
University of Groningen DNA nanotechnology as a tool to … · DNA nanotechnology as a tool to...
128
University of Groningen DNA nanotechnology as a tool to manipulate lipid bilayer membranes Meng, Zhuojun IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite from it. Please check the document version below. Document Version Publisher's PDF, also known as Version of record Publication date: 2017 Link to publication in University of Groningen/UMCG research database Citation for published version (APA): Meng, Z. (2017). DNA nanotechnology as a tool to manipulate lipid bilayer membranes. [Groningen]: University of Groningen. Copyright Other than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons). Take-down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons the number of authors shown on this cover page is limited to 10 maximum. Download date: 21-05-2020
Transcript of University of Groningen DNA nanotechnology as a tool to … · DNA nanotechnology as a tool to...

University of Groningen
DNA nanotechnology as a tool to manipulate lipid bilayer membranesMeng, Zhuojun
IMPORTANT NOTE: You are advised to consult the publisher's version (publisher's PDF) if you wish to cite fromit. Please check the document version below.
Document VersionPublisher's PDF, also known as Version of record
Publication date:2017
Link to publication in University of Groningen/UMCG research database
Citation for published version (APA):Meng, Z. (2017). DNA nanotechnology as a tool to manipulate lipid bilayer membranes. [Groningen]:University of Groningen.
CopyrightOther than for strictly personal use, it is not permitted to download or to forward/distribute the text or part of it without the consent of theauthor(s) and/or copyright holder(s), unless the work is under an open content license (like Creative Commons).
Take-down policyIf you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediatelyand investigate your claim.
Downloaded from the University of Groningen/UMCG research database (Pure): http://www.rug.nl/research/portal. For technical reasons thenumber of authors shown on this cover page is limited to 10 maximum.
Download date: 21-05-2020

DNA nanotechnology as a tool to
manipulate lipid bilayer membranes
Zhuojun Meng

DNA nanotechnology as a tool to manipulate lipid bilayer membranes
Zhuojun Meng
PhD thesis
University of Groningen
October 2017
Zernike Institute PhD thesis series 2017-22
ISSN: 1570-1530
ISBN: 978-90-367-9976-8 (printed version)
ISBN: 978-90-367-9975-1 (electronic version)
The research described in thesis was carried out in Polymer Chemistry and
Bioengineering group at Zernike Institute for Advanced Materials,
University of Groningen, The Netherlands. This work was financially
supported by the Chinese Scholarship Council (CSC), the University of
Groningen and the Netherlands Organization for Science Research (NWO).
Cover design by: Zhuojun Meng
Printed by: Ridderprint BV

DNA nanotechnology as a tool to manipulate lipid bilayer membranes
PhD thesis
to obtain the degree of PhD at the University of Groningen on the authority of the
Rector Magnificus Prof. E. Sterken and in accordance with
the decision by the College of Deans.
This thesis will be defended in public on
Friday 13 October 2017 at 16.15 hours
by
Zhuojun Meng
born on 5 May 1987 in Henan, China

Supervisor
Prof. A. Herrmann
Assessment committee
Prof. S. Vogel
Prof. A. M. van Oijen
Prof. D. J. Slotboom

Dedicated this book to myself
and my best friend
Qing Liu


Contents
Chapter 1
Functionalization of Lipid Bilayer Membranes ......................................................... 9
1. 1 Lipid bilayer membranes ................................................................................... 10
1.2 Classification and Preparation of Liposomes .............................................. 12
1.3 Modification and Applications of liposomes ............................................... 14
1.4 Motivation and Thesis Overview ...................................................................... 24
References ......................................................................................................................... 26
Chapter 2
Stability Study of Lipid-DNA on the Liposomal Membrane ............................... 31
2.1 Introduction .............................................................................................................. 32
2.2 Results and Discussion ......................................................................................... 35
2.3 Conclusion ................................................................................................................. 42
2.4 Experimental Section ............................................................................................ 42
References ......................................................................................................................... 48
Chapter 3
Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA .. 51
3.1 Introduction .............................................................................................................. 52
3.2 Results and Discussion ......................................................................................... 54
3.3 Conclusion ................................................................................................................. 63
3.4 Experimental Section ............................................................................................ 65
References ......................................................................................................................... 69

Chapter 4
DNA Replacement and Hybridization Chain Reaction on the Surface of
Liposome Membrane ......................................................................................................... 73
4.1 Introduction .............................................................................................................. 74
4.2 Results and Discussion ......................................................................................... 76
4.4 Experimental Section ............................................................................................ 84
References ......................................................................................................................... 88
Chapter 5
Performing DNA Nanotechnology Operations on a Zebrafish Surface ......... 91
5.1 Introduction .............................................................................................................. 92
5.2 Results and Discussion ......................................................................................... 94
5.3 Conclusion ................................................................................................................ 101
5.4 Experiment Section .............................................................................................. 103
References ....................................................................................................................... 105
Summary ............................................................................................................................. 108
Samenvatting .................................................................................................................... 114
Acknowledgements ....................................................................................................... 119

Chapter 1
Functionalization of
Lipid Bilayer Membranes

Chapter 1
10
1. 1 Lipid bilayer membranes
Lipids play an important role in the physiology and pathophysiology of
living systems why they are produced, transported, and recognized by the
concerted actions of numerous enzymes, binding proteins, and receptors.1
Micelles are formed by the aggregation of single-chain lipids in a polar
solvent (such as water) beyond a particular concentration, known as
Critical Micelle Concentration (CMC) (Fig. 1.1A). Therefore, the micelle
formation and stability are highly dependent on the lipid concentration and
solvent composition (Fig. 1.1B).
Two-chain lipids can hardly be packed into micelles due to the bulky
hydrophobic part. They usually form a lipid bilayer membrane, which is a
thin polar sheet made of two layers of lipid molecules and is characterized
by hydrophobic tails facing inwards towards each other and hydrophilic
head groups facing outwards to associate with aqueous solution.2 At this
moment, the hydrophobic parts of the molecules are still in contact with
water, which leads to an energetically unfavorable state of the bilayer. This
is overcome through folding of the bilayer membrane into a liposome with
closed edges (Fig. 1.1C).3,4

Functionalization of Lipid Bilayer Membranes
11
Fig. 1.1 (A) Surface tension as a function of the surfactant concentration. Schematic structure
of a micelle (B) and a liposome (C). (Fig. 1.1 C was adapted from reference 4)

Chapter 1
12
1.2 Classification and Preparation of Liposomes
Depending on the number of bilayers, liposomes can be classified into two
categories: unilamellar vesicles (ULV) and multilamellar vesicles (MLV).
Unilamellar vesicles can also be classified into three categories on the basis
of their sizes, which can vary from nanometer to micrometer range: small
unilamellar vesicles (SUV), large unilamellar vesicles (LUV) and giant
unilamellar vesicles (GUV). GUVs also include other morphologies such as
multilamellar vesicles (MLV), which consist of SUVs or multiple concentric
bilayers (Fig. 1.2).
Fig. 1.2 Schematic structure of unilamellar and multilamellar liposomes.
There are four classical methods to prepare liposomes, differing in the way
how the lipids are dried from organic solvent and then redispersed in
aqueous buffer.5 These steps can be performed individually or jointly.6
These four methods are:
1. Hydration of a Thin Lipid Film.7
2. Reverse-Phase Evaporation Technique. 8
3. Solvent (Ether or Ethanol) Injection Technique.9,10
4. Detergent Dialysis.11
Functionalization of Lipid Bilayer Membranes
13
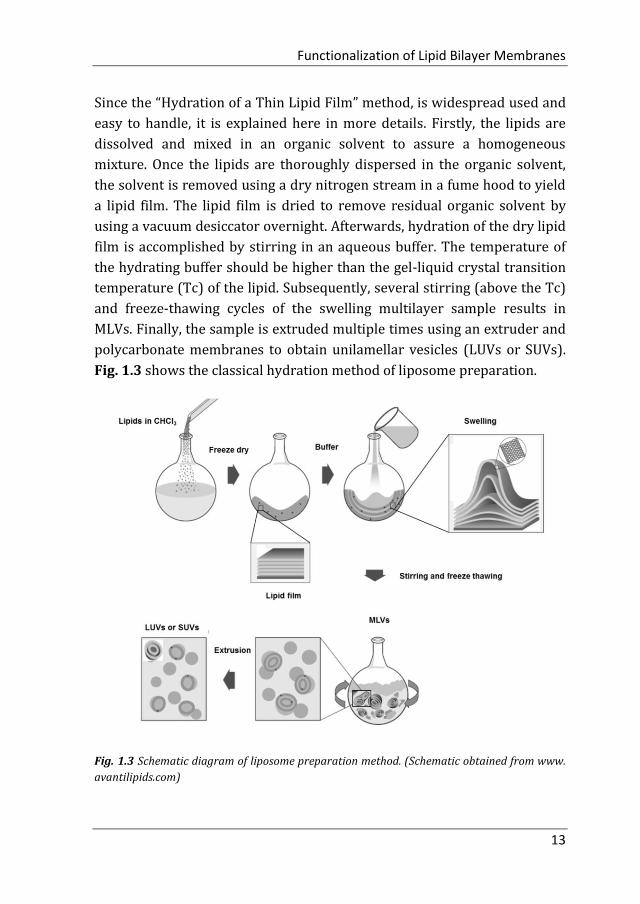
Since the “Hydration of a Thin Lipid Film” method, is widespread used and
easy to handle, it is explained here in more details. Firstly, the lipids are
dissolved and mixed in an organic solvent to assure a homogeneous
mixture. Once the lipids are thoroughly dispersed in the organic solvent,
the solvent is removed using a dry nitrogen stream in a fume hood to yield
a lipid film. The lipid film is dried to remove residual organic solvent by
using a vacuum desiccator overnight. Afterwards, hydration of the dry lipid
film is accomplished by stirring in an aqueous buffer. The temperature of
the hydrating buffer should be higher than the gel-liquid crystal transition
temperature (Tc) of the lipid. Subsequently, several stirring (above the Tc)
and freeze-thawing cycles of the swelling multilayer sample results in
MLVs. Finally, the sample is extruded multiple times using an extruder and
polycarbonate membranes to obtain unilamellar vesicles (LUVs or SUVs).
Fig. 1.3 shows the classical hydration method of liposome preparation.
Fig. 1.3 Schematic diagram of liposome preparation method. (Schematic obtained from www.
avantilipids.com)

Chapter 1
14
1.3 Modification and Applications of liposomes
The first discovery of liposomes in 1964 by A. D. Bangham12 was the
starting point for these self-assembled containers to become a
multifunctional tool in biology, biochemistry and medicine today. Because
of the structure, charge, chemical composition and colloidal size can be well
controlled by preparation methods, liposomes can be useful in various
applications. Vesicles can also be prepared from natural substances and are
therefore in many cases nontoxic, biodegradable, biocompatible, targetable
and non-immunogenic.13 Due to these properties, liposomes can be used as
drug14-16 protein, plasmid17 and gene18-21 delivery vehicles in medicine and
diagnosis.
1.3.1 Loading and surface modification
Molecular interactions between the cargo and the lipid bilayer membrane
play an important role on liposome formation and cargo encapsulation.22
Liposomes consist of an aqueous core surrounded by a lipid bilayer,
sectioning off two separate inner areas. They can carry hydrophobic
molecules in their hydrocarbon tail region (between the phospholipid
bilayer), or hydrophilic molecules in the core and direct the cargo to the
required diseased site in the body with some targeting moieties on the
surface.23 The thickness of the lipid bilayer is around 4 to 10 nm, which is a
natural barrier for many substances such as sugars and proteins.24 But
small hydrophilic substances such as water, gases, ammonia and glycerol
can penetrate freely through the bilayer.25-27 Some large hydrophilic
substances can be encapsulated in the water core of the liposome during
liposome preparation using the common thin layer hydration method.
Cationic liposomes, which are made of positively charged lipids, appear to
be better suited for DNA delivery due to the natural charge-charge
interaction between the positively charged lipid head groups and the
negatively charged phosphate groups of the DNA-backbone.28,29 Due to
their favorable interactions with negatively charged DNA and cell
membranes,30-33 cationic liposome–DNA complexes are increasingly being
researched for their use in gene therapy and nucleic acid release.34,35 In
order to increase liposomal drug accumulation in the desired cells and

Functionalization of Lipid Bilayer Membranes
15
tissues, the use of targeted liposomes with surface modification has been
suggested.
Surface modification of liposomes with controlled propertied requires the
chemical conjugation of peptides, DNA, antibodies or other targeting
molecules. Moreover, some “smart” vesicle designs allow the release of the
encapsulated cargo by incorporation of transport channels.36-39 Both
chemical attachment and physical interactions can be used to achieve
surface modification (Fig. 1.4A).
Fig. 1.4 (A) Schematic representation of liposomes surface modifications. (B) Interaction of the
particle with cell surface antigens and receptors.40 (C) Scheme of tetrac tagged liposome and
enhanced delivery by the ligand-mediated targeting strategy.41 (Fig. 1.4 B was adapted from
reference 40. Fig. 1.4 C was adapted from reference 41)
To realize active targeting, the liposome surface can be coated with ligands
or antibodies that will confer cell type-specificity to ensure that the
liposomes are internalized and that their content is released, improving the
efficacy and reducing side effects over non-targeted cells (Fig. 1.4B).40 For
instance, tetraiodothyroacetic acid (tetrac), a small molecule which binds

Chapter 1
16
to integrin αvβ3, was used for the surface modification of liposomes and
successfully enhanced the tumor-targeting ability of PEGylated liposome
(Fig. 1.4C).41 Although the physical properties of liposomes were not
significantly changed, tetrac-tagged liposomes showed significantly higher
cancer cell localization than the unmodified PEGylated liposome, and
tumor growth was effectively retarded. The ligand-mediated targeting
strategy could provide better therapeutic effects with more accurate
delivery of nanoparticles.
1.3.1.1 Membrane fusion
Surface modification of lipid bilayers can also be used for membrane fusion
which is an essential process of life resulting in the highly regulated
transport of bio-molecules both between and within cells.42-44 Membrane
fusion is an essential but not a spontaneous process as free energy is
required to overcome the electrostatic and steric repulsions between two
merging membrane surfaces and to break the hydration shell.45,46 A highly
conserved protein machinery, known as SNARE proteins (soluble N-
ethylmaleimide sensitive factor attachment protein receptors), facilitates
the communication within a cell.47-49 The SNAREs from synaptic vesicles
interact with the SNAREs from the target membrane to form a coiled-coil
bundle of four helices, pulling the membranes tightly together and
initiating fusion.
Design and construction of simplified artificial model systems mimicking
natural systems are one of the most promising approaches for studying
complex biological mechanisms.50 Several of these systems have been
reported for realizing membrane fusion, such as DNA51-53, peptides54, 55,
enzymes56 and polymers57. Yang et al. designed an artificial biorthogonal
targeting system that was able to target liposomes and other nanoparticles
efficiently to the tissue of interest by using coiled coil forming peptides,
E4[(EIAALEK)4] (E4) and K4[(KIAALKE)4] (K4) (Fig. 1.5C), which are
known to trigger liposomal membrane fusion when tethered to lipid
vesicles in the form of lipopeptides.58 The same group proved that E4
peptide-modified liposomes could deliver far-red fluorescent dye TOPRO-3
iodide (E4-Lipo-TP3) and doxorubicin (E4-Lipo-DOX) into HeLa cells

Functionalization of Lipid Bilayer Membranes
17
expressing K4 peptide (HeLa-K) on the surface. Then, E4-Lipo-TP3 and E4-
Lipo-DOX were injected into zebrafish xenografts of HeLa-K (Fig. 1.5A, B).
The results showed that E4-liposomes delivered TP3 to the implanted
HeLa-K cells (Fig. 1.5D), and E4-Lipo-DOX could suppress cancer
proliferation in the xenograft when compared to nontargeted conditions.
These data demonstrated that coiled-coil formation enables drug
selectivity and efficacy in vivo.
Fig. 1.5 Drug Delivery by E4/K4 Coiled-Coil Formation in Cells (A) and Zebrafish (B). (C)
Schematic representation of coiled-coil structure between peptides E and K. (D) E4/K4 coiled-
coil formation allows delivering the content in the liposome to cancer cells in the xenograft
zebrafish.58 (This figure was reproduced with permission from reference 58)
1.3.1.2 Controlled release
Conventional liposomes (Fig. 1.6A) are easily recognized by the
mononuclear phagocyte system and are rapidly cleared from the blood
stream.59 Many methods have been suggested to achieve long circulation of
liposomes in vivo by modification of the liposomal surface with hydrophilic

Chapter 1
18
polymers to delay the elimination process, such as coating the surface of
liposomes with biocompatible polymers like poly(ethylene glycol) (PEG)
linked phospholipids. These can be incorporated into the liposomal bilayer
to form a hydrophilic polymer shield over the liposome surface, protecting
the liposome from penetration or disintegration by plasma proteins60-65
(Fig. 1.6B). While many varieties have been synthesized by using
chemically modified forms of PEG, in some cases it’s necessary to make the
liposomes shed their cloak of modified PEG molecules when they reach
their target (Fig. 1.6C). In this way they can interact with the target and
release their payload. Using imaging technologies, visual evidence of the
effect of PEGylation on the circulation kinetics of the liposomes was
provided (Fig. 1.6D).66 The images clearly demonstrate that PEGylation
significantly enhances the persistence of liposomes in the blood stream. At
the same time, the uptake of PEGylated liposomes in organs (liver and
spleen) responsible for particle clearance decreased.
Fig. 1.6 Schematic representation of (A) conventional liposome, (B) PEG-liposome and (C)
chemically modified PEG-liposome. (D) The effect of PEGylation on the circulation persistence
of liposomes. The liposomes were labeled with Tc-99m, administered in rats, and the rats were
imaged with a gamma camera over 24 h. As is evident from the heart (H) image signal, the
PEG-liposomes remained in circulation even 24 h post-injection. The accumulation in the liver
(L) and the spleen (S) was also lower in the case of PEG-liposomes, as compared to the plain
liposomes.66 (Fig. 1.6 D was adapted from reference 66)

Functionalization of Lipid Bilayer Membranes
19
Ligands conjugated with hydrophobic molecules form amphiphiles. The
hydrophobic part can insert into the liposome bilayer, exposing the ligand
outside of the liposomes for being recognized or for other interactions. For
instance, DNA-b-polypropyleneoxide (DNA-ppo) has proven to be stably
anchored into the lipid membrane for over at least 24 h. In this way, the
containers are encoded with sequence information. The DNA-ppo present
on the surface was used for anchoring a photosensitizer by hybridization.
Upon light irradiation the PPO was oxidized leading to cargo release (Fig.
1.7).67
Fig. 1.7 Illustration of selective cargo release from DNA block copolymer (DBC) -decorated
phospholipid vesicles. (1) DNA-ppo is stably anchored in unilamellar lipid vesicles; (2) DBC-
decorated vesicles are functionalized with conjugated DNA-photosensitizers by hybridization;
(3) singlet oxygen is generated by light irradiation; and (4) selective cargo release is induced
by the oxidative effect of singlet oxygen.67 (This figure was reproduced with permission from
reference 67)
1.3.2 Stimuli-responsive liposomes
Liposomes can suspend cargos with their peculiar solubility properties and
act as a sustained-release system for microencapsulated molecules. After
modification, liposomes can be used as stimuli-responsive nanoparticles,
which are visionary concepts to deliver and release a drug exactly where it
is needed.68,69 There are several ways to trigger cargo release, such as
light,70 temperature71,72 and magnetism.73 Often two or more triggers need
Chapter 1
20
to be combined to appropriately improve the cargo release kinetics and
distribution to reduce side effects.
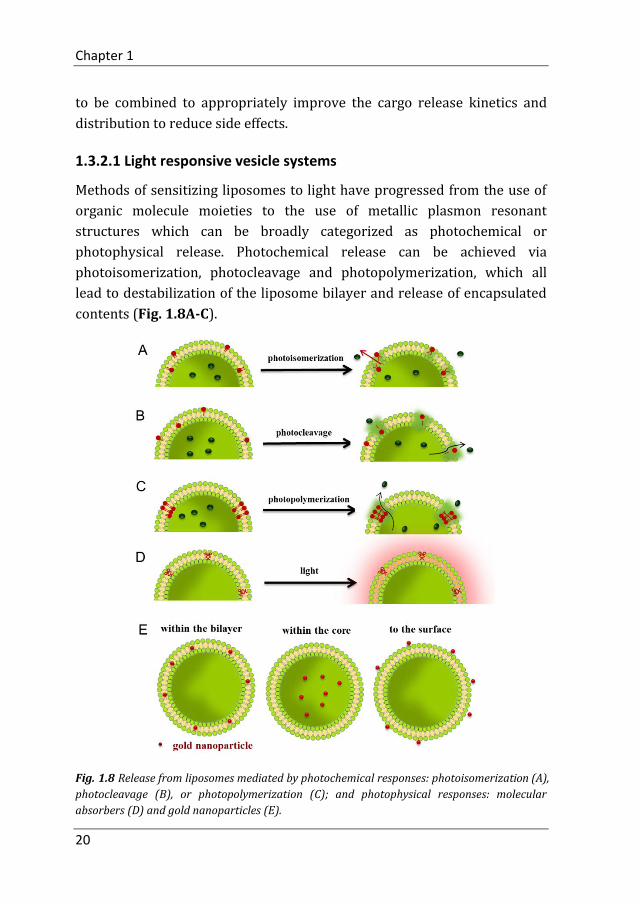
1.3.2.1 Light responsive vesicle systems
Methods of sensitizing liposomes to light have progressed from the use of
organic molecule moieties to the use of metallic plasmon resonant
structures which can be broadly categorized as photochemical or
photophysical release. Photochemical release can be achieved via
photoisomerization, photocleavage and photopolymerization, which all
lead to destabilization of the liposome bilayer and release of encapsulated
contents (Fig. 1.8A-C).
Fig. 1.8 Release from liposomes mediated by photochemical responses: photoisomerization (A),
photocleavage (B), or photopolymerization (C); and photophysical responses: molecular
absorbers (D) and gold nanoparticles (E).

Functionalization of Lipid Bilayer Membranes
21
On the other hand, photophysical release from liposomes does not rely on
any chemical changes of structures within or associated with the bilayer
membrane. Examples of photophysical release discussed here take
advantage of photothermal conversion of absorbed light with ensuing
thermal and/or mechanical processes in the lipid membrane and the
surrounding medium. The methods for achieving photophysical release are
developed around molecular absorbers (Fig. 1.8D) or gold nanoparticles
(Fig. 1.8E).70
1.3.2.2 Temperature responsive vesicle systems
Temperature-responsive liposomes are classified into two types:
traditional temperature-responsive liposomes and liposomes modified
with temperature-responsive polymers. Traditional temperature-
responsive liposomes which are composed of temperature-responsive
lipids show the greatest permeation of the lipid membrane at its gel-to-
liquid crystalline phase transition temperature.
Moreover, liposomes modified with temperature-responsive polymers
exhibit a lower critical solution temperature (LCST) behavior. These
polymers are soluble in an aqueous solution below this temperature but
dehydrate and aggregate if heated above the LCST. This behavior induces
the release of a drug within a polymer-modified liposome. For instance, a
temperature-responsive polymer, poly (N-isopropylacrylamide)-co-N,N'-
dimethylaminopropylacrylamide (P(NIPAAm-co-DMAPAAm)) was
synthesized and used for liposome modification. This research showed that
the polymer underwent dehydration and aggregation above 40 °C and that
temperature-responsive polymer-modified liposomes had faster cellular
uptake and release compared to non-modified liposomes (Fig. 1.9).74

Chapter 1
22
Fig. 1.9 Liposomes modified with temperature-responsive polymers are used for cellular
uptake. The copolymer displayed a thermosensitive transition at a lower critical solution
temperature (LCST) that is higher than body temperature. Above the LCST, the temperature-
responsive liposomes started to aggregate and release their content. The liposomes showed a
fixed aqueous layer thickness (FALT) at the surface below the LCST, and the FALT decreased
with increasing temperature. Above 37°C, cytosolic release from the temperature-responsive
liposomes was higher than that from the PEGylated liposomes, indicating intracellular
uptake.74 (This figure was adapted from reference 74)
1.3.2.3 Magnetic responsive vesicle systems
Magnetoliposomes are composed of a lipid bilayer surrounding
superparamagnetic iron oxide nanoparticles. Due to the biocompatibility,
size, material-dependent physicochemical properties and potential
applications as alternative contrast enhancing agents for magnetic
resonance imaging, magnetoliposomes are ideal candidates to achieve a
spatial and temporal control over drug release.75,76 Superparamagnetic iron
oxide nanoparticles (SPION) can be guided to their site of action using an
externally applied magnetic field. The subsequent accumulation of SPION
in the target site can be exploited for simultaneous drug delivery, MR
imaging or hyperthermia therapy of cancer (Fig. 1.10).

Functionalization of Lipid Bilayer Membranes
23
Fig. 1.10 Superparamagnetic iron oxide nanoparticles can be guided to the site of action using
an externally applied magnetic field.77 (This figure was adapted from reference 77)
In the beginning, liposomes were studied only for their physicochemical
properties as models of membrane morphology. Today, they are used as
delivery devices to encapsulate cosmetics, drugs, fluorescent detection
reagents, and as vehicles to transport nucleic acids, peptides, and proteins
to specific cellular sites in vivo. Advances in therapeutic applications of
liposomes have been achieved through surface modifications. With these
surface modifications, their biological stability could be increased, which
includes reduced constituent exchange and leakage as well as reduced
unwanted uptake by cells of the mononuclear phagocytic system.78
Targeting components such as antibodies can be attached to liposomal
surfaces and were used to create large antigen-specific complexes. In this
sense, liposomal derivatives are being used to target cancer cells in vivo, to
enhance detectability in immunoassay systems.

Chapter 1
24
1.4 Motivation and Thesis Overview
The overall goal of the work described in this thesis was to use DNA
nanotechnology as a tool to manipulate lipid bilayer surfaces. Our group
synthesized and characterized a new family of DNA amphiphiles containing
modified nucleobases. The modification is introduced in uracil and consists
of hydrophobic moieties. Through solid phase synthesis, the modified
nucleotides can be incorporated in any desired position and several
modifications per DNA strands can be introduced.79 The resulting DNA
sequences still undergo specific Watson-Crick base pairing. This property
combined with the amphiphilic nature of this lipid-DNA qualifies the
material as appealing candidate to interact with and manipulate biological
membrane structures.
In chapter 2, a powerful new approach was introduced by modifying DNA
with lipid chains at four nucleobases to tightly anchor the nucleotide to the
lipid membrane. This strategy allows highly stable incorporation of DNA
into the liposomal bilayer, thereby limiting dissociation. Several assays
were employed proving the incorporation and stable anchoring in the
phospholipid bilayer. These measurements involve small vesicles and
fluorescence energy transfer. These experiments allow to measure how
long the DNA amphiphiles remain in the bilayer.
In chapter 3, efficient fusion of liposomes was studied using lipid-DNA
introduced in the chapter before. While the orientation of DNA
hybridization played a significant role in the efficacy of full fusion of DNA-
grafted vesicles, the number of anchoring units was found to be a crucial
factor as well. As compared to vesicles functionalized with single-anchored
or double-anchored DNA, liposomes containing quadruple-anchored
oligonucleotides were found to be highly fusogenic, achieving considerable
full fusion of up to 29% without notable leakage. This study demonstrates
the importance of the DNA-anchoring strategy in hybridization-induced
vesicle fusion, as not only the structural properties of the unit itself, but
also the number of anchoring units determines its favorable fusion-
inducing properties. Several fluorescence assays, dynamic light scattering
and cryogenic transmission electron microscopy were utilized to prove
these results.

Functionalization of Lipid Bilayer Membranes
25
In chapter 4, we expand the functionality of DNA encoded vesicles
significantly. It was demonstrated that strand replacement can be carried
out. In this chapter it will be outlined what sequences and what DNA
amphiphiles are needed to reach this goal, i.e. changing the surface
functionalities of liposomes by the simple addition of oligonucleotides.
Moreover, it will be detailed how such a surface modification can be
amplified by a simple DNA-triggered supramolecular polymerization.
In chapter 5, we investigated whether it is possible to insert the lipid-
modified DNA sequences into the membrane of live zebrafish to function as
artificial receptor. We demonstrate that oligonucleotides functionalized
with a membrane anchor can be immobilized on a zebrafish. Protruding
single-stranded DNA atop the fish was functionalized by Watson-Crick base
pairing employing complementary DNA sequences. In this way, small
molecules and liposomes were guided and attached to the fish surface. The
anchoring process can be designed to be reversible allowing exchange of
surface functionalities by simple addition of DNA sequences. To achieve
this on a fish surface, the strand exchange experiments established in
chapter 4 on simple vesicles as model were crucial. Finally, a DNA based
amplification process was performed atop of the zebrafish enabling the
multiplication of surface functionalities from a single DNA anchoring unit.

Chapter 1
26
References
1. Fahy, E.; Subramaniam, S.; Brown, H. A.; Glass, C. K.; Merrill, A. H. Jr.; Murphy, R. C.; Raetz, C. R.; Russell, D. W.; Seyama, Y.; Shaw, W.; Shimizu, T.; Spener, F.; Van Meer, G.; Van Nieuwenhze, M. S.; White, S, H.; Witztum, J. L.; Dennis, E. A.; A comprehensive classification system for lipids. Journal of Lipid Research 2005, 46, 839-861.
2. Margineanu, D. G.; Equilibrium and non-equilibrium approaches in biomembrane thermodynamics. Archives Internationales de Physiologie et de Biochimie 1987, 95, 381-422.
3. Svetina, S.; Zeks, B.; Shape behavior of lipid vesicles as the basis of some cellular processes. The Anatomical Record 2002, 268, 215-225.
4. Lasic, D. D.; Liposomes in Gene Delivery. 1997, March 13, by CRC Press.
5. Mozafari, M. R.; Liposomes: An Overview of Manufacturing Techniques. Cell Mol. Biol. Lett. 2005, 10, 711-719.
6. Laouini, A.; Jaafar-Maalej, C.; Limayem-Blouza, I.; Sfar, S.; Charcosset, C.; Fessi, H.; Preparation, Characterization and Applications of Liposomes: State of the Art. J. Colloid Sci. Biotechnol. 2012, 1, 147-168.
7. Bangham, A.; Gier, J. D.; Greville, G.; OSMOTIC PROPERTIES AND WATER PERMEABILITY OF PHOSPHOLIPID LIQUID CRYSTALS. Chem. Phys. Lipids 1967, 1, 225-246.
8. Szoka, F. J.; Papahadjopoulos, D.; Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 4194-4198.
9. Szebeni, J.; Breuer, J. H.; Szelenyi, J. G.; Bathori, G.; Lelkes, G.; Hollan, S. R.; Oxidation and denaturation of hemoglobin encapsulated in liposomes. Biochim. Biophys. Acta 1984, 798, 60-67.
10. Batzri, S.; Korn, E. D.; Single bilayer liposomes prepared without sonication. Biochim. Biophys. Acta 1973, 298, 1015-1019.
11. Zumbuehl, O.; Weder, H. G.; Liposomes of controllable size in the range of 40 to 180 nm by defined dialysis of lipid/detergent mixed micelles. Biochim. Biophys. Acta 1981, 640, 252-262.
12. Bangham, A. D.; Horne, R.W.; Negative staining of phospholipids and their structural modification by surface active agents as observed in the electron microscope. J Mol Biol 1964, 8, 660-668.
13. Lasic, D. D.; Applications of Liposomes. Handbook of Biological Physics 2005, chapter 10.
14. Chen, Y.; Sen, J.; Bathula, S. R.; Yang, Q.; Fittipaldi, R.; Huang, L.; molecular pharmaceutics 2009, 6, 696-705.
15. Dutta, D.; Pulsipher, A.; Luo, W.; Yousaf, M. N.; Synthetic Chemoselective Rewiring of Cell Surfaces: Generation of Three-Dimensional Tissue Structures J. Am. Chem. Soc. 2011, 133, 8704-8713.

Functionalization of Lipid Bilayer Membranes
27
16. Chen, H.; Kim, S.; Li, L.; Wang, S.; Park, K.; Cheng, J.; Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Förster resonance energy transfer imaging. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 6596-6601.
17. Samad, A.; Sultana, Y.; Aqil, M.; Liposomal Drug Delivery Systems: An Update Review. Current Drug Delivery 2007, 4, 297-305.
18. Li, S.; Huang, L.; In vivo gene transfer via intravenous administration of cationic lipid-protamine-DNA (LPD) complexes. Gene Therapy 1997, 4, 891-900.
19. Torchilin, V. P.; Levchenko, T. S.; Rammohan, R.; Volodina, N.; Sternberg, B. P.; D’Souza, G. G. M.; Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome–DNA complexes. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1972-1977.
20. Dzau, V. J.; Mann, M. J.; Morishta, R.; Kaneda, Y.; Fusigenic viral liposome for gene therapy in cardiovascular diseases. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11421-11425.
21. Shi, N.; Pardridge, W. M.; Noninvasive gene targeting to the brain. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 7567-7572.
22. Villasmil-Sánchez, S.; Rabasco, A. M.; González-Rodríguez, M. L.; Thermal and 31P-NMR studies to elucidate sumatriptan succinate entrapment behavior in Phosphatidylcholine/Cholesterol liposomes. Comparative 31P-NMR analysis on negatively and positively-charged liposomes. Colloids and Surfaces B: Biointerfaces 2013, 105, 14-23.
23. Mozafari, M. R.; Liposomes: an overview of manufacturing techniques. Cell Mol. Biol. Lett. 2005, 10, 711-719.
24. Gubernator, J.; Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 567-582.
25. Cohen, B. E.; Bangham, A. D.; Diffusion of small nonelectrolytes across liposome membranes. Nature 1972, 236, 173-174.
26. Cullis, P. R, Hope, M. J.; Bally, M. B.; Influence of pH gradients on the transbilayer transport of drugs, lipids, peptides and metal ions into large unilamellar vesicles. Biochim. Biophys. Acta 1997, 1331, 187-211.
27. Lasic, D. D.; Liposomes: from physics to aplications. Biophysical Jounal 1994, 67, 1358-1362.
28. Chonn, A.; Cullis, P. R.; Devine, D. V.; THE ROLE OF SURFACE CHARGE IN THE ACTIVATION AND ALTERNATIVE PATHWAYS OF COMPLEMENT BY LIPOSOME. J Immunol 1991, 46, 4234-4241.
29. Eastman, S. J.; Siegel, C.; Tousignant, J.; Smith, A.E.; Cheng, S.H.; Scheule, R.K.; Biophysical characterization of cationic lipid: DNA complexes. Biochim. Biophys. Acta 1997, 1325, 41-62.
30. Leventis, R.; Silvius, J. R.; Interactions of mammalian cells with lipid dispersions containing novel metabolizable cationic amphiphiles. Biochim. Biophys. Acta 1990, 1023, 124-132.
31. Felgner, P. L.; Gadek, T. R.; Holm, M.; Roman, R.; Chan, H. W.; Wenz, M.; Northrop, J. P.; Ringold, G. M.; Danielson, M.; Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 7413-7417.
32. Templeton, N. S.; Cationic liposome-mediated gene delivery in vivo. Biosci. Rep. 2002, 22, 283-295.

Chapter 1
28
33. Wrobel, I.; Collins, D.; Fusion of cationic liposomes with mammalian cells occurs after endocytosis. Biochim. Biophys. Acta 1995, 1235, 296-304.
34. Shim, G.; Kim, M.; Park, J. Y.; Oh, Y.; Application of cationic liposomes for delivery of nucleic acids. ASIAN JOURNAL OF PHARMACEUTICAL SCIENCES 2013, 8, 72-80.
35. Kang, S. H.; Cho, H.; Shim, G.; Lee, S.; Kim, S.; Choi, H.; Kim, C.; Oh, Y.; Cationic Liposomal Co-delivery of Small Interfering RNA and a MEK Inhibitor for Enhanced Anticancer Efficacy. Pharm Res. 2011, 28, 3069-3078.
36. Dudia, A.; Koҫer, A.; Subramaniam, V.; Kanger, J. S.; Biofunctionalized Lipid-Polymer Hybrid Nanocontainers with Controlled Permeability. Nano Lett. 2008, 8, 1105-1110.
37. Cisse, I.; Okumus, B.; Joo, C.; Ha, T.; Fueling protein–DNA interactions inside porous nanocontainers. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12646-12650.
38. Birkner, J. P.; Poolman, B.; Koҫer, A.; Hydrophobic gating of mechanosensitive channel of large conductance evidenced by single-subunit resolution. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12944-12949.
39. Louhivuori, M.; Risselada, H. J.; Giessen, van der E.; Marrink, S. J.; Release of content through mechano-sensitive gates in pressurized liposomes. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 19856-19860.
40. Yhee, J. Y.; Lee, S.; Kim, K.; Advances in targeting strategies for nanoparticles in cancer imaging and therapy. Nanoscale 2014, 6, 13383.
41. Lee, S.; Kim, J.; Shim, G.; Kim, S.; Han, S. E.; Kim, K.; Kwon, I. C.; Choi, Y.; Kim, Y. B.; Kim, C.; Oh, Y.; Tetraiodothyroacetic acid-tagged liposomes for enhanced delivery of anticancer drug to tumor tissue via integrin receptor. J Control Release 2012, 164, 213-220.
42. Ma, M.; Bong, D.; Controlled Fusion of Synthetic Lipid Membrane Vesicles. Acc Chem Res. 2013, 46, 2988-2997.
43. Kumar, P.; Guha, S.; Diederichsen, U.; SNARE protein analog-mediated membrane fusion. J. Pept. Sci. 2015, 21, 621-629.
44. Kong, L.; Askes, S. H. C.; Bonnet, S.; Kros, A.; Campbell, F.; Temporal Control of Membrane Fusion through Photolabile PEGylation of Liposome Membranes. Angew. Chem. Int. Ed. 2016, 55, 1396-1400.
45. Chernomordik, L. V.; Kozlov, M. M.; Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 2003, 72, 175-207.
46. Cohen, F.S.; Melikyan, G. B.; The energetics of membrane fusion from binding, through hemifusion, pore formation and pore enlargement. J. Membr. Biol. 2004, 199, 1-14.
47. Weber, T.; Zemelman, B. V.; Mcnew, J. A.; Westermann, B.; Gmachl, M.; Parlati, F.; Söllner, T. H.; Rothman, J. E.; SNAREpins: minimal machinery for membrane fusion. Cell 1998, 92, 759-772.
48. Jahn, R.; Scheller, R. H.; SNAREs–engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631-643.
49. Hong, W. J.; Lev, S.; Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014, 24, 35-43.
50. Kumar, P.; Guha, S.; Diederichsen, U.; SNARE protein analog-mediated membrane fusion. J. Pept. Sci. 2015, 21, 621-629.

Functionalization of Lipid Bilayer Membranes
29
51. Stengel, G.; Zahn, R.; Höök, F.; DNA-induced programmablefusionof phospholipidvesicles. J. Am. Chem. Soc. 2007, 129, 9584-9585.
52. Chan, Y. H. M.; van Lengerich, B.; Boxer, S. G.; Lipid-anchored DNAmediates vesicle fusion as observed by lipid content mixing. Biointerphases 2008, 3,17-21.
53. Chan, Y. H. M.; van Lengerich, B.; Boxer, S. G.; Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotide. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 979-984.
54. Zheng, T.; Voskuhl, J.; Versluis, F.; Zope, H. R.; Tomatsu, I.; Marsden, H. R.; Kros, A. Controlling The Rate of Coiled Coil Driven Membrane Fusion. Chem. Commun. 2013, 49, 3649-3651.
55. Kong, L.; Askes, S. H.; Bonnet, S.; Kros, A.; Campbell, F. Temporal Control of Membrane Fusion through Photolabile PEGylation of Liposome Membranes. Angew. Chem. Int. Ed. 2016, 55, 1396-1400.
56. Mukai, M.; Sasaki, Y.; Kikuchi, J.; Fusion-Triggered Switching of Enzymatic Activity on an Artificial Cell Membrane. Sensors 2012, 12, 5966-5977.
57. Su, W.; Luo, Y.; Yan, Q.; Wu, S.; Han, K.; Zhang, Q.; Gu, Y.; Li, Y.; Photoinduced Fusion of Micro-Vesicles Self-Assembled from Azobenzene-Containing Amphiphilic Diblock Copolymers. Macromol. Rapid Commun. 2007, 28, 1251-1256.
58. Jian Yang, Yasuhito Shimada, René C. L. Olsthoorn, B. Ewa Snaar-Jagalska, Herman P. Spaink, and Alexander Kros, Application of Coiled Coil Peptides in Liposomal Anticancer Drug Delivery Using a Zebrafish Xenograft Model. ACS Nano. 2016, 10, 7428-7435.
59. Nag, O. K.; Awasthi, V.; Surface Engineering of Liposomes for Stealth Behavior. Pharmaceutics 2013, 5, 542-569.
60. Papahadjopoulos, D.; Allen, T. M.; Gabizon, A.; Sterically stabilized liposomes-improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 11460-11464.
61. Pashin, Y. V.; Bakhitova, L. M.; Bentkhen, T. I.; Antimutagenic activity of simple phenols and its dependence on the number of hydroxyl groups. Bull Exp Biol Med. 1986, 102, 1121-1123.
62. Woodle, M. C.; Lasic, D. D.; Sterically stabilized liposomes. Biochim. Biophys. Acta 1992, 1113, 171-199.
63. Woodle, M. C.; Newman, M. S.; Cohen, J. A.; Sterically stabilized liposomes: physical and biological properties. J Drug Target 1994, 2, 397-403.
64. Woodle, M. C.; Newman, M. S.; Collins, L. R.; Efficient evaluation of long circulating or stealth liposomes by studies of in vivo blood-circulation kinetics and final organ distribution in rats. Biophys J. 1990, 57, A261.
65. Çağdaş, M.; Sezer, A.D.; Bucak, S.; Liposomes as Potential Drug Carrier Systems for Drug Delivery. Application of Nanotechnology in Drug Delivery. 2014 Chapter 1.
66. Nag, O. K.; Yadav, V. R.; Hedrick, A.; Awasthi, V.; Post-modification of preformed liposomes with novel non-phospholipid poly(ethylene glycol)-conjugated hexadecylcarbam -oylmethyl hexadecanoic acid for enhanced circulation persistence in vivo. Int. J. Pharm. 2013, 446, 119-129.

Chapter 1
30
67. Rodríguez-Pulido, A.; Kondrachuk, A. I.; Prusty, D. K.; Gao, J.; Loi, M. A.; Herrmann, A.; Light-Triggered Sequence-Specific Cargo Release from DNA Block Copolymer–Lipid Vesicles. Angew. Chem. Int. Ed. 2013, 52, 1008-1012.
68. Chiu, H. C.; Lin, Y. W.; Huang, Y. F.; Chuang, C. K.; Chern, C. S.; Polymer Vesicles Containing Small Vesicles within Interior Aqueous Compartments and pH-Responsive Transmembrane Channels. Angew. Chem. Int. Ed. 2008, 47, 1875-1878.
69. Volodkin, D. V.; Skirtach, A. G.; Möhwald, H.; Near-IR Remote Release from Assemblies of Liposomes and Nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 1807-1809.
70. Leung, S. J.; Romanowski, M.; Light-Activated Content Release from Liposomes. Theranostics 2012, 2, 1020-1036.
71. Li, L.; Hagen, ten T. L.M.; Hossann, M.; Süss, R.; Rhoon, van G. C.; Eggermont, A. M.M.; Haemmerich, D.; Koning, G. A.; Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J Control Release 2013, 168, 142-150.
72. Tai, L.; Tsai, P.; Wang, Y.; Wang, Y.; Lo, L.; Yang, C.; Thermosensitive liposomes entrapping iron oxide nanoparticles for controllable drugrelease. Nanotechnology 2009, 20, 1-9.
73. Nappini, S.; Bombelli, F. B.; Bonini, M.; Nord, B.; Baglioni. P.; Magnetoliposomes for controlled drug release in the presence of low-frequency magnetic field. Soft Matter 2010, 6, 154-162.
74. Wang, J.; Ayano, E.; Maitani, Y.; Kanazawa, H.; Tunable Surface Properties of Temperature-Responsive Polymer-Modified Liposomes Induce Faster Cellular Uptake, ACS Omega 2017, 2, 316-325.
75. Monnier, C. A.; Burnand, D.; Rutishauser, B. R.; Lattuada, M.; Petri-Fink, A.; Magnetoliposomes: opportunities and challenges. Eur J Nanomed. 2014, 6, 201-215.
76. Amstad, E.; Kohlbrecher, J.; Müller, E.; Schweizer, T.; Textor, M.; Reimhult, E.; Triggered Release from Liposomes through Magnetic Actuation of Iron Oxide Nanoparticle Containing Membranes. Nano Lett. 2011, 11, 1664-167.
77. Laurent, S.; Saei, A.A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M.; Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: opportunities and challenges. Expert Opin Drug Deliv. 2014, 11, 1449-1470.
78. Woodle, M. C.; Surface-modified liposomes: assessment and characterization for increased stability and prolonged blood circulation. Chem. Phys. Lipids 1993, 64, 249-262.
79. Anaya, M.; Kwak, M.; Musser, A. J.; Müllen, K.; Herrmann, A.; Tunable Hydrophobicity in DNA Micelles: Design, Synthesis, and Characterization of a New Family of DNA Amphiphiles. Chem. Eur. J. 2010, 16, 12852-12859.

Chapter 2
Stability Study of Lipid-DNA on
the Liposomal Membrane
Parts of this chapter were published in: Chem. Eur. J. 2017, 23, 9391-9396.

Chapter 2
32
2.1 Introduction
Deoxyribonucleic acid (DNA) is a macro molecule that carries hereditary
information of all known living organisms and many viruses. Its double-
stranded helix structure was discovered by Watson & Crick in 1953,1 which
has greatly fueled many technologies dealing with DNA and hence
revolutionized modern science. In recent years DNA has become a valuable
functional building block and tool in nanotechnology and material science
due to the unique nature and properties of DNA and DNA hybrid materials.
A wide variety of products and applications have been realized using DNA
technologies among which is incorporating DNA with a functional group
and utilizing its information-carrying capability to develop DNA detection
systems. For instance, fluorescent dye-labeled DNA was used as probe
monitor in PCR2 or for sequence analysis.3,4 Additionally, coupling DNA
strands with moieties like polymers or nanoparticles changes the
morphological structure and introduces new functionalities, which are

Stability Study of Lipid-DNA on the Liposomal Membrane
33
different from conventional polymers. For instance, DNA conjugated gold
nanoparticles were used in DNA microarray technology.5-7 Another
functional moiety chemically conjugated with DNA consists of hydrophobic
molecules, such as long alkyl chains, cholesterol, or fatty acids, resulting in
amphiphiles, which spontaneously form nanoparticles in solution and
enhance the pharmacokinetic behavior and trans-membrane delivery.
Their amphiphilic nature arises from the hydrophilic DNA backbone
containing charged phosphodiester bonds and the hydrophobicity of
attached alkyl chains.8 These nanoparticles can be further functionalized
through hybridization of a modified complimentary DNA or internalization
of payloads in the hydrophobic core.9
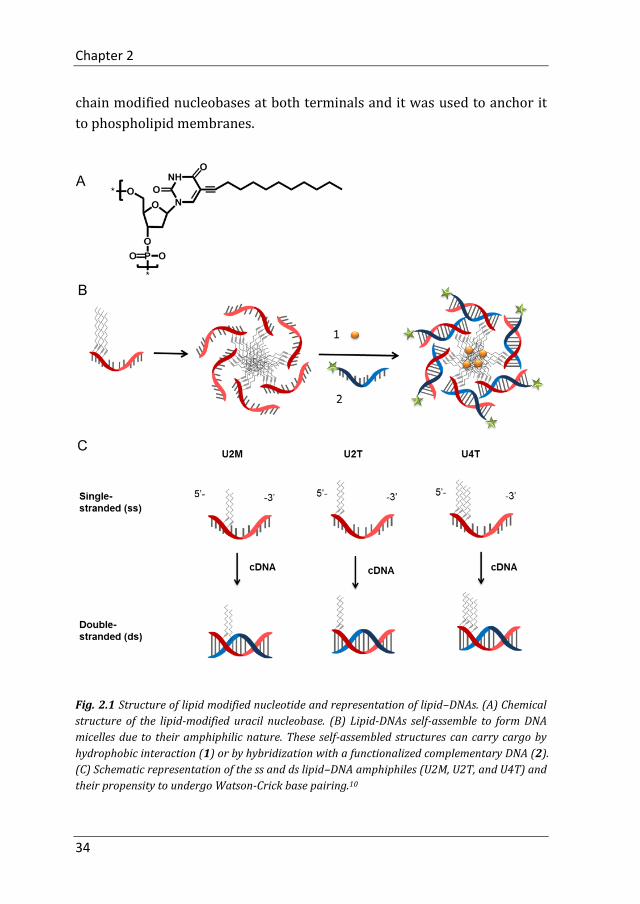
Our group reported the synthesis and characterization of a family of DNA
amphiphiles containing hydrophobically modified nucleobases.10,11
Specifically, 1-dodecyne (C12H22) was attached to a uracil base which was
further attached to the 5’ or 3’ position of a DNA sequence (Fig. 2.1A). In
aqueous environment, due to their amphiphilic nature, lipid-DNA self-
assembles into micelles whereby the hydrophilic DNA strands shield the
hydrophobic lipid core. These DNA micelles can be loaded with cargo by
hydrophobic interactions or hybridization with functionalized
complementary DNA (Fig. 2.1B). The aggregation properties of lipid-DNA
can be relatively easy manipulated by changing the length of the lipid part
or the number and position of the modified uracil bases within the DNA
sequence. Fig. 2.1C shows three different lipid-DNAs. U2M and U2T are
lipid DNA with two modified uracil bases either in the middle or at the
terminus and U4T represents lipid DNA with four modified uracil bases at
the 5’ end.10
Because of the amphiphilic and sequence specific properties, lipid-DNA can
be used for liposome surface modification by insertion of the hydrophobic
part into the membrane while the hydrophilic DNA is exposed to the
aqueous medium. Compared with existing terminal modifications, our
design allows the precise and easy introduction of hydrophobic units at
arbitrary positions and numbers in a DNA sequence through conventional
solid-phase synthesis. In this chapter, DNA was modified with four lipid
Chapter 2
34
chain modified nucleobases at both terminals and it was used to anchor it
to phospholipid membranes.
Fig. 2.1 Structure of lipid modified nucleotide and representation of lipid–DNAs. (A) Chemical
structure of the lipid-modified uracil nucleobase. (B) Lipid-DNAs self-assemble to form DNA
micelles due to their amphiphilic nature. These self-assembled structures can carry cargo by
hydrophobic interaction (1) or by hybridization with a functionalized complementary DNA (2).
(C) Schematic representation of the ss and ds lipid–DNA amphiphiles (U2M, U2T, and U4T) and
their propensity to undergo Watson-Crick base pairing.10

Stability Study of Lipid-DNA on the Liposomal Membrane
35
2.2 Results and Discussion
2.2.1 Lipid-DNA design and characteristics
To obtain stable incorporation of DNA into the liposomal bilayer, we use
lipid-DNA (U4T-18), which has been designed to contain four modified
uracil nucleobases at the 5’ position of a 18-mer oligonucleotide (including
the 4 lipid modified uracil bases). CrU4T-18 is complementary to U4T-18
with the lipid anchor at the opposite terminus (i.e. the 3’ position). Cr-
ATTO488 is a 14-mer DNA complementary to U4T-18 and was covalently
attached an ATTO488 dye to the 3’ end (Table 2.1).
Table 2.1 Sequences of modified DNA.
Name Sequence (5’→ 3’)*
U4T-18 UUUUGCGGATTCGTCTGC
CrU4T-18 GCAGACGAATCCGCUUUU
14mer GCGGATTCGTCTGC
Cr-ATTO488 GCAGACGAATCCGC-ATTO488
*: U represents the lipid-modified uracil base.
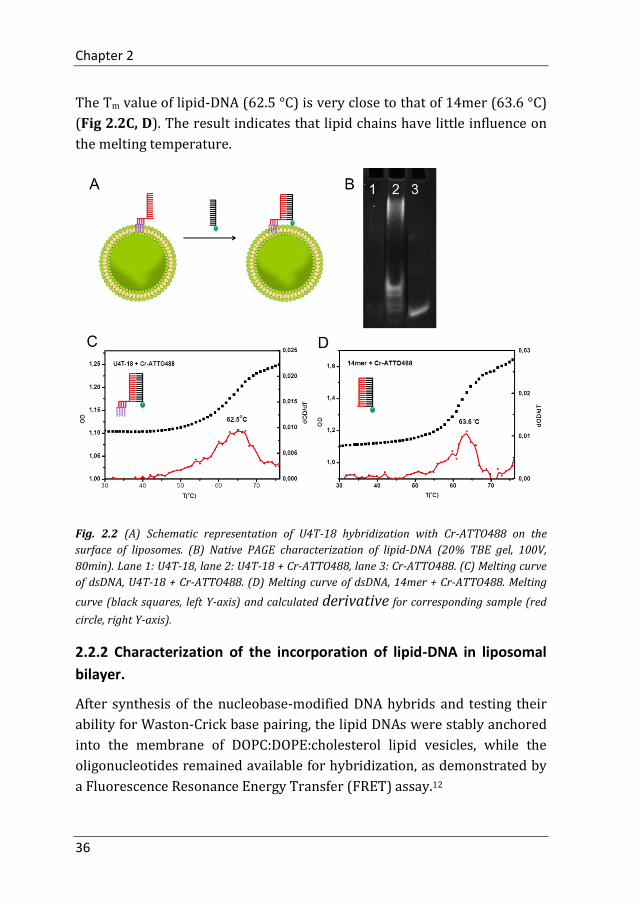
U4T-18 can be attached to the liposome surface by insertion of four lipid-
modified nucleobases into the lipid membrane while the remaining 14mer
DNA part is protruding into the aqueous medium. This DNA unit can
hybridize with the DNA part from CrU4T-18 or Cr-ATTO488 (Fig. 2.2A).
According to the results from polyacrylamide gel electrophoresis (PAGE), a
lower electrophoretic mobility of hybridized lipid-DNA (lane 2) is observed
compared to ssDNA controls (lane 1 and lane 3), indicating successful
Watson-Crick base pairing (Fig. 2.2B).
After confirming hybridization, the melting temperature (Tm) of the ds-
lipid-DNA (U4T-18+Cr-ATTO488) was determined. The ds-lipid-DNA and
ds14mer (14mer+Cr-ATTO488) were heated at 0.5 °C/min while
measuring the absorption at 260 nm. Afterwards the first derivative of the
curve was calculated and Tm of the ds DNA was taken at maximum slope.
Chapter 2
36
The Tm value of lipid-DNA (62.5 °C) is very close to that of 14mer (63.6 °C)
(Fig 2.2C, D). The result indicates that lipid chains have little influence on
the melting temperature.
Fig. 2.2 (A) Schematic representation of U4T-18 hybridization with Cr-ATTO488 on the
surface of liposomes. (B) Native PAGE characterization of lipid-DNA (20% TBE gel, 100V,
80min). Lane 1: U4T-18, lane 2: U4T-18 + Cr-ATTO488, lane 3: Cr-ATTO488. (C) Melting curve
of dsDNA, U4T-18 + Cr-ATTO488. (D) Melting curve of dsDNA, 14mer + Cr-ATTO488. Melting
curve (black squares, left Y-axis) and calculated derivative for corresponding sample (red
circle, right Y-axis).
2.2.2 Characterization of the incorporation of lipid-DNA in liposomal
bilayer.
After synthesis of the nucleobase-modified DNA hybrids and testing their
ability for Waston-Crick base pairing, the lipid DNAs were stably anchored
into the membrane of DOPC:DOPE:cholesterol lipid vesicles, while the
oligonucleotides remained available for hybridization, as demonstrated by
a Fluorescence Resonance Energy Transfer (FRET) assay.12

Stability Study of Lipid-DNA on the Liposomal Membrane
37
Since ATTO488 and rhodamine dyes show energy transfer when there is a
sufficiently short distance between them,13 ATTO488 was covalently
attached to the 3’ end of a 14-mer DNA complementary to U4T-18 (Cr-
ATTO488) to act as a donor. In parallel, rhodamine-functionalized
phospholipid (Rh-DHPE) was incorporated in the liposomal bilayer to
function as an acceptor (Fig. 2.3A). To observe the dynamic emission
changes of donor and acceptor after adding Cr-ATTO488, fluorescence
emission spectra with excitation at 470 nm of Cr-ATTO488 (donor,
emission maximum 520 nm) and Rh-DHPE (acceptor, emission maximum
592 nm) were recorded over 30 min (Fig. 2.3B). The fluorescence of donor
significantly decreased by adding Cr-ATTO488 and the fluorescence of
acceptor slightly increased at the same time, illustrating that FRET is
induced by DNA hybridization.
Fig. 2.3 (A) Schematic of FRET assay demonstrating that oligonucleotides anchored into
liposomal bilayers via lipid-DNA remain available for hybridization. (B) Fluorescence emission
of Cr-ATTO488 (donor, emission maximum 520 nm) and Rh-DHPE (acceptor, emission
maximum 592 nm) followed over 30 min after adding Cr-ATTO488.
Meanwhile, as demonstrated by the increase in the maximum intensity
ratio I592/I520 (acceptor/donor peak) (Fig. 2.4D), hybridization only
occurred upon mixing of Cr-ATTO488 with U4T-18-grafted Rh-DHPE-
containing vesicles, positioning both dyes sufficiently close to each other to
achieve FRET (Fig. 2.4A), whereas for vesicles containing non-
complementary lipid-DNA, CrU4-18, (Fig. 2.4B) or no lipid-DNA at all (Fig.
2.4C), no FRET was observed.

Chapter 2
38
Fig. 2.4 Anchoring of lipid-DNA in the membrane and hybridization on the vesicle surface
leads to Fluorescence Resonance Energy Transfer (FRET) upon hybridization of donor-
modified complementary DNA with DNA-functionalized, acceptor-containing vesicles. (A)
FRET is achieved when complementary Cr-ATTO488 DNA hybridizes with U4T-18 and brings
the donor close to the acceptor, rhodamine, positioned in the membrane. If hybridization is not
possible, either due to mismatch of the two DNA strands (B) or the absence of membrane-
grafted DNA (C) FRET does not occur. (D) Fluorescence spectra of systems capable of FRET
(red) and non-FRET controls, either due to DNA mismatch (blue) or absence of membrane-
grafted DNA (green).
Disruption of vesicles by addition of Triton X-100 to a final concentration of
0.3% (v/v) resulted in a drop in FRET in the U4T-18 vesicles hybridized
with Cr-ATTO488 (Fig. 2.5A vs Fig 2.4D), confirming that FRET was
indeed caused by bringing the donor in close vicinity to the acceptor dye
located in the liposomal membrane. As expected, in two control non-FRET
systems in which DNA hybridization could not occur, either due to absence
of DNA in the membrane (Fig. 2.5B) or the presence of non-
complementary DNA (Fig. 2.5C) energy transfer from donor to acceptor
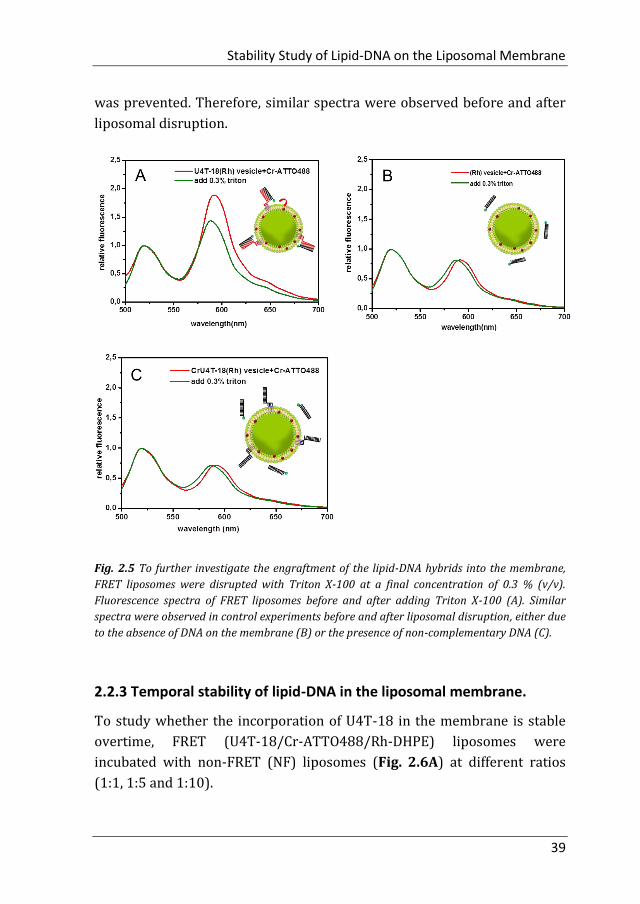
Stability Study of Lipid-DNA on the Liposomal Membrane
39
was prevented. Therefore, similar spectra were observed before and after
liposomal disruption.
Fig. 2.5 To further investigate the engraftment of the lipid-DNA hybrids into the membrane,
FRET liposomes were disrupted with Triton X-100 at a final concentration of 0.3 % (v/v).
Fluorescence spectra of FRET liposomes before and after adding Triton X-100 (A). Similar
spectra were observed in control experiments before and after liposomal disruption, either due
to the absence of DNA on the membrane (B) or the presence of non-complementary DNA (C).
2.2.3 Temporal stability of lipid-DNA in the liposomal membrane.
To study whether the incorporation of U4T-18 in the membrane is stable
overtime, FRET (U4T-18/Cr-ATTO488/Rh-DHPE) liposomes were
incubated with non-FRET (NF) liposomes (Fig. 2.6A) at different ratios
(1:1, 1:5 and 1:10).

Chapter 2
40
Fig. 2.6 Measurement of stability of lipid-DNA in liposomes over time. FRET (U4T-18/Cr-
ATTO488/Rh-DHPE) liposomes were incubated with non-FRET (NF) liposomes (A) at different
ratios (1:1, 1:5 and 1:10), and the relative Rh-DHPE/ATTO488 (IA/ID) emission intensity ratio
was monitored over 24 h after mixing (B). Fluorescence spectra of Cr-ATTO488/Rh-DHPE pair
in FRET liposomes mixed with NF liposomes at different ratio (v/v): 1:1(red line), 1:5(blue line),
1:10(green line) (C). Solid and dashed lines represent the spectra of the mixed systems before
and after adding Triton X-100, respectively.
The relative Rh-DHPE/ATTO488 (IA/ID) emission intensity ratio of the
three systems was monitored over 24 h after mixing (Fig. 2.6B). If lipid-
DNA redistributes from FRET liposomes to NF liposomes, a decrease in
relative fluorescence of acceptor peak would be observed. After 24 h, some
of the acceptor intensity had dropped, but the relative fluorescence IA/ID of
the mixture remained at a similar value as that during the initial
measurement before non-FRET liposomes were added. The results
demonstrate that the lipid–DNA is stably anchored in the liposomes over at
last 24 hours. Fig. 2.6C shows the fluorescence spectra of Cr-ATTO488/Rh-
DHPE pair in FRET liposomes mixed with NF liposomes at different ratio
before and after liposomal disruption.

Stability Study of Lipid-DNA on the Liposomal Membrane
41
Moreover, lipids were mixed with U4T-18 at different molar ratios (5000,
1000, 100, 62.5). The final concentration of Cr-ATTO488 and lipid-mixture
(DOPC+DOPE) were kept at 7.32 µM and 0.45 mM, respectively, in all FRET
experiments. The results show the I592/I520 ratio increased markedly with
higher U4T-18 densities in the membrane (Fig. 2.7, Table 2.2). These
results demonstrate that when more lipid DNA is incorporated into the
membrane more DNA strands can be attached to this vesicle surface by
hybridization (Table 2.2).
Fig. 2.7 U4T-18/Rh-DHPE fluorescence spectra of FRET liposomes mixed with Cr-ATTO488 at
different lipid/U4T-18 ratios. The inset shows a zoom-in of the acceptor Rh-DHPE peak. Solid
lines and dashed lines represent the spectra of the FRET system before and after adding Triton
X-100, respectively. Lipids were mixed with U4T-18 at different molar ratios (5000, 1000, 100,
62.5).
Table 2.2 The acceptor/donor fluorescence intensity ratios (I592/I520) at different lipid/U4T-18
ratios.
Lipid : U4T-18
ratio
U4T-18:liposome
ratio I592/I520 FRET system
5000 8 0.22
1000 38 0.24
100 380 0.31
62.5 608 0.44

Chapter 2
42
2.3 Conclusion
In conclusion, we proposed a powerful new approach employing lipid-DNA
which contains four lipid chains modified nucleobases to tightly anchor the
nucleotide to the lipid membrane. The incorporation and stability of lipid-
DNA on the liposomal membrane were proved by FRET. FRET was
achieved when the hybridization occurred between Cr-ATTO488 and U4T-
18, which brought the donor (Cr-ATTO488) close to the acceptor
(rhodamine) that was positioned in a U4T-18 functionalized membrane.
Meanwhile, the I592/I520 (acceptor/donor peak) ratio increased markedly
with higher U4T-18 densities in the membrane, and disruption of vesicles
by addition of Triton X-100 resulted in a drop of FRET vesicles system,
confirming that FRET was indeed caused by bringing the donor in close
vicinity to the acceptor dye located in the liposomal membrane. Finally, the
lipid–DNA remained stably anchored in the liposomes for at least 24 hours.
2.4 Experimental Section
2.4.1 Materials
Cholesterol (Chol), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)
and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased
from Avanti Polar Lipids (Alabaster, USA) (Fig 2.8A-C, purity >99%) and
used without further purification. Headgroup-labeled phospholipid,
Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-
phosphoethanolamine (triethylammonium salt) (Rh-DHPE) was purchased
from Invitrogen (Amsterdam, Netherlands), and used as received (Fig.
2.8D). The DNA-dye conjugate Cr-ATTO488 was purchased from
Biomers.net GmbH (Ulm, Germany). Trition X-100 (10% in water), and
Tris/HCl buffer were purchased from Sigma-Aldrich (St. Louis, United
States). Anhydrous CHCl3 was purchased from Acros Organics (Geel,
Belgium) and stored over molecular sieves. For all experiments, ultrapure
water (specific resistance > 18.4 MΩ cm) was obtained by a Milli-Q water
purification system (Sartorius).
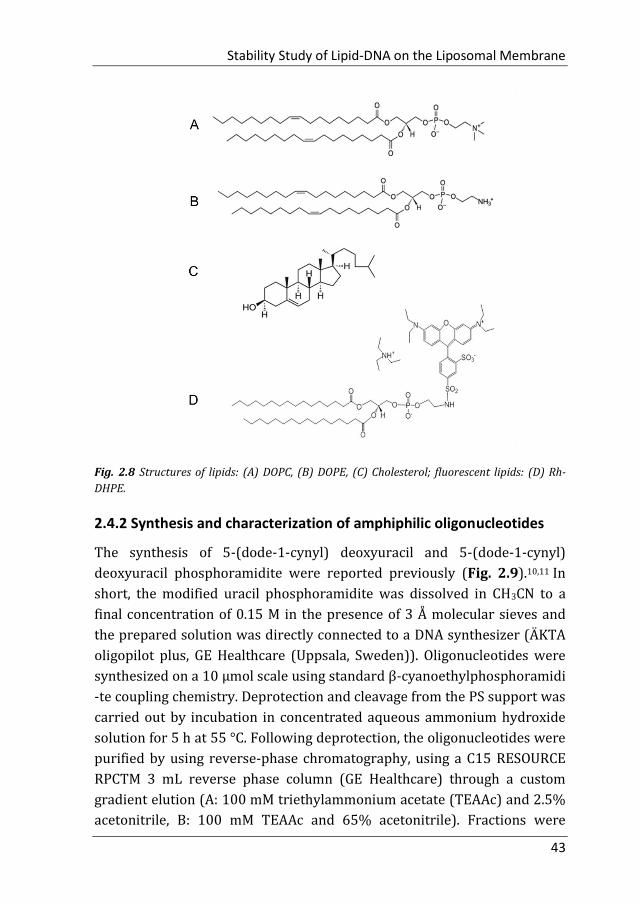
Stability Study of Lipid-DNA on the Liposomal Membrane
43
Fig. 2.8 Structures of lipids: (A) DOPC, (B) DOPE, (C) Cholesterol; fluorescent lipids: (D) Rh-
DHPE.
2.4.2 Synthesis and characterization of amphiphilic oligonucleotides
The synthesis of 5-(dode-1-cynyl) deoxyuracil and 5-(dode-1-cynyl)
deoxyuracil phosphoramidite were reported previously (Fig. 2.9).10,11 In
short, the modified uracil phosphoramidite was dissolved in CH3CN to a
final concentration of 0.15 M in the presence of 3 Å molecular sieves and
the prepared solution was directly connected to a DNA synthesizer (ÄKTA
oligopilot plus, GE Healthcare (Uppsala, Sweden)). Oligonucleotides were
synthesized on a 10 μmol scale using standard β-cyanoethylphosphoramidi
-te coupling chemistry. Deprotection and cleavage from the PS support was
carried out by incubation in concentrated aqueous ammonium hydroxide
solution for 5 h at 55 °C. Following deprotection, the oligonucleotides were
purified by using reverse-phase chromatography, using a C15 RESOURCE
RPCTM 3 mL reverse phase column (GE Healthcare) through a custom
gradient elution (A: 100 mM triethylammonium acetate (TEAAc) and 2.5%
acetonitrile, B: 100 mM TEAAc and 65% acetonitrile). Fractions were

Chapter 2
44
desalted using centrifugal dialysis membranes (MWCO 3000, Sartorius
Stedim). Oligonucleotide concentrations were determined by UV
absorbance using extinction coefficients. Finally, the identity and purity of
the oligonucleotides was confirmed by RPC-HPLC (Fig. 2.10) and MALDI-
TOF mass spectrometry (Fig. 2.11).
Fig. 2.9 Synthesis of 5-(dode-1-cynyl) deoxyuracil 2 and 5-(dode-1-cynyl) deoxyuracil
phosphoramidite 3.
Fig. 2.10 MALDI-TOF mass spectra of lipid-DNAs. (A) U4T-18, (B) CU4T-18 and (C) CrU4T-18.
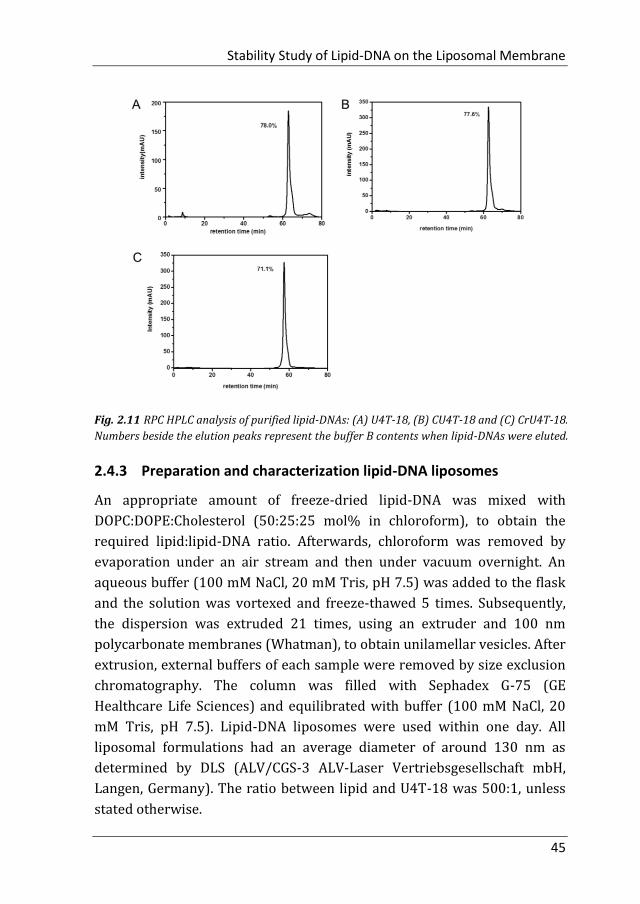
Stability Study of Lipid-DNA on the Liposomal Membrane
45
Fig. 2.11 RPC HPLC analysis of purified lipid-DNAs: (A) U4T-18, (B) CU4T-18 and (C) CrU4T-18.
Numbers beside the elution peaks represent the buffer B contents when lipid-DNAs were eluted.
2.4.3 Preparation and characterization lipid-DNA liposomes
An appropriate amount of freeze-dried lipid-DNA was mixed with
DOPC:DOPE:Cholesterol (50:25:25 mol% in chloroform), to obtain the
required lipid:lipid-DNA ratio. Afterwards, chloroform was removed by
evaporation under an air stream and then under vacuum overnight. An
aqueous buffer (100 mM NaCl, 20 mM Tris, pH 7.5) was added to the flask
and the solution was vortexed and freeze-thawed 5 times. Subsequently,
the dispersion was extruded 21 times, using an extruder and 100 nm
polycarbonate membranes (Whatman), to obtain unilamellar vesicles. After
extrusion, external buffers of each sample were removed by size exclusion
chromatography. The column was filled with Sephadex G-75 (GE
Healthcare Life Sciences) and equilibrated with buffer (100 mM NaCl, 20
mM Tris, pH 7.5). Lipid-DNA liposomes were used within one day. All
liposomal formulations had an average diameter of around 130 nm as
determined by DLS (ALV/CGS-3 ALV-Laser Vertriebsgesellschaft mbH,
Langen, Germany). The ratio between lipid and U4T-18 was 500:1, unless
stated otherwise.

Chapter 2
46
2.4.4 Calculation of lipid-DNA/liposome ratio.
The amount of lipid-DNAs per liposome was calculated using the equation:
where Φ is the number of lipids per liposome which can be calculated from
geometrical considerations:
where Souter and Sinner are the outer and inner surface area of the spherical
liposomes. Assuming the thickness of the lipid bilayer is 5 nm.14,15 α is the
average cross-sectional area of the lipid headgroups, which is assumed to
be (2*80+65)/3=75 Å for DOPC:DOPE(2:1 molar ratio).16 Router is the
averaged radius of spherical liposomes, which was determined by DLS.
2.4.5 Characterization of lipid-DNA incorporation in liposomes
measured by Fluorescence Resonance Energy Transfer (FRET) assay
Fluorescence emission spectra of Cr-ATTO488 (donor) and Rh-DHPE
(acceptor) in the 500–700 nm region were recorded with excitation at 470
nm using a SPECTRAMAX M2 (Molecular Devices) fluorescence
spectrophotometer. Measurements were carried out at constant
temperature of 25.0 °C, using a 100 mM NaCl, 20 mM Tris, pH 7.5 buffer.
2.4.6 FRET assay via DNA hybridization
U4T-18 was incorporated in Rh-DHPE/(DOPC+DOPE) (3:97 molar ratio)
liposomes to obtain U4T-18 liposomes with a lipid to U4T-18 ratio of 500:1.
Subsequently, an aliquot of these liposomes was mixed with a small
amount of Cr-ATTO488 such that [U4T-18] = [Cr-ATTO488] = 0.906 μM
and with a final lipid (DOPC+DOPE) concentration of 0.45 mM. Then, U4T-
18 and Cr-ATTO488 were hybridized using an Eppendorf Mastercycler
(Germany). The protocol consisted of heating the mixture 15 min to 40 °C

Stability Study of Lipid-DNA on the Liposomal Membrane
47
and slowly cooling to 4 °C over a period of 140 min. Afterwards, the
emission spectra of Cr-ATTO488/Rh-DHPE pair were measured.
Author contributions
Meng Z designed and conducted the experiments, performed data analysis
and wrote the manuscript. Liu Q and de Vries JW synthesized lipid-DNA.
Herrmann A supervised the project.

Chapter 2
48
References
1. Watson, J. D.; Crick, F. H.; Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737-738.
2. Kapandis, A. N.; Weiss, S.; Fluorescent probes and bioconjugation chemistries for single-molecule fluorescence analysis of biomolecules. J. Chem. Phys. 2002, 117, 10953-10964.
3. Demidov, V. V.; PNA and LNA throw light on DNA. TRENDS in Biotechnology 2003, 4-7.
4. Liu, Z.; Liu, B.; Ding, J.; Liu. J.; Fluorescent sensors using DNA-functionalized graphene oxide. Anal Bioanal Chem 2014, 406, 6885-6902.
5. Cho, H.; Jung, J.; Chung, B. H.; Scanometric analysis of DNA microarrays using DNA intercalator-conjugated gold nanoparticle. Chem. Commun. 2012, 48, 7601-7603.
6. Niemeyer, C. M.; Ceyhan, B.; Noyong, M.; Simon, U.; Bifunctional DNA–gold nanoparticle conjugates as building blocks for the self-assembly of cross-linked particle layers. Biochem Biophys Res Commun. 2003, 311, 995-999.
7. Park, S. Y.; Lytton-Jean, A. K. R.; Lee, B.; Weigand, S.; Schatz, G. C.; Mirkin, C. A.; DNA-programmable nanoparticle crystallization. Nature 2008, 451, 553-556.
8. Kwak M.; Herrmann, A.; Nucleic acid amphiphiles: synthesis and self-assembled nanostructures. Chem. Soc. Rev. 2011,40, 5745-5755.
9. Kwak, M.; Musser, A. J.; Lee, J.; Herrmann, A.; DNA-functionalised blend micelles: mix and fix polymeric hybrid nanostructures. Chem. Commun. 2008, 29, 326.
10. Anaya, M.; Kwak, M.; Musser, A. J.; Müllen, K.; Herrmann, A.; Tunable Hydrophobicity in DNA Micelles: Design, Synthesis, and Characterization of a New Family of DNA Amphiphiles. Chem. Eur. J. 2010, 16, 12852-12859.
11. Kwak, M.; Minten, I. J.; Anaya, D. M.; Musser, A. J.; Brasch, M.; Nolte, R. J. M.; Müllen, K.; Cornelissen, J. J. L. M.; Herrmann, A.;Virus-like Particles Templated by DNA Micelles: A General Method for Loading Virus Nanocarriers. J. Am. Chem. Soc. 2010, 132, 7834-7835.
12. Rodríguez-Pulido, A.; Kondrachuk, A. I.; Prusty, D. K.; Gao, J.; Loi, M. A.; Herrmann, A.; Light-Triggered Sequence-Specific Cargo Release from DNA Block Copolymer–Lipid Vesicles. Angew. Chem. Int. Ed. 2013, 52, 1008-1012.
13. Alfonta, L.; Singh, A. K.; Willner, I.; Liposomes Labeled with Biotin and Horseradish Peroxidase: A Probe for the Enhanced Amplification of Antigen-Antibody or Oligonucleotide-DNA Sensing Processes by the Precipitation of an Insoluble Product on Electrodes. Anal. Chem. 2001, 73, 91-102.
14. Leonenko, Z. V.; Finot, E.; Ma, H.; Dahms, T. E. S.; Cramb, D. T.; Investigation of Temperature-Induced Phase Transitions in DOPC and DPPC Phospholipid Bilayers Using Temperature-Controlled Scanning Force Microscopy. Biophys J. 2004, 86, 3783-3793.

Stability Study of Lipid-DNA on the Liposomal Membrane
49
15. Gramse, G.; Perez, A. D.; Edwards, M. A.; Fumagalli, L.; Gomila, G.; Nanoscale Measurement of the Dielectric Constant of Supported Lipid Bilayers in Aqueous Solutions with Electrostatic Force Microscopy. Biophys J. 2013, 104, 1257-1262.
16. Wiethoff, C. M.;Gill, M. L.; Koe, G. S.; Koe, J. G.; Middaugh, C. R.; The Structural Organization of Cationic Lipid-DNA Complexes. J. Biol. Chem. 2002, 277, 44980-44987.


Chapter 3
Efficient Fusion of Liposomes by Nucleobase
Quadruple-Anchored DNA
Parts of this chapter were published in: Chem. Eur. J. 2017, 23, 9391-9396.

Chapter 3
52
3.1 Introduction
Liposomes are a particularly effective class of nanocontainers, being able to
encapsulate and protect both small molecules and bio-macromolecules,
such as proteins or DNA.1-3 The engineering of liposomes has advanced to a
level that enables the manipulation of their surfaces with specific ligands in
order to improve their functionality. For instance, proteins, carbohydrates
and vitamins have been used as targeting units to improve the cellular
specificity of these nanocontainers. Moreover, some “smart” vesicle designs
allow the release of the encapsulated cargo through physicochemical
responses of the liposomal membrane to external stimuli4,5 or by
incorporation of transport channels.6-9 Another strategy by which
liposomes can deliver their payload to cells, is via membrane fusion,10-12
which has previously been demonstrated for drug13-16 and gene delivery17-
20 applications.
In many cellular processes, including exocytosis, endocytosis, and the
transfer of membrane proteins between cellular compartments, membrane
fusion plays a crucial role.21,22 Most membrane fusion events follow a
similar order:docking, hemifusion and full fusion. As part of the docking
process, membranes are brought into close proximity, which can cause the
outer layers to merge while the inner layers stay separated, resulting in
hemifusion. Full fusion is achieved when the outside and inside layers of
both membranes merge and content mixing occurs. Recently, several

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
53
groups have reported hemifusion and full fusion of liposomes by exploiting
Watson-Crick base pairing of complementary membrane-anchored
oligonucleotides. In these studies, DNA was grafted onto the liposomal
surface using cholesterol- or fatty acid-derivatives conjugated at the 5’- or
3’-end of the DNA oligomers.23-26 However, full fusion induced by these
systems was only achieved to a limited extent, i.e. below 4%,25,27 or with a
significant degree of content leakage.28 These limitations may be related to
DNA duplex formation and/or linkers separating the two membrane
surfaces, thereby inhibiting further membrane contact and preventing full
fusion. However, the design of the hydrophobic anchor employed to graft
the DNA into the lipid membrane could play a crucial role as well. Once two
vesicles are brought close enough for full fusion, insufficient affinity of the
hydrophobic domain of the DNA-conjugate for the bilayer or weak
mechanical coupling between the anchor and the oligonucleotides may
disable further fusion (Fig. 3.1A).
Here, we report of a powerful new approach for anchoring DNA on a
membrane and to achieve vesicle-vesicle fusion by employing DNA that is
modified with lipid chains at four nucleobases (Fig. 3.1B, C). This strategy
achieved a highly stable incorporation of DNA into the liposomal bilayer,
thereby limiting dissociation and keeping the base-pairing nucleotides
close to the surface and allowing for a markedly more efficient full fusion as
compared to other, previously reported, anchoring strategies.
Fig. 3.1 Schematic representation of vesicle fusion using lipid-modified oligonucleotides. An
oligonucleotide anchored with a single unit might be pulled out of the membrane after
hybridization and aggregation of two vesicles, which hinders full fusion (A). In the strategy
presented here, highly efficient vesicle fusion was induced by DNAs that were modified at the
nucleobases, enabling stable grafting of quadruple anchored oligonucleotides capable of non-
zipper-oriented (B) and zipper-oriented hybridization of complementary strands (C).

Chapter 3
54
3.2 Results and Discussion
Table 3.1. Sequences of DNA modified with lipid-nucleobases, poly(propylene oxide) and
cholesterol.
Name Sequence (5’→ 3’)*
U4T-18 UUUUGCGGATTCGTCTGC
CU4T-18 UUUUGCAGACGAATCCGC
CrU4T-18 GCAGACGAATCCGCUUUU
Cr-ATTO488 GCAGACGAATCCGC-ATTO488
U2T-16 UUGCGGATTCGTCTGC
CrU2T-16 GCAGACGAATCCGCUU
22PPO poly(propylene oxide)-5'-CCTCGCTCTGCTAATCCTGTTA-3'
Cr22PPO 5'-TAACAGGATTAGCAGAGCGAGG-3'-poly(propylene oxide)
14Chol Cholesterol-5'-GCGGATTCGTCTGC-3'
Cr14Chol 5'-GCAGACGAATCCGC-3'-Cholesterol
*: U represents the lipid-modified uracil base.
In the approach hereto achieve fusion employing novel anchoring units,
complementary oligonucleotides containing four uracil (U) bases modified
with dodec-1-yne (C12H22) at 3’ or 5’ position of DNA oligomers were
employed29: enabled by the previously published phosphoramidite
building block and automated DNA synthesis, U4T-18 has been fabricated
to contain four modified uracil nucleobases at the 5’ position of the 18-mer
oligonucleotide (Table 3.1), whereas CU4T-18 is complementary to U4T-
18 with the lipid anchor at the same terminus (i.e. the 5’ position) as U4T-
18.
Upon hybridization, the lipid functionalities are oriented in the DNA double
helix in a so-called ‘non-zipper’-like arrangement (Fig. 3.2). In contrast,
CrU4T-18, which is also complementary to U4T-18, was prepared with the

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
55
lipid anchor on the opposite terminus (i.e. the 3’ position) and therefore
allows for a ‘zipper’-like orientated hybridization.
Fig. 3.2 Schematic representations of lipid-modified DNA hybrids in non-zipper and zipper like
arrangements.
3.2.1 Docking of Liposomes Grafted with Quadruple-Anchored DNA
After establishing that lipid-modified oligonucleotides remained stably
incorporated into phospholipid bilayers for extended period of times, their
functionality for hybridization-induced vesicle-vesicle interaction was
explored. The fusion of lipid bilayers is a three-step process: docking,
hemifusion and full fusion. DNA hybridization allows docking of vesicles by
overcoming the repulsive hydration forces between the lipid-headgroups,
i.e. bringing the lipid bilayers of the liposomes functionalized with
complementary DNA into close proximity to each other. Liposomal docking
was observed when U4T-18 vesicles were incubated in a 1:1 ratio with
vesicles decorated with the complementary DNA sequence (CrU4T-18 or
CU4T-18), each formulation with an average diameter of around 130 nm.
After 5 hours, the average liposomal diameter, as determined by dynamic
light scattering (DLS), increased from 130 nm to around 350 nm and 300
nm, for the zipper and non-zipper orientated hybridization, respectively,
while the diameter of the U4T-18 vesicles alone did not change notably
(Fig. 3.3). This indicates that DNA hybridization and vesicle aggregation
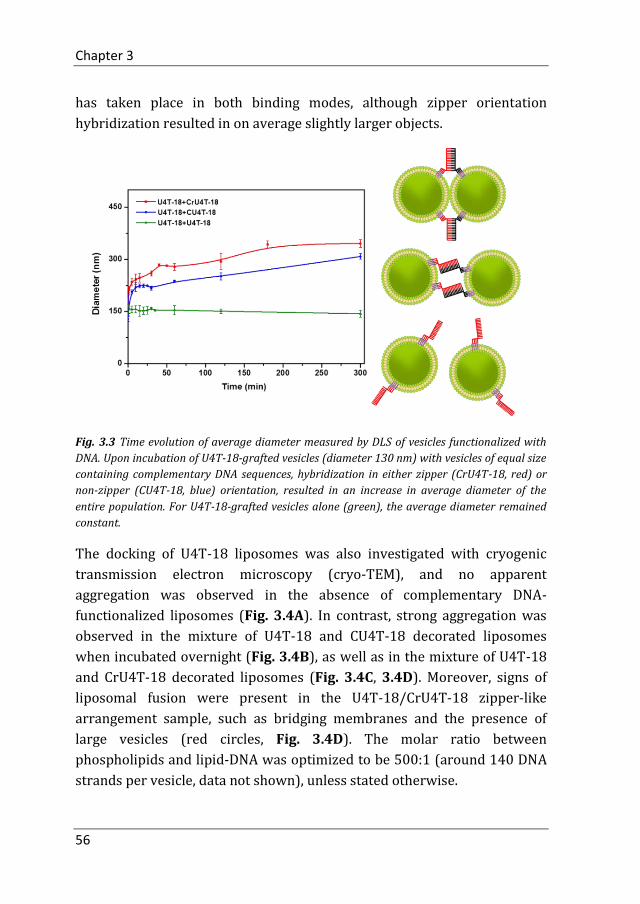
Chapter 3
56
has taken place in both binding modes, although zipper orientation
hybridization resulted in on average slightly larger objects.
Fig. 3.3 Time evolution of average diameter measured by DLS of vesicles functionalized with
DNA. Upon incubation of U4T-18-grafted vesicles (diameter 130 nm) with vesicles of equal size
containing complementary DNA sequences, hybridization in either zipper (CrU4T-18, red) or
non-zipper (CU4T-18, blue) orientation, resulted in an increase in average diameter of the
entire population. For U4T-18-grafted vesicles alone (green), the average diameter remained
constant.
The docking of U4T-18 liposomes was also investigated with cryogenic
transmission electron microscopy (cryo-TEM), and no apparent
aggregation was observed in the absence of complementary DNA-
functionalized liposomes (Fig. 3.4A). In contrast, strong aggregation was
observed in the mixture of U4T-18 and CU4T-18 decorated liposomes
when incubated overnight (Fig. 3.4B), as well as in the mixture of U4T-18
and CrU4T-18 decorated liposomes (Fig. 3.4C, 3.4D). Moreover, signs of
liposomal fusion were present in the U4T-18/CrU4T-18 zipper-like
arrangement sample, such as bridging membranes and the presence of
large vesicles (red circles, Fig. 3.4D). The molar ratio between
phospholipids and lipid-DNA was optimized to be 500:1 (around 140 DNA
strands per vesicle, data not shown), unless stated otherwise.

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
57
Fig. 3.4 Cryo-TEM images of (A) U4T-18 decorated liposomes, (B) a mixture of U4T-18 and
CU4T-18 decorated liposomes, and (C, D) a mixture of U4T-18 and CrU4T-18 decorated
liposomes. The red circles in (D) indicate vesicles that are suggestive of hemifusion. (All the
samples were incubated at 4 °C overnight.)
3.2.2 Hemifusion of Liposomes Grafted with Quadruple-Anchored DNA
To investigate the second step of vesicle fusion, i.e. hemifusion, a lipid
mixing assay based on FRET was conducted.30 Similar to a procedure
reported previously,31 the membranes of liposomes decorated with U4T-18
were stained with 0.5 mol% NBD-DHPE (donor) and 0.5 mol% Rh-DHPE
(acceptor) (FRET liposomes), while complementary DNA-functionalized
vesicles, grafted with CrU4T-18 or CU4T-18, were prepared without
fluorescently-labeled lipids (non-fluorescent liposomes). Lipid mixing
between FRET and non-fluorescent liposomes would increase the average
distance between donor and acceptor dyes, thereby attenuating FRET and
consequently increasing donor emission. Both zipper orientated and non-
zipper orientated hybridization were able to induce lipid mixing to a
similar exent (± 40%, Fig. 3.5), suggesting that hemifusion occurs
irrespective of the orientation of DNA hybridization.

Chapter 3
58
Fig. 3.5 Lipid mixing between U4T-18 grafted vesicles loaded with 0.5 mol% NBD-DHPE and
0.5 mol% Rh-DHPE and CrU4T-18 (zipper, red) or CU4T-18 (non-zipper, blue) decorated
vesicles measured by an increase in NBD emission due to a reduction in FRET efficiency. For
NBD/Rh loaded vesicles incubated with unloaded vesicles that contained non-complementary
DNA (U4T-18), no reduction in FRET efficiency was observed (green). The NBD emission of
vesicles prepared with 0.25 mol% of NBD-DHPE and 0.25% Rh-DHPE was considered full
(100%) lipid mixing (These data represent the average of three experiments).
3.2.3 Full Fusion of Liposomes Grafted with Quadruple-Anchored DNA
The concluding step of vesicle fusion consists of content mixing, i.e. the
merging of the aqueous compartments of both liposomes. This process was
evaluated by a content mixing assay, employing a protocol as reported
previously.31 In short, the fluorescent dye sulforhodamine B was
encapsulated at a self-quenching concentration (10 mM) into U4T-18
functionalized liposomes, while CrU4T-18 or CU4T-18 functionalized
liposomes were prepared without any dye. Full fusion of the U4T-18 vesicle
with its complementary counterpart would lead to content mixing and
Sulforhodamine B dilution, thereby dequenching its fluorescence resulting
in an increase in emission.
Upon exposure of U4T-18-decorated Sulforhodamine B-containing
liposomes to complementary DNA-decorated unloaded liposomes, there
was a prominent increase of sulforhodamine B emission. The mixing
induced by DNA hybridization in the zipper orientation was markedly
higher (29%, after 1 hour) than that by DNA hybridized in non-zipper

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
59
orientation (18%) (Fig. 3.6), while for liposomes grafted with the same,
and therefore non-complementary, U4T-18 lipid-DNA, only a negligible
amount of dequenching occurred (2%).
Fig. 3.6 Content mixing between liposomes decorated with U4T-18 and loaded with
sulforhodamine B and unloaded liposomes functionalized with CrU4T-18 (zipper, red) or
CU4T-18 (non-zipper, blue). Content mixing was measured as an increase in sulforhodamine B
emission due to dequenching, suggesting DNA-induced full fusion. U4T-18-grafted
sulforhodamine B-loaded liposomes mixed with unloaded U4T-18 decorated liposomes, which
could not hybridize, were used as a control (green). The fluorescence intensity upon maximal
dequenching of sulforhodamine B by disruption of liposomes in 0.3% (w/v) Triton X-100 was
considered 100% content mixing (These data represent the average of three experiments).
3.2.4 Leakage Test
Leakage of the aqueous content of vesicles into the surrounding medium
during the fusion process, possibly due to pore formation, has shown to be
a significant hurdle in DNA-induced vesicle fusion.28 To distinguish clean
fusion from leaky fusion in the dye dequenching-based content mixing
assay employed here, U4T-18-grafted vesicles incubated with either CU4-
18- or CrU4T-18-grafted vesicles were precipitated using an
ultracentrifuge and the fluorescence intensity of the supernatants analyzed.
Supernatants of liposomes fused in either orientation, as well as that of
U4T-18 before fusion, displayed a very similar fluorescent intensity (Fig.
3.7), demonstrating that full fusion was achieved with minimal leakage.
The leakage was calculated to be below 2% for both DNA configurations.
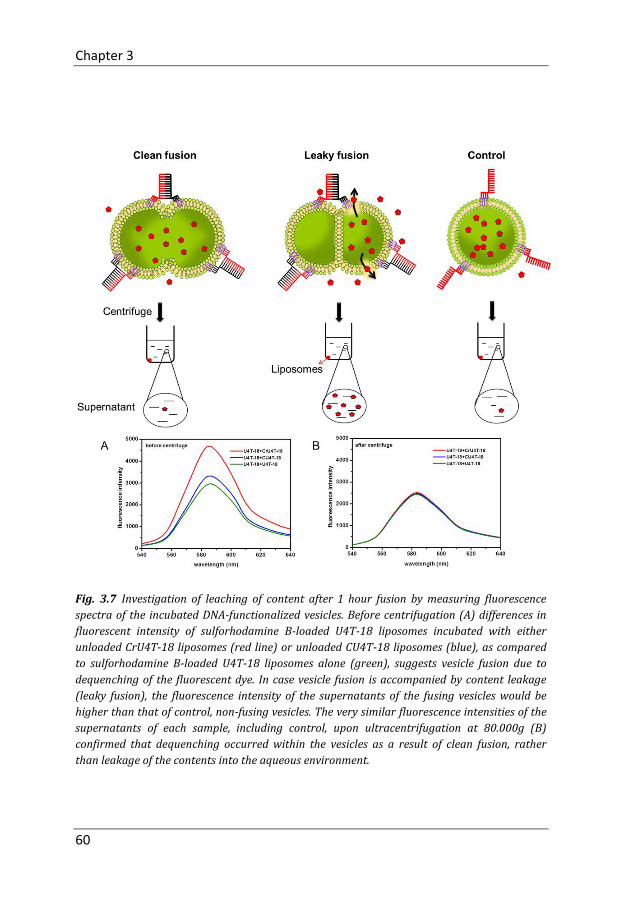
Chapter 3
60
Fig. 3.7 Investigation of leaching of content after 1 hour fusion by measuring fluorescence
spectra of the incubated DNA-functionalized vesicles. Before centrifugation (A) differences in
fluorescent intensity of sulforhodamine B-loaded U4T-18 liposomes incubated with either
unloaded CrU4T-18 liposomes (red line) or unloaded CU4T-18 liposomes (blue), as compared
to sulforhodamine B-loaded U4T-18 liposomes alone (green), suggests vesicle fusion due to
dequenching of the fluorescent dye. In case vesicle fusion is accompanied by content leakage
(leaky fusion), the fluorescence intensity of the supernatants of the fusing vesicles would be
higher than that of control, non-fusing vesicles. The very similar fluorescence intensities of the
supernatants of each sample, including control, upon ultracentrifugation at 80.000g (B)
confirmed that dequenching occurred within the vesicles as a result of clean fusion, rather
than leakage of the contents into the aqueous environment.

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
61
3.2.5 Influence of Number of Anchoring Units on Efficacy of DNA-
Induced Full Fusion
To evaluate whether the strategy by which the DNA is anchored into the
lipid bilayer, and specifically the number of anchoring units, is a
determining factor in hybridization-induced vesicle fusion, double
anchored variants of U4T-18 comprising the same (complementary)
sequence for hybridization, but modified with only two, rather than four,
lipid-modified uracil nucleobases (U2T-16, CrU2T-16, Table 3.1), were
synthesized and evaluated. As compared to the quadruple-anchored DNAs,
incubation of vesicles functionalized with complementary U2T-16
oligonucleotides resulted in markedly lower full fusion efficacy (8%, Fig.
3.8).
Fig. 3.8 Content mixing between liposomes decorated with U2T-16 and loaded with
sulforhodamine B and unloaded liposomes functionalized with CrU2T-16. Content mixing was
measured as an increase in sulforhodamine B emission due to dequenching (red), indicating
full fusion induced by zipper-oriented hybridization. U2T-16-grafted sulforhodamine B-loaded
liposomes mixed with unloaded U2T-16 decorated liposomes, which could not hybridize, were
used as a control (green). The fluorescence intensity upon maximal dequenching of
sulforhodamine B by disruption of liposomes in 0.3% (w/v) Triton X-100 was considered 100%
content mixing (These data represent the average of three experiments).
Moreover, for vesicles that contained single anchored oligonucleotides, that
consisted of single-stranded DNA modified with poly(propylene oxide)
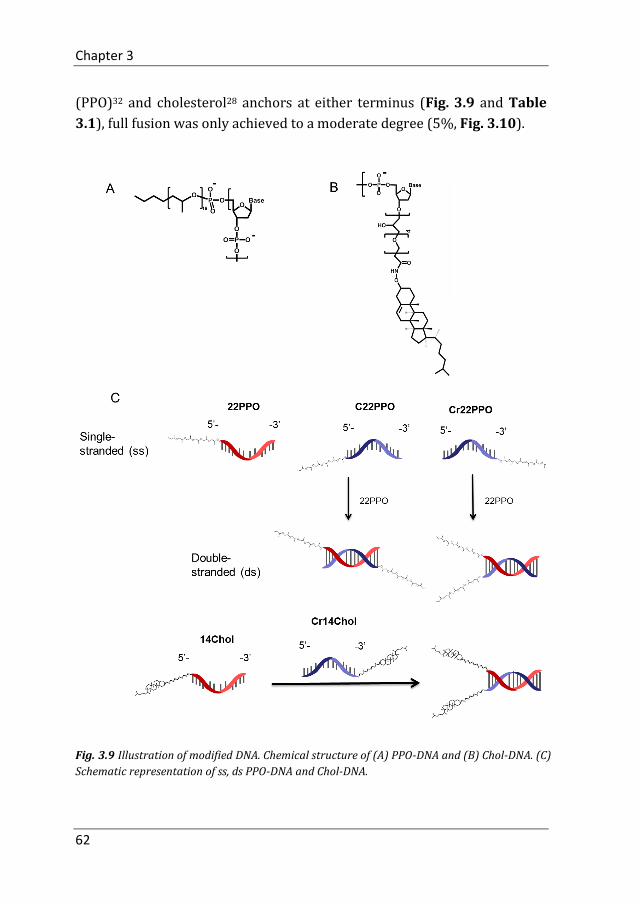
Chapter 3
62
(PPO)32 and cholesterol28 anchors at either terminus (Fig. 3.9 and Table
3.1), full fusion was only achieved to a moderate degree (5%, Fig. 3.10).
Fig. 3.9 Illustration of modified DNA. Chemical structure of (A) PPO-DNA and (B) Chol-DNA. (C)
Schematic representation of ss, ds PPO-DNA and Chol-DNA.

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
63
Fig. 3.10 Content mixing between liposomes decorated with 22PPO/Cr22PPO (A) and
14Chol/Cr14Chol (B). Content mixing was measured by increase in sulforhodamine B emission
due to dequenching (These data represent the average of three experiments).
3.3 Conclusion
These results demonstrate that, besides zipper or non-zipper orientation of
hybridization, the extent of full fusion in DNA hybridization-induced vesicle
fusion is highly dependent on the anchoring strategy of the hybridizing
nucleotides. Previously, other research groups have studied vesicle fusion
using lipid-anchored DNA. Höök et al. were the first to exploit the unique
properties of polynucleotides to induce controllable vesicle fusion via
complementary hybridization.23 In their approach, sticky-ended, double-
stranded DNA constructs were used, which were grafted into the liposomal
bilayer by means of two cholesterol anchors, each conjugated via a PEG-
linker to the termini of the double-stranded DNA anchors.33 The double-
stranded, bivalent cholesterol-anchored DNA was much more efficient in
inducing vesicle fusion than single-stranded, monovalent cholesterol-
anchored DNA, which only resulted in around 5% content mixing after 1
hour, indicating insufficient grafting stability of a monovalent anchor to
withstand the strain during DNA hybridization and bilayer reorganization.
Bivalent single-stranded oligonucleotides, i.e. two cholesterol moieties
conjugated to a single DNA, were evaluated as well,28 and although only the
efficiency regarding hemifusion, rather than full fusion, was reported,
hemifusion of vesicles grafted with complementary single-stranded,

Chapter 3
64
bivalent cholesterol-anchored DNA was similarly effective as that of their
bivalent double-stranded counterparts.
A second DNA-mediated vesicle fusion strategy, reported by Boxer et al.;
also utilized double anchored oligonucleotides. Single-stranded
complementary DNA modified with a C18 diglyceride at either terminus
was used,24 which, besides a longer chain length, are structurally relatively
similar to the U2T-16 lipid-DNAs used in the current study. The hemifusion
of vesicles functionalized with complementary diglyceride-anchored DNA
was highly efficient, illustrated by lipid mixing ratios of up to 80%,
depending on number of DNAs per vesicle24 and the presence and length of
non-hybridizing, linking sequences.25 Remarkably, however, full fusion of
vesicles grafted with the double anchored diglyceride-modified DNA
remained quite limited, with content mixing of around 2-3% for non-
repeating DNA sequences.24,25 Also taking into account the markedly
reduced full fusion achieved with the double anchored U2T-modified DNAs
as compared to the quadruple-anchored U4T-modified oligonucleotides, it
is conceivable that the number of anchoring moieties, is an important
factor in the design of lipid-DNAs and that a multivalent anchor is an
important prerequisite for efficient vesicle fusion.
Variations in experimental setup commonly obscure any comparison of
results produced in different studies, in particular of those performed in
different research groups. In order to bring the results of the current study
into context with previously reported data, cholesterol-anchored DNAs
used by Höök et al. were synthesized and evaluated in vesicles using the
content mixing assay that was also used for the U4T-18-grafted vesicles.28
Upon obtaining an extent of full fusion that was quite similar to that
reported previously by Höök et al. (Fig. 3.10B), it could be concluded that
U4T-anchored DNA indeed possesses highly favorable fusogenic properties
when incorporated into liposomal membranes, and that its remarkable
efficiency was not merely related to experimental factors.
In this study, we have established a new anchoring strategy for
oligonucleotides in vesicle membranes enabled by attaching a hydrophobic
unit to the nucleobase. The membrane anchors are incorporated into the

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
65
oligonucleotide by automated solid phase synthesis allowing precise
control over the position and number of hydrophobic units within a DNA
sequence. Therewith, this strategy overcomes structural limitations in the
context of terminal labeling with lipid moieties. With a zipper configuration
and four anchoring units close to 30% full fusion was achieved, which
might be related to the higher affinity of a quadruple lipid anchor to the
membrane, as compared to a double or single lipid anchor. We speculate
that strong anchoring limits (partial) dissociation during fusion, thereby
preventing leakage due to pore formation, keeping the double-stranded
DNA close to the vesicle surface, and consequently bringing docked vesicles
in close proximity to enhance full fusion. This ‘proximity effect’ is further
supported by the observation that zipper-orientated hybridization is more
efficient than non-zipper-orientated hybridization. In addition, a
conformational change of the lipid-modified DNA during hybridization
could induce a reorientation of the lipid anchors, disrupting the
arrangement of lipids around the lipid-modified nucleobases, and thereby
facilitating membrane fusion.
In the future, we will investigate DNA sequences with nucleobase mediated
anchoring of different designs, such as multiple anchoring regions within a
single strand, allowing to further improve the efficacy of the DNA-induced
vesicle fusion.
3.4 Experimental Section
3.4.1 Materials
Cholesterol (Chol), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)
and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased
from Avanti Polar Lipids (Alabaster, USA) (purity >99%) and used without
further purification. The DNA-dye conjugate Cr-ATTO488 was purchased
from Biomers.net GmbH (Ulm, Germany). Trition X-100 (10% in water),
Sulforhodamin B and Tris/HCl buffer were purchased from Sigma-Aldrich
(St. Louis, United States). Anhydrous CHCl3 was purchased from Acros
Organics (Geel, Belgium) and stored over molecular sieves. Preparation of

Chapter 3
66
liposomes was performed in double deionized water (Super Q Millipore
system).
3.4.2 Preparation and characterization lipid-DNA liposomes
An appropriate amount of freeze-dried lipid-DNA was mixed with
DOPC:DOPE:Cholesterol (50:25:25 mol% in chloroform), to obtain the
required lipid:lipid-DNA ratio. For lipid mixing experiments, 0.5 mol%
NBD-DHPE and 0.5 mol% Rh-DHPE were included. Afterwards, chloroform
was removed by evaporation under an air stream and then under vacuum
overnight. An aqueous buffer (100 mM NaCl, 20 mM Tris, pH 7.5) was
added to the flask and the solution was vortexed and freeze-thawed 5 times.
10 mM sulforhodamine B was encapsulated in U4T-18 decorated liposomes
for content mixing. Subsequently, the dispersion was extruded 21 times,
using an extruder and 100 nm polycarbonate membranes (Whatman), to
obtain unilamellar vesicles. After extrusion, external buffers of each sample
were removed by size exclusion chromatography. The column was filled
with Sephadex G-75 (GE Healthcare Life Sciences) and equilibrated with
buffer (100 mM NaCl, 20 mM Tris, pH 7.5). Lipid-DNA liposomes were used
within one day. All liposomal formulations had an average diameter of
around 130 nm as determined by DLS (ALV/CGS-3 ALV-Laser
Vertriebsgesellschaft mbH, Langen, Germany). The ratio between lipid and
U4T-18 was 500:1, unless stated otherwise.
3.4.3 Cryo TEM
Liposomes (total lipid concentration 2 mg/mL) were deposited on a glow-
discharged holey carbon-coated grid (Quantifoil 3.5/1, QUANTIFOIL Micro
Tools GmbH). The excess of solution was blotted off with a filter paper. The
grid was vitrified in liquid ethane using a Vitrobot (FEI) and stored in liquid
nitrogen before being transferred to a Philips CM 120 cryo-electron
microscope equipped with a Gatan model 626 cryo-stage, operating at 120
kV. Images were taken in low-dose mode using slow-scan CCD camera.
3.4.4 Lipid mixing
Fluorescence measurements were performed on a Tecan Plate Reader
Infinite M1000 (Männedorf, Switzerland). NBD emission was measured

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
67
continuously, at 530 nm for 3500 s, upon mixing fluorescent U4T-18
decorated liposomes with non-fluorescent CU4T-18 or CrU4T-18 decorated
liposomes. The 0% value (F0) was determined by measuring NBD emission
of U4T-18 decorated liposomes, which were added to an equal volume of
U4T-18 decorated liposomes at t=0. The 100% value of lipid mixing (F100%)
was determined by measuring NBD emission of liposomes which contained
0.25mol% NBD-DHPE and 0.25% Rh-DHPE. The percentage of lipid mixing
was determined by the fluorescence (NBD) increase, %F(t). %F(t)=(F(t)-
F0)/(F100%-F0) where F(t) is the fluorescence intensity of NBD measured at
time t.
3.4.5 Content mixing
10 mM sulforhodamine B was encapsulated into liposomes decorated with
U4T-18. CU4T-18 or CrU4T-18 was grafted onto non-fluorescent liposomes.
Liposomes with encapsulated sulforhodamine B were separated from non-
encapsulated dye using Sephadex G-75 size exclusion columns equilibrated
with 100 mM NaCl, 20 mM Tris buffer, pH 7.5. After mixing two liposome
formulations, the percentage of content mixing was determined by the
increase in emission of the sulforhodamine B, %F(t)=(F(t)-F0)/(F100%-F0)
where F(t) is the fluorescence intensity of sulforhodamine B measured at
time t. The fluorescence intensity at 580 nm was monitored in a continuous
fashion for 3600 s. Measurements were performed on a Tecan Plate Reader
Infinite M1000 (Männedorf, Switzerland) at room temperature. F0 was the
fluorescence intensity measured at the time when two liposome
populations were mixed together. The 100% value (F100%) was the
fluorescence intensity measured after disruption of liposomes in 0.3%
(w/v) Triton X-100 to obtain 100% release. The fluorescence intensity of
U4T-18 decorated Sulforhodamine B liposomes mixed with U4T-18
decorated non-fluorescent liposomes was used as a negative control.
3.4.6 Evaluation of fusion-induced leakage
U4T-18-decorated liposomes loaded with sulforhodamine B, unloaded
CrU4T-18- and CU4T-18-grafted liposomes were prepared as described
above, and incubated for 1h. The dispersions were subsequently
centrifuged during 20 min at 80.000 g, at 4 °C, using OptimaTM TLX

Chapter 3
68
Ultracentrifuge (Beckman Coulter) to precipitate the liposomes.
Fluorescence emission spectra of supernatants, before and after
centrifugation in the 540–640 nm region were recorded with excitation at
520 nm using a SPECTRA max M2 (Molecular Devices) fluorescence
spectrophotometer. Measurements were carried out at constant
temperature of 25 °C.
Author contributions
Meng Z designed and conducted the experiments, performed data analysis
and wrote the paper. Yang J assisted in designing and performing the lipid
and content mixing experiments. Liu Q and de Vries JW synthesized lipid-
DNA. Gruszka A performed cryo TEM experiments. Rodríguez-Pulido A and
Crielaard BJ interpreted the data and prepared the manuscript. Kros A and
Herrmann A supervised the project. All authors edited the manuscript.

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
69
References
1. Rodríguez-Pulido, A.; Ortega, F.; Llorca, O.; Aicart, E.; Junquera, E.; A Physicochemical Characterization of the Interaction between DC-Chol/DOPE Cationic Liposomes and DNA. J. Phys. Chem. B 2008, 112, 12555-12565.
2. Liu, J.; Jiang, X.; Ashley, C.; Brinker, C. J.; Electrostatically Mediated Liposome Fusion and Lipid Exchange with a Nanoparticle-Supported Bilayer for Control of Surface Charge, Drug Containment, and Delivery. J. Am. Chem. Soc. 2009, 131, 7567-7569.
3. Peer, D.; Park, E. J.; Morishita, Y.; Carman, C. V.; Shimaoka, M.; Systemic Leukocyte-Directed siRNA Delivery Revealing Cyclin D1 as an Anti-Inflammatory Target. Science 2008, 319, 627-630.
4. Chiu, H. C.; Lin, Y. W.; Huang, Y. F.; Chuang, C. K.; Chern, C. S.; Polymer Vesicles Containing Small Vesicles within Interior Aqueous Compartments and pH-Responsive Transmembrane Channels. Angew. Chem. Int. Ed. 2008, 47, 1875-1878.
5. Volodkin, D. V.; Skirtach, A. G.; Möhwald, H.; Near-IR Remote Release from Assemblies of Liposomes and Nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 1807-1809.
6. Dudia, A.; Koҫer, A.; Subramaniam, V.; Kanger, J. S.; Biofunctionalized Lipid-Polymer Hybrid Nanocontainers with Controlled Permeability. Nano Lett. 2008, 8, 1105-1110.
7. Cisse, I.; Okumus, B.; Joo, C.; Ha, T.; Fueling protein–DNA interactions inside porous nanocontainers. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12646-12650.
8. Birkner J. P.; Poolman B.; Koҫer A (2012) Hydrophobic gating of mechanosensitive channel of large conductance evidenced by single-subunit resolution. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12944-12949.
9. Louhivuori, M.; Risselada, H. J.; Giessen, van der E.; Marrink, S. J.; Release of content through mechano-sensitive gates in pressurized liposomes. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 19856-19860.
10. Ma, M.; Bong, D.; Controlled Fusion of Synthetic Lipid Membrane Vesicles. Acc Chem Res. 2013, 46, 2988-2997.
11. Kumar, P.; Guha, S.; Diederichsen, U.; SNARE protein analog-mediated membrane fusion. J. Pept. Sci. 2015, 21, 621-629.
12. Kong, L.; Askes, S. H. C.; Bonnet, S.; Kros, A.; Campbell, F.; Temporal Control of Membrane Fusion through Photolabile PEGylation of Liposome Membranes. Angew. Chem. Int. Ed. 2016, 55, 1396-1400.

Chapter 3
70
13. Chen. Y.; Sen, J.; Bathula, S. R.; Yang, Q.; Fittipaldi, R.; Huang, L.; Novel Cationic Lipid That Delivers siRNA and Enhances Therapeutic Effect in Lung Cancer Cells. Mol pharm. 2009, 6, 696-705.
14. Dutta, D.; Pulsipher, A.; Luo, W.; Yousaf, M. N.; Synthetic Chemoselective Rewiring of Cell Surfaces: Generation of Three-Dimensional Tissue Structures. J. Am. Chem. Soc. 2011, 133, 8704-8713.
15. Chen, H.; Kim, S.; Li L.; Wang, S.; Park, K.; Cheng, J,. Release of hydrophobic molecules from polymer micelles into cell membranes revealed by Förster resonance energy transfer imaging. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 6596-6601.
16. Mora, N. L.; Bahreman, A.; Valkenier, H.; Li, H.; Sharp, T. H.; Sheppard, D. N.; Davis, A. P.; Kros, A.; Targeted anion transporter delivery by coiled-coil driven membrane fusion. Chem. Sci. 2016, 7, 1768-1772.
17. Li, S.; Huang, L.; In vivo gene transfer via intravenous administration of cationic lipid–protamine–DNA (LPD) complexes. Gene Therapy 1997, 4, 891-900.
18. Torchilin, V. P.; Levchenko, T. S.; Rammohan, R.; Volodina, N.; Papahadjopoulos-Sternberg B.; D’Souza G. G. M.; Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome–DNA complexes. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1972-1977.
19. Dzau, VJ.; Mann, M. J.; Morishta, R.; Kaneda, Y.; Fusigenic viral liposome for gene therapy in cardiovascular diseases. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11421-11425.
20. Shi, N.; Pardridge, W. M.; Noninvasive gene targeting to the brain. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 7567-7572.
21. Chen, Y. A.; Scheller, R. H.; SNARE-MEDIATED MEMBRANE FUSION. Nat. Rev. Mol. Cell Biol. 2001, 2, 98-106.
22. Brunger, A. T.; Structure and functionof SNARE and SNARE-interacting proteins. Quarterly Reviews of Biophysics 2006, 38, 1-47.
23. Stengel, G.; Zahn, R.; Höök, F.; DNA-Induced Programmable Fusion of Phospholipid Vesicles. J. Am. Chem. Soc. 2007, 129, 9584-9585.
24. Chan, Y. H. M.; Lengerich, van B.; Boxer, S. G.; Lipid-anchored DNA mediates vesicle fusion as observed by lipid and content mixing. Biointerphases 2008, 3, 17-21.
25. Chan, Y. H. M.; Lengerich, van B.; Boxer, S. G.; Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 979-984.
26. Lengerich, B.; Rawle, R. J.; Bendix, P. M.; Boxer, S. G.; Individual Vesicle Fusion Events Mediated by Lipid-Anchored DNA. Biophys J 2013, 105, 409-419.
27. Xu, W.; Wang, J.; Rothman, J. E.; Pincet, F.; Accelerating SNARE-Mediated Membrane Fusion by DNA–Lipid Tethers. Angew. Chem. Int. Ed. 2015, 54, 14388-14392.

Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA
71
28. Stengel, G.; Simonsson, L.; Campbell, R. A.; Höök, F.; Determinants for Membrane Fusion Induced by Cholesterol-Modified DNA Zippers. J. Phys. Chem.B 2008, 112, 8264–8274.
29. Anaya, M.; Kwak, M.; Musser, A, J.; Müllen, K.; Herrmann, A.; Tunable Hydrophobicity in DNA Micelles: Design, Synthesis, and Characterization of a New Family of DNA Amphiphiles. Chem. Eur. J. 2010, 16, 12852-12859.
30. Marsden, H. R.; Tomatsu, I.; Kros, A.; Model systems for membrane fusion. Chem. Soc. Rev. 2011, 40, 1572-1585.
31. Versluis, F.; Voskuhl, J.; Kolck, van B.; Bremmer, M.; Albregtse, T.; Kros, A.; In Situ Modification of Plain Liposomes with Lipidated Coiled Coil Forming Peptides Induces Membrane Fusion. J. Am. Chem. Soc. 2013, 135, 8057-8062.
32. Alemdaroglu, F. E.; Ding, K.; Berger, R.; Herrmann, A.; DNA-Templated Synthesis in Three Dimensions: Introducing a Micellar Scaffold for Organic Reactions. Angew. Chem. Int. Ed. 2006, 45, 4206-4210.
33. Pfeiffer, I.; Höök, F.; Bivalent Cholesterol-Based Coupling of Oligonucletides to Lipid Membrane Assemblies. J. Am. Chem. Soc. 2004, 126, 10224-10225.


Chapter 4
DNA Replacement and Hybridization Chain
Reaction on the Surface of Liposome Membrane
This chapter has been submitted for publication.

Chapter 4
74
4.1 Introduction
Synthetic biology and cell surface engineering techniques in-vitro and in-
vivo have resulted in novel tools for the development of membrane
biology,1 offering promising membrane-based devices that may enable new
types of artificial tissues,2,3 biosensors,4,5 drug delivery approaches,6,7 3D
bio-printing and the study of lipid metabolism.8 To boost the development
of these technologies, there is a growing need to enhance surface
engineering techniques of membranes under in-vitro and in-vivo conditions
with particular emphasis on exploiting artificial surface receptors9 and

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
75
designing novel biomaterials guided by natural processes,10 such as self-
assembling peptides, proteins and DNA oligonucleotides. Especially the
latter class of biomacromolecules is very appealing to fabricate complex
architectures because the sequence specific base pairing of
oligonucleotides allows the prediction of the resulting structure based on
the sequence composition, qualifying nucleic acids as indispensible
building blocks in soft matter nanotechnology.11 In conjunction with
advances in solid phase DNA synthesis methods,12 a plethora of
programmed 2- and 3-dimensional self-assembled architectures was
achieved.13,14
The facile chemical modification of oligonucleotides with hydrophobic
anchors also allowed the fabrication of DNA-based functional
membranes.15 In the context of liposomes, DNA hybridization-induced
vesicle aggregation,16 and fusion were realized.17 Photoresponsive DNA-
lipid assemblies, fabricated by either anchoring DNA with a azobenzene
moiety18 or hybridization of a photosensitizer mediated the cargo release
from liposomes.19 While these functions relied on simple DNA amphiphiles
that were inserted in the membrane, vesicle deformation and even
destruction of these containers was achieved with immobilizing and
polymerizing more complex DNA origami structures.13,20 Further extension
of these concepts led to a DNA-based atomistically determined molecular
valve capable of controlling transport of small molecules across a biological
membrane.21
In this chapter, we implement such membrane engineering related DNA
nanotechnology on the surface of a phospholipid bilayer. We performed
studies to establish the anchoring of DNA amphiphiles in such a bilayer,
subsequent hybridization, strand replacement and DNA hybridization
chain reaction (HCR). For that purpose, vesicles served as a model system.
Previously, hybridization of DNA on a liposome surface was
demonstrated,22 however, strand replacement and DNA HCR, to the best of
our knowledge, has never been performed in liposomal systems.
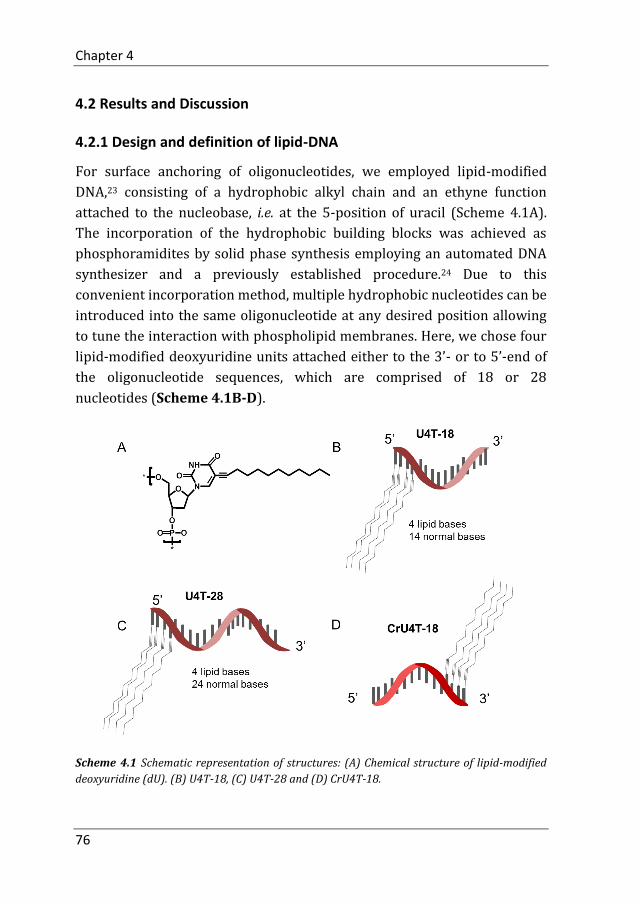
Chapter 4
76
4.2 Results and Discussion
4.2.1 Design and definition of lipid-DNA
For surface anchoring of oligonucleotides, we employed lipid-modified
DNA,23 consisting of a hydrophobic alkyl chain and an ethyne function
attached to the nucleobase, i.e. at the 5-position of uracil (Scheme 4.1A).
The incorporation of the hydrophobic building blocks was achieved as
phosphoramidites by solid phase synthesis employing an automated DNA
synthesizer and a previously established procedure.24 Due to this
convenient incorporation method, multiple hydrophobic nucleotides can be
introduced into the same oligonucleotide at any desired position allowing
to tune the interaction with phospholipid membranes. Here, we chose four
lipid-modified deoxyuridine units attached either to the 3’- or to 5’-end of
the oligonucleotide sequences, which are comprised of 18 or 28
nucleotides (Scheme 4.1B-D).
Scheme 4.1 Schematic representation of structures: (A) Chemical structure of lipid-modified
deoxyuridine (dU). (B) U4T-18, (C) U4T-28 and (D) CrU4T-18.

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
77
They are abbreviated as UxTy, where x represents the number of lipid-
modified uracils at the terminus while y denotes the overall number of
nucleotides of the sequence (Table 4.1). The four consecutive hydrophobic
anchoring units guarantee stable incorporation into a phospholipid
membrane of vesicles for at least 24 h as proven by a fluorometric assay,
which was described in Chapter 2. 24
Table 4.1 Sequences and modifications of DNA.
DNA Sequence ( 5’→3’) *
U4T-18 5’-UUUUGCGGATTCGTCTGC-3’
CrU4T-18 5’-GCAGACGAATCCGCUUUU-3’
U4T-28 5’-UUUUAATTGGGTGCGGCTTAGGATCTGA-3’
C488 5’-GCAGACAGGTCCGC-3’-ATTO488
C594 5’-GCAGACAGGTCCGCGTTTGT-3’-ATTO594
20-mer 5’-ACAAACGCGGATTCGTCTGC-3’
M1 5’-GTGCGGCTTAGGATCTGATGAAA
CTCAGATCCTAAGCCGCACCCAATT-3’
M1-FAM 6-FAM-5’-GTGCGGCTTAGGATCTGATGAAACTCA
GATCCTAAGCCGCACCCAATT-3’
M2 5’-GTTTCATCAGATCCTAAGCCGCACAAT
TGGGTGCGGCTTAGGATCTGA-3’
M2-Cy3 Cy3-5’-GTTTCATCAGATCCTAAGCCGCACAAT
TGGGTGCGGCTTAGGATCTGA-3’
*: U represents the modified uracil base. ATTO594, ATTO488, 6-FAM, Cy3 represent fluorescent dyes covalently bound to the DNA oligonucleotides.
4.2.2 DNA hybridization and replacement on the surface of liposomes
Firstly, U4T-18 was stably anchored into the membrane of vesicles
(diameter 120 nm) by in situ modification, i.e. by addition of a U4T-18
solution to plain liposomes (DOPC:DOPE:Cholesterol, 2:1:1, molar ratio).
Followed by 1h incubation at 50 °C. In situ modification spontaneously

Chapter 4
78
occurred due to the hydrophobic units part of lipid-DNA piercing into the
interior of the phospholipid bilayer. Then two Fluorescence Resonance
Energy Transfer (FRET) systems, Rhodamine Rh-DHPE/C594 and
C488/Rh-DHPE, were used to demonstrate the availability of the anchor
sequence for hybridization and DNA replacement (Fig 4.2A).
Fig. 4.2 (A) Schematic representation of DNA replacement on the surface of liposomes. (B)
Fluorescence spectra (λEX = 470 nm) of FRET system with U4T-18/Rh-DHPE/C594. FRET is
achieved when C594 hybridizes with U4T-18 to bring the donor Rh closer to the acceptor C594
(dashed purple line). Afterwards, C594 was peeled off from U4T-18 by hybridizing with 20-mer
(black line). Disruption of liposomes by addition of Triton X-100 (0.3% (v/v)) results in
termination of FRET (dashed red line). (C) After C594 was removed (black line Fig. 4.2B), U4T-
18 remained on the liposome and maintained the ability to hybridize with C488, which leads to
FRET between donor C488, and acceptor Rh-DHPE (black line). Liposome disassembly after the
addition of 0.3% (v/v) Triton X-100 results in an increase of C488 donor emission (dashed red
line).
In the Rh-DHPE/C594 system, ATTO594 was covalently attached to the 3’
end of a 20mer DNA (C594) to act as an acceptor. In parallel, rhodamine-
functionalized phospholipid (Rh-DHPE) was incorporated into the
liposomal bilayer to function as a donor (Fig 4.2A, step 1). As
demonstrated by a clear emission peak at 624 nm (C594, acceptor
fluorescence peak), hybridization only occurred upon mixing of C594 with

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
79
U4T-18-grafted Rh-DHPE-containing vesicles, positioning both dyes
sufficiently close to each other to achieve FRET (Fig 4.2B, dashed purple
line). Afterwards, a 20-mer DNA oligonucleotide that is fully
complementary to C594 was introduced to peel off C594 from U4T-18 (Fig
4.2A, step 2), which resulted in a higher signal of the donor emission of Rh-
DHPE (592 nm) and lowering of the acceptor emission (C594, 624 nm)
signal (Fig 4.2B, black line). Then, a 14-mer DNA which is complementary
to U4T-18 was covalently attached to ATTO488 and hybridized with free
U4T-18 (Fig 4.2A, step 3). In this case, C488 acted as donor for Rh-DHPE
which forms a second FRET system: C488/Rh-DHPE. Disruption of
liposomes by addition of Triton X-100 resulted in an increased donor peak
(C488, 520 nm) and slightly decreased acceptor peak (Rh-DHPE, 592 nm)
(Fig 4.2C), confirming that replacement by hybridization on the surface of
the vesicles was successfully achieved.
Fig. 4.3 Absence of FRET when liposomes are decorated with CrU4T-18. (A) Fluorescence
spectra (λEX = 470 nm) of non-FRET system with CrU4T-18/Rh-DHPE/C594 (dashed purple
line). After 1h incubation, 20-mer was added to the system (black line). Disruption of liposomes
by addition of Triton X-100 (0.3% (v/v)) results in termination of FRET (dashed red line). (B)
Fluorescence spectra of non-FRET system with CrU4T-18/C488/Rh-DHPE before (black line)
and after (dashed red line) addition of Triton X-100.
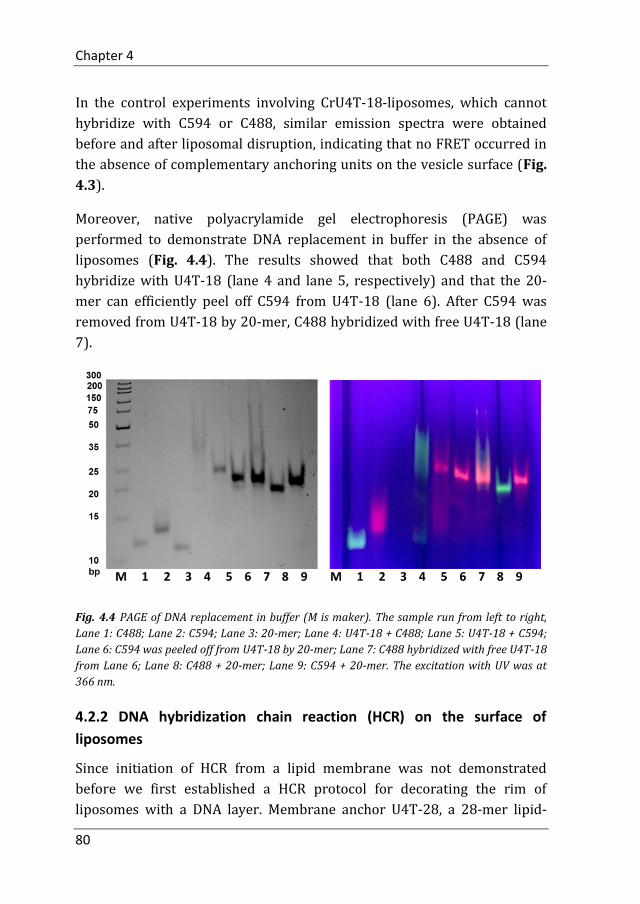
Chapter 4
80
In the control experiments involving CrU4T-18-liposomes, which cannot
hybridize with C594 or C488, similar emission spectra were obtained
before and after liposomal disruption, indicating that no FRET occurred in
the absence of complementary anchoring units on the vesicle surface (Fig.
4.3).
Moreover, native polyacrylamide gel electrophoresis (PAGE) was
performed to demonstrate DNA replacement in buffer in the absence of
liposomes (Fig. 4.4). The results showed that both C488 and C594
hybridize with U4T-18 (lane 4 and lane 5, respectively) and that the 20-
mer can efficiently peel off C594 from U4T-18 (lane 6). After C594 was
removed from U4T-18 by 20-mer, C488 hybridized with free U4T-18 (lane
7).
Fig. 4.4 PAGE of DNA replacement in buffer (M is maker). The sample run from left to right,
Lane 1: C488; Lane 2: C594; Lane 3: 20-mer; Lane 4: U4T-18 + C488; Lane 5: U4T-18 + C594;
Lane 6: C594 was peeled off from U4T-18 by 20-mer; Lane 7: C488 hybridized with free U4T-18
from Lane 6; Lane 8: C488 + 20-mer; Lane 9: C594 + 20-mer. The excitation with UV was at
366 nm.
4.2.2 DNA hybridization chain reaction (HCR) on the surface of
liposomes
Since initiation of HCR from a lipid membrane was not demonstrated
before we first established a HCR protocol for decorating the rim of
liposomes with a DNA layer. Membrane anchor U4T-28, a 28-mer lipid-

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
81
DNA with 4 modified lipid bases, was introduced to liposomes. Next,
hairpin strands M1 (partially complementary to U4T-28) and M2 (partially
complementary to M1) were added. Hybridization of M1 to U4T-28 results
in liberation of its loop that subsequently can hybridize with M2.25 Opening
of the M2 hairpin exposes a sequence that binds to a new M1 monomer
from the solution. In turn, opening of the M1 hairpin exposes a sequence
that can bind new M2. This effectively triggers the “supramolecular
polymerization” of M1 and M2 with surface anchor U4T-28 as initiator (Fig.
4.5).
Fig. 4.5 Schematic representation of lipid-DNA initiated HCR . a’, b’, and c’ are regions which
are complementary to regions a, b, and c, respectively. Hairpin M1 can be unfolded by
hybridization with initiator U4T-28 or open M2, while hairpin M2 can be unfolded by
hybridization with open M1, resulting in growing DNA strands.
Agarose gel electrophoresis analysis was used to prove U4T-28 initiated
HCR with M1 and M2 (Fig. 4.6). The hairpin sequences M1 and M2 (Fig. 4.6,
lane 1 and 2, respectively) did not hybridize in the absence of U4T-28 (Fig.
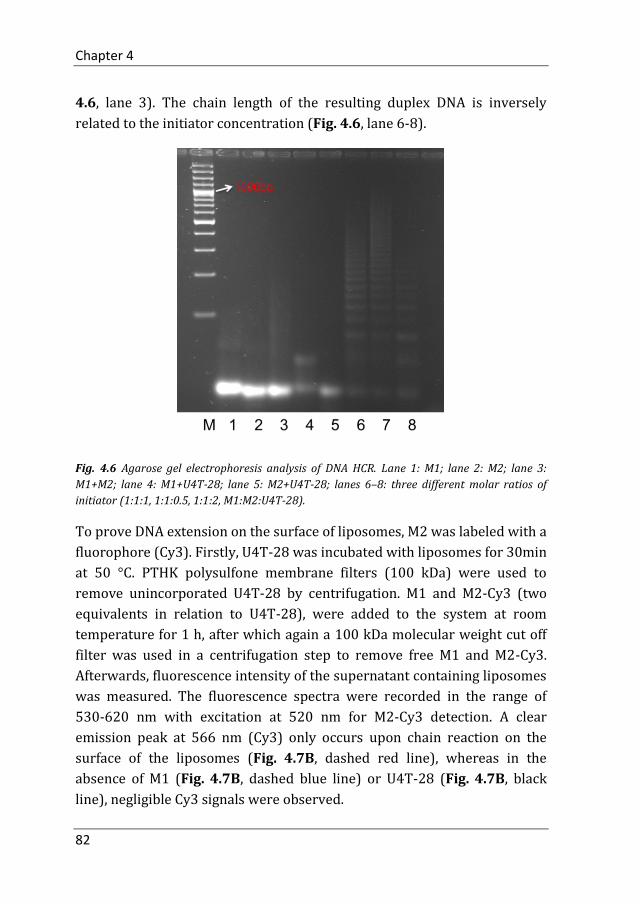
Chapter 4
82
4.6, lane 3). The chain length of the resulting duplex DNA is inversely
related to the initiator concentration (Fig. 4.6, lane 6-8).
Fig. 4.6 Agarose gel electrophoresis analysis of DNA HCR. Lane 1: M1; lane 2: M2; lane 3:
M1+M2; lane 4: M1+U4T-28; lane 5: M2+U4T-28; lanes 6–8: three different molar ratios of
initiator (1:1:1, 1:1:0.5, 1:1:2, M1:M2:U4T-28).
To prove DNA extension on the surface of liposomes, M2 was labeled with a
fluorophore (Cy3). Firstly, U4T-28 was incubated with liposomes for 30min
at 50 °C. PTHK polysulfone membrane filters (100 kDa) were used to
remove unincorporated U4T-28 by centrifugation. M1 and M2-Cy3 (two
equivalents in relation to U4T-28), were added to the system at room
temperature for 1 h, after which again a 100 kDa molecular weight cut off
filter was used in a centrifugation step to remove free M1 and M2-Cy3.
Afterwards, fluorescence intensity of the supernatant containing liposomes
was measured. The fluorescence spectra were recorded in the range of
530-620 nm with excitation at 520 nm for M2-Cy3 detection. A clear
emission peak at 566 nm (Cy3) only occurs upon chain reaction on the
surface of the liposomes (Fig. 4.7B, dashed red line), whereas in the
absence of M1 (Fig. 4.7B, dashed blue line) or U4T-28 (Fig. 4.7B, black
line), negligible Cy3 signals were observed.

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
83
Fig. 4.7 DNA HCR on the surface of liposomes. (A) Schematic representation of HCR for
decorating the outer layer of liposomes with a DNA shell. Liposomes (DOPC/DOPE/Chol = 2:1:1
mol%) decorated with U4T-28, and incubated with M1 and M2-Cy3 for 1 h, lead to DNA HCR
(I). After centrifugation and filtration, the fluorescence of supernatant containing liposomes
was measured. (B) Fluorescence spectra of Cy3 in HCR system (I, dashed red line). In the
absence of M1 (II, dashed blue line) or U4T-28 (III, black line), no fluorescence was observed.
4.3 Conclusion
In this chapter, we invented new concepts for the DNA functionalization of
a liposome surfaces. We explored DNA hybridization and the dynamic
exchange of DNA sequences on the surface of liposomes with two FRET
systems. As an anchoring unit a DNA amphiphile was utilized. Hydrophobic
units were incorporated into nucleobases, which pierce into the interior of
the phospholipid bilayer. DNA hybridization on the surface of liposomes
was proved by FRET between C594 (acceptor) and U4T-18-grafted Rh-
DHPE-containing (donor) vesicles, positioning both dyes sufficiently close
to each other to achieve FRET. After that, a 20-mer DNA oligonucleotide
that is fully complementary to C594 was introduced to peel off C594 from
U4T-18, which was confirmed by the increase of donor signal. Then, C488
hybridized with free U4T-18 acted as a donor for Rh-DHPE. The results
demonstrated that the hybridization process can be designed to be

Chapter 4
84
reversible allowing exchange of surface functionalities by simple addition
of DNA sequences. Finally, a DNA based amplification process was
performed atop of the liposome enabling the multiplication of surface
functionalities from a single DNA anchoring unit. A DNA probe, M2-Cy3,
was employed to detect the DNA HCR on the liposome’s surface. Compared
with control experiments, which were lacking DNA oligonucleotide
monomer for HCR or of the initiator, a significant stronger fluorescence
intensity of Cy3 was observed which can only be rationalized when
multiplication of DNA occurs on surface of liposomes. The hybridization
chain reaction preformed in this chapter allows accumulation of multiple
cargoes or signals on the liposomal surface by using anchored single DNA
strands. The experiments shown in this chapter significantly extend the
functionality of liposomal system regarding loading and decorating the
vesicle surface. This might be exploited in the fields of drug delivery or
diagnostics.
4.4 Experimental Section
4.4.1 Materials
Cholesterol (Chol), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)
and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased
from Avanti Polar Lipids (Alabaster, USA) (purity >99 %) and used without
further purification. Headgroup-labeled phospholipid, lissamine rhodamine
B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (triethylammoni
-um salt) (Rh-DHPE) was purchased from Invitrogen (Amsterdam,
Netherlands), and used as received. PTHK polysulfone membrane filters
with a NMWL of 100 kDa were purchased from Sigma-Aldrich. The DNA-
dye conjugates C488, C594 and M2-Cy3 were purchased from Biomers.net
GmbH (Ulm, Germany). Triton X-100 (10% in water) was purchased from
Sigma-Aldrich (St. Louis, United States). Anhydrous CHCl3 was acquired
from Acros Organics (Geel, Belgium) and stored over molecular sieves. For
all experiments, ultrapure water (specific resistance > 18.4 MΩ cm) was
obtained by a Milli-Q water purification system (Sartorius).

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
85
4.4.2 Synthesis and characterization of amphiphilic oligonucleotides
Fig. 4.8 MALDI-TOF mass spectra of (A) U4T-18, (B) U4T-28 and (C) CrU4T-18.
Fig. 4.9 RPC HPLC analysis of purified lipid-DNAs: (A) U4T-18, (B) U4T-28 and (C) CrU4T-18.
Numbers beside the elution peaks represent the buffer B contents when lipid-DNAs were eluted.

Chapter 4
86
The amphiphilic oligonucleotides were synthesized and purified as
reported previously (see Chapter 2).23,26 The identity and purity of the
oligonucleotides were confirmed by RPC-HPLC and MALDI-TOF mass
spectrometry (Fig. 4.8 and Fig. 4.9).
4.4.2 Preparation and characterization lipid-DNA liposomes
Firstly, chloroform was removed from lipid mixture (DOPC:DOPE:
Cholesterol, 2:1:1, molar ratio) by evaporation under an air stream and
then under vacuum overnight. The dried lipid mixture was dissolved in an
aqueous PBS buffer (150 mM NaCl, 15 mM K2HPO4, 5 mM KH2PO4) by 5
cycles of vortexing and freeze-thawing. Subsequently, the sample was
dispersed by extruding 21 times using an extruder and 100 nm
polycarbonate membranes (Whatman) to obtain large unilamellar vesicles
(LUVs), after which the liposomes with lipid-DNA were incubated at 50 °C
for 1h. All lipid-DNA liposomes were used within one day, and had an
average diameter of around 120 nm as determined by DLS (ALV/CGS-3
ALV-Laser Vertriebsgesellschaft mbH, Langen, Germany). The molar ratio
between lipid and U4T-18 was 500:1.
4.4.3 DNA replacement on liposomes measured by Fluorescence
Resonance Energy Transfer (FRET) assay
U4T-18 was incorporated in Rh-DHPE/(DOPC+DOPE) (3:97 molar ratio)
liposomes to obtain U4T-18 liposomes with a lipid to U4T-18 ratio of 500:1.
Subsequently, an aliquot of these liposomes was mixed with a small
amount of Cr-ATTO488 or CrATTO594 such that [U4T-18] = [Cr-ATTO488]
= [Cr-ATTO594] = 0.906 μM and with a final lipid (DOPC+DOPE)
concentration of 0.45 mM. Fluorescence emission spectra of
donor/acceptor, CrATTO488/Rh-DHPE or Rh-DHPE/CrATTO594 in the
500–700 nm region, were recorded with excitation at 470 nm using a
SpectraMax M3 (Molecular Devices) fluorescence spectrophotometer.
Measurements were carried out at a constant temperature of 25 °C.

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
87
4.4.4 Native polyacrylamide gel electrophoresis to detect DNA
replacement
Native polyacrylamide gel electrophoresis was performed using a 15% gel
made with TBE buffer (90 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.0)
and run at 120 V for 100 min.
4.4.5 Agarose gel electrophoresis to monitor HCR
The 2 % agarose gel was prepared by using SB buffer (10 mM NaOH, pH
adjusted to 8.5 with boric acid) 27 and run at 90 V for 3 h. After
electrophoresis, the agarose gel was stained with ethidium bromide and
visualized under UV light.
4.4.6 DNA hybridization chain reaction on the surface of liposomes
After incubation of 0.45 mM liposomes with 0.906 μM U4T-28 at 50 °C for
30 min, PTHK polysulfone membrane filters (100 kDa) were used to
remove unincorporated U4T-28 by centrifugation at 1000 rpm (rotor: FA-
45-18-11) for 30 min. M1 and M2-Cy3 (two equivalents in relation to U4T-
28) were added to the system at RT for 1 h, after which again a 100 kDa
molecular weight cut off filter was used in a centrifugation step to remove
free DNA (1000 rpm, 30 min). After re-suspension and centrifugation, the
supernatant was washed twice with PBS buffer. Afterwards, the
fluorescence spectra were recorded in the range of 530-620 nm with
excitation at 470 nm for M2-Cy3, using a SpectraMax M3 (Molecular
Devices) fluorescence spectrophotometer. Measurements were carried out
at 25 °C.
Author contributions
Meng Z designed, conducted the experiments and performed data analysis.
Meng Z and Yang J prepared the manuscript. Liu Q synthesized lipid-DNA.
Kros A and Herrmann A supervised the project.

Chapter 4
88
References
1. Saeui, C. T.; Mathew, M.P.; Liu, L.; Urias, E.; Yarema, K. J.; Cell Surface and Membrane Engineering: Emerging Technologies and Applications. J. Funct. Biomater. 2015, 6, 454-485.
2. Cukierman, E.; Pankov, R.; Stevens, D. R.; Yamada, K. M.; Taking cell matrix adhesions to the third dimension. Science 2001, 294, 1708-1712.
3. Revzin, A.; Rajagopalan, P.; Tilles, A. W.; Berthiaume, F.; Yarmush, M. L.; Toner, M.; Designing a hepatocellular microenvironment with protein microarraying and poly(ethylene glycol) photolithography. Langmuir 2004, 20, 2999-3005.
4. Shear, J. B.; Fishman, H. A.; Allbritton, N. L.; Garigan, D.; Zare, R. N.; Scheller, R. H.; Single cells as biosensors for chemical separations. Science 1995, 267, 74-77.
5. Rider, T. H.; Petrovick, M. S.; Nargi1, F. E.; Harper, J. D.; Schwoebel, E. D.; Mathews1, R. H.; Blanchard, D. J.; Bortolin, L. T.; Young, A. M.; Chen, J.; Hollis, M. A.; A B cell-based sensor for rapid identification of pathogens. Science 2003, 301, 213-215.
6. Yang, J.; Shimada, Y.; Olsthoorn, R. C. L.; Snaar-Jagalska, B. E.; Spaink, H. P.; Kros, A.; Application of Coiled Coil Peptides in Liposomal Anticancer Drug Delivery Using a Zebrafish Xenograft Model. ACS Nano. 2016, 10, 7428-7435.
7. Yang, J.; Bahreman, A.; Daudey, G.; Bussmann, J.; Olsthoorn, R. C. L.; Kros, A.;.; Drug Delivery via Cell Membrane Fusion Using Lipopeptide Modified Liposomes. ACS Cent. Sci. 2016, 2, 621-630.
8. Han, G. S.; O'Hara, L.; Carman, G. M.; Siniossoglou, S.; An unconventional diacylglycerol kinase that regulates phospholipid synthesis and nuclear membrane growth. J. Biol. Chem. 2008, 283, 20433-20442.
9. Mrksich, M.; What can surface chemistry do for cell biology? Curr. Opin. Chem. Biol. 2002, 6, 794-797.
10. Zhang, S.; Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003, 21, 1171-1178.
11. Seeman, N. C.; DNA in a material world. Nature 2003, 421, 427-431.
12. Caruthers, M. H.; Gene synthesis machines: DNA chemistry and its uses. Science 1985, 230, 281-285.
13. Kocabey, S.; Kocabey, S.; Kempter, S.; List, J.; Xing, Y.; Bae, W.; Schiffels, D.; Shih, W. M.; Simmel, F. C.; Liedl, T.; Membrane-Assisted Growth of DNA Origami Nanostructure Arrays. ACS Nano. 2015, 9, 3530-3539.

DNA Replacement and Hybridization Chain Reaction on the Surface of Liposome Membrane
89
14. Todhunter, M.E.; Jee, N. Y.; Hughes, A. J.; Coyle, M. C.; Cerchiari, A.; Farlow, J.; Garbe, J. C.; LaBarge, M. A.; Desai, T. A.; Gartner, Z. J.; Programmed synthesis of three-dimensional tissues. Nat. Methods 2015, 12, 975-981.
15. Yoshina-Ishii, C.; Boxer, S.G.; Arrays of Mobile Tethered Vesicles on Supported Lipid Bilayers. J. Am. Chem. Soc. 2003,125, 3696-3697.
16. Jakobsen, U.; Simonsen, A.C.; Vogel, S.; DNA-Controlled Assembly of Soft Nanoparticles. J. Am. Chem. Soc. 2008, 130, 10462-10463.
17. Stengel, G.; Zahn, R.; Höök, F.; DNA-Induced Programmable Fusion of Phospholipid Vesicles. J. Am. Chem. Soc. 2007, 129, 9584-9585.
18. Hernández-Ainsa, S.; Ricci, M.; Hilton, L.; Aviñó, A.; Eritja, R.; Keyser, U. F.; Controlling the Reversible Assembly of Liposomes through a Multistimuli Responsive Anchored DNA. Nano Lett. 2016, 16, 4462-4466.
19. Rodríguez-Pulido, A.; Kondrachuk, A. I.; Prusty, D. K.; Gao, J.; Loi, M. A.; Herrmann, A.; Light-Triggered Sequence-Specific Cargo Release from DNA Block Copolymer–Lipid Vesicles. Angew. Chem. Int. Ed. 2013, 52, 1008-1012.
20. Czogalla, A.; Czogalla, A.; Kauert, D. J.; Franquelim, H. G.; Uzunova, V.; Zhang, Y.; Seidel, R.; Schwille, P.; Amphipathic DNA Origami Nanoparticles to Scaffold and Deform Lipid Membrane Vesicles. Angew. Chem. Int. Ed. 2015, 127, 6601-6605.
21. Burns, J. R.; Seifert, A.; Fertig, N.; Howorka, S.; A biomimetic DNA-based channel for the ligand-controlled transport of charged molecular cargo across a biological membrane. Nat. Nanotechnol. 2016, 11, 152-156.
22. Kurz, A.; Bunge, A.; Windeck, A.; Rost, M.; Flasche, W.; Arbuzova, A.; Strohbach, D.; Müller, S.; Liebscher, J.; Huster, D.; Herrmann, A.; Lipid-Anchored Oligonucleotides for Stable Double-Helix Formation in Distinct Membrane Domains. Angew. Chem. Int. Ed. 2006, 45, 4440-4444.
23. Anaya, M.; Kwak, M.; Musser, A. J.; Mullen, K.; Herrmann, A.; Tunable hydrophobicity in DNA micelles: design, synthesis, and characterization of a new family of DNA amphiphiles. Chem. Eur. J. 2010, 16, 12852-12859.
24. Meng, Z.; Yang, J.; Liu, Q.; de Vries J. W.; Gruszka, A.; Rodríguez-Pulido, A.; Crielaard, B J.; Kros, A.; Herrmann, A.; Efficient Fusion of Liposomes by Nucleobase Quadruple-Anchored DNA. Chem. Eur. J. 2017, DOI: 10.1002/chem.201701379.
25. Dirks, R. M.; Pierce, N. A.; Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275-15278.
26. Kwak, M.; Minten, I. J.; Anaya, D. M.; Musser, A. J.; Brasch, M.; Nolte, R. J. M.; Müllen, K.; Cornelissen, J. J. L. M.; Herrmann, A.;Virus-like Particles Templated by DNA Micelles: A General Method for Loading Virus Nanocarriers. J. Am. Chem. Soc. 2010, 132, 7834-7835.
27. Brody, J. R.; Kern, S. E.; Sodium boric acid: a Tris-free, cooler conductive medium for DNA electrophoresis. BioTechniques 2004, 36, 214-216.


Chapter 5
Performing DNA Nanotechnology Operations
on a Zebrafish Surface
This chapter has been submitted for publication.

Chapter 5
92
5.1 Introduction
Zebrafish is an ideal model vertebrate system with robust, external, and
transparent development allowing sophisticated imaging and experimental
techniques. In just a few days, embryos develop from single cells to
remarkably complex body structures and organ systems (Fig. 5.1).
Zebrafish embryos exhibit the great feature that they develop as "see
through" embryos, meaning that, all internal development can be clearly
observed from the outside in the living system.1
Fig. 5.1 Timeline of zebrafish embryonic development during 72 hours.
Fluorescent probes, such as synthetic organic dyes,2,3 fluorescent
proteins,4,5 and quantum dots,6-8 have been used extensively to monitor
biomolecules and biologically relevant species in vitro and in vivo. Due to
their transparent properties, zebrafish embryos have received great
attention for live vertebrate imaging due to the possibility to conduct high
resolution in vivo imaging. Compared to mammals, many structures and
processes are similar in zebrafish such as the brain and spinal cord.
Because of its outstanding suitability for imaging and transgenesis
approaches, the developing zebrafish has become a leading vertebrate
model for studies in drug discovery and a variety of human diseases.9,10 In
many cases, molecular imaging using fluorescent probes has been carried
out within cells. However, live cell imaging is not completely acceptable for
obtaining detailed information about the biological effects of analytes in a
tissue context. In contrast, images of the insides of live animals provide a
more informative view of these effects. Moreover, fluorescent imaging

Performing DNA Nanotechnology Operations on a Zebrafish Surface
93
studies using organisms that are genetically close to humans have become
highly attractive (Fig. 5.2).11
Fig. 5.2 Live cell and animal imaging using fluorescent probes. The trend for detection of
analytes has changed from live cell imaging to whole animal imaging.11
Owing to its small size, optical transparency, external fertilization and easy
manipulation, zebrafish are perhaps the most suitable vertebrate model
animals for in vivo imaging using fluorescent probes.
In this chapter, we implemented a membrane engineering related DNA
nanotechnology on the surface of a living animal. We investigated whether
it was possible to insert the lipid-modified DNA sequences into the
membrane of live zebrafish to function as an artificial receptor. Firstly, the
immobilization of membrane-anchor-functionalized oligonucleotides on a
zebrafish was demonstrated. Then, functionalization of protruding single-
stranded DNA atop the fish was realized by Watson-Crick base pairing
employing complementary DNA sequences. In this way, small molecules
and liposomes were guided and attached to the fish surface. The anchoring
process can be designed to be reversible allowing exchange of surface
functionalities by simple addition of DNA sequences. Finally, a DNA based
amplification process was performed atop of the zebrafish enabling the
multiplication of surface functionalities from a single DNA anchoring unit.

Chapter 5
94
5.2 Results and Discussion
5.2.1 DNA hybridization on a zebrafish surface and strand replacement
A DNA-based receptor can be used to modify the cell membrane with new
functions, such as for immobilization of surface probes or other payloads
for targeted delivery onto or through the lipid bilayer. For that purpose, 1
µM lipid-DNA U4T-18 was utilized and incubated with zebrafish embryos
of 1 day post-fertilization (dpf) for 1 h. Previously the group of Irvine12
performed cell membrane insertion of oligonucleotides carrying diacyl-
lipid (C18) or cholesterol anchors using an incubating time of 30 min.
Because electrostatic repulsion might delay incorporation into the
membrane, we prolonged the time for incubation of lipid-DNA with
zebrafish membrane to 1 h.
Subsequently, the 20mer oligonucleotide C594, which is partially
complementary to U4T-18 and contains the red emitting fluorophore
ATTO594, was added to hybridize to the lipid-modified surface anchor.
Fluorescence microscopy showed staining of the fish surface as evidenced
by the red emission (Fig. 5.3A) indicating that U4T-18 was successfully
incorporated into the fish skin and that the protruding single-stranded
DNA chain can selectively undergo sequence specific hybridization.
Next, we investigated if it is possible to dynamically exchange the red label
by a removal strand. This strategy of strand replacement was previously
introduced in the context of a DNA fueled molecular machine.13 Here, we
employed this strategy for the reversible and gentle labeling of the skin of a
living animal. Therefore, 2 µM 20-mer DNA oligonucleotide that is fully
complementary to C594 was introduced to peel off C594 from U4T-18. The
removal of C594 was proven by disappearance of red fluorescence on the
fish surface (Fig. 5.3B). After removal of C594, U4T-18 remained on the
skin and kept the ability to hybridize with other complementary DNA
sequence such as C488, a 14mer DNA oligonucleotide complementary to
U4T-18 and labeled with the green emitting fluorophore ATTO488. Clear
evidence for the strand replacement was the green fluorescence observed
on the exterior of the fish (Fig. 5.3C). Control experiments in which U4T-18
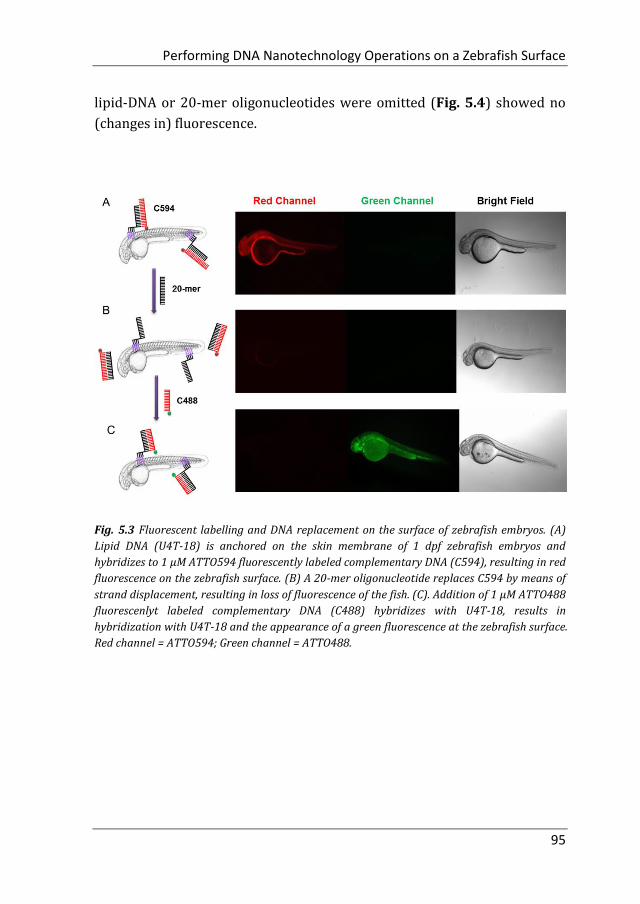
Performing DNA Nanotechnology Operations on a Zebrafish Surface
95
lipid-DNA or 20-mer oligonucleotides were omitted (Fig. 5.4) showed no
(changes in) fluorescence.
Fig. 5.3 Fluorescent labelling and DNA replacement on the surface of zebrafish embryos. (A)
Lipid DNA (U4T-18) is anchored on the skin membrane of 1 dpf zebrafish embryos and
hybridizes to 1 µM ATTO594 fluorescently labeled complementary DNA (C594), resulting in red
fluorescence on the zebrafish surface. (B) A 20-mer oligonucleotide replaces C594 by means of
strand displacement, resulting in loss of fluorescence of the fish. (C). Addition of 1 µM ATTO488
fluorescenlyt labeled complementary DNA (C488) hybridizes with U4T-18, results in
hybridization with U4T-18 and the appearance of a green fluorescence at the zebrafish surface.
Red channel = ATTO594; Green channel = ATTO488.

Chapter 5
96
Fig. 5.4 Fluorescence images of two control experiments: (A) untreated 2dpf zebrafish without
DNA anchoring unit was incubated with 1 µM ATTO594 labeled complementary DNA (C594)
for 1h. No fluorescence labelling was detected. (B) 2dpf zebrafish was decorated with U4T-18,
incubated with 1 µM C594 for 1h, washed three times with egg water, and subsequently
exposed to solution containing 1 µM ATTO488 labeled complementary DNA (C488). Although
the green labelled DNA was added to the fish, red fluorescence was detected on its surface. This
is due to lack of the removal strand. After all treatments of the fish with DNA, washing three
times with egg water was performed. Red channel = ATTO594; Green channel = ATTO488.
5.2.2 Loading larger containers to the zebrafish surface by Watson-
Crick base pairing
After demonstrating that base pairing is a very efficient tool to attach small
oligonucleotides to the live animal surface, we attempted to load larger
cargo to the zebrafish membrane. Therefore, phospholipid bilayer of
liposomes of 120 nm diameter was loaded with rhodamine-functionalized
phospholipid (Rh-DHPE), which is characterized by a red emission.
Likewise, the surface of the vesicles was decorated with lipid-modified
DNA that is complementary to that on the zebrafish (Fig. 5.5A). Proof of
successful loading of the fish surface by supramolecular bonds was
provided by fluorescence microcopy showing characteristic red
fluorescence originating from the liposomes, which are bound to the fish
surface (Fig. 5.5B). This result opens the way for potential DNA-mediated
delivery of liposomal cargo. These experiments demonstrate that the DNA
hybridization overcomes the repulsive hydration forces between the lipid
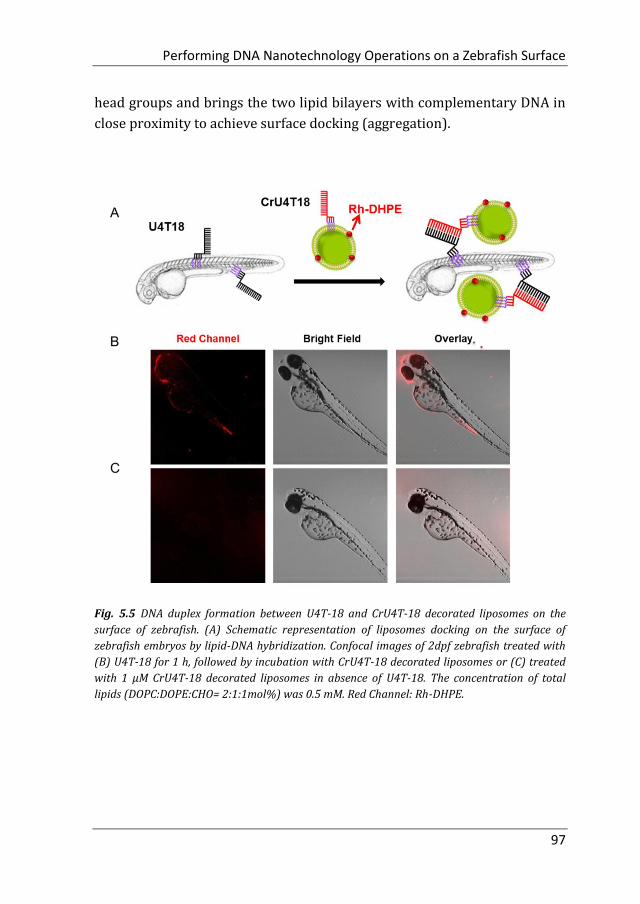
Performing DNA Nanotechnology Operations on a Zebrafish Surface
97
head groups and brings the two lipid bilayers with complementary DNA in
close proximity to achieve surface docking (aggregation).
Fig. 5.5 DNA duplex formation between U4T-18 and CrU4T-18 decorated liposomes on the
surface of zebrafish. (A) Schematic representation of liposomes docking on the surface of
zebrafish embryos by lipid-DNA hybridization. Confocal images of 2dpf zebrafish treated with
(B) U4T-18 for 1 h, followed by incubation with CrU4T-18 decorated liposomes or (C) treated
with 1 µM CrU4T-18 decorated liposomes in absence of U4T-18. The concentration of total
lipids (DOPC:DOPE:CHO= 2:1:1mol%) was 0.5 mM. Red Channel: Rh-DHPE.

Chapter 5
98
5.2.3 Nucleic acid mediated amplification process on live fish surface
To demonstrate the broad versatility of zebrafish surface engineering
enabled by lipid-DNA, we performed a DNA-based amplification process on
the animal, i.e. hybridization chain reaction (HCR). Previously, this method
was employed for augmenting the signal during nucleic acid detection.14
Later, this technique was utilized for surface modification with DNA
hydrogels.15 Here, we demonstrate that this supramolecular
polymerization can be performed on the exterior of the living animal.
Since initiation of HCR from a lipid membrane was not demonstrated
before we first established a HCR protocol for decorating the rim of
liposomes with a DNA layer (see chapter 4). Then, the optimized DNA
anchors and sequences were employed for modification of the fish surface
(Fig. 5.6). Compared to the previous experiments, for membrane anchoring
U4T-28, a 28mer lipid-DNA with 4 modified lipid bases, was bound to
zebrafish skin. Next, hairpin strands M1 (partially complementary to U4T-
28) and M2 (partially complementary to M1) were added (sequences see
chapter 4).16 Hybridization of M1 to U4T-28 results in liberation of its loop
that subsequently can hybridize with M2. Opening of the M2 hairpin
exposes a sequence that binds to a new M1 monomer from the solution. In
turn, opening of the M1 hairpin exposes a sequence that can bind new M2.
This effectively triggers the “supramolecular polymerization” of M1 and M2
with surface anchor U4T-28 as initiator, leading to extended DNA on the
zebrafish membrane (Fig. 5.6A). The realization of HCR was investigated
on the membrane of 1 dpf zebrafish embryo. As shown schematically in Fig.
5.6B, U4T-28 at the concentration of 1 µM was exposed to zebrafish
embryos for 1 h, followed by the incubation with a mixture of 2 µM M1-
FAM and 2 µM M2-Cy3 for 2 h. Both monomers were labeled with two
different fluorophores (FAM and Cy3). Green and red fluorescence could be
clearly observed due to progression of polymerization of M1-FAM and M2-
Cy3 from the initiator (Fig. 5.6C).

Performing DNA Nanotechnology Operations on a Zebrafish Surface
99
Fig. 5.6 DNA hybridization chain reaction (HCR) on the surface of zebrafish embryos. (A)
Schematic representation of DNA HCR on the surface of zebrafish embryos. a’, b’, and c’ are
regions that are complementary to regions a, b, and c, respectively. Hairpin M1 can be
unfolded by hybridization with initiator U4T-28, resulting in growing DNA strands. (B)
Addition of M1-FAM and M2-Cy3 to the U4T-28 pre-treated zebrafish resulted in DNA HCR and
concomitant increase in fluorescence. (C) Fluorescence images of 1 dpf zebrafish embryos after
incubation with U4T-28 for 1 h, and subsequent exposure to 1 µM M1-FAM and M2-Cy3 for 1 h.
Green channel: 6-FAM; Red channel: Cy3. (D) Normalized fluorescence intensity of attached
DNA on the surface of zebrafish embryos. Fluorescence intensities of images (C) and Fig.5.7A
were calculated by Image J and plotted as a percentage relative to the fluorescence of M1-FAM
or M2-Cy3 of Fig. 5.6C. The intensities of Fig. 5.6C were set to 100%.

Chapter 5
100
Two control experiments were performed to prove the HCR was initiated
on the membrane of zebrafish (Fig. 5.7). As shown in Fig. 5.7A, free M1-
FAM was washed away before the addition of M2-Cy3. In this case, the
fluorescent signals of M1-FAM and M2-Cy3 were 10 and 20 times lower,
respectively, than those obtained in the presence of HCR (Fig. 5.6D). Also,
when the monomer M1-FAM was omitted the HCR could not proceed and
consequently no fluorescence could be detected on the fish surface (Fig
5.7B).
Fig. 5.7 HCR on zebrafish skin depends on DNA hybridization between U4T-28, M1 and M2.
Fluorescence images of 1 dpf zebrafish (A) that were first incubated with U4T-28 and M1-FAM
for 2 hours, subsequently washed 3 times with egg water before the addition of M2-Cy3, or (B)
similar to (A) but without M1-FAM. Green channel: 6-FAM; Red channel: Cy3.
The signal increase by HCR was also clearly demonstrated by an
experiment involving non-fluorescent M2 and M1-FAM. In case of HCR
approximately 10-fold stronger green fluorescence was observed on the
surface of 2 dpf zebrafish embryos (Fig. 5.8A) compared to labeling with a
single fluorophore per anchor unit (Fig. 5.8B).

Performing DNA Nanotechnology Operations on a Zebrafish Surface
101
Fig. 5.8 In vivo DNA HCR enhances the fluorescence intensity of labeling. (A) Fluorescence
images of 2dpf zebrafish embryos that were first decorated with U4T-28, followed by 3 times
washing with egg water, incubation with M1-FAM for 1 h and M2 for another 1 h. (B) Only
FAM fluorescent labeled M1 (M1-FAM) was added after anchoring of U4T28 on the fish and
washing three times with egg water. Green channel: M1-FAM.
5.3 Conclusion
Previously, oligonucleotides were covalently attached to live cells by
metabolic oligosaccharide engineering allowing the introduction of
orthogonal chemical handles on the cell surface for DNA anchoring without
dependence on endogenous receptors.17 Besides oligosaccharides, cell-
surface proteins were exploited for the chemical modification of cells with
DNA.18,19 Similarly, cell-surface proteins were decorated with DNA by non-
covalent interactions.20 An alternative strategy for introducing artificial
DNA receptors on live cell surfaces represents the utilization of
oligonucleotides carrying hydrophobic membrane anchors, as described in
this study.21 Based on such an anchoring strategy, Watson-Crick base
pairing was exploited for the programmed synthesis of three-dimensional
tissues.22 The examples above demonstrate that anchored DNA in a lipid
bilayer developed into a powerful tool for realizing exciting functionalities
in the context of synthetic and natural membranes, even including live cells.
On the other hand, DNA nanostructures were employed in higher
organisms in the context of functional in-vivo imaging23 and for the

Chapter 5
102
targeted delivery of siRNA.24 To the best of our knowledge, there is no
experiment about DNA-based membrane engineering in a living animal
involving a wide variety of functions yet.
In this chapter, we demonstrated that lipid-DNA sequences with four
anchoring units could be readily incorporated in the surface layer of
zebrafish embryos. The single-stranded DNA present on the surface can be
functionalized by Watson-Crick base pairing enabling the sequence specific
functionalization of the live animal with small molecules or larger cargos,
for example, liposomal systems. The payloads connected by the
supramolecular tether DNA can be reversibly removed employing a
removal strand, which represents a very mild stimulus just requiring the
addition of a DNA sequence not affecting the life of the fish. Finally, we
successfully demonstrated the performance of a DNA mediated
amplification process on the fish skin. The hybridization chain reaction
allows attachment of multiple moieties on a single anchored DNA strand
allowing multiplication of cargoes or signals on the surface. Moreover, it
was shown that surface modification of model membranes in form of
liposomes by various DNA nanotechnology procedures could be easily
transferred to the live animal. This allows establishment of DNA based
surface functionalization procedures and their facile and fast
implementation in zebrafish. However, challenges in the application of
lipid-DNA in the transition of zebrafish to higher mammals remain. For
instance, the DNA part in lipid-DNA is susceptible to the deoxyribonuclease
in the circulation system of mammals. On the other hand, unlike zebrafish,
mammals are not clear and transparent, which makes the direct
visualization of fluorescently labeled tissues impossible. Nevertheless, due
to the broad application of zebrafish as animal model in drug development,
toxicology and nanoparticles characterization,25 we believe that the
platform presented here allows amalgamation of DNA nanotechnology
tools with live animals and enables efficient bio-barcoding as well as in vivo
tracking.

Performing DNA Nanotechnology Operations on a Zebrafish Surface
103
5.4 Experiment Section
5.4.1 Materials
Cholesterol (Chol), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)
and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased
from Avanti Polar Lipids (Alabaster, USA) (purity >99%) and used without
further purification. Headgroup-labeled phospholipid, lissamine rhodamine
B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (triethylammoni
-um salt) (Rh-DHPE) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-
dihexadecanoyl-sn-glycero-3-phosphoethanolamine (triethylammonium
salt) (NBD-DHPE) were purchased from Invitrogen (Amsterdam,
Netherlands), and used as received. The DNA-dye conjugates C488, M1-
FAM and M2-Cy3 were purchased from Biomers.net GmbH (Ulm, Germany).
Anhydrous CHCl3 was purchased from Acros Organics (Geel, Belgium) and
stored over molecular sieves.
5.4.2 Zebrafish strain, husbandry, and egg collection
Wildtype zebrafish were used in this study, and were maintained and
handled according to the guidelines from http://zfin.org. Fertilization was
performed by natural spawning at the beginning of the light period, and
eggs were raised at 28 °C. All experimental procedures were conducted in
compliance with the directives of the animal welfare committee of the
Leiden University.
5.4.3 Microscopy images
Zebrafishes were seeded in a glass bottom flask with egg water. After
incubation for 1 h with lipid-DNA, they were washed three times and then
incubated with other DNA oligonucleotides for 1 h. For live imaging,
zebrafish embryos were anaesthetized with 0.003% tricaine and mounted
on 0.6% low-melting agarose. Fluorescent images were acquired using a
Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems,
Wetzlar, Germany) and merged with Leica application suite advanced
fluorescence software (Leica Microsystems) or ImageJ software (National
Institutes of Health, Bethesda, MD, USA). A Leica MZ16FA stereo
microscope was used for stereo images. Images were adjusted for

Chapter 5
104
brightness and contrast using ImageJ. The wavelength settings for C488
were Ex/Em: 495/520nm (Ex laser: 488 nm), for C594 Ex/Em: 601/627
nm (Ex laser: 532 nm), for M1-FAM Ex/Em: 494/518 nm (Ex laser: 488
nm), and M2-Cy3 Ex/Em: 550/570 nm (Ex laser: 532 nm).
Author contributions
Yang J conducted the experiments and performed data analysis. Yang J and
Meng Z designed the experiments and prepared the manuscript. Liu Q
synthesized lipid-DNA. Yasuhito S produced the zebrafish embryo. Herman
S and René CLO interpreted the data. Kros A and Herrmann A supervised
the project. All authors edited the manuscript.

Performing DNA Nanotechnology Operations on a Zebrafish Surface
105
References
1. Kimmel, C. B.; Sepich, D. S.; Trevarrow B.; Development of segmentation in zebrafish. Development 1988, 104, 197-207.
2. Shafizadeh, E.; Huang, H.; Lin, S.; Transgenic zebrafish expressing green fluorescent protein. Methods Mol. Biol. 2002, 183, 225–223.
3. Ju, B.; Xu, Y.; He, J.; Liao, J.; Yan, T.; Hew, C. L.; Lam, T. J.; Gong, Z.; Faithful expression of green fluorescent protein (GFP) in transgenic zebrafish embryos under control of zebrafish gene promoters. Dev. Genet. 1999, 25, 158–167.
4. Farber, S. A.; Pack, M.; Ho, S. Y.; Johnson, I. D.; Wagner, D. S.; Dosch, R.; Mullins, M. C.; Hendrickson, H. S.; Hendrickson, E. K.; Halpern, M. E.; Genetic analysis of digestive physiology using fluorescent phospholipid reporters. Science 2001, 292, 1385–1388.
5. Laughlin, S. T.; Baskin, J. M.; Amacher, S. L.; Bertozzi, C. R.; In vivo imaging of membrane-associated glycans in developing zebrafish. Science 2008, 320, 664–667.
6. Dubertret, B.; Skourides, P.; Norris, D. J.; Noireaux, V.; Brivanlou, A. H.; Libchaber, A.; In vivo imaging of quantum dots encapsulated in phospholipid micelles. Science 2002, 298, 1759–1762.
7. Rieger, S.; Kulkarni, R. P.; Darcy, D.; Fraser, S. E.; Koster, R. W.; Quantum dots are powerful multipurpose vital labeling agents in zebrafish embryos. Dev. Dyn. 2005, 234, 670–681.
8. Son, S. W.; Kim, J. H.; Kim, S. H.; Kim, H.; Chung, A. Y.; Choo, J. B.; Oh, C. H.; Park, H.C.; Intravital imaging in zebrafish using quantum dots. Skin Res Technol. 2009, 15, 157–160.
9. Santana, S.; Rico, E. P.; Burgos. J. S.; Can zebrafish be used as animal model to study
Alzheimer's disease? Am J Neurodegener Dis. 2012, 1, 32-48.
10. Yang, J.; Shimada, Y.; Rene, C. L.; Olsthoorn, B.; Ewa Snaar-Jagalska, Herman P. Spaink, Alexander Kros, Application of Coiled Coil Peptides in Liposomal Anticancer Drug Delivery Using a Zebra fish Xenograft Model. ACS Nano. 2016, 10, 7428−7435.
11. Ko, S.; Chen, X,; Yoon, J.; Shin, I.; Zebrafish as a good vertebrate model for molecular imaging using fluorescent probes. Chem. Soc. Rev. 2011,40, 2120–2130.
12. Liu, H.; Kwong, B.; Irvine, D. J.; Membrane anchored immunostimulatory oligonucleotides for in-vivo cell modification and localized immunotherapy. Angew. Chem. Int. Ed. 2011, 50, 7052-7055.
13. Yurke, B.; Turberfield, A. J.; Mills, A. P.; Simmel, F. C.; Neumann, J. L.; A DNA-fuelled molecular machine made of DNA. Nature 2000, 406, 605-608.

Chapter 5
106
14. Dirks, R. M.; Pierce, N. A.; Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275-15278.
15. Kahn, J. S. Trifonov, A.; Cecconello, A.; Guo, W.; Fan, C.; Willner, I.; Integration of Switchable DNA-Based Hydrogels with Surfaces by the Hybridization Chain Reaction. Nano Lett. 2015, 15, 7773-7778.
16. Dirks, R. M.; Pierce, N. A.; Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275-15278.
17. Chandra, R. A.; Douglas, E. S.; Mathies, R. A.; Bertozzi, C. R.; Francis, M. B.; Programmable Cell Adhesion Encoded by DNA Hybridization. Angew. Chem. Int. Ed. 2006, 45, 896-901.
18. Hsiao, S. C.; Shum, B. J.; Onoe, H.; Douglas, E. S.; Gartner, Z. J.; Mathies, R. A.; Bertozzi, C. R.; Francis, M. B.; Direct Cell Surface Modification with DNA for the Capture of Primary Cells and the Investigation of Myotube Formation on Defined Patterns. Langmuir 2009, 25, 6985-6991.
19. Zhao, W.; Loh, W.; Droujinine, I. A.; Teo, W.; Kumar, N.; Schafer, S.; Cui, C. H.; Zhang, L.; Sarkar, D.; Karnik, R.; Karp, J. M.; Mimicking the inflammatory cell adhesion cascade by nucleic acid aptamer programmed cell-cell interactions. FASEB J 2011, 25, 3045-3056.
20. Bailey, R. C.; Kwong, G. A.; Radu, C. G.; Witte, O. N.; Heath, J. R.; DNA-Encoded Antibody Libraries: A Unified Platform for Multiplexed Cell Sorting and Detection of Genes and Proteins. J. Am. Chem. Soc. 2007, 129, 1959-1967.
21. Selden, N. S.; Todhunter, M. E.; Jee, N. Y.;Liu, J. S.; Broaders, K. E.; Gartner, Z. J.; Chemically Programmed Cell Adhesion with Membrane-Anchored Oligonucleotides. J. Am. Chem. Soc. 2012, 765-768.
22. Todhunter, M. E.; Jee, N. Y.; Hughes, A. J.; Coyle, M. C.; Cerchiari, A.; Farlow, J.; Garbe, J. C.; LaBarge, M. A.; Desai, T. A.; Gartner, Z. J.; Programmed synthesis of three-dimensional tissues. Nat Methods. 2015,12, 975-981.
23. Bhatia, D.;Surana, S.; Chakraborty, S.; Koushika, S. P.; Krishnan, Y.; A synthetic icosahedral DNA-based host-cargo complex for functional in vivo imaging. Nat Commun. 2011, 2, 339.
24. Lee, H.; Lytton-Jean, A. K. R.; Chen, Y.; Love, K. T.; Park, A. I.; Karagiannis, E. D.; Sehgal, A.;Querbes, W.; Zurenko, C. S.; Jayaraman, M.; Peng, C. G.; Charisse, K.; Borodovsky, A.; Manoharan, M.; Donahoe, J. S.; Truelove, J.; Nahrendorf, M.; Langer, R.; Anderson, D. G.; Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012, 7, 389-393.
25 Evensen, L.; Johansen, P. L.; Koster, G.; Zhu, K.; Herfindal, L.; Speth, M.; Fenaroli, F.; Hildahl, J.; Bagherifam, S.; Tulotta, C.; Prasmickaite, L.; Mælandsmo, G. M.; Snaar-Jagalska, E.; Griffiths, G.; Zebrafish as a model system for characterization of nanoparticles against cancer. Nanoscale 2016, 8, 862-877.
https://www.ncbi.nlm.nih.gov/pubmed/?term=LaBarge%20MA%5BAuthor%5D&cauthor=true&cauthor_uid=26322836

Summary

Summary
108
Summary
Great efforts have been dedicated to use DNA as a building block in
nanotechnology because of its versatile properties, such as high specificity
and programmability to form complex structures. The rapid development
of chemical methodology has been proven to be useful not only for the
preparation of pristine DNA and RNA, but also to make base, or backbone
modifications of the oligonucleotides as well as many analogues. The
structural variety has been further expanded from 2D-patterns or 3D-nano-
objects, which are basically fabricated with pristine DNA, to DNA hybrids,
such as DNA-nanoparticle hybrids including inorganic or soft materials.
Specially, the phosphoramidite chemistry has been adapted for the
modification of DNA with hydrophobic polymers or lipids, yielding DNA
amphiphiles. These DNA amphiphiles can be used to perform surface
modifications of biological membranes.
In Chapter 2, we have established a new strategy for anchoring
oligonucleotides in vesicle membranes enabled by attaching hydrophobic
units to the nucleobase. The membrane anchors are incorporated into the
oligonucleotide by automated solid phase synthesis allowing precise
control over the position and number of hydrophobic units within a DNA
sequence. A FRET system was used to prove the incorporation and stability
of lipid-DNA on the liposomal membrane. Single-stranded DNA
functionalized with four lipid-modified nucleobases was stably grafted
onto the membrane of lipid vesicles for at least 24 hours.

Summary
109
After demonstrating the stability of lipid-DNA in the liposomal bilayer,
lipid-DNA induced liposome fusion was studied in Chapter 3. The result
demonstrates the importance of the DNA-anchoring strategy in
hybridization-induced vesicle fusion, as not only the structural properties
of the unit itself, but also the number of anchoring units determine the
fusogenic properties. It was found that the orientation of DNA
hybridization and the number of anchoring units played a crucial role in
liposomal fusion. The zipper-orientated hybridization is more efficient than
non-zipper-orientated hybridization, which supports that double-stranded
DNA close to the vesicle surface could bring the docked vesicles in close
proximity to enhance full fusion. In the zipper orientation, the
hybridization event including vesicles with complementary sequences
brings their membranes in close proximity than in the non-zipper
configuration. Meanwhile, compared to vesicles functionalized with single-
anchored or double-anchored DNA, liposomes containing quadruple-
anchored oligonucleotides were proved to be highly fusogenic, achieving
considerable full fusion of up to 29% without notable leakage, which might
be related to the higher affinity of a quadruple lipid anchor to the
membrane. With this fusion system the most efficient fusogenic DNA
probes were produced known to date.
To further extend the functionality of DNA-based vesicles, the study
presented in Chapter 4 was focused on the functionalization of DNA
amphiphiles in a phospholipid bilayer. We explored DNA hybridization and
the dynamic exchange of DNA sequences on the surface of liposomes by
simple addition of DNA sequences with two FRET systems. FRET between
C594 (acceptor) and U4T-18-grafted Rh-DHPE-containing (donor) vesicles
demonstrated that DNA hybridization was achieved on the surface of
liposome. Subsequently, a 20-mer DNA oligonucleotide was introduced to
replace U4T-18 from C594 due to full hybridization. The disassembly of
C594 from the vesicle surface was realized since C594 was attached to the
liposome employing 14 complementary nucleotides while the removal
strand formed a duplex involving 20 nucleotides. The hybridization energy
for the latter is larger than for a 14mer duplex structure. Afterwards, the
free U4T-18 hybridized with C488, which is a donor for Rh-DHPE-
containing vesicles. Moreover, a DNA based amplification process was

Summary
110
performed on the surface of liposomes with a DNA probe, M2-Cy3. A
remarkable fluorescence intensity of Cy3 was obtained due to the DNA
hybridization chain reaction, which confirmed the capability of the
multiplication of surface functionalities from a single DNA anchoring unit
on the vesicle surface.
At the end of the thesis, a more complex membrane system was
functionalized with DNA. In Chapter 5, the incorporation of nucleobase
quadruple-anchored DNA in the surface layer of zebrafish embryos was
evaluated. The payloads connected by the supramolecular tether DNA can
be reversibly removed employing a removal strand, which represents a
very mild stimulus just requiring the addition of a DNA sequence not
affecting the life of the fish. Similar as on the vesicle surface, we
successfully demonstrated the performance of a DNA mediated
amplification process on the fish skin. The hybridization chain reaction
allows attachment of multiple moieties on a single anchored DNA strand
allowing multiplication of cargoes or signals on the surface. Moreover, it
was shown that surface modification of model membranes in form of
liposomes by various DNA nanotechnology procedures could be easily
transferred to the live animal. This allows establishment of DNA based
surface functionalization procedures and their facile and fast
implementation in zebrafish. Due to the broad application of zebrafish as
animal model in drug development, toxicology and nanoparticles
characterization in living systems, we believe the platform presented here
allows amalgamation of DNA nanotechnology tools with live animals and
enables efficient bio-barcoding as well as in vivo tracking.
Overall, this thesis has shown that chemical synthesis is a valuable tool to
produce functional DNA molecules to increase the complexity of membrane
engineering approaches. This was achieved by modification of nucleobases
by hydrophobic units and solid phase synthesis to fabricate amphiphilic
DNA strands. The piercing of hydrophobic units on the DNA into the inner
part of phospholipid membranes leads to stable anchoring. With this as a
starting point, the dynamic process of vesicle fusion was achieved, which
might be important in the future for synthesizing minute amount of
compounds by exploiting content mixing of liposomes filled with different

Summary
111
reactants. Similarly, the fusion approach might be utilized in the context of
actively transporting vesicle payloads in cells. In this case, the DNA
hybridization will fuel the fusion of vesicle with cell membranes to deliver
cargoes in the cytosol without being dependent on endocytosis processes.
One could even think of transferring this concept to live animals as
demonstrated in this thesis for zebrafish. It is without any doubt that DNA
amphiphiles bear lots of future potential for sophisticated DNA
nanotechnology functions in the realm of synthesis biology.


Samenvatting

Samenvatting
114
Samenvatting
Grote inzet is toegewijd aan het gebruik van DNA als bouwsteen in de
nanotechnologie door zijn veelzijdige eigenschappen, zoals hoge
specificiteit en programmeerbaarheid om complexe structuren te vormen.
De snelle ontwikkeling van chemische methodologie is bewezen nuttig te
zijn, niet alleen voor de bereiding van ongerepte DNA en RNA, maar ook om
basen- of ruggengraatmodificaties van de oligonucleotiden evenals vele
analogen te maken. De structurele verscheidenheid is verder uitgebreid
van 2D-patronen of 3D-nano-objecten, welke hoofdzakelijk zijn
vervaardigd met ongerept DNA, naar DNA-hybriden, zoals hybriden van
DNA-nanodeeltjes waaronder anorganische of zachte materialen. In het
bijzonder is de fosforamidiet-chemie aangepast voor de modificatie van
DNA met hydrofobe polymeren of lipiden, waardoor DNA-amphiphiles
worden verkregen. Deze DNA-amphiphiles kunnen worden gebruikt om
oppervlakte modificaties op biologische membranen uit te voeren.
In Hoofdstuk 2 hebben we een nieuwe strategie vastgesteld voor het
verankeren van oligonucleotiden in vesikelmembranen door middel van
hydrofobe eenheden aan de nucleobase te bevestigen. De membraanankers
worden in de oligonucleotide geïncorporeerd door geautomatiseerde
vastefasesynthese waardoor nauwkeurige controle over de positie en het
aantal hydrofobe eenheden binnen een DNA-sequentie mogelijk is. Een
FRET-systeem werd gebruikt om de integratie en stabiliteit van lipide-DNA
op het liposomale membraan te bewijzen. Enkelstrengs DNA

Samenvatting
115
gefunctionaliseerd met vier lipide-gemodificeerde nucleobasen was
gedurende ten minste 24 uur stabiel geënt op het membraan van
liposomen.
Nadat de stabiliteit van lipide-DNA in de liposomale dubbellaag was
aangetoond, werd lipide-DNA geïnduceerde liposoomfusie bestudeerd in
Hoofdstuk 3. Het resultaat laat het belang zien van de DNA-
verankeringsstrategie bij hybridisatie geïnduceerde vesikelfusie, aangezien
niet alleen de structurele eigenschappen van de eenheid zelf, maar ook het
aantal verankeringseenheden de fusogene eigenschappen bepalen. Er werd
gevonden dat de oriëntatie van DNA hybridisatie en het aantal
verankeringseenheden een cruciale rol speelden bij liposomale fusie. De
rits-georiënteerde hybridisatie is efficiënter dan de niet-rits georiënteerde
hybridisatie, wat ondersteunt dat dubbelstrengs DNA vlakbij het
vesikeloppervlak de verbonden vesikels in nabijheid zou kunnen brengen
om volledige fusie te verbeteren. In de rits-oriëntatie brengt de
hybridiseringsgebeurtenis vesikels met complementaire sequenties in hun
membranen dichter bij elkaar dan in de non-ritsconfiguratie. In
tegenstelling tot vesikels welke gefunctionaliseerd zijn met enkelvoudig
verankerd of dubbel verankerd DNA, bleken liposomen welke viervoudig-
verankerde oligonucleotiden bevatten zeer fusogeen zijn, waardoor een
aanzienlijke volledige fusie tot 29% werd bereikt zonder opmerkelijke
lekkage, wat mogelijk verband houdt met de hogere affiniteit van een
viervoudige-lipide anker in het membraan. Met dit fusiesysteem werden de
meest efficiënte fusogene DNA probes tot nu toe geproduceerd.
Om de functionaliteit van DNA-gebaseerde vesikels verder uit te breiden,
was de studie in Hoofdstuk 4 gericht op de functionalisatie van DNA-
amphiphiles in een fosfolipide dubbellaag. We onderzochten DNA
hybridisatie en de dynamische uitwisseling van DNA sequenties op het
oppervlak van liposomen door middel van de toevoeging van DNA
sequenties met twee FRET systemen. FRET tussen C594 (acceptor) en
U4T18-geënte Rh-DHPE-bevattende (donor) vesikels toonde aan dat DNA
hybridisatie op het liposoomoppervlak werd bereikt. Vervolgens werd een
20-mer DNA oligonucleotide geïntroduceerd om U4T18 van C594 te
vervangen door volledige hybridisatie. De demontage van C594 uit het

Samenvatting
116
vesikeloppervlak werd gerealiseerd aangezien C594 aan het liposoom was
verbonden met 14 complementaire nucleotiden, terwijl de
verwijderingsstreng een duplex vormde waarbij 20 nucleotiden betrokken
waren. De hybridisatie-energie voor deze laatste is groter dan voor een 14-
mer duplexstructuur. Daarna werd de vrije U4T18 gehybridiseerd met
C488, welke een donor is voor Rh-DHPE-bevattende vesikels. Verder werd
een DNA-gebaseerd amplificatieproces uitgevoerd op het oppervlak van
liposomen met een DNA-probe, M2-Cy3. Een opmerkelijke fluorescentie-
intensiteit van Cy3 werd verkregen door de DNA-hybridisatie ketenreactie,
welke de mogelijkheid van vermenigvuldiging van oppervlaktefuncties uit
een enkele DNA-verankeringseenheid op het vesikeloppervlak bevestigde.
Aan het eind van het proefschrift werd een complexer membraansysteem
gefunctionaliseerd met DNA. In hoofdstuk 5 werd de opname van
nucleobase viervoudig-verankerd DNA in de oppervlaktelaag van zebravis
embryo's geëvalueerd. De lading verbonden door de supramoleculaire
DNA-ketting kunnen omkeerbaar verwijderd worden door gebruik te
maken van een verwijderingsstreng, welke door enkel de toevoeging van
een DNA-sequentie een zeer milde stimulus vertegenwoordigt wat de
levensduur van de vis niet beïnvloedt. Net als op het vesikeloppervlak,
hebben we met succes de prestatie van een DNA gemedieerde
amplificatieproces op de vissenhuid aangetoond. De
hybridisatieketenreactie maakt het mogelijk om meerdere delen op een
enkele verankerde DNA-streng toe te voegen, waardoor vermenigvuldiging
van ladingen of signalen op het oppervlak mogelijk is. Bovendien toonde
het aan dat oppervlakmodificatie door verschillende DNA-nanotechnologie
procedures aan modelmembranen in de vorm van liposomen gemakkelijk
op het levende dier kon worden overgedragen. Dit maakt het mogelijk om
DNA-gebaseerde oppervlakfunctionalisatie procedures en hun eenvoudige
en snelle implementatie in zebravis te bepalen. Door de brede toepassing
van zebravis als diermodel in de ontwikkeling van geneesmiddelen,
toxicologie en nanodeeltjes karakterisatie in levende systemen, geloven wij
dat het hier gepresenteerde platform de combinatie van DNA-
nanotechnologie met levende dieren in staat stelt en efficiënte bio-
barcoding en in-vivo tracking mogelijk maakt.

Samenvatting
117
Globaal gezien heeft dit proefschrift aangetoond dat chemische synthese
een waardevol hulpmiddel is om functionele DNA-moleculen te produceren
om de complexiteit van membraan aanpassingsbenaderingen te verhogen.
Dit werd bereikt door modificatie van nucleobasen door hydrofobe
eenheden en vastefase synthese om amfifiele DNA strengen te fabriceren.
Het doordringen van hydrofobe eenheden op het DNA in het binnenste
gedeelte van fosfolipide membranen leidt tot stabiele verankering. Hiermee
werd het dynamische proces van vesikelfusie bereikt, wat in de toekomst
belangrijk kan zijn voor het synthetiseren van kleine hoeveelheden door
het gebruik van de inhoudsmenging van liposomen gevuld met
verschillende reactanten te exploiteren. Op dezelfde manier zou de
fusiebenadering gebruikt kunnen worden in de context van het actief
transporteren van beladen vesikels in cellen. In dit geval zal de DNA
hybridisatie leiden tot de fusie van vesikels met celmembranen om
ladingen in het cytosol te leveren zonder afhankelijk te zijn van
endocytische processen. Men zou zelfs kunnen denken over het
overbrengen van dit concept op levende dieren, zoals reeds aangetoond in
dit proefschrift voor zebravissen. Het is zonder twijfel dat DNA-amfifielen
een groot toekomstpotentieel hebben voor geavanceerde DNA-
nanotechnologie functies op het gebied van synthetische biologie.


Acknowledgements

Acknowledgements
120
Acknowledgements
So happy to finish this thesis! 2017 has been the most important year to me
up to now. This is not only because I finished my PhD study in Groningen
University but also a lovely new member joined our family, my lovely
daughter Ruby. Looking back to the past five years, I am filled with all sorts
of feelings and memories. It would not be possible to finish this thesis
without the help of many kind people surrounding me. I would like to
gratefully acknowledge those who have contributed to this thesis and
supported me during my PhD study.
First and foremost, I would like to give my deepest gratitude to my
supervisor, Prof. Andreas Herrmann, who gave me the opportunity to
conduct PhD study in Netherlands and be a part of our group! I still
remember how excited I was when I received the offer letter from you. I
learned a lot from the discussion with you in your office, where you always
patiently explained the ideas and mechanisms to me. I learned a lot from
your broad knowledge, inspiring ideas and enthusiasm in scientific
researches. Also, I would like to thank you for your encouragement which
was supportive for my first two years when I was painful and lost because
of the failing results. And I also remember how delighted and relieved I was
when I finally got the fruitful time and finished all the manuscripts. Those
experiences trained me to be a person with independent thought and an
open mind which is helpful for my future study and work. Also thank you

Acknowledgements
121
so much for offering me the researcher contract when I was pregnant,
which was a tremendous help to our life and study.
I also would like to thank the members of the reading committee: Prof. S.
Vogel, Prof. A. M. van Oijen, and Prof. D. J. Slotboom for the time and the
evaluation on my thesis. I want to express my special thanks to you for
your valuable comments which helped me improve the manuscript.
I am deeply grateful to Prof. Alexander Kros in Leiden University. It was a
great experience and fruitful collaboration with your group. Thank you
very for your patience and dedicated time on the experimental design, data
analysis and paper writing. At the meantime, I would like to thank your
group member: Dr. Jian Yang. Your great efforts helped me on my work
and I learned a lot from the work together with you.
I would like to thank other collaborators involved in my projects. Prof. B.
Poolman, Prof. A. Kocer, Prof. S. J. Marrink, Prof. A.M. van Oijen, Dr.
Duygu, Dr. Gemma and Rianne. I am really grateful to all of you for the
valuable discussions on my projects, which helped a lot to improve my
scientific work. I want to thank Jelle for dedicated time to do TIRFM
measurements together with me. The discussions with you are always
inspiring and joyful.
Also, I take this opportunity to thank China Scholarship Council (CSC).
Thanks for the scholarship for my four years’ PhD study, which gives me
the opportunity to go abroad and study. Meanwhile, I would like to show
my gratitude to my Alma mater, Zhengzhou University (ZZU), which gave
me full support when I applied for the CSC scholarship. So far as I know,
ZZU is the only University who establishes a free English course for CSC
applicants and pays for their IELTS or TOEFL test fees. To help us better
understand and communicate with foreign universities, ZZU encouraged
and subsidized us to go to Beijing to join the CSC meeting. When I went
abroad and started my PhD study, I got a suitcase form ZZU like a gift which
is full of encouragement and expectation. Thanks to Prof. K. Y. Tang and
Prof. X. J. Zheng in Zhengzhou University. Both of you gave me great
support during my application of CSC scholarship and allowed me to have
my Master defense ahead of schedule, which made my PhD study abroad

Acknowledgements
122
possible. I am very grateful to both of you for your encouragement and
comfort when I felt confused and loss about my PhD study during the first
two years.
Thanks to everyone in the PCBE group for being always nice and helpful.
Very special thanks to Bart for your valuable discussions and dedicated
time on my manuscript polishing. Karin, many thanks to you for your help
with all the paper work. I remember last time I gave you my claim for the
stay in Leiden but found two receipts were lost. I was going to pay by
myself since it was just 30 euro. But you told me that a student shouldn’t
pay for that and you would help me argue for it if the finance department
questioned me, which made me feel fully supported! Ursula, thanks so
much for your efforts on the submission of my thesis. Without your help, it
cannot go so smooth! And you are so efficient and enthusiastic. Every time
I go to your office and ask if you can help me do something, you always
gave me positive answers! Wish you will have a wonderful stay in China!
Special thanks to Evgeny for your invaluable technical support on my
project.
Special thanks are given to Alberto, my first daily supervisor. Thanks for
your patience and understanding with me when my English was so poor to
communicate. Your Spanish enthusiasm made me feel warmly welcomed
and thanks so much for teaching me everything, not only the experimental
operation, but also the data analysis and PowerPoint design during my first
days in our lab. And also thank you and your wife for your warm hospitality
when Qing and I were in Madrid. Agnieszka, you are a “walking heater” to
me. When I was lost and had no idea about writing the introduction of my
thesis, you helped me clarify my thoughts and pinpoint the right direction.
You gave so much help to my experiments and my personal life. Thanks for
your delicious Polish food, the cute sweater to my little Ruby and the
wonderful “girls’ night” in your sweet apartment. Jan Willem, you are
always positive, smiling and helping everyone. You took me to the stationer
to get all the stationery and taught me how to use the instruments in our
lab. Whenever I got a problem and came to you, you always sat down and
answered my questions. Alina, so many thanks for your helping my

Acknowledgements
123
experiments and you showed me a different perspective to look into the life.
I remember when I told you I didn’t want to use EtBr because of its toxicity
and you said you can do it for me if I was so worried. To be honest, I was
shocked because I would never do something like that. I really enjoyed all
the fun time with you in and out of the lab, like the first time for me to have
a relaxing time in a hot spring and we went to the painting shop to find a
dyestuff to cover the flaw on my door. Thanks also to Alessio and Diego for
your discussions and fun time in the lab and office. Dear Mark, thanks a lot
for your efforts on my thesis and your kindly help in Qing’s and my life, like
making a Dutch phone call or reading a Dutch letter. Wish you find a
girlfriend ASAP! Eliza, it’s so nice to have you in life! You gave me so many
advices when I needed your help like our trip to Poland. We had a
wonderful journey with your detailed suggestions! Hongyan, thanks very
much to be my Paranymph and it’s surprisingly good to live together with
you for the last two months! Wish you will have more high quality papers
and find your soulmate! Wei, Jun & Pei, Avishek and Pavlo, you are
always willing to help me in my experiments.
My great thanks go to Jingyi for all your warm help during my five years
life in Groningen. Whenever I meet a problem, you are the first person I
prefer to ask for help because I know I will get detailed suggestions and
comfort from you. Countless delicious dishes and food were taken in your
apartment with your excellent cooking skill. I would never forget the good
time when you, Shuo, Qing and I watched the Chinese show in your giant
TV and enjoyed the hotpot in the cold winters. I always feel the quality of
my life was reduced since you got pregnant… I wish Shou & you all the best
and every success! Lei, I really appreciate your efforts on our collaborated
experiments. You are open to share your working experiences and it’s very
nice to work together with you. There is so much fun time with you in and
out of the lab, like we went to Paris for holiday together. We were worried
to get robbed in Sacré-Cœur and discussed so many notes on security. And
finally found we were over-worried and had a nice time there. Wish
Zhongtao and your work went well in China.
I sincerely like to express my appreciations to Lifei and Kai. Both of you are
like “second professors” to me. Lifei, your rich specialty base and multi

Acknowledgements
124
discipline knowledge always amazed me when I discussed with you. Your
analysis capability is so brilliant that you gave me lots experiment
suggestions even the project had no relation with yours. I’ll treasure the
good time that we shared one apartment and lived together. Your excellent
cooking skill gave me lots of wonderful “delicious” memories. Qing and I
will be your first customers if your noodle restaurant was open, which
however would be a huge loss for science world. May Qinhong, Lulu & you
live a happy life! Kai, I really appreciate your advice, the fruitful
discussions and our collaborations. Your limitless compassion for study
and experiment always remind me that I can do better. You are like a “gold
digger” that you can find flashpoints from a lot experimental results which
is dependent on your diligent literature reading. Without you, the studies
could not be performed, and we could not have such nice publications.
Wish Juanjuan & you have a good time in USA.
Jing, I really appreciate so many pleasant times with you and Jiaying, like
we went to Keukenhof to enjoy the beauty of tulips and picked up the
Chinese chestnut when I was pregnant. Also there were lots of good
memories of our trip to Portugal and we were a good team. I was the guide.
Qing was the bodyguard. Jiaying was the map. You were a very competent
sentry since you had such a high level of vigilance. And thanks very much
for the nice food and taking care of us when we came back to Groningen
after a long train travel. Yu, Pengkun, Shuaidong, Miancheng, Xintong,
Gurudas, Karolin and Kseniya thanks very much for being my colleagues
in PCBE group. It is indeed a very wonderful experience to work together
with all of you.
Dear friends, Qiuyan, thank you so much for your kindly help during my
life in Groningen. I was so afraid and nervous when I the first time arrived
in Schiphol airport. It was so nice of you to take a 3 hours train to Schiphol
to pick me up which was a big relief to me. You took me to the supermarket
and helped me to get my first bicycle in Groningen which made my first day
here much easier. You always asked me to go outings with you and your
friends, which made me not feel lonely. What impressed me most was
when you knew I was sad and crying for the death of my hamster, you came
to comfort me and buried my hamster together with me. You are really a

Acknowledgements
125
good friend to me and I wish you all the best in the future! Tao & Wenjun, I
really enjoy the Majiang nights with you guys and hopefully we can do it
again in the future. Yu, thanks so much for giving me advise and comfort
when I was scared of the delivery. And you always give me detailed
information when I ask you questions, like Ruby’s first rash. Wish June,
Hein & you a happy life! Tiancai & Yang, I am missing your yummy noodle
and looking forward to meeting you and your little girl at your defense. Jin,
thanks very much for your help when I was confused about my TEM results
and you are always patient to answer my questions. Chao & Xin, thanks
very much for taking care of dundun when Qing and I was on holiday. Wish
you all the best in the future! Guowei, thank you for giving me the
opportunity and trust to design your thesis cover. It’ll not be free next time
(*^__^*).
Meanwhile, I would like to express my appreciation to Groningen
University (RUG). RUG is a fantastic and amazing university, and I am so
fortunate to be part of it. RUG is at the forefront in providing freedom and
equality for all the students. When we first came here, we had
1200euro/month scholarship from CSC and found the other PhDs have
much higher salary than us. We felt kind of wronged since we did the same
work. So we wrote a joint letter and sent it to the headmaster nervously.
This was the first and only time in my life (until now) to sign a joint letter. I
felt quite perturbed and worried because I wasn’t taught to express my
demands. It’s inspiring that the university discussed over our letter in a
committee meeting and finally decided to give every CSC student 400euro
per month as a housing allowance. You cannot image how jealous they are
for CSC PhD from the other university. ↖(^ω^)↗
Also, thanks for the American sitcom, How I Met Your Mother, which
brings Qing and me so many joys and happiness. Although it is a comedy,
HIMYM taught us how to deal with failure, hold on to dreams, and even face
the death. There were countless times that we were tired and exhausted
after one day work, we watched HIMYM to get relaxed, like talking to some
old friends.

Acknowledgements
126
Finally, I would like to give my gratitude to my family. 首先感谢我的爸爸妈
妈,感谢您们这么多年对我学习和生活上的支持。每当我困惑失落的时候,
你们总能给我鼓励和支持。感谢黄妈妈,感谢您来荷兰帮我们照看小宝宝,
让我们可以放心的进行工作和学习。My little girl, Ruby 小梦安, I always
feel sweet and implausible when I look at you. Here is a quote from
“Spider-man” which could accurately represent my feeling to you. “When I
look in your eyes, and you’re looking back in mine, everything feels not quite
normal. Because I feel stronger and weaker at the same time, I feel excited
and at the same time terrified. The truth is I don’t know what I feel, except I
know what kind of man I want to be.”
Qing, my dear husband and best friend. I am so lucky to meet you at the
right place and right time. Life in a foreign country is not easy. Without you
I cannot finish my PhD study. There were so many times that I was
confused, self-doubted and about to give up because of the endless and
hopeless failing experiment results, you always gave me tremendous
encouragement and support. Your patience, respect, tolerance and
understanding helped me be a better person. When we were traveling in
Hallstatt, we got trapped in the big snow and couldn’t find the hotel. It was
dark and cold in the night, but I felt so safe and warm because you were
there. You are like some kind of superhero that could solve any problem
and protect me. Life is like unknown journeys, and I expect the next one
together with you.

Publications
127
Publications
1. Z. Meng, J. Yang, Q. Liu, J. W. de Vries, A. Gruszka, A. Rodrίguez-Pulido, A.
Kros, A. Herrmann. Efficient Fusion of Liposomes by Nucleobase Quadruple-
Anchored DNA. Chem. Eur. J. 2017, 23, 9391-9396.
2. Z. Meng, Q. Liu, J. Sun, M. Loznik, K. Liu, A. Herrmann, Highly stiff and
stretchable DNA liquid crystalline organogels with fast self-healing and
magnetically responsive behaviors. Nat. Commun. Submitted.
3. J. Yang, Z. Meng, Q. Liu, Y. Shimada, R. C. L. Olsthoorn, H. Spaink, A.
Herrmann, A. Kros, Employing DNA hybridization for Zebrafish Surface
Engineering. Angew. Chem. Int. Ed. Submitted.
4. Z. Meng, K. Liu, Q. Liu, P. Zhao, A. Herrmann, Study of the Hybridization
Properties of DNA-Surfactant Complex in Organic Phase. Manuscript in
preparation.
5. L. Zhang, L. Zheng, Z. Meng, K. Balinin, M. Loznik, A. Herrmann, Accelerating
Chemical Reactions by Molecular Sledding. Chem. Commun. 2017, 53: 6331-
6334.
6. A. S. Lubbe, Q. Liu, J. W. de Vries, J. C.M. Kistemaker, A. H. de Vries, I. F. Plo, Z.
Meng, W. Szymanski, A. Herrmann and B. L. Feringa. Photoswitching of DNA
hybridization using a molecular motor. J. Am. Chem. Soc. Submitted.
7. Q. Liu, Z. Meng, H. Fang, H. Li, K. Liu, A. Herrmann, Fluorescence properties
of lipid-DNA in liquid and liquid crystal states. Manuscript in preparation.
8. Q. Liu, A. Gruszka, J. Hurst, J. W. de Vries, F. Fröß, U. Hage, Z. Meng, K. U. B.
Schmidt, S. Schnichels, A. Herrmann, M. Spitzer, Lipid modified aptamers as
vehicles for ophthalmic drug delivery. Controlled Release. Submitted.


















