soc. Jpn., 1999, 72, 2011.48 (a) Y. Takano, K. Hiraki, H. M. Yamamoto, T. Nakamura and
T. Takahashi, Synth. Met., 2001, 120(1–3), 1081; (b) S. Moroto,K. Hiraki, Y. Takano, Y. Kubo, T. Takahashi, H. M. Yamamotoand T. Nakamura, J. Phys. IV, 2004, 114, 339.
49 S. A. Baudron, P. Batail, C. Rovira, E. Canadell and R. Cl�erac, Chem.Commun., 2003, 1820.
50 S. Aoyagi, K. Kato, A. Ota, H. Yamochi, G. Saito, H. Suematsu,M. Sakata and M. Takata, Angew. Chem. Int. Ed., 2004, 43, 3670.
51 T. Kakiuchi, Y. Wakabayashi, H. Sawa, T. Takahashi andT. Nakamura, J. Phys. Soc. Jpn., 2007, 76, 113702.
52 M. Watanabe, Y. Noda, Y. Nogami and H. Mori, J. Phys. Soc. Jpn.,2004, 73, 921.
This journal is ª The Royal Society of Chemistry 2009
Top Related