
UNIVERSITÁ DEGLI STUDI DI PISA FACOLTÁ DI SCIENZE …core.ac.uk/download/pdf/14693930.pdf · The...
69
UNIVERSITÁ DEGLI STUDI DI PISA FACOLTÁ DI SCIENZE M.F.N. CORSO DI LAUREA IN SCIENZE BIOLOGICHE Tesi di Laurea: VARIAZIONE DI MARKERS MOLECOLARI IN PAZIENTI CON ANEURISMA CEREBRALE Relatore Candidato Professore Luca Giovannini Leonardo Settimini Anno Accademico 2006-2007
Transcript of UNIVERSITÁ DEGLI STUDI DI PISA FACOLTÁ DI SCIENZE …core.ac.uk/download/pdf/14693930.pdf · The...

UNIVERSITÁ DEGLI STUDI DI PISA FACOLTÁ DI SCIENZE M.F.N.
CORSO DI LAUREA IN SCIENZE BIOLOGICHE
Tesi di Laurea:
VARIAZIONE DI MARKERS MOLECOLARI IN PAZIENTI
CON ANEURISMA CEREBRALE
Relatore Candidato Professore Luca Giovannini Leonardo Settimini
Anno Accademico 2006-2007

2
RIASSUNTO
INTRODUZIONE
L’aneurisma è una patologia dell’apparato vascolare che si manifesta come una dilatazione
abnorme di un vaso che può colpire qualsiasi vena o arteria del corpo. In particolare un
aneurisma cerebrale è localizzato nel punto di biforcazione delle arterie cerebrali che
costituiscono il poligono di Willis; la sua insorgenza può dipendere da fattori congeniti o
acquisiti. La prima manifestazione clinica, nella maggior parte dei casi è rappresentata da
un’emorragia, dovuta alla rottura della parete dei vasi, a livello dello spazio subaracnoideo.
Studi condotti su pazienti affetti da tale patologia hanno evidenziato la presenza di fenomeni
infiammatori associati a variazioni di marker biologici specifici sia in campioni tissutali che
plasmatici. La componente infiammatoria è associata a fenomeni di rimodellamento della
parete vasale, in particolare a livello della tonaca media.
SCOPO DEL LAVORO
Lo studio, effettuato in collaborazione con la clinica di Neurochirurgia dell’Ospedale “Santa
Chiara” di Pisa, prevede la valutazione del coinvolgimento di alcuni markers biologici nello
sviluppo dell’aneurisma cerebrale e le possibili interazioni tra i markers coinvolti.
MATERIALI E METODI
I volontari appartengono ai seguenti gruppi sperimentali: Controllo (n=10), Aneurisma non
sanguinante (n=10) e Aneurisma sanguinante (n=16). I campioni di plasma prelevati sono
stati utilizzati per la valutazione della concentrazione dei markers biologici (MMP-9, TIMP-1,
IL-1β, IL-18 e VEGF) attraverso l’utilizzo di saggi immunoenzimatici (ELISA-sandwich).

3
RISULTATI
I livelli di MMP-9 aumentano significativamente sia nel gruppo con aneurisma non
sanguinante (223.38 ± 31.6 vs 92.8 ± 21.7 ng/ml con p < 0.01) rispetto al controllo sia nel
gruppo con aneurisma sanguinante (304.16 ± 48.6 vs 92.8 ± 21.7 ng/ml con p < 0.05).
Non si è invece osservata una significativa differenza tra i gruppi con aneurisma sanguinante
e non sanguinante. I livelli plasmatici del TIMP-1 rimangono pressoché invariati nei tre
gruppi presi in esame: nel gruppo controllo si ha una media di 120.17 ng/ml (SD = 15.21), nel
gruppo con aneurisma non sanguinante la media è di 135.73 ng/ml (SD = 21.91) mentre nel
gruppo con aneurisma sanguinante è di 128.69 ng/ml (SD = 13.37).
Nel rapporto MMP-9/TIMP-1 si ha un aumento significativo rispetto al gruppo controllo, sia
nel gruppo dell’aneurisma non sanguinante (3.27 ± 0.76 vs 0.96 ± 0.33 ng/ml con p < 0.01)
che nel gruppo dell’aneurisma sanguinante (3.38 ± 0.63 vs 0.96 ± 0.33 ng/ml con p < 0.05).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante.
I livelli di IL-1β mostrano un aumento significativo di tale parametro rispetto al controllo sia
nel gruppo con aneurisma non sanguinante (74.5 ± 21.27 vs 19.67 ± 3.78 pg/ml con p < 0.05)
che nel gruppo con aneurisma sanguinante (58.06 ± 16.5 vs 19.67 ± 3.78 pg/ml con p < 0.05).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante (p = 0.053).
I livelli di IL-18 non evidenziano un aumento significativo rispetto al gruppo controllo, sia nel
gruppo con aneurisma non sanguinante (215.6 ± 55.96 vs 104.8 ± 21.80 pg/ml con p = 0.065)
che nel gruppo con aneurisma sanguinante (305.4 ± 95.48 vs 104.8 ± 21.80 pg/ml con p =
0.075).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante.

4
Infine i livelli di VEGF hanno mostrato un aumento significativo di tale parametro sia nel
gruppo con aneurisma non sanguinante (45.60 ± 6.03 vs 26.08 ± 4.86 pg/ml con p < 0.01) che
nel gruppo con aneurisma sanguinante (94.71 ± 36.18 vs 26.08 ± 4.86 pg/ml con p < 0.05)
rispetto al controllo.
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante.
Sono state inoltre osservate delle correlazioni tra i markers indagati:
L’IL-1β ha evidenziato una correlazione positiva significativa con l’MMP-9 (p = 0.00215) e
non significativa con il VEGF (p = 0.28).
L’IL-18 ha una correlazione positiva significativa con l’MMP-9 (p = 0.0024) e con l’IL-1β
(p = 0.0017) e non significativa con VEGF (p = 0.62).
Infine il VEGF presenta una correlazione positiva significativa solo con l’MMP-9 (p = 0.011).
CONCLUSIONI
Sulla base dei dati emersi da questo studio è possibile affermare che tra i meccanismi
maggiormente coinvolti nello sviluppo di un aneurisma vi è l’aumento di citochine
proinfiammatorie e l’upregulation del sistema dell’MMP, si può inoltre ipotizzare l’esistenza
di segnali precoci la cui espressione viene downregolata con il procedere della patologia.
Infine, possibili fattori coinvolti nello sviluppo dell’aneurisma sono molecole ad attività
modulatoria e proinfiammatoria indiretta come IL-18.

5
ABSTRACT
INTRODUCTION
The aneurysm desease is a vascular patology characterized by an abnormal blood vessel wall
dilatation. In particular, cerebral aneurysm is located at the bifurcation of the arteries that
form the Willis’s cerebral circulation. The first clinical evidence is usually a subarachnoide
spaces haemorrhage caused by lesion of blood vessel wall. Many clinical studies have
shown the presence of inflammatory activities in plasma and tissue samples as well as the
presence of specific biological markers for cerebral aneurysm.
AIM OF THE WORK
This study was done in collaboration whit “Neurochirurgia dell’Ospedale Santa Chiara di
Pisa” with the aim to evaluate possible biological markers involved in the development of the
cerebral aneurysm.
MATERIALS AND METHODS
36 volunteers were recruited and divided in 3 groups as follow: Control (n=10), unruptured
cerebral aneurysm (n=10), ruptured cerebral aneurysm (n=16). Plasma samples were collected
and the concentration of different biological markers (MMP-9, TIMP-1, IL-1β, IL-18 e
VEGF) was assaied using an ELISA- sandwich method.
RESULTS
MMP-9 levels were significantly increased in unruptured cerebral aneurysm (223.38 ± 31.6 vs
92.8 ± 21.7 ng/ml con p < 0.01) and in ruptured cerebral aneurysm (304.16 ± 48.6 vs 92.8 ±
21.7 ng/ml con p < 0.05) respect to the control group. TIMP-1 levels were found unchanged
in all experimental groups. MMP-9/TIMP-1 ratio was significantly increased in unruptured
cerebral aneurysm (3.27 ± 0.76 vs 0.96 ± 0.33 ng/ml con p < 0.01), and in ruptured cerebral
aneurysm (3.38 ± 0.63 vs 0.96 ± 0.33 ng/ml con p < 0.05) respect the control group.

6
IL-1β levels were significantly increased in unruptured cerebral aneurysm (74.5 ± 21.27 vs
19.67 ± 3.78 pg/ml con p < 0.05) and in ruptured cerebral aneurysm (58.06 ± 16.5 vs 19.67 ±
3.78 pg/ml con p < 0.05) respect the control group, while no significant difference was found
beetween unruptured and ruptured groups (p=0.053). IL-18 levels did not show any
significant increase beetween unruptured cerebral aneurysm (215.6 ± 55.96 vs 104.8 ± 21.80
pg/ml con p = 0.065) and ruptured cerebral aneurysm (305.4 ± 95.48 vs 104.8 ± 21.80 pg/ml
con p = 0.075) respect the control group. Moreover, the plasmatic levels of VEGF were
increased in unruptured cerebral aneurysm (45.60 ± 6.03 vs 26.08 ± 4.86 pg/ml con p < 0.01)
and in ruptured cerebral aneurysm (94.71 ± 36.18 vs 26.08 ± 4.86 pg/ml con p < 0.05)
respect the control group. Concernig the relationship between these markers, we have found a
positive correlation bbetween IL-1β and MMP-9 (p = 0.00215) but not with VEGF (p =
0.28). IL-18 shows a significantly positive correlation with MMP-9 (p = 0.0024) and with IL-
1β (p = 0.0017) but not with VEGF (p = 0.62). Finally, VEGF shows a significantly positive
correlation only with MMP-9 (p = 0.011).
CONCLUSIONS
In our study we observed an increase of proinflammatories cytokines and the up-regulation of
MMP-9 during the development of cerebral aneurysm; moreover we can hypotesize the
presence of precocius signs that decrease with pathology progression.
In conclusion, involvment of biological markers in cerebral aneurysm have shown their role
as modulators of the inflammatory process.

7
INDICE
INTRODUZIONE 8
Aneurisma cerebrale 9
1. Aneurisma 9
2. Aneurisma cerebrale 11
Processo infiammatorio nell’aneurisma cerebrale 16
1. Infiammazione 16
2. Infiammazione ed aneurismi cerebrali 23
Parametri biologici analizzati 27
1. Sistema delle metalloproteasi 27
2. Citochine proinfiammatorie 29
3. fattore di crescita dell’endotelio vascolare 30
SCOPO RICERCA 32
PROTOCOLLO SPERIMENTALE 35
MATERIALI E METODI 37
RISULTATI 43
DISCUSSIONE E CONCLUSIONI 52
BIBLIOGRAFIA 57
RINGRAZIAMENTI

8
INTRODUZIONE

9
ANEURISMA CEREBRALE
ANEURISMA.
L’aneurisma è una dilatazione localizzata, abnorme, di un vaso sanguigno dovuta
all’indebolimento,alla perdita di integrità strutturale della parete od all’aumento della
pressione intravascolare, come nell’ipertensione arteriosa. Può manifestarsi in qualsiasi
arteria o vena del corpo, ma più frequentemente nell’aorta o nel cuore. Gli aneurismi possono
essere “veri” o “falsi” : l’aneurisma vero è delimitato, generalmente, dall’intera parete
arteriosa, che risulta spesso assottigliata e strutturalmente alterata sotto il profilo quantitativo
e qualitativo. L’aneurisma falso, invece, è costituito da un ematoma extravascolare che
comunica con il lume vasale. La creazione di una breccia nella parete del vaso determina la
formazione del sacco aneurismatico, delimitato dagli strati più esterni dell’arteria o dal tessuto
periarterioso (1-2).
Localizzazione. Le principali localizzazioni dell’aneurisma sono nel tratto intratoracico e
in quello addominale dell’aorta, nei rami dell’aorta addominale, nelle arterie cerebrali, in
quelle periferiche e nella arteria polmonare (3).
Classificazione. Gli aneurismi possono essere classificati in base alle caratteristiche
morfologiche macroscopiche in due gruppi : gli aneurismi sacciformi sono limitati
esclusivamente ad una parte della parete vasale e aventi dimensioni variabili tra i 5 ed i 20 cm
di diametro; gli aneurismi fusiformi invece si formano in seguito ad una progressiva, ma
graduale dilatazione dell’intera circonferenza del vaso e raggiungono dimensioni variabili, sia
nel diametro (oltre 20 cm ) che nella lunghezza (3).

10
In base alla causa scatenante si distinguono aneurismi aterosclerotici , luetici, micotici,
congeniti e traumatici (1-2) :
• Aneurismi aterosclerotici. L’aterosclerosi può dare luogo alla rottura della tunica
media con conseguente formazione di un aneurisma . Questi aneurismi si sviluppano
prima dei 50 anni e sono di gran lunga più frequenti nei soggetti maschili. Alcuni
autori ritengono che alterazioni genetiche in grado di compromettere elementi
strutturali del tessuto connettivo, coinvolti nella resistenza dei vasi sanguigni,
possano rappresentare un substrato particolarmente sensibile all’ azione di
indebolimento svolta dall’aterosclerosi o dall’ipertensione sulla parete aortica (4-5-
6).
• Aneurismi luetici. È stata esagerata l’importanza della sifilide nella genesi degli
aneurismi. Per molti anni la maggior parte delle lesioni è stata considerata di natura
luetica. Anche in questo caso si ha la rottura della tunica media .
• Aneurismi micotici. Nel corso di setticemie e soprattutto di endocardite lenta,
possono comparire aneurismi numerosi e piccoli, chiamati micotici.
• Aneurismi congeniti. Sono caratterizzati da una precoce comparsa e da molteplicità.
La loro localizzazione più frequente è quella intracranica.
• Aneurismi traumatici. Sono distinti tra quelli da sforzo, che quasi sempre
presuppongono la preesistenza di una lesione luetica , aterosclerotica o congenita, e
quelli da ferite da armi da taglio o da fuoco.

11
ANEURISMA CEREBRALE.
Epidemiologia e localizzazione. Un aneurisma cerebrale è una lesione facilmente
riscontrabile in un esame autoptopico, con una percentuale pari all’1 -6 % in una serie di
autopsie effettuate su individui adulti (7-8). Molti di questi aneurismi , comunque, sono di
piccole dimensioni e la loro presenza, in un campione di adulti che si sono sottoposti ad
angiografia cerebrale, è nell’ordine dello 0.5-1 % (9). Gli aneurismi endocranici sono abnormi
dilatazioni della pareti vasali comunemente localizzate al punto di diramazione delle maggiori
arterie che scorrono attraverso lo spazio subaracnoideo alla base del cervello e che
costituiscono il poligono di Willis. I siti più colpiti (80-85 %) , situati nella circolazione
anteriore, sono a) la giunzione dell’arteria carotide interna e della arteria comunicante
posteriore, b) l’arteria comunicante anteriore, c) la maggiore biforcazione dell’arteria media
cerebrale. Gli aneurismi della circolazione posteriore sono localizzati d) alla biforcazione
della arteria basilare o e) alla giunzione della arteria vertebrale e della arteria cerebellare
ipsilaterale inferiore posteriore (2-10-11-12). Aneurismi intracranici multipli, nel numero di
due o tre, sono ricontrati nel 20-30% dei pazienti (13).
FIG. 1 Rappresentazione schematica del poligono arterioso di Willis ( dalRobbins) .

12
Diagnosi. In un ridotto numero di casi, cefalee ricorrenti possono precedere di giorni o
settimane l’evento emorragico, talora associate a sintomi premonitori quali nausea e vomito.
Questi sintomi possono essere causati o dalla massa di un aneurisma “gigante”(14), oppure da
una “perdita premonitrice” causata da una piccola fuoriuscita di sangue nella parete o nello
spazio subaracnoideo (15-16). Spesso questi sintomi premonitori non sono riconosciuti come
tali da parte del medico, che erroneamente li identifica come emicranie, sinusiti o influenza
(17). Nella maggior parte dei casi però è l’evento emorragico a livello dello spazio
subaracnoideo la prima manifestazione di un aneurisma cerebrale. In questo caso un violento,
severo, mal di testa a “colpo di pugnale”, localizzato in prevalenza nella regione nucale, è
accompagnato da possibili disturbi della coscienza con obnubilazione del sensorio, coma e,
talora, da segni neurologici locali. Successivamente, ma non prima di qualche ora, appaiono
segni di irritazione meningea quali vomito, rigidità nucale, fotofobia. Sintomi a focolaio di
varia entità possono comparire nel caso di emorragie provocate da aneurismi arteriosi: questa
sintomatologia può essere di tipo centrale e derivare o da una lesione encefalica provocata
dalla rottura dell’aneurisma (ematoma intraparenchimale) o dall’ischemia secondaria allo
spasmo arterioso che complica spesso il versamento subaracnoideo. Esistono casi, infine, in
cui la perdita di coscienza non è preceduta dalla cefalea iniziale (11,18-19).
Eziologia. Questo tipo di lesioni della parete vasale sono provocate, oltre che da lesioni
acquisite meno frequenti di natura aterosclerotica o infettiva (aneurismi fusiformi), da
alterazioni strutturali congenite della parete, formate durante il rimaneggiamento
ontogenetico della rete circolatoria cerebrale, che realizzano “luoghi di minore resistenza”,
specialmente nei punti in cui si dipartono i rami collaterali (aneurismi sacculari). Per tale
ragione, nel poligono di Willis gli aneurismi si localizzano in corrispondenza degli sbocchi
dei canali anastomotici, vale a dire nei punti di contatto fra le varie sezioni arteriose del
poligono (18).

13
Questi aneurismi, definiti “a bacca”, si formano a causa di difetti dello sviluppo della lamina
elastica interna dei vasi. Lo stress delle onde sistoliche causa una erniazione dell’ intima, con
formazione degli aneurismi sacculari (20,21).
Gli aneurismi che sorgono dalle arterie intracraniche sono più comuni di quelli che sorgono
dalle arterie extracraniche di dimensioni equivalenti. Si è ipotizzato che ciò avvenga perché le
arterie intracraniche hanno un’ attenuata tunica media e mancano di una lamina elastica
esterna. Ad un esame microscopico, un aneurisma a bacca ha la tunica media molto sottile o
mancante, e la lamina elastica interna risulta assente o severamente frammentata. Così la
parete dell’aneurisma è generalmente composta dalla tonaca intima e da quella avventizia,
con una variabile quantità di sostanza fibroialina interposta tra questi due strati che costituisce
un “locus minoris resistentiae”e predispone alla fessurazione o rottura della parete stessa
(18,22).
Fattori di rischio.
• Fattori di rischio congeniti: a supporto del ruolo dei fattori genetici nella patogenesi
dell’aneurisma cerebrale sono state trovate correlazioni tra la presenza
dell’aneurisma e la presenza di malattie ereditarie del tessuto connettivo, nonché la
familiarità (23,24). I disordini del tessuto connettivo più coinvolti nell’ insorgere di
questo tipo di lesioni sono la sindrome di Ehlers-Danlos di tipo IV, la
neurofibromatosi di tipo I, la sindrome di Marfan e la malattia autosomica dominante
renale policistica (23,24). Non è conosciuta l’estensione di questi disordini in una
popolazione di pazienti aventi un aneurisma cerebrale, in uno studio condotto su 100
pazienti con aneurisma cerebrale, 5 risultavano aver avuto malattie del tessuto
connettivo (25).

14
Tuttavia si ottiene che la vera frequenza debba essere più alta in quanto in primo
luogo questi disordini spesso rimangono non diagnosticati a causa della variabilità
nella loro espressione fenotipica, e secondariamente, la storia familiare è
frequentemente negativa poiché la malattia è causata da una nuova mutazione.
• Fattori di rischio acquisiti: Parecchi studi hanno fatto supporre che i fattori acquisiti
giocano un ruolo importante nella patogenesi degli aneurismi intracranici. Per
esempio, gli aneurismi intracranici sono molto rari nei bambini, e sebbene l’età
media dei pazienti colpiti da emorragia subaracnoidea sia di circa 50 anni ,
l’incidenza di questo disturbo tende ad aumentare con l’età fino all’ottava decade di
vita (26). Inequivocabili fattori di rischio acquisiti sembrano essere il fumo di
sigaretta, l’assunzione di alcol, l’ipertensione, l’età di insorgenza ed anche il sesso
femminile (10,27-34) : il fumo di sigaretta è stato l’unico fattore identificato in tutte
le popolazioni studiate nonché quello più facilmente prevenibile (35-37); un abuso
nell’assunzione di alcol appare incrementare il rischio di un’emorragia subaracnoidea
(28,29); l’ipertensione è il fattore di rischio più studiato per lo sviluppo e la rottura di
un aneurisma, ed appare associato ad entrambi i fenomeni (26,35,37); il primo
episodio di emorragia subaracnoidea colpisce in maggioranza soggetti compresi tra i
40 ed i 60 anni con una prevalenza nel 4° decennio per il sesso maschile mentre nel
quinto è più colpito quello femminile (10,18,38-40).
Rottura dell’aneurisma ed emorragia subaracnoidea. Essendo la diagnosi di
aneurisma cerebrale molto difficile, a volte la sua prima manifestazione è rappresentata da
un’emorragia essenzialmente a livello dello spazio subaracnoideo. Sebbene questa emorragia
sia molto comune durante periodi di stress e di sforzo può manifestarsi in qualsiasi momento
(41).

15
Un emorragia subaracnoidea è un evento devastante associato ad un alto tasso di morbosità e
mortalità: circa il 12% dei pazienti muore prima di avere ricevuto attenzioni mediche, il 40%
dei pazienti ospedalizzati muore entro un mese dopo l’evento e più di un terzo di coloro che
sopravvivono hanno importanti deficit neurologici.
Pazienti che hanno avuto un SAH hanno una maggior probabilità di sviluppare un nuovo
aneurisma: ogni anno nuovi aneurismi insorgono nel 2% dei pazienti colpiti da un episodio
precedentemente diagnosticato di SAH, ed in questo gruppo di pazienti l’incidenza della
rottura dell’aneurisma è di 6 casi su 10.000 all’anno, maggiore rispetto all’incidenza nella
popolazione generale (42-45). Nonostante i passi in avanti effettuati nelle ultime decadi nella
diagnosi e nella pratica chirurgica di questa malattia il tasso di vittime e l’incidenza di questa
malattia non sono cambiati(46-51).
Aneurisma non sanguinante ed aneurisma sanguinante. L’alta incidenza di soggetti
con aneurisma e l’elevata mortalità ha reso necessario lo studio di meccanismi fisiopatologici
che portano allo sviluppo e alla rottura dell’aneurisma. In uno studio, effettuato su pazienti
in cui erano diagnosticati sia casi di rottura sia di aneurismi non sanguinanti,sono stati
documentati danni all’endotelio, cambiamenti nella struttura della parete nonché una
significativa invasione di cellule infiammatorie nell’aneurisma sanguinante rispetto ad un
aneurisma non sanguinante. Si è ipotizzato che la fragilità della parete dell’aneurisma
sanguinante fosse dovuta all’infiltrazione nella parete stessa di macrofagi e linfociti con
conseguente perdita di proteine della matrice extracellulare e di cellule muscolari lisce e
comparsa di sostanza fibroialina (51). I pazienti con aneurisma non sanguinante che
presentano un quadro patologico simile a quello presente per gli aneurismi sanguinanti (danno
endoteliale, parete modificata ed infiltrazione di cellule infiammatorie) si pensa abbiano un
rischio maggiore di rottura. Comunque la bassa probabilità di rottura di un aneurisma non
sanguinante (49-51) si pensa sia dovuta all’assenza di tale quadro patologico.

16
PROCESSO INFIAMMATORIO NELL’ANEURISMA CEREBRALE
INFIAMMAZIONE.
Processo generale dell’infiammazione. (2,52) L’infiammazione è fondamentalmente
una risposta protettiva il cui obiettivo finale è l’eliminazione della causa iniziale di danno
cellulare e delle sue conseguenze. La risposta infiammatoria è strettamente connessa al
processo di riparazione, dato che mette in moto una serie di eventi che sono finalizzati alla
guarigione e alla sostituzione del tessuto danneggiato. D’altronde occorre considerare che l’
infiammazione e la riparazione sono processi potenzialmente dannosi, come nel caso delle
reazioni di ipersensibilità a punture di insetto, a farmaci e tossine, oltre alle comuni malattie
croniche quali l’artrite reumatoide, l’aterosclerosi e la fibrosi polmonare. La risposta
infiammatoria ha luogo nel tessuto connettivo vascolarizzato, che comprende il plasma, le
cellule circolanti (neutrofili, monociti, eosinofili, linfociti, basofili e piastrine), i vasi
sanguigni ed i componenti cellulari e del tessuto connettivo ( i mastociti, i fibroblasti, i
macrofagi e la matrice extracellulare composta prevalentemente da proteine fibrose strutturali
(collagene ed elastina), glicoproteine adesive (fibronectina, laminina, collagene non cellulare
ed altre) e proteoglicani.
FIG. 2: Processo infiammatorio (Robbins)

17
Schematicamente si distinguono due versioni del processo infiammatorio:
• Infiammazione acuta: è la risposta immediata e precoce ad uno stimolo lesivo. È un
processo di breve durata con tempi che vanno da pochi minuti o qualche ora, fino a
pochi giorni. Andando ad analizzare lo svolgimento del processo, dopo una breve e
transitoria vasocostrizione si osserva una vasodilatazione a livello del microcircolo
arterioso che provoca un aumento del flusso ematico, un aumento della permeabilità
vascolare e una fuoriuscita di liquidi e proteine dai vasi del microcircolo ( “essudato”);
questo fenomeno è alla base della formazione dell’edema. La perdita di liquidi porta
all’addensamento degli eritrociti nei vasi del microcircolo e determina un aumento
della viscosità del sangue, che si riflette nella presenza di piccoli vasi dilatati
contenenti eritrociti impaccati. All’instaurarsi di tale condizione, denominata “stasi”, i
leucociti, inizialmente soprattutto i neutrofili, tendono ad assumere un orientamento
periferico avvicinandosi all’ endotelio vascolare ( “marginazione”). Successivamente i
leucociti aderiscono all’endotelio, e poco dopo oltrepassano la parete dei vasi
(“diapedesi”) e migrano nei tessuti interstiziali verso la sede colpita dal danno
attraverso un processo chiamato “chemiotassi” definito come movimento orientato
lungo un gradiente chimico costituito da uno stimolo chemiotattico.
Successivamente si ha la fagocitosi dell’agente lesivo. Durante la chemiotassi e la
fagocitosi, i leucociti attivati possono rilasciare metaboliti tossici e proteasi
nell’ambiente extracellulare causando danni tessutali. Se vi è stata una distruzione
tessutale di scarsa entità e se le cellule parenchimali sono in grado di rigenerare, una
risposta infiammatoria si può risolvere, una volta neutralizzato lo stimolo, con il
ritorno alla normalità nella zona colpita dall’infiammazione(2-52).
• Infiammazione cronica.L’infiammazione cronica si può considerare un’infiammazione
di lunga durata ( settimane o mesi ) in cui l’infiammazione attiva, la distruzione
tessutale ed i tentativi di riparazione procedono simultaneamente. Anche se

18
l’infiammazione cronica può seguire quella acuta, più frequentemente la causa è da
ricercare nella mancata eradicazione dell’ agente lesivo, con persistente attivazione di
processi reattivi atti ad eliminare l’agente, ma che rappresentano una concausa del
danno tessutale, e di processi di riparo tessutale. Diversamente dall’infiammazione
acuta, in quella cronica si ha una netta predominanza di cellule macrofagiche ed un
contributo di altri tipi cellulari quali linfociti, plasmacellule ed eosinofili, variabile a
seconda dell’ agente lesivo che riflette la presenza di una reazione persistente allo
stimolo lesivo. Le reazioni vascolari si attenuano, mentre si assiste, soprattutto nelle
prime fasi, alla proliferazione di piccoli vasi (angiogenesi), ed una reazione
fibroblastica determina processi di rimaneggiamento tessutale che determinano la
progressiva sostituzione del parenchima danneggiato con tessuto connettivo
(fibrosi)(2,52).
Eventi cellulari. Una funzione fondamentale dell’infiammazione è di permettere ai
leucociti di giungere nella sede del danno dove uccidono gli agenti lesivi, i batteri ed altri
microrganismi, degradano il tessuto necrotico ma possono anche prolungare l’infiammazione
e indurre danno tessutale attraverso il rilascio di enzimi, mediatori chimici e radicali tossici
dell’ossigeno(2,52).
• Rotolamento, adesione e diapedesi. L’adesione ed il fenomeno della diapedesi sono
determinate dal legame tra molecole di adesione complementari presenti sulla
superficie dei leucociti e delle cellule endoteliali; i mediatori chimici come le
chemochine ed alcune citochine influiscono su questo processo modulando
l’espressione di superficie o l’avidità delle molecole di adesione.
• Chemiotassi. Sostanze esogene ed endogene possono agire come agenti chemiotattici,
responsabili del movimento cellulare. I mediatori endogeni comprendono componenti
del sistema del complemento, prodotti della via lipossigenasica e chemochine.

19
• Attivazione dei leucociti. Molti fattori chemiotattici, tra cui citochine secrete da
leucociti sensibilizzati, se presenti in concentrazioni elevate, inducono anche una serie
di risposte, che nel loro insieme prendono il nome di “attivazione dei leucociti”. Nei
neutrofili queste risposte comprendono una produzione di metaboliti dell’acido
arachidonico; degranulazione, secrezione di enzimi lisosomiali ed attivazione
dell’esplosione ossidativa; modulazione di molecole di adesione dei leucociti. Il
macrofago, una volta attivato, oltre alle risposte sopra elencate per i neutrofili ,
produce sia molecole (citochine e fattori chemiotattici) che regolano la migrazione di
altre cellule, sia molecole che inducono la proliferazione dei fibroblasti, la deposizione
di collagene e l’angiogenesi (fattori di crescita). Nell’infiammazione cronica, linfociti
e macrofagi sono in stretta reciproca relazione: i linfociti possono essere attivati dal
contatto con l’antigene o da citochine proinfiammatorie e, una volta attivati producono
“linfochine”, che possono contribuire ad attivare i monociti ed i macrofagi . Le
citochine prodotte dai macrofagi attivati, a loro volta attivano i linfociti.
• Fagocitosi. La fagocitosi è un processo importante, che avviene ad opera dei
macrofagi, responsabile dell’uccisione o degradazione del materiale ingerito.
• Rilascio di prodotti da parte dei leucociti e relativo danno tessutale. Le modificazioni
della membrana e del metabolismo del leucocita durante la chemiotassi, l’attivazione e
la fagocitosi provocano il rilascio di una serie di prodotti non solo nei fagolisosomi,
ma talvolta anche negli spazi extracellulari. Le sostanze più importanti rilasciate dai
neutrofili sono mediatrici del danno endoteliale e tessutale che possono amplificare
l’effetto dello stimolo infiammatorio iniziale(2,52).
Riparazione e angiogenesi. Una tappa importante in questo processo di guarigione
consiste nello sviluppo del tessuto di granulazione contraddistinto dalla formazione di nuovi

20
vasi sanguigni (angiogenesi) e dalla proliferazione di fibroblasti, deputati alla deposizione di
nuova matrice extracellulare (ECM):
1. Angiogenesi. I vasi preesistenti producono gemmazioni da cui germogliano nuovi
vasi verso lo stimolo angiogenetico. Questa fase è controllata dalle interazioni tra
fattori di crescita ( il fattore di crescita dell’endotelio vascolare (VEGF), il fattore di
crescita dei fibroblasti (bFGF) ed il fattore di crescita endoteliale (EGF), cellule
vascolari e proteine della matrice extracellulare: le cellule vascolari si muovono e
migrano verso lo stimolo angiogenetico; le metalloproteasi destabilizzano
l’interazione cellula-matrice partecipando al rimodellamento che si verifica durante
l’invasione endoteliale e permettendo, in seguito alla degradazione della (ECM), la
liberazione di fattori di crescita che regolano l’angiogenesi (49,50).
2. Proliferazione di fibroblasti. E’ innescata da diversi fattori di crescita,
principalmente TGF-β. Col progredire della riparazione aumenta l’attività di sintesi
dei fibroblasti che aumentano la deposizione di ECM; inoltre si assiste ad una
regolazione della degradazione della ECM.
Mediatori chimici dell’infiammazione.
Durante il processo infiammatorio si assiste alla sintesi di un ampio spettro di sostanze
chimiche che vengono liberate e svolgono compiti importanti nel condizionare vari aspetti
dell’ infiammazione. I mediatori di origine plasmatica (come il complemento) sono presenti in
circolo sotto forma di precursori che devono essere attivati, attraverso una serie di tagli
proteolitici, per acquisire le loro proprietà biologiche specifiche. I mediatori di origine
cellulare sono normalmente sequestrati all’interno di granuli intracellulari, prima di essere
secreti oppure sono sintetizzati “de novo” in risposta ad uno stimolo. Le fonti principali dei
mediatori di origine cellulare sono le piastrine, i neutrofili, i monociti/macrofagi ed i
mastociti, anche se le cellule di origine mesenchimale possono essere indotte a produrre

21
alcuni dei mediatori. Per quanto riguarda la loro attività, alcuni dei mediatori hanno un’
azione enzimatica diretta , altri causano danno ossidativo, mentre la maggior parte dei
mediatori agisce tramite il legame a specifici recettori sulle cellule bersaglio. Un mediatore
chimico può stimolare il rilascio di altri mediatori chimici secondari da parte delle cellule
bersaglio. Questi mediatori secondari consentono l’attivazione di meccanismi volti ad
amplificare od a controbilanciare l’azione del mediatore iniziali(2,52).
Segue una classificazione dei principali mediatori coinvolti nell’infiammazione:
o Amine vasoattive. In particolare le due amine istamina e serotonina sono tra i primi
mediatori ad essere rilasciati durante l’infiammazione. Entrambe sono coinvolte nella
dilatazione delle arteriole e nell’aumento della permeabilità vascolare delle venule.
o Proteasi plasmatiche. Fanno parte di tre sistemi strettamente correlati tra di loro: a) Il
sistema del complemento, formato da 20 proteine, che è coinvolto nell’azione di
difesa contro i microrganismi e che da origine ad una serie di prodotti coinvolti
nell’aumento della permeabilità vascolare, nella chemiotassi e nell’opsonizzazione.
o Il sistema delle chinine, che genera la formazione di peptidi vasoattivi a partire dà
proteine plasmatiche chiamate chininogeni per mezzo di proteasi specifiche chiamate
callicreine; è responsabile del il rilascio finale della bradichinina, un potente
modulatore della permeabilità vascolare, della vasodilatazione e della contrazione
della muscolatura liscia. c) Il sistema della coagulazione, che consta di due vie
convergenti che culminano nell’attivazione della trombina con conseguente
formazione di fibrina.
o Metaboliti dell’acido arachidonico. Sono sintetizzati da due principali classi di
enzimi: le ciclossigenasi (prostaglandine e trombossani) e le lipossigenasi
(leucotrieni e lipossine). Questi metaboliti possono intervenire in ogni tappa del
processo infiammatorio, infatti sono presenti nell’essudato infiammatorio e la loro
sintesi aumenta nelle sedi dell’infiammazione.

22
o Fattore attivante le piastrine (PAF). E’un altro mediatore bioattivo di derivazione
fosfolipidica che, oltre alla stimolazione delle piastrine, causa vasocostrizione,
broncocostrizione, aumento dell’adesione leucocitaria all’endotelio, la chemiotassi e,
a concentrazioni bassissime, l’aumento della permeabilità delle venule.
o Citochine e chemochine. Le citochine sono proteine, prodotte principalmente da
linfociti e da macrofagi attivati, che modulano le funzioni di altre cellule. La
secrezione di questi mediatori è transitoria e strettamente regolata. Queste proteine
possono influenzare la sintesi o l’azione di altre citochine e sono multifunzionali,
poiché una singola di esse può avere azioni modulatorie sia positive che negative.
Esplicano i loro effetti legandosi a specifici recettori presenti sulle cellule bersaglio,
la cui espressione può essere regolata da vari segnali esogeni ed endogeni.
Le citochine possono essere raggruppate in varie classi sulla base della funzione
principale o in relazione al tipo di cellula bersaglio: a) citochine che regolano
l’attivazione, la proliferazione ed il differenziamento dei linfociti (IL-2, IL-4, IL-10,
TGF-β); b ) citochine implicate nell’immunità naturale (TNF-α, IL-1β, IFN-α, IL-6);
c) citochine che attivano le cellule dell’infiammazione (IFN-γ, TNF-α, IL-5 , IL-10,
IL-12, IL-18); d) chemochine, che hanno azione chemiotattica nei confronti di vari
leucociti (IL-8); d) citochine che stimolano l’ emopoiesi (IL-3, IL-7).
o Ossido di azoto (NO). E’un gas solubile prodotto dalle cellule endoteliali e dai
macrofagi capace sia di indurre una vasodilatazione, per il suo effetto di rilassamento
sulle cellule muscolari lisce dei vasi, sia di regolare il reclutamento dei leucociti.
o Costituenti lisosomiali dei leucociti. Sia i neutrofili che i monociti possiedono granuli
lisosomiali contenenti idrolisi acide, collagenasi, gelatinasi ed elastasi particolarmente
attive nelle reazioni infiammatorie croniche. Queste pericolose proteasi sono però
tenute sotto controllo da un sistema di inibitori delle proteasi presente nel siero e nei
liquidi tessutali.

23
o I radicali liberi dell’ossigeno. Sono rilasciati dai leucociti nell’ambiente
extracellulare in seguito ad esposizione ad agenti chemiotattici, ad immunocomplessi
o durante la fagocitosi. Il loro rilascio extracellulare può aumentare l’espressione di
chemochine, di citochine e di molecole d’adesione endoteliali e leucocitarie,
amplificando la cascata che scatena la risposta infiammatoria.
La sostituzione del tessuto di granulazione con tessuto cicratiziale comporta una variazione
nella composizione della ECM: alcuni dei fattori di crescita che stimolano la sintesi del
tessuto connettivo modulano anche la sintesi e l’attivazione delle metalloproteasi che
degradano la ECM. Il bilancio netto tra la sintesi e la degradazione definisce il
rimodellamento del tessuto connettivo.

24
INFIAMMAZIONE ED ANEURISMI CEREBRALI.
L’infiammazione nella patogenesi dell’aneurisma cerebrale.
In letteratura vi è un cospicuo numero di studi che confermano il coinvolgimento del processo
infiammatorio nello sviluppo di un aneurisma aortico (elencane); studi clinici su pazienti con
aneurisma hanno documentato la presenza di fenomeni connessi al processo infiammatorio,
quali il danno endoteliale, la perdita di cellule muscolari lisce e di fibre collagene a livello
della tonaca media della parete e la presenza di un infiltrato cellulare infiammatorio, oltre al
rilevamento di mediatori chimici dell’infiammazione in campioni tissutali ed ematici
(51,53,54).
In particolare è stata evidenziata la presenza della componente infiltrante a livello di
aneurismi intracranici andati incontro a rottura (56).
Se pure tali osservazioni in passato avevano fatto pensare a cambiamenti legati
all’aterosclerosi (55), Chyatte et al. hanno trovato che nell’aneurisma cerebrale la presenza di
cellule infiltranti non è solo limitata alla tonaca intima, che è la zona della parete più soggetta
a cambiamenti aterosclerotici, ma è piuttosto una reazione generalizzata.
Markers molecolari dell’infiammazione ed aneurismi cerebrali.
Il coinvolgimento dell’infiammazione negli aneurismi intracranici è supportato da studi che
hanno analizzato molecole correlate all’infiammazione in campioni ematici di pazienti con
aneurismi intracranici. Livelli sierici di elastasi e collagenasi sembrano essere elevati in
pazienti con aneurismi intracranici; questi dati sono confermati da studi che hanno
evidenziato alti livelli tessutali di collagenasi ed elastasi (57-62). Le principali collagenasi
coinvolte appartengono alla classe di enzimi delle metalloproteasi e sono l’MMP-2 e l’MMP-
9 (63-65): è documentato che queste molecole sono prodotte da cellule infiammatorie,
specialmente macrofagi attivati, in altre patologie dell’apparato vascolare come gli aneurismi
addominali aortici (66,67).

25
La concentrazione plasmatica e tessutale del fattore di crescita dell’endotelio vascolare
(VEGF), in pazienti con aneurismi cerebrali sanguinanti e non, risulta elevata rispetto a quella
di controlli sani (68,69).
Stress emodinamici, rimodellamento vascolare, infiammazione ed aneurismi
cerebrali.
Il rimodellamento della parete vascolare è strettamente associato agli stress emodinamici: un
vaso sanguigno qualsiasi è un organo attivo che può essere soggetto a processi dinamici di
rimodellamento in risposta a cambiamenti nel flusso sanguigno, nella pressione del sangue e
nel metabolismo (70,71). Questo rimodellamento indotto da un elevato flusso sanguigno è un
processo adattativo della parete del vaso che serve a ridurre gli stress da taglio, esercitati sulla
parete, a livelli fisiologici di base incrementando il diametro del lume sanguigno attraverso
piccole diminuzioni dello spessore del vaso (72-74).
Gli aneurismi intracranici sono localizzati presso le biforcazioni tra le arterie che
costituiscono il “poligono di Willis”: a livello di queste biforcazioni, si creano anomali stress
emodinamici (ad esempio turbolenze nel flusso) che portano ad un aumento della pressione
esercitata sulla parete dal flusso sanguigno, a cui fa seguito un rimodellamento della parete
del vaso che può giocare un ruolo importante nella formazione, nello sviluppo e nella
conseguente rottura di un aneurisma (75-79).
Gli stress emodinamici possono indurre un processo infiammatorio attivando le cellule
endoteliali e leucocitarie con conseguente aumento dell’espressione di molecole di adesione e
di chemochine (80-84). Queste molecole attraggono neutrofili e monociti circolanti
facilitando la loro invasione all’interno della parete del vaso dove secerneranno citochine e
proteasi ( metalloproteasi ed elastasi) promuovendo il rimodellamento della parete (73,83).
Mentre un normale rimodellamento in risposta ad un incremento nel flusso sanguigno è un
processo fisiologico, adattativo, con cui il vaso cerca di limitare gli stress da taglio, lo

26
sviluppo di un aneurisma, legato ad un anomalo rimodellamento, è una manifestazione
patologica dovuta al fallimento del tentativo di ristabilire l’omeostasi.
La formazione di un aneurisma intracranico può quindi iniziare come un normale processo
fisiologico di rimodellamento che diventa anomalo, asimmetrico e focalizzato a seguito della
particolare geometria dei vasi coinvolti. Successivamente si ha la formazione di un
microaneurisma che crescendo sarà soggetto a più elevati stress da taglio (76). In quest’area si
avrà sia il danneggiamento che l’attivazione delle cellule endoteliali che causeranno un
accumulo di cellule infiammatorie. Quando l’infiammazione sarà intensa, la maggior parte dei
componenti strutturali della parete del vaso sarà distrutta e ciò guiderà alla rottura del vaso
stesso(20).

27
PARAMETRI BIOLOGICI ANALIZZATI
Sulla base dell’evidente natura flogistica dell’aneurisma, abbiamo concentrato la nostra
attenzione sul ruolo di alcune citochine proinfiammatorie, del fattore di crescita dell’endotelio
vascolare e del sistema delle metalloproteasi che sono i parametri che abbiamo investigato nel
corso del nostro studio.
SISTEMA DELLE METALLOPROTEASI.
MMP-9.
Questa molecola appartiene alla famiglia delle metalloproteasi della matrice (MMPs), una
famiglia di endopeptidasi zinco-dipendenti deputata alla degradazione delle proteine della
matrice extracellulare (85-87). Queste molecole sono secrete come zimogeni (pro-MMPS),
che vengono attivati, tramite un taglio proteolitico, da una varietà di proteasi; sono inibite
dagli inibitori tissutali specifici delle metalloproteasi (TIMPS) e dalla α2-macroglobulina (85-
87). La regolazione dell’attività delle MMPs è importante nei processi di rimodellamento
tissutale, nell’infiammazione, nella crescita tumorale e nello sviluppo delle metastasi (87,88).
L’MMP-9 umano, conosciuto come gelatinasi B, è secreto come uno zimogeno di 92 kDa
(pro-MMP-9), attivato tramite un taglio proteolitico nei pressi del residuo 87, che produce una
molecola attiva di 82 kDa. Il pro-MMP-9 può essere attivato dall’ MMP-3, da certe proteinasi
batteriche (85,87). L’MMP-9 è inibito dall’α2-macroglobulina o dal TIMP-1, che si legano sia
alla forma inattiva che a quella attiva (87).
Il pro-MMP-9 è secreto da monociti, macrofagi, neutrofili, cheratinociti, fibroblasti,
osteoclasti, cellule endoteliali e varie cellule tumorali (85,88-90) mentre la sua espressione è
regolata positivamente da TGF-β1, IL-1β, TGF-α, TNF-α, VEGF e l’IL-1α (88,89,90,96).
I substrati per l’ MMP-9 includono gelatina, collageni di tipo IV,V,VII,X e XI, il fibrinogeno
e l’IL-1β (87).

28
L’ MMP-9 è coinvolta nell’infiammazione, nel rimodellamento tissutale, nella guarigione
delle ferite, nella mobilizzazione di fattori di crescita legati alla matrice come il VEGF (90-
92) ed a condizioni patologiche come l’anormale deposizione di collagene, associata al cancro
al pancreas, gli aneurismi cerebrali, gli aneurismi del tratto addominale dell’aorta e
l’aterosclerosi dell’arteria carotidea (63,64,66,92).
TIMP-1.
La famiglia di inibitori tissutali delle metalloproteasi (TIMPs) è composta da molecole che
formano, con un rapporto di 1:1, complessi non covalenti con le MMPs, bloccando l’accesso
dei substratial sito catalitico di questi enzimi (93).
Il TIMP-1 è una proteina glicosilata, inducibile, composta da un dominio N-terminale,
contenente i siti di legame per la zona che lega i substrati dell’MMP-9, e da un dominio C-
terminale che si lega esternamente a questo sito, aumentando l’affinità totale per l’MMP-9.
Il TIMP-1 lega con alta affinità anche la forma inattiva dell’ MMP-9, formando un complesso
in cui TIMP-1 mantiene la capacità di inibire l’attività di un'altra molecola di MMP-9 (94).
TIMP-1 è sintetizzato diffusamente da molte cellule e tessuti e la sua trascrizione è indotta da
citochine proinfiammatorie, TGF-β1 e da esteri del forbolo (93,94). Molte delle funzioni
fisiologiche del TIMP-1 sono strettamente a quelle dell’ MMP-9, ed un improprio
bilanciamento della produzione di queste due molecole è associato a condizioni patologiche
come le artiti, la crescita tumorale, le metastasi, aneurismi cerebrali e malformazioni
arterovenose cerebrali (63-64,93). Vanno ricordati poi le attività distinte dall’ MMP-9, quali
l’attività eritroide potenziante, ed il ruolo svolto nella sopravvivenza e nella crescita cellulare
(95).

29
CITOCHINE PROINFIAMMATORIE.
L’INTERLEUCHINA 1 β (IL-1β).
I membri della famiglia del gene dell’IL-1 favoriscono un incremento dell’espressione di
geni proinfiammatori, come i geni della cicloossigenasi-2, della sintasi inducibile dell’ossido
nitrico, e l’espressione di molecole d’adesione sulla superficie di cellule endoteliali. Tali
proprietà promuovono l’infiltrazione di cellule infiammatorie ed immunocompetenti nello
spazio extravascolare (96).
Sono state individuate sette molecole appartenenti a questa famiglia di cui tre sono state
profondamente studiate per il loro ruolo in patologia (IL-1α, IL-1β e IL-1 Ra), mentre le
restanti quattro sono state recentemente scoperte ed il loro ruolo in patologia non è
documentato.
L’IL-1β è spesso riscontrato nella circolazione sanguigna e alti livelli di questa molecola è
spesso, ma non sempre, correlata con la severità di una patologia ( 98); è sintetizzato da parte
di monociti attivati come un precursore, mancante di peptide leader, che rimane all’interno del
citosol fino a quando non subisce un taglio, da parte dell’enzima convertente l’ IL-1β ( ICE ),
presso una coppia di amminoacidi (acido aspartico-alanina) situati nei residui 116-117, a cui
fa seguito il trasporto all’ esterno della cellula (96). Se il pro-IL-1β è localizzato
nell’ambiente extracellulare, in seguito a necrosi, ci sono proteasi extracellulari, tra cui le
MMPs , che possono formare la forma attiva della molecola (96). È documentato il suo ruolo
in patologie legate all’ infiammazione (96) e nello sviluppo degli aneurismi cerebrali. Inoltre
favorisce l’induzione dell’espressione dell’ MMPs (97).

30
L’INTERLEUCHINA 18 (IL-18).
Questa citochina è stata per la prima volta descritta nel 1989 come un fattore inducente l’
IFN-γ (98). Così come per il pro-IL-1β, il precursore dell’IL-18 (pro-IL-18) subisce un taglio
da parte dell’ICE, in un residuo di acido aspartico localizzato nella sequenza aminoacidica
dell’ N-terminale. La forma matura dell’IL-18 così formata presenta un’elevata omologia con
la forma matura dell’IL-1β.
L’azione svolta dall’IL-18 nel processo proinfiammatorio è dovuta sia all’induzione, con la
compartecipazione di IL-12 come molecola costimolante, dell’IFN-γ e di altre citochine nelle
cellule T e nelle cellule natural killer (NK) (98), sia alla modulazione di metalloproteasi della
matrice e alla migrazione di cellule natural killer all’interno del sito infiammatorio (98).
FATTORE DI CRESCITA DELL’ENDOTELIO VASCOLARE VEGF.
Il VEGF, conosciuto anche come fattore di permeabilità vascolare (VPF), è una glicoproteina
omodimerica legante l’eparina, con potenti attività angiogeniche, fitogeniche e di aumento
della permeabilità vascolare, specifiche per le cellule endoteliali (99). Il VEGF è espresso in
numerose cellule tumorali umane e di topo . Nei tessuti normali l’espressione del VEGF è
stata trovata in macrofagi attivati, cheratinociti, cellule mesangiali e dell’epitelio viscerale del
glomerulo renale , epatociti, cellule muscolari lisce(100). L’espressione del VEGF è regolata
positivamente dagli esteri del forbolo, dal TGF-β,da condizioni di ipossia (6-10,18) mentre il
suo rilascio all’interno della matrice extracellulare è favorito dalle MMPs (91).
In letteratura è stato riportato che citochine proinfiammatorie, come l’IL-6 e l’IL-1β,
inducono l’espressione di VEGF in varie cellule. Il gene per il VEGF umano, organizzato in
otto esoni, come risultato di un processo di splicing alternativo, produce 4 trascritti che
codificheranno quattro forme mature di VEGF ognuna preceduta da un peptide segnale di 26
aminoacidi.(99).

31
In vitro il VEGF induce la proliferazione di cellule endoteliali, il rilascio del fattore di von
Willebrand dalle cellule endotelialie l’espressione dell’attività di fattori tissutali in cellule
endoteliali e monociti. È stata inoltre vista un’azione chemiotattica per i monociti e
osteoblasti (99).
In vivo il VEGF può indurre l’angiogenesi così come l’incremento della permeabilità
microvascolare favorendo lo stravaso del fibrinogeno plasmatici e il conseguente deposito di
fibrina che altera la matrice extracellulare. La modificazione della matrice extracellulare
promuove la migrazione di macrofagi, fibroblasti e cellule endoteliali. Queste proprietà
implicano un ruolo importante nell’infiammazione e nell’angiogenesi normale e patologica,
un processo associato alla guarigione delle ferite, alla crescita embrionale e alla crescita e
metastasi di tumori in forma solida. Elevati livelli di VEGF sono stati osservati nei fluidi
sinoviali di pazienti con artrite reumatoide , ed in campioni di plasma di pazienti colpiti da
cancro, dalla sindrome di Kawasaki, da aneurismi intracranici (69).

32
SCOPO DEL LAVORO

33
La rottura di un aneurisma cerebrale, con la conseguente emorragia a livello dello spazio
subaracnoideo, ancora oggi presenta un alto tasso di morbosità e mortalità a causa
dell’evoluzione pressoché asintomatica della patologia.
In collaborazione con la clinica di neurochirurgia dell’ospedale Santa Chiara di Pisa, abbiamo
pertanto allestito un protocollo sperimentale per valutare il coinvolgimento di alcuni markers
biologici, associati al processo infiammatorio, nello sviluppo di un aneurisma cerebrale
(MMP-9, TIMP-1, IL-1β e VEGF).
Particolare interesse abbiamo posto nel valutare il possibile coinvolgimento, nella patogenesi
di questa patologia della citochina proinfiammatoria IL-18, dato che si tratta di una molecola
di recente scoperta.
Inoltre abbiamo finalizzato lo studio alla costruzione di un ipotetico network di attivazione di
segnali coinvolti nella regolazione dello sviluppo dell’aneurisma e nella sua rottura.

34
PROTOCOLLO SPERIMENTALE

35
Sono stati reclutati, presso la clinica di neurochirurgia dell’ospedale Santa Chiara di Pisa, 36
volontari suddivisi nei seguenti tre gruppi sperimentali:
1. gruppo controllo: composto da 10 persone in cui è stata riscontrata la presenza
né di aneurismi intracranici, né di altre patologie cerebrovascolari;
2. gruppo con aneurisma non sanguinante: composto da 10 pazienti in cui è stata
riscontrata la presenza di aneurismi prima di una loro eventuale rottura;
3. gruppo con aneurisma sanguinante: composto da 16 pazienti in cui si è
manifestata una emorragia a livello dello spazio subaracnoideo.
I pazienti, una volta ottenuto il loro consenso per la partecipazione a questo studio, sono stati
sottoposti a prelievo di campioni ematici durante le normali procedure clinico-ospedaliere.
Una volta che tali campioni sono stati centrifugati a 10.000 G per 10 minuti, si è provveduto
alla raccolta dei surnatanti, successivamente conservati a – 80 °C.
Si è poi proceduto alla valutazione delle concentrazioni plasmatiche delle citochine
proinfiammatorie, IL-1β ed IL-18, del fattore di crescita dell’endotelio vascolare (VEGF),
della metalloproteasi 9 (MMP-9) e dell’inibitore tissutale della metalloproteasi (TIMP-1).

36
MATERIALI E METODI

37
La valutazione dei parametri studiati, nei campioni di plasma raccolti, è stata effettuata
utilizzando saggi immunoenzimatici (o “ELISA”, EnzimeLinkedImmunoSorbentAssay).
TEST ELISA
Questo metodo permette la rilevazione e la quantificazione di sostanze come peptidi, proteine,
anticorpi e ormoni. In un ELISA, un antigene deve essere immobilizzato su una superficie
solida in modo da poterlo facilmente identificare attraverso un anticorpo coniugato ad un
enzima. La rivelazione è ottenuta incubando questo complesso enzimatico con un substrato
che produce un prodotto rilevabile. L'elemento fondamentale della strategia di rilevazione è
pertanto l'altissima specificità dell'interazione tra anticorpo e antigene.
FIG. 3: micropiastra da 96 pozzetti per dosaggio ELISA (www.bendermedsystems.com)
I saggi ELISA sono solitamente eseguiti in micropiastre da 96 o 384 pozzetti che legano
passivamente anticorpi e proteine. Il fatto che i reagenti dell'ELISA siano immobilizzati sulla
superficie della micropiastra facilita enormemente la separazione del materiale legato da
quello non legato durante il saggio.
I saggi ELISA non competitivi garantiscono elevata sensibilità poiché, anche a bassissime
concentrazioni di analita, un'alta frazione di esso reagirà con l'anticorpo in eccesso.

38
Sono però meno specifici dei saggi competitivi, anche se l'impiego di anticorpi monoclonali e
del metodo "sandwich" ne ha notevolmente aumentato la specificità. Gli enzimi di uso più
comune sono la perossidasi (generalmente quella estratta da radice di rafano o Horse Radish
Peroxidase, HRP) che catalizza la scissione di H2O2 e la fosfatasi alcalina (AP) che rimuove il
fosfato da molecole fosforilate. Sono impiegati anche altri enzimi, pure se meno diffusamente,
come la b-galattosidasi (bGal), che idrolizza il lattosio. È disponibile un'ampia gamma di
substrati per ELISA basati sia su coniugati HRP sia AP. La scelta del substrato dipende dal
grado di sensibilità di rivelazione necessario e dalla strumentazione disponibile
(spettrofotometro, fluorimetro o luminometro).
FIG. 4: tipologie di ELISA (www.bendermedsystems.com)

39
Il saggio che abbiamo utilizzato nel nostro studio è L’ELISA sandwich (fig.4) che è, tra i
saggi immunoenzimatici, il più usato e prevede la cattura degli antigeni dal campione sulla
parete di microcellette ricoperte da anticorpi, detti anticorpi di cattura.
L'antigene catturato viene evidenziato ricoprendolo con un secondo strato di anticorpi
specifici - gli anticorpi di rivelazione - con o senza ulteriori fasi di amplificazione.
L'anticorpo secondario di rilevazione è spesso coniugato a un enzima e la digestione del
substrato è la dimostrazione della presenza di un determinato antigene: questa tecnica è nota
come ELISA "sandwich".Il sistema sandwich è il più sensibile e specifico per il rilevamento
di basse quantità di antigene.
Nei sistemi sandwich si possono usare come anticorpi di cattura e di rilevazione sia anticorpi
monoclonali sia policlonali. I monoclonali (Mab) sono specifici verso un singolo epitopo,
permettendo la rilevazione fine di piccole differenze quantitative di antigene. Spesso si usa un
anticorpo policlonale come anticorpo di cattura per riuscire a trattenere la maggior quantità
possibile di antigene e poi si usa un monoclonale come anticorpo di rilevazione per aumentare
la specificità.
Una considerazione importante quando si progetta un saggio ELISA sandwich è che
l'anticorpo di cattura e quello di rilevazione devono riconoscere due epitopi non sovrapposti o
comunque non troppo vicini.
Anticorpi di cattura e di rilevazione che non interferiscono uno con l'altro e che si possono
legare contemporaneamente al proprio bersaglio sono considerati una "coppia" e sono adatti
per un saggio ELISA sandwich. Entrambi gli anticorpi hanno il frammento F(ab')2: questo
sistema permette di eliminare l'interferenza con anticorpi eterofili presenti in campioni
biologici come siero e plasma. Questi kit permettono una procedura 'two step' oppure 'one
step' se si usano piastre pronte già rivestite di avidina.

40
PROTOCOLLO STANDARD DEL SAGGIO ELISA SANDWICH
Nel nostro studio abbiamo utilizzato cinque kit :
• “Quantikine TIMP-1 Immunoassay” kit, della “R&D Systems”;
• “Quantikine MMP-9 Immunoassay” kit, della “R&D Systems”;
• “Quantikine VEGF Immunoassay” kit, della “R&D Systems”;
• “Human IL-18 Instant ELISA” kit, della “Bender MedSystems”;
• “Human IL-1β CytoSetTM” kit, della “BioSource”.
Si riporta di seguito il protocollo standard del saggio ELISA-sandwich utilizzato nella
valutazione dei parametri analizzati:
(il fondo di ogni pozzetto della piastra è già rivestito dall’anticorpo monoclonale di cattura
specifico per la molecola ricercata ).
1. preparazione di tutti i reagenti e degli standards,
2. rimozione delle strisce di pozzetti dalla struttura della piastra,
3. aggiunta degli standard e dei campioni nei pozzetti (vengono effettuate diverse
diluizioni con assay buffer a seconda della molecola ricercata),
4. i pozzetti si mettono ad incubare per 2 ore a temperatura ambiente (per il
TIMP-1 e l’MMP-9 è richiesta un’agitazione di 500 ± 50 rpm),
5. dopo avere svuotato i pozzetti, vengono effettuati 3-4 lavaggi con 400 µl di
wash buffer, per lavare via le sostanze non legate,
6. vengono aggiunti 200 µl di coniugato - anticorpo policlonale legante un
enzima - per ogni pozzetto,

41
7. si lasciano ad incubare i pozzetti per un’ora a temperatura ambiente (2 ore nel
caso del VEGF; per il TIMP-1 e l’MMP-9 è richiesta un ‘agitazione di 500±
50 rpm),
8. ripetizione del passaggio 5,
9. vengono aggiunti 200 µl della Soluzione Substrato con successiva incubazione
per 20-30 minuti a temperatura ambiente – in questa fase và evitata
l’esposizione alla luce,
10. aggiungere 50 µl della Soluzione di Stop,
11. misurare l’assorbanza di ogni pozzetto, entro 30 minuti, usando un lettore di
piastra settato a 450 nm di lunghezza d’onda.
L’“Instant ELISA”, usato per valutare i livelli di IL-18 è una tipologia di saggio
immunoenzimatico innovativa che prevede un protocollo di minor durata in quanto sia
l’anticorpo monoclinale di cattura che il coniugato sono presenti all’interno dei pozzetti prima
dell’aggiunta dei campioni e degli standards.

42
RISULTATI
43
SISTEMA DELLE METALLOPROTEASI
VALORI PLASMATICI DI MMP-9.
MEDIA ERR.ST
Controllo 92,76 21,67
Aneurisma non sanguinante 223,38 31,59
Aneurisma Sanguinante 304,16 48,60
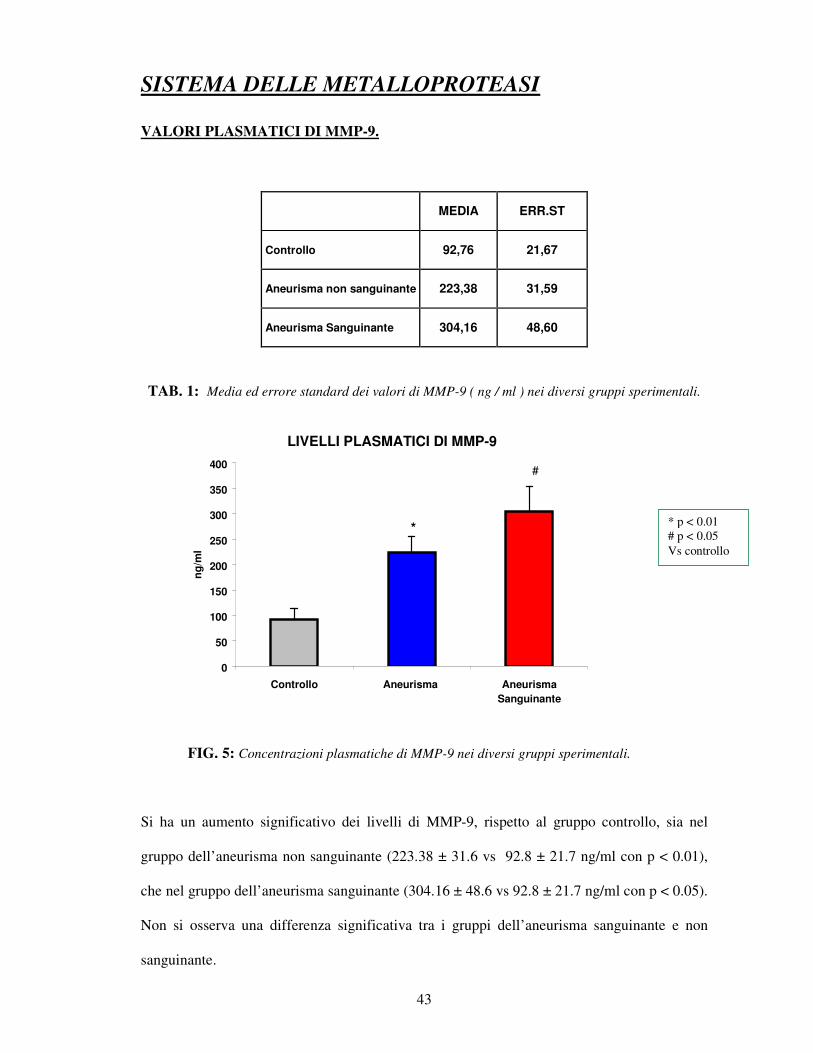
TAB. 1: Media ed errore standard dei valori di MMP-9 ( ng / ml ) nei diversi gruppi sperimentali.
LIVELLI PLASMATICI DI MMP-9
0
50
100
150
200
250
300
350
400
Controllo Aneurisma Aneurisma
Sanguinante
ng
/ml
*
#
FIG. 5: Concentrazioni plasmatiche di MMP-9 nei diversi gruppi sperimentali.
Si ha un aumento significativo dei livelli di MMP-9, rispetto al gruppo controllo, sia nel
gruppo dell’aneurisma non sanguinante (223.38 ± 31.6 vs 92.8 ± 21.7 ng/ml con p < 0.01),
che nel gruppo dell’aneurisma sanguinante (304.16 ± 48.6 vs 92.8 ± 21.7 ng/ml con p < 0.05).
Non si osserva una differenza significativa tra i gruppi dell’aneurisma sanguinante e non
sanguinante.
* p < 0.01 # p < 0.05 Vs controllo
44
VALORI PLASMATICI DEL TIMP-1.
MEDIA ERR.ST.
Controllo 120,17 15,21
Aneurisma non sanguinante 135,72 21,91
Aneurisma Sanguinante 128,69 13,37
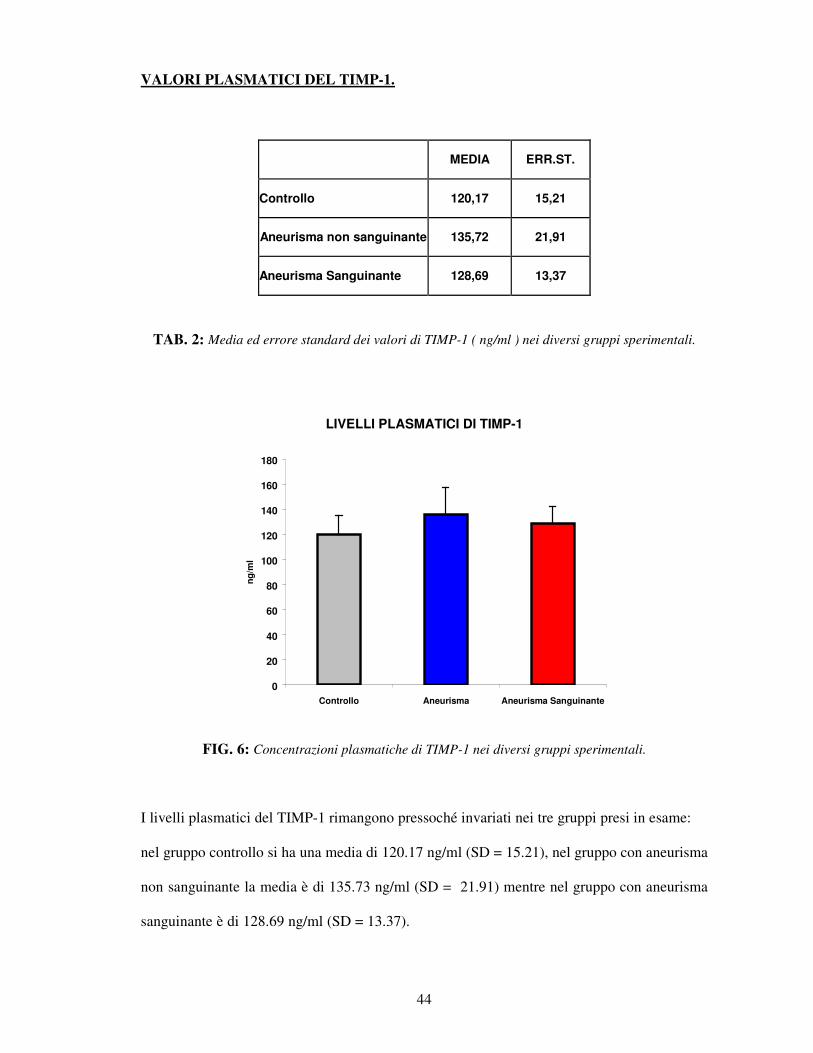
TAB. 2: Media ed errore standard dei valori di TIMP-1 ( ng/ml ) nei diversi gruppi sperimentali.
LIVELLI PLASMATICI DI TIMP-1
0
20
40
60
80
100
120
140
160
180
Controllo Aneurisma Aneurisma Sanguinante
ng
/ml
FIG. 6: Concentrazioni plasmatiche di TIMP-1 nei diversi gruppi sperimentali.
I livelli plasmatici del TIMP-1 rimangono pressoché invariati nei tre gruppi presi in esame:
nel gruppo controllo si ha una media di 120.17 ng/ml (SD = 15.21), nel gruppo con aneurisma
non sanguinante la media è di 135.73 ng/ml (SD = 21.91) mentre nel gruppo con aneurisma
sanguinante è di 128.69 ng/ml (SD = 13.37).
45
RAPPORTO MMP-9/TIMP-1
MEDIA ERR ST
Controllo 0,96 0,32
Aneurisma non sanguinante 3,27 0,76
Aneurisma sanguinante 3,38 0,62
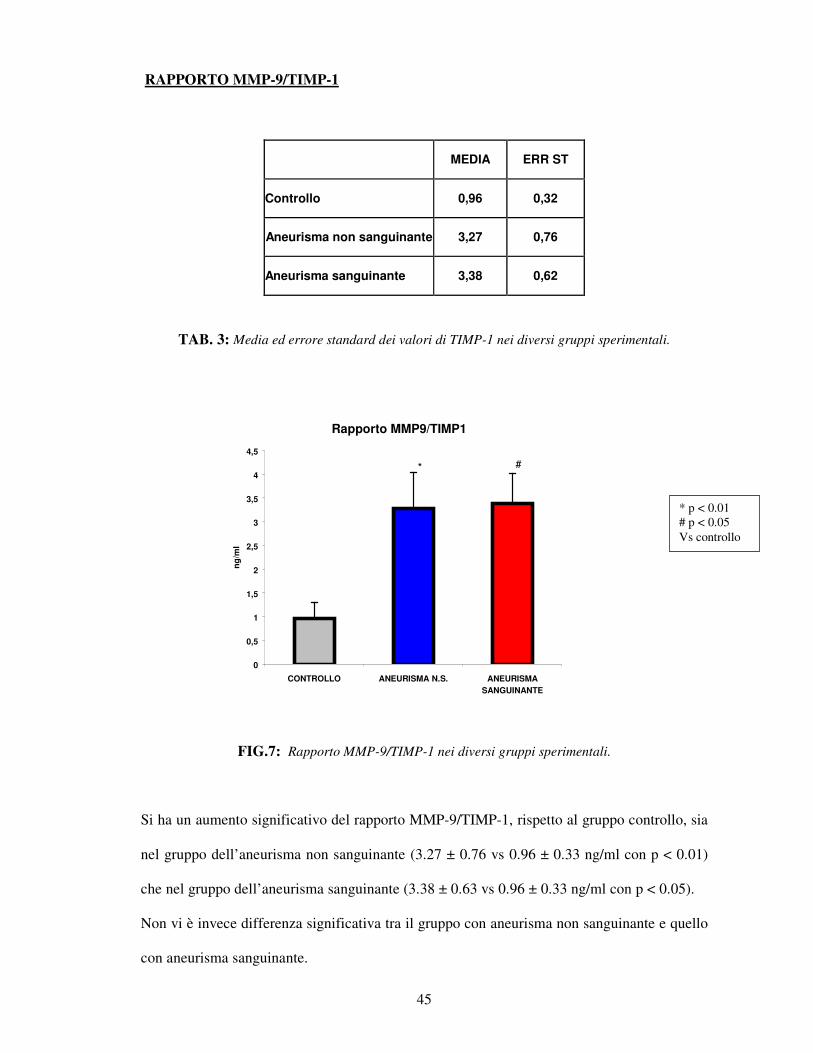
TAB. 3: Media ed errore standard dei valori di TIMP-1 nei diversi gruppi sperimentali.
Rapporto MMP9/TIMP1
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
CONTROLLO ANEURISMA N.S. ANEURISMA
SANGUINANTE
ng
/ml
* #
FIG.7: Rapporto MMP-9/TIMP-1 nei diversi gruppi sperimentali.
Si ha un aumento significativo del rapporto MMP-9/TIMP-1, rispetto al gruppo controllo, sia
nel gruppo dell’aneurisma non sanguinante (3.27 ± 0.76 vs 0.96 ± 0.33 ng/ml con p < 0.01)
che nel gruppo dell’aneurisma sanguinante (3.38 ± 0.63 vs 0.96 ± 0.33 ng/ml con p < 0.05).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante.
* p < 0.01 # p < 0.05 Vs controllo
46
CITOCHINE PROINFIAMMATORIE
VALORI PLASMATICI DI IL-1ββββ.
MEDIA ERR. ST.
Controllo 19,66 3,76
Aneurisma non sanguinante 74,50 21,27
Aneurisma sanguinante 58,05 16,51
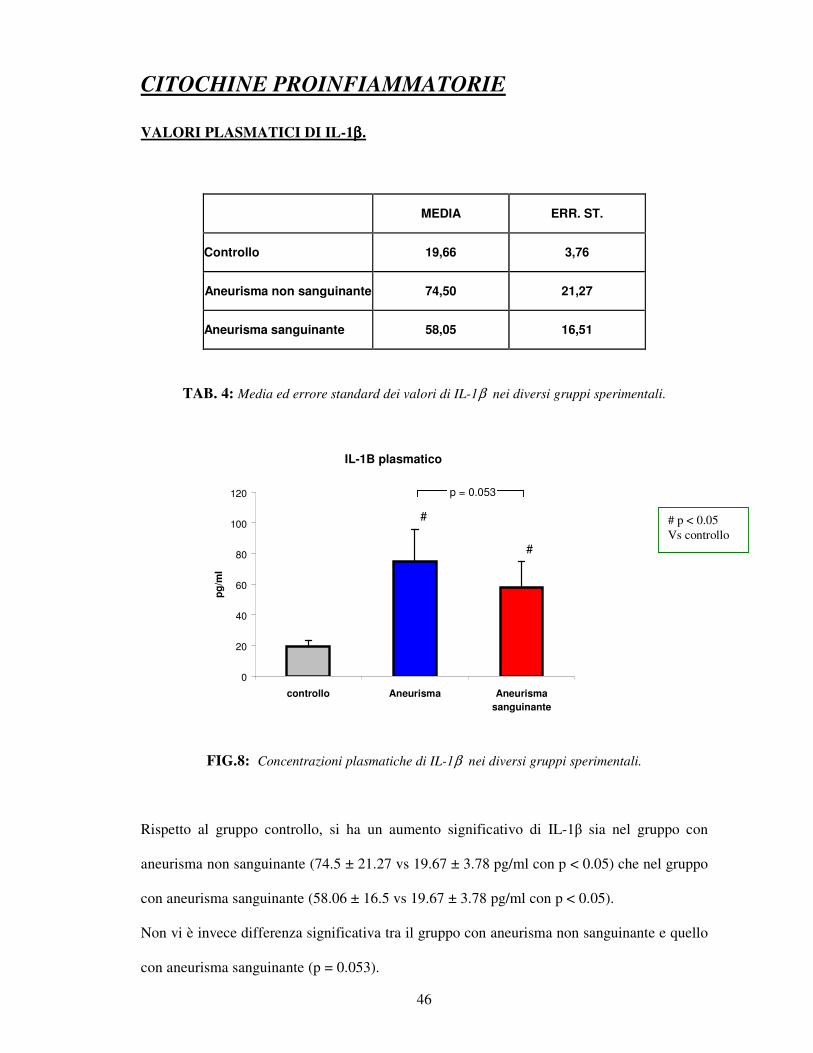
TAB. 4: Media ed errore standard dei valori di IL-1β nei diversi gruppi sperimentali.
IL-1B plasmatico
0
20
40
60
80
100
120
controllo Aneurisma Aneurisma
sanguinante
pg
/ml
#
#
p = 0.053
FIG.8: Concentrazioni plasmatiche di IL-1β nei diversi gruppi sperimentali.
Rispetto al gruppo controllo, si ha un aumento significativo di IL-1β sia nel gruppo con
aneurisma non sanguinante (74.5 ± 21.27 vs 19.67 ± 3.78 pg/ml con p < 0.05) che nel gruppo
con aneurisma sanguinante (58.06 ± 16.5 vs 19.67 ± 3.78 pg/ml con p < 0.05).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante (p = 0.053).
# p < 0.05 Vs controllo
47
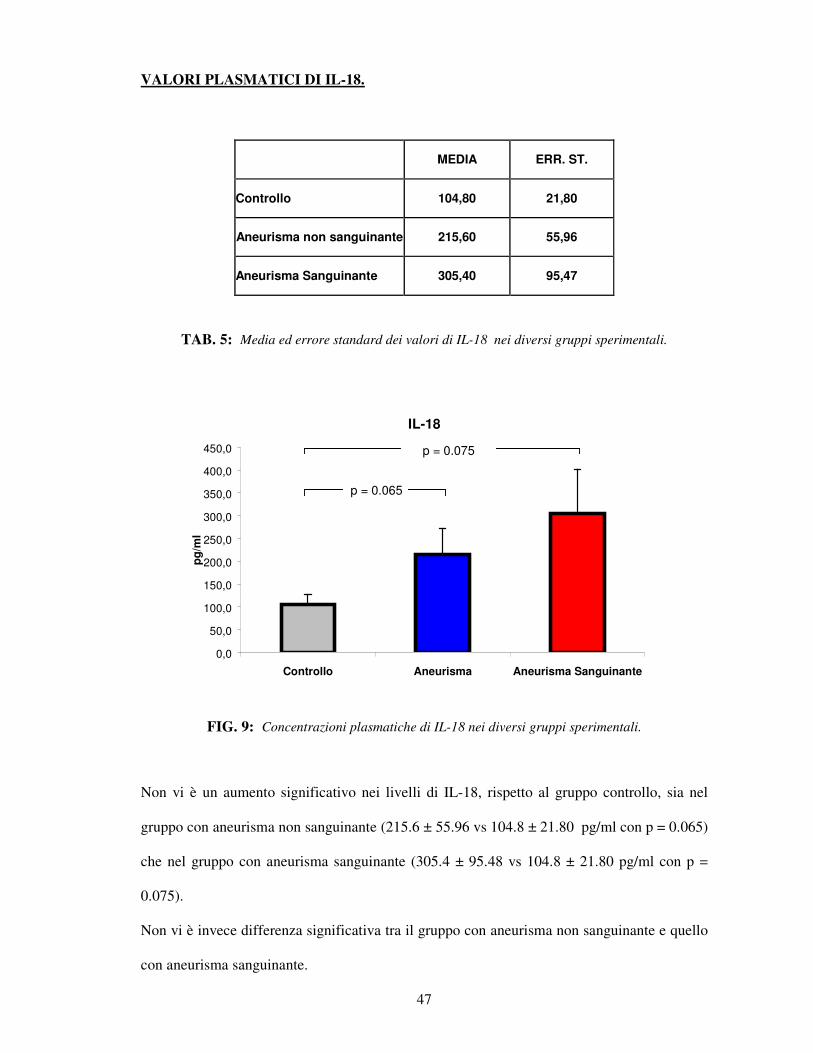
VALORI PLASMATICI DI IL-18.
MEDIA ERR. ST.
Controllo 104,80 21,80
Aneurisma non sanguinante 215,60 55,96
Aneurisma Sanguinante 305,40 95,47
TAB. 5: Media ed errore standard dei valori di IL-18 nei diversi gruppi sperimentali.
IL-18
0,0
50,0
100,0
150,0
200,0
250,0
300,0
350,0
400,0
450,0
Controllo Aneurisma Aneurisma Sanguinante
pg
/ml
p = 0.075
p = 0.065
FIG. 9: Concentrazioni plasmatiche di IL-18 nei diversi gruppi sperimentali.
Non vi è un aumento significativo nei livelli di IL-18, rispetto al gruppo controllo, sia nel
gruppo con aneurisma non sanguinante (215.6 ± 55.96 vs 104.8 ± 21.80 pg/ml con p = 0.065)
che nel gruppo con aneurisma sanguinante (305.4 ± 95.48 vs 104.8 ± 21.80 pg/ml con p =
0.075).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante.
48
FATTORE DI CRESCITA DELL’ENDOTELIO VASCOLARE
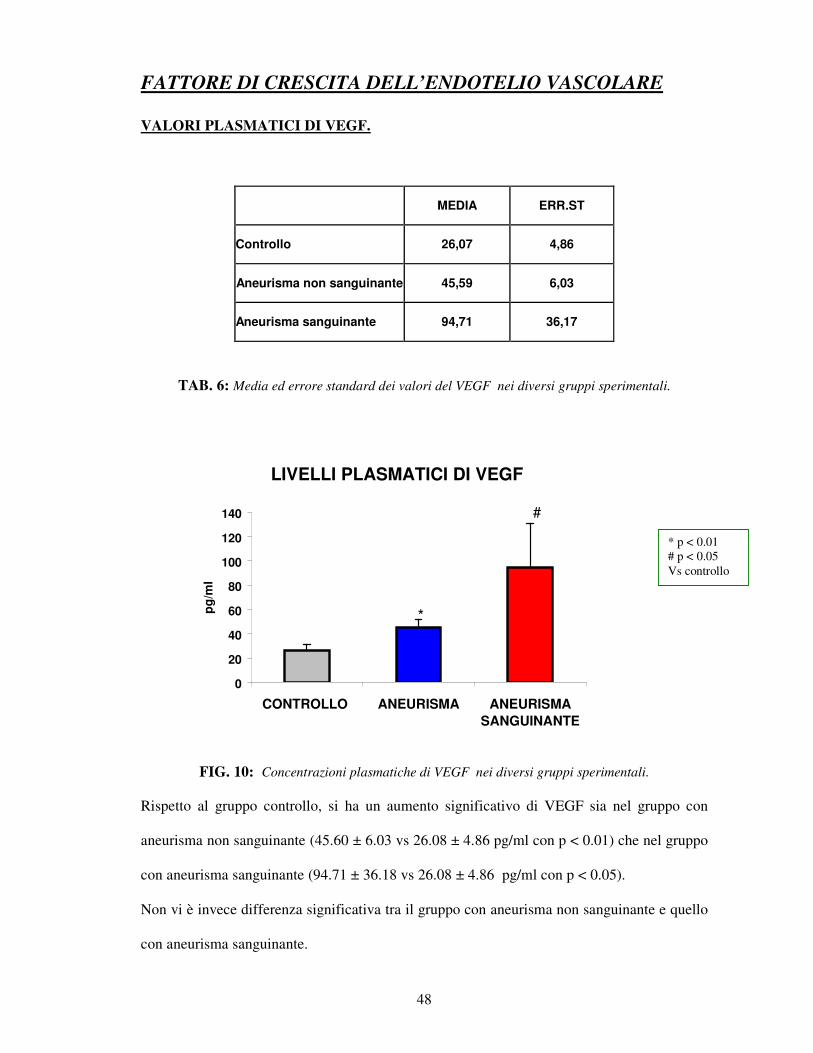
VALORI PLASMATICI DI VEGF.
MEDIA ERR.ST
Controllo 26,07 4,86
Aneurisma non sanguinante 45,59 6,03
Aneurisma sanguinante 94,71 36,17
TAB. 6: Media ed errore standard dei valori del VEGF nei diversi gruppi sperimentali.
LIVELLI PLASMATICI DI VEGF
0
20
40
60
80
100
120
140
CONTROLLO ANEURISMA ANEURISMA
SANGUINANTE
pg
/ml
*
#
FIG. 10: Concentrazioni plasmatiche di VEGF nei diversi gruppi sperimentali.
Rispetto al gruppo controllo, si ha un aumento significativo di VEGF sia nel gruppo con
aneurisma non sanguinante (45.60 ± 6.03 vs 26.08 ± 4.86 pg/ml con p < 0.01) che nel gruppo
con aneurisma sanguinante (94.71 ± 36.18 vs 26.08 ± 4.86 pg/ml con p < 0.05).
Non vi è invece differenza significativa tra il gruppo con aneurisma non sanguinante e quello
con aneurisma sanguinante.
* p < 0.01 # p < 0.05 Vs controllo
49
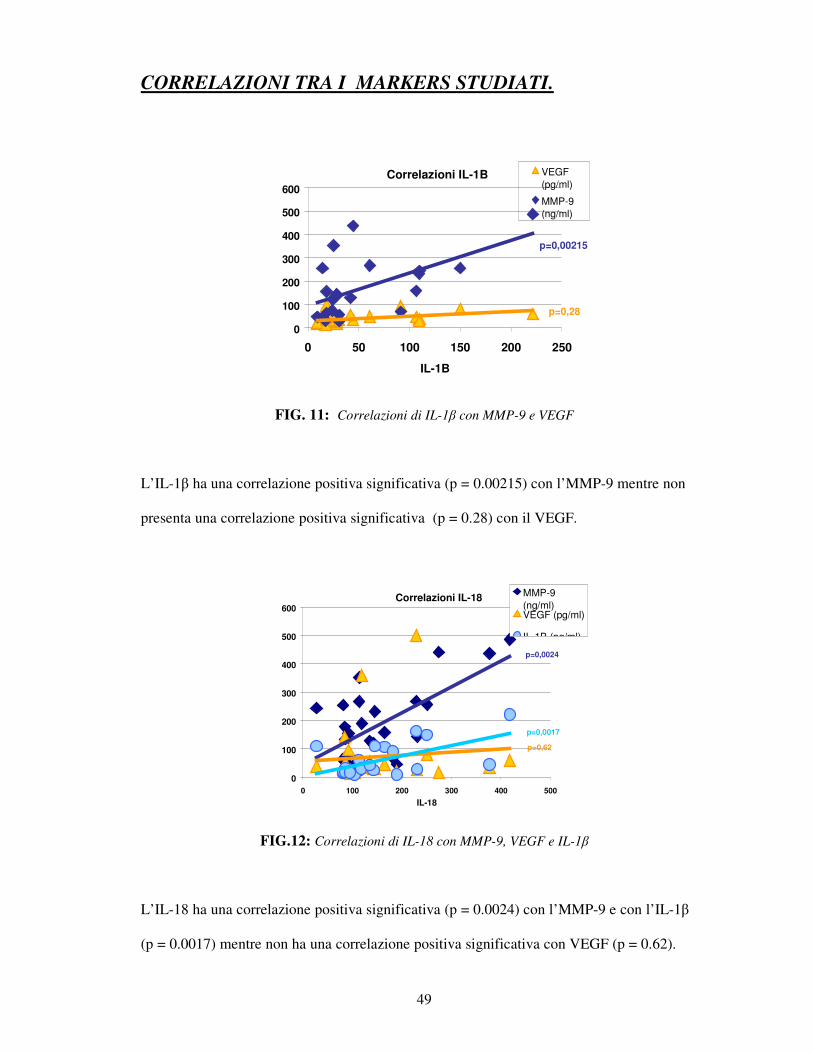
CORRELAZIONI TRA I MARKERS STUDIATI.
Correlazioni IL-1B
p=0,28
p=0,00215
0
100
200
300
400
500
600
0 50 100 150 200 250
IL-1B
VEGF
(pg/ml)
MMP-9
(ng/ml)
FIG. 11: Correlazioni di IL-1β con MMP-9 e VEGF
L’IL-1β ha una correlazione positiva significativa (p = 0.00215) con l’MMP-9 mentre non
presenta una correlazione positiva significativa (p = 0.28) con il VEGF.
Correlazioni IL-18
p=0,0024
p=0,62
p=0,0017
0
100
200
300
400
500
600
0 100 200 300 400 500
IL-18
MMP-9
(ng/ml)VEGF (pg/ml)
IL-1B (pg/ml)
FIG.12: Correlazioni di IL-18 con MMP-9, VEGF e IL-1β
L’IL-18 ha una correlazione positiva significativa (p = 0.0024) con l’MMP-9 e con l’IL-1β
(p = 0.0017) mentre non ha una correlazione positiva significativa con VEGF (p = 0.62).

50
Correlazioni VEGF
p=0,011
0
100
200
300
400
500
600
0 20 40 60 80
VEGF
MMP-9 (pg/ml)
FIG.13: Correlazioni di VEGF con MMP-9
Il VEGF presenta una correlazione positiva significativa solo con l’MMP-9 (p = 0.011).

51
DISCUSSIONE E CONCLUSIONI

52
DISCUSSIONE DEI RISULTATI.
VARIAZIONE DEI LIVELLI PLASMATICI DEI MARKERS STUDIATI.
I risultati ottenuti comparando campioni plasmatici provenienti da soggetti con aneurisma,
sanguinante e non, con quelli provenienti da una popolazione di controllo, permettono di
confermare il coinvolgimento di alcuni markers biologici nella patologia dell’aneurisma
cerebrale. In particolare per il sistema delle metalloproteasi abbiamo dimostrato un aumento
del rapporto MMP-9/TIMP-1 dovuto al fatto che mentre si osserva un aumento significativo
della concentrazione di MMP-9, non vi sono variazioni di rilievo nei livelli di TIMP-1.
Questo aspetto risulta in linea con quanto documentato in letteratura (60,61,63-65,100) a
conferma dell’ipotesi di un anomalo rimodellamento della parete vascolare dell’arteria
cerebrale associato all’indebolimento della parete stessa (20).
Il fattore di crescita dell’endotelio vascolare (VEGF) è uno dei più potenti fattori
angiogenetici, tuttavia alcuni studi di immunoistochimica hanno dimostrato un
coinvolgimento del VEGF nel favorire lo sviluppo di malformazioni vascolari cerebrali
attraverso un aumento della sua espressione locale (102;103). La presenza nella parete
dell’aneurisma di leucociti può causare il rilascio di questo fattore di crescita che, insieme al
fattore di crescita basico dei fibroblasti (b-FGF), è coinvolta nella formazione di un
aneurisma; infatti la presenza di questi due fattori di crescita è stata evidenziata
immunoistochimicamente nella parete di aneurismi cerebrali (69). Recentemente uno studio
condotto su pazienti con aneurismi non sanguinanti ha riportato elevati livelli di VEGF in
questi ultimi rispetto ai controlli sani suggerendo un suo potenziale contributo nella
formazione e nella crescita di aneurismi intracranici (68).

53
I dati ottenuti dal nostro studio sono pertanto perfettamente in linea con la letteratura
confermando l’aumento significativo del VEGF in aneurismi non sanguinanti (p<0.01) e
sanguinanti (p<0.05), rispetto a controlli sani, ottenuto nel nostro studio.
L’aumento significativo dei livelli plasmatici della citochina proinfiammatoria IL-1ββββ negli
aneurismi sanguinanti e non sanguinanti rispetto a controlli sani conferma quanto pubblicato
da Moriwaki et al. (2006): in topi knockout per il gene dell’ IL-1β (IL-1β-/-), nei quali
veniva indotta la formazione di un aneurisma cerebrale, i cambiamenti associati ad una fase
più avanzata dell’aneurisma cerebrale erano pressoché assenti, permettendo di ipotizzare che
IL-1β giochi un ruolo importante nella progressione di un aneurisma cerebrale (105).
A tal proposito risulta interessante quanto emerso dal nostro studio: i pazienti con aneurisma
presentano una concentrazione plasmatica di IL-1β superiore rispetto al gruppo con aneurisma
sanguinante; se pure il dato emerso non risulti ancora significativo (p=0.053) è tuttavia
possibile ipotizzarne una evoluzione in tal senso ampliando la numerosità dei campioni, infatti
il gruppo con aneurisma sanguinante (n=16) presenta un numero di soggetti reclutati superiore
a quello con aneurisma non sanguinante (n=10); questo è dovuto alla difficoltà nel reperire
pazienti con aneurisma non sanguinante rispetto a pazienti con aneurisma sanguinante.
Partendo da questo presupposto è possibile ipotizzare un coinvolgimento abbastanza precoce
di questa citochina nella progressione di un aneurisma cerebrale ed un eventuale spegnimento
del segnale nelle fasi tardive.
Un aspetto innovativo del nostro studio, non essendo ancora stati documentati studi a
riguardo, è stata la valutazione dell’eventuale coinvolgimento dell’IL-18 nel processo di
sviluppo di un aneurisma cerebrale. Il dato emerso indica un aumento, non ancora
significativo, della concentrazione plasmatica di tale citochina sia negli aneurismi non
sanguinanti (p = 0.065) che nei sanguinanti (p = 0.075) rispetto ai controlli sani.

54
In analogia con quanto precedentemente ipotizzato per l’IL-1β, a riguardo dell’esiguo numero
di campioni plasmatici del gruppo con aneurisma non sanguinante, è lecito supporre anche in
questo caso che, disponendo di un campione più numeroso e riducendo la diversa quantitativa
dei due gruppi, sia possibile ottenere un aumento significativo della concentrazione dell’IL-18
solo nel gruppo con aneurisma non sanguinante. questo aspetto confermerebbe l’ipotesi circa
la presenza di segnali precoci nello sviluppo dell’aneurisma cerebrale, con il segnale,
associato a molecole come l’IL-18 e l’IL-1β, che dopo un progressivo aumento si attenua
lentamente mano a mano che ci si avvicina alla rottura dell’aneurisma.
Questo aspetto ha particolare significato se si rammenta che l’azione di queste citochine è
associato alla regolazione di molteplici eventi a valle che costituiscono un network di
regolazioni. Dal nostro studio appare infatti che i livelli di IL-18 sono significativamente
correlati nel corso di un aneurisma cerebrale con l’IL-1β e con l’MMP-9; per quanto riguarda
l’MMP-9 va sottolineata l’ulteriore correlazione con il VEGF. Pertanto è possibile affermare
che l’IL-18 è anch’essa coinvolta nell’infiammazione associata ad aneurisma in quanto
citochina in grado di indurre l’espressione di altre molecole coinvolte in tale processo (96).
Dato che non vi è una correlazione diretta tra l’IL-18 e il VEGF è possibile ipotizzare che
l’influenza di questa citochina venga mediata dall’intermediazione dell’ MMP-9.
Infatti il sistema delle metalloproteasi, oltre a regolare il rimodellamento della matrice
extracellulare, svolge un ruolo chiave nell’angiogenesi, favorendo il rilascio di fattori di
crescita tissutali, durante la loro azione di degradazione della matrice extracellulare (91).

55
Concludendo, sulla base dei dati emersi da questo studio è possibile affermare che:
• L’aneurisma è una patologia caratterizzata da una componente infiammatoria.
• Tra i meccanismi maggiormente coinvolti nello sviluppo di un aneurisma vi sono
l’aumento di citochine proinfiammatorie e l’upregulation del sistema dell’MMP.
• Si può ipotizzare l’esistenza di segnali precoci che attivano cascate di eventi e la cui
espressione viene downregolata con il procedere della patologia.
• Possibili fattori coinvolti nello sviluppo dell’aneurisma sono molecole ad attività
modulatoria e proinfiammatoria indiretta come IL-18 .
Studi futuri verranno effettuati ampliando il numero dei pazienti nei vari gruppi sperimentali.
Sarebbe inoltre interessante andare a valutare l’eventuale coinvolgimento di altri marker
biologici associati all’infiammazione come ad esempio altre citochine proinfiammatorie (IL-
12) o alcune chemochine (IL-8).

56
BIBLIOGRAFIA

57
1. Michael B. Gravanis. Fisiopatologia cardiovascolare. Ed. MC GRAW-HILL.
2. Ramzi S. Cotrn, Collins T, Kumar V. – Robbins : le basi patologiche delle malattie - Ed.
PICCIN, 2000.
3. Maranon G . – Diagnostica eziologia differenziale -. Società editrice universo.1966.
4. Kuivaniemi H, Tromp G, Prockop DJ. Genetic causes of aortic aneurysms. Unlearning at
least part of what the textbooks say. J Clin Invest. 1991 Nov;88(5):1441-4.
5. Prockop DJ. Mutations in collagen genes as a cause of rare and perhaps common diseases
of connective tissue. Acta Paediatr Scand Suppl. 1991;379:55-7; discussion 58.
6. Knox JB, Sukhova GK, Whittemore AD, Libby P.Evidence for altered balance between
matrix metalloproteinases and their inhibitors in human aortic diseases. Circulation. 1997
Jan 7;95(1):205-12.
7. MCCORMICK WF, NOFZINGER JD. SACCULAR INTRACRANIAL ANEURYSMS:
AN AUTOPSY STUDY. J Neurosurg. 1965 Feb;22:155-9.
8. Inagawa T, Hirano A. Autopsy study of unruptured incidental intracranial aneurysms.
Surg Neurol. 1990 Dec;34(6):361-5.
9. Atkinson JL, Sundt TM Jr, Houser OW, Whisnant JP. Angiographic frequency of anterior
circulation intracranial aneurysms.J Neurosurg. 1989 Apr;70(4):551-5.
10. Schievink WI.Intracranial aneurysms.N Engl J Med. 1997 Jan 2;336(1):28-40. Review.
11. Kassell NF, Torner JC, Jane JA, Haley EC Jr, Adams HP.
12. Pasqualino G.L. Panattoni. Anatomia umana.2002.
13. Rinne J, Hernesniemi J, Puranen M, Saari T.Multiple intracranial aneurysms in a defined
population: prospective angiographic and clinical study. Neurosurgery. 1994
Nov;35(5):803-8.

58
14. Raps EC, Rogers JD, Galetta SL, Solomon RA, Lennihan L, Klebanoff LM, Fink ME.
The clinical spectrum of unruptured intracranial aneurysms. Arch Neurol. 1993
Mar;50(3):265-8.
15. Leblanc R.The minor leak preceding subarachnoid hemorrhage. J Neurosurg. 1987
Jan;66(1):35-9.
16. Ostergaard JR. Headache as a warning symptom of impending aneurysmal subarachnoid
haemorrhage. Cephalalgia. 1991 Feb;11(1):53-5.
17. Schievink WI, van der Werf DJ, Hageman LM, Dreissen JJ. Referral pattern of patients
with aneurysmal subarachnoid hemorrhage. Surg Neurol. 1988 May;29(5):367-71.
18. Macchi G. Malattie del sistema nervoso. ED.PICCIN.
19. Teasdale G, Jennett B.Assessment of coma and impaired consciousness. A practical scale.
Lancet. 1974 Jul 13;2(7872):81-4.
20. Hashimoto T, Meng H, Young WL. Neurol. Res. 2006 Jun;28(4):372-80. Intracranial
aneurysms: links among inflammation, hemodynamics and vascular remodeling.
21. Krings T, Piske RL, Lasjaunias.PL.Intracranial arterial aneurysm vasculopathies: targeting
the outer vessel wall. Neuroradiology.2005Dec;47(12):931-7.Epub 2005Sep1. Review.
22. Austin.G, FisherS, Dickson D, AndersonD, Richardson S.The significance of the
extracellular matrix in intracranial aneurysms.Ann Clin Lab Sci.1993MarApr;23(2):97-
105.
23. Schievink WI, Michels VV, Piepgras DG. Neurovascular manifestations of heritable
connective tissue disorders. A review. Stroke. 1994 Apr;25(4):889-903. Review.
24. Schievink WI, Schaid DJ, Rogers HM, Piepgras DG, Michels VV. On the inheritance of
intracranial aneurysms. Stroke. 1994 Oct;25(10):2028-37. Review.
25. Schievink WI, Prakash UB, Piepgras DG, Mokri B. Alpha 1-antitrypsin deficiency in
intracranial aneurysms and cervical artery dissection. Lancet. 1994 Feb 19;343(8895):452-
53.

59
26. Phillips LH 2nd, Whisnant JP, O'Fallon WM, Sundt TM Jr.The unchanging pattern of
subarachnoid hemorrhage in a community.Neurology. 1980 Oct;30(10):1034-40.
27. Juvela S. Prehemorrhage risk factors for fatal intracranial aneurysm rupture. Stroke. 2003
Aug;34(8):1852-7. Epub 2003 Jun 26.
28. Longstreth WT Jr, Nelson LM, Koepsell TD, van Belle G. Cigarette smoking, alcohol use,
and subarachnoid hemorrhage. Stroke. 1992 Sep;23(9):1242-9.
29. Juvela S, Hillbom M, Numminen H, Koskinen P. Cigarette smoking and alcohol
consumption as risk factors for aneurysmal subarachnoid hemorrhage. Stroke. 1993
May;24(5):639-46.
30. Teunissen LL, Rinkel GJ, Algra A, van Gijn J. Risk factors for subarachnoid hemorrhage:
a systematic review. Stroke. 1996 Mar;27(3):544-9.
31. Weir BK, Kongable GL, Kassell NF, Schultz JR, Truskowski LL, Sigrest A. Cigarette
smoking as a cause of aneurysmal subarachnoid hemorrhage and risk for vasospasm: a
report of the Cooperative Aneurysm Study.
32. Lanzino G, Kassell NF, Germanson TP, Kongable GL, Truskowski LL, Torner JC, Jane
JA. Age and outcome after aneurysmal subarachnoid hemorrhage: why do older patients
fare worse?
33. Kleinpeter G, Lehr S. Is hypertension a major risk factor in aneurysmal subarachnoid
hemorrhage? Wien Klin Wochenschr. 2002 May 15;114(8-9):307-14.
34. Juvela S. Alcohol consumption as a risk factor for poor outcome after
aneurysmalsubarachnoid haemorrhage. BMJ. 1992 Jun 27;304(6843):1663-7.
35. Sacco RL, Wolf PA, Bharucha NE, Meeks SL, Kannel WB, Charette LJ, McNamara PM,
Palmer EP, D'Agostino R. Subarachnoid and intracerebral hemorrhage: natural history,
prognosis, and precursive factors in the Framingham Study.Neurology. 1984
Jul;34(7):847-54.

60
36. Petitti DB, Wingerd J. Use of oral contraceptives, cigarette smoking, and risk of
subarachnoid haemorrhage.Lancet. 1978 Jul 29;2(8083):234-5.
37. Adamson J, Humphries SE, Ostergaard JR, Voldby B, Richards P, Powell JT. Are
cerebral aneurysms atherosclerotic? Stroke. 1994 May;25(5):963-6.
38. Barrow DL, Reisner A. Natural history of intracranial aneurysms and vascular
malformations. Clin Neurosurg. 1993;40:3-39.
39. Camarata PJ, Latchaw RE, Rüfenacht DA, Heros RC. Intracranial aneurysms. Invest
Radiol. 1993 Apr;28(4):373-82.
40. Ostergaard JR, Reske-Nielsen E, Buhl J. Deficiency of reticular fibers in cerebral arteries.
On the etiology of saccular aneurysms in childhood. Br J Neurosurg. 1989;3(1):113-5.
41. Schievink WI, Karemaker JM, Hageman LM, van der Werf DJ. Circumstances
surrounding aneurysmal subarachnoid hemorrhage. Surg Neurol. 1989 Oct;32(4):266-72.
Review.
42. Rinne JK, Hernesniemi JA. De novo aneurysms: special multiple intracranial aneurysms.
Neurosurgery. 1993 Dec;33(6):981-5. Review.
43. Chambers WR, Harper BF Jr, Simpson JR. Familial incidence of congenital aneurysms of
cerebral arteries: report of cases of ruptured aneurysms in father and son.J Am Med
Assoc. 1954 May 22;155(4):358-9.
44. Norrgård O, Angquist KA, Fodstad H, Forsell A, Lindberg M . Intracranial aneurysms
and heredity .Neurosurgery. 1987 Feb;20(2):236-9.
45. Ronkainen A, Hernesniemi J, Ryynänen M. Familial subarachnoid hemorrhage in east
Finland, 1977-1990. Neurosurgery. 1993 Nov;33(5):787-96; discussion 796-97.
46. Schievink WI, Wijdicks EF, Parisi JE, Piepgras DG, Whisnant JP. Sudden death from
aneurysmal subarachnoid hemorrhage. Neurology. 1995 May;45(5):871-4.

61
47. Longstreth WT Jr, Nelson LM, Koepsell TD, van Belle G. Clinical course of spontaneous
subarachnoid hemorrhage: a population-based study in King County, Washington.
Neurology. 1993 Apr;43(4):712-8.
48. Schievink WI, Wijdicks EF, Piepgras DG, Chu CP, O'Fallon WM, Whisnant JP. The poor
prognosis of ruptured intracranial aneurysms of the posterior circulation. J Neurosurg.
1995 May;82(5):791-5.
49. Winn HR, Almaani WS, Berga SL, Jane JA, Richardson AE. The long-term outcome in
patients with multiple aneurysms. Incidence of late hemorrhage and implications for
treatment of incidental aneurysms. J Neurosurg. 1983 Oct;59(4):642-51.
50. Heiskanen O. Risk of bleeding from unruptured aneurysm in cases with multiple
intracranial aneurysms. J Neurosurg. 1981 Oct;55(4):524-6.
51. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and
inflammatory response of ruptured cerebral aneurysms. A comparative study between
ruptured and unruptured cerebral aneurysms. Stroke. 1999 Jul;30(7):1396-401.
52. Pontieri-Russo-Frati. Patologia generale. 2005
53. Cajander S, Hassler O. Enzymatic destruction of the elastic lamella at the mouth of
cerebral berry aneurysm? An ultrastructural study with special regard to the elastic tissue.
Acta Neurol Scand. 1976 Mar;53(3):171-81.
54. Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms.
Neurosurgery. 1999 Nov;45(5):1137-46; discussion 1146-7.
55. Kosierkiewicz TA, Factor SM, Dickson DW. Immunocytochemical studies of
atherosclerotic lesions of cerebral berry aneurysms. J Neuropathol Exp Neurol. 1994
Jul;53(4):399-406.
56. FrÃsen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J,
Jääskeläinen J. Remodeling of saccular cerebral artery aneurysm wall is associated

62
with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004
Oct;35(10):2287-93. Epub 2004 Aug 19.
57. Gaetani P, Grazioli V, Tancioni F, Casari E, Tartara F, Rodriguez y Baena R.
Abnormalities of collagen cross-linkage in posterior communicating artery aneurysms: a
preliminary study. Neurol Res. 1996 Dec;18(6):541-5.
58. Baker CJ, Fiore A, Connolly ES Jr, Baker KZ, Solomon RA. Serum elastase and alpha-1-
antitrypsin levels in patients with ruptured and unruptured cerebral aneurysms.
Neurosurgery. 1995 Jul;37(1):56-61; discussion 61-2.
59. Chyatte D, Lewis I. Gelatinase activity and the occurrence of cerebral aneurysms. Stroke.
1997 Apr;28(4):799-804.
60. Todor DR, Lewis I, Bruno G, Chyatte D. Identification of a serum gelatinase associated
with the occurrence of cerebral aneurysms as pro-matrix metalloproteinase-2. Stroke.
1998 Aug;29(8):1580-3.
61. Gaetani P, Rodriguez y Baena R, Tartara F, Messina AL, Tancioni F, Schiavo R, Grazioli
V. Metalloproteases and intracranial vascular lesions. Neurol Res. 1999 Jun;21(4):385-90.
62. Bruno G, Todor R, Lewis I, Chyatte D. Vascular extracellular matrix remodeling in
cerebral aneurysms. J Neurosurg. 1998 Sep;89(3):431-40.
63. Aoki T, Kataoka H, Moriwaki T, Nozaki K, Hashimoto N. Role of TIMP-1 and TIMP-2
in the Progression of Cerebral Aneurysms.
64. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix
metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke.
2007 Jan;38(1):162-9. Epub 2006 Nov 22.Stroke. 2007 Jun 14;
65. Kim SC, Singh M, Huang J, Prestigiacomo CJ, Winfree CJ, Solomon RA, Connolly ES Jr.
Matrix metalloproteinase-9 in cerebral aneurysms. Neurosurgery. 1997 Sep;41(3):642-66;
discussion 646-7.

63
66. Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation
of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal
aneurysms. Circulation. 2001 Jul 17;104(3):304-9.
67. Thompson RW, Parks WC. Role of matrix metalloproteinases in abdominal aortic
aneurysms.Ann N Y Acad Sci. 1996 Nov 18;800:157-74. Review.
68. Sandalcioglu IE, Wende D, Eggert A, Regel JP, Stolke D, Wiedemayer H. VEGF plasma
levels in non-ruptured intracranial aneurysms. Neurosurg Rev. 2006 Jan;29(1):26-9. Epub
2005 Aug 25.
69. Skirgaudas M, Awad IA, Kim J, Rothbart D, Criscuolo G. Neurosurgery. 1996
Sep;39(3):537-45; discussion 545-7. Expression of angiogenesis factors and selected
vascular wall matrix proteins in intracranial saccular aneurysms.
70. Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med.
1994 May 19;330(20):1431-8. Review. No abstract available.
71. De Mey JG, Schiffers PM, Hilgers RH, Sanders MM. Toward functional genomics of
flow-induced outward remodeling of resistance arteries. Am J Physiol Heart Circ Physiol.
2005 Mar;288(3):H1022-7. Review.
72. Kamiya A, Togawa T. Adaptive regulation of wall shear stress to flow change in the
canine carotid artery. Am J Physiol. 1980 Jul;239(1):H14-21.
73. Tronc F, Mallat Z, Lehoux S, Wassef M, Esposito B, Tedgui A. Role of matrix
metalloproteinases in blood flow-induced arterial enlargement: interaction with NO.
Arterioscler Thromb Vasc Biol. 2000 Dec;20(12):E120-6.
74. Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, Li S, Chien S, Schwartz MA.
Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal
reorganization and effects on gene expression. EMBO J. 2002 Dec 16;21(24):6791-800.
75. Ellegala DB, Day AL. Ruptured cerebral aneurysms. N Engl J Med. 2005 Jan
13;352(2):121-4.

64
76. Hoi Y, Meng H, Woodward SH, Bendok BR, Hanel RA, Guterman LR, Hopkins LN.J.
Effects of arterial geometry on aneurysm growth: three-dimensional computational fluid
dynamics study. Neurosurg. 2004 Oct;101(4):676-81.
77. Gonzalez CF, Cho YI, Ortega HV, Moret J. Intracranial aneurysms: flow analysis of their
origin and progression. AJNR Am J Neuroradiol. 1992 Jan-Feb;13(1):181-8.
78. Foutrakis GN, Yonas H, Sclabassi RJ. Saccular aneurysm formation in curved and
bifurcating arteries. AJNR Am J Neuroradiol. 1999 Aug;20(7):1309-17.
79. Jou LD, Wong G, Dispensa B, Lawton MT, Higashida RT, Young WL, Saloner D.
Correlation between lumenal geometry changes and hemodynamics in fusiform
intracranial aneurysms. AJNR Am J Neuroradiol. 2005 Oct;26(9):2357-63.
80. Shyy YJ, Hsieh HJ, Usami S, Chien S. Fluid shear stress induces a biphasic response of
human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc
Natl Acad Sci U S A. 1994 May 24;91(11):4678-82.
81. Hoefer IE, van Royen N, Rectenwald JE, Deindl E, Hua J, Jost M, Grundmann S,Voskuil
M, Ozaki CK, Piek JJ, Buschmann IR.Arteriogenesis proceeds via ICAM-1/Mac-1-
mediated mechanisms. Circ Res. 2004 May 14;94(9):1179-85. Epub 2004 Apr 1.
82. Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in
endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment.
EMBO J. 2001 Sep 3;20(17):4639-47.
83. Shyy JY, Chien S. Role of integrins in cellular responses to mechanical stress and
adhesion. Curr Opin Cell Biol. 1997 Oct;9(5):707-13. Review.
84. Li S, Kim M, Hu YL, Jalali S, Schlaepfer DD, Hunter T, Chien S, Shyy JY. Fluid shear
stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. J.
Biol. Chem. 1997 Nov 28;272(48):30455-62.
85. Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg GI. SV40-
transformed human lung fibroblasts secrete a 92-kDa type IV collagenase which is

65
identical to that secreted by normal human macrophages. J Biol Chem. 1989 Oct
15;264(29):17213-21.
86. Matrisian LM. The matrix-degrading metalloproteinases. Bioessays. 1992 Jul;14(7):455-
63. Review.
87. Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo
A, Engler JA. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med.
1993;4(2):197-250. Review.Lyons
88. JG, Birkedal-Hansen B, Pierson MC, Whitelock JM, Birkedal-Hansen H.Interleukin-1
beta and transforming growth factor-alpha/epidermal growth factor induce expression of
M(r) 95,000 type IV collagenase/gelatinase and interstitial fibroblast-type collagenase by
rat mucosal keratinocytes. J Biol Chem. 1993 Sep 5;268(25):19143-51.
89. Iwata H, Kobayashi S, Iwase H, Masaoka A, Fujimoto N, Okada Y. Production of matrix
metalloproteinases and tissue inhibitors of metalloproteinases in human breast
carcinomas. Jpn J Cancer Res. 1996 Jun;87(6):602-11.
90. Gress TM, Muller-Pillasch F, Lerch MM, Friess H, Buchler M, Adler G. Expression and
in-situ localization of genes coding for extracellular matrix proteins and extracellular
matrix degrading proteases in pancreatic cancer. Int J Cancer. 1995 Aug 9;62(4):407-13.
91. Ueda Y, Imai K, Tsuchiya H, Fujimoto N, Nakanishi I, Katsuda S, Seiki M, Okada Y.Am.
Matrix metalloproteinase 9 (gelatinase B) is expressed in multinucleated giant cells of
human giant cell tumor of bone and is associated with vascular invasion J. Pathol. 1996
Feb;148(2):611-22.
92. Goetzl EJ, Banda MJ, Leppert D J. Matrix metalloproteinases in immunity.Immunol. 1996
Jan 1;156(1):1-4.
93. Westbrook CA, Gasson JC, Gerber SE, Selsted ME, Golde DW. Purification and
characterization of human T-lymphocyte-derived erythroid-potentiating activity. J Biol
Chem. 1984 Aug 25;259(16):9992-6.

66
94. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe
P,Itohara S, Werb Z, Hanahan D.Matrix metalloproteinase-9 triggers the angiogenic
switch during carcinogenesis. Nat Cell Biol. 2000 Oct;2(10):737-44.
95. Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of
metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol. 1997
Oct;74(2):111-22. Review.
96. Kolkenbrock H, Orgel D, Hecker-Kia A, Zimmermann J, Ulbrich N. Generation and
activity of the ternary gelatinase B/TIMP-1/LMW-stromelysin-1 complex. Biol Chem
Hoppe Seyler. 1995 Aug;376(8):495-500.
97. Charles A. Dinarello MD. Proinflammatory and anti-inflammatory cytokines in
rheumatoid arthritis. 2000
98. Bugno M, Witek B, Bereta J, Bereta M, Edwards DR, Kordula T. Reprogramming of
TIMP-1 and TIMP-3 expression profiles in brain microvascularendothelial cells and
astrocytes in response to proinflammatory cytokines.FEBS Lett. 1999 Apr 1;448(1):9-14.
99. Dinarello CA. Interleukin-18. Methods. 1999 Sep;19(1):121-32. Review.
100. Ferrara N. Vascular endothelial growth factor and the regulation of angiogenesis. Recent
Prog Horm Res. 2000;55:15-35; discussion 35-6. Review.
101. Nagase H. Activation mechanisms of matrix metalloproteinases. Biol Chem. 1997 Mar-
Apr;378(3-4):151-60. Review.
102. Josko J. Cerebral angiogenesis and expression of VEGF after subarachnoid hemorrhage
(SAH) in rats. Brain Res. 2003 Aug 15;981(1-2):58-69.
103. Koizumi T, Shiraishi T, Hagihara N, Tabuchi K, Hayashi T, Kawano T. Expression of
vascular endothelial growth factors and their receptors in and around intracranial
arteriovenous malformations Neurosurgery. 2002 Jan;50(1):117-24; discussion 124-6

67
104. Sure U, Butz N, Schlegel J, Siegel AM, Wakat JP, Mennel HD, Bien S,Bertalanffy H.
Endothelial proliferation, neoangiogenesis, and potential de novo generation of
cerebrovascular malformations.J Neurosurg. 2001 Jun;94(6):972-7
105. Moriwaki T, Takagi Y, Sadamasa N, Aoki T, Nozaki K, Hashimoto N. Impaired
progression of cerebral aneurysms in interleukin-1beta-deficient mice. Stroke. 2006
Mar;37(3):900-5. Epub 2006 Jan 26.

68
RINGRAZIAMENTI

69
Vorrei ringraziare il professor Luca Giovannini per l’opportunità di svolgere la tesi nel suo
laboratorio oltre che per l’indubbia disponibilità mostrata nel periodo dell’internato.
Vorrei inoltre ringraziare Claudio, per la pazienza che ha mostrato giorno dopo giorno


















