
Xin Xia - DiVA portalsu.diva-portal.org/smash/get/diva2:716217/FULLTEXT02.pdf · Xin Xia, Chunfang...
80
DISSOLVING THE ROCKS Solubility Enhancement of Active Pharmaceutical Ingredients using Mesoporous Silica Xin Xia
Transcript of Xin Xia - DiVA portalsu.diva-portal.org/smash/get/diva2:716217/FULLTEXT02.pdf · Xin Xia, Chunfang...

D I S S O L V I N G T H E R O C K S S o l u b i l i t y E n h a n c e m e n t o f A c t i v e P h a r m a c e u t i c a l I n g r e d i e n t s u s i n g M e s o p o r o u s S i l i c a Xin Xia


Dissolving the Rocks Solubility Enhancement of Active Pharmaceutical Ingredients using Meso-porous Silica
Xin Xia

Doctoral Thesis 2014
This thesis is based on work performed at Stockholm University and Nano-logica AB, Stockholm, Sweden. Faculty opponent Associate Professor Göran Frenning Department of Pharmacy Uppsala University, Sweden Evaluation committee Associate Professor Ulla Elofsson SP Technical Research Institute of Sweden Sweden Associate Professor Andreas Nyström Institute of Environmental Medicine Karolinska Institutet, Sweden Dr. Robert Corkery Joma International AS Norway Substitute Associate Professor German Salazar-Alvarez Department of Materials and Environmental Chemistry Stockholm University, Sweden
©Xin Xia, Stockholm University 2014 ISBN 978-91-7447-924-9 Printed in Sweden by US-AB, Stockholm 2014 Distributor: Department of Materials and Environmental Chemistry, Stockholm University

Abstract
Poor aqueous solubility is one of the greatest barriers for new drug candi-dates to enter toxicology studies, let alone clinical trials. This thesis focuses on contributing to solving this problem, evaluating the oral toxicity of meso-porous silica particles, and enhancing the apparent solubility and bioavaila-bility of active pharmaceutical ingredients in vitro and in vivo using mesopo-rous silica particles. Toxicological studies in rats showed that two types of mesoporous silica particles given by oral administration were well tolerated without showing clinical signs of toxicity. Solubility enhancement, including in vivo bioavail-ability and in vitro intracellular activity, has been evaluated for selected drug compounds. Mesoporous silica was shown to effectively increase drug solu-bility by stabilizing the amorphous state of APIs, such as itraconazole (anti-fungal), dasatinib (anti-cancer), atazanavir (anti-HIV) and PA-824 (anti-tuberculosis). Itraconazole was successfully loaded into a variety of porous silica materials showing a distinct improvement in the dissolution properties in comparison to non-porous silica materials (and the free drug). Mi-croporosity in SBA-15 particles has advantages in stabilizing the supersatu-ration state of dasatinib. Small pore sizes show better confinement of ataza-navir, contributing to a higher dissolution of the drug compound. In the in vivo animal studies, NFM-1 loaded with atazanavir shows a four-fold in-crease in bioavailability compared to free crystalline atazanavir. PA-824 has a higher dissolution rate and solubility after loading into AMS-6 mesoporous particles. The loaded particles show similar antibacterial activity as the free PA-824. This thesis aims at highlighting some of the important factors enabling the selection of adequate mesoporous structures to enhance the pharmacokinetic profile of poorly water-soluble compounds, and preparing the scientific framework for uncovering the effects of drug confinement within mesopores of varying structural properties. Keywords: mesoporous silica, drug delivery, solubility enhancement, active pharmaceutical ingredients, oral toxicity, confinement, crystallization, phar-maceutical excipients, bioavailability.


Abstract in Chinese 论文摘要 利用介孔二氧化硅增加药物活性成分的溶解度 口服药中的药物活性成分在水中的溶解度是影响其在体内吸收的重要因素。低水溶性使很多候选药物在进行毒性实验和临床实验时被
淘汰。本论文主要研究了介孔二氧化硅的口服耐受度并利用其增加难
溶药物的表观溶解度。 实验表明大鼠对所研究的两种介孔二氧化硅颗粒具有良好的口服耐受度。本论文中研究的四种药物:伊曲康唑(抗真菌),达沙替尼
(抗癌),阿扎那韦(抗艾滋病),和 PA-824(抗结核病)均为难溶药物。介孔二氧化硅颗粒可以有效的提高这些药物的溶解度及溶解
速率。伊曲康唑被负载到不同的多孔二氧化硅中,这些材料均不同程
度的提高了药物的溶解度,但是无孔的二氧化硅却没有表现出类似的
作用。SBA-15 的微孔结构可以帮助稳定达沙替尼的超饱和状态,这对维持药物在体内保持有效浓度是非常有意义的。较小的孔(2.5nm)比大孔(7nm)对增加阿扎那韦的溶解更有帮助。当阿扎那韦被负载到NFM-1 颗粒中时,其生物利用度是未负载的药物晶体的四倍。将 PA-824负载到 AMS-6颗粒中也增加了药物的溶解度和溶解速率。在细胞培养实验中,AMS-6颗粒成功的将药物输送到了巨噬细胞,并展示了和晶体药物相似的抗结核活性。 本论文着重强调了为难溶药物选择合适介孔材料的影响因素,旨在揭示介孔材料的结构因素对药物在孔内结晶限制的影响。 关键词:介孔二氧化硅,药物输送,增溶,药物活性成分,口服毒性,
(结晶)限制,结晶,药物辅料,生物利用度。


To my beloved family


List of papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I In vivo oral toxicological evaluation of mesoporous silica particles§ Natalia Kupferschmidt, Xin Xia, Roberto Hanoi Labrador, Rambabu Atluri, Lluis Ballell, and Alfonso E. Garcia-Bennett, Nanomedicine, 2013, 8, 57-64.
II Systematic study of itraconazole in solubility enhancement and solid dispersion state using different nanoporous silica particles Xin Xia, Chunfang Zhou, and Adam Feiler, Submitted to In-ternational Journal of Pharmaceutics.
III Influence of microporosity in SBA-15 on the release proper-ties of anticancer drug dasatinib Tomas Kjellman*, Xin Xia*, Viveka Alfredsson, and Alfon-so E. Garcia-Bennett, Submitted to Journal of Material Chemistry-B.
IV In vivo enhancement in bioavailability of atazanavir in the presence of proton-pump inhibitors using mesoporous mate-rials§ Xin Xia, Chunfang Zhou, Lluis Ballell, and Alfonso E. Gar-cia-Bennett, ChemMedChem, 2012, 1, 43-48.
V Encapsulation of anti-tuberculosis drugs within mesoporous silica and intracellular antibacterial activities Xin Xia, Kevin Pethe, Ryangyeo Kim, Lluis Ballell, David Barros, Jonathan Cechetto, HeeKyoung Jeon, Kideok Kim, Alfonso E. Garcia-Bennett, Submitted to Nanomaterials.
*I shared the first author position with Tomas Kjellman in paper III. § Reprints were made with permission from the respective publishers.

Other relevant publications
VI Toxicology of nanomaterials and their uses in medicine Xin Xia, Natalia Kupferschmidt and Alfonso E. Garcia-Bennett, in Frontiers in Nanobiomedical Research: Hand-book of Nanobiomedical Research. In press, Release October 2014, World Scientific Publishing, ed. Vladimir Torchilin, ISBN: 978-981-4520-64-5.
VII Patent: Super-saturating delivery vehicles for poorly water-soluble pharmaceutical and cosmetic active ingredients Inventor: Chunfang Zhou, Xin Xia, and Alfonso E. Garcia-Bennett. WO2012035074 A1, PCT/EP2011/065959, Publica-tion date 22nd March 2012, International Filing Date 14th Sep-tember 2011.
My contribution to the papers included in this thesis was:
I I took part in planning the study. I performed the synthesis and characterization of mesoporous silica. I took part in the writing of the manuscript.
II I was responsible for the planning and execution of the ex-perimental work (except synthesis of mesoporous silica). I analyzed the data and was responsible for writing the manu-script.
III I took part in planning the study. I performed the experi-mental part regarding drug loading and release. I was respon-sible for characterizations (except small angle X-ray diffrac-tion and TEM). I took part in the writing of the manuscript.
IV I was responsible for the planning and execution of the ex-
perimental work (except synthesis of mesoporous silica, TEM, and in vivo study). I analyzed the data and was re-sponsible for writing the manuscript.
V I was responsible for the planning and execution of the ex-
perimental work regarding drug loading and release. I ana-lyzed the data and was responsible for writing the manu-script.

Contents
1 Introduction ........................................................................................................ 17
2 Aim of the thesis ................................................................................................. 19
3 Background ......................................................................................................... 20 3.1 Drug dissolution and bioavailability ................................................................................ 20 3.2 Formulation approaches for enhancing the solubility of poorly water-soluble drugs ..... 22
3.2.1 Chemical methods ................................................................................................... 22 3.2.2 Physical methods .................................................................................................... 23 3.2.3 Carriers and stabilizers ............................................................................................ 25
3.3 Mesoporous silica ............................................................................................................. 27 3.3.1 Synthesis mechanism .............................................................................................. 27 3.3.2 Classification of mesoporous silica from their template system ............................ 28 3.3.3 Classification of mesoporous silica according to their pore structure .................... 30 3.3.4 Toxicology .............................................................................................................. 32 3.3.5 Application in life science ...................................................................................... 33
4 Experimental ....................................................................................................... 35 4.1 Materials ........................................................................................................................... 35
4.1.1 Mesoporous silica particles ..................................................................................... 35 4.1.2 APIs used in this thesis ........................................................................................... 36
4.2 API loading methods ........................................................................................................ 36 4.3 Analytical methods ........................................................................................................... 37
4.3.1 Powder X-ray diffraction (XRD) ............................................................................ 37 4.3.2 Small angle X-ray scattering (SAXS) ..................................................................... 38 4.3.3 Scanning electron microscopy (SEM) .................................................................... 38 4.3.4 Transmission electron microscopy (TEM) ............................................................. 38 4.3.5 Nitrogen adsorption/desorption isotherms .............................................................. 39 4.3.6 Thermogravimetric analysis (TGA) ........................................................................ 40 4.3.7 Differential scanning calorimetry (DSC) ................................................................ 40 4.3.8 Dynamic light scattering (DLS) .............................................................................. 40
4.4 Dissolution testing ............................................................................................................ 41 4.5 In vivo oral administration ............................................................................................... 41
4.5.1 Oral tolerance .......................................................................................................... 41 4.5.2 Administration of atazanavir with omeprazole ....................................................... 42
4.6 Intracellular study ............................................................................................................. 42

5 Oral toxicity of mesoporous silica ...................................................................... 43
6 Improving drug dissolution ................................................................................ 45 6.1 Comparison of porous silica particles in the dissolution of itraconazole ......................... 45 6.2 Influence of microporosity of SBA-15 in the release of dasatinib ................................... 49 6.3 In vivo enhancement in bioavailability of atazanavir ....................................................... 51 6.4 Intracellular antibacterial activities of anti-tuberculosis drug encapsulated into mesoporous silica ........................................................................................................................ 54
7 Discussion ........................................................................................................... 57 7.1 Physical properties of APIs .............................................................................................. 57 7.2 Loading state .................................................................................................................... 59 7.3 Supersaturation ................................................................................................................. 62 7.4 Important factors in the selection of optimal mesoporous silica ...................................... 63
8 Conclusions ........................................................................................................ 64
9 Future work ........................................................................................................ 66
Acknowledgements ................................................................................................... 67
References ................................................................................................................. 69

Abbreviations
2D, 3D 2-dimentional, 3-dimentional AMP Amorphous mesoporous silica particles AMS Anionic surfactant-templated mesoporous silica API Active pharmaceutical ingredients APES 3-aminopropyl triethoxysilane ATV Atazanavir AUC Area under the curve (pharmacokinetics) BCS Biopharmaceutics Classification System BET Brunauer-Emmett-Teller CSDA Co-structure directing agent CT Confirmatory test CTAB Cetyltrimethylammonium bromide DAS Dasatinib DFT Density functional theory DRF Dose range finding test DSC Differential scanning calorimetry EVK Evonik fumed silica particles FDA U.S. Food and drug administration HAART Highly Active Antiretroviral Therapy HIV Human immunodeficiency virus HPMC Hydroxypropyl methylcellulose ITZ Itraconazole MPTS 3-mercaptopropyl triethoxysilane MTB Mycobacterium tuberculosis MTD Maximum tolerated dose NFM Nanoporous folic acid-templated material SBA Santa Barbara amorphous mesoporous silica SEM Scanning electron microscopy SLNs Solid lipid nanoparticles STA St. Andrews University material P123 Poly(ethylene glycol)-block-poly(propylene glycol)-block-
poly(ethylene glycol) with a ratio 20-70-20. P104 Poly(ethylene glycol)-block-poly(propylene glycol)-block-
poly(ethylene glycol) with a ratio 27-61-27. PBS Phosphate buffered saline PVP Polyvinylpyrrolidone

PEG Polyethylene glycol SAXS Small angle X-ray scattering SGF Simulated gastric fluid SIF Simulated intestinal fluid TEM Transmission electron microscopy TEOS Tetraethyl orthosilicate TMOS Tetramethyl orthosilicate TGA Thermogravimetric analysis USP United States Pharmacopeial Convention XRD X-ray diffraction
17
1 Introduction
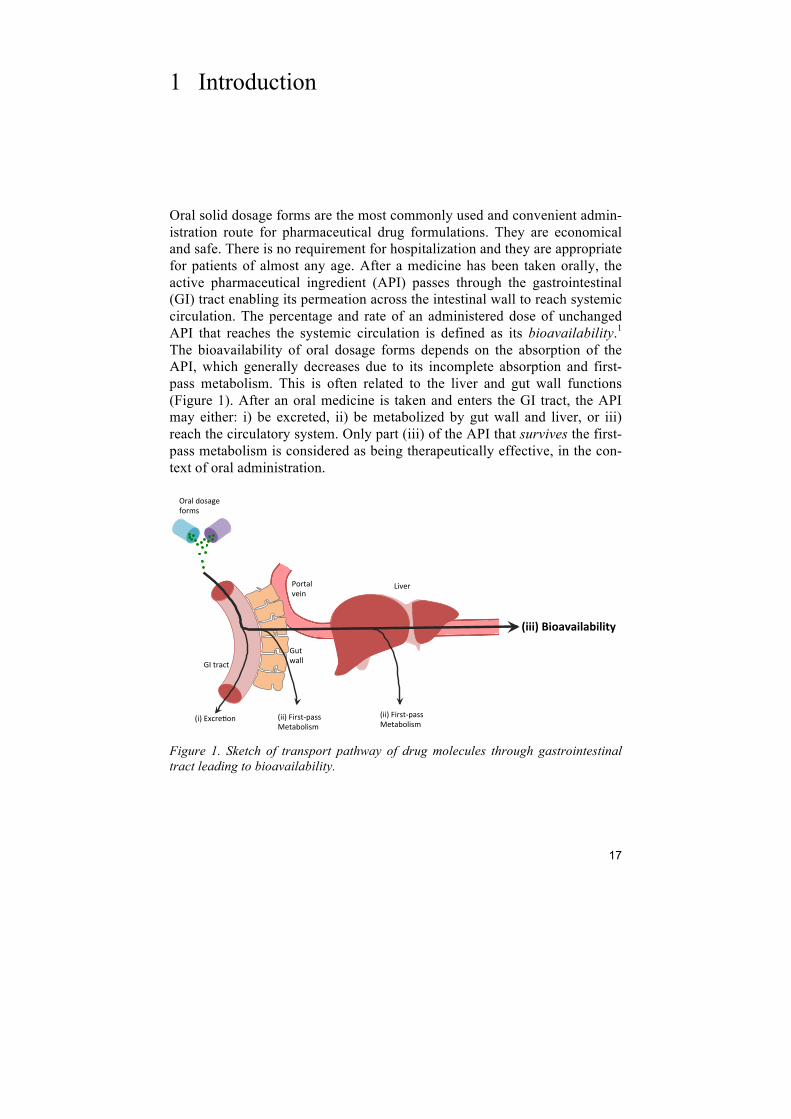
Oral solid dosage forms are the most commonly used and convenient admin-istration route for pharmaceutical drug formulations. They are economical and safe. There is no requirement for hospitalization and they are appropriate for patients of almost any age. After a medicine has been taken orally, the active pharmaceutical ingredient (API) passes through the gastrointestinal (GI) tract enabling its permeation across the intestinal wall to reach systemic circulation. The percentage and rate of an administered dose of unchanged API that reaches the systemic circulation is defined as its bioavailability.1 The bioavailability of oral dosage forms depends on the absorption of the API, which generally decreases due to its incomplete absorption and first-pass metabolism. This is often related to the liver and gut wall functions (Figure 1). After an oral medicine is taken and enters the GI tract, the API may either: i) be excreted, ii) be metabolized by gut wall and liver, or iii) reach the circulatory system. Only part (iii) of the API that survives the first-pass metabolism is considered as being therapeutically effective, in the con-text of oral administration.
Figure 1. Sketch of transport pathway of drug molecules through gastrointestinal tract leading to bioavailability.
!"#$%&'$#
!($#)&**#
+%&*#,-.&/0#1-%2.#
345#67'%08-9# 3445#:4%.$;<&..#=0$&>-*4.2#
3445#:4%.$;<&..#=0$&>-*4.2#
?-%$&*#@049#
A4@0%#
!"""#$%"&'('")'*")"+,$

18
The physical properties of a drug molecule (e.g. aqueous solubility and per-meability), have been correlated with its absorption and oral bioavailability.2 In drug discovery, more than half of new drug candidates are poorly water soluble, leading to inconsistent and limited systemic exposure i.e. low bioa-vailability.3 Low bioavailability may result in the precipitation of high doses of an API in order to reach the therapeutic level. This is associated with tox-icity issues and high manufacturing and therapeutic cost. The main focus of the pharmaceutical field in drug formulation is to improve bioavailability by improving drug solubility and dissolution rate. Various techniques have been developed to improve the aqueous solubility of drugs: particle size reduction4, solid lipid dispersion5, amorphous drug formulations6,7, etc. Many of these methods, however, are costly and do not show significant enhancement in solubility. In recent years, scientists in the field of nanotechnology have been motivated to develop nanostructured ma-terials for poorly water-soluble drug delivery. Mesoporous silica materials were proposed to be good candidates for drug delivery in 2001, exploiting the possibility of their use in sustained or controlled release strategies.8 Mesoporous silica materials are fully-porous amorphous silica particles with ordered pore structures and with pore sizes in the range of 2 - 50 nanome-ters.9,10 As a potential drug delivery carrier it has the following advantageous physical properties:11
i) high surface and pore volume to carry sufficient amount of API; ii) particles are composed of amorphous silicon dioxide. The same
chemical composition has been approved by the U.S. Food and drug administration (FDA) as a drug excipient and food addi-tive;
iii) sharp pore size distribution gives the possibility to design the pore size to fit different sizes of drug molecules;
iv) ordered pore networks can be tailored from 2-dimentional to 3-dimentional connectivity, which may control the dissolution rate;
v) tailorable particle size for different administration routes; vi) compatible with tableting.
Studies indicate this type of material can be used for: enhancing the solubili-ty of hydrophobic drugs with high partition coefficient,12 controlled and tar-geted release of drugs, 13,14 diagnostic and theranostic applications.15

19
2 Aim of the thesis
Over the last two decades a number of studies have established a functional role for mesoporous materials in drug delivery applications. The main aim in this thesis was to investigate the application of mesoporous silica for solubil-ity enhancement of APIs. The topic was approached from several perspec-tives in the following articles: Paper I - Toxicity To evaluate in vivo oral toxicity of mesoporous silica particles. Paper II – Comparison of different porous silica particles To understand how different mesoporous silica particles and commercial porous silica particles contribute to the solubility enhancement of itracona-zole. Paper III – Fine tuning of structural properties To investigate the influence of microporosity in mesoporous silica particles over drug release and drug recrystallization. Paper IV – Actual pharmaceutical problems To understand the solubility and in vivo oral bioavailability enhancement of atazanavir released from mesoporous silica particles in the presence of a proton-pump inhibitor. Paper V – Intracellular delivery To study the intracellular activity of anti-tuberculosis drugs, when loaded into mesoporous silica particles.

20
3 Background
3.1 Drug dissolution and bioavailability Solid oral dosage formulations have been the major administration route for almost a century. It was not until 1951 that Edwards16 discovered that the absorption of oral dosage forms is related to the dissolution process of that drug, though the fundamental research of dissolution processes has been developing since the end of 19th century.17 In today’s research, drug dissolution is considered as one of the most im-portant physical properties related to drug absorption and formulation devel-opment. In the case of solid drugs, dissolution is a process whereby the drug solid disintegrates and goes into the solution phase, becoming a homogene-ous molecular dispersion. Drug molecules can be absorbed, distributed, me-tabolized and excreted by the body. We use the term bioavailability to de-scribe the extent of an administered dose that reaches the systemic circula-tion, i.e. how much drug is effectively absorbed. The link between drug dissolution and bioavailability became well recog-nized when Amidon et al. developed the Biopharmaceutics Classification system (BCS) in 1995.2 BCS classifies drug substances into four types based on their aqueous solubility and permeability (Figure 2): high solubility/high permeability (Class I), low solubility/high permeability (Class II), high solu-bility/low permeability (Class III), and low solubility/low permeability (Class IV).
Figure 2. Biopharmaceutics Classification System.

21
The solubility of a solid in a liquid is defined in quantitative terms as the concentration of a solid (solute) in a saturated solution at a certain tempera-ture. It is an intrinsic material property that can be altered only by chemical modification of the molecule. The apparent solubility is the concentration of the solute at apparent equilibrium (supersaturation) that can be improved by modification of the crystal structure of the molecule. In this thesis, solubility enhancement refers to an apparent solubility enhancement. It is now widely accepted that the solubility of a drug compound and its dis-solution rate determines the drug’s appearance in the body for BCS class II drug compounds. Formulation development helps these drug compounds to achieve much higher bioavailability by increasing their aqueous solubility. A large proportion of lead compounds discovered by pharmaceutical compa-nies have poor water solubility, resulting in poor bioavailability, which is the main obstacle for them to enter clinical trails. Serajuddin18 reported approx-imately one-third of new drug candidates synthesized by pharmaceutical industries have an aqueous solubility of less than 10µg/ml, another one-third have a solubility from 10 to 100 µg/ml, and the rest is above 100 µg/ml. Hence, one of the major barriers for solid oral dosage forms in pharmaceuti-cal formulations is to increase the aqueous solubility of drug substances so that they can reach clinical trials and eventually the market and patients. Some drug compounds have excellent aqueous solubility at certain pHs, in other words, their solubility is pH-dependent. Drug compounds that have good solubility at low pH have less formulation problems if their primary absorption occurs in the stomach. However, some drug compounds have poor stability in acidic conditions requiring formulation development for protecting them from gastric acidity and improving their dissolution in neu-tral pH (intestine, pH 6 – 7.4). Furthermore, patient non-compliance may be associated with pH-dependent solubility. A typical example is antiretroviral drugs used in Highly Active Antiretroviral Therapy (HAART) for the treat-ment of Human immunodeficiency virus (HIV). APIs used in this treatment are often chemically modified to their acidic forms to increase their bioavail-ability, e.g. atazanavir. However, the frequent and high doses of several acidic drugs may increase the acidity in patients’ stomach and result in nega-tive symptoms, such as stomach pain and heartburn.19 In fact the FDA issued a warning against co-administration of proton-pump inhibitors to treat these symptoms since the bioavailability may decrease significantly (as much as 78% for atazanivir), as these medicines induce higher stomach pH.20

22
3.2 Formulation approaches for enhancing the solubility of poorly water-soluble drugs
Formulation is the main strategy to improve the absorption of class II drugs. It is very risky and costly to develop class IV compounds, since no in vitro/in vivo correlations are expected due to their poor water solubility and poor permeability. These drugs have to go back to the lead optimization phase of drug development, and require structural modeifications.21 Howev-er, enhancing solubility is the main concern of this thesis, focusing on the formulation of class II drugs in oral administration.
3.2.1 Chemical methods
3.2.1.1 Crystalline salt forms In the 1950s, Nelson investigated the salt forms of several weakly acidic compounds showing higher dissolution rate and aqueous solubility than their free acid form.22,23 Crystalline salt forms of weak bases or acids often show better dissolution compared with their corresponding acid or base forms. This has been reported to be a good approach for both liquid formulations24 and solid dosage forms.22,23 Although salt forms are the most common approach of increasing solubility for oral solid dosage forms, it is not always an option. For example, the sol-ubility of a hydrochloride salt in the stomach may be limited by the high concentration of Cl-.25 Even if the salt form has higher solubility, a potential risk is that it may affect the pH in the GI tract, which may be a cause of pa-tient non-compliance. Many prescribed medical therapies fail to achieve the desired outcomes due to patient non-compliance, where side effects are one of the main reasons.26
3.2.1.2 Chemical structure design It is often feasible to increase the solubility of a drug compound already in drug design. Chemical structure modification in order to enhance solubility includes: i) addition of ionizable groups that will be charged in buffers, such as basic amines or carboxylic acids; ii) adding hydrogen donors and accep-tors; iii) adding polar groups; iv) reducing molecular weight; and v) out-of-plane substitution, e.g. adding ethyl groups to shift the planarity of a mole-cule hence interfering with crystal formation to achieve a more soluble high energy crystal form.27 However, these methods have a lot of restrictions in practice. A drug candidate must have both hydrophilic and lipophilic proper-ties since it must be polar to be soluble in aqueous conditions and interact with molecular targets, and must be fatty to cross cell membrane and avoid rapid excretion. These types of modifications may also affect the drugs po-tency and mode of action.

23
3.2.2 Physical methods
3.2.2.1 Particle size reduction Various physical formulation strategies are based on the Noyes-Whitney equation (Eq.1), which describes the parameters that affect the diffusion-controlled dissolution rate of solids,28
!"!"= !"(!!!!)
!! (1)
where the dissolution rate of a drug solid dC/dt is related to: D - the diffusion coefficient of the drug in solution, A - surface area of the interface between the drug solid and the solvent, Cs - the saturation solubility of the drug in solution in the diffusion layer, C - the concentration of the drug in the solu-tion, V is the volume of the dissolution medium, and h - the thickness of the diffusion layer. According to the equation, increasing the effective surface area may enhance the dissolution rate. Reducing particle size is the most straightforward way to increase surface area. The equation also describes the relationship between dissolution rate and solubility. The dissolution rate will increase if the saturation solubility is enhanced to reach a supersaturation state. This is a solution containing more solute than can be dissolved under normal circumstances. Solubility is also a function of particle size according to the Kelvin equation (Eq. 2):29 ! = !! ∙ exp(!!∙!!
!∙!") (2)
In this equation, S is the apparent solubility as a function of particle radius r, S∞ is the solubility of the crystalline material, γ is the interfacial tension be-tween drug and solvent, VM is the molar volume of the drug, R is the gas constant and T is the temperature. This equation indicates that solubility increases enormously with the reduction of particle size. Figure 3 shows the solubility increase (solubility according to Eq. 2 divided by the solubility of the crystalline material) as a function of particle size for typical organic compounds.

24
Figure 3. Solubility increase as a function of particle size for typical organic com-pounds (derived from reference 29).
Conventional methods of particle size reduction use mechanical stress to disintegrate the drug solid. Nanomilling is one of the most common methods to produce nanosized drug particles in industrial scale. In the process, the generated high energy and shear forces provide the necessary energy input to disaggregate the drug’s micron size (or even larger) particles to nanoparti-cles. The milling process is carried out with a milling medium, water or a suitable buffer, the drug and a stabilizer.30 It is necessary to introduce stabi-lizers in the system, since nanoparticles may very easily re-agglomerate. The selection of suitable stabilizers is a crucial parameter in the process. A fine nanomilling process takes a period ranging from hours to several days, de-pending on the drug’s hardness, quantities and target particle size.4,31 This method can be very energy and time consuming for drugs with high hard-ness.
3.2.2.2 Amorphous drug formulation Most drugs are synthesized in crystalline forms. In a drug crystal, the mole-cules are arranged in long-range order extending in all three dimensions. In contrast, amorphous solids lack the long-range order in crystals. To move a solute molecule into solution, two kinds of interactions must be broken: i) the interactions among solute molecules in the crystal (lattice energy), and ii) the interactions among the solvent molecules in the space required to ac-commodate the solute molecule (cavitation energy).32 Therefore, the amor-phous form of a drug may provide a much better energetic path for solubility since there are no or only weak lattice barriers in comparison to the crystal-line form. However, the resulting poor physical and chemical stability is the major challenge in the development of pharmaceutical products containing amorphous APIs, since they are less thermodynamically stable.33
1
10
100
1000
1 10 100 1000
Sol
ubili
ty in
crea
se
Particle diameter (nm)

25
There are several methods for preparing drug amorphous formulations, such as melt methods and spray drying. In melt methods, the drugs are heated above the melting point and cooled either slowly or rapidly in order to con-trol the drugs crystallization behavior.34 In the melt quenching method, the drug melts and cools rapidly to avoid crystal growth.35,36 Feeding drugs to-gether with a stabilizer into a thermo-controlled screw system to create a solid dispersion is called hot melt extrusion.37,38 These methods are not ap-propriate for thermolabile drug compounds since they need to be heated above their melting point. With the help of stabilizers, the spray-drying method has been used in the production of solid dispersions (amorphous drug forms) in large scale manu-facturing.39,40 Drugs are prepared in solution or slurry with solvent and a stabilizer before the spray drying process. The spray nozzle disperses the solution or slurry into atomized droplets. The solvent within the droplet is evaporated under hot gas flow.
3.2.3 Carriers and stabilizers Most of the physical methods enhance API solubility by preparing thermo- dynamically unstable (high energy) forms, since these require less energy to solubilize. Hence there is a compromise between all of these factors during the formulation of the drug compound. Various carriers have been intro-duced to the system as stabilizer. Moreover, some carriers have been devel-oped for solubility enhancement and as stabilizers, wetting agents or precipi-tation inhibitors.41
Figure 4. Sketch of (a) surfactant, (b) HPMC, (c) solid-lipid nanoparticles, and (d) dendrimers used as solubility enhancer or stabilizer for drug formulation.
3.2.3.1 Surfactants Surfactants are amphiphilic molecules that consist of at least one polar head group (hydrophilic) and one non-polar tail group (hydrophobic). They dif-fuse in water, accumulate at an interface (air-water or oil-water), and subse-quently modify the surface properties of the interface. Another characteristic

26
of surfactants is that the hydrophobic tail groups tend to aggregate to reduce the contact with water and subsequently form micelles in aqueous solution. The minimum concentration required to form micelles is called the critical micelle concentration (CMC), which is different for each surfactant. The driving force of these characteristics is to reduce the free energy of the sys-tem (e.g. the interface between a solid and a liquid). Surfactants in solution can form different liquid crystal phases including isotropic micellar, micellar cubic, hexagonal columnar, bicontinuous cubic and lamellar phases depend-ing on the concentration and temperature of the system.42 In the synthesis of mesoporous materials, this property is taken into consideration when design-ing different mesoporous structures (see section 3.3). Surfactants are often used to improve the wetting properties of solid drugs and increase the rate of disintegration of solids to finer particles. The interac-tion between the hydrophobic tail groups and the hydrophobic surface of the drug molecule may increase the solubility of drugs in water. They are also used as stabilizers by preventing the drug molecules from recrystallization.43
3.2.3.2 Polymer matrices Polymer matrices are the most common excipients for stabilizing either mi-cro/nano drug particles or amorphous states. In the conventional methods, a hydrophilic carrier matrix of one or more polymers are used, for example, polyvinylpyrrolidone (PVP), polyethylene glycol (PEG), and cellulose de-rivatives.33,44,45 Polymer matrices stabilize the dispersed amorphous drug particles or domains through vitrification. In some cases, the carriers also increase the wettability and the surface area of the dispersed amorphous drugs.46 Hydroxypropyl methylcellulose (HPMC) is one of the most commonly used drug excipient for solubility and stability enhancement. For example, the formulation of commercially available itraconazole capsule (Sporanox®) is a molecular dispersion of itraconazole in a HPMC matrix coated on inert sugar spheres. HPMC in this formulation is believed to enhance the solubility of itraconazole, increase the wettability and prevent itraconazole from precipi-tation in the stomach and in the intestine.47
3.2.3.3 Solid lipid nanoparticles Solid lipid nanoparticles (SLNs) are a type of colloidal* carrier reported to enhance the dissolution rate of poorly water-soluble drugs and control the release of encapsulated drugs.5,48–50 SLNs have a mean particle size between 50 to 1000 nm, are composed of physiological lipids dispersed in water or in
* Colloidal particles or colloids have typical sizes in the range between 1 and 1000 nm and are charac-terized by having clear phase boundaries and forming stable homogenous dispersions in other substances (e.g. a solid phase dispersed in a liquid).

27
an aqueous surfactant solution. The main advantages of SLNs are: i) they are synthesized from physiologically tolerated lipid components that have good biocompatibility,51,52 ii) SLN formulations can be stable for many years,53,54 and iii) their production is rapid and effective.51 Limited drug loading ca-pacity and drug leaching during storage are the main drawbacks of this carri-er.55
3.2.3.4 Dendrimers Dendrimers are repetitively branched molecules with core units, branching dendrons and surface ligands surrounding the core (Figure 4).56 Their unique structure makes them possible to entrap hydrophobic drug molecules by electrostatic interactions, hydrogen bonding, or polar forces. The formation of molecular dispersions of APIs within the dendrimer is one rationale for solubility enhancement.57 The high synthetic costs and complex purification of dendrimers are the main obstacles for the implementation of this technol-ogy. However, Starpharma Holdings Limited have successfully commercial-ized their dendrimer product, DEP™ docetaxel, for solubility enhancement of APIs.58
3.3 Mesoporous silica Porosity provides a remarkable increase in surface area that may give higher reactivity and efficiency. Ordered mesoporous silica were first synthesized in 1992 and are a family of nanostructured porous particles with pore widths between 2 to 50 nm.9,10 Their sharp pore size distribution, high surface area (often >900m2/g), and large pore volume were soon recognized as suitable properties for carrying and releasing drug payloads. Amorphous colloidal (non-porous) silicon dioxide is generally recognized as safe (GRAS), and stated as having “negligible toxicity when ingested”. Silicon dioxide and silica gel have received exemptions from tolerances and clearances for food commodities in USA.59 Furthermore the ease of functionalization of silanol-containing surface allows better control of drug release as well as adequate compatibility of the particles with many formulations and biological media. All these advantageous properties make mesoporous silica good candidates for pharmaceutical excipients in drug delivery.
3.3.1 Synthesis mechanism The most common synthesis of mesoporous silica materials is based on sur-factant template reaction using a combination of surfactant and sol-gel chemistry, where organic micellar-templated mesophases direct the silica monomers into a mesoscopically ordered amorphous structure. The template molecules form micelles in a self-assembling process. In aqueous solution,

28
micelles can form different liquid crystal phases like 1D-lamellar, 2D-hexagonal and 3D-cubic depending on the template molecule properties and solution conditions. A silica precursor is added to form silica walls surround-ing the micelles. Tetraethyl orthosilicate (TEOS) is one of the most com-monly used silica precursors. In water, TEOS hydrolyzes replacing the alkoxide groups (-OR) with hydroxyl groups (-OH), producing silanol groups (≡Si-OH). The following condensation reactions involving the si-lanol groups lead to siloxane bonds (≡Si-O-Si≡). After a suitable condensa-tion period, the silica-organic framework is filtered and the organic template is removed by calcination or by solvent extraction (see Figure 5).
Figure 5. A generalized synthesis route for mesoporous silica materials in a surfac-tant template type mechanism.
3.3.2 Classification of mesoporous silica from their template system
Mesoporous silica particles can be classified by their template system. Typi-cal template systems reported for mesoporous silica syntheses are: surfac-tants, triblock copolymers, and other molecules that can form structured meso-units through supramolecular hydrogen bonding, e.g. folic acid.60 Cationic surfactant templated mesoporous silica Surfactants can form different liquid crystal phases above their critical mi-cellar concentration (CMC). Depending on their head group charge, surfac-tants are classified as anionic, cationic, non-ionic (neutral) and zwitterionic. Cationic surfactants were used for the synthesis of the first mesoporous silica MCM-41.9,10 The synthesis mechanism for cationic surfactant template mes-oporous silica involves the interaction of the surfactant head group (S+) and silica species (I-) under basic conditions.
Micelles and silica precursor
Organic and inorganic phase, as-synthesized mesoporous silica
Mesoporous silica
SiOC2H5
C2H5O OC2H5
OC2H5Si(OH)4
H2OCH3CH2OHH2O
SiO2

29
Anionic surfactant templated mesoporous silica The synthesis of mesoporous silica using anionic surfactants as templates (e.g. N-Lauroyl-L-alanine) is conducted under alkaline conditions where the charges of the silica species are negative, I-. When using anionic surfactants as the template, a co-structure directing agent (CSDA) is applied to create a bridge between the negatively charged surfactant head group and the nega-tively charged silica species: S-N+I-.61 One of the most common CSDA used is 3-aminopropyl triethoxysilane (APES, shown in Figure 6).
Figure 6. Schematic illustration of the interaction between the head group of anion-ic surfactant and APES.
Triblock copolymer templated mesoporous silica In 1998, Zhao et al.62 synthesized ordered mesoporous silica particles by using triblock copolymer, Poly(ethylene glycol)-block-poly(propylene gly-col)-block-poly(ethylene glycol) (P123), as the template. P123 is nonionic triblock copolymer composed of a central hydrophobic chain of polyoxypro-pylene (PPO) flanked by two hydrophilic chains of polyoxyethylene (PEO). The synthesis is performed in strong acidic conditions. The synthesis mech-anism is based on the electrostatic interaction between the hydrophilic head group(S0), proton(H+), anions (Cl- in this case) and silica species(I+): (S0H+)(Cl-I+). Non-surfactant (folic acid) templated mesoporous silica Some biological molecules such as nucleotides, vitamins and their deriva-tives (DNA, RNA, etc.) can also form supramolecular structures in aqueous solutions, through hydrogen bonding and other supramolecular interactions. For example, folic acid (FA) consists of a pterin group, a p-aminobenzoyl group and a glutamic acid moiety,63 where the pterin groups form planar tetramers and subsequently self-aggregate to hexagonal liquid crystal phases

30
through π-π interaction(Figure 7). The folic acid negatively charged carbox-ylate groups are exposed towards the exterior of the stack. The amino group of the CSDA, 3-aminopropyl triethoxysilane (APES), is used to interact with the exposed carboxylate group. The mechanism of folic acid templated mes-oporous silica can be considered to be FA-N+I-.
(a) (b)
Figure 7. (a) Structures of organic template molecules used in this thesis. (b) Su-pramolecular self-assembled tetramers formed by folic acid molecules.
3.3.3 Classification of mesoporous silica according to their pore structure
Ordered mesoporous silica particles have been prepared with a variety of mesostructures ranging from 1D-lamellar to 3D-connected cage type struc-tures governed by cubic or hexagonal close packing of spherical micelles. Structures with cylindrical pores have uniform pore diameters (e.g. MCM-489, AMS-664, NFM-165, SBA-1562), structures with interconnected cage- type pores possess spherical or ellipsoidal cages that are connected by small-er pores (window), such as FDU-166, SBA-1666–68 and AMS-869. The pore structure can be determined by low angle X-ray diffraction (2θ between 0.5° and 6°) and transmission electron microscopy (TEM). In this work two types of mesostructures were investigated, possessing hex-agonal cylindrical pores of two sizes (large: SBA-15 and small: NFM-1) and cubic interconnected cylindrical pores (large: STA-11 and small: AMS-6) to compare the influence of pore sizes and pore structures on the drug dissolu-tion behavior. The mesoporous materials used in this thesis are:
R
N
N
NN
ON
HH
HN
N
N
N
O
NH
H
H
N
N
NN
O N
HHH
NN
N
N
O
N H
H
H
OHO
O
OHO
HN
NH
N
HN N
N
O
H2N
CHNCH
COOH
CH3O
HO CH2CH2O CH2CHO
CH3CH2CH2O H20 70 20
N-Lauroyl-L-alanine
P123
Folic acid
R
R
R
R

31
SBA-15 Santa Barbara amorphous-15 (SBA-15) is first synthesized by Zhao et al. using triblock copolymer P123 as template.62 The SBA-15 particles have 2D hexagonal pore symmetry, where the pore size is approximately 8 nm. This material also possesses micropores within the pore wall of less than 2 nm. In paper III of this thesis, two versions of SBA-15, with or without microporos-ity, have been used to compare the release of an anti-cancer drug dasatinib. STA-11 This material has 3D-bicontinuous cubic pore structure. The synthesis of St. Andrews University material-11 (STA-11) is derived from SBA-15, using P123 as a template.70 In the synthesis, 3-mercaptopropyl triethoxysilane (MPTS) is added as a CSDA. The thiol groups in MPTS penetrate into the P123 micelles and induce a phase transformation from a 2D-hexagonal to a 3D-bicontinous cubic structure by swelling the surfactant. The particles are spherical with a size around 4.8 µm (measured by SEM). AMS-6 AMS-6 belongs to the group of anionic surfactant-templated mesoporous silica materials (AMS-n). Its synthesis uses N-lauroyl-L-alanine as template, which is negatively charged in the synthesis solution and APES as the CSDA creating an electrostatic interaction between the anionic surfactant (S-
) and silica framework (I-). AMS-6 particles have a 3D cubic cylindrical pore structure with a pore size of approximately 4.5 nm, depending on specific synthesis conditions (see papers). The particle size is approximately 300 nm measured by scanning electron microscopy (SEM). NFM-1 The synthesis of NFM-1 mesoporous silica particles is based on the supra-molecular self-assembly of folic acid molecules. NFM-1 particles have a pore size of 2.5 nm with a hexagonal pore structure. The particles morpholo-gy is varied from spheroids, to rods, and to fibers, depending on the synthe-sis conditions.71 AMP-1 Amorphous mesoporous silica particles (AMP-1) particles are derived from the synthesis of MCM-41 using CTAB as templates. The particles are syn-thesis under alkaline condition and have worm-like pore structure (More details described in paper II).

32
Figure 8. Pore structure unit cell models showing the pore structures of (a) 3D cubic structure (AMS-6 and STA-11), and (b) 2D hexagonal structure (SBA-15 and NFM-1).
3.3.4 Toxicology Toxicology studies of mesoporous silica have been conducted in order to ascertain safety in their use in drug delivery. It is well known that silicosis* is derived from breathing crystalline silica dust. However, it is of importance to differentiate the toxicity of crystalline and amorphous silica families. Amor-phous silica is reported to have a better lung tolerability compared to crystal-line silica, probably due to its higher surface area and amorphous state that leads to faster dissolution and removal from the alveoli.72 The toxicity of mesoporous silica needs to be carefully assessed before in-troducing it as a functional excipient. The administration route is crucial for toxicological assessment. Hudson et al. reported that doses of 40 mg/kg of mesoporous silica (MCM-41, SBA-15 and mesocellular foam particles† with particle size of 29-140 nm, 800 nm, and 4 µm with pore size of 3, 7, 16 nm respectively) are safe through intraperitoneal (i.p.) and intravenous (i.v.) administration to mice. Doses of 1.2 g/kg were lethal or lead to euthanasia of mice. No toxicity was observed when administered intramuscularly or sub-cutaneously.73 The effect of particle shape is reported by Huang et al.,74 which indicates that short-rod mesoporous silica particles after intravenous administration have a faster clearance rate than long-rod particles with both excretion routes. Another study also demonstrated that a single dose of 500 mg/kg and daily doses of 80 mg/kg for 14 days showed no sign of toxicity, with a complete particle clearance from the organism completed after 4 weeks of intravenous administration.75 Mesoporous silica has the same chemical composition as amorphous colloi-dal silica which is approved for use as a food additive (E551 in EU regula-
* Silicosis is a form of occupational lung disease which is an irreversible condition. The symptoms in-clude shortness of breath, cough, fever, chest pain, etc. † Mesocellular foam is a material with a larger, more disordered pore system.
(a) (b)

33
tions).76 In this thesis, we reported an in vivo oral toxicology study with two different types of mesoporous silica particles (see paper I and Chapter 5).
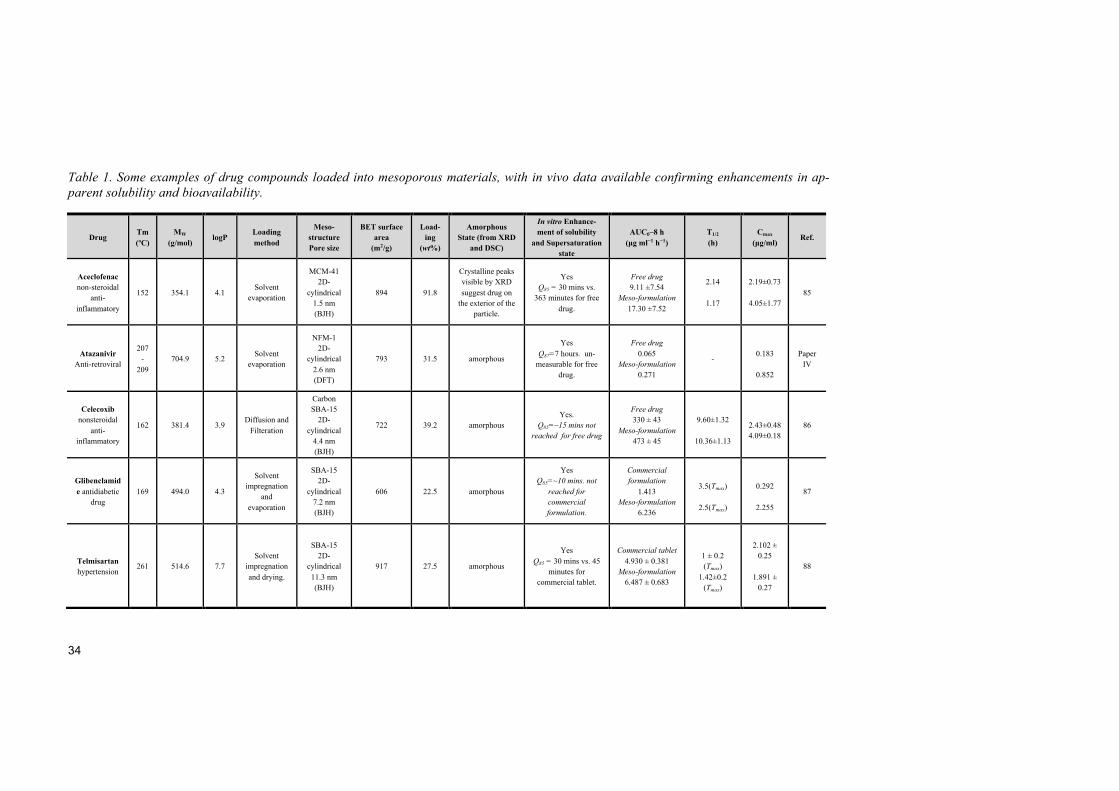
3.3.5 Application in life science It was not until 10 years after the discovery of mesoporous silica that its application in drug delivery was proposed.8 In 2001, Vallet-Regi et al. pro-posed MCM-41 particles to be used to deliver drug compounds. Mesoporous silica attracts lots of attention for targeted drug delivery in cancer therapy. Zink and his co-workers increased the accumulation of mesoporous silica nanoparticles in tumor to 12 % leading to an enhanced tumor inhibition rate.77 Multifunctional mesoporous silica particles incorporating iron oxide nanocrystals, anti-cancer drugs and folic acid have been reported as a strate-gy for simultaneous imaging and therapeutic applications.78 Another good example of potential uses of mesoporous silica is to contribute to solving multidrug resistance. Chen et al. reported that co-delivery of anti-cancer drug, doxorubicin and Bcl-2 siRNA by mesoporous silica could enhance the efficacy of doxorubicin in drug-resistant cancer cells.79 Vallhov et al. report-ed that mesoporous silica could be used as adjuvants in immunotherapies.80 Mesoporous silica has also been found useful to tailor the release kinetics of APIs, DNA, siRNA, nutraceuticals and growth factors from the internal pore space of mesopourous materials.81 It also has been found to be a potentially versatile vehicle for local delivery of neurotrophic factors, where sustained release of soluble factors was achieved after loading into mesoporous silica.82,83 Among all potential applications of mesoporous silica, solubility and bioa-vailability enhancement is the one receiving most attention from industry, since bioavailability enhancement is the major goal in oral drug formula-tions.12,14,84 In vivo studies on bioavailability enhancement when loaded with mesoporous silica are compared in Table 1.85–88 Many poorly water-soluble drugs show increased bioavailability after loading into mesoporous silica compared to the free drugs or commercial formulations. Mesoporous silica particles show good potential in stabilizing the amorphous state of these APIs. The main focus of this thesis is to explore the impact of applying suit-able mesoporous materials to enhance the pharmacokinetic profile of poorly water-soluble compounds.
34
Table 1. Some examples of drug compounds loaded into mesoporous materials, with in vivo data available confirming enhancements in ap-parent solubility and bioavailability.
Drug Tm (ºC)
MW (g/mol)
logP Loading method
Meso-structure Pore size
BET surface area
(m2/g)
Load-ing
(wt%)
Amorphous State (from XRD
and DSC)
In vitro Enhance-ment of solubility
and Supersaturation state
AUC0–8 h (µg ml−1 h−1)
T1/2
(h) Cmax
(µg/ml) Ref.
Aceclofenac non-steroidal
anti-inflammatory
152 354.1 4.1 Solvent
evaporation
MCM-41 2D-
cylindrical 1.5 nm (BJH)
894 91.8
Crystalline peaks visible by XRD suggest drug on
the exterior of the particle.
Yes Q85 = 30 mins vs.
363 minutes for free drug.
Free drug 9.11 ±7.54
Meso-formulation 17.30 ±7.52
2.14
1.17
2.19±0.73
4.05±1.77 85
Atazanivir Anti-retroviral
207-
209 704.9 5.2 Solvent
evaporation
NFM-1 2D-
cylindrical 2.6 nm (DFT)
793 31.5 amorphous
Yes Q87=7 hours. un-
measurable for free drug.
Free drug 0.065
Meso-formulation 0.271
-
0.183
0.852
Paper IV
Celecoxib nonsteroidal
anti-inflammatory
162 381.4 3.9 Diffusion and
Filteration
Carbon SBA-15
2D-cylindrical
4.4 nm (BJH)
722 39.2 amorphous Yes.
Q85=~15 mins not reached for free drug
Free drug 330 ± 43
Meso-formulation 473 ± 45
9.60±1.32
10.36±1.13
2.43±0.48 4.09±0.18
86
Glibenclamide antidiabetic
drug 169 494.0 4.3
Solvent impregnation
and evaporation
SBA-15 2D-
cylindrical 7.2 nm (BJH)
606 22.5 amorphous
Yes Q85=~10 mins. not
reached for commercial formulation.
Commercial formulation
1.413 Meso-formulation
6.236
3.5(Tmax)
2.5(Tmax)
0.292
2.255
87
Telmisartan hypertension
261 514.6 7.7 Solvent
impregnation and drying.
SBA-15 2D-
cylindrical 11.3 nm (BJH)
917 27.5 amorphous
Yes Q85 = 30 mins vs. 45
minutes for commercial tablet.
Commercial tablet 4.930 ± 0.381
Meso-formulation 6.487 ± 0.683
1 ± 0.2 (Tmax)
1.42±0.2 (Tmax)
2.102 ±
0.25
1.891 ± 0.27
88

35
4 Experimental
4.1 Materials
4.1.1 Mesoporous silica particles Six different types of mesoporous silica particles have been used in this the-sis. For the synthesis of various mesoporous silica materials, anionic surfac-tants N-lauroyl-L-alanine, triblock copolymers Pluronic® P123 and P104, and folic acid have been used as templates. P123 and P104 differ in the ratio of PEO:PPO:PEO (P123, 20:70:20, P104, 27:61:27). All surfactants used form micelles in aqueous solution under alkaline or acidic conditions (details in Table 2). APES has been used as CSDA in the synthesis of NFM-1 and AMS-6. MPTS has been used as CSDA in the synthesis of STA-11. Tetrae-thyl orthosilicate (TEOS) and tetramethyl orthosilicate (TMOS, only in pa-per III) were used as silica source in this thesis. Chemicals were purchased from Sigma-Aldrich except for N-lauroyl-L-alanine that was purchased from Nanologica AB, Sweden. Unless stated chemicals were used as received without further purification. The as-synthesized materials were calcined at 500 - 550°C for 6 hours to remove the templates.
Table 2. Synthesis details of the mesoporous silica materials included in this thesis.
* This material was with additional NaI at a final concentration of 1M.
Material Synthesis route Template Pore size
(nm) Pore structure Synthesis conditions Publication
NFM-1 FA-N+I- FA 2-3 2D-hexagonal alkaline I, II, IV
AMS-6 S-N+I- C12Ala 4-5 3D-bicontinuous alkaline I, II, IV, V
AMP-1 (S+I-) CTAB 6-7 Worm-like alkaline II
SBA-15 (S0H+)(X-I+) P123 7-8 2D-hexagonal acidic II
SBA-15(micro-meso) (S0H+)(X-I+) P104 7-8 2D-hexagonal acidic III
SBA-15(meso)* (S0H+)(X-I+) P104 7-8 2D-hexagonal acidic III
STA-11 (S0H+)(X-I+) P123 7-8 3D-bicontinuous acidic IV
36
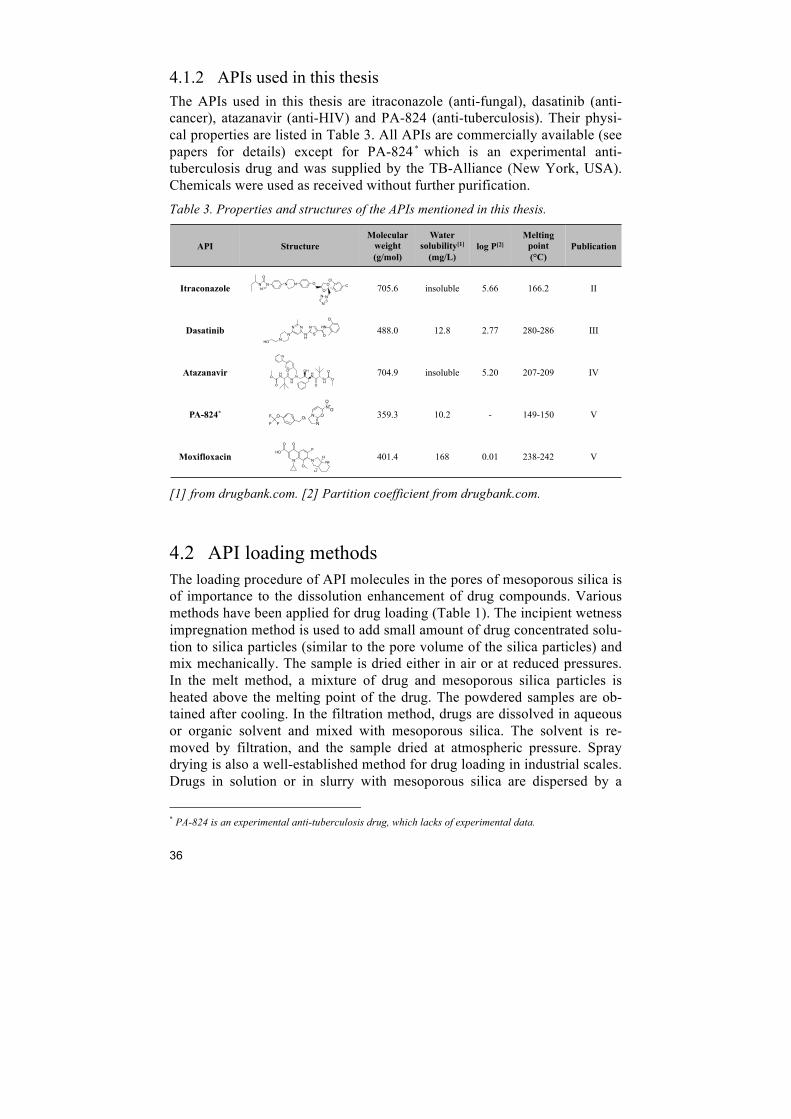
4.1.2 APIs used in this thesis The APIs used in this thesis are itraconazole (anti-fungal), dasatinib (anti-cancer), atazanavir (anti-HIV) and PA-824 (anti-tuberculosis). Their physi-cal properties are listed in Table 3. All APIs are commercially available (see papers for details) except for PA-824 * which is an experimental anti-tuberculosis drug and was supplied by the TB-Alliance (New York, USA). Chemicals were used as received without further purification.
Table 3. Properties and structures of the APIs mentioned in this thesis.
[1] from drugbank.com. [2] Partition coefficient from drugbank.com.
4.2 API loading methods The loading procedure of API molecules in the pores of mesoporous silica is of importance to the dissolution enhancement of drug compounds. Various methods have been applied for drug loading (Table 1). The incipient wetness impregnation method is used to add small amount of drug concentrated solu-tion to silica particles (similar to the pore volume of the silica particles) and mix mechanically. The sample is dried either in air or at reduced pressures. In the melt method, a mixture of drug and mesoporous silica particles is heated above the melting point of the drug. The powdered samples are ob-tained after cooling. In the filtration method, drugs are dissolved in aqueous or organic solvent and mixed with mesoporous silica. The solvent is re-moved by filtration, and the sample dried at atmospheric pressure. Spray drying is also a well-established method for drug loading in industrial scales. Drugs in solution or in slurry with mesoporous silica are dispersed by a
* PA-824 is an experimental anti-tuberculosis drug, which lacks of experimental data.
API Structure Molecular
weight (g/mol)
Water solubility[1]
(mg/L) log P[2]
Melting point (°C)
Publication
Itraconazole 705.6 insoluble 5.66 166.2 II
Dasatinib 488.0 12.8 2.77 280-286 III
Atazanavir 704.9 insoluble 5.20 207-209 IV
PA-824* 359.3 10.2 - 149-150 V
Moxifloxacin 401.4 168 0.01 238-242 V
HN
O
O
O
NH
N
N
HN
O
NH
O
O
OH
ON
NOO
FF
F
N+O
O-
N
O
HO
O
O
F
NHNH
H
S
N
O
HN
NH
NN
NNHO
Cl
NN N
O
N N O O
ONN
N
ClCl

37
spray nozzle into atomized droplets. The solvent in the droplets is evapo-rated under hot gas flow. In this thesis, APIs were loaded into mesoporous silica particles by wetness impregnation followed by solvent evaporation, since it is practical for la-boratory scale control of the loading amount. APIs were dissolved in organic solvent forming a concentrated drug solution. After adding calcined mesopo-rous silica particles in the concentrated drug solution, the suspension was brought to equilibrium under gentle stirring for 2 hours. The solvent was removed by rotary evaporation under reduced pressure. Samples were left to dry open to air over night.
4.3 Analytical methods
4.3.1 Powder X-ray diffraction (XRD) Powder X-ray diffraction is used for two purposes in this thesis: i) to deter-mine the mesophases structure, and ii) to investigate the crystallinity of APIs after loading into mesoporous silica. XRD is a fundamental technique to determine the atomic and molecular structure of a crystal. X-rays can be considered waves of electromagnetic radiation. When an incident X-ray beam hits a crystal with regular array of atoms, it scatters forming a regular array of waves. These waves interfere constructively with each other along specific directions, determined by Bragg’s law (Eq. 3): 2!"#$% = !" (3) Here d is the interplanar distance, θ is the scattering angle, n is any integer, and λ is the wavelength of the incident X-ray beam. The incident X-ray radi-ation produces peaks at certain scattering angles and gives a diffraction pat-tern, which contains the structural information of the sample.
4.3.1.1 Low angle X-ray diffraction The periodicity of mesoporous silica materials arises from their pore struc-ture, and is typically larger than 2 nm resulting in large unit cell parameters with diffraction peaks occurring at low scattering angles. Low angle X-ray diffraction is used here to investigate the pore structure of mesoporous silica materials. Ordered mesoporous silica particles give reflections in X-ray dif-fraction at two regions: the periodicity of the mesopores give sharp peaks between 0.5° to 6° (2θ), and a broad peak between 20° to 24° (2θ) is con-sistent with the amorphous silica framework. Mesostructures of this kind can

38
be described by conventional crystallographic space group symmetries. However, the structural determination of mesoporous materials is rather difficult due to insufficient diffraction peaks. For a detailed structure analy-sis, transmission electron microscopy (TEM) is a good complementary tech-nique. Low angle XRD patterns presented in this thesis were collected by a powder diffractometer (PAnalytical X’Pert alpha1, Almelo, the Netherlands) using CuKα radiation (λ = 1.5418 Å) at 40 kV and 40 mA. The diffraction patterns were recorded between 1° and 6°.
4.3.1.2 High angle X-ray diffraction XRD is also used to determine the crystallinity of API loaded into mesopo-rous silica. The XRD patterns for loaded samples were collected in the same instrument mentioned in Low angle XRD with different settings. The 2θ is in a range of 5° to 50°.
4.3.2 Small angle X-ray scattering (SAXS) Small angle X-ray scattering is an X-ray diffraction based technique that is used to determine the structure of calcined SBA-15 samples in paper III. The SAXS patterns were recorded by a Ganesha SAXS system (SAXSLAB, Denmark) with 2θ between 0.1° to 4°. The powder samples were placed in quartz capillaries sealed with wax.
4.3.3 Scanning electron microscopy (SEM) Scanning electron microscopy (SEM) is used to study the morphology and topography of the particles and their surfaces as well as their size. In a scan-ning electron microscope a filament emits a beam of electrons that interact with atoms in the sample producing various signals. The signals (secondary electrons, backscattered electrons, characteristic X-rays, cathodolumines-cence, etc.) that can be detected contain information about the sample’s sur-face topography and composition. In this thesis, all the SEM images were obtained by a JEOL JSM-7401F SEM operating at an accelerating voltage of 1-2kV without sputtered coating. In some SEM images in this thesis, an in-lens detector was used to collect upper secondary electron signals.
4.3.4 Transmission electron microscopy (TEM) Transmission electron microscopy (TEM) is used to determine the pore structure of mesoporous silica particles complementing low angle XRD, since low angle XRD provides limited structural information that is not suf-ficient to refine the structure of a mesoporous material. TEM provides the crystallographic structural information of the sample in atomic scale using a

39
high-energy electron beam. The electron beam generated by an electron gun, typically a tungsten filament, interacts and transmits through an ultra-thin specimen after being accelerated and focused by electromagnetic lenses. Electrons are scattered or transmitted through the sample, which produces a non-uniform distribution of electrons containing structural and chemical information of the sample. An image is obtained by detecting electron transmitted through the specimen on a fluorescent screen, a layer of photo-graphic film or a charge-coupled device (CCD) camera. In this thesis, TEM images were collected using a JEOL-3010 microscope, operating at 300 kV (Cs 0.6 mm, resolution 1.7 Å). Images were recorded using a CCD camera with specialized imaging shadow-graph (SIS) analysis (size=1024×1024megapixels, pixel size=23.5×23.5 µm; model: Keen View, Olympus Soft Imaging Solutions, Munster, Germany) using low-dose condi-tions on calcined and loaded samples.
4.3.5 Nitrogen adsorption/desorption isotherms Gas adsorption is the most practical and reliable method to determine the porosity of mesoporous materials. The textural information of a solid materi-al including surface area, pore volume and pore size distribution can be giv-en by the adsorption of gases on a porous solid at various relative pressures and fixed temperature.89 For silica based mesoporous materials, nitrogen is the most common adsorbate for porosity measurements. In a typical meas-urement, a calibrated volume of gas is added into a known amount of meso-porous silica sample in a tube of pre-determined volume immersed in liquid nitrogen. An adsorption isotherm can be obtained by plotting the amount of adsorbed gas against the relative pressure after a reasonable equilibration time, which is the pressure difference in the sample tube after the addition of known amount of nitrogen. The porosity information, such as surface area, pore volume and pore size distribution, can be calculated from the nitrogen adsorption isotherm curves. The surface area in this thesis is given by the amount of gas molecules needed to cover the surface with a complete mono-layer based on the Brunauer-Emmett-Teller (BET) theory developed in 1938.90 The result is calculated in the relative pressure (p/p0) range of 0.05-0.3. The total pore volume is obtained by the amount of gas adsorbed at a relative pressure close to 0.94 from adsorption. The calculation of pore size distribution in this thesis is based on density functional theory,91,92 taking into consideration various pore geometries. The equilibrium distribution of the adsorbate in the pores can be presented as a function of the density of adsorbed fluid.93 In this work, nitrogen adsorption/desorption isotherm was used to i) deter-mine the porosity of calcined mesoporous silica particles, and ii) to confirm the presence of loaded drug compounds within the pores, from the changes

40
in pore volume and surface area. All the isotherm data were obtained at 77K using a Micromeritics Tristar II 3020 apparatus.
4.3.6 Thermogravimetric analysis (TGA) Thermogravimetric analysis (TGA) is a method for measuring the change in weight of samples as a function of increasing temperature with a constant heating rate, which allows obtaining information about the physical and chemical properties of a material. In this thesis, the measurement is per-formed using a TGA-7 instrument (PerkinElmer, MA, USA) with a tempera-ture ramp between 20 and 1000°C and a heating rate of 20°C/min in air at-mosphere. In a mesoporous silica – API system, silica is thermally stable under 1000°C, while the API is an organic compound that is decomposed during the measurement. It is used in this thesis to determine the amount of API loaded into mesoporous silica materials. The decomposition temperature of loaded API shifts to lower temperatures compared to free drug compound since it is amorphous within the pores and less thermodynamically stable.
4.3.7 Differential scanning calorimetry (DSC) Differential scanning calorimetry (DSC) measures the difference in the amount of heat required to increase the temperature of a sample and refer-ence, as a function of temperature. In a DSC analysis, the temperature of the sample and reference holder is nearly the same increasing linearly as a func-tion of time. When the sample undergoes a physical transformation such as melting or crystallization, it will either absorb or release heat to the envi-ronment. In order to maintain the same temperature as the reference, more or less heat flows to the sample, where the amount of heat flow can be meas-ured. In this thesis, DSC is used to study the crystallinity level of the loaded sample. By analyzing API loaded mesoporous silica sample in DSC, the ratio of API in its amorphous and crystalline states can be calculated. A DSC Q2000 (TA Instruments, NJ, USA) has been used for the work in this thesis. The loaded sample (4 to 6 mg) added in an open aluminum sample pan and heated from 20°C to maximum 400°C (depend on the API melting point) at a temperature ramping rate of 10 °C/min.
4.3.8 Dynamic light scattering (DLS) Dynamic light scattering (DLS) is a technique to determine the size distribu-tion of particles in suspension. Particles in suspension undergo Brownian motion, where the distance between particles is constantly changing with time. When light hits the particles in solution, it scatters in all directions at different intensities. The particle size information can be calculated by these intensity fluctuations using Stokes-Einstein theory (Eq. 4):

41
! = !"!!"!!
(4) Here D is the diffusion constant, k is Boltzmann’s constant, T is the absolute temperature, η is the viscosity of the fluid, and dH is the hydrodynamic diam-eter of the particle. In this thesis, DLS is conducted by a Mastersizer 2000 particle size analyzer (Malvern instruments Ltd., UK) for determining the particle size distribution of mesoporous silica particles in water or buffer.
4.4 Dissolution testing Dissolution testing was used in this thesis to obtain the kinetic release curves of free and loaded API. The technique is also routinely used to provide in vitro drug release (kinetic) information in the pharmaceutical industry. To mimic the conditions in vivo in the dissolution testing, buffers with simi-lar pH as stomach and intestine fluids were used in the experiments. The dissolution media used in this thesis are simulated gastric fluid (SGF, pH 1.2), simulated intestinal fluid (SIF, pH 6.8), and phosphate buffered saline (PBS, pH 7.4). According to the United States Pharmacopeial Convention (USP), there are four standard apparatuses: i) USP Dissolution Apparatus 1 – Basket (37°C), ii) USP Dissolution Apparatus 2 – Paddle (37°C), iii) USP Dissolution Ap-paratus 3 – Reciprocating Cylinder (37°C), iv) USP Dissolution Apparatus 4 – Flow-Through Cell (37°C).94 For oral solid dosage forms, the most com-monly used apparatus is USP 2, which was also used in this thesis. The re-lease profile was obtained by UV/Vis spectrophotometer (Cecil, CE3021, UK).
4.5 In vivo oral administration
4.5.1 Oral tolerance Studies were performed on Sprague Dawley rats (6 – 7 weeks in age). The animals were administered with either NFM-1 or AMS-6 by oral gavage formulated in PBS. Animals administered with PBS alone were used as the control group. A single dose stepwise procedure was performed for a dose range finding test (DRF). Four animals were administered particles at the starting doses of 30 mg/kg for NFM-1 and 40 mg/kg for AMS-6. The ab-sence or presence of particle-related clinical signs indicated the next dose level. (For more dosage details refer to Table 2 of paper I.) The following

42
confirmatory test (CT) was performed on different groups of animals (five males and five females in each group). The dose of NFM-1 and AMS-6 in the CT was 2000 mg/kg and 1200 mg/kg, respectively, for 7 consecutive days. More experimental details can be found in paper I.
4.5.2 Administration of atazanavir with omeprazole Studies were performed on Sprague Dawley rats (6 – 7 weeks in age). All rats were dosed orally with 100 mg/kg of omeprazole 5 hours prior to the administration of atazanavir (10 mg/kg). From literature, stomach acid reaches the lowest acidity 5 hours after the administration of omeprazole.95 The dose of atazanavir loading into mesoporous silica is equivalent to free atazanavir calculated by the loading amount (based on TGA results). All powders were administered by oral gavage as a suspension in phosphate buffered saline. The time points for blood extraction were 0.5 h, 1 h, 2 h, 4 h, 8 h and 24 h after atazanavir administration. The whole blood samples were analyzed by High-performance liquid chromatography (more experimental details given in paper IV).
4.6 Intracellular study The High-content screening assay in infected macrophages was performed based on the protocol developed by Christophe et al.96 Briefly, 384-well Evotec plates (#781058) were first pre-plated with 0.5 ml of drug com-pounds or drug-loaded mesoporous silica particles or controls dispensed by an automated dispensing system EVOBird (Evotec) in 10 µl RPMI 1640 (Gibco) supplemented with 10% heat-inactivated fetal calf serum (FCS, Gibco). Raw 264.7 cells (American Type Culture Collection TIB-71) were infected with Mycobacterium tuberculosis H37Rv-GFP at a multiplicity of infection of 2:1 and dispensed into 384-well plates. After 5 d of incubation, macrophages were stained with Syto 60, 5µm (invitrogen, S11342) for 1 hour at 37°C. Image acquisition was performed on an EVOscreen Mark III platform integrated with Opera. Bacterial load and macrophage number were quantified using proprietary image analysis software. Isoniazid and DMSO has been used as positive and negative controls, respectively.
Figure 9. Scheme of high-content screening assay in infected macrophages.
Infection 2h
Add to cell culture plate
Drug solution or controls
Incubated for 5 days
Analysis

43
5 Oral toxicity of mesoporous silica
Adequate toxicological and biocompatibility profiles of mesoporous silica are a prerequisite for their application in drug delivery. Toxicological studies of different mesoporous silica particles have been conducted in several ad-ministration routes. For instance, Lu et al. 97 reported that 100 mg/kg dose of mesoporous silica particles (particle size 50 – 100 nm) for 10 days are well tolerated by female nude mice administered intravenously. In 2012, the max-imum tolerated dose (MTD) after intravenous administration of mesoporous silica and nonporous silica with different morphologies was determined to be 30-65 mg/kg and 450 mg/kg, respectively.98 However, no reports regarding the toxicological properties of mesoporous silica particles under oral admin-istration have been published. In this work, we evaluated the oral toxicological properties of two types of mesoporous silica, AMS-6 (average 230 nm particle size by DLS) and NFM-1 (2168 nm particle size by DLS, see Table 1, paper I). The MTD was de-termined by oral administration. The silica particles were administered by oral gavage. The study was divided to two steps: dose range finding test (DRF) and confirmatory test (CT). In the DRF, the animals were adminis-tered with a range of doses respectively and monitored to detect any signs of discomfort or toxicity. The MTD was not reached at a dose of 2000 mg/kg and 1200 mg/kg of NFM-1 and AMS-6 particles, respectively. No mortality was recorded associated with any of the NFM-1 and AMS-6 doses: 30 to 2000 mg/kg for NFM-1, and 40 to 1200 mg/kg for AMS-6. The animals exhibited normal behavior and normal bodyweight development. It is worth noting that the dispersion properties of the particles in PBS made it difficult to approach higher doses. The highest dose in the DRF was administered in the subsequent confirmatory test (CT). In the CT, animals were administered mesoporous silica daily for 7 consecu-tive days. No relevant clinical signs were observed in response to the parti-cles in all the animals that reached the end of the study. The bodyweight evolution of both groups that were administered with NFM-1 and AMS-6 was normal throughout (Figure 10). There was no difference in hematology or biochemistry parameters from animals treated with mesoporous silica compared with the control animals. Minor negative signs of toxicity, sporad-ic rhinorrhea (runny nose), were observed in both test and control group,

44
indicating that they are associated with the administration technique. Ab-sence of food in intestine was observed in one animal (NFM-1 group) prob-ably due to a blockage in the stomach caused by the silica particles. Breath-ing difficulties and a white fluid in the lungs were observed, which may be associated with toxic effects. However, these signs could result from acci-dental aspiration of dosed formulation as a result of reflux after oral gavage administration. The toxic effects were caused by the particles formulation reaching the lungs, not by oral administration of the particles per se.
Figure 10. Normal bodyweight development throughout the confirmatory test. The animals were administered by oral gavage for 7 consecutive days with NFM-1 and AMS-6 formulated in PBS. The control group was administered with PBS alone. M=male, F=female animals.
Our results appear to be consistent with those observed for other toxicologi-cal studies using different type of solid (nano)particles. Polystyrene particles were not absorbed through the GI tract neither, when the particles size was above 300 nm in a similar rat study.99 In a previous toxicology study using non-porous amorphous silica particles (with the same chemical composition as mesoporous silica), these were found to be well tolerated by rats at an oral dose of 500 mg/kg per day, for a period of 6 months. In an even longer term study, a daily dose of 100 mg/kg of amorphous silica was given to 20 male and 20 female Wistar rats for 2 years without noting any adverse effects on behavior, clinical signs and weight gain.100 Neither NFM-1 nor AMS-6 is absorbed by the GI tract probably due to their large particle size, contrib-uting to their oral tolerance. This work showed both NFM-1 and AMS-6 mesoporous silica particles are well tolerated in rats under oral administra-tion even at high doses, which is strong support for the development of mes-oporous silica as oral drug delivery vehicles. However, these results suggest that drug targeting may be limited in the context of oral administration, or at very least be only attainable with particles below 300 nm in size.

45
6 Improving drug dissolution
6.1 Comparison of porous silica particles in the dissolution of itraconazole
Various studies have shown that drugs loaded into mesoporous silica have higher apparent solubility and bioavailability (Table 1). In this work, we compared the dissolution of a model drug, itraconazole, when it is loaded into a range of different mesoporous silica particles, commercially available silica particles and non-porous silica particles. Itraconazole is a lipophilic drug with a log P of 5.66. Mellaerts and coworkers have studied itraconazole loaded into mesoporous silica particles both in vitro and in vivo, suggesting that mesoporous silica particles (SBA-15) can enhance the solubility and bioavailability of itraconacole.12,101 It is of great interest to conduct a system-atic study for better understanding of the mechanism of solubility enhance-ment and the degree of crystallization of the drugs within the pores. NFM-1, SBA-15 and Evonik fumed silica (EVK) particles were loaded with itraconazole at 30 wt%, 50 wt% and 70 wt%, respectively. Details of silica particles used in this work are shown in Table 1 in paper II. The solid disper-sion state of itraconazole after loading was analyzed by DSC, XRD and ni-trogen isotherm. Figure 11(a) shows that free itraconazole in the crystallite phase melts at between 166 and 170 °C. From the DSC curves in Figure 11 (b) and (d), the loaded samples at 30 wt% are fully amorphous, whereas at 50 wt% and 70 wt% a clear melting peak suggests the presence of crystalline drug. NFM-1 particles at 30 wt% show a small melting peak, indicating re-crystallization occured at this loading amount. However, in Figure 11 (d), drug loaded in EVK particles have a melting peak at 153 °C at 50 wt% load-ing, and two melting peaks are observed for the sample at 70 wt% loading. The melting peak at 153 °C corresponds to a polymorphic phase of itracona-zole.102 Evonik particles induce the drug to form a different polymorphic phase at 50 wt%, and a mixture of at least two polymorphic phases at 70 wt% loading. Non-porous silica particles do not prevent the drug from re-crystallization. XRD results are consistent with DSC, which also suggests that evaporation of the drug onto the surface of non-porous micron-sized particles results in high crystallinity (see paper II, Figure 2).

46
Figure 11. (a) DSC curves of free itraconazole and itraconazole (ITZ) loaded into non-porous silica particles, and DSC curves of itraconazole loaded into (b) SBA-15, (c) NFM-1, (d) Evonik fumed silica (EVK), at 30 wt%, 50 wt% and 70 wt% loading. (The DSC curves were normalized based on weight of itraconazole in the loaded samples.)
Table 4 shows that the pore volume and BET surface area decreases signifi-cantly after loading with varying amounts of itraconazole compared with the unloaded particles. SBA-15 particles have relatively large surface areas (773.7 m2/g) and high pore volume (1.01 cm3/g) whilst NFM-1 particles have similar surface area but much lower pore volume (0.42 cm3/g). EVK particles have very high pore volume of 1.96 cm3/g but low surface area of 265.0 m2/g, due to its broad distribution of large pores. SBA-15 particles showed a systematic decrease in pore volume and surface area with increas-ing loading amount. At 30 wt% loading, the pores of NFM-1 particles are fully occupied by drug molecules, since it shows the most significant de-crease in surface area from 777.0 to 21.0 m2/g and its pore volume reduced to almost zero. EVK particles posses a large remaining pore volume after loading at 70 wt%. However, both DSC and XRD suggest EVK with 70 wt% loading shows high drug crystallinity.
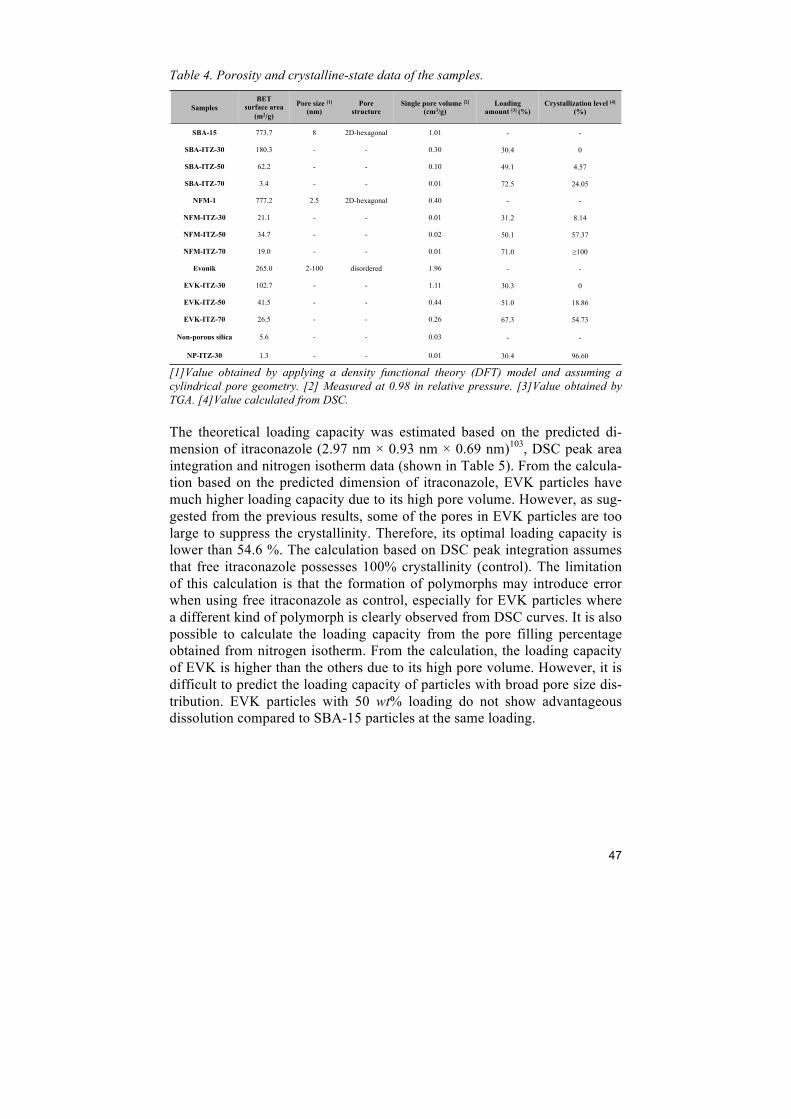
47
Table 4. Porosity and crystalline-state data of the samples.
[1]Value obtained by applying a density functional theory (DFT) model and assuming a cylindrical pore geometry. [2] Measured at 0.98 in relative pressure. [3]Value obtained by TGA. [4]Value calculated from DSC. The theoretical loading capacity was estimated based on the predicted di-mension of itraconazole (2.97 nm × 0.93 nm × 0.69 nm)103, DSC peak area integration and nitrogen isotherm data (shown in Table 5). From the calcula-tion based on the predicted dimension of itraconazole, EVK particles have much higher loading capacity due to its high pore volume. However, as sug-gested from the previous results, some of the pores in EVK particles are too large to suppress the crystallinity. Therefore, its optimal loading capacity is lower than 54.6 %. The calculation based on DSC peak integration assumes that free itraconazole possesses 100% crystallinity (control). The limitation of this calculation is that the formation of polymorphs may introduce error when using free itraconazole as control, especially for EVK particles where a different kind of polymorph is clearly observed from DSC curves. It is also possible to calculate the loading capacity from the pore filling percentage obtained from nitrogen isotherm. From the calculation, the loading capacity of EVK is higher than the others due to its high pore volume. However, it is difficult to predict the loading capacity of particles with broad pore size dis-tribution. EVK particles with 50 wt% loading do not show advantageous dissolution compared to SBA-15 particles at the same loading.
Samples BET
surface area (m2/g)
Pore size [1]
(nm) Pore
structure Single pore volume [2]
(cm3/g) Loading
amount [3] (%) Crystallization level [4]
(%)
SBA-15 773.7 8 2D-hexagonal 1.01 - -
SBA-ITZ-30 180.3 - - 0.30 30.4 0
SBA-ITZ-50 62.2 - - 0.10 49.1 4.57
SBA-ITZ-70 3.4 - - 0.01 72.5 24.05
NFM-1 777.2 2.5 2D-hexagonal 0.40 - -
NFM-ITZ-30 21.1 - - 0.01 31.2 8.14
NFM-ITZ-50 34.7 - - 0.02 50.1 57.37
NFM-ITZ-70 19.0 - - 0.01 71.0 !100
Evonik 265.0 2-100 disordered 1.96 - -
EVK-ITZ-30 102.7 - - 1.11 30.3 0
EVK-ITZ-50 41.5 - - 0.44 51.0 18.86
EVK-ITZ-70 26.5 - - 0.26 67.3 54.73
Non-porous silica 5.6 - - 0.03 - -
NP-ITZ-30 1.3 - - 0.01 30.4 96.60

48
Table 5. Estimated loading capacity (wt%) based on calculations from: i) ! =!!!!
!!!!!!!!! (P- loading percentage, VM – pore volume of silica particles, VD – vol-
ume of single drug molecule, MW – molecular weight of drug molecule, NA - Avoga-dro's constant); ii) crystallization level from DSC curves.! = !!(!!!!"#$%)
!!!!!!"#$%, where PL
is the loading percentage obtained from TGA, PCryst is the ratio integrated peak area of loaded sample and free drugs; and iii) the decreases of pore volume at different loading amounts. ! = !!
!!!!!! , PL is the loading percentage obtained from TGA, PF is
the percentage of filled pores calculated from remained pore volume.
Drug release studies were performed in simulated gastric fluid (SGF) with 0.4 wt% cetyltrimethylammonium bromide (CTAB) as a wetting agent. The kinetic release curves (shown in Figure 12) suggests all the loaded samples have a faster dissolution and higher solubility than the free drug except ITZ loaded onto non-porous silica particles, owing to the more thermodynamical-ly stable crystalline phase formed onto non-porous silica particles with lim-ited solubility. Samples with 30 wt% loading showed the highest release compared with higher loadings, since excess drug forms hydrophobic crys-tals which inhibits the dissolution of amorphous drug from the pores. SBA-15 and EVK particles showed similar solubility enhancement at 30 wt%, however, SBA-15 at 50 wt% loading showed an enhanced release kinetic profile in comparison to EVK sample at the same loading amount.
Samples From (i) From (ii) From (iii)
SBA-15 38.3% 47.9% 38.0%
NFM-1 19.7% 29.3% <30%
Evonik 54.6% 45.9% 42.9%
Non-porous silica !0 !0 !0
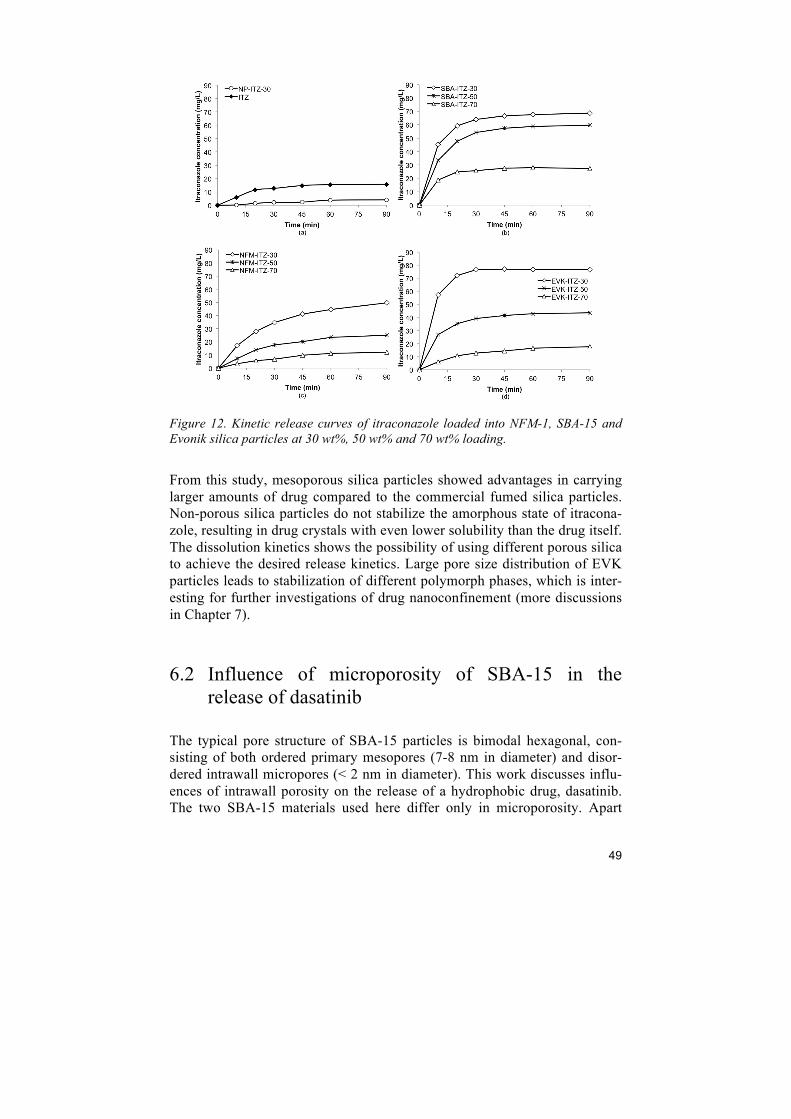
49
Figure 12. Kinetic release curves of itraconazole loaded into NFM-1, SBA-15 and Evonik silica particles at 30 wt%, 50 wt% and 70 wt% loading.
From this study, mesoporous silica particles showed advantages in carrying larger amounts of drug compared to the commercial fumed silica particles. Non-porous silica particles do not stabilize the amorphous state of itracona-zole, resulting in drug crystals with even lower solubility than the drug itself. The dissolution kinetics shows the possibility of using different porous silica to achieve the desired release kinetics. Large pore size distribution of EVK particles leads to stabilization of different polymorph phases, which is inter-esting for further investigations of drug nanoconfinement (more discussions in Chapter 7).
6.2 Influence of microporosity of SBA-15 in the release of dasatinib
The typical pore structure of SBA-15 particles is bimodal hexagonal, con-sisting of both ordered primary mesopores (7-8 nm in diameter) and disor-dered intrawall micropores (< 2 nm in diameter). This work discusses influ-ences of intrawall porosity on the release of a hydrophobic drug, dasatinib. The two SBA-15 materials used here differ only in microporosity. Apart
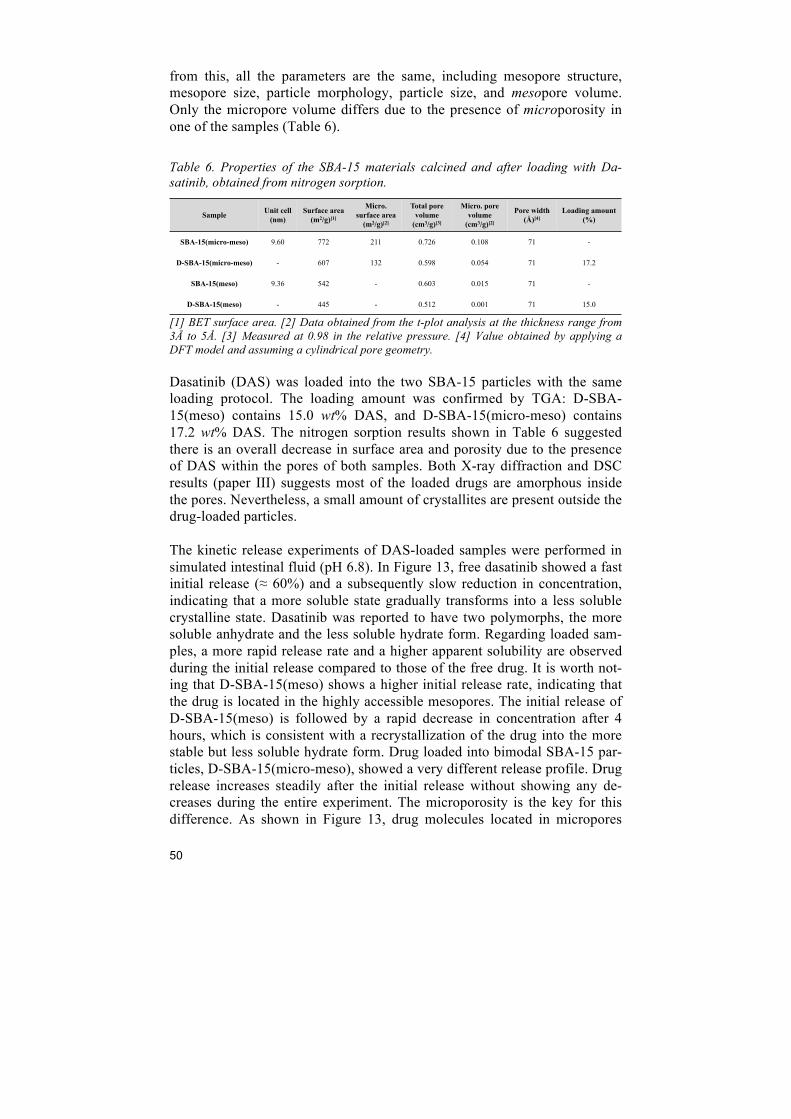
50
from this, all the parameters are the same, including mesopore structure, mesopore size, particle morphology, particle size, and mesopore volume. Only the micropore volume differs due to the presence of microporosity in one of the samples (Table 6). Table 6. Properties of the SBA-15 materials calcined and after loading with Da-satinib, obtained from nitrogen sorption.
[1] BET surface area. [2] Data obtained from the t-plot analysis at the thickness range from 3Å to 5Å. [3] Measured at 0.98 in the relative pressure. [4] Value obtained by applying a DFT model and assuming a cylindrical pore geometry. Dasatinib (DAS) was loaded into the two SBA-15 particles with the same loading protocol. The loading amount was confirmed by TGA: D-SBA-15(meso) contains 15.0 wt% DAS, and D-SBA-15(micro-meso) contains 17.2 wt% DAS. The nitrogen sorption results shown in Table 6 suggested there is an overall decrease in surface area and porosity due to the presence of DAS within the pores of both samples. Both X-ray diffraction and DSC results (paper III) suggests most of the loaded drugs are amorphous inside the pores. Nevertheless, a small amount of crystallites are present outside the drug-loaded particles. The kinetic release experiments of DAS-loaded samples were performed in simulated intestinal fluid (pH 6.8). In Figure 13, free dasatinib showed a fast initial release (≈ 60%) and a subsequently slow reduction in concentration, indicating that a more soluble state gradually transforms into a less soluble crystalline state. Dasatinib was reported to have two polymorphs, the more soluble anhydrate and the less soluble hydrate form. Regarding loaded sam-ples, a more rapid release rate and a higher apparent solubility are observed during the initial release compared to those of the free drug. It is worth not-ing that D-SBA-15(meso) shows a higher initial release rate, indicating that the drug is located in the highly accessible mesopores. The initial release of D-SBA-15(meso) is followed by a rapid decrease in concentration after 4 hours, which is consistent with a recrystallization of the drug into the more stable but less soluble hydrate form. Drug loaded into bimodal SBA-15 par-ticles, D-SBA-15(micro-meso), showed a very different release profile. Drug release increases steadily after the initial release without showing any de-creases during the entire experiment. The microporosity is the key for this difference. As shown in Figure 13, drug molecules located in micropores
Sample Unit cell (nm)
Surface area (m2/g)[1]
Micro. surface area
(m2/g)[2]
Total pore volume
(cm3/g)[3]
Micro. pore volume
(cm3/g)[2]
Pore width (Å)[4]
Loading amount (%)
SBA-15(micro-meso) 9.60 772 211 0.726 0.108 71 -
D-SBA-15(micro-meso) - 607 132 0.598 0.054 71 17.2
SBA-15(meso) 9.36 542 - 0.603 0.015 71 -
D-SBA-15(meso) - 445 - 0.512 0.001 71 15.0
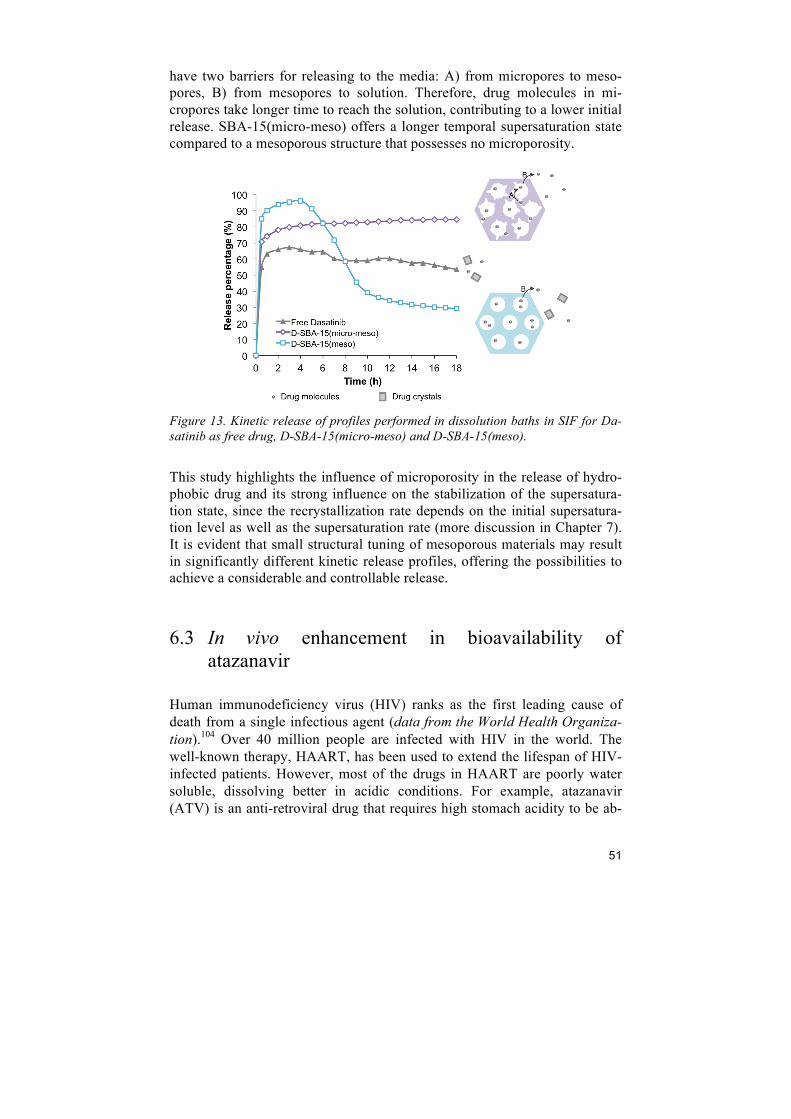
51
have two barriers for releasing to the media: A) from micropores to meso-pores, B) from mesopores to solution. Therefore, drug molecules in mi-cropores take longer time to reach the solution, contributing to a lower initial release. SBA-15(micro-meso) offers a longer temporal supersaturation state compared to a mesoporous structure that possesses no microporosity.
Figure 13. Kinetic release of profiles performed in dissolution baths in SIF for Da-satinib as free drug, D-SBA-15(micro-meso) and D-SBA-15(meso).
This study highlights the influence of microporosity in the release of hydro-phobic drug and its strong influence on the stabilization of the supersatura-tion state, since the recrystallization rate depends on the initial supersatura-tion level as well as the supersaturation rate (more discussion in Chapter 7). It is evident that small structural tuning of mesoporous materials may result in significantly different kinetic release profiles, offering the possibilities to achieve a considerable and controllable release.
6.3 In vivo enhancement in bioavailability of atazanavir
Human immunodeficiency virus (HIV) ranks as the first leading cause of death from a single infectious agent (data from the World Health Organiza-tion).104 Over 40 million people are infected with HIV in the world. The well-known therapy, HAART, has been used to extend the lifespan of HIV-infected patients. However, most of the drugs in HAART are poorly water soluble, dissolving better in acidic conditions. For example, atazanavir (ATV) is an anti-retroviral drug that requires high stomach acidity to be ab-

52
sorbed well. Its bioavailability decreases by 78% if co-administered with proton-pump inhibitors, typically used to treat the symptoms of heartburn and stomach pains that are common side effects of HIV medication. The decrease in bioavailability results from the reduced acidity in the patients’ stomach after administration with the proton-pump inhibitor. In this work, three kinds of mesoporous silica particles,NFM-1, AMS-6, and STA-11, have been investigated for solubility enhancement. The pore size of the two 3D-cubic structured samples, AMS-6 and STA-11, are 43 Å and 78 Å, respectively. NFM-1 has a 2D-hexagonal pore structure pos-sessing 26 Å pores. Atazanavir was loaded into silica particles via an evapo-ration protocol using methanol as a solvent. The loading amounts were de-termined by TGA to be 31.5 wt%, 32.8 wt%, and 28.2 wt% for NFM-1-ATV, AMS-6-ATV, and STA-11-ATV, respectively. Figure 14 (a) shows in vitro dissolution curves performed in SIF (pH 6.8), for free and loaded atazanavir. The free drug shows a poor solubility profile due to the neutral pH. Drug loaded in all kinds of mesoporous silica show a significant enhancement in dissolution, both dissolution rate and apparent solubility. Stabilization of the amorphous state of drug compounds can be described by equation 5 (Section 7.2). Below a certain critical pore diameter (d*), drug molecules are more likely to disperse in the pore than to form crystals. The absence of peaks in X-ray diffractograms is evidence that the drugs are in their amorphous state (Figure 3 in Paper IV). STA-11-ATV samples reached its highest dissolution percentage after four hours, which release faster than AMS-6-ATV and NFM-1-ATV probably due to its larger pore size. The release percentage of loaded atazanavir decreased significant-ly after 8 hours due to metastable supersaturation state, among which NFM-1-ATV showed the slowest decreases. It indicates that smaller pores in NFM-1 particles have beneficial effect on the stabilization of supersaturation state. This is similar with what is observed in bimodal SBA-15 particles loaded with dasatinib in the previous section. NFM-1 particles possesses pores of 26 Å which is in the same range as the intrawall porosity in SBA-15(micro-meso).

53
(a) (b)
Figure 14. (a) Kinetic release curves of free and loaded atazanavir. (b) Dissolution curves of NFM-1-ATV at different doses of loaded atazanavir: 5.6 mg/L, 11.3 mg/L, 22.6 mg/L and 45.1 mg/L.
To further investigate the stability of the supersaturation state, NFM-1-ATV was released at different ATV doses: 5.6 mg/L, 11.3 mg/L, 22.6 mg/L and 45.1 mg/L. Figure 14 (b) shows the kinetic release curves in SIF of NFM-1 loaded with ATV. The supersaturation state is stable for 24 hours at the dose of 5.6 mg/L. A small decrease occurred at dose of 11.3 mg/L. When the dose increased to 22.6 mg/L and 45.1 mg/L, the released drug reached the maxi-mum concentration of 13 mg/L within 5 hours, followed by significant de-creases in concentration. The highest dose maintained the shortest supersatu-ration state which has the lowest concentration after 24 hours. High dose results in faster dissolution rate and supersaturation rate, inducing a sudden nucleation and recrystallization. An in vivo pharmacokinetic study was conducted on Sprague Dawley rats for validation of the observed solubility enhancement of atazanavir. Ataza-navir was co-administered with proton-pump inhibitor, omeprazole, which lowers the stomach acidity.95 NFM-1-ATV and free atazanavir were given to rats at a single oral dose equivalent to 10 mg/kg of atazanavir for both sam-ples 5 hours after the administration of omeprazole. The pharmacokinetic profile of atazanavir concentration in whole-blood plasma is shown in Figure 15. NFM-1-ATV showed a statistically significant improvement in bioavail-ability compared to free drug. The area under the curve (AUC) of NFM-1-ATV is 271.91 ng h/mL, which is a four-fold enhancement than free ATV.

54
Figure 15. Pharmacokinetic profile for ATV in whole blood samples for NFM-1-ATV and free ATV co-administered with omeprazole to Sprague Dawley rats. Mean ± standard deviation. N=3 animals per time point. Cmax: 18.35 ng/ml (ATV) and 85.25 ng/ml (NFM-1-ATV). AUC: 65.84 ng h/ml (ATV) and 271.91 ng h/ml (NFM-1-ATV).
This work compared the solubility enhancement of atazanavir in different mesoporous silica particles. Materials with small pore size show benefits in the maintenance of the supersaturation state. The enhancement was repro-duced in vivo, where the AUC increased as high as 4 times. This work sug-gests a potential use of mesoporous silica in solving formulation and phar-macokinetic problems associated with multiple drugs and incompatibilities, which is a severe problem in HIV medication, such as atazanavir, fosampre-navir, indinavir, and delavirdine.
6.4 Intracellular antibacterial activities of anti-tuberculosis drug encapsulated into mesoporous silica
Tuberculosis (TB) is a lethal infectious disease mainly caused by Mycobac-terium tuberculosis (MTB). MTB is taken up by alveolar macrophages when they attack the lungs. However, macrophages are not able to digest MTB in which the bacteria reproduce unchecked. Mesoporous silica particles have been reported to be engulfed by macrophages effectively.105 Clemens et al.106 utilized mesoporous silica nanoparticles with a polyethyleneimine coating to release rifampin and with cyclodextrin-based pH-operated valves that open only at acidic pH to release isoniazid into MTB-infected macrophages. In this work, we investigated the possibility of using mesoporous silica parti-cles as drug carriers to deliver anti-tuberculosis agents into macrophages.
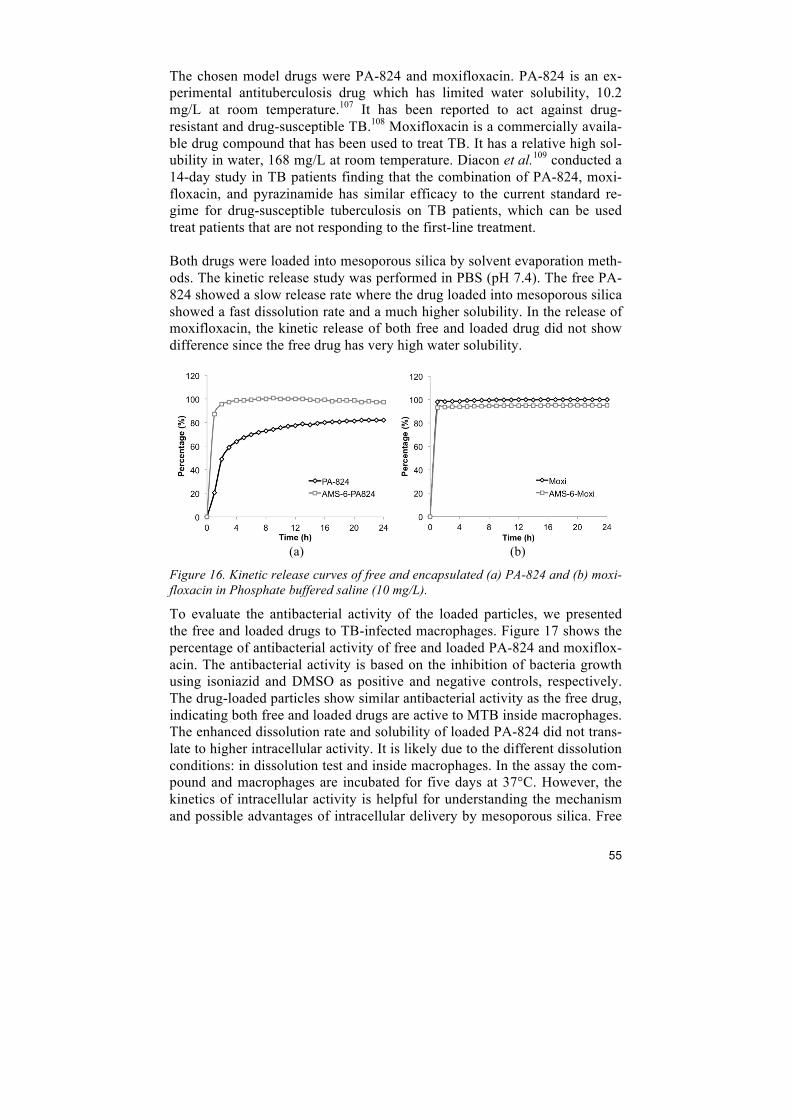
55
The chosen model drugs were PA-824 and moxifloxacin. PA-824 is an ex-perimental antituberculosis drug which has limited water solubility, 10.2 mg/L at room temperature.107 It has been reported to act against drug-resistant and drug-susceptible TB.108 Moxifloxacin is a commercially availa-ble drug compound that has been used to treat TB. It has a relative high sol-ubility in water, 168 mg/L at room temperature. Diacon et al.109 conducted a 14-day study in TB patients finding that the combination of PA-824, moxi-floxacin, and pyrazinamide has similar efficacy to the current standard re-gime for drug-susceptible tuberculosis on TB patients, which can be used treat patients that are not responding to the first-line treatment. Both drugs were loaded into mesoporous silica by solvent evaporation meth-ods. The kinetic release study was performed in PBS (pH 7.4). The free PA-824 showed a slow release rate where the drug loaded into mesoporous silica showed a fast dissolution rate and a much higher solubility. In the release of moxifloxacin, the kinetic release of both free and loaded drug did not show difference since the free drug has very high water solubility.
(a) (b)
Figure 16. Kinetic release curves of free and encapsulated (a) PA-824 and (b) moxi-floxacin in Phosphate buffered saline (10 mg/L).
To evaluate the antibacterial activity of the loaded particles, we presented the free and loaded drugs to TB-infected macrophages. Figure 17 shows the percentage of antibacterial activity of free and loaded PA-824 and moxiflox-acin. The antibacterial activity is based on the inhibition of bacteria growth using isoniazid and DMSO as positive and negative controls, respectively. The drug-loaded particles show similar antibacterial activity as the free drug, indicating both free and loaded drugs are active to MTB inside macrophages. The enhanced dissolution rate and solubility of loaded PA-824 did not trans-late to higher intracellular activity. It is likely due to the different dissolution conditions: in dissolution test and inside macrophages. In the assay the com-pound and macrophages are incubated for five days at 37°C. However, the kinetics of intracellular activity is helpful for understanding the mechanism and possible advantages of intracellular delivery by mesoporous silica. Free

56
and loaded moxifloxacin did not show difference in intracellular antibacteri-al activity since the drug is highly water-soluble and probably releases in the cell culture medium.
Figure 17. Antibacterial activity percentage of (a) PA-824, (b) Moxifloxacin, (c) AMS-6-PA824, and (d) AMS-6-Moxi (Concentration in logarithm scale).
In a subsequent ongoing study on the intracellular activity of AMS-6 parti-cles loaded with another anti-TB agent, we clearly observed that AMS-6 particles were taken up by macrophages (Figure 18). The green fluorescence results from the extracted AMS-6 particles. These particles were functional-ized with amine groups, which can be taken up in greater numbers than cal-cined silica particles. Additionally, amine groups will strongly influence the nature of the adsorbed protein corona from the cell culture media.110 The adsorption of proteins on the particle surface depends on the electrostatic interaction and may have positive effects on their uptake by macrophages.111
(a) (b)
Figure 18. Fluorescence microscopy images of (a) untreated TB infected macro-phages; (b) AMS-6 particles (green) with anti-tuberculosis agent treated TB infected macrophages.
Concentration (µM)0.001 0.1 10
0
20
40
60
80
100
Concentration (µM)0.001 0.1 10
0
20
40
60
80
100
Concentration (µM)0.001 0.1 10
0
20
40
60
80
100
Concentration (µM)0.001 0.1 10
0
20
40
60
80
100
Per
cent
age
Act
ivity
(%)
Per
cent
age
Act
ivity
(%)
Per
cent
age
Act
ivity
(%)
Per
cent
age
Act
ivity
(%)
(a) (b)
(d) (c)

57
7 Discussion
Overall, the studies designed in this thesis suggest mesoporous silica can be used as a drug delivery carrier for solubility enhancement of poorly water-soluble drugs and are particularly suitable for oral administration of APIs. However, understanding the relationship between pore structures, loading state of the drugs, confinement effects in mesopores of different size and geometry, and ultimately the dissolution behavior of APIs released in vivo requires further investigation in order to be able to provide a generic plat-form for drug delivery. In this chapter some reflections based on the work conducted are considered with the aim to answer critical questions regarding how improving drug solubility may be approached.
7.1 Physical properties of APIs Different poorly water-soluble drugs loaded into mesoporous silica particles have been studied in this thesis (Table 7). There is no single answer from the work conducted on how one may select the most appropriate silica particles (pore structures, pore size, pore geometries or functional surfaces) to achieve the desired pharmacokinetic properties for a specific API. For example from the work presented, smaller pores offer benefits to the release of atazanavir, while 6 to 7 nm pores were shown to be more appropriate for the solubility enhancement of itraconazole. Whilst these two drug compounds have some similar physical and chemical properties, as molecular weight, log P etc.; other properties such as the number of hydrogen acceptor groups and crystal-lization behavior are considerably different. Needless to say that the desired and optimal pharmacokinetic release profile in vivo is also different for the aforementioned drugs. One may use the well-known Lipinski’s rule of 5 to assess the physical and chemical properties of a drug compound vis a vis its solubility.112 This rule states that generally for a drug compound to be orally active (have sufficient bioavailability) it must not violate more than one of the following criteria:
• No more than 5 hydrogen bond donors (nitrogen or oxygen atoms with one or more hydrogen atoms);
• Not more than 10 hydrogen bond acceptors (nitrogen or oxygen at-oms);

58
• A molecular mass less than 500 g/mol; • A partition coefficient (log P) not greater than 5.
One can note that most of the compounds studied in this thesis violate more than one of these rules. Whilst this may be a useful parameter for pharma-ceutical chemists it does not allow us to decide if a particular drug com-pound will benefit from the use of mesoporous materials as a vehicle for solubility enhancement, what structure and pore size should be used or what type of release profiles (or extent of supersaturation) can be achieved. From our work on the dasatanib and itraconazole compounds, which form a rich variety of polymorphs with different solubility profiles, one can con-clude that understanding the crystallization behaviour of the individual drug compounds may indeed be a viable route to predicting the outcome of en-capsulation within mesoporous materials. One can argue that compounds that form crystalline states rapidly and with high lattice energy (e.g. niclos-amide, Table 7) are likely to be more difficult to encapsulate within nanopo-rous materials, whilst those that form crystalline structures with low lattice energy will be easily encapsulated and achieve stable amorphous states with ease. Hence one may talk about “strong” glass formers (good packing effi-ciency) or “fragile” glass formers (poor packing efficiency), invoking the glass transition temperature, Tg, which is directly proportional to the bond strength in the compound as well as other thermodynamic and supramolecu-lar properties. The approach to use Tg as a suitable parameter to predict the compatibility of APIs with mesoporous materials may be helpful, and has been used previously.113 Another helpful approach may be to relate the crys-tallization behaviour to fundamental properties such as the boiling point Tb and melting point Tm of the compounds. The ratio of Tb / Tm can also be used to quickly assess the lattice strength of crystalline phases of a particular compound. This has previously been used to correlate the formation of glassy phases by organic and inorganic liquids.114 These approaches may allow us to assess which drug compounds can or cannot be easily used to-gether with mesoporous silica materials.

59
Table 7. Physical properties of APIs studied.
[1] predicted boiling point at 760 mmHg from ChemSpider.com. [2] predicted boil-ing point of moxifloxacin hydrochloride.
7.2 Loading state In classical nucleation theory, the formation of nuclei is accompanied by a free energy change, ΔGcryst, which is the sum of the change in volume free energy, ΔGV, and unfavorable surface free energy, ΔGA. Volume free ener-gy, corresponding to the interatomic (or intermolecular) bonding energy, is always negative and stabilizing, whilst surface free energy, resulting from the creation of an interface between the nucleus and the surrounding medi-um, is always positive and destabilizing. The crystal properties of a com-pound can be affected dramatically by changing the surface area to volume ratio. As shown in Figure 19 (a), the maximum free energy change, ΔGcrit, corresponds to a critical radius, rcrit. Nuclei smaller than rcrit will dissolve spontaneously, whereas larger nuclei will grow.115 The critical size of organ-ic crystal nuclei are in the range of nanometers to tens of nanometers,116 which is in the same range of the pore size of mesoporous silica. Different polymorphs have their distinct free energy profiles, ΔGcrit, and rcrit. Figure 19 (b) shows free energy profiles of a more stable polymorph B in bulk and a metastable polymorph A that is more stable at the critical size and slightly beyond. Thus, confinement provides an alternative route to conventional
Drug Structure Molecular
weight (g/mol)
Solubility (ml/L) log P
Melting point
(°C)
Boiling point[1]
(°C)
Unit cell (Å)
Number of Hydrogen acceptor (donor)
Atazanavir (ATV) 704.9 insoluble 5.20 207-209 995.5 9.86*29.245
*8.327 7 (5)
Dasatinib (DAS) 488.0 12.8 2.77 280-286 572.1 14.14*8.18*
22.14 8 (3)
Itraconazole (ITZ) 705.6 insoluble 5.66 166.2 850.0 8.62*20.15*
21.04 9 (0)
Ketoconazole (KTZ) 531.4 0.087 4.35 146 753.4 - 6 (0)
PA-824 359.3 10.2 4.3 149-150 462.3 14.70*5.77*17.70 2 (0)
Moxifloxacin (MOX) 401.4 168 0.01 238-242 636.4[2] - 7 (2)
Niclosamide 327.1 1.6 4.49 230 424.5 - 4 (2)
HN
O
O
O
NH
N
N
HN
O
NH
O
O
OH
S
N
O
HN
NH
NN
NNHO
Cl
NN N
O
N N O O
ONN
N
ClCl
NNOO
O
NN
Cl
Cl O
ON
NOO
FF
F
N+O
O-
N
O
HO
O
O
F
NHNH
H
O
NH
HO
N+
O
-O
ClCl
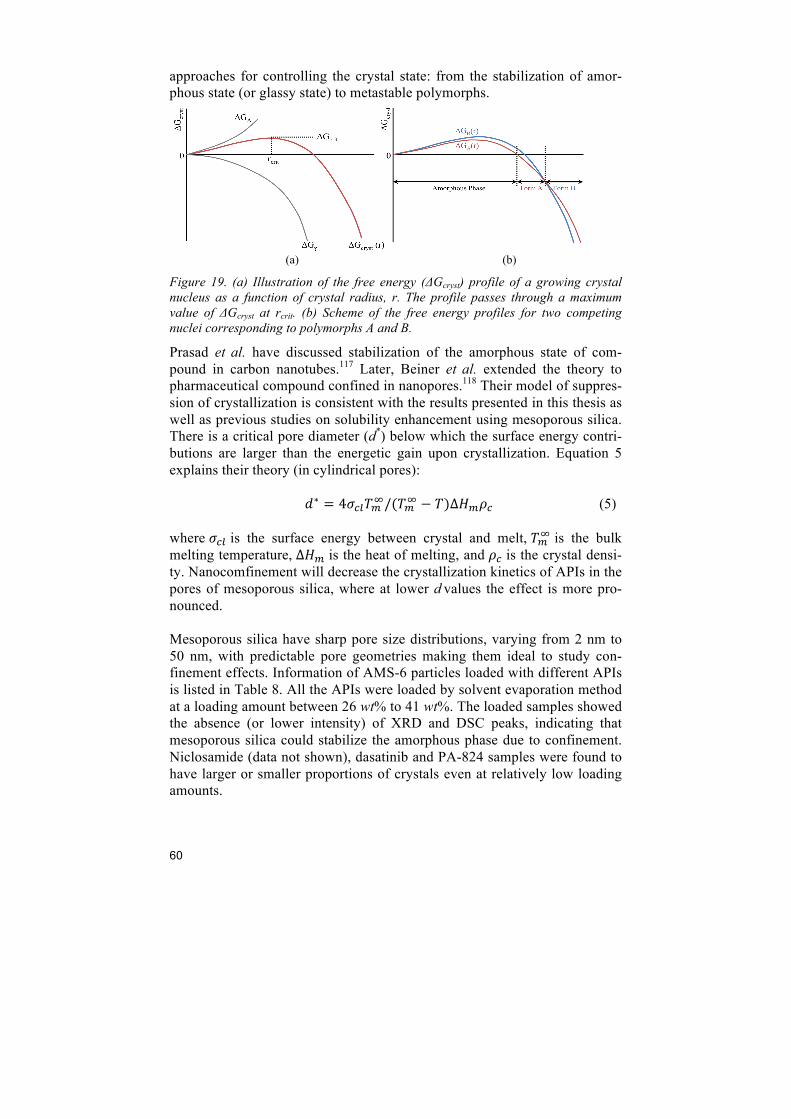
60
approaches for controlling the crystal state: from the stabilization of amor-phous state (or glassy state) to metastable polymorphs.
(a) (b)
Figure 19. (a) Illustration of the free energy (ΔGcryst) profile of a growing crystal nucleus as a function of crystal radius, r. The profile passes through a maximum value of ΔGcryst at rcrit. (b) Scheme of the free energy profiles for two competing nuclei corresponding to polymorphs A and B.
Prasad et al. have discussed stabilization of the amorphous state of com-pound in carbon nanotubes.117 Later, Beiner et al. extended the theory to pharmaceutical compound confined in nanopores.118 Their model of suppres-sion of crystallization is consistent with the results presented in this thesis as well as previous studies on solubility enhancement using mesoporous silica. There is a critical pore diameter (d*) below which the surface energy contri-butions are larger than the energetic gain upon crystallization. Equation 5 explains their theory (in cylindrical pores): !∗ = 4!!"!!!/(!!! − !)∆!!!! (5) where !!" is the surface energy between crystal and melt, !!! is the bulk melting temperature, ∆!! is the heat of melting, and !! is the crystal densi-ty. Nanocomfinement will decrease the crystallization kinetics of APIs in the pores of mesoporous silica, where at lower d values the effect is more pro-nounced. Mesoporous silica have sharp pore size distributions, varying from 2 nm to 50 nm, with predictable pore geometries making them ideal to study con-finement effects. Information of AMS-6 particles loaded with different APIs is listed in Table 8. All the APIs were loaded by solvent evaporation method at a loading amount between 26 wt% to 41 wt%. The loaded samples showed the absence (or lower intensity) of XRD and DSC peaks, indicating that mesoporous silica could stabilize the amorphous phase due to confinement. Niclosamide (data not shown), dasatinib and PA-824 samples were found to have larger or smaller proportions of crystals even at relatively low loading amounts.
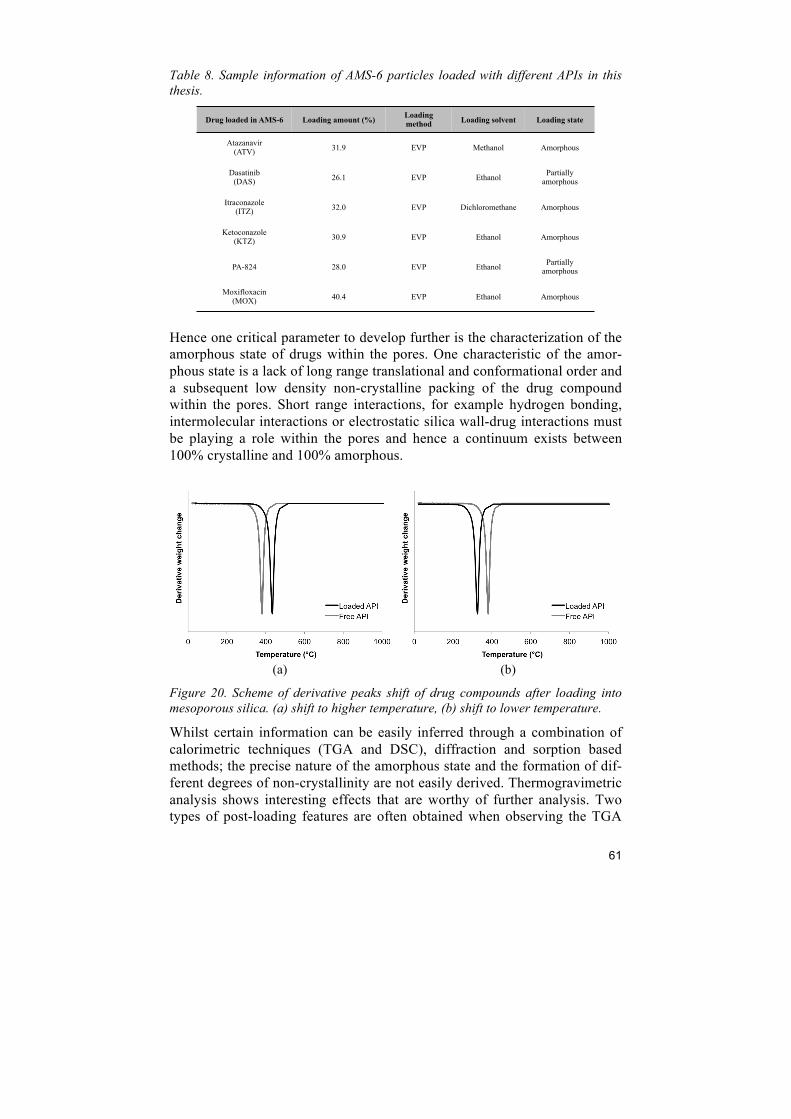
61
Table 8. Sample information of AMS-6 particles loaded with different APIs in this thesis.
Hence one critical parameter to develop further is the characterization of the amorphous state of drugs within the pores. One characteristic of the amor-phous state is a lack of long range translational and conformational order and a subsequent low density non-crystalline packing of the drug compound within the pores. Short range interactions, for example hydrogen bonding, intermolecular interactions or electrostatic silica wall-drug interactions must be playing a role within the pores and hence a continuum exists between 100% crystalline and 100% amorphous.
(a) (b)
Figure 20. Scheme of derivative peaks shift of drug compounds after loading into mesoporous silica. (a) shift to higher temperature, (b) shift to lower temperature.
Whilst certain information can be easily inferred through a combination of calorimetric techniques (TGA and DSC), diffraction and sorption based methods; the precise nature of the amorphous state and the formation of dif-ferent degrees of non-crystallinity are not easily derived. Thermogravimetric analysis shows interesting effects that are worthy of further analysis. Two types of post-loading features are often obtained when observing the TGA
Drug loaded in AMS-6 Loading amount (%) Loading method Loading solvent Loading state
Atazanavir (ATV) 31.9 EVP Methanol Amorphous
Dasatinib (DAS) 26.1 EVP Ethanol Partially
amorphous
Itraconazole (ITZ) 32.0 EVP Dichloromethane Amorphous
Ketoconazole (KTZ) 30.9 EVP Ethanol Amorphous
PA-824 28.0 EVP Ethanol Partially amorphous
Moxifloxacin (MOX) 40.4 EVP Ethanol Amorphous

62
derivative curves(Figure 20), either; (a) a shift to a higher decomposition temperature, or (b) a shift towards lower temperatures. The former is typical of a more “ordered” or more stable phase being encountered post-loading together with the insulating properties of the silica matrix and is typical of as-synthesized samples containing surfactants.119 The latter, which is indica-tive of an amorphous loading results from the formation of amorphous state that decomposes at lower temperature. Hence this peak shift may be helpful in correlating the degree of amorphous state, together with other techniques.
7.3 Supersaturation Poorly soluble drugs in their stabilized amorphous form within mesoporous silica exhibited highly supersaturated solution concentrations (i.e. apparent solubility), and always higher than the saturation concentration of the crys-talline form. For example, atazanavir showed more than 50 times increase in solubility after loading into mesoporous silica. Increased solubility in vitro has been shown to be related to increased bioa-vaiability in paper IV as well as in other in vivo studies.101,120 In a supersatu-ration system, especially for amorphous drugs, typical dissolution profiles consist of a rapid initial buildup of drug supersaturation and subsequent re-duction in concentration due to recrystallization. In paper III, the different dissolution profiles from two versions of SBA-15 particles suggest that the decline in drug concentration could be retarded to different degrees by fine tuning the pore structure. The gradual, diffusion-controlled drug release from SBA-15 with microporosity prevented a rapid buildup of supersaturation in the dissolution bath, subsequently avoiding drug recrystallization later in the release profile and in comparison to the sample loaded into SBA-15 without micropores. These results indicate that the rate of supersaturation generation affects the dissolution profile as well as the inhibition of recrystallization. This effect was also observed for the dissolution of atazanavir in paper IV, where a high dose (or loading amount) resulted in a high degree of supersat-uration, but also a faster decline of drug concentration in solution due to the sudden onset of recrystallization. Thus an additional conclusion from this thesis is that the stabilization of the supersaturation state is directly related to the dose (or loading level) for any fixed mesoporous material with a specific diffusion path. By slowing diffusion by careful selection of pore structures and pore features (such as the introduction of microporosity) one may in-crease the supersaturation stability.

63
7.4 Important factors in the selection of optimal mesoporous silica
To fully understand the optimal selection of mesoporous silica structures for a certain type of API and achieve the desired pharmacokinetic profile, both the glass transition temperature and the Tb / Tm ratio may useful in the predic-tion of the loading state of the drug compound and therefore its potential pharmacokinetic profile. The critical pore diameter d* is a good initial refer-ence for the loading state. The availability of well-ordered mesostructures with sharp pore size distribution makes these materials ideal for further in-vestigation of confinement effects. Fine tuning of the pore structure and mesopore size are key for the optimization of the supersaturation state and pharmacokinetic profile. These points are schematically summarized in Fig-ure 21.
Figure 21. Important factors in the selection of optimal mesoporous silica for the highest solubility enhancement.
API
Supersaturation Loading state
Tb / Tm Solubility
Dose, Loading amount
Material selection

64
8 Conclusions
Mesoporous silica particles were evaluated as drug carriers for the solubility enhancement of poorly water-soluble active pharmaceutical ingredients. In the oral toxicity study, two types of mesoporous silica particles were admin-istered at single doses, indicating that these particles are well tolerated for oral administration. This study is the basis for the further development of mesoporous silica as drug carriers. Paper II discussed mesoporous silica particles with sharp pore size distribu-tion compared to commercial porous silica particles and non-porous silica particles in the context of drug crystallinity and dissolution enhancement of poorly water-soluble drug, itraconazole. By studying the kinetic release of drug compound loaded into mesoporous silica, we have a better understand-ing of crystallization and polymorphism of drug compounds. Itraconazole was stabilized in amorphous state in all porous silica particles used in the study, showing a significant increase in its dissolution in aqueous solutions. Non-porous silica did not show any advantage when loaded with itracona-zole, revealing that porosity is the key factor in solubility enhancement. Low loading percentage (30 wt%) contribute to a faster dissolution rate and higher solubility compared to higher loadings (50 and 70 wt%). To understand more about the influence of internal pore structure on the drug release, two SBA-15 particles with and without microporosity were loaded with hydrophobic drug, dasatinib, at similar amounts. The mi-croporosity does not affect the loading state of the encapsulated drug. How-ever, these two types of particles showed significant differences in the kinet-ic release of encapsulated dasatinib. The absence of micropores in SBA-15 particles results in reaching a critical supersaturation level rapidly, followed with recrystallization of the drug. The SBA-15 particles with micropores kept a longer supersaturation state due to its slower kinetic release. In the study of paper IV, the solubility of atazanavir was enhanced when loaded into mesoporous silica. In the in vivo study, the bioavailability of atazanavir was 4-fold higher than the free drug when co-administered with proton-pump inhibitor, which is a widely used medicine to treat the side effect from the HIV therapies. This study shows the versatility of mesopo-

65
rous silica particles in solving drug-drug interactions, which can improve patient compliance. Interactions between mesoporous particles and macrophages were investi-gated in paper V. An experimental anti-tuberculosis drug, PA-824, shows an advantageous kinetic release profile after loading into AMS-6 particles. Mesoporous silica successfully delivered anti-tuberculosis agent to macro-phages. This thesis has studied the solubility enhancement when drugs are loaded into mesoporous silica, and discussed the influence of different structural factors, as well as the translation from in vitro dissolution study, to in vitro intracellular study, and to in vivo pharmacokinetic study. In conclusion mes-oporous silica are well tolerated orally in rats, and can be used to enhance the solubility of poorly water-soluble active pharmaceutical ingredients.

66
9 Future work
To use as a functional excipient, long-term studies are required to fully eval-uate the oral toxicity of mesoporous silica, though the current results showed that high doses of NFM-1 and AMS-6 particles do not lead to bodyweight decreases or other clinical symptoms associated with toxicity due to the presence of particles. Having said this, Kupferschmidt et al. reported that SBA-15 particles may have interactions with digestive processes and con-tribute to weight loss in obese mice.121 These interactions must be carefully assessed in particular if mesoporous materials are to be administered with other therapies (where they could cause an interference due to adsorption of a secondary drug within their pores), i.e. do mesoporous materials have any contraindications? Regarding the solubility enhancement, further understanding of the underly-ing theory will guide the selection of optimal silica particles for various types of APIs. A study revealing how the physical and chemical properties of API related to porosity and surface chemistry of silica will be appreciated. In vivo studies on the loaded particles for the understanding of in vitro – in vivo correlation will promote mesoporous materials for further clinical stud-ies. It is worth noting that the enhanced solubility may change the effective drug doses required for a particular treatment, and hence it needs further development. The encouraging results of solubility enhancement of poorly water-soluble drugs through encapsulation in mesoporous silica should be given serious consideration in the pharmaceutical industry as credible ena-bling technology, and warrants further animal studies with good promise for human use.

67
Acknowledgements
I would like to express my deepest gratitude to my supervisor Dr. Alfonso Garcia-Bennett. Thank you for encouraging my research! Your enthusiasm to science, sharing the knowledge, guiding and supports allow me to grow as a scientist. I also would like to give great acknowledgement to my co-supervisor Prof. Xiaodong Zou, for your concern and help. Working in your group is a great experience. I greatly appreciate the Nanologica team: Andreas, Adam, Krim, Chunfang, Natalia, Malin, Moa, Hanoi, Rambabu, Naeem, Erik, Adrian, Olga, Sara, Sana, René, Maggan, Arnav, Aishah, Jeffery, and Daryl. I spent most of my time in this big and growing family. I was so lucky to be a part of you. The pharma girls, Chunfang and Natalia, I had a great time with you in all the projects and sharing office. Thank you for your company and help! To my colleagues in Stockholm University, I had a very good time as a member in the academic group. I want to thank the former and current group members: Junliang, Feifei, Tom, Wei, Jie, Haoquan, Yifeng, Fabian, Yun-xiang, Leifeng, Hong, Peng, Ana, Hani, Elina, Fei, Yunchen, Ning, Liang, Qingpeng, Changjiu, Ken, Alexandra, Qingxia, Mikaela, Zhengyang, Da-liang, Changhong, Zhengbao, Tan, Farlan,…. I appreciate the help from Kjell, Lars G, Lars E, Jekabs, and Cheuk-Wai for the instruments. I want to thank all the administration staffs: Karin, Anna-Karin, Ann-Britt, Ann, Han-na, and Anita. Lluis, thank you for so many inspirational discussions! I really enjoy the collaborations with you. Prof. Viveka Alfredsson and Dr. Tomas Kjellman, it was a nice collaboration, thank you for your support and help. Dr. Kevin Pethe, Ryangyeo, Neeraj, Jonathan, Heekyoung, and Kideok, I am grateful for your help and valuable discussions! Prof. Jöns Hilborn and Jan Bohlin, I appreciate your support in the DSC instrument and discussions! To SP team, Karin, Eva, Annika, Tony, Rositsa, thank you for your help and company! I appreciate the support from the FP7-ITN project, DENDREAMER. I had a great time with all the partners and made friends with the professors and

68
students. I established great collaborations within ORCHID project. It was an impressive experience. Travel grants from C F Liljevalch J:ors stipend-iefond is acknowledged. I am grateful for my wonderful friends in Sweden and China. Special thanks to, Chenchen, Bjarne, Shuai Shi, Youjia, Ning, Jiao Yang, Chennan, Ying Liu, Vivianne, Alvaro, Yihui, Wei Li, Fei Wang, Jianing, Cao Cao, Qianyue, etc etc… for all the fun times we spent together. I am happy to have all of you on my side! Finally, I am deeply gratitude to my parents for the unwavering love and support, and my dearest husband, Chang, for the best time of my life to be together with you (^^)!

69
References
1. Halbert, G. in Textb. pharamaceutical Med. (Griffin, J. P.) 81–100 (Blackwell Publishing Ltd., 2009). doi:10.1002/9781444317800
2. Amidon, G. L., Lennernäs, H., Shah, V. P. & Crison, J. R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12, 413–420 (1995).
3. Fahr, A. & Liu, X. Drug delivery strategies for poorly water-soluble drugs. Expert Opin. Drug Deliv. 4, 403–416 (2007).
4. Peltonen, L. & Hirvonen, J. Pharmaceutical nanocrystals by nanomilling: critical process parameters, particle fracturing and stabilization methods. J. Pharm. Pharmacol. 62, 1569–79 (2010).
5. Mukherjee, S., Ray, S. & Thakur, R. S. Design and evaluation of itraconazole loaded solid lipid nanoparticulate system for improving the antifungal therapy. Pak. J. Pharm. Sci. 22, 131–8 (2009).
6. Hancock, B. C. & Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 17, 397–404 (2000).
7. Blagden, N., de Matas, M., Gavan, P. T. & York, P. Crystal engineering of active pharmaceutical ingredients to improve solubility and dissolution rates. Adv. Drug Deliv. Rev. 59, 617–630 (2007).
8. Vallet-Regi, M., Ramila, A., del Real, R. P. & Perez-Pariente, J. A New Property of MCM-41 : Drug Delivery System. Chem. Mater. 13, 308–311 (2001).
9. Beck, J. S. et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 114, 10834–10843 (1992).

70
10. Kresge, C. T., Leonowicz, M. E., Roth, W. J., Vartuli, J. C. & Beck, J. S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 359, 710–712 (1992).
11. Garcia-Bennett, A. E. Synthesis, toxicology and potential of ordered mesoporous materials in nanomedicine. Nanomedicine (Lond). 6, 867–877 (2011).
12. Mellaerts, R. et al. Enhanced release of itraconazole from ordered mesoporous SBA-15 silica materials. Chem. Commun. (Camb). d, 1375–7 (2007).
13. Vallet-Regí, M., Balas, F. & Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed. Engl. 46, 7548–58 (2007).
14. Lu, J., Liong, M., Zink, J. I. & Tamanoi, F. Mesoporous silica nanoparticles as a delivery system for hydrophobic anticancer drugs. Small 3, 1341–6 (2007).
15. Vallhov, H., Gabrielsson, S., Strømme, M., Scheynius, A. & Garcia-Bennett, A. E. Mesoporous silica particles induce size dependent effects on human dendritic cells. Nano Lett. 7, 3576–3582 (2007).
16. Edwards, L. J. The dissolution and diffusion of aspirin in aqueous media. Trans. Faraday Soc. 47, 1191–1210 (1951).
17. Noyes, A. A. & Whitney, W. R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 19, 930–934 (1897).
18. Serajuddin, A. T. M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 59, 603–616 (2007).
19. Sosnik, A., Chiappetta, D. a & Carcaboso, A. M. Drug delivery systems in HIV pharmacotherapy: what has been done and the challenges standing ahead. J. Control. Release 138, 2–15 (2009).
20. Briston-Myers Squibb. Prescribing information for atazanavir sulfate (Reyataz) capsules. Princeton, NJ, USA (2004).
21. Kerns, E. H. & Di, L. Pharmaceutical profiling in drug discovery. Drug Discov. Today 8, 316–323 (2003).

71
22. Nelson, E. Solution rate of theophylline salts and effects from oral administration. J. Am. Pharm. Assoc. 46, 607–614 (1957).
23. Nelson, E. Comparative dissolution rates of weak acids and their sodium salts. J. Am. Pharm. Assoc. 47, 297–299 (1958).
24. Sweetana, S. & Akers, M. J. Solubility principles and practices for parenteral drug dosage form development. PDA J Pharm Sci Technol. 50, 330–342 (1996).
25. Li, S. et al. Investigation of solubility and dissolution of a free base and two different salt forms as a function of pH. Pharm. Res. 22, 628–635 (2005).
26. Jin, J., Sklar, G. E., Oh, V. M. Sen & Li, S. C. Factors affecting therapeutic compliance: A review from the patient’s perspective. Ther. Clin. Risk Manag. 4, 269–286 (2008).
27. Kerns, E. H. & Di, L. in Drug-like Prop. Concepts, Struct. Des. Methods - from ADME to Toxic. Optim. 56–85 (Elsevier, 2008).
28. Dokoumetzidis, A. & Macheras, P. A century of dissolution research: from Noyes and Whitney to the biopharmaceutics classification system. Int. J. Pharm. 321, 1–11 (2006).
29. Reintjes, T. Solubility Enhancement with BASF Pharma Polymers - Solubilizer Compendium. (2011).
30. Merisko-Liversidge, E., McGurk, S. L. & Liversidge, G. G. Insulin nanoparticles: a novel formulation approach for poorly water soluble Zn-insulin. Pharm. Res. 21, 1545–1553 (2004).
31. Gao, L., Zhang, D. & Chen, M. Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J. Nanoparticle Res. 10, 845–862 (2008).
32. Sinko, P. J. & Singh, Y. Martin’s Physical Pharmacy and Pharmaceutical Sciences. (Lippincott Williams & Wilkins, 2011). doi:10.1097/00000441-196101000-00040
33. Serajuddin, A. T. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 88, 1058–66 (1999).

72
34. Chiou, W. L. & Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 60, 1281–1302 (1971).
35. Andronis, V., Yoshioka, M. & Zografi, G. Effects of sorbed water on the crystallization of indomethacin from the amorphous state. J. Pharm. Sci. 86, 346–51 (1997).
36. Watanabe, T. et al. Stability of amorphous indomethacin compounded with silica. Int. J. Pharm. 226, 81–91 (2001).
37. Wilson, M., Williams, M. A., Jones, D. S. & Andrews, G. P. Hot-melt extrusion technology and pharmaceutical application. Ther. Deliv. 3, 787–797 (2012).
38. Repka, M. A. et al. Applications of hot-melt extrusion for drug delivery. Expert Opin. Drug Deliv. 5, 1357–1376 (2008).
39. Yonemochi, E. et al. Physicochemical properties of amorphous clarithromycin obtained by grinding and spray drying. Eur. J. Pharm. Sci. 7, 331–338 (1999).
40. Takeuchi, H., Nagira, S., Yamamoto, H. & Kawashima, Y. Solid dispersion particles of amorphous indomethacin with fine porous silica particles by using spray-drying method. Int. J. Pharm. 293, 155–164 (2005).
41. Brouwers, J., Brewster, M. E. & Augustijns, P. Supersaturating Drug Delivery Systems : The Answer to Solubility-Limited Oral Bioavailability? J. Pharm. Sci. 98, 2549–2572 (2009).
42. Holmberg, K., Jönsson, B., Kronberg, B. & Lindman, B. Surfactants and Polymers in Aqueous Solutions, 2nd edn. 471 (John Wiley & Sons, Ltd, 2002).
43. Overhoff, K. a et al. Effect of stabilizer on the maximum degree and extent of supersaturation and oral absorption of tacrolimus made by ultra-rapid freezing. Pharm. Res. 25, 167–75 (2008).
44. Leuner, C. & Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 50, 47–60 (2000).
45. Vasconcelos, T., Sarmento, B. & Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 12, 1068–1075 (2007).

73
46. Craig, D. Q. M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 231, 131–144 (2002).
47. Brewster, M. E. et al. Comparative interaction of 2-hydroxypropyl-beta-cyclodextrin and sulfobutylether-beta-cyclodextrin with itraconazole: phase-solubility behavior and stabilization of supersaturated drug solutions. Eur. J. Pharm. Sci. 34, 94–103 (2008).
48. Yang, S., Zhu, J., Lu, Y., Liang, B. & Yang, C. Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm. Res. 16, 751–757 (1999).
49. Demirel, M., Yazan, Y., Müller, R. H., Kiliç, F. & Bozan, B. Formulation and in vitro-in vivo evaluation of piribedil solid lipid micro- and nanoparticles. J. Microencapsul. 18, 359–371 (2001).
50. Yang, S. C. et al. Body distribution in mice of intravenously injected camptothecin solid lipid nanoparticles and targeting effect on brain. J. Control. Release 59, 299–307 (1999).
51. Mehnert, W. & Mäder, K. Solid lipid nanoparticles: production, characterization and applications. Adv. Drug Deliv. Rev. 47, 165–196 (2001).
52. Müller, R. H., Maaβen, S., Weyhers, H., Specht, F. & Lucks, J. S. Cytotoxicity of magnetite-loaded polylactide, polylactide/glycolide particles and solid lipid nanoparticles. Int. J. Pharm. 138, 85–94 (1996).
53. Freitas, C. & Müller, R. H. Correlation between long-term stability of solid lipid nanoparticles (SLN) and crystallinity of the lipid phase. Eur. J. Pharm. Biopharm. 47, 125–132 (1999).
54. Freitas, C. & Müller, R. H. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLNTM) dispersions. Int. J. Pharm. 168, 221–229 (1998).
55. Ramteke, K. H., Joshi, S. A. & Dhole, S. N. Solid Lipid Nanoparticle: A Review. IOSR J. Pharm. 2, 34–44 (2012).
56. Astruc, D., Boisselier, E. & Ornelas, C. Dendrimers designed for functions: from physical, photophysical, and supramolecular

74
properties to applications in sensing, catalysis, molecular electronics, photonics, and nanomedicine. Chem. Rev. 110, 1857–959 (2010).
57. Gupta, U., Agashe, H. B., Asthana, A. & Jain, N. K. Dendrimers: novel polymeric nanoarchitectures for solubility enhancement. Biomacromolecules 7, 649–58 (2006).
58. http://www.starpharma.com/drug_delivery access on 7th May, 2014.
59. Generally recognized as safe determination for silicon dioxide when added directly and/or indirectly to human food. (Lewis & Harrison, LLC, 2009).
60. Ciuchi, F. et al. Self-recognition and self-assembly of folic acid salts: columnar liquid crystalline polymorphism and the column growth process. J. Am. Chem. Soc. 116, 7064–7071 (1994).
61. Che, S. et al. A novel anionic surfactant templating route for synthesizing mesoporous silica with unique structure. Nat. Mater. 2, 801–5 (2003).
62. Zhao, D. et al. Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores. Science (80-. ). 279, 548–552 (1998).
63. Mastropaolo, D., Camerman, A. & Camerman, N. Folic acid: crystal structure and implications for enzyme binding. Science (80-. ). 210, 334–336 (1980).
64. Garcia-Bennett, A. E., Terasaki, O., Che, S. & Tatsumi, T. Structural Investigations of AMS-n Mesoporous Materials by Transmission Electron Microscopy. Chem. Mater. 16, 813–821 (2004).
65. Atluri, R., Hedin, N. & Garcia-Bennett, A. E. Nonsurfactant supramolecular synthesis of ordered mesoporous silica. J. Am. Chem. Soc. 131, 3189–91 (2009).
66. Zhao, D., Huo, Q., Feng, J., Chmelka, B. F. & Stucky, G. D. Nonionic Triblock and Star Diblock Copolymer and Oligomeric Surfactant Syntheses of Highly Ordered, Hydrothermally Stable, Mesoporous Silica Structures. J. Am. Chem. Soc. 120, 6024–6036 (1998).

75
67. Yu, T. et al. Pore Structures of Ordered Large Cage-Type Mesoporous Silica FDU-12s. J. Phys. Chem. B 110, 21467–21472 (2006).
68. Yu, C., Yu, Y. & Zhao, D. Highly ordered large caged cubic mesoporous silica structures templated by triblock PEO–PBO–PEO copolymer. Chem. Commun. 575–576 (2000). doi:10.1039/b000603n
69. Garcia-Bennett, A. E., Miyasaka, K., Terasaki, O. & Che, S. Structural Solution of Mesocaged Material AMS-8. Chem. Mater. 16, 3597–3605 (2004).
70. Hodgkins, R. P. In situ functionalisation of mom-ionic block copolymer templated mesoporous silicas. (2005).
71. Zhou, C. F., Kunzmann, A., Rakonjac, M., Fadeel, B. & Garcia-Bennett, A. Morphological properties of nanoporous folic acid materials and in vitro assessment of their biocompatibility. Nanomedicine 7, 327–334 (2012).
72. Borm, P. J. a, Schins, R. P. F. & Albrecht, C. Inhaled particles and lung cancer, part B: paradigms and risk assessment. Int. J. cancer 110, 3–14 (2004).
73. Hudson, S. P., Padera, R. F., Langer, R. & Kohane, D. S. The biocompatibility of mesoporous silicates. Biomaterials 29, 4045–4055 (2008).
74. Huang, X. et al. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano 5, 5390–5399 (2011).
75. Liu, T. et al. Single and repeated dose toxicity of mesoporous hollow silica nanoparticles in intravenously exposed mice. Biomaterials 32, 1657–1668 (2011).
76. Scientific Opinion of the Panel on Food Additives and Nutrient Sources added to Food. Calcium silicate and silicon dioxide/silicic acid gel added for nutritional purposes to food supplements. EFSA J. 1132, 1–24 (2009).
77. Meng, H. et al. Use of Size and a Copolymer Design Feature To Improve the Biodistribution and the Enhanced Permeability and Retention Effect of Doxorubicin-Loaded Mesoporous Silica

76
Nanoparticles in a Murine Xenograft Tumor Model. ACS Nano 5, 4131–4144 (2011).
78. Liong, M. et al. Multifunctional Inorganic Nanoparticles for Imaging, Targeting, and Drug Delivery. ACS Nano 2, 889–896 (2008).
79. Chen, A. M. et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small 5, 2673–7 (2009).
80. Vallhov, H. et al. Adjuvant properties of mesoporous silica particles tune the development of effector T cells. Small 8, 2116–24 (2012).
81. Rosenholm, J. M., Sahlgren, C. & Lindén, M. Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles--opportunities & challenges. Nanoscale 2, 1870–83 (2010).
82. Garcia-Bennett, A. E. et al. In vitro generation of motor neuron precursors from mouse embryonic stem cells using mesoporous nanoparticles. Nanomedicine (Lond). in press, (2014).
83. Garcia-Bennett, A. E. et al. Delivery of Differentiation Factors by Mesoporous Silica Particles Assists Advanced Differentiation of Transplanted Murine Embryonic Stem Cells. Stem Cells Transilational Med. 2, 906–15 (2013).
84. Ambrogi, V. et al. Improvement of dissolution rate of piroxicam by inclusion into MCM-41 mesoporous silicate. Eur. J. Pharm. Sci. 32, 216–22 (2007).
85. Kumar, D., Sailaja Chirravuri, S. V & Shastri, N. R. Impact of surface area of silica particles on dissolution rate and oral bioavailability of poorly water soluble drugs: a case study with aceclofenac. Int. J. Pharm. 461, 459–68 (2014).
86. Wang, F., Barnes, T. J. & Prestidge, C. a. Celecoxib confinement within mesoporous silicon for enhanced oral bioavailability. Mesoporous Biomater. 1, 1–16 (2013).
87. Van Speybroeck, M. et al. Preventing Release in the Acidic Environment of the Stomach via Occlusion in Ordered Mesoporous

77
Silica Enhances the Absorption of Poorly Soluble Weakly Acidic Drugs. J. Pharm. Sci. 100, 4864–4876 (2011).
88. Zhang, Y. et al. Spherical mesoporous silica nanoparticles for loading and release of the poorly water-soluble drug telmisartan. J. Control. Release 145, 257–63 (2010).
89. Lowell, S., Joan, E. S., Martin, A. T. & Matthia, T. in Charact. porous solids powders Surf. area, pore size, density 42, (Kluwer Academic Publishers, 2004).
90. Brunauer, S., Emmett, P. H. & Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 60, 309–319 (1938).
91. Lastoskie, C., Gubbins, K. E. & Quirke, N. Pore size distribution analysis of microporous carbons: a density functional theory approach. J. Phys. Chem. 97, 4786–4796 (1993).
92. Ravikovitch, P. I., Domhnaill, S. C. O., Neimark, A. V, Schueth, F. & Unger, K. K. Capillary Hysteresis in Nanopores: Theoretical and Experimental Studies of Nitrogen Adsorption on MCM-41. Langmuir 11, 4765–4772 (1995).
93. Landers, J., Gor, G. Y. & Neimark, A. V. Density functional theory methods for characterization of porous materials. Colloids Surfaces A Physicochem. Eng. Asp. 437, 3–32 (2013).
94. <711> DISSOLUTION. U.S. Pharmacopeial Conv. 1, (2011).
95. Takehara, Y. et al. Evidence that endogenous GRP in rat stomach mediates omeprazole-induced hypergastrinemia. Am. J. Physiol. 271, G799–G804 (1996).
96. Christophe, T. et al. High Content Screening Identifies Decaprenyl-Phosphoribose 2’ Epimerase as a Target for Intracellular Antimycobacterial Inhibitors. PLOS Pathog. 5, 1–10 (2009).
97. Lu, J., Liong, M., Li, Z., Zink, J. I. & Tamanoi, F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small 6, 1794–1805 (2010).
98. Yu, T., Greish, K., McGill, L. D., Ray, A. & Ghandehari, H. Influence of geometry, porosity, and surface characteristics of silica

78
nanoparticles on acute toxicity: their vasculature effect and tolerance threshold. ACS Nano 6, 2289–301 (2012).
99. Jani, P., Halbert, G. W., Langridge, J. & Florence, A. T. Nanoparticle uptake by the rat gastrointestinal mucosa: quantitation and particle size dependency. J. Pharm. Pharmacol. 42, 821–826 (1990).
100. http://www.inchem.org/documents/jecfa/jecmono/v05je04.htm access on 10th May.
101. Mellaerts, R. et al. Increasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silica. Eur. J. Pharm. Biopharm. 69, 223–30 (2008).
102. Doty, M., Kipp, J., Rebbeck, C., Werling, J. & Wong, J. Polymorphic form of itraconazole, US20030100568A1. (2003).
103. Mellaerts, R. et al. Physical state of poorly water soluble therapeutic molecules loaded into SBA-15 ordered mesoporous silica carriers: a case study with itraconazole and ibuprofen. Langmuir 24, 8651–9 (2008).
104. Tubercolosis WHO global tuberculosis report 2013. http://www.who.int/tb/publications/factsheet_global.pdf?ua=1 (2013).
105. Witasp, E. et al. Efficient internalization of mesoporous silica particles of different sizes by primary human macrophages without impairment of macrophage clearance of apoptotic or antibody-opsonized target cells. Toxicol. Appl. Pharmacol. 239, 306–19 (2009).
106. Clemens, D. L. et al. Targeted intracellular delivery of antituberculosis drugs to Mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrob. Agents Chemother. 56, 2535–45 (2012).
107. Li, X. et al. Synthesis and antitubercular activity of 7-(R)- and 7-(S)-methyl-2-nitro-6-(S)-(4-(trifluoromethoxy)benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazines, analogues of PA-824. Bioorg. Med. Chem. Lett. 18, 2256–62 (2008).

79
108. Stover, C. K. et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–6 (2000).
109. Diacon, A. H. A. et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet 6736, 1–8 (2012).
110. Lynch, I., Salvati, A. & Dawson, K. A. Protein-nanoparticle interactions: What does the cell see? Nat. Nanotechnol. 4, 546–7 (2009).
111. Arai, T. & Norde, W. The behavior of some model proteins at solid-liquid interfaces 1. Adsorption from single protein solutions. Colloids and Surfaces 51, 1–15 (1990).
112. Lipinski, C. a, Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 (2001).
113. Mahlin, D., Ponnambalam, S., Höckerfelt, M. H. & Bergström, C. a S. Toward in silico prediction of glass-forming ability from molecular structure alone: a screening tool in early drug development. Mol. Pharm. 8, 498–506 (2011).
114. Cohen, M. H. & Turnbull, D. Molecular Transport in Liquids and Glasses. J. Chem. Phys. 31, 1164 (1959).
115. Jiang, Q. & Ward, M. D. Crystallization under nanoscale confinement. Chem. Soc. Rev. 43, 2066–79 (2014).
116. Bernstein, J. Polymorphism in Molecular Crystals. 429 (Oxford University Press, 2002).
117. Prasad, B. R. & Lele, S. Stabilization of the amorphous phase inside carbon nanotubes: Solidification in a constrained geometry. Philos. Mag. Lett. 70, 357–361 (1994).
118. Rengarajan, G. T., Enke, D., Steinhart, M. & Beiner, M. Stabilization of the amorphous state of pharmaceuticals in nanopores. J. Mater. Chem. 18, 2537 (2008).

80
119. Margolese, D., Melero, J. a., Christiansen, S. C., Chmelka, B. F. & Stucky, G. D. Direct Syntheses of Ordered SBA-15 Mesoporous Silica Containing Sulfonic Acid Groups. Chem. Mater. 12, 2448–2459 (2000).
120. Van Speybroeck, M. et al. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur. J. Pharm. Sci. 41, 623–30 (2010).
121. Kupferschmidt, N., Csikasz, R. I., Ballell, L., Bengtsson, T. & Garcia-Bennett, A. E. Large pore mesoporous silica induced weight loss in obese mice. Nanomedicine 1–9 (2014). doi:10.2217/nnm.13.138











![Persistent Homology for the Quantitative Prediction of ...Persistent Homology for the Quantitative Prediction of Fullerene Stability Kelin Xia,[a] Xin Feng,[b] Yiying Tong,*[b] and](https://static.fdocuments.in/doc/165x107/5e411ea16250f04f8f3fe475/persistent-homology-for-the-quantitative-prediction-of-persistent-homology-for.jpg)






