
Authors: Alexey Lastovetsky, Xin Zuo, Peng Zhao Speaker: Xin Zuo
Upload
anabel-hinesCategory
view
217download
0
Refinement: Refinement: A Crucial Step to Approach A Crucial Step to Approach
Accurate PredictionsAccurate Predictions
Xin Gao
PhD student
2006.11.6

Outline• Traditional Protein Structure Prediction
Introduction Methods Review Experimental Results
• Refinement Motivation Methods Review Proposed Research Plan

Outline• Traditional Protein Structure Prediction
Introduction Methods Review Experimental Results
• Refinement Motivation Methods Review Proposed Research Plan

Traditional Protein Structure Prediction — Introduction
• WHY do we study protein structure prediction problem?
• WHAT determines protein structures?
• HOW can we know protein structures?

Traditional Protein Structure Prediction — Introduction
• WHY? One of the most significant “grand
challenges” in Science. Key problem in Proteomics, the next step
in understanding life processes after the Human Genome Project are successfully completed.
Necessary step in studying protein functions. Improve, or even revolutionize human medicine and health care.

Traditional Protein Structure Prediction — Introduction
• WHAT? Inference of Structure from Sequence
Observation: Structure of a protein is uniquely determined by its amino acid sequence according to both energy and kinematics. (exceptions exist)

Traditional Protein Structure Prediction — Introduction
Inference of Function from Structure
Observation:
1) Proteins perform functions through their structures.
2) Proteins in the same fold usually have similar functions.
3) Proteins with novel, not yet observed, folds are rarely discovered recently.

Traditional Protein Structure Prediction — Introduction
• HOW? Experimental Methods
X-ray Crystallography
Nuclear Magnetic Resonance Spectroscopy (NMR)
Shortage: Costly and time consuming.
Computational Methods Have been studied for 3 decades. Great process
has been made.

Outline• Traditional Protein Structure Prediction
Introduction
Methods Review Experimental Results
• Refinement Motivation Methods Review Proposed Research Plan

Traditional Protein Structure Prediction — Methods Review
• Basic hypothesis Anfinsen’s (1973) thermodynamic hypothesis:
Proteins are not assembled into their native structures by a biological process, but folding is a purely physical process that depends only on the specific amino acid sequence of the protein.
Anfinsen’s hypothesis implies that in principle protein structure can be predicted if a model of the free energy is available, and if the global minimum of this function can be identified.

Traditional Protein Structure Prediction — Methods Review
• Computational Methods Ab Initio Methods Comparative Modeling Methods Fold-recognition Methods Consensus-based Methods Other Methods
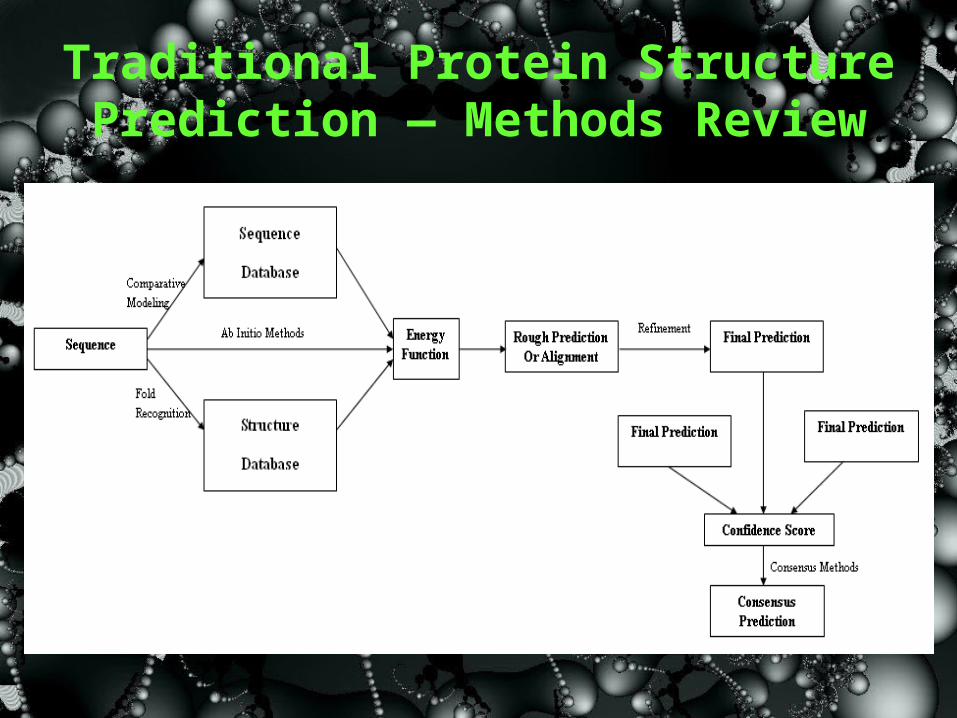
Traditional Protein Structure Prediction — Methods Review

Traditional Protein Structure Prediction — Methods Review
Ab Initio Methods (Template-free Modeling)
1) Basic Idea:
According to Anfinsen’s (1973) thermodynamic hypothesis, such methods attempt to identify the structure with the minimum free energy by solely using the first principles: energy and kinematics.

Traditional Protein Structure Prediction — Methods Review
Ab Initio Methods (Template-free Modeling)
2) Major Steps: Choose a first principle
based energy function. Apply an algorithm to
generates all possible
conformations. Use a search strategy to
search for the conformation
that minimizes the energy
function.
Traditional Protein Structure Prediction — Methods Review
Ab Initio Methods (Template-free Modeling)
3) Advantages: Do not depend on any template databases. Can be used when other methods fail. Can be used as a complementary approach for others,
e.g., loop modeling.
4) Limitations: Computationally demanding.

Traditional Protein Structure Prediction — Methods Review
Ab Initio Methods (Template-free Modeling)
5) Famous Servers: Folding@Home (A distributed computing project-people
from through out the world download and run software to band together).
6) Current Development: Becoming more and more important to deal with hard targets or hard parts of targets; hybrid servers with other methods are preferred.

Traditional Protein Structure Prediction — Methods Review
Comparative Modeling Methods
1) Basic Idea:
Aim to predict the structures of a target protein, when a clear evolutionary relationship between the target and a protein of known structure can be easily detected from the sequence.
Based on the observation that when two proteins have more than 30% sequence identity, the structures of them are very similar.

Traditional Protein Structure Prediction — Methods Review
Comparative Modeling Methods
2) Major Steps: Choose a template
database and a scoring
matrix or profile. Do sequence-sequence
alignment on each template
in the database, and select
the one best aligned. Refine side chains and
regions of low sequence
identity.

Traditional Protein Structure Prediction — Methods Review
Comparative Modeling Methods
3) Advantages: If there is indeed a homologous template in the database,
the prediction result can be very accurate, usually with rmsd<4A.
Can do prediction very fast.
4) Limitations: Database dependent. Can only generate good predictions for easy targets,
which have homologous templates in the database.

Traditional Protein Structure Prediction — Methods Review
Comparative Modeling Methods
5) Famous Servers: SAM-T02, FFAS03.
6) Current Development: Structure dependent score and gap penalty, and profile-profile alignment techniques are being used to deal with targets with distant homology from templates.

Traditional Protein Structure Prediction — Methods Review
Fold Recognition Methods
1) Basic Idea:
Aim to predict the structure of a target protein even if no sequence similarity can be detected.
Based on the notion that structure is evolutionary more conserved than sequence.

Traditional Protein Structure Prediction — Methods Review
Fold Recognition Methods
2) Major Steps: Choose a reasonable structure database and an energy
function. Do sequence-structure alignment on each template in the
database, and select the one best aligned. Refine side chains and non-aligned regions.

Traditional Protein Structure Prediction — Methods Review
Fold Recognition Methods
3) Advantages: Can detect distant homology. Can predict protein structures even if they have no
sequence similarity or they are evolutionarily unrelated.
4) Limitations: Database dependent. The predictions generated are usually medium
resolution.

Traditional Protein Structure Prediction — Methods Review
Fold Recognition Methods
5) Famous Servers: RAPTOR, SPARK, PROSPECTOR, FUGUE.
6) Current Development: Different profile extracting methods are being tested. Fragment assembly and mini-threading techniques are used to improve the accuracy.

Traditional Protein Structure Prediction — Methods Review
Consensus Based Methods
1) Basic Idea:
Based on the observation that different servers usually generate good predictions for different targets. Why not combine their strength together?

Traditional Protein Structure Prediction — Methods Review
Consensus Based Methods
2) Major Kinds: Selection-only consensus methods: Try to choose the
best predictions from the input prediction set. Can not do better on a target than the best input server.
Hybrid consensus methods: Try to combine different regions extracted from different input predictions to construct a new and hopefully better prediction.

Traditional Protein Structure Prediction — Methods Review
Consensus Based Methods
3) Famous Servers: ACE, Pcons, Pmodeller, 3D-SHOTGUN.
4) Current Development: Bad quality predictions are sometimes supported by many servers, and are then selected. New techniques are being used to eliminate the input server correlation to overcome this problem.

Traditional Protein Structure Prediction — Methods Review
Other Methods
Combine different methods together. Fragment assembly is usually used. Famous servers including ROSETTA, TOUCHSTONE.

Outline• Traditional Protein Structure Prediction
Introduction Methods Review
Experimental Results• Refinement
Motivation Methods Review Proposed Research Plan

Traditional Protein Structure Prediction — Experimental Results
Critical Assessment of Protein Structure
Prediction (CASP)
• Began in 1994 (CASP1)
• Held every two years
• The most objective assessment in the field.
• In CASP7 (May-Aug, 2006), 98 automated servers and 204 human expert servers are registered.
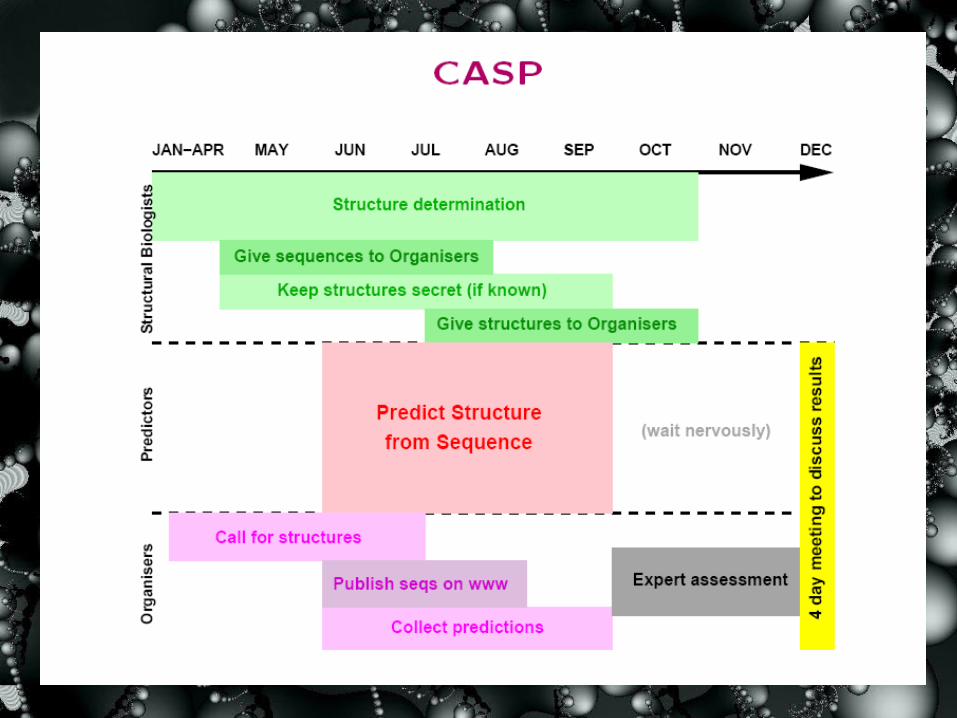

Traditional Protein Structure Prediction — Experimental Results
Best servers:
TASSER, ROSETTA, RAPTOR-ACE, RAPTOR, PModeller, SPARK.
Observation: 1) Consensus servers usually outperform individual servers.
2) There are more and more hybrid servers.
3) Most servers can generate good predictions or at least good regions for many targets. Thus, refinement is urgently needed.

Outline• Traditional Protein Structure Prediction
Introduction Methods Review Experimental Results
• Refinement Motivation Methods Review Proposed Research Plan

Refinement — Motivation
• What is refinement? Goal:
To make predictions to be more accurate.
No formal definition. My definition:
Given a set of reasonably good predictions, construct a prediction that is more close to the native structure.

Refinement — Motivation
Reasonably good:
The whole structure is close to native, or there are good regions in the structure that are close to those regions in native,
Close to native:
One of the most controversial problems in the field. No measure is considered to be perfect.
Here, rmsd or GDT score is better than some thresholds.

Refinement — Motivation
• Why is refinement possible?
Data are taken from SBC evaluation, on 2006.10.30, of 86 targets.
http://www.pdc.kth.se/~bjornw/casp7/targets/results/
Server Name
Sum GDT of Top1
Average GDT of Top1
Sum GDT of Best of
Top 5
Average GDT of Best
of Top5
TASSER 52.17 0.607 54.19 0.630
ROBETTA 48.86 0.568 51.61 0.600RAPTOR 47.31 0.550 50.27 0.585

Refinement — Motivation
Quick notes about GDT:
1) Zemla et al, Global Distance Test
2) Defined as the average coverage of the target sequence of the substructures with the four different distance thresholds (1, 2, 4, and 8A).
3) Weakness: Since the GDT score focuses only on the size of the substructures, the detailed match information of models and native structures is partially missed.

Refinement — Motivation Some Instances:
For T0198 of CASP6, RAPTOR predicted two good regions, but the orientation of them is wrong, which got a low score.
T0198 by RAPTOR T0198 Native

Refinement — Motivation
Taken from Zhang Yang’s online evaluation server.
http://zhang.bioinformatics.ku.edu/TM-score/

Outline• Traditional Protein Structure Prediction
Introduction Methods Review Experimental Results
• Refinement Motivation
Methods Review Proposed Research Plan

Refinement — Methods Review
• Two Major Categories of Methods
Partial Structure Refinement
Whole Structure Refinement• Ab Initio Methods• Template-Based Methods• Consensus-Based Methods

Refinement — Methods Review
• Partial Structure Refinement Based on the assumption that backbone
structures of core regions are good. Aim to refine other regions.
Loop modeling methods: LOOPY (Honig Lab, ab initio method
to generate initial conformations, random tweak method to close conformations)
Side chain packing methods:SCWRL (Dunbrack Lab, graph theory)
SCATD (Jinbo Xu, tree decomposition)

Refinement — Methods Review
• Whole Structure Refinement Ab Initio Methods:
Basic Idea: Assume the structure is roughly good, just need to “shake” a little bit to achieve a conformation with lower energy.
Server: RAPTORESS (Xin Gao et al., integer linear programming based backbone refinement)

Refinement — Methods Review
Template-Based Methods:
Basic Idea: Extract information from a set of particularly chosen templates, and refine the structure according to such information
Server: MODELLER (Andrej Sali, try to optimize probability density function for each of the restraint features of the model); SEGMOD (Michael Levitt, a segment match modeling using a database of known protein X-ray structures).

Refinement — Methods Review
Consensus-Based Methods:Basic Idea: Suppose we can get an input
prediction set, each structure of which contains some close to native regions, try to combine them together and get a hybrid but closer to native structure.
Server: TASSER (Zhang Yang, hyperbolic Monte Carlo sampling method to assemble continuous template fragments); POPULUS (Marc Offman et al., “move-set” based genetic algorithm to reshuffle and repack structural components).

Refinement — Methods Review• TASSER (Threading/ASSEmbly/Refinement)
Steps:1) Thread the sequence through a representative template
library (35% pairwise sequence identity cutoff) by PROSPECTOR.
2) Split target sequence into threading template aligned and unaligned regions, parallel hyperbolic Monte Carlo sampling is exploited to assemble full-length protein models by rearranging the continuous aligned fragments (building blocks) excised from threading templates.During assembly, building blocks are kept rigid and off-lattice to retain their geometric accuracy, unaligned regions are modeled on a cubic lattice by an ab initio procedure.
Performance:Ranked number one in CASP7, much better than any other servers, even including consensus servers.

Refinement — Methods Review
• TASSER (Threading/ASSEmbly/Refinement)

Refinement — Methods Review
• POPULUSMove set:
X = single crossover
XX = double crossover
C = coil mutation
H = helix mutation
CCD = Cyclic Coordinate Descent Algorithm

Refinement — Methods Review
Move set:
Protein Mutation

Refinement — Methods Review
Flowchart:CASP6 submitted models
Ratio: 2:1:1:1
Energy based scoring scheme, top 25
D(Ave, Best) < 0.0001, Sum(Cur)=Sum(Previous), D(Si, Sj) < 0.04, N(rounds) > 20
Top 20 structures returned

Refinement — Methods Review
Performance:1) Did not attend CASP7 with POPULUS server2) Show Move-set is good
Use (GDT+Maxsub+TMscore)/3 as scoring function, assume already known native structure. Average is around 80%.
3) Energy based scoring function is good Aim: score the native structure as the global
minimum. Use native structures as input, the output
structures are stable with the input (20/23 cases, rmsd<0.6A).
Score(native)<Score(Lowest output) (21/23 cases)

Outline• Traditional Protein Structure Prediction
Introduction Methods Review Experimental Results
• Refinement Motivation Methods Review
Proposed Research Plan

Refinement — Proposed Research Plan
• Basic Idea: By assembling good and long fragments, the
traditional search space can be greatly reduced. An efficient energy function is used to direct assembly process.
• Subproblems:
1) How to find good fragments?
2) What is the assembly process?3) What is the energy function?


Refinement — Proposed Research Plan
How to find good fragments?
The main task of my project.
Basic Idea: Develop a confidence score which can evaluate the confidence for an aligned region.
Goal: Try to increase the sensitivity (recognize good regions as good) as much as possible, while keeping a high specificity (recognize bad regions as bad).

Refinement — Proposed Research Plan
Preliminary Experiments:
1) Statistical Alignment Coverage (RAPTOR results for CASP7)
#100%
#
aligned residuesCoverage
residues in sequence

Refinement — Proposed Research Plan
Analysis:
There is a huge gap between how much the alignment covers in the target and how much the good regions cover in the target. So we have to filter out about half alignment parts which are aligned but bad regions.
Server Name Top1 Coverage
Top1 Ave GDT
Best Top5 Coverage
Best Top5 Ave GDT
RAPTOR 90.17% 55.0% 92.0% 58.5%

Refinement — Proposed Research Plan
Preliminary Experiments:
2) Measure Local Quality Directly by CHARMM Energy Function
Server Name Ave # Regions per Target
Difference Between Best Energy and Worst
RAPTOR 6.7 <2%
bond angle dihedral angle improper torsionangle van der Waals electrostaticE E E E E E E
For aligned regions supported by all the top 5 models, with length at least 10 amino acids in length.

Refinement — Proposed Research Plan
Analysis:
(a) This energy function is not good enough for separating locally good regions and locally bad regions, because it works better on the whole protein structure rather than the parts, and in aligned regions, those terms are usually similar, very close to the standard values.
(b) Small number of gaps in a relatively long region may be tolerable to reduce the effect of alignment errors, and to reduce the number of such regions with increasing their length.

Refinement — Proposed Research Plan
Preliminary Experiments:
3) How much can good regions cover and their locations.
(In Process)
Server Name Total Coverage of Good Regions in
Top5
Relative Locations of Good Regions
RAPTOR ? ?

Refinement — Proposed Research Plan
Preliminary Experiments:
4) Different Information Influences The Local Quality
(a) Local alignment quality: alignment score for the region.
(b) Consensus information: how much do other servers support this region.
(c) Local energy: not enough to determine region quality, but will be helpful.
(d) Server related information: average coverage, different servers will prefer different alignments.
(e) Target information: target length, target categories (easy/medium/hard).
(f) Region information: region length, region position.

Refinement — Proposed Research Plan
Possible Strategies:
(a) Neural Network: take all kinds of information as input value, train a neural network to predict how good an input region is.
(b) SVM: extract all kinds of information as features, describe a region as a vector of these features, train an SVM to classify whether an input region is good or not.
(c) Linear Programming: suppose the confidence score is the linear combination of all kinds of information, try to optimize the weights and maximize the confidence gap between good and bad regions.

Refinement — Proposed Research Plan
Importance:
1) Protein motif discovery or functional site and active site study.
2) Contrast to traditional prediction quality criteria, this can be used for researchers as a blind (without knowing native structure) prediction quality criteria.
3) Can be used to improve the accuracy of consensus based, and fragment assembly based methods.

Refinement — Proposed Research Plan
What is the assembly process?
Future work.Idea: Try some possible algorithms to
assemble the good fragments selected with high priority, the 9-mer fragments selected in the candidate set (Shuaicheng’s work) with medium priority, the 3-mer or single residue with low priority.
Goal: For well-aligned targets, exact search strategy can be used due to the small search space; otherwise, search space will also be significantly reduced, some heuristic algorithm can be used.

Refinement — Proposed Research Plan
What is the energy function?
The vital problem in all the methods described. The most important problem in current stage.
The work will be meaningless with a wrong objective!
A universal energy function:
By combining all existing energy functions, optimize their terms to make sure close to
native structures always have lower energy than decoy structures. (Joint work with Shuaicheng Li, Dongbo Bu)

Summary
Previous study has shown refinement is an indispensable step to solve protein structure prediction problem.
Refinement can be done based on the current methods and current PDB database.
CASP can provide an objective evaluation.

Thank youQuestions & Comments


















