







Languages
Pages
Legal


Algebraic method for treating the multiphoton selective excitationof the linear triatomic molecule HCN in intense laser fields
Ying Daia,* , Shiliang Dingb
aCollege of Chemistry and Molecular Engineering, State Key Laboratory of Rare Earth Materials Chemistry and Applications,Peking University, Beijing 100871, People’s Republic of China
bDepartment of Chemistry, Shandong University, Jinan, 250100, People’s Republic of China
Received 18 March 1999; received in revised form 4 June 1999; accepted 10 November 1999
Abstract
In this paper, the multiphoton transition Hamiltonian for the linear triatomic molecule HCN is given by using the quadraticanharmonic model and Sudden approximation. From this Hamiltonian we find a dynamic algebrah4. The bond-selectivevibrational transition probability of HC and the long-time averaged energy are calculated.q 2000 Elsevier Science B.V. Allrights reserved.
Keywords: Lie algebra; Multiphoton transition; Selective vibrational excitation
1. Introduction
There has been an increased interest in studying thephenomenon of multiphoton transition in recent yearsby the development of high-powered laser. Thestudies using Lie Group and Lie algebraic method tosolve the multiphoton question have increased [1–4].The Lie algebraic method is characterized by theexplicit expressions of the time evolution operatorU�t; t0�, which are equivalent to gaining the wavefunctions and the physical parameters of the system.In principle, using this approach, we can obtain theexact solutions to the Schro¨dinger equation. But thatis invalid when the dynamic Lie algebra cannot befound [5]. In this article, the bond-selective transitionfor HCN is discussed by using Lie algebra method.
Levine [1] suggested that the Hamiltonian of ananharmonic Morse oscillator can be represented as a
quadratic form:
H � "v0P2
21
Q2
2
!�1�
wherev0 is the frequency of the oscillator.P, Q areoperators. The equivalent form of the Hamiltonian is:
H � "v0 A1A2 112
ı0
� ��2�
whereı0 is an operator tending to the identity operatoronly in the anharmonicity parameter
x0 � 1N� v0
4D! 0
limit. (N is twice the number of bound states,D is thedissociation energy of the molecule.) The relationsamong the operatorsP, Q and the creation and
Journal of Molecular Structure (Theochem) 528 (2000) 85–90
0166-1280/00/$ - see front matterq 2000 Elsevier Science B.V. All rights reserved.PII: S0166-1280(99)00474-1
www.elsevier.nl/locate/theochem
E-mail address:[email protected] (Y. Dai).

annihilation operatorsA1, A2 are:
A^ � 1��2p �Q 7 iP� �3�
The relations among the operators ofA1, A2 and ı0are:
�A1; A2� � 2ı0; �ı0; A^� � 72x0A^ �4�
Then the spectrum of the Hamiltonians (1) and (2) is:
HuN; yl � "v0�y 2 x0y2�uN; yl �5�
whereuN; yl is the normalized eigenfunction andy thevibrational quantum number.
In the previous papers [5,6], we have studied themultiphoton excitation for the diatomic moleculesusing this model. In this contribution, we apply themodel to study the selective excitation of lineartriatiomic molecule HCN.
2. The Hamiltonian and the dynamic algebra forthe linear triatomic molecule HCN
We consider the linear triatomic molecule HCNplaced in intense laser field. The Hamiltonian of themolecular system is represented as two coupled quad-ratic anharmonic oscillators:
H1 �"
"v0
P2
21
Q2
2
!#HC
1
""v0
P2
21
Q2
2
!#CN
2PHCPCN
MC�6�
The first two terms represent the oscillators of HC andCN bond, respectively. The last term indicates thecoupling between the two oscillators,MC is the Catomic mass.
Since its dissociation energy and mass are large, theCN bond is not easily excited through HC pumping[6]. According to the Sudden approximation, the CNbond can be frozen and its momentum can be see as aconstant. Then the Hamiltonian are derived by using
Eqs. (2) and (3):
H1 � "v0 A1A2 1ı02
� �� �HC
1�"v0�y 2 x0y2��CN
2PCN
MC
i��2p �A1 2 A2�HC �7�
wherey is a vibrational quantum number. We assumethat the molecule and the laser fieldsE�t�areconstrained to one plane. The time-dependentmolecule–laser field interaction Hamiltonian isexpressed in the semiclassical dipole approximationas:
H 0 � 2m�kRHC; kRCN�E�t� �8�
where kRHC and kRCN are the internuclear distances ofthe HC and CN, respectively.m�kRHC; kRCN� is thedipole moment function, it can be expanded to firstorder inR (the distance of the H to the center of massof CN) at the equilibrium nuclear configurationR0 � 1:70 A as [7]:
m�kRHC; kRCN� � 2:871 0:76�~R2 ~R0�
� 2:871 0:76�~RHC 1 a~RCN 2 �~R0HC 1 akR0
CN��
� 2:871 0:76�kRHC 2 kR0HC�1 0:76a�kRCN 2 kR0
CN�� 2:871 0:76xHC 1 0:76axCN �9�
in Debye units. Here a� MC=�MC 1 mN�,xHC � kRHC 2 kR0
HC, xCN � kRCN 2 kR0CN, kR0
HC and kR0CN
are nuclear equilibrium separations of HC and CN,respectively.xCN is taken as a constant with the CNbond frozen [5,6] and
xHC ��������
"
mv0
s1��2p �A1 1 A2�
" #HC
wherem is the reduced mass of HC.The total Hamiltonian of HCN with the frozen CN
Y. Dai, S. Ding / Journal of Molecular Structure (Theochem) 528 (2000) 85–9086

in the laser field is expressed as:
H � H1 1 H 0 �"
"v0
A1A2 1
ı02
!#HC
1 �"v0�y 2 x0y2��CN 2
PCN
MC
i��2p �A1 2 A2�HC
2
(�2:871 0:76ax2 1 0:76
�" �������
"
mv0
s1��2p �A1 1 A2�
#HC
)
�E�t� � "v0�A1A2�HC 1 { �"v0�y 2 x0y2��CN
2 �2:871 0:76ax2�E�t�} 1
"v0
ı02
!HC
2
"0:76��
2p
�������"
mv0
s�A1 1 A2�
#HC
�E�t�2PCN
MC
i��2p �A1 2 A2�HC
� "v0�A1A2�HC 1 �"v0�y 2 x0y2��CN 1 d0E0
1
"v0
2
!HC
ı0 2d1��
2p A1 2
d2��2p A2 � H0 1 V
�10�Here
d1 � 0:76
�������"
mv0
s0@ 1AHC
E�t�1iPCN
MC�11�
d2 � 0:76
�������"
mv0
s0@ 1AHC
E�t�2iPCN
MC�12�
d0 � 2�2:871 0:76ax2�E�t� �13�
H0 � "v0�A1A2�HC 1 �"v0�y 2 x0y2��CN �14�
V � d0E0 1"v0
2
� �HC
ı0 2d1��2p A1 2
d2��2p A2 �15�
whereE0 is an identity operator (ı0 is equivalent to itonly in the harmonic oscillator).
In the interaction picture,V is transformed into
VI �t� � e�iH0t="�V e2�iH0t="� � d0T0 1"v0
2ı0
2d1��2p eiv0t2x0 eiv0tı0A1 2
d2��2p e2iv0tı0A2
� d0T0 1"v0
2ı0 2
d1��2p Ba1 2
d2��2p a2�
Xi
ai ai
�16�The subscriptI denotes the interaction picture.
ai �i � 0; 1;2;3� are E0, ı0, a1 � eiv0tı0A1,a2 � e2iv0tı0A2, respectively;.ai �i � 0;1;2; 3� are a0 � d0, a1 � "v0=2, a2 �2d1
��2p
B, a3 � 2d2=��2p
, respectively.
Here B� ei2v0x0t. They have the commutationrelations:
�ı0; E0� � 0; �E0; a1� � 0; �E0; a2� � 0;
�ı0; a1� � 22x0a1;
�ı0; a2� � 2x0a2; �a2; a1� � Bp ı0
�17�
According to expressions (16) and the relations (17),we can construct the dynamic Lie algebrah4:
E0; ı0; a1; a2
Then we can useh4 to obtain the time evolutionoperator.
3. The time evolution operators
In the interaction picture, the time evolution opera-tor is defined as:
UI �t� � eZ0E0 eZ1ı0 eZ2a1 eZ3a2 �18�whereZi are complex parameters andi � 0;1;2; 3. Itis important to guarantee that the time evolution is anunitary operator. According to Eqs. (17) and (18) theunitary condition is [5]:
Zp0 � 2Z0 �19�
Y. Dai, S. Ding / Journal of Molecular Structure (Theochem) 528 (2000) 85–90 87

Zp2B 2 Zp
2Zp1x0B 1 Z3 1 Z3Z1x0 1
13
Z23Z2Z1Bpx2
0
213
Z23Z2Bpx0 � 0 �20�
Zp1 2
12
Z p3Z p
2B 1 Z1 112
Zp3Zp
2Zp1Bx0
112
Z3Z2Z1Bpx0 212
Z3Z2Bp � 0 �21�In order to obtain the solution of the time evolutionoperator, we put Eq. (18) into the dynamic equation:
i"dUI �t; t0�
dt� VI�t�UI�t; t0� �22�
UI�t; t0� � 1 �23�and obtain the set of coupled nonlinear differentialequation group:
i" _Z0 � d0 �24�
i" _Z1 � "v0
22
d��2p Z2e22x0Z1Bp
2 2"v0Z2Z3x0Bp �1 2 x0�y 2 1��y1 2 2x0y
1 1� �
�25�
i" _Z2 � 2d��2p Z2e22x0Z1BpZ2
2x0 2d��2p Be2x0z1
1 "v0Z2�1 2 2x0�y 1 1��
1 "v0Z3Z22x0�4x0 1 2�1 2 2x0y��Bp �26�
i" _Z3 � 2d��2p e22x0Z1 2 "v0�1 2 2x0�y 2 1��Z3 �27�
with initial condition:
Zi � 0; i � 0;1; 2;3: �28�Combining the Eqs. (19)–(21) and (24)–(28) andsolving this differential equation group by numericalmethod, we can obtain the complex parametersZi andtime-evolution operatorUI �t�. Then the vibrationaltransition probability of the HC bond from the groundstatey0 � 0 to the excited stateyn:
P0!yn�t� � ukN; ynuUI�t; 0�uN;0lu2 �29�
The averaged energy is defined by:
e�t�h i �X27
n�0
Pn�t�en �30�
the time-averaged energy of excitation is defined by:
�e�v;T� � limT!∞
1T
ZT
0ke�t�l dt �31�
with ke�t�l determined at the frequencyv . T is theoptical period.
4. Results and discussion
Atomic units are used in the calculation. The valuesof parameters for HCN are given in the Table 1 [6].
SupposexCN � RCN 2 RCN0and the momentum of
the CN is equal to root-mean square:
PCN �������P2
CN
q�32�
because
�P2CN � N; y j2 1
2�A1 2 A2�2jN; y
� �
� N; y j2 12�A2
1 1 A22 2 A1A2 2 A2A1�jN; y
� �
� 12
kN; y uA1A2 uN; yl 112
kN; y uA2A1uN; yl
� 12
{ �1 2 x0�y 2 1��y 1 �1 2 x0y��y 1 1�}
� y 2 x0y2 1
12
�33�
Y. Dai, S. Ding / Journal of Molecular Structure (Theochem) 528 (2000) 85–9088
Table 1Values of parameters of HCN in au units
HC CN
m� 1693 m� 11857a � 0:9697 a � 1:2643D � 0:2133 D � 0:3609x0 � 0:01804 x0 � 0:00683v0 � 0:01539 v0 � 0:009858Mc � 22020

then
PCN �������P2
CN
q�
������������������y 2 x0y
2 112
r�34�
4.1. Transition probability
Fig. 1 represents the curves of the transition prob-ability versus laser frequency from the ground to thefive vibrational states for the HC bond when the CNbond is frozen in yCN � 0, yCN � 1, yCN � 2,yCN � 5, respectively. The laser intensity isE0 �0:009 au (I < 2:844 T W=cm2). It shows that thereare different kinds among the peaks at both the
position and the height. That is, there is little influenceon the vibrational excitation of the HC bond on whichstate the CN bond stays.
Fig. 2 gives the curves of the vibrational transitionprobabilities of the HC bond from the ground state tothe different excited states whenyCN � 0. P1 repre-sents the curve from the ground state to the firstexcited state, the peak values are atv � 0:898v0
andv � 0:992v0. They belong to single photon tran-sition.P3 represents the curve from the ground state tothe third excited state, the peak values are atv �0:836v0 and v � 0:890v0. They belong to three-photon transitions.P5 represents the curve from theground state to the fifth excited state, the peak valuesare atv � 0:796v0 , v � 0:758v0. It belongs to thesix photons transition atv � 0:758v0. P7 representsthe curve from the ground state to the seventh excitedstate, the multiphoton peak values are atv � 0:686v0 , v � 0:704v0. These show that asthe vibrational quantum number increases, the energylevel interval decreases and the energy state densityincreases.
4.2. Time behavior
At E0 � 0:009 au (I < 2:844 T W=cm2), we calcu-lated the long time-averaged energy�e�v;T�. Fig. 3gives the curves of�e�v;T� as a function of optical
Y. Dai, S. Ding / Journal of Molecular Structure (Theochem) 528 (2000) 85–90 89
Fig. 1. The curves of the transition probability with frequency fromthe ground to the five states: (a)yCN � 0, (b)yCN � 1, (c)yCN � 2,(d) yCN � 5.
Fig. 2. The probabilities with frequency from the ground to the 1, 2,5, 7 states withE0 � 0:009 au for HC bond atyCN � 0.
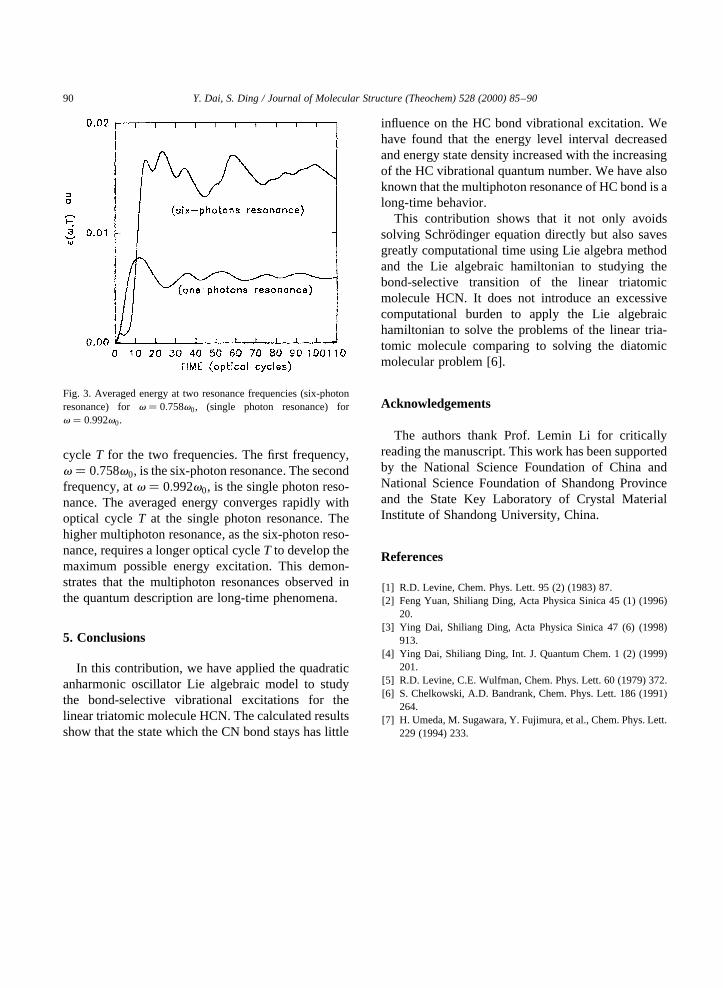
cycle T for the two frequencies. The first frequency,v � 0:758v0, is the six-photon resonance. The secondfrequency, atv � 0:992v0, is the single photon reso-nance. The averaged energy converges rapidly withoptical cycleT at the single photon resonance. Thehigher multiphoton resonance, as the six-photon reso-nance, requires a longer optical cycleT to develop themaximum possible energy excitation. This demon-strates that the multiphoton resonances observed inthe quantum description are long-time phenomena.
5. Conclusions
In this contribution, we have applied the quadraticanharmonic oscillator Lie algebraic model to studythe bond-selective vibrational excitations for thelinear triatomic molecule HCN. The calculated resultsshow that the state which the CN bond stays has little
influence on the HC bond vibrational excitation. Wehave found that the energy level interval decreasedand energy state density increased with the increasingof the HC vibrational quantum number. We have alsoknown that the multiphoton resonance of HC bond is along-time behavior.
This contribution shows that it not only avoidssolving Schro¨dinger equation directly but also savesgreatly computational time using Lie algebra methodand the Lie algebraic hamiltonian to studying thebond-selective transition of the linear triatomicmolecule HCN. It does not introduce an excessivecomputational burden to apply the Lie algebraichamiltonian to solve the problems of the linear tria-tomic molecule comparing to solving the diatomicmolecular problem [6].
Acknowledgements
The authors thank Prof. Lemin Li for criticallyreading the manuscript. This work has been supportedby the National Science Foundation of China andNational Science Foundation of Shandong Provinceand the State Key Laboratory of Crystal MaterialInstitute of Shandong University, China.
References
[1] R.D. Levine, Chem. Phys. Lett. 95 (2) (1983) 87.[2] Feng Yuan, Shiliang Ding, Acta Physica Sinica 45 (1) (1996)
20.[3] Ying Dai, Shiliang Ding, Acta Physica Sinica 47 (6) (1998)
913.[4] Ying Dai, Shiliang Ding, Int. J. Quantum Chem. 1 (2) (1999)
201.[5] R.D. Levine, C.E. Wulfman, Chem. Phys. Lett. 60 (1979) 372.[6] S. Chelkowski, A.D. Bandrank, Chem. Phys. Lett. 186 (1991)
264.[7] H. Umeda, M. Sugawara, Y. Fujimura, et al., Chem. Phys. Lett.
229 (1994) 233.
Y. Dai, S. Ding / Journal of Molecular Structure (Theochem) 528 (2000) 85–9090
Fig. 3. Averaged energy at two resonance frequencies (six-photonresonance) forv � 0:758v0, (single photon resonance) forv � 0:992v0.
Top Related