
WTRP-Wireless Token Ring Protocol
15
Direct Selection of Antibodies from Complex Libraries with the Protein Fragment Complementation Assay Holger Koch, Nico Gra ¨fe, Ralph Schiess and Andreas Plu ¨ ckthun * Biochemisches Institut der Universita ¨t Zu ¨ rich, Winterthurerstr. 190 CH-8057 Zu ¨ rich Switzerland The aim of the present study was to develop the protein fragment complementation assay (PCA) for the intracellular selection of specific binding molecules from the fully synthetic HuCAL w antibody library. Here, we describe the first successful selections of specific antibodies by PCA, and we discuss the opportunities and limitations of this approach. First, we enriched an antibody specific for the capsid protein D of bacteriophage lambda (gpD) by ten successive rounds of competitive liquid culture selection. In an independent approach, we selected a specific antibody for the c-Jun N-terminal kinase 2 (JNK2) in a single-step selection setup. In order to obtain specific antibodies in only a single PCA selection round, the selection system was thoroughly investigated and several strategies to reduce the amount of false positives were evaluated. When expressed in the cytoplasm of Escherichia coli, the PCA-selected scFv antibody fragments could be purified as soluble and monomeric proteins. Denaturant-induced unfolding experi- ments showed that both antibody fragments are stable molecules, even when the disulfide bonds are reduced. Furthermore, antigen-specificity of the PCA- selected antibody fragments is demonstrated by in vivo and in vitro experiments. As antigen binding is retained regardless of the antibody redox state, both PCA-selected antibody fragments can tolerate the loss of disulfide bridge formation. Our results illustrate that it is possible to select well-expressed, stable, antigen-specific, and intracellular functional anti- bodies by PCA directly. q 2005 Elsevier Ltd. All rights reserved. Keywords: intrabodies; protein fragment complementation assay; HuCAL w antibody library; in vivo selection; cytoplasmic expression *Corresponding author Introduction An extensive list of DNA sequences, coding for proteins with unknown functions, is being gene- rated by the ongoing worldwide genome sequen- cing programs. 1 Elucidating the functions of these proteins will be one of the most important challenges in the years to come. 2 Therefore, one important task of biotechnology is to generate specific binding molecules against a wide variety of targets with reasonable resources. Although a variety of selection systems have proven success in generating such binding molecules and are thus commonly used, it is unlikely that they will provide the required throughput without additional com- ponents and development. In most display tech- niques the purification and immobilization of the target molecules has to be worked out individually, and binders against every target have to be selected in separate experiments. Consequently, there is great demand for a method that would allow 0022-2836/$ - see front matter q 2005 Elsevier Ltd. All rights reserved. Present addresses: N. Gra ¨fe, Roche Diagnostics TR-BC1, Nonnenwald 2, D-82377 Penzberg, Germany; R. Schiess, Institute for Molecular Systems Biology, ETH Ho ¨nggerberg HPT, CH-8093 Zu ¨ rich, Switzerland. Abbreviations used: BSA, bovine serum albumin; CDR, complementary-determining region; DHFR, dihydrofolate reductase; ELISA, enzyme-linked immunosorbent assay; GdnHCl, guanidine hydrochloride; gpD, capsid protein D of bacteriophage lambda; HuCAL, human combinatorial antibody library (MorphoSys AG, Munich); IPTG, isopropyl-b-D- thiogalactopyranoside; JNK2, c-Jun N-terminal kinase 2; mDHFR, murine dihydrofolate reductase; PCA, protein fragment complementation assay; RU, resonance unit; scFv, single-chain antibody fragment consisting of the variable domains of the heavy (V H ) and light chain (V L ) connected by a peptide linker; SPR, surface plasmon resonance; TBS, Tris-buffered saline; TMP, trimethoprim; wt, wild-type. E-mail address of the corresponding author: [email protected] doi:10.1016/j.jmb.2005.12.043 J. Mol. Biol. (2006) 357, 427–441
Transcript of WTRP-Wireless Token Ring Protocol

doi:10.1016/j.jmb.2005.12.043 J. Mol. Biol. (2006) 357, 427–441
Direct Selection of Antibodies from Complex Librarieswith the Protein Fragment Complementation Assay
Holger Koch, Nico Grafe, Ralph Schiess and Andreas Pluckthun*
Biochemisches Institut derUniversitat Zurich,Winterthurerstr. 190CH-8057 ZurichSwitzerland
0022-2836/$ - see front matter q 2005 E
Present addresses: N. Grafe, RochTR-BC1, Nonnenwald 2, D-82377 PSchiess, Institute for Molecular SystHonggerberg HPT, CH-8093 Zurich
Abbreviations used: BSA, bovineCDR, complementary-determiningdihydrofolate reductase; ELISA, enimmunosorbent assay; GdnHCl, ghydrochloride; gpD, capsid proteinlambda; HuCAL, human combinalibrary (MorphoSys AG, Munich);thiogalactopyranoside; JNK2, c-JunmDHFR, murine dihydrofolate redfragment complementation assay;scFv, single-chain antibody fragmevariable domains of the heavy (V(VL) connected by a peptide linkeplasmon resonance; TBS, Tris-buffetrimethoprim; wt, wild-type.
E-mail address of the [email protected]
The aim of the present study was to develop the protein fragmentcomplementation assay (PCA) for the intracellular selection of specificbinding molecules from the fully synthetic HuCALw antibody library. Here,we describe the first successful selections of specific antibodies by PCA, andwe discuss the opportunities and limitations of this approach. First, weenriched an antibody specific for the capsid protein D of bacteriophagelambda (gpD) by ten successive rounds of competitive liquid cultureselection. In an independent approach, we selected a specific antibody forthe c-Jun N-terminal kinase 2 (JNK2) in a single-step selection setup. In orderto obtain specific antibodies in only a single PCA selection round, the selectionsystem was thoroughly investigated and several strategies to reduce theamount offalse positives were evaluated. When expressed in the cytoplasm ofEscherichia coli, the PCA-selected scFv antibody fragments could be purified assoluble and monomeric proteins. Denaturant-induced unfolding experi-ments showed that both antibody fragments are stable molecules, even whenthe disulfide bonds are reduced. Furthermore, antigen-specificity of the PCA-selected antibody fragments is demonstrated by in vivo and in vitroexperiments. As antigen binding is retained regardless of the antibodyredox state, both PCA-selected antibody fragments can tolerate the loss ofdisulfide bridge formation. Our results illustrate that it is possible to selectwell-expressed, stable, antigen-specific, and intracellular functional anti-bodies by PCA directly.
q 2005 Elsevier Ltd. All rights reserved.
Keywords: intrabodies; protein fragment complementation assay; HuCALw
antibody library; in vivo selection; cytoplasmic expression
*Corresponding authorlsevier Ltd. All rights reserve
e Diagnosticsenzberg, Germany; R.ems Biology, ETH, Switzerland.serum albumin;region; DHFR,zyme-linked
uanidineD of bacteriophage
torial antibodyIPTG, isopropyl-b-D-N-terminal kinase 2;uctase; PCA, proteinRU, resonance unit;nt consisting of the
H) and light chainr; SPR, surfacered saline; TMP,
ing author:
Introduction
An extensive list of DNA sequences, coding forproteins with unknown functions, is being gene-rated by the ongoing worldwide genome sequen-cing programs.1 Elucidating the functions of theseproteins will be one of the most importantchallenges in the years to come.2 Therefore, oneimportant task of biotechnology is to generatespecific binding molecules against a wide varietyof targets with reasonable resources. Although avariety of selection systems have proven success ingenerating such binding molecules and are thuscommonly used, it is unlikely that they will providethe required throughput without additional com-ponents and development. In most display tech-niques the purification and immobilization of thetarget molecules has to be worked out individually,and binders against every target have to be selectedin separate experiments. Consequently, there isgreat demand for a method that would allow
d.

428 Direct in Vivo Selection of Intrabodies by PCA
substantially faster selection of binding mole-cules.3,4 Since the protein fragment complemen-tation assay (PCA) might have the potential tobecome a robust but very simple selection technol-ogy, and even allows parallel selection againstseveral targets in the same experiment, the aim ofthe present study was to test and develop PCA forthe intracellular selection of specific binding mole-cules from two different single-chain antibodylibraries. The use of PCA to select from naıveprotein libraries directly has not been describedbefore.
The PCA strategy described here is based on thegenetic dissection of the murine enzyme dihydro-folate reductase (mDHFR).5–7 In this approach, twointeracting partners (here, antibody and antigen) aregenetically fused to the two halves of the dividedmDHFR. When Escherichia coli is co-transformed withboth plasmids, the fusion partners can dimerize witheach other and thereby reassemble the dissectedenzyme from its individual fragments. Since bacterialDHFR is inhibited specifically through the antibiotictrimethoprim and thus cell division cannot occur, thereassembled mDHFR restores the biosyntheticreactions required for bacterial propagation. Anti-body–antigen interaction is thereby linked directly tobacterial survival and is detectable simply by colonyformation. For carrying out PCA selections, only thegenes of the target and of the binding molecule haveto be available. Therefore, a very rapid identificationfrom libraries and thus “generation” of specificbinding molecules is conceivable, and the work ofpurifying and immobilizing the antigen of interest isnot required, at least before more detailed investi-gations of the binders are carried out. In summary,PCA requires only transformation of plasmids,functional expression of the fusion proteins, andanalysis of bacterial cells. Consequently, we believethat this technology may offer great potential in termsof speed, simplicity, and future automation.
In order to test whether the PCA system would besuitable for use with antibodies, several scFvfragments were tested in previous model experi-ments.8 In these experiments, all evaluated anti-bodies were able to reassemble the separatedmDHFR domains into a functional enzyme throughspecific binding to its cognate target. In addition,every antibody that had been used in this studyinteracted either specifically with its antigen orshowed no reactivity at all. This was an importantobservation, since it suggests that selection ofantibodies should be feasible with PCA.
Nevertheless, utilization of recombinant antibodyfragments in the reducing environment of thecytoplasm regularly leads to folding and stabilityproblems due to restricted formation of their intra-chain disulfide bond.9 Consequently, low levels ofexpression of soluble protein and limited half-livesof antibody domains, which are either aggregatingor being degraded proteolytically, are usuallyobserved when expressed in the cytoplasm.Furthermore, incorrectly folded molecules mayengage in undesired and unspecific interactions
and thereby greatly complicate the selection pro-cess.
To overcome the problem of incomplete antibodyfolding in the bacterial cytoplasm, severalapproaches had been suggested: either the anti-bodies were expressed as fusions to a very solubleprotein,10 which leads to enhanced solubility of thefused antibody domains, albeit not necessarily to acompletion of the folding process, or the antibodieswere produced in modified E. coli strains that allowmore efficient oxidation of the cytoplasmicallyexpressed antibodies.11–13 In alternative strategies,the optimization of individual antibodies wascarried out in order to enhance the thermodynamicstability or the solubility and expression level of aparticular molecule.14–19 Such subsets of antibodieshave been expressed in the bacterial cytoplasm in astable and functional form, even though someresidual aggregation or soluble aggregate formationmay well occur. Additionally, the stability-engine-ered antibodies were used as frameworks to whichother specificities have been grafted.20,21
Even though such single-framework antibodylibraries appear suitable for direct intracellularselection, they have been usually applied to in vitro(most often phage display) selections, rather than inintracellular selections from the complex library.22,23
Instead, the intracellular performance of the selectedantibodies was usually evaluated in subsequentexperiments, separate from the selection itself. Sinceit is not ensured that the phage-selected antibodies,which contain disulfides during the selection, bindtheir antigen also in the reducing cytoplasm of a cell, ithas become a routine procedure to perform a yeasttwo-hybrid screen, subsequent to an initial phagedisplay selection round.24,25 This two-step selectionstrategy allows the isolation of antibodies based ontheir in vivo binding activity, and by transformingonly a small pool of phage-selected binders, theproblem of low transformation efficiency of yeast isovercome. Nevertheless, this two-step procedureremains laborious, and the yeast two-hybrid methodis used only for an evaluation of binders, rather than aselection strategy itself. In contrast, the high trans-formation efficiency of E. coli should allow the directselection of antibodies in the bacterial cytoplasm.Thus, utilization of PCA would allow the combi-nation of both antibody selection and antibodyevaluation in only one process and might, therefore,greatly improve the throughput of selection.
Although it has been possible to increase thestability of individual antibodies, it might be morepromising to apply designed antibody libraries inwhich a high proportion of molecules remainfunctional under reducing conditions of the bacterialcytoplasm. For this purpose, we assembled twoantibody libraries in the scFv format. In order tostart the PCA selections from well-expressed anti-body frameworks, suitable for cytoplasmicexpression, we utilized a restricted group of verystable HuCALw (Human Combinatorial AntibodyLibrary, MorphoSys AG, Munich) master frameworkcombinations. Since the stability, the expression yield
Direct in Vivo Selection of Intrabodies by PCA 429
and the aggregation behavior of all human variableantibody domains has been investigated recently in acomprehensive study,26 only the domains possessingthe most favorable properties for an intracellularapplication were included in our scFv antibodylibrary construction. Fortunately, the modular designof the HuCALw antibody library27 allowed us tocombine the most stable heavy chain (VH3) with allseven types of light chain domains in our first scFvantibody library. In this library, complementary-determining regions (CDR) CDR-H3 and CDR-L3are diversified as described,27 while CDR1 and CDR2of both heavy and light chain are consensussequences according to the subgroup. In the secondapproach, the constructed scFv antibody libraryresulted from a combination of both the most stableheavy chain domain (VH3) with the most stable lightchain (Vk3) domain. In this case, the complexity of thelatter, so-called “single-framework” antibody libraryresulted from diversification of all six CDR cassettesof both the heavy chain domains and the light chaindomains (S. Urlinger, C. Rothe et al., Morphosys A.G.,unpublished results). Both types of antibody librarieswere constructed with a complexity of more than 109
transformants. The performance of both antibodylibraries in the reducing environment of the bacterialcytoplasm has been investigated thoroughly, and thedirect selection of functional intracellular antibodiesfrom the assembled libraries is reported. We criticallydiscuss the opportunities and current limitations ofdirect PCA selection.
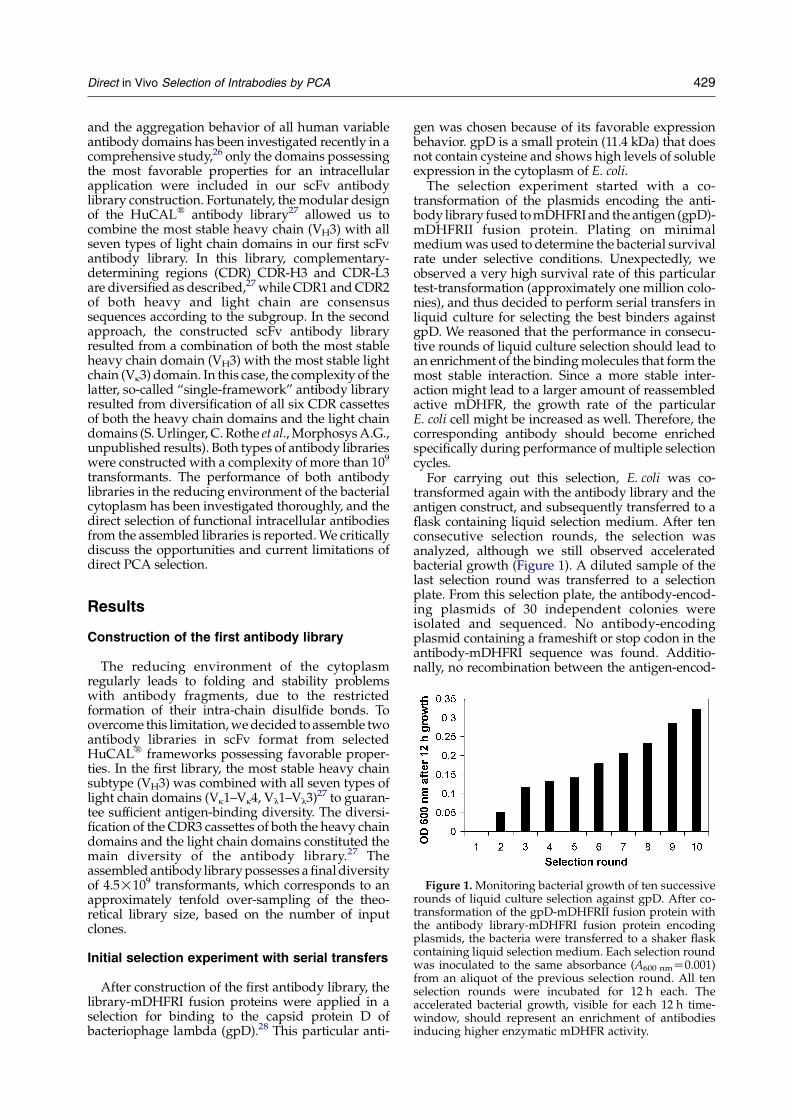
Figure 1. Monitoring bacterial growth of ten successiverounds of liquid culture selection against gpD. After co-transformation of the gpD-mDHFRII fusion protein withthe antibody library-mDHFRI fusion protein encodingplasmids, the bacteria were transferred to a shaker flaskcontaining liquid selection medium. Each selection roundwas inoculated to the same absorbance (A600 nmZ0.001)from an aliquot of the previous selection round. All tenselection rounds were incubated for 12 h each. Theaccelerated bacterial growth, visible for each 12 h time-window, should represent an enrichment of antibodiesinducing higher enzymatic mDHFR activity.
Results
Construction of the first antibody library
The reducing environment of the cytoplasmregularly leads to folding and stability problemswith antibody fragments, due to the restrictedformation of their intra-chain disulfide bonds. Toovercome this limitation, we decided to assemble twoantibody libraries in scFv format from selectedHuCALw frameworks possessing favorable proper-ties. In the first library, the most stable heavy chainsubtype (VH3) was combined with all seven types oflight chain domains (Vk1–Vk4, Vl1–Vl3)27 to guaran-tee sufficient antigen-binding diversity. The diversi-fication of the CDR3 cassettes of both the heavy chaindomains and the light chain domains constituted themain diversity of the antibody library.27 Theassembled antibody library possesses a final diversityof 4.5!109 transformants, which corresponds to anapproximately tenfold over-sampling of the theo-retical library size, based on the number of inputclones.
Initial selection experiment with serial transfers
After construction of the first antibody library, thelibrary-mDHFRI fusion proteins were applied in aselection for binding to the capsid protein D ofbacteriophage lambda (gpD).28 This particular anti-
gen was chosen because of its favorable expressionbehavior. gpD is a small protein (11.4 kDa) that doesnot contain cysteine and shows high levels of solubleexpression in the cytoplasm of E. coli.
The selection experiment started with a co-transformation of the plasmids encoding the anti-body library fused to mDHFRI and the antigen (gpD)-mDHFRII fusion protein. Plating on minimalmedium was used to determine the bacterial survivalrate under selective conditions. Unexpectedly, weobserved a very high survival rate of this particulartest-transformation (approximately one million colo-nies), and thus decided to perform serial transfers inliquid culture for selecting the best binders againstgpD. We reasoned that the performance in consecu-tive rounds of liquid culture selection should lead toan enrichment of the binding molecules that form themost stable interaction. Since a more stable inter-action might lead to a larger amount of reassembledactive mDHFR, the growth rate of the particularE. coli cell might be increased as well. Therefore, thecorresponding antibody should become enrichedspecifically during performance of multiple selectioncycles.
For carrying out this selection, E. coli was co-transformed again with the antibody library and theantigen construct, and subsequently transferred to aflask containing liquid selection medium. After tenconsecutive selection rounds, the selection wasanalyzed, although we still observed acceleratedbacterial growth (Figure 1). A diluted sample of thelast selection round was transferred to a selectionplate. From this selection plate, the antibody-encod-ing plasmids of 30 independent colonies wereisolated and sequenced. No antibody-encodingplasmid containing a frameshift or stop codon in theantibody-mDHFRI sequence was found. Additio-nally, no recombination between the antigen-encod-

Figure 2. Test of two strategies to reduce unspecificbacterial growth. (a) Influence of the interaction strengthof mDHFRI and mDHFRII on specific and unspecificantibody–antigen interaction pairs. Bacterial growthresulting from co-transformations utilizing the wild-type (wt) gpD-mDHFRII fusion protein (black bars) was
430 Direct in Vivo Selection of Intrabodies by PCA
ing and the antibody-encoding plasmids wasdetected. However, in two out of 30 constructsinvestigated, a deletion of the antibody genes wasfound. Yet, these observed occasional deletions aremost probably not present in the cell but rather resultfrom the isolation strategy of the antibody-encodingplasmids (see Materials and Methods). None of thesequenced antibodies was found multiple times.
Since the growth rate was still increasing fromround to round (Figure 1), liquid culture selectionwas continued. After another five rounds (a total of15 serial transfers of the E. coli culture), the growthrate did reach a plateau value. Sequencing of someof the clones from the 15th selection round showedthat the antibody diversity was only slightlyreduced (of 15 antibodies, 12 were different),when compared to the diversity of the tenthselection round (of 30 antibodies, all were different).This result shows that a diverse number ofantibodies could meet the selection criteria, eventhough only one specifically interacting pair wasfound (see the next section).
In order to simplify the specificity verification ofPCA-selected antibodies, we expressed the selectedantibodies in the mDHFRI fusion format directlyand performed an antigen enzyme-linked immuno-sorbent assay (ELISA) with crude extracts of theantibody expression cultures. Finally, out of the 30randomly chosen antibody fusions from the tenthselection round, one antibody specifically bindingthe antigen gpD (antibody D10) was identified inour ELISA setup (data not shown). This antibodywas retained after the 15th serial transfer.
D10 represents thus the first specific antibody thathas been selected successfully by PCA from a naiveantibody library. Although its functionality wasshown in the reducing intracellular environment, itcontained no mutation in its constituent VH3-Vl3antibody domains. This implies that the antibody D10retains functionality in the reducing environment ofthe bacterial cytoplasm, even though its intra-chaindisulfide bonds are not formed. The further charac-terization of D10 is described below.
Strategies to reduce unspecific bacterial growth
From the large number of antibodies that hadbecome enriched in the initial PCA selection (allcorrect in sequence), it became clear that in mostcases unspecific antibody–antigen interactionevents led to bacterial growth. Unspecific bacterialgrowth did not result from genetic instability of thePCA constructs. In order to reduce unspecificmDHFR activity, the influence of several para-meters on bacterial growth under selective condi-tions was evaluated.
set to 100% and compared to bacterial growth resultingfrom co-transformations of the mDHFRII I114A mutant(grey bars). (b) Influence of induction conditions onspecific and unspecific antibody–antigen interactionpairs. Bacterial growth resulting from induction with0.2 mM IPTG was set to 100% (black bars) and comparedto bacterial growth resulting from induction with only0.1 mM IPTG (grey bars).
Utilization of a mDHFRII point mutant
In this co-transformation experiment, two differenttypes of mDHFRII fragments (fusion partner of theantigen) were utilized: the wild-type (wt) mDHFRIIfragment, which has been used in our initial PCA
selection against gpD, and an mDHFRII variantcontaining the I114A point mutation.5 By utilizingthis latter, weakly associating mDHFRII fragment, thestringency of selection should be increased. Wereasoned that only binding molecules that form arather stable interaction would compensate thereduced ability of the mDHFRII I114A fragments toassociate into the active enzyme. In order to verify theeffect of the mDHFRII mutant, the gpD-specificantibody D10 and ten gpD unspecific antibodiesresulting from the initial PCA selection against gpDwere co-transformed with both the wt gpD-mDHFRIIfusion protein and the gpD-mDHFRII I114A mutantconstruct. For all of the chosen antibodies, bacterialgrowth was reduced significantly if the mDHFRIII114A mutant was co-transformed (Figure 2(a)).However, since this effect was identical for both thegpD specific antibody D10 and all unspecificantibodies tested, utilization of the mDHFRII I114Amutant does not seem to improve the PCA selection

Direct in Vivo Selection of Intrabodies by PCA 431
system in rewarding cognate interactions over non-cognate ones.
Influence of reduced induction strength
The initial PCA selection against gpD wasperformed at a high induction level (1 mM isopro-pyl-b-D-thiogalactopyranoside (IPTG)) to induce theexpression of the mDHFR fusion proteins. As thishigh induction strength may have induced the fusionproteins to aggregate, a simple reduction of theexpression level might reduce the proportion ofmisfolded antibodies, and unspecificbacterial growth might become more limited. Toinvestigate the effect of reduced induction strength,we co-transformed the gpD-specific antibody D10and ten gpD-unspecific antibodies with the wt gpD-mDHFRII antigen construct and incubated thetransformed cells on selection plates containingdifferent concentrations of IPTG. Reduction of theconcentration of IPTG from 1 mM to 0.2 mM hadalmost no effect on all investigated interaction pairs.Neither the number of colonies nor their size wasreduced. However, a completely different result wasobserved upon a further decrease of the concentrationof IPTG to 0.1 mM IPTG (Figure 2(b)). Under theseconditions, almost no viable bacterial colonies weredetected on the selection plates for nine out of tenunspecific antibodies. In contrast, there was noinfluence on the specific antibody–antigen pair thathad been selected by PCA before. Thus, we concludedthat the reduction of induction strength might be apromising strategy to refine the PCA selectionsystem.
Construction of the second antibody library
Sequence analysis of unselected library membershad shown an unbiased distribution of all differentlight chain subgroups (data not shown). In contrast,sequencing of the antibody fusions, which had beenenriched in our initial PCA selection against gpD,revealed that approximately 90% of the unspecificantibodies were comprised of the light chain domainsVl2 and Vl3, which when unpaired are least stable.26
This led us speculate that these antibodies might notbe able to fold properly in the bacterial cytoplasm,
Table 1. Summary of the PCA selection experiments
Applied selection strategy Library usedSuse
GpD fused to wt mDHFRII, competitionselection, ten selection rounds, 1 mMIPTG/25 8C
VH3-7VL
GpD fused to wt mDHFRII, single-stepselection, 0.1 mM IPTG/25 8C
VH3-Vk3
JNK2 fused to wt mDHFRII, single-stepselection, 0.2 mM IPTG/25 8C
VH3-Vk3
a Specificity verification by ELISA (antibody-mDHFRI fusions, crub Specificity verification by utilization of PCA (co-transformation of
antigen).
and might therefore lead to mDHFR reconstitutionthrough unspecific interactions with the antigen and/or the DHFR fragment. To reduce bacterial growththat is not dependent on cognate antibody–antigeninteractions, the design of the scFv antibody librarywas then revised, focusing on a more restricted groupof stable antibody frameworks. Thus, in a secondapproach, the modular design of the HuCALw Goldantibody library was used to combine the most stableheavy chain (VH3) with only the most stable lightchain (Vk3) domain. The binding diversity of this newsingle-framework antibody library resulted from adiversification of all six CDR cassettes. Constructionof the particular scFv antibody library finally yieldeda diversity of 3.8!109 transformants, which rep-resents, in contrast to our first antibody library, noover-sampling of the theoretical library size.
Selection experiments under improvedconditions
In order to verify whether the proposed improve-ments (library and reduced induction strength) leadto a PCA selection of specific clones more rapidly, anew selection was carried out. In contrast to ourfirst selection experiment, two antigens wereutilized in this second series of selections. Besidesusing gpD, again for reasons of high solubleexpression, we additionally used the c-JunN-terminal kinase-2 (JNK2)29 as antigen, a proteinthat has attracted considerable scientific interest asa potential drug target.30 JNK2 is approximatelyfive times larger than gpD (48.2 kDa versus11.4 kDa) and contains, in contrast to gpD, tenreduced cysteine residues.
With the new library and at lower inductionstrength, the bacterial survival under selectionconditions was reduced dramatically with bothantigens, compared to the results from the first setof experiments (Table 1). In order to determine theimpact of each of the implemented improvementsteps (new antibody library and lower expressionlevel), several additional co-transformations andexpression tests were performed (data not shown).A decrease of bacterial growth (one order ofmagnitude) was observed when the VH3-Vk3antibody library was co-transformed instead of
rvival rate underlective conditions
No. of antibodiestested for specificity
Specific antibodiesidentified
1.0!106 cfu 30a D10
1.0!103 cfu 50a None
1.2!102 cfu 80b J21
de cell extracts).the selected antibodies with the specific antigen and an unrelated

Table 2. Amino acid sequences of the PCA selected antibodies D10 and J21
VH Subtype CDR1 CDR2 CDR3
D10a VH3 GFTFSSYAMS AISGSGGSTYYADSVKG FSYVSGMDYJ21a VH3 GFTFSSYGMS NISSDGSNTNYADSVKG TYIQDF
VL Subtype CDR1 CDR2 CDR3D10a Vl3 SGDALGDKYAS DDSDRPS QSYDNDFYGTJ21a Vk3 RASQSVNSFLA DASNRAT QQYNSYPF
a Framework sequences of both VH and VL domains correspond to the HuCALw master genes.27
432 Direct in Vivo Selection of Intrabodies by PCA
the initial antibody library. As this antibody library-dependent effect was observed reproducibly forevery antigen-mDHFRII fusion protein utilized, itsuggests that we were able to reduce the proportionof misfolded antibodies by using this new antibodylibrary. Furthermore, the expression level of thegpD-mDHFRII fusion protein is significantly higherthan the expression level of the JNK2-mDHFRIIfusion protein (data not shown), and this mayprovide another factor why co-transformation ofJNK2-mDHFRII with both types of antibodylibrary-mDHFRI fusion yielded one order ofmagnitude fewer bacterial colonies than the corre-sponding co-transformations of gpD-mDHFRII.Finally, we found that reduction of the inductionstrength had the greatest influence on bacterialgrowth. A tenfold reduction of the concentration ofIPTG (from 1 mM to 0.1 mM) yielded a 100-foldimpaired bacterial survival rate. In summary, weconclude that bacterial survival under selectionconditions depends strongly on the expression levelof the mDHFR fusion proteins, and on the qualityand stability of the binding molecule library used.
As we aimed to establish PCA for single-stepselections, clones from selections with both anti-gens were characterized directly, and no rounds ofconsecutive liquid culture selection were per-formed. From the 1000 bacterial colonies of oursecond gpD selection, we directly expressed 50 ofthe selected antibodies in the mDHFRI fusionformat and performed again an antigen ELISAwith crude extracts of the antibody expressioncultures. From this set, no gpD-specific antibodywas identified (data not shown). We cannotexclude that it would have been possible toidentify gpD-specific antibodies by simply screen-ing more clones.
The specificity of the antibodies resulting fromJNK2 selection was tested in the PCA systemdirectly (data not shown). For this purpose, theisolated plasmids of putative JNK2-binding anti-bodies were co-transformed with the unspecificgpD-mDHFRII antigen construct. We expected nobacterial growth in these control transformations ifa particular antibody was specific for JNK2. Incontrast, if a particular antibody was an unspecificbinder, it probably would induce bacterial growthwith every antigen utilized. Out of the 78 JNK2selected antibodies, 75 induced bacterial growthwith the unspecific antigen gpD. Nevertheless,three candidates showed no bacterial growthwhen co-transformed with gpD-mDHFRII, of
which only one (antibody J21) was able to inducebacterial growth after repeated co-transformationwith JNK2-mDHFRII. Thus, similar to the initialPCA selection against gpD, one specific antibodyresulted from the selection against JNK2 (Table 2).
Expression and protein purification of thePCA-selected scFv antibody fragments
To determine the biophysical and binding pro-perties of the PCA-selected antibodies in vitro, thescFv fragments D10 and J21 were re-cloned in avector that corresponds to the PCA selectionplasmid but contains no DHFR fusion. Both scFvantibody fragments were then expressed andpurified from the E. coli cytoplasm by immobilizedmetal ion affinity chromatography (Figure 3), eitherin the presence of 2-mercaptoethanol (to avoidoxidation and obtain the reduced form) or in theabsence of reducing agent. For the latter, oxidizing,purification strategy, the formation of disulfidebonds was subsequently induced by air-oxidation,catalyzed by addition of CuSO4. Following thisexpression and purification protocol, we were ableto purify 5–8 mg of the scFv fragment D10 and8–12 mg of the scFv fragment J21 in functional formfrom 1 l shake-flask bacterial expression cultures.
In addition, the PCA-selected scFv antibodyfragments were re-cloned for expression in theperiplasm of E. coli. Both scFv antibody fragmentswere found to be soluble upon periplasmicexpression where disulfide formation can occur(Figure 3(b)).
Finally, the purified scFv antibody fragments,which resulted from cytoplasmic expression, werecharacterized by gel chromatography, equilibriumdenaturation experiments, ELISA, and Biacoremeasurements (see below).
In vivo and in vitro specificity verification ofthe PCA-selected scFv antibody fragments
To determine the in vivo specificity of the PCA-selected scFv antibody fragments, the plasmidsencoding the gpD-specific antibody D10 and theJNK2-specific antibody J21 were co-transformedwith four different antigen-mDHFRII fusion con-structs. Besides the respective specific antigens gpDand JNK2, two unrelated antigens (the leucinezipper GCN431 and the peptidyl-prolyl cis/transisomerase FkpA32) were tested. After separate co-transformation of both antibody-encoding plasmids

Figure 3. Purification and redox-state analysis of thePCA-selected scFv antibody fragments D10 and J21.Proteins were expressed both in the cytoplasm and inthe periplasm of E. coli and analyzed by SDS-PAGE (15%polyacrylamide gel). The size marker is indicated in kDa.(a) Redox-state analysis of the cytoplasmically expressedantibody samples, immobilized metal ion affinity chro-matography-purified in either the presence of 2-mercap-toethanol to avoid oxidation (reduced samples) or in theabsence of any reducing agent. For the latter samples, thedisulfide formation was subsequently induced by air-oxidation catalyzed by addition of CuSO4. Samples ofsoluble elution fractions were separated in the presence(reducing) or in the absence (non-reducing) of 2-mercap-toethanol in the SDS-PAGE loading buffer (LB). Note thatunder non-reducing conditions the oxidized form of thescFv antibody fragment D10 migrates faster than thecorresponding reduced antibody sample, whereas bothredox states of the scFv antibody fragment J21 migratealmost identically. (b) Western blot analysis of theperiplasmically expressed scFv antibody fragments D10and J21 (lane 1, insoluble fraction of crude expressionlysate; lanes 2 and 3, soluble fraction of crude expressionlysate in either reducing or non-reducing loading buffer).Expressed antibody fragments were detected with ananti-FLAG tag M1 antibody. Note that the periplasmicallyexpressed scFv antibody fragments migrate at a highermolecular mass than the cytoplasmically expressed scFvantibody fragments because of their additional FLAG tags(see Materials and Methods).
Direct in Vivo Selection of Intrabodies by PCA 433
with all four antigen-encoding plasmids, thetransformed E. coli cells were incubated in parallelunder selective conditions. After three days ofincubation we could show that both D10 and J21exclusively induced bacterial growth with theirspecific antigen, and no background bacterialgrowth with any of the unspecific antigens wasdetectable (Figure 4).
To demonstrate specificity of the PCA-selectedscFv antibody fragments in vitro as a function oftheir redox state, we performed ELISA experiments.Binding of D10 and J21 in either reduced oroxidized form to biotinylated gpD, biotinylated
JNK2 and bovine serum albumin (BSA) wascompared. In this ELISA, both scFv antibodyfragments clearly favored binding to their cognateantigen over binding to any unspecific antigen.Interestingly, all applied antibody fractions retainedcomparable antigen binding regardless of theantibody redox state (Figure 5).
To test whether the antigen binding of the PCA-selected antibody fragments is indeed comparablefor both the reduced and oxidized scFv antibodyfragments, we additionally determined the disso-ciation constant of the gpD-specific antibody D10by Biacore (Figure 6). As expected from our ELISAexperiments, the value of the dissociation constant(KD) obtained for either the oxidized (28.7 mM) orthe reduced (30.5 mM) antibody fragment D10 isalmost identical. The affinity of the scFv antibodyfragment J21 could not be determined accurately byBiacore, but was estimated to be of the order of50–100 mM.
Analytical gel-filtration chromatography
After purification of the cytoplasmicallyexpressed antibodies, the PCA-selected scFv anti-body fragments D10 and J21 in either reduced oroxidized form were subjected to analytical gel-filtration chromatography (Figure 7). Both scFvantibody fragments were determined to be mono-meric, independent of their redox state.
Equilibrium denaturation experiments
The thermodynamic stability of the PCA-selectedscFv antibody fragments was examined by guani-dine hydrochloride (GdnHCl) equilibrium dena-turation experiments (Figure 8). Unfolding of thescFv antibody fragments under both non-reducingand reducing conditions was monitored by the shiftof the fluorescence emission maximum as afunction of the concentration of denaturant afterexcitation at 280 nm. As the denaturant-inducedunfolding was not fully reversible for all theproteins investigated (data not shown), no DGN-U
values are reported; instead, the midpoint of thetransition of denaturation are given (Table 3), whichis a semi-quantitative measure for the stability ofthe scFv antibody fragments.
With the knowledge of the denaturation proper-ties of both the isolated VH and VL domains and thecombinations of these domains in scFv antibodyfragments,26 the resulting antibody fragments canbe grouped into different classes.33 If the intrinsicstability of one domain is significantly higher thanthe total stability (intrinsic plus interface stabili-zation) of the other domain, a visible step in theunfolding curve is observed. Both redox states ofthe scFv antibody fragment D10 showed such anunfolding behavior. The two transitions observedare thus assigned to the unfolding of the separateVH and VL domains. If, in contrast, the intrinsicstability of one domain is in the same range as thetotal stability of the other domain, no step will be
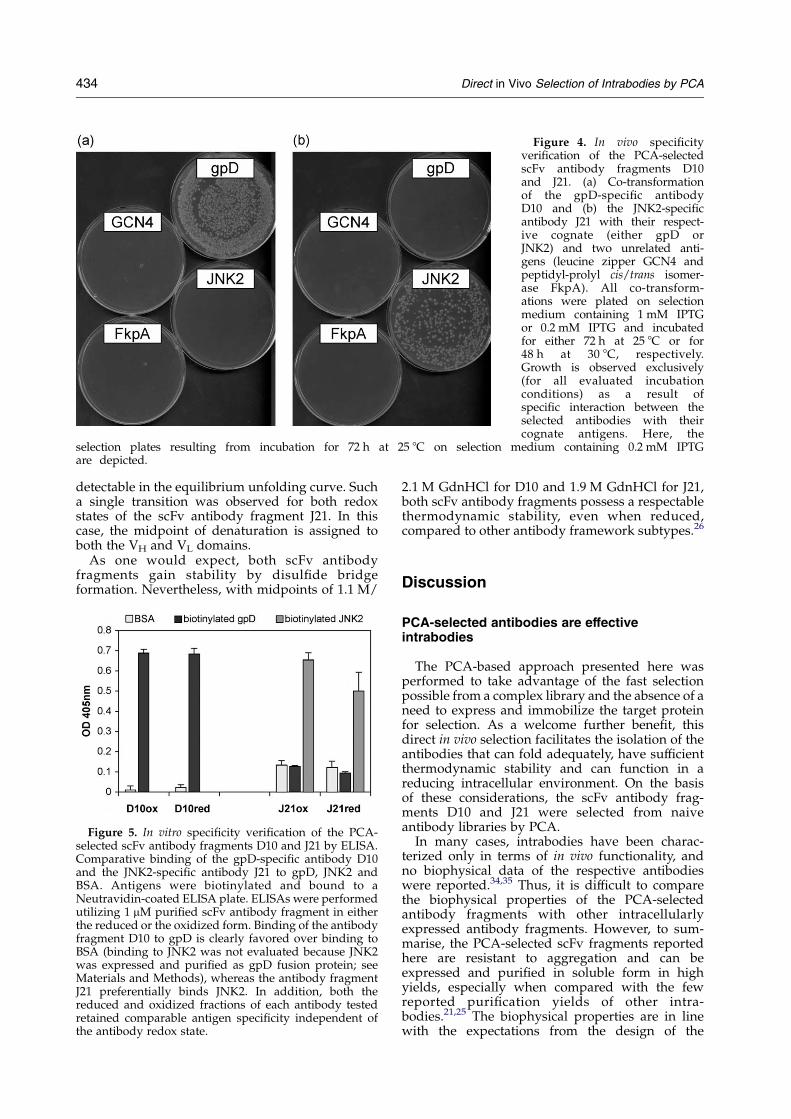
Figure 4. In vivo specificityverification of the PCA-selectedscFv antibody fragments D10and J21. (a) Co-transformationof the gpD-specific antibodyD10 and (b) the JNK2-specificantibody J21 with their respect-ive cognate (either gpD orJNK2) and two unrelated anti-gens (leucine zipper GCN4 andpeptidyl-prolyl cis/trans isomer-ase FkpA). All co-transform-ations were plated on selectionmedium containing 1 mM IPTGor 0.2 mM IPTG and incubatedfor either 72 h at 25 8C or for48 h at 30 8C, respectively.Growth is observed exclusively(for all evaluated incubationconditions) as a result ofspecific interaction between theselected antibodies with theircognate antigens. Here, the
selection plates resulting from incubation for 72 h at 25 8C on selection medium containing 0.2 mM IPTGare depicted.
434 Direct in Vivo Selection of Intrabodies by PCA
detectable in the equilibrium unfolding curve. Sucha single transition was observed for both redoxstates of the scFv antibody fragment J21. In thiscase, the midpoint of denaturation is assigned toboth the VH and VL domains.
As one would expect, both scFv antibodyfragments gain stability by disulfide bridgeformation. Nevertheless, with midpoints of 1.1 M/
Figure 5. In vitro specificity verification of the PCA-selected scFv antibody fragments D10 and J21 by ELISA.Comparative binding of the gpD-specific antibody D10and the JNK2-specific antibody J21 to gpD, JNK2 andBSA. Antigens were biotinylated and bound to aNeutravidin-coated ELISA plate. ELISAs were performedutilizing 1 mM purified scFv antibody fragment in eitherthe reduced or the oxidized form. Binding of the antibodyfragment D10 to gpD is clearly favored over binding toBSA (binding to JNK2 was not evaluated because JNK2was expressed and purified as gpD fusion protein; seeMaterials and Methods), whereas the antibody fragmentJ21 preferentially binds JNK2. In addition, both thereduced and oxidized fractions of each antibody testedretained comparable antigen specificity independent ofthe antibody redox state.
2.1 M GdnHCl for D10 and 1.9 M GdnHCl for J21,both scFv antibody fragments possess a respectablethermodynamic stability, even when reduced,compared to other antibody framework subtypes.26
Discussion
PCA-selected antibodies are effectiveintrabodies
The PCA-based approach presented here wasperformed to take advantage of the fast selectionpossible from a complex library and the absence of aneed to express and immobilize the target proteinfor selection. As a welcome further benefit, thisdirect in vivo selection facilitates the isolation of theantibodies that can fold adequately, have sufficientthermodynamic stability and can function in areducing intracellular environment. On the basisof these considerations, the scFv antibody frag-ments D10 and J21 were selected from naiveantibody libraries by PCA.
In many cases, intrabodies have been charac-terized only in terms of in vivo functionality, andno biophysical data of the respective antibodieswere reported.34,35 Thus, it is difficult to comparethe biophysical properties of the PCA-selectedantibody fragments with other intracellularlyexpressed antibody fragments. However, to sum-marise, the PCA-selected scFv fragments reportedhere are resistant to aggregation and can beexpressed and purified in soluble form in highyields, especially when compared with the fewreported purification yields of other intra-bodies.21,25 The biophysical properties are in linewith the expectations from the design of the
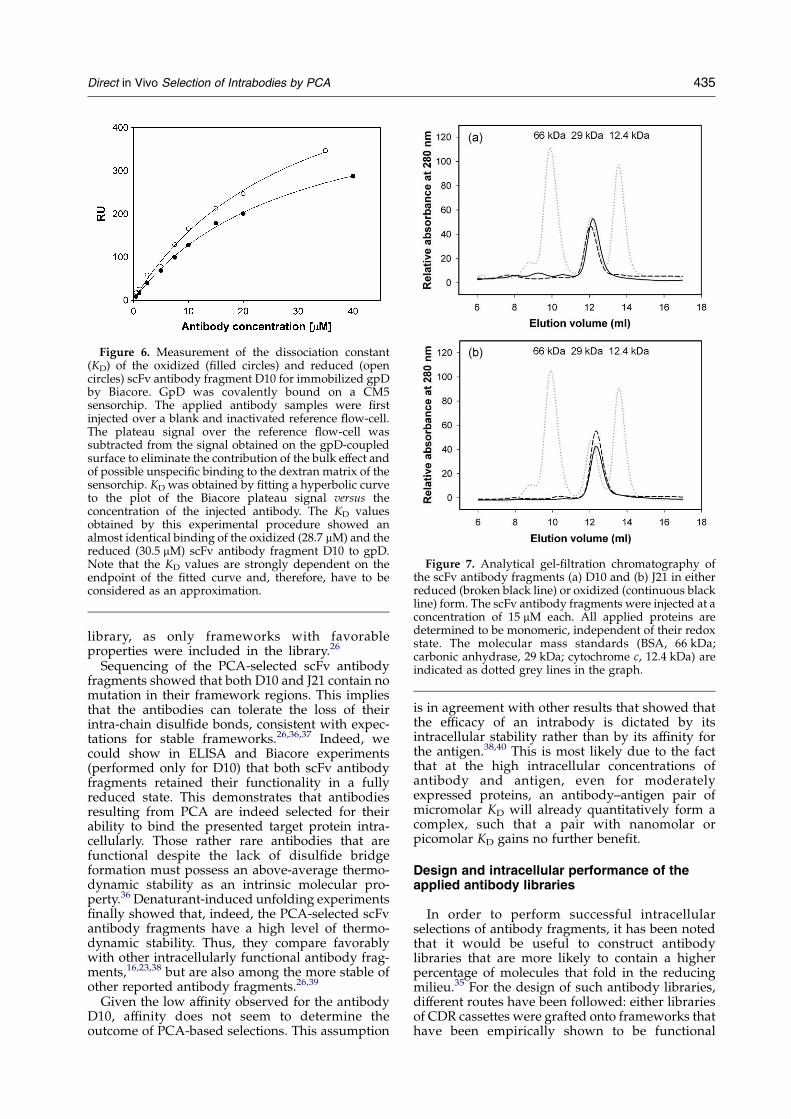
Figure 6. Measurement of the dissociation constant(KD) of the oxidized (filled circles) and reduced (opencircles) scFv antibody fragment D10 for immobilized gpDby Biacore. GpD was covalently bound on a CM5sensorchip. The applied antibody samples were firstinjected over a blank and inactivated reference flow-cell.The plateau signal over the reference flow-cell wassubtracted from the signal obtained on the gpD-coupledsurface to eliminate the contribution of the bulk effect andof possible unspecific binding to the dextran matrix of thesensorchip. KD was obtained by fitting a hyperbolic curveto the plot of the Biacore plateau signal versus theconcentration of the injected antibody. The KD valuesobtained by this experimental procedure showed analmost identical binding of the oxidized (28.7 mM) and thereduced (30.5 mM) scFv antibody fragment D10 to gpD.Note that the KD values are strongly dependent on theendpoint of the fitted curve and, therefore, have to beconsidered as an approximation.
Figure 7. Analytical gel-filtration chromatography ofthe scFv antibody fragments (a) D10 and (b) J21 in eitherreduced (broken black line) or oxidized (continuous blackline) form. The scFv antibody fragments were injected at aconcentration of 15 mM each. All applied proteins aredetermined to be monomeric, independent of their redoxstate. The molecular mass standards (BSA, 66 kDa;carbonic anhydrase, 29 kDa; cytochrome c, 12.4 kDa) areindicated as dotted grey lines in the graph.
Direct in Vivo Selection of Intrabodies by PCA 435
library, as only frameworks with favorableproperties were included in the library.26
Sequencing of the PCA-selected scFv antibodyfragments showed that both D10 and J21 contain nomutation in their framework regions. This impliesthat the antibodies can tolerate the loss of theirintra-chain disulfide bonds, consistent with expec-tations for stable frameworks.26,36,37 Indeed, wecould show in ELISA and Biacore experiments(performed only for D10) that both scFv antibodyfragments retained their functionality in a fullyreduced state. This demonstrates that antibodiesresulting from PCA are indeed selected for theirability to bind the presented target protein intra-cellularly. Those rather rare antibodies that arefunctional despite the lack of disulfide bridgeformation must possess an above-average thermo-dynamic stability as an intrinsic molecular pro-perty.36 Denaturant-induced unfolding experimentsfinally showed that, indeed, the PCA-selected scFvantibody fragments have a high level of thermo-dynamic stability. Thus, they compare favorablywith other intracellularly functional antibody frag-ments,16,23,38 but are also among the more stable ofother reported antibody fragments.26,39
Given the low affinity observed for the antibodyD10, affinity does not seem to determine theoutcome of PCA-based selections. This assumption
is in agreement with other results that showed thatthe efficacy of an intrabody is dictated by itsintracellular stability rather than by its affinity forthe antigen.38,40 This is most likely due to the factthat at the high intracellular concentrations ofantibody and antigen, even for moderatelyexpressed proteins, an antibody–antigen pair ofmicromolar KD will already quantitatively form acomplex, such that a pair with nanomolar orpicomolar KD gains no further benefit.
Design and intracellular performance of theapplied antibody libraries
In order to perform successful intracellularselections of antibody fragments, it has been notedthat it would be useful to construct antibodylibraries that are more likely to contain a higherpercentage of molecules that fold in the reducingmilieu.35 For the design of such antibody libraries,different routes have been followed: either librariesof CDR cassettes were grafted onto frameworks thathave been empirically shown to be functional
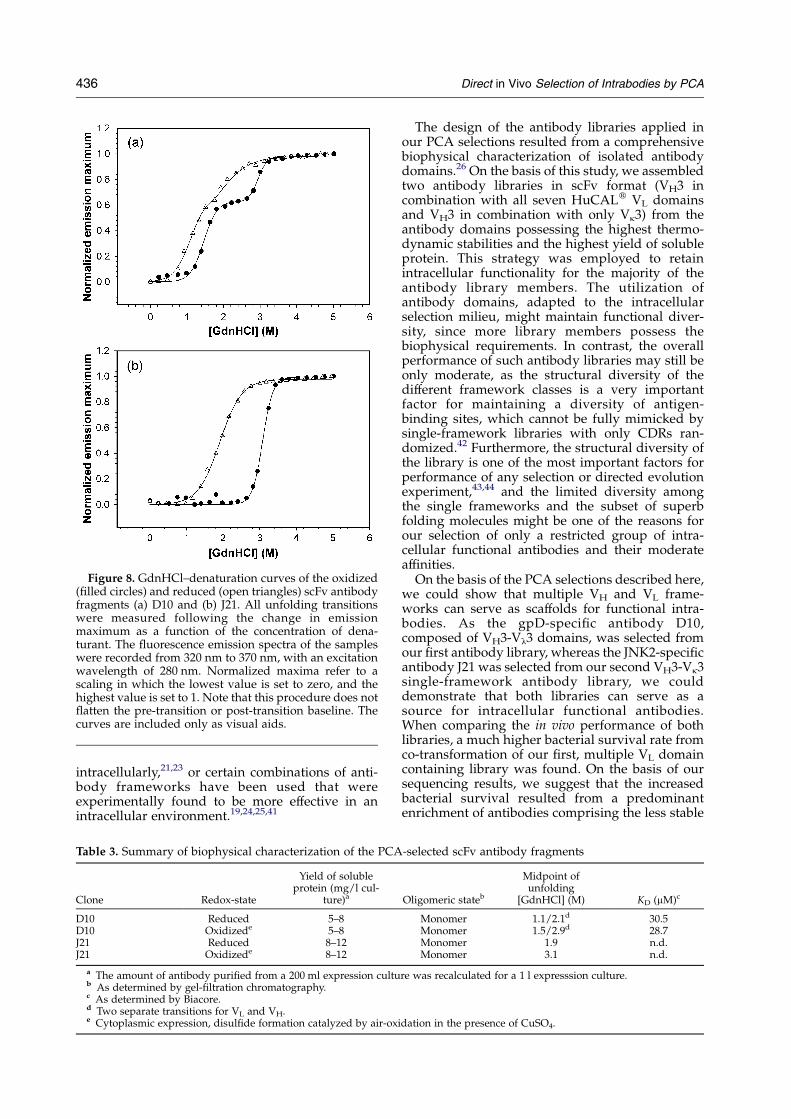
Figure 8. GdnHCl–denaturation curves of the oxidized(filled circles) and reduced (open triangles) scFv antibodyfragments (a) D10 and (b) J21. All unfolding transitionswere measured following the change in emissionmaximum as a function of the concentration of dena-turant. The fluorescence emission spectra of the sampleswere recorded from 320 nm to 370 nm, with an excitationwavelength of 280 nm. Normalized maxima refer to ascaling in which the lowest value is set to zero, and thehighest value is set to 1. Note that this procedure does notflatten the pre-transition or post-transition baseline. Thecurves are included only as visual aids.
436 Direct in Vivo Selection of Intrabodies by PCA
intracellularly,21,23 or certain combinations of anti-body frameworks have been used that wereexperimentally found to be more effective in anintracellular environment.19,24,25,41
Table 3. Summary of biophysical characterization of the PCA
Clone Redox-state
Yield of solubleprotein (mg/l cul-
ture)a
D10 Reduced 5–8D10 Oxidizede 5–8J21 Reduced 8–12J21 Oxidizede 8–12
a The amount of antibody purified from a 200 ml expression cultub As determined by gel-filtration chromatography.c As determined by Biacore.d Two separate transitions for VL and VH.e Cytoplasmic expression, disulfide formation catalyzed by air-ox
The design of the antibody libraries applied inour PCA selections resulted from a comprehensivebiophysical characterization of isolated antibodydomains.26 On the basis of this study, we assembledtwo antibody libraries in scFv format (VH3 incombination with all seven HuCALw VL domainsand VH3 in combination with only Vk3) from theantibody domains possessing the highest thermo-dynamic stabilities and the highest yield of solubleprotein. This strategy was employed to retainintracellular functionality for the majority of theantibody library members. The utilization ofantibody domains, adapted to the intracellularselection milieu, might maintain functional diver-sity, since more library members possess thebiophysical requirements. In contrast, the overallperformance of such antibody libraries may still beonly moderate, as the structural diversity of thedifferent framework classes is a very importantfactor for maintaining a diversity of antigen-binding sites, which cannot be fully mimicked bysingle-framework libraries with only CDRs ran-domized.42 Furthermore, the structural diversity ofthe library is one of the most important factors forperformance of any selection or directed evolutionexperiment,43,44 and the limited diversity amongthe single frameworks and the subset of superbfolding molecules might be one of the reasons forour selection of only a restricted group of intra-cellular functional antibodies and their moderateaffinities.
On the basis of the PCA selections described here,we could show that multiple VH and VL frame-works can serve as scaffolds for functional intra-bodies. As the gpD-specific antibody D10,composed of VH3-Vl3 domains, was selected fromour first antibody library, whereas the JNK2-specificantibody J21 was selected from our second VH3-Vk3single-framework antibody library, we coulddemonstrate that both libraries can serve as asource for intracellular functional antibodies.When comparing the in vivo performance of bothlibraries, a much higher bacterial survival rate fromco-transformation of our first, multiple VL domaincontaining library was found. On the basis of oursequencing results, we suggest that the increasedbacterial survival resulted from a predominantenrichment of antibodies comprising the less stable
-selected scFv antibody fragments
Oligomeric stateb
Midpoint ofunfolding
[GdnHCl] (M) KD (mM)c
Monomer 1.1/2.1d 30.5Monomer 1.5/2.9d 28.7Monomer 1.9 n.d.Monomer 3.1 n.d.
re was recalculated for a 1 l expresssion culture.
idation in the presence of CuSO4.

Direct in Vivo Selection of Intrabodies by PCA 437
light chain domains Vl2 and Vl3. These antibodiesmay not fold properly in the bacterial cytoplasm,therefore leading to bacterial growth throughunspecific interactions with the antigen-mDHFRIIfusion protein. For this reason, the VH3-Vk3 single-framework antibody library appears a betterstarting point for forthcoming PCA selections.
Conclusions
Here, we have presented the first successfulapplication of the protein fragment complementa-tion assay (PCA) for the intracellular selection ofantibodies from a complex naıve library. Twospecific scFv antibody fragments for two indepen-dent antigens have been enriched from complexlibraries by different selection strategies. Theantibody fragments generated show a high solubleexpression yield and possess high thermodynamicstability. Furthermore, we could show that antibodyfunctionality does not require disulfide bondformation, but it is also not impeded by it. Withthese results, a direct in vivo selection system forgenerating binding molecules, and particularlyintrabodies, has become available. Nevertheless,its development and application is still at an earlystage, with non-specific intracellular binding andlack of reward for high-affinity interactions asremaining challenges to be overcome. The identi-fication of specific binding molecules in only asingle PCA selection round would be highlyattractive for proteomics projects, and furtherimprovements may bring this technology towardsthis goal.
Materials and Methods
Construction of mDHFR fusion proteins
Design of the first antibody library
This antibody library has been assembled in the scFvformat through combination of four HuCAL (MorphoSysAG, Munich) sublibraries,27 in order to combine the moststable heavy chain domain (VH3)26 with all seven lightchain domains. In this library design, only the CDR3cassettes of both the heavy chain domains and the lightchain domains were diversified. The starting point for theconstruction of the antibody library was the scFvmaster gene sublibraries of HuCALw in the orientationVH-(Gly4Ser)4-VL.27 As recipient vector, the PCA antibodyplasmid pHK36, which already contained the HuCAL-derived scFv anti-FkpA, 7B2, was used.8 First, a fragmentof approximately 1 kb was removed from pHK36 bydigestion with BsmI. This was done in order to removeseveral restriction sites, which would be incompatiblewith the modular design of the HuCALw antibody library.Then, the modified vector pHK36 as well as the HuCALw
master gene sublibraries were digested with Eco52I andEcoRI. After ligation of the gel-purified scFv antibodyfragments in pHK36, the ligation mix was electroporatedin E. coli XL1-Blue cells (Stratagene). Finally, the con-structed antibody library (pHK44) possesses a diversity of
4.5!109 transformants, which corresponds to a tenfoldover-sampling of the theoretical library size.
Design of the refined antibody library
The second antibody library was assembled in the scFvformat through combination of two HuCALw sublibrariesin order to combine the most stable heavy chain domain(VH3) with the most stable light chain domain (Vk3). In thisparticular library, all six CDR cassettes of both the heavychain domains and the light chain domains were diversi-fied. The starting point for construction of the secondantibody library was the Fab master gene sublibraries ofHuCAL Gold (MorphoSys AG, Munich). As recipientvector the BsmI-modified PCA antibody plasmid pHK36(see construction of the first antibody library), whichalready contained the HuCAL-derived scFv anti-FkpA,7B2 was used.8 First, the anti-FkpA antibody, 7B2, present inthe vector, was removed from pHK36 and replaced by aHuCALw-derived dummy VH3-(Gly4Ser)4-Vk3 scFv anti-body fragment. Additionally, the restriction site BspEI,which would have been incompatible with the intendedcloning strategy, was removed by site-directed mutagenesisfrom the linker between the scFv and the mDHFRIfragment. All listed modifications of pHK36 finally yieldedthe vector pHK46, which is fully compatible with theflanking restriction sites of the HuCALw Gold VH3 and theVk3 sublibraries. First, both pHK46 and the HuCALw GoldVk3 sublibrary were digested with PstI and MscI. Afterligation of the gel-purified Vk3 sublibrary in pHK46, theligation mix was electroporated in E. coli XL1-Blue cells(Stratagene). This particular cloning step finally yielded thevector pHK47, which still contained the VH3 dummydomain plus the Vk3 sublibrary with a diversity of 5.4!107
transformants. After construction of the Vk3 sublibrary thevector pHK47 and the HuCALw Gold VH3 sublibrary weredigested with BspEI and Bpu1102I. Similar to the construc-tion of the Vk3 sublibrary, the gel-purified VH3 sublibrarywas first ligated to pHK47, and afterwards electroporated inE. coli XL1-Blue cells (Stratagene). Finally, the constructedsingle-framework (VH3-Vk3) antibody library (pHK48)possesses a diversity of 3.8!109 transformants, whichrepresents no over-sampling of the theoretical library size.
Design of the antigen constructs
First, the b-lactamase resistance cassette of the antigen-encoding plasmid pHK88 was replaced by an optimizedchloramphenicol resistance cassette45 (engineered toeliminate several restriction sites), yielding the plasmidpHK40. Thus, both the antibody and antigen-encodingplasmids contain different resistance genes in order toallow an efficient separation of the plasmids after aselection has been performed.
In order to exchange the antigen of the antigen vectorpHK40, gpD was PCR-amplified from the plasmidpAT101,28 using the following oligonucleotides: forward5 0-GGA TCC GCA TGC TTG CGA GCA AAG AAA CCTTTA CC-3 0 and backward 5 0-CAT CTT TAT AGT CGAATT CAA CGA TGC TGA TTG CCG TTC C-3 0. Afterdigestion of both the resulting PCR fragment and thevector pHK40 with SphI and EcoRI the antigen gpD-encoding fragment was gel-purified and ligated inpHK40, finally yielding the plasmid pHK43. JNK2a2was PCR amplified from the plasmid pAT222_JNK246
using the following oligonucleotides: forward 5 0-GCTCAG GCA TGC TTT CCG ACT CTA AAT GTG ACA G-3 0
and backward 5 0-GCT GCA GAA TTC TCG ACA GCC

438 Direct in Vivo Selection of Intrabodies by PCA
TTC AAG GGG-3 0. After digestion of both the resultingPCR fragment and the vector pHK43 with SphI and EcoRIthe antigen JNK2-encoding fragment was gel-purifiedand ligated in pHK43, finally yielding the plasmidpHK92.
Protein fragment complementation assay
Electro-competent E. coli BL21/pRep4 cells (transform-ation efficiencyZ1!109/mg of DNA for selection experi-ments and at least R3!108/mg of DNA for specificityverification) were co-transformed with 100 ng of each theantibody-mDHFRI and the antigen-mDHFRII fusionprotein-encoding plasmids. After incubation for 1 h at37 8C (no selection pressure), the cells were washed withM9 minimal medium and either transferred to M9medium containing 50 mg/ml of kanamycin (kanR,repressor plasmid pRep4), 100 mg/ml of ampicillin(ampR, antibody library-encoding plasmid), 10 mg/ml ofchloramphenicol (camR, antigen-encoding plasmid),2 mg/ml of trimethoprim (to inhibit bacterial DHFR andselect for functional mDHFR), and various concentrations(100 mM to 1 mM) of IPTG (induction of both mDHFRfusion proteins). By adding 5% (w/v) of Casamino acids(Difco) to the standard composition of M9 minimalmedium, the incubation time on solid selection mediumwas reduced to 72 h at 25 8C or 48 h at 30 8C.
For performance of successive selection rounds inliquid culture, 75 ml of selection medium (compositionas above) was inoculated from the respective precultureto a final A600Z0.001. Expression of the mDHFR fusionproteins was induced directly by addition of IPTG to afinal concentration of 1 mM. Each of the ten selectionrounds was performed for 12 h at 25 8C.
For in vivo specificity determination of both scFvantibody fragments D10 and J21, selection plates contai-ning 1 mM or 0.2 mM IPTG were utilized. Theseparticular selection plates were incubated for 72 h at25 8C and 48 h at 30 8C, respectively.
Isolation of antibody-encoding plasmids
After performance of a PCA selection experiment, theantibody-encoding plasmids had to be isolated to verifythe antigen-specificity of each individual antibody. Sinceovernight cultures inoculated from the selection plateswere expected to contain three plasmids, the antibody-encoding plasmid had to be separated from the repressorplasmid pRep4 and the antigen-encoding plasmid. Afterpreparation of DNA from the selection plate, a smallsample of the miniprep elution fraction was heavily over-digested with either RsrII (gpD selection) or Bsp120I(JNK2 selection). These restriction sites were presentexclusively in the repressor plasmid pRep4 and theantigen-encoding plasmid. Therefore, the antibody-encoding plasmids remain undigested. In contrast, if aselected antibody gene would contain also one (ormultiple) restriction site (n) of the chosen enzymes, aparticular antibody clone would be lost. This might havebeen the reason for the two observed antibodygene deletions in the gpD-selection experiment. Afterre-transformation of the digested DNA sample into E.coliXL-10 cells (Stratagene), the transformation mix wasplated on LB-agar plates (2% (w/v) glucose; 100 mg/ml ofampicillin), and one resulting bacterial colony of eachindividual antibody construct was used for repeatedDNA isolation. Finally, the purity of the antibody-
encoding plasmid was verified by EcoRI/SphI restrictiondigestion of a proportion of the latter DNA isolation.
Expression of selected antibody-mDHFRI fusionproteins
After isolation of the antibody-encoding plasmid,E. coli BL21/pRep4 (Qiagen) cells were transformedwith the vectors. 50 ml of 2YT medium containing50 mg/ml of kanamycin and 100 mg/ml of ampicillinwas inoculated with an overnight preculture to a finalA600Z0.1. Expression was induced at an A600 ofapproximately 0.8 by addition of IPTG to a finalconcentration of 1 mM. Expression was performed for4 h at 25 8C.
Antigen preparation for in vitro specificity verification
For expression of the antigen constructs the vectorspAT222 (GenBank accession no. AY327137) andpAT222_JNK2 were used. Expression from these vectorsyields a fusion protein comprising an N-terminal avi-tagfor in vivo biotinylation, bacteriophage lambda proteingpD, followed by JNK2 (in the case of the vectorpAT222_JNK2), and a C-terminal His6 tag for purification(avi-gpD-(JNK2)-His6). Both gpD and JNK2 wereexpressed, biotinylated and purified as described.46 Thepurity of the samples was checked by SDS-PAGE analysisand the concentration was determined by measuring theabsorbance at 280 nm.
In vitro specificity verification of PCA-selectedantibodies by ELISA
Biotinylated gpD or biotinylated JNK2 (100 ml/well;10 mg/ml) was bound to a neutravidin-coated (100 ml/well; 5 mg/ml) ELISA plate (MaxiSorp). After beingwashed with Tris-buffered saline (TBS; 20 mM Tris–HCl(pH 7.5), 150 mM NaCl) containing 0.05% (v/v) Tween,the plates were blocked for 1 h with 300 ml/well of TBS,0.5% (w/v) BSA. ELISAs were performed either withcrude extract supernatant (150 ml/well) of the antibody-mDHFRI fusion proteins (initial PCA selection, screen forgpD binders) or with 1 mM purified scFv antibodyfragment (redox state-dependent binding of D10 andJ21). Binding of the PCA-selected scFv antibody frag-ments was analyzed by detection of their N-terminalRGS-His6 tag of either the antibody-mDHFRI fusionproteins or the purified antibody fragments asdescribed.46
Construction of the cytoplasmic expression vectors
The PCA-selected scFv antibody fragments D10 and J21were cloned via SphI and HindIII into the standardexpression vector pQE32 (Qiagen). The vector pQE32basically corresponds to the PCA selection plasmid butdoes not contain any mDHFR fusion protein. It containsan N-terminal RGS-His6 tag, which was used forsubsequent purification of the scFv antibody fragments.Both antibodies were amplified by PCR using thefollowing oligonucleotides: forward (used for the amplifi-cation of both antibodies) 5 0-GGA TCC GCA TGC TTGAAG TGC AAT TGG TGG AAA G-3 0 and backward (forthe antibody D10) 5 0-GAG GAT CCA AGC TTC TAT TACTGG CCA AGA ACG GTT AAC TTC-3 0 (or the antibodyJ21) 5 0-GAG GAT CCA AGC TTC TAT TAC GTA CGTTTA ATT TCA ACT TTC G-3 0.

Direct in Vivo Selection of Intrabodies by PCA 439
Cytoplasmic expression of the PCA-selected scFvantibody fragments
All expression experiments were carried out usingE. coli BL21/pRep4 (Qiagen) cells. Medium (1 l of 2YTmedium in a 5 l flask without baffles, 150 rpm) containing50 mg/ml of kanamycin and 100 mg/ml of ampicillin wasinoculated with an overnight preculture to a final A600Z0.1. Cytoplasmic expression of the scFv antibody frag-ments D10 and J21 was induced at an A600 of approxi-mately 0.8 by addition of IPTG to a final concentration of0.5 mM. Expression was allowed to continue for 4 h at25 8C. Expression cultures were harvested by centrifu-gation in portions of 200 ml each. Resulting cell pelletswere stored at K80 8C.
Construction of the periplasmic expression vectors
The PCA-selected scFv antibody fragments D10 and J21were cloned via BspEI and EcoRI into the expressionvector pMx7 (Morphosys). In pMx7, the scFv antibodyfragments are expressed under control of the inducible lacpromoter/operator and secreted to the periplasm. Thefinal expression cassette consists of a phoA signalsequence, the short FLAG tag (DYKD), the scFv antibodyfragment in the orientation VH-(Gly4Ser)4-VL, the longFLAG tag (DYKDDDD) and a His6 tag.
Periplasmic expression of the PCA-selected scFvantibody fragments
All expression experiments were carried out usingE. coli SB536 cells.47 Medium (1 l of 2YT medium in a 5 lflask without baffles, 150 rpm) containing 25 mg/ml ofchloramphenicol and 50 mM K2HPO4 was inoculatedwith an overnight pre-culture to a final A600Z0.1.Periplasmic expression of the scFv antibody fragmentsD10 and J21 was induced at an A600 of approximately 0.8by addition of IPTG to a final concentration of 0.5 mM.Expression was allowed to continue for 4 h at 25 8C.Expression cultures were harvested by centrifugation inportions of 200 ml each. Resulting cell pellets were storedat K80 8C until Western blot analysis was performed.
Immobilized metal ion affinity chromatographypurification of the cytoplasmically expressed scFvantibody fragments
After cell lysis by French press, the crude extracts of thePCA-selected scFv antibody fragments D10 and J21 werecentrifuged at 20500 rpm in an SS34 rotor for 60 min at4 8C. After passing the supernatant through a 0.22 mmpore size filter, 20 ml of French press lysate supernatantwas loaded onto an equilibrated Protinow Ni-2000column (Machery-Nagel) to purify the scFv antibodyfragments via their N-terminal RGS-His6 tag. To removeunspecific binding proteins, the columns were washedwith ten column volumes of lysis buffer (50 mMNa2HPO4 (pH 8.0), 300 mM NaCl) and five columnvolumes of high-salt buffer (50 mM Na2HPO4 (pH 8.0),900 mM NaCl). The scFv antibody fragments were elutedby adding two column volumes each of 20 mM imidazole,50 mM imidazole and 100 mM imidazole. Both scFvantibody fragments were purified following the sameprotocol. In the case of the reduced antibody samples,20 mM 2-mercaptoethanol was included in all buffers toavoid oxidation of the scFv antibody fragments.16 Theformation of disulfide bonds in the purification of the
oxidized scFv antibody fragments was catalyzed by air-oxidation in the presence of 10 mM CuSO4.37 The purity ofthe samples was checked by SDS-PAGE analysis and theconcentration was determined by measuring absorbanceat 280 nm. The purification was always done for only200 ml of the 1 l expression culture. The amount of scFvantibody fragment, which was purified from 200 ml of theexpression culture, was finally recalculated to refer thetotal protein yield to that from a 1 l expression culture.
Western blot analysis
For immunoblots, the crude extracts of the periplasmi-cally expressed scFv antibody fragments were analyzedon SDS/15% (w/v) polyacrylamide gels and electro-transferred onto nitrocellulose membranes (Millipore).The PCA-selected antibodies were detected with an anti-FLAG tag M1 antibody (Sigma). An alkaline phosphatase-conjugated goat anti-mouse antibody (Sigma) was usedas secondary antibody. The immunoblots were developedusing the BCIP/NBT color development solution (Biorad)according to the instructions of the supplier.
Gel-filtration chromatography
Samples of purified scFv antibody fragments wereanalyzed on a Superdex-75 gel-filtration column (AKTA,Amersham-Pharmacia) at a concentration of 15 mM each,with 300 mM NaCl, 50 mM Na2HPO4 (pH 7.5) and either10 mM CuSO4 in the case of the oxidized antibodies or2 mM DTT for the reduced antibodies as running buffer.Proteins were injected in a volume of 100 ml, and thecolumn was run with a flow-rate of 500 ml minK1.Cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa),and BSA (66 kDa) were used as molecular massstandards. Elution was followed by detection of theabsorbance at 280 nm.
Equilibrium denaturation experiments
Fluorescence spectra were recorded at 20 8C with a PTIAlpha Scan spectrofluorimeter (Photon Technologies,Inc.). Protein/GdnHCl mixtures (1.5 ml) containing afinal protein concentration of 0.5 mM and denaturantconcentrations ranging from 0 M to 5 M GdnHCl wereprepared from freshly purified protein and a GdnHClstock solution (6 M GdnHCl in 50 mM Na2HPO4 (pH 7.5),150 mM NaCl; in the case of the reduced antibodies,2 mM DTT was added). Each final concentration ofGdnHCl was determined from its refractive index. Afterincubation for two days at 4 8C, the fluorescence emissionspectra of the samples were recorded from 320 nm to370 nm, with an excitation wavelength of 280 nm. Thefluorescence emission maximum, which was determinedby fitting the fluorescence emission spectrum to aGaussian function was plotted versus the concentrationof GdnHCl. To compare the denaturation curves of theoxidized or reduced scFv fragments in one plot, theemission spectra were normalized by setting the highestvalues to 1 and the lowest to zero.
Surface plasmon resonance
Surface plasmon resonance (SPR) was measured usinga BIAcore 3000 instrument (BIAcore). The running bufferwas 10 mM Hepes (pH 7.5), 150 mM NaCl, 3 mM EDTA,0.005% (v/v) Tween 20 (in the case of the reduced scFvantibody fragment, 2 mM DTT was added). Biotinylated

440 Direct in Vivo Selection of Intrabodies by PCA
gpD was immobilized on a CM5 chip (BIAcore) to 320resonance units (RU) by amine-coupling as described inthe BIAapplications Handbook. The interactions weremeasured at a flow rate of 5 ml minK1. Pulses (10 ml) ofeither oxidized or reduced scFv antibody fragment D10were injected in various concentrations (0.5 mM to 40 mM),followed by a dissociation time of 900 s for the oxidizedscFv antibody fragment and 1200 s for the reduced scFvantibody fragment to allow regeneration. The signal at theplateau was calculated by subtracting the signal obtainedon a deactivated control surface from the signal obtainedon the gpD-coupled surface. Each data point was theaverage of two measurements. The dissociation constantwas obtained by plotting (SigmaPlot 2001, SPSS Inc.) thesignal (in RU) against the concentration of the injectedantibody (AB), and fitting to a hyperbolic curve:
RU Z RUmax½AB�=ðKD C ½AB�Þ
where RUmax is the maximal value at saturation.
Acknowledgements
The authors thank Dr Patrik Forrer, Dr KasparBinz, Daniel Steiner, and Dr Patrick Amstutz for theprovision of purified gpD and JNK2, and Mor-phoSys AG, Munich for collaboration on theHuCALw antibody library. H.K. was a recipient ofa fellowship from the Ernst Schering ResearchFoundation, and N.G. received a travel grant(European Exchange Program) from the SwissFederal Office for Education and Science. Thiswork was supported by grants from the DeutschesBundesministerium fur Bildung und Forschung,the European Union (EULIP) and SchweizerischeNational fonds.
References
1. Broder, S. & Venter, J. C. (2000). Sequencing the entiregenomes of free-living organisms: the foundation ofpharmacology in the new millennium. Annu. Rev.Pharmacol. Toxicol. 40, 97–132.
2. Chanda, S. K. & Caldwell, J. S. (2003). Fulfilling thepromise: drug discovery in the post-genomic era.Drug Discov. Today, 8, 168–174.
3. Hayhurst, A. & Georgiou, G. (2001). High-throughputantibody isolation. Curr. Opin. Chem. Biol. 5, 683–689.
4. Bradbury, A., Velappan, N., Verzillo, V., Ovecka, M.,Chasteen, L., Sblattero, D. et al. (2003). Antibodies inproteomics I: generating antibodies. Trends Biotechnol.21, 275–281.
5. Pelletier, J. N., Campbell-Valois, F. X. & Michnick, S. W.(1998). Oligomerization domain-directed reassembly ofactive dihydrofolate reductase from rationally designedfragments. Proc. Natl Acad. Sci. USA, 95, 12141–12146.
6. Pelletier, J. N., Arndt, K. M., Pluckthun, A. &Michnick, S. W. (1999). An in vivo library-versus-library selection of optimized protein-protein inter-actions. Nature Biotechnol. 17, 683–690.
7. Remy, I., Wilson, I. A. & Michnick, S. W. (1999).Erythropoietin receptor activation by a ligand-induced conformation change. Science, 283,990–993.
8. Mossner, E., Koch, H. & Pluckthun, A. (2001). Fastselection of antibodies without antigen purification:adaptation of the protein fragment complementationassay to select antigen-antibody pairs. J. Mol. Biol. 308,115–122.
9. Biocca, S., Ruberti, F., Tafani, M., Pierandrei-Amaldi, P.& Cattaneo, A. (1995). Redox state of single chain Fvfragments targeted to the endoplasmic reticulum,cytosol and mitochondria. Biotechnology (NY), 13,1110–1115.
10. Bach, H., Mazor, Y., Shaky, S., Shoham-Lev, A.,Berdichevsky, Y., Gutnick, D. L. & Benhar, I. (2001).Escherichia coli maltose-binding protein as a molecularchaperone for recombinant intracellular cytoplasmicsingle-chain antibodies. J. Mol. Biol. 312, 79–93.
11. Proba, K., Ge, L. & Pluckthun, A. (1995). Functionalantibody single-chain fragments from the cytoplasmof Escherichia coli: influence of thioredoxin reductase(TrxB). Gene, 159, 203–207.
12. Jurado, P., Ritz, D., Beckwith, J., de Lorenzo, V. &Fernandez, L. A. (2002). Production of functionalsingle-chain Fv antibodies in the cytoplasm ofEscherichia coli. J. Mol. Biol. 320, 1–10.
13. Venturi, M., Seifert, C. & Hunte, C. (2002). High levelproduction of functional antibody Fab fragments inan oxidizing bacterial cytoplasm. J. Mol. Biol. 315, 1–8.
14. Proba, K., Worn, A., Honegger, A. & Pluckthun, A.(1998). Antibody scFv fragments without disulfidebonds made by molecular evolution. J. Mol. Biol. 275,245–253.
15. Auf der Maur, A., Escher, D. & Barberis, A. (2001).Antigen-independent selection of stable intracellularsingle-chain antibodies. FEBS Letters, 508, 407–412.
16. Martineau, P. & Betton, J. M. (1999). In vitro foldingand thermodynamic stability of an antibody fragmentselected in vivo for high expression levels inEscherichia coli cytoplasm. J. Mol. Biol. 292, 921–929.
17. Ohage, E. & Steipe, B. (1999). Intrabody constructionand expression. I. The critical role of VL domainstability. J. Mol. Biol. 291, 1119–11128.
18. Wirtz, P. & Steipe, B. (1999). Intrabody constructionand expression III: engineering hyperstable VH
domains. Protein Sci. 8, 2245–2250.19. Auf der Maur, A., Zahnd, C., Fischer, F., Spinelli, S.,
Honegger, A., Cambillau, C. et al. (2002). Directin vivo screening of intrabody libraries constructedon a highly stable single-chain framework. J. Biol.Chem. 277, 45075–45085.
20. Jung, S. & Pluckthun, A. (1997). Improving in vivofolding and stability of a single-chain Fv antibodyfragment by loop grafting. Protein Eng. 10, 959–966.
21. Donini, M., Morea, V., Desiderio, A., Pashkoulov, D.,Villani, M. E., Tramontano, A. & Benvenuto, E. (2003).Engineering stable cytoplasmic intrabodies withdesigned specificity. J. Mol. Biol. 330, 323–332.
22. Soderlind, E., Strandberg, L., Jirholt, P., Kobayashi, N.,Alexeiva, V., Aberg, A. M. et al. (2000). Recombininggermline-derived CDR sequences for creating diversesingle-framework antibody libraries. Nature Biotech-nol. 18, 852–856.
23. Desiderio, A., Franconi, R., Lopez, M., Villani, M. E.,Viti, F., Chiaraluce, R. et al. (2001). A semi-syntheticrepertoire of intrinsically stable antibody fragmentsderived from a single-framework scaffold. J. Mol. Biol.310, 603–615.
24. Visintin, M., Settanni, G., Maritan, A., Graziosi, S.,Marks, J. D. & Cattaneo, A. (2002). The intracellular

Direct in Vivo Selection of Intrabodies by PCA 441
antibody capture technology (IACT): towards aconsensus sequence for intracellular antibodies.J. Mol. Biol. 317, 73–83.
25. Tanaka, T. & Rabbitts, T. H. (2003). Intrabodies basedon intracellular capture frameworks that bind theRAS protein with high affinity and impair oncogenictransformation. EMBO J. 22, 1025–1035.
26. Ewert, S., Huber, T., Honegger, A. & Pluckthun, A.(2003). Biophysical properties of human antibodyvariable domains. J. Mol. Biol. 325, 531–553.
27. Knappik, A., Ge, L., Honegger, A., Pack, P., Fischer, M.,Wellnhofer, G. et al. (2000). Fully synthetic humancombinatorial antibody libraries (HuCAL) based onmodular consensus frameworks and CDRs random-ized with trinucleotides. J. Mol. Biol. 296, 57–86.
28. Yang, F., Forrer, P., Dauter, Z., Conway, J. F., Cheng, N.,Cerritelli, M. E. et al. (2000). Novel fold and capsid-binding properties of the lambda-phage display plat-form protein gpD. Nature Struct. Biol. 7, 230–237.
29. Kallunki, T., Su, B., Tsigelny, I., Sluss, H. K., Derijard,B., Moore, G. et al. (1994). JNK2 contains a specificity-determining region responsible for efficient c-Junbinding and phosphorylation. Genes Dev. 8,2996–3007.
30. Manning, A. M. & Davis, R. J. (2003). Targeting JNKfor therapeutic benefit: from junk to gold? Nature Rev.Drug Discov. 2, 554–565.
31. Berger, C., Weber-Bornhauser, S., Eggenberger, J.,Hanes, J., Pluckthun, A. & Bosshard, H. R. (1999).Antigen recognition by conformational selection.FEBS Letters, 450, 149–153.
32. Missiakas, D., Betton, J. M. & Raina, S. (1996). Newcomponents of protein folding in extracytoplasmiccompartments of Escherichia coli SurA, FkpA andSkp/OmpH. Mol. Microbiol. 21, 871–884.
33. Worn, A. & Pluckthun, A. (1999). Different equili-brium stability behavior of ScFv fragments: identifi-cation, classification, and improvement by proteinengineering. Biochemistry, 38, 8739–8750.
34. Tse, E., Lobato, M. N., Forster, A., Tanaka, T.,Chung, G. T. & Rabbitts, T. H. (2002). Intracellularantibody capture technology: application to selection ofintracellular antibodies recognising the BCR-ABLoncogenic protein. J. Mol. Biol. 317, 85–94.
35. Visintin, M., Tse, E., Axelson, H., Rabbitts, T. H. &Cattaneo, A. (1999). Selection of antibodies forintracellular function using a two-hybrid in vivosystem. Proc. Natl Acad. Sci. USA, 96, 11723–11728.
36. Worn, A. & Pluckthun, A. (1998). An intrinsicallystable antibody scFv fragment can tolerate the loss ofboth disulfide bonds and fold correctly. FEBS Letters,427, 357–361.
37. Proba, K., Honegger, A. & Pluckthun, A. (1997). Anatural antibody missing a cysteine in VH: con-sequences for thermodynamic stability and folding.J. Mol. Biol. 265, 161–172.
38. Rajpal, A. & Turi, T. G. (2001). Intracellular stability ofanti-caspase-3 intrabodies determines efficacy inretargeting the antigen. J. Biol. Chem. 276,33139–33146.
39. Worn, A. & Pluckthun, A. (2001). Stability engineer-ing of antibody single-chain Fv fragments. J. Mol. Biol.305, 989–1010.
40. Worn, A., Auf der Maur, A., Escher, D., Honegger, A.,Barberis, A. & Pluckthun, A. (2000). Correlationbetween in vitro stability and in vivo performance ofanti-GCN4 intrabodies as cytoplasmic inhibitors.J. Biol. Chem. 275, 2795–2803.
41. Gennari, F., Mehta, S., Wang, Y., St Clair Tallarico, A.,Palu, G. & Marasco, W. A. (2004). Direct phage tointrabody screening (DPIS): demonstration by iso-lation of cytosolic intrabodies against the TES1 site ofEpstein Barr virus latent membrane protein 1 (LMP1)that block NF-kappaB transactivation. J. Mol. Biol. 335,193–207.
42. Ewert, S., Honegger, A. & Pluckthun, A. (2004).Stability improvement of antibodies for extracellularand intracellular applications: CDR grafting to stableframeworks and structure-based framework engin-eering. Methods, 34, 184–199.
43. Perelson, A. S. (1989). Immune network theory.Immunol. Rev. 110, 5–36.
44. Ostermeier, M. (2003). Synthetic gene libraries: insearch of the optimal diversity. Trends Biotechnol. 21,244–247.
45. Reece, K. S. & Phillips, G. J. (1995). New plasmidscarrying antibiotic-resistance cassettes. Gene, 165,141–142.
46. Binz, H. K., Amstutz, P., Kohl, A., Stumpp, M. T.,Briand, C., Forrer, P. et al. (2004). High-affinity bindersselected from designed ankyrin repeat proteinlibraries. Nature Biotechnol. 22, 575–582.
47. Bass, S., Gu, Q. & Christen, A. (1996). Multicopysuppressors of prc mutant Escherichia coli include twoHtrA (DegP) protease homologs (HhoAB), DksA, anda truncated R1pA. J. Bacteriol. 178, 1154–1161.
Edited by F. Schmid
(Received 31 October 2005; accepted 5 December 2005)Available online 6 January 2006


















