Update on Dystonia, Chorea and Tics
9
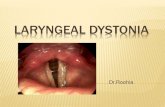
1 Update on Dystonia, Chorea, and Tics Joseph Jankovic, MD Professor of Neurology, Distinguished Chair in Movement Disorders, Director, Parkinson's Disease Center and Movement Disorders Clinic, Department of Neurology, Baylor College of Medicine, Houston, Texas Phenomenology and classification of dystonia: A consensus update. Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK. Mov Disord 2013;28:863-73 • Dystonia is defined as a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both. • Dystonic movements are typically patterned and twisting. • Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation. • Some forms of dystonia, such as blepharospasm and laryngeal dystonia, are not associated with postures, but are characterized by focal involuntary contractions that interfere with physiological opening or closing of the eyelids or the larynx. • Dystonia is classified along two axes: 1. Clinical characteristics, including age at onset, body distribution, temporal pattern and associated features (additional movement disorders or neurological features) 2. Etiology, which includes nervous system pathology and inheritance. The prevalence of primary dystonia: A systematic review and meta‐analysis Steeves et al. Mov Disord 2012;27:1789-96 Genetic Classification of Dystonias Primary Dystonias Classifica tion Chromosome Gene mutation Gene product Pattern of inheritance Onset Distribution, additional features Origin Comment DYT1 9q34, GAG deletion, TOR1A/TorsinA AD C Distal limbs, generalized Penetrance: 30% AJ, 70% NJ DYT2 NM AR Spanish gypsies, Iranian Jews DYT6 8q21-22 THAP1 AD A, C Cervical, cranial, brachial German-American Mennonite-Amish DYT7 18p AD A Cervical, cranial, spasmodic dysphonia, hand tremor German DYT13 1p36.13-32 AD A, C Cranial-cervical and upper limb Italian DYT17 20p11.22-q13.12 AR C Cervical dystonia, dysphonia, segmental, generalized Lebanese DYT21 2q14.3-q21.3 AD A Late-onset Sweden DYT23 9q34.11,CIZ1 AD A Cervical Caucasians DYT24 3, ANO3 AD A Cranial-cervical-laryngeal, tremor, myoclonus European DYT25 18p, GNAL AD A Cervical>cranial>arm European A = adult onset; AJ = Ashkenazi Jewish; AD = autosomal dominant; AR = autosomal recessive; C = childhood onset; NJ = Non-Jewish; NM = not mapped yet Genetic Classification of Dystonias Dystonia Plus Syndromes Classifica tion Chromosome Gene mutation Gene product Pattern of inheritance Onset Distribution, additional features Origin Comment DYT3 Xq TAF1 XR A Parkinsonism Filipinos (Lubag) mosaic striatal gliosis DYT4 19p13.3-p13.2 TUBB4 (β-tubulin 4a) AD C,A Whispering dysphonia, cranial, cervical, limb, facial atrophy, ptosis, edentulous Australian DYT5a 14q22.1 GCH1/GTP cyclohydrolase I AD C Gait disorder, parkinsonism, myoclonus, spasticity Dopa-responsive dystonia, diurnal fluctuation DYT5b 11p15.5 tyrosine hydroxylase AR C Gait disorder, parkinsonism, myoclonus, spasticity Dopa-responsive dystonia, diurnal fluctuation DYT11 7q21-31 SGCE ε-sarcoglycan AD C Myoclonus, behavioral and psychiatric problems Alcohol-responsive DYT12 19q12-13.2 ATP1A3 Na/K-ATPase α3 subunit AD A, C Cranial, upper limbs, parkinsonism, depression, seizures Rapid-onset, triggered by stress DYT15 18p11 AD C Myoclonus Canadian DYT16 2q31.2 PRKRA protein kinase, interferon-inducible double-stranded RNA-dependent activator AR C Dystonia, axial, oromandibular, laryngeal, parkinsonism Brazilian, German
Transcript of Update on Dystonia, Chorea and Tics

1
Update on Dystonia,
Chorea, and Tics
Joseph Jankovic, MD Professor of Neurology, Distinguished Chair in Movement Disorders,
Director, Parkinson's Disease Center and Movement Disorders Clinic,
Department of Neurology, Baylor College of Medicine, Houston, Texas
Phenomenology and classification of dystonia: A consensus update.
Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK.
Mov Disord 2013;28:863-73
• Dystonia is defined as a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both.
• Dystonic movements are typically patterned and twisting. • Dystonia is often initiated or worsened by voluntary action
and associated with overflow muscle activation. • Some forms of dystonia, such as blepharospasm and
laryngeal dystonia, are not associated with postures, but are characterized by focal involuntary contractions that interfere with physiological opening or closing of the eyelids or the larynx.
• Dystonia is classified along two axes: 1. Clinical characteristics, including age at onset, body distribution, temporal pattern and associated features (additional movement disorders or neurological features)
2. Etiology, which includes nervous system pathology and inheritance.
The prevalence of primary dystonia: A systematic review and meta‐analysis
Steeves et al. Mov Disord 2012;27:1789-96
Genetic Classification of Dystonias Primary Dystonias
Classification
Chromosome Gene mutation Gene product
Pattern of inheritance
Onset
Distribution, additional features
Origin
Comment
DYT1 9q34, GAG deletion,
TOR1A/TorsinA
AD C Distal limbs, generalized Penetrance: 30% AJ, 70% NJ
DYT2 NM AR Spanish gypsies, Iranian Jews
DYT6 8q21-22
THAP1 AD A, C Cervical, cranial, brachial German-American
Mennonite-Amish
DYT7 18p AD A Cervical, cranial, spasmodic dysphonia, hand tremor German
DYT13 1p36.13-32 AD A, C Cranial-cervical and upper limb Italian
DYT17 20p11.22-q13.12 AR C Cervical dystonia, dysphonia, segmental, generalized Lebanese
DYT21 2q14.3-q21.3 AD A Late-onset Sweden
DYT23 9q34.11,CIZ1 AD A Cervical Caucasians
DYT24 3, ANO3 AD A Cranial-cervical-laryngeal, tremor, myoclonus European
DYT25 18p, GNAL AD A Cervical>cranial>arm European
A = adult onset; AJ = Ashkenazi Jewish; AD = autosomal dominant; AR = autosomal recessive; C = childhood onset; NJ = Non-Jewish; NM = not mapped yet
Genetic Classification of Dystonias Dystonia Plus Syndromes
Classification
Chromosome Gene mutation Gene product
Pattern of inheritance
Onset
Distribution, additional features
Origin
Comment
DYT3 Xq TAF1
XR A Parkinsonism Filipinos (Lubag) mosaic striatal
gliosis
DYT4 19p13.3-p13.2 TUBB4 (β-tubulin 4a)
AD C,A Whispering dysphonia,
cranial, cervical, limb, facial atrophy, ptosis, edentulous
Australian
DYT5a
14q22.1
GCH1/GTP cyclohydrolase I
AD
C Gait disorder, parkinsonism, myoclonus, spasticity
Dopa-responsive dystonia, diurnal
fluctuation
DYT5b 11p15.5 tyrosine hydroxylase
AR C Gait disorder, parkinsonism,
myoclonus, spasticity Dopa-responsive dystonia, diurnal
fluctuation
DYT11 7q21-31 SGCE
ε-sarcoglycan AD C Myoclonus, behavioral and
psychiatric problems Alcohol-responsive
DYT12 19q12-13.2
ATP1A3 Na/K-ATPase α3
subunit
AD A, C Cranial, upper limbs,
parkinsonism, depression, seizures
Rapid-onset, triggered by stress
DYT15 18p11 AD C Myoclonus Canadian
DYT16
2q31.2 PRKRA
protein kinase, interferon-inducible
double-stranded RNA-dependent
activator
AR C Dystonia, axial,
oromandibular, laryngeal, parkinsonism
Brazilian, German

2
Paroxysmal Dyskinesias
PKD DYT10/19
PNKD DYT8/20
PED DYT9/18
16p11.2-q12.1 (IC)
proline-rich transmembrane protein 2 gene (PRRT2)
Infantile convulsions, migraine, episodic ataxia
Anticonvulsants
2q33-35 (PNKD)
10q22
(α-subunit of a Ca-sensitive K channel, KCNMA1)
Clonazepam
16p12-q12 (ICCA)
1p35-p31
glucose transporter type 1 (GLUT1) (SLC2A1)
Anticonvulsants, levodopa, BoNT
The genetics of dystonia: new twists in an old tale. Charlesworth et al. Brain 2013;136:2017-37
• In the past few years, with the advent of new sequencing
technologies, there has been a step change in the pace of
discovery in the field of dystonia genetics.
• In just over a year, four new genes have been shown to
cause primary dystonia (CIZ1, ANO3, TUBB4A and GNAL)
• PRRT2 has been identified as the cause of paroxysmal
kinesigenic dystonia
• Other genes, such as SLC30A10 and ATP1A3, have been
linked to more complicated forms of dystonia or new
phenotypes.
A workable strategy for identifying the likely genetic basis for some of the major forms of dystonia
Charlesworth et al. Brain 2013;136:2017-37
Genetics and Pathophysiology of Neurodegeneration with
Brain Iron Accumulation (NBIA) Schneider SA, Dusek P, Hardy J, Westenberger A, Jankovic J, Bhatia KP.
Curr Neuropharmacol 2013;11:59-79
Condition (Acronym) Synonym Gene Chromosomal
position
PKAN NBIA1 PANK2 20p13
PLAN NBIA2, PARK14 PLA2G6 22q12
FAHN SPG35 FA2H 16q23
MPAN -- C19orf12 19q12
Kufor-Rakeb disease PARK9 ATP13A2 1p36
Aceruloplasminemia -- CP 3q23
Neuroferritinopathy -- FTL 19q13
BPAN (SENDA)
-- n.k. n.k.
Idiopathic late-onset
cases
-- Probably
heterogene
ous
Probably
heterogeneous
Distinguishing features in different types of NBIA. aCP = aceruloplasminemia; BPAN = beta-propeller protein-associated neurodegeneration;
FAHN = fatty acid hydroxylase-associated neurodegeneration; INAD = infantile neuroaxonal
dystrophy; KRD = Kufor-Rakeb disease; MPAN = mitochondrial membrane protein-associated
neurodegeneration; PKAN = pantothenate kinase associated neurodegeneration; WSS =
Woodhouse-Sakati syndrome.
Horvath. Brain 2013;136:1687-91
T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation.
McNeill et al. Neurology 2008;70:1614-9
PKAN
17 y/o
Infantile
Neuroaxonal
Dystrophy
9 y/o
Neuro-
ferritinopathy
69 y/o
Acerulo-
plasminemia
55 y/o
T2* MRI Scan

3
PKAN
17 y/o
Infantile
Neuroaxonal
Dystrophy
9 y/o
Neuro-
ferritinopathy
69 y/o
Acerulo-
plasminemia
55 y/o
Fast Spin Echo (FSE)
T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation.
McNeill et al. Neurology 2008;70:1614-9
Levodopa
Anticholinergics
Tetrabenazine
Benzodiazepines
Selective
Peripheral
Denervation
Myectomy Pallidotomy
Deep Brain Stimulation
(Globus Pallidus)
Central
Surgeries
Peripheral
Surgeries
Botulinum
Toxin B
Botulinum
Toxin A
Physical and Occupational Therapy
Constraint Induced Therapy, rTMS
Dystonia
Oral Medications
Chemodenervation Surgical Therapies
Other Modalities
Jankovic J. Lancet Neurol 2009;8:844-56
Baclofen Oral, IT
Mov Disord 2013;28:1001-12
Huntington Disease
Movement Disorders
Behavioral Symptoms
Cognitive Decline
First Symptoms of Huntington Disease
Chorea 877 28
Trouble walking 262 9
Unsteadiness/imbalance 260 8
Difficult to get along with 167 5
Depression 165 5
Clumsiness 154 5
Speech difficulty 149 5
Memory loss 93 3
Trouble holding an object 62 2
Lack of motivation 53 2
Suspicions/paranoia 48 2
Intellectual decline 44 1
Changes in sleep 30 1
Hallucinations 23 1
Weight loss 20 0.6
Sexual problems 7 0.2
Other mental 363 12
Other physical 297 10
% of observed
Symptom # of Pts. * symptoms **
*Number of patients from a total of 1,901 with initial symptom information.
**Percent of all observed symptoms from a total of 3,086. Patients could report up to three initial symptoms.
Foroud et al. JNNP 1999;66:52-56 Ross and Tabrizi. Lancet Neurol 2011;10:83-98
Progression of Huntington Disease

4
Birth 10 20 30 40 50 60 70 0
Juvenile/adolescent onset
Early onset
Mid-life onset
Late onset
Clinical onset
Dis
ease
pro
gre
ssio
n
Age
Age-dependent Progression of HD
Birth 10 20 30 40 50 60 70
Age
Huntington Disease
5 – 15 years 15 – 30 years Duration
Dementia, dysarthria, abnormal eye movements, tremor, seizures, ataxia, myoclonus
Dementia, dysarthria,
abnormal eye
movements, dystonia,
rigidity
Late features
Personality changes,
rigidity, bradykinesia,
dystonia
Chorea, personality
changes
Initial features
AD (from the father) AD Inheritance
< 15 35 – 55 Age at onset
Juvenile onset Adult onset
CAG Repeats 37 – 50 >50
Normal HD Harris et al. Ann Neurol 1992;31:69-75
Normal HD
Atrophy: Putamen > Caudate
Medium-Sized Spiny Neuron
Projecting neurons
Interneurons (spared)
1
1
SNc SNr
GPe
GPi
2
3 GABA
Tachykinin
GABA
Substance P
GABA
Tachykinin
Ach, Somatostatin
• <40% loss of neurons in SN
• Neuronal intranuclear inclusions formed by
aggregation of the N-terminal huntingtin fragments
GABA
Enkephalin
STRIATUM
Pathology of Huntington Disease
(CAG)n
27-35 CAG repeats - “intermediate”
26 CAG repeats – “normal allele”
40 CAG repeats – “inevitable” HD
> ~70 - juvenile onset HD
n
70
27
40
exon
tel
~210 Kb
cen
1 2 67 Encodes a 348 kD, 3,144 AA protein, “huntingtin“, Htt
In HD the polyglutamine segment near the NH2 is elongated
Huntingtin Gene Htt (IT15) – 4p16.3
36-39 CAG repeats - “reduced penetrance” 36

5
Genetic Diagnostic Testing Autopsy-Proven Huntington Disease with 29 Trinucleotide Repeats
Kenney C, Powell S, Jankovic J. Mov Disord 2007;22:127-30
• 65 y/o man with a 5-year history of cognitive decline, chorea, inability to sustain tongue protrusion, gait and balance problems, MMSE 27/30
• 29/20 CAG repeats
• Additional studies were normal including CK, anti-cardiolipin Ab, ESR, thyroid function, blood smear for acanthocytes, and DNA analysis for dentatorubral-pallido-luysian atrophy
• Patient died after a fall during which he suffered SAH
• Autopsy: acute SAH in the right Sylvian fissure; thinning of the cortical ribbon, moderate gliosis and neuronal loss in the caudate and putamen, ubiquitin-positive neuronal intranuclear inclusions in the cortical and other areas
• Patient’s father, who died at age 79, had cognitive decline and involuntary movements. No other FHx of neurologic disorders
• The patient’s “asymptomatic”, 38 y/o, son has 32/19 CAG repeats
Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS.
Killoran A, Biglan KM, Jankovic J, Eberly S, Kayson E, Oakes D, Young AB,
Shoulson I. Neurology 2013;80:2022-7
• The Prospective Huntington At Risk Observational Study (PHAROS)
enrolled adults at risk for HD, assessed every 9 months with the
UHDRS by investigators unaware of participants' gene status.
• 50 (5.1%) of the 983 participants had an intermediate allele (IA) (27-35
CAG repeats).
• The IA subjects were significantly worse on apathy and suicidal
ideation and on 5 of the 9 other behavioral items and on total behavior
scores than controls and expanded participants.
• CONCLUSIONS: In a cohort at risk for HD, the IA was associated with
significant behavioral abnormalities but normal motor and cognition.
This behavioral phenotype may represent a prodromal stage of HD,
with the potential for subsequent clinical manifestations, or be part of a
distinct phenotype conferred by pathology independent of the CAG
expansion length.
Mov Disord 2012;27:1714-7
A Randomized, Double-Blind, Placebo-Controlled Trial of Tetrabenazine as Antichorea Therapy in
Huntington’s Disease (TETRA-HD)
• N = 84 HD subjects randomized, 16 centers
• Tetrabenazine (n = 54) or placebo (n = 30)
• Dosage increased over 7 weeks up to 8 tablets (100 mg) daily, until the desired antichoreic effect or intolerable side effects occurred
• During the last 5 weeks of the study, the dosage remained constant (unless reduced because of intolerable adverse effects)
• Primary outcome: change from baseline in UHDRS chorea score
• Secondary outcomes: CGI, the UHDRS total motor score, functional scales, gait score, tolerability, and safety
Huntington Study Group. Mov Disord 2006;11:136-142
Mean Change in UHDRS Total Maximal Chorea Score (Primary Study Endpoint: From Baseline to Week 12)
TETRA-HD
ANCOVA=analysis of covariance; ITT-LOC=intent-to-treat analysis.
There was blinded washout of study drug at week 12. Change favors tetrabenazine
(P=0.0001; ANCOVA, ITT-LOC). Chorea Scale range: 0 to 28
Week
Ch
ore
a
-2 0 2 4 6 8 10 12 14 16
-6
-5
-4
-3
-2
-1
0
1
Placebo
Tetrabenazine
N = 84 (TBZ = 54, placebo = 30)
Mean score decline (UHDRS units): 5.0 (TBZ) vs 1.5 (placebo) (p < 0.0001)
Huntington Study Group. Mov Disord 2006;11:136-142

6
Expert Opinion on Orphan Drugs 2013
• Tetrabenazine (TBZ) is a centrally-acting, dopamine depleting drug that has
been used for treatment of hyperkinetic movement disorders but was not
commercially available in the United States until 2008, when the Food and Drug
Administration (FDA) approved TBZ for the management of chorea associated
with Huntington’s disease (HD) under an orphan drug designation.
• While sedation, insomnia, mood changes, parkinsonism, and restlessness may
occur, these adverse effects can be managed effectively with appropriate
titration and monitoring. A black box warning against depression and
suicidality warrants careful patient selection, close monitoring and judicious
use of antidepressants.
• TBZ possesses a unique mechanism of action as a presynaptic dopamine
depletor that offers possible advantages over dopamine receptor blocking
drugs.
• TBZ has a strong potential for application in other hyperkinetic movement
disorders, particularly tardive dyskinesia and Tourette syndrome, but
randomized, controlled clinical trials are lacking.
Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol 2009;8:844-56
Experimental Therapeutics • Pridopidine (ACR16; Huntexil,
“dopaminergic stabilizer”)
(MermaiHD and HART trials)
• Ethyl-EPA (Miraxion)
• Latrepirdine (Dimebon)
(DIMOND and HORIZON trials)
• Creatine, CoQ10, Lithium
• Selististat (sirtuin deacetylase),
• Histone deacetylase inhibitors
(HDACi 4b)
• Phosphodiesterase PDE10A
inhibitors (TP-10)
• PBT2
• Ganglioside GM1
• Rhes inhibitors
• Neurturin
• Anti-htt Ab (intrabody)
• Small interfering (si) RNA
• Antisense oligonucleotides
• Cell replacement (fetal cells,
embryonic stem cells or
induced pluripotent cells)
Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol 2009;8:844-56
N Engl J Med 2012;367:1753-4 Phenocopies of HD
• HDL1 – AD, seizures (prion protein, PRNP; 20p12)
• HDL2 – AD, no seizures (junctophilin, JPH3; 16q24.3)
• HDL3 – AR (4p16.3)
• DRPLA – AD (c-Jun NH-terminal kinase, JNK, 12p)
• Neuroacanthocytosis – AR (VPS13A, 9q21)
• McLeod syndrome – X-linked (HK, Xp21)
• Mitochondrial encephalomyopathies
• Benign hereditary chorea (NKX2-1/TITF-1, 14q13.1-q21.1)
• Ataxia-Chorea:
• SCA1(ataxin-1, CAG expansion, 6p23)
• SCA2 (ataxin-2, CAG expansion, 12q24.1)
• SCA17 (TATA-Box binding protein, CAG expansion, 6q27)
• Friedreich’s ataxia (frataxin, GAA expansion, 9p13)
• Ataxia telangiectasia (protein kinase PI-3, ATM gene, 11q22)
• Neurodegeneration with brain iron accumulation (NBIA)
• Psychogenic chorea

7
ADHD
Tics
OCD
Behavioral problems poor impulse control, self-injurious behavior,
and other behavioral problems
TS
PLUS
Jankovic. Tourette's Syndrome. N Engl J Med 2001;345:1184-92
Tic Characteristics • Simple or complex movements (motor) or sounds (phonic)
• Jerk-like (clonic), dystonic, tonic, or blocking
• Premonitory feelings or sensations
• Intermittent
• Repetitive (stereotypic)
• Temporary suppressibility
• Suggestibility
• Increase with stress
• Increase during relaxation after stress
• Decrease with distraction and with concentration
• Waxing and waning, transient remissions
• Persist during sleep
Malignant Tourette Syndrome Cheung MC, Shahed J, Jankovic J. Mov Disord 2007;22:1743-50
• Malignant TS defined as ≥ 2 emergency room visits or ≥ 1
hospitalizations for TS symptoms or its associated
behavioral co-morbidities
• Of 332 TS patients evaluated during the three-year period,
17 (5.1%) met criteria for malignant TS
• Compared to patients with non-malignant TS, those with
malignant TS were significantly more likely to have a
personal history of obsessive compulsive
behavior/disorder, complex phonic tics, coprolalia,
copropraxia, self-injurious behavior, mood disorder,
suicidal ideation, and poor response to medications
In one patient with a “whiplash” tic causing compressive cervical myelopathy, we were able to reverse the neurological deficit with botulinum toxin injections into the cervical muscles. This treatment modality can be particularly effective and even life-saving in patients with tics manifested by severe, repetitive, neck extension. Such tics, if left untreated could results in secondary compressive myelopathy and quadraparesis.
Malignant Tourette Syndrome Cheung MC, Shahed J, Jankovic J. Mov Disord 2007;22:1743-50
Krauss JK, Jankovic J. Severe motor tics causing cervical myelopathy in Tourette’s syndrome. Mov Disord 1996;11:563-6
Age When Tic and OCD Symptoms Are at Their Worst
(N = 46)
(N = 19)
Bloch et al. Arch Pediatr Adolesc Med 2006;160:65-9
46 children with TS
Structured interview
at a mean age of 11.4
years and again
at 19.0 years
Tic and OCD Severity at Initial Assessment in Childhood (time 1) and Follow-up in Early Adulthood (time 2)
Tic Severity (N = 46) measured by the Yale Global Tic Severity Scale (YGTSS),
OCD (N = 19) by the Children’s Yale-Brown Obsessive Compulsive Scale (CY-BOCS)
1/3 → remission IQ → OCD
Bloch et al. Arch Pediatr Adolesc Med 2006;160:65-9

8
• We reviewed medical records of all new TS patients, ≥ 19 y/o on
initial evaluation, referred to our Movement Disorders Clinic over the
past 5 years, and compared them with 100 TS patients ≤18 y/o
• The mean age of 43 adult TS patients was 58.8 ± 6.7 years; the
mean age at initial visit of children with TS was 12.9 ±2.0 years
• Of the adult TS patients, 35 (81.4%) had a history of tics with onset
before age 18 (mean age at onset 8.5 ± 3.4 years); 8 (18.6%) reported
first occurrence of tics after age 18 (mean age at onset 37.8 ± 13.2
years); only 2 (4.7%) patients reported tic onset after age 50
• Conclusion: Adult TS largely represents re-emergence or
exacerbation of childhood-onset TS. During the course of TS,
phonic and complex motor tics, self-injurious behaviors, and ADHD
tend to improve, but facial, neck and trunk tics dominate the adult
TS phenotype.
Tourette Syndrome in Adults Jankovic J, Gelineau-Kattner R, Davidson A. Mov Disord 2010;25:2171-5
Widespread abnormality of the GABA-ergic system in Tourette syndrome.
Lerner et al. Brain 2012;135:1926-36
Decreased binding of [11C]flumazenil: bilateral ventral striatum (VS), bilateral thalamus (Th), right insula (Ins) and bilateral amygdala (Amg)
Increased binding of [11C]flumazenil: bilateral SN, left periaqueductal grey (PAG), right posterior cingulate cortex (PCC) (Cing) and bilateral cerebellum, dentate
nuclei (CB).
The bereitschaftspotential in jerky movement disorders.
van der Salm et al. J Neurol Neurosurg Psychiatry 2012;83:1162-7
6/14 TS patients had a BP prior to tics, two of which
were late BPs
TOURETTE SYNDROME Therapeutic Strategies
TICS
First Line Guanfacine
Tetrabenazine
Second Line
Fluphenazine, Risperidone Atypical Antipsychotics
Clonazepam Topiramate
BotulinumToxin Habit Reversal
Third Line Deep Brain Stimulation
OCD
First Line Cognitive Behavioral
Therapy
SSRI’s
Second Line Atypical Antipsychotics
ADHD
First Line Behavioral Therapy
Guanfacine
Second Line Clonidine
Methylphenidate
Other CNS stimulants
Atomoxetine
Third Line Deep Brain Stimulation
Jankovic, Kurlan.
Mov Disord 2011;26:1149-56
Deep Brain Stimulation for TS – Target Selection Viswanathan A, Jimenez-Shahed J, Baizabal-Carvallo F. Jankovic J.
Stereotact Funct Neurosurg 2012;90:213-24
Bilateral GPi DBS in TS Baylor College of Medicine

9
www.jankovic.org
Parkinson’s Disease Center and Movement Disorders Clinic
THANKS