







Languages
Pages
Legal


19/12/2018 1
Aline DECHANET – Chef de Projets - URC PNVS

2
IMPLIQUANT LA PERSONNE HUMAINE (RIPH) N’IMPLIQUANT PAS LA
PERSONNE HUMAINE
LOI NATIONALE INFORMATIQUE ET LIBERTE (CNIL)
LOI JARDE CODIFIEE / CODE DE LA SANTE PUBLIQUE
RECHERCHE DITE « HORS
CHAMP » (n’entrant pas dans le
champ de la loi Jardé)
CATEGORIE 1 CATEGORIE 2 CATEGORIE 3 CATEGORIE 4
INTERVENTIONNELLE
INTERVENTIONNELLE A
RISQUES ET CONTRAINTES
MINIMES (RIRCM)
� liste des interventions
relevant de cette catégorie
fixée par arrêté
NON INTERVENTIONNELLE
� aucun risque ; aucune
contrainte
� observationnelle
réutilisation 2aire
de données
(et/ou collections) déjà acquises
ou d’un registre agréé, ou de
dossiers médicaux sans que de
nouvelles infos soient collectées
auprès des participants
� pas de nécessité de revenir au
participant
INFORMATION
INDIVIDUELLE
spécifique au projet
INFORMATION
INDIVIDUELLE
spécifique au projet
INFORMATION INDIVIDUELLE
OU COLLECTIVE
spécifique au projet
INFORMATION INDIVIDUELLE OU
COLLECTIVE
CONSENTEMENT ECRIT CONSENTEMENT ECRIT OU
EXPRES NON OPPOSITION* NON OPPOSITION*
AVIS FAVORABLE CPP AVIS FAVORABLE CPP AVIS FAVORABLE CPP AVIS ETHIQUE POUR
PUBLICATION (IRB)
ASSURANCE ASSURANCE
AUTORISATION ANSM INFORMATION ANSM INFORMATION ANSM
MR-001
OU Autorisation CNIL
MR-001
OU autorisation CNIL MR-003
OU autorisation CNIL MR-004
OU CEREES + Autorisation CNIL
MR / recherche « interne » : Inscription sur le registre interne du DPO (Data
Protection Officer)
MR / recherche « interne » :
Registre interne DPO
MR004 : Répertoire public INDS

Constitution des dossiers pour soumission
aux autorités
19/12/2018 3
� Documents pour le dépôt des dossiers ANSM / CPP
Type de recherche Documents
Recherches
Interventionnelles (RI)
Formulaires pour l’ANSM
et le CPP disponibles sur le
site de l’ANSM
Recherches
Interventionnelles à
Risques et Contraintes
Minimes (RIRCM)
Formulaire pour le CPP :
même formulaire que
pour les recherches
interventionnelles,
disponibles sur le site de
l’ANSM
Recherches non
interventionnelles (RNI)
Contenu défini dans
l’arrêté du 9/12/2016

Formulaires CPP / ANSM
RIPH
RI RIRCM
Médicaments - Courrier de demande d’autorisation
initiale ANSM
- Formulaire de demande d’autorisation
initiale ANSM /CPP
- Courrier de demande d’autorisation de
modification substantielle ANSM
- Formulaire de demande de modification
substantielle ANSM / CPP
NA
19/12/2018 4
https://www.ansm.sante.fr/Activites/Medicaments-et-produits-biologiques/Avis-aux-
promoteurs-Formulaires/(offset)/3

Formulaires CPP / ANSM RIPH
RI RIRCM
Dispositifs
médicaux
- Courrier de demande d’autorisation
initiale ANSM
- Formulaire de demande
d’autorisation initiale ANSM / CPP
- Courrier de demande d’autorisation
de modification substantielle ANSM
- Formulaire de demande de
modification substantielle ANSM/ CPP
- CPP : Même formulaire de
demande d’autorisation
initiale que pour les
recherches interventionnelles
- CPP : Même formulaire de
demande de modification
substantielle que pour les
recherches interventionnelles.
19/12/2018 5

Formulaires CPP / ANSM RIPH
RI RIRCM
Hors produits
de santé
- Courrier de demande d’autorisation
ANSM
- Formulaire de demande
d’autorisation ANSM / CPP
- Courrier de demande d’autorisation
de modification substantielle
ANSM/CPP (pas de formulaire)
- CPP : Même formulaire de
demande d’autorisation
initiale que pour les
recherches interventionnelles
- CPP : Même courrier que pour
les recherches interventionnelles
(pas de formulaire)
19/12/2018 6

ANSM: Agence Nationale de Sécurité du
Médicament
� Autorité compétente en charge d’autoriser, surveiller et inspecter les
RIPH mentionnées au 1° de l’article L. 1121-1 du CSP et dispose d’un
pouvoir de police sanitaire, notamment en cas de risque pour la
santé publique.
� Pour autoriser un essai clinique, l’ANSM se prononce au regard de la
sécurité des personnes qui se prêtent à l’essai en considérant
notamment la sécurité et la qualité des produits utilisés au cours de
la recherche, leurs conditions d’utilisation, et la sécurité des
personnes au regard des actes pratiqués, des méthodes utilisées et
des modalités prévues pour le suivi de ces personnes.
19/12/2018 7

Document de référence ANSM : Avis aux promoteurs
� Ce document s’adresse aux promoteurs d’essais cliniques, aux demandeurs
d’autorisation d’essai clinique et autres sociétés prestataires de service
mandatées par ces promoteurs, ainsi qu’à toute personne ou organisme
susceptible d’être concerné par ce dispositif.
� Objectif de faciliter la lecture du dispositif législatif et réglementaire applicable à
ces recherches en France et d’apporter des informations pratiques en termes de
procédure, format et contenu des dossiers relatifs aux essais cliniques de
médicaments transmis à l’ANSM.
� 2 tomes :
� Tome I :
� La demande d’autorisation d’essai clinique à l’ANSM,
� La déclaration du début de l’essai,
� Les modifications de l’essai,
� La fin d’essai
� Tome II : Vigilance des essais cliniques de médicament.19/12/2018
8

19/12/20189

19/12/2018 10

Demande d’autorisation d’essai clinique de
médicament auprès de l’ANSM
� Préalablement à la mise en place d’un essai clinique de médicament, le
demandeur doit effectuer les étapes suivantes :
� Obtenir un numéro d’identification de l’essai correspondant au numéro
d’enregistrement de l’essai dans la base européenne des essais cliniques de
médicaments (EudraCT) : https://eudract.ema.europa.eu
� Transmettre un dossier de demande d’autorisation d’essai clinique de médicament
(AEC) à l’ANSM
� Transmettre un dossier de demande d'avis au CPP
� Avant la demande d’avis au CPP
� En même temps que la demande d’avis au CPP
� Après avoir déposé la demande d’avis au CPP ou après avoir obtenu l’avis du CPP
L’essai ne peut débuter qu’après obtention à la fois de l’autorisation d’essai
accordée par l’ANSM et de l’avis favorable rendu par le CPP.
19/12/2018 11

EudraCT� Le numéro EudraCT
� Numéro unique attribué à chaque essai.
� Identifiant unique des essais cliniques ayant au moins un site dans la
Communauté
� Doit être inclus dans toutes les demandes d’autorisation d’essais cliniques dans
la Communauté, et, en tant que de besoin, dans les autres documents relatifs
aux essais (ex : déclarations d’effets graves inattendus ou SUSARS).
� Base de données des essais cliniques EudraCT
� Base de données de tous les essais cliniques interventionnels portant sur des
médicaments dans la Communauté européenne, soumis aux Comités de
protection des personnes (en France : CPP) et à l’autorité compétente (ANSM) à
partir du 1er mai 2004.
Elle a été établie conformément à la Directive 2001/20/CE.
� Portail donne accès aux promoteurs à l’application et la documentation qui s’y
rapporte et permet aussi :
� d’obtenir un numéro EudraCT
� de compléter, sauvegarder et imprimer une version papier et/ou électronique du
formulaire de demande d’autorisation d’essai clinique19/12/2018 12

19/12/2018 13
Documents RIPH
� Pièce requise pour tout type de dossier
� Pièce requise pour certains types de dossiers
Courrier de demande
d’autorisation d’essai clinique
(AEC)
� Modèle de l’ANSM recommandé
Formulaire de demande
d’AEC
� Formulaire de demande d’autorisation d’essai clinique issu de
l’application EudraCT (FAEC) Format PDF texte ou Word
� FAEC issu de l’application EudraCT « full data set » Format XML
Protocole de l’essai clinique � Protocole
�
Fr
Résumé du protocole
� Avis d’une association de patients
� Informations complémentaires pour les essais de phase précoce
telles que demandées à l’annexe 6 si non décrites dans le protocole
� Charte du comité de surveillance indépendant (DSMB) si applicable
Brochure pour l’investigateur
actualisée (ou document qui
la remplace)
� Brochure pour l’investigateur (BI) et/ou résumé des caractéristiques
du produit (RCP) [médicament(s) expérimenté(s) et médicament(s)
de référence]
� Information de référence sur la sécurité (IRS) permettant de
déterminer le caractère attendu /inattendu d’un effet indésirable

19/12/2018 14
Documents RIPH
� Pièce requise pour tout type de dossier
� Pièce requise pour certains types de dossiers
Dossier du médicament
expérimental (DME)
� DME complet ou simplifié
� Copie autorisation d’ouverture de l’établissement de fabrication du
ME
� Copie des autorisations des PUI de réalisation de préparations
rendues nécessaires par les recherches impliquant la personne
humaine ou de préparation de médicaments radiopharmaceutiques
� Copie autorisation d’ouverture de l’établissement importateur du ME
� Certificat établi par la personne qualifiée dans l’UE que la fabrication
est conforme à des BPF aux moins équivalentes aux BPF en vigueur
dans l’UE
� Attestation de conformité aux BPL des études non cliniques
mentionnées dans le DME
� Attestation de conformité aux BPC des études cliniques mentionnées
dans le DME
� Certificat d’analyse du ME
� Autorisation du tiers propriétaire des données relatives au ME
Dossier du médicament
auxiliaire (DMA)
� DMA complet ou simplifié

19/12/2018 15
Documents RIPH
� Pièce requise pour tout type de dossier
� Pièce requise pour certains types de dossiers
Autres documents � Copie de l’avis CPP dès que disponible
� Copie de l’avis scientifique rendu par un Etat membre ou l’EMA ou de
son résumé, si disponible
� Copie décision d’approbation du PIP initial (J120) ou en cours
d’évaluation « opinion PIP », du rapport d’évaluation « summary
report PIP » par l’EMA et le cas échant, copie des décisions
d’approbations des modifications du PIP et des rapports d’évaluation
de celles-ci, si l’essai s’inscrit dans un plan d’investigation pédiatrique
(PIP)
�
Fr
Contenu de l’étiquetage des ME
� Dossier technique relatif à tout autre produit qu’un médicament
utilisé au cours de l’essai
�
Fr
Formulaire de demande d’attestation en vue d’une importation de
médicaments nécessaires à la réalisation de la recherche

Courrier de demande d’autorisation d’essai clinique
(AEC) � Il doit obligatoirement être signé préciser les informations suivantes :
� en objet du courrier :
� le titre de l’essai , le numéro de protocole attribué par le promoteur , le numéro EudraCT
� les particularités de l’essai
� les caractéristiques spécifiques des personnes susceptibles de participer à l’essai
� s’il s’agit d’une première administration à l’Homme d’une nouvelle substance active
� s’il existe des avis scientifiques sur l’essai ou sur le médicament expérimental (ME)
� si l’essai s’inscrit ou est destiné à s’inscrire dans un plan d’investigation pédiatrique
(PIP)
� si le ME ou le médicament auxiliaire est classé comme stupéfiant ou substance
psychotrope
� la localisation des informations pertinentes dans le dossier de demande d’AEC
� la localisation des informations de référence sur la sécurité (IRS) dans le dossier de
demande d’AEC, permettant d’évaluer le caractère attendu ou inattendu de tout
effet indésirable survenant au cours de l’essai
19/12/2018 16

Formulaire de demande d’autorisation d’essai clinique
(FAEC) � Le formulaire de demande d'AEC (FAEC) doit être saisi à partir du site Internet
de la base EudraCT (base européenne des essais cliniques)
� Le FAEC au format PDF texte doit être signé. La signature du demandeur
confirme que le promoteur s’est assuré que :
� les informations fournies sont complètes,
� les documents joints au dossier sont fidèles aux informations disponibles,
� l’essai clinique sera conduit conformément au protocole,
� l’essai clinique sera conduit et les suspicions d’effets indésirables graves et
inattendus et autres informations relatives aux résultats seront communiquées
conformément à la législation applicable.
19/12/2018 17

Protocole et résumé du protocole � Protocole daté (version) et signé
� Résumé :
� Rédigé en français tel que prévu lors de la soumission de la demande d’avis auprès
du CPP concerné.
� Doit comprendre au moins les informations suivantes :
� titre de la recherche,
� nom du/des médicaments étudié(s) ainsi qu’utilisé(s) comme comparateur(s),
� rationnel de la recherche,
� objectifs,
� critères d’évaluation,
� critères de sélection des patients ou des sujets,
� type d’étude (randomisée ou non par exemple),
� nombres de sujets à inclure,
� déroulement de la recherche.
19/12/2018 18

La brochure pour l’investigateur (BI) � Document qui rassemble l’ensemble de données non cliniques et, le cas
échéant, cliniques concernant le médicament expérimental et qui sont
pertinentes pour l’essai clinique concerné par la demande.
� Objectif est de fournir aux investigateurs et autres personnes impliquées dans
la conduite de l’essai, les informations qui leur permettront de comprendre
plus facilement la raison d’être des principales caractéristiques du protocole et
de mieux s’y conformer, par exemple la dose, la fréquence ou l’intervalle
d’administration, le mode d’administration et les procédures de surveillance
de la sécurité.
� Si le médicament dispose d’une autorisation de mise sur le marché (AMM), la BI
peut être remplacée par un autre document qui peut
� le résumé des caractéristiques du produit (RCP) du ME, si ce médicament est utilisé
conformément à l’AMM
� le RCP du ME complété par un résumé des données cliniques et/ou non cliniques
pertinentes étayant l’utilisation du ME dans l’essai lorsque celui-ci est utilisé dans des
conditions différentes de celles de l’AMM.
19/12/2018 19

Dossier du médicament expérimental (DME) � Le DME donne des informations sur la qualité du ME, sa fabrication, ses contrôles
ainsi que des données issues d’études non cliniques et de l’utilisation clinique du
ME.
� PARTIE 1 DONNEES RELATIVES A LA QUALITE PHARMACEUTIQUE, CHIMIQUE ET BIOLOGIQUE
� PARTIE 2 DONNEES NON CLINIQUES
� PARTIE 2 DONNEES CLINIQUES
� Un DME doit être versé pour chaque ME qu’il s’agisse :
� du médicament expérimenté (testé)
� du médicament de référence (comparateur)
� DME simplifié si
� Le ME dispose d’une AMM dans :
� un Etat membre (EM), un pays ICH , une ATUc
� Une autre forme pharmaceutique ou autre dosage du ME dispose d’une AMM dans un EM
ou pays ICH tt le ME est fourni par le titulaire de l’AMM u placebo
� Le ME a été l’objet d’un précédent dossier d’AEC (ou AMS) autorisé par l'ANSM [2] ET le ME
n’est pas modifié19/12/2018
20

Dépôt du dossier � Uniquement par voie électronique sur une boite mail dédiée : aec-
� L’objet de l’e-mail devra comprendre les mentions suivantes : « Type de
soumission (cf. tableau 1)/ Type d’essai (cf. tableau 2) / N° Phase d’essai
/ N° EudraCT / Domaine thérapeutique »
� Envoi de la réponse du promoteur => boite mail dédiée : aec-
� L’objet de l’e-mail devra comprendre les mentions suivantes: « Type de
réponse (cf. tableau 3) » / référence de la demande à l’ANSM / N°
EudraCT »
19/12/2018 21

19/12/2018 22

19/12/2018 23
Etapes Recherche Interventionnelle
Dossier de demande Le promoteur envoi le dossier à l’ANSM
Notification
(recevabilité)
A réception du dossier complet : notification par l’ANSM de la
date de réception du dossier + date d’autorisation en l’absence
d’autorisation expresse
Si dossier incomplet : Liste des documents envoyée par l’ANSM
délai fixé par l’ANSM pour l’envoi des documents
Evaluation 60 jours
Demande
d’informations
complémentaires
Demande possible – délai fixé par l’ANSM pour l’envoi des
réponses
Envoi par l’ANSM au CPP pour information
Notification
(autorisation)
60j maximum même si questions formulées par l’ANSM
Si pas de retour = Autorisation
Envoi de l’autorisation par le Promoteur au CPP
Délai de 2 ans pour démarrer la recherche sinon autorisation
caduque. Possibilité de prorogation sur justificatif
Délai de traitement du dossier

CPP
� Les Comités de Protection des Personnes (CPP) :
� Sont charger d’émettre un avis préalable sur les conditions de validité de toute
recherche impliquant la personne humaine, au regard des critères définis par l’article
L 1123-7 du Code de la Santé Publique (CSP).
� Sont visés par les articles L 1121-1 à L 1126-11 du Code de la Santé Publique.
� Se prononcent sur les conditions dans lesquelles le promoteur de la recherche
assure la protection des personnes et notamment des participants, sur le bien-fondé
et la pertinence du projet de recherche et sur sa qualité méthodologique.
� 39 CPP répartis sur 7 interrégions
19/12/2018 24

CPP� Sont agréés par le Ministre chargé de la santé pour une durée de 6 ans et ont
une compétence régionale. Les membres sont nommés par le directeur général
de l’Agence Régionale de Santé pour une période de 3 ans renouvelable, ils
exercent leurs fonctions bénévolement. Leur composition pluridisciplinaire
comprend 14 membres titulaires et 14 suppléants répartis sur deux collèges.
� Les membres sont soumis à l’obligation de déclaration publique d’intérêts (article
L 1123-3 du CSP) mentionnant leurs liens directs ou indirects avec les promoteurs
ou investisseurs de recherches. Ces déclarations d’intérêts visent à s’assurer de
l’impartialité des membres appelés à se prononce
19/12/2018 25

Composition d’un CPP� Les CPP sont composés de deux collèges (article R.1123-4 du code de la santé publique)
� Premier collège : Personnes ayant une qualification et une expérience approfondie
en matière de recherche biomédicale (médecins notamment pédiatres, psychiatres)
et des personnes qualifiées en biostatistique ou épidémiologie :
� des médecins généralistes ;
� des pharmaciens hospitaliers ;
� des infirmier(e)s.
� Deuxième collège : personnes qualifiées en matière éthique ;
� des psychologues ;
� des travailleurs sociaux ;
personnes qualifiées en matière juridique ;
représentants d’associations agréées de malades et d’usagers du
système de santé.
� Chaque CPP désigne son Président, élu à la majorité des membres titulaires présents le
jour du scrutin. S’il appartient au premier collège, le Vice-Président, élu dans les mêmes
conditions, appartiendra au deuxième collège (article R.1123-10 du code de la santé
publique).
19/12/2018 26

Missions d’un CPP
� Les CPP ont pour missions d’émettre les avis suivants :
� un avis délibératif sur les projets de recherche impliquant la
personne humaine (projets de recherche initiale et
amendements sur recherche en cours) ;
� un nouvel avis délibératif dans le cadre du second examen après
avis défavorable d’un premier CPP ;
� un avis consultatif sur l'utilisation d'éléments et de produits du
corps humain à des fins scientifiques
� un avis consultatif sur les projets de recherche visant à évaluer
les soins courants
19/12/2018 27

Arrêté du 2 décembre 2016 fixant le contenu, le format et les modalités de
présentation du dossier de demande d'avis au comité de protection des
personnes sur un projet de recherche mentionnée au 1° de l'article L. 1121-1
du code de la santé publique portant sur un médicament à usage humain
� Le dossier de demande d’avis mentionné à l’article 2 comprend (article 3) :
� Un dossier administratif
� Courrier daté et signé mentionnant :
� Le titre de l’étude
� N°EudraCT (RIPH1 med)
� N° ID RCB
� Nom du promoteur
� Les coordonnées du contact promoteur (CP)
� Justification de la catégorie de l’étude
� La liste des documents fournis avec n° de version
� Un dossier sur la recherche
19/12/2018 28

� Avant de déposer un dossier de demande d’autorisation et/ou d’avis sur une
recherche impliquant la personne humaine, les promoteurs doivent obtenir un
numéro d’enregistrement de la recherche : ID-RCB qui permet d' identifier chaque
recherche réalisée en France.
19/12/2018 29
ID-RCB : RIPH (sauf médicament)

19/12/2018 30
Documents RIPH
RI RIRCM RNI
Médicament DM HPS DM HPS
Protocole daté et comportant un
numéro de version X X X X X X
Le résumé (en français) du
protocole, daté et comportant
un numéro de version
X X X X X X
Formulaire de demande initiale
d’autorisation ANSM ou d’avis
CPP
X X X X X X
Le document additionnel à la
demande d’avis au comité de
protection des personnes décrit
X X X X X
Supports susceptibles d’être
utilisés en vue du recrutement
des personnes ;
X X X X X
la copie de la ou des
autorisations de lieux de
recherches (L. 1121-13 du CSP).
X X X
19/12/2018 31
Documents RIPH
RI RIRCM RNI
Médicament DM HPS DM HPS
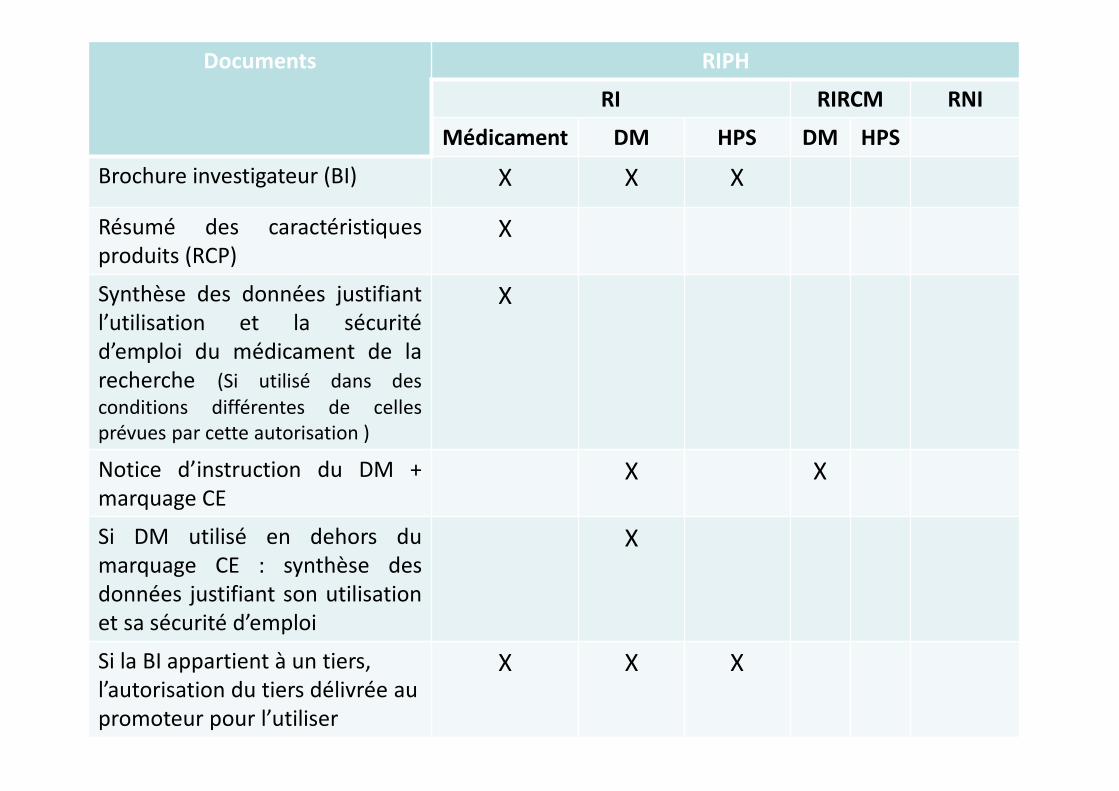
Brochure investigateur (BI) X X X
Résumé des caractéristiques
produits (RCP)X
Synthèse des données justifiant
l’utilisation et la sécurité
d’emploi du médicament de la
recherche (Si utilisé dans des
conditions différentes de celles
prévues par cette autorisation )
X
Notice d’instruction du DM +
marquage CEX X
Si DM utilisé en dehors du
marquage CE : synthèse des
données justifiant son utilisation
et sa sécurité d’emploi
X
Si la BI appartient à un tiers,
l’autorisation du tiers délivrée au
promoteur pour l’utiliser
X X X

19/12/2018 32
Documents RIPH
RI RIRCM RNI
Médicament DM HPS DM HPS
Le document d’information
destiné aux personnes qui se
prêtent à la recherche (L. 1122-1
du CSP)
X X X X X X
Le formulaire de recueil du
consentementX X X X* X*
Si le médicament dispose d’une
AMM, le dossier comprend une
comparaison et la description et la
justification des divergences
pertinentes en terme de sécurité
des personnes entre le document
d’information destiné aux
personnes qui se prêtent à la
recherche et la notice prévue à
l’article R. 5121-148 du CSP, au
regard des contre-indications et
des effets indésirables graves ou
des mises en garde ou précautions
d’emploi particulières
X

19/12/2018 33
Documents RIPH
RI RIRCM RNI
Médicament DM HPS DM HPS
Si DM marqué CE, le dossier
comprend une comparaison et
une justification des divergences
pertinentes en terme de sécurité
des personnes entre le document
d’information de la recherche et la
notice d’instruction du DM
X
La copie de l’attestation
d’assurance (L. 1121-10 du CSP)X X X X X
Une justification de l’adéquation
des moyens humains, matériels et
techniques au projet de recherche
et de leur compatibilité avec les
impératifs de sécurité des
personnes qui s’y prêtent
X X X X X
Les curriculum vitae du ou des
investigateursX X X X X X
La liste des investigateurs X X X X X X

19/12/2018 34
Documents RIPH
RI RIRCM RNI
Médicament DM HPS DM HPS
Décision de l’ANSM si disponible X X X
Cahier d’observation +/- questionnaires X
Document attestant que l’étude a
été demandée par l’ANSM, la HAS, le
ministère chargé de la santé ou
l’agence européenne des
médicaments
X
Description de l’utilisation (exclusive ou
non) de données extraites de systèmes
d’information existants ou de bases
d’études déjà réalisées
X
Origine et nature des données nominatives
recueillies, justification de recours à celles-
ci, mode de circulation des données,
destinataires des données personnelles
traitées, durée de conservation des
données, et le cas échant, transfert en
dehors de l’union européenne.
X
Déclaration de conformité à la MR003 ou
autorisation de la CNILX

Tirage au sort du CPP � La loi dispose que le CPP compétent pour examiner un projet de recherche,
préalablement à sa mise en œuvre, est désigné aléatoirement (article L. 1123-6
du CSP).
� A compter du 2 juillet 2018, les échanges entre les promoteurs et les CPP sont
désormais gérés par le système d’information des recherches impliquant la
personne humaine (SI RIPH) : https://cnriph.sante.gouv.fr/
19/12/2018 35
Un promoteur ou un mandataire
peut directement créer un compte
Une fois le compte activé, le
promoteur ou mandataire reçoit un
email avec ses accès au service web
et un mot de passe temporaire.
Il devra changer son mot de passe à
la première connexion

Création du dossier sur le SI RIPH � Créer un dossier
19/12/2018 36
Cliquer sur « Créer un dossier »
dans le menu de droite
Connexion en tant que
mandataire :
- Possibilité de choisir un
promoteur, si déjà rentré,
dans la base à l’aide du
menu déroulant.
- Si non, on peut enregistrer
directement un promoteur
en remplissant les champs
situés en-dessous

Création du dossier sur le SI RIPH � Créer un dossier
19/12/2018 37
Renseigner les champs liés à
l’identification de la
recherche.
On valide le dossier en cliquant
sur le bouton
« Demander une création de
dossier »
- Soumission du dossier de recherche au tirage au sort une fois que toutes les pièces
constitutives du dossier sont déposées
- Cliquer sur le bouton « tirage au sort »
- Réception d’un message électronique vous indiquant le CPP tiré au sort.
- SI RIPH sera l’interface pour communiquer avec le CPP désigné � Messagerie interne

Création du dossier sur le SI RIPH � Suivre le dossier
19/12/2018 38
Fiche dossier, :
-Informations relatives au
parcours de ce dernier.
- Les dates de :
> soumission du dossier
> de recevabilité
> de commission à laquelle
passera le dossier
> de début et de fin d’étude
> de fin de suspension d’étude
> la chronologie du dossier et les
dates de passage
d’un statut à l’autre
On peut aussi consulter l’avis
donné par le CPP en
charge du dossier suite aux
délibérations en
commission.

Création du dossier sur le SI RIPH � En attente : (brouillon, avant tirage au sort)
� Nouveau
� Incomplet
� Incompétent : le dossier sort des compétences juridiques d’un CPP. Aucune
action n’est donc possible puisque le dossier ne pourra être instruit par aucun
CPP.
� Recevable
� Non Recevable
� Demande d’informations ou de modifications
� Avis Favorable
� Avis Défavorable
A chaque changement de statut un e-mail de notification est envoyé au
promoteur. 19/12/2018 39

19/12/2018 40
Etapes Recherche Interventionnelle RIRCM/RNI
Demande d’avis initial SI RIPH
Dossier de demande Promoteur envoi le dossier au CPP désigné sans délai � Dossier papier en 4
exemplaires
Notification (recevabilité) 10 jours suivant réception du dossier complet
Si dossier incomplet : Liste des docs manquants envoyée par le CPP qui fixe
un délai pour la réception des documents
Sans réponse : Le promoteur renonce à sa demande
Evaluation 45 jours maximum 45 j
Mais procédure allégée – en
Comité restreint
Demande d’informations
complémentaires
1 seule demande possible du CPP
Envoi par le Promoteur à l’ANSM
pour info
Sur demande du CPP le
Promoteur,
accompagné de l’IC, peut être
entendu par le CPP.
Réponse promoteur Le promoteur n’est pas contraint par un délai de réponse mais l’horloge
s’arrête et ne reprend que lorsque le CPP a reçu les réponses.
Notification (avis) 45 j pour l’évaluation du dossier
15 j pour l’examen des réponses
Délai maximal CPP: 60 jours
45j
La validité de l’avis favorable a été augmentée à 2 ans et le délai de dépôt d’un recours en cas d’avis
défavorable porté à 1 mois vs 15 jours antérieurement

CPP : Avis défavorable
� Le promoteur, s’il le désire, dans le cas
d’un avis défavorable, peut bénéficier
d’un réexamen par un autre CPP en
soumettant son dossier à nouveau
� Dans le cas d’un avis défavorable : «
demander un second avis »
� Reprise du numéro initial et le dossier est
pré rempli
� Uniquement ajouter les pièces
complémentaires
� Soumettre au tirage au sort
� Dès lors, un nouveau CPP (excepté le CPP
qui a rendu l’avis) sera tiré un sort pour
l’étude du dossier.
19/12/2018 41

Normes pour le nomming des documents
� ETI - Etiquettes
� DOC - Autres documents
� AUT - Autorisation
� ASS - Assurance
� JUS - Justification lieux de
recherche
� CVI - CV investigateurs
� LIS - Liste investigateurs
� QUE - Echelles/questionnaires
PUB - Publicité/affiches
� NOT - Notice d’utilisation
� MCE - Marquage
� CE COU - Courrier
� CRF - CRF
� RCP - RCP
� BRO - Brochure investigateur
� INF - Doc Information
� RES - Résumé
� PRO - Protocole
� LET - Lettre
� SOI - Doc soins courants
� ADD - Doc additionnel
� DEM - Demande autorisation.
19/12/2018 42

INDS
Institut national des données de santé
� Groupement d'intérêt public, l'Institut national des données de santé a
succédé en avril 2017 à l’Institut des données de santé (IDS), créé en 2007.
� Evolution dans le cadre de la loi de modernisation du système de santé du 26
janvier 2016
� Volonté de renforcer et d’accroître le périmètre d’action de l’Institut
� Acteur central dans les relations entre producteurs de données et utilisateurs.
� Composé de 24 membres
� Mission : Favoriser le dialogue entre tous les acteurs afin de répondre au
mieux à leurs besoins et faire progresser l’ensemble du système de santé.
19/12/2018 43

Institut national des données de santé� L'utilisation des données de santé à caractère personnel est soumise à un
cadre législatif et règlementaire.
� Le traitement des données à caractère personnel pour une recherche, une
étude ou évaluation dans le domaine de la santé doit répondre à un motif
d'intérêt public en application de l'article 54 de la loi Informatique et Libertés.
� L'INDS assure le secrétariat unique dans le processus d'autorisation des
études, recherches et évaluation dans le domaine de la santé n'impliquant
pas la personne humaine et non éligibles à une méthodologie de
référence.
� L’INDS est aussi chargé de recevoir les demandes de modifications relatives
à des recherches, études ou évaluations dans le domaine de la santé en
cours (mise en conformité)
� Seules les modifications substantielles qui ont un impact sur le traitement des
données à caractère personnel sont concernées par cette démarche
19/12/2018 44

Champ d’action � Concerne les recherches dans le domaine de la santé qui relèvent du chapitre IX
de la loi Informatique et Libertés et qui n’appartiennent pas aux recherches
impliquant la personne humaine.
� En particulier les projets reposant sur la réutilisation de données de santé à
caractère personnel (par exemple celles issues de dossiers médicaux, d’un registre
agréé, de cohortes existantes ou du Système National des Données de Santé (SNDS)
et ses composantes) qui ne seraient pas éligibles à la méthodologie de référence
MR-004.
� Ne concerne pas les recherches interventionnelles ou non interventionnelles en
santé impliquant la personne humaine au sens de l’article R. 1121-1 du code de
la santé publique.
� Ces recherches sont prises en charge par la Commission Nationale des Recherches
Impliquant la Personne Humaine (CNRIPH) avant transmission à un Comité de
Protection des Personnes (CPP)
19/12/2018 45

Dépôt du dossier � L’ensemble des éléments du dossier doit être déposé sur la plateforme de dépôt
de dossier accessible depuis le site de l’INDS : http://www.indsante.fr/
� Les experts du CEREES doivent pouvoir disposer d’un délai minimal de 14 jours avant
la session mensuelle de ce comité
� Ajouter le délai d’enregistrement de l’INDS et de saisine du CEREES.
19/12/2018 46

Pièces constitutives du dossier
� Formulaire en ligne complété et signé
19/12/2018 47

Pièces constitutives du dossier� Un résumé de l'étude, recherche ou évaluation selon la grille retenue par le
CEREES
� Ce résumé doit impérativement être rédigé en français (5 pages maximum)
19/12/2018 48

Pièces constitutives du dossier� Un protocole scientifique incluant au moins les précisions demandées dans le
résumé.
� Le protocole peut être soumis en anglais
� La / les déclaration(s) d’intérêt du(es) responsable(s) de traitement et/ou du
responsable de la recherche
� Conformément à l’article 193 de la loi de modernisation du système de santé du 26 janvier 2016
et son décret d’application n°2016-1872 du 26 décembre 2016, le responsable de traitement et le
responsable scientifique de la recherche doivent remplir une déclaration d’intérêts en lien avec
l’objet de la recherche.
19/12/2018 49

Pièces constitutives du dossier
� La demande d’autorisation CNIL pré-remplie
19/12/2018 50

Pièces constitutives du dossier
� La lettre / notice d’information aux personnes concernées, de non opposition
et/ou de consentement le cas échéant
� La liste des financeurs de l’étude, le cas échéant
� L’avis émis par le comité scientifique et/ ou éthique qui a / ont évalué le projet et
la composition de ce(s) comité(s), le cas échéant
� Attention : Le résumé de l’étude, la déclaration d’intérêt publique, la demande
d’autorisation CNIL doivent être téléchargé et au format demandé par l’INDS
19/12/2018 51

Qui doit déposer la demande � La demande doit être déposée et signée soit par le responsable de traitement
de l’étude, recherche ou évaluation, soit par le responsable de sa mise en
œuvre pour le compte du responsable de traitement dans le cas d’une étude
sous-traitée à ce dernier ou un service mandaté à cet effet.
� Qui est le responsable de traitement ?
� C’est l’équivalent du « promoteur » pour les recherches impliquant la personne humaine
� Il détermine l’objectif du projet et les moyens de l’atteindre mais cela ne veut pas
nécessairement dire qu’il accède aux données et qu’il réalise lui-même la recherche. C’est lui
qui est juridiquement responsable de l’étude. L’autorisation de la CNIL, le cas échéant, sera
libellée à son attention.
� Qui est le responsable de la mise en œuvre ?
� C’est l’équivalent de « l’investigateur coordonnateur » pour les recherches impliquant la
personne humaine
� C’est généralement la personne physique qui rédige le protocole scientifique et supervise la
manipulation des données.
19/12/2018 52

Dépôt standard CEREES-CNIL� L’INDS vérifie la conformité de votre dossier ; il l’envoie
� Transmis sous 7 jours au Comité d’expertise pour les recherches, les études et
les évaluations dans le domaine de la santé (CEREES) pour examen.
� Le CEREES se réunit une fois par mois :
� Evalue la méthodologie de la demande
� La nécessité de recourir à des données à caractère personnel
� La pertinence de celles-ci par rapport à la finalité du traitement
� Si besoin, la qualité scientifique du projet.
� Le comité transmet ensuite son avis à l’INDS qui en informe le demandeur. Faute
d’avis dans un délai d’1 mois, ce dernier est réputé favorable.
� Si l'avis émis par le CEREES est favorable, avec ou sans recommandations, l'INDS
transmet le dossier à la CNIL, seule habilitée à délivrer une autorisation de
traitement des données.
� Si l'avis émis par le CEREES est réservé ou défavorable, le demandeur peut soit:
� Compléter son dossier en vue d'un nouvel examen par le Comité
� Demander à l'INDS de saisir la CNIL pour autorisation.19/12/2018 53

Rôle de la CNIL � CNIL : Commission Nationale de l’Informatique et des Libertés.
La CNIL a pour mission générale de veiller à ce que l’informatique ne porte
atteinte ni à l'identité humaine, ni aux droits de l’homme, ni à la vie privée, ni
aux libertés individuelles ou publiques. Dans le cadre des traitements de
données à caractère personnel ayant une finalité d'intérêt public d’étude, de
recherche ou d'évaluation dans le domaine de la santé, la CNIL a compétence
pour autoriser ou refuser leur mise en œuvre, après avis rendus par le CEREES
voire l’INDS. Pour chaque demande de traitement la Commission vérifie les
garanties présentées par le demandeur pour l'application des dispositions de
la loi Informatique et libertés, la conformité de la demande aux missions du
demandeur et elle statue sur la durée de conservation des données
nécessaires au traitement.
19/12/2018 54

55
Merci de votre attention
55
Des questions ?
19/12/2018
Top Related