
UNIVERSIDADE DESÃO PAULO INSTITUTO DE QUIMICA · 2015-05-04 · clean-upfollowed by MEKC in high...
260
UNIVERSIDADE DE SÃO PAULO INSTITUTO DE QUIMICA DESENVOLVIMENTO DE ESTRATEGIAS DE PRE- CONCENTRAÇÃO ELETROFORESE CAPILAR (CE) VISANDO A ANÁLISE DE PESTICIDAS EM FRUTAS E LEGUMINOSAS Clóvis Lúcio da Silva Tese de Doutorado Marina F. M. Tavares São Paulo ' 2003 .
Transcript of UNIVERSIDADE DESÃO PAULO INSTITUTO DE QUIMICA · 2015-05-04 · clean-upfollowed by MEKC in high...

UNIVERSIDADE DE SÃO PAULO~
INSTITUTO DE QUIMICA
~ ~
DESENVOLVIMENTO DE ESTRATEGIAS DE PRE-
CONCENTRAÇÃOP~ ELETROFORESE CAPILAR
(CE) VISANDO A ANÁLISE DE PESTICIDAS EM
FRUTAS E LEGUMINOSAS
Clóvis Lúcio da Silva
Tese de Doutorado
Marina F. M. TavaresSão Paulo '
2003 .

Ficha CatalográficaElaborada pela Divisão de Biblioteca e
Documentação do Conjunto das Químicas da USP.
Silva, Clóvis Lúcio daS586d Desenvolvimento de estratégias de pré-concentração para
eletroforese capilar (CE) visando a análise de pesticidas emfrutas e leguminosas / Clóvis Lúcio da Silva. -- São Paulo,2003.
219p.
Tese (doutorado) - Instituto de Química da Universidadede São Paulo. Departamento de Química Fundamental.
Orientador: Tavares, Marina Franco Maggi
1. Eletroforese capilar: Química analítica 2. Pesticida:Agricultura I. T. 11. Tavares, Marina Franco Maggi,orientador.
543.0871 CDD


A 13á~ Li+'fuv pelo- aA11bY,~
C<iVYÍN\ho-~co-wq:JlM1he..r~~~~~
v~ t'\.aI USP.

'Ntio- éteA1fyaq~ o-{o-vtc!/ qU-el~{o-vt~ o- fvcu;o!'
AbretJta,JIl/L~

A~~Oi'
- A D~ewlIpr~&L~.
- A~~p~
- À ProF DrQ. Mari.t1.cvFrCU'\,C.&M~Tevvar~
peUv oríet'\tet.ÇCt& ~ oport~ ~
":~~~Lrcibalho:
- À FAPESP peUv~~~tAilo1rconceiUdcv
(prOGeMJ&Yl.i. 99(05082 -6)
- A~~ do- Lace-- - LaboY~ório- ~
cr~o-srr~~Eld"roforeJJe'CcqJí1aY"
- A L~ qfA.e/ CO"V\tr~CM11I dire:t"Ct/ OU!
~et"~~ parCt/ Ct/~~~
LVcibalho:

Lista de abreviaturas
À - comprimento de onda
y - ganho na pré-concetração do analito
IJ. - mobilidade eletroforética
<p - relação entre o volume das micelas e o volume da fase líquida
v - velocidade
11 -viscosidade
lJ.a - mobilidade aparente
13-CO - beta ciclodextrina
y-CO - gama ciclodextrina
lJ.eo - mobilidade do fluxo eletrosmótico
ACN - Acetonitrila
BGS - Eletrólito de sepração
BuOH - Butanol
CO - Ciclodextrina
CE - Eletroforese capilar
CGE - Eletroforese em gel
CIEF - focalização isoelétrica capilar
CITP - Isotacoforese capilar
CM(BGS) - concentração de micela na zona do eletrólito de separação
CM(S) - concentração das micelas na zona da amostra
cmc- concentração micelar crítica
cmt - temperatura micelar crítica
CN - ciano fase ligada
CPE - extração por cloud-point
Cs - concentração do tensoativo em solução
CV - coeficiente de variação
CZE - eletroforese capilar de zona
OAO - detector de arranjo de diodos
Om-13-CO - dimetil beta ciclodextrina

EOF - fluxo eletrosmótico
EtOAC - acetato de etila
EtOH - Etanol
Fe - força eletrostáticav
FESI - injeção com amplificação de campo
FESI-RMN - Injeção de campo amplificado com migração reversa das micelas
Ff - Força friccionai
FSCE - Eletroforese em solução livre
GCB - carbono grafitizado
GC-ECO - Cromatografia gasosa com detector de captura de elétrons
GC-MS - Cromatografia gasosa acoplada à espectrometria de massa
GPC - extração cromatográfica com permeação em gel
GS - Grau de substituição
HLB - balanço hidrofílico e lipofílico
HPLC - Cromatografia líquidas de alto desempenho
K - fator de retenção
LO -limite de detecção
linj - comprimento de plug da amostra injetado
LLE - extração em fase líquida
LQ -limite de quantificação
Isweep - comprimento da zona da amostra após o sweeping
MECK - Cromatografia eletrocinética micelar capilar
MeOH - Metanol
MRLs - nível Maximo de resíduo
MRMs - método multiresíduo
MSPO - dispersão da amostra na fase sólida
M-SRMM - stacking com migração reversa das micelas modificado
M-SRW - stacking usando a migração reversa das micelas e um plug de água
modificado
N - Número de agregação
NH2 - amino fase ligada

NP -fase normal
NSM - stacking em modo normal
NTE - Esterase neurotóxica
OE - oxietileno
PAHs - hidrocarboneto policíclicos aromáticos
PCBs - bifenil policlorados
PCDFs - dibenzenofuranos
PrOH - Propanol
q - Carga •
r - raio
REPSM - stacking com polaridade reversa do eletrodo
RP - fase reversa
S - zona da amostra injetada
SOS - dodecil sulfato de sódio
SFC - Fluido supercrítico
SNA - Sistema nervoso assimpático
SPC - clean-up em fase sólida
SPE - extração em fase sólida
SPME - microextratores em fase sólida
SRMM - Stacking com migração reversa das micelas
SRMs - Método de resíduo simples
SRW - stacking usando a migração reversa das micelas e um plug de água
TBS - Tetraborato de sódio (B4Na207.10H20) pH 9,3
TF - Tampão fosfato (H3POJNaH2P04) pH 2,5
Tm-f3-CO - trimetil beta ciclodextrina
UV - Ultra-violeta

"DESENVOLVIMENTO DE ESTRATÉGIAS DE PRÉ-CONCENTRAÇÃO PARA
ELETROFORESE CAPILAR (CE) VISANDO A ANÁLISE DE PESTICIDAS EM
FRUTAS E LEGUMINOSAS"
O uso de pesticidas constitui um importante aspecto na agricultura
moderna, com inquestionável beneficio na produção agrícola. Porém a
contaminação dos alimentos por pesticidas constitui um sério risco a saúde do
consumidor. A determinação de resíduo de pesticidas em alimentos envolve
procedimentos laboriosos, com elevado tempo de análise e várias etapas de pré
concentração.
Neste trabalho, procedimentos alternativos de extração e pré-concentração
para analise multiresíduo de pesticida em água, frutas e tubérculos foram
desenvolvidos.
A eletroforese capilar em seu modo MEKC em condições de alto e baixo
fluxo eletrosmótico foi empregado para a otimização da separação de diferentes
classes de pesticidas (triazinas, organofosforado, carbendamidazóis, feniluréia e
carbamatos). A composição do eletrólito de separação otimizada para condições
de alto EOF foi: 10 mmol L-1 de tetraborato de sódio (pH 9,3),50 mmol L-1 de SDS
e 5% etanol e 5% propanol, enquanto que para condições de baixo EOF foi: 10
mmol L-1 de tampão fosfato (pH 2,5), 25 mmol L-1 de SDS e 10% metano!.
Estratégias de pré-concetração on-line conhecida como sweeping (SW) e
stacking nos modos de migração reversa das micelas (SRMM) e migração reversa
com um plug de água (SWR) bem como as suas versões modificadas foram
avaliadas, obtendo fatores de pré-concetração de variaram de 2,6 a 19 para o SW,
2,9 a 15 para o SRW e de 5,5 a 15 para o método SRMM modificado.
Varias metodologias de extração envolvendo extração em fase sólida (SPE)
e extração líquido-líquido (LLE) foram testada. A estratégia de extração por cloud
point foi aplicada a uma amostra de abacaxi. O procedimento denominado
dispersão da matriz em fase sólida (sigla inglesa MSPD), que minimiza o uso de
solventes orgânicos e é de fácil implementação foi aplicado a amostras de
cenoura.

A combinação do SPE off-line e das estratégias de pré-concentração
anteriormente mencionadas permitiram a determinação de alguns pesticidas na
concentração de 0,1 I1g L-1 em amostras de água potável.
O método de extração e clean-up MSPD seguida da análise de MEKC em
alto EOF foi otimizado e algumas figuras de mérito foram estabelecidas baseados
em protocolos de validação para análise de pesticidas (IAEA-FAO). Boa
linearidade (r > 0,99) foi obtido para todos os pesticidas estudados, exceto para
linuron e dimetoato. A precisão do método foi estimada através de testes de
recuperação. Dois níveis de fortificação foram utilizados para a avaliação, foram
obtidos recuperações de 51 a 89 % para o nível mais baixo e 67 a 100% para o
maior nível. Foi obtida uma boa precisão intraensaio (CV < 15%).
O método otimizado foi aplicado para análise multiresíduo de cenouras.
Uma amostra adquirida no comércio local foi quantificada encontrando-se 0,88 mg
kg-1 de simazina, 0,13 mg kg-1 de atrazina e 0,08 mg kg-1 de propazina.

"OEVELOPMENT OF PRECONCENTRATION STRATEGIES FOR
PESTICIOES ANALYSIS BY CAPILLARY ELECTROPHORESIS (CE) IN
FRUITS ANO TUBERS".
The use of pesticides constitutes an important aspect of modem
agriculture, with unquestionable impact on crop production. However, food
contamination by pesticide residues is a serious risk for the consumer. The
determination of pesticide residues in food usually involves laborious
procedures, with time consuming sample clean up and preconcentration steps
prior to the analysis.
In this work, alternative methodologies for extraction, pre-concentration
and analysis of pesticide multi-residue in water, fruits and tubers were
developed.
Capillary electrophoresis in its micellar mode (micellar electrokinetic
chromatography, MEKC) under low and high electroosmotic flow (EOF)
conditions was used for the separation of pesticides from different chemical
classes (triazines, organophosphorous, carbendamidazols, phenilurea and
carbamates). Optimized electrolyte compositions were: high EOF - 10 mmol L-1
tetraborate (pH 9.3), 50 mmol L-1 SOS, 5 % ethanol and 5 % propanol; low EOF
- 10 mmol L-1 phosphate buffer (pH 2.5), 25 mmol L-1 SOS and 10 % methanol.
On-line preconcentration strategies for MEKC known as sweeping (SW)
and stacking with reverse migrating micelles with (SRW) and without (SRMM) a
plug of water prior to the sample plug as well as modified versions of SRW and
SRMM were evaluated and contrasted in terms of signal enhancement factor
(peak height ratios) Signal enhancement factors for SW varied from 2,6 to 19
for SRW from 2,9 to 15, whereas for modified-SRMM from 5,5 to 15.

Among the extraction methodologies, several procedures involving solid
phase extraction (SPE) and liquid-liquid extraction (LLE) were tested. It is worth
mentioning a strategy based upon cloud point extraction, which was applied to
pineapple samples and a procedure denominated matrix solid-phase dispersion
(MSPD), which combines low cost, saves in solvents and easy implementation,
applied to carrots. The combination of off-line SPE and the above mentioned
on-line preconcentration strategies allowed the determination of selected
pesticides in the 0.1 Jl9/L levei (drinking water sample).
A complete methodology involving MSPD for extraction and sample
clean-up followed by MEKC in high EOF was optimized and a few figures of
merit were established based on method validation protocols for pesticide
analysis (IAEA-FAO). Good linearity (r>0.99) was obtained for ali pesticides
under investigation, except for linuron and dimetoate. The method accuracy
was estimated by recovery tests. Two levei standard spiking were conducted
with recoveries of 51 to 89 % for the lowest levei and 67 to 100 % for the
highest leveI. Acceptable intra-day precision was obtained (CV < 15 %).
The optimized method was applied to the analysis of multi-residue
pesticides in carrots. In a sample acquired in a local grocery store an unusual
amount of triazines was found: simazine (O,~8 mglkg), atrazine (0,13 mglkg)
and propazine (O,088mg/kg).

íNDICE GERAL,
If1dice de tabelas~
Indice de Figuras
~apítulo 1
1 - Introdução,1.1 - Histórico
1.2 - Considerações gerais sobre pesticidas
~ .3- Efeitos tóxicos dos pesticidas
tI.4 - Pesticidas e seus mecanismos de ação tóxica
1.4.1- Organofosforados e carbamatos
:1.4.2 - Pesticidas Organoclorados
1.4.3 - Herbicidas Triazinas
1.5 - Técnicas utilizadas para identificação e quantificação de pesticidas
Capítulo 2
2 - Fundamentos da Eletroforese capilar (CE)
2.1 - Fluxo eletrosmótico
. 2.2 - Migração eletroforética
. 2.3 - Modo de separação utilizado em análise de pesticidas por eletroforese
capilar
2.3.1 - Cromatografia eletrocinética micelar capilar
2.4 - Sistemas micelares
2.4.1 -Tensoativos
2.4.2 - Classificação dos tensoativos
2.4.3 - Propriedades gerais dos tensoativos
2.4.3.1 - Processo de agregação dos tensoativos em água
2.4.3.2 - Número de agregação
2.4.3.3 - Kraft Point
2.4.3.4 - Fatores que influenciam o valor da cmc
111
1
1
1
7
15
20
20
23
24
25
28
29
31
32
34
34
38
39
39
41
41
48
48
49

3 - Objetivos 54
Capítulo 4 -Instrumentação analítica, materiais, reagentes e amostras 55
4 - Materiais e reagentes 55
4.1 - Instrumentação 55
4.2 - Materiais utilizados na preparação de soluções e amostras 56
4.3 - Reagentes e soluções 57
Capítulo 5 - Otimização da composição do eletrólito de separação 61
5.- Otimização da separação em pH 9,3 61
5.2 - Otimização da separação em pH 2,5 77
Capítulo 6 - Estratégias de pré concentração 85
6 - Introdução. 85
6.1 - Avaliação das estratégias de pré-concentração on-line na ausência de 87
EOF
6.1.1 - Sweeping de analitos neutros em MEKC 87
6.1.2 - Stacking com migração reversa das micelas (SRMM) e (M-SRMM1 91
6.1.2.1 - Variação da estratégia SRMM 94
6.1.3 - Stacking com migração reversa das micelas e um plug de água (SRW) 98
6.1.4 - Junção das estratégias de pré-concentração off-line e on-line 103
. 6.2 - Avaliação das estratégias de pré-concentração on-line na presença de 109
alto de EOF.
6.2.1 - Sweeping 109
6.2.2 - Stacking em baixo campo elétrico (alta condutividade da amostra) 113
Capítulo 7 - Métodos de extração e clean up 120
7 - Introdução 120
1227.1- Algumas considerações sobre extração e clean-up
7.1.1- Extração Líquido-Líquido (LLE) 122

1237.1.2 - Extração em fase sólida
7.2 - Procedimento utilizado para os estudos preliminares em SPE (solid
phase extration)
7.3 - Métodos de Extração e clean-up
7.3.1 - Método I
7.3.2 - Método 11
7.3.3 - Método 111
7.3.4 -Método IV
7.3.5 - Método V - Extração por dispersão da matriz em fase sólida (MSPD)
128
138
138
142
145
149
152
1547.3.5.1 - Procedimento de extração
7.3.6 - Avaliação das metodologias 111 e V aplicadas a amostras (cenoura e 162
abacaxi) adquiridas no comércio local
7.3.6.1 Avaliação do método 111 de extração 162
7.3.7 - Avaliação do Método V de extração 164
7.4 - Avaliação da estabilidade térmica dos pesticidas. 166
1697.5 - Extração por cloud point - CPE (sigla inglesa para Cloud Point Extration)
7.5.1 - Condições experimentais para a realização da OPE
7.5.2 - Emprego do CPE como técnica analítica
7.5.3 - Emprego de CPE em análise de pesticidas
7.5.4 - Otimização dos parâmetros experimentais da metodologia CPE
7.5.4.1 - Parâmetro 1: composição do eletrólito de separação
7.5.5 - Parâmetro 2: concentração do Triton-X114 empregado na CPE
7.5.6 - Aplicação da metodologia otimizada em amostra real
175
176
178
181
181
191
193

Capítulo 8 - Figuras de mérito
8 - Introdução
8.1 - Método analítico otimizado
8.2 - Preparação das amostras
8.3 - Linearidade
8.4 - Limite de detecção e quantificação
8.5 - Recuperação
8.6 - Precisão dos testes de recuperação da análise de pesticidas em
amostras de cenoura fortificadas em dois níveis de concentração
8.6.1 - Repetibilidade (precisão) de tempo e áreas de pico para a mistura
padrão de pesticidas estudada
8.7.7 - Quantificação de amostra
8.8 - Conclusões
Capítulo 9 - Conclusões Gerais e Perspectivas futuras
9 - Conclusões
9.1 - Perspectivas futuras
Súmula Curricular
Apêndice - Protocolos de validação
196
196
199
200
200
205
207
208
209
212
215
216
216
219

íNDICE DE TABELAS
Tabela 1.1 - Controle de pestes, doenças e ervas daninhas. 6
Tabela 1.2 - Algumas das estruturas dos pesticidas da classe dos 9
herbicidas clorados
Tabela 1.3 - Algumas das estruturas dos pesticidas da classe feniluréia 10
Tabela 1.4 - Algumas estruturas pertencentes aos pesticidas da classe 11
dos organofosforados
Tabela 1.5 - Algumas estruturas dos pesticidas da classe das triazinas. 12
Tabela 1.6 - Algumas estruturas dos pesticidas da classe dos 13
organoclorados.
Tabela 1.7 - Algumas estruturas dos pesticidas da classe dos carbamatos 14
Tabela 1.8 - Consumo de pesticidas no Brasil (1997-1999) 19
Tabela 2.1 - Estruturas de alguns tensoativos 40
Tabela 6.1 - Comparação entre as alturas de picos encontradas para é 97
estratégias de pré-concetração (SRMM e M-SRMM)
Tabela 6.2 - Comparação entre as alturas de picos encontradas para cada 102
estratégias de pré-concetração (SRW e M-SRW)
Tabela 6.3 - Avaliação comparativa entre as estratégias de pré- 108
concetração
Tabela 6.4 - Comparação entre as alturas de picos encontradas para cada 112
tempo de injeção utilizado durante o sweeping, e suas alturas relativas à
injeção sem emprego da estratégia.
Tabela 6.5 - Alturas de picos encontradas para cada concentração de sal 118
adicionado à amostra, e suas alturas relativas a injeção sem NaCI
Tabela 7.1 - Processo de retenção dos analitos 124
Tabela 7.2 - Características dos solventes utilizados em SPE 127
Tabela 7.3 - Recuperação para os pesticidas estudados na extração em fa 131
sólida.
Tabela 7.4 - Valores de porcentagem de recuperação obtidos para os 12 134
pesticidas utilizando C18 como fase estacionária.
Tabela 7.5 - Porcentagens de recuperação para as eluições em cascata. 135

Tabela 7.6 - Recuperação dos padrões de pesticidas, para 0,8 mL de mistL 137
padrão 5 mg/L, utilizando como eluente 8 mL de MeOH:CH2CI2 (1 :99 v/v)
Tabela 7.7 - Valores de recuperação para os métodos de extração 158
avaliados.
Tabela 7.8 - Análise da perda porcentual para cada pesticida empregado. 168
Tabela 7.9 - Propriedades aquosas de alguns sistemas de Tensoativos 174
não-iônicos e zwitteriônicos que têm sido empregados em extrações por
Cloud Point
Tabela 8.1 - Definição dos termos requeridos para validação de 197
metodologias
Tabela 8.2 - Curvas analíticas obtidas para os pesticidas utilizados 204
Tabela 8.3 - Valores de limite de detecção e quantificação os pesticidas 206
estudados.
Tabela 8.4 - Porcentagem de recuperação obtida para a mistura de pestici< 207
estudada na fortificação de amostras de cenoura
Tabela 8.5 - Precisão intra-ensaio (% CV) obtidos para a análise de 208
pesticidas em cenoura em dois níveis de fortificação
Tabela 8.6 - Valores de área e tempo de migração para avaliação da 210
repetibilidade
Tabela 8.6 - Valores de área e tempo de migração para avaliação da 211
repetibilidade
Tabela 8.6 - Valores de concentração de pesticidas encontrados nas 214
amostras de cenoura analisadas
11

íNDICE DE FIGURAS
figura 1.1 - Relação de apllcaçãoe datas aproximadas de introdução
paraospestitiàasrepresentativos, Uustrando o progresshro.aumento 00
atividade.
Figura. t.2 - Pracesscr de transacetUação' da acetitcotina pet'a enzU'rr8'
colinesterase
.. Figura 1..3 -Processo .de inativação .da cofinester'ase:por'pestiooas
organofostorados.
Figura 2.t - Formação: do. ftuxo.eletrosmótica no ,menor de·um· capüar.de
sitica fundida após aplicação de um campo eiétrico
.Figura 2.2 ~FÓfrnutaestruturat do SOS
Figura 2.3 - Estrutura química e modelo esquemático das ciclodextrinas:
a-CO·, t5-CD e y-CD.
Figura 2.4 - Esquema para uma separação eletroforética no modo MEKC
Figurã 2~5 - MEKCem condição óe· baixo·pH (supressão do SOr).
Figura 2.6 - Esquema de uma micela iônica baseada no modelo de
Hattey.
Figura 2.7 - Algumas estruturas formadas por tensoativos
figura 2~B-Müdartças ·das caracteristicas ffsrco..qlíimicas ·de UJT1a
solução de tensoativo após ter atingido o CMC.
Figura 2.9- Dfagrarttd de rõsesdoKraft po'int
Figura 4.1 - Equipamento de CE utHizado para a reaiização das anáiises....... ... 5'" .!--....n. A.' ,. ,_~,.,.,.,.{, • "",,,,",.,.' ..-r'l9uta ·.:1 - :nmueneta:oasoCl\,J.uCA.LfmaS ucnula1STla separaçao
eletroforética de uma mistura de pesticidas.
Figura 52 - Influência das- CUs. detw.auas fIa: sep"'dtaçátl eietroforéttca de
uma mistura de pesticidas
Figura 5..2 - Efeiiü 00 :6'fij 35 :sottre :â 1>--eprdl:açãceietlofo1étlcá rleuma
mistura de pesticidas.
Figura 5.4 - inJ}uênaa na' separação.e':e1nlfotélica:·pera: ·crdição:ae-tO: %. de
diferentes solventes ao 'eletróHto de separação.
rlgUla 5~5 ~ :infiuêftCT-dfla .~aração.eiettoforética peiaadição .·tia
misturas binárias de soiventes ao eietróHto de' separação
15
32
37
38
44
46
47
48
55
62
70
In

Figura 5.6 - Melhores separações utilizando misturas binárias de álcoois 71
no eIetróflto.
,;,
Figura 5.7 - Gráfico de mobilidade em função da espécie e concentração
- -dos-solventes -orgâNcos-empregados para'etetrótitos 'en1 pH 9,3.
Figura 5.8 -Avaliação da influência do Brij 35@ na separação
efetroforética de uma mistura de pesndda.
Figura 5.9 -Degradação do carbaril como resultado de sua hidrólise em
me1eatcatioo.Figura 5.10 - Influência na separação eletroforética pela- adição- de 5% de
dtferentes solventes ao eJetrólito de separação
Figura--S.11 - Influência na -separação -eletroforética pela adição 0010%
de ·dlferentes-sotventesao-eletTófito de separação
Figura- 5.12 - - Influência- na separação eletroforética pela adição de
misturas- binárias de soWentes ao- efetrófito de separação
figura 5.13 - Gráfico de mobilidade em função da especle e
ooncentração dos SOiventes orgânicos-empregados -para êtetróMus em
pH2,5.
Figura 6.1 - Representaçãoesquemattza do sweeping.
Figura 6.2 - Sweeping (baixo EOF) para uma mistura -padrão de
pesticida nacoocentraçãode 0,5 mgll
Figura 6.3 - Stacking sob campo elétrico elevado (baixo EOF).
frgutcl 6.4 - Representação esquemática par:a a estratégia M-SRMM.
Figura 6.5 - Comparação entre as estratégias SRMM e M-SRMM para
uma mistura de pesticidas, na -concentr-açãode {),5 -mgll.
Figura'6.6 - Representação esquemática para as estratégias M-SRW.
Frgura, 6,7 - Comparação entre- as estratégias SRW e M-SRW para uma
mistura de pesticidas, na concentraçãode1mg/l.
Figura '6.8 - Avatiaçãoda junção das estratégias -de :pré-"concentração
on--Hne e- off,;,/ine- para análise de amostra de água fortificada com uma
mistura de- 9 padrões- de- pesticrdas na concentração de- O, l ~g/t.
Figura 6.9 - Aplicação das -estratégias de pré-concentração em uma
72
74
76
78
80
82
83
88
90
92
94
96
104
106
IV

amostra de cenoura fortificada com 2,5 1J.9/L de uma mistura padrão de 9
pesticidas
Figura 6.10 - Representação esquemática para o sweeping 110
Figura 6.11- Sweepingem meio homogêneo (alto EOf) para uma 111
mistura padrão de pesticidas.
Figura 6.12 - Stacking com alta concentração de sal na amostra. 114
Figura 6.13 - Stacking com alta concentração de sal na amostra (alto 117
EOF) para uma mistura padrão de pesticidas
Figura 7.1 - Esquema de extração para o Método I 140
Figura 7.2 - Método I aplicado à amostra de cenoura. 141
Figura 7.3 - Esquema de extração para o Método 11. 143
Flgura 7.4 - ·Método ·U -de -extração aplicado à amostras de cenoura 144
Figura 7.5 - Esquema de extração para o Método 111. 147
Figura 7.6 - Método 111 de extração aplicado à amostras de cenoura 148
F1gura 7.7 - Esquema ·de extração para o Método ·tv 150
Figura 7.8 - Método IV de extração aplicado à amostras de cenoura 151
Figura 7.9 - Esquema para o Método· MSPD- 154
Figura 7.10 - Método V (MSPD) de extração aplicada a amostras de 156
cenoura
Figura 7.11 - Gráfico de recuperação para os métodos avaliados 159
Figura 7. "'2 - AvaHação· do Método m· para amostras- de cenoura e- 163
abacaxi
Figufa 7.13 - Avaliação·do·Método V -para-amostras de·cenoura 1-65
Figura 7.14 - Avaliação da estabilidade térmica dos pesticidas 167
Figura 7.15 - Representação- esquemática de- uma micela- de- forma 170-
rugosa de um tensoativo não-iônico
figufa 7:t6-Uiagrama defases parao1ensoativo Tmon X-114 171
Figura 7.17 - Representação esquemática dos passos envolvidos em 176
uma extfaçãopof'cfouà point (CPE)
Figura 7.18 - esquema de extração utilizado para a metodologia de cloud 182
polnt.
v

Figura 7.19 - Separação em meio não-aquoso de pesticidas após CPE. 183
Figura 7.20 - Avaliação comparativa das adições de SOS ao eletr6lito de 185
separação (NACE)
figura 7.21 - Eletroterogramas obtidos ajustando-se a condutividade do 187
eletrólito de separação
Figura 7.22 - Avaliação comparativa entre SDS e octil sufato de sódio 188
como tensoativos do eletrólito de separação
Figura 7.23 - Avaliação comparativa entre dodecanot e SOS 190
Figura 7.24 - Variação da concentração de tensoativo (Triton X-114) 192
para CPE em amostra sintética
Figura 7.25 - Esquema de CPE aplicado à amostra de abacaxi. 194
Figura 7.26 - Realização de CPE para uma amostra de abacaxi 195
Figura 8.1 - Curvas analíticas obtidas para dimetoato, carbendazim, 202
simazina e atrazina
Figura 8.2 - Curvas analíticas obtidas para carbaril, linuron, ametrina e 203
-propazina
Figura 8.3 - Curva analítica obtida para o propoxur 204
Figura 7.4 - Avaliação do Método V para amostras de- cenoura 213
VI

CAPÍTULO 1

1 - Introdução
1.1 - HistóricoOs pesticidas vêm sendo utilizados desde da Antiguidade. Há
registros de papiros Hebreus datados de aproximadamente 1550 A.C. onde
consta uma lista de formulações que eram utilizadas para inibir ataque de
pulgas em residências da época1. Em 900 O.C. os chineses já usavam arsênio
para controle de pragas de jardim. No mundo ocidental o primeiro uso do
arsênio como inseticida data de 1669, era misturado ao mel para ser usado
como iscas para a eliminação de formigas. O uso de tabaco como inseticida de
contato foi mencionado mais tarde no mesmo século. No século XVIII foram
poucos os materiais introduzidos com a finalidade de controle ou eliminação de
pragas. No início do século XIX o desenvolvimento de novos compostos ainda
era pequeno. A partir de 1870, o número de compostos aumentou
gradativamente. Equipamentos para aplicação de pesticidas em culturas
agrícolas começaram a ser desenvolvidos.
A segunda grande guerra e diversificações de atividades empresariais
foram fatores responsáveis pela intensificação na pesquisa e desenvolvimento
de novos pesticidas. A guerra levou à pesquisa de agentes químicos com fins
militares, favorecendo a síntese de muitos inseticidas orgânicos. A
diversificação das indústrias alemãs foi o segundo fator,. pois passaram a atuar
neste ramo dos pesticidas a partir de intermediários químicos utilizados na
produção de corantes.
1C.p , Bryan, The papyrus Hebreus, translated from the german version. O. Appleton and
company, New York, 1931
I

Ainda na década de 40 (séc. XX) verificou-se a introdução e produção de
compostos organo-sintéticos, em substituição aos produtos inorgânicos. Isto
significou uma ruptura na base tecnológica antes conhecida, que gerou
inovações sobre as quais se deu a expansão e estruturação da indústria de
agroquímicos em nível mundial. Em 1939 foi descoberto a atividade pesticida do
DDT (organoclorado), que apresentava um alto poder de controle sobre
diversas pragas, custo reduzido e aparente eficácia. Nesta época foram também
introduzidos no mercado fungicidas, sendo os principais os ditiocarbamatos e
cloranil. Neste período, os fungicidas sofriam a concorrência de produtos
inorgânicos, tais como, sulfato de cobre, cal, enxofre, que eram baratos e
eficientes. Os primeiros fungicidas organo-sintéticos decorreram da produção
dos organoclorados. Os principais foram os halogenados, depois vieram 05
mercuriais, os heterocíclicos, os nitrogenados, 05 ditiocarbamatos e os
propriconazóis (fungicidas sistêmicos).
Em relação aos herbicidas, em 1932 se desenvolveu o primeiro deles, que
era organo-sintético e se chamava DNOC. Era um herbicida não seletivo e por
esta razão não foi bem aceito.
Sem dúvida, o DDT influenciou as pesquisas realizadas nos anos 40.
Novos clorados foram colocados no mercado, tais como: TOE, metaxiclor, BHC,
ciclodienos clorados. Ao final desta década os organoclorados já apresentavam
obsolência tecnológica na medida em que já apareciam insetos resistentes a
tais produtos. Portanto, o fenômeno da resistência gerado pela aplicação de tais
produtos aparece já no final dos anos 40.
Com relação aos herbicidas, nos anos 40 foram descobertos os
fenoxiácidos, de caráter seletivo. Isto foi um marco na ciência de controle de
ervas daninhas. Estes novos produtos deveriam ser preferencialmente seletivos,
2

~ .
l'
pois, caso contrário, iria requerer do agricultor, conhecimentos complexos a
cerca de práticas agronomas que levassem ao controle químico das ervas
daninhas sem prejudicar a cultura principal. Em função disto, já em 1944 novos
herbicidas foram colocados no mercado: MCPA e 2,4-0 e em 1945 os auxílicos:
TCA, profam e disoneb. Também foi desenvolvido nesta época o primeiro
herbicida carbamato, o IPC.
Nos anos 50 dois fatores concorreram para que a inovação se desse de
maneira intensa na indústria de pesticidas agrícolas. Os fatores foram: as novas
tecnologias de síntese orgânica e a expansão do mercado a que se destinavam
os pesticidas. Os organofosforados que apresentavam maior rapidez na ação,
passam a ser produzidos em larga escala, mediante a exploração de matéria
prima intermediária comum à produção dos clorados. A partir daí os produtos
inorgânicos deixaram de prevalecer nas vendas dos produtos fitossanitários.
Mas, estes produtos que de início foram soluções, começaram a apresentar
problemas de intoxicações e mortes de mamíferos, apontando mais uma vez
para a periculosidade dos pesticidas.
No Brasil, no início da década de 1950, a introdução de pesticidas
fosforados para substituir o uso do OOT, veio acompanhada de um
procedimento cruel. Foi ensinado que para misturar o OOT, formulado como pó
solúvel na água, o agricultor deveria usar o braço, com a mão aberta girando
meia volta em um e outro sentido, para facilitar a dissolução do produto. Como
o OOT tem uma dose letal alta (demanda uma alta absorção do produto para
provocar a morte), somente cerca de 15 anos depois os problemas de saúde
apareceriam. Contudo, quando o agricultor tentava repetir a técnica com o
3

parathion, o primeiro pesticida fosforado introduzido no Brasil, caía morto,
fulminado; fato que se repetiu em diversas regiões do país. 2
Nos anos 50, a modernização da agricultura, principalmente nos EUA,
abriu excelente mercado para os herbicidas, incentivando a intensificação de
pesquisas. Foram lançadas várias famílias: uréias, carbamatos, anilinas, todos
de pré-emergência.
Um fato histórico muito importante também correlacionado com o uso
desses produtos foi a Guerra do Vietnã, ocorrida entre os anos de 1954 a 1975.
O país se dividiu em duas metades: o Vietnã do Norte, apoiado pelos soviéticos
e chineses e o Vietnã do Sul, fortemente armado pelos norte-americanos que
para lá levaram milhares de soldados. Dentre todas as armas de guerra
presentes, destacavam-se os herbicidas desfolhantes (o mais famoso ficou
conhecido como "agente laranja"), que foram utilizados pelos norte-americanos
pela seguinte razão: como a resistência vietnamita era composta por bandos de
guerrilheiros que se escondiam nas florestas, formando tocaias e armadilhas
para os soldados americanos, a aspersão de nuvens de herbicidas por aviões
fazia com que as árvores perdessem suas folhagens, dificultando a formação de
esconderijos. Contudo, essa operação militar, aparentemente bem sucedida,
trouxe conseqüências ambientais e de saúde catastróficas para a população
local, que foram:
- Contaminação ambiental generalizada da fauna, flora e de todos os recursos
minerais do ecossistema local por período superior a 10 anos sem se estimar os
efeitos tóxicos pela bioacumulação no ambiente;
- Deformações em recém-nascidos por vários anos além de outros efeitos
teratogênicos.
21nformaçóes retiradas do site: \.'lVr''1.planetii~ll'ID-~nico~cºrn tJr
4

Fatos como estes remeteram toda a sociedade a outro tema muito
importante na história desses produtos: a criação de toda uma ciência voltada
ao estudo tóxico desses produtos para seres humanos e ambiente.
Foi nos anos 60 que houve o lançamento do primeiro fungicida sistêmico.
Nesta época foram desenvolvidos os inseticidas piretróides sintéticos, menos
tóxicos e mais eficientes. Os carbamatos, menos perigosos aos seres humanos,
cresceram em substituição aos clorados. Foi também um período de introdução
no mercado de vários organofosforados. Quanto aos herbicidas houve um
desenvolvimento nas inovações, que redundou no lançamento dos herbicidas
pós-emergentes que interferem na ação fotossintética das ervas daninhas.
Essa década também foi um marco para os movimentos ambientalistas,
que começavam a denunciar problemas relativos à resistência adquirida por
algumas espécies de insetos e os efeitos da bioacumulação em função do uso
de pesticidas. Tais movimentos lutavam contra o uso dos pesticidas
organoclorados.
Nos anos 70 houve uma grande queda no número de novos produtos
colocados no mercado. Isto se deu em função da dificuldade e dos custos de
obtenção de novas moléculas químicas, que se destinavam para este fim. As
exigências tornaram-se maiores, pois, deveriam atender a no~os padrões de
segurança, saúde e meio ambiente. Nesta época os movimentos ambientalistas
se fortaleceram e realizaram novas denúncias sobre as conseqüências da
utilização dos pesticidas. Foi dado enfoque aos problemas da poluição das
águas subterrâneas por herbicidas e fungicidas e das propriedades
cancerígenas de alguns pesticidas. O resultado prático da ação dos
ambientalistas foi a substituição de grande parte dos inseticidas organoclorados
pelos inseticidas organofosforados nos países desenvolvidos.
5

Outra novidade no seguimento dos inseticidas foi a introdução de alguns
feromônios, ou seja, hormônios sexuais responsáveis pela atração entre os
insetos, que acabaram por servir como um componente de manejo de diferentes
pestes, vindo a auxiliar no uso mais racional dos pesticidas químicos.
Nos anos 80 houve a introdução de alguns herbicidas dos grupos: difenil,
sulfonil-uréia, imidazolimonas, cicloexamedionas, oxi-fenoxi, quinoleno e ácido
carbaxílico.
Na tabela 1.1 é sumarizada uma seqüência dos maiores descobrimentos
na indústria de pesticidas, com suas respectivas datas aproximadas de
desenvolvimento.
Tabela 1.1 - Controle químico de pestes, doenças e ervas daninhas: uma
seqüência dos maiores descobrimentos.
Data aproximada de introdução19301942194519451945194819561956195819671975
pesticidasFungicida ditiocarbamato
DDTHerbicida Ac. Fenoxialcanóico
BHCInseticida organofosforado sistêmico
Inseticidas ciclodienosInseticidas carbamatos
Herbicidas triazinasHerbicidas bipiridilium
Fungicidas benzoimidazóisInsetic.idas píretróides fotoestavéis
Atualmente existe uma extensa e polêmica discussão acerca da introdução
de organismos geneticamente modificados (trangênicos) na agricultura visando
a diminuição do uso de pesticidas3. Os trangênicos são seres vivos cuja
estrutura genética - a parte da célula onde está armazenado o código da vida -
foi alterado pela inserção de genes de outros organismos, de modo a atribuir ao
3 El>.1raido do site: http://transgenicos.vilabol.uol.com.br
6

receptor características não programadas pela natureza. Desta forma é possível
inserir determinados genes em plantas e estas passam a produzir toxinas,
impedindo o ataque de determinadas pragas. Pode-se também proporcionar à
planta resistência a herbicidas ou ainda produzir grãos acrescidos de vitaminas
e sais minerais que sua espécie não possuía. Porém. existe controvérsia quanto
a efeitos que estas plantas trangênicas poderiam causar à saúde humana. Uma
linha de pensamento julga organismos geneticamente modificados como um
risco eminente à saúde humana e ao meio ambiente. Apesar destes
argumentos as grandes corporações detentoras da tecnologia garantem que os
trangênicos são organismos seguros ao consumo humano.
1.2 - Considerações gerais sobre pesticidas
Os pesticidas formam um grupo constituído por um grande número de
compostos orgânicos. muito dos quais são isômeros posicionais. geométricos e
óticos4. Os pesticidas também diferem quanto grau de ionização, polaridade e
solubilidade em água. Abrangem uma ampla classe de compostos. que podem
ser divididos em três grupos: biológico. inorgânico e orgânico. O ultimo grupo é o
mais amplamente usado com pronunciada atividade fisiológica; é constituído
principalmente por organoclorados. organofosforados. carbamatos e triazinas.
entre outros. de uso crescente na agricultura5: Nas Tabelas de 1.2 a 1.7 é
apresentada a estrutura de alguns pesticidas representantes de cada uma destas
classes.
4 K.H. Buchel, (editor) and Hohnwood, G.M. (translator); ·Chemistry of Pesticides", John Wiley &Sons, New York, 1983.5 F. Barlow; Chemistry and Formulafion. In, Pesficide Applicafion : Principies and practice, P.T.Haskel (Ed.), Oxford University Press, Oxford (1985), pp 1 - 34.
7

o termo pesticida é usado para designar todos os produtos químicos que
atuam no controle, prevenção, eliminação de pragas que atacam plantações e
rebanhos bem como vetores de doenças para os seres humanos. Dependendo da
sua utilização estes são classificados como, herbicidas, inseticidas, fungicidas,
acaricidas, nematicidas, etc.
Devido ao seu largo emprego na agricultura, aliado a sua persistência e
toxidade, estes compostos são fonte de contaminação ambiental apresentando
sérios riscos à saúde humana, pela incorporação de seus resíduos em águas,
solos e alimentos. O destino de pesticidas no meio ambiente é determinado por
fatores como adsorção, absorção, Iixiviação e volatilização, assim como por
transformações químicas, fotoquímicas e microbiológicas. A importância de cada
fator para um dado pesticida depende de suas propriedades físico-químicas, tais
como estrutura, pressão de vapor, hidrofilicidade, coeficiente de partição,
estabilidade térmica e fotoquímica, em conjunto com suas interações com o meio
(solo, sedimento, água), que por sua vez depende da biomassa, conteúdo de
matéria orgânica, pH, capacidade de troca iônica, textura e hidrogeologia.
8

Tabe
la1.
2-
Alg
umas
estr
utur
asdo
spe
stic
idas
dacl
asse
dos
herb
icid
ascl
orad
os.
~'.~.
...:.:
;.,.
-:
Fór
mul
a
estr
utur
al
nom
en
xy
zR 1
R 2R 3
R 4R s
2,4
-D
I1
IH
IH
IC
OO
HI
CI
IH
IC
II
HI
H
2,4
-D
BI
3I
HI
HI
CO
OH
IC
II
HI
CI
IH
IH
MC
PA
I1
IH
IH
IC
OO
HI
CH
3I
HI
CI
IH
IH
R4R
ií
2,4,
5-
T3
HH
CO
OH
CH
3H
CI
HI
H
*/;O-,.(C
)n-z
2,4
,5-T
P1
CH
3H
CO
OH
CI
HC
IC
IH
.I2,
4-
1C
H3
HC
OO
HC
IH
CI
CI
HR2
R1y
DP
MC
PP
2,4
-CP
PA
2,3
-C
PP
A
2,2
-C
PP
A
2-
PP
A
dica
mba
dicl
ofop
1 1 1 1 1 1
CH
3
CH
3
CH
3
CH
3
H H
H H H H H
CH
3
CO
OH
CO
OH
CO
OH
CO
OH
H
CO
OH
CI
CH
3
H H CI H
H CI H H H H
CI H H H CI
CI
H H H H H H 9
H H H H
CO
OH
CI

Ta
be
la1
.3-
Alg
um
as
est
rutu
ras
do
sp
est
icid
as
dacl
ass
efe
nilu
réia
No
me
R1
R2
R3
XY
Mo
nu
ron
HC
IH
CH
3C
H3
Flu
om
etu
ron
CF
3H
HC
H3
CH
3
Me
tob
rom
uro
nH
Br
HO
CH
3C
H3
Diu
ron
HC
IC
IC
H3
CH
3
Linu
ron
CI
CI
HC
H3
OC
H3
~o
Clo
rto
ruro
nC
IC
H3
HC
H3
CH
3
-11
Fe
nu
ron
HH
HC
H3
CH
3R
~/,
~-C-I-X
Iso
pro
turo
nH
(CH
3hC
HH
CH
3C
H3
R1
Hy
Me
toxu
ron
CI
OC
H3
HC
H3
CH
3
Mo
no
linu
ron
HC
IH
CH
3O
CH
3
Ne
bu
ron
HC
IC
IC
4H9
CH
3
Clo
roxu
ron
HH
CH
3C
H3
-0
-o
-C
I
Sid
uron
HH
H
-yH
H3C
10

Tab
ela
1.4
-A
lgu
ma
se
stru
tura
sp
ert
en
cen
tes
ao
sp
est
icid
as
da
cla
sse
do
so
rga
no
fosf
ora
do
s
No
me
Xy
ZR
1R
2R
3R
4R
sR
s
Fe
na
mifó
sO
NH
CH
(CH
3hO
CH
2CH
3H
CH
3S
CH
3H
H-
Iso
fen
fós
SN
HC
H(C
H3h
OC
H2C
H3
HH
HH
(a)
-
*~x
Rue
lene
OO
CH
3N
HC
H3
CI
H(b
)H
H-
'I'\
RS
-O
-P
--.
Clo
rfe
nvi
fos
OO
C2H
sO
C2H
sC
IH
CI
HH
(c)
-\
y
R2
R1Z
Par
atio
nS
OC
2HS
OC
2Hs
HH
N0
2H
HH
'ialif
osM
alat
íon
sf~
C'°
S
C2 H
S °"-...,!-S-CH~
CH
30,li P-S
-iH
-C
OO
CH
/",
;/
25
C2H
SO°
CH
30C
H-C
OO
CH
22
5
°°
Dia
zino
n
OM'I-
-ÁH3
Clo
rpiri
f6s
O--((
CIH
3C
-o ,,1
1II
~o
__/
~I
P-S
-C
H2
-C
-N
H-C
H3
H3 C
-O
//"
'I'\
CH
30,.
1P",
-CI
OM
e°
N
/'<D
imet
oato
N=
\
CH
3C
H(C
H3)2
(a)
CO
OC
H(C
H3h
(b)
C(C
H3)
z
(c)
C=C
CI
11

Tab
ela
1.5
-A
lgum
ases
trut
uras
dos
pest
icid
asda
clas
seda
stri
azin
as.
Est
rutu
rage
ral
Nom
eR
1R
2R
3
Atra
zina
CI
CH
(CH
3hC
H2C
H3
Sim
azin
aC
IC
H2C
H3
CH
2CH
3
Pro
pazi
naC
IC
H(C
H3h
CH
(CH
3hT
ertib
utila
zina
CI
C(C
H3h
CH
2CH
3
Cia
nazi
naC
IC
H2C
H3
C(C
H3h
CN
Ter
butri
nS
CH
3C
H(C
H3h
CH
2CH
3
R1de
smet
rina
SC
H3
CH
(CH
3hC
H3
NÁ
NS
imet
rina
SC
H3
CH
2CH
3C
H2C
H3
R3
,)l
A/R
2P
rom
etrin
aS
CH
3C
H(C
H3h
CH
(CH
3hN
NN
Am
etrin
aS
CH
3C
H(C
H3h
CH
2CH
3
II
Atra
tona
OC
H3
CH
(CH
3hC
H2C
H3
HH
Pro
met
ona
OC
H3
CH
(CH
3hC
H(C
H3h
Met
aból
itos
OH
--
hidr
oxila
dos
das
tria
zina
sac
ima
Met
aból
itos
de-
--
H
alqu
ilado
sda
s
tria
zina
sac
ima
12

Tab
ela
1.6
-A
lgu
ma
se
stru
tura
sd
os
pe
stic
ida
sd
acl
ass
ed
os
org
an
ocl
ora
do
s.
Est
rutu
raN
ome
R1
R2
R3
R4
R5
R6
R7
R8
R9
R10
X
Ger
al.
P,p
'-DD
TH
HC
IH
HH
HC
IH
HC
CI 3
.,·,
x*
O,p
'-D
DT
HH
HH
CI
HH
CI
HH
CC
b
-I
-P
,p'-D
DD
HH
CI
HH
HH
CI
HH
CH
Ch
*CH
"/,.8 O
,p'-
DD
DH
HH
HC
IH
HC
IH
HC
HC
h
R4R5
R1D
R9P
,p'-D
DE
HH
CI
HH
HH
CI
HH
CC
b
O,p
'-DD
EC
IH
HH
HH
HC
IH
HC
Cb
13

Tabela 1.7 - Algumas estruturas dos pesticidas da classe dos carbamatos.
Estrutura Geral Nome R1 R2
Carbofuran - CH3
Propoxur~yc~
CH3
H
I 0x)R~N-C-O-R1
IIO Metomil H CH2CH3I
" ~c" ~H:3NA~c S-CH:3
Tiofanato ,,#8 CH3c
metil o s I~ II 1I)r)
./C" ./~N~"N ~~c o H H
Aldicarb'" ~C", /C~
CH3
N/C", /CH3H C S
3Carbaril CH3
Co~ h
14

1.3- Efeitos tóxicos dos pesticidas
Com o decorrer do tempo novos agentes químicos foram criados, onde
sua atividade e especificidade aumentaram, juntamente com sua toxicidade.
No começo século passado os pesticidas mais empregados eram os
inorgânicos, que apresentavam modesta atividade e eram necessárias altas
relações de uso normalmente 10 100 kg/ha6. O subseqüente
desenvolvimento dos pesticidas orgânicos sintéticos, particularmente nos
últimos 60 anos, mudou radicalmente esta situação. Os níveis de atividade
aumentaram drasticamente como ilustra a Figura 1.1.
6 ç;jz-. o Herbicidas Im Inseticidas • Fungicidas
5
o2~ 3co-ºlicoco 2"DO'lo
.....J
..o~o.. .
oióli~
.1--"',.:I)),'".~
,- ._,1---
..'
4la.
Figura 1.1 - Relação de aplicação e datas aproximadas de introdução
para os pesticidas representativos, ilustrando o progressivo aumento de
atividade?
6 J.1. Grahan-Sryce: "Pesticides research for the improvement of human welfare". PesticidesChemistry: Human welfare and environmenl. Vol 1.Perganon Press. (Mivamato,J. andKearny,P.C.,Eds) 1983, pp.21-49.7J.L Grahan-Sryce; Proc. 8th Sul. Insectic. Fungic. Conto 3 (1975) 901
15

Em 1962 foi publicada uma das primeiras obras detalhando os efeitos
adversos da utilização de pesticida, iniciando um debate acerca das
implicações da atividade humana sobre o ambiente e o custo ambiental do uso
generalizado de pesticidas.8 Com a publicação deste livro o debate público
sobre o uso de pesticidas tomou-se uma realidade, pois até então essa
questão era restrita aos círculos acadêmicos e publicações técnicas, e também
se tomou a primeira obra a denunciar em escala pública o risco e os danos
ambientais causados pelo uso indiscriminado de pesticidas, tais como o DDT
que é um pesticida tóxico e letal ao ser humano.
Com o aumento de toxicidade, aumE;lnta-se a preocupação dos riscos
que estes agentes químicos provocariam aos mamíferos, especialmente aos
seres humanos. Alguns agentes específicos utilizados na agricultura são
responsabilizados por aumentar os riscos de câncer na língua, estômago,
cérebro, próstata, tecido conectivo e linfático, sistema hematopoiético entre os
fazendeiros 6,9,10. Em alguns estudos enfocando a exposição da mulher a
pesticidas, apresentaram alto índice de câncer no ovário, aquelas que tiveram
contato prolongado com os herbicidas da classe das triazinas11 e câncer de
mama, quando em contato com inseticidas12,13 e múltiplos mielomas14 e linfoma
de non-Hodgkin 15,16.
8 RCarson, Primavera Silenciosa; Houghton Miffin publisher, New Vorle, 1962.9 A .Blair, H. Malker, K.P.,. J. Work Environ. Health 11(1985), 397 - 407.1oN. Pearce, J.S .Ruf., Am. J. Ind. Med. 18 (1990), 133 -142.11 A. Donna, P Crosignani, F. Bobiette, J. Work. Environ. Health 15 (1989), 47 - 5312 F. Falck, A. River, M.S. Wolff, and S. Golbolds;. Arch. Environ. Health 47 (1992),143 -146.131.8. Wolfe, P.G. Toniolo, , L.W. Lu, River, M, and HDubin. J Natl. Cancer Inst. 85(1993),648 - 652.14 S.H. Zahm, and A Blair, J. Ind. Med. 49 (1992) 815.15 A. R Folson, S. Zhang, Sellers, T. A. Zheng, L.H. Kushi and J.R Cerhan,. J. OCCup.Environ. Med. 38 (1996) 1171.--
16

A exposição a alguns pesticidas foi associada a efeitos crônicos ou
agudos sobre o sistema nervoso, renal, respiratório e inclusive ao sistema
reprodutor tanto do homem quanto da mulher. 17
Estudos recentes nos Estados Unidos, mostraram que a exposição de
larvas de sapos machos a doses 30 vezes menores do que as legalmente
permitidas de um tipo de herbicida, a atrazina, estimula o hermafroditismo
nestes animais em desenvolvimento. A atrazina, herbicida mais vendido nos
Estados Unidos em sua categoria, também afeta sapos machos adultos:
diminui em dez vezes a taxa de testosterona, o principal hormônio masculino.
No Brasil estudos feitos pela Profa. Vara Almeida da Faculdade de
Medicina Veterinária e Zootecnia (FMZV-USP), demonstrou que o fenvalerato,
inseticida da classe dos piretróides 11, causa a feminilização de ratos. 18
A intoxicação exógena aguda por inseticidas carbamatos e
organofosforados tem sido um problema freqüente nos serviços de emergência
dos grandes hospitais, seja por ingestão acidental ou por tentativa de suicídio.
No estado do Rio de Janeiro, principalmente no Grande Rio, há um
importante problema de saúde pública relacionado a esses inseticidas: a
utilização irregular de carbamatos e organofosforados como raticida,
principalmente o carbamato Aldicarb, classificado como extremamente tóxico, e
comercializado ilegalmente com os nomes de "Chumbinho" e "Japan", entre
outros. Tais intoxicações têm causado inúmeros óbitos no Rio de Janeiro.
16 S.H. Zahn, 0.0. Weisenberg, R.C. Saal, J.B.Vaught, PA Babbitt, and A. Blair; Pesticideuse, genetics suceptibility, and non-Hodgkin Iymphoma in women, In'Ihird InternationalSynposium: Issues in Health, Safety, and agriculture- (Mcdufie, H.M., Oosman, J.A., Senchuk,K.M., and Okenchock, H.M., Eds. (1995), pp. 127 -134, Lewis, Michigan.17 IA Greaves; Agricultural Health, Health Environ. Digest. 4 (1992), 1 - 4.18 R. Zorzetto, S. Mendes; Pesquisa FAPESP, 79 (2002) 50.
17

o LDso é a quantidade de substância tóxica necessária para eliminar
50% de uma população de teste dentro de um período estabelecido. Ele é
usado para estimar a toxicidade oral ou dérmica, em termos de quantidade de
substância tóxica por unidade de massa corporal (mg/kg). Por exemplo,
pesticidas semelhantes ao ácido 2,4-dicloroacético (2,4-D), Dicamba, Parathion
e Carbofuran são incluídos entre outros agentes de alto grau de toxicidade
(LDso O- 50 mg/kg). O LDso somente não é eficiente para avaliar a toxicidade,
porque ele dá informações somente sobre a toxicidade aguda de um composto
em relação a uma certa espécie de animal e não descreve possíveis interações
devido à prolongada exposição ao composto (ex. toxicidade crônica). A
toxicidade crônica refere-se a capacidade de causar envenenamento por
exposições de longo tempo e baixos níveis destas substâncias. 19 Dados de
animais e humanos submetidos a estes testes indicaram inúmeros exemplos
de toxicidade crônica, incluindo carcinogenicidade causadas por compostos
como: (2,4-D), triazinas, Endrin, Linuron, Rotenone, Captan, Hidrazina malêica;
Teratogenicidade por Cianazina, Carbaryl, Endrin, Rotenone e Benomil;
Mutagenicidade por Dilato, Trifluralin, Dimetoato, Carbaril, Rotenone, Benomil e
hidrazina malêica; Neurotoxidade por fosfato de Etil (p-nitrofenil) tiobenzeno
(EPN) e efeitos sobre a reprodutividade por Dibromocloropropano (DBCP),
Dimetoato, Rotenone e Benomil.
O uso abusivo e desordenado de pesticidas causa sérios problemas de
natureza ecológica e de saúde pública2o. Alguns destes problemas são:
aparecimento de pestes mais resistentes à destruição de insetos polinizadores,
19 C. Bbornard, S.DaberKow, M.E.Smith, and N.N. Uri; Sei. Total Environ. 203 (1997) 229
20 B. Suganavan;. J. Environ. Sei. Heaith B 31(1996),307 - 323.
18

bioacumulação e a existência de resíduos na atmosfera, litosfera e
hidrosfera21 ,22.
o consumo mundial de agrotóxico sofreu um rápido aumento na última
metade do século passado. Entre os anos de 50 e 80 o crescimento se deu a
uma taxa anual de cerca de 10%.
Mais da metade da produção mundial de agrotóxico é consumida nos
Estados Unidos e na Europa Ocidental, regiões estas que abrigam cerca de
25% das terras globais ocupadas por culturas. Por outro lado cerca de 20% dos
agrotóxicos são consumidos nos países em desenvolvimento que contam com
55% das terras cultivadas.
Na tabela 1.8 são apresentados os valores em toneladas
comercializados no Brasil.
Tabela 1.8 - Consumo de pesticidas no Brasil (1997-1999) - valor em
toneladas. Fonte: Sindicato Nacional da Industria de Defensivos Agrícolas -
SINDAG...,- ...' -.....-_-~...... -~."~'-""~'-'-"'" - ---...,...,....._..... _" ..-
Pesticidas 1997 1998 1999- .
Herbicida 132.574 151.095 142.855
Inseticida 64.420 79.398 68.158
Fungicida 40.133 47.154 46.826
Acaricida 14.014 11.280 13.655
Outros* 14.099 17.875 16.581
total 265.240 306.802 288.075
* - reguladores de crescimento e espalhantes adesivos.
21 V.G. Zuin, F.P.S. Airoldi" N.R. Nascimento, M.D. Landgraf, and M.O.O. Rezende, J,Braz.Chem. Soe. 10 (1999), 25 - 30.22 J.R. Plemmer. J. Environ. Sei. Health B 31 (1996) 645 - 670.
19

1.4 - Pesticidas e seus mecanismos de ação tóxica
1.4.1- Organofosforados e carbamatos
Os organofosforados fazem parte do grupo químico formado por ésteres
de ácido fosfórico e outros ácidos à base de fósforo. Em relação aos pesticidas
clorados e carbamatos, os organofosforados são considerados mais tóxicos
(em termos ,de toxicidade aguda), porém se degradam rapidamente no
ambiente. Depois de absorvidos, os organofosforados e seus produtos de
biotransformação são rapidamente distribuídos por todos os tecidos. Os
compostos mais lipofílicos podem alcançar concentrações significativas no
tecido nervoso, ou outros tecidos ricos em lipídios. A sua absorção poderá
ocorrer por via respiratória, cutânea ou oral.
Os pesticidas carbamatos (compostos por ésteres de ácido
metilcarbônico ou dimetilcarbônico) utilizam vias metabólicas que rapidamente
os degradam em oximas . sulfóxidos , sulfo e acetonitrilas. Os carbamatos são
considerados de toxicidade aguda média, sendo degradados rapidamente e
não se acumulam em tecidos gordurosos. A maioria dos carbamatos, em geral,
não causa sintomatologia exuberante no sistema nervoso central (SNC) e,
quando esses sinais estão presentes, são considerados sinais de gravidade.
Os compostos organofosforados e carbamatos são inibidores da
colinesterase, impedindo a inativação da acetilcolina23, permitindo assim, a
ação mais intensa e prolongada do mediador químico nas sinapses
colinérgicas, na membrana pós-simpática.
23 A .F. Midio, SE Da Silva, "Inseticida-Acaricida Organofosforados e Carbamatos" EditoraROCA, 1995 pp. 35-39
20

A acetilcolina é sintetizada no neurônio a partir da acetilcoenzina A e da
colina24. É inativada por hidrólise sob ação da acetilcolinesterase, através da
transacetilação (uma forma de transesterificação) com a formação de colina e
ácido acético que, por sua vez, são reutilizados para a formação da
acetilcolina. A Figura 1.2 mostra este processo.
Acetilcolina-
?J +A--HJC'7'O~ "-
_/ -o~( rz~· ;t- Glll
+'I! ó'~
Sítio esterásico
- Colinestera
Sítio aniônico
Figura 1.2 - Processo de transacetilação da acetilcolina pela enzima
colinesterase
A acetilcolina é o mediador químico necessário para a transmissão do
impulso nervoso em todas as fibras simpáticas pré ganglionares do SNA
(sistema nervoso assimpático), todas as fibras parassimpáticas pós-
ganglionares e algumas fibras simpátias pós-ganglionares. Ainda é o
transmissor neuro-humoral do nervo motor do músculo estriado (placa
mioneural) e algumas sinapses interneurais do SNC. Para que ocorra a
transmissão sináptica é necessário que a acetilcolina seja liberada na fenda
sináptica e ligue-se a um receptor pós-sináptico. Em seguida, acetilcolina
disponível é hidrolizada pela colinesterase.
24 F. Matsumura, "Toxicology of Pesticides", 2a ed., Plenum Press, New York and London,1985.
21

Quando há a inibição da acetilcolinesterase, ocorre um acúmulo de
acetilcolina na fenda, levando a uma hiperestimulação colinérgica.
Na superfície da colinesterase existe um centro para inativação da
acetilcolina que contém um sítio aniônico e um esterásico. A inibição da
colinesterase se dá através da ligação do composto com o centro esterásico da
enzima diferindo apenas o tipo de ligação (fosforilação em organofosforados e
carbamilação em carbamatos). A Figura 1.3 ilustra este processo de ínativação
da colinesterase, para os pesticidas organofosforados.
organofosforado,--- x
RD,p'RD-'Y. ~/~
$€-r 1--::,.
-
-HY--
.- Colinesterase
Figura 1.3 - Processo de inativação da colinesterase por pesticidas
organofosforados.
A colinesterase fosforilada é posteriormente hidrolisada e a enzima
regenerada. Se isso não ocorrer, supõe-se que uma forma fosforilada muito
estável tenha sido produzida pela perda de um grupo alquil. Este fenômeno
denomina-se "envelhecimento" da enzima e quando corre, esta não mais se
regenera. Este fato é importante na terapêutica, pois dele depende a utilização
ou não de oximas (reativação da enzima). Os organofosforados são ditos como
inibidores irreversíveis da acetilcolinesterase, enquanto os carbamatos são
ditos inibidores reversíveis (sofrem hidrólise in vivo em 12 a 48 horas).
22

A ação neurotóxica tardia de alguns organofosforados nos nervos
periféricos é independente da inibição da colinesterase, mas parece estar
intimamente relacionada à fosforilação de uma esterase específica no tecido
nervoso. Este processo conhecido como esterase neurotóxica (NTE), causa
também o "envelhecimento" da enzima, ou seja, o estabelecimento de ligações
covalentes estáveis entre a enzima e o grupamento fosfato do organofosforado,
que provoca ataxia e agrava o desenvolvimento da doença. A NTE pode ser
também encontrada em linfócitos periféricos humanos (ainda não estabelecidos
como indicadores biológicos de efeito), no fígado, em plaquetas, entre outros
tecidos. A função fisiológica dessa enzima é ainda desconhecida bem como a
cadeia de eventos que culmina na polineuropatia em até 2 semanas após a
exposição ao organofosforado. Parece que o efeito neurotóxico ocorre pela
inibição na produção da enzima mais do que pela inibição de sua atividade, ou
seja, parece interferir nos processos de transporte axoplasmático, manutenção
e reparo de integridade das células nervosas (corpo celular e axônio). A
síndrome aparece após exposição sucessiva, mas não necessariamente
constante aos organofosforados. A presença de compostos fosfinados ou
carbamatos bloqueia a reação.
1.4.2 - Pesticidas Organoc/orados
Os pesticidas organoclorados, (ex. o 1,1,1-tricloro-2,2-di-(4-c1orofenil)
etano (OOT)), constituem um extenso grupo de compostos, largamente usado
na agricultura como inseticida. A problemática de seu uso concentra-se na sua
alta taxa de acúmulo nas cadeias alimentares e no meio ambiente. Os
23

pesticidas organoclorados são Iipofílicos, podendo causar sérios danos à
saúde. Embora sejam menos tóxicos (em termos de toxicidade que provoca
morte imediata) que outros organo-sintéticos, são também mais persistente no
corpo e no ambiente, causando efeitos patológicos a longo prazo. Devido a
este fato o uso destes inseticidas foi muito combatido, porém ainda hoje são
utilizados em culturas de frutas e leguminosas25.
Aldrin, Dieldrin e Endrin são considerados os compostos que mais
comumente causam envenenament026. Em geral os sinais de envenenamento
por diferentes organoclorados são similares e são expressos por uma
hiperatividade neural ou hiperativação do sistema nervoso central. Existe
considerável evidência que os inseticidas organoclorados agem
eletrofisiologicamente e, associado às propriedades enzimáticas das
membranas das células nervosas, causam mudanças na cinética de troca de
íons Na+ e ~ e no seu fluxo através da membrana. Distúrbios no transporte de
cálcio ou na atividade da Mg2+e Ca2+-ATPase também estão relacionados com
estes pesticidas, bem como a atividade da fosfoquinase27.28.29. Os
organoclorados foram largamente usados na agricultura e nos programas de
combate à malária nos anos de 1940 a 1960 com sensíveis efeitos benéficos.
1.4.3 - Herbicidas Triazinas
Dos herbicidas da classe das triazinas, a Atrazina tem sido o pesticida
de maior enfoque, pois é um dos contaminantes mais comumente encontrados
25 R.K. Juhler, M.R. Chrestensen, and G. Hilbert, J AOAC Int. 82 (1999) 337 - 352.26 V.G. Zsuin, and J.H. Velegas. Phytother. Res. 14 (2000) 73 - 88.27 F. Matsumura and K.C. Patil, Science 166 (1969) 121 -122.280.W.End, RACarchman and W.L. Oewey, J. Toxicol- Environ. Health 8 (1981) 707 -718.29 O.L. Shankland, Neurobehav. Toxicol. Tetratol. 4 (1982) 805 - 811.
24

em matrizes ambientais29. A Atrazina é um dos pesticidas mais empregados
em todo o mundo, em especial nos Estados Unidos30.
A Atrazina é metabolizada em quatro compostos hidroxiatrazina e três
clorotriazinas, e na conjugação destes compostos. Os compostos
hidroxitriazinas são metabólitos predominantemente encontrados em plantas,
enquanto que os clorotriazinas são encontrados em tecidos animais, solos e
A triazina age nas plantas inibindo a fotossíntese. Apesar dos herbicidas,
incluindo Atrazina, não estarem associados a distúrbios do sistema nervoso,
pesquisadores da Universidade de Sassari na Itália demonstraram que a
atrazina exerce um efeito tóxico sobre o sistema nervoso central. 31 Ratos ao
receberem doses de Atrazina apresentaram um decréscimo na atividade
elétrica nas células cerebrais, da parte do cérebro que controla os músculos
motores, resultando em perda de equilíbri032. A Atrazina altera a produção de
duas substâncias químicas produzidas no sistema nervoso central, a dopamina
e norepinefrina33. Ambas transmitem impulsos através das células nervosas, e
agem como hormônios. A deficiência na produção destas substâncias altera o
nível de dois hormônios a prolactina e a luteinizina32. Os produtos resultantes
da metabolização também influenciam na produção de dopamina e
norepinefrina34. Alem destes homônimos, estudos demonstraram que a
Atrazina também influencia na produção de outros hormônios como:
30 Fonte : www.epa.gov/oppsrrd1/reregistration/atrazina/index.htm31 M.V.Podda, Pharmaeol. Res. 36 (1997)19932 CancerWeb, 1995-1998. The on-Iine medicaI dictionary. www.graylab.ac.uk33 P.C.Oas, W.K.McElroy,R.L.Cooper, Toxieol. Sei. 56 (2000) 32434 P.C.Oas, W.K.McElroy,R.L.Cooper, Toxieol. Sei. 59 (2001) 127
25

progesterona35, testosterona36, estrogêni037 e os provenientes da tireóide38
.
Estudos realizados mostraram que atrazina apresenta efeitos sobre a
resistência imunológica. Experiências realizadas com ratos monstraram que,
três semanas após os ratos terem recebido doses diárias de 100 mg/kg de
Atrazina, estes desenvolveram uma linfopenia (redução do número de glóbulos
brancos)39. A exposição de células sanguíneas humanas ao herbicida triazina
apresentou uma diminuição na produção de interleucina, uma proteína
regulatória do sistema imunológico e interferon uma proteína que combate
infecções virais40.
1.5 - Técnicas utilizadas para identificação e quantificação de pesticidas
Os métodos oficialmente adotados para a determinação de pesticidas
nas mais diversas matrizes, utilizam a cromatografia gasosa com detector de
captura de elétrons (GC-ECO)41, cromatografia gasosa acoplada à
espectrometria de massas (GC-MS)42 e cromatografia líquido de alta eficiência
(HPLC) com detecção U~·44.45. Sendo que em alguns casos reações de
derivatização são necessárias objetivando inclusive a determinação via análise
35 RL.Cooper, Repro. Toxicol. 10 (1996) 25736 J.Kniewald, J. Appl. Toxieol. 15 (1995) 21537 J.T. Sanderson, Toxieol. Sei. 54 (2Mo) 12138I.N.Kornilovskaya, Eur. J. Endoerinol. 130 (1994) 12939 J.G.Vos, EL.Krajnic, Immunotoxicity of pesticides. In Hayes, A .W., RC. Schenell, T.S. Miya(eds.). Developments in the science and practice of toxicology. Proceeding of the 3rd
International congress toxicology. San Diego CA, USA. Aug.28 - Sept.3, 1983. Amsterdam, TheNetherlands: Elsevier Scientific Publishes. pp. 229-240.40 RJ.Hooghe, S.Devos" EL.Hooghe-Peters. Life Sei. 66 (2000) 251941 A. Venant, EM. Neste, J.Mallet.; Food Addíot. Contm., 71 (1990) 117.42 B. Koppen, J.AOAC Int., 77 (1994) 81043 J. Mao, M.K. Erstfed, P.H. Fockler, J. Agrie. Food Chem., 41(1993) 596 - 601.44 RA Chaoman J. Chromatogr. 258 (1983) 175 - 18245 F.G.Sanchez, AN. Diaz, AG.Pareza J. Chromatogr. A 754 (1996) 97 -102.
26

por fluorescência46. A cromatografia com fluido supercrítico (SFC), também tem
sido utilizada para a determinação de pesticidas.47
A determinação de vários pesticidas e seus metabólitos a nível de
traços em matrizes ambientais requer métodos de separação de alto poder de
resolução, eficiência e seletividade única. Entre os métodos analíticos de
separação e identificação de substâncias que estão rapidamente crescendo e
se desenvolvendo nas últimas duas décadas destaca-se a eletroforese capilar
(CE). A CE teve um rápido avanço e atualmente é uma das técnicas de
separação analítica largamente empregada nos setores farmacêutico48 e
bioquímic049 e a cada dia vem ganhando destaque em diversas áreas. A
ampla divulgação da CE decorre do fato de ser uma técnica de fácil
implementação e que oferece uma variedade de modos de separação que
podem ser efetuados em uma única coluna capilaro, onde compostos oriundos
de diversas matrizes podem ser analisados51. Separações com os modos CZE
(eletroforese de zona) e MEKC (cromatografia eletrocinética micelar) permitem
determinar desde moléculas neutras às ionizadas. A seletividade e resolução
da separação são diretamente controladas por diferentes aditivos adicionados
ao eletrólito de separação (solventes, agentes complexantes, polímeros,
ciclodextrinas,etc.). Em artigos de revisão recentemente publicados, a
potencialidade da técnica é divulgada para uma grande gama de compostos
46 s.o. Mcgavey J chromatogr. B. 659 (1994) 243 - 257
47 0 .E.Knowles, B.E. Ricther, M.S. Wygant, L. Nixon, M.R.Anderson J. Assoc. Oft. Anal.
Chem 71 (1988) 451.
48 J.Yang, H.Long, H.Liu, A .Huamg, Y. Sun ,J.Chromatogr. A. 811(1998)27449 J.Chapman, J.Hobbs, LC-GC 5 (1999) 44350 K. o. Altria; Capillary Electrophoresis Guidebook.principles, instrumentation, operation, andapplications; Methods in Molecular Bio/ogy, Series 52; Series Editor J. M. Walker; HumanaPress, 1995.51 J. P. Landers,; Handbook of High Performance Capillary Electrophoresis; CRC Press, 1993.
27

em matrizes ambientais, incluindo a análise de pesticidas52•53
, compostos
orgânicos 54e inorgânicos55. A CE vem sendo utilizada para a análise e
quantificação de pesticidas em águas naturais, com grande aceitação pelos
pesquisadores. Uma série de classes de pesticidas vem sendo analisada
como, organofosforados, carbamatos, triazinas entre outras, com níveis de
detecção da ordem de 0,1 fJ.g/L. Para se alcançar estes níveis de detecção
alçou-se mão de técnicas de pré-concentração como a extração em fase
52 F.Menzinger, Ph. Schimitt-Kopplin, D. Freitag, A .Kettrup, J.Chromatogr. A, 891(2000)4553 D.T. Eash, R.J. Bushway, J. Chromatogr. A, 880 (2000) 28154 A .Karcher, Z.EI Rassi, Electrophoresis 20(1999)328055 A .R.Timerbaev. E.Dabek-Zlotozynska, M.A. van den Hoop, Analyst 6(1999)81156 Y.S.Wu, S.F.Y. Lu J. Microco/. Sep., 10 (1998), 525 - 535
28

~~u
-~
~
i
~
u
~
~~9
'B~
u~.....--.. -.)
~
CAPÍTULO 2

29
2 - Fundamentos da Eletroforese capilar (CE)
A eletroforese é uma técnica de separação baseada na migração
diferenciada de compostos iônicos ou ionizáveis, em um campo elétrico1.
o uso da eletroforese como técnica de separação foi introduzida por
Tiselius2em 1937, que obteve a separação parcial de algumas proteínas
constituintes de soro sanguíneo em solução livre em tubos verticais em formato
de "U". Em 1967, Hjérten3 descreveu o primeiro trabalho com o uso de campo
elétrico elevado empregando tubos de quartzo com dimensões de 300 j..lm de
diâmetro interno e 36 cm de comprimento, com voltagem de 2,5 a 3,0 kV. Em
1979, Everaerts et al4 demonstraram o uso de tubos de teflon com diâmetros
interno de 200 j..lm, para a separação de 16 ânions, constituindo a primeira
publicação de eletroforese capilar. Na década de 80, Jorgenson e Lukacs5
avançaram com a técnica, com o uso de capilares de sílica fundida de 75 j..lm
de diâmetro interno.
A eletroforese capilar (CE) é uma técnica analítica que engloba uma
família de técnicas, com mecanismos de separação singulares e seletividade
característica, como a eletroforese em solução livre (FSCE), cromatografia
eletrocinética micelar (MEKC), eletroforese em gel (CGE), focalização
isoelétrica capilar (CIEF) e isotacoforese capilar (CITP).6.7 Em eletroforese
1 Electrophoresis - Theory, methods and application; Beer, M., Ed; Acadenic Press Inc., J.
Chromatogr.Lib., Vol 18, Elsevier, Amsterdan (1970).
2 A Tiselius; A New Apparatus for Electrophoresis Analysis of Colloidal Mixtures, Trans.Faraday Soc., 33 (1937) 5243S. Hjérten, High Performence Zone Electrophoresis, Chromatogr. Rev., 9 (1967)1224 F.M. Everaerts, F.E.P. Mikkers, T.P.E.M. Verheggen, ,J.Chromatogr., 11 (1979) 115 J. Jorgenson, K.D. Lukacs, ,Anal. Chem 53 (1981) 12986 R. Kuhn; S. Hoffstetter-kuhn; "Capillary electrophoresis: principies and pratice': New YorK:Springer Verlag, 1993, 375.7 M.F.M. Tavares, Quím. Nova 5 (1997) 20

capilar, a separação é conduzida em tubos de dimensões capilares, de 15 a
100 !-lm de diâmetro interno, e 50 a 100 cm de comprimento, usualmente
preenchidos com solução tampão. O uso do capilar oferece muitas vantagens
sobre os outros meios utilizados para eletroforese (placas de gel, papel, etc.).
Devido a fatores geométricos (a relação entre a área superficial interna e o
volume é apreciavelmente grande), um capilar possibilita a dissipação eficiente
do calor gerado pela passagem da corrente elétrica (efeito Joule). Além disso,.a alta resistência elétrica do capilar permite o estabelecimento de campos
elétricos elevados (100 a 500 V/cm), resultando em separações de alta
eficiência (geralmente excede 105 pratos), resolução inigualável e tempos de
análise apreciavelmente curtos.
As separações conduzidas por CE são realizadas em capilares muito
estreitos (capilares com diâmetros diminutos), com mínimo consumo de
amostra (quantidade nL) e, muitas vezes, proporcionam uma resolução do
analito que é superior a muitas técnicas cromatográficas tradicionais como
HPLC.
Na eletroforese capilar as amostras podem ser introduzidas no capilar
por métodos eletrocinéticos ou hidrodinâmicos. Na injeção eletrocinética, um
gradiente de potencial é estabelecido ao longo do comprimento do capilar por
um período de tempo conhecido, enquanto que na injeção hidrodinâmica
utiliza-se um gradiente de pressão. O gradiente de pressão pode ser
estabelecido por diferentes mecanismos: pressurização ou vácuo em um dos
reservatórios de solução, ou por gravidade onde um dos reservatórios é
elevado em relação ou outro e a amostra é inserida no capilar por sifonagem.
30

Uma variedade de detectores tem sido usado na eletroforese capilar,
incluindo: UVNis, fluorescência, condutividade, amperométrico, espectrometria
de massas, etc. O detector mais amplamente utilizado é o UVNis, destacando
se o com arranjo de diodos (diode array detector- DAD), que tem a vantagem
de detecção simultânea em múltiplos comprimentos de ondas e a habilidade de
fornecer dados espectrais.
2.1 - Fluxo e/etrosmótico
Um importante fenômeno que ocorre na eletroforese capilar é o
transporte eletrosmótico de espécies em solução. Este fenômeno tem origem
na parede interna do capilar de sílica fundida não desativada, devido à
presença de grupos silanóis (SiOH) que se ionizam a silanoato (SiO-) numa
faixa de pH entre 2 e 8,58. Como mostrado na Figura 2.1, é formada
inicialmente uma primeira camada de cátions do tampão a fim de estabilizar a
carga negativa da parede do capilar a qual, por atração eletrostática,
permanece imobilizada, sendo denominada camada fixa. Devido ao excesso de
cargas negativas, uma segunda camada de cátions, esta última móvel, forma
se logo após a camada fixa, dando origem ao que se convencionou chamar de
dupla camada elétrica de cátions. No momento da aplicação do campo elétrico,
os cátions hidratados da camada móvel migram em direção ao eletrodo
negativo arrastando todo o volume da solução, este movimento da solução é o
gerador do fluxo eletrosmótico.
8 M.F.M Tavares, Quim. Nova 19 (1996)173-18131

a· .. no... do "''''-q' . ~ ...... -~ I.i:\ \ !
\tJ si ~"~u.~-"t'-'''' ' ·r-----·~~''''''---v· · · : \!r \ .. r ~.· ·· I \ .'. '\,i! ,'/ \1/ \L '" p~rf.ltíu dQ t: ~fp il ;$t
Si Si Si Si . . ... . .. .
· 10 ç~ l)~.' . . '~.' .' .. " '''.. " . ...• O·.;;;.r. __ .. _d\..' u tI ;. X Z-j .. .. .. .. .. <tJ
tXH1<.:tt,iô"" môv~j : .•. @
I lt l o@ b(iJ b0 b~
@ @ (f) @ , • ~ .. '" '< ,"" . , _. • ' . ~ • - • ~ <. , .~,.. , ,
~ -$ !~~, .. ~. @
Fluxo Eletroosmótko (EOf) ... .......... .. ... . ...•.
Qtodo
o
Figura 2.1 - Formação do fluxo eletrosmótico no interior de um capilar
de sílica fundida após aplicação de um campo elétrico
o fluxo eletrosmótico é caracterizado por um perfil linear de velocidade,
não contribuindo, portanto, para o alargamento das bandas. Esta peculiaridade
distingue eletroforese capilar dos métodos cromatográficos em fase líquida,
que apresentam um perfil de velocidade parabólico, característico do fluxo
induzido por pressão. Além disso, o fluxo eletrosmótico, em geral de grande
magnitude, é responsável pela condução dos sOlutos, sem distinção de carga,
em direção ao detector, permitindo assim a análise simultânea de amostras
contendo solutos catiônicos, neutros e aniônicos.
2.2 - Migração eletroforética
Quando um eletrólito no interior do capilar sofre a aplicação de um
campo elétrico, os íons presentes em solução são atraídos em direção aos
eletrodos de sinais contrários aos de suas cargas (q) pela ação de uma força
eletrostática (F e) descrita por:
32

(1)
À medida que o íon é acelerado de seu estado de equilibrio até uma
velocidade v, surge uma força friccionai (Ff) que tende a retardá-lo. A força Ff
pode ser expressa pela Lei de Stoke:
Ff =6n llrv (2)
e é função da viscosidade da solução (r1>, do raio iônico da espécie (r)
(considerada esférica), e da velocidade com a qual o íon se move em solução
(v).
Depois de atingido o equilíbrio entre as duas forças (quase
instantaneamente), os íons na solução movem-se com uma velocidade
constante, dada por
v =JlE (3)
A constante de proporcionalidade I-l é chamada de mobilidade
eletroforética e é característica de um íon em um dado conjunto de condições
(meio e temperatura). Combinando-se as equações 1, 2 e 3 , obtem-se a
relação
Jl = q =_v_ (cm2V-1s-1) (4)
6rptr E
Além da mobilidade eletroforética, um outro valor de grande importância em CE
é a mobilidade aparente (J...la). A mobilidade aparente leva em consideração a
contribuição da mobilidade do fluxo eletrosmótico (J..I.eo) em relação à
mobilidade eletroforética dos íons (J..I.e). A mobilidade aparente de um íon é
dada pela expressão:
33

J..la =J..leo + J..le (5)
Pode-se também ser calculada através de parâmetros experimentais,
onde é dada pela equação (6).
(6)
onde:
/la = Ileo + Ile
v = Voltagem aplicada
Llol = comprimento total do capilar
t =tempo de migração
Ldet =comprimento efetivo do capilar (até o detector)
2.3 - Modo de separação utilizado em análise de pesticidas por
e/etroforese capilar
2.3.1 - Cromatogratia e/etrocinética micelar capilar
Uma das desvantagens da separação eletroforética em solução livre é a
incapacidade de resolver misturas de solutos neutros, o que foi solucionado por
Terabe e colaboradores com a introdução da" cromatografia eletrocinética
micelar capilar, MEKC. 9
o princípio básico da MEKC é a partição diferencial dos analitos entre a
fase aquosa e a micelar, que recebe o nome de fase "pseudo -estacionária",
uma vez que são agregados de moléculas carregadas e, como tal, apresentam
9 S. Terabe; K. Otsuka; K. Ichikawa; A. Tsuchiya; T. Ando; Anal. Chem., 56 (1984) 111
34

mobilidade eletroforética e estão sujeitas à ação do campo elétrico. O agente
formador de micelas mais utilizado em MEKC é o dodecil sulfato de sódio,
50S, cuja fórmula estrutural é mostrada a seguir.
CH3-(CH2h1-0-S03Na
Figura 2.2 - Fórmula estrutural do 50S
Para que ocorra a formação da micela em solução, o detergente deve
estar presente em uma determinada concentração mínima, a qual é
denominada concentração micelar crítica, cmc, que para o 50S é 8,27 mmolL-1
(em água).
Um analito insolúvel em água, adicionado a uma solução contendo
micelas, se alojará no interior hidrofóbico da micela, passando a maior parte do
tempo de análise desta forma. Já um composto altamente hidrofílico,
praticamente não apresentará interação com a porção hidrofóbica da micela,
interagindo com a parte externa da micela composta pelos grupos hidrofílicos.
Porém este processo de interação do analitos e a micela é um processo
dinâmico, desenvolvendo um equilíbrio de partição entre a solução aquosa e a
micela. A MEKC pode ter seu desempenho melhorado pela adição de outras
substâncias no tampão de análise, como é o caso dos solventes orgânicos (ex.
metanol, etanol ou acetonitrila) ou seletores quirais (ciclodextrinas)1O. As
ciclodextrinas são substâncias neutras formadas por ciclo oligossacarídeos
10 Ph. Schimitt, ,A.W. Garrison, O. Freitag, A Kettrup,; J. Chromatogr. A., 792 (1997) 419
429.
35

naturais, compostas de várias unidades glicopiranose. Na Figura 2.3 é
mostrada a estrutura das ciclodextrinas.
a-I A-tvpeHidroxila primária -A Ligação glicosídica
~m~-o- I 6ctl~otl(:. _ . <t
4 ... I If -1 '1 . -OM
0.p I~ '~t
6 Hidroxil as se cund árias
t
Q ~..p n
n = 1, ~-ciclodextrina
n = 2, )'-ciclodextrina.
a... cic10 dex trina tem apenas 6 Utlidades de ~cose
o14.6 A
o15.4 A
o17.5 A
"Ic =- >
t\IW 972 'IW 11.35 MW 1297
Figura 2.3 - Estrutura química e modelo esquemático das ciclodextrinas:
a-CO, 13-CO e y-CO. 11
A cavidade interna das ciclodextrinas apresenta características
hidrofóbicas, enquanto que a sua parte externa características hidrofílicas,
devido aos grupos hidroxila ali presentes. A melhora na separação ocorre
11 J.H.T.Luong, A.L.Nguyen J. Chromatogr. A 792 (1997) 431 -444.36

devido à interação do grupo hidrofóbico do analito no interior de cavidade
hidrofóbica da CO e também às interações externas com os grupos hidroxila
que estão dispostos na estrutura das COs, interações do tipo dipolo-dipolo. As
ciclodextrinas possuem diferentes graus de afinidade pelo analito, desta forma,
o analito terá seu tempo de migração alterado podendo assim, ser diferenciado
de seu par crítico. Na Figura 2.4 é ilustrada uma separação MEKC, na
ausência (A) e presença de seletores quirais (8), em elevado EOF.
Detecção t tlnjeçãO
< '0 0 0 0'0· 0e 0Vosm r::-v... A"· - ~ 5 5
V '-.:.J '- ..o ~'tJ .-"~ G). . -.<9&fR, .. ) Q.__ .. G A
SOLUT
B
Figura 2.4 - Esquema para uma separação eletroforética no modo MEKC, na
ausência (A) e na presença (B) de seletores quirais. Detalhe mostrando
monômero e micelas de SOS, ciclodextrinas e a interação de diversos solutos
neutros com ambas as fases. Um soluto não retido pela micela migra com
velocidade de fluxo eletrosmótico, Vosrn (VEOF). A velocidade eletroforética da
micela é Vem. enquanto a sua velocidade aparente é Vm.
37
+

Existe a possibilidade de realização de MEKC em condições de pH
baixo, onde o EOF é suprimido. Um esquema exemplificando o mecanismo de
separação na condição de ausência de EOF é mostrado na Figura 2.5.
INJEÇÃO _.ij,. .ij. DETECÇAO
o G G G Q Q Q Q~
~ VEOF
+
Figura 2.5 - MEKC em condição de baixo pH (supressão do EOF).
Quando o potencial é aplicado micelas que partem do lado catódico (-)
migram em direção ao eletrodo de carga oposta, capturando o analito e
conduzindo-o para a janela de detecção.
2.4 - Sistemas micelares
o MEKC é normalmente o modo de separação selecionado para a
determinação de pesticidas, pois em sua maioria são compostos neutros. Para
se atingir seletividade e resolução é comum adicionar ao eletrólito de
38

separação aditivos orgânicos (álcoois ou acetonitrila), que ocasionam
mudanças na estrutura micelar no meio de separação, alterando as
propriedades físico-químicas, entre elas: viscosidade do meio, constante
dielétrica do meio e na superfície da micela, número de agregação micelar e
cmc. Nos próximos tópicos serão feitas abordagens a respeito de ambientes
micelares.
2.4.1 -Tensoativos
As moléculas anfifílicas, ou os tensoativos são conhecidas por
possuírem, dentro de sua estrutura molecular, regiões de polaridade distintas,
uma parte hidrofílica (ou polar) unida a uma parte hidrofóbica (ou apoiar).
Essas moléculas possuem a capacidade de autoagregar-se em solução
aquosa e, dependendo de sua estrutura e concentração, formam micelas,
vesículas ou cristais líquidos liotrópicos. Todos estes sistemas apresentam
propriedades interessantes de solubilização e são capazes de afetar a
velocidade de reações químicas, deslocar equilíbrios, controlar
regiosseletividade e modificar o comportamento de espécies geradas
fotoquimicamente 12,13,14,15.
2.4.2 - Classificação dos tensoativos
12J.H. Fendler , "Membrane Mimetic Chemistry", Wiley: New York (1982)
13L.S. Romsted, In:"Surfactantes in Solution" (K.L.Mital e B.Lindman, Eds.) Plenum Press: NewYork; (1984) Vo12, pp. 1015.
14CA Bunton, G. Savelii" Adv. Phys.Org.Chem., 22 (1986) 213
39

Um grande número de tensoativos com várias combinações de grupos
hidrofílicos e hidrofóbicos é comercialmente disponível. Dependendo da carga
da parte polar hidrofílica, os tensoativos são classificados como aniônico, não
iônico e zwitteriônicos. Na tabela 2.1 são apresentados alguns representantes
de cada classificação.
Tabela 2.1 - Estruturas de alguns tensoativos.
classe fórmula nome
Aniônico CH3(CH2)110S03-Na+ Dodecilsulfato de sódio
(SDS)
Catiônico CH2(CH2)1sN+(CH3).B( Brometo de hexadecil
trimetilamônio (CTAB)
Não-iônico CH3(CH2)11 (OCH2CH2h 3OH Poli-oxietileno dodecil
éter (BRIJ 35)
Zwitteriônico 3-(hexadecildimetil-CH3(CH2hsN+(CH3h(CH2hS03-
amônio)-Propano-1-
sulfato (CDAPS)
Os tensoativos aniônicos, não-iônicos e zwitteriônicos são de grande
interesse econômico devido a sua aplicação como detergente, emulsificante,
dispersante e umectante. A propriedade dos tensoativos não-iônicos em
solução é uma função da porcentagem em peso de polioxietileno (OE). Assim,
o número de unidades de óxidos de etileno introduzidas na molécula durante a
síntese do tensoativo depende das características desejadas do produto final.
40

o balanço hidrofílico e lipofílico (HLB)16é um bom exemplo de caracterização
dos não-iônicos:
HLB=%OE
5 (7)
41
onde % OE é dada em peso. Esta escala varia de O, produto de partida, não
contendo OE, a 20, o próprio polioxietileno glicol. Produtos com HLB maior que
10 são mais solúveis em água do que em óleo e os com HLB menor que 10 o
contrário17. As formulações de detergentes aquosos empregam os não-iônicos
com 50 % em peso de OE (HLB 10) e ou acima, sendo que as melhores
propriedades de detergência são encontradas entre 55 a 75 % de OE
(respectivamente, HLB 11 e 15).
Os tensoativos catiônicos, do tipo sais de amônio quaternário,
possuem atividade germicida/bactericida e são empregados em composição
anti-sépticas e desinfetantes de uso doméstico, industrial e hospitalar. Os
catiônicos também são bastante utilizados tanto como amaciantes de roupas
como na formulação de creme rinse.
2.4.3 - Propriedades gerais dos tensoativos
2.4.3.1 - Processo de agregação dos tensoativos em água
16 P. Becker, , in "Nonionic Surfactants" Vo1.1. (Schick, M.J., ed.) Marcel Dekker, Inc., NewYork,1966.
17A.W. Adamson, , "Physical Chemistry of surfaces·, 2nd ed., JohnWiley and Sons, Inc., NewYork (1967)

Os tensoativos, em solução de baixa concentração (aproximadamente
10-5 moIL-1), encontram-se na forma de monômero, comportando-se como um
eletrólito forte, no caso de um monômero iônico, ou simples soluto orgânico,
estando completamente dissociados. Conforme se aumenta a concentração do
tensoativo a solubilização de sua parte hidrofóbica torna-se energeticamente
desfavorável devido a dois fatores: alta tensão interfacial água/hidrocarboneto,
diminuição da entropia do sistema causada pela estruturação das moléculas de
água ao redor dos hidrocarbonetos.
Numa tentativa de reduzir a energia interfacial do sistema, a cadeia de
hidrocarbonetos do monômero (ou moléculas) do tensoativo na água
apresentam-se enroladas e medem aproximadamente 70 % do comprimento
da molécula completamente estendida 18.
Ainda nesta faixa de concentração, um pouco acima de 10-5 molL-1, o
balanço energético está equilibrado. A perda de entropia devido à estruturação
da água e a existência da grande energia interfacial iguala-se ao ganho em
entropia devido à dissociação da cabeça iônica, solvatação pelo grupo
hidrofílico e entropia configuracional da cabeça polar dos não-iônicos.
Aumentando-se a concentração de tensoativo, ocorre um aumento na energia
livre do sistema.
Para minimizar a energia livre total ocorre o fenômeno de adsorção
dos monômeros na interface ar-água, com as cadeias dos hidrocarbonetos
voltadas para fora da solução e os grupos hidrofílicos presentes na interface
18P.H. Elworthy, J.Chem. Soc., 388 (1963)42

43
aquosa19. Isto reduz a energia interfacial água-hidrocarboneto e aumenta a
entropia liberando as moléculas de água presas ao redor da cadeia carbônica.
Neste fenômeno de adsorção ocorre a substituição de moléculas de
água presente na interface, pelo grupo hidrofílico do tensoativo. Como as
forças intermoleculares de atração entre molécula de água e um grupo não
polar são menores do que entre duas moléculas de água, reduz-se o poder de
contração da superfície, ou tensão superficial.
A partir do momento que ocorre uma saturação na interface água-ar, e
aumentando-se a concentração do tensoativo, os monômeros começam a
associar-se em dímeros, trímeros, tetrâmeros, diminuindo a energia livre do
sistema.20
o aumento da concentração do tensoativo continua a aumentar a
energia livre do sistema. Como conseqüência final, ocorre a formação de um
agregado chamado micela no qual a parte hidrofóbica monomérica se agrupa
formando um núcleo (ou miolo) micelar e a parte hidrofílica se dispõe na
superfície voltando-se para o solvente majoritário (água). Na figura 2.6 é
apresentado o esquema de uma micela aquosa de um tensoativo aniônico.
19p .H. Elworthy, A.T. Florence, C.B. Macfarlane, ·Solubilization by Surface Active Agents"Chapman and Hall Ltd., London, 1968.
20 P. Mukerjee, Adv. Colloid Interface Sei., 1 (1967) 241

- Camadade Stem -
Dupla camadade Gouy-Chapman
44
Figura 2.6 - Esquema de uma micela iônica baseada no modelo de Hatley.
A micela é envolvida pela camada de Stem que contém os grupos
iônicos e também 50 a 80 % dos contra-íons, concedendo à micela uma carga
residual positiva ou negativa em função da carga das cabeças.
A camada de Stem é envolvida por uma dupla camada elétrica difusa,
denominada de dupla camada de Gouy-Chapman, e que contém os restantes
dos contra-íons solvatados65.
Apesar da controvérsia a respeito de modelos para a estrutura de
micelas, a maioria dos pesquisadores está de acordo com uma forma
aproximadamente esférica67,21 ,22. As cadeias hidrocarbônicas estão dispostas
de maneira aproximadamente radial, mas com alguma desorganização,
21 D. Attwood & A T. Florence, Surfactant Systems. ehapman and Hall, London, New York,1983.22 e.A. Bunton, F. Nome, F.H. Quina, & L.S. Romsted, íon Binding and reactivity at changedaqueous interfaces. Aee, Chem. Res., 24 (1991)357.

conferindo ao núcleo uma natureza Iíquida66. A penetração da água pode
ocorrer até os quatro primeiros grupos metilênicos próximos ao grupo de
cabeça (camada palasídica). Há interações entre os contra-íons e moléculas de
água, na camada de Stem. A dupla camada de Gouy-Chapman contém os
contra-íons restantes e se estende radialmente até a fase aquosa23. O modelo
de Hartley é o mais citado na literatura, devido ao fato de ser o que melhor
concorda com essa exposição64. Dependendo da concentração do tensoativo
no meio micelar, do tipo do tensoativo utilizado, se o meio é aquoso ou
orgânico, bem como a força iônica do meio, modificações estruturais ocorrem
como pode ser verificado através da Figura 2.7.
No caso de tensoativos zwitteriônicos, a estrutura micelar é diferente.
Supõe-se que os monômeros orientem-se radialmente no agregado micelar.
Isto faz com que o miolo seja rodeado por uma camada positiva e esta é
rodeada por uma camada negativa. Esta estrutura pode ser comparada a
micelas catiônicas não dissociadas24. No caso de tensoativos não-iônicos, o
miolo micelar encontra-se envolvido pelas cabeças solvatadas de cadeias poli-
oxietilênicas65.
23 A .W. Adamson, Physical Chemistry of surfaces. 3ed. Wiley -Interscience Publication, NewYork,1976.
24M.S. Baptista, L Cuccovia, ,H.Chaimovich, ,MJ. Politi, , W.F.,Reed,. J.Phys. Chem. 96 (1992) 644~5

Monômeros
Mícela tubular
Mícela Esférica
Microemulsão AJO .
Micela reversa
Microemulsão OIA
Sonicação)o
Vesícula multilamelar Vesícula uni/amelar
Figura 2.7 - Algumas estruturas formadas por tensoativos
A concentração mínima de tensoativo necessária para a formação de
uma micela simples é denominada de concentração micelarcrítica (cmc).
Conhecendo-se o valor do cmc, a concentração do detergente e o número de
agregação é possível calcular a concentração de micela em moles por litro,
usando a seguinte formula:
Cs- cmc[micela] =--N-- (9)
46

Onde, Cs é a concentração do tensoativo em solução e N é o número de
agregação. Quando o valor de cmc é atingido, observa-se uma brusca variação
das propriedades físico-químicas da solução do tensoativo. Na figura 2.8 são
apresentadas as diversas variações das características da solução.
Pressão osmótica
Q).......cQ)
Ucoo..([)Q)''-ou(J)
uC\Juc='
cmc
~~--r--- Ressonânciamagnética
Solubilização
Tensão superficial~::::::::--..~-
Condutividadeequivalente
Concentração do tensoativo
Figura 2.8 - Mudanças das características físico-químicas de uma solução de
tensoativo após ter atingido o cmc.
2.4.3.2 - Número de agregação
Este é o número de moléculas de monômero do tensoativo presentes
em uma micela simples. O número de agregação pode ser obtido pela divisão
da massa molecular da micela pela massa molecular do monômero. A massa
molecular da micela pode ser obtida por várias técnicas incluindo filtração em
gel, espalhamento de luz, equilíbrio de sedimentação e espalhamnento de raio-
X em baixos ângulos (small-angle X-ray scattering). Como o tamanho micelar,
o número de agregação pode ser também influenciado pela força iônica.
47

2.4.3.3 - Kraft Point
A baixas temperaturas, os tensoativos permanecem no estado de
cristais insolúveis e estão em equilíbrio com uma pequena quantidade de
monômero dissolvido. A medida que a temperatura da solução aumenta a
quantidade de monômero em solução também aumenta até atingir a cmc,
formando então micelas. A temperatura no qual o monômero atinge a cmc é
chamada de temperatura micelar crítica (cmt). A temperatura onde todas as
três fases -cristalina, micelar e monomérica - coexistem é chamada de kraft
point (Figura 2.9). A esta temperatura a solução do tensoativo toma-se límpida
e a concentração do tensoativo atinge a cmc. Para muitos tensoativos, o kraft
point é idêntico ao cmt.
2'E
~.~
o(J)
c:::.<go-oo'ro(jo
~ã5uc:::o()
cmtCristais dotensoativo
Micelas
Monõmero dotensoativo
.Temperatura, °C
cmc
Figura 2.9 - Diagrama de fases do Kraft point.
48

2.4.3.4 - Fatores que influenciam o valor da cmc
Os valores de cmc dos tensoativos dependem de vários fatores como
o comprimento da cadeia hidrofóbica do detergente, natureza do grupo polar,
temperatura, presença de sais e outros solutos25.
De uma maneira geral, o aumento da cadeia carbônica (grupo
hidrofóbico), fixando as demais variáveis, provoca um aumento da
hidrofobicidade do monômero, diminuindo o valor de cmc e aumentando o
tamanho das micelas. A variação da natureza do grupo hidrofílico, para
tensoativos que possuam a mesma cadeia hidrofóbica, não provoca grandes
alterações no valor de cmc, comparado com outros fatores26. Entretanto, a
natureza do grupo hidrofílico é um fator importante na determinação do
tamanho micelar e na reatividade de reações catalisadas por soluções
micelares aquosas.
Grupos hidrofílicos pequenos permitem que os contra-íons se
aproximem da interface micelar, fazendo com que o grau de dissociação seja
pequeno. Isto diminui a carga efetiva da micela, o que resulta em um aumento
do número de agregação micelar. Por outro lado, grupos hidrofílicos muito
grandes não permitem a aproximação dos contra-íons, aumentando o grau de
dissociação e diminuindo o número de agregação micelar7,28.
25D. Attwood, A.T. Florence, "Surfactants Systems. Their Chemistry, Pharmacy and Biology",Chapman and Hall, London, 1983.
26 K. Shinoda, T. Nakagawa, B. Tamamushi, T. Isemura, "Colloidal Surfactants" AcademicPress, Inc., New York, 1963.
27R. Zana J. Colloid interface Sei., 78 (1980) 330
28p. Lianos, R. Zana, J. Colloid Interface Sei., 88 (1982) 59449

Com referência ao aumentá da temperatura e pressão, pode-se dizer
que com o aumento da temperatura assume-se a possibilidade de um
decréscimo da cmc em função da desidratação dos monômeros. Por outro
lado, o aumento excessivo da temperatura provoca um aumento na cmc,
causado pela quebra da estrutura da água ao redor da cadeia carbônica, que
se opõe ao processo de micelização83. Com relação à pressão, o valor de cmc
aumenta com o aumento do valor de pressão. Este comportamento tem sido.explicado como função de uma solidificação do interior micelar, ou de um
aumento da constante dielétrica da água induzida pela pressão84.
Com relação à incorporação de aditivos à solução de tensoativo, pode-
se dividir estes aditivos de acordo com o principal efeito causado sobre o
sistema. 1) eletrólitos pequenos, cujo efeito é essencialmente eletrostático e
afetam principalmente as micelas, como o NaCI, LiCI; 2) aditivos que atuam
principalmente na estrutura das moléculas de água, como a uréia e 3) aditivos
não-iônicos que afetam a interface por adsorção, como os álcoois.
A presença de eletrólitos pequenos na solução de tensoativos iônicos
reduz a repulsão eletrostática entre os grupos hidrofílicos que se localizam na
interface micela-água, isto então provoca uma diminuição no valor de cmc e um
aumento no número de agregação. Dependendo do tensoativo, o aumento na
concentração de eletrólito pode provocar a transição de geometria micelar de
esferas para cilindros, como no caso de haletos de tetradeciltrimetilamônio com
haleto de sódi029. Estas mudanças normalmente estão associadas a aumento
na viscosidade da solução, motivo pelo qual, na maioria dos casos, a
29T. Imae, S. Ikeda, J. Chem. Phys., 90 (1986) 521650

viscosidade de detergentes líquidos para uso doméstico é ajustada pela adição
de sais inorgânicos como NaCl, Na2S04 e MgS04.
A adição de compostos que quebram a estrutura da água (por
exemplo, a uréia) aumenta o valor de cmc30. Para os não-iônicos, ocorre
também um aumento do ponto de turvação e diminuição do valor da cmc com a
adição deste tipo de aditivo.
Os efeitos da adição de n-álcoois em sistemas micelares podem ser
avaliados de acordo com o tamanho da cadeia alquil do álcool31,32. Adição de
álcoois de cadeias pequenas (entre 1 a 3 carbonos) em uma primeira instância
provoca diminuição do valor da cmc de tensoativos iônicos, com conseqüente
aumento na ionização da micela (grau de dissociação). Efeito mais
pronunciado é obtido com o aumento do hidrofobicidade do álcool adicionado.
Porém, à medida que a concentração do álcool de cadeia pequena aumenta
em solução, um aumento no valor de cmc é notado, porém o grau de ionização
não diminui. Este fato pode ser explicado da seguinte maneira: as moléculas de
álcool com cadeias de 1 a 4 átomos de carbono são dissolvidas
predominantemente na fase aquosa, apresentando um processo dinâmico, no
qual as moléculas de álcool trocam do meio aquoso para o meio micelar
continuamente. Os álcoois de cadeias pequenas associam-se na camada
palasídica da micela. As moléculas de água, presentes nesta camada, são
deslocadas pelas moléculas de álcool, como conseqüência, a constante
dielétrica da camada de barreira diminui (micela-solução). Este efeito aumenta
a repulsão entre os grupos hidrofílicos iônicos (cabeça do tensoativo),
3Op. Mukerjee, Adv. Golfoid Interface Sei., 67 (1962) 19031 R.Zana, S.Yiv, C.Strazielle, P.Lianos, J. Golf. Int. Sei., 80 (1981) 20832 S.Yiv, R.Zana, W.Ulbricht, H.Hoffman, J.Golf. Int. Sei., 80(1981) 224
51

desestabilizando a micela e levando a dissociação de um certo número de íons
de tensoativo. Com isso, o número de agregação diminui e a associação dos
monômeros do tensoativo ocorre em uma concentração menor do que aquela
na ausência de álcool. Se a concentração de álcool for continuamente
aumentada ocorrerá um aumento da cmc do sistema micelar, pois o álcool
presente na solução micelar solvata a cadeia alquil do monômero ionizado do
tensoativo, diminuindo a energia livre e favorecendo a permanência do
monômero em solução. Para a formação de micela é necessária uma
quantidade maior de tensoativo, com isso aumenta o cmc.
Para álcoois de cadeias mais longas de 4 a 6 carbonos, a diminuição
da cmc e aumento da ionização da micelas são também evidenciados. Estes
álcoois possuem a capacidade de associar-se mais integralmente com a micela
(ficam "ancorados"), esta capacidade aumenta com o tamanho da cadeia do
álcool. Baseado nesta informação dois fenômenos ocorrem conjuntamente, um
de ordem estérica e outro eletrostática.
1) Efeito estérico: as moléculas de álcool ficam intercaladas entre os íons do
tensoativo na micela, aumentando a média da distância entre elas. Isto resulta
em um decréscimo na densidade de carga na superfície da micela e um
aumento na ionização, com conseqüente diminuição da cmc.
2) Efeito eletrostático: este efeito diz respeito à mudança da constante
dielétrica da camada palasídica. Medidas espectroscópicas mostraram que a
constante dielétrica na superfície da micela em água apresenta valores entre
40 a 50. Estes valores são relativamente altos, levando em consideração que a
camada palasídica contém água, grupos iônicos do tensoativo e alguns contra-
52

íons que vão até o terceiro ou quarto grupo metilênico da cadeia alquil do
tensoativo. O álcool quando se associa ("ancora") à micela substituindo a água
por ele mesmo, diminui a constante dielétrica e, como conseqüência, aumenta
a repulsão entre os grupos polares do tensoativo, aumentando a ionização das
micelas e diminuindo a cmc33.
Para tensoativos não-iônicos, álcoois como metanol e etanol,
aumentam o valor da cmc devido ao enfraquecimento da hidratação da cadeia
carbônica. O n-propanol exibe um efeito intermediário, baixas concentrações
causam uma diminuição do valor da cmc. Já álcoois como butanol e pentanol
diminuem o valor da cmc como conseqüência da penetração dos mesmos na
camada palasídica da micela formando uma micela mista. 34
A adição de solventes, no caso acetonitrila, também ocasiona
alterações no valor da cmc. A solubilização de acetonitrila em soluções
anfifílicas causa um rompimento na estrutura da água, com adições de
pequenas quantidades deste solvente orgânico e a formação de novas pontes
de hidrogênio entre moléculas de acetonitrila e água, onde as moléculas de
acetonitrila ficam "engaioladas" pelas moléculas de água. Isto causa um
rearranjo das moléculas de água diminuindo o efeito hidrofóbico. A acetonitrila
tem a facilidade de solvatar os monômeros do tensoativo fazendo com que
uma quantidade maior de tensoativo seja necessária para formar uma micela,
aumentando, portanto o número de agregação e a cmc. As micelas de SOS
formadas em misturas acetonitrila-água são menores, mais dissociadas e
apresenta maior fluidez.
33S.Backlund, K.Rundt, K.S.Birdi, S. Dalager, J. Colloid Interface Sci. 79 (1981) 578
34 F.A .Green, J. Colloid Interface Sci. 41 (1972) 12453

CAPÍTULO 3

3- Objetivos
Foram objetivos deste projeto de pesquisa:
1) Estabelecer um eletrólito apropriado para a separação de pesticidas das
classes triazinas, carbamatos, carbendamidazóis, organofosforados e feniluréia
via eletroforese capilar (CE).
2) Avaliação das estratégias de pré-concentração on-line descritas em literatura e·
propor estratégias alternativas para análise de pesticidas via CE.
3) Avaliação e estabelecimento de uma metodologia de extração e clean-up,
visando à determinação de pesticidas em matrizes vegetais.
4) Avaliação do emprego de ambientes micelares (extração por cloud-point - CPE)
na extração e análise de pesticidas em frutas.
5) Determinar figuras de méritos para o método analítico otimizado e aplicá-lo a
uma amostra real.
54

~~U
HJ~
~ CAPÍTULO li-
~
u
~t~, ,s~ 'B
u1~-J

4 - Materiais e reagentes
4.1 - Instrumentação
Equipamento de e/etroforese capilar
Para a realização das análises por CE foi utilizado um equipamento de
eletroforese capilar Agilent TechnoJogies (Paio Alto. CA, EUA) equipado com
detector de arranjo de diodos (DAD) de 190 a 600 nm. fonte de alta tensão de O±
30kV. controlador de temperatura por circulação de ar, e software para controle
do equipamento e aquisição de dados (Chemistation CE versão 5.0). Na Figura
3.1 é mostrada uma foto do equipamento utilizado.
Figura 4.1 - Equipamento de CE utilizado para a realização das análises.
55

Banho-maria
Um banho-maria modelo 550 (Fisaton, SP, Brasil), com controlador de
temperatura de 30-110 cC foi utilizado.
pHmetro
Um pHmetro DM 21 (Digimed, SP, Brasil) com eletrodo de vidro combinado
Ag/AgCI foi utilizado para a determinação de pH das soluções preparadas.
Centrífuga
Centrífuga Quimis 222 T28 (Quimis, São Paulo, Brasil) foi utilizada na preparação
de amostras.
Manifold
Manifold Supelco Vispred™ DL de 12 portas (Supelco, Sigma-Aldrich Family, São
Paulo, Brasil), para a realização dos SPE.
Estufa
Estufa Fanem 515 C (Fanem, Guarulhos, Brasil)
4.2 - Materiais utilizados na preparação de soluções e amostras
a) micropipetas volumétricas da Gilson: 250 !J.L; 200 - 1000 !J.L e 1000 - 5000 !J.L.
b) funil de separação de 150 mL.
c) coluna de vidro de 25 cm x 1 em
d) balões volumétricos de 5, 10,25,50 e 100 mL.
56

e) cartuchos para extração em fase sólida (SPE) com fases estacionárias: CN
(ciano), amino (NH2), C18 (octadecil sílica) (Strata, Phenomenex, Labtron,
Brasil), com 1 g de sorvente .
f) barras magnéticas para agitação.
g) vía/s de 0,5 a 1 mL.
h) unidades filtrantes Millex (Or. Millex, Millipore, SP, Brasil) de 0,22 J-lm e
0,45 J-lm.
i) seringas descartáveis.
j) tubos eppendorf de 2 mL.
k) capilares de sílica fundida (Polymicro Technologies, Phoenix, AZ, EUA) de 75
J-lm de diâmetro interno e 360 J-lm de diâmetro externo.
4.3 - Reagentes e soluções
Os solventes orgânicos utilizados para a preparação de soluções padrão
de pesticidas, empregados nos testes de extração foram todos de grau HPLC,
obtidos da Merck.
Foi utilizada água desionizada (Milli-Q system, Millipore, Bedford, MA,
EUA) para a preparação de soluções de ácidos e bases, assim como das
soluções tampão. A seguir são descritas as soluções utilizadas durante a
realização deste trabalho.
~ Solução concentrada de NaOH, 1 moVL
Foram dissolvidos 4 g de NaOH (Reagente PA - Aldrich) em 100 mL de
água desionizada e a solução foi armazenada em frasco de polietileno.
57

=> Solução tampão de ácido fosfóricoHosfato de sódio monobásico
(H3PO./NaH2P041
Esta solução será identificada a partir desta seção pela sigla TF.
Foram preparados 250 mL de uma solução estoque TF a 100 mmol/L com
pH 2,5. Estas soluções foram preparadas através da diluiçãol dissolução
adequadas dos reagentes H3P04 (Merck, grau PA 't = 85 % e d = 1,95 g/mL)
NaH2P04 (Sigma-Aldrich, grau PA ) segundo a equação de Henderson
Hasselbach. O pH foi medido com o uso de um pHmetro. Foram utilizados 2,09 g
de NaH2P04e 0,5 mL de H3P04concentrado; a solução obtida foi acondicionada
em frasco de polietileno e mantida sob refrigeração.
=> Solução tampão de Tetraborato de sódio (Na28407.10H20)
Esta solução será identificada a partir desta seção através da sigla TBS.
Foi preparada uma solução estoque de TBS 100 mmollL a partir da
dissolução de 9,53 g de Na2B4Ü7.10H20 (Sigma-Aldrich, grau PA) em 250 mL de
H20; a solução foi armazenada em um frasco de polietileno e mantida sob
refrigeração.
=> Eletrólito para separações em meio não aquoso (NACE)
Eletrólito utilizando-se SOS (dodecilssulfato de sódio) ou NaCI solubilizado
em mistura de solventes metanol:acetonitrila (1: 1, v/v) acrescida de 50 !-LL ácido
clorídrico concentrado para 50 mL (PA Merck, 't = 32%, d = 1,16 g/L), foi
empregada nas separações via NACE.
58

~Solução de dodecilssuNato de sódio (SDS), ciclodextrinas
Foram preparados 100 mL de solução 100 mmol/L de SOS através da
dissolução de 2,88 g do reagente padrão (SOS grau PA, Merck); a solução obtida
foi acondicionada em frasco de polietileno.
Foram utilizadas ciclodextrinas de diferentes espécies e graus de
substituição (13 e y-ciclodextrinas) de diferentes procedências (Merck e Beckman).
Como tratam-se de reagentes caros e vendidos em pequenas quantidades (250
mg a 1 g), os eletrólitos de corrida que utilizaram estes reagentes foram
preparados em pequeno volume (5 mL), pesando-se a massa necessária (em mg)
para a concentração requerida diretamente no balão de preparação do eletrólito.
~ Tensoativos e fases sorventes
Foram utilizados os seguintes tensoativos nas extrações por Cloud Point:
- Triton X114 (Fluka, Sigma-Aldrich Family, São Paulo, SP), lote n°. 402781/1.
Foram utilizadas os seguintes sorventes para a realização de estudos de
extração de pesticidas em frutas e leguminosas:
Florisil (silicato de magnésio ativado), da Sigma (Sigma-Aldrich Family, São
Paulo, SP), lote n°. 118H0303.
Celite 545 AW (Supelco, Sigma-Aldrich Family, São Paulo, SP).
Carvão ativado (Sigma, Sigma-Aldrich Family, São Paulo, SP).
~ Padrões de pesticidas
Padrões de diferentes classes de pesticidas (aldicarb, dimetoato,
carbendazin, simazina, atrazina, propazina, ametrina, clorpirifós, pirazofós, diuron,
59

Iinuron, propoxur, carbaril, carbofuran) foram adquiridos junto à Riedel - de Haen
(Sigma-Aldrich Family, São Paulo, SP). Estes padrões são comercializados sob a
forma de pó, acondicionados em frascos de vidro âmbar com cerca de 250 mg.
Soluções padrão estoques destes pesticidas foram preparadas na concentração
de 1000 mg/L. Porém, para simazina e carbendazim a solução estoque foi
preparada na concentração de 300 mg/L. O solvente utilizado para a dissolução
do carbendazim foi acetona, a simazina foi solubilizada em etanol e os demais
pesticidas em metano\. Estas soluções foram acondicionadas em frascos âmbar
de 5 mL, mantidas sob refrigeração (-12 °C), e preparados através da diluição
adequada antes do uso.
=> Amostras de frutas e leguminosas
Amostras de frutas (abacaxi) e leguminosa (cenoura) foram adquiridas
no comércio local. Amostras testemunho foram adquiridas em locais onde é
praticada a cultura orgânica.
=> Condicionamento de capilares
Colunas capilares novas antes de serem utilizadas foram lavadas
previamente com NaOH 1,0 mol/L por 25 min, água desionizada por 20 min e
por último com o eletrólito de corrida por 25 mino Durante a realização das
análises, a coluna capilar era lavada com o eletrólito de corrida por 3 min a
cada análise efetuada.
60

CAPÍTULO 5

5 - Otimização da composição do eletrólito de separação
Foram realizados experimentos em dois níveis de pH, um em tampão TBS
(pH 9,3) e outro em tampão TF (pH 2,5). Para as separações em pH 9.3 foram
avaliadas as potencialidades das ciclodextrinas, dos solventes orgânicos e a
formação de micelas mistas pela adição de um tensoativo não-iônico ao eletrólito,
para se atingir a seletividade e resoluções adequadas para a separação de uma
mistura padrão de pesticidas. Já para as separações em pH 2,5 não houve a
necessidade da inclusão das ciclodextrinas no eletrólito de separação, para que
resolução satisfatória fosse alcançada. Apenas com a influência dos solventes
orgânicos no eletrólito, foi atingida resolução até a linha de base.
5.1 - Otimização da separação em pH 9,3
A concentração de solvente orgânico adicionado ao eletrólito de separação
foi mantida constante, como também as concentrações TBS e SOS. Foram
avaliadas várias espécies de ciclodextrinas (CO), naturais (P e y CO) e
derivatizadas (dimetil-p-CO, hidroxipropil-p-CO e trimetil-p-CO). Em ensaios
preliminares ficou evidente que à medida que se aumentava a concentração de
CO no eletrólito de separação, a resolução melhorava passando por um ótimo de
concentração que estava em torno de 20 mmol L-1. Na figura 5.1 são mostrados os
eletroferogramas obtidos na separação de 12 pesticidas de diferentes classes
(aldicarb, dimetoato, carbendazim, carbofuram, simazina, atrazina, carbaril,
propazina, diuron, linuron, pirazofós, clorpirifós) em um eletrólito contendo 45
mmol L-1 de SOS, 10 % metanol, 10 mmol L-1 de tetraborato de sódio (TBS) e 20
61

mmol L-1 de cada uma das CO naturais.
45
23
7
10
I I I i I I I2 4 6 8 10 1 2 14
Tempo, min
6
4+5 710
9
2+3 812
I2
I4
I6
I8
i10
Tempo, min
i1 2
i14
I16
i18
Figura 5.1 - Influência das ciclodextrinas naturais na separação eletroforética de
uma mistura de pesticidas. Composição do eletrólito: 10% metanol, 45 mmol r1 de
SOS, 10 mmol L-1 TBS, (A) 20mmol L-1 de y-CO e (B) 20 mmol L-1 de (3-CO. À =220 nm, capilar de sílica fundida de 50 cm (efetivo), 27kV, 25°C, inj. = 8 s/2,5kPa.
1) aldicarb, 2) dimetoato, 3) carbendazim, 4) carbofuram, 5) simazina, 6) atrazina,
7) carbaril, 8) propazina, 9) diuron, 10) linuron, 11) pirazofós e 12) c1orpirifós.
Entre as COs naturais, a y-CO proporcionou uma maior resolução entre os
componentes da mistura. Na figura 5.2 são apresentados os eletroferogramas62

obtidos para as ciclodextrínas derivatizadas. Através dos eletroferogramas obtidos,
é possível avaliar a influência das ciclodextrinas derivatizadas na separação da
mesma mistura de pesticidas mostrada anteriormente. A trimetil-J3-CO apresentou
a melhor separação para a mistura de pesticidas. Este fato pode estar relacionado
com o grau de substituição (GS) das ciclodextrinas utilizadas. ü grau de
substituição da trimetil-J3-CO é 3,0 enquanto que para a hidroxipropil-J3-CO é 0,6 e
o dimetil-J3-CO é 1,8. ü grau de substituição é definido como o número médio de
grupos hidroxilas substituídos pelos grupos funcionais em cada unidade
glicopiranose da ciclodextrina (0< GS <3). Quanto maior o GS maior será a
capacidade de complexação por inclusão da ciclodextrina1,2, favorecendo a
formação de complexos mais estáveis. Talvez por essa razão o tempo de análise
seja mais rápido quando a trimetil-J3 CO é utilizada no eletrólito de corrida. A
trimetil-J3 CO é neutra e durante a separação é conduzida através do capilar pelo
EüF. Quando o analito particiona com a micela de SOS ele é desacelerado, pois a
micelas apresentam carga negativa que resiste ao EüF. Enquanto o analito estiver
complexado com a CO, ele será arrastado pelo EüF, adquirindo maior velocidade
em relação aos analitos que interagirem com as micelas.
I M.C.Vescina, A .M.Fernúer, Y. Guo, JChromatogr. A, 973 (2002) 1872 K.L.Larsen, W.ZinunemJalln, JChromatogr. A, 836 (1999) 3
63

3+4
5
7 9
6
A 2 8 103+4
5 7 8
I2
10
r I I I I2 4 6 8 10
10Tempo, min 6 92
5+6+7
B 12
3+4 Tempo, mino8
1+2 9
c 1112
I2
I4
I6
Tempo, mino
I8
I10
64
Figura 5.2 -Influência das COs derivatizadas na separação eletroforética de uma
mistura de pesticidas.Composição do eletrólito: 10% MeOH, 45 mmol L-1 de
SOS,10 mmol L-1 de T8S (A) trimetil-f3 CO (8) dimetil-f3-CO (C) Hidroxipropil-f3-CO
e, 1..=220, capilar de sílica fundida de 50 cm (efetivo), 27 kV, 25°C, inj. = 8 s/2,5
kPa. 1) aldicarb, 2 )dimetoato, 3) carbendazim, 4) carbofuram, 5) simazina, 6)
atrazina, 7) carbaril, 8) propazina, 9) diuron, 10) linuron, 11) pirazofós e 12)
clorpirifós.

Apesar da trimetil-fl CO (Fig. 5.2 A) ter apresentado melhor resolução e
uma separação em tempos menores, ainda persiste a co-eluição entre o
carbofuran e a simazina (picos 3 e 4). Tensoativos neutros têm sido usados
conjuntamente com tensoativos iônicos em separações eletroforéticas para
aumentar a seletividade e a resolução. O efeito do brij 35® sobre a separação da
mistura de pesticidas foi avaliado. Os eletroferogramas obtidos são apresentados
na Figura 5.3. É possível avaliar através dos eletroferogramas que o brij 35®
proporcionou melhora na resolução com sensível diminuição no tempo de análise,
pois a separação ocorreu em tempo menor que 8 minutos (Figura 5.3 A). O brij®
35 altera a estrutura micelar do SOS, pois os monômeros do brij 35® agregam-se
com as micelas de SOS, aumentando o seu tamanho, que por conseqüência
diminui a sua densidade de carga. Com a diminuição da densidade de carga da
micela, esta apresenta menor atração pelo eletrodo de carga oposta (ânodo) e por
conseqüência exerce menor resistência ao EOF. Outro fato que contribui para a
melhoria na separação é a diminuição na constante dielétrica da superfície da
micela de SOS, possibilitando que as interações entre o analito e a superfície das
micelas possam ser maximizadas. Juntando-se a isso o efeito benéfico que a
trimetil- fl-CO já imprimia ao sistema de separação, obteve-se uma separação para
todos os pesticidas. Outro fato a ser destacado é que a concentração de trimetil-
fl-CO no eletrólito de separação foi diminuída de 20 para 12 mmol L-l.
65
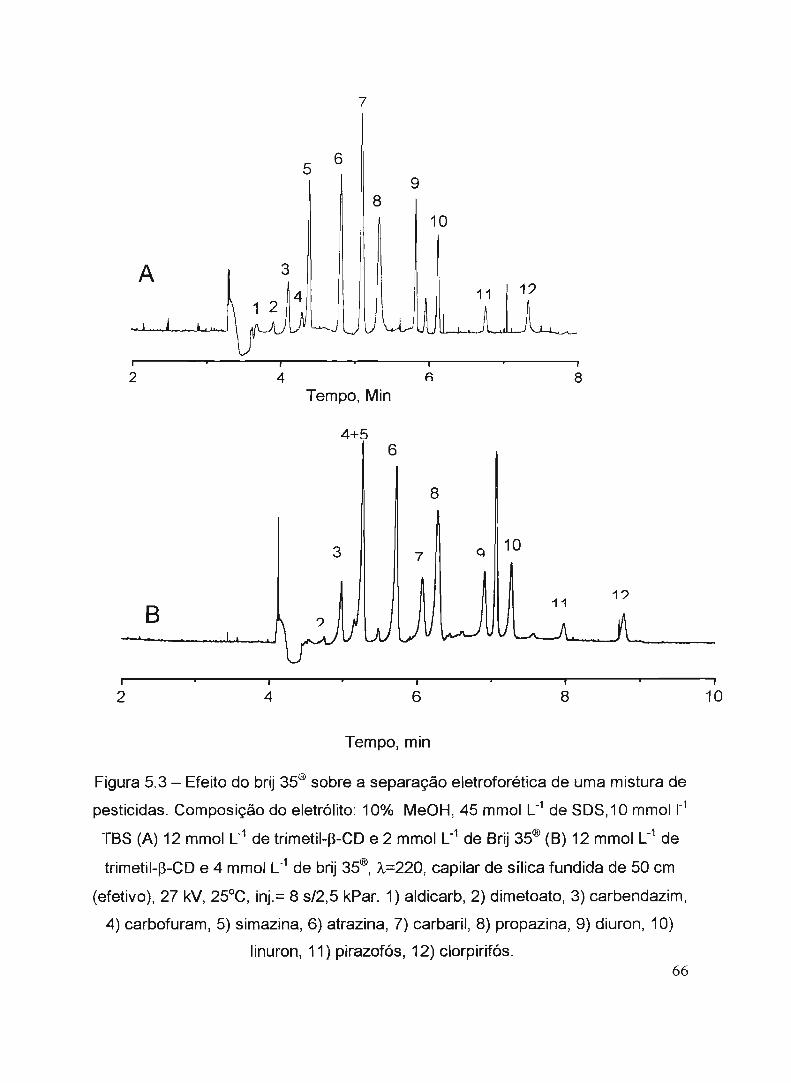
A
56
7
89
10
2 4 8
2
B
4
Tempo, Min
4+56
3 7
6
8
Q10
11
8
1?
10
Tempo, min
Figura 5.3 - Efeito do brij 35® sobre a separação eletroforética de uma mistura de
pesticidas. Composição do eletrólito: 10% MeOH, 45 mmol L-1 de SOS, 1Ommol r1
TBS (A) 12 mmol L-1 de trimetil-f3-CO e 2 mmol L-1 de Brij 35® (8) 12 mmol L-1 de
trimetil-f3-CO e 4 mmol L-1 de brij 35®, À=220, capilar de sílica fundida de 50 cm
(efetivo), 27 kV, 25°C, inj.= 8 s/2,5 kPar. 1) aldicarb, 2) dimetoato, 3) carbendazim,
4) carbofuram, 5) simazina, 6) atrazina, 7) carbaril, 8) propazina, 9) diuron, 10)
Iinuron, 11) pirazofós, 12) clorpirifós.66

Embora tenha sido obtida uma boa separação utilizando como eletrólito 45
mmol L-1 SOS, 10 % metanol, 12 mmol L-1 trimetil- f3 CO, 2 mmol L-1 Brij 35®, para
a mistura de pesticidas estudada, este eletrólito não apresentou bons resultados
quando foram realizados testes de repetibilidade, provavelmente em virtude de
sua complexa composição. Além disso, o fator que mais contribuiu para a não
utilização deste eletrólito de separação foi que, em virtude da adição de Brij 35® ,a
densidade de carga das micelas de SOS se torna menor, motivo que impossibilita
o uso deste eletrólito no emprego de estratégias de pré-concentração on-line,
estratégias estas que serão explanadas no capítulo 6.
Considerando estes fatores, novas alternativas para a separação destes
pesticidas foram avaliadas. Estudos sobre a influência dos solventes orgânicos no
desempenho das separações eletroforéticas foram realizados. Solventes como
metanol, etanol, propanol, butanol e acetonitrila foram avaliados, bem como
misturas entre estes solventes. Para a realização destes estudos foi empregada
uma mistura de pesticidas na concentração de 10 mg/L que continham; dimetoato,
carbendazim, atrazina, simazina, carbaril, ametrina, propazina, diuron, propoxur e
linuron.
Inicialmente avaliou-se a influência dos solventes individualmente, onde
foram preparados vários eletrólitos de separação contendo 10% de cada um dos
álcoois, e manteve-se constante a concentração dos outros componentes do
eletrólito de separação, que consistia de 50 mmol L-1 de SOS e 10 mmol L-1 do
tampão TBS. Na figura 5.4 são mostrados os eletroferogramas obtidos.
67

5
3 47 8 6
6 4+* 79 3
8A 2 2
9B10
I I I I I I iI I I I I4 6 8 10 12 2 4 6 8 10 12 14
Tempo, mino Tempo, mino
5
*~
6
3 7
810
C 2
I10
I8
I6
Tempo, mino
I10
I8
I6
Tempo, mino
I I I I I I2 6 8 10 12 14
Tempo, mino 7
4 6
3 8 9
* \ /5 10
E
I4
I2
Figura 5.4 - Influência na separação eletroforética pela adição de1 O % de
diferentes solventes ao eletrólito de separação. Composição do eletrólito: 50 mmol
L-1 SOS e 10 mmol L-1 TBS, (A)10% ACN, (B) 10% MeOH, (C)10% EtOH, (O) 10%
BuOH e (E) 10% PrOH. )..=220, Ldet= 50 em, 27 kV, 25°C, inj.= 8 s/2,5 kPa. (1)
dimetoato, 2) carbendazim, 3) simazina, 4) atrazina, 5) carbaril, 6) ametrina, 7)
propazina, 8) diuron, 9) propoxur e 10) linuron e *) desconhecido.
68

Percebe-se que o pico desconhecido co-eluiu com todos os eletrólitos
avaliados. Nota-se também que o eletroferograma onde o n-butanol foi utilizado
(figura 5.4 D) o perfil foi bastante alterado. Isto pode ser indicativo de que nesta
concentração (10%) o n-butanol esta favorecendo a dissociação dos monômeros
de SDS da micela, causando mudanças na sua estrutura micelar prejudicando a
estabilidade do eletrólito de separação. É possível notar através da Figura 5.4 A
que a ordem de eluição para o diuron varia quando a acetonitrila é adicionada ao
eletrólito de separação.
A seguir na Figura 5.5 serão mostrados os eletroferogramas obtidos para
as misturas binárias na proporção de 5 % para cada solvente: PrOH:MeOH,
ACN:MeOH, ETOH:MeOH, BuOH:MeOH, BuOH:ACN. É possível notar que as
misturas binárias onde o butanol esta presente, foi obtido melhores efeitos sobre a
separação. A maioria dos álcoois utilizados (exceção feita ao butanol) estão
dissolvidos predominantemente na fase aquosa, em um processo dinâmico no
qual as moléculas de álcool trocam do meio aquoso para o meio micelar
continuamente, podendo contribuir para uma diminuição da cmc das micelas,
decorrente da diminuição da constante dielétrica do meio onde as micelas estão
contidas. Dependendo da interação entre as micelas e as moléculas dos solventes
não se descarta a possibilidades dos solventes diminuírem a constante dielétrica
da superfície das micelas, devido à desidratação da camada palasídica. Estes
motivos que podem estar ocorrendo isolados ou em conjunto possam estar
contribuindo para melhor solubilização dos analitos no meio ou na superfície das
micelas, proporcionando melhoria na separação.
3 R.Zana, S. Yiv, C. Strazielle, P. Lianos, J. Co". Int. Sei, 80 (1981) 208.
69

4+* *66
7 7+94
3 5 3
A 8 B
L]U~9
2 2
~ ~~I I I I i I I i I I
2 4 6 8 10 5 2 4 6 8 10
Tempo, mino Tempo, mino
63 4 7
C* 8 10
2
J ;-~ U'-'5 6 8+7I I I I I I 42 4 6 8 10 12
Tempo, mino93
*3 104
56D 7
~ UJ I~2 8
_~J * 10 L~L-,~
I I I I I I I I I I i I I2 4 6 8 10 12 14 2 4 6 8 10 12
Tempo, mino Tempo, mino
Figura 5.5 -Influência na separação pela adição de misturas binárias de solventes ao
eletrálito de separação. Composição do eletrálito: 50 mmol L-1 SOS, 10 mmol L-1, (A)
5% ACN, 5% MeOH, (B) 5% EtOH, 5% MeOH, (C) 5% BuOH, 5% MeOH, (O) 5%
BuOH, 5% ACN e (E) 5% PrOH, 5% MeOH. À = 220 nm, Ldel= 50 em, 27 kV, 25°C,
inj.= 8 s/2,5 kPa. 1) dimetoato, 2) carbendazim, 3) simazina, 4) atrazina, 5) carbaril, 6)
ametrina, 7) propazina, 8) diuron, 9) propoxur, 10) linuron e *) desconhecido.
70

Na figura 5.6 são mostrados os eletroferogramas que apresentaram
separação na linha de base para a mistura dos 10 pesticidas, utilizando misturas
binárias de solventes.
8 10 9
I2
B
2
1
I6
*
I8
I10
I12
Tempo, mino
Figura 5.6 - Melhores separações utilizando misturas binárias de álcoois no
eletrálito. Composição do eletrálito: 50 mmol L-1 de SOS, 10 mmol L-1 TBS, (A) 5%
PrOH e 5% EtOH (B) 5% BuOH e 5% EtOH. Inj.= 8 s/2,5kPa, 1..=220, Ldet. = 50 cm,
27 kV, 25°C. 1) dimetoato, 2) carbendazim, 3) simazina, 4) atrazina, 5) carbaril, 6)
ametrina, 7) propazina, 8) diuron, 9) propoxur, 10) linuron e *) desconhecido
71

A mistura BuOH:EtOH apresentou boa separação, porém não é um
eletrólito estável. Em injeções consecutivas observou-se deformação dos picos
nos eletroferogramas obtidos. O butanol apresenta forte interação com a micela de
SOS, possivelmente contribui para um aumento no seu grau de dissociação. Por
este motivo, com o decorrer do tempo as micelas de SOS sofrem mudanças em
sua estrutura, diminuindo a sua capacidade de interagir com o analito. Na figura
5.7 é mostrado um gráfico de mobilidade dos pesticidas em função da
concentração e o solvente orgânico adicionado ao eletrólito de separação.
~6) (7) (8) (9) (10) (11 ) (12) (13)
~:::c :::c
O :::c o~ O::::J o ::::J o :::c
co « I!I co I!I « ~ o(3) (4) (5) ::R ::R ::R ::R ::R I!I
(1 ) (2)o ::R o ::RI,(') o I,(') o ::RI,(') o o I,(') o
I,(') I,(') I,(') o-- -- I,('):::c :::c :::c :::c --O o :::c o o :::c :::c o :::c :::c~~ ~
o~ ~
o o~
o o ~« lI] ~ ~::::J ::::J li:li: co co li:
cft. ::R ::R ::R ::R ::R ::R ::R ::R ::R::R o o o o ::R o ::Ro o o I,(') o o o o oo I,(') o I,(') I,(') I,(') I,(') oI,(') ..- ..- ..- ..- ..- I,(') I,(')
O,OOE+OO ,---,--,
-2,OOE-06 -.-dimetoato
-a- carbendazin~ -4,OOE-06'li)
simazina
;. -6,OOE-06 " atrazinaN
E -S OOE-06~carbaril
o ' --- ametrina-eu-1,OOE-OS't:J ---;- - propazinaeu
't:J-1,20E-OS --diuron
:co _. propoXJr:E -1,40E-OS , , linuron
-,....--1,60E-OS
"-1,SOE-OS
Figura 5.7 - Gráfico de mobilidade em função da espécie e concentração dos
solventes orgânicos empregados para eletrólitos em pH 9,3.
72

Várias composições de eletrólitos proporcionaram resolução na linha de
base, como: (2), (6), (7), (9), (10) e (13) que estão representados em vermelho na
Figura 5.7, desconsiderando a co-eluição do pico desconhecido identificado por
(*). Porém o eletrálito de separação que apresentou melhor desempenho quanto à
estabilidade e repetibilidade na separação dos pesticidas estudados foi: 50 mmol
L-1 de SOS, 10 mmol L-1 de tetraborato de sódio, 5% PrOH, 5 % EtOH em pH 9,3.
representado pelo número 13 na figura 5.7. Aos eletrálitos que foram adicionados
o butanol, após 2 ou 3 injeções da amostra este perdia completamente a
capacidade de proporcionar a separação dos pesticidas estudados, deformando
os picos.
Foi avaliada também a influência da adição de um tensoativo neutro (brij
35~ ao eletrólito de separação otimizado. Para a realização do experimento
utilizou-se uma mistura padrão contendo 10 pesticidas na concentração de 10 mg
L-1. Concentrações crescentes de Brij 35® foram adicionadas ao eletrólito de
separação. Na figura 5.8 são apresentados os eletroferogramas obtidos.
73

5
3 467
35
4 6 7
A2
i i i I I246 8
Tempo, mino
i10
I12
B 8 1092
* *
~Ül ~J '----''--'..J
I i I I I I I2 4 6 8 10 12
Tempo, min
6+7 6+7+8
3 4 5 34
*+10 5
/10
8 *C O2 9 2
L-V '-' \..-I ...) ~i I I i I I I I i I I i2 4 6 8 10 12 2 4 6 8 10 12
Tempo, mino Tempo, mino
Figura 5.8 -Avaliação da influência do Brij 35® na separação eletroforética de uma
mistura de pesticida. Composição do eletrólito: 50 mmol L-1 de SOS, 10 mmol L-1
TBS, 5% PrOH e 5% EtOH, (A) sem Brij 35® ,(B) com 0,5 mmol L-1 de Brij 35®, (C)
com 1,5 mmol L-1 de Brij 35® ,(O) com 3,0 mmol L-1 de Brij 35®, Inj.= 8 s/25 kPa,
À=220, Ldet= 50 em, 27 kV, 25°C. 1) dimetoato, 2) Carbendazim, 3) simazina, 4)
atrazina, 5) carbaril,6) ametrina, 7) propazina, 8)diuron, 9) propoxur, 10) linuron.
(*) provável produto de degradação de algum pesticida.
74

Nota-se pronunciada influência sobre o tempo e resolução da separação, à
medida que a concentração do Brij 35® é aumentada. A interação do Brij 35® com
SDS promove a formação de uma micela mista de maior volume, com isso sua
densidade de carga diminui. Desta forma a micelas não terão mais tanta atração
pelo eletrodo de carga oposta, sendo arrastadas mais facilmente pelo EOF.
É prática comum em eletroforese capilar diluir o analito em uma solução de
condutividade semelhante ao do eletrólito de separação, para minimizar efeitos de
alargamento de picos. Até então as misturas de pesticidas foram diluídas em
solução de TBS 30 mmollL, para que atingisse a mesma condutividade do
eletrólito de separação. Porém houve o surgimento de um novo pico identificado
por um asterisco (Fig. 5.6) e a concomitante diminuição do pico do carbaril (pico
5). Este fato é decorrente da hidrólise do carbaril, que gradativamente se
transforma em 1-Naftol.4 Foram preparadas duas amostras contendo carbaril na
concentração de 10 mg L-1, onde uma foi diluída em TBS 30 mmol L-1 e outra em
uma solução 20% de MeOH em água. Entre as injeções foi dado um intervalo de
tempo de 60 minutos. Os eletroferogramas obtidos são apresentados na figura
5.9.
75
4 I.C.MacRae. "Reviews ofEnvironmental Contamination and Toxicologt, 1989. 109; p. 1-87.

A
1a injeção
5 B
1a injeção
5
Área: 64,0
I
2a injeçãoapós 60 min
2a injeçãoapós 60 min
Área: 39,4
IÁrea: 22,3
,/
Io
I2
I6
Tempo, mino
I8
I10
I12
Io
I2
I4
I6
Tempo, mino
I8 10 12
Figura 5.9 -Degradação do carbaril como resultado de sua hidrólise em meio
alcalino. Composição do eletrólito: 50 mmol L-1 de SDS, 10 mmol L-1 T8S, 5%
PrOH e 5% ETOH. Carbaril diluído em 20% MeOH em água (A) e em 30 mmol L-1
de T8S (8). À=220nm, Ldet=50 em, 27 kV, 25 °c, inj. = 8 s/25 kPa. (5) Carbaril, (*)
provável presença de 1-Naphtol.
Através da Figura 5.9 (8) nota-se o aumento do pico do 1-naphtol, em
contra partida a diminuição do pico do carbaril.
76

o carbaril em pH 7,0 leva de 24 a 30 dias para a degradação total, em pH
8,0 de 2 a 3 dias, enquanto em pH 9,0 apenas1 dias.
5.2 - Otimização da separação em pH 2,5
Para avaliar a influência da concentração de solvente orgânico na
separação eletroforética de uma mistura padrão de pesticidas, as concentrações
de tampão TF e SOS foram mantidas constantes. A mistura padrão de pesticidas
na concentração de 10 mg L-1 era composta de: ametrina, carbendazim,
propazina, propoxur, atrazina, linuron, diuron, simazina e carbaril. A mistura de
pesticida foi preparada em uma solução de 25 mmol L-1 de tampão TF.
Inicialmente foram avaliadas individualmente as influências de cada solventes
orgânico sobre a separação eletroforética. Para isso, cada eletrólito de separação
continha 5% de cada um dos solventes orgânicos selecionados. Os solventes
empregados foram, EtOH, MeOH, ACN e PrOH. Na figura 5.10 são mostrados os
eletroferogramas obtidos.
5 http://ohioline.osu.edulb672/storin~sticides.html. 77

9
A
7+5 9
3 !,8
1 4
IB2li
1
1 3 6+8
7
4+5
i2
I I4 6
Tempo, mino
i8
i
9 2
I4
i6
Tempo, mino
I8
i10
c
9 I2
i4
I6
I8
9
Tempo, mín
i2
o
i4
1 3 52
4 6
Tempo, mino
i6
8
7
i8
1 3 5 82
74 6E
i III i I i2 4 6 8
Tempo, mino
Figura 5.10 - Influência na separação eletroforética pela adição de 5% de
diferentes solventes ao eletrólito de separação. Composição do eletrólito: 25 mmol
L-1 de SOS, 20 mmol L-1 de tampão TF, (A) 5% MeOH, (8) 5% PrOH, (C) sem
solvente, (O) 5% ETOH e (E) 5% ACN. À=220 nm, Ldet= 50 em, tensão = - 26 kV,
25°C, inj = 8 s/25 kPa. 1) ametrina, 2) carbendazim, 3) propazina, 4) propoxur, 5)
atrazina, 6) linuron, 7) diuron, 8) simazina e 9) carbaril.
78

No modo MEKC em condições de baixo pH a mobilidade eletroforética das
micelas é a única responsável pela migração dos analitos através do capilar, pois
não há mais a influência do EOF. Por este motivo qualquer pequena mudança na
constante dielétrica do eletrólito de separação provoca modificação significativas
na separação. É possível notar que a separação para a mistura padrão de
pesticidas foi resolvida quando EtOH ou ACN (Fig. 5.10 O e E) era adicionado ao
eletrólito de sepa~ação. Quando o propanol (Fig. 5.10 B) foi usado houve pequeno
aumento no tempo de eluição dos pesticidas, aproximadamente 2 minutos. Isto
possivelmente se deve à diminuição da densidade de carga na superfície das
micelas, causada pela solubilização parcial do propanol na micela de SOS. Por
apresentar menor densidade de carga sofrerá menos atração pelo anodo,
diminuindo sua velocidade. A concentração dos solventes adicionados ao
eletrólito de separação foi dobrada, sendo agora de 10 %. Os efeitos deste
aumento de concentração dos solventes sobre a separação são apresentados na
figura 5.11.
79

99
8 83
5 7
13
12 7 26
4 46
l_A 8
"J
I I I I I I I I i I i2 4 6 8 10 2 4 6 8 10 12
Tempo, mino Tempo, mino
9 9
3 5 8 3+4 81 5
2 1 277 64 6
C O
............~~ Ul-.JLJ~U----
i I I I I I I I I2 4 6 8 10 2 4 6 8
Tempo, mino Tempo, mino
Figura 5.11 - Influência na separação eletroforética pela adição de 10% de
diferentes solventes ao eletrólito de separação. Composição do eletrólito: 25 mmol
L-1 de SOS, 20 mmol L-1 de tampão TF, (A) 10% MeOH, (8) 10% PrOH, (C) 10%
EtOH e (0)10% ACN. Â=220 nm, Ldet= 50 em, - 26 kV, Inj. = 8 s/2,5 kPa. 1)
ametrina, 2) carbendazim, 3) propazina, 4) propoxur, 5) atrazina, 6) linuron, 7)
diuron, 8) simazina, 9 )carbaril.
80

Com exceção da ACN, todos os álcoois empregados na proporção de 10%
resolveram os componentes da mistura padrão de pesticidas.
Também foi avaliada as misturas binárias entre MeOH:ACN, EtOH:PrOH e
ACN:PrOH. A proporção de cada solvente na mistura foi de 5%. Na figura 5.12
são mostrados os eletroferogramas obtidos.
81

8+9
I2
I4
I6
I8
I10
I12
Tempo, mino
Figuar 5.12 - Influência na separação eletroforética pela adição de misturas
binárias de solventes ao eletrólito de separação. Composição do eletrólito: 25
mmol L-1 de SOS, 20 mmol L-1 de tampão TF, (A) 5% MeOH, 5% ACN, (8) 5%
PrOH, 5% EtOH, (C) 5 %ACN, 5%PrOH. À=220 nm,Ldet = 50 em, - 26 kV, 25 DC,
Inj. = 8 s/25 kPa. 1) ametrina, 2) carbendazim, 3) propazina, 4) propoxur, 5)
atrazina, 6) linuron, 7) diuron, 8) simazina e 9) carbaril.
82

Para misturas binárias dos solventes estudados, somente a mistura
EtOH:PrOH promoveu a separação dos componentes da mistura padrão de
pesticidas. Na figura 5.13 é apresentado um gráfico das mobilidade em função
dos solventes e suas misturas utilizadas.
2,OOE-OS --.------------------------------
1 ,80E-OS -+-~----------------------------+- ametrina
- carbendazin
propazina
propoxur
~atrazina
_Iinuron
---'-- diuron
--simazina
carbaril
IZ I Z
I I I I I Z Z~ o ~o o o o o ~ ~
.....Q.l ..... ..... - Q.l a..~ a.. a.. w
~~ ~
~ ~o '$. o
~;:F. ~ ~
~o 1.0 1.0o o
1.0o1.0
o 1.0 oo 1.0 o o I Io I o
\:3 ) ( •.j.) ( : ) (b) 17) (x) (9)Q.l o .....~ - a..w~ ~
~oo o 1.01.0 1.0
(IO)dl) ( 12 J
--If)í I I
.---.~
CJ) 1 ,60E-OS -~>
N 1,40E-OSE()
~ 1,20E-OS"O
~ 1,OOE-OS -+------":
:.õ~ 8,OOE-06E
6,OOE-06 --
4,OOE-06 -,--.,---,----,-------,---,--,-----,-----,------,---,--,
Figura 5.13 - Gráfico de mobilidade em função da espécie e concentração dos
solventes orgânicos empregados para eletrálitos em pH 2,5.
Fica mais claro avaliar a influência dos solventes e suas misturas na
separação eletroforética através da Figura 5.13. É possível perceber que a ordem
de eluição para alguns pesticidas é alterada conforme se muda o solvente e a
concentração adicionada ao eletrálito de separação. Isso possivelmente se deve
a mudanças na constante dielétrica do meio de separação, propiciando maior ou
83

menor solubilização dos pesticidas no meio. Outro ponto a ser levantado é a
mudança estrutural que por ventura tais solventes possam ocasionar a micela,
mudanças estas como: alteração na cmc e por conseqüência no número de
agregação. Se alguns destes solventes se solubilizar preferencialmente na micela,
estes podem alterar significativamente a constante dielétrica na superfície das
micelas, através da desidratação da camada palasídica, que como conseqüência
diminuiria a cOl'Jstante dielétrica da superfície das micelas favorecendo a
solubilização dos pesticidas nesta superfície.
84

~~U
kJ~
~
~
u
~t~, ,~~ ,5
ÇJl~-J
CAPÍTULO 6

6 - Introdução.
A eletroforese capilar (CE) tem se mostrado uma ferramenta analítica
poderosa para separação de espécies em diferentes amostras, devido
principalmente a vantagens, como alta eficiência de coluna, análises rápidas e
consumo de pequenas quantidades de amostras e solventes em comparação
como HPLC. Para a separação de compostos neutros a cromatografia
eletrocinétiça micelar capilar (MEKC) é o modo de eletroforese mais
apropriado.
o maior desafio para a eletroforese capilar é a detecção de analitos em
baixas concentrações. A baixa detectibilidade da técnica decorre do pequeno
volume do caminho óptico que capilares com diâmetros internos em geral de
75 ~m proporcionam. Técnicas de pré-concentração off-line ou on-line são
sempre utilizadas quando existe a necessidade de determinação de analitos
em baixas concentrações. Uma variedade de estratégias de injeção e
manipulação de amostra para pré-concentração de analitos tem sido
desenvolvida para proporcionar melhores valores de limite de detecção (LO)
em MEKC, entre elas o sweeping 1.2 e o stacking 3,4. Estas estratégias
permitem a injeção de uma grande quantidade de amostra no capilar, sem que
a eficiência da coluna seja comprometida. Ambas as estratégias estão
baseadas na manipulação da condutividade do meio onde a amostra é inserida
em relação à condutividade do eletrólito de separação.
1 J.P. Ouirino, S. Terabe, Scíence, 16 (1998) 4652 J.P. Ouirino., S Terabe., Anal. Chem., 71 (1999) 16383 R. Weinberger, E. Sapp, S. Moring, J. Chromatogr. 516 (1990) 2714 Y. Sera, N. Matsubara, K. Otsuda, S. Terabe, Electrophoresis 22 (1988) 3509
85

No sweeping a condutividade da amostra em relação a do eletrólito de
separação é semelhante, não promovendo mudanças na velocidade de
migração das micelas ao penetrarem na zona da amostra.
No stacking a velocidade de migração das micelas é modificada com o
aumento ou diminuição da condutividade da amostra em relação à do eletrólito
de separação. Quando a condutividade é menor, estabelece-se um alto campo
elétrico na zona da amostra após aplicação do potencial, desta forma as
micelas experimentam um aumento em suas velocidades de migração, ao
penetrarem na zona da amostra. Situação contrária é obtida quando a
condutividade da amostra é maior ·em relação à do eletrólito de separação.
Na literatura são encontrados vários modos de stacking, entre eles,
modo normal (NSM, do inglês normal stacking mode)5.6, stacking usando
polaridade reversa (REPSM, do inglês reversed electrode polarity stacking
mode)?, stacking com migração reversa das micelas (SRMM, do inglês stacking
with reverse migrating micelles)8, stacking com migração reversa das micelas e
um plug de água (SRW, do inglês stacking using reverse migrating micelles and
a water plug)9, amplificação da injeção da amostra por campo (FESI do inglês
Field-enhanced sample injection)1O, amplificação da injeção da amostra por
campo e migração reversa das micelas (FESI-RMM, do inglês Field-enhenced
sample injection with reverse migrating micelles)11, stacking com alta
concentração de sal na amostra12, entre outras. No presente trabalho foram
avaliadas as técnicas de sweeping e stacking nos modos, SRMM, SRW e alta
5 J.P.Quirino, S. Terabe, J. Chromatogr. A 791 (1997) 1196 W. Shi,C.P. Landers, J. Sep. Sei. 25 (2002) 2157 J.P.Quirino, S. Terabe, J. Chromatogr. A 791 (1997) 2558 J.P.Quirino, S. Terabe, Anal. Chem. 70 (1998)1499 J.P.Quirino, S. Terabe, J. Chromatogr.B 714 (1998) 2910 J.P.Quirino, S. Terabe, J. Chromatogr. A 714 (1998) 2911 J.P.Quirino, S. Terabe, Anal. Chem. 70 (1998) 189312 J.Palmer. J.P. Landers. A .M. Stalcup, J. Chromatogr. A 71 (1999) 369
86

concentração de sal na amostra. As estratégias de pré-concentração sweeping
e alta concentração de sal na amostra foram avaliadas em condições de alto
EOF, pH 9,3, enquanto as estratégias SRMM, SRW foram avaliadas em
condições de baixo EOF, pH 2,5. Baseando-se na estratégia SRMM foi
proposta neste presente trabalho uma variação desta estratégia de pré
concetração on-line que foi denominada M-SRMM (stacking modificado com
migração reversa das micelas). Considerações mais detalhadas a respeito de
todas as estratégias citadas são dadas a seguir.
6.1 - Avaliação das estratégias de pré-concentração on-line na ausência
de EOF
6.1.1 - Sweeping de analitos neutros em MEKC
A técnica de sweeping em CE envolve a interação das micelas contidas no
eletrólito de separação e as moléculas do analito contidas na zona da amostra,
que por sua vez está isenta de micelas. A condição básica para que o processo de
sweeping seja realizado é que a amostra a ser injetada esteja livre de aditivo
(isto inclui micelas e ciclodextrinas e altas quantidades de solventes orgânicos). O
sweeping em MEKC é definido como a captura e acumulação de analitos por
micelas carregadas contidas no eletrólito de separação, sendo que, estas
penetram na zona da amostra durante a aplicação da tensão. Separações
utilizando ciclodextrinas sulfatadas ou microemulsão também podem ser
empregadas para a execução desta estratégia de concentração dos analitos13.
13 IP. Quirino, S. Terabe, K. Otsuka, B Vicent, G. Vich, J. Chromatogr. A 838 (1999) 3
87

Mantendo-se a resistência constante ao longo do capilar através da
preparação da amostra com condutividade similar em relação à do eletrólito de
separação (BGS), estabelece-se a condição para a realização do sweeping.
o comportamento da zona do analito em MEKC sob um processo de
sweeping na zona da amostra é mostrado na figura 6.1 .
• ' Injeção Detecção
B
A
Zona sem micelas
Moléculas do analito sofrendosweeping
5
Sweeping completado
c
Figura 6.1 - Representação esquemática do sweeping
Na Figura 6.1 (A) é mostrada uma longa injeção de um plug de amostra
(S) em um capilar preenchido com BGS. Os analitos são neutros, não possuem
cargas, e por isso não migram por si mesmos em direção ao detector a não ser
quando interagem com as micelas. Na Figura 6.1 (8) é então aplicado o
potencial; as micelas do lado catódico (-) migram em direção ao eletrodo de
carga oposta, penetrando assim na zona do analito capturando-o e levando-o
88

em direção ao detector. A região marcada em preto na Figura 6.1 (8) indica a
acumulação do analito pelas micelas. Neste processo a velocidade das
micelas livres é sempre maior do que a velocidade das micelas que
incorporaram as moléculas do analito. Após um certo período todo o analito é
conduzido e acumulado pelas micelas, o que foi representado na figura 6.1 (C);
a partir deste instante, os analitos acumulados são separados via MEKC. A
velocidade de migração das micelas quando aos analitos estão adsorvidos a
ela é menor do que as micelas livres, desta forma, o analito nunca entrará na
zona de ausência de micelas1(região marcada em branco).
O analito que apresentar maior fator de retenção (k), apresentará
maiores fatores de pré-concentração. O fator de retenção ou capacidade (k) pode
ser expresso como:
k =Kcj> (1)
onde K é o coeficiente de partição ou distribuição, que é a relação entre
concentração de analito incorporado às micelas e a concentração do analito livre
na fase líquida, e cj> é a relação entre o volume das micelas e o volume da fase
líquida.
Quantitativamente o comprimento da zona da amostra após o sweeping (I sweep)
pode ser aproximadamente previsto pela equação1:
I sweep = linj. 1/(1+k) (2)
onde hnj é o comprimento da zona do analito, e k é o fator de retenção do analito.
Avaliando-se a equação 2 é possível notar que o sweeping é dependente do fator
de retenção (k) e do comprimento do plug de analito injetado (Iil)l'). A equação
89

também prediz que para analitos com grande afinidade pelas micelas, ou seja,
altos k, serão obtidos maiores fatores de pré-concentração.
Para a realização dos experimentos uma solução padrão contendo 9
pesticidas na concentração de 0,5 mg L-1 foi utilizada. A amostra foi diluída em
uma solução contendo 20% metanol e 25 mmol L-1 de tampão TF, atingindo
condutividade semelhante ao eletrólito de separação (2,35 mS.cm-1). Na figura 6.2
são mostrados os eletroferogramas obtidos.
1 82015 4 6
:) 10 C 9~ 5
O
25 2 4 6 8
208
15 B:)
~105O
2 4 6 8
2015 A 7
:) 10~ 5 8 9
1 2 345O
2 3 4 5 6 7 8
Tempo, minoFigura 6.2 - Sweeping (baixo EOF) para uma mistura padrão de pesticida na
concentração de 0,5 mg L-1. Amostra foi diluída em 25 mmol/de tampão TF,
condutividade da amostra similar ao do BGS. (A) injeção = 10 s/2,5 kPA, (B)
injeção =100 s/2,5 kPA, (C) injeção =150 s/2,5 kPA.Composição do eletrólito: 25
mmol L-1 de TF, 10 % MeOH, 50 mmollL SOS..À = 220 nm, capilar de sílica
fundida 50 cm (efetivo), -25 kV, 25°C. 1) ametrina, 2) carbendazim, 3) propazina,
4) propoxur, 5) atrazina, 6) linuron, 7) diuron, 8) simazina, 9) carbaril.
90

Analisando a Figura 6.2 (A) e (B), nota-se que o aumento no tempo de
injeção de 10 para 100 (20% do comprimento do capilar) proporcionou um
sensível aumento na intensidade dos picos, preservando a resolução. Elevando
se o tempo de injeção de 100 para 150 segundos (Fig. 6.2 C), o aumento da
intensidade de picos ainda é percebido para alguns pesticidas, principalmente
para ametrina, propoxur e linuron (picos 1, 4 e 6, respectivamente). Para os picos
7,8 e 9 notam-se severas deformações.
A eficiência do sweeping está intimamente relacionada com a capacidade
que o analito possui em interagir com a micela (k). Para valores maiores de k, o
ganho de pré-concentração é maior em relação aos analitos que possuem
menores k. A deformação dos picos poderia ser resultado de um pronunciado
efeito de dispersão, pelo fato da baixa interação entre estes pesticidas (propoxur,
linuron e carbaril) e a micela. Outro fator que contribui para este acontecimento é
que injetando 150 s de amostra no capilar (30% do comprimento do capilar), o
percurso de separação no capilar será encurtado, proporcionando um
comprometimento da separação, principalmente para compostos com menor fator
capacidade (k).
6.1.2 - Stackinq com migração reversa das micelas (SRMMJ e fM-SRMMJ
No SRMM a amostra é dissolvida em um meio de baixa condutividade
em relação ao BGS, que após a aplicação de um potencial irá gerar uma região
de campo elétrico elevado. O ganho na pré-concetração do analito (y) é igual a
relação entre as condutividades do BGS e da zona da amostra. O
comportamento da zona do analito em MEKC sob um processo de stacking
com campo elétrico elevado na zona da amostra é mostrado na Figura 6.3.
91

llnjeção lDetecção
sMoléculas sofrendo stacking
Zona sem micelas
A
B
Stacking intermediário
Stacking completado
Figura 6.3 - Stacking sob campo elétrico elevado (baixo EOF).
Na figura 6.3 (A) é representada a injeção da amostra em um longo plug,
em um capilar preenchido com BGS. Quando a tensão é aplicada, as micelas do
lado catódico (-) entram no capilar14• sendo aceleradas em direção ao eletrodo de
carga oposta (ânodo (+». A concentração das micelas que entram na zona da
amostra (CM(S» é menor do que aquelas no BGS (CM(BGS», prevista pela
equação:
14 J.P. Quirino, S. Terabe, J. Chromatogr. A. 781 (1997) 119
92
c
D

CM(S) = ~(BGS) (3)
Y
onde y é a relação entre a condutividade do BGS e a condutividade da zona da
amostra11. O analito então é incorporado e acumulado pelas micelas que
penetram na zona da amostra (Figura 6.3 B e C). No início do processo de
stacking a concentração das micelas na zona da amostra é pequena, então a
interação entre as moléculas do analito e as micelas é parcial, não contribuindo
para a formação de uma banda estreita (Figura 6 C). Com o aumento da
concentração de micelas na zona de amostra o processo de stacking é
concluído (Figura 6.3 D).
A finalização da estratégia é atingida quando a concentração de micelas
na zona da amostra é igual a do BGS. O aumento na concentração das micelas
permite um aumento no k para cada analito; com isso ocorre uma focalização
da zona da amostra, diminuindo o comprimento da zona do analito (Figura 6.3
D).
A mudança da velocidade eletroforética efetiva na interface que divide a
zona da amostra e o BGS é característico do stacking15,16 . A velocidade
eletroforética efetiva das micelas na zona da amostra é muito maior do que
aquela quando se aproxima da interface que divide a zona da amostra e a zona
do BGS (lado anódico). causando assim um gradiente de campo elétrico na
extensão da zona da amostra.
15 R.L. Chien, D. S. Burgi, Anal. Chem. 64 (1992) 489Aló IP. Quirino, S. Terabe, J. High Resolut. Chromatogr. 22 (1999) 367
93

6.1.2.1 - Variação da estratégia SRMM
Uma variação da estratégia SRMM é proposta no presente trabalho.
Com a variação, a estratégia passou a ser chamada de M-SRMM I para
distinção da estratégia original. Através da estratégia proposta a eficiência com
a qual as micelas capturam os analitos é aumentada. Na Figura 6.4 é mostrada
uma representação esquemática das etapas do M-SRMM.
Injeção 1 1Detector
A
+,'B
~"''''~':'::''''',II"---. ------- ,.. "" ": ,
Figura 6.4 - Representação esquemática para a estratégia M-SRMM,
94
c
o

A mudança em relação à estratégia SRMM é a aplicação de um potencial
normal «+) no lado de injeção e (-) no lado do detector), por um determinado
tempo, após a amostra ter sido injetada no capilar. Agora as micelas do lado
catódico (lado do detector) migram em direção ao ânodo (lado da injeção),
penetrando na zona da amostra, capturando e acumulando os analitos. Passado
este tempo, aplica-se um potencial invertido «-) no lado de injeção e (+) no lado do
detector) e as micelas do lado catódico, agora lado da injeção, migram em direção
ao anodô (lado do detector), capturando mais moléculas dos analitos. O aumento
de sinal resultante desta técnica vem do fato dos analitos serem empilhados
(stacking) duas vezes. Este processo é representado pelas Figuras 6.4 (8) e (C) e,
a partir daí, o processo se desenvolve como no SRMM, descrito anteriormente.
Para a realização deste experimento uma amostra contendo 9 pesticidas na
concentração de 0,5 mg/L foi utilizada. A amostra foi diluída em solução contendo
20% MeOH. Na Figura 6.5 são apresentados os eletroferogramas obtidos,
comparando as duas estratégias de stacking, o SRMM e o M-SRMM.
95

9
30 -
25 - C2 O -
::J 15 -
«E 1 O -
5 -
I
-5
30
25 B20
::J15«
E1 O
5
O
2
30
A25
20mAU
15
10
5
2
1 2
4
4
1
6
9
6
6Te~.min
2
5
3
64 7 8
8
8
10
10
10
Figura 6.5 - Comparação entre as estratégias SRMM e M-SRMM para uma
mistura de pesticidas, na concentração de 0,5 mg L-', (A) injeção = 10 s./2,5 kPA,
B) injeção de 100 s/2,5 kPA, (C) injeção de 100 s/2,5 kPA, seguido de aplicação
de potencial (+25 kV) por 2 min.. Composição do eletrólito: 25 mmol L-1 de TF, 10
% MeOH, 50 mmol L-1 SDS..À = 220 nm, capilar de sílica fundida 50 cm (efetivo),
25 kV, 25 oCo 1) ametrina, 2) carbendazim, 3) propazina, 4) propoxur, 5) atrazina,
6) linuron, 7) diuron, 8) simazina, 9) carbaril.
96

Através da Figura 6.5 é possível notar um sensível aumento de sinal quando
a estratégia M-SRMM foi utilizada (Figura 6.5 C). Outro fator a ser evidenciado é
que os picos 7,8 e 9 não apresentaram alargamento ou deformações tanto para a
SRMM quanto para a M-SRMM. Este fato havia sido verificado quando o sweeping
como estratégia de pré-concentração foi emprego (Fig. 6.2).
Na Tabela 6.1 são apresentados os valores comparativos de alturas de pico
para as estratégias SRMM e M-SRMM, em relação a uma situação arbitrária de
referência.
Tabela 6.1 - Comparação entre as alturas de picos encontradas para cada
estratégias de pré-concetração (SRMM e M-SRMM)
SRMM M-SRMM
Pesticida Altura do pico Altura do ArJAR Altura do Ap/AR
de pico pico
referência(AR)
ametrina 1,35 1,26 0,93 8,34 6,1
carbendazim 0,80 1,10 1,3 3,88 4,9
propazina 1,67 6,13 3,6 18,28 11,0
propoxur 0,92 2,76 3,0 8,73 9,5
atrazina 2,51 7,81 ~,1 21,58 8,6
linuron 1,00 3,49 3,4 9,93 10,0
diuron 1,25 4,48 3,5 10,18 8,1
simazina 2,21 6,56 2,9 11,89 5,4
carbaril 4,49 13,09 2,9 31,49 7,0
Ap Altura do pico (com a estratégia aplicada)
AR altura do pico de referência. (mistura padrão de pesticidas na concentração
de 0,5 mg/L, inj 10s/2,5 kPA)
97

Epossível notar através da Tabela 6.1 que a estratégia proposta (M-SRMM)
proporcionou aumento na intensidade dos picos para todos os pesticidas
estudados, quando comparada com a técnica de SRMM convencional. O ganho de
sinal é da ordem de 4 - 11 vezes.
6.1.3 • Stacking com migracão reversa das micelas e um plug de água (SRW)
Nesta estratégia de pré-concetração as amostras também são preparadas
em meio de baixa condutividade em relação ao BGS. Um tensoativo (SOS) é
adicionado à amostra em concentração pouco maior que seu cmc, para aumentar a
solubilidade dos analitos. Um plug de água é injetado antes da injeção da amostra.
O plug de água proporciona uma zona de baixa condutividade, que após a
aplicação do potencial gera um elevado campo elétrico no local, comparado ao do
eletrólito de separação (BGS). As micelas do lado catódico (injeção) migram em
direção ao anodo (detector), entram na zona da amostra capturando e conduzindo
o analito para o detector. Quando as micelas atravessarem a zona de amostra e
encontrarem a zona de água, estas são aceleradas, devido ao elevado campo
elétrico da zona da água, e são empilhadas na interface que divide a zona de água
e o BGS do lado anódico (detector).
O efeito do stacking é causado pela mudança abrupta na velocidade de
migração das micelas ao encontrarem a zona de água. Após estabelecido o
equilíbrio, a zona de água é expulsa do capilar e a separação é conduzida por
MEKC convencional.
A mesma modificação realizada para a SRMM foi também realizada para
esta estratégia de pré-concentração que consistia em aplicar uma polaridade
98

normal «+) injeção, (-) detector), após a injeção da água e amostra. Foi notado um
aumento na intensidade dos picos para os pesticidas estudados. Na Figura 6.6 é
mostrada uma representação esquemática do mecanismo de como a zona do
analito se comporta nesta estratégia.
1Injeção 1Detector
w A
lona da amostra Micelas aceleradasI I +-Etapa 1
Micelas acumuladas
Stacking completo
Figura 6.6 - Representação esquemática para as estratégias M-SRW.
B
c
D
Inicialmente um plug de água é injetado antes da injeção de um longo
plug de amostra. Uma polaridade normal é aplicada por 2,5 minutos (Figura 6.6
A). As micelas do lado catódico (Etapa 1) migram em direção a zona da amostra,
ao encontrarem a zona da água estas são aceleradas, até atingirem a zona da
amostra, onde sofrem desaceleração, porém continuamente penetram na zona da
99

amostra capturando e acumulando as moléculas dos analitos (figura 6.6 B).
Posteriormente polaridade invertida é aplicada e as micelas do lado catódico
(Etapa 2) migram em direção ao eletrodo de carga oposta, penetrando na zona da
amostra capturando os analitos, e levando-os em direção ao detector (figura 6.6
C). As micelas atingem novamente a zona da água, sofrendo uma nova
aceleração, que por sua fez são desaceleradas quando atingem o BGS do lado
catódico proporcionando a compressão da banda da amostra (Figura 6.6 D).
Para esta técnica inicialmente é injetado um plug de água e posteriormente
a amostra. Melhores resultados foram obtidos quando a proporção de água e
amostra foi de 1:2.
Para a realização desta técnica uma mistura padrão contendo 9 pesticidas
na concentração de 1 mg L-1 foi utilizado. A mistura foi diluída em uma solução
contendo 20% metano!. As estratégias foram comparadas e os eletroferogramas
obtidos são apresentados na figura 6.7.
100

2 5 5 9
2 O 4
t 5 C 3 7:::l 6<t:
E t O8
1 2
Figura 6.7- Comparação entre as estratégias SRW e M-SRW para uma
mistura de pesticidas, na concentração de 1 mg L-1. (A) Injeção de 10 sego /2,5
kPA, B) Injeção de 50 s H20 seguida de 100 s amostra.l2,5 kPA, (SRW), (C)
injeção de 50 seg H20 seguida de 100 sego amostra.l2,5 kPA, e tensão +25 kV
durante 2,5 min (M-SRW). Composição do eletrólito: 25 mmol L-1 de TF, 10 %
MeOH, 50 mmol L-1 SOS..1..= 220 nm, capilar de sílica fundida, 50 cm (efetivo), -25
kV, 25°C. 1) ametrina, 2)carbendazim, 3)propazina, 4)propoxur, 5)atrazina,
6)linuron, 7) diuron, 8) simazina, 9)carbaril.
101

Na tabela 6.2 são apresentados os valores comparativos das alturas de
picos encontradas para cada estratégia (SRW e M-SRW).
Tabela 6.2 - Comparação entre as alturas de picos encontradas para cada
estratégias de pré-concetração (SRW e M-SRW)
Ap Altura do piCO (com a estrategla aplicada)
AR altura do pico de referência. (mistura padrão de pesticidas na concentração
de 1 mg/L, inj. 10s/2,5 kPA).
SRW M-SRW
Pesticida Altura do pico Altura do AplAR Altura do Ap/AR
de pico pico
referência(AR)
ametrina 0,8 1,76 2,2 6,45 8,0
carbendazim 0,6 4,68 7,8 7,81 13,0
propazina 1,01 1,31 1,3 17,43 17,2
propoxur 0,63 2,74 4,3 5,43 8,6
atrazina 1,24 5,47 4,3 24,55 19,7
linuron 0,73 3,17 4,3 11,08 15,1
diuron 1,45 5,12 3,5 14,44 10,0
simazina 0,69 3,25 4,8 7,12 10,3
carbaril 2,69 10,91 4,0 24,35 9,0..
Nota-se avaliando a Tabela 6.2 que a estratégia M-SRW proporciona
sensível aumento na intensidade dos picos de todos os pesticidas estudados. A
inversão de polaridade após a injeção demonstrou ser uma estratégia viável para
pré-concentrações em condições de ausência de EOF. A dupla migração das
micelas proporciona uma melhor acumulação dos analitos.
102

Iiff,\íl.~I
\I
6.1.4 - Junção das estratégias de pré-çonçentração off-line e on-line
Dentre as estratégias de pré-concentração off-line destaca-se a extração
em fases sólida SPE (do inglês solid phase extraction). A SPE é uma técnica
simples de preparação de amostra baseada nos conceitos da cromatografia
líquida17. Recentemente vários pesquisadores tem utilizada a SPE como técnica de
pré-concentração de analitos em amostras de água18,19.
Após verificada a potencialidade das estratégias de pré-concentração on-line
em MEKC em baixo EOF, avaliou-se a junção das estratégias off-line e on-line para
propiciar melhores limites de detecção.
Promoveu-se uma SPE para uma mistura de pesticidas em água, na
concentração de 0,1 IJ.g L-1 em um volume total de 250 mL. Percolou-se esta
solução por um cartucho C18 previamente condicionado (10 mL de metanol e 10 mL
de água). Os analitos foram eluídos do cartucho com 6 mL de metanol, e a solução
foi evaporada até a secura. Este procedimento foi realizado 3 vezes e os analitos
foram ressuspendido em três soluções diferentes: 1) 1 mL de 25 mmollL de tampão
fosfato (condutividade similar ao BGS), para a realização do sweeping em meio
homogêneo, 2) 1mL de solução contendo 4 mmol L-1de tampão fosfato e 10 mmol
L-1da SOS, para realização do estratégia SRW e 3) 1 mL de solução 20 % metanol
em água, para realização da estratégia M-SR~M. Na Figura 6.8 são mostrados os
eletroferogramas obtidos.
17 J.S.Fritz and M.Macka, J. Chromatogr. A, 902 (2000) 137-16618 Ehogendoorn, Piet Van Zoonen, J. Chromatogr. A, 892 (2000) 43519 C. Aguiar, I. Ferrer, F. Borrull, R.M. Marcé, O. Barceló, Anal. Chim. Acta 386 (1999) 237
103

5
4
1:::l 3«
C 5E 2 72 9
oA~
-1
3 5 6 8
9
B
7
5
9
1 O
, o
86
Tem p o • m in
-,6
5
4
:::l3«
E2
A
o
4
Figura 6.8 - Avaliação da junção das estratégias de pré-concentração on
fine e off-line para análise de amostra de água fortificada com uma mistura de 9
padrões de pesticidas na concentração de O, 1 ~g/L. (A) SRW com inj = 50seg H20
e 1,6 min amostra, (8) M-SRMM com injeção de 1,6 mino da amostra e 2 min, +25
kVe (C) Sweeping com inj. = 1,6 mino Todas com 2,5 kPA. Composição do
eletrólito: 25 mmol L-1 de TF, 10 % MeOH, 50 mmol L-1 SOS. À = 220 nm, capilar de
sílica fundida 50 cm (efetivo), -25 kV, 25 oCo 1) ametrina, 2) carbendazim, 3)
propazina, 4) propoxur, 5) atrazina, 6) linuron, 7) diuron, 8) simazina, 9) carbaril.
104

r.
As estratégias de pré-concentração foram também avaliadas para uma
amostra de cenoura fortificada com 2,5 f.1g/L de uma mistura padrão contendo 9 de
pesticidas. O procedimento de extração e c1ean-up utilizado foi o proposto por
Minelli et aI. 20, procedimento que será explanado detalhadamente no capítulo 7.
Foram realizadas 3 extrações e as amostra foram ressuspendidas em 1mL de: 1)
solução de 20 % de metanol em água, para a estratégia modificada de stacking
com migração reversa das micelas M-SRMM, 2) 1 mL de uma solução contendo 4
mmollL 'de tampão fosfato (pH 2,5) e 10 mmol L-1 de SDS, para a estratégia
stacking com migração reversa das micelas e um plug de água (SRW) e 3) 1 mL
de solução tampão fosfato 25 mmol L-1., para a estratégia de sweeping. Os
eletroferogramas obtidos para as estratégias SRW, M-SRMM e sweeping são
apresentados na Figura 6.9.
20 E. Minelli, A .Angioni, M. Melis, F.M. Pirisi, P. Cabras, J. AOAC international80 (1997) 1315
105

2 0-T
15
::> ~ 10 8 E
5
O
~ 2 4 6 a 10_ 1:.2
40
30
20
::> 10 ~
E O
-10
-2 0
~ 2 4 6 8 10
30
9 25
-ZU
::> 15 ~
E 10
5 --
O
-5 2 4 6 8 10 12
Tem p o , m in _
FiguraS:9 - Aplicação das estratégIas de pré-uoncentração em uma amostra de
cenoura fortificada com 2,5 J..I.9/L de uma mistura padrão de 9 pesticidas. (A) M-SRMM com
inj_ de 1,6-min-e 2- min, +25 KV~ (9). Sweepingcom inj. de 1,6min daamostra-e-(C)SRMM,
50s. de H20 seguido de 1,6 min de amostra, todas com 2,5 kPA. Composição do eletrólito:
- 25 mmofl-1 de TF,10 % -MeOH, 50 mmolL"1 '80S .À=220 nm, capilar de síficafundlda 50
cm (efetivo), -25 kV, 25°C. 1) ametrina, 2) carbendazim, 3) propazina, 4) propoxur, 5)
afrazina, 6}linuron, 7) diuron, _ 8) simazina, 9)carbafiL_
106

..,III
lI,,.
\
fI\
A estratégia que permitiu a identificação de maior número de pesticidas foi a
estratégia modificada de stacking com migração reversa das micelas M-SRMM. Na
tabela 6.3 são feitas avaliações comparativas para as estratégias de sweeping,
SRW e M-SRMM com base em seus limites de detecção e altura dos picos.
Das estratégias de pré-concetração avaliadas as estratégias denominadas
stacking de uma maneira geral apresentaram melhores fatores de pré-
concentração. Outro ponto a ser destacado é que nas estratégias de stacking basta
a diluição da amostra em meio de baixa condutividade, enquanto que para o
sweeping é necessário ajustar a condutividade da amostra à condutividade do
SGS. Porém, o sweeping apresenta melhor repetibilidade entre injeções
consecutivas, fato não observado quando a estratégia utilizada era o stacking,
principalmente para o SRW e M-SRW. As estratégias M-SRMM e M-SRW
apresentaram maiores LO em relação as estratégias originais.
107

.-'ªA".]_t4.1_'!,i#A....-;~~ ?Jf}D %i)€._",\(W~P ;,âfW4;<;y. J$ljW·?4'- F"'.~i!';t~;q:><r .:wv:.trifVw:s:..~_\"'"'.~....-::.~-~...-ro, __~"r.-·'''''#'·r·_ .~.~.
Tabela 6.3 - Avaliação comparativa entre as estratégias de pré-concetração
PESTICIDA REFERÊNCIA SWEEPING SRW M SRMMLO LO YLD YPKH LO YLD YPKH LO YLD YPKH
L-1 L-1 L-1 L-1119 ~._ IlQ Ilg
Ametrina
Carbendazim
Atrazina
Propoxur
Propazina
Oiuron
Linuron
Simazina
Carbaril
93
170
71
110
56
100
80
18
22
5.2 18 19
43 4.0 4.1
28 2.5 2.6
8.1 13 15
17 3.2 3.4
6.6 15 16
15 5.1 5.2
3.6 4.9 5.0
5.6 4.0 4.1
27 3.4
36 4.7
46 1.6
15 7.5
7.6 7.4
20 5.3
20 5.0
6.6 2.6
5.3 4.2
6.3
8.7
2.9
15
14
9.6
7.2
4.9
7.7
17 5.5
35 4.8
6.5 11
9.6 12
4.4 13
10 10
11 7.1
3.5 5.0
2.7 8.4
6.3
5.5
12
14
15
11
8.0
5.6
9.6
YLOD = LOreferência/LOestratégia
YPH =PKHreferência/PKHestratégiaCondição de referência: inj. 10 s 125 mBar (1 mBar =100 Pa).
108

iI
ll-
It
ttIi
A estratégia M-SRMM apresentou menores limites de detecção em relação à
estratégia SRW para todos os pesticidas avaliados. Comparando-se a estratégia M-
SRMM com o sweeping, esta apresenta limites de detecção maior para a ametrina,
propoxur e diuron, porém limites de detecção menores para os demais pesticidas
estudados.
6.2 - Avaliação das estratégias de pré-concentração on-line na presença de alto
de EOF.
6.2.1 - Sweeping
Nesta estratégia a amostra é preparada em um meio de condutividade similar
ao do eletrólito de separação.
109

t injeção
s
t Detector
A
EOF • I I micelas catódicas
Migração das Acumulação do.-micelas ..-Analito pela Micelas
I Sweeping completo.. das moléculas
Figura 6.10 - Representação esquemática para o sweeping em alto EOF.
8
c
Na Figura 6.10 (A) é mostrada uma longa injeção de um plug de amostra (S)
em um capilar preenchido com BGS. Após a aplicação do potencial normal, as micelas
de SDS do lado catódico migram em direção ao eletrodo de carga oposta, penetrando
na zona da amostra (S), capturando e acumulando os analitos na zona que divide a
amostra e o BGS do lado anódico (Figura 6.10 B), os analitos acumulados depois são
separados via MEKC. O EOF é o responsável por conduzir os analitos em direção ao
detector.
110

Para a realização dos experimentos, uma solução padrão contendo 9 pesticidas
na concentração de 1 mg L-1 foi utilizada. A amostra foi preparada em uma solução
contendo 20% metanol e 30 mmollL de NaCI, atingindo condutividade semelhante ao
eletrólito de separação (4,08 mS.cm-1). Na figura 6.11 são mostrados os
eletroferogramas obtidos.
16
16
16
9
14
14
14
8
12
12
12
10
10
10
4
56
4
3
3
8
8
3 4
2
8
Tempo, mino
2
6
6
6
2
4
4
4
2
2
2
A
B
c10864
::J 2'ª -~ -+-------.-4-6-+----,r____r_-~---.-...--~-_.__---,r____r_-~__,_-.______r-_.__-._____,
10 o864
::J 2'ª o-+---------,-2
-4+----r-.,...-----,r----r--r-----r--"'T"""'"""---r-.,...-----,r----r---r------r-....----.--,
10 o8642o-1------ '--_'--_...J\-__-II--JI._-.II...J'.-___._"----
-2 r------,r----.--r---T--'T"""""--r--r-----,r----r--r----.--...--~-_.__---,r____,
o
Figura 6.11- Sweeping em meio homogêneo (alto EOF) para uma mistura padrão de
pesticidas. (A) inj. 10 s/2,5 kPA, (B) 100 s/2,5 kPA, (C) 150 s/2,5 kPA. Composição do
eletrólito de separação: 50 mmol L-1 de SOS, 10 mmol L-1 de TBS (pH 9,3),5% PrOH,
5% EtOH. À = 220 nm, capilar de sílica fundida 50 cm (efetivo), 27 kV, 25 oCo 1)
carbendazim, 2) simazina, 3) atrazina, 4) carbaril, 5) ametrina, 6) propazina, 7) diuron,
8) propoxur, 9) linuron.
111

II
Através da Figura 6.11 (8) é possível verificar que foram obtidos
aumentos nas alturas e nas áreas dos picos com o aumento no tempo de injeção,
sem perdas significativas de resolução na separação eletroforética. No entanto,
quando o tempo de injeção foi aumentado para 150 s (Fig. 6.11 C), apesar de
terem sido obtidos aumentos ainda mais significativos, pode-se verificar co-
eluições e alargamento de picos. Na tabela 6.4 são relacionadas as alturas de
picos para injeções de 100 e 150 segundos, contra a altura dos picos sem o
emprego da estratégia de sweeping. Esta relação indica quantas vezes houve o
aumento da altura do pico resultante da aplicação da estratégia.
Tabela 6.4 - Comparação entre as alturas de picos encontradas para cada tempo
de injeção utilizado durante o sweeping, e suas alturas relativas à injeção sem
emprego da estratégia.
100 s de injeção 150 s.de injeção
Pesticida Altura do pico Altura do AplAR Altura do Ap/AR
de pico (Ap) pico (Ap)
referência(AR)
carbendazim 0,14 1,1 7,5 0,5 4,0
simazina 0,80 5,1 6,4 5,8 7,3
atrazina 0,33 3,9 12,0 7,7 24,0
carbaril 1,10 8,3 7,5 8,1 7,3
ametrina 0,15 4,2 28,2 9,6 64,0
propazina 0,41 1,1 2,6 10,0 24,3
diuron 0,06 1,6 27,3 3,3 55,1
propoxur 0,23 1,3 5,6 3,7 16,2
linuron 0,12 1,1 8,7 2,0 16,6
Ap Altura do pico (com a estratégia aplicada)AR altura do pico de referência (mistura padrão de pesticidas na concentração de1 mg L-1
, inj 10 s/2,5 kPA).112

6.2.2 - Stacking em baixo campo elétrico (alta condutividade da amostra)
o uso de alta concentração de sal na amostra conferindo a ela uma alta
condutividade, foi primeiramente proposto por Palmer et aI.21, utilizando esta condição
para pré-concentração on-Iine de amostras em separações MEKC. É adicionado NaCI
à amostra a fim de atingir duas ou três vezes a condutividade do eletrólito de
separação (BGS) e a separação é promovida sob alto fluxo eletrosmótico. Segundo o
mecanismo proposto, as micelas que penetram na zona da amostra, que partem do
lado catódico (lado do detector) experimentam uma redução de sua velocidade
eletroforética, (devido a alta condutividade da zona da amostra) capturando o analito e
levando-o em direção ao eletrodo de carga oposta (anodo, lado da injeção) e, por fim,
acumulando o analito na zona limítrofe entre a zona da amostra e o BGS do lado
anódico (+), promovendo uma compressão na zona da amostra responsável pelo
aumento do sinal analítico. Porém, Quirino et al.22 demonstraram que o procedimento
apresentado por Palmer apresentava um processo de desempilhamento (destacking)
alargando novamente a banda da amostra, comprometendo a pré-concentração. O
comportamento da zona da amostra em MEKC, sob a ação do stacking com baixo
campo elétrico na zona da amostra é mostrado na figura 6.12.
211. Palmer, N. J. Munro, 1. P.Landers, Anal. Chem. 71(1999) 167922 1..P. Quirino, S. Terabe, P. Bocek, Anal. Chem. 72 (2000) 1934
113

~ Injeção ~ Detecção
A
B
c
~ Ausência de micelas
H Stack:ing completado
Começo do stack:ing
5
o
Alargamento de bandaf----t causado pelo destack:ing
I IDestack:ing das
micelas
Figura 6.12 - Stacking com alta concentração de sal na amostra.
I
I Para efeito de simplificação, considere que o EOF seja inexistente. A alta
concentração de sal na matriz produz um reduzido campo elétrico na zona da amostra
e comparativamente um elevado campo elétrico no BGS. O aumento do campo pode
l ser quantitativamente descrito por um aumento do fator y', o qual é a relação entre a,r condutividade da zona da amostra S e a do BGS23
. O valor de y' é sempre maior queI 1. Na figura 6.12 (A) é mostrada a injeção de um plug de amostra com alta
~ condutividade em um capilar preenchido por BGS. Quando a tensão é aplicada
23 R.-L Chien" D.S.Burgi, Anal. Chem. 64 (1992) 489A.
114

· 1
: l
(Figura 6.12 B) as micelas aniônicas do lado catódico (lado da injeção) entram na
zona da amostra empilhando os analitos na zona de alta condutividade. Na figura 6.12
B a região marcada em preto indica o inicio do stacking.
A região imediatamente antes apresentada da zona da amostra na figura 6,12
(B) apresenta uma alta concentração de micelas que se acumulam, pois a micelas
sofrem uma desacerelação quando encontram a zona da amostra S de maior
condutividaqe10. A concentração das micelas que são desacelerada pela zona S
(CM(S» é maior do que aquela presente na zona BGS (CM(BGS», e pode ser
calculada através da equaçã0 11.24:
As micelas que foram acumuladas em uma estreita banda decorrente da
desaceleração das mesmas entre a zona da amostra e o BGS, penetram na zona da
amostra capturando e acumulando as moléculas do analito (Figura 6.12 B e C). O
processo de captura dos analitos pelas micelas é denominado sweeping e nesta etapa
a pré-concentração é favorecida, devido a maior concentração de micelas que
penetram na zona da amostra causada pelo stacking inicial das micelas. A diminuição
da velocidade eletroforética das micelas quando penetram na zona da amostra,
proporcionada pela alta condutividade nesta zona, permite que as micelas tenham
maior interação com as moléculas do analito.
Quando as micelas que capturaram e acumularam as moléculas do analito
atingirem a zona limítrofe entre a zona do analito e o BGS (lado anódico) estas
experimentarão uma aceleração, devido a condutividade do BGS ser menor
24 J.P. Quirino, S. Terabe, P. Bocek,Anal. Chem. 72 (2000) 1934
115

configurando assim um destacking (Figura 6.12 D). A concentração das micelas
na zona do destacking pode ser aproximadamente prevista por:
CM(destacked) =CM(S)/y' (6)
As micelas que partem em direção a zona do BGS promovem um
diminuição da concentração das micelas na zona da amostra, diminuindo o fator
retenção para cada um dos analitos. Esse destacking proporciona um alargamento
de banda, pois as micelas que deixam a zona da amostra apresentam moléculas
do analito.
Para a realização do experimento uma mistura padrão contendo 9
pesticidas na concentração de 1 mg L-1 foi empregada. Foram preparadas 3
amostras, cada uma continha concentrações crescentes de NaCl, 30, 60 e 120
mmol L-1, respectivamente. Foi também preparada uma amostra diluída em 20 %
MeOH em água, e injetado por 10 s a 2,5 kPa. As amostras diluídas em
concentrações crescentes de NaCI foram injetados hidrodinamicamente por
50s/2,5 kPa para cada amostra. Os eletroferogramas obtidos são apresentados na
figura 6.13.
116

5
9
~ 10
vJLJL
12 14 16
6 7
8
10
5
4
8
3
642
o
15
2
8
6
-4 +--r--r--T---'r-..........,,..........,---r---r-r-----..--.....----.----r----.-----.o
-2
14
10
12
1614
8 9
12
:::>
~ 4
10
~~ ~Ll o
43
8 10
Terrpo, mn.
2
4 62
c
o
15
10
14
12
:::>~ 5
f10
8A 5
10
B 3 4 76
Figura 6.13 - Stacking com alta concentração de sal na amostra (alto EOF) para uma
mistura padrão de pesticidas. Amostras dissolvidas em (A) 20% MeOH em água, 10
s/2,5 kPa, (B) 30 mmol L-1 de NaCI, 50 s/2,5 kPa (C) 60 mmol L-1 de NaCI 50 s/2,5
kPa, (0)120 mmol L-1 de NaCl, 50 s/2.5 kPa. Composição do eletrólito de separação:
50 mmol L-1 de SOS, 10 mmol L-1 de TBS (pH 9,3), 5% PrOH, 5% ETOH. Â= 220 nm,
capilar de sílica fundida 50 cm (efetivo), 27 kV, 25°C. 1) dimetoato 2) carbendazim, 3)
simazina, 4) atrazina, 5) carbaril, 6) ametrina, 8) propazina, 8) Oiuron, 9) propoxur, 10)
Linuron.
8
~L166 8 10 12 14
Terrpo, mn
2
42o6 8 10 12 14 16
Terrpo, mn
42o
4
2
o
-2
t
II,r
117

o maior fator de pré-concentração foi atingido para a concentração de
sal de 60 mmol L-1 (Figura 6.12 B). Quando a concentração de sal adicionada foi
de 120 mmol/L, houve pequeno decréscimo na intensidade dos picos dos
pesticidas. Na Tabela 6.5 são mostradas as alturas dos picos para cada estratégia
de injeção realizada.
Tabela 6.5 - Alturas de picos encontradas para cada concentração de sal
adicionado à amostra, e suas alturas relativas a injeção sem NaCI.
30 mmol L-1 60 mmol L-I 120 mmol L-1
Pesticida Altura do pico Altura do AplAR Altura do Ap/AR Altura do Ap/AR
de pico (Ap) pico(Ap) pico(Ap)
referência(AR.)
dimetoato 0,2 0,4 2,2 0,5 2,63 0,3 1,6
carbendazim 0,6 3,5 5,7 5,3 8,59 3,0 5,0
simazina 2,2 8,1 3,6 9,0 4,06 8,1 3,6
atrazina 1,9 8,6 4,5 10,0 5,38 8,3 4,4
carbaril 3,4 14,7 4,3 15,2 4,43 10,0 3,0
ametrina 1,9 8,0 4,2 9,4 5,06 7,6 4,0
propazina 1,6 5,0 3,0 9,5 5,96 7,6 4,7
diuron 0,7 3,6 5,0 3,9 5,24 5,1 7,0
propoxur 0,5 3,0 6,0 3,7 7,38 3,1 4,2
linuron 0,3 1,9 6,0 2,2 7,16 1,4 4,7
Ap Altura do pico
AR altura do pico de referência. (mistura padrão de pesticidas na concentração de
1 mg/L, inj 10 s/2,5 kPa)
118

"
Analisando a Tabela 5.5 verifica-se que o aumento da concentração de
sal na amostra de 30 para 60 mmol L-1 proporciona um aumento no ganho na
altura dos picos quando comparado com os picos da injeção referência (Ap/AR).
No entanto, quando a concentração de sal foi aumentada para 120 mmol L-1,
houve decréscimo na altura dos picos quando relacionados aos picos da injeção
de referência, exceção feita ao diuron, que alcançou um aumento de 5,2 para 7,0.
A hipótese de perda de eficiência descrita por Quirino et aI.23 causada pelo
desempilhamento (destacking) do analito na zona da amostra pôde ser
confirmada.
5.2.2 - Conclusões para este capítulo
Após a realização destas estratégias de pré-concetração on-line é
possível ressaltar alguns pontos importantes, tais como:
- As estratégias quando realizadas em pH 2,5 apresentaram melhor capacidade
de pré-concentrar em relação às estratégias realizadas em pH 9,3.
-A estratégia M-SRW apresentou grande irreprodutibilidade entre replicatas.
Talvez pelo fato de que o equilíbrio não era atingido no mesmo ponto entre as
injeções, pois o percurso da amostra e do plug .de água eram grande. O controle
da pressão da injeção e da tensão aplicada no processo da estratégia é ponto
crucial para se atingir razoável precisão entre injeções consecutivas.
-A estratégia M-SRMM apresentou maiores fatores de pré-concetração quando
comparados ao a estratégia convencional.
- O stacking de uma maneira geral é mais fácil de implementar, quando
comparado com o sweeping.119

~~u
*tl4J~
i
~
u
~t.,.., ,~
~ 'B
u1~-J
CAPÍTULO 7

"
7 - Introdução
o uso de pesticidas promove inquestionável aumento na produção agrícola.
Porém os resíduos que permanecem em frutas e vegetais constituem um risco ao
consumidor1. Este fato estimulou o estabelecimento de diretrizes legais para o
controle de seus níveis através do Maximun Residue Leveis (MRLs). Além disso,
estimulou o desenvolvimento de pesticidas com baixa persistência e toxicidade
.para o ser humano. Assim sendo, o número de pesticidas registrados e ou
recomendados aumentou extraordinariamente, gerando como conseqüência
dificuldades analíticas para o seu controle2.
Para controlar os níveis de resíduos de pesticidas em produtos agrícolas
cada país tem uma agência governamental, a qual monitora os resíduos s levando
em consideração dois fatores complementares: monitoramento regular, focalizado
no produto agrícola in natura, determinando níveis individuais dos pesticidas e
observando os níveis aceitáveis (MRLs),3 e o estudo da dieta total, o qual consiste
em avaliar os níveis de pesticidas encontrados em frutas e vegetais, na forma em
que serão consumidos.
Métodos analíticos são necessários para quantificar e confirmar a presença
de resíduos de pesticidas em frutas e vegetais. Métodos multirresíduos (MRMs) e
de resíduo simples (SRMs) são métodos analíticos para o tratamento das
amostras e determinação de pesticidas, porém o MRM apresenta a capacidade de
determinar diferentes resíduos (classes) de pesticidas em uma única análise.
1 H.8.S. Conacher and J. Mes ; Food Addit. Contam. 10 (1993) 5.2 E. Papadopoulou-Mourikou, J. Assoc. Off. Anal. Chem., 74 (1991) 745.3 Food Drugs Administration, J. Assoc. Off. Anal. Chem. Int., 77(1994) 1.
120

Entre os métodos multirresíduos mais comumente usados estão os propostos por
Mills 4 , Mills et al5 , Storherr a16, Luke et. al7
, Sawyer et. al8 e Krauser 9. Existe uma
infinidade de métodos multirresíduos, a escolha adequada depende da matriz a
ser analisada (sólido ou líquido) da sua composição (quantidade de gordura,
açúcar, água, etc.) ou até mesmo da espécie de pesticidas que será identificado10,
Em geral estes métodos são laboriosos e despendem grande quantidade de
solventes orgânicos. Recentemente significativas evoluções foram evidenciadas
na extração e determinação de resíduo de pesticidas em frutas e vegetais.
Principal atenção é dada à simplificação, miniaturização, e maior eficiência
nos métodos de extração da amostra e clean-up. Procedimentos utilizando
extração em fase sólida (SPE)11, microextratores em fase sólida (SPME)12 e clean-
up em fase sólida (SPC), em cartuchos contendo as mais diversas fases sólidas
(polares ou apoiares), vêm sendo usados com o intuito de se aprimorar os
métodos de extração, bem como a extração por fluido supercrítico (SFE)13.
4 P. A . MiIIs, J. Assoe. Off. Anal.Chem., 42 (1959) 7345 P. A. Mills,J.H. Onley and R A. Gaither, J. Assoe. Off. Ana/.Chem., 46 (1963) 1866 R W. Storherr, M.E. Getzand RR Watts, J. Assoe. Off. Ana/.Chem., 47 (1964)10877 M.A. Luke, J.E. Froberg and H.T. Masumoto, J. Assoe. Off. Ana/.Chem., 58 (1975) 10208 L.D. Sawyer, B.M. Mc Mahon, W.H.Newsome and G.A. Parker, in Helrich(Editor), Official methodsof analysis, association of Official chemist Agriculture, Chemistry, contamination,Drugs, Volume1.VA, USA,1990, p.274.9 RT. Krauser, J. Assoe. Off. Ana/.Chem., 63 (1980) 1114.10 US food and drug administration, Pesticides Analytical Methods, Vol. 1, Ch.3 e 4, 1992.Transmital No. 200/1(10/1999), form FDA 290a (6/92)11 G.S. Nunes, M.L.Ribeiro, L.Polese, D. Barceló, J. Chromatogr. A, 795 (1988) 43-5112 K. Hiroyuki, L.L. Heather and P. Jusz, J. Chromatogr. A, 880 (2000) 35-6213 S.J. Lehotay, J. Chromatogr. A. 785 (1997) 289-312.
121

7.1- Algumas considerações sobre extração e c/ean-up
7.1.1- Extração Liquido-Líquido fLLE)
Em LLE, constituintes da amostra são extraídos por solventes imiscíveis na
fase onde as amostras estão contidas. Vários solventes orgânicos voláteis são
utilizados, incluindo pentano, hexano, éter etílico, acetato de etila, clorofórmio e
diclorometano. No caso dos métodos multiresíduos, o solvente extrator terá que
ser propício para extrair compostos com um grande intervalo de polaridade, de~'
várias matrizes contendo quantidades diferentes de água, gordura, açúcar e
outras substâncias. O caminho usual para extrair os 'resíduos de amostras é a
desintegração da matriz em homogeneizadores de alta velocidade (ex. Ultra
Turrax ou equivalente), na presença de solvente ou mistura de solventes. Os
solventes mais utilizados são acetona, acetonitrila (ACN) e o metanol (MeOH). A
vantagem da ACN é que compostos muito lipofílicos presentes nas matrizes, como
gordura, não são extraídos, diminuindo a quantidade de co-extratores. As
desvantagens da ACN são seu alto preço e toxicidade. A acetona é largamente
usada para a extração de diferentes tipos de pesticidas, e é escolhida pelas
vantagens que apresenta em relação a ACN, MeOH ou acetato de etila (EtOAC),
que são, baixa toxicidade, fácil purificação, evaporação e baixo custo. No entanto,
por apresentar baixo ponto de ebulição (56,5 °C) pode, por causa da evaporação,
causar erros de quantificação. Após a homogeneização da amostra com os
solventes adequados, o extrato é filtrado e o procedimento LLE é aplicado,
visando isolamento e pré-concentração, bem com um clean-upparcial dos analitos
de interesse.
122

o clean-up é um procedimento realizado que visa a remoção dos
compostos co-extraídos, após a maceração da matriz com um solvente orgânico
apropriado. A não realização deste procedimento pode interferir em determinações
cromatográficas ou ocasionar danos à instrumentação analítica, no caso de
colunas cromatográficas. Este procedimento envolve um ou mais passos
utilizando LLE, extração cromatográfica com permeação em gel (GPC), extração
cromatográfi~ sobre colunas empacotadas com diferentes sorventes ou extração
em fase sólida (SPE).
7.1.2 - Extração em fase sólida
Após o uso de LLE para isolar e pré-concentrar os componentes da
amostra, o uso de uma extração em um sistema de fase sólido-líquido é
necessário. Na literatura ele é freqüentemente denominado como extração em
fase sólida (SPE).
SPE é uma técnica simples de preparação de amostra baseada nos
conceitos da cromatografia líquida14.
Os analitos de interesse são retidos na fase sólida conforme é descrito na
Tabela 7.1.
~. 14 J.S.Fritz and M.Macka, , J. Chromatogr. A, 902 (2000) 137-166
~ 123

Tabela 7.1 - Processo de retenção dos analitos.
1) Fase Reversa (fase móvel polar, fase estacionária não polar)
Interações hidrofóbicas do tipo:
não-polar/não-polar
van der Waals ou forças de dispersão
2) Fase Normal ( fase móvel não-polar, fase estacionária polar)
Interações do tipo hidrofilica
polar/polar
pontes de hidrogênio
1[-1[
dipolo-dipolo
dipolo induzido/dipolo
3) Troca Iônica
Atrações eletrostáticas dos grupos carregados do analito com os grupos carregados
da superfície do material da fase estacionária.
4) Adsorção
Interações do tipo hidrofóbica-hidrofílica podem ocorrer e dependem da natureza
química da fase estacionária
Separações (ou extrações) em fase reversa envolvem um soluto polar
(geralmente aquoso) ou moderadamente polar e uma fase estacionária não-polar.
O analito de interesse é moderadamente não-polar. Vários dos materiais que
constituem a fase reversa são sílicas do tipo alquil ou aril (como por exemplo, C1a>
Ca). Os grupos silanóis hidrofílicos que constituem a superfície da sílica usada no
recheio da coluna (geralmente com tamanho de poro de 60 Acom partículas de 40
124

Ilm de tamanho) são modificados quimicamente com grupamentos funcionais de
natureza hidrofóbica como os grupos alquil e aril, conforme a seguinte reação:
CH3 CH3
-Si-oH+CI-Si-618H37--.~ -Si-o-Si-618H37 + HCI
bH3 bH3
A retenção dos analitos orgânicos na fase estacionária é devida
primeiramente às forças de interação entre as ligações carbono-hidrogênio do
analito e os grupos funcionais da superfície da sílicp. Estas interações não-
polar/não-polar envolvem interações do tipo forças de van der Walls. Para eluir o
analito que ficou retido na fase estacionária deve-se usar um solvente
moderadamente não-polar ou polares para romper as interações feitas entre a
fase estacionária e o analito de interesse,
Separações (ou extrações) em fase normal envolvem um analito polar a
" moderadamente não-polar e uma fase estacionária polar. Sílicas com
grupamentos funcionais (ciano, amino e diol), constituem um meio polar de
adsorção (Florisil, Alumina, etc.) e são tipicamente utilizadas sob condições de
fase normal de eluição. A retenção de um analito em fase normal é devida às
interações entre os grupos funcionais polares do analito com os grupos polares da
fase estacionária. Estas interações incluem pontes de hidrogênio, interações do
tipo 1t - 1t, dipolo-dipolo e dipolo induzido entre outros. Um composto adsorvido
através destes mecanismos é eluído com um solvente mais polar que o solvente
original da matriz.
125

As sílicas com grupamentos amino, ciano e diol possuem grupamentos
alquil que estão ligados aos grupamentos funcionais polares da superfície da
sílica. Estas sílicas, devido a estes grupamentos funcionais são muito mais
hidrofílicas do que as sílicas utilizadas em fase reversa. Assim como as sílicas de
fase normal, estes recheios podem ser utilizados para adsorver compostos polares
de matrizes não-polares. Estes materiais podem ser usados sob condições de
fase reversa (qom amostras aquosas), para explorar as propriedades hidrofóbicas
das pequenas cadeias alquil de geometria cadeira ligadas aos grupos funcionais.
Na Tabela 7.2 algumas características dos solventes orgânicos comumente
utilizados em SPE são mostradas.
126

NãoNão
SimMuito pouco
SimSimSimSimSimSimSimI fortefracaPolar
Não-Polar
Tabela 7.2 - Características dos solventes utilizados em SPE_, ._... ..... ~ _ ..... , ._.....~_.__ r · __ _ ••__
Polaridade Fase reversa Fase normal Solventes Miscível em...._ __ _ __.. _. .._.. .. .._..__..__._ __._.~.9.~!i'~ .
forte fraca Hexano nãoIsooctano Não
Tetracloreto de Nãocarbono
ClorofórmioCloreto demetileno
(diclorometano)Tetraidrofurano
Éter dietílicoAcetato de etila
AcetonaAcetonitrilaIsopropanol
MetanolÁgua
Ácido acético
Muitas extrações MRMs possuem um passo de clean-up usando colunas
adsorventes10 com as seguintes fases estacionárias: Florisil, Alumina e Sílica gel.
Muitas colunas adsorventes apresentam boa eficiência para promover clean-up,
somente quando são eluídas com solventes de baixa polaridade. Aumentando-se
a polaridade do solvente maior será a quantidade de interferentes eluídos e menor
será a eficiência do clean-up.
Florisil e Alumina são bastante utilizados nas análises de resíduos de
pesticidas, em matrizes que apresentem altos teores de gordura, pois retém
preferencialmente alguns lipídios.
Em geral, sílica gel não é tão eficiente quanto Florisil e Alumina no que diz
respeito à separação dos componentes co-extraídos da matriz de interesse15.
15 A. Ambrus and H.P.Thier. Pure Appl. Chem., 58 (1986)1035
127

't
"
Muitos autores16,17,18 usaram colunas ou mini-colunas de sílica gel para c1ean-up
de extratos de amostras.
Misturas de sorventes também são usadas, combinando sorventes com
propriedades hidrofílicas e lipofílicas, como é o caso das colunas de sílica e
carbono ou misturas de carbono com MgO e celite. Petersen et a1. 19, usaram uma
coluna de clean-up contendo carvão ativado - MgO - Celite, para a determinação
por GC (cromatografia gasosa), de pesticidas organofosforados em chá preto,
após serem extraídos em acetato de etila. Nunes et al11 realizaram comparações
entre vários procedimentos de extração incluindo mistura de fases, para amostras
de cenoura e batata, para pesticidas das classes dos carbamatos,
Na literatura também são apresentados trabalhos com sorvente a base de
carbon02o,21. O GCB (do inglês, graphitized carbon black) é o maior representante
destes sorventes. Torres et al.22 realizaram um trabalho de comparação entre
octadecilsílica e GCB como material para extração em fase sólida de fungicidas e
inseticidas em frutas e vegetais.
7.2 - Procedimento utilizado para os estudos preliminares em SPE (solid
phase extration)
Foram avaliadas as seguintes fases: CN, NH2, Florisil, C18, Charcoal:celite
(1:4 mIm). Os cartuchos de SPE contendo fase reversa (NH2, C18) e fases
normais (CN, NH2, Florisil), foram adquiridos junto à Phenomenex. Todos os
16 F.J. Schenck and R.Wagner., Food Addit. Contam., 12 (1995) 53517 P.Armishaw and R.G.Millar, J. AOAC Int., 76 (1993) 131718 G.E.Miliadis, P.A. Siskos and G.S.Vasilikiotis, J. AOAC, 73 (1990) 43519 J.H.Petersen and K.G.Jensen, Z. Lebensm. Unters. Forsch., 182 (1986) 48920 M.battista, A. di Corcia and M. Marchetti, Anal. Chem., 61 (1989) 93521 A. Lagana, A. Marino, G.Fago and B.P.Martinez, Analusis, 22 (1994) 6322 C.M. Torres, Y.Picós and J.Manes, J. Chromatog. A., 778 (1997) 127-137
128

cartuchos continham 1 9 dos adsorventes. A mistura de sorventes Charcoal:celite
foi feita em um almofariz com o auxilio de um pistilo, usando-se a proporção de 1
para 4 em massa, respectivamente. Foram usados 1,5 9 da mistura para a
realização dos experimentos. Transferiu-se a mistura para uma coluna de vidro
(25 cm x 1,0 em), contendo uma frita de pirex de 1 em e 1 cm de sulfato de sódio
anidro foi adicionado no fundo da coluna. Sobre a mistura adicionou-se mais 4 9
de sulfato de sódio anidro e cobriu-se com lã de vidro. As fases consideradas não
polares C18 e NH2 foram condicionadas com 20 mL de MeOH e 10 mL de H20
desionizada. As fases polares NH2, CN, Florisil e Charcoal:celite foram
condicionadas com 20 mL de éter de petróleo, ambas a uma vazão de 3 mL min-1.
Sobre os cartuchos e colunas pré-condicionadas adicionou-se 1 mL de uma
solução padrão contendo os 12 pesticidas (1-metomil, 2-aldicarb, 3-carbendazin,
4-carbofuran, 5-simazina, 6-atrazina, 7-carbaril, 8- propazina, 9-diuron, 10-linuron,
11-pirazofós, 12-clorpirifós) na concentração de 10 mg/L. Para a eluição dos
pesticidas em cada uma das fases sorventes foram utilizados 2 tipos de eluentes
diferentes:
1) MeOH
2) 2) MeOH:CH2Cb (1 :99 v/v)
Para cada fase sorvente testada foram realizados três ensaios e em cada
ensaio, separadamente, utilizou-se um dos solventes de eluição, exceto para a
fase charcoal:celite que foi eluida somente com o eluente 2. O volume utilizado
dos solvente para eluição foi de 7 mL, a uma vazão de 3 mL min-1. Após a
passagem da mistura de pesticidas pelo cartucho ou coluna, este foi seco, através
de um fluxo de nitrogênio por 5 mino Após a eluição, os eluatos foram evaporados
129

até a secura e ressuspendidos em 1 mL de uma solução contendo 20% de MeOH
em água. As separações eletroforética foram realizadas em um eletrólito de
separação composto por 45 mmol/L de SOS, 10 mmol/L de TBS, 2 mmol L-1 Brij
35®, 10 % MeOH e 12 mmol L-1 de tm-f3-CO. O tempo de injeção foi de 10 s /2,5
kPa. Na Tabela 6.3 são mostrados os resultados de recuperação obtidos para
cada fase sorvente.
130

.,..
Tabela 7.3 - Recuperação para os pesticidas estudados na extração em fase sólida.
Material SE Recuperação (%)Idsorvente metomil aldicarb carbofuran carbaril carbendazin sunazma atraziana propazina diuron linuron pirazofós clorpirifós
! (NP) 1 59,6 90,1 59,1 30,0 29,0 34,2 37,0 38,1 52,5 44,9 6,30 nd2 34,17 99,1 57,4 25,5 31,1 34,5 41,6 41,5 52,6 47,1 5,01 nd
! (RP) 1 21,3 57,6 33,8 15,1 20,9 21,3 24,9 27,9 34,7 31,2 4,50 nd2 47,3 82,4 55,9 42,0 31,1 31,6 35,6 27,3 44,0 38,8 4,01 nd
(NP) 1 23,2 71,4 42,7 19,0 nd .28,3 33,3 38,6 36,7 33,0 nd nd2 51,6 93,2 54,4 18,2 nd 35,6 45,3 48,8 53,2 46,8 nd nd
'Ísil 1 62,2 62,7 38,4 9,7 35,0 25,0 29,7 32,4 35,6 31,5 nd nd2 44,1 75,1 41,3 13,1 16,7 13,0 22,8 21,0 37,9 20,69 nd nd
rcoal:celite 2 100 100 80,8 49,8 nd 70,4 77,4 nd nd nd nd nd1 29,9 100 98,1 45,6 16,0 57,7 62,9 73,2 76,6 70,5 5,20 nd2 69,5 100 93,3 30,8 11,4 29,5 48,7 47,1 82,8 69,0 4,20 nd
SE, Solução eluente: (1) MeOH; (2) MeOH:CH2Ch (1:99 v/v); nd : não detectado
1 ':11

É possível notar através da Tabela 7.1 que a pior performance na
recuperação ficou a cargo dos pesticidas da classe do organofosforados (pirazofós
e clorpirifós), ficando com valores de recuperação abaixo de 6,5%, sendo que o
clorpirifós não foi recuperado em nenhuma das fases e eluentes avaliados. A
porcentagem recuperada para os pesticidas da classes das triazinas (simazina,
atrazina e propazina) ficou entre 13 e 77,4 %, onde o menor resultado foi obtido,
quando o Florisil foi usado como fase adsorvente tendo como eluente o
MeOH:CH2Cb. O charcoal:celite apresentou o melhor resultado para as triazinas
(70,4 % para a simazina e 77,4 % para a atrazina), porém não houve recuperação
para a propazina. Para os pesticidas da classe do carbamatos (metomil, aldicarb,
carbofuran e carbaril), os melhores resultados foram obtidos para o aldicarb para
as fases sólidas Charcoal:celite e C18, onde este foi 100% recuperado. Para o
metomil o melhor resultado foi obtido quando fase charcoal:celite foi usada (100%
recuperado). Carbofuran obteve resultados satisfatórios de recuperação para C18e
chacoal:celite. Para o carbaril em geral as recuperações foram baixas oscilando
entre 9,7 % a 49,8. O pior resultado foi obtido quando se usou Florisil com fase
sorvente e metanol com eluente; 49,8% foi obtido para charcoal:celite como fase
sorvente. O Carbendazim, da classe dos benzoimidazóis, apresentou resultados
de recuperação bem abaixo do esperado, oscilando entre 11,4 % e 35,0%, não
sendo detectado quando se usou CN e charcoal:celite como fases sorventes. Para
diuron e linuron, da classe da uréia, foram obtidos os melhores resultados quando
se usou C18 como fase sorvente.
132

o charcoal:celite apresentou bom resultados quando comparado a outras
fases sólidas, entretanto foi somente possível a recuperação da metade dos
pesticidas existentes na mistura padrão contento os 12 pesticidas.
De uma maneira geral (salvo algumas exceções), a maioria das
porcentagens de recuperações obtidas estão abaixo de 70%. Este problema
poderia estar ligado ao fato de que a quantidade de eluente utilizado não foi
suficiente para eluir todos os pesticidas da fase sorvente. Então optou-se em
realizar um novo experimento onde, a quantidade de eluente utilizado seria duas
vezes maior do a utilizada até então. Utilizou-se C18 como fase sorvente,
previamente condicionado e MeOH:CH2Ch (1 :99 v/v) como eluente. Adicionou-se
1,0 mL de uma solução padrão contendo os 12 pesticidas citados anteriormente,
na concentração ~e 10 mg/L. Na Tabela 7.4 são apresentados os resultados de
recuperações obtidos.
133

Tabela 7.4 - Valores de porcentagem de recuperação obtidos para os 12
pesticidas utilizando C18 como fase estacionária.
Pesticidas Recuperação (%)
7 mL 14 mL
metomil 69,5 62,3
aldicarb 100 94,5
carbendazin 11,4 22,0
carbofuran 93,3 93,1
simazina 29,5 32,3
atrazina 48,7 65,3
carbaril 30,8 45,2
propazina 47,1 67,5
diuron 82,8 84,5
linuron 69,0 82,5
pírazofós 4,2 8,60
Através da Tabela 7.4 é possível notar que com o aumento do volume do
eluente, a quantidade de pesticida recuperado aumentou para quase todos os
pesticidas investigados. Porém para os pesticidas carbendazim, simazina,
atrazina, carbaril, propazina e pirazofós continuam apresentando valores de
recuperação relativamente baixos.
Outra hipótese levantada para explicar a baixa recuperação foi a possível
saturação da fase sorvente pelos pesticidas. Esta hipótese foi avaliada realizando
um experimento onde um cartucho NH2 foi acoplado em "cascata" a um cartucho
de CN. Foram utilizados os cartuchos Sep-Pak® Plus CN e NH2 (750 mg) da
marca Waters, devido ao seu formato permitir uma fácil conexão entre os
cartuchos; além disto CN e NH2 eram as únicas fases sólida com este formato
134

disponíveis no laboratório para realização deste teste. Já os cartuchos da marca
Phenomex, em formato de seringa, não permitiam tais adaptações. Os cartuchos
NH2 (parte superior) e o CN (parte inferior) foram condicionados previamente com
20 mL de CH2Cb, e em seguida adicionou-se 1,0 mL de solução padrão contendo
os 12 pesticidas na concentração de 10 mg/L ao topo da "cascata". Os cartuchos
foram eluidos separadamente com 7 mL de MeOH:CH2Cb (1 :99 v/v). Os eluatos
foram evaporados e ressuspendidos em solução contendo 20% de metanol em
água.
Na Tabela 7.5 são mostradas as porcentagens de recuperação para os
pesticidas, para cada etapa da eluição em cascata.
Tabela 7.5 - Porcentagens de recuperação para as eluições em cascata.
Pesticidas Recuperação (%)
NH2(SUP) CN(inf) Fração não Total
retida
metomil 48,1 25,4 8,0 81,5
aldicarb 65,6 36,1 nd 100
carbendazin 43,6 25,1 0,2 68,7
carbofuran 49,3 32,5 3,7 85,5
simazina 53,1 25,3 7,1 85,4
atrazina 50,5 25,2 7,0 82,7
carbaril 62,9 30,2 7,0 99,5
propazina 46,9 24,2 7,2 78,3
diuron 54,9 25,8 6,2 86,9
linuron 51,0 24,6 7,0 82,6
pirazofos 8,0 nd nd 8,0
nd - não detectado
135

Analisando a Tabela 7.5 fica evidenciado a saturação das fases
adsorventes, onde a parte superior da cascata (NH2) apresentou os maiores
valores de recuperação, diminuindo para a fase subseqüente.
Baseado nas evidências da experiência anterior foram feitas novos ensaios,
agora adicionando-se 0,5 mL de uma solução 5 mg L-1 sobre as fases sorventes
(não utilizando o artifício da cascata). Foram utilizadas como fases sorventes o C18
(fase reversa - pré-condicionada com 20 mL MeOH e 5 mL de água) e NH2 (fase
normal - pré-condicionada com 20 mL de CH2Cb). Cartuchos da marca Phenomex
com 1 g de fase sólida foram utilizados para este teste. Os pesticidas foram
eluídos com 8 mL de MeOH:CH2Cb (1 :99 v/v). Os eluatos foram evaporados até a
secura e ressuspendidos em 0,5 mL de uma solução contendo 20% de metanol
em água.
Na Tabela 7.6 são apresentados os valores de recuperação obtidos em
duplicata de realizações.
136

Tabela 7.6 - Recuperação dos padrões de pesticidas, para 0,5 mL de mistura
padrão 5 mg/L, utilizando como eluente 8 mL de MeOH:CH2Cb (1 :99 v/v).
pesticidas Recuperação (%)
C18 NH2
metomil 95,7±3,5 82,0±4,2
aldicarb 72,8±4,0 92,0±4,2
carbendazin 64,1±2,5 79,1±1,2
. carbofuran 79,3±O,7 78,O±2,8
simazina 76,6±1,2 71 ,5±2,1
atrazina 82,5±3,5 74,O±10,O
carbaril 85,1±1,2 72,0±5,8
propazina 77,0±6,8 77,6±5,O
diuron 83,1±2,6 86,7±4,0
linuron 82,0±4,1 70,5±6,2
pirazofos 8,O±O,8 12,O±O,9
clorpirifos nd nd
nd - não detectado
Analisando a Tabela 7.6 e comparando-a com a tabela 7.3 é possível notar
sensível aumento na porcentagem de recuperação para os pesticidas analisados,
após a diminuição da quantidade de amostra inserida nos cartuchos.
Com base nos dados apresentados nesta seção nota-se a vulnerabilidade
dos cartuchos SPE, para uso em procedimentos de clean-up, pois ao se tratar de
amostras reais além dos compostos de interesse a matriz apresenta também
compostos que apresentam afinidade pela fase estacionária, podendo
137

potencializar o efeito da saturação. É necessário cuidado redobrado para avaliar a
quantidade e volume de amostra adicionados aos cartuchos.
7.3 - Métodos de Extração e clean-up
Foram selecionadas 5 metodologias de extração e c1ean-up da literatura.
Para a escolha, levou-se em consideração a aplicabilidade, simplicidade e tempo
de extração. Alguns procedimentos de extração sofreram modificações sendo as
principais modificações: (i) Massa de amostra, (ii) Quantidade de solvente utilizado
para as extrações, (iii) Quantidade de solvente utilizado na etapa de extração
líquido-líquido, (iv) Em alguns casos, substituição da fase estacionária para
realização de clean-up. A seguir serão descritos os procedimentos utilizados. Foi
empregada para a avaliação das extrações uma amostra "testemunho" de
cenoura. Para todas as metodologias de extração avaliadas utilizou-se da mesma
amostra de cenoura.
7.3.1 - Método I
Extração
Este procedimento baseou-se na metodologia proposta por G.S.Nunes et
al. 11 Inicialmente a amostra foi congelada (-20°C) e reduzida a pó por intermédio
de um ralador doméstico e, posteriormente, homogeneizada. Pesou-se 10 g do
material homogeneizado, adicionou-se 20 mL de MeOH e 10 mL de CH3CN,
agitando-se vigorosamente no Turrax por 5 min cada etapa (22000 rpm).
Centrifugou-se, e o extrato orgânico foi levado a um funil de separação, e a este
adicionou-se 0,5 g de NaCI e extraiu-se com duas alíquotas de 10 mL de éter de
138

petróleo (ETP), guardou-se a fase etérea. O extrato orgânico foi reextraido com
duas alíquotas de 10 ml de CH2CI2, adicionando-se a este 10 mL de solução
saturada de NaCI. Coletou-se a fase contendo o CH2Cb. Evaporou-se até
aproximadamente 1 mL, com fluxo de nitrogênio e em banho de aquecimento a 35
oCo
C/ean-up
Cartuchos contendo 1 g de fase estacionária C18 foram utilizados nos
procedimentos de clean-up. Os cartuchos foram adaptados ao sistema Manyfold
Supelco Visipred™ DL, 12 portas. Os cartuchos foram condicionados com 20 mL
de metanoI e posteriormente com 5 mL de H20, a uma vazão de 3 mUmin. Nesta
etapa é imprescindível que a fase sólida do cartucho não seque. A amostra
evaporada na etapa anterior até cerca de 1 mL foi adicionada aos cartuchos. Após
o término da passagem da amostra através do cartucho, este foi seco com um
fluxo de nitrogênio por 5 minutos. Os compostos de interesse foram eluídos do
cartucho com 6 mL de CH2Cb:MeOH (99: 1 v/v). O eluato foi evaporado até a
secura e ressuspendido em 1 mL de uma solução de 20 % de MeOH em água.
Na Figura 7.1 é mostrado um esquema sumarizando cada etapa.
139

10 g Amostra+
10 mLACN20 mL MeOH
Homogeneização (5 mino cI Turrax)Centrifugação
IFase orgânica(sobranadante)
II Resíduo I
II 10 + 10 mL ETP
IDescarte
ILLE 0I
1 10 + 10 mL CH2Cb I
N2 até 1 mLSPE C18Eluir cl6 mL CH2CI2:MeOH (99:1 v/v)
IEluato IN2 até secura1mL MeOH/água (20:80 v/v)
I Análise
Figura 7.1 - Esquema de extração para o Método I.
Na Figura 7.2 são mostrados os eletroferogramas obtidos para a extração
de uma amostra de cenoura, fortificada com 150 llL de uma mistura padrão
contendo (dimetoato, carbendazim, atrazina, carbaril, ametrina, propazina, linuron
e propoxur), na concentração de 50 mg/L. Foi utilizado como eletrólito de
separação: 50 mmol/L de SDS, 10 mmol/L TBS, 5% PrOH e 5% ETOH.
140

10
8 5
6 3 44 8::J
~ 2
O
-2
-4
10 O 2 4 6 8 10 12 14 16
8
6
:::> 4
~#
2
O
-2
O 2 4 6 8 10 12 14 16
Tempo, min
Figura 7.2 - Método I aplicado à amostra de cenoura. (A) Cenoura in natura, (B)
Cenoura fortificada com 0,15 mL de mistura de pesticida padrão (50 mg L-1).
Eletrólito de separação: 50 mmol L-1 de SDS, 10 mmol L-1 TBS, 5 % MeOH e 5%
PrOH. Outras condições: À =220 nm, tempo 25 DC, comprimento efetivo do capilar:
50 cm. Tensão aplicada: 27 kV, Inj. 1Os/2,5 kPa.1) dimetoato, 2) carbendazim, 3)
simazina, 4) atrazina, 5) carbaril, 6) ametrina, 7) propazina, 8) linuron I 9) propoxur
e #) não identificado
141

7.3.2 - Método 11
Extração
Este procedimento de extração foi baseado no Bulletim 900B - Supelco23.
Em um béquer de 250 mL pesou-se 50 g de amostra e homogeneizou-se com 100
ml de ACN, usando Turrax por 5 minutos, em velocidade máxima (22000 rpm).
Logo após, adicionou-se 8 g de NaCI e homogeneizou-se novamente por 5
minutos. Transferiram-se 13 mL do sobrenadante para um tubo de centrífuga com
capacidade de 15 mL, e adicionaram-se 3 g de sulfato de sódio anidro granular·
para desidratação, agitou-se manualmente. Centrifugou-se por 5 minutos em alta
velocidade (3500 rpm). Transferiram-se 15 mL do centrifugado para um tubo de
ensaio e evaporou-se até um volume aproximado de 0,5 mL, sob fluxo de
nitrogênio.
Clean-up
Este volume reduzido foi transferido para um cartucho C18, previamente
condicionado com 20 mL de MeOH e 5 mL de H20. A amostra passou através do
cartucho a uma vazão de 3mLlmin (o cartucho estava adaptado ao sistema
Manyfold supelco Visipred™ DL). A amostra foi eluída com 6 mL de MeOH:CH2Ch
(1 :99 v/v). O eluato foi evaporado até a secura e ressuspendido em 1 mL de uma
solução contendo 20% metanol em água.
Na Figura 7.3 é mostrado um esquema sumarizando cada etapa.
23 Sigma-Aldrich (SUPELCO)- Bulletin 900B - Tl96900B - 1997 - (http:\sigmaaldrich.com/saws.nsf/supproducts?openframeset)
142

50g amostra+
100 mL ACN
Fase orgânica(sobrenadante)
3g de Na2S04 anidroagitação manualcentrifugação
Fase orgânica(sobrenadante)
N2 até 0,5 mLSPE C18Eluir cl7 mL CH2CI2:MeOH (99:1 v/v)
I eluato IN2até secura1 mL MeOH/água (20:80 v/v)
.--1---,IAnálise I I
II resíduo I
IIresíduo I
Figura 7.3 - Esquema de extração para o Método 11.
Na Figura 7.4 são mostrados os eletroferogramas obtidos para a extração
de uma amostra de cenoura, fortificada com 1,14 mL de uma mistura padrão
contendo (dimetoato, carbendazim, atrazina, carbaril, ametrina, propazina, linuron
e propoxur), na concentração de 50 mg L-1. Foi utilizado como eletrólito de
separação 50 mmol L-1 de SOS, 10 mmol L-1 TBS, 5% PrOH e 5% ETOH.
143

15
10
B 5
161412108642o
o 2 4 6 8 10 12 14 16 1815
#10
::>5 A
~o
Tempo, min
Figura 7.4 - Método 11 de extração aplicado à amostras de cenoura. (A) Cenoura
in natura, (B) Cenoura fortificada com 1,14 mL da mistura de pesticida padrão (50
mg L-1). Eletrálito de separação: 50 mmol L-1 de SOS, 10 mmol L-1 TBS, 5 %
MeOH e 5% PrOH. Outras condições: À =220 nm, tempo 25°C, comprimento
efetivo do capilar: 50 cm, Tensão aplicada: 27 kV, Inj. 10s/2,5 kPa. #)
desconhecido, 3)simazina, 4) atrazina, 5) carbaril, 6)ametrina, 7) propazina, 8)
linuron e 9) propoxur
144

7.3.3 - Método 111
Extração
Este procedimento de extração baseou-se na metodologia proposta por
Minelli et al.24 Pesou-se 15 g da porção homogeneizada da amostra, adicionou-se
30 mL de uma mistura de solventes contendo acetona, éter de petróleo e
diclorometano na proporção de 1:1:1 (v/v) e 4 g de NaCI. A mistura foi sonificada
por 15 minutos. A extração foi realizada mais uma vez. As fases orgânicas foram
transferidas para 4 frascos de centrífuga, de 15 mL cada frasco; foram
adicionados 2 g de NaS04 anidro granular para desidratação, agitou-se
manualmente. O material foi centrifugado por 3 minutos na velocidade máxima
(3500 rpm). Uma alíquota 20 mL do extrato orgânico seco foi transferido para um
tubo de ensaio com tampa, de capacidade de 25 mL, e evaporado até próximo de
1 mL, sob fluxo de nitrogênio e em banho de aquecimento a 35°C.
Clean-up
O extrato evaporado foi colocado em cartucho contendo 1 g de fase
estacionária NH2, previamente condicionado com 25 mL de CH2Ch (adaptado ao
sistema manyfold). Após a passagem da amostra pelo cartucho, este foi seco com
um fluxo de nitrogênio. Os compostos de interesse foram eluídos com 7 mL de
CH2Ch:MeOH (99:1). O eluato foi evaporado até a secura e ressuspendido em 1
mL de uma solução contendo 20 % metanol em água.
No procedimento descrito por Minelli et aI.25, os autores sugerem a agitação
da amostra com a mistura de solventes para a extração dos compostos. Mas
24 E.Y.MineUi, A. Angioni, M.Melis, F.M.Flippo andP.Cabras, J AOAC Inter. 80 (1997) 1315
145

segundo Babic et al,25 o processo de sonificação da amostra com o solvente é
mais eficiente. Outras alterações foram realizadas como na pesagem da amostra
que era de 5 g foi alterada 15 g, a quantidade de mistura de solventes extratores
que era de 15 mL passou a 30 mL, bem como a fase sólida sugerida pelos autores
que era cianopropil, foi substituída por aminopropil. A etapa na qual adiciona-se
Na2S04 para secagem dos solventes de extração também não existia no
procedimento original. Na Figura 7.5 é apresentado um esquema sumarizando as
etapas da extração.
25 S.Babic, M.Petrovic and M.Kastean-Macan, ,1. Chromatog. A, 823 (1998) 3-9
146

15 de amostra+
30 mL de acetona:CH2CI2:ETP (1:1:1 v/v)+
4g de NaCI
Sonificação, 15 minocentrifugação
IFase orgânica(sobrenadante)
1
8g de Na2S04 anidrocentrifugação
Fase orgânica(sobrenadante)
II resíduo I
II resíduo I
N2 até 1 mLSPE NH2
Eluir c/ 7 mL CH2Cb:MeOH (99:1 v/v)
I eluato IN2 até secura1 mL MeOH/água (20:80 v/v)
I Análise I
Figura 7.5 - Esquema de extração para o Método 111.
Na Figura 7.6 são mostrados os eletroferogramas obtidos para a extração
de uma amostra de cenoura, fortificada com 150 IlL de uma mistura padrão
contendo (dimetoato, carbendazim, atrazina, carbaril, ametrina, propazina, linuron
e propoxur), na concentração de 50 mg L-1. Foi utilizado como eletrólito de
separação 50 mmol/L de SOS, 10 mmol L-1 TBS, 5% PrOH e 5% ETOH.
147

10
8 56
3 4 67
:J4 8
<:( 2E
o-2
-4
10 O 2 4 6 8 10 12 14 16
8
6
::::> 4 A<:( 2E
o-2
-4O 2 4 6 8 10 12 14 16
Tempo, min
Figura 7.6 - Método 111 de extração aplicado à amostras de cenoura. (A) Cenoura
in natura, (B) Cenoura fortificada com 0,15 mL de mistura de pesticida padrão (50
mg L-1). Eletrólito de separação: 50 mmol L-1 de SOS, 10 mmol L-1 TBS, 5 %
MeOH e 5% PrOH. Outras condições: À =220 nm, tempo 25 DC, comprimento
efetivo do capilar: 50 em. Tensão aplicada: 27 kV, Inj. 10s/2,5 kPa, 1)dimetoato, 2)
carbendazim, 3)simazina, 4) atrazina, 5) carbaril, 6)ametrina, 7) propazina, 8)
linuron e 9) propoxur
148

7.3.4 -Método IV
Extração
Este procedimento de extração baseou-se na metodologia proposta por
Niessner et al26 . Pesou-se 10 g da porção homogeneizada da amostra,
adicionou-se 66 mL de acetona, e homogeneizou-se com o auxílio do Turrax (5
min), água foi adicionada para obter um volume de 100 mL e a mistura foi
novamente homogeneizada com o Turrax (+ 5 min). Adicionou-se 3 g de celite não
silanizada e o extrato foi filtrado em funil de buchner (filtrado em papel rápido), 1O
mL do filtrado foi evaporado para 1 mL.
Clean-up
A amostra final foi passada através de um cartucho C18, previamente
condicionada com 20 mL de MeOH e 5 mL de H20. Os compostos de interesse
foram eluídos com 6 mL de CH2Ch:MeOH (99:1). O eluato foi evaporado até a
secura e ressuspendido em 1 mL de uma solução contendo 20% metanol em
água. Na Figura 7.7 é apresentado um esquema sumarizando as etapas de
extração.
26 G.Niessner, W.Buchberger and R. Eckerstorfer, 1. Chromatog. A 846 (1999) 341-348
149

10g de amostra+
66 mL de acetona+
44 mL de água
Homogeneização c/ Turrax& (10 min)3g de celitefiltração
IFase orgânica
(filtrado)
Secar até 1 mLSPE C18Eluir c/ 6 mL CH2CI2:MeOH (99:1 v/v)
I eluato I
N2 até secura1 mL MeOH/água (20:80 v/v)
,-----L.....--,
IAnálise I
Iresíduo I
Figura 7.7 - Esquema de extração para o Método IV.
Na Figura 7.8 são mostrados os eletroferogramas obtidos para a extração
de uma amostra de cenoura, fortificada com 1,5 mL de uma mistura padrão
contendo (dimetoato, carbendazim, atrazina, carbaril, ametrina, propazina, linuron
e propoxur), na concentração de 50 mg L-1. Foi utilizado como eletrólito de
separação 50 mmollL de SDS, 10 mmol/L TBS, 5% PrOH e 5% ETOH.
150

15 # 5
10 # 3 467
# # 8::J
~5
O
20 O 2 4 6 8 10 12 14 16
15
10::J
~ 5
O
-5O 2 4 6 8 10 12 14 16
Tempo, min
Figura 7.8 - Método IV de extração aplicado à amostras de cenoura. (A) cenoura
in natura, (B) Cenoura fortificada com 1,5 mL de mistura de pesticida padrão (50
mg L-1). Eletrólito de separação: 50 mmol L-1 de SDS, 10 mmol L-1 TBS, 5 %
MeOH e 5% PrOH. Outras condições: Â = 220 nm, tempo 25 DC, comprimento
efetivo do capilar: 50 cm: Tensão aplicada: 27 kV, Inj. 10s/2,5 kPa.. #)
desconhecido, 3)simazina, 4) atrazina, 5) carbaril, 6)ametrina, 7) propazina, 8)
linuron e 9) propoxur
151

7.3.5 - Métcidu V - Extraçãa Par dispersão. da matriz em fase sólida (MSPD)
A MSPD é uma técnica de clean-up, que está sendo desenvolvida
para análise de MRMsem frutas e vegetars· 27 ,28. A MSPD é um procedimento
simplificado, que permite a eliminação da etapa de LLE. Neste processo a amostra
é homogeneizada com a adição de pequena quantidade de água, quando
necessário. Logo após Tetira.;.se uma pequena parte'do'tlomogeneizado'e agre'ga-
se a este uma quantidade pré-determinada de adsorvente, particularmente Ca, C1a
ou Rorisil, e homogeneiza-se com auxilio de um almofariz e piStilo29. A mistura é
conduzida a uma coluna de vidro e eluída com solventes ou mistura de solventes
apropriados. Muitos procedimentos 'MSPDtambém ·~nvolvem uma coluna
adicional para obter um novo fracionamento e clean-up. O material desta coluna
(f1orisil ou sílica, por exemp1o} pode·' ser' coiocad:o: no, fundo: da:· mesma· coJuna- de
MSPD ou pode ser usado em uma coluna externa adaptada à coluna do MPDS.
.Neste processo, a extração, ·fracionamento e-purificação são -realizadas em uma
única etapa.
Kandenczki et aI.30 demonstraram a apHcabilidade do· MSPD para um
grande número de matrizes de frutas e vegetais visando análise de resíduos de
.pesticidas.
7.3.5'.1 - Procedimento de extração
Este procedimento de extração baseou-se na metodologia proposta por
Ling e Huangl31 traía-se de uma metodologia MSPD. Pesaram-se
27A.di Corcia, SMarchese and R.Samprei, 1 Chromatog. A.,642(1993)16328 A. t Valenzuela, R. LOrenzini, M.J. Redondo, G.Font, J. Chromatogr. A, 839(1999)101-10729 S. A. Baker, lI. Chromatog. A, 885(2000)115-12730 L. Kandenczki, Z.Arpad, 1 Gardi, A. Ambrus, L.Gyorfi, GReese and W.Ebing, , I. AüAC Int,75(1992)53
152

100 9 de amostra. A amostra foi- 1:10mogene~zada em blender até obter-se um
material homogêneo; 2,5 9 da porção homogeneizada da amostra foi transferida
para um almofariz de vidro. Nesta etapa a amostra foi fortificada quando
necessário. Adicionaram-se 5 9 de F1orisi1 previamente ativado (150°C, 24 horas),
e misturou-se gentilmente por aproximadamente 2 minutos. Transferiu-se o
homogeneizqdo para uma co1una de vidro (25 cm-xt;Ocm), contendo uma disco
de v-idrosinterizado de 0,3 mm e sulfato de -sódio anidro foi adicionado no fundo
da coluna até preencher cerca de 1cm. Adicionou-se uma quantidade de Florisil
no fundo da coluna (1,0 9,) sobreposta ao suffato de sódio, que foi condicionada
com 10 mL de diclorometano. Em seguida a amostra foi introduzida. O conteúdo
:dacolunafoi -pressionado com um -embolo de seringa, para uma suave
compactação. Finalmente adicionou-se 0,2 cm de sulfato de sódio anidro sobre o
material empacotado. e cobriu- com lã de vidro. Os compostos de interesse foram
eluídos com 30 mL CH2Cb:MeOH (99:1), sob um fluxo de 3 mL min-1. O eluato foi
evaporado até a secura e ressuspendido em1 mlde uma solução contendo ·20%
metanol em água.
Torres et al.32 exploraram a possibilidade de usar o- MSPD para a
determinação de pesticidas organoclorados e organofosforados em laranja,
usando diferentes fases adsorventes (C18, C8, C2 , CN, Sflica, ftorisH, Alumina).
Stafford e Lin33 descreveram uma metodologia para a determinação de
resíduos de oxamil e metomil em maças e laranjas usando C18 como fase
sorvente.
31 .Y.C.Lmg and I.P.Huang, J. Chromatogr. A, 695(1.995)75-82
32 C.M.Torres, Y.picós, M.J.Redondo and J.Manes, , J. Chromatogr. A, 719(1996)95
153

Este método constitui em uma boa opção de extração, demandando
menores tempos de análise e quantidades de solventes em relação aos métodos
convencionais de extração. As principais vantagens do MSPO são:
(i) Apresenta menor tempo de análise e maior interação entre a matriz e a
amostra. (ii) Evita a formação de emulsão ao redor da partícula do adsorvente
possibilitando maior extração e reprodutibilidade. (iii) Por requerer menores
quantidades de amostra, não há a necessidade de grandes quantidades de
solventes para executar as eluições. Na Figura 7.9 estão sumarizadas as etapas
de extração para o MSPO.
:MeOH (99:1 v/v)
ual com pistilo
0:80 v/v)
2,5 9 de amostra é colocada em almofariz+
5g de Florisil ativada (150 °C/24 Hs)
Homogeneização man
Coluna de vidro (25 x 1 em) preenchida com:1) Na2S04 anidro (3 g)2) Florisil ativado (1 g)
Condicionamento cf
I Adição da amostra à coluna IEluir cf 30 mL CH2CI2
I eluato IN2 até secura1 mL MeOH/água (2
I Análise I
Figura 7.9 - Esquema para o Método MSPO.
154

Na Figura 7.10 são mostrados os eletroferogramas obtidos para a extração
de uma amostra de cenoura, fortificada com 150 IlL de uma mistura padrão
contendo dimetoato, carbendazim, atrazina, carbaril, ametrina, propazina, linuron e
propoxur, na concentração de 50 mg/L. Foi utilizado como eletrólito de separação:
50 mmollL de SOS, 10 mmollL TBS, 5% PrOH e 5% ETOH.
155

15
510
3 46 7
::> 5«E 8 9
O
15 O 2 4 6 8 10 12 14
10
::> 5«E
O
o 2 4 6 8
Tempo, min
10 12 14
Figura 7.10 - Método V (MSPD) de extração aplicada à amostra de cenoura. (A)
cenoura in natura, (B) Cenoura fortificada com 0,15 mL de mistura padrão de
pesticida (50 mg L-\ Eletrólito de separação: 50 mmol L-1 de SOS, 10 mmol L-1
TBS e 5 % MeOH e 5% PrOH.Outras condições: À = 220 nm, tempo 25 DC,
comprimento efetivo do capilar: 50 cm. Tensão aplicada: 27 kV, .Inj. 1Os/2,5 kPa.1)
dimetoato, 2) carbendazim, 3) simazina, 4) atrazina, 5) carbaril, 6) ametrina, 7)
propazina, 8) linuron e 9) propoxur
As metodologias de extração apresentadas neste capítulo foram
comparadas com base nos valores de recuperação. Foram realizadas
recuperações em dois níveis de fortificação (83 ppb e 0,24 ppm). Os resultados
obtidos para cada metodologia de extração são apresentados na Tabela 7.7. Foi
156

realizada duplicata para cada nível de fortificação. Também na Figura 7.11 são
mostrados os gráficos das recuperações obtidas para melhor visualização.
157

Tab
ela
7.7
-V
alor
esde
recu
pe
raçã
opa
raos
mét
odos
dee
xtra
ção
aval
iado
s.
Pes
ticid
asM
étod
oI
Mé
tod
oII
Mét
odo
IIIM
étod
oIV
Mét
odo
V
N1
N2
N1
N2
N1
N2
N1
N2
N1
N2
limet
oato
51
,7±
1,8
48,6
±11
,2-
-90
,5±O
,37
2,1
±8
72,0
±8,O
61
,3±
10
,256
,6±
8,7
100,
4±4,
4
~arbendazim
29,4
±9,
251
,5±
5,5
--
58,0
±5
27,3
±1,
1-
55
,7±
8,0
52,O
±3,3
72,1
±7,
5
iimaz
ina
39,O
±4,
145
,3±
2,8
52,O
±1,8
52,2
±4,
757
,0±
3,7
47
,6±
3,5
60,5
±7,
06
4,2
±5
,389
,0±
7,1
62,8
±5
üra
zin
a74
,6±
7,3
51
,02
,578
,O±2
,459
,5±
3,2
76,4
±163
,16±
163
,0±
11,2
89
,6±
15
,382
,0±
7,2
73,6
±5,
6
~arbaril
93
,6±
156
,6±
3,6
75,1
±6,
563
,2±
10,1
69,5
±1,
56
8,3
±2
,864
,5±
13,2
82,6
±16
,387
,O±7
,185
,5±
2,9
3met
rina
42,4
±8,
245
,2±
282
,8±
3,5
56,7
±21
,261
,5±
4,3
66
,0±
1,7
59,2
±5,
67
0,5
±9
,380
,3±
4,2
66,7
±4,1
)rop
azin
a66
,5±
6,7
50,2
±19
,110
1,0±
10,7
58,2
±4,
471
,2±
0,3
63,5
±1
52,2
±2,
36
9,0
±5
,051
,1±
4,9
67,2
±4
inur
on50
,9±
2,2
47,5
±6,
183
,85±
3,6
77,4
±4,
265
,O±7
,96
9,0
±1
,345
.,0±1
5,3
92
,4±
12
,351
,0±
1,8
84,2
±6,
7
xop
oxu
r63
,4±
9,7
52,1
8±13
,484
,2±
7,2
47,5
±14
,584
,4±
4,3
58,1
±13
,261
,3±
6,3
60
,4±
10
,654
,1±
5,9
76,7
±4,1
N1
83pp
bN
20,
24pp
m
158


Para o Método I os valores de recuperação para o menor nível de
fortificação ficou entre 29,4 % (carbendazin) e 93,6 %(carbaril), enquanto para o
maior nível de fortificação as recuperações ficam em torno dos 50 % para todos os
pesticidas analisados. O Método I de extração apresenta várias etapas de
extração não sendo viável para análises de rotina.
Para o Método 11 não foi possível a recuperação para os pesticidas
dimetoato e carbendazin. As recuperações para os demais pesticidas ficam entre
52,0 % a 101% para ao menor nível de fortificação, enquanto que para o maior
nível os valores de recuperação ficaram entre 47,5 (propoxur) e 77,4% (Iinuron).
O método 111 de extração foi de fácil implementação, onde os extratos
obtidos apresentaram um aspecto límpido. A etapa de evaporação antes do clean
up é fácil, pois o solvente extrator é composto de uma mistura de acetona, éter de
petróleo e diclorometano, que são solventes com baixo ponto de ebulição. Os
valores de recuperação para o menor nível de fortificação ficaram entre 57,0 a
90,5 % e 27,3 a 72,1 % para o maior nível de fortificação. A diminuição nos
valores de recuperação quando o nível de fortificação é aumentado é
provavelmente causado pela saturação do cartucho de SPE. Os valores de %CV
para ambos os níveis de fortificação são relativamente baixos « 15%).
O Método IV de extração apresenta poucas etapas de extração, porém a
etapa de evaporação do extrato antes do clean-up é demasiadamente lenta pelo
fato do solvente extrator ser composto de um mistura de 60 % de acetona em
água. Os valores de recuperação para o menor nível de fortificação ficou entre
160

45,0 % para o linuron e 72,0 % para o dimetoato, porém não foi possível a
recuperação para o carbendazim. Para o maior nível de fortificação os valores
ficaram entre 55,7% para o carbendazin e 92,4 % para o linuron.
O Método V de extração dentre os avaliados é o método mais fácil de ser
implementado. Em média o tempo para se realizar o procedimento de extração
para este método é em torno de 30 minutos, entre homogeneização da amostra
com Florisil e a eluição dos compostos da coluna. As recuperações para o menor
nível de fortificação ficou entre 51,0 % para o linuron e 89 % para a simazina. Já
para o maior nível de fortificação os valores de recuperação ficaram entre 62,8 %
para a simazina e 100,4 % para o dimetoato.
Dada a complexibilidade da matriz analisada e a larga faixa de polaridade
dos pesticidas estudados, os métodos 111 e V apresentaram resultados mais
robustos, além disto são de fácil implementação podendo ser utilizados em
análises de rotina. Segundo o guia de validação elaborado pela
AOAC/FAO/IAENIUPAC (apêndice 1), para concentrações de fortificação que
variam de 0,1 a 1 mg kg-1, o valor de recuperação deve estar compreendido entre
70 - 110 %. O método de extração com MSPD é o que mais se aproxima dos
valores de recuperação estipulado.
161

7.3.6 - Avaliação das metod%gias 111 e V aplicadas a amostras (cenoura e
abacaxi) adquiridas no comércio local
7.3.6.1 Avaliação do método 111 de extração
Amostras de cenoura e abacaxi foram utilizadas para avaliação do método
de extração e clean-up. As amostras de cenoura e abacaxi foram adquiridas junto
ao comércio local. Para este método optou-se em realizar análises da casca e do
interior da cenoura e do abacaxi, analisando-se separadamente cada fração da
amostra. Na Figura 7.12 são mostrados os eletroferogramas obtidos para as
amostras de abacaxi e cenoura para o Método 111.
162

25 25
20 20
15 A 1 15 C1
1010
L~~2 ::::>
« 55 E
OO
-5-5
-10
-10O 2 4 6 8 O 2 4 6 8
Tempo, min Tempo, min
25 25
20 20
15 8 15 O 110
*10
~::::>
5 ~ 5
O O
-5 -5
-10 -10O 2 4 6 8
O 2 4 6 8
Tempo, min Temp, min
Figura 7.12 - Avaliação do Método 111 para amostras de cenoura e abacaxi. (A)
Miolo da cenoura, (8) Casca da cenoura, (C) Casca do Abacaxi e (O) Polpa do
abacaxi. Amostras ressuspendidas em solução de 20% MeOH e 30 mmol L-1 de
NaCI em água. Eletrólito de separação: 50 mmol L-1 de SOS, 10 mmol L-1 de T8S,
5%/5% (PrOH: ETOH), Inj: 20 s/2,5 kPa. Temp. 25°C, comprimento do capilar: 50
cm (efetivo), 1.,=200 nm. 1) dimetoato, 2) carbendazim e * 1- Naftol (produto de
degradação do carbaril)
163

É possível notar através da Figura 6.11 que o interior de cenoura
apresentou 1,7 vezes mais dimetoato do que a casca (relação entre áreas);
também detectou-se a presença do carbendazim no miolo, o que não foi
identificado na casca. A presença de uma concentração maior de pesticidas no
interior da cenoura do que na casca poderia ser explicado pelo fato que o miolo é
o caminho pelo qual os nutrientes (sais minerais) da cenoura percorrem até
chegar às f0l.has - diferente da fotossíntese - desta forma ocorre, um efeito de pré
concentração no interior da cenoura. No caso do abacaxi tal fenômeno não foi
observado; detectou-se uma quantidade semelhante de dimetoato tanto na casca
quanto na polpa do abacaxi.
7.3.7- Avaliação do Método V de extração
_As extrações foram realizadas para amostra adquiridas também no
mercado local, porém em dias diferentes. Os eletroferogramas obtidos são
mostrados na Figura 7.13. Para estas avaliações as amostras de cenoura foram
utilizadas inteiras.
164

8
6
4:::>
~ 2
O
-2
10 2
8
6:::>
~ 4
2
2
4
4
1
2
* 6
2
6
Tempo, min
8
8
A
B
10
10
Figura 7.13 - Avaliação do Método V para amostras de cenoura. (A) Cenoura-
amostra 1, (8) Cenoura - amostra 2. Amostras ressuspendidas em solução de
20% MeOH e 30 mmol L-1 de NaCI em água. Eletrólito de separação: 50 mmol
L-1 de SOS, 10 mmol L-1 de T8S, 5%/5% (PrOH: ETOH), Inj: 20 s/2,5 kPa,
Temp. 25 aC, comprimento do capilar: 50 cm (efetivo), À=200 nm. 1) simazina,
2)atrazina, 3)propazina, 4) pirazofós , (*) 1- Naftol e (#) não identificado.
165

Na Figura 7.13 foi possível a identificação de simazina, atrazina, propazina,
em pelo menos uma das amostras, fato inusitado, pois segundo a Agência
Nacional de Vigilância sanitária 34, simazina e propazina não são registradas para
o uso em culturas de cenouras. Além disso, uma das amostras apresentou o
produto de decomposição do carbaril.
7.4 - Avaliação da estabilidade térmica dos pesticidas.
Esta análise visou avaliar se o tempo e a temperatura de cozimento
normalmente usado para cozinhar legumes (ex. cenoura) é suficiente para a
degradação dos pesticidas contidos neles. Para a avaliação realizou-se o seguinte
procedimento: em um béquer, adicionaram-se 199 mL de água desionizada e 0,2
mL de uma mistura padrão contendo 10 pesticidas na concentração de 1000 mg/L,
atingindo uma concentração final aproximada de 1 mg/L. Os pesticidas
empregados foram: dimetoato, aldicarb, carbendazin, carbofuran, atrazina,
simazina, carbaril, propazina, diuron e linuron. Aqueceu-se a solução por
intermédio de um bico de Bunsen, até a ebulição por um período de 25 minutos.
Logo após esfriou-se a solução até temperatura ambiente, ajustou-se o volume
novamente para 200 mL, retirou-se uma alíquota de 30 mL e levou-a para um funil
de separação com capacidade de 60 mL. Realizaram-se duas extrações
consecutivas com duas alíquotas de 10 mL de diclorometano. A parte orgânica foi
evaporada até a secura sob fluxo de nitrogênio e ressuspendida em uma solução
contendo 20 % MeOH em água. Realizou-se o mesmo procedimento para uma
solução que não sofreu aquecimento. Na Figura 7.14 são mostrados os
34 Agência Nacional de vigilância sanitária. Portaria~ 10/SNVS de 08 de março de 1985. DOU 14/03/85
166

eletroferogramas obtidos para a solução que sofreu aquecimento e outra sem
aquecimento.
50 -
40 -9
30 -#
::J20 -
~10
B10 - 61} 3 5 ~ 8
O '-'
I I I I I I
50 2 3 4 5 6 7 8
7 940
1030 6
::J20
~ A 8 #10
O
2 4 6 8
Tempo, min
Figura 7.14 - Avaliação da estabilidade térmica dos pesticidas.Amostra sem
aquecimento (A) e amostras após o aquecimento (B). Eletrólito de separação: 45
mmol L-1 SOS, 10 % MeOH, 15 mmol L-1 trimetil-(3-ciclodextrina, 15mmol L-1
tetraborato de sódio, 2 mmol L-1 Brij 35, 27°C, 27kV. Tempo de injeção 8 s /2,5
kPa, À =220 nm (1) dimetoato, (2) aldicarb, (3) carbendazim, (4) carbofuram, (5)
simazina, (6) atrazina, (7) carbaril, (8) propazina, (9) diuron (#) não identificado e
(10) linuron.
167

Na Tabela 7.8 a seguir, são mostrados os valores de áreas antes e depois
do aquecimento, bem como a perda percentual para cada pesticida
Tabela 7.8 - Análise da perda porcentual para cada pesticida empregado.
pesticidas Area sem Area após Perda
aquecimento aquecimento porcentual
1 - Dimetoato 4,90 3,52 28,10
2 - Aldicarb 6,75 5,15 23,60
3 - Carbendazin 6,67 1,89 71,50
4 - Carbofuran 13,50 - 100,0
5 - Simazina 11,70 2,05 82,40
6 - Atrazina 40,39 12,10 70,01
7 - Carbaril 71,03 - 100,0
8 - Propazina 17,28 3,30 80,91
9 - Diuron 61,42 49,11 20,02
10- linuron 49,67 21,85 56,04
Observa-se através da Tabela 7.8 que os pesticidas carbofuram e carbaril
são totalmente degradados pela ação do calor. Os pesticidas carbendazim,
atrazina, propazina e simazina degradam acima de 70%, linuron atinge 56% de
degradação, enquanto que o dimetoato, aldicarb e diuron sofrem degradação de
apenas 20 % em média.
168

7.5 - Extração por cloud point - CPE (sigla inglesa para Cloud Point Extration)
Tensoativos podem se agregar em solução aquosa para formar estruturas
coloidais como clusters que são conhecidos como micelas (micelas normais). Alguns
aspectos estruturais dos agregados micelares (grau de rugosidade ou
irregularidades da superfície da micela, penetração de água no interior da micela,
etc.) ainda são objeto de muita discussão na comunidade científica.35 Porém,
dependendo .da espécie do tensoativo e das condições da solução onde este se
encontra, as micelas podem adotar uma variedade de formas desde esferas até
elipsóides. Em ambos os casos, a região interna da micela contém grupamentos
químicos da molécula do surfactante com características hidrofóbicas e a superfície
externa é constituída de grupamentos hidrofílicos, hidratados por ligações com
moléculas de água.36
No caso de micelas de tensoativos não-iônicos polioxietilênicos, a região
externa é comprimida por espirais.37 Uma representação esquemática de um
agregado micelar típico de um surfactante não-iônico é mostrado na Figura 7.15.
Muitos tensoativos como, por exemplo, C1OE4 e C12Es e ainda tensoativos
zwiteriônicos como Ca -lecitina, formam micelas altamente polidispersas.38
35 F. H. Quina; W. L. Hinze; Ind. Eng. Chem. Res., 38 (1999) 4150.36 E. Pramuro; E. Pelizzetti; in Surfactants in Analytical Chemistry: Applications of OrganizedAmphiphilic Media; Elsevier: Amsterdam, 1996.37 M. J. Rosen; Surfactants and Interfacial Phenomena; Wiley-Interscience: New York, 1978.38 C. L. Liu; Y. J. Nikas; D. Blankschtein; Biotechnol. Bioeng.; 52 (1996) 185.
169

D - Cadeia hidrofóbica
~ Unidade de oxielileno
A - Molécula de água
-~
Figura 7.15 - Representação esquemática de uma micela de forma rugosa de um
tensoativo não-iônico. No caso é o Brij-35 (polioxietileno(23)dodecanol), o
agregado micelar é formado por cerca de 40 moléculas individuais de Brij-35.
Soluções aquosas de determinados tensoativos exibem um comportamento
de separação de fases com a mudança da temperatura do meio. Este fenômeno é
utilizado em estudos de separação para o desenvolvimento de um processo de
extração, purificação e pré-concentração3:} dos analitos desejados e é conhecido
como cloud point (ponto de névoa). Um diagrama de fases para um tensoativo
não-iônico é apresentado na figura 7.16.
39 W.Hinze, E. Praumauro, Cri/o Rev. Anal. Chem. 24 (1993) 133
170

40,-- .....
2L30
Temp .fe
20
L
10
2 4 6 8 10 12
% Triton X -114
Figura 7.16 - Diagrama de fases para o tensoativo Triton X-11435.
Inicialmente quando o tensoativo é adicionado à solução aquosa, estando
em concentração acima da sua cmc, o aspecto da solução é límpido e
transparente (L). Após a mudança da temperatura do meio micelar, duas fases
isotrópicas são formadas e a solução fica turva (2L), devido à diminuição da
solubilidade do tensoativo no meio. A separação de ambas pode ser acelerada por
centrifugação e, como conseqüência duas fases líquidas transparentes serão
obtidas: uma incluindo um precipitado hidrofóbico, chamado de fase rica em
tensoativo, contendo o analito de interesse e outra, pobre em tensoativo.
O mecanismo pelo qual a separação de fases ocorre ainda é motivo de
discussão entre várias linhas de pensamento. Alguns autores propõem que a
separação de fases seria devido a um aumento no número de agregação micelar
171

(um aumento no tamanho da micela) quando a temperatura é aumentada40,41.
Outros sugerem que o mecanismo de separação de fases seria causado por
mudanças na interação micelar, no qual é repulsiva a baixas temperaturas e
predominantemente atrativa a altas temperaturas42.O fato que a presença de sal
favorece a separação de fases tem sido interpretado como sendo devido a
diminuição do efeito de repulsão eletrostática. Outros autores explicam o
fenômeno d~ cloud poínt com base no processo de desidratação que ocorre na
camada externa das micelas de tensoativos não-iônicos quando a temperatura
aumenta. A temperatura de cloud poínt de alguns tensoativos é apresentada na
Tabela 7.9. A temperatura de cloud point depende da estrutura e concentração
dos tensoativos43,44. Esta temperatura pode ser modificada também pela presença
de sais, álcalis, ácidos, polímeros, uréia45 e outros tensoativos46. Em geral,
tensoativos não-iônicos de cadeias longas necessitam de altas temperaturas para
atingir o seu cloud point, evidenciando maior capacidade de hidratação.
Kato47 tem chamado a atenção dos estudiosos da área a respeito do fato de
que a forma da micela podem sofrer mudanças dramáticas com a alteração das
condições da solução onde se encontra (concentração de tensoativo na solução,
aditivos, natureza destes aditivos e temperatura).
40B. Lindman, H. Wennerstrom, J. Phys. Chem.95(1991)605341M.Corti, V.DeGiorgio, J.B.Hayter, M.Zulanf, Chem. Phys. Lett. 109(1984)57942V.Degiorgio, R.Piazza, M.Corti, C.Minero, J. Chem. Phys. 82(1984)102543S.Kawamorita, H. Watanabe, K.Haraguehi, Anal. Sei., 1 (1985)4544M.E.Femández Laespada, J.L. Pérez-Pavón, B.Moreno-Cordero, Analyst, 118(1993)20945 A. M.AIGhamdi, H.A. NasrEiDin, Colloid Surfaees A 125 (1997) 546 W.L.Henze, E.Pramauro, CRC Crit. Rev. Anal. Chem. 24 (1993)13347 T. Kato; Mieroestruture of Nonionie Surfaetants, in Strueture-Performanee Relationships ínSurfaetants; Mareei Dekkeer: New York, 1997, Chapter 8, pp. 325.
172

Os parâmetros micelares críticos, isto é, cmc e número de agregação (número
de moléculas de tensoativo por micela), para diferentes surfactantes não-iônicos e
zwiteriônicos têm sido utilizados em diferentes estudos de separação e pré
concentração de diferentes analitos. Parâmetros de interesse dos tensoativos mais
utilizados em estudos de separação e pré-concentração são também citados na
Tabela 7.9
173

452585 10-2
02
-20
65 30
-40
19-2
142 3
2-3
82
3-3
235
-42
64
-69
2-7
10
,6
50
0
8416
012
7
4030
3,0
0,0
49
-0,0
65
0,0
10
0,0
00
6
0,6-
0,8
0,0
5
5,9-
7,5
Ta
be
la7.
9-
Pro
pri
ed
ad
es
aq
uo
sas
deal
guns
sist
em
as
de
Te
nso
ativ
os
nã
o-i
ôn
ico
se
zwitt
eri
ôn
ico
sq
ue
têm
sido
em
pre
ga
do
sem
ext
raçõ
es
po
rC
lou
dP
oínt
14
Su
rfac
tan
te(a
bre
viaç
ão)
.~-_..
cmc
a(m
mo
l[:
1)N
b'
---
..._
._
.,.
--C
iàii
élpo';n~(OC)
Tri
ton
X-1
00
(TX
-10
0)
0,1
7-0
,30
12
0-1
40
64
-65
Tri
ton
X-1
14
(TX
-114
)0
,20
-0,3
5-
23
-25
PO
NP
E-7
,50
,08
5-
5-20
PO
NP
E-1
00
,07
-0,0
85
10
06
2-6
5C
eH17
C6H
4(O
CH
2CH
2h,s
OH
,-
-48
-52
Igep
alC
O-6
30
CH
3(C
H2h
(OC
H2C
H2h
OH
(CeE
3)(C
H3h
CH
(CH
2ho(
OC
H2C
H2)
1S0H
,G
en
ap
ol
X-1
50
CH
3(C
H2)
g(O
CH
2CH
2)40
HC
10E
4)(C
H3h
CH
(CH
2)10
(OC
H2C
H2)
eOH
,G
en
ap
ol
X-8
0C
H3(
CH
2)11
(OC
H2C
H2)
40H
(C1
2E4)
0,0
2-0
,06
Bri
j-30
1:1
C12
E4:
C12
EeC
H3(
CH
2)11
(OC
H2C
H2)
SO
H(C
12E
s)C
H3(
CH
2)13
(OC
H2C
H2)
60
H(C
14E
6)C
16H
31(O
CH
2CH
2)10
0H(C
16E
10)
Bri
j-56
C1e
H3s
(OC
H2C
H2)
100H
Brij
-97
Oct
il~-D-tioglucose
(OT
G)
9,0
Oct
il~-D-glucosídeo
(OG
)2
0,0
-25
,0C
g -A
PS
04
45
,0T
rito
nX
-11
4-C
iba
cro
nA
zul
afin
ida
de
0,5
5S
urf
act
an
te
Plu
ron
ícL6
1(E
02P
031
E0
2)P
luro
nic
P105
(E06
eP06
gE03
4)C
e-Ie
citin
a
a-
conc
entra
ção
mic
elar
crfti
ca;
b-
N=
núm
ero
deag
rega
ção
mic
elar
;c
-te
mpe
ratu
rade
clou
dpo
int
(pon
tode
névo
a).
174

As propriedades termodinâmicas de soluções de tensoativos não-iônicos têm
sido modeladas teoricamente por diferentes grupos de pesquisa utilizando-se
softwares. Como exemplo podem ser citados os programas PREDICT e MIX 48.
Estes programas fornecem informações a respeito do formato das mice/as, tamanho,
tensão superficial, etc.
7.5.1 - Condições experimentais para a realização da CPE
Uma representação esquemática de como é feita uma extração via CPE é
ilustrada pela Figura 7.17. Primeiramente, o tensoativo não-iônico ou zwiUeriônico é
adicionado a uma solução aquosa onde esteja presente o analito de interesse. A
concentração final do tensoativo deve ser maior que a sua cmc, para garantir a
presença de micelas na solução. Um equilíbrio de partição é estabelecido na
solução e o analito é incorporado pelas micelas presentes em solução. No próximo
estágio, a temperatura da solução em questão é alterada; nesta etapa ocorrerá uma
separação de fases na solução. Quando são utilizadas amostras constituídas por
fluidos biológicos, é necessária a presença de sal para induzir a separação de fases
com uma maior eficiência49. Após deixar que sistema bifásico separe em duas fases,
por gravidade ou centrifugação rápida, os analitos são concentrados em um
pequeno volume (tipicamente da ordem de 50 a 400 ~L) que é uma fase rica em
tensoativo.50 Dependendo da densidade desta fase em relação à fase aquosa, ela
poderá estar acima ou abaixo da fase aquosa. Através da adição de sal ao sistema
48 S. Puwada; D. Blankschtein; J. Chem. Phys., 32 (1990) 3710.49 S. R. Sirimanne; D. G. Patterson; L. Ma; J. B. Justice; J. Chromatogr., 716 (1998) 129.50 B. M. Cordera; J. L. P. Pavom; C. G. Pinto; M. E. F. Laespada; Ta/anta, 40 (1993) 1703.
175

a densidade da fase aquosa pode ser ajustada51, fazendo com que a fase rica em
tensoativo esteja localizada abaixo ou acima da solução, tornando desta forma o
procedimento mais adequado de acordo com a necessidade.
Tensoativo diluído emsolução, aspecto límpido.
Aspecto turvo
c~nlrifu~e
ou
·~,i-;·· \ /
\ Fase aquosa
Fase micelar concentrada
000Oco
roO
6->
Figura 7.17 - Representação esquemática dos passos envolvidos em uma
extração por cloud point (CPE)
7.5.2 - Emprego do CPE como técnica analítica
A primeira separação de fases baseada no fenômeno de cloud point refere-
se à extração de complexos de íons metálicos solúveis em água. A eficiência do
processo depende da hidrofobicidade do ligante e do complexo formado. Este tipo
51 G. Stangl; R. Niessner; Mikrochim. Acia, 113 (1994) 1.
176

de extração por cloud point foi inicialmente descrito por Watanabe e Tanaka 52.
Para a pré-concentração de Zn(lI) foi usado como ligante 1-(2-piridil azo) naftol
(PAN) e PONPE 7.5, como extrator. Posteriormente esta metodologia foi aplicada
para a determinação de diferentes íons metálicos em diferentes tipos de
amostra53.54,55.
Outra aplicação para a CPE relaciona-se ao isolamento e purificação de
espécies biológicas, principalmente proteínas. Esta metodologia foi utilizada por
Bordier'para a separação de proteínas hidrofílicas em membranas biológicas,
usando uma solução de Triton X-114 como extrator. Neste campo de bio-
separações a CPE encontra a sua principal aplicação, levando em conta o volume
de literatura relacionado à extração e purificação de proteínas de membranas e
outros biomateriais57.58
.
A CPE também vem sendo utilizada para a extração de compostos
orgânicos, como é o caso dos hidrocarbonetos policíclicos aromáticos (PAHs), e
posterior determinação por HPLC. O uso de tensoativos não-iônicos foi proposto
por muitos autores59,OO.61. Uma das limitações desta técnica é a alta absorção
destes tensoativos em região do UV. Uma possível maneira de superar esta
limitação é o emprego de tensoativos que não absorvam no UV, como os iônicos e
52 H.Watanabe, H. Tanaka, Talanta 25 (1978) 58553 M.C. Cerrato-oliveros, O. Jiménez de Blas, J.L. Pérez-Pavón, B. Moreno-Cordero,J. Anal. Atom.Spectrom.13 (1998) 54754 C.Garcia-Pinto, J.L. Pérez-Pavón, B. Moemo-Cordero, E. Romero-Beato, S. Graça-Sánchez, J.Anal.Atom. Spectrom. 11 (1996) 37 \.~
55 K.Watanabe, N.Omae, M.UJtagaki, Bunseki Kagaku 48 (1999) 215956 C.Bordier, J.Biol. Chem. 256 (1981) 160457 A Sanches-Ferrer, R.Bru, F. Garcia-Carmona, Crit.Rev. Biochem. Mal. Biol. 29 (1994) 27558 N.H.Heegaard, D.R.Jakobsen, D.Klattschou, Anal. Bochem. 253 (1997) 25959 A. Boeckelen, R Niessner, Fresenius J. Anal. Chem. 346 (1993) 43560 C. García-Pinto, J.L. Pérez-Pavón, B. Moreno-Cordero, Anal.Chem. 66(1994)87461 E.R. Brouwer, A. N.J. Hermans, H. Lingerman, UA .Th. Brinkman, J. Chromatogr. 669 (1994) 45
177

os zwitteriônicos62,63. Outra maneira é o emprego de detecção por fluorescência43
.
Ferrer et al.64 propuseram um procedimento de clean-up, onde a amostra (após
CPE), era percolada em uma coluna de sílica-gel e os PAHS eram eluidos da
coluna com uma mistura de solventes, possibilitando sua detecção sem a
interferência do tensoativo não-iônico.
Os bifenil policlorados (PCBs) são compostos orgânicos que exibem boa
estabilidade térmica, podendo a pré-concentração por CPE ser feita usando
tensoativos com a temperatura de cloud point maiores que 75°C. Eiguren-
Fernández et aI. usaram tensoativos não-iônicos para a pré-concentração de
PCBsffi e dibenzonofuranos (PCDFs)66, em amostras de água do mar e posterior
determinação por HPLC. A pré-concentração de dibenzo-p-dioxinas em amostras
de soro humano usando Triton X-100 a 50°C foi p~oposta e implementada com
7.5.3 - Emprego de CPE em análise de pesticidas
A determinação de pesticidas em água e solo tem sido investigada usando
CPE. O uso de Genapol X-OSO e Genapol150 combinados e analisados via HPLC
com detecção fluorométrica foi aplicado para a detecção do herbicida
napropamida em águas naturais68. Pesticidas organofosforadosoo, como metil e eUI
paration, paraoxon e fenitrotion foram determinados em amostras de água de rio,
62 D.Sicilia, S.Rubio, D.Pérez-Bendito, N.Mariasso, E.A .G. Zagatto, Anal. Chim. Acta 392 (1999) 2963 T. Saitoh, W.L.Hinze, Anal. Chem. 63 (1991) 252064 R Ferrer, 1.L. Beltran, 1. Guiteras, Anal. Chim. Acta 330 (1996) 19965 A .Eigueren-Fernández, Z.Sosa-Ferrera, J.1. Santana-Rodriguez, Anal. Chim. Acta 358 (1998) 14566 A .Eigueren-Fernández, Z.Sosa-Ferrera, 1.1. Santana-Rodriguez, Analyst 124 (1999) 48767 S.RSirimanne, 1.R Barr, D.G.Patterson, L. Ma,Anal. Chem. 68 (1996) 155668 G.Stangl, RNiessner, J.Albaiges, Int. Environ.Anal. Chem. 58 (1995) 1569 C.García-Pinto, lL.Pérez-Pavón, B.Moreno-Cordero, Anal.Chem. 67 (1995) 2696
178

usando Triton X-114 como extrator e a sua separação feita por HPLC (com
detecção eletroquímica), atingindo limites de detecção na ordem de 0,03 ng mL-1
para o fenitrotion e 0,08 ng mL-1 para o paraoxon. A metodologia poderia ser
utilizada para a determinação destes pesticidas em água para consumo, pois a
concentração máxima permitida pela União Européia é de 0,1 Ilg L-1 para cada
substância. Outro processo baseado em CPE com tensoativo não-iônicos (Triton
X-114), foi desenvolvido para a determinação dos fungicidas Folpet, Captan e
Captafol70 em amostras de água e posterior determinação via HPLC.
Evdokimov e von Wandruszka71 avaliaram a possibilidade da eliminação de
resíduos de DDT em solo através de uma mistura de dois tensoativos (Igepal ICO-
630 e Triton X-114). A porcentagem de extração obtida foi acima de 83 % quando
o solo em questão foi tratado com 3 % da mistura dos tensoativos por um período
de 2 h. A eletroforese capilar em meio não aquoso foi usada por Carabias-
Martinez et aI. 72 para a determinação de herbicidas da classe da triazinas em
águas naturais, utilizando um tensoativo não-iônico (Triton X-114) como extrator.
A técnica de eletroforese capilar, nos seus mais diversos modos de
separação, apresenta-se como uma ferramenta analítica viável para a realização
de experimentos que envolvam ambientes micelares.
O uso de sistemas micelares para a extração e pré-concentração de
compostos orgânicos é uma alternativa extremamente vantajosa, devido a várias
características como:
70 R.Carabias-Martinez, E.Rodriguez-Gonzalo, M.G. García-Jimenez, C.García-Pinto, J.L.Pérez-Pavón.J.Herandez-mendez, J Chromatogr. 754 (1996 )8571 E.Evdokimov, R.von Wandruszka, Ana/. Lett. 31 (1998) 228972 R Carabias-Martinez, E.Rodriguez-Gonzalo, J.Dominguez-Alvares, J.Hernández-Méndez, Anal. Chem. 71(1999) 2468
179

~ Alta capacidade de pré-concentrar analitos de diferentes polaridades.
~ O fator de pré-concentração pode ser otimizado por modificações do tipo e
da concentração do tensoativo utilizado, bem como as condições de
análise em que esta extração ocorrerá.
~ Os tensoativos possuem baixíssima toxidez e são mais baratos quando
comparados aos solventes de uso comum em extrações líquido-líquido.
~ Os tensoativos mais comuns são facilmente encontrados no comércio
especializado.
~ Não é necessário evaporar solventes, minimizando assim perda por
evaporação do analito.
).- As operações experimentais envolvidas nesta metodologia são fáceis, e a
fase rica em tensoativo é compatível com várias técnicas analíticas (HPLC,
MEKC, CEC, FSCE, etc.)
Apesar de todas estas vantagens a CPE vem sendo negligenciada no uso
de extrações em matrizes vegetais, em especial à extração de pesticidas. As
extrações convencionais e clean-up usadas para a determinação de pesticidas
são laboriosas, e por conter várias etapas a perda de analito é inevitável,
promovendo baixos valores de recuperações. Desenvolver técnicas de extração
capaz de diminuir estas limitações são contribuições inestimáveis para o
aprimoramento do processo.
180

7.5.4 - Otimização dos parâmetros experimentais da metodologia CPE
7.5.4.1 - Parâmetro 1: composição do eletrólito de separação
Pelo fato dos tensoativos neutros empregados na CPE absorverem
fortemente no UV, a separação eletroforética em meio aquoso de pesticidas após
o emprego da metodologia fica prejudicada. Como os tensoativos e as moléculas
de pesticidas são neutros, estes apresentam interação pelas micelas carregadas
de SDS, nãp propiciando a discriminação entre elas. Assim sendo, a janela de
detecção do tensoativo sobrepõe a do analito encobrindo o seu sinal. Segundo
Canabias-Martinez aI. 73 algumas triazinas podem ser protonadas em meio
orgânico e pH aparente menor que 1,5. Desta forma as triazinas carregadas
positivamente migram mais rápido em direção ao detector do que os monômeros
dos tensoativos neutros, propiciando separação entre ambos.
Esta proposta foi avaliada realizando um procedimento CPE para uma
mistura contendo 5 pesticidas (ametrina, simazina, carbendazim, propazina e
atrazina). Na figura 7.18 é apresentado um esquema do procedimento CPE
utilizado. Em um balão volumétrico de capacidade 25 mL adicionou-se 0,5 mL de
uma mistura padrão dos cinco pesticidas na concentração de 50 mg L-1 e 1% (m/v)
do tensoativo Triton X-114, cuja a temperatura de cloud point nestas condições é
aproximadamente 39 oC. A solução foi aquecida por 15 minutos em banho-maria
a uma temperatura de 45 oCo Terminado este tempo a solução (turva) foi
transferida para tubos de centrífuga e centrifugada por 5 minutos (3500 rpm). A
parte superior do centrifugado foi descartada (fase pobre em tensoativo) e a parte
73 R Carabias-Martinez, E.Rodriguez-Gonzalo, J.Domingues-Alvares, , J. Hernandez-Mendez, , JChromatogr. A, 869 (2000) 451
181

inferior (fase rica em tensoativo) foi seca com auxílio de um secador de cabelos.
Os analitos foram ressuspendidos em 1 mL de uma mistura de solventes
composta de ACN:MeOH (70 : 30 v/v) e 5 mmol L-1 de HCI. O meio de separação
empregado foi ACN:MeOH (1:1 vlv) e 10 mmol L-1 de HCI.
25 mL solução padrão 1 mg L-1 + 1% triton X114 (m/v)
Aquecimento, 15 min, 45°C
I Centrifugação, 5 min I
Separação de fases
IFase rica em tensoativo
Contendo o analito
Evaporação e ressuspensão emACN:MeOH 70:30 (v/v) e 5 mmol L-1 HCI
I Análise I
Fase pobre em tensoativo,Sobrenadante
Figura 7.18 - Esquema de extração utilizado para a metodologia de cloud point.
Na figura 7.19 é mostrado o eletroferograma obtido para a mistura padrão
de pesticida.
182

25 -
20 -
15 -
::J 10-«E
5-
0
-5O
I
2
2
I
4I
6
Triton X-114
I
8I
10I
12I
14
Tempo, mino
Figura 7.19 - Separação em meio não-aquoso de pesticidas após CPE.
Composição do eletrólito: ACN:MeOH (1: 1 v/v) e 10 mmol L-1 de HCI, injeção
eletrocinética : 5kV/10s. À = 220 nm, comprimento efetivo do capilar: 50 em,
temperatuta: 25°C, Tensão aplicada: 25 kV.1) carbendazim, 2) ametrina, 3)
simazina, 4) propazina e 5) atrazina.
Nota-se através da Figura 7.19 que houve a diferenciação entre os analitos
de interesse e tensoativo utilizado na CPE, tornando-se viável a determinação dos
pesticidas.
Foi avaliado o efeito que o SDS poderia ocasionar na separação
eletroforética em meio não aquoso. Esperava-se que a adição do SDS ao eletrólito
de separação promovesse um maior atraso na saída do tensoativo, devido a
183

interações solfofóbicas entre o tensoativos. Porém o resultado encontrado foi um
aumento na intensidade dos picos de pesticidas e a aproximação do pico do
tensoativo aos picos dos pesticidas. Para o experimento foram adicionados ao
eletrólito de separação 10 e 20 mmol L-1 de SOS. Na Figura 7.20 são
apresentados os eletroferogramas obtidos.
184

35
230 6
o"i- ..l.------.JIUWYl.......J~.JUl./
25
20
::J« 15E
10
5
c1
34
56
jLl
Or------'-_...IJUl1-.JWUl-._-
35
30.25
20
::J« 15E
10
5
35
30
25
20
::J« 15E
10
5
O
o
B
A
2
2
4 6 8
Tempo, mino
4 6 8
Tempo, mino
10
10
12
12
O -l-------...__--"l'UUlJL..- --
o 2 4 6
Tempo, mino
8 10 12
Figura 7.20 - Avaliação comparativa das adições de SOS ao eletrólito de
separação (NACE). Composição do eletrólito: ACN:MeOH (1:1 v/v) e 10 mmol L-1
de HCI , A) sem SOS, 8) 10 mmol L-1 de SOS e C) 20 mmol L-1 de SOS, injeção
eletrocinética: 5 kV/10 s, À =220 nm, comprimento efetivo do capilar: 50 em,
temperatura: 25 aC, Tensão aplicada: 25 kV. 1) carbendazim, 2) ametrina,
3)simazina, 4)propazina e 5)atrazina 6) tensoativo.
185

Através da Figura 7.20 nota-se um aumento na intensidade dos picos de
pesticidas a medida que se aumenta a concentração do SOS no eletrólito de
separação. Porém o pico do tensoativo CPE se aproximou dos picos dos analito
de interesse. O aumento na intensidade dos picos dos pesticidas é ocasionado
porque com a adição de SOS ao eletrólito de separação este adquiriu uma
condutividade maior em relação à amostra injetada proporcionando um stacking
no momento da injeção eletrocinética. Para verificar este fato foi confeccionado
um eletrólito de separação com NaCI, porem tomou-se cuidado para que a
condutividade fosse idêntica ao eletrólito contendo SOS. Na Figura 7.21 são
mostrados os eletroferogramas obtidos.
186

25 - 2
20 - 315 - 4
:::l 10 - C 1 5«E 5-
~ lA0--5 I I I I
O 2 4 2 6 8 1025-20 - 3
15 - lh:::l 10 - B« 1E 5-
~0--5 I I I I I
25-0 2 4 6 8 10
20-15 -
23
:::l 10 - A4«U~E 5- 1
O ~
I I I I I
O 2 4 6 8 10
Tempo, min
Figura 7.21 - Eletroferogramas obtidos ajustando-se a condutividade do eletrólito
de separação. Composição do eletrólito: ACN:MeOH (1:1 vlv) e 10 mmol L-1 de
HCI, (A) Eletrólito sem adição de NaCI ou SOS (cond. 1362j.lS cm-1), (8) Eletrólito
com SOS (cond. 2,74 mS cm-1) e (C) Eletrólito com NaCI (cond. 2,71 mS cm-1
). ,
injeção eletrocinética: 5 kV/10 s, À =220 nm, comprimento efetivo do capilar: 50
cm, temperatura: 25 cC, Tensão aplicada: 25 kV. 1) carbendazim, 2) ametrina,
3)símazina, 4)(propazina e 5) atrazina.
Mantendo-se a condutividade e o tempo de injeção constante, as áreas dos
picos dos pesticidas foi idêntica quando SOS ou NaCI foram adicionados ao
eletrólito de separação, obtendo aumento na intensidade dos picos em relação ao
eletrólíto sem NaCI ou SOS.
187

Outro fator avaliado foi o tamanho da cadeia do tensoativo utilizado. Para a
realização do experimento utilizou-se octil sulfato de sódio (aniônico). Foi
preparada uma mistura padrão dos 5 pesticidas, na concentração de 10 mg/L. Os
eletroferogramas comparando o efeito do octil sulfato de sódio em relação ao SOS
são mostrados na Figura 7.22.
35 8 130.25
20
:::> 2« 1 5E
1 o 435
o
2 4 6 8 1 o35 Tempo, min
30
A25 1
20
:::>« 1 5E 2
1 O
3 455
O
2 3 4 5 6 7 8 9 1 O
Tem p o, m in.
Figura 7.22 - Avaliação comparativa entre SOS e octil sufato de sódio como
tensoativos do eletrólito de separação. Composição do eletrólito: ACN: MeOH (1: 1
v/v) e 10 mmol L-1 de HCI com, A) 20 mmol L-1 de octil sulfato e B) 20 mmol L-1 de
SOS. Injeção eletrocinética: 5kV/10s, À, =220 nm, comprimento efetivo do capilar:
50 cm, temperatura: 25°C, Tensão aplicada: 25 kV. 1) carbendazim, 2) ametrina,
3) simazina, 4) propazina e 5) atrazina.
188

Nota-se através da Figura 7.22 que o comprimento da cadeia hidrofóbica do
tensoativo utilizado no eletrólito influência no tempo de eluição dos compostos
bem como na intensidade que apresentam os picos.
Novo questionamento surgiu a respeito da possibilidade de hidrólise do
SOS (éster para álcool) nestas condições drásticas de pH. Se este fato ocorresse
não seria possível a amplificação das alturas dos picos dos pesticidas estudados,
pois o álcool gerado da hidrólise não apresentaria carga (dodecanol). A fim de
sanar esta dúvida, foram preparados eletrólitos de separação com 30 e 60
mmol L-1 de dodecanol e os eletroferogramas obtidos comparado a um eletrólito
contendo 20 mmol/L de SOS. Foi injetada uma mistura de padrões contendo
(carbendazim, ametrina, propazina, atrazina e simazina), na concentração de 10
mg L-1. Na Figura 7.23 são apresentados os eletroferogramas obtidos para cada
eletrólito de separação.
189

25 25
20 20
A 2 B15 15 2
::J 3 ::J ,.,Ei 10 « 10 .)
1 45E
1 45
5 5
OO
O 2 4 6 8 O 2 4 6 8
Tempo, mino Tempo, mino
252
25
20 OC 20
15 32 15
1::J ::J 4Ei 10
«,., E.) 10 5
1 45 5 5
O l~ I o ~'-- L....
o 2 4 6 8 O 2 3 4 5 6 7 8
Tempo, min Tempo, mino
Figura 7.23 - Avaliação comparativa entre dodecanol e SOS. Composição do
eletrólito: ACN: MeOH (1: 1 v/v) e 10 mmol L-1 de HCI, A) sem aditivos, 8)30 mmol
L-1 de dodecanol, C) 60 mmol L-1 de dodecanol e O) 20 mmol L-1 de SOS. Injeção
eletrocinética: 5 kV/10 s, . À =220 nm, comprimento efetivo do capilar: 50 em,
temperatura: 25°C, Tensão aplicada: 25 kV. 1) carbendazim, 2) ametrina, (3
)simazina, 4) propazina e 5) atrazina.
190

Através da Figura 7.23 fica evidenciado que se houvesse a degradação do
SDS na condição de pH do eletrólito de separação, haveria sensível perda na
capacidade de pré-concentração por parte do SDS diluído no eletrólito de
separação.
7.5.5 - Parâmetro 2: concentração do Triton-X114 empregado na CPE
A fim de se otimizar a quantidade de tensoativo (triton X-114) usado para a
extração dos pesticidas, foi realizada uma extração utilizando-se uma solução
padrão de pesticidas na concentração de 1 mg L-1. Foram utilizadas diferentes
concentrações de tensoativo que variaram de 0,25 a 1,5 % (m/v). Na Figura 7.24
são mostrados os eletroferogramas obtidos.
191

20 20 2
A B15 15
210 10
=> =>« «E E
5 5 4 51
1 3 4 5O O
2 4 6 8 10 2 4 6 8 10
Tempo, mino Tempo, mino
108
34rl
2
I
1
6
Tempo, mino
4
20
2 O15
4
5 10
3 =>«E
1 5
_LA '------., ~ L.----O
4 6 8 10 2
Tempo, mino
c
2
01--1
'---------'
5
20
15
10
Figura 7.24 - Variação da concentração de tensoativo (Triton X-114) para CPE em
amostra sintética. (A) CPE com 0,25%. (8) CPE com 0,5%. (C) CPE com 1,0% e
(O) CPE 1,5 %. Condições de análise: 20 moi L-1 SOS, 10 mmol L-1 HCI,
ACN:MeOH (1: 1 v/v). À = 220 nm, comprimento efetivo do capilar: 50 em,
temperatura: 25 °C, Tensão aplicada: 25 kV. 1) carbendazim, 2) ametrina, 3)
simazina, 4) propazina, 5) atrazina.
192

Fica evidente observando-se a Figura 7.24, que a quantidade de pesticida
extraído é dependente da concentração de tensoativo empregado na extração,
pois à medida que aumenta a concentração do tensoativo, a interação entre o
analito e a micela é favorecida, facilitando a incorporação do analito pela micela. O
fator de pré-concentração é outro fato que se pode observar. O volume inicial da
amostra antes do c10ud point é de 25 mL, após a realização da extração este
volume fica reduzido a aproximadamente 1 mL, obtendo assim um fator de pré
concentração da ordem de 25 vezes. Baseado nestes experimentos optou-se em
utilizar para as próximas extrações a concentração de 1% de tensoativo, pois
observou-se também que concentrações acima de 1 %, o volume final da fase rica
em tensoativo após a extração aumenta demasiadamente, prejudicando pré
concentração. Mesmo porque, um volume grande de fase rica em tensoativo
levaria horas para que o processo de evaporação fosse concluído, inviabilizando
análises em rotina.
7.5.6 - Aplicação da metodologia otimizada em amostra real.
Após a otimização de parâmetros, foi realizado um CPE para uma amostra
de abacaxi, adquirida no comércio local. O esquema da metodologia utilizado é
apresentado na Figura 7.25. Na figura 7.26 são mostrados os eletroferogramas
para uma amostra de abacaxi.
193

50 g de Abacaxi+
100g de água
Turrax 5 minFiltração
I 25 mL do suco + 1% Triton X114 (m/v) IAquecimento, 15 min,40°C
I Centrifugação, 5 min ISeparação de fases
IFase rica em tensoativo
Contendo o analito
Evaporação e ressuspensão emACN:MeOH 70:30 (v/v) e 5 mmol L-1 HCI
I Análise I
Fase pobre em tensoativo,Sobrenadante
Figura 7.25 - Esquema de CPE aplicado à amostra de abacaxi.
194

10
8
6
:::> 4c:(E 2
O A-2
10 O 2 4 6
8
6
:::> 3c:(
4
E 2
O B-2
O 2 4 6
Tempo, min
Figura 7.26 - Realização de CPE para uma amostra de abacaxi. (A) Amostra in
natura, (8) amostra fortificada com 1O~L de mistura padrão de pesticida (50 mg
L-1) .. Composição do eletrólito: ACN: MeOH (1:1 v/v) e 10 mmol L-1 de HCI .
Injeção eletrocinética: 5 kV/10 s. 1) ametrina, 2) atrazina, 3) propazina. ').." = 220
nm, comprimento efetivo do capilar: 50 cm, temperatura: 25 °C, Tensão aplicada:
25 kV.
Com a metodologia CPE foi possível a identificação de ametrina, atrazina e
propazina em uma amostra de abacaxi. A metodologia mostrou-se de simples
implementação e barata, quando comparada à extração em fase sólida e mais
rápida e menos dispendiosa quando comparado com a extração líquido-líquido.
195

~~u
~
tkJ~
i:
~
u
~t~, ,9~ 'B
u1"""'-'-J
CAPÍTULO 8

8 - Introdução
A validação de um método é o processo usado para estabelecer os
parâmetros de desempenho analítico que são adequados para a aplicação para a
qual é destinado1 ,2,3.
Os principais parâmetros da validação de um método estão definidos na
Tabela.8.1
1 F. Bressole; M. Audran; T. N. Pham; J. J. Vallon; J. Chromatogr. B, 687 (1996), 303.
2 R. Causon; J. Chromatogr. B, 689 (1997), 175.3 Quality assurance principies for analylical laboratories , Frederick M. Garfield by AOACinternational, 1991, Library of congress catalog card number 91-22409, ISBN 0-935584-46-3, USA
196

No
me
Line
arid
ade
Rec
uper
ação
Lim
itede
dete
cção
(LO
)
Lim
ited
eQ
uant
ifica
ção
(LQ
)
Exa
tidão
Pre
cisã
o
Pre
cisã
oIn
term
ediá
ria
Rep
etib
ilida
de
Rep
rodu
tibili
dade
Rob
uste
z
Sen
sibi
lidad
eE
spec
ifici
dade
Tabe
la8.
1-
Def
iniç
ãodo
ste
rmos
requ
erid
ospa
rava
lidaç
ãode
met
odol
ogia
s
Def
iniç
ões
.-
-.
--..
-..-
_.-.
Cor
resp
onde
àca
paci
dade
dom
étod
ode
forn
ece
rre
sulta
dos
dire
tam
ente
prop
orci
onai
sà
conc
entr
ação
dasu
bstâ
ncia
emex
ame
dent
rode
uma
dete
rmin
ada
varia
ção.
Rel
ação
entr
ea
quan
tidad
ede
amos
tra
med
ida
ead
icio
nada
aum
ade
term
inad
am
atriz
após
umpr
oces
sode
extr
ação
.R
epre
sent
aa
mai
sba
ixa
conc
entr
ação
dasu
bstâ
ncia
emex
ame
que
pode
ser
dete
ctad
aco
mum
cert
olim
itede
conf
iabi
lidad
eut
iliza
ndo
umde
term
inad
opr
oced
imen
toex
perim
enta
l.R
epre
sent
aa
mai
sba
ixa
conc
entr
ação
dasu
bstâ
ncia
emex
ame
que
pode
ser
quan
tific
ada
com
umlim
itede
conf
iabi
lidad
eut
iliza
ndo
umde
term
inad
opr
oced
imen
toex
perim
enta
l.É
ogr
aude
conc
ordâ
ncia
entr
eos
resu
ltado
sin
divi
duai
sen
cont
rado
se
umva
lor
acei
toco
mo
refe
rênc
ia.
Rep
rese
nta
ogr
aude
repe
tibili
dade
entr
eos
resu
ltado
sde
anál
ises
indi
vidu
ais
quan
doo
proc
edim
ento
éap
licad
odi
vers
asve
zes
emum
am
esm
aam
ostr
aho
mog
ênea
emco
ndiç
ões
idên
ticas
dean
ális
e.E
xpre
ssa
oef
eito
das
vari
açõe
sde
ntro
dola
bora
tório
devi
doa
even
tos
com
odi
asdi
fere
ntes
dean
ális
e,an
alis
tas,
equi
pam
ento
s,et
c.C
orre
spon
deao
sre
sulta
dos
obtid
osat
ravé
sde
vári
asre
prod
uçõe
sde
umm
étod
ose
mpr
ena
sm
esm
asco
ndiç
ões
expe
rimen
tais
dean
ális
eR
efer
e-se
aos
resu
ltado
sdo
ses
tudo
sde
cola
bora
ção
entr
ela
bora
tório
sC
orre
spon
deà
capa
cida
dede
umm
étod
ode
não
ser
afet
ado
por
uma
pequ
ena
ede
liber
ada
mod
ifica
ção
emse
uspa
râm
etro
s.S
lope
(incl
inaç
ão)
dacu
rva
deca
libra
ção.
Rep
rese
nta
aca
paci
dade
dom
étod
ode
senv
olvi
dode
ava
liar
defo
rma
ineq
uívo
caa
subs
tânc
iaem
exam
ena
pres
ença
deco
mpo
nent
esqu
epo
dem
vir
ain
terf
erir
nasu
ade
term
inaç
ãonu
ma
mis
tura
cO~J?I~xa:
.
197

Todos os termos definidos na Tabela 8.1 são conhecidos como
parâmetros de performance analítica e algumas vezes como figuras analíticas
de mérito. Muitos desses termos são familiares e usados diariamente no
laboratório. Existem na literatura vários protocolos de validação propostos tais
como as normas USp4 e as normas ICH5 que são voltados a para as análises
de fármacos e afins. Já no âmbito ambiental são utilizados os protocolos de
validação baseados nas diretrizes da AOAC, FAO, IAEA e IUPAC (Training and
Reference Centre for Food and Pesticide Control)6, diretrizes estas que foram
aplicadas no presente trabalho.
Foram avaliados os seguintes termos para a validação da metodologia
de extração MSPO (método V): Linearidade, recuperação, limite de detecção e
quantificação, exatidão e precisão. O método de extração V foi escolhido pela
sua facilidade de implementação e por se tratar de uma metodologia pouco
explorada para a extração e identificação de pesticidas em matrizes vegetais.
8.1 - Método analítico otimizado
As separações eletroforéticas foram conduzidas usando como eletrólito
de separação: tampão tetraborato de sódio 10 mmollL, 50 mmollL SOS, 5%
etanol, 5% propanol. As condições analíticas foram as seguintes: potencial
aplicado durante as análises foi de 25 kV (polaridade normal), 25°C de
temperatura, a detecção foi fixada em 220 nm e a injeção da amostra foi feita
por 10 s a 25 mBar. A coluna capilar, com ltot =58,3 cm e Liet =50 cm, foi
lavada previamente com NaOH 1,0 mollL durante 20 min, seguido por água
4 United States Pharmacopeia (USP), US Pharmacopoeial Conventional Inc.; Rockville, MD1995, pp 1982.&International Conference on Harmonisation of Technical Requirements for Registration ofPharmaceuticals for Human Use, Guidelines, www.ich.ora
199

durante 20 min e por último com o tampão de corrida por 25 mino Entre as
corridas foram feitas lavagens com o tampão de corrida durante 4 mino
8.2 - Preparação das amostras
Foi preparada uma mistura de solução padrão de pesticidas contendo:
dimetoato, carbendazin, simazina, atrazina, propoxur, carbaril, ametrina,
propazina, diuron e linuron na concentração de 50 mg/L (usada como solução
estoque) para a avaliação de linearidade e repetibilidade. Para a realização dos
testes de recuperação para a metodologia MSPD, foi realizado o procedimento
descrito no capitulo 7, seção 7.3.5.1 (Figura 7.9). Recuperações foram
realizadas em dois níveis de fortificação (83 e 250 Ilg/L).
8.3 - Linearidade
A linearidade de um procedimento analítico é definida como sendo a
capacidade de gerar resultados diretamente proporcionais à concentração da
substância em exame. A calibração é um dos mais importantes estágios na
análise química. Sem um bom procedimento de calibração, precisão e exatidão
não podem ser obtidas. Para a maioria das técnicas analíticas cromatográficas,
assim como para a CE, uma relação de primeira ordem (linear) é observada
entre a resposta do detector (y, área ou altura dos picos) e a concentração (x)
do analito nas amostras, podendo ser descrita pela equação de regressão
linear: y =ax + b, onde o parâmetro a é a inclinação da reta e o parâmetro b
são a intersecção do gráfico de calibração. Idealmente (a) deve ser reprodutível
de ensaio para ensaio e (b) não deve ser significativamente diferente de zero.
Ó www.iaea.org.aUtrc/pest-qa_val~de.pdf
200

A regressão linear deve também apresentar um alto coeficiente de correlação (r
> 0,99). O coeficiente de correlação tem um valor ideal de unidade.
Para avaliação da linearidade foram preparadas misturas padrões de
pesticidas nas concentrações de: 0,5; 2; 4; 6; 8; 10 mg/L, partindo-se da
solução estoque 50 mglL descrita no item 8.2. Nas figuras 8.1 a 8.3 são
mostradas as curvas analíticas obtidas para cada pesticida.
201

;i.
. ~ ,;
.4.
m 3 · ,~
~! -.
1 ;
,) " O
,-···-·-----.. -7-·---... -'" -~·----,· ··.,.·----· .. <· ...... ·--
DiOletoato
•
•
A 1:1
R
•
V,I .JEI Errar
.0, 2~2 0,0,1822 0,56;! 0,(lSf!78
SO N p
0'/7826 Q,ol'.99
.~ .-...---.-r--",. --_r_:--r'.-;.. ... ;~~._ . ....__--r:-... , .. ,..
•
0,00383
2 4 . 6 8 10
Concl,mtraçlão, mg/~
.• ----.,.-_ ...... ----~ .... ,...----)< .. ,.""._----, _.:.-._---~.':"" '--
50
40·
30·
m -4: 20-
1Cl •
C, ·
• Sima;z.ina
•
. ./' Par~ Vinue Erra'
A -Q.7iJej~& 1,63!)64 B 4,97~; o,~ --.---:-~-.-._-:--.,.---_.
50
• 0,99263 ~'1519 6 .;i,0:01 __ ,~~w'._. __ ,_ . ..., •• _
1-': .. 'r, i .,..'--,----.,.,.-'-,-,.,.~'-----,------..
O 2 4 6 8 10
COncentraçÉio, mg/L
ti . , Carbend;;tzirjl
/
• ~; -:1
d·"J 11
co ~ ~h
'<x: < ...... ,,, . \ ... ":..~-.;.,-,-_. ~ ~'.._;~-;;Ba;7
~ O,Se1 (~04231 • _ ...... _ ... "'._'..--_. -..,...~""'""--
~~ SI)
0,992" Q,26761 8,3582&.
; O"~ --:---;-" ..... -----'.---"'-,~ , ... "..,.-_.
• (l-, • .. · ... .....------r···~·~·r- .. ,,-.,-.-.- ',
2 4 ii Ei 10
concetra!;ão, mg/L 8() ,~ ._--, .•... ---,.,.-----> .. ".--_., .... ----.,.,,---
70
6{),
50
CO 40 Q)
<3Q 2Q
1(j
Ó
Atrazlna
/ /
/ ~ . -i- . i ' ...... 2 .~
4
•
•
Paremeter Veitje Errcr
A -1,~. 2,7~1
B 7,15068 0,'<&!2 ___ o _ ' , _,~_--;_~"' . __ • __
($O p -_.'- ....;.. .. ---_ ...... :--.......... _-0,99231 :) &1167 <l10oo1 --""---:-_.---.--~ ... . ~_.
~._...,--:-r'<-.-""'--
6 8 10
(loncetraçi§9, mg/L
Figura 8.1 - Curvas analíticas obtil:jas para, dimetoato, c~:irbendE~~im, simazina e atrazina.
202
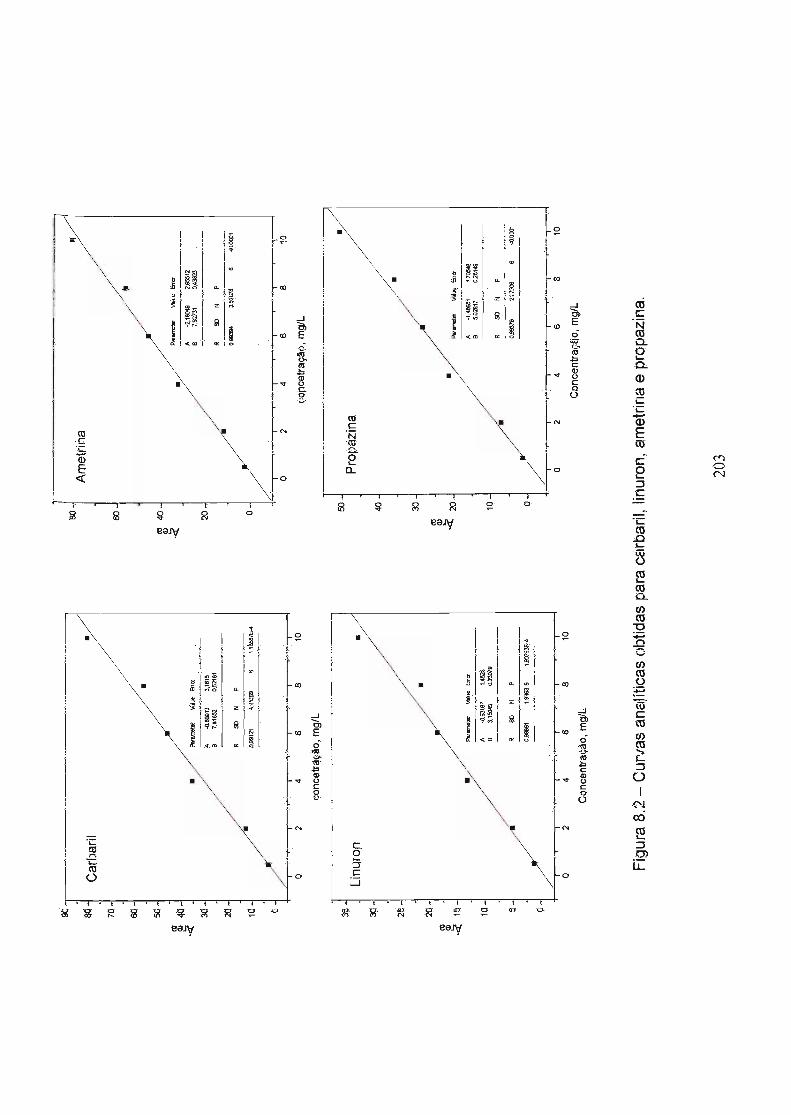

40
35 Propoxur •
30
2S •20 •111
~ 15 """""".. Value Em::r"
•A -1.42993 1.38491B 3.61746 0.22058
10
R '" N P
5 • 0.99211 1.8458:2 6 <0000'
Oa·
O 2 4 6 8 10
Concetração, rrg/L
Figura 8.3 - Curva analítica obtida para o propoxur.
Na Tabela 8.2 são descritas as equações das curvas analíticas obtidas
assim como o valor de robtido.
Tabela 8.2 - Curvas analíticas obtidas para os pesticidas utilizados
-----_ ...._... - -_._-------_._-- - --- .-... _- -------_.
Pesticida
Dimetoato
Carbendazin
Simazina
Atrazina
Carbaril
Ametrina
Linuron
Propazina
Propoxur
Equação da curva
analítica obtida
y = -0,28 + 0,56 x
y =-0,22 + 0,58 x
y = -0,70 + 4,97 x
y=-1,62+7,15x
y = -0,89 + 7,81 x
y = - 2,18 + 7,92 x
Y=-0,63+ 3,15 x
Y= -1,48 + 5,02 x
y = -1,42 + 3,61 x
r (coeficiente de
correlação da reta)
0,978
0,992
0,992
0,992
0,991
0,993
0,988
0,993
0,992
204

Foram obtidos curvas analíticas com coeficiente de correlação
satisfatórios, maiores que 0,99; exceção feita ao dimetoato (r= 0,978) e Iinuron
(r= 0,988).
8.4 - Limite de detecção e quantificação
o Limite de detecção revela a concentração mínima da substância em
análise presente na amostra que pode ser detectada com um certo limite de
confiabilidade através de um certo procedimento experimental. O limite de
quantificação indica a concentração mínima da substância em exame presente
na amostra que pode ser quantificada com precisão e exatidão aceitáveis por
um procedimento experimental. Tanto LO quanto LO podem ser obtidos a partir
dos parâmetros de ajuste da curva analítica (desvio padrão na regressão do
intercepto e da inclinação da reta).
Neste trabalho optou-se por calcular LO e LO com base na relação sinal
ruído (S/r), sendo que LO é a concentração que proporciona um S/R = 3 e LOO
é a concentração que proporciona um S/R = 10. No procedimento utilizado,
uma solução de concentração de 1 mg/L foi considerada e tanto o sinal quanto
o ruído (1/5 da altura do sinal de linha base, Ep-p) foram medidos diretamente
do eletroferograma obtido. O LO e o LO foram então estimados por regra de
três de acordo com as expressões:
Média do sinal do pico (3 injeções)3 x Ep-p/5
Média do sinal do pico (3 injeções)10 x Ep-p/5
1 mg/LLOO
1 mg/LLOQ
205

Foram avaliados os LO e LQ para uma mistura de pesticidas contendo 9
pesticidas na concentração de 1 mg/L. Duas misturas foram avaliadas, onde
uma foi diluída em uma solução contendo 20% metanol e água e outra em 20%
metanol e 30 mmollL de NaCI em água. Este procedimento foi utilizado para a
realização de uma análise comparativa entre a utilização da estratégia de
sweeping em alto EOF e a amostra sem sofrer o processo de pré-concentração
on-line. Na tabela 8.3 são apresentados os valores de LO e LQ para os
pesticidas estudados.
Tabela 8.3 - Valores de limite de detecção e quantificação os pesticidas
estudados.
o valor do rUldo na linha de base para ambas as tecOIcas fOI de 0,02
Pesticidas Sem estratégia de pré- Sweeping
concentração
Alt. pico LO LQ Alt. pico LO LQ
(mg/L) (mg/L) (mg/L) (mg/L)
Oimetoato 0,46 0,13 0,43 1,54 0,04 0,13
Carbendazim 1,15 0,052 0,17 5,73 0,01 0,04
Simazina 0,75 0,080 0,27 4,77 0,01 0,04
Carbaril 1,57 0,038 0,13 8,96 0,01 0,02
Ametrina 0,87 0,069 0,23 4,57 0,01 0,04
Propazina 0,77 0,078 0,26 4,43 0,01 0,05
Diuron 0,42 0,14 0,48 2,27 0,03 0,09
Propoxur 0,45 0,13 0,44 1,84 0,03 0,11
linuron 0,29 0,21 0,69 1,44 0,04 0,14. .
Os valores de limite detecção variaram de 0,038 mg/L a 0,21 mg/L
quando a estratégia de pré-concentração não foi utilizada. Quando a estratégia
de pré-concetração foi empregada os LO variaram entre 0,01 a 0,04 mg/L.
206

8.5 - Recuperação
A recuperação absoluta (R, em %) foi calculada através da equação 1 :
[1 ]
Recuperação absoluta =concentração obtida X 100
concentracão real
Na Tabela 8.4 são mostrados os valores de recuperação obtidos,
calculados a partir da triplicata de injeção para cada amostra.
Tabela 8.4 - Porcentagem de recuperação obtida para a mistura de pesticidas
estudada na fortificação de amostras de cenoura
Pesticidas Nível 1 de fortificação Nível 2 de fortificação
(83 ~g/L) (240 ~g/L)
Dimetoato 57 ± 9 100 ± 4
Carbendazin 52 ± 3 72 ± 7
Simazina 89 ± 7 63 ± 5
atrazina 82 ± 7 74 ±6
Carabril 87 ±7 86 ± 3
Ametrina 80 ±4 67 ±4
Propazina 51 ± 5 67 ± 4
Linuron 51 ± 2 84 ±7
Propoxur 54 ±6 77 ±4
Os valores de recuperação obtidos variaram de 51 a 89 % no nível 1 de
fortificação. E para o nível 2 de fortificação os valores de recuperação obtidos
variaram de 67 a 100 %. Segundo a IAEA6 são considerados aceitáveis
valores de recuperação entre 70 a 110 % para os níveis de fortificação
estudados.
207

8.6 - Precisão dos testes de recuperação da análise de pesticidas em
amostras de cenoura fortificadas em dois níveis de concentração
A precisão é o parâmetro que indica o quanto se repetiram os resultados
da análise sendo esta realizada em uma mesma amostra diversas vezes, sob
as mesmas condições. As repetições podem ser feitas em um único dia
(precisão intra-ensaio) ou em dias diferentes (precisão intermediária ou inter-
ensaio). A precisão é calculada pelo desvio padrão relativo (RSD) ou
coeficiente de variação (CV), expresso pelo a estimativa do desvio padrão da
média (área ou concentração) dividido pela média, ou seja:
CV = (Desvio padrão/Média) x 100
Na Tabela 8.5 são mostrados os valores de % de CV obtidos para
a análise de pesticidas em amostras de cenoura fortificadas em dois níveis de
concentração (83 e 240 Ilg/L).
Tabela 8.5 - Precisão intra-ensaio (% CV) obtidos para a análise de pesticidas
em cenoura em dois níveis de fortificação'-. ". ---_ - - -_ ~p ~- ,", - ~- .~." ,." ...
Pesticidas % CV - Nível 1 de fortificação % CV - Nível 2 de fortificação
(83 Ilg/L) (240 Ilg/L).. --_.'--~---- - --- -- .- - - _._--~ -~---_. -_ .. - _..._---- _._--- ---_.- ..~ _._---
Dimetoato 15 4
Carbendazin 6 10
Simazina 8 8
atrazina 9 8
Carabril 8 3
Ametrina 5 6
Propazina 10 6
Linuron 4 8
Propoxur 11 5
.. --- _. 'Seg'ündÕ'õ' prôtocoio'da -iAEAs-ã' preCisãó"inter~ensaiô' é-'satisfatória nas
seguintes circunstâncias:
208

Faixa de concentração
10 - 100 ~g/kg
100 - 1000 ~g/kg
> 1 mg/kg
CV,%
20
15
10
Portanto, os valores de % CV obtidos para ambos os níveis de
fortificação apresentados na Tabela 8.4 estão abaixo dos limites de % CV
estipulados pela IAEA.
8.6.1 - Repetibilidade (Precisão) de tempo e áreas de pico para a mistura
padrão de pesticidas estudada
Na tabela 7.6 são mostrados os valores de média, desvio padrão e %
CV para as áreas e tempo de migração para os constituintes de uma mistura
padrão contendo 9 pesticidas na concentração de 10 mg/L. Foram
considerados os resultados obtidos de 23 injeções consecutivas.
209

Tab
ela
8.6
-V
alor
esde
área
ete
mpo
de
mig
raçã
opa
raa
valia
ção
dare
petib
ilida
de
--~-~---"'"'''''''''''~--''''~--''-''''''--'''----''--'''''-'''''''''''''-...,.~-_._--~,--_
_.--
----,,~_...
.,--_._
_...-.._~-
_--_._
._..--
__-
----
-_--
_._._._._.~
amet
rina
carb
aril
._-.
-_..'
-'
Car
bend
azin
sim
azin
aat
razi
na•
_••••••
--.
_.--
__
,_
,"._
._
._
._
•••.•••••••
_._
_""'_
.-."_
"_
_~
•••••
_._
_...._
._
.0
.0•••
'"_
_•_
_
tem
poár
eate
mpo
área
tem
poár
eate
mpo
área
tem
poár
eate
mpo
5,18
4,93
16,9
75,
3364
,38
6,67
67,6
48,
4111
1,34
9,13
72,3
510
,33
5,4
4,84
17,1
25,
2365
,17
6,56
66,2
48,
2911
3,88
9,01
872
,58
10,2
35,
464,
8417
,36
5,22
65,0
56,
5666
8,3
114,
159,
039
72,3
510
,26
5,08
4,85
16,9
85,
2364
,41
6,58
65,9
88,
3311
5,49
9,08
70,5
510
,32
5,97
4,86
16,9
45,
2464
,25
6,6
66,4
48,
3711
3,92
9,13
271
,01
10,3
86,
234,
8716
,75
5,25
65,1
16,
5866
,38
8,3
114,
059,
039
71,2
810
,23
5,23
4,81
16,7
45,
1965
,37
6,51
66,6
78,
2211
4,99
8,93
71,8
310
,144
5,58
4,8
16,9
75,
1863
,94
6,51
165
,83
8,22
112,
318,
9471
,43
10,1
65,
134,
7916
,55
5,16
64,9
46,
4966
,38
8,21
112,
548,
9371
,21
10,1
636,
344,
7717
,68
5,14
64,9
36,
4765
,58
8,19
114,
818,
9170
,210
,148
5,93
4,76
17,0
95,
1364
,86,
4366
,41
8,09
811
2,98
8,77
70,8
19,
955,
434,
7117
,06
5,08
63,7
46,
3766
,32
8,03
113,
918,
707
72,2
79,
95,
24,
717
,36
5,07
365
,26
6,37
67,4
18,
044
115,
898,
7169
,27
9,92
5,2
4,66
17,2
45,
013
65,3
46,
266
,19
7,73
111,
838,
3870
,44
9,58
6,43
4,63
17,0
894,
9764
,05
6,18
465
,19
7,76
113,
578,
3670
,43
9,58
6,23
4,65
17,0
5.5
64,0
86,
1965
,83
7,76
112,
618,
3671
,03
9,56
5,93
4,63
16,6
84,
9865
,59
6,17
65,1
47,
7511
5,12
8,35
70,2
9,57
5,67
4,63
17,7
74,
9863
,44
6,18
65,1
77,
7811
5,62
8,38
67,5
89,
615,
224,
6217
,52
4,97
63,4
76,
1866
,26
7,79
113,
88,
470
,27
9,65
6,09
4,63
16,8
34,
9765
,02
6,19
65,6
47,
8111
5,54
8,41
70,1
89,
685,
054,
6117
,87
4,96
64,6
6,16
65,7
7,74
110,
328,
3370
,85
9,53
5,06
4,59
17,1
54,
9764
,95
6,15
64,9
37,
7311
2,15
8,32
69,5
39,
555,
834,
616
,79
4,94
64,1
86,
1665
,39
7,76
114,
388,
3569
,95
9,59
5,60
4,7
217
,11
5,09
64,6
16.
3666
,03
8.02
113,
708,
6970
,76
9,91
0,45
0.10
0.34
0,12
0,62
0,18
0,67
0,25
1,48
0.31
1,14
0,31
_._.~~~_._?:?~~
?,04~
..__.
__?,?~
..__.
,0,9
7_.
__2;..~.
__w.".:!.:.?.~
..__,,_
_~-:.}J_
_..__
..~9..._
.~.
:.~]_
_._.1
,61_
..].:
27
dim
etoa
to.,
"_.
_,_o
_.
área
Méd
iaD
esvi
oP
adrã
oC
V,%
210
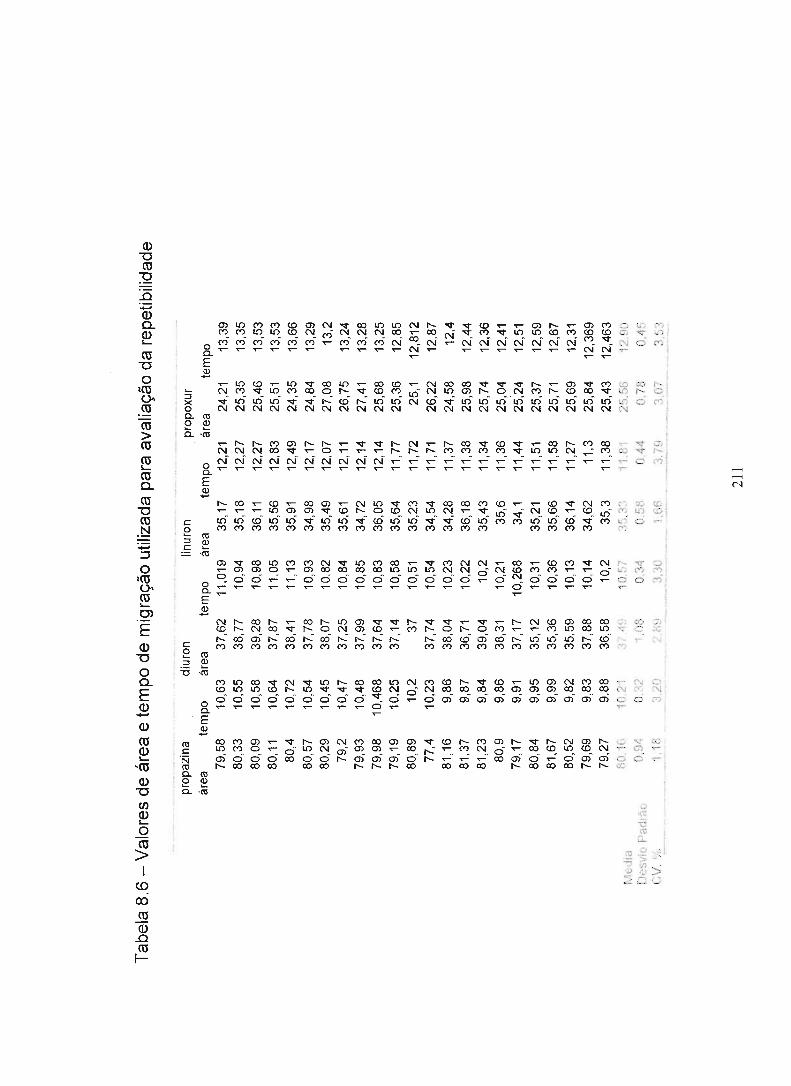
Tabe
la8
.6-
Val
ores
deár
eae
tem
pode
mig
raçã
out
iliza
dapa
raav
alia
ção
dare
petib
ilida
de
-..
0',0
._
or.......
prop
azin
ad
iuro
nlin
uron
prop
oxur
área
tem
po
área
tem
po
área
tem
po
área
tem
po
79,5
810
,63
37,6
211
,019
35,1
712
,21
24,2
113
,39
80,3
310
,55
38,7
710
,94
35,1
81
2,2
725
,35
13,3
580
,09
10,5
839
,28
10,9
836
,11
12
,27
25
,46
13,5
380
,11
10,6
437
,87
11,0
535
,56
12
,83
25,5
113
,53
80,4
10,7
238
,41
11,1
335
,91
12
,49
24,3
513
,66
80,5
710
,54
37,7
810
,93
34,9
81
2,1
724
,84
13,2
980
,29
10,4
538
,07
10,8
235
,49
12
,07
27,0
813
,279
,210
,47
37,2
510
,84
35,6
112
,11
26,7
513
,24
79,9
31
0,4
837
,99
10,8
534
,72
12
,14
27,4
113
,28
79,9
810
,468
37,6
410
,83
36,0
512
,14
25
,68
13,2
579
,19
10,2
537
,14
10,5
835
,64
11
,77
25,3
612
,85
80,8
910
,237
10,5
135
,23
11
,72
25,1
12,8
1277
,410
,23
37,7
410
,54
34,5
411
,71
26,2
212
,87
81,1
69
,86
38,0
410
,23
34,2
81
1,3
724
,58
12,4
81,3
79,
8736
,71
10,2
236
,18
11
,38
25,9
812
,44
81,2
39,
8439
,04
10,2
35,4
31
1,3
425
,74
12,3
68
0,9
9,8
638
,31
10,2
13
5,6
11
,36
25,0
412
,41
79,1
79,
9137
,17
10,2
6834
,11
1,4
425
,24
12,5
180
,84
9,95
35,1
210
,31
35,2
111
,51
25,3
712
,59
81,6
79
,99
35,3
610
,36
35,6
61
1,5
825
,71
12,6
780
,52
9,8
235
,59
10,1
33
6,1
41
1,2
725
,69
12,3
179
,69
9,83
37,8
810
,14
34,6
21
1,3
25,8
412
,369
79,2
79,
8836
,58
10,2
35
,311
,38
25,4
312
,463
ME
:Jlé
l80
.1G
10
21
31'
«1f1
10.5
735
.33
11.8
12
5.5
612
.9C
1
De
svio
Par
jr;:jo
0,94
0.3
2í.
OS
0,34
0.5
80.
440
,78
0,4
5C
V.
'/;,
1.1e
3.Z
02,H~
3'}
P1
,6f'
3.7
93
07
3,5
3,o
JV
--...
....
....
._.
,....
.."
211

Os coeficiente de variação expressos para avaliar o parâmetro de
repetibilidade do método encontram-se entre os valores de 0,97 % a 8,19% que
demonstra que o método apresenta boa repetibilidade.
8.7 - Quantificação de amostra
Foram feitas quantificações para amostras de cenoura adquiridas no
mercado local, porém em dias diferentes. Os eletroferogramas obtidos são
mostrados na Figura 8.4. Para estas avaliações as amostras de cenoura foram
utilizadas inteiras (casca e miolo).
212
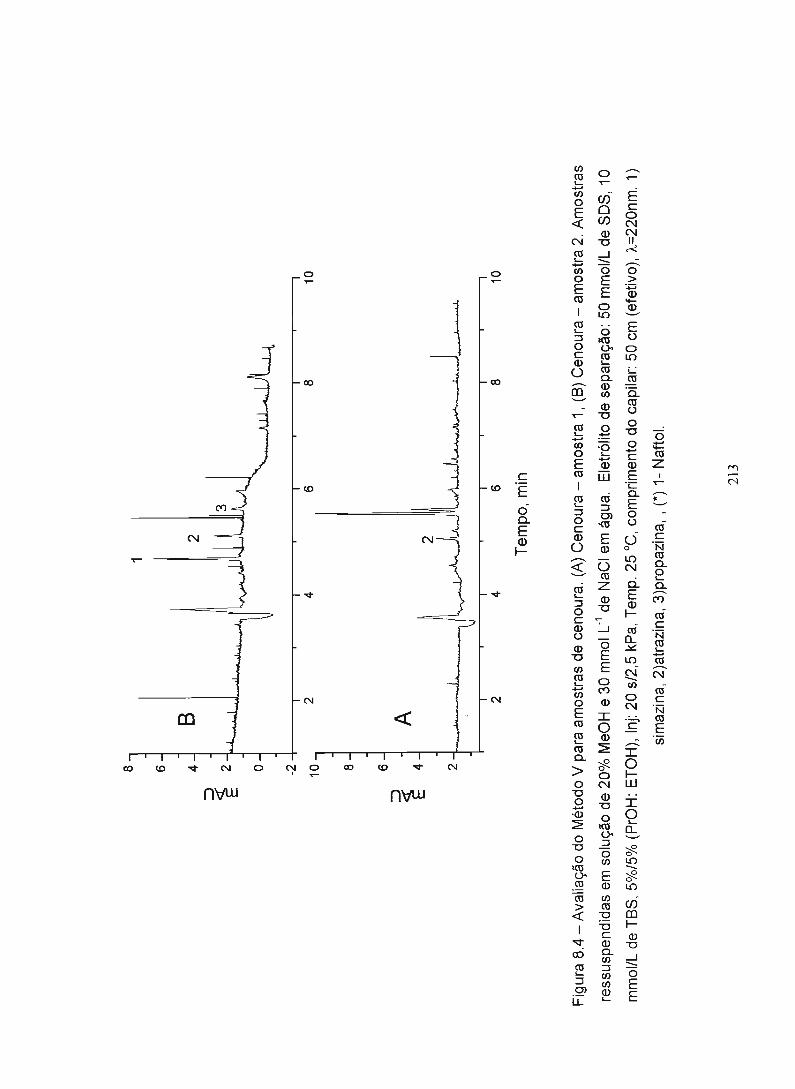
:1I
1
81
~~
2:::> ~
2 O -2,
,,
,
102
46
810
8 6:::>
A~
4 2_..
-_...
--_
.--
~-~
,
24
68
10
Te
mp
o,
min
Fig
ura
8.4
-A
valia
ção
doM
étod
oV
para
amos
tras
dece
nour
a.(A
)C
enou
ra-
amos
tra
1,(8
)C
enou
ra-
amos
tra
2.A
mos
tras
ress
uspe
ndid
asem
solu
ção
de20
%M
eOH
e30
mm
olL-
1de
NaC
Iem
água
.E
letr
ólito
dese
para
ção:
50m
mol
lLde
SD
S,
10
mm
ol/L
deT
8S,
5%/5
%(P
rOH
:E
TO
H),
Inj:
20s/
2,5
kPa,
Tem
p.2
5°C
,co
mpr
imen
todo
capi
lar:
50cm
(efe
tivo)
,À
=220
nm.
1)
sim
azin
a,2)
atra
zina
,3)
prop
azin
a,,(
*)1-
Naf
tol.
213

Para os extratos de cenoura analisados e identificados na Figura 8.4 A
e B foram quantificados os seguintes pesticidas: símazina, atrazina e
propazina. Na quantificação destas amostras foram utilizadas as curvas
analíticas mostras nas Figuras 8.1 a 8.3. Na tabela 8.6 são mostrados os
valores de concentração para cada pesticida identificado.
Tabela 8.6 - Valores de concentração de pesticidas encontrados nas amostras
de cenoura analisadas
Pesticidas
Amostras Simazina Atrazina Propazina
(mg k-1g) (mg kg-1) (mg kg-1
)
Amostra 1 0,88 0,13 0,08
Amostra 2 0,08
* média de duplicata de injeção
214

8.8 - Conclusões
Foram obtidos valores satisfatórios de linearidade (r> 0,99) exceção feita
ao dimetoato (r= 0,978) e Linuron (r = 0,988). Os valores de limite detecção
variaram de 0,038 mg/L a 0,21 mg/L quando a estratégia de prã-concentração
não foi utilizada. Quando a estratégia de prã-concentração foi empregada os
LD variaram entre 0,01 a 0,04 mg/L. Os valores de recuperação obtidos
variaram de 51 a 89 % no níve 1 de fortificação. Para o nível 2 de fortificação
os valores de recuperação obtidos variaram de 67 a 100 %. Segundo a IAEA6
são considerados aceitáveis valores de recuperação entre 70 a 120 % para os
níveis de fortificação estudados. Os valores de % CV calculados para a
recuperação ficaram abaixo de 15% em ambos os níveis de fortificação,
estando dentro do especificado pelo IAEA. Os valores de % CV para 20
injeções consecutivas ficaram abaixo de 3,8% para os componentes da mistura
padrão, exceção feita ao dimetoato onde seu % CV ficou em 8 %.
215

~~U
kJ~
t~.
1~
u
~t~, ,~~ 'B
u1......... -J
CAPÍTULO 9

9 - Conclusões
As separações eletroforéticas conduzidas tanto em condições de alto
EOF (pH 9,3) e baixo EOF (pH 2,5), foram otimizadas, para uma mistura
padrão de pesticidas de diferentes classes. Verificou-se que os n-alcoóis
proporcionam mudanças na estrutura das micelas presentes no eletrólito de
separação, mudando características modo: emc, constante dielétrica da
camada palasídica das micelas, número de agregação e ionização. A medida
que o tamanho da cadeia do álcool aumenta as modificações nas estruturas
são maximizadas, pois estes álcoois interagem fortemente com as micelas, e
ficam "ancorados" entre os monômeros dos tensoativos, aumentando a
repulsão eletrostáticas das cabeças polares, contribuindo para a ionização do
agregado.
Os métodos de pré-concentração on-line empregados em MEKC,
principalmente quando realizados em baixo pH (2,5) apresentam melhores
limites de detecção quando comparados as técnicas em altas condições de pH.
O m-SRMM técnica híbrida da SRMM apresentou valores de limites de
detecção entre 2,7 a 35 IJ.Ug, para a mistura de pesticidas padrão composta
por : ametrina, carbendazim, atrazina, propoxur, propazina, diuron, linuron,
simazina e carbari!. Estes valores foram comparados com as estratégias de
sweeping e SRW, os limites de detecção encontrados para estas estratégias
variam de 5,2 a 43 para o sweeping e 5,3 a 46 para o SRW. Estas estratégias
216

de pré-concentração foram empregadas em amostras de água e cenoura. Foi
possível atingir níveis de concentração da ordem de 0,1 ppb unindo as
estratégias on-Iine e off-line de pré-concetração para amostras de água. Já
para amostras de cenoura utilizando como metodologia de extração o método
111, foi possível a detecção da ordem de 2,5 ppb.
Várias fases estacionárias foram avaliadas com o intuito de realizar o
clean-up nas extrações de pesticidas em frutas e leguminosas. As fases que
apresentaram os melhores resultados de recuperação foram C18 (RP) e NH2
(NP). Porém o NH2 apresentou melhor performance para a realização de clean
up em amostras reais. O Charcoal:celite foi específico para um pequeno
números de pesticidas estudados. Foram empregados dois sistemas de eluição
onde os eluente eram MeOH, MeOH:CH2Cb (1 :99). Para os eluente MeOH e
MeOH:CH2Cb, os resultados foram satisfatórios e similares, porém optou-se
pela utilização de MeOH:CH2Cb para a realização das demais eluições em
amostras reais, pelo fato deste apresentar menor temperatura de ebulição,
sendo assim muito mais propício para a evaporação.
A metodologia de extração MSPD apresentou boa seletividade e baixo
nível de ruído na linha base dos eletroferogramas obtidos, indicando que o
processo de clean-up foi eficiente. Valores de recuperações variaram entre
52,6 e 89,0 % para extrações no nível mais baixo de fortificação e 62,8 a 100,4
% para o nível mais alto de fortificação. Para o MSPD, dois itens devem ser
217

levados em consideração para se obter melhores resultados: o adsorvente
deve ser pré-lavado para remover potenciais interferentes e a co-coluna de
clean-up deve ser pré-condicionado com um solvente orgânico apropriado. O
método MSPD apresentou várias vantagens em relação aos outros métodos de
extração e c1ean-up avaliados, pois este método é mais simples, permite uma
interação maior entre os analitos e a fase adsorvente empregada, o consumo
de solventes é reduzido e neste procedimento de extração a formação de géis
ao redor da fase adsorvente é bastante minimizado em relação aos métodos
avaliados. Evidenciou-se que o processo de c1ean-up realizado com cartuchos
de fases adsorventes atinge a saturação muito facilmente, porém no método
MSPD a quantidade de adsorvente adicionados à amostra é uma variável,
podendo ser alterada e, portanto, o processo de saturação pode ser evitado,
adicionando-se a amostra quantidades maiores de fases adsorventes.
O procedimento de extração em meio micelar mostrou ser um
ametodologia bastante atrativa:
~ Alta capacidade de pré-concentrar analitos de diferentes polaridades.
'ji> O fator de pré-concentração pode ser otimizado por modificações do
tipo e da concentração do tensoativo utilizado, bem como as condições
de análise em que esta extração ocorrerá.·
'ji> Os tensoativos são possuem baixíssima toxidez e são mais baratos
quando comparados ao uso de extrações líquido-líquido.
218

~ Os tensoativos mais comuns são facilmente encontrados no comércio
especializado.
> Não é necessário evaporar solventes, minimizando assim perda por
evaporação do analito.
~ As operações experimentais envolvidas nesta metodologia são fáceis, e
a fase rica em tensoativo é compatível com várias técnicas analíticas
(HPLC, CEC, CE, etc.)
Porém a maioria dos tensoativos empregados absorvem na região do
UV, o que impossibilitaria a sua utilização para a extração de uma gama maior
de pesticida. Este problema pode ser contornado, pela utilização de tensoativos
zwiUeriônicos ou polímeros que não absorvam na região do UV.
A CE mostrou-se ser uma técnica de separação alternativa viável para
emprego em análises de pesticidas em amostras de água, frutas e tubérculos
com tempos de análise reduzidos quando comparados à técnica de
cromatografia líquida de alto desempenho (HPLC) e cromatografia gasosa
(CG). Além de ser uma técnica bastante versátil onde separações utilizando-se
o meio aquoso ou solventes é passível de realização.
9.1 - Perspectivas futuras
Explorar as técnicas de extração em meio micelar, empregando
tensoativos zwitteriônicos, iônicos e outras espécies de neutros. Estudar a
219

influência da adição de sais inorgânicos e solventes orgânicos na performance
de extração e pré-concetração. Estender o emprego desta técnica (cloud point)
para um maior número de frutas e pesticidas. Unir a extração por cloud point às
estratégias de pré-cocentração estudadas nesta tese de doutoramento.
220

~~U
l4J~
i
~
u
~t~, ~~~ 'B
'-.)1.....--.. -J
ANEXO 1

GUIDELINES FOR SINGLE-LABORATORY VALIDATION DF ANALYTICALMETHODS FOR TRACE-LEVEL CONCENTRATIONS OFORGANIC
CHEMICALS
Table of Contents
~~~ 1
Table 1 Summary of parameters to be assessed for method validation 5TabIe 2 Within laboratóry performance criteria for quantitative organic trace
an~y~s 1Table 3 Suitability of detection methods for theeonfrrmatorydetermination
of substances at trace leveIs 1Figure 1 Overview of Method Validation 9Figure 2 Verification of Analyte Stability 9
Annex 1 Method v~idation for pesticide residues 10Annex 2 Methodvalidationfor residues ofveterinary drugs 25Annex 3 Worked example of method validation, as applied to a method for
pesticides residues . 39
Annex 4 Statistical methods applicable to validation of analytic~ methods 51Annex 5 GIossary and list of abbreviations 64
Annex 6 DeveIopment of principIes for assuring reliability ofanalytical results 10
Overview
1 Whatis method validation?
An analytical method is the series of procedures fiom receipt of a sample to the production ofthe fmal resulto Validatien· is- the process ofverifying thata method is fit for purpose.
2 Purpose of this document
Method v~idation guideIines for use in trace an~ysis have been proposed by various authorsbut there is little consistency in the recommended approaches. The general guidelines formethods of chemical analysis proposedby standards orgamsations are impractical for use inIaboratories involved in monitoring of trace leveI organic chemicals.The validity of methods has often been established on the basis af inter-Iaboratory studies ofperformance but that process can be uneconomic and toa slow or restricted· in scope.Vali~ation in a single laboratory can provide a practical and cost-effective alternative (orintermediate}. approach.
The guidelines were elaborated by the AOAC/FAOIIAEAlIUPAC Expert consultation with-thepartidpatiofiofL. Ald-er, AHill,P.T.-Holland, J; Lamas, S.M. Lee,-J. D; MacNeil, 1.--0 'Rangers;P.vanZoonen, and A.Ambrus (Scientific Secretary) The background of the elaboration of the Guidelines is described inA __ ~•• ~

OrgilDit Chemieals
These Guidelines identify adequate minimum requirements for the validation of methods afana1ySis foc"trace leveIs" iH ''6fgaftkdremica1s,m1}r~toverify thatamethodis'tltf1}fpurpose, according to the current general requirements for the reliability of analytical data.
Tliis document afms to assist analysts to perform method' validation in a scientifieaIry sound-and practical manner, and national authorities and accreditation bodies in assessing.acceptability.ofiliemethods .developedcand·3pplied in testing Jabor-atories.
The requirements idenüfred here Me intended· fOfguidanee, t{)-·be- applied- pragmatieallYFatherthag" prescrlptively. 'Ih€!: analyst .slmuld address.- only tlmse.: requirements.: appropriate to, lhe..method and the purposes for which the method will be used. They may be modified oradapted, 'as necessaryfor the 'specitlc purpose. .
3 Relationship between method validation, method development, quality control,proflciency testin~ and reportina ofuncertain~
The method may be deveIoped in-house, taken from the literature or otherwise obtained froma thirdj>3l1y. The method ma'y .then be adapted ormodifled tomatchtherequirements.andcapabilities of the Iaboratory and/or the purpose for which the method will be used.
TypicaHy, validation follows completion of the' developrnent of a method and it is assumedthat requirements- such·as- calibratfon., system· suitatiili'ty, analYte stabilHy; etc., liave- ·hemestablished satisfactorily. When validating and using a method of analysis, measurementsmust be made within the calibrated range of the detection ~tem used. In,general, validation
. WI11 precede practic3l appncation ofthe method to lhe analySis 6f samples'btit siíbsequent.internal quality controI (IQC. refcrred to in the present document as performance verification)is. an im~()rtant continuing,aspect of the. process.. Re«pJire1UeJlts. foI: penor-mance veclficationdata1
,2,3, ,5 have been identifled as a subset ofthose required for method validation.
Proficiency testing-4·6,1,8 (or other inter-laboratory testing procedures9,lO,l1), where practicable,prevides anitnportant means for -verifying the 'general accuracyofresults ·getterated"byamethod, and provides information on the between-Iaboratory variability of the resuIts.However, proficiency testing generally does not address analyte stability or homogeneity andextractability of analytes in the processed sample.
Where 1IIlcertainty data must be reportedto-1he"client·(the· purchaser 'Of llSer of-resuhs), this:'informanon sbould -incerporatepetformanct! -venflCaâondata and not --rely"sôlélronmeth6dvalidation data.
4 Fitness forllurpose-ofmethods and-results
Analytical methods, and the validation required for them, must be appropriate to the purposefor which the results are likeIy to be used. In general, method "performance and validationreqtiirementS slmuld'OO agreed' -between -the arialyst aild the client"{lhe puréhaset/user ofresuIts, as appropriate). Method performance requirements have been defined by
tal 4-t.. ~p~n 12,13,14,15 db A 'O' A 'C T_4- ti al5,16govemmen .a,UUlQ'[.lI;J.J;S. an . y~ \t:"\I . .I.l.U,ema on .. .-

General guidelines for method validation ·have been developed by. various'OI'ganisatíons.4•5.;17.UI;l9;l9.20.21 Speclnc iguidelines 4mve;,been ~developec!t .for single:-laboratoryvalidation of methods for pesticide residues22 and are under development for residues ofveterinary drugs in food and animal feeds23
•
Methods and analytical results are often classified loosely as quantitative, semi-quantitative or. .qualitative,(screen~g).. These Mlt~ories do .not have well.,defmed oruniversally a~edbooodaries. The method..performance criteria given in theseGuidelines indicate .aeceptabilityfor quantitative data, but the boundaries to be applied in practice should be agreed between theana1yst and~ the~:client..
... 5 Limits to the scope ofvalidation
Extensions of methods to new analytes, matrices (eg.,. additional tissues or commoditygroups), much lower concentrations, much smaller test po~ions, use of the method in otherlaboratories, etc., shoulo be vafidated' as ihdícated' In tlUs document. Minor cnanges Ül
methods may be validated through performance verification but, where the change leads to.unsaüsfacto~ performance,..the metOOd maY ,requife,modification ,and re-:validation, asindicated here.
For multi-analyte and/or multhmatrix methods, it is likely. tob~ impr~ctical to. validate amethotlfor allcomDBia~ofanaJyt~ coneematitm'and·-tJpe otsample"matmt tÍlaf may..be--.·encountered in subsequent use of the method. InitiaI validation should incorporate as many ofthe target analytes and matrices as practicable. However it is proposed that a method may bevalidated'iriiüãl1y'for "'represerttative matrices'" arid, "'represerttàtive ana1ytes" . This can limit.the costand time required before samples are arialysed, while establishing the keyperformance characteristics of the method. Subsequently, performance validation data must begenerated to establish method performance for alI other analytes and matrices for whichresults will bereported.. Where the performance verifiçation data indicate that method-per.formanceis -·notadequate.the,methodmaybemodified.as·:apprnpriateandsubsequentlyvalidated. If the modification is not successful, an alternative method is necessary.
6 Requirements for validation
Diagrams of lhe decision processes (Figures I, 2) and parameters to be assessed (Table I) indetermining overall validation requirements are presented in this document.
The amount of effort allocated to the validation of a method may vary considerably. Whereappropriate. data, are already available,. it may; not be nece~. for. the analyst. to, perfonn al1the tests. However, alI required information must be included or referred to in the validationreport.
. 6.. 1. Parameters to be assessed should be restricted to those which are appropriate both tothe rnethod and to the purpose for which the particular method is to be applied. Inmany cases, perfonnancecharacteristicswitlírespecttoseveralparameters maybe' '.obtained simultaneously using a single experiment. Test designs where differentfactors are changed at the same time under statistical control (factori~l1 experiment

6.2. The criteria to be applied are likely to be specific tothe purpose forwhich the methodis te'be uSed. EXamples are given. m, Alméxes t andZ.,. descrlDmg- valiUation. oImethods for the determination of residues of pesticides and veterinary drugs.respectively. Different criteria may be defined according to the purpose. If the
·0 d·· d -ét1t-.'~':Sm...~1J '.-h-oi ~.n.. , ct3ti.o-o· oo -b ..t ih ~ ••-ilti-r~, 'foo-;;tori"oo oiIeo ÇSJre .. ena C,Q,WAI 00 e~~ ç.J..JJ.JQ~ 0_0 •• 0 QDS a .Oi.fl. ,.f q~l-'y Q ""p~ mus:t o
revised or an improved method must be sought.. 00 o oooo o .0
00
• o o o
7. General-criteFia-- fOF~' multi~analyte' metbo~,for' pestieide-and- veterinary- drug o
residues o
7.1 lhe generáI performance criteria required Ior analysis of pesticide and veterinary drugresidues are summarised in Table 2. Deviation from these criteria may be acceptablefor certain.analytes aJid:.matrkes..wheDo~owith:appráPriatejusfifi€ation~:o.
The analytical techniques or their combination listed in Table o3 are generally suitable for thecORfirmatioIl'Qfa1la1ytes'intést~ples;oNatioDa1authorltiesimay-setoparticJ;llai:reqUirements
for specified compounds. Analysts should be aware of such requirements when conductingmethod validations to demonstrate "fitness for purpose". .
A

Tab1
e1.
Sunu
nary
ofpa
ram
eter
sto
beas
sess
edfo
rm
etho
dva
lldat
ion
-Ex
istin
g~alytkal
inet
hod,
forw
lúch
prev
ious
test
sof
theparamet~
have
show
nth
atit
isva
lidfo
ron
eor
mor
ean
alyt
e/m
atrix
cbm
bina
tions
Mod
ifkat
ion
ofáI
lN
ewm
etho
d ino
tyet
Expe
rimen
ttyp
esPa
ram
eter
sto
bete
sted
exis
ting
met
hod
valid
àtéd
whi
chm
aybl
;lco
tnbi
ned
Perf
onna
nce
Add
ition
al'A
dditi
oila
lM
uchl
owet
Aqo
ther
vetificatio~*
mat
rixan
alyt
eco
ncen
tratio
illa
bora
tory
ofan
alvt
e,Sp
ecif
idty
(sho
wth
atN
ó(p
ro-
Yes
,ifi
nter
-Y
esY~:
ifin
ter-
Rig
otou
sY
esor
No.
Rig
or·
Ye~.
Rig
orou
sch
ecks
the
dete
Cte
dsi
gpal
isvi
ded
crite
-fer
enc:{
!fro
mfe
renc
efro
mchec~
not
ousch~cks
may
bem
aybe
nece
ssar
yif
due
totli
éan
alyt
e.no
tria
form
a-m
atrlX
isap
-~trixisap~
nece
8sar
yif
nece
ssro
rift
heth
e~etennimition
anot
her
com
poun
d)tr
ixb1
anks
pare
nein
QC
pafe
ntin
QC
the
PEtrf
Ol'I&
dete
rmW
atio
nsYs~
sys~em
isdi
ffer
ento
t,
and
conf
ir-an
ce(I
fthe
tem
isfl,
Uld
amen
-w
here
the
éxte
ntof
Irlat
ion
ofde
eern
unat
ion
tally
diff
eren
tor
inte
rfer
ence
sfro
mth
ean
ályt
ear
esy
stem
isw
here
tbe
exte
ntof
rria
~ces
are
tiílc
erta
in.
met
)sim
ilétf
orin
terf
eren
ces
trom
coni
pare
dw
ithex
ist-
bette
rth
ema~
isW
l-in
gm
etho
dsce
rtain
Ana
lytic
alR
ange
,Y
esY
esY
esY
esY
esY
es"
'
Ye~
Cal
ibrá
üon
rang
!3-
Rec
over
ythr
ough
anal
ytic
alra
nge-
extra
ctid
rt,cl
ean-
up,
LO
OItO
Q-I
tlatri
xde
rivat
i$at
ion
and
effe
ct.
mea
sure
j:J;le
ntC
alib
ratió
nra
nge
for
NoN
oY
esY
esY
es,f
orre
p-Y
es,f
Q.rr
epresert~
Y~,
forr
epre
sent
ativ
eLi
near
ity,r
epro~
dete
rmirí
atio
nof
aJla-
rese
ntat
ive
tativ
e~alytes
anal
ytes
duci
bfl;j
tyan
ds~g-
Me
,an
alyt
esna
l/noi
seL
OD
andL
OQ
,"
NoY
es,(
parti
alY
es,p
artia
lY~
Yes
",Y
esYe~
Low
estc
alib
rate
dif
mat
tixis
for
rept
e-le
vei,
ând
Iow
leve
Ifr
omá
repr
e-se
nted
spik
eie
cove
ryd,
atase
nted
clas
s)an
alvt
esR
epor
tirlli
Lim
it.LC
LY
áSN
oN
oN
oN
oN
oN
oA
naly
test
abili
tyin
NoY
es.ü
nles
sY
es.m
tless
Yli$
No
No.
unle
ssexttac~
Yes
.ife
xtta
ctlo
nlfm
alth
ean
aiyt
etio
nlfiÚ
atso
lveó
tIs
..~
SélII
lJlle
extra
cts*
tmà
mxis
from
solv
enti
sdi
fl'ér
ent
are
ptes
ente
dis
repr
e-di
fl'er
ent,
ortll
efro
Ijtth
atus
edin
ancl
ass
sene
edc1
ean-
4Pis
Iess
exi~ting
mét
hod,
orth
estrin~ent
c1~n-up
isIe
ssst
rin-
5

,Exi
sting
anal
ytic
alm
etho
d,fo
i'w
hich
prev
ious
test
sof
the
para
met
erha
vesh
own
that
it18
valid
for
one
orm
ore
anal
yte/
mat
rixtó
mbi
natio
nsM
odifi
çatio
nof
anN
ewm
etho
d.no
tyet
Exp
enm
entt
yP,es
,Pa
ram
eter
sto
bete
sted
exis
ting
met
hod
valid
atêd
whi
Chm
aybe
com
bine
dPe
rfor
man
ceA
dditi
onal
Add
itibn
alM
uchl
ower
Ano
ther
verif
icat
ion*
mat
rix
anaI
yte
cO,n
cent
ratio
illa
b,Q
rato
ryof
anal
vte
gent
,com
pare
dw
ith.
exist
inJ!
met
hQds
used
.A
naly
test
abili
tydu
r-Y
esY
esY
es,
Idea
llyN
oN
oN
o'in
gsa
mpl
est
orag
e.o
"
"
Extra
ctio
nef
ficie
ili::y
.+N
oId
éally
Idea
llyId
eally
No
No,
UI)1
essd
iffer
-YeSiunlesspteviou~y
ente
xtra
ctio
nco
o-te
sted
extra
ctib
ndi
tions
~Iilployed"
proc
edur
e1s
Use
d.H
omog
enei
ty.o
f"
Yes~
No.
unle
ssth
eN
oNo
No,
\tJüe
ssth
eN
o,un
J.ess
the
Yes
,unl
ess
á.pr
evi-
'"
See
belo
wan
alyt
icaI
sample~
mat
ri"15
equi~JIlent
isequip~ntis
ousl
yte
sted
sam
ple
subs
tant
ially
chan
ged
,change~
proc
essi
ngpr
oced
ure
diff
eren
tis
used
'Ana
lyte
stab
ility
illN
oY
es,u
nIes
sa
Yes
,w1l
ess
Idei
illy
No
No,
unJ~ss
proce~
No,
únle
sspr
oced
ure
sam
ple
proc
essin
&*
repr
esen
ted
are
ptec
dure
invo
lves
invo
lves
high
erte
m-
Rep
eata
bilit
y,re
-m
atri"
sent
edhi
gher
tllm
pera
tlire
,per~ture,
lóng
ertim
e.pr
odud
bilit
yan
alyt
e10
nger
Um
e,fin~r
conu
ninu
tion,
coar
serc
omminu~
etc.
than
valid
ated
tion,
etc.
proç
edur
es.
*On-
goio
gqu
ality
cont
rol
•If
rele
vant
info
rmat
ion
isno
tava
ilabl
e,
tR
epre
setlt
ativ
eap
alyt
esma
ybe
chos
enon
the
basi
sof
hydr
olys
iS.o
xida
tion
and
phdt
olys
isch
arac
teris
dcs
oSt
abili
tyda
tain
Jon
repr
esen
tativ
eco
mm
oditi
essh
ould
prov
ide
suff
icie
ntin
form
atio
o.A
dditi
'?pal
test
sar
ere
quire
d,fo
reX
ampl
e,wh
f,!re
:(a
)sa
mpl
esar
est
ored
beyo
ndth
eU
me
perio
dte
sted
(eg.
stabi
liíY
test
edup
to4
wee
ksan
dm~urable
aita
lyte
loss
occu
rsdu
ring
this
perio
d,sa
mpl
esqQ
tana
lyze
dun
til6
~,
~"
(b)
stab
ility
test
sW
ere
pert'
orm
edat
:Ç-1
8°e
,but
the
sam
ples
are
stor
edin
the
labó
ratil
ryat
::;;5
°C;
(c)
'sam
ples
are
norm
ally
sior
edat
::;;.,.
.15°
C,bu
tsto
rage
~emperatlÚ'e
rises
to+5
°C).
•In
foln
latio
non
effic
ienc
yof
extra
ctio
nm
aybe
avai
labl
efro
mth
em
anuf
açtu
rero
rcom
pany
'Vhi
chiS
regi
ster
ing
1he
com
potm
d.~
Occ
asió
nally
~th
repe
ated
analys~
ofte
stpô
rtion
sof
posi
tive
siunp
les.
",.
6

Org.anic Chemicals
Table 2. Within LaboratoryMethod Valid.ation Criteria for Analysis ofpesticide_ -r-esiduesand vet~dnar-y-dr-ugsl
Concentration Repeatability Reproducibility Truenessl/:·
CVA%3 CVL%4 CVA%j CVL%4 Range of mean%recovery
~1 M'kg 35 36 53 54 50-120
> 1J.Lg!kg ~ 0.01 mglkg 30 32- 45 46 60-120-
> 0.01 mglkg ~ 0.1 mglkg 20 22 32 34 10-120
> 0.1 mgllçg ~ 1 uyy'Içg 15 18 23 25 70-110
> 1mglkg 10 14 16 19 10-110
1. With multi-residue methods. there may be certain analytes where these quantitative performance criteriacannot be strictly met. The acceptabllity of data produced under these conditions will depend on the purposeof the analyses e.g. when checking for MRL compliance the indicated criteria should be fulfilled as far asteclmiccilly possibl~. While any data well-below the' MRL may-be acceptãble With the-higber uncertéiinty.
2. These recovery ranges are appropriate for multi-residue methods. Stricter criteria may be necessary forsome purposes e.g. methods for single analytes or veterinary drug residues (seeCodexV3. 1996).
3~ CVA: Coefficient-ofvariation- for-analysis excludingsample processing. Tbe parameter can be estimated.from tests performed with reference iIlaterials or ana1ytical portions spiked before extraction. Areferencematerial preparedin thelaboratorymay.be used in tbeabsenceofa certified reference material.
4. CV:L: Overall coefficlent Dfvariation ofa laboratory resulto allowmg tlP to 10% variabllity ofsampleprocesSing. -
Table 3. Examples of detection methods suitable for the confirmatory analysis ofsubstIDlEes-
Detection method Criterion
LC or GC and Mass spectrometry if sufticient number of fragment ions are monitored
.LC-DAD if the UV spectrum is characteristic
LC - fluorescence in combination with other techniques
··2-D TLC - (&pectr~photometry) - in combination with other techniques
GC-ECD. NPD. FPD only if combined with two or more separation_techniques1
Derivatisation if it was not the first choice method
-L'C-imffiUIlQgl'aRl in combination with other techniques
'~ç-úVNIS (single wavelength) in combination with other techniques'-
1. Otber chroma1ographic systems (applying stationary and/or mobile phases of dilferent selectivity) or othertechniques. ' .
7

8. References1. CodexÃJimentarius. Fm>d::and,Agricultur.eOr.ganisatiooeftlle,UDitedNations. Rome.
1993. voI. 2, pp. 405-415.2. AnnualRepor! oflhe WorlángParty on Pesticide Residues: 1992, Supplement to The
Pestiddes:R-egister-. HMStàtioneryOffke; Loooooi 199'3; pp. 12:.~~3. Codex Alimentarius Volume 3 - 1995. Residues ofVeterinary Drugs in foods.4. Thompson, M., & Wóod, R., Pure & Appl. Chem., 6.7; 1995, .649.
, 5. AOAC Official Methods Program, Quíck & EasyAssociateReferees Manual onDeveIopmen(, Study, Review and ApprovaJProcess, 1995. AOAC International, 481
6.. PockUngiM. w~ D:, P,i1re & Appt Chem.., Vot 62~No.1.. pp. t49- 162.,199f:L7. Thompson, M., & Wood, R., Pure & AppI. Chem., 65,1993,2123.8. Thompson, M., & Wood, R., j. of AOAC International, 76, 1993, 926-940, 1993.9. Uorwitz,W., P.ure & AppLCbem.• iO, 1988.iã5-.864.10. Horwitz. W., Pure & AppI. Chem.• 66, 1994, 1903.11. Horwitz, W .• Pure & Appl. Chem" 67,1995,331. , ,12. Anonymous. CiJuncil1jitec(jve 96/23/ECof2!/Apri/19!Jô, GlNo. L lZS·Z3.5:96,p ItT
3213. Hill, A.R.e.• Qualíty controIprocedures forpesticide residues ana!ysis -guideJines for
residues monitoringin the European Union. Document78261VU97, EuropeanCommission, Brussels.1997.
l4- European Commission,. 1999., Guidançe DoCJ.llDellt (U,l:R'esidue. A~ytical met;hods.. VI~"II.1 8064NI197-rev 4 15.12.98. " '" ~ , , .
15. US Environmental Protection Agency Office ofPesticide Programme, Tolerence MethodV.alidatiQn SQP: At ']'MV-l
16. AOAC Peer Verified Methods Policies andProcedures, 1993, AOAC Intemational; 2200Wilson Blvd, Suite 400, Arlington, Virginia 22201-3301 USA North Frederick Avenue,Sune 500., Gaithersburg. MD 2.()S.11...24'11 USA
17. AOAC Peer-Verified Methods Programme Manual on Policies and Procedures,Checklistfor Method DevelQpment, Characterization and Rugedness Tes~g for Peer-Verified'MetOOdStibm:ission - Methoôs forChemical Residues 'in 'Foods
18. EURACHEM, The fitness for Purpose of Analytica1 Methods - A Laboratory Guide toMethod Validation and Related Topics Guide 3.0, prepared by David Holcomb.
19. Wood. R., Harmonised GLfor in-house methodvalidation (lUPAG; inpreparation)20. Bravenboer ], Putten A. v.d., Verwaal W., van Bavel-Salm M., Validation ofmethods,
,Inspectorate_for.Health,Protection.:Report No..95,.D01,NL~ .199:521. Nordic Committee on Food Analysis, NMKL, Validation oí cheniical anatytical methods,
NMKL Procedure No. 4.,1996 issued in February 1991. '22. HiU·A. R.e.• Reynolds S.L.. 1999, Guidelines. for m-HOuse Validation of Analytical
Methods for Pesticide Residues in Food and Animal Feeds, Analyst, 1999, 124, 953-958.23. European Commission: Commission Decision laying down analytical methods to be used
'for detetting cettâin stibstances and residuesthereofinliveammáls,aad animal proouctsaccording to Council Directive 96/23/EC, Final Version: approved by CRLs
24, Jülicher, B., Gowik, P. and Uhlig, S., 1999, Analyst, 124,537 - 545.25. Gowik, P., ]ülicher, B. and Uhlig, 5.,]. 1998, Chromatogr. B, 716, 221 - 232~
26. ]ülicher, B., Gowik, P. and Uhlig, S., 1998, Analyst, 123, 113 - 119.2"1- Youden.&Steiner,(19.75)StatisticalManualoftheAOAC~AOAC International.
Gaithersburg, MD.
8


















