
UPDATE ON PSORIATIC ARTHRITIS: EPIDEMIOLOGY, … · T cells and the Interleukin-23/17 pathway, 2)...
35
Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59 http://www.fmv-uba.org.ar/ 25 UPDATE ON PSORIATIC ARTHRITIS: EPIDEMIOLOGY, PATHOGENESIS, CLINICAL ASSESSMENT AND TREATMENT Daniel E. Furst Address: 1000 Veteran Ave., Rehabilitation Center Room 32-59, Los Angeles, California, 90095-1670. Telephone: 310-794-9504. Fax: 310-206-8606 E-mail: [email protected] Sangmee Bae James Louie Key Words: Psoriatic arthritis, Immunopathogenesis, Diagnosis, Clinical Features, Measurement, Treatment
Transcript of UPDATE ON PSORIATIC ARTHRITIS: EPIDEMIOLOGY, … · T cells and the Interleukin-23/17 pathway, 2)...

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
25
UPDATE ON PSORIATIC ARTHRITIS: EPIDEMIOLOGY, PATHOGENESIS, CLINICAL ASSESSMENT AND TREATMENT Daniel E. Furst Address: 1000 Veteran Ave., Rehabilitation Center Room 32-59, Los Angeles, California, 90095-1670. Telephone: 310-794-9504. Fax: 310-206-8606 E-mail: [email protected] Sangmee Bae James Louie Key Words: Psoriatic arthritis, Immunopathogenesis, Diagnosis, Clinical Features, Measurement, Treatment

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
26
ABSTRACT Psoriasis is a chronic inflammatory skin disease that affects 2-3% of the general population. Among these patients, 6-42% will also have psoriatic arthritis (PsA), an inflammatory arthropathy associated with psoriasis. About 5% will, however, present without psoriasis. PsA can present as peripheral arthritis, axial, involvement or both—often with enthesitis. The deformity and damage that occurs results in significant functional limitations and detrimental effects on the patients‘ quality of life. In contrast to rheumatoid arthritis (RA), the erosive joint destruction in patients with PsA is more often oligoarticular and asymmetrical, and disease progression is often more extensive. Recent pathogenetic studies of PsA emphasize several areas: 1) immunologic factors including T cells and the Interleukin-23/17 pathway, 2) abnormal bone remodeling that involves bone erosion and bone proliferation; a unique pattern seen in PsA, 3)genetics and 4)environmental factors. The heterogeneity of clinical presentation has hindered the development of universal diagnostic criteria and a validated measure of treatment response. However, recent observational studies showed that the CASPAR criteria have high specificity and sensitivity for diagnosing PsA. Although standard DMARDs like sulfasalazine are used, the increased understanding of PsA has engendered the development of more effective therapeutic options, mainly TNF inhibitors.
INTRODUCTION Psoriatic arthritis (PsA) is a chronic autoimmune inflammatory process that begins in the enthesis, where tendons and ligaments attach to the bone, and then spreads to the synovium. The various clinical subsets of PsA present as: (1) peripheral joint pain, swelling, bony resorption and reparative bony formation, resulting in deformity, in an asymmetric pattern (oligo) or a symmetrical pattern (polyarticular) or restricted to in the distal interphalangeal (DIP) joints. The arthritis may be of such severity that bone resorbs on both the proximal and distal parts of the joint (mutilans): (2) axial pain in sacroiliac and spinal ligamentous attachment sites: (3) an admixture of both peripheral and axial involvement .These clinical subsets were first described by Moll and Wright and still remain to be characterized by genetic and immunologic methods. The disease course is unpredictable but the deformity and damage in more than half of the patients can lead to significant functional limitations. Because enthesitis is the initiating site of inflammation, PsA is classified under the disease group ‗spondyloarthropathies (SpA)‘, in contrast to rheumatoid arthritis (RA). Both PsA and RA demonstrate erosive peripheral joint damage and there may be substantial difficulties differentiating the two. This chapter will describe the epidemiology and pathogenesis of PsA, its clinical presentation (including the differences between PsA and RA) and update the data on its treatment.
EPIDEMIOLOGY Due to the absence of universal diagnostic criteria, studies depicting the epidemiology of this disease describe widely varying estimations of incidence and prevalence. There are wide ethnic and geographic variations in the prevalence and incidence of PsA (see Table 1-2). The prevalence of psoriasis in the general population is 2-3%, but in patients with inflammatory arthritis the prevalence is 5-20.2%; and in patients with psoriasis it is 6-42% [2]. The overall incidence and prevalence estimates of PsA also varies widely among different countries around the world: from 1 per 100,000 in a Japanese study to 420 per 100,000 in an Italian study (median 180)[3](see Table 2). Some authors speculate that PsA is more common in the colder north (e.g. European countries) than the warmer tropics. A recent study supports this conclusion,

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
27
suggesting a significant geographic variation of occurrence of PsA[4]. Because the definition of PsA is not uniform, these are likely underestimations of the true disease frequency. Also, the lack of studies in Africa, Eastern Europe and South America limits an understanding of geographical variations of PsA. Although this geographic variability may reflect incomplete reporting, it may also reflect real genetic or environmental factors The onset of psoriasis typically precedes the development of the arthritic component. 85% of patients develop psoriasis prior to arthritis, 5-10% of patients experience both components concurrently and 5-10% demonstrate arthritis before skin involvement [2, 5]. While some studies suggest the severity of psoriasis is associated with the development of arthritis, it is not predictive of the severity of arthritis[5]. Four retrospective [6-9] and two prospective studies [10, 11] found a wide variation in the male/female ratio, from 0.4 to 1.3(median 0.9). In contrast to RA which has a female predominance, PsA is considered an equigender disease. Age at diagnosis is more uniform than in RA, with a median of 47.7 years (range 40.7-52.0). Geoepidemiological differences of PsA may be confounded by genetic and environmental factors. While HLA-B27 is known to be associated with Ankylosing Spondylitis and related spondyloarthropathies, HLA-Cw*6, among others (see below ‗Genetic susceptibility‘) is associated with PsA. In addition, associations with IL12B and IL23R have recently been confirmed [12]. Certain environmental factors, including HIV infection, trauma and psychological stress seem in some way related to PsA. Many additional genetic and environmental risk factors remain to be identified.
PATHOGENESIS Recent imaging (e.g. MRI) studies suggest the primary site of inflammation in PsA centers about the enthesis, the site where tendons and ligaments attach to bone. The close proximity of the enthesitis and inflamed synovium suggests that the synovitis of PsA is associated with release of proinflammatory and immunologic mediators from closely related entheses [13]. Although the pathogenesis of PsA remains unclear, the roles of immunologic, genetic, and environmental factors have been studied.
THE ROLE OF IMMUNE CELLS Immune reactivity in PsA is probably related to antigen driven CD8+ T lymphocyte responses. Both the skin and joints of patients with PsA display a predominantly lymphocytic infiltrate and the primary composition of the perivascular cellular infiltrate is activated T cells, later migrating to dermal papillae of skin, the sublining stroma in the joint and the inflamed entheses[14, 15]. A large number of B lymphocytes forming primitive germinal centers are also present in the synovium of patients with PsA, but the significance of this B cell abundance is not clear[15]. CD8+ T cells are the predominant cell type in epidermis, synovial fluid and enthesis, while CD4+ T cells are more abundant in the tissues. In the synovial fluid the majority of CD8+ T cells are activated(HLA-DR+) and mature(CD45RO+) memory T cells[16]. Although the T cell subsets in peripheral blood from PsA patients have similar CD4:CD8 ratios as psoriasis patients and normal controls, the CD3+ T cells of patients with PsA are more adherent and CD11, CD44,CD 25 and HLA-DR markers are also slightly enhanced[17]. T cell activation requires two signals for the initial activation; 1) the binding of T cell receptor(TCR) with class I or II MHC of antigen presenting cells(predominantly class I in PsA), 2) the interaction of costimulatory molecules of the T cell and APC[18](see Table3). The positive effect of T cell targeted drugs on psoriasis and PsA supports the concept that T cell plays an important role in disease pathogenesis. The T cell repertoire consists of self peptides that may induce self MHCs to enhance autoreactivity. Several studies suggested pathogenetic, oligoclonal expansions in the CD8+ TCR

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
28
repertoire of synovial fluid and peripheral blood cells [19, 20]. In addition, highly homologous CD3 regions at the amino acid level are present in skin and synovium , suggesting an antigen-driven response. The monoclonal IgG1 antibody, efalizumab, is directed against lymphocyte function antigen (LFA)-1 CD 11a which binds to CD54 (ICAM-1) on endothelial cells, disrupts the binding of T cells to endothelial cells and thereafter blocks downstream events which down regulate the secretion of cytokines and proinflammatory mediators. Additional interactions between NK receptors on memory effector CD8+ T cells have been proposed. NK cells possess both inhibitory and activating killer immunoglobulin-like receptors(KIR), and the presence of activating KIRs (KIR2DS1 and KIR2DS2) increases the susceptibility to PsA[21, 22]. This combination of innate immune signals triggers the T cell clones to express autoreactivity to self MHC.
ABNORMAL BONE REMODELLING Bone erosion Bone remodeling is highly dysregulated in PsA and aggressive bone erosions are recognized radiographically. Osteoclastogenesis begins with osteoclast precursors (OCPs) receiving two different contact signals: 1) macrophage colony stimulating factor (MCSF) binding to c-fms, and 2) RANKL binding to RANK. It has been suggested that the increased expression of RANKL and relative downregulation of osteoprotegerin (OPG), can neutralize the activity of RANKL, and promote osteoclastogenesis in PsA. Immunohistochemical analysis of the subchondral bone and synovium of PsA patients show strong expression of RANK on perivascular mononuclear cells and osteoclasts. There is intense staining of RANKL in the synovial lining, but OPG staining is restricted to the endothelial cells away from the site of bone erosion [23]. These results suggest that osteoclast precursors migrate to the inflamed area and are exposed to RANKL; when unopposed by OPG, this may lead to osteoclastogenesis in the involved synovial lining. The large number of osteoclasts detected in PsA bone is not only a result of active osteoclast differentiation driven by RANK. Peripheral blood mononuclear cells (PBMCs) obtained from PsA patients vs. normal controls show that a large number of PBMCs are TRAP (T-cell receptor activating protein)- positive osteoclast precursors in patients; these precursors mature to osteoclasts without RANKL or MCSF stimulation[23]. These PBMCs also express higher levels of CD11b, CD14, CD16, CD51/CD61 and RANK, all established markers of OCPs[24]. Ritchlin and his colleagues found that osteoclasts derived from CD 16+ cells are the main destructors in PsA , and that increased CD16 expression is associated with more bone erosion, suggesting CD 16 as a potential OCP marker in PsA[25]. The TNF-α which is produced by PsA PBMCs also enhances osteoclastogenesis. In mouse models, TNF-α directly increases the number of osteoclast precursors; anti- TNF-α therapy in PsA patients substantially reduces OCP frequency as early as two weeks after starting therapy. This highlights the fact that TNF-α plays a substantial role in the pathogenesis of the bony disease in PsA. Osteoclast precursors migrate to the psoriatic joint and encounter unopposed RANKL and TNF-α, leading to differentiation and activation of osteoclasts[26]. Thus, the erosions seen in PsA are the result of RANK, osteoclast precursor, and TNF-α driven events.
Bone Proliferaton
Not only bone erosion, but also new bone formation is characteristic of PsA[27] . Two protein families have been suggested as critical regulators of bone remodeling; the wingless(Wnt) glycoprotein family and the Dickkopf-1(DKK-1)protein family [28] (see Figure 1). DKK-1 antagonizes the Wnt pathway by binding Wnt co-receptor LRP5 and cell surface coreceptor Kremen-1/2 which is then internalized and inactived. Studies indicate that DKK-1 is a major contributor to bone resorption. Patients with inflammatory arthritis express high levels of DKK-1

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
29
in serum as well as in the inflamed joints; blocking DKK-1 reduces the numbers of osteoclasts and increases osteophytes in animal models[29, 30]. Cytokines such as TNF-α induce DKK-1 expression of the inflamed joints, which explain an aspect of TNF-α mediated effects where bony proliferation does not seem to be inhibited[31].
SOLUBLE MEDIATORS Synovial tissue in PsA predominantly expresses Th1 cytokines, including tumor necrosis factor(TNF)- α, interferon-γ, IL-1β,IL-2, IL-6, IL-10, IL-12 and IL15 but not IL-4 or IL-5[16, 32]. Synovial inflammation, proliferation, angiogenesis and matrix metalloproteinase (MMP) release occur in response to cytokines released by T cell activation. Angiogenesis upregulation, with expression of vascular endothelial growth factor (VEGF) and its receptors Flt-1 and KDR and angiopoietin-1 and-2, is observed in PsA synovium[33].
TNF-α High levels of TNF-α are seen by immunostaining PsA synovial tissue. TNF-α is osteoclastogenic, increasing the numbers of osteoclast precursors and promoting mesenchymal cell expression of RANKL and M-CSF with the help of IL-1(also induced by TNF activity). Cartilage degradation is enhanced by TNF-α induced production of MMPs by synovial fibroblasts. TNF-α also facilitates marrow macrophage, osteoclast precursor trafficking, lymphocyte migration through endothelial cells, and abnormal vasculogenesis. This is done by expression of adhesion molecules such as ICAM-1, E-selectin, and growth factors (eg VEGF and transforming growth factor (TGF)- β). Anti TNF-α agents effectively control all aspects of the disease.
IL-23-IL-17 pathway A subset of CD4+ T cells that are characterized by the production of IL-17 and IL-22( but not IL-4 or IFN-ϒ) has recently been identified in animal models of human autoimmune diseases[34, 35]. These Th17 cells are thought to be involved in the adaptive immunity to specific pathogens and evidence is emerging that abnormal Th17 responses are associated with autoimmune disorders. IL-23 is a proinflammatory cytokine and new member of the IL-12 family. It is produced by dendritic cells and macrophages, serving as a survival factor for Th17 cells. Elevated Th17 cells were correlated with clinical rheumatic disease activity; they mediate disease onset and contribute to sustained synovial inflammation[36]. The IL-17 secreted by Th17 cells exerts potent joint-destructive activities by stimulating the production of IL-1 and TNF-α and inducing RANKL on mesenchymal cells[28]. Currently, it is suggested that the balance of the inductive effect of IL-17 and inhibitory effect of IFN-ϒ is important[37]. Moreover, Yago and colleagues have found that IL-23 directly induced osteoclastogenesis from cultures of human PBMC in the absence of RANKL and OPG. Anti-IL-17 and anti TNF agents suppress IL-23 induced osteoclastogenesis[37]. Blockade of the p40 subunit of human IL-12/23 has significantly reduced signs and symptoms of psoriatic arthritis[38]. In patients who receive anti TNF agents for psoriasis, only responders show downregulated IL-17 pathway cytokines. This indicates that suppression of the Th17 immune response, by reducing inflammatory dendritic cell products, is necessary for response to anti-TNF agents [39].
GENETIC SUSCEPTIBILTY Heredity markedly influences the propensity for psoriasis and PsA, as evidenced by familial clustering of the disease. About 15% of first degree relatives of PsA patients will also have PsA, and an additional 30 to 40% will have psoriasis. Moreover, the odds ratio for recurrence of PsA in affected siblings was estimated to be greater than 27 [2, 40], higher than in psoriasis 4 to

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
30
11)[41]. Twin studies reveal an increased frequency in monozygotic and dizygotic twins[42]. Additional studies suggest that PsA is probably a polygenic, multifactorial disease with many susceptiblility loci. Among identified genes, HLA genes within the major histocompatibility complex (MHC), especially class I genes are the most important. Linkage studies (see Table 4) posit an increased frequency of HLA-B13, B17(and its subtypes HLA-B38 and B39), Cw6, Cw7, DR4, and DR7, with Cw*0602 loci accounting for one-third of genetic susceptibility to psoriasis through strong linkage to PSORS1[43]. Cw*0602 loci are also significantly increased in patients with PsA[44], and a recent retrospective cohort suggested that HLA-Cw*06 carriers were associated with longer intervals of time between onset of psoriasis and development of PsA(RR 0.54, p=0.001)[45]. Ongoing studies indicate MHC alleles may be associated with disease phenotype, as patients with B27 have more spinal involvement whereas patients with B38 and B39 have more peripheral polyarthritis [46]. HLA-B39, B27 in the presence of DR7 and DQw3 increases the risk for disease progression, while B22 has a protective effect [47-49]. The recently elucidated IL23-TH17 pathway has also been studied at the genetic level, and suggests association with both psoriasis and PsA [50, 51]. IL-23 has two subunits; IL-23p19(encoded by IL23A) and IL-12p40(encoded by IL12B), recognized by the receptor which is encoded by IL23R and IL12RB1, respectively. IL-12 also is a heterodimer of IL-12p40 and IL-12p35[52]. The upregulation of these cytokines plays a key role in a growing number of autoimmune disorders, including psoriasis (see pathogenesis). Also, the effects of anti-TNF agents have aroused interest in TNF polymorphisms. Certain TNF promoter regions such as positions -238 or -308 are associated with psoriasis and PsA and are also associated in linkage disequilibrium with Cw0602. TNF promoter site -857T may be a risk factor independent of the PSORS1 allele[53]. Association studies have also identified Killer cell immunoglobulin like receptor (KIR) genes on chromosome 19, related to NK cell activity [22, 54]. As sequencing the human genome and genome wide association scans(GWAS) became possible, single nucleotide polymorphisms(SNPs) have been used to detect subtle differences in DNA(see table 4). A single GWAS for PsA has been published, using 233 cases of psoriasis (including 91 with PsA) and 519 controls, genotyped for 311,398 SNPs[51]. This confirmed class I MHC as the most strongly associated with PsA. Susceptibility has also been associated with IL-23 receptor (IL23R) and IL-12p40 (IL12B) genes for IL-2 and 21(chromosome 4q27), as well as seven additional regions. Other susceptibility loci have been studied, but their roles in the development of PsA are not yet clear.
ENVIRONMENTAL FACTORS Infectious agents (viral and bacterial) and physical trauma have been suggested as potential risk factors for the onset of PsA. Psoriatic lesions arising from traumatic areas, known as the Koebner phenomenon, provide the hypothesis of ―deep Koebner phenomena‖ as an explanation for posttraumatic PsA [55] and a history of injury requiring medical consultation had an odds ratio of 2.53. Other factors that were associated with development of inflammatory arthritis in patients with psoriasis included: rubella vaccination, recurrent oral ulcers, fractured bone requiring hospital admission[55] and HIV infection[56]. There is also emerging evidence that PsA patients show an enhanced response to gram positive infection by amplifying the inflammatory cascade [57, 58]. Significantly elevated T cell reactivity levels to bacteria in serum and enhanced proliferative response to staphylococcal superantigens occur in patients with PsA. Smoking and PsA have an interesting relationship. Smoking prior to psoriasis onset decreases time to onset of PsA, but smoking after psoriasis onset increases time to development of PsA[4].

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
31
CLINICAL ASSESSMENT CLINICAL FEATURES Peripheral joints Peripheral joint inflammation is the most typical presentation of PsA, presenting as an oligoarthritis(in former studies, this was referred to as asymmetric involvement). This gradually progresses to a polyarticular, more symmetric disease. Unlike rheumatoid arthritis (RA), joint involvement in PsA is more common in the distal interphalangeal joints, with fewer joints involved [59]. The joint distribution tends to affect multiple joints in a single digit rather than occurring in the same joints on both sides, explaining the asymmetry that is characteristic of PsA[2]. Asymmetric knee involvement has been reported in 40% of cases at PsA onset, and DIP arthritis is seen in 1-59% of cases(62-64) Interestingly, the joints are typically less tender than in patients with RA (60,61).The erythema pattern in PsA is usually a reddish/purplish discoloration over the inflamed joint [60, 61]. The more destructive arthritis mutilans is characterized by osteolysis of the phalanges, metatarsals, and metacarpals and occurs in 1 to 5% of patients. The presence of five or more involved joints at entry to clinic, late onset, the number of actively inflamed joints at each clinic visit and the degree of damage are predictors of progression of clinical and radiological damage. A low erythrocyte sedimentation rate (ESR) is associated with less progression and severity [62-64].
Axial joints PsA patients have an increased frequency of spondylitis- chiefly sacroiliitis- with an estimated frequency between 5% to 36%[65]. Spinal involvement is another feature that helps distinguish PsA from RA. Inflammatory spinal disease presents as back pain, usually occurring at rest and with immobility but improving with activity. This may be hard to differentiate from ankylosing spondylitis(AS), but there are distinguishable features. The severity of pain, stiffness and sacroiliitis is much greater in AS[62, 63]; in contrast, spondylitis in PsA may be limited to one area of the spine, and sacroiliitis may be completely asymptomatic. The higher frequency of concomitant peripheral joint involvement in PsA can also serve as a discriminator.. In prospective studies, axial PsA has a better prognosis and less functional damage compared to AS [66, 67].
Enthesitis Over the last 10 years, advances in imaging modalities have elucidated entheses as the primary lesion of PsA [68]. Clinically, enthesitis may be asymptomatic even when ultrasound and MRI identify inflammation at the attachment of ligament and tendon to bone. Both common and uncommon clinical variants (Table 5) of PsA show considerable association with radiographic features of enthesitis[13]. Entheseal involvement is more common in the lower limbs than upper limbs and particularly common in the heel. Also, pain and swelling at femoral condyles, epicondyles and tendon insertions to pelvis/thorax/spine are common symptoms[65]. McGonagle et al. state that several clinical features suggest an enthesitis-related pathology, beside obvious clinical and/or radiographic enthesitis: DIP disease, spinal inflammation, mono/oligoarthritis/dactylitis [13].
Dactylitis Sausage-shaped digits, or dactylitis, has been recognized as a typical clinical feature of PsA and occurs in 5-53% of PsA patients[65]. Inflammation of the flexor tendons and occasionally the

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
32
interphalangeal joints lead to diffuse swelling and tenderness of the entire digit, defined as dactylitis. Some suggest there is a strong association between peripheral enthesitis and dactylitis, suggesting that enthesitis at multiple sites of the digit is the primary cause of dactylitis[13], but no MRI studies have specifically assessed enthesopathic insertion sites to confirm this hypothesis. The presence of dactylitis indicates active disease and should be differentiated from other conditions that may mimic digit swelling, such as tuberculosis, syphilis, sarcoidosis, sickle cell anemia etc. A prospective study of 260 patients with dactylitis revealed an association with greater radiological joint damage(p<0.0001)[69].
Other clinical features The typical scenario seen in 75 to 85% of patients demonstrates that psoriasis precedes the onset of arthritis by an average of 10 years; in 5-10%, psoriasis may occur simultaneously or follow the onset of arthritis[2, 64, 70]. A positive family history in a first degree relative may serve as an important clue to search the ―hidden‖ sites of psoriasis in the finger and toe nails, at the hairline, under the breasts, around the umbilicus or at the intergluteal cleft [71]. Psoriasis vulgaris is the major form of psoriasis associated with PsA, but other variants include pustular, guttate, flexural and erythrodermic psoriasis. Nail lesions, most commonly nail pitting (but sometimes onycholysis), occur more commonly in patients with PsA than patients with uncomplicated psoriasis(87% vs. 40%)[72]. In a population study, other psoriasis features associated with a higher likelihood of PsA were scalp lesions (hazard ratio[HR] 3.89) and intergluteal/perianal lesions(HR 2.35)[73, 74]. Other extra-articular manifestations include features common to the spondyloarthropathies: conjunctivitis and iridocyclitis is the most common, occurring in 2-25% of cases; mucous membrane lesions, urethritis and aortic root dilatation[2]. Recent studies raise the possibility of an increased prevalence of subclinical atherosclerosis in PsA patients, leading to cardiovascular complications such as hypertension and insulin resistance [75-79].
RADIOLOGIC FEATURES Conventional radiographs Hallmarks of PsA on conventional radiographs are both resorptive and proliferative changes. Typical destructive changes include joint erosions, usually asymmetric and marginal osteolysis leading to pencil-in cup deformities or acro-osteolysis and arthritis mutilans. Proliferative features include periarticular periostitis, spur formation, and ivory phalanx, seen in phalangeal tufts and soft tissue [80, 81]. Axial disease in PsA present as asymmetrical sacroiliitis, non-marginal/asymmetrical syndesmophytes and paravertebral ossification. Involvement of the cervical spine with relative sparing of lumbar segments is more typical of PsA than AS[82].
Magnetic resonance imaging MRI provides information on the extent and severity of joint damage. Many studies have described characteristic MRI features of synovitis, tenosynovitis, dactylitis, enthesitis, bone marrow edema, periostitis, onychopathy, spondylitis and asymptomatic disease. MRI is very sensitive for the early detection of sacroiliitis and axial PsA in the pre-erosive phase[83]. Imaging reveals subclinical axial or peripheral joint involvement and information about the underlying pathology. MRI studies of periarticular soft tissue was the first to describe enthesitis as a distinctive feature of PsA[84], and fat suppressed T2 weighted images show enthesitis as the initiating lesion in PsA [85]. Bone edema, especially when in the diaphysis, is pathognomonic in PsA, and is important in the sacroiliitis of axial PsA. No MRI studies have been done specifically

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
33
on PsA spondylitis. Initially, synovitis and bone erosion findings are frequently indistinguishable from RA [86, 87].
Ultrasonography High frequency transducers and Doppler ultrasonography has been useful in the assessment of enthesitis and dactylitis associated with PsA[88], and have shown higher sensitivity than MRI in detecting early enthesitis[89, 90]; detection of synovitis was lower than when using MRI[91].
Other imaging studies Bone scintigraphy is used to confirm inflammation that is not seen radiographically, but it has largely been replaced by MRI and ultrasonography due to its low specificity[92]. Computer tomography may be useful in the assessment of erosions in sacroiliac joints, but it is less effective in distinguishing synovial inflammation of the peripheral joints[93].
DIAGNOSTIC CRITERIA Early classification criteria were developed by Moll and Wright. They defined PsA as an inflammatory arthritis, in the presence of psoriasis, and in the absence of rheumatoid factor. However, these criteria have low specificity and are insufficient to differentiate PsA from RA. Subsequent studies by Bennett, Gladman et al, Vasey and Espinoza, the European Spondyloarthropathy Study Group (ESSG), McGonagle et al, and Fournie have proposed different classification criteria[94]. In a study that compared the test performance characteristics of these proposed criteria, the most sensitive criteria were those of Vasey and Espinoza, McGonagle and Gladman (99%) with Vasey and Espinoza having the best combination of sensitivity and specificity [95]. Bennett and ESSG criteria had inadequate sensitivity, and Fournie had good sensitivity but low feasibility.
CASPAR criteria The Classification Criteria for Psoriatic Arthritis(CASPAR) study group developed criteria based on observational clinical data from 588 cases and 536 controls(Table 6)[27]. The CASPAR criteria are easy to use with a high specificity of 98.7%, but less sensitivity than the criteria of Vasey and Espinoza (91.4 vs 97.2%). Development of diagnostic criteria with high specificity allows enrollment of appropriate patients for therapeutic interventions. A recent study demonstrates that the CASPAR criteria have high sensitivity in early PsA as well as in patients with longstanding disease [96, 97]. Neither of these diagnostic criteria separate the psoriatic arthritis variants.
MEASURES OF RESPONSE There is no fully validated measure to assess disease activity in PsA and many tools that are used have been borrowed from RA, psoriasis and ankylosing spondylitis. PsA disease activity and treatment response must assess a larger number of peripheral joints and include axial joints as well as enthesitis, dactylitis and skin and nail psoriasis[98-104]. Adequate treatment will also lead to improvement in functional class and quality of life. Several radiographic methods originally developed for patients with RA are used to evaluate radiographic joint changes seen in patients with PsA. Other imaging modalities such as ultrasonography and MRI provide better imaging of enthesitis and axial PsA as well as peripheral joint involvement, at least compared to conventional radiography. Although studies prove that these methods show adequate responsiveness, they are generally not validated in PsA, and properly using such measures in

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
34
the assessment of PsA requires further study. At the OMERACT 8 meeting of 2007, a set of core measures for clinical trials of PsA were agreed upon, as were a set of important but not mandatory measures(see Table 7)[105]
PERIPHERAL JOINT ACTIVITY ACR Response Criteria (modified for PsA) This combined index is a modification of the ACR Response Criteria for RA, with which all rheumatologists are already familiar. For PsA, the tender and swollen joint criteria also includes the distal interphalangeal phalngeal joints. This criterion performed better than the PsARC(see below) in 2 randomized , double blind, controlled trials of TNFi for PsA[106].
PsARC The Psoriatic Arthritis Response Criteria This is a combined index requiring response in 2 of 4 criteria, one of which must be tender joint or swollen joint counts. The tender joint or swollen joint count must improve by at least 30%, while the patient or physician global outcome measure must improve by at least 20%(1of 5 on a Likert scale)[106].
DAS/DAS28 Like the ACR Response criteria, this measure is familiar to rheumatologists. The DAS28 and its variations are taken directly from rheumatoid arthritis. The DAS28 comprises tender joint and swollen joint count, either ESR or CRP and a patient global health measure in a complex mathematical formula. This criterion responded better than PsARC or ACR response criteria in two analyzed studies of TNFi in PsA [106]. It must be pointed out that the ACR Response criteria and DAS, while fully validated in RA and responsive in PsA, are not fully validated in the latter. Nevertheless, they are recommended as one of the core measures in PsA.
SKIN ACTIVITY PASI (Psoriasis Area Severity Index) This fully validated scale scores the erythematosus, indurated, plaques and adherent silver scales that characterize psoriasis vulgaris.[107]. Using the PASI, investigators distinguish erythema, induration and scaling and grade by percent of body affected as mild (1-3%), moderate (3-10%) and severe lesions (greater than 10%).
PATIENT GLOBAL AND PAIN Visual Analogue Scales/Numerical Rating Scales/Likert Scales: All are patient response measures, variously laid out from 100 mm scales to 10 point scales to 5-7 point scales. Among the core measures, pain and patient global measures are included within the DAS, ACR Response and PsARC[105]. They have been tested and all are responsive, reliable and feasible[108]
FUNCTION/HEALTH RELATEDQUALITY OF LIFE MEASURES The Health Assessment questionnaire Disability Index (HAQ-DI) and Short Form-36 (SF-36) are known and carefully described and validated instruments in numerous rheumatic diseases. The HAQ-DI consists of 20 questions on a 0-3 ordinal scale in 8 daily functional domains. The SF-36 comprises 36 questions on a 0-3 ordinal scale in 8 domains (4 physical health scales and 4 mental health scales) spanning measures of general quality of life.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
35
The Dermatology Life Quality Index(DLQI) is a validated measure in psoriasis[109]; it consists of 10 questions on a 0-3 scale to assess how much the skin affects the patient‘s life. While they are not specifically validated for PsA, these patient response outcomes have been applied in PsA with good reliability and responsiveness [108, 110]. The other measures noted in Table 7 are clearly applicable in PsA but either awaits validation and/or definitive use. They were considered important measures which may be used but are not core/mandatory measures in PsA trials [105]. They will be briefly described. Several specific measures of dactylitis and enthesitis have been developed and are being actively tested [111-113]. Among the dactylitis indices, Healy and Helliwell showed that the Leeds Dactylitis Index (LDI) has been validated by OMERACT criteria[111, 112]. Likewise, they examined several enthesitis indices, feeling that the Leeds Enthesitis Index would be ―robust and reliable‖[113]. The widespread use and testing of these or other dactylititis and enthesitis indices in PsA are underway and they may well become core measures in the future. Nail indices apply to psoriasis and, of course, can be used in PsA. Unfortunately, despite an abundance of different nail measurement tools, none is fully validated or standardized[114]. According to Fernandez-Sueiro et al, standard spinal movement measures such as the modified Schober test, chest expansion and cervical rotation measurements performed well in the spinal involvement of PsA[115]. In 2009, Lubrano et al developed and validated the Psoriatic Arthritis Radiology Index for radiologic assessment of axial involvement in PsA and this index awaits widespread use[116]. A study of a potential synovial biomarker, CD3 has recently been published by Pontifex et al. This study demonstrates the change in CD3 correlates with the change of DAS28 scores and MRI scores, suggesting CD3 as a biomarker for treatment response in PsA[117].
RESEARCH MEASURES Radiologic procedures are turning to ultrasound (US) and magnetic resonance imaging (MRI) as more sensitive measures of inflammation before the structural change in bone are detected by radiographs. The international Outcome Measures in Rheumatology Clinical Trials(OMERACT) has developed the Psoriatic Arthritis MRI scoring system(PsAMRIS) to evaluate inflammatory destructive changes in PsA[118, 119]. This scoring system has acceptable reliability for bone erosion, edema, synovitis, tenosynovitis and bone proliferation (ICCs 0.84-0.91)[120]. MRI was used in studies of anti-TNF agents to detect the effects on synovial inflammation and bone edema, shown by significant reduction in gadolinium uptake following treatment. In these studies, the extent of inflammation corresponded well with the intensity of the bone edema signal and MRI synovitis[121, 122].It is anticipated that MRI changes will be used more frequently in clinical trials of PsA. Ultrasonography, like MRI, is more sensitive to inflammatory and destructive changes of the joints in PsA than X-ray or clinical examination [91]. A study of 30 patients with psoriasis showed evidence that severe entheseal involvement in patients with psoriasis was associated with a more aggressive clinical course and increased risk of developing PsA[123], and suggested early detection of PsA in asymptomatic patients with psoriasis may lead to preferable prognosis and disease outcome.Unlike, MRI, though, there has been no published validation of this technique in PsA.
TREATMENT There have been a number of therapies shown to be successful for psoriasis [124-126]. However, this review will concentrate solely on the treatment of psoriatic arthritis.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
36
DMARDs Among the non-biologic DMARDs, there has been a small amount of evidence supporting the use of methotrexate, sulfasalazine, leflunomide and cyclosporine for the treatment of psoriatic arthritis. Table 8 outlines the double-blind, randomized placebo-controlled trials regarding these drugs in psoriatic arthritis. There have been two small double-blind randomized trials using methotrexate. In a very early study(1964), using 2 mg per kilogram IV methotrexate weekly and a poorly defined "joint index", psoriatic arthritis responded to the methotrexate compared to placebo ( "statistically significant")[127]; in comparison, low dose oral methotrexate(7.5-15 mg po weekly) did not improve PsA (P =0.39-0.89)[128]. Five double-blind randomized studies used sulfasalazine. Among those five, three showed that sulfasalazine was effective and two did not. Although the quality and size of these studies varied, the larger studies did not demonstrate efficacy [129, 130]. Finally there has been one study of cyclosporine and one of leflunomide, each showing statistically significant efficacy for the treatment of psoriatic arthritis [131, 132]. Overall, then, the data supporting the use of non-biologic DMARDs for psoriatic arthritis are weak although these drugs are used.
BIOLOGICS There has been much research published regarding the use of biologics in PsA. This work cannot be thoroughly reviewed here secondary to space limitations. Instead we have chosen to review selected studies that demonstrate exemplary work on each of the biologics.
Anti TNF-α agents Inhibitors of TNF-α, including adalimumab, etanercept and infliximab have been approved by the USA FDA for PsA [133-135].
Etanercept Etanercept is a soluble p75 TNF-α receptor fusion protein that was initially used to treat RA. In PsA, several studies compared etanercept (25mg twice weekly subcutaneous) to placebo for skin and joint disease (see Table 9). A randomized, double-blind 12 week study of 60 PsA patients assessed the efficacy and safety of etanercept[136]. Patients had active PsA, defined as ≥3 swollen joints and ≥3 tender or painful joints at the time of enrollment. Patients on stable methotrexate(MTX; 47% of patients) were allowed to continue MTX, while other DMARDs were discontinued before beginning the study. Etanercept-treated patients had significantly better clinical outcome compared to the placebo group (Table 9). There were no serious adverse events and no adverse events occurred significantly more in the etanercept group. A larger, phase III, multicenter study of 205 patients showed significant improvement in the signs and symptoms of PsA and psoriasis(Table 9, Figure 2), and clinical responses were sustained throughout a 48 week open label phase[137]. Quality of life measured by HAQ and SF-36, were also significantly improved in the etanercept group. Radiographic progression was measured by modified Sharp score (mTSS), and over one year of observation, the etanercept group showed a mean change of -0.03U compared to +1.00U progression in the placebo group(p=0.0001; Figure 3). Patients who continued in a subsequent open label extension with etanercept showed sustained inhibition of radiologic progression in both groups [138]. Higher initial doses have been suggested to have greater benefit in skin manifestations but in a trial of 752 patients, 50mg twice weekly for 12 weeks did not show better response than 25 mg twice weekly for the arthritic component[139].

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
37
Infliximab Similar study designs have been used in trials using infliximab, a human/mouse chimeric monoclonal antibody. There have been three double-blind trials conducted [140-142]; the larger two were placebo controlled (see Table 9). The dose used in these studies was 5mg/kg, administered intravenously at weeks 0, 2, 6 followed by infusions every 8 weeks thereafter. In the Infliximab Multinational Psoriatic Arthritis Controlled Trials (IMPACT and IMPACT2), 104 and 200 patients who had failed prior DMARD treatment were randomized into infliximab or placebo groups. IMPACT demonstrated a significantly higher proportion of patients in the infliximab-treated group who achieved the primary endpoint joint responses at the end of the randomized control trial (RCT) compared to the placebo group (see Table 9). Skin, dactylitis and enthesitis improved as well [141]. A second phase III study(IMPACT2) also showed the effectiveness of infliximab for PsA(Table 9, Figure 4) as well as for skin, enthesitis and QOL[142]. Radiographic progression measured during open-label extension (using Sharp-van der Heijde scores) showed no mean progression in either placebo/infliximab or infliximab/infliximab groups over 50 weeks[143], indicating that delaying infliximab for 14 weeks still can inhibit radiological progression. Clinical efficacy is maintained at one year [144] and two years [145].
Adalimumab Adalimumab, a fully human, anti-TNF monoclonal antibody has been evaluated for its short and long term efficacy and safety in PsA [146-149]. The largest phase III study was the Adalimumab Effectiveness in Psoriatic Arthritis Trial(ADEPT); a double-blind, randomized placebo-controlled study for 24weeks comprised of 313 patients with moderate to severe PsA[146]. Significant difference in response was demonstrated during the double-blind period (Table 9, Figure 5), and response rate did not differ between patients taking methotrexate along with the adalimumab(50% of the patients). Radiographic progression using the modified total Sharp score was significantly inhibited; the mean change was -0.2 in the adalimumab group versus +1.0 in the patients treated with placebo (p<0.001). Quality of life measures also demonstrated significant improvement (mean ± SD change in HAQ-DI were -0.4±0.5 in adalimumab group compared to -0.1±0.4 in placebo group at week24; p<0.01). Adalimumab was generally well tolerated, with similar numbers of serious adverse events and infections between the two groups(Table 10). During 48 and 102 week open label extensions, continued clinical and radiographic responses were noted in those remaining on therapy. Adverse events profiles in the longer term extensions did not differ from the 24 week data. Further analysis demonstrated the level of baseline systemic inflammation, measured by serum C-reactive protein(CRP) was a predictor of radiographic progression[150]. Univariant analysis indicated elevated baseline CRP was strongly associated with greater changes in mTSS, joint erosion, and joint space narrowing scores in the placebo group(CRP≥1.0mg/dl; odds ratio 3.28, p<0.001). In the adalimumab treated patients baseline CRP≥2.0 was associated with greater improvement in mTSS. An additional multicenter double-blind RCT of 100 patients, conducted in 2007, supported the previous study [148].
Golimumab There has been one double-blind, placebo-controlled trial regarding the efficacy and safety of golimumab in patients with active PsA. The GO-REVEAL study randomized 405 patients into subcutaneous injections of placebo (n=113), golimumab 50mg (n=146) or 100mg (146) every 4 weeks through week 20[151]. Patients treated with golimumab demonstrated statistically significant improvement over placebo for PsA joints (Table 9, Figure 7). QOL measures (mean ± SD changes in HAQ-DI were 0.33±0.55 for golimumab 50mg, 0.39±0.50 for 100mg and -

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
38
0.01±0.49 for placebo), enthesitis, dactylitis and psoriatic skin and nail disease also showed significant improvement with golimumab compared to the placebo group. There was no evidence that higher doses (100 mg monthly) improved outcome in the arthritis components, and underlying MTX did not increase the benefit. Skin/nail outcomes showed numerically higher numbers of response in the higher treatment dose. There were no differences in the type or frequency of adverse events between the two golimumab doses.
NEW DRUGS Ustekinumab Based on the pathophysiological importance of the IL-17 pathway and their cytokines, the inhibition of IL12/23 was suggested as a possible therapeutic option for PsA. Ustekinumab is a human monoclonal antibody that inhibits the receptor binding of IL12/23 thru the p40 subunit. There has been a single phase II study[38] assessing the efficacy and safety of ustekinumab in PsA. It randomized 146 patients with active PsA who had inadequate response to DMARDs, NSAIDs and antiTNF agents. Group 1(n=76) received either 90mg or 63mg/wk ustekinumab for 4 weeks and group 2(n=70) was assigned to placebo. At week 12, ACR20 was achieved by 42% in group1 compared to 14% in group2 (p=0.0002; Figure 8), ACR50 responses were 25% vs. 7% and ACR70 responses were 7% vs. 0% (all p<0.01). There were statistically significant improvements in dactylitis and enthesopathy in the ustekinumab treated group; HAQ-DI also improved compared to baseline (median change from baseline -0.25 vs. 0.00, p=0.0005). The response was maintained for an additional 26 weeks in group 1 after the final dose of ustekinumab. There were similar proportions of adverse events and infections in both groups and a small number of serious adverse events occurred in the placebo group.
REFERENCES 1. Goldring, S.R. and M.B. Goldring, Eating bone or adding it: the Wnt pathway decides. Nat Med, 2007. 13(2): p. 133-4. 2. Gladman, D.D., et al., Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii14-7. 3. Alamanos, Y., P.V. Voulgari, and A.A. Drosos, Incidence and prevalence of psoriatic arthritis: a systematic review. J Rheumatol, 2008. 35(7): p. 1354-8. 4. Chandran, V. and S.P. Raychaudhuri, Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun, 2010. 34(3): p. J314-21. 5. Gladman, D.D., Psoriatic arthritis. Rheum Dis Clin North Am, 1998. 24(4): p. 829-44, x. 6. Kaipiainen-Seppanen, O., Incidence of psoriatic arthritis in Finland. Br J Rheumatol, 1996. 35(12): p. 1289-91. 7. Shbeeb, M., et al., The epidemiology of psoriatic arthritis in Olmsted County, Minnesota, USA, 1982-1991. J Rheumatol, 2000. 27(5): p. 1247-50. 8. Hukuda, S., et al., Spondyloarthropathies in Japan: nationwide questionnaire survey performed by the Japan Ankylosing Spondylitis Society. J Rheumatol, 2001. 28(3): p. 554-9. 9. Alamanos, Y., et al., Epidemiology of psoriatic arthritis in northwest Greece, 1982-2001. J Rheumatol, 2003. 30(12): p. 2641-4. 10. Soderlin, M.K., et al., Annual incidence of inflammatory joint diseases in a population based study in southern Sweden. Ann Rheum Dis, 2002. 61(10): p. 911-5. 11. Savolainen, E., et al., Total incidence and distribution of inflammatory joint diseases in a defined population: results from the Kuopio 2000 arthritis survey. J Rheumatol, 2003. 30(11): p. 2460-8.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
39
12. Cargill, M., et al., A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet, 2007. 80(2): p. 273-90. 13. McGonagle, D., P.G. Conaghan, and P. Emery, Psoriatic arthritis: a unified concept twenty years on. Arthritis Rheum, 1999. 42(6): p. 1080-6. 14. Veale, D., et al., Reduced synovial membrane macrophage numbers, ELAM-1 expression, and lining layer hyperplasia in psoriatic arthritis as compared with rheumatoid arthritis. Arthritis Rheum, 1993. 36(7): p. 893-900. 15. Veale, D.J., et al., Immunohistochemical markers for arthritis in psoriasis. Ann Rheum Dis, 1994. 53(7): p. 450-4. 16. Costello, P. and O. FitzGerald, Disease mechanisms in psoriasis and psoriatic arthritis. Curr Rheumatol Rep, 2001. 3(5): p. 419-27. 17. Dunky, A., J. Neumuller, and J. Menzel, Interactions of lymphocytes from patients with psoriatic arthritis or healthy controls and cultured endothelial cells. Clin Immunol Immunopathol, 1997. 85(3): p. 297-314. 18. Myers, W., M. Opeola, and A.B. Gottlieb, Common clinical features and disease mechanisms of psoriasis and psoriatic arthritis. Curr Rheumatol Rep, 2004. 6(4): p. 306-13. 19. Tassiulas, I., et al., Clonal characteristics of T cell infiltrates in skin and synovium of patients with psoriatic arthritis. Hum Immunol, 1999. 60(6): p. 479-91. 20. Costello, P.J., et al., Psoriatic arthritis joint fluids are characterized by CD8 and CD4 T cell clonal expansions appear antigen driven. J Immunol, 2001. 166(4): p. 2878-86. 21. Ritchlin, C.T., Pathogenesis of psoriatic arthritis. Curr Opin Rheumatol, 2005. 17(4): p. 406-12. 22. Nelson, G.W., et al., Cutting edge: heterozygote advantage in autoimmune disease: hierarchy of protection/susceptibility conferred by HLA and killer Ig-like receptor combinations in psoriatic arthritis. J Immunol, 2004. 173(7): p. 4273-6. 23. Ritchlin, C.T., et al., Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest, 2003. 111(6): p. 821-31. 24. Suda, T., et al., Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev, 1999. 20(3): p. 345-57. 25. Chiu, Y.G., et al., CD16 (FcRgammaIII) as a potential marker of osteoclast precursors in psoriatic arthritis. Arthritis Res Ther, 2010. 12(1): p. R14. 26. Lam, J., et al., TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest, 2000. 106(12): p. 1481-8. 27. Taylor, W., et al., Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum, 2006. 54(8): p. 2665-73. 28. Schett, G. and S.L. Teitelbaum, Osteoclasts and Arthritis. J Bone Miner Res, 2009. 24(7): p. 1142-1146. 29. Morvan, F., et al., Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res, 2006. 21(6): p. 934-45. 30. Diarra, D., et al., Dickkopf-1 is a master regulator of joint remodeling. Nat Med, 2007. 13(2): p. 156-63. 31. Schett, G., et al., Tumor necrosis factor alpha and RANKL blockade cannot halt bony spur formation in experimental inflammatory arthritis. Arthritis Rheum, 2009. 60(9): p. 2644-54. 32. Danning, C.L., et al., Macrophage-derived cytokine and nuclear factor kappaB p65 expression in synovial membrane and skin of patients with psoriatic arthritis. Arthritis Rheum, 2000. 43(6): p. 1244-56. 33. Fearon, U., et al., Angiopoietins, growth factors, and vascular morphology in early arthritis. J Rheumatol, 2003. 30(2): p. 260-8.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
40
34. Harrington, L.E., et al., Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol, 2005. 6(11): p. 1123-32. 35. Park, H., et al., A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol, 2005. 6(11): p. 1133-41. 36. Leipe, J., et al., Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum, 2010. 62(10): p. 2876-85. 37. Yago, T., et al., IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther, 2007. 9(5): p. R96. 38. Gottlieb, A., et al., Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomised, double-blind, placebo-controlled, crossover trial. Lancet, 2009. 373(9664): p. 633-40. 39. Zaba, L.C., et al., Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol, 2009. 124(5): p. 1022-10 e1-395. 40. Karason, A., T.J. Love, and B. Gudbjornsson, A strong heritability of psoriatic arthritis over four generations--the Reykjavik Psoriatic Arthritis Study. Rheumatology (Oxford), 2009. 48(11): p. 1424-8. 41. Bhalerao, J. and A.M. Bowcock, The genetics of psoriasis: a complex disorder of the skin and immune system. Hum Mol Genet, 1998. 7(10): p. 1537-45. 42. Pedersen, O.B., et al., On the heritability of psoriatic arthritis. Disease concordance among monozygotic and dizygotic twins. Ann Rheum Dis, 2008. 67(10): p. 1417-21. 43. Nair, R.P., et al., Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet, 2006. 78(5): p. 827-51. 44. Gladman, D.D., et al., HLA-C locus alleles in patients with psoriatic arthritis (PsA). Hum Immunol, 1999. 60(3): p. 259-61. 45. Eder, L., et al., HLA-Cw*06 Allele Increases the Duration of Time between the Onset of Psoriasis and Psoriatic Arthritis. 2010 Arthritis & Rheumatism abstract, 2010. 62(10): p. S220-21. 46. Gladman, D.D. and V.T. Farewell, HLA studies in psoriatic arthritis: current situation and future needs. J Rheumatol, 2003. 30(1): p. 4-6. 47. Castelino, M. and A. Barton, Genetic susceptibility factors for psoriatic arthritis. Curr Opin Rheumatol, 2010. 22(2): p. 152-6. 48. Korendowych, E. and N. McHugh, Genetic factors in psoriatic arthritis. Curr Rheumatol Rep, 2005. 7(4): p. 306-12. 49. Gladman, D.D., et al., HLA markers and progression in psoriatic arthritis. J Rheumatol, 1998. 25(4): p. 730-3. 50. Huffmeier, U., et al., Genetic variants of the IL-23R pathway: association with psoriatic arthritis and psoriasis vulgaris, but no specific risk factor for arthritis. J Invest Dermatol, 2009. 129(2): p. 355-8. 51. Liu, Y., et al., A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet, 2008. 4(3): p. e1000041. 52. Nograles, K.E., R.D. Brasington, and A.M. Bowcock, New insights into the pathogenesis and genetics of psoriatic arthritis. Nat Clin Pract Rheumatol, 2009. 5(2): p. 83-91. 53. Reich, K., et al., TNF polymorphisms in psoriasis: association of psoriatic arthritis with the promoter polymorphism TNF*-857 independent of the PSORS1 risk allele. Arthritis Rheum, 2007. 56(6): p. 2056-64. 54. Williams, F., et al., Activating killer cell immunoglobulin-like receptor gene KIR2DS1 is associated with psoriatic arthritis. Hum Immunol, 2005. 66(7): p. 836-41. 55. Pattison, E., et al., Environmental risk factors for the development of psoriatic arthritis: results from a case-control study. Ann Rheum Dis, 2008. 67(5): p. 672-6.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
41
56. Njobvu, P. and P. McGill, Psoriatic arthritis and human immunodeficiency virus infection in Zambia. J Rheumatol, 2000. 27(7): p. 1699-702. 57. Muto, M., et al., Significance of antibodies to streptococcal M protein in psoriatic arthritis and their association with HLA-A*0207. Tissue Antigens, 1996. 48(6): p. 645-50. 58. Yamamoto, T., I. Katayama, and K. Nishioka, Peripheral blood mononuclear cell proliferative response against staphylococcal superantigens in patients with psoriasis arthropathy. Eur J Dermatol, 1999. 9(1): p. 17-21. 59. Myers, W.A., A.B. Gottlieb, and P. Mease, Psoriasis and psoriatic arthritis: clinical features and disease mechanisms. Clin Dermatol, 2006. 24(5): p. 438-47. 60. Jajic, I., Blue coloured skin in psoriatic arthritis. Clin Exp Rheumatol, 2001. 19(4): p. 478. 61. Buskila, D., et al., Patients with rheumatoid arthritis are more tender than those with psoriatic arthritis. J Rheumatol, 1992. 19(7): p. 1115-9. 62. Gladman, D.D., V.T. Farewell, and C. Nadeau, Clinical indicators of progression in psoriatic arthritis: multivariate relative risk model. J Rheumatol, 1995. 22(4): p. 675-9. 63. Queiro-Silva, R., et al., A polyarticular onset predicts erosive and deforming disease in psoriatic arthritis. Ann Rheum Dis, 2003. 62(1): p. 68-70. 64. McHugh, N.J., C. Balachrishnan, and S.M. Jones, Progression of peripheral joint disease in psoriatic arthritis: a 5-yr prospective study. Rheumatology (Oxford), 2003. 42(6): p. 778-83. 65. Cantini, F., et al., Psoriatic arthritis: a systematic review. International Journal of Rheumatic Disease, 2010(13): p. 300-317. 66. Hanly, J.G., M.L. Russell, and D.D. Gladman, Psoriatic spondyloarthropathy: a long term prospective study. Ann Rheum Dis, 1988. 47(5): p. 386-93. 67. Chandran, V., et al., Axial psoriatic arthritis: update on a longterm prospective study. J Rheumatol, 2009. 36(12): p. 2744-50. 68. McGonagle, D., et al., Histological assessment of the early enthesitis lesion in spondyloarthropathy. Ann Rheum Dis, 2002. 61(6): p. 534-7. 69. Brockbank, J.E., et al., Dactylitis in psoriatic arthritis: a marker for disease severity? Ann Rheum Dis, 2005. 64(2): p. 188-90. 70. Mease, P. and B.S. Goffe, Diagnosis and treatment of psoriatic arthritis. J Am Acad Dermatol, 2005. 52(1): p. 1-19. 71. Moll, J.M. and V. Wright, Familial occurrence of psoriatic arthritis. Ann Rheum Dis, 1973. 32(3): p. 181-201. 72. Gladman, D.D., et al., HLA antigens in psoriatic arthritis. J Rheumatol, 1986. 13(3): p. 586-92. 73. Wilson, F.C., et al., Incidence and clinical predictors of psoriatic arthritis in patients with psoriasis: a population-based study. Arthritis Rheum, 2009. 61(2): p. 233-9. 74. Griffiths, C.E., et al., A classification of psoriasis vulgaris according to phenotype. Br J Dermatol, 2007. 156(2): p. 258-62. 75. Kimhi, O., et al., Prevalence and risk factors of atherosclerosis in patients with psoriatic arthritis. Semin Arthritis Rheum, 2007. 36(4): p. 203-9. 76. Gonzalez-Gay, M.A., et al., Subclinical atherosclerosis in patients with psoriatic arthritis. J Rheumatol, 2008. 35(10): p. 2070-1; author reply 2071. 77. Gonzalez-Juanatey, C., et al., High prevalence of subclinical atherosclerosis in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum, 2007. 57(6): p. 1074-80. 78. Gonzalez-Juanatey, C., et al., Endothelial dysfunction in psoriatic arthritis patients without clinically evident cardiovascular disease or classic atherosclerosis risk factors. Arthritis Rheum, 2007. 57(2): p. 287-93. 79. Tam, L.S., et al., Subclinical carotid atherosclerosis in patients with psoriatic arthritis. Arthritis Rheum, 2008. 59(9): p. 1322-31.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
42
80. Ory, P.A., D.D. Gladman, and P.J. Mease, Psoriatic arthritis and imaging. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii55-7. 81. van der Heijde, D., et al., Psoriatic arthritis imaging: a review of scoring methods. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii61-4. 82. Helliwell, P.S., P. Hickling, and V. Wright, Do the radiological changes of classic ankylosing spondylitis differ from the changes found in the spondylitis associated with inflammatory bowel disease, psoriasis, and reactive arthritis? Ann Rheum Dis, 1998. 57(3): p. 135-40. 83. McQueen, F.M., N. Dalbeth, and A. Doyle, MRI in psoriatic arthritis: insights into pathogenesis and treatment response. Curr Rheumatol Rep, 2008. 10(4): p. 303-10. 84. Jevtic, V., et al., Distinctive radiological features of small hand joints in rheumatoid arthritis and seronegative spondyloarthritis demonstrated by contrast-enhanced (Gd-DTPA) magnetic resonance imaging. Skeletal Radiol, 1995. 24(5): p. 351-5. 85. McGonagle, D., Imaging the joint and enthesis: insights into pathogenesis of psoriatic arthritis. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii58-60. 86. Peterfy, C.G., Magnetic resonance imaging of the wrist in rheumatoid arthritis. Semin Musculoskelet Radiol, 2001. 5(3): p. 275-88. 87. McQueen, F., M. Lassere, and M. Ostergaard, Magnetic resonance imaging in psoriatic arthritis: a review of the literature. Arthritis Res Ther, 2006. 8(2): p. 207. 88. Kane, D., et al., Ultrasonography in the diagnosis and management of psoriatic dactylitis. J Rheumatol, 1999. 26(8): p. 1746-51. 89. Olivieri, I., et al., Retrocalcaneal bursitis in spondyloarthropathy: assessment by ultrasonography and magnetic resonance imaging. J Rheumatol, 1998. 25(7): p. 1352-7. 90. Weiner, S.M., et al., Ultrasonography in the assessment of peripheral joint involvement in psoriatic arthritis : a comparison with radiography, MRI and scintigraphy. Clin Rheumatol, 2008. 27(8): p. 983-9. 91. Wiell, C., et al., Ultrasonography, magnetic resonance imaging, radiography, and clinical assessment of inflammatory and destructive changes in fingers and toes of patients with psoriatic arthritis. Arthritis Res Ther, 2007. 9(6): p. R119. 92. Grigoryan, M., et al., Imaging in spondyloarthropathies. Curr Rheumatol Rep, 2004. 6(2): p. 102-9. 93. Puhakka, K.B., et al., Imaging of sacroiliitis in early seronegative spondylarthropathy. Assessment of abnormalities by MR in comparison with radiography and CT. Acta Radiol, 2003. 44(2): p. 218-29. 94. Helliwell, P.S. and W.J. Taylor, Classification and diagnostic criteria for psoriatic arthritis. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii3-8. 95. Taylor, W.J., et al., A comparison of the performance characteristics of classification criteria for the diagnosis of psoriatic arthritis. Semin Arthritis Rheum, 2004. 34(3): p. 575-84. 96. D'Angelo, S., et al., Sensitivity of the classification of psoriatic arthritis criteria in early psoriatic arthritis. J Rheumatol, 2009. 36(2): p. 368-70. 97. Chandran, V., C.T. Schentag, and D.D. Gladman, Sensitivity of the classification of psoriatic arthritis criteria in early psoriatic arthritis. Arthritis Rheum, 2007. 57(8): p. 1560-3. 98. Mease, P.J., Assessment tools in psoriatic arthritis. J Rheumatol, 2008. 35(7): p. 1426-30. 99. Mease, P.J., et al., Psoriatic arthritis assessment tools in clinical trials. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii49-54. 100. Feldman, S.R. and G.G. Krueger, Psoriasis assessment tools in clinical trials. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii65-8; discussion ii69-73. 101. Mease, P.J. and M.A. Menter, Quality-of-life issues in psoriasis and psoriatic arthritis: outcome measures and therapies from a dermatological perspective. J Am Acad Dermatol, 2006. 54(4): p. 685-704.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
43
102. Gladman, D.D., et al., International spondyloarthritis interobserver reliability exercise--the INSPIRE study: II. Assessment of peripheral joints, enthesitis, and dactylitis. J Rheumatol, 2007. 34(8): p. 1740-5. 103. Gladman, D.D., et al., International spondyloarthritis interobserver reliability exercise--the INSPIRE study: I. Assessment of spinal measures. J Rheumatol, 2007. 34(8): p. 1733-9. 104. Chandran, V., et al., International multicenter psoriasis and psoriatic arthritis reliability trial for the assessment of skin, joints, nails, and dactylitis. Arthritis Rheum, 2009. 61(9): p. 1235-42. 105. Gladman, D.D., et al., Consensus on a core set of domains for psoriatic arthritis. J Rheumatol, 2007. 34(5): p. 1167-70. 106. Fransen, J., et al., Performance of response criteria for assessing peripheral arthritis in patients with psoriatic arthritis: analysis of data from randomised controlled trials of two tumour necrosis factor inhibitors. Ann Rheum Dis, 2006. 65(10): p. 1373-8. 107. Nestle, F.O., D.H. Kaplan, and J. Barker, Psoriasis. N Engl J Med, 2009. 361(5): p. 496-509. 108. Kwok, T. and J.E. Pope, Minimally important difference for patient-reported outcomes in psoriatic arthritis: Health Assessment Questionnaire and pain, fatigue, and global visual analog scales. J Rheumatol, 2010. 37(5): p. 1024-8. 109. Shikiar, R., et al., Validity and reliability of patient reported outcomes used in psoriasis: results from two randomized clinical trials. Health Qual Life Outcomes, 2003. 1: p. 53. 110. Tan, J.Y., et al., Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: a meta-analysis. J Dermatolog Treat, 2010. 111. Ritchlin, C.T., Therapies for psoriatic enthesopathy. A systematic review. J Rheumatol, 2006. 33(7): p. 1435-8. 112. Healy, P.J. and P.S. Helliwell, Measuring dactylitis in clinical trials: which is the best instrument to use? J Rheumatol, 2007. 34(6): p. 1302-6. 113. Healy, P.J. and P.S. Helliwell, Measuring clinical enthesitis in psoriatic arthritis: assessment of existing measures and development of an instrument specific to psoriatic arthritis. Arthritis Rheum, 2008. 59(5): p. 686-91. 114. Augustin, M. and A. Ogilvie, Methods of outcomes measurement in nail psoriasis. Dermatology, 2010. 221 Suppl 1: p. 23-8. 115. Fernandez-Sueiro, J.L., et al., Evaluation of ankylosing spondylitis spinal mobility measurements in the assessment of spinal involvement in psoriatic arthritis. Arthritis Rheum, 2009. 61(3): p. 386-92. 116. Lubrano, E., et al., Psoriatic arthritis spondylitis radiology index: a modified index for radiologic assessment of axial involvement in psoriatic arthritis. J Rheumatol, 2009. 36(5): p. 1006-11. 117. Pontifex, E.K., et al., Change in CD3 positive T-cell expression in psoriatic arthritis synovium correlates with change in DAS28 and magnetic resonance imaging synovitis scores following initiation of biologic therapy - a single centre, open-label study. Arthritis Res Ther, 2011. 13(1): p. R7. 118. Ostergaard, M., et al., The OMERACT psoriatic arthritis magnetic resonance imaging scoring system (PsAMRIS): definitions of key pathologies, suggested MRI sequences, and preliminary scoring system for PsA Hands. J Rheumatol, 2009. 36(8): p. 1816-24. 119. McQueen, F., et al., Developing a magnetic resonance imaging scoring system for peripheral psoriatic arthritis. J Rheumatol, 2007. 34(4): p. 859-61. 120. McQueen, F., et al., Testing an OMERACT MRI scoring system for peripheral psoriatic arthritis in cross-sectional and longitudinal settings. J Rheumatol, 2009. 36(8): p. 1811-5. 121. Antoni, C. and B. Manger, Infliximab for psoriasis and psoriatic arthritis. Clin Exp Rheumatol, 2002. 20(6 Suppl 28): p. S122-5.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
44
122. Marzo-Ortega, H., et al., Efficacy of infliximab on MRI-determined bone oedema in psoriatic arthritis. Ann Rheum Dis, 2007. 66(6): p. 778-81. 123. Girolomoni, G. and P. Gisondi, Psoriasis and systemic inflammation: underdiagnosed enthesopathy. J Eur Acad Dermatol Venereol, 2009. 23 Suppl 1: p. 3-8. 124. Greaves, M.W. and G.D. Weinstein, Treatment of psoriasis. N Engl J Med, 1995. 332(9): p. 581-8. 125. Weger, W., Current status and new developments in the treatment of psoriasis and psoriatic arthritis with biological agents. Br J Pharmacol, 2010. 160(4): p. 810-20. 126. Lowes, M.A., A.M. Bowcock, and J.G. Krueger, Pathogenesis and therapy of psoriasis. Nature, 2007. 445(7130): p. 866-73. 127. Black, R.L., et al., Methotrexate Therapy in Psoriatic Arthritis; Double-Blind Study on 21 Patients. JAMA, 1964. 189: p. 743-7. 128. Willkens, R.F., et al., Randomized, double-blind, placebo controlled trial of low-dose pulse methotrexate in psoriatic arthritis. Arthritis Rheum, 1984. 27(4): p. 376-81. 129. Clegg, D.O., et al., Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis. A Department of Veterans Affairs Cooperative Study. Arthritis Rheum, 1996. 39(12): p. 2013-20. 130. Combe, B., et al., Sulphasalazine in psoriatic arthritis: a randomized, multicentre, placebo-controlled study. Br J Rheumatol, 1996. 35(7): p. 664-8. 131. Fraser, A.D., et al., A randomised, double blind, placebo controlled, multicentre trial of combination therapy with methotrexate plus ciclosporin in patients with active psoriatic arthritis. Ann Rheum Dis, 2005. 64(6): p. 859-64. 132. Kaltwasser, J.P., et al., Efficacy and safety of leflunomide in the treatment of psoriatic arthritis and psoriasis: a multinational, double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum, 2004. 50(6): p. 1939-50. 133. Gottlieb, A., et al., Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol, 2008. 58(5): p. 851-64. 134. Menter, A., et al., Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol, 2008. 58(5): p. 826-50. 135. Mease, P.J. and C.E. Antoni, Psoriatic arthritis treatment: biological response modifiers. Ann Rheum Dis, 2005. 64 Suppl 2: p. ii78-82. 136. Mease, P.J., et al., Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet, 2000. 356(9227): p. 385-90. 137. Mease, P.J., et al., Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheum, 2004. 50(7): p. 2264-72. 138. Mease, P.J., et al., Continued inhibition of radiographic progression in patients with psoriatic arthritis following 2 years of treatment with etanercept. J Rheumatol, 2006. 33(4): p. 712-21. 139. Sterry, W., et al., Comparison of two etanercept regimens for treatment of psoriasis and psoriatic arthritis: PRESTA randomised double blind multicentre trial. BMJ, 2010. 340: p. c147. 140. Van Den Bosch, F., et al., Randomized double-blind comparison of chimeric monoclonal antibody to tumor necrosis factor alpha (infliximab) versus placebo in active spondylarthropathy. Arthritis Rheum, 2002. 46(3): p. 755-65. 141. Antoni, C.E., et al., Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheum, 2005. 52(4): p. 1227-36. 142. Antoni, C., et al., Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann Rheum Dis, 2005. 64(8): p. 1150-7.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
45
143. Kavanaugh, A., et al., The Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT): results of radiographic analyses after 1 year. Ann Rheum Dis, 2006. 65(8): p. 1038-43. 144. Kavanaugh, A., et al., Infliximab maintains a high degree of clinical response in patients with active psoriatic arthritis through 1 year of treatment: results from the IMPACT 2 trial. Ann Rheum Dis, 2007. 66(4): p. 498-505. 145. Antoni, C.E., et al., Two-year efficacy and safety of infliximab treatment in patients with active psoriatic arthritis: findings of the Infliximab Multinational Psoriatic Arthritis Controlled Trial (IMPACT). J Rheumatol, 2008. 35(5): p. 869-76. 146. Mease, P.J., et al., Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum, 2005. 52(10): p. 3279-89. 147. Gladman, D.D., et al., Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the adalimumab effectiveness in psoriatic arthritis trial. Arthritis Rheum, 2007. 56(2): p. 476-88. 148. Genovese, M.C., et al., Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J Rheumatol, 2007. 34(5): p. 1040-50. 149. Mease, P.J., et al., Impact of adalimumab on symptoms of psoriatic arthritis in patients with moderate to severe psoriasis: a pooled analysis of randomized clinical trials. Dermatology, 2010. 220(1): p. 1-7. 150. Gladman, D.D., et al., Risk factors for radiographic progression in psoriatic arthritis: subanalysis of the randomized controlled trial ADEPT. Arthritis Res Ther, 2010. 12(3): p. R113. 151. Kavanaugh, A., et al., Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheum, 2009. 60(4): p. 976-86. 152. Rahman, P., et al., Association between the interleukin-1 family gene cluster and psoriatic arthritis. Arthritis Rheum, 2006. 54(7): p. 2321-5. 153. Mease, P.J., et al., Adalimumab for long-term treatment of psoriatic arthritis: 2-year data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann Rheum Dis, 2009. 68(5): p. 702-9.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
46
Table 1. Incidence of psoriatic arthritis[3]
Country Study Incidence(/100,000/year)(95% CI)
Finland Kaipiainen-Seppanen, 1996 6.1 (4.6–7.6)
Sweden Soderlin, 2002 8 (4–15)
Greece Alamanos, 2003 3.0 (1.6–4.5)
Finland Savolainen, 2003 23.1 (13.2–37.5)
USA Shbeeb, 2000 6.6 (5.0–8.2)
Japan Hukuda, 2001 0.1
Table 2. Prevalence of psoriatic arthritis[3]
Country Study Prevalence /100,000(95% CI)
Europe
Sweden Hellgren, 1969 20 (9–40)
Netherlands Van Romunde, 1984 40 (6–80)
Greece Alamanos, 2003 57 (50–63)
Greece Trontzas, 2005 170 (100–240)
France Saraux, 2005 190 (80–350)
Italy Salaffi, 2005 420 (310–610)
Norway Madland, 2005 195 (180–210)
America
USA Shbeeb, 2000 101 (81–121)
USA Gelfand, 2005 250 (180–310)
Asia
Japan Hukuda, 2001 1
China Zeng, 2008 10-100
Australia Minaur, 2004 470

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
47
Table 3. Costimulatory signals
T cell Antigen Presenting cells (APC)
CD 2 LFA-3 LFA-1 ICAM-1 CD 28, CTLA-4 CD 80, CD 86 VLA-4 VCAM-1

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
48
Table 4. Loci associated with PsA(linkage association studies)[52] Gene Chromosome Odds ratio(p value) Study
IL12B 5q33 1.1(0.0013) 1.5(4.72×10
-7)
[51] [50]
IL23R 1p31 1.70(8×10-4
) 1.59(0.002)
[51] [50]
IL2/IL21 4q27 1.37(0.002) [51] IL-1 gene cluster 2q13 1.82 [152] TNF 6p21 1.96(0.0025) [53] HLA class 1 6p21 2.4(6.9×10-11)
NR [51] [44]
KIRs 19q/6p21 NR [22]

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
49
Table 5. Clinical features of PsA [13] Common features *Polyarthritis *Spinal inflammation *Peripheral enthesitis *DIP arthritis *Monoarthritis/oligoarthritis *Dactylitis(―sausage digits‖)
Uncommon features *Palmar plantar pustulosis *SAPHO Syndrome-synovitis, acne, pustulosis, hyperostosis, and osteolysis *Spondylodiscitis *Arthritis mutilans *Onycho-pachydermo-periostitis *Chronic mulifocal recurrent osteomyelitis

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
50
Table 6. The CASPAR criteria
Inflammatory articular disease (joint, spine, or entheseal) with 3 points from the following : 1. Evidence of Psoriasis (a)Current psoriasis : psoriatic skin or scalp disease present today as judged by a rheumatologist or dermatologist† (b)History of psoriasis: A history of psoriasis that may be obtained from a patient, family physician, dermatologist, rheumatologist, or other qualified health care provider (c)Family history of psoriasis : A history of psoriasis in a first- or second-degree relative according to patient report 2. Nail dystrophy Typical psoriatic nail dystrophy including onycholysis, pitting, and hyperkeratosis observed on current physical examination 3. A negative test for RF By any method except latex but preferably by enzyme-linked immunosorbent assay or nephelometry, according to the local laboratory reference range 4. Dactylitis (a)Current dactylitis: Swelling of an entire digit (b)History of dactylitis: recorded by a rheumatologist 5. Radiographic evidence of juxtaarticular new bone formation Ill-defined ossification near joint margins (but excluding osteophyte formation) on plain radiographs of the hand or foot † Current psoriasis is assigned a score of 2; all other features are assigned a score of 1.

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
51
Table 7. Domains to be used in clinical trials of PsA Core domains Important measures
Peripheral joint activity
Skin activity
Patient global
Pain
Physical function
HRQoL
Enthesitis
Dactylitis
Spine
Fatigue
Nails
Physician global
Radiology
Acute phase reactants

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
52
Table 8. Randomized control trials of DMARDs in PsA
Study
N (Drug/ Placebo)
Dose RCT duration, months
Measure of response Outcome Drug Placebo
P Value
MTX (crossover)
Black, 1964 21/21 IV 2mg/kg x3 wkly
2 ―Joint index‖ Response
- ―Stat avg‖
MTX Willkens, 1984 21/16 7.5-15mg Po wkly
3 Joint Tenderness/ Joint Swelling (―Important Change‖)
9/5 10/2 0.87 + 0.39
SSZ Clegg, 1996 109/112 2.0gm 9 Joint Tenderness/ Joint Swelling (% Improvement)
58.7/59.6 47.3/51.8 0.22 + 0.43
SSZ M Farr, 1990 12/12 2.0gm 6 Joint Tenderness (% Improvement)
58% 0% ―Significantly improved‖
SSZ SM Fraser, 1993
19/20 40mg/kg 3 Joint Tenderness (% Improvement)
65% 70% <0.02 For both
SSZ Gupta AK, 1996 10/14 3gm/d 2 Joint Tenderness(% Improvement)
57% 9% 0.061
Patient Global(Improvement) -1.1 +0.3 0.003
SSZ Combe, 1996 60/60 2.0gm/d 6 Joint Tenderness (Improvement) -4.5 -5.6 0.16
CSA Fraser, 2005 38/34 2.5mg/kg/d →4mg/kg/d
12 Ultrasound synovitis(reduced) 33% 6% <0.05
LEF Kaltwasser, 2004
95/95 100mg 6 PsARC 58.9% 29.7% <0.001
*abbreviations RCT randomized control trial; MTX methotrexate; SSZ sulfasalazine; CSA cyclosporine; LEF leflunomide
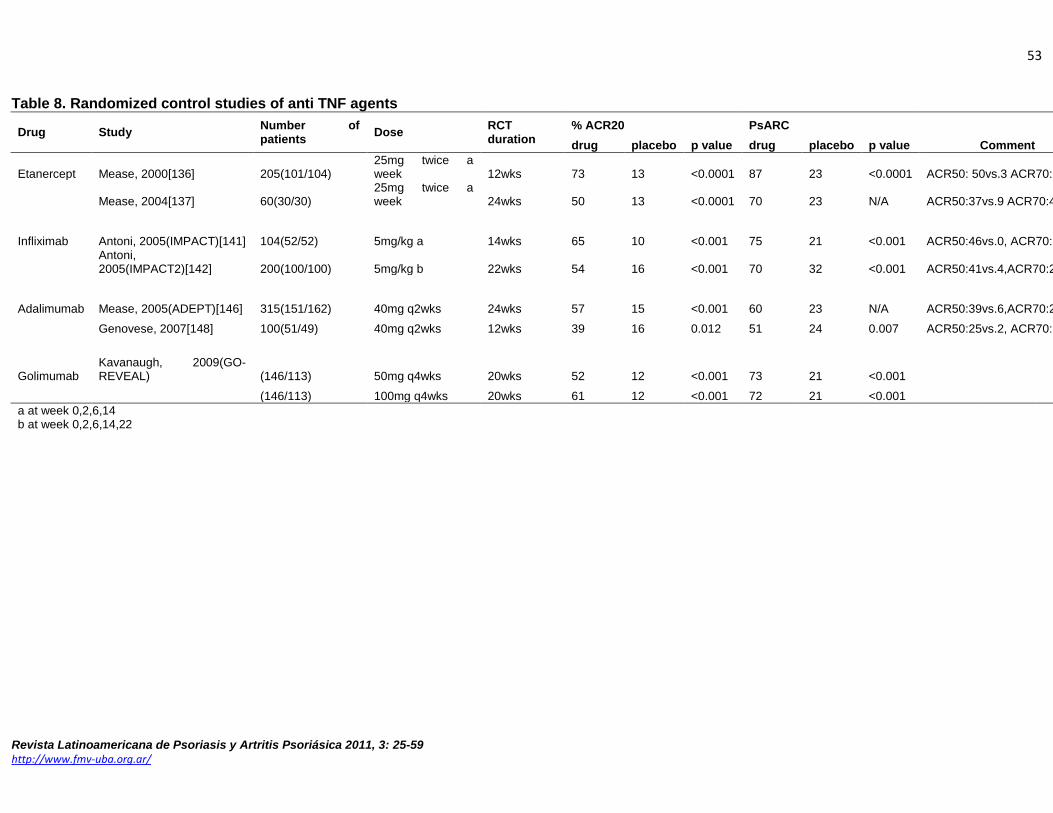
Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
53
Table 8. Randomized control studies of anti TNF agents
Drug Study Number of patients
Dose RCT duration
% ACR20 PsARC
Comment drug placebo p value drug placebo p value
Etanercept Mease, 2000[136] 205(101/104) 25mg twice a week 12wks 73 13 <0.0001 87 23 <0.0001 ACR50: 50vs.3 ACR70:13vs.0
Mease, 2004[137] 60(30/30) 25mg twice a week 24wks 50 13 <0.0001 70 23 N/A ACR50:37vs.9 ACR70:4vs.1
Infliximab Antoni, 2005(IMPACT)[141] 104(52/52) 5mg/kg a 14wks 65 10 <0.001 75 21 <0.001 ACR50:46vs.0, ACR70:29vs.0
Antoni, 2005(IMPACT2)[142] 200(100/100) 5mg/kg b 22wks 54 16 <0.001 70 32 <0.001 ACR50:41vs.4,ACR70:27vs.2
Adalimumab Mease, 2005(ADEPT)[146] 315(151/162) 40mg q2wks 24wks 57 15 <0.001 60 23 N/A ACR50:39vs.6,ACR70:23vs.1
Genovese, 2007[148] 100(51/49) 40mg q2wks 12wks 39 16 0.012 51 24 0.007 ACR50:25vs.2, ACR70:14vs.0
Golimumab Kavanaugh, 2009(GO-REVEAL) (146/113) 50mg q4wks 20wks 52 12 <0.001 73 21 <0.001
(146/113) 100mg q4wks 20wks 61 12 <0.001 72 21 <0.001
a at week 0,2,6,14 b at week 0,2,6,14,22

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
54
Table 9. Adverse events in randomized control studies
Drug Study N SAE, N(%) SIE, N(%) D/C d/t AE Death Lost f/u
(drug/placebo) Drug Placebo Drug Placebo Drug placebo drug placebo drug placebo
Etanercept Mease, 2000 60(30/30) 0(0) 1(3) 0(0) 0(0) 0(0) 0(0) 0(0) 0(0) 0(0) 0(0)
Mease, 2004 205(101/104) 4(4) 4(4) 0(0) 1(1) 1(1) 1(1) 0(0) 1(1) 1(1) 3(3)
Infliximab Antoni, 2005(IMPACT) 104(52/52) 1(2) 1(2) 0(0) 0(0) 2(4) 1(2) 0(0) 0(0) 1(2) 1(2)
Antoni, 2005(IMPACT2) 200(100/100)* 13(9)^ 6(6) 0(0) 0(0) 6(4)^ 1(1) 0(0) 0(0) 2(2) 1(1)
Adalimumab Mease,2005(ADEPT) 315(151/162) 5(3) 7(4) n/a n/a 3(2) 1(1) 0(0) 0(0) 0(0) 1(1)
Genovese, 2007 100(51/49) 1(2) 2(4) 0(0) 1(2) 1(2) 2(4) 0(0) 0(0) 0(0) 0(0)
Golimumab Kavanaugh, 2009(GO-REVEAL) (343/113)** 7(2) 7(6) 2(<1) 4(4) 6(2) 4(4) 0(0) 0(0) 1(0) 1(1)
*45patients in the placebo and 9 of the infliximab group entered early escape at wk16 ^value of combined group including all patients randomized to placebo who entered early escape at week 16 **51patients in the placebo and 28 in the Golimumab 50mg group entered cross over to golumumab 50mg and golimumab 100mg at week 16

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
55
Figure 1. Wnt signaling pathway[1]
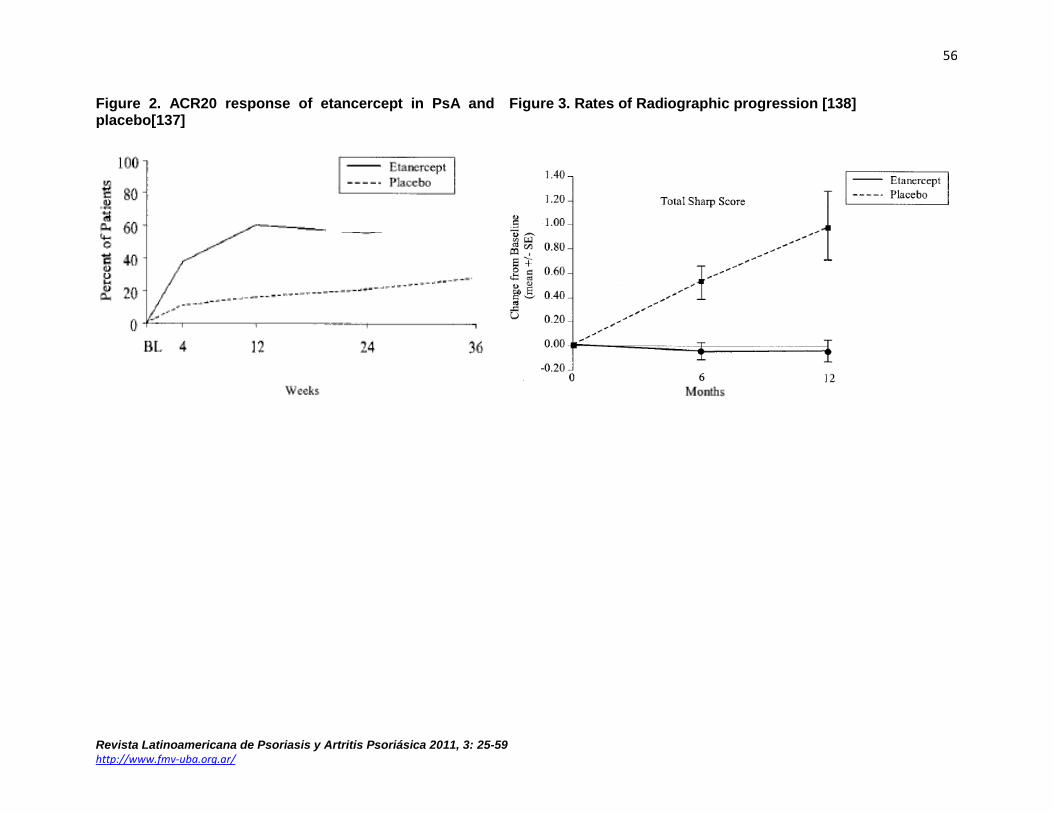
Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
56
Figure 2. ACR20 response of etancercept in PsA and placebo[137]
Figure 3. Rates of Radiographic progression [138]

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
57
Figure 4. ACR response to infliximab in PsA and Placebo[142]

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
58
Figure 5. ACR response with adalimumab in PsA and Placebo[146]
Figure 6. ACR response with sustained adalimumab in PsA[153]

Revista Latinoamericana de Psoriasis y Artritis Psoriásica 2011, 3: 25-59
http://www.fmv-uba.org.ar/
59
Figure 8. ACR20 responses for ustekinumab in PsA and Placebo[38]
Figure 7. ACR20 response to golimumab in PsA and Placebo[151]


















