







Languages
Pages
Legal


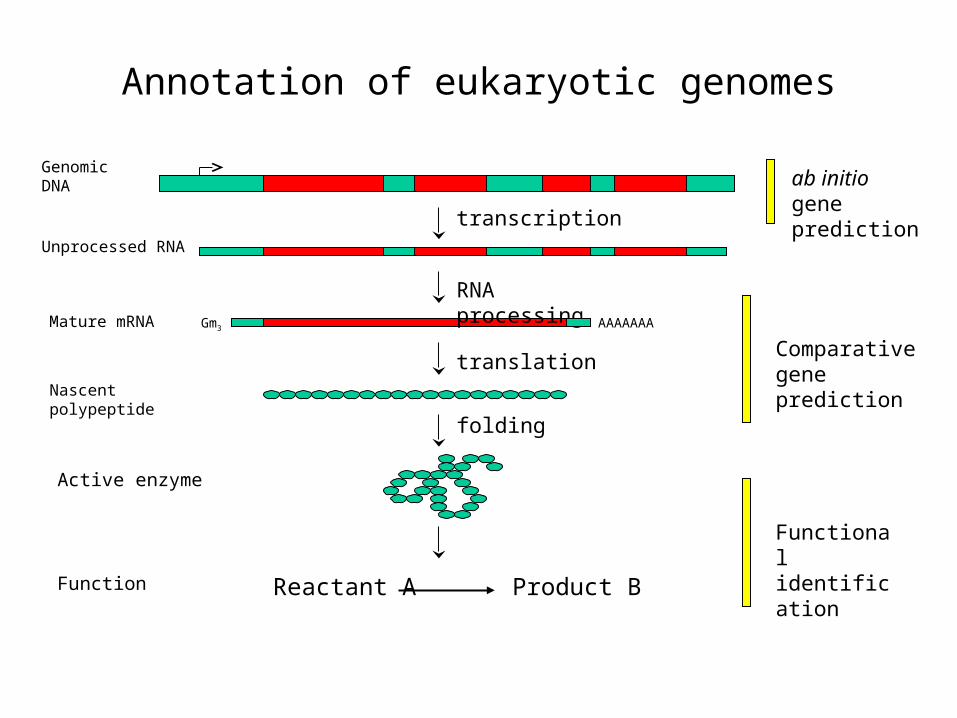
Annotation of eukaryotic genomes
transcription
RNA processing
translation
AAAAAAA
Genomic DNA
Unprocessed RNA
Mature mRNA
Nascent polypeptide
folding
Reactant A Product BFunction
Active enzyme
ab initio gene prediction
Comparative gene prediction
Functional identification
Gm3

Genome analysis overview: C.elegans

Gene finding: ab initio• What features of a ORF can we use?
• Size - large open reading frames
• DNA composition - codon usage / 3rd position codon bias
• Other features:
• Kozak sequence CCGCCAUGG
• Ribosome binding sites
• Termination signal (stops)
• Splice junction boundaries

Gene finding: comparative
• Use knowledge of known coding sequences to identify region of genomic DNA by similarity
• transcribed DNA sequence
• peptide sequence
• related genomic sequence
Annotation of eukaryotic genomes
transcription
RNA processing
translation
AAAAAAA
Genomic DNA
Unprocessed RNA
Mature mRNA
Nascent polypeptide
folding
Reactant A Product BFunction
Active enzyme
ab initio gene prediction
Comparative gene prediction
Functional identification
Gm3


Artemis display for S.pombe cosmid



Methods for searching• Pairwise alignments: matching a query sequence against a database of subject sequences
• Needleman & Wunsch - global alignment
• Smith-Waterman - local alignment
• FastA
• BLAST
• Others: SSAHA, WABA
• see Chapter 7 Developing Bionformatics Computer Skills

BLAST - local similarity searches• BLAST (Basic Local Alignment Search Tool) is the workhorse of genome annotation due to it’s early optimisation for the UNIX platform
• Underlies most of the web-based servers world-wide
• Comes in many flavours:• BLASTN - DNA against DNA
• BLASTX - DNA against Protein
• BLASTP - Protein against Protein
• TBLASTN - Protein against DNA
• TBLASTX - DNA against DNA at the peptide level

BLAST - results• BLAST returns high-scoring pairs (HSPs) with a score and p-value. Blast output files can be large and difficult to interpret.
• Hence we need tools to make sense of the data - both to filter/process the file and to visualise the resulting multiple sequence alignments.
• MSPcrunch - a post-processor for BLAST with a number of different output types.
• BioPerl - modules for handling sequences and BLAST output

Standard similarity searches for first-pass annotation
• genomic DNA v transcript data
• BLASTN / EST_GENOME
• TBLASTX
• genomic DNA v genomic DNA
• BLASTN
• TBLASTX
• genomic DNA v non-redundant protein data
• BLASTX

Data for gene prediction
• EST/mRNA - intra-species matches
• TBLASTX - inter-species matches
• BLASTX - intra-species matches
• BLASTX - inter-species matches
• Coding measures - genefinder, hexamer
• Splice sites - consensus sequences


Multiple Sequence alignments in ACEDB

Manual review of gene predictions
• Check concordance with transcript data
• Check concordance with peptide similarity data
• Check splice site usage (intron / exon boundaries)
• Set of human appraised gene predictions. The translations of the CDS sequences are used for protein feature analysis and initial assignment (ID, function)

Top Related