
Universidade de Lisboa Faculdade de Farmácia Departamento de...
244
Universidade de Lisboa Faculdade de Farmácia Departamento de Farmácia Galénica e Tecnologia Farmacêutica Development of a Melanoma Therapeutic Vaccine Candidate by the Coencapsulation of Melanoma Antigen Peptides and Toll-like Receptor Ligands as Adjuvants into Polymeric Nanoparticles Joana Catarina Mendão Azeitão da Silva Doutoramento em Farmácia Especialidade em Tecnologia Farmacêutica 2014
Transcript of Universidade de Lisboa Faculdade de Farmácia Departamento de...

Universidade de Lisboa
Faculdade de Farmácia
Departamento de Farmácia Galénica e Tecnologia Farmacêutica
Development of a Melanoma Therapeutic Vaccine Candidate by
the Coencapsulation of Melanoma Antigen Peptides and Toll-like Receptor Ligands as Adjuvants into Polymeric
Nanoparticles
Joana Catarina Mendão Azeitão da Silva
Doutoramento em Farmácia
Especialidade em Tecnologia Farmacêutica
2014


Universidade de Lisboa
Faculdade de Farmácia
Departamento de Farmácia Galénica e Tecnologia Farmacêutica
Development of a Melanoma Therapeutic Vaccine Candidate by
the Coencapsulation of Melanoma Antigen Peptides and Toll-like Receptor Ligands as Adjuvants into Polymeric
Nanoparticles
Joana Catarina Mendão Azeitão da Silva
Tese orientada pela Prof.ª Doutora Helena Isabel Fialho Florindo Ferreira, especialmente elaborada para a obtenção do grau de doutor no ramo de Farmácia, especialidade de Tecnologia Farmacêutica

This thesis was supervised by:
Prof. Dr. Helena F. Florindo (supervisor)
Assistant Professor
Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia,
Universidade de Lisboa, Lisbon, Portugal.
Prof. Dr. Mafalda Videira (co-supervisor)
Assistant Professor
Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia,
Universidade de Lisboa, Lisbon, Portugal.
Prof. Dr. Véronique Préat (co-supervisor)
Vice-Rector
Louvain Drug Research Institute, Advanced Drug Delivery & Biomaterials,
Université Catholique de Louvain, Brussels, Belgium.
This work was developed at:
Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia,
Universidade de Lisboa, Lisbon, Portugal.
&
Louvain Drug Research Institute, Advanced Drug Delivery & Biomaterials,
Université Catholique de Louvain, Brussels, Belgium.
Joana M. Silva was financially supported by Fundação para a Ciência e a Tecnologia,
Ministério da Educação e Ciência, Portugal (PhD Grant SFRH/BD/64295/2009). The work
reported in this thesis was financially supported by PTDC/SAU-FAR/119389/2010, Pest-
OE/SAU/UI4013/2011 and by Fonds de la Recherche Scientifique Médicale, Belgium.

Ao João
À minha família


Preface
v
Preface
“There is plenty of room at the bottom” was a lecture given by Richard Feynman at an
American Physical Society meeting at Caltech on December 29, 1959. Feynman considered
the possibility of direct manipulation of individual atoms as a more powerful form of
synthetic chemistry than those used at the time. The talk is considered to be a seminal event in
the history of nanotechnology, as it inspired the conceptual beginnings of the field decades
later. More than half a century before that (1890), the New York surgeon William B. Coley
successfully treated some patients with sarcoma using bacterial toxins, the so called Colley’s
toxins. Other medical pioneers including Robert Koch, Louis Pasteur, and Emil von Behring,
had recorded observations of erysipelas infection coinciding with cancer regression. In 1909,
Paul Ehrlich successfully carried out immunization in animals with tumor cells and suggested
that tumors are kept under control by the immune system. Paul Ehrlich's “magic bullet”
theory has inspired many generations of scientists to explore numerous molecular cancer
therapeutics.
Nowadays, more than a century after the important observations made by our scientist
ancestors, society is still struggling to beat cancer and both cancer immunotherapy and
nanomedicines are far from being at their highest potential. This research project results from
the desire to make a contribution to solve this public health problem through the convergence
of both fields.
This research work was developed between January 2010 and June 2014 in two main
research laboratories: Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade
de Farmácia, Universidade de Lisboa, Lisbon, Portugal; and Louvain Drug Research Institute,
Advanced Drug Delivery & Biomaterials, Université Catholique de Louvain, Brussels,
Belgium. The present research project resulted from the will to transpose a long expertise and
successful achievements in both the nanotechnology and vaccine fields to cancer vaccine
research. At that time, Dr. Helena Florindo had recently completed her PhD work where she
could demonstrate the successful use of biodegradable polymeric particulate systems as safe
adjuvants for a vaccine against the horse condition known as strangles. Dr. Véronique Préat
had conducted several research projects on particulate drug delivery systems and also on
mucosal and dermal vaccine delivery. As so, a very favorable scenario in terms of expertise
and scientific background was established to embrace a new challenge and start the research
in the cancer vaccine field.

vi
Apart from the official institutions previewed at the moment of approval of the
doctoral grant of Joana M. Silva, the necessity to collaborate with other research groups from
other institutions emerged. Dr. Christine Jérôme and Dr. Hélène Freichels from the Center for
Education and Research on Macromolecules, Université de Liège, Liège, Belgium, provided
part of the functionalized polymers, without which the formulation of the nanoparticles would
not be possible. Dr. Carlos Afonso and Dr. Catarina Rodrigues from the Bioorganic
Chemistry group of iMed.UL synthesized a fluorophore-grafted polymer that allowed part of
the fluorescence studies with nanoparticles. Dr. Ana Viana from Centro de Química e
Bioquímica, Faculdade de Ciências da Universidade de Lisboa, Lisbon, Portugal, provided
further insight on the physicochemical characterization of the produced nanoparticles through
her expertise on atomic force microscopy. Dr. Manuel Prieto from Centro de Química-Física
Molecular & Institute of Nanoscience and Nanotechnology, Instituto Superior Técnico,
Universidade de Lisboa, Lisbon, Portugal, allowed Joana M. Silva to use the confocal
microscopy facilities of his laboratory, which were a crucial tool for great part of the studies
conducted on nanoparticle characterization. Moreover, one of his co-worker, Dr. Sandra
Pinto, provided important expertise and help in the development of the confocal microscopy
studies. Finally, Dr. Luís Graça and Dr. Vanessa Oliveira from Instituto de Medicina
Molecular, Faculdade de Medicina da Universidade de Lisboa, Lisbon, Portugal, provided
access to their facilities to develop in vitro and in vivo immunological characterization studies
and also contributed with valuable expertise on cellular immunology.
All the mentioned researchers and institutions were essential in the development of
this research. The interdisciplinary knowledge of the researchers involved contributed to the
enrichment of the developed work, proving once again that the success of science depends on
our effort to collaborate and our capacity to envision and accept different perspectives.

Abstract
vii
Abstract
Biodegradable polymeric nanoparticles (NPs) are promising vaccine delivery systems
due to their capacity to entrap antigens and immunoadjuvants, providing specific targeting to
antigen-presenting cells (APCs), protection from in vivo degradation and sustained release
over time. We hypothesized that the coentrapment of several melanoma-associated antigens
(MAAs) along with Toll-like receptor (TLR) ligands in mannose-functionalized aliphatic
polyester-based NPs could be an effective antitumor strategy. Untargeted PEGylated PLGA-
based and mannose-grafted NPs were formulated and physicochemically characterized.
Sperical 140 to 190 nm NPs were produced displaying mannose residues available for binding
at the surface. Different NP internalization patterns by immortalized and primary APCs were
verified. Multiple endocytic pathways demonstrated to be involved in NP internalization. NPs
demonstrate both endo-lysosomal and cytosolic localizations and a tendency to accumulate
nearby the endoplasmic reticulum. High entrapment efficiencies of several antigens and TLR
ligands adjuvants in NPs were obtained.
The type of immune response induced by NPs loaded with TLR ligands and the model
antigen ovalbumin (OVA) was primarily characterized in OT II mice. The coentrapment of
OVA and the TLR ligands Poly(I:C) and CpG was crucial to induce high levels of IFN-γ and
IL-2, as well as high IgG2c/IgG1 ratios. Mannose-functionalization of NPs potentiated the
Th1 immune response. The antitumor efficacy of the vaccine was then tested in a murine
melanoma model using therapeutic and prophylactic settings. Immunization of mice with the
NPs loaded with melanoma antigen peptides and the TLR ligands decreased the growth rate
of murine B16F10 melanoma tumors in both therapeutic and prophylatic settings. The
combination of MHC class I- and class II-restricted melanoma antigens in different mannose-
functionalyzed NPs containing TLR ligands induced the highest tumor growth delay. Overall,
the present work shows that the combination of the multifuctional properties in terms of
antigen and adjuvant delivery and active targeting of the immune system translate the great
potential of the developed NPs to be used as a cancer vaccine.
Keywords
Cancer vaccine, nanoparticles, PLGA, mannose receptor targeting, antigen presenting cells, melanoma.


Resumo
ix
Resumo
A utilização de nanopartículas (NPs) poliméricas biodegradáveis como sistemas de
veiculação de vacinas para cancro é uma estratégia promissora devido à capacidade destes
para veicular antigénios e adjuvantes, proporcionando a vetorização destas moléculas para as
células apresentadoras de antigénios, evitando a sua degradação in vivo e permitindo a
libertação controlada destas moléculas ao longo do tempo. O presente projeto de investigação
baseou-se na hipótese de que a veiculação simultânea de vários antigénios de melanoma e
ligandos dos recetores Toll-like em NPs de poliésteres alifáticos funcionalizadas com resíduos
de manose poderia exercer um efeito antitumoral.
Os poliésteres alifáticos ácido poliláctico-co-glicólico (PLGA) e policaprolactona,
assim como as suas formas Peguiladas (PEG-b-PLGA e PEG-b-PCL), foram utilizados para
formular NPs pelo método de dupla emulsão evaporação de solvente. Foram também
preparadas NPs funcionalizadas com resíduos de manose com o objetivo de vetorizar
ativamente as NPs para as células apresentadoras de antigénios através dos seus recetores de
manose. Para tal, o polímero PEG-b-PCL foi substituído na formulação pela sua forma
manosilada (man-PEG-b-PCL). Para a realização de ensaios em que se revelou necessária a
marcação das NPs com fluorescência foi introduzido na formulação o polímero PLGA
marcado com um fluoróforo (um derivado de fluoresceína ou um derivado de rodamina).
A caracterização físico-química das NPs preparadas consistiu na determinação do seu
diâmetro hidrodinâmico, potencial zeta e análise da morfologia por microscopia de força
atómica. Foi desenvolvido um ensaio de ligação de lectina para verificar a disponibilidade
para ligação dos resíduos de manose à superfície das NPs. A fim de analisar a interação das
NPs com células apresentadoras de antigénios, foram utilizadas duas linhas celulares
imortalizadas de células de murganho (células dendríticas e macrófagos) assim como células
dendríticas primárias derivadas de células da medula óssea de murganho. A viabilidade
celular em presença de crescentes concentrações de NPs foi avaliada através dos ensaios MTT
e AlamarBlue®. O perfil de internalização das NPs pelas diferentes células apresentadoras de
antigénios foi analisado a diferentes tempos de incubação e concentrações de NPs recorrendo
a citometria de fluxo e microscopia confocal. A identificação das vias endocíticas utilizadas
para a internalização das NPs foi realizada recorrendo à utilização de inibidores específicos de
cada via endocítica e analisando o seu efeito na internalização das NPs por citometria de fluxo
e microscopia confocal. O tráfego intracelular das NPs após a sua internalização foi analisado

x
por imunofluorescência em microscopia confocal, analisando o grau de colocalização das NPs
com organelos marcados.
Foram preparadas NPs contendo vários antigénios e adjuvantes. Como antigénios
foram utilizados dois péptidos com especificidade para o complexo major de
histocompatibilidade (MHC) classe I (Melan-A:26-35(27L) e gp100:209-217(2M))
correspondentes aos epítopos mais frequentemente encontrados nos melanomas humanos,
com afinidade aumentada para MHC classe I devido à modificação de um aminoácido. Foi
também testado um péptido específico para MHC classe II (gp100:44-59). A proteína
ovalbumina (OVA) foi utilizada como antigénio modelo. A capacidade de loading dos
antigénios nas NPs foi determinada utilizando o ensaio MicroBCA® e confirmada para a
OVA por HPLC. Como adjuvantes, foram testados dois ligandos dos recetores Toll-like, CpG
e Poly(I:C). Estas moléculas mimetizam os ácidos nucleicos bacterianos e virais e são
ligandos dos recetores Toll-like 3 e 9, respetivamente. A ativação destes recetores na
membrana endosomal de células dendríticas induz a sua ativação e a produção de interferões
do tipo 1, conduzindo a uma resposta do tipo T auxiliar 1 (Th1). A capacidade de loading
destas moléculas nas NPs foi avaliada através do ensaio Oligreen®.
Foram realizados vários ensaios em murganhos para avaliar o potencial antitumoral da
vacina preparada e avaliar o impacte de vários fatores na eficácia da mesma, como sejam (i) a
importância da utilização de NPs como veículo da vacina; (ii) o impacte da funcionalização
das NPs com manose; (iii) o possível efeito sinérgico resultante da associação de dois
ligandos dos recetores Toll-like; (iv) o impacte da combinação de dois antigénios peptídicos
na mesma NP ou em NPs separadas; e (v) o efeito da combinação de NPs contendo péptidos
específicos para MHC classe I ou MHC classe II. Foi realizado um ensaio preliminar no
modelo murino transgénico OT II para avaliar o perfil da resposta imunitária induzida pela
vetorização dos adjuvantes Poly(I:C) e CpG assim como do antigénio modelo OVA pelas
NPs, com ou sem funcionalização de manose. Para avaliar a eficácia antitumoral das NPs,
foram realizados dois ensaios utilizando um modelo murino de melanoma. As NPs contendo
os diferentes antigénios de melanoma e os ligandos dos recetores Toll-like, com ou sem
funcionalização de manose, foram administradas a murganhos antes ou após a administração
das células de melanoma B16F10. Foram realizadas três imunizações intervaladas de duas
semanas, antes da administração do tumor. A velocidade de crescimento tumoral foi avaliada
através da medição do volume tumoral. Após 21 dias da injeção do tumor, os animais foram
sacrificados e os baços e tumores foram recolhidos e pesados. Os esplenócitos foram
colocados em cultura e reestimulados com os péptidos durante 72 h após as quais os

Resumo
xi
sobrenadantes das culturas foram recolhidos para a quantificação das citocinas IFN-γ, IL-2,
IL-4, IL-5, IL-6 e IL-10 por ELISA. A produção de Granzima B foi igualmente avaliada por
ELISA após 4 h de reestimulação. A capacidade proliferativa dos esplenócitos foi avaliada
pelo ensaio AlamarBlue® e a percentagem de células CD3+CD4+ e CD3+CD8+ foi avaliada
por citometria de fluxo. Num esquema terapêutico, foram realizadas três imunizações com
intervalos de uma semana, após 7 dias da administração do tumor. A velocidade de
crescimento tumoral foi avaliada assim como a produção de citocinas pelos esplenócitos após
reestimulação antigénica.
As NPs foram preparadas de forma reprodutível com diâmetros hidrodinâmicos entre
140 e 190 nm, baixos índices de polidispersão (entre 0,11 e 0,28) e potencial zeta próximo da
neutraliade a pH 7,4. A observação das NPs por microscopia de força atómica revelou
morfologia esférica e superfície lisa e uniforme. O ensaio de ligação de lectina revelou que os
resíduos de manose estão à superfície das NPs disponíveis para ligação. Foram observados
diferentes perfis de internalização das NPs pelas linhas celulares imortalizadas e células
dendríticas primárias, sendo que as últimas revelaram internalizar as NPs de forma mais
imediata e independente do tempo de incubação e da concentração. A funcionalização das
NPs com manose aumentou a internalização das NPs pelas linhas celulares imortalizadas mas
não pelas células dendríticas primárias, provavelmente devido à elevada capacidade de
internalização destas. Macropinocitose, endocitose mediada por clatrina e endocitose
dependente de caveolina e de “rafts” lipídicos demonstraram mediar a internalização das NPs.
Uma vez internalizadas, as NPs demonstraram localização endolisosomal e citosólica e
tendência para acumulação próximo do retículo endoplasmático ao longo do tempo. Este
perfil de tráfego intracelular é favorável à indução da resposta imunitária desejada uma vez
que, não só permite o acesso aos recetores Toll-like 3 e 9 localizados na membrana
endosomal, assim como permite a apresentação de antigénios pelas vias MHC classe I e
classe II, uma vez que permite o acesso ao retículo endoplasmático e aos lisosomas,
respectivamente.
As NPs revelaram elevadas capacidades de loading dos vários antigénios e adjuvantes
testados (20 a 40 µg de antigénio e 7 a 11 µg de adjuvante por mg de polímero). O ensaio no
modelo murino transgénico OT II demonstrou que a utilização de NPs para a veiculação de
OVA e de ambos os adjuvantes CpG e Poly(I:C) é crucial para a indução de elevados níveis
de IFN-γ e IL-2, assim como elevadas razões IgG2c/IgG1. A funcionalização das NPs com
manose contribuiu para a indução da resposta imunitária Th1. Nos ensaios utilizando o
modelo murino de melanoma, comparando com a utilização dos antigénios e adjuvantes em

xii
solução, todas as vacinas baseadas em NPs testadas reduziram a velocidade de crescimento
tumoral, quer no regime profilátio quer no terapêutico. A maior redução do crescimento
tumoral foi obtida em animais imunizados com uma combinação de NPs manosiladas
contendo o péptido Melan-A:26-35(27L) (MHC classe I) ou gp100:44-59 (MHC classe II)
associado a ambos os adjuvantes Poly(I:C) e CpG.
Os resultados dos ensaios in vivo realizados, tanto de velocidade de crescimento
tumoral como do perfil de produção das citocinas, de Granzima B e de capacidade de
proliferação dos esplenócitos, conduziram às seguintes conclusões:
• A indução de uma resposta duradoura do tipo Th1 depende da veiculação
simultânea de antigénios e adjuvantes na mesma NP;
• A funcionalização das NPs com manose demonstrou melhorar a eficácia das
mesmas relativamente à capacidade de indução da resposta Th1/antitumoral;
• Os adjuvantes CpG e Poly(I:C) demonstraram exercer um efeito sinérgico quando
veiculados na mesma NP;
• A imunização com dois antigénios específicos para MHC classe I veiculados
separadamente em NPs demonstrou ser superior à utilização de apenas um péptido;
• A veiculação simultânea de dois péptidos específicos para MHC classe I (Melan-
A:26-35(27L) e gp100:209-217(2M)) na mesma NP demonstrou um efeito negativo na
resposta antitumoral, possivelmente devido à competição de ambos os péptidos para as
moléculas MHC classe I;
• A combinação de NPs funcionalizadas com manose contendo ambos os adjuvantes
CpG ou Poly(I:C) e um péptido específico para MHC classe I (Melan-A:26-35(27L) ou classe
II (gp100:44-59) demonstrou a resposta antitumoral mais potente, sugerindo a importância da
ativação simultânea das células T CD4+ e CD8+ na eficácia da resposta antitumoral.
Em suma, o presente trabalho demonstra o potencial da utilização de NPs em
imunoterapia para cancro devido à combinação das suas propriedades de vectorização ativa
para o sistema imunitário assim como da sua capacidade de veiculação simultânea de diversos
antigénios tumorais e adjuvantes da resposta imunitária.
Palavras-chave
Vacinas para cancro, nanopartículas, PLGA, recetor da manose, células apresentadoras de
antigénios, melanoma.

Acknowledgments/Agradecimentos
xiii
Acknowledgments/Agradecimentos
Há pequenos momentos que mudam as nossas vidas para sempre. O momento em que
decidi responder ao anúncio de uma vaga para um aluno de doutoramento no Departamento
de Tecnologia Farmacêutica e Farmácia Galénica da Faculdade de Farmácia da Universidade
de Lisboa foi um desses momentos. Na altura pouco sabia sobre nanotecnologia ou sobre o
grupo de investigação e já tinha passado mais de um mês da data limite de resposta ao
anúncio. Mas um impulso levou-me a tentar a minha sorte e decidi escrever à prof. Doutora
Helena Florindo a perguntar se haveria alguma hipótese de ainda considerarem a minha
candidatura. Parece que o meu email chegou no momento certo.
Ao longo do meu doutoramento tive a oportunidade de conhecer pessoas fantásticas
que me marcaram e que vou recordar para sempre. Foi uma jornada que, como todas as outras
na vida, me proporcionou momentos muito bons e momentos muito difíceis. Os momentos
bons vão ser recordados para sempre com muito carinho e saudade. Os momentos mais
difíceis vão ser recordados com orgulho, pois certamente contribuíram para o meu
crescimento pessoal e profissional. Na reta final desta etapa sinto que me tornei uma pessoa
mais resiliente, confiante, tolerante e preserverante. Não podia deixar de agradecer a todas as
pessoas que contribuíram para o sucesso deste meu percurso.
As minhas primeiras palavras de agradecimento não poderiam deixar de ser para a
minha orientadora e amiga prof. Doutora Helena Florindo. Quero agradecer-lhe por ter
aceitado a minha candidatura no verão de 2009 para ser a sua primeira aluna de
doutoramento. Não podia estar mais agradecida por ter tido o privilégio de a ter como minha
orientadora. Quero agradecer-lhe pelo facto de nunca ter ouvido da sua parte uma palavra
negativa. Sempre que as dificuldades apertaram e o caminho pareceu mais tenebroso, ou
mesmo quando cometi os erros mais injustificáveis por distração ou teimosia, sempre recebi
palavras de encorojamento e preserverança. Quero agradecer-lhe o facto de me ter dado
espaço e liberdade para tentar e errar, para decidir e arriscar, sem julgamentos ou acusações.
Sempre me senti confortável com as minhas decisões porque a Helena assim o permitiu. Não
poderia estar mais agradecida por poder ter trabalhado consigo e vou recordar para sempre
com carinho e saudade estes anos de trabalho e amizade.
I am also very grateful for having prof. Véronique Préat as my co-supervisor. I would
like to express my gratitude for having been supervised by such an outstanding scientist and

xiv
to have had the opportunity to work in such an amazing team. I would like to thank you for all
the fruitful discussions and wise advices and also for the recognition that I am receiving for
working with you. I also thank you for the encouragement to continue my carrier as a
researcher.
Quero agradecer à minha co-orientadora prof. Doutora Mafalda Videira por me ter
acompanhado nos meus primeiros passos no laboratório. Recordo especialmente o mês de
agosto de 2010 em que trabalhámos juntas na bancada como duas colegas.
Agradeço também à prof. Doutora Matide de Castro e ao prof. Doutor José Morais na
qualidade de directora e ex-director da Faculdade de de Farmácia da Universidade de Lisboa
por possibilitarem a realização do meu doutoramento nesta nobre casa.
Quero agradecer ao prof. Doutor Rogério Gaspar na qualidade de líder do grupo
Intracell_ADD e ex-líder do grupo Nanomedicines and Drug Delivery Systems pelo
dinamismo que implementou em ambos os grupos de investigação e por despertar o nosso
espírito crítico.
Agradeço também a todos os investigadores que me ajudaram e possibilitaram a
realização deste projecto. Agradeço à investigadora Liana Silva pelos conselhos e orientação e
por possibilitar a colaboração entre o nosso grupo de investigação e o grupo de Molecular
Biophysics and Biological Fluorescence do Centro de Química-Física Molecular & Institute
of Nanoscience and Nanotechnology do Instituto Superior Técnico. Agradeço ao prof. Doutor
Manuel Prieto por me possibilitar o acesso às instalações de microscopia confocal do Centro
de Química-Física Molecular & Institute of Nanoscience and Nanotechnology do Instituto
Superior Técnico. Agradeço à Sandra Pinto por todo o tempo que despendeu em ensinar-me
tudo o que sei sobre microscopia confocal e estudos de colocalização e por estar sempre
disponível para esclarecer as minhas dúvidas. Agradeço a todo o grupo de Biophysics and
Biological Fluorescence do Centro de Química-Física Molecular & Institute of Nanoscience
and Nanotechnology do Instituto Superior Técnico pelo interesse no meu trabalho, pelas
críticas e sugestões e por se mostrarem disponíveis em me ajudar. Agradeço à Doutora
Manuela Gaspar por toda a ajuda que me prestou nos ensaios in vivo, sempre com muita
energia e boa disposição. Agradeço à prof. Doutora Ana Viana pela ajuda no microscópio de
força atómica e pela disponibilidade e prontidão em ajudar-me. Agradeço ao prof. Doutor
Luís Graça da Unidade de Imunologia Celular do Instituto de Medicina Molecular por aceitar
colaborar connosco e me receber no seu laboratório, colocando à minha disposição todos os
recursos necessários para a realização de parte do meu trabalho experimental. Agradeço
também o interesse no meu trabalho e a disponibilidade demonstrada por todos os elementos

Acknowledgments/Agradecimentos
xv
do seu grupo (Ana Água-Doce, Sílvia Almeida, Marta Monteiro, Alexandre, Raquel e em
especial Vanessa Oliveira) em me ajudarem e me transmitirem os seus conhecimentos de
imunologia. Agradeço ao prof. Doutor Carlos Afonso e à Catarina Rodrigues do grupo
Bioorganic Chemistry group do iMed.UL aceitarem colaborar connosco e sintetizarem tão
prontamente o polímero marcado com fluorescência, sem o qual parte dos estudos não
poderiam ter sido realizados.
I would like also to acknowledge Dr. Christine Jérôme and Dr. Hélène Freichels from
the Center for Education and Research on Macromolecules, Université de Liège, Liège,
Belgium, for providing part of the functionalized polymers, without which the formulation of
the nanoparticles would not be possible.
I would like to acknowledge the team at FARG for receiving me so well and helping
me in everything they could. Thank you for making me feel that I was part of the team and
never miss home and for everything I have learnt. I will never forget how Bernard, Nathalie
L., Malory, Jean Pierre, Eduardo, Anne des Rieux, Julie T., Salomé, Eloise, Edith, Claúdia,
Nicholás, Régis, Damien, Nathalie S., Aude, Patrick, Vanessa, Pauline, and specially Gaëlle
welcome me. Un grand MERCI à tous!
Não poderia deixar de agradecer aos meus colegas e amigos do Intracell_ADD. Foi
com muita alegria que vos fui vendo chegar ao grupo e que juntos construímos esta equipa
onde foi tão agradável trabalhar independentemente de todas as dificuldades que possamos ter
passado. Sinto-me uma sortuda por poder ter partilhado estes anos da minha vida convosco!
Não posso deixar de agradecer a cada um de vós. Raquel, obridaga por me contagiares com a
tua alegria, energia e curiosidade cinetífica e por estares sempre disponível para ouvires as
minhas lamúrias. Eva, obrigada pela tua amizade, por estares sempre disponível para me
ajudar e por teres sempre uma palavra amiga e positiva nos momentos mais difícieis. Carina,
obrigada pela boa energia que trouxeste ao laboratório e pelo bom ambiente que manténs, por
estares sempre disponível para ajudar e pela tua amizade. Obrigada às minhas meninas
cassulas Ana Matos e Melissa pela frescura que trouxeram ao laboratório e amizade que
demonstraram em tão pouco tempo. Obrigada aos meninos do laboratório João, Nuno e André
(são poucos mas bons) por nos farerem rir e por acartarem o material pesado. Agradeço
também à Andreia, Ana Carreira e Vanessa, porque apesar de ter tido menos contacto
convosco, não deixámos de partilhar vários bons momentos que vou recordar com carinho.
Ana Saraiva, obrigada pela amizade, pelos teus ensinamentos sábios e por ser sempre tão
agradável estar contigo. Natércia, obrigada pela tua alegria e espontaneadade.

xvi
Não posso deixar de agradecer aos colegas que me receberam quando cheguei ao
laboratório Sara, Joana Marto, Andreia Ascenso, Rui Lopes, Lara, Giuliana e Gonçalo.
Obrigada por me receberem tão bem e pelos bons momentos que partilhámos ao longo destes
anos. Deixo também um agradecimento especial à Ana Salgado, à Carla, à D. Fernanda e à
Alexandra pelo apoio e disponibilidade que sempre demonstraram em me ajudar no que fosse
preciso.
Deixei para o fim as pessoas mais importantes da minha vida, a minha família e o
João. Pai e mãe, qualquer agradecimento que eu escreva aqui nunca será suficiente para
exprimir todo o orgulho que tenho em ser vossa filha e o quão vos estou agradecida pela
educação que me deram e por todo o esforço que ao longo das vossas vidas fizeram para
garantir o melhor para mim. Se hoje estou a terminar esta tese devo-lo a vocês e aos valores
que me ensinaram. Muito obrigada por tudo. Mana, tu sabes que és e sempre serás o modelo
que eu tento seguir. Tal como os pais tu educaste-me e ensinaste-me tudo o que sei hoje.
Muito obrigada por estares sempre presente e teres sempre um conselho sabio para me dar.
Cunhadinho, tu sabes que és como um irmão para mim e o quanto me inspiraste em cumprir
esta etapa da minha carreira. Muito obrigada pelos teus ensinamentos e conselhos e pela tua
amizade. Muito obrigada às minhas queridas sobrinhas Andreia e Ritinha por tantos
abracinhos e beijinhos que derretem o coração da tia. Desculpem se a tia não esteve tão
disponível para as nossas brincadeiras e pinturas nos últimos tempos.
Por fim, a pessoa mais especial com quem partilhei os últimos treze anos da minha
vida. João, não tenho palavras que possam exprimir o quanto te quero agradecer por estares
sempre ao meu lado e me apoiares incondicionalmente nas minhas decisões. Obrigada por
rires comigo nos bons momentos. Obrigada por chorares comigo nos momentos mais difíceis
e por enxugares as minhas lágrimas e me mostrares o caminho. Obrigada pela tua panciência
quando eu me torno insuportável. Obrigada por esperares por mim.

List of abbreviations
xvii
List of abbreviations
α-GalCer Alpha-galactosylceramide
β-BL β-butyrolactone
ε-CL ε-caprolactone
ABC Amphiphilic block copolymers
Abs Antibodies
ADO Adenosine
AFM Atomic force microscopy
AICD Activation-induced cell death
APCs Antigen-presenting cells
APLs Altered peptide ligands
BSA Bovine serum albumin
CD1 Cluster of differentiation 1
CLRs C-type lectin receptors
CMPs Carbohydrate mimetic peptides
Con A Concanavalin A
CSC Cancer stem cells
CTLs Cytotoxic T lymphocytes
CTLA-4 Cytotoxic T-lymphocyte antigen-4
DCM Dichloromethane
DCs Dendritic cells
DC-SIGN Dendritic cell-specific intracellular adhesion molecule-3-grabbing non-
integrin
DD Differential display
DDSs Drug delivery systems
DLS Dynamic light scattering
DMSO Dimethilsulfoxide
DPBS Phosphate buffer containing Ca2+ and Mg2+
dsRNA Double stranded RNA
DT Diptheria and Tetanus
DXO 1,5-dioxepan-2-one
EMA European Medicines Agency
EPR Enhanced permeability and retention
ER Endoplasmic reticulum
F-PLGA Fluorescein derivative-grafted poly(lactic-co-glycolic) acid
FDA Food and Drug Administration
FITC Fluorescein isothiocyanate
GA Glycolide/glycolic acid

xviii
GM-CSF Granulocyte macrophage colony-stimulating factor
Grz B Granzyme B
IAP Inhibitor of apoptosis proteins
IFN-γ Interferon gamma
HA Hemagglutinin
HAV Hepatitis A virus
HBsAg Hepatitis B surface antigen
HBV Hepatitis B virus
HCV Hepatitis C virus
HIB Haemophilus influenza type B
HLA Human leukocyte antigen
HPLC High performance liquid chromatography
HPV Human papilloma virus
HRP Horseradish peroxidase
HSV Herpes simplex virus
hTERT Human telomerase reverse transcriptase
i.d. Intradermal
IFN-α Interferon alpha
IL Interleukin
i.m. Intramuscular
iDCs Immature dendritic cells
IRF3 Interferon regulatory factor 3
ISCOM Immune stimulating complexes
LA Lactide/lactid acid
LCs Langerhans cells
LDV Laser Doppler Velocimetry
LNs Lymph nodes
LPS Lipopolysaccharide
MALT Mucosal-associated lymphoid tissue
man-PEG-b-PCL Mannose-grafted poly(ε-caprolactone-b-ethylene glycol)
mDCs Mature dendritic cells
MDSCs Myeloid-derived suppressor cells
MHC Major histocompatibility complex
MIIC Major histocompatibility complex class II compartment
MPLA Monophosphoryl lipid A
MPs Microparticles
MPS Mononuclear phagocytic system
MR Mannose receptor
MSs Microspheres
MTT 3-(4,5-dimethylthiazol-2-yl)-2,3-(4,5-dimethylthiazol-2-yl)-2,5-

List of abbreviations
xix
diphenyltetrazolium bromide
MUC1 Human milk mucin
NF-κB Nuclear factor-κB
NK Natural killer
NKT Natural killer T
NOD Nucleotide oligomerization domain
NP-F Fluorescent NPs in which one tenth of the PLGA mass was replaced by
fluorescein derivative-grafted PLGA
NP-R Fluorescent NPs in which one tenth of the PLGA mass was replaced by
rhodamine 6G derivative-grafted PLGA
NPs Nanoparticles
NSs Nanospheres
ODN Oligodeoxynucleotides
OVA Ovalbumin
P3HB Poly(3-hydroxybutyrate)
PALS Phase Analysis Light Scattering
PAMPs Pathogen-associated molecular patterns
PAP Prostatic acid phosphatase
PBMCs Peripheral blood mononuclear cells
PCL Polycaprolactone
PD-1 Programmed death-1
pDCs Plasmacytoid DCs
PdI Polydispersity Index
PDLA Poly(D-lactic acid)
PDLLA Poly(D,L-lactic acid)
PDXO Poly(1,5-dioxepan-2-one)
PEA Polyethylene adipate
PEG Poly(ethylene glycol)
PEG-b-PLGA Poly(D,L-lactic-co-glycolide-b-ethylene glycol)
PEI Polyethylenimine
PEST Penicillin - streptomycin
PGA Polyglycolide/polyglycolic acid
PHBV Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)
PLA Polylactide/polylactic acid
PLGA Poly(lactic-co-glycolide) or poly(lactic-co-glycolic) acid
PLGA-α-Cl-ε-PCL Poly(lactide-co-glycolide-co-α-cloro-ε-caprolactone)
PLLA Poly(L-lactic acid)
PPF Poly(propylene fumarate)
PGE2 Prostaglandin E2
PRRs Pattern recognition receptors

xx
PSA polysialic acid
PTMC Poly(trimethylene carbonate)
R-PLGA Rhodamine 6G derivative-grafted poly(lactic-co-glycolide)
RDA Representational difference analysis
RES Reticuloendothelial system
RGD Arg-Gly-Asp
RIG Retinoic-acid inducible gene
RLRs Retinoic-acid inducible gene-like receptors
ROP Ring opening polymerization
SAGE Serial analysis of gene expression
s.c. Subcutaneous
scFv Single-chain Fv antibody
SeM Streptococcus equi M-like protein
Siglec Sialic acid-binding immunoglobulin-like lectin
siRNA Small interfering RNA
SSH Suppression subtractive hybridization
ssRNA Single-stranded RNA
S. equi Streptococcus equi
TAAs Tumor-associated antigens
TACA Tumor associated carbohydrate antigens
TAP Transporter associated with antigen processing
TCR T cell receptor
TGF Tumor growth factor
TILs Tumor-infiltrating lymphocytes
Tg Glass transition temperature
Tm Melting temperature
TMC N-trimethyl chitosan
Th T helper
TICAM Toll-interleukin 1 receptor domain-containing adaptor molecule
TIR Toll-interleukin 1 receptor domain
TLRs Toll-like receptors
TRP2 Tyrosinase-related protein 2
TT Tetanus toxoid
TNF Tumor necrosis factor
VLP Virus-like particle
VSSP Very small size proteoliposome
WGA Wheat germ agglutinin
W/O Water-in-oil
W/O/W Water-in-oil-in-water

List of figures
xxi
List of figures
Figure 1.1. Recommended primary and adjuvant treatment for melanoma (stage 0 to IV),
according to National Comprehensive Cancer Network (NCCN) guideline,
v1.2014. The box in the lower right presents the systemic therapy recommended
for metastatic or advanced melanoma (widespread stage IV). Adapted from
National Comprehensive Cancer Network (NCCN) Guideline for Patients,
v1.2014.
6
Figure 1.2. Antigen presentation by dendritic cells to T cells. A. Activated DCs presenting
extracellular antigens on MHC class II complexes to CD4+ T cells induce the
differentiation of CD4+ T cells into T helper (Th) 1 or Th2 CD4+ T cells,
depending on the cytokines produced by DCs upon activation. IL-2, IL-3, IFN-γ,
TNF-α and GM-CSF induce a Th1 profile while IL-4, IL-5, IL-6, IL-10 and IL-13
induce a Th2 profile. Other secondary Th profiles, which are not represented in
the figure, can also be induced (Th17, Th9, Th22, Treg). Th1 cells enhance the
activation of CD8+ T cells and CTL functions through the secretion of cytokines
(such as IL-2, IL-12 and IFN-γ) and Th2 stimulate the secretion of antibodies by
B cells. The recruitment of cells of the innate immune system, such as NK cells,
granulocytes and macrophages, plays also an important role in the immune
response against both extracellular and intracellular pathogens. B. Intracellular
antigens are presented through MHC class I to CD8+ T cells inducing the
differentiation of CTLs, which are the main effector cells in pathogen-infected
cell lysis and tumor cell lysis. CD8+ T cells can also develop a memory
phenotype that will allow a prompt response in the case of a second infection or
tumor relapse or metastasis. Extracellular antigens can also induce this kind of
response through their cross-presentation by the MHC class I pathway to CD8+ T
cells.
10
Figure 1.3. DC role in T cell activation and DC-T cell synapse. DCs play a major role in
immunosurveillance and immune response initiation, placing a bridge between
the innate and the adaptive branches of the immune system. Upon contact with
pathogens or tumor cells/apoptotic tumor bodies, iDCs start the processing of
antigens for further presentation to T cells. The recognition of pathogen
associated molecular patterns (PAMPs) from pathogens by the pattern recognition
receptors (PRRs) on DCs plays an essential role for proper DC maturation. After
migration from the periphery to the lymph nodes, mDCs have contact to naïve T
cells. In the DC-T cell synapse, three signals are essential for T cell activation.
Signal 1 is the antigen-specific signal and results from the presentation of MHC-
12

xxii
antigen complexes by DCs and their recognition by the antigen-specific TCR.
Signal 2 is the co-stimulatory signal, mainly mediated by the trigger of CD28 and
CD40L on T cells by CD80, CD86 and CD40 expressed by mDCs. Signal 3 is the
cytokine priming signal which is mediated by many cytokines and interferons
secreted by mDCs and determines polarization of the activated T cells and
consequently the profile of the initiated immune response.
Figure 1.4. Desired immune response elicited by a therapeutic cancer vaccine. Cancer
vaccines must target antigens and immunoadjuvants to iDCs either in the skin or
in the lymph nodes. After processing, antigens will be presented through MHC
class I or MHC class II complexes to CD8+ or CD4+ T lymphocytes, respectively.
Simultaneously, immunoadjuvants promote the activation and maturation of DCs,
which consequently express costimulatory factors and secrete cytokines, such as
INF-γ and IL-12, which are crucial in the effective stimulation of T cells and in
determining the CD4+ phenotype. CD8+ lymphocytes acquire a cytotoxic
phenotype, known as CTLs, able to cause tumor cell lysis directly. Some of these
CD8+ lymphocytes also acquire a memory phenotype which is crucial to the
maintenance of immunity. Th1 cells enhance the action of CTLs by enhancing
clonal expansion at the tumor site and promoting the generation and maintenance
of the memory phenotype. Th1 cells secrete IFN-γ, which can further sensitize
tumor cells to CTL action by upregulating MHC class I and other components of
the antigen-processing machinery and promoting the recruitment of cells from the
innate immune system such as the NK cells, granulocytes or macrophages that
also contribute to tumor cell lysis. Adapted from Silva J.M. et al. (2013) J Control
Release; 168:179–199.
14
Figure 1.5. Melanoma altered peptide ligands Melan-A:26-35(27L) and gp100:209-217(2M).
The substitution of the native alanine by lysine in the position 2 of the peptide
Melan-A:26-35 renders it more immunogenic and more resistent to peptidases.
The substitution of threonine in the wild type form of the peptide gp100:209-217
by methionine in the position 2 improves the binding of the peptide to the MHC
class I molecule groove, increasing its immunogenicity.
20
Figure 1.6. TLR cellular location and respective ligands. Mammalian TLRs are classified into
several groups based on the type of PAMP they recognize. TLR1, 2, 4 and 6
recognize lipids, such as lipopolysaccharide (LPS) from Gram-negative bacteria,
lipoteichoic acid from Gram-positive bacteria and lipoarabinomannan from
mycobacteria. TLR5 senses bacterial flagellin. TLR3, 7, 8 and 9 detect nucleic
acids derived from viruses and bacteria. TLR3 recognize double stranded RNA
(dsRNA), which is produced by many viruses during replication. TLR7
recognizes synthetic imidazoquinoline-like molecules, guanosine analogs such as
loxoribine, viral single-stranded RNA (ssRNA) and small interfering RNA
29

List of figures
xxiii
(siRNA). Human TLR8, which has high homology to mouse TLR7, participates
in the recognition of imidazoquinolines and ssRNA. TLR9 recognizes non-
methylated CpG DNA motifs present in bacterial and viral genomes, as well as
non-nucleic acids, such as hemozoin from the malaria parasite. TLRs can also be
divided into two subfamilies distinguished by their subcellular localization:
TLR1, 2, 4, 5, 6 and also TLR10 in humans and TLR11 in mice are expressed at
the cell surface while TLR3, 7, 8 and 9, are localized in the endosomal
membrane. From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers
and Polymeric Biomaterials, Taylor & Francis Editorial Group, London, UK,
2014 [In Press].
Figure 1.7. TLR signaling. Following stimulation, all TLRs except TLR3 recruit MyD88,
IRAKs and TRAF6 to activate the Ubc13/TAK1 pathway, allowing NF-ĸB to
translocate to the nucleus. TAK1 also activates the MAPK pathway, which
mediates AP-1 activation. IRF5 is recruited by the MyD88-IRAK4-TRAF6
complex, phosphorylated and translocated to the nucleus. NF-ĸB, AP-1 and IRF5
control the expression of genes encoding inflammatory cytokines. TIRAP is
recruited by TLR4, TLR1/2 and TLR2/6, activating the MyD88-dependent
pathway. TRIF is recruited by TLR3 and TLR4, and interacts with TBK1 and
IKKi, which mediate phosphorylation of IRF3. Phosphorylated IRF3 dimerizes
and is translocated to the nucleus to induce expression of type I IFN and IFN-
inducible genes. TRAF3 forms a complex with TBK1 and IKKi. TRIF interacts
with TRAF6 and RIP1, which mediate NF-ĸB activation. TLR4, but not TLR3,
utilizes TRAM for activation of the TRIF-dependent pathway. Adapted from
Kawai T. & Akira S. (2007) Semin Immunol; 19: 24–32.
30
Figure 1.8. Mechanisms of bulk and surface erosion of polymers. From Peppas N. and
Narasimah B. (2014) J Control Release; pii: S0168-3659(14)00450-7.
47
Figure 1.9. Typical cargo release curve from aliphatic polyester-based particulate vaccine
delivery systems. An exemplificative curve of the release behavior of a
macromolecule from an aliphatic polyester-based particulate delivery system is
represented. The release from aliphatic polyester particulate delivery systems
normally happens in three phases. It starts with a “burst release phase” (1.) that
can function as the vaccine priming dose. The following “lag phase” (2.) and the
final “sustained final phase” (3.) can provide controlled release of the antigens
and adjuvants and mimic vaccine boosting doses. From Silva JM. et al. (2014)
Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor &
Francis Editorial Group, London, UK, 2014 [In Press].
49
Figure 1.10. Schematic representation of a functionalized particle intended to function as a
vaccine delivery system. NPs can be functionalized with antibodies directed to
receptors expressed on cells of the immune systems (e.g. anti-DEC205 to target
57

xxiv
DCs). Carbohydrates are frequently used for active targeting of NPs. Simple
carbohydrate residues, such as mannose or fucose, are used to target C-type
lectins expressed on DCs. More complex carbohydrates can be also used, such as
hyaluronic acid which targets CD44 receptors on several tumor cell types. NPs
can be equipped with an immunogenic coating in order to increase their
recognition by the immune system (e.g. coating with immunogenic polymers such
as chitosan or pluronic block copolymers). The cargo to be delivered by NPs can
be either adsorbed at NP surface or/and entrapped in the polymeric matrix.
Figure 1.11. Desired immune responses against extracellular and intracellular pathogens. A.
Once recognized and internalized by DCs, extracellular pathogens are digested in
endo-lysosomal organelles and resultant peptides are loaded on MHC class II
molecules. MHC-antigen complexes are presented on DC surface to CD4+ T
cells. The recognition of the antigens happens through the TCR on T cells. The
costimulatory environment resultant from pathogen infection and DC maturation
causes T cell activation, differentiation and proliferation. Upon extracellular
pathogen infection, the cytokine priming by DCs favors a Th2 profile, which
results in the activation of B cells and secretion of antibodies against the
pathogen. These antibodies can act through direct pathogen neutralization,
complement activation and/or signaling the pathogen to phagocytic cells through
opsonization. The recruitment of cells of the innate immune system, such as NK
cells, granulocytes and macrophages, plays also an important role in the immune
response against extracellular pathogens. Memory Th1 and Th2 cells are also
induced and rapidly reacquire their lineage-specific effector functions upon
antigen reencounter. B. Intracellular pathogens seem to need a much more
complex immune response with the involvement of cellular and humoral branches
of the immune system. Antigens from intracellular pathogens are presented
through MHC class I complexes to CD8+ T cells inducing the differentiation of
CTLs, which are the main effector cells in pathogen-infected cell lysis. CD8+ T
cells can also develop a memory phenotype that will allow a prompt response in
the case of a second infection. The presentation of pathogen antigens through
MHC class II to CD4+ T cells is crucial for the enhancement of CTL functions
through the auxiliary action of Th cells. Th1 cells enhance CTL functions through
the secretion of cytokines (such as IL-2, IL-12 and IFN-γ) and Th2 cells stimulate
the secretion of antibodies by B cells. Players from the innate immune system,
such as NK cells, granulocytes and macrophages, are also recruited in the
destruction of intracellular pathogens.
63
Figure 2.1. Zeta potential of NPs and man-NPs at three pH values mimicking intracellular
compartments: DPBS at pH 7.4 (mimicking cytosol) and in 200 mM
phosphate/citrate buffer at pH 6.0 (mimicking early endosomes) or pH 5.0
84

List of figures
xxv
(mimicking lysosomes). Mean ± SD; N = 3, n = 3. Statistics: one-way ANOVA
and Tukey’s post test. Relative to zeta potential at pH 7.4: ** p < 0.01. NS,
statistically non-significant. Two-way ANOVA and Bonferroni’s post test were
used to compare NPs and man-NPs.
Figure 2.2. Analysis of NP morphology and size by atomic force microscopy. 3D images of
NPs (A) and man-NPs (C), as well as section analysis of NPs (B) and man-NPs
(D) where mean diameters are presented. Mean diameters ± SD were calculated
from fifty individual NPs from section analysis of five different areas imaged for
each type of NP.
85
Figure 2.3. Detection of mannose residues at man-NP surface using a lectin recognition
assay. A. Schematic representation of man-NP aggregation in the presence of Con
A due to the binding of mannose residues to one of the four binding sites in Con
A tetramers. B. NP size distribution represented as frequency curves of Intensity
(%) in function of diameter determined by DLS for man-NPs (upper graph) and
NPs (lower graph), before and after the incubation with Con A. C. Confocal
microscopy images of the NP-Con A-FITC aggregates (scale bars = 20 µm). D.
Fluorescence given in arbitrary units (a.u.) of NP-Con A-FITC aggregates. Mean
± SD; N = 3, n = 3. Statistical analysis: one-way ANOVA and Tukey’s post test.
86
Figure 2.4. The effect of temperature (4 °C, 25 °C and 37 °C) in DPBS (A), the effect of pH
(DPBS at pH 7.4 or phosphate/citrate buffer 200 mM at pH 6.0 or 5.0) at 37 °C
(B) and the effect of fluid constitution (DPBS or cell culture medium, CCM) at 37
°C on NP size are represented as the overlay of frequency curves of Intensity (%)
as a function of the diameter over time determined by DLS (histograms). The
variation of mean hydrodynamic diameter and zeta potential of NPs over time is
represented in the graphics on the right. The analysis of a representative batch of
NPs out of four is presented. (N = 4, n = 3). Statistics: two-way ANOVA and
Bonferroni’s post test. * p < 0.05, *** p < 0.001.
87
Figure 2.5. Determination of cell viability by the MTT assay of JAW SII DCs and J744.1
macrophage-like cells incubated for 42 h with man-NPs and NPs, respectively
(A). Determination of cell viability by the AlamarBlue® assay of BMDCs
incubated for 24, 42 and 48 h with NPs (B). Cell culture medium and 0.5 % (v/v)
triton X-100 were used as negative and positive control, respectively. Mean ± SD;
N = 3, n = 3. Statistics: one-way ANOVA and Tukey’s post test. Relative to cell
culture medium: * p < 0.05, ** p < 0.01 and *** p < 0.001. Two-way ANOVA
and Bonferroni’s post test were used to compare JAW SII and J744.1 cell lines
(A) and incubation times (B). ### p < 0.001.
88
Figure 2.6. Internalization of NPs by JAW SII murine immature DCs (A), J744.1 murine
macrophage-like cells (B) and BMDCs (C), expressed by percentage of positive
89

xxvi
cells in the population sorted by a flow cytometer. Mean ± SD; N = 3, n = 3. One-
way ANOVA and Tukey’s post test were used to compare uptake at any time
point to 3 h of incubation in the respective NP concentration: NS, statistically
non-significant; * p < 0.05, ** p < 0.01 and *** p < 0.001. Two-way ANOVA
and Bonferroni’s post test were used to compare uptake at 100 µg/ml vs. 250
µg/ml: # p < 0.05, ## p < 0.01 and ### p < 0.001.
Figure 2.7. Confocal microscopy images obtained after 18 h of incubation of BMDCs with
three different concentrations of NPs (250, 500 and 1000 µg/ml). Plasma
membrane was stained with WGA-Alexa Fluor® 594. Representative images of
three independent experiments are shown. Scale bars = 15 µm.
90
Figure 2.8. Effect of endocytic inhibitors on the internalization of NPs by BMDCs. A.
Energy-dependency of NP internalization by BMDCs. Clathrin-mediated
endocytosis (CME) involvement was analyzed by confocal microscopy (B) and
flow cytometry (C) after treatment with dynasore, chlorpromazine and hypertonic
sucrose solution. For colocalization analysis of NPs and OVA in BMDCs (D),
NP-R (green) were added to BMDCs at 500 µg/ml and incubated for 30 min or 18
h. OVA-Alexa Fluor® 647 (red) were simultaneously added at 10 µg/ml and
incubated for 30 min. Hoescht® 342 was used for nucleus contrast (blue).
Colocalized pixels from red and green channels, obtained through WCIF ImageJ
software, appear in white (right). This software was also used to quantify the
percentage of colocalization of the NPs with OVA (%ColNP) and OVA with NPs
(%ColOVA), the Mander’s coefficients for both entities (MNPs and MOVA) and the
Pearson’s correlation coefficient (Rr). Mean ± SD from at least 25 cells from six
independent images is represented. Macropinocytosis involvement analyzed by
confocal microscopy (E) and flow cytometry (F) after treatment with rottlerin and
cytochalasin D. G. Caveolin and lipid raft involvement analyzed by flow
cytometry after treatment with genistein and nystatin. DMSO was tested as the
vehicle control. Scale bars = 25 µm. Results from flow cytometry analysis are
expressed as normalized median fluorescence intensity relative to untreated
samples (mean ± SD; N = 3, n = 3). Statistics: one-way ANOVA and Tukey’s
post test. *** p < 0.001. Two-way ANOVA and Bonferroni’s post test were used
to compare uptake at 4 °C and 37 °C at different time points (A).
92
Figure 2.9. Intracellular trafficking and colocalization analysis of NPs in BMDCs. A.
Confocal microscopy images obtained after 30 min, 1, 2, 3 and 18 h of BMDCs
incubation with NP-R (red). Cells were incubated with rabbit anti-mouse anti-
EEA1 for early endosomes (EE), anti-Rab7a for EE and late endosomes (EE +
LE), anti-LAMP1 for LE and lysosomes (LE + Lys) and anti-calnexin for the
endoplasmic reticulum (ER) and then labeled with goat anti-rabbit IgG Alexa
Fluor® 488 (green). Hoescht® 342 was used for nuclei contrast (blue). Scale bars
94

List of figures
xxvii
= 15 µm. Colocalized pixels from red and green channels, obtained through
WCIF ImageJ software, appear in white below the respective image. This
software was also used to quantify the percentage of colocalization of NPs with
each organelle-labeling protein (B), the Mander’s coefficient values for the red
channel (M1) (C) and the Pearson’s correlation coefficient (Rr) (D). Mean ± SD
from at least 25 cells from six independent images is represented.
Figure 2.10. Percentage of colocalization of organelles (green pixels) with NPs (red pixels)
(A). Mander’s coefficient of green channel (M2) (B). Mean ± SD from at least 25
cells from six independent images is represented.
95
Figure 2.11. Comparison of the cellular uptake of mannose-grafted NPs containing 15 %
(w/w) (man(15%)-NPs) or 30 % (w/w) (man(30%)-NPs) of mannose-grafted
PEG-b-PCL polymer with non-functionalized NPs (NPs). NPs at 100 or 250
µg/ml were added to JAW SII DCs (A) or BMDCs (C) and incubated for 24 h.
NPs at 250 µg/ml were added to J744.1 macrophage-like cells and incubated for 3
h (B). NP internalization levels are expressed by percentage of positive cells in
the population sorted by a flow cytometer (mean ± SD, N = 3, n = 3). Statistics:
two-way ANOVA and Bonferroni’s post test. Relative to non-functionalized NPs:
NS, statistically non-significant; * p < 0.05, *** p < 0.001. One-way ANOVA
and Tukey’s post test were used in B.
96
Figure 2.12. Surface expression of the mannose receptor (MR/CD206) and the costimulatory
molecule CD80 by BMDCs before and after treatment with LPS. Black lines:
untreated BMDCs. Colored lines: BMDCs treated with 100 ng/ml LPS from
E.coli for 24 h. Tinted curves: isotype antibody control. Numbers correspond to
median fluorescence intensity levels. One representative experiment out of three
is represented.
97
Figure 2.13. Schematic representation of the hypothetical intracellular trafficking of the
developed NPs in DCs. NPs are internalized by DCs and contained within early
endosomes (EE), where NPs start to degrade and release some of their cargo.
Immunoadjuvants with receptors located in the endosomal membrane might get
access to them. As endosomes start to mature, their pH drops and NPs become
neutral or positively charged, being able to interact with endo-lysosomal
membranes and escape to cytosol. Here, NPs accumulate nearby the endoplasmic
reticulum (ER), where the loading of antigens on MHC class I molecules
happens. MHC class I-peptide complexes are transported to the cell surface in
exocytic vesicles to be presented to CD8+ T cells. Some NPs continue to degrade
in the lysosomes, releasing their cargo. Here, peptides can be loaded on MHC
class II molecules. MHC class II-peptide complexes are then transferred to cell
surface to be presented to CD4+ T cells.
101

xxviii
Figure 3.1. SDS-PAGE (12 % gel) of NPs entrapping OVA and the adjuvants Poly(I:C) and
CpG. Lanes: (1) Standard molecular weight markers; (2) Standard OVA solution
(4 µg); (3) NP blank; (4) NP[OVA]; (5) NP[OVA+CpG]; (6)
NP[OVA+Poly(I:C)] and (7) NP[OVA+Poly(I:C)+CpG]. A representative image
of three independent experiments is presented.
122
Figure 3.2. Anti-OVA specific immunoglobulin sera titers from OT II mice immunized with
OVA- and adjuvant-loaded NPs. OT II mice were immunized with OVA and the
adjuvants CpG and Poly(I:C) in solution, NP[OVA], NP[OVA+P+C] or man-
NP[OVA+P+C] on weeks 0, 2 and 4. The production of anti-OVA total IgG (A),
IgG1 (B) and IgG2c (C) in sera collected on weeks 1,3,5,7,9 and 12 after the first
immunization were evaluated by ELISA. Sera from naїve OT II mice were used
as control. The titres reported are the reciprocal of serum dilution that gave an
optical density 5 % higher than the control at the same dilution. D. Th1/Th2
indexes in OT II mice immunized with OVA and adjuvant-loaded NPs, calculated
as a ratio of the obtained mean IgG2c and IgG1 titers for each group at each time
point. The grid line at Th1/Th2 = 1 separates the Th1-biased indexes (> 1) from
the Th2-biased ones (< 1). The bars correspond to mean ± SD; n = 3. Statistics:
two-way ANOVA and Bonferroni’s post test. Relative to levels determined on
naїve mice: * p < 0.05, ** p < 0.01 and *** p < 0.001.
123
Figure 3.3. Cytokine levels in supernatants of cultured splenocytes from immunized OT II
mice. Twelve weeks after the first immunization mice were sacrificed, spleens
were collected and splenocytes were put in culture and restimulated with 20
μg/ml of OVA for 72 h. Cytokine titers (A. IL-2, B. IFN-γ, C. IL-4, D. IL-6) were
determined by ELISA and compared to the respective standard. Mean ± SD; n =
3. Statistics: one-way ANOVA and Tukey’s post test. Relative to levels
determined on naїve mice: * p < 0.05, ** p < 0.01 and *** p < 0.001.
125
Figure 3.4. Antitumor therapeutic efficacy of the nanoparticulate vaccines. A. Schedule of the
therapeutic assay. B. Mean tumor growth curves given by mean tumor volume
over time, determined by V = ½ × (L × W2), where L (length) is the longest
dimension and W (width) is the perpendicular dimension to the length. C. Mean
final tumor weight per 100 g of final body weight. D. Mean final spleen weight
per 100 g of initial body weight. Mean ± SD, N = 6, n = 3. Statistics: two-way
ANOVA and Bonferroni’s post test (B) or one-way ANOVA and Tukey’s post
test (C & D); * p < 0.05, ** p < 0.01 and *** p < 0.001.
126
Figure 3.5. Cytokine levels in the supernatants of cultured splenocytes from mice in the
therapeutic study. Splenocytes were restimulated with 20 μg/ml of Melan-A:26
(A) or gp100:209 (B) for 72 h, and cytokines were measured in culture
supernatants by ELISA. Mean ± SD, n = 3. Statistics: one-way ANOVA and
Tukey’s post test. Relative to the saline control group: * p < 0.05, ** p < 0.01 and
127

List of figures
xxix
*** p < 0.001.
Figure 3.6. Efficacy of the nanoparticulate vaccines in the protection against tumor challenge.
A. Schedule of the prophylactic assay. B. Mean tumor growth curves given by
tumor volume. C. Representative images of excised tumors from each treatment
group by the end of the assay. D. Mean final tumor weight per 100 g of final body
weight. E. Mean final spleen weight per 100 g of initial body weight. Mean ± SD,
N = 6, n = 3. Statistics: one-way ANOVA and Tukey’s post test. Relative to the
saline control group: * p < 0.05, ** p < 0.01 and *** p < 0.001.
129
Figure 3.7. Cytokine levels in the supernatants of cultured splenocytes from mice in the
prophylactic study. Splenocytes were restimulated with 20 μg/ml of Melan-A:26
(A) or gp100:209 or gp100:44 for group (9) (B) for 72 h, and cytokines were
measured in culture supernatants by ELISA. Mean ± SD, n = 3. Statistics: one-
way ANOVA and Tukey’s post test. Relative to the saline control group: * p <
0.05, ** p < 0.01 and *** p < 0.001.
130
Figure 3.8. Relative granzyme B (Grz B) production by splenocytes collected from mice in
the protection study. Splenocytes (effector cells, E) were cultured with B16F10
cells (target cells, T) or breast cancer MDA-MB-231 cells as unspecific target
control (unsp T). After 4 h of incubation, Grz B levels were measured in culture
supernatants by ELISA. Relative ratios of the Grz B production were determined
by comparing the production in the presence or absence of target cells. Mean ±
SD, n = 3. Statistics: one-way ANOVA and Tukey’s post test. Relative to levels
of the saline control group: * p < 0.05, ** p < 0.01 and *** p < 0.001.
131
Figure 3.9. Splenocyte proliferation assay from mice in the protection study. Splenocytes
were restimulated with 20 µg/ml of each antigen (Melan-A:26, gp100:209 or
gp100:44), 1 μg/ml of the mitogen LPS from E. coli, Triton X-100 0.025 % (v/v)
as a negative control, or left untreated for 48 h. Splenocyte proliferation was
measured using the AlamarBlue® reagent. Stimulation Index (SI) was calculated
as the ratio of the proliferation in each sample to the proliferation of the untreated
splenocytes for each immunization group. Mean ± SD, n = 3. Statistics: two-way
ANOVA and Bonferroni’s post test; * p < 0.05, ** p < 0.01 and *** p < 0.001.
132
Figure 3.10. Representative flow cytometry histograms of the percentages of CD3+ versus
CD4+ and CD3+ versus CD8+ T cells for each group in the protection study, after
restimulation with Melan-A:26 (A), gp100:209 (C) or gp100:44 (D). Mean
percentages of each T cell subset after restimulation Melan-A:26 (B), gp100:44
(E) or gp100:209 (F) are presented. Mean ± SD, n = 3. Statistics: one-way
ANOVA and Tukey’s post test. Relative to the saline control group: * p < 0.05,
** p < 0.01 and *** p < 0.001.
133
Figure 3.11. Tolerability to the vaccines. The tolerability to vaccine formulations was 134

xxx
evaluated by monitoring body weight of the animals throughout the therapeutic
(A) and the protection (B) studies, given by the mean ratio of body weight to the
initial body weight of all animals. Mean ± SD, N = 6, n = 3.
Figure 3.12. Expression of the costimulatory molecules CD40 and CD86 by BMDCs upon
incubation with NPs. BMDCs were incubated for 24 h with OVA 2.5 µg/ml, LPS
100 ng/ml, Poly(I:C) 5 µg/ml, NP[blank], NP[OVA], NP[OVA+P],
NP[OVA+P+C] and man-NP[OVA+P+C)] or left untreated. After staining with
anti-mouse fluorescently labeled monoclonal antibodies for flow cytometry (PE-
CD11c, FITC-CD86 and APC-CD40) 10,000 cells were analyzed in a flow
cytometer. A. Upregulation of CD40 and CD86 presented in histograms. Each
histogram presents the isotype control (blue line), untreated cells (red line) and
treated cells (orange or green line for CD40 or CD86, respectively). B. Median
fluorescence intensity translating the expression of CD40 or CD86 for each
condition tested. Mean ± SD, N = 3, n = 3. Statistics: one-way ANOVA and
Tukey’s post test. * p < 0.05, ** p < 0.01, *** p < 0.001, NS, statistically non-
significant.
141

List of tables
xxxi
List of tables
Table 1.1. Pros and cons of universal antigen-based and personalized cancer vaccines. 17
Table 1.2. Main barriers to cancer vaccine efficacy and possible strategies for their
overcoming.
18
Table 1.3. Classification, mechanism of action and current application of several adjuvants
for human vaccines.
25
Table 1.4. Examples of studies demonstrating superiority in the combination of several TLR
ligands (A), in the association of TLR ligands to antigens (B), and in the use of
nanoparticulate vaccines (C).
33
Table 1.5. Examples of synthetic aliphatic polyesters and their manufacturing methods. 40
Table 1.6. Main advantages and disadvantages of PLGA as a particulate vaccine delivery
system component.
43
Table 1.7. Schematic representation of the main classes of particulate vaccine delivery
systems that can be prepared with aliphatic polyesters.
45
Table 1.8. Polymer properties related with in vivo degradation. 46
Table 1.9. Inherent adjuvanticity of aliphatic polyester particulate delivery systems for
vaccines.
50
Table 1.10. Pattern Recognition Receptors (PRRs) targeted by functionalized PLGA NPs. 58
Table 1.11. Examples of aliphatic polyester particulate delivery systems applied to
prophylactic vaccination, cancer therapeutic vaccines and immunotherapy.
65
Table 2.1. Nanoparticle size and polydispersity index (PdI) (mean ± SD; N ≥ 5, n = 3). 84
Table 3.1. Nanoparticle size, polydispersity index (PdI) and zeta potential (mean ± SD; N ≥
5, n = 3).
119
Table 3.2. Entrapment Efficiency (EE %) and Loading Capacity (LC µg/mg) of antigens and
immunoadjuvants in NPs (mean ± SD; N ≥ 5, n = 3).
121


Aim and outline of the thesis
xxxiii
Aims and outline of the thesis
The main goal of the research work presented in this thesis was to develop a
melanoma therapeutic vaccine by using biodegradable polymeric nanoparticles to deliver
melanoma-associated antigens and immunoadjuvant molecules and induce an antitumor
immune response.
To achieve the main goal of this research, several questions were addressed:
1. Is it possible to produce biodegradable polymeric nanoparticles with the desired
physicochemical characteristics in a reproducible way?
2. Can biodegradable polymeric nanoparticles entrap melanoma-associated antigens
and immunoadjuvant molecules?
3. Can biodegradable polymeric nanoparticles target and be internalized by antigen-
presenting cells?
4. Which endocytic mechanisms are involved in the internalization of the
nanoparticles?
5. Once internalized by antigen-presenting cells, which is the intracellular pathway
and final fate of the nanoparticles?
6. Which is the profile of the immune response induced by nanoparticles entrapping
antigens and immunoadjuvants?
7. Can nanoparticles induce an antitumor immune response?
8. Which is the impact of the coentrapment of different melanoma-associated
antigens and immunoadjuvant molecules in the antitumor response induced by the
nanoparticles?
The developed research work sought to answer these questions in order to provide
sufficient information to achieve the main goal of this research. Some of these questions were
well established from the beginning of the research. Others were raised during the
development of this project as a consequence of the observations made and the reading of
peer reviewed international publications from another scientific research groups that were
published in the meanwhile.
The developed research work can be divided in two main parts, presented in this thesis
in chapter 2 and chapter 3. The first part (chapter 2) is directed to the development of

xxxiv
biodegradable polymeric nanoparticles as a platform for vaccine delivery. A mechanistic
approach was taken in order to characterize the produced nanoparticles in terms of
physicochemical properties, cellular interaction profiles, endocytic mechanisms involved in
nanoparticle internalization as well as the intracellular traffic followed. This work has been
accepted to be published in the special issue “Nanotechnology for Vaccine Development” of
the peer review international journal Nanomedicines (IF 2012 5.26).
The second part of the research work (chapter 3) sought to apply the produced and
characterized nanoparticles as a cancer vaccine delivery system in vivo in murine models. In a
preliminary phase, a transgenic murine model was used in order to characterize the profile of
the immune response induced by the nanoparticulate vaccine. In a second phase, nanoparticles
entrapping melanoma-associated antigens and immunoadjuvant molecules were used in a
murine melanoma model in order to verify the antitumor efficacy of the developed vaccine.
Both therapeutic and prophylactic settings were tested. In all the in vivo assays performed,
several factors that were hypothesized to affect the antitumor performance of the vaccine were
evaluated, including the nanoparticle functionalization with targeting moieties, the
combination of two different immunoadjuvant molecules, the combination of different
melanoma-associated antigens and the impact of the delivery of the antigens in separated
nanoparticles. This second part of the research work has been submitted in the peer review
international publication Journal of Controlled Release (IF 2012 7.63).
The research work presented in chapter 2 and chapter 3 is preceded by a general
introduction that sets the scenario to the reader concerning several important aspects that
served as the foundation for the development of this research work. Chapter 4 consists on an
integrative discussion where the present research work is integrated in an historical context.
Also, the answers to the questions presented in this section are summarized and discussed.
Finally, a conclusion and future perspective remark is presented on chapter 5, where a balance
of the difficulties for cancer vaccine efficacy that have been overcome and those that remain
to be solved is presented, including the regulatory issues regarding this area.

Table of contents
xxxv
Table of contents
Preface vi
Abstract vii
Resumo ix
Acknowledgments/Agradecimentos xiii
List of abbreviations xvi
List of figures xxi
List of tables xxxi
Aims and outline of the thesis xxxiii
Table of contents xxxv
Chapter 1 - General introduction 1
1. Cancer immunotherapy: towards a cancer vaccine 3
1.1. Cancer immunotherapy 3
1.2. Immunotherapy of melanoma 5
2. Desired immune response for a cancer vaccine 7
2.1. Cancer vaccines must activate cytotoxic T lymphocytes (CTLs) and T
helper (Th) 1 cells
7
2.2. Reaching CTLs and Th1 cells through dendritic cells (DCs) – the
bridge between adaptive and innate immune system
8
3. Vaccine basic components 14
3.1. Antigens 15
3.1.1. Peptide cancer antigens 15
3.1.2. Non-peptide cancer antigens 21
3.2. Adjuvants 24
3.2.1. TLR ligands 28
3.3. Delivery system 36
4. Reaching the immune system with nanoparticles as a cancer vaccine 38
4.1. Aliphatic polyesters-based particulate delivery systems: classification
and characterization of the different aliphatic polyesters
38
4.2. Biocompatibility and biodegradability of aliphatic polyesters:
degradation and elimination mechanisms
46
4.3. Inherent adjuvanticity 50

xxxvi
5. The influence of physicochemical factors on NP performance 51
5.1. Influence of size 51
5.2. Influence of chemical constitution and hydrophilicity/hydrophobicity 54
5.3. Influence of charge 54
5.4. Influence of shape 55
6. Targeting NPs to the immune system 55
6.1. Passive targeting 55
6.2. Active targeting 52
7. Current status of the use of aliphatic polyester-based NPs for cancer
vaccines
61
8. Translational aspects and regulatory requirements 68
Chapter 2 - Development of Functionalized Nanoparticles for Vaccine
Delivery to Dendritic Cells: a Mechanistic Approach
71
Abstract 75
Keywords 75
1. Introduction 77
2. Materials and Methods 78
2.1. Materials 78
2.2. Preparation of NPs 79
2.3. Physicochemical characterization of NPs 79
2.4. Detection of mannose on functionalized NPs by Lectin Recognition
Assay
80
2.5. Cell culture conditions 81
2.6. Generation of murine bone marrow derived DCs 81
2.7. In vitro cell viability in the presence of NPs 81
2.8. Nanoparticle uptake by flow cytometry analysis 81
2.9. Confocal microscopy imaging analysis 82
2.10. Study of endocytic pathways involved in NP internalization 83
2.11. Statistics 83
3. Results 83
3.1. Physicochemical characterization of NPs 83
3.2. Detection of mannose residues at man-NP surface by lectin-
recognition assay
85

Table of contents
xxxvii
3.3. Effect of pH, temperature and medium composition on NP stability 86
3.4. Effect of NPs on viability of APCs 88
3.5. Uptake of NPs by APCs 89
3.6. Endocytic pathways involved in NP internalization 90
3.7. Intracellular trafficking followed by NPs in BMDCs 93
3.8. In vitro targeting of APCs with mannose-grafted NPs 95
4. Discussion 97
5. Conclusions & future perspective 101
6. Executive Summary 102
Acknowledgments 103
Financial & competing interests disclosure 103
Ethical conduct of research 103
Chapter 3 - Combination of peptides and adjuvants in mannose-
functionalized nanoparticles for melanoma immunotherapy
105
Abstract 109
Graphical abstract 109
Keywords 109
1. Introduction 111
2. Materials and methods 112
2.1. Materials 112
2.2. Mice 113
2.3. Preparation of NPs 114
2.4. Physicochemical characterization of NPs 114
2.5. Determination of the loading of antigens and adjuvants 114
2.6. Integrity of the NP-entrapped OVA 115
2.7. Cell line culture conditions 115
2.8. Characterization of the immune response induced by the
nanoparticulate vaccines in OT II mice
116
2.9. Therapeutic efficacy of the nanoparticulate vaccines 116
2.10. Protection efficacy of the nanoparticulate vaccines against tumor
challenge
116
2.11. Cytokine and Granzyme B quantification 117
2.12. Splenocyte proliferation assay 118

xxxviii
2.13. Analysis of spleen T lymphocyte subsets 118 2.14. Statistics 118
3. Results 119
3.1. NP physicochemical characterization 119
3.1.1. NP size and zeta potential 119
3.1.2. Loading of antigens and adjuvants 120
3.1.3. Ovalbumin integrity after NP formulation 121
3.2. Immune response characterization induced in OT II mice by the
nanoparticulate vaccines
122
3.2.1. Antibody production 122
3.2.2. Cytokine secretion 124
3.3. Antitumor therapeutic efficacy of the nanoparticulate vaccines 125
3.3.1. Tumor growth 125
3.3.2. Cytokine secretion 127
3.4. Protection efficacy of the nanoparticulate vaccine against tumor
challenge
128
3.4.1. Tumor growth 128
3.4.2. Cytokine secretion 129
3.4.3. Granzyme B production 130
3.4.4. Splenocyte proliferation assay 131
3.4.5. Spleen T lymphocytes subset analysis 132
3.5. Vaccine tolerability 134
4. Discussion 134
5. Conclusions 137
Acknowledgments 138
Appendix 139
1. Induction of DC maturation by NPs 139
1.1. Method 139
1.2. Results 139
2. Results & conclusions 141
Chapter 4 - Integrative discussion 145
Chapter 5 - Concluding remarks & future perspectives 157
References 163

Table of contents
xxxix
Curriculum vitae 193


Chapter 1

Part of this chapter is adapted from the following published manuscripts:
Scientific publications in peer review international journals:
1. Silva JM, Videira M, Gaspar R, Préat V, Florindo HF, Immune System Targeting by
Nanoparticles for Cancer Vaccines. J Control Release, 2013, 168(2):179-99.
http://dx.doi.org/10.1016/j.jconrel.2013.03.010. (IF 2012 7.63)
2. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V, PLGA-based
nanoparticles: an overview of biomedical applications, J Control Release, 2012, 161(2):505-
22. http://dx.doi.org/10.1016/j.jconrel.2012.01.043. (IF 2012 7.63)
Book chapters:
1. Silva JM, Zupancic E, Peres C, Silva LC, Gaspar R, Préat V and Florindo HF,
Immunomodulation against microbial pathogens through dendritic cells, In: Microbial
pathogens and strategies for combating them: science, technology and education (A. Méndez-
Vilas, Ed., Formatex Research Center, Badajoz, Spain, 2013, pp. 1686-1705. ISBN (13)
Volume 3: 978-84-942134-1-0.
2. Silva JM, Préat V, Florindo HF, Role of the Aliphatic Polyesters as Particulate
Vaccine Delivery Systems, Encyclopedia of Biomedical Polymers and Polymeric
Biomaterials, Taylor & Francis Editorial Group, London, UK, 2014 [In Press].

General introduction
3
Chapter 1 – General introduction
Sseveral strategies are being used by immunologists and engineers to design new
materials, molecules and carriers to manipulate the type of immune response for therapeutic
and prophylactic purposes, but also to study and comprehend the immune system. Some
examples include the engineering of microenvironments to clarify the interaction kinetics
between mature DCs and naïve T cells, the modification of material surfaces to induce or
prevent complement activation and the engineering of adjuvants, antigens and carriers to
promote host immune responses (1). These strategies not only will contribute to prophylactic
vaccine development against infectious diseases but also are likely to contribute to the
development of immunotherapeutics for cancer, allergies and autoimmune diseases.
In the last decades, convergence of many scientific disciplines exploring
nanotechnology and nanomedicine has successfully driven the first generation of
nanomedicines to the clinic (2). A great number of promising nano-sized tools for biomedical
applications have been emerging. Among them, biodegradable nanoparticles (NPs) are
gaining increased attention for their ability to serve as specialized biocompatible delivery
systems of a multitude of molecules, including proteins, peptides, nucleic acids and
oligonucleotides that usually constitute the main vaccine components. There are a
considerable number of advantages in using biodegradable NPs as vaccine delivery systems
that will be presented throughout this chapter. One aspect that stands out is their unique
capacity of targeting the immune system and co-deliver antigens and adjuvants
simultaneously to the same cell, enabling the generation of effective immune responses (3).
1. Cancer immunotherapy: towards a cancer vaccine
1.1. Cancer immunotherapy
It is well known that the immune system is able to mount antitumor responses that can
be enhanced using a number of strategies (4). Cancer immunotherapy takes advantage of the
specificity of the immune system to provide a more efficacious and better tolerated therapy
than the conventional treatments, such as chemotherapy and radiotherapy. Current approaches
to cancer immunotherapy can be categorized as those that attempt to enhance the host’s own
immune response to a tumor, such as the development of prophylactic and therapeutic cancer
vaccines, cytokine therapy, administration of immune activating antibodies and
radioimmunotherapy, and those that use adoptive cell therapies from a non-tumor-bearing

Chapter 1
4
syngeneic or, more often, allogeneic donor (5). The identification of defined T cell tumor-
associated antigens (TAAs) at the molecular level and the demonstration that T cells of well-
defined specificity can mediate the complete elimination of large solid tumors have promoted
interest in cancer vaccine development (6).
Despite the challenges in the development of cancer vaccines and the many
mechanisms by which tumors can evade or subvert immune responses (which will be
discussed in section 3.), immunotherapy is an attractive strategy for cancer therapy and cancer
vaccines are one of the most ambitious but promising tools in this field. In contrast to
vaccines that induce protective immunity against viral pathogens associated with the
induction of cancer, such as high risk serotypes of the human papillomavirus (HPV) and the
hepatitis B virus (HBV), it is envisioned that general cancer vaccines would be administered
after the onset or detection of disease. Thus, they would be considered therapeutic rather than
prophylactic vaccines. Another critical aspect of cancer vaccines is that they are more
effective in the adjuvant setting for the treatment of minimal residual disease than in the
treatment of larger bulky primary tumors. Human leukocyte antigen (HLA) expression can
also influence the efficacy of cancer vaccines, which must, therefore, either have sufficient
antigen complexity to provide broad immunological coverage of the treated patient population
either be personalized according to the host’s HLA profile (7). Most important, cancer
vaccines must be constructed taking into account the desired immune response that will
effectively enhance patient’s defenses against the tumor and also abrogate the mechanisms of
tumor evasion and tolerance.
Some of the clinical trials of the first cancer vaccines have provided evidence of
clinical benefit thus encouraging the development of other vaccines. Diverse vaccine
platforms evaluated in clinical trials for cancer therapy include injection of peptides and
proteins in adjuvants, recombinant viruses or other recombinant microorganisms, delivery of
killed tumor cells, or infusion of protein- or peptide-activated DCs to the patient (8). US Food
and Drug Administration (FDA) and European Medicines Agency (EMA) have recently
approved the Sipuleucel-T vaccine (Provenge®, Dendreon Corp.) for the therapy of asymp-
tomatic metastatic castrate-resistant prostate cancer (9). This vaccine consists of leukapheresis
and reinfusion of APCs obtained from peripheral blood mononuclear cells (PBMCs), after
incubation with the prostate antigen prostatic acid phosphatase (PAP) fused to granulocyte
macrophage colony-stimulating factor (GM-CSF). The requirement of leukapheresis and cell
culture processing of PBMCs are pointed out as drawbacks of this strategy, since a limited
number of vaccinations can be used.

General introduction
5
Challenges to the development of cancer vaccines still include the identification and
characterization of the antigen(s) to be targeted, the definition of the desired immune response
to be elicited by the vaccine, and the choice of the appropriate vaccine delivery system.
1.2. Immunotherapy of melanoma
Malignant melanoma is an aggressive, therapy-resistant malignancy of melanocytes,
the skin cells responsible for the production of the skin pigment melanin (10). Melanoma
starts in the epidermis but it can easily grow to the dermis, reach the lymphatic system and
spread to other organs such as the lymph nodes, brain, lungs, liver and bones (11). Incidence
rates for melanoma have been rising in the last 30 years, resulting in an increasing public
health problem. The American Cancer Society estimates that about 76,100 new melanoma
cases will be diagnosed in 2014. It is one of the most common cancers in young adults and the
5-year survival rate for stage IV is range from 15 % to 20 % (12). In a near past, treatment
options for patients with advanced or metastatic melanoma were limited by modest response
rates and failure to improve overall survival (13,14). The scenario slightly changed in 2011
with the approval of ipilimumab and vemurafenib, both of which improved overall survival in
phase III clinical trials. More recently, the targeted inhibitors dabrafenib and trametinib have
demonstrated similar therapeutic profiles and have also been approved. Ipilimumab, a fully
human monoclonal antibody, exploits the natural ability of the immune system to eradicate
primary cancer cells by inhibiting the binding of cytotoxic T-lymphocyte antigen-4 (CTLA-4)
to its ligands, thereby potentiating antitumor T-cell response. Vemurafenib and dabrafenib
inhibit the BRAF V600 mutation which affects up to 50 % of melanoma patients. These drugs
prevent oncogenic activities such as uncheck proliferation and evasion of immune response.
Trametinib, another targeted-therapy approach, inhibits the MEK protein, the only known
substrate of the BRAF V600 protein, which leads to decreased cell signaling and proliferation
in cancer cells (15). Although these new agents have demonstrated long-term improvement in
patient outcome, the inclusion in clinical trials is always encouraged as first choice treatment
for melanoma patients in stages IB to IV after surgery in the hope of providing advantages
from new combinatory regimens or innovative therapies (Fig. 1.1.) (11). Thus, there is still
critical need of new therapeutic strategies for melanoma.
Melanoma is one of the most immunogenic tumors and has been one of the main
targets of cancer immunotherapy research (16). In fact, there are strong arguments to expect
good outcomes from therapeutic melanoma vaccines, including the absence of the primary

Chapter 1
6
melanoma in 5 % of patients with metastatic melanoma, documented spontaneous remissions,
lymphocytic infiltration of tumors, expression of developmental and melanoma-specific
antigens on tumor tissue, and responses to immunotherapeutic agents, such as interleukin
(IL)-2, interferon alpha (IFN-α) and ipilimumab (17–20). Clinical trials have been ongoing for
decades testing several vaccination strategies, including whole tumor cells, cell lysates, and
proteins or specific peptide fragments (21,22). Nevertheless, the therapeutic benefit of these
strategies remains unclear since few clinical trials have demonstrated significant benefit (23).
Figure 1.1. Recommended primary and adjuvant treatment for melanoma (stage 0 to IV), according to
National Comprehensive Cancer Network (NCCN) guideline, v1.2014. The box in the lower right presents
the systemic therapy recommended for metastatic or advanced melanoma (widespread stage IV). Adapted
from National Comprehensive Cancer Network (NCCN) Guideline for Patients, v1.2014.

General introduction
7
2. Desired immune response for a cancer vaccine
2.1. Cancer vaccines must activate cytotoxic T lymphocytes (CTLs) and T
helper (Th) 1 cells
Most TAAs are not cell surface proteins but rather intracellular ones. Peptide
fragments of these antigens are bound by newly synthesized class I major histocompatibility
complex (MHC) molecules, or HLA in humans, and shuttled to the cell surface for
immunological recognition. Thus, in order to be effective, it is critical that cell-mediated
immunity, particularly cytotoxic T lymphocytes (CTLs), is the focus of the immune response
elicited by a cancer vaccine (7). In the presence of the appropriate stimulation, CD8+ T cells
undergo proliferation and differentiate into CTLs (24). Activated CTLs can migrate to
peripheral tissues and exert two main effector functions: direct contact-mediated cytotoxicity
and secretion of effector cytokines, such as interferon gamma (IFN-γ) and tumor necrosis
factor (TNF)-α, which can mediate local inflammation. Another essential function of
activated CD8+ T cells is the acquisition of memory. Memory CD8+ T cells can be maintained
for long periods of time without antigenic stimulation and potentiate a better and faster
immune response upon relapse or metastasis (25).
In recent years, it has become evident that CD4+ T cells also play a critical role in the
development of effective antitumor immunity. Indeed, CD8+ lymphocytes can fail to maintain
functionality in the absence of CD4+ T helper cells. These cells can enhance antitumor CTL
responses by increasing clonal expansion at the tumor site, promoting the generation and
maintenance of memory CTL phenotypes, preventing activation-induced cell death (AICD)
and functioning as antigen-presenting cells (APCs) for CD8+ T cells (26). Classically, effector
CD4+ cells are considered to differentiate into T helper (Th)1 or Th2 cells, even that other
subsets have been described such as the IL-17-secreting Th17, the IL-9-secreting Th9, the IL-
22-secreting Th22 and the tumor growth factor (TGF)-β-producing regulatory T (Treg) cells
are now also well described (27). Although Th1 and Th2 subtypes can both mediate
anticancer functions, IFN-γ-secreting Th1 cells are more effective in this role (28). Becker
(2006) reviewed the relationship of the Th1/Th2 imbalance with tumor presence and
prognosis, highlighting that tumor cells induce increased Th2 cytokine serum levels, namely
IL-4, IL-6 and IL-10, which suppress the production of Th1 cytokines, such as IL-2, IL-12
and IFN-γ, resulting in the prevention of the activation of CD8+ cytotoxic T cell precursors
(29). Acting together in the process of tumor eradication, the combined action of CD8+ and

Chapter 1
8
IFN-γ-secreting Th1CD4+ T cells starts with activation of tumor-specific CD8+ T cells that
can kill tumor cells directly. The survival and persistence of CD8+ T cells as memory cells are
also regulated by tumor-specific CD4+ T cells. Both CD8+ and, especially, Th1 CD4+ T cells,
secrete IFN-γ, which can further sensitize tumor cells to CD8+ T cells by upregulating MHC
class I and other components of the antigen-processing machinery. This promotes the
recruitment of cells from the innate immune system such as natural killer (NK) cells,
granulocytes or macrophages, and interfering with crucial functions of the tumor stroma,
namely, angiogenesis (30).
2.2. Reaching CTLs and Th1 cells through DCs – the bridge between
adaptive and innate immune system
Naïve CD8+ T cells, located in secondary lymphoid organs, constantly survey and
sample APCs searching for cognate peptide-MHC complexes for which their T cell receptors
(TCRs) have specific affinity. APCs, DCs by excellence, but also macrophages and B cells,
are known for their specialized capacity for antigen presentation to T cells, leading to
activation of innate and adaptive immune responses or tolerance to innocuous antigens (31).
Among APCs, DCs are considered as the main APC population due to several intrinsic
characteristics that are not present on macrophages or B cells. DCs take advantage of their
strategic location in peripheral tissues, such as skin and mucosal surfaces, wich are the most
frequent routes of pathogen entry (32). Moreover, after internalization of antigens, DCs are
able to migrate towards T cell-rich areas, such as sentinel lymph nodes (LNs), where they can
activate naїve T cells in an antigen-specific manner, placing a bridge between innate and
adaptive immune system. This activation comprises the presentation of antigen-MHC
complexes, and the upregulation of costimulatory and soluble molecules, such as cytokines
and chemokines (e.g., IL-12 and type I IFNs) which affect T cells in a paracrine way (33). In
fact, type I IFN production by host DCs is an important step during the innate immune
recognition of tumors and has demonstrated to correlate with positive prognostics in early
stage disease (34).
Compared to macrophages, DCs have a similar capacity for endocytosis and an
equivalent number of lysosomes but less proteolytic activity, which limits full degradation of
antigens, and increases the effectiveness of antigen presentation (35). DCs, therefore,
constitute a preferential target of vaccines since they can function as a route of entry to the
immune system and a means of controlling both innate and adaptive immune responses.

General introduction
9
Immature DCs (iDCs), located in peripheral tissues, function as sentinel cells of the
immune system by detecting and incorporating pathogens and becoming activated in response
to specific signals or proinflammatory cytokines in the microenvironment (36). Once DCs
have taken up antigens, they start to migrate in a chemokine-dependent manner into naïve T-
cell regions. DCs begin to express a lymph node-homing chemokine receptor, CCR7, which
enables them to enter afferent lymphatic vessels and migrate into LNs, where CCL19 and
CCL21, the ligands for CCR7, are expressed (37). During their migration, DCs will process
the antigens they have taken up and transfer them to surface complexes by one of three
pathways: through MHC class I (or HLA-A, -B or -C in humans), MHC class II (or HLA-D in
humans), or lipid antigen-presenting cluster of differentiation 1 (CD1) molecules (38).
Extracellular antigens are taken up by APCs by one of the endocytic mechanisms - receptor-
dependent endocytosis (clathrin-mediated, caveolae-mediated, lipid-raft-mediated or
phagocytosis) or receptor-independent endocytosis (fluid phase
endocytosis/macropynocytosis) - and placed into a membrane-delimited compartment of the
endocytic pathway (39). This compartment undergoes a series of modifications and finally
fuses with lysosomes or MHC class II compartments (MIIC). Peptide-loaded MHC class II
molecules are then transported to the cell surface where they engage antigen-specific CD4+ T
cells. Antigenic peptides derived from cytosolic proteins (endogenously synthesized antigens)
are processed in the proteasome into peptides and transported from the cytoplasm into the
endoplasmic reticulum (ER) via the transporter associated with antigen processing (TAP)
molecular complex, where they associate with nascent produced MHC class I molecules and
are delivered to the cell surface, becoming available to be recognized by CD8+ T cells (1).
The antigen presentation by DCs to T cells is schematically represented in Figure 1.2..

Chapter 1
10
Figure 1.2. Antigen presentation by dendritic cells to T cells. A. Activated DCs presenting
extracellular antigens on MHC class II complexes to CD4+ T cells induce the differentiation of CD4+
T cells into T helper (Th) 1 or Th2 CD4+ T cells, depending on the cytokines produced by DCs upon
activation. IL-2, IL-3, IFN-γ, TNF-α and GM-CSF induce a Th1 profile while IL-4, IL-5, IL-6, IL-10 and
IL-13 induce a Th2 profile. Other secondary Th profiles, which are not represented in the figure,
can also be induced (Th17, Th9, Th22, Treg). Th1 cells enhance the activation of CD8+ T cells and
CTL functions through the secretion of cytokines (such as IL-2, IL-12 and IFN-γ) and Th2 stimulate

General introduction
11
the secretion of antibodies by B cells. The recruitment of cells of the innate immune system, such
as NK cells, granulocytes and macrophages, plays also an important role in the immune response
against both extracellular and intracellular pathogens. B. Intracellular antigens are presented
through MHC class I to CD8+ T cells inducing the differentiation of CTLs, which are the main
effector cells in pathogen-infected cell lysis and tumor cell lysis. CD8+ T cells can also develop a
memory phenotype that will allow a prompt response in the case of a second infection or tumor
relapse or metastasis. Extracellular antigens can also induce this kind of response through their
cross-presentation by the MHC class I pathway to CD8+ T cells.
An alternative antigen presentation pathway, designated cross-presentation or cross-
priming, has also been described. The term cross-presentation describes the process whereby
APCs acquire exogenous soluble or cell-associated antigens (from infected or tumor cells)
and then present them bound to MHC class I molecules. This mechanism insures the
activation of CD8+ T cells in response to a virus that does not infect APCs or against tumor
cells, and consequently is highly relevant for antitumor vaccines. Cross-presentation seems to
occur through retrotranslocation of antigens from endosomal compartments into the cytosol,
followed by re-importation of proteasomally processed peptides back into endosomes via
TAP, where peptide loading of recycled MHC class I molecules occurs (1,40). In the absence
of stimulus, steady-state APCs constantly present self-antigens to T cells, inducing T cell
tolerance to these antigens in order to avoid autoimmune reactions. However, in the presence
of a foreign antigen, additional stimuli, apart from the simple presentation of the antigen, are
necessary to induce T cell expansion and acquisition of effector functions.
For T cells to be effectively activated by DCs, three signals are provided in a DC-T
cell synapse (Fig. 1.3.). Firstly, T cells need to recognize the processed antigen in the context
of MHC class I (for CD8+ T cells) or MHC class II (for CD4+ T cells). Each T cell carries a
unique TCR and only those cells that recognize the antigens presented by DCs in the form of
a MHC-antigen complex will become activated. Secondly, during maturation, DCs also
upregulate other cell surface molecules, such as MHC class I and II, and costimulatory
molecules, such as CD40, the receptor for CD40L expressed by naïve T cells, and also CD80
and CD86 (B7-1 and B7-2 in humans, respectively), which are the ligands for the
immunoglobulin superfamily member CD28, also expressed by naïve T cells. This is the
reason why iDCs in the periphery cannot efficiently activate T cells. Thirdly, T cells need
cytokines and chemokines, such as IL-12 and type I IFNs, which are produced by DCs and

Chapter 1
12
directly affect T cells in a paracrine fashion (41,42). These three signals promote T cell
expansion, survival and acquisition of effector functions.
Figure 1.3. DC role in T cell activation and DC-T cell synapse. DCs play a major role in
immunosurveillance and immune response initiation, placing a bridge between the innate and the
adaptive branches of the immune system. Upon contact with pathogens or tumor cells/apoptotic tumor
bodies, iDCs start the processing of antigens for further presentation to T cells. The recognition of
pathogen associated molecular patterns (PAMPs) from pathogens by the pattern recognition receptors
(PRRs) on DCs plays an essential role for proper DC maturation. After migration from the periphery to
the lymph nodes, mDCs have contact to naïve T cells. In the DC-T cell synapse, three signals are essential
for T cell activation. Signal 1 is the antigen-specific signal and results from the presentation of MHC-
antigen complexes by DCs and their recognition by the antigen-specific TCR. Signal 2 is the costimulatory
signal, mainly mediated by the trigger of CD28 and CD40L on T cells by CD80, CD86 and CD40
expressed by mDCs. Signal 3 is the cytokine priming signal which is mediated by many cytokines and
interferons secreted by mDCs and determines polarization of the activated T cells and consequently the
profile of the initiated immune response.
As mentioned, cancer vaccines should predominantly activate CD8+ T cells. A
schematic representation of the desired immune response that a cancer vaccine must induce is
shown in Figure 1.4.. CD8+ T cells predominantly recognize endogenous intracellular

General introduction
13
antigens that are presented by MHC class I molecules, which are ubiquitously expressed by
the majority of normal and tumor tissues. Unfortunately, cancer cells are notoriously
unreliable as antigen presenters. They lack activating costimulatory molecules, produce
immunosuppressive soluble factors, and have unstable genomes that are prone to losing key
molecules required for antigen processing and presentation (43). On the other hand, most
conventional vaccines elicit antibody and CD4+ helper T cells responses rather than CTL
responses. For a vaccine to induce CTL responses, the antigen of interest needs to reach the
cytoplasm of DCs to be cross-presented through MHC class I to CD8+ T cells. As key players
orchestrating immunity, DCs are a natural focus for immunotherapy. Basically, two strategies
have been studied to target these cells and to use them in cancer immunotherapy:
administration of ex vivo tumor-peptide pulsed DCs or in vivo direct delivery of tumor
peptides (38). For both strategies, there are a number of vaccine delivery systems that are
being evaluated to more efficiently load antigens into DCs, including recombinant viral
vectors, virus-like particles, liposomes, microparticles (MPs) and NPs to name a few (7).
Figure 1.4. Desired immune response elicited by a therapeutic cancer vaccine. Cancer vaccines must
target antigens and immunoadjuvants to iDCs either in the skin or in the lymph nodes. After processing,
antigens will be presented through MHC class I or MHC class II complexes to CD8+ or CD4+ T
lymphocytes, respectively. Simultaneously, immunoadjuvants promote the activation and maturation of
DCs, which consequently express costimulatory factors and secrete cytokines, such as INF-γ and IL-12,
which are crucial in the effective stimulation of T cells and in determining the CD4+ phenotype. CD8+
lymphocytes acquire a cytotoxic phenotype, known as CTLs, able to cause tumor cell lysis directly. Some

Chapter 1
14
of these CD8+ lymphocytes also acquire a memory phenotype which is crucial to the maintenance of
immunity. Th1 cells enhance the action of CTLs by enhancing clonal expansion at the tumor site and
promoting the generation and maintenance of the memory phenotype. Th1 cells secrete IFN-γ, which can
further sensitize tumor cells to CTL action by upregulating MHC class I and other components of the
antigen-processing machinery and promoting the recruitment of cells from the innate immune system
such as the NK cells, granulocytes or macrophages that also contribute to tumor cell lysis. Adapted from
Silva J.M. et al. (2013) J Control Release; 168:179–199.
3. Vaccine basic components
Vaccines have had a significant impact in global heath as they permitted the control of
many infectious diseases and the eradication of smallpox (44,45). After the observation by
Edward Jenner in 1796 that milkmaids were generally immune to smallpox, he tested a
primordial way of vaccination by installing subcutaneously material from milkmaids’ lesions
containing cowpox in a healthy individual and confirmed protection against smallpox (46).
Later, Pasteur developed techniques for inactivation of microorganisms and demonstrated for
the first time that the rationale behind smallpox vaccination could be extended to several other
infectious diseases (47). Since then, remarkable progresses have been made and many tools
are now available for the production of successful vaccines. The idea of employing vaccines
to treat cancer arose one century later with the work of Paul Ehrlich and William Coley.
Ehrlich attempted to generate immunity against cancer by injecting weakened tumor cells,
however with no success (48). Coley developed a mixture of heat-killed Streptococcus
pyogenes and Serratia marcescens bacteria (the Coley’s toxin), and tested it in cancer patients
obtaining successful results due to the adjuvant effect of the toxin in the immune response
(49). However, it soon became clear that anticancer vaccines would have to be therapeutic
rather than prophylactic and the poor induction of immune responses by tumor cells had to be
circumvented (50).
Three critical elements are considered to be essential in the composition of an
effective vaccine: antigen, adjuvant, and delivery system (51). Antigens are molecules
(proteins, peptides, lipids, etc) recognized by the immune system that are able to induce an
adaptive immune responses. An adjuvant is a factor that is able to stimulate the innate
immune system and influence the profile of the elicited immune response. A vaccine delivery
system is defined as a platform that insures optimal delivery of both antigen and adjuvant for
the effective activation of both innate and adaptive branches of the immune system. It has
been demonstrated that antigens and adjuvants must act concomitantly on the same APC to
obtain its full activation (3). Indeed, the administration of an isolated antigen can induce a

General introduction
15
Th2-predominant immune response or even tolerance to the antigen. As a result, the efficacy
of a cancer vaccine formulation can be considerably improved by the development of delivery
systems able to protect and deliver antigens and adjuvants simultaneously (52).
3.1. Antigens
3.1.1. Peptide cancer antigens
The selection of the appropriate TAAs is one of the main hurdles of cancer vaccine
development. In a general way, TAAs for anticancer vaccines must fulfill the following
criteria: (i) include peptide sequences that bind to MHC class I, (ii) be processed by tumor
cells and become available for binding to MHC class I molecules, (iii) be recognized by the T
cell repertoire in a MHC class I-restricted way, and (iv) drive the expansion of CTL
precursors expressing TAA-specific TCRs (53). Since the molecular cloning of the first gene
reported to encode a CTL-defined human tumor antigen (MAGE-A1), and with the
development of new technologies, many other TAAs that are recognized by T cells on human
cancers have been identified and characterized (54,55). This process has clearly beneficiated
from the integration of molecular cloning by screening tumor-derived cDNA libraries with
autologous tumor-specific T lymphocytes with novel strategies such as (i) reverse
immunology (epitope prediction on the basis of known HLA-binding motifs performed by
dedicated software); (ii) biochemical methods associated with chromatography and mass
spectrometry to fractionate TAA peptides naturally expressed on tumor cells in the context of
HLA molecules, and (iii) DNA microarray technology which allows comparison of gene
expression profiles in normal and tumor tissues, such as representational difference analysis
(RDA), differential display (DD), suppression subtractive hybridization (SSH), and serial
analysis of gene expression (SAGE) (55). Apart from the epitopes recognized by CD8+ or
CD4+ T cells, other types of tumor antigens have been identified or created. These include
analogs or artificially modified epitopes, antigens identified by antibodies (mimotopes),
immunogenic fusion proteins (new T-cell epitopes resultant from translocation of
chromosomes during carcinogenesis resulting in fusion of distant genes), splicing aberrations,
point mutations, fusion junctions in epitopes and antigens detected by the SEREX technology
(55–57).
TAAs can be broadly classified in two categories: (i) shared tumor antigens, and (ii)
unique tumor antigens. The first group is expressed by many tumor types and its expression
on normal tissues is either absent or quantitatively and qualitatively different. Examples are
the cancer-testis antigens (MAGE, GAGE and NY-ESO1). The second group comprises

Chapter 1
16
antigens that are expressed uniquely by individual tumors and correspond to new epitopes that
are products of point mutations or splicing alterations (58,59). An additional group of TAAs
includes those that might be expressed by cancer stem cells (CSCs), a minor population of
tumor cells which shows the features of stemness and, therefore, may represent an important
target for most tumors (60). TAAs can also be defined as dispensable and indispensable for
tumor growth and survival. Indispensable TAAs, also called universal TAAs, have the
remarkable feature of being necessary for tumor growth and progression. These antigens are
overexpressed by neoplastic and fetal cells, but weakly expressed or expressed in a phase-
specific way in normal cells. Normally they have limited generation of antigen-loss variants
and, therefore, they are attractive targets for cancer vaccines (53). Examples are the inhibitors
of apoptosis proteins (IAP) and the human telomerase reverse transcriptase (hTERT) (61,62).
Studies on universal TAAs demonstrate that such antigens are, more than any other TAA,
frequently expressed in many different human tumors and, therefore, they apparently provide
the most common neoplasia-related and easily identifiable immune target. Besides their
attractiveness as antigens for cancer vaccines, universal TAAs have several drawbacks that
obviate this hypothesis. For instance, it remains to be established whether they are more
immunogenic when compared with other TAAs obtained from the same tumor (63). Also, the
frequency of peptide-specific CTL precursors in PBMCs, which is determinant for the
generation of the final number of effector T cells, can be very low and not overcome tumor
escape from immune surveillance (64).
An alternative strategy to overcome the inherent problems of using universal antigens
is the application of personalized vaccination. This approach takes into account the
immunological diversity of individual patients by measuring CTL precursors in PBMCs prior
to vaccination to select the antigens with higher CTL precursor frequency (65,66). Unique
tumor antigens constitute a growing class of T cell-defined epitopes that exhibit strong
immunogenicity. Some of these antigens, which often derive from mutation of genes that
have relevant biological functions, are less susceptible to immunoselection and may be
retained even in advanced tumors. Immunogenicity and constitutive expression of unique
tumor antigens provide strong incitement for the design of novel patient-tailored therapies that
target such determinants (67,68). Pros and cons of both strategies of cancer vaccination are
listed on Table 1.1.. The next step in the development of vaccines is to identify biomarkers of
the immune response that allow not only in the identification of vaccine responders but also
new vaccine development. It is not certain if personalized vaccinology will lead us to abandon
the universal empiric vaccine approach but it is likely that the future of vaccinology passes by

General introduction
17
more accurate prediction of vaccine responses, adverse reactions, number of doses needed to
induce an effective response, and direct us toward a new paradigm of vaccine development
(69).
Table 1.1. Pros and cons of universal antigen-based and personalized cancer vaccines.
UNIVERSAL ANTIGEN-BASED CANCER VACCINES
Pros Cons
• Frequently expressed in many different human tumors;
• can be easily identified; • since they are indispensable for tumor growth
and progression, the generation of antigen-loss variations is limited or even absent and the tumor growth control is more efficient.
• Antigen processing-related problems; • antigen-specific T cells have low frequencies; • risk of low immunogenicity; • in some cases, these molecules might be more
easily targeted by antibodies rather than by antigen-specific T cells as some antigens are normal molecules involved in migration and cell adhesion;
• delayed and weak primary CTL responses.
PERSONALIZED CANCER VACCINES Pros Cons
• Generally highly immunogenic antigens; • memory CTLs are able to start rapid and
strong secondary responses; • reduced chances of inducing regulatory T
(Treg) cells.
• Their production is time-consuming and expensive;
• the primary response is absent; • the selection of high CTL precursor frequencies
does not ensure tumor regression; • risk of failure of antigen presentation.
From Silva J.M. et al. (2013) J Control Release; 168:179–199.
Apart from the problems related to different vaccine modalities, cancer vaccines have
additional barriers deeply related to tumor immunology that need to be overtaken. Clinical
trials of cancer vaccines demonstrated very low overall objective response rates (21,22).
Table 1.2. presents some of the barriers that can contribute to explain these disappointing
results. These can be either tumor escape mechanisms and/or difficulties related to T-cell
constraints (70,71). Such processes can compromise the efficacy of any vaccine targeting
shared or unique tumor antigens. Another set of hurdles in the development of therapeutic
vaccines as a treatment modality for cancer includes difficulties in the selection the optimal
dose and schedule and the validation of methods for the evaluation of the efficacy of the
vaccine. Obstacles in the patentability of many immunotherapeutic strategies, which have
resulted in a lack of interest and a subsequent lack of funding by the pharmaceutical industry,
have also hampered clinical development (72).

Chapter 1
18
Table 1.2. Main barriers to cancer vaccine efficacy and possible strategies for their overcoming.
DESCRIPTION HOW TO OVERCOME T-CELL BIOLOGY-RELATED MECHANISMS
Low Affinity T-cell receptor (TCR)
“Self- reactive” T cells that are not centrally deleted are kept in periphery with low affinity to the “self-antigen” (73).
Improve affinity of the TCR through appropriate substitution and use of T cells grafted with high-affinity engineered TCR (74).
Ignorance of self-antigens
Self-reactive T cells that have gained access to the periphery remain ignorant and do not respond to their respective antigens. Tumor cells themselves might keep them nonreactive (75).
The threshold of ignorance can be overcome by T cell activation and ignorant T cells can be made to exhibit effector responses in the form of cytolysis or cytokine synthesis (76).
Activation-induced cell death (AICD)
Suicidal death of a fraction of the responding cells at the amplification phase compromising the immune response (77).
A c-Jun N-terminal kinase inhibitor has demonstrated to rescue CTLs from AICD (78).
T-cell exhaustion
The chronic presence of tumor antigens can induce T cell exhaustion by activation of the programmed death-1 (PD-1) molecule on T cells that ultimately leads to T cell death (79).
PD-1 and CTLA-4 blockade are examples of how T cell exhaustion can be reversed and exhausted T cells can be rejuvenated (80).
Regulation of the immune system
Cell-mediated regulation of antitumor immunity involves multiple players and many different modes of action. Treg and myeloid-derived suppressor cells (MDSCs) are the major responsible in the promotion of T cell dysfunction in the tumor microenvironment (81).
Daclizumab is a FDA-approved humanized anti-CD40 antibody for the regulation of Treg cells (82). Another potential strategy is disarming the adenosine (ADO)/prostaglandin E2 (PGE2) pathway (involved in the inhibition of effector T cells by Treg) (83).
Helpless CTL
CD4+ helper T cells are essential for the generation of a robust and long-lasting memory CTL response. In their absence, “helpless” CTL are not long lasting and they die early (26).
Use MHC class II–determined helper epitopes as “cognate help” for CD8+ activation (84). CD8+ as well as CD4+ T cells can be engineered to express a MHC class I–restricted TCR of interest (85). PLGA particles have demonstrated to be able to induce antigen presentation through both MHC class I and class II complexes (86).
TUMOR CELL-RELATED MECHANIMS
MHC class I down-regulation
The most frequent mechanism of tumor evasion consists in MHC class I-down regulation on tumor cells, decreasing the possibilities of tumor antigen recognition by T cells (87,88).
This is one of the main reasons to use particulate delivery systems for cancer immunotherapy as a way to obviate the low presentation of tumor antigens by tumor cells (52).
T-cell relevant antigen down-regulation
Tumor cells can edit their immunologic print to avoid immunosurveillance and recognition by T cells (87).
Use personalized vaccination by measuring CTL precursors in PBMCs of individual patients in pre-vaccination to select the antigens with higher CTL precursor frequency (65,66).
Expression of immunosuppressive
molecules
Tumor cells can secrete immunosuppressive factors such as IL-10, TGF-β, galectin-1,gangliosides, PGE2 and other molecules to counteract effective immune responses (89).
TGF-β blockade, for instance through the overexpression of sTβRII (a soluble TGF-β type II receptor) or by using a TGF-β neutralizing antibody (1D11), significantly decreased tumor growth and metastasis in two orthotopic mammary carcinoma models (90).
Tolerization of host T cells by cross-
presentation of tumor-derived antigens.
Tumor cells can present directly their antigens to T cells and induce tolarization of T cells due to the lack of effective costimulation (75).
Transfer genes encoding costimulatory molecules into tumor cells to improve their ability to provide costimulation during antigen presentation to activate T cells (91).
Creation of hostil tumor microenvironment
Tumor microenvironment in solid tumors is an immunoregulatory network composed by malignant cells surrounded by a tumor-conditioned stroma that contains extracellular matrix and a variety of nonmalignant populations, including myeloid cells, lymphocytes, fibroblasts, and endothelial cells (71).
Effector cell recruitment into metastases, transduction of tumor cells to express specific chemokines, ligands that target tumor-expressed receptors or antibody conjugates complexed with specific chemokines that selectively recognize tumor markers are potential strategies to get access to the tumor microenvironment (92).
From Silva J.M. et al. (2013) J Control Release; 168:179–199.

General introduction
19
Many melanoma antigens, which are recognized by T cells in the context of class I and
II MHC molecules, have been molecularly characterized. These antigens are mostly normal
proteins which include the following:
• differentiation antigens (e.g. Melan-A/MART-1, tyrosinase, gp100) expressed
both on normal and neoplastic melanocytes;
• the so-called cancer/testis antigens, expressed on different tumors and on
spermatocytes/spermatogonia of testis (MAGE-A, BAGE and GAGE);
• ubiquitous antigenic proteins;
• tumor-restricted antigens derived from point mutations (individual antigens) or
splicing alterations which generate new epitopes (93).
A systematic screening has been reported by Banlalam et al., who examined tumor-
infiltrating T lymphocytes (TILs) of 59 metastatic melanoma lesions. The authors concluded
that the two antigens most frequently recognized by TILs of patients having the appropriate
HLA-I allele were Melan-A/MART-1 (mainly in association with HLA-A*0201) and gp100
(associated with several HLA contexts: HLA-A*0101, *0201, *0301, *1101, *6801; HLA-
B*3501; HLA-Cw*0501, *0602 and *0802), while tyrosinase and MAGE were much less
frequently targeted. Melan-A was clearly the MAA most frequently recognized by HLA-
A*0201 TILs, with recognition observed by 30% of these TIL lines directed against the
epitopes Melan-A:26–35 and/or Melan-A:27–35. Other studies reinforce these facts
(16,94,95).It has been reported that Melan-A is expressed by 90 % of primary melanomas and
80 % of metastatic ones. The antigen gp100 is a glycoprotein expressed at low levels in
melanocytes but also strongly expressed in most melanomas (96) As so, these antigens are
attractive targets to anti-melanoma vaccine.
Analogs of immunogenic peptides modified at TCR contact sites but, nevertheless,
maintaining the capacity of activating some TCR-mediated functions are defined as altered
peptide ligands (APLs). A particular class of APLs, termed superagonist peptides, has been
identified as capable of enhancing T-cell stimulation by inducing immunological functions
not detected with the cognate wild type peptide (97). Different tumor-optimized peptide
analogs have been identified and can be divided in three major groups on the basis of the
strategy used for their identification: (i) epitopes bearing amino acid substitutions associated
with improved TCR interaction, such as conservative/semiconservative substitutions at odd
number positions in the middle of peptide for HLA-A*0201-restricted epitopes or in some
cases in position 1 or 6; (ii) epitopes designed to have a better HLA binding affinity through

Chapter 1
20
introduction of specific amino acid substitutions in positions involved in class I HLA binding
(mostly positions 2 and 9); (iii) epitopes modified for enhancing stability in biological
systems (93). In this context, a modified form of the Melan-A:26-35 peptide, in which lysine
substitutes for the native alanine (Melan-A:26-35(27L); ELAGIGILTV), has been noted to be
more immunogenic than the native form by eliciting tumor-reactive CTLs more efficiently
both in vitro and in vivo, and also to have higher peptidase resistance (98,99). Similarly, an
equivalent modified peptide has also been developed for the gp100:209-217 antigen (Fig.
1.5.). In a pilot study in which patients with metastatic melanoma were vaccinated with either
the native class I-restricted gp100:209-217 peptide or a peptide with a methionine substitution
for threonine at the second position (gp100:209-217(2M); IMDQVPFSV), the modified
peptide was superior to the native form in eliciting immune reactivity (91 % vs. 25 % of
patients, P = 0.006) (100). The increased immunogenicity of both Melan-A:26-35(27L) and
gp100:209-217(2M) peptides can be explained by the incorporation of one of the two
dominant anchor amino acid residues (leucine or methionine) at position 2, which can lead to
enhanced stability of peptide-MHC complexes (101).
Figure 1.5. Melanoma altered peptide ligands Melan-A:26-35(27L) and gp100:209-217(2M). The
substitution of the native alanine by lysine in the position 2 of the peptide Melan-A:26-35 renders it more
immunogenic and more resistent to peptidases. The substitution of threonine in the wild type form of the
peptide gp100:209-217 by methionine in the position 2 improves the binding of the peptide to the MHC
class I molecule groove, increasing its immunogenicity.
Both Melan-A:26-35(27L) and gp100:209-217(2M) antigen peptides have been being
successfully studied in preclinical and clinical trials, as part of several immunization
strategies against melanoma. The analysis of 684 consecutive patients with metastatic
melanoma treated with a high-dose intravenous bolus of IL-2, either alone or in combination
with a variety of melanoma vaccines, showed that patients who received the gp100:209-
217(2M) peptide plus IL-2 showed a stronger trend to increased objective responses

General introduction
21
compared with IL-2 alone (22 % vs. 12.8 %; p= 0.009) and also compared with patients who
received a variety of vaccines that did not include this immunogenic peptides (13.8 %; p =
0.009) (102). A study where 35 HLA-A2+ patients with completely resected stage I-III
melanoma were vaccinated multiple times over 6 months with gp100:209-217(2M),
emulsified in the adjuvant montanide, demonstrated by direct ex vivo tetramer analysis and
IFN-γ cytokine flow cytometry quantification a significant antigen-specific CD8+ T-cell
immune response with a functional intact memory component (103). Immunological
responses were also obtained with DNA vaccines encoding for the antigenic peptides in
question (104–106).
Most MAAs that have been defined and have so far been moved to translational
research are self-epitopes that are predominantly restricted to MHC class I molecules (HLA-
A, -B or -C). Even that high levels of CD8+ T cells might be achieved with the immunization
of MHC class I-restricted peptides, the immune responses induced might not be enough to
eradicate the tumor as they will fail on the induction of CD4+ T lymphocytes. Several MHC
class II-restricted epitopes from melanoma antigens have been identified and have
demonstrated to induce CD4+ T cell-responses (107). The combination of MHC class I- and
class II-restricted peptides might contribute to the development of stronger antitumor immune
responses. The use of multiple peptides can also help to overcome the HLA-independent
heterogeneity of immune responses to individual peptides and antigens in patients with the
same haplotype, and maximize the proportion of responding patients (108).
3.1.2. Non-peptide cancer antigens
For a long time, proteins and peptides were seen as the only primary targets of
adaptive immune responses, and so were called T cell-dependent antigens, whereas
carbohydrates and lipids were considered not to be recognized by the complete adaptive
machinery and seen as T cell-independent antigens (109). Now it is known that lipid antigen
recognition is also an important component of host defense. APCs also display a pathway of
antigen presentation for lipid-based antigens where those are recognized by members of the
MHC class I-like CD1 family. The CD1 family, which is constitutively expressed on APCs,
consists of five isoforms that, based on sequence similarity, have been classified into three
groups: group 1 (CD1a, CD1b and CD1c) and group 2 (CD1d), both involved in antigen
presentation, and group 3 (CD1e), involved in lipid processing and trafficking (110,111).
Similar to what happens with peptide antigens, lipid antigens can be synthesized outside
APCs by microorganisms or by other cells and be internalized, or can be synthesized within

Chapter 1
22
APCs as self- or microbial lipids generated by invading pathogens. Exogenous lipids gain
access to the organelles of the endocytic pathway after an internalization process facilitated by
lipoproteins or by fusion with the plasma membrane (112). These lipids are then transported
into the cellular compartments where CD1 molecules traffic to form antigenic complexes.
Group 1 CD1 molecules present lipid antigens to clonally diverse T cells, either CD4+, CD8+
or CD4-CD8 double negative T cells, and preferentially release different types of cytokines
according to their profile. In contrast, group 2 CD1d molecules present lipid antigens to
natural killer T (NKT) cells, a unique subset of the T lineage that coexpress TCRs and
receptors of the NK cell lineage (110). Stimulation of NKT cells induces rapid secretion of
large amounts of immune regulatory Th1 and Th2 cytokines, such as IFN-γ and IL-4,
respectively (113). A wide range of self and foreign lipid antigens is known to be presented
by CD1 molecules, including lipopeptides, diacylglycerolipids, sphingolipids, mycolates and
osphomycoketides (114). The most extensively studied CD1d antigen is alpha-
galactosylceramide (α-GalCer). This synthetic glycolipid is an extremely potent stimulatory
ligand eliciting the secretion of cytokines by NKT cells, leading these cells to enhance the
activation of DCs and up-regulate costimulatory molecules and cross-priming of tumor and
viral antigens, thus acting as a strong adjuvant for Th1 and CTL responses (115).
Aberrant glycosylation is known to be a hallmark of cancer cell transformation
defining stage, direction and tumor progression (116). As such, another class of tumor
antigens that is a promising target in cancer immunotherapy is the Tumor Associated
Carbohydrate Antigens (TACA). A number of TACA have been identified and tested as
tumor vaccine targets, including mucin-related (O-linked) (Tn, Sialyl Tn, and Thomsen–
Friedenreich antigens), blood group Lewis-related (LewisY, Sialyl LewisX, Sialyl LewisA, and
LewisX), glycosphingolipids [Globo H, stage-specific embryonic antigen-3 (SSEA-3)], and
sialic acid containing glycosphingolipids or gangliosides [GM2, GD2, GD3, fucosyl GM1,
the NeuGcGM3 variant of GM3 and the polysialic acid (PSA)] [reviewed in (117)]. A great
advantage of targeting TACA is the potential to broaden the spectrum of antigens recognized
by the immune response, and thereby reduce the risk of developing resistant tumors because
of antigen loss or mutation of a given protein or lipid antigen. The expression of TACA on
normal tissues and the low immunogenicity of the carbohydrate antigens are the main
drawbacks in the use of TACA as cancer vaccine targets because these effects can result in
tolerance or autoimmunity responses (118). Despite the poor immunogenicity of pure
carbohydrates, as they seem not to be able to recruit an adaptive immune response, a new
generation of TACA-based vaccines that can produce not only antitumor antibodies but also

General introduction
23
cellular immune responses is emerging, based on three main approaches: (i) glycoprotein- and
glycopeptide-based vaccines to facilitate antitumor cellular responses; (ii) targeting of pattern
recognition receptors (PRRs) to enhance antigen internalization and DC activation, as
molecules containing carbohydrates (e.g., lipopolysaccharide) are recognized by some Toll-
like receptors (TLRs) and C-type lectins; and (iii) substitution of carbohydrate epitopes with
protein or peptide surrogates to overcome the T-cell independent nature of the “anti-
carbohydrate” response, for example, by using Carbohydrate Mimetic Peptides (CMPs),
which can induce antitumor antibodies and contribute to cellular immune responses
(118,119). One example of a glycoprotein that can be modified to yield highly immunogenic
TACA is the human milk mucin (MUC1), which is one of most promising TAAs since it is
overexpressed on tumor cells with an altered glycosylation pattern (120). Since these
endogenous structures are usually tolerated by the immune system and owing to the biological
micro-heterogeneity of glycoproteins, MUC1 isolated from tumor cells is not suitable for
vaccination (121). Synthetic glycopeptide antigens have been conjugated to immune
stimulating components in order to generate sufficient immunogenicity. Fully synthetic two-
component vaccines either with T-cell peptide epitopes or with TLR ligands or three-
component vaccines with both these stimulants have been synthesized (122). For instance,
MUC1 glycopeptide antigens have been coupled to tetanus toxoid (TT), resulting in the
induction of a strong and highly selective immune response (123). Later on, it has been
demonstrated that bovine serum albumin (BSA) could replace the very expensive TTox as the
immune-stimulating carrier protein in exploratory immunization studies of synthetic MUC1
antitumor vaccines (124).
TACA-based cancer vaccines can also be used with nanoparticulate delivery systems
to enhance their efficacy. For example, Mazorra et al. (2008) demonstrated that the
conjugation of GM3, a ubiquitous antigen overexpressed in several epithelial tumor types, to
very small size proteoliposomes (VSSP) derived from Neisseria meningitides membrane
proteins, induced protective antitumor antibodies in a mice melanoma model, which were
dependent on the activation of CD8+ T cells, showing the direct involvement of the cellular
immune response in the induced antitumor protection (125). Pejawar-Gaddy et al. (2010) used
virus-like particles (VLP) decorated with MUC1, and obtained MUC1-specific CD8+
cytotoxic T cells that significantly delayed tumor growth in immunized mice (59).
Overall, the selection of a cancer vaccine target antigen should be based not only on
the presence of the antigen in a variety of tumor tissues but also on the role that this antigen

Chapter 1
24
plays in tumor growth and metastasis. As such, lipid and carbohydrate-based tumor antigens
may be suitable alternatives to peptide tumor antigens.
3.2. Adjuvants
Adjuvants have been simply defined as components that are added to vaccine antigens
to make them more immunogenic. The imprecision of this definition has lead to the inclusion
of several different agents and strategies in this class, from “antigen delivery systems” to
“immune potentiators” (126). Only a small number of approved adjuvants for human vaccines
is currently available, besides not being able to elicit always the desired protective and
sustained immune response. In fact, for many years, aluminum salts were the only acceptable
adjuvants and are present in many marketed vaccines. Apart from the good track record of
safety, these adjuvants have some limitations such as local reactions, augmentation of IgE
responses, ineffectiveness for some antigens and inability to augment cell-mediated immune
responses, especially CTL responses which are crucial for the development of cancer
therapeutic vaccines and vaccines against intracellular pathogens (127). As so, the
development of new adjuvants is needed. Lower antigen dose in vaccine, increase breadth of
response, enable complex combination vaccine by overcoming antigenic competition,
overcome limited immune response in some populations such as the elderly, young children,
chronic disease and immunocompromised patients, increase effector T cell response and
antibody titers, induce protective responses more rapidly, extend the duration of response by
enhancing memory B and T cell responses and shift the immune response toward a specific T
cell phenotype to mount the appropriate response against defined pathogens are some of the
reasons for including an adjuvant in a vaccine formulation (126,128). Some of the known
adjuvants either licensed or under investigation for human vaccines, are presented in Table
1.3..

General introduction
25
Table 1.3. Classification, mechanism of action and current application of several adjuvants for human
vaccines.
Adjuvant Class Mechanism of action Application Licensed adjuvants
Alum Aluminium salt
• Induces Th2 immune responses and effective humoral immunity;
• increases stability and immunogenicity of antigens due to their adsorption onto the alum particles creating a depot effect;
• induces activation of the innate immune system with influx of neutrophils, eosinophils, NK cells, CD11b+ monocytes and DCs to the site of injection;
• induces activation of several molecular mechanisms in DCs and macrophages, such as activation of cytosolic phospholipase A2 with release of araquidonic acid, activation of the NLRP3 inflammasome through uric acid release and induction of the release of dsDNA that activates monocytes (127).
The only adjuvant available for more than 80 years. Is currently present in several human prophylactic vaccines: hepatitis A virus (HAV), hepatitis B virus (HBV), human papiloma virus (HPV), Diptheria and Tetanus (DT), Haemophilus influenza type B (HIB) and pneumococcal conjugate vaccines.
MF59 Oil-in water (O/W) emulsion
• This squalene-based O/W emulsion induces a local immunostimulatory environment at the injection site characterized by the up-regulation of cytokines, chemokines and other innate immunity genes, which promote the recruitment of CD11b+ and MHCII+ cells; • enhances antigen uptake by DCs.
Flu/Pandemic flu (EU)
Virosomes Lipid vesicles
• Virosomes are virus-like particles (VLPs) that retain the cell binding and membrane fusion properties of the native virus but lack the viral genetic material;
• virosomes interact efficiently with APCs resulting in activation of T cells;
• the repetitive arrangement of the viral envelope on the virosomal surface mediates a co-operative interaction with IgG receptors on B lymphocytes, stimulating strong antibody responses;
• fusion of the virosomes with the endosomal membrane getting access to the MHC class I presentation pathway, priming also CTL activity and a balanced Th1/Th2 response;
• possibility of inclusion of lipophilic adjuvants (129).
HAV and Flu (EU)
AS03
O/W emulsion +
Squalene +
α-tocopherol
• Triggers a transient production of cytokines at the injection site and in the draining lymph nodes; • enhances recruitment of granulocytes
and antigen-loaded monocytes in the draining lymph nodes; • α-tocopherol increases antibody
response, modulates cytokine expression, increases antigen loading and the recruitment of granulocytes to the draining lymph nodes (130).
Pandemic flu (EU)
AS04 MPL + Alum
• Consists in a combinatory adjuvant composed by monophosphoryl lipid A (MPLA) adsorbed to alum; • MPLA is a detoxified derivative of
lipopolysaccharide isolated from Gram-negative bacteria that acts as an agonist of TLR4; • TLR4 activation by MPLA induces local
activation of NF-kB activity and cytokine production, thus providing an innate immune
HBV and HPV (EU, USA)

Chapter 1
26
signal for optimal activation of APCs; • the combination of MPLA with alum in
this adjuvant system prolongs its effect in the site of injection (131)
AS01
MPLA +
Liposome +
QS21
• QS21 is a highly purified saponin extracted from the bark of a South American tree, Quillaja saponaria Molina; • combination of QS21 with emulsion or
liposome has the objective of decreasing its intrinsic lytic activity while retaining its adjuvanticity; • QS21 is a potent adjuvant for CTL
induction and promotes Th1 cytokine secretion (IL-2 and IFN-γ) and antibodies of the IgG2a isotype; • QS21 improves antigen presentation by
APCs and therefore optimize T cell response; • QS21 also improves B cell response
although it remains to be established if this is through a direct effect or via APC/T cell stimulation; • the combination of MPLA and QS21
induces both cellular and humoral immune responses (132).
HAV and Flu (EU)
Adjuvants under clinical development
Montanide W/O emulsion
• W/O emulsion where the oil phase can be from mineral (Montanide ISA 51) or non-mineral (Montanide ISA720) nature; • a multiple water-in-oil-in-water
(W/O/W) emulsion is also under study (Montanide 206D); • depot effect where the type of emulsion
controls the antigen release rate; • protection of the antigen from
degradation; • inflammation at the site of injection; • enhances uptake of antigens by APCs
due to interactions of the surfactant and the cellular membrane; • specific cytokines can be induced
according to the type of emulsion; • microdiffusion of the oil droplets to the
draining lymph nodes with stimulation of the lymphocyte accumulation and alteration of their recirculation, facilitating cell association; • have been extensively used as a
veterinary vaccine adjuvant (133).
Cancer, Malaria
AS02
MPLA +
W/O emulsion +
QS21
• Similar to the approved adjuvant AS01 with the difference that in AS02, MPLA and QS21 are transported in a W/O emulsion instead of a liposome (132).
Malaria
Immune-stimulating complexes (ISCOM)
Saponins +
cholesterol
+ phospholipids
• Spherical particles of ~ 40 nm diameter with remarkable stability, held together by the strong affinity between saponin and cholesterol; • based on the principle of coformulation
of antigen and adjuvant; • manufacturing difficulties related to the
physical incorporation of the antigen, mainly with recombinant DNA proteins; • the amount of saponin required for the
actual construction is often considerably higher than that needed to achieve a potent adjuvant effect with risk of side effects.
Approved for veterinary use.
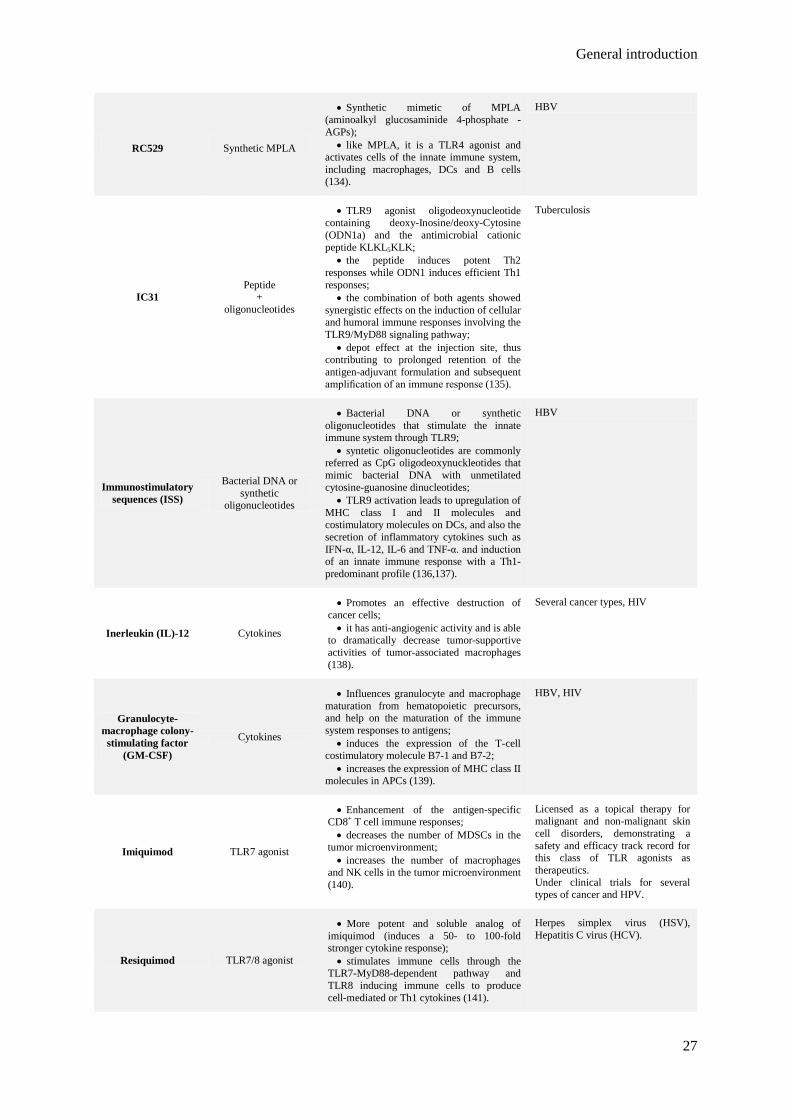
General introduction
27
RC529 Synthetic MPLA
• Synthetic mimetic of MPLA (aminoalkyl glucosaminide 4-phosphate - AGPs);
• like MPLA, it is a TLR4 agonist and activates cells of the innate immune system, including macrophages, DCs and B cells (134).
HBV
IC31 Peptide
+ oligonucleotides
• TLR9 agonist oligodeoxynucleotide containing deoxy-Inosine/deoxy-Cytosine (ODN1a) and the antimicrobial cationic peptide KLKL5KLK;
• the peptide induces potent Th2 responses while ODN1 induces efficient Th1 responses;
• the combination of both agents showed synergistic effects on the induction of cellular and humoral immune responses involving the TLR9/MyD88 signaling pathway;
• depot effect at the injection site, thus contributing to prolonged retention of the antigen-adjuvant formulation and subsequent amplification of an immune response (135).
Tuberculosis
Immunostimulatory sequences (ISS)
Bacterial DNA or synthetic
oligonucleotides
• Bacterial DNA or synthetic oligonucleotides that stimulate the innate immune system through TLR9;
• syntetic oligonucleotides are commonly referred as CpG oligodeoxynuckleotides that mimic bacterial DNA with unmetilated cytosine-guanosine dinucleotides;
• TLR9 activation leads to upregulation of MHC class I and II molecules and costimulatory molecules on DCs, and also the secretion of inflammatory cytokines such as IFN-α, IL-12, IL-6 and TNF-α. and induction of an innate immune response with a Th1-predominant profile (136,137).
HBV
Inerleukin (IL)-12 Cytokines
• Promotes an effective destruction of cancer cells; • it has anti-angiogenic activity and is able
to dramatically decrease tumor-supportive activities of tumor-associated macrophages (138).
Several cancer types, HIV
Granulocyte-macrophage colony-stimulating factor
(GM-CSF)
Cytokines
• Influences granulocyte and macrophage maturation from hematopoietic precursors, and help on the maturation of the immune system responses to antigens;
• induces the expression of the T-cell costimulatory molecule B7-1 and B7-2;
• increases the expression of MHC class II molecules in APCs (139).
HBV, HIV
Imiquimod TLR7 agonist
• Enhancement of the antigen-specific CD8+ T cell immune responses; • decreases the number of MDSCs in the
tumor microenvironment; • increases the number of macrophages
and NK cells in the tumor microenvironment (140).
Licensed as a topical therapy for malignant and non-malignant skin cell disorders, demonstrating a safety and efficacy track record for this class of TLR agonists as therapeutics. Under clinical trials for several types of cancer and HPV.
Resiquimod TLR7/8 agonist
• More potent and soluble analog of imiquimod (induces a 50- to 100-fold stronger cytokine response); • stimulates immune cells through the
TLR7-MyD88-dependent pathway and TLR8 inducing immune cells to produce cell-mediated or Th1 cytokines (141).
Herpes simplex virus (HSV), Hepatitis C virus (HCV).

Chapter 1
28
Poly(I:C) TLR3 agonist
• Synthetic analog of double-stranded RNA;
• TLR3 is the only TLR known which signaling cascade does not start with MyD88 but with TIR domain-containing adaptor (TRIF), which finally activates IRF3, NF-κB and AP-1 transcription factors;
• TLR3 is expressed in myeloid DCs but not in plasmacytoid DCs, which raises the possibility of a key role in cross-presentation and anti-viral responses;
• induction of type I IFN and the production of inflammatory cytokines that can activate NK cells, promote DC maturation and enhance antigen-specific CD8+ T cell responses (142)
Several types of cancer
Flagellin TLR5 agonist
• Major structural protein of Gram-negative flagella; • induction of proinflammatory cytokines
and chemokines by lymphoid and nonlymphoid cells, • generalized recruitment of T and B
lymphocytes to secondary lymphoid sites, DC activation, and direct activation of T cells (143).
The verification that heterologous peptide sequences could be inserted into flagellin to create vaccines that were effective at eliciting humoral immunity in the absence of an adjuvant conducted to the generation of a range of flagellin–antigen fusion proteins, mainly with viral antigens, and have proven to be effective vaccines in animal models.
From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials,
Taylor & Francis Editorial Group, London, UK, 2014 [In Press].
A strategy currently used to develop new adjuvants results from the recognition that
the innate immune system is the first line of defense against pathogens. Several PRRs, which
are involved in the initiation, promotion and execution of immune responses, have been
identified (33). TLRs, nucleotide oligomerization domain (NOD)-like receptors, scavenger
receptors, retinoic-acid inducible gene (RIG)-like receptors (RLRs) and C-type lectin
receptors recognize some conservative elements exclusively expressed on pathogens, known
as pathogen-associated molecular patterns (PAMPs), including unique lipids, lipoproteins,
proteins, carbohydrates and nucleic acids (144). Recognition of PAMPs by PRRs activates
intracellular signaling pathways that induce activation of DCs, which thus acquire a mature T
cell-stimulating phenotype culminating in the secretion of inflammatory cytokines,
chemokines, IFNs and upregulation of costimulatory molecules (145).
3.2.1. TLR ligands
In mammals, the family of TLRs (the main members are represented on Figure 1.6.) is
the largest and most extensively studied PRR class. TLR activation can engage not only the
innate immune system but can also induce adaptive immune responses, either through direct

General introduction
29
engagement of TLRs with their ligands on B and T cells, or through indirect mechanisms
mediated by TLR-activated DCs (146).
Figure 1.6. TLR cellular location and respective ligands. Mammalian TLRs are classified into several
groups based on the type of PAMP they recognize. TLR1, 2, 4 and 6 recognize lipids, such as
lipopolysaccharide (LPS) from Gram-negative bacteria, lipoteichoic acid from Gram-positive bacteria
and lipoarabinomannan from mycobacteria. TLR5 senses bacterial flagellin. TLR3, 7, 8 and 9 detect
nucleic acids derived from viruses and bacteria. TLR3 recognize double stranded RNA (dsRNA), which is
produced by many viruses during replication. TLR7 recognizes synthetic imidazoquinoline-like molecules,
guanosine analogs such as loxoribine, viral single-stranded RNA (ssRNA) and small interfering RNA
(siRNA). Human TLR8, which has high homology to mouse TLR7, participates in the recognition of
imidazoquinolines and ssRNA. TLR9 recognizes non-methylated CpG DNA motifs present in bacterial
and viral genomes, as well as non-nucleic acids, such as hemozoin from the malaria parasite. TLRs can
also be divided into two subfamilies distinguished by their subcellular localization: TLR1, 2, 4, 5, 6 and
also TLR10 in humans and TLR11 in mice are expressed at the cell surface while TLR3, 7, 8 and 9, are
localized in the endosomal membrane. From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers
and Polymeric Biomaterials, Taylor & Francis Editorial Group, London, UK, 2014 [In Press].
Recognition of ligands by TLRs induces recruitment of different cytoplasmic adaptor
molecules and can initiate two major independent but complementary signaling pathways
(Fig. 1.7.): (i) recruitment of the adaptor MyD88 upon activation of TLR2, 4, 7, 8, and 9 or of
Toll-interleukin 1 receptor domain (TIR)-containing adaptor molecule (TICAM)-1 upon

Chapter 1
30
activation of TLR3, both resulting in activation of the nuclear factor-κB (NF-κB) and
production of inflammatory cytokines, such as IL-1, IL-6, TNF-α, and IL-12; and (ii)
activation of IFN regulatory factor 3 (IRF3) through TRIF–TICAM-1 recruitment by TLR3
and TLR4 engagement leading to type I IFN production and subsequently induction of IFN-
responsive genes, such as antiviral genes and the CXC–chemokine, IP-10/CXCL10 (147,148).
The involvement with the humoral branch for the production of antigen-specific antibodies
has also been reported (149). From these observations, one can predict that TLR ligands may
act as very potent immune modulators and vaccine adjuvants and, given the preferential Th1-
profile involved, these molecules could have a huge impact on cancer immunotherapy. Some
examples of TLR agonists that are under investigation for use as adjuvants in human vaccines
were presented on Table 1.3..
Figure 1.7. TLR signaling. Following stimulation, all TLRs except TLR3 recruit MyD88, IRAKs and
TRAF6 to activate the Ubc13/TAK1 pathway, allowing NF-ĸB to translocate to the nucleus. TAK1 also
activates the MAPK pathway, which mediates AP-1 activation. IRF5 is recruited by the MyD88-IRAK4-
TRAF6 complex, phosphorylated and translocated to the nucleus. NF-ĸB, AP-1 and IRF5 control the
expression of genes encoding inflammatory cytokines. TIRAP is recruited by TLR4, TLR1/2 and TLR2/6,
activating the MyD88-dependent pathway. TRIF is recruited by TLR3 and TLR4, and interacts with

General introduction
31
TBK1 and IKKi, which mediate phosphorylation of IRF3. Phosphorylated IRF3 dimerizes and is
translocated to the nucleus to induce expression of type I IFN and IFN-inducible genes. TRAF3 forms a
complex with TBK1 and IKKi. TRIF interacts with TRAF6 and RIP1, which mediate NF-ĸB activation.
TLR4, but not TLR3, utilizes TRAM for activation of the TRIF-dependent pathway. Adapted from Kawai
T. & Akira S. (2007) Semin Immunol; 19: 24–32.
Even that TLR activation is prone to the induction of a Th1 profile, structural
differences on TLR agonists seem to induce slightly distinct immune responses. This
differences are mainly related to the induction of antibody secretion, the activation of NK and
CTLs and the secretion of different cytokines [reviewed by Duthie et al. (2011) (150)].
Engagement of TLR2, 4 and 5 seems to correlate to the enhancement of the suppressive
function of Treg which might be related to the expression of these PRR on CD4+CD25+ Treg
cells, while the other TLRs have a broader expression on CD4+ T cells and are related to the
reversion of the suppressive action of Treg (151). A well known example of the dependence
on an agonist structure is the case of the TLR9 agonists CpG oligodeoxynucleotides (ODN),
where different classes can be defined according to their structural characteristics. Class A
CpG ODN are generally 20-21 bases in length with a single CpG motif within a hair-pin-
forming palindromic phosphodiester region and induce IFN-γ production by plasmacytoid
DCs (pDCs), by NK cells, and monocyte maturation into DCs, but are poor inducers of B cell
activation. In contrast, class B CpG ODN, which incorporate one or more CpG motifs within
a fully phosphorothioate backbone, fail to induce IFN-γ production by pDCs but trigger their
expression of costimulatory molecules and TNF-α secretion, B cell proliferation, and IgM
production, as well as up-regulation of activation markers and IL-6 production by both B cells
and monocytes (152). Class C CpG ODN consist of a stimulatory hexameric CpG motif
linked by a T spacer to GC-rich palindromic sequences and are very strong in the stimulation
of B cells as well as type I IFN secretion (153).
Synthetic molecules mimicking original PAMPs, for example the TLR9 ligand
unmethylated CpG ODN and the TLR3 ligand polyribosinic:polyribocytidic acid [Poly(I:C)],
are also recognized by TLRs, which increases their potential for clinical investigation (154).
Molecular dynamic simulation using computational approaches has been demonstrated to be
an important tool to improve understanding of the interactions between TLRs and their
ligands and to identify key structural issues that potentiate the rational design of new
molecules with pre-defined agonist or antagonist action on these receptors (155,156). Several
TLR ligands have been tested as adjuvants of cancer vaccines in clinical trials, including for

Chapter 1
32
melanoma (157), lymphoma (158), glioblastoma (159), breast, prostate, ovarian and lung
cancers (160), with quite promising results, supporting the broad conclusion that they can be
safe and effective vaccine adjuvants [reviewed by Engel et al. (2011) (161)].
Combined TLR ligation on DCs has been demonstrated to trigger distinct signaling
pathways that resulted in activation of these cells in a synergistic manner in terms of
proinflammatory cytokine and chemokine production and also expression of costimulatory
factors (Table 1.4.A). For example, Warger et al. (2006) observed that DCs activated by the
combined use of TLR7 and TLR3 ligands induced CD4+ and CD8+ T cells that were
insensitive to the inhibitory effects of Treg. These DCs demonstrated a faster and more
sustained secretion of proinflammatory cytokines and expression of costimulatory molecules,
resulting in a marked increase in CTL effector functions in wild-type mice (162). Gautier et
al. (2005) demonstrated that the different TLR signaling pathways cooperate in the secretion
of IL-12p70 through an autocrine loop of type I IFN, and that different TLR ligands are likely
to be necessary for inducing an optimal response to pathogens (148). Kasturi et al. (2011)
demonstrated that immunization of mice with synthetic NPs containing antigens and ligands
for TLR4 (MPLA) and TLR7 (R837) induced synergistic increases in antigen-specific
neutralizing antibodies compared to immunization with a single TLR ligand. Moreover, these
investigators demonstrated that direct triggering of TLRs on B cells was essential for antibody
responses and that co-activation of both TLRs on the same B cell was needed (163). The
synergistic effect observed with the simultaneous stimulation of MyD88-independent and
MyD88-dependent pathways by distinct TLR ligands can be explained by an enhanced
activation of DCs in terms of proinflammatory cytokine and chemokine production and also
expression of costimulatory molecules resulting from the greater induction of gene expression
because two independent signaling pathways are activated in parallel (164).

General introduction
33
Table 1.4. Examples of studies demonstrating superiority in the combination of several TLR ligands (A),
in the association of TLR ligands to antigens (B), and in the use of nanoparticulate vaccines (C).
A. SUPERIORITY OF COMBINATION OF SEVERAL TLR LIGANDS
Nanocarrier Compared groups Study design Outcome Ref.
PLGA NPs NP[OVA+Poly(I:C)a] or NP[OVA+CpGb] vs NP[OVA+Poly(I:C)] + NP[OVA+CpG] In vitro
• ~2-fold increase in MHC class I-restricted antigen presentation; • ~3-fold increase antigen-specific
CD8+ T cell proliferation.
(165)
PLGA NPs NP[Agc+MPLAd] or NP[Ag+R837e] vs NP[Ag] + NP[Ag+MPLA+R837]
Preclinical (mice)
• ~2-fold increase in antigen-specific IgG titers with ~1.5-fold increase in avidity; • enhanced and more sustained
germinal center response; • 2 to 3-fold increase in
frequencies of antigen specific memory CD4+ and CD8+ T cells.
(163)
None
Poly(I:C) vs R848f vs LPSg vs CpG vs P3Ch
vs R848+P3C vs R848+LPS vs Poly(I:C)+R848 vs Poly(I:C)+LPS vs
Poly(I:C)+CpG vs Poly(I:C)+P3C
In vitro
• All TLR combinations induced higher secretion of IL-6 and RANTES and enhanced upregulation of CD40, CD86, CD70 and Ox40 ligand; • most prominent synergy for
the combination of Poly(I:C)+R848.
(162)
SIINFEKLi+Poly(I:C) or R848 vs SIINFEKL+Poly(I:C)+R848
In vitro
• ~3-4-fold faster release of IL-6 and RANTES; • ~4-fold higher levels of IL-6
after 20 h of TLR agonist removal; • higher CD4+ and CD8+ T cell
proliferation; • ~3-4-fold increase in IFN-γ
production; • ~2.5-fold increase in resistance
to Treg inhibitory action.
Preclinical (mice)
• Higher CD4+ and CD8+ T cell proliferation; • Higher target cell lysis and IFN-γ
production.
None LPS vs R848 vs Poly(I:C) vs
LPS+R848 vs LPS+Poly(I:C) vs R848+Poly(I:C)
In vitro • All TLR combinations induced
higher IL-12p40 and IL-12p70 secretion.
(148)
PEI or C32 NPs
NP[pSP-D-CD40Lj+CpG] vs NP[pSP-D-CD40L+CpG+Poly(I:C)]
Preclinical (mice)
• Lower tumor growth; • higher tumor-free survival.
(166)
B. SUPERIORITY OF ANTIGEN + TLR LIGANDS OVER ANTIGEN ALONE
Nanocarrier Compared groups Study design Outcome Ref.
PLGA NPs NP[TRP2] vs NP[TRP2+7-acyl lipid Ak] Preclinical (mice)
• 12-fold increase in IFN-γ-secreting CD8+ T cells; • 2-fold increase in the percentage
of mice with controlled tumor growth.
(167)
PLGA NPs NP[OVA] vs NP[OVA+LPS] In vitro
• ~1.4-fold increase in IFN-γ secretion by antigen-specific CD8+ T cells. (168)
Preclinical
(mice) • ~2-fold increase in antigen-
specific IgG titer and IFN-γ secretion.
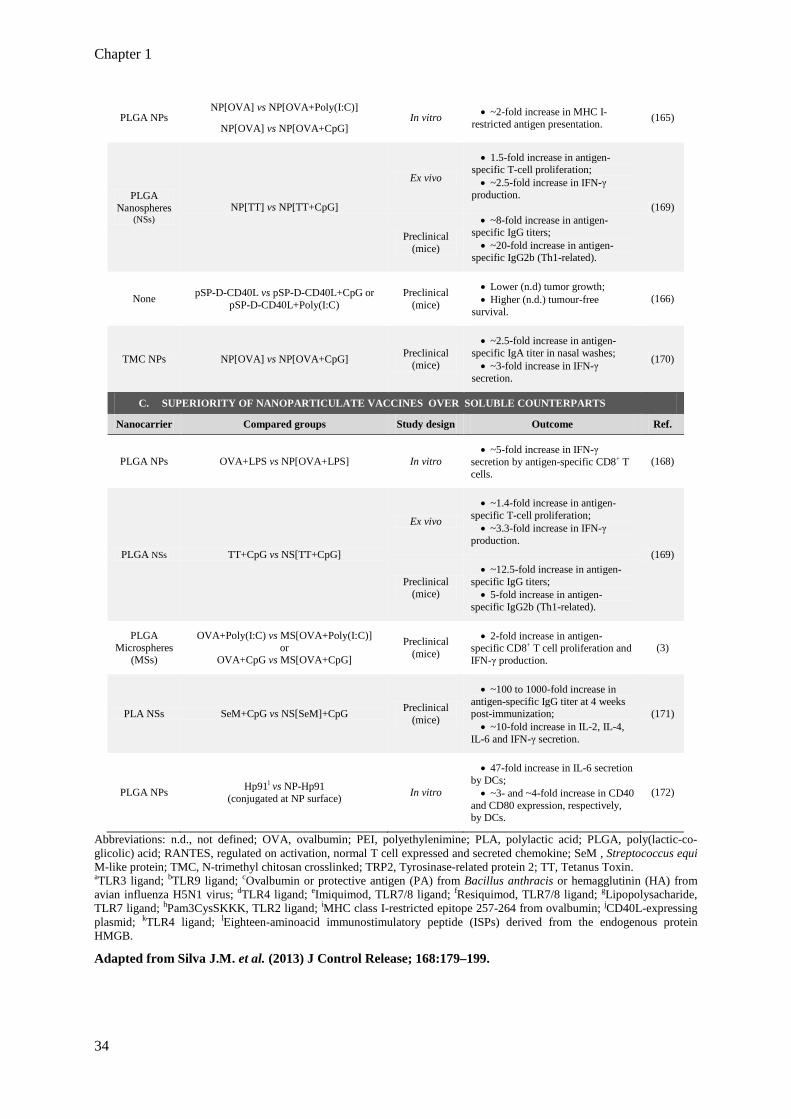
Chapter 1
34
PLGA NPs NP[OVA] vs NP[OVA+Poly(I:C)]
In vitro • ~2-fold increase in MHC I-restricted antigen presentation. (165)
NP[OVA] vs NP[OVA+CpG]
PLGA Nanospheres
(NSs) NP[TT] vs NP[TT+CpG]
Ex vivo
• 1.5-fold increase in antigen-specific T-cell proliferation; • ~2.5-fold increase in IFN-γ
production. (169)
Preclinical (mice)
• ~8-fold increase in antigen-specific IgG titers; • ~20-fold increase in antigen-
specific IgG2b (Th1-related).
None pSP-D-CD40L vs pSP-D-CD40L+CpG or pSP-D-CD40L+Poly(I:C)
Preclinical (mice)
• Lower (n.d) tumor growth; • Higher (n.d.) tumour-free
survival. (166)
TMC NPs NP[OVA] vs NP[OVA+CpG] Preclinical (mice)
• ~2.5-fold increase in antigen-specific IgA titer in nasal washes; • ~3-fold increase in IFN-γ
secretion.
(170)
C. SUPERIORITY OF NANOPARTICULATE VACCINES OVER SOLUBLE COUNTERPARTS
Nanocarrier Compared groups Study design Outcome Ref.
PLGA NPs OVA+LPS vs NP[OVA+LPS] In vitro • ~5-fold increase in IFN-γ
secretion by antigen-specific CD8+ T cells.
(168)
PLGA NSs TT+CpG vs NS[TT+CpG]
Ex vivo
• ~1.4-fold increase in antigen-specific T-cell proliferation; • ~3.3-fold increase in IFN-γ
production. (169)
Preclinical (mice)
• ~12.5-fold increase in antigen-specific IgG titers; • 5-fold increase in antigen-
specific IgG2b (Th1-related).
PLGA Microspheres
(MSs)
OVA+Poly(I:C) vs MS[OVA+Poly(I:C)] or
OVA+CpG vs MS[OVA+CpG]
Preclinical (mice)
• 2-fold increase in antigen-specific CD8+ T cell proliferation and IFN-γ production.
(3)
PLA NSs SeM+CpG vs NS[SeM]+CpG Preclinical (mice)
• ~100 to 1000-fold increase in antigen-specific IgG titer at 4 weeks post-immunization; • ~10-fold increase in IL-2, IL-4,
IL-6 and IFN-γ secretion.
(171)
PLGA NPs Hp91l vs NP-Hp91 (conjugated at NP surface) In vitro
• 47-fold increase in IL-6 secretion by DCs; • ~3- and ~4-fold increase in CD40
and CD80 expression, respectively, by DCs.
(172)
Abbreviations: n.d., not defined; OVA, ovalbumin; PEI, polyethylenimine; PLA, polylactic acid; PLGA, poly(lactic-co-glicolic) acid; RANTES, regulated on activation, normal T cell expressed and secreted chemokine; SeM , Streptococcus equi M-like protein; TMC, N-trimethyl chitosan crosslinked; TRP2, Tyrosinase-related protein 2; TT, Tetanus Toxin. aTLR3 ligand; bTLR9 ligand; cOvalbumin or protective antigen (PA) from Bacillus anthracis or hemagglutinin (HA) from avian influenza H5N1 virus; dTLR4 ligand; eImiquimod, TLR7/8 ligand; fResiquimod, TLR7/8 ligand; gLipopolysacharide, TLR7 ligand; hPam3CysSKKK, TLR2 ligand; iMHC class I-restricted epitope 257-264 from ovalbumin; jCD40L-expressing plasmid; kTLR4 ligand; lEighteen-aminoacid immunostimulatory peptide (ISPs) derived from the endogenous protein HMGB.
Adapted from Silva J.M. et al. (2013) J Control Release; 168:179–199.

General introduction
35
One of the main hurdles in cancer immunotherapy is the difficulty to abrogate the
immunosuppressive tumor environment. There are a number of mechanisms by which tumors
may actively evade or suppress an immune response: deletion of immune effector cell by
expression of death-inducing ligands by tumor cells, induction of tolerance of tumor-reactive
T cells induced by tumor cells, suppression of tumor-reactive T cells by Treg cells or MDSCs,
ignorance of tumor as a result of spatial separation of T and tumor cells, and induction of
tolerance of host T cells by cross-presentation of tumor-derived antigens (75,173).
Despite the vital importance of functional Treg in balancing inflammatory responses
and preventing autoimmune diseases, these cells have been frequently found to comprise the
majority of TILs and to correlate with poor survival in cancer patients (174). Regulating the
powerful immune-modulating activity of Treg is anticipated to become an important tool in
cancer immunology and has been demonstrated to increase T-cell activation and infiltration in
large tumors (175). Along with costimulatory molecules and cytokines, TLR ligands are one
of the major classes of molecules that regulate Treg and alter their function, through direct or
indirect effects. Indirect effects result from the activation of DCs by TLR ligands. TLR-
activated DCs secrete cytokines, mainly IL-6, IFN-γ and IL-4, which make T cells refractory
to the suppressive actions of Treg (176). Warger et al. (2006) demonstrated that distinct
combinations of two TLR ligands resulted in synergistic activation of DCs that converted to
superior CD4+ and CD8+ T-cell responses while abrogating Treg-mediated suppression (162).
However, in the absence of the TLR stimulus, antigen-loaded DCs were shown to induce Treg
cell proliferation with retention of their immunosuppressive activity, demonstrating that the
capacity of DCs to reverse Treg immunosuppressive function is highly dependent on TLR-
mediated activation (177). Treg cells have been shown to selectively express a multitude of
TLRs, namely TLR4, 5, 7 and 8, which activation induces upregulation of several activation
markers and enhances their proliferation and survival, thus reversing their
immunosuppressive function (178). On the other hand, TLR2 engagement on Treg has been
shown to augment proliferation and survival, enhancing their suppressive function (179).
Olivier et al. (2011) tested a large panel of TLR ligands in combination with a model antigen
covalently linked to an inert delivery system and observed that the negative modulation of
effector CD4+ T cells exerted by Tregs could not be circumvented, whichever TLR ligand was
used as adjuvant (180). Given the contradictory results obtained for the role of TLR
engagement on Treg, it has been hypothesized that TLR ligands can regulate T cell-mediated
immune responses by multiple approaches, possibly including: (i) TLR-mediated activation of
DCs resulting in secretion of cytokines and costimulatory factors that stimulate effector T

Chapter 1
36
cells; (ii) direct effects on effector T cells enhancing their functions and clonal expansion
rendering them resistant to Treg suppressive actions; and (iii) expansion of the Treg cell
population with a transient loss of immunosuppressive function in the early response stage,
and reacquisition at the late stage of immune response to regulate the expanded effector T
cells following clearance of the TLR ligands. At this phase, TLR-mediated enhanced Treg
function can then play a critical role in the prevention of immune pathology or in maintaining
low levels of pathogens, effects that are needed for the maintenance of immunologic memory
(179). In some types of cancers, tumoral Treg infiltration have been shown to correlate with a
better control of the tumor, possibly through the production of pro-inflammatory cytokines,
and because intratumoral accumulation of Tregs is often associated with the overall level of
tumor infiltration by immune cells, including CTLs (181).
Another important action of TLR ligands in reversing tumor immunosuppression is
exerted on MDSCs. MDSCs accumulate in tumor-bearing hosts and cancer patients and play a
major role in tumor-related immunosuppression, hampering successful immunotherapy
through inhibition of CD4+ and CD8+ T cell function and control of NK cell cytotoxicity
(173). Zoglmeier et al. (2011) showed for the first time that short-term activation with the
TLR9 ligand CpG blocked the suppressive function of MDSCs in tumor-bearing mice,
promoting MDSC differentiation and restoring the disturbed balance of mature and immature
myeloid cells through the induction of IFN-α (182).
In summary, developing effective immunotherapy for cancer requires two steps:
targeted, antigen-specific immune stimulation through effective DC activation; and
concurrent reversal of tumor-driven immune suppression. TLR ligands have been shown to
play important roles in both steps and promise to become essential tools for effective cancer
immunotherapy.
3.3. Delivery system
Live attenuated virus- or bacterial toxoid-based vaccines generally induce excellent
effector and memory cell formation, whereas the safer and currently preferred peptide
immunization or subunit vaccines elicit poor cellular immune responses. It can thus be
anticipated that mimicking the signals provided during invasion with pathogens should
significantly improve subunit vaccination efficacy (183). On the other hand, the efficacy of
cancer vaccination strategies depends on the targeted delivery of TAAs to APCs and
consequent presentation of TAAs to T cells in an immunostimulatory environment. As
mentioned in the previous section, this can be achieved by using molecules with known

General introduction
37
adjuvant potential, such as the TLR ligands (184). However, additional strategies are needed
in order to prevent their prompt in vivo degradation, to simultaneously target antigens and
adjuvants to APCs and to promote their sustained and modulated release in those cells.
The uptake of antigens by DCs depends on several antigen-associated properties, such
as size, shape, surface charge, hydrophobicity/hydrophilicity, as well as receptor interactions.
Particulate vaccines, such as whole cell vaccines, virosomes, virus-like particles or antigens
formulated in particulate adjuvants have large surfaces, which provide improved electrostatic
or receptor interactions with DCs when compared with soluble antigens (185). Several studies
have demonstrated that nanoparticulate delivery systems could enhance the uptake of antigens
and adjuvants by APCs and that these systems are associated with better immune responses
than those obtained with the soluble counterparts (Table 1.4.C) (3,168,169,171,172). Also,
nanocarriers can provide protection to the encapsulated molecules, avoiding the prompt
degradation of vaccine components such as proteins, peptides, nucleic acids and
oligonucleotides, which would otherwise suffer immediate degradation when administered
(186,187). Several immunoadjuvant molecules, such as some TLR ligands, can cause a life-
threatening septic shock-like state in higher doses (188). As such, these molecules not only
benefit from protection by nanocarriers but their safety is also improved because lower doses
are needed and their systemic distribution is restricted (189). Moreover, nanocarriers can
provide direct intracellular access, facilitating engagement of the intracellular TLR3, 7, 8 and
9 by their ligands, improving their efficacy as vaccine adjuvants (190). Nanocarriers are able
to target the immune system and co-deliver antigens and adjuvants simultaneously to the
same APC, enabling its coordinated activation (3). As mentioned before, it has been verified
that the association of TLR ligands induce superior immune responses over the antigen used
alone (Table 1.4.B). In addition to the advantage of delivering antigens and adjuvants
simultaneously in the same particulate delivery system, the combination of several adjuvants
can also have a positive effect on the activation of the immune system, has verified for the
combined TLR ligation on DCs (Table 1.4.A). Furthermore, biodegradable polymeric NPs
have the additional advantage of providing sustained release profiles to the entrapped
molecules according to the polymer’s physicochemical properties (191). Finally, NP
functionalization further allows the active targeting of APCs improving the vaccine outcome
(192). All these advantageous properties confer intrinsic adjuvanticity to biodegradable NPs
and make them promising vaccine delivery systems (193). The aliphatic polyesters-based
particulate delivery systems will be under focus in the next sections.

Chapter 1
38
4. Reaching the immune system with nanoparticles as a cancer vaccine
4.1. Aliphatic polyesters-based particulate delivery systems: classification
and characterization of the different aliphatic polyesters
The first studies on aliphatic polyesters for biomedical applications date from the
1960s and leaded to the development of materials for surgical implants and tissue repair, such
as the resorbable sutures from polyglycolide (Dexon®) and poly(L-lactide-co-glycolide)
(PLGA) (Vicryl®). Since then, the aliphatic polyesters have been considered in a variety of
medical applications such as prosthetics, artificial skin, dental implants, vascular grafts, pins,
bone screws, stents, and plates for temporary internal fracture fixation (194). The exploration
of these polymers as vehicles for controlled drug delivery started on the 1970s. After that,
these composites have been explored to deliver a wide range of active molecules, such as
contraceptive steroids (195–197), anticancer agents (198), local anesthetics (199) non-steroid
anti-inflammatory drugs (NSAIDs) (200) as well as macromolecular biological therapeutic
agents with short half-life (201). The LHRH agonist leuprorelin acetate (Lupron®) and
goserelin acetate (Zoladex®), both based on poly(lactic-co-glycolic) acid (PLGA), intended
for endometriosis and prostate cancer therapy, are examples of successful commercially
available drug delivery systems (DDSs) (202–204). Aliphatic polyesters have also found
potential for biomedical application as controlled release antigen delivery systems. Advances
in biotechnology allowed the development of subunit vaccines that, apart from the several
advantages over the traditional vaccines, have as general drawback the need for repeated
immunizations due to poor immunogenicity (205,206). Aliphatic polyester-based delivery
systems emerged as promising tools to increase antigen immunogenicity.
In the past three decades, synthetic biodegradable polymers have been increasingly
used for different biomedical applications as an attempt to avoid most of the problems
associated with natural polymers, such as higher costs, lack of reproducibility and
questionable purity (207). Synthetic polymers can now be easily produced with tailored
mechanical and degradative properties and high uniformity in terms of molecular weight
distribution, monomer orientation, sequence, stereo-regularity, polymer shape, morphology
and chemical functionality. This uniformity is required by regulatory entities and thus allows
their application as integral components in final dosage forms, DDSs and in implantable
devices (202–204,208). Poly(amides), poly(aminoacids), poly(alkyl-α-cyanoacrylates),
poly(esters), poly-(orthoesters), poly(urethanes), and poly(acrylamides) have been used to
prepare polymeric devices for biomedical applications (209). Among the synthetic
biodegradable polymers, the thermoplastic aliphatic polyesters have generated tremendous

General introduction
39
interest because of their excellent biocompatibility, biodegradability, and mechanical strength,
which is reflected on their long history of biomedical applications such as sutures, bone
screws, tissue engineering scaffolds and DDSs. Simple formulation methods can be used to
modify those polymers in order to design carrier systems able to deliver a variety of drug
classes such as peptides, proteins, and nucleic acids. Most importantly, they are approved by
FDA and EMA for drug delivery in humans (186,210,211).
The synthesis of aliphatic polyesters can follow three major routes. The first route is
polycondensation of a hydroxyacid or a diol and a diacid, a method which drawbacks include
the low degree of polymerization resulting in low molecular weight polymers and the
impossibility of synthesis of block copolymers (212). The second route is ring opening
polymerization (ROP) of lactones and other cyclic diesters which under proper conditions can
result in polyesters with high molecular weights and limited side reactions such as
racemization (213,214). The third route is enzymatic polymerization, which is carried out
under mild conditions avoiding the use of toxic reagents and with the possibility to recycle the
catalyst, being its major drawback the relatively low molecular weight of the obtained
polymers (215,216).
The aliphatic polyesters mainly used for biomedical applications are those derived
from the monomers glycolide/glycolic acid (GA), lactide/lactid acid (LA), β-butyrolactone (β-
BL), ε-caprolactone (ε-CL), 1,5-dioxepan-2-one (DXO) and trimethylene carbonate (TMC).
They can be classified as homopolymers or copolymers depending on their homogenous or
heterogeneous composition on repeating units, respectively (213). The molecular structures of
the main aliphatic polyester homo- and copolymers as well as their synthesis mechanisms
starting from the correspondent monomers are represented in Table 1.5..
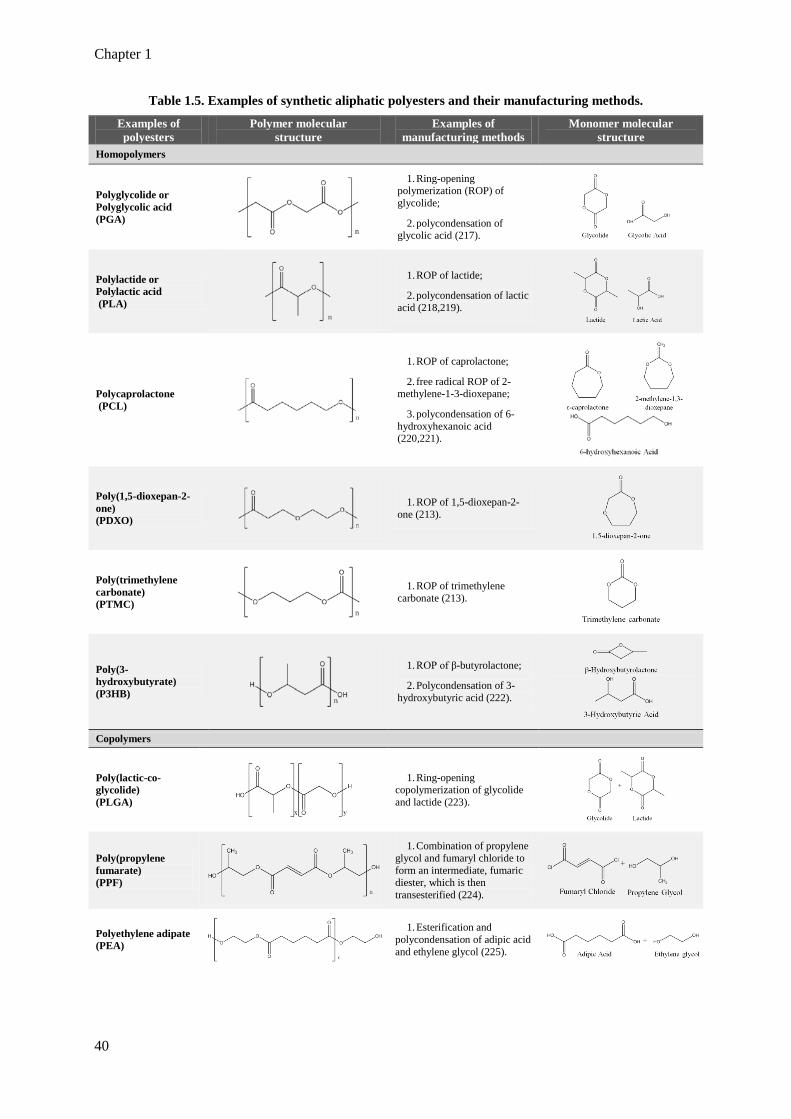
Chapter 1
40
Table 1.5. Examples of synthetic aliphatic polyesters and their manufacturing methods.
Examples of polyesters
Polymer molecular structure
Examples of manufacturing methods
Monomer molecular structure
Homopolymers
Polyglycolide or Polyglycolic acid (PGA)
1. Ring-opening polymerization (ROP) of glycolide;
2. polycondensation of glycolic acid (217).
Polylactide or Polylactic acid (PLA)
1. ROP of lactide;
2. polycondensation of lactic acid (218,219).
Polycaprolactone (PCL)
1. ROP of caprolactone;
2. free radical ROP of 2-methylene-1-3-dioxepane;
3. polycondensation of 6-hydroxyhexanoic acid (220,221).
Poly(1,5-dioxepan-2-one) (PDXO)
1. ROP of 1,5-dioxepan-2-one (213).
Poly(trimethylene carbonate) (PTMC)
1. ROP of trimethylene carbonate (213).
Poly(3-hydroxybutyrate) (P3HB)
1. ROP of β-butyrolactone;
2. Polycondensation of 3-hydroxybutyric acid (222).
Copolymers
Poly(lactic-co-glycolide) (PLGA)
1. Ring-opening copolymerization of glycolide and lactide (223).
Poly(propylene fumarate) (PPF)
1. Combination of propylene glycol and fumaryl chloride to form an intermediate, fumaric diester, which is then transesterified (224).
Polyethylene adipate (PEA)
1. Esterification and polycondensation of adipic acid and ethylene glycol (225).

General introduction
41
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)
1. ROP of butyrolactone and valerolactone (oligomeric aluminoxane as a catalyst);
2. Copolymerization of 3-hydroxybutyric acid and 3-hydroxypentanoic acid (226).
Poly(lactide-co-glycolide-co-α-cloro-ε-caprolactone) (PLGA-α-Cl-ε-PCL)
1. Ring-opening copolymerization of α-cloro-ε-caprolactone, glycolide and lactide. The chloride can be then substituted to further functionalization (227).
n, number of repeated units; x, number of units of lactic acid; y, number of units of glycolic acid; k, number of units of 3-hydroxybutyric acid; z, number of units of 3-hydroxypentanoic acid; r, number of units of α-cloro-ε-caprolactone
From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor &
Francis Editorial Group, London, UK, 2014 [In Press].
Polylactic acid (PLA)-based polymers were largely commercialized as fiber materials
for resorbable sutures. After this, different prosthetic devices have been developed.
Nowadays, PLA resins are approved by the FDA and EMA for food and surgical applications
such as DDSs (228). Because the monomer lactide exists in three diastereomeric forms, PLA
possesses chiral molecules and can be found in four forms: poly(L-lactic acid) (PLLA),
poly(D-lactic acid) (PDLA), poly(D,L-lactic acid) (PDLLA) - a racemic mixture of PLLA and
PDLA - and meso-poly(lactic acid). When polymerized, atactic, heterotactic and isotactic
chains exhibit strikingly different crystallinity, thermal properties and degradability profiles.
Considering its use in biomedical research, only PLLA and PDLLA have shown promise and
thus have been extensively studied (229). Polyglycolic acid (PGA) polyesters are readily
degraded and apart from its use in absorbable sutures, they have found less use as
homopolymers, appearing predominantly as copolymers with lactide and trimethylene
carbonate monomers (230).
During the resorbable polymer “boom” of the 1970s and 1980s, polycaprolactone
(PCL) and its copolymers were used in a number of DDSs due to their numerous advantages
over other biopolymers in use at that time: (i) tailorable degradation kinetics and mechanical
properties; (ii) ease of manufacture and shaping, enabling appropriate pore sizes conducive to
tissue in-growth; (iii) controlled delivery of entrapped drugs; (iv) functionalization to render
the polymer more hydrophilic or adhesive; and (v) superior rheological and viscoelastic
properties, such as low melting (Tm) and glass transition temperatures (Tg) (231).
Polyhydroxyalkanoates are biodegradable and biocompatible polyesters that can be
produced by bacterial and synthetic routes. The most common polymer is poly(3-
hydroxybutyrate) (P3HB), a semi-crystalline isotactic polymer that undergoes surface erosion

Chapter 1
42
due to the hydrophobicity of the backbone and its crystallinity (222). It has been used to
produce particulate DDSs (232,233) and also other composites for non-biomedical
applications (234).
Poly(propylene fumarate) (PPF) is a high-strength polymeric biomaterial that while
technically a polyester, possesses the unique ability to be cross-linked through the unsaturated
bonds in its backbone. As a result, its degradation is dependent on molecular weight, cross-
linker and cross-linking density. PPF is injected as a liquid that becomes solid upon cross-
linking. Therefore it is considered promising for biomedical applications, such as filling bone
defects and long-term delivery of ocular drugs. PPF-based scaffolds allow the design of
structures that may not be attainable from non-crosslinkable degradable polymers (235,236).
To our knowledge, it has never been used to formulate particulate delivery systems.
Physical properties such as Tg, Tm, and crystallinity can be significantly affected by
copolymerization (237). Random, block, and graft copolymerization can yield products with
significantly different properties and be used to obtain a product with a particular combination
of desirable properties. For instance, in the 1970s it had already been recognized that PCL
was particularly amenable to form polymer blends (231). Completely amorphous polymers
like poly(1,5-dioxepan-2-one) (PDXO) are also very valuable for copolymerization to
improve the elasticity of the polymer (238). PLA toughness limits its applications, rendering
it a good candidate for copolymerization (239). Another example is the case of P3HB, which
reduced stability makes it a poor candidate for controlled delivery applications, being most
commonly used as a copolymer with 3-hydroxyvalerate to create PHBV (240). PHBV is less
crystalline than P3HB with a lower Tg and Tm, widely used to tissue engineering, for
instance, to produce polymer composites with high porosity and controlled pore scaffolds
(241). It has also been used to produce particulate DDSs (242,243).
Random copolymers can be easily synthesized by polymerization of different
monomers by the synthesis methods mentioned before. The final copolymer composition
depends on the composition of the monomer feed as well as the reactivity ratio of the
monomers. Normally, copolymers are represented as AB products, where A and B represent
constituent blocks of different nature. The most well-known example of a random polyester
copolymer is PLGA, the copolymer of the LA and GA monomers (186). PLGA can be
produced with different stoichiometric ratios of its monomers and several molecular weights,
two parameters that are of determinant influence on the hydrophobicity and degradation
kinetics of the polymer. The forms of PLGA are usually identified by the monomers’ ratio
used. Accordingly, PLGA 50:50 identifies a copolymer whose composition is 50 % LA and

General introduction
43
50 % GA. The ratio 50:50 is the most widely used of the PLGA copolymers in
nanotechnology (244). PLGA has been widely used to formulate different biodegradable
devices, such as MPs and NPs, implants and in situ-formed devices (245). Because of their
ability to protect molecules from the aggressive in vivo environment, biodegradable PLGA
particles have been investigated for sustained and targeted delivery of various agents,
including hydrophilic and hydrophobic drugs, proteins and peptides, plasmid DNA and
siRNA (246,247). It has also been extensively tested as vaccine particulate delivery systems
to encapsulate a variety of antigens such as proteins, peptides, lipopeptides, cell lysates,
viruses or plasmid DNA (172,248–252). Some advantages and disadvantages of PLGA as a
particulate vaccine delivery system are presented in Table 1.6., some of which are transversal
to all aliphatic polyester-based vaccine particulate delivery systems.
Table 1.6. Main advantages and disadvantages of PLGA as a particulate vaccine delivery system
component. ADVANTAGES
• Biodegradability and biocompatibility; • approval for parenteral use by regulatory authorities around the world; • commercially available with different physicochemical properties (several molecular weights and lactide:glycolide ratios); • drug release profile can be tailored by selecting PLGA polymers with the appropriate properties. A pulsatile release is also possible,
which provides more effective immune responses as it can avoid the risk of tolerance and substitute the need of several boosting administrations typically required to induce protective immunity (253); • protection of encapsulated biomolecules from in vivo degradation in the blood stream, gastro-intestinal tract and nasal mucosa
(254,255); • delivery of antigens to be presented by both MHC class I and MHC class II pathways; • transportation of more than one molecule concomitantly in the same particulate delivery system (e.g. several antigens or co-
encapsulation of antigen and adjuvant) increasing the probability of coordinated and synergistic action (3); • capacity to induce strong T cell responses containing very low doses of antigens and adjuvants, allowing the use of lower doses of these
molecules minimizing the potential side effects often associated with the use of adjuvants (256); • possibility of blending or co-polymerization with other materials; • can be stably stored and easily scaled up for pharmaceutical manufacture.
DISADVANTAGES
• Environment acidification upon degradation compromising cargo’s stability; • risk of denaturation of the incorporated biomacromolecules by the organic solvents and/or shear stress used during particle formulation; • low encapsulation efficiencies of some macromolecules; • lack of suitable functional groups for efficient covalent bioconjugation of bioactive ligands or the adsorption of negatively charged
therapeutics such as nucleic acids; • required sterilization for clinical use; • potential expensive DDS.
From Silva J.M. et al. (2013) J Control Release; 168:179–199.
Advances in synthetic polymer chemistry have unleashed seemingly unlimited
strategies for producing block copolymers beyond the established AB copolymers, with
arbitrary numbers and types of repeating units. Combining chemically distinct blocks in ABA
or BAB triblock, multiblock, star-block and graft copolymers offers unparalleled
opportunities for designing new nanostructured materials with enhanced functionality and

Chapter 1
44
properties, often without adding substantially costs of production (257,258). AB-diblock and
multiblock copolymers possess amphiphilic properties (they are also known as amphiphilic
block copolymers, ABCs) due to their two regions of distinct chemical nature that undergo
phase separation as a result of chain association in solvents that selectively dissolve one of the
blocks. This process results in the formation of nanoscopic supramolecular core/shell
structures - polymeric micelles - through interactions between the hydrophilic and
hydrophobic blocks with one another and the surrounding medium. The chemical nature of
the core-forming block may allow drug incorporation via chemical, physical, and/or
electrostatic means (259). These systems offer several advantages for drug delivery as it may
be possible to choose appropriate block copolymers for specific purposes, such as prolonged
circulation time, introduction of targeting moieties, and modification of the drug-release
profile (260). Poly(ethylene glycol) (PEG)-block-poly(ester)s are one class of the most
frequent ABCs used to polymeric micelle formation. For instance, triblock copolymer
micelles of PLA–PEG–PLA and PLGA–PEG–PLGA were used to encapsulate plasmid DNA
encoding for a multiple-epitope antigen gene of the hepatitis C virus (HCV) (261). As
controlled-release gene coatings, these triblock micelles had shown high DNA loading
efficiency, low cytotoxicity, sustained-release profile and satisfying immune responses in
vivo. A new poly(lactide-co-glycolide-co-ε-caprolactone)-graft mannosylated poly(ethylene
oxide) copolymer was synthesized by combination of “clip” and “click” chemistries and
demonstrated potential to formulate micelles with DC- and macrophage-targeting capacity
(227).
Aliphatic polyesters can also form star polymers taking advantage of synthesis by
ROP of cyclic esters in a “core-first” approach (262). Star polymers are branched, multi-
armed polymeric materials in which the branches radiate from a central core. They offer
increased concentration of functional end groups comparing to polymers of equal molecular
weight, with improved solubility and distinct hydrodynamic volumes. They often have lower
melt viscosities, distinct thermal properties and improved physical processing, while other
properties are more influenced by arm molecular weight than the total molecular weight of the
polymer star. These polymers have been prepared from the monomers LA, GA, β-BL, ε-CL,
and TMC as well as from their copolymers (263). Aliphatic biodegradable star polymers have
found particular utility as controlled release DDSs and in nanotechnology applications, such
as molecular imaging. Their hydrophobic interiors and hydrophilic coronas have the capacity
to accommodate hydrophobic drugs within their interior, providing a way to transport the
payload through aqueous media to the desired physiological target and control the rate of drug

General introduction
45
release (264,265). Star polymers have also demonstrated the capacity to accommodate water-
soluble molecules in their hydrophilic arms (266). It is noteworthy that immunogens (antigens
and adjuvants) are either hydrophilic or hydrophobic molecules, making star polymers
promising tools for vaccine delivery systems. Unfortunately, these structures are still under-
explored as vaccine delivery systems.
Among the different forms of aliphatic polyester-based DDSs, the most common are
micro/nanospheres, micro/nanocapsules, polymeric micelles, nanogels, star copolymers,
films, cylinders, in situ forming implants, scaffolds, and foams (267,268). Some of these
DDSs are relevant for vaccine applications and are schematically represented on Table 1.7..
Table 1.7. Schematic representation of the main classes of particulate vaccine delivery systems that can be
prepared with aliphatic polyesters.
DDS Definition Main formulation methods
Micro/nanospheres
Particle composed by a polymeric matrix in which the cargo is dispersed. Molecules can also be adsorbed at the surface.
Solvent evaporation and solvent extraction processes (single or multiple emulsion); phase separation (coacervation); spray-drying; dialysis (233,245).
Micro/nanocapsules
Vesicular systems in which the drug is confined to an aqueous or oily cavity surrounded by a single polymeric membrane, being able to function as a reservoir.
Nanoprecipitation; emulsion–diffusion, double emulsification; emulsion-coacervation; polymer-coating and layer-by-layer (269).
Micelles
Supramolecular core/shell structures with a hydrophobic core stabilized by a hydrophilic shell. Can function as nanoscopic depots or stabilizers for poorly water-soluble compounds.
Self-assembly of block copolymers (260)
Star polymers
Supramolecular branched, multi-armed polymeric materials in which the hydrophilic branches radiate from a hydrophobic central core. Can accommodate hydrophobic molecules in the core and/or hydrophilic molecules associated with their branches.
Controlled polymerization by ring-opening polymerization (ROP) of cyclic esters in a “core-first” approach (263).
Nanogels
Swollen polymeric networks with tunable chemical and physical structures. Promising tool to overcome major biological barriers due to physicochemical properties manipulation after its injection into body fluids.
Physical self-assembly of interactive polymers, polymerization of monomers in a homogeneous phase or in a micro- or nanoscale heterogeneous environment; crosslinking of preformed polymers; template-assisted nanofabrication (267).
From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor &
Francis Editorial Group, London, UK, 2014 [In Press].

Chapter 1
46
4.2. Biocompatibility and biodegradability of aliphatic polyesters:
degradation and elimination mechanisms
Biocompatibility refers to the ability of a material to perform with an appropriate host
response in a specific application. Interest in the aliphatic polyesters is derived primarily from
this property. These polymers are readily hydrolyzed to form the corresponding hydroxyacids,
many of which are byproducts of various metabolic pathways in the body under normal
physiological conditions and are easily metabolized in the body via the Krebs cycle and
physiologically eliminated (270).
Vert el al. (1992) had defined several terms related to biodegradation that should not
be confounded (Table 1.8.) (271). The biodegradability of a polymer does not guarantee its
biocompatibility since it does not indicate if the degradation products are inert and/or if they
are eliminated from the body. As so, the fact that a polymer is bioresorbable is as important as
its biodegradability. The homopolymers PLA, PGA and PCL and their copolymers are known
to be bioresorbable (213,272). During this section, PLGA degradation will be the primary
focus as an exemplificative case.
Table 1.8. Polymer properties related with in vivo degradation.
Biodegradable Polymer suffer break down due to macromolecular degradation with in vivo dispersion, but no proof for the elimination from the body.
Bioresorbable
Polymer undergoes bulk degradation and elimination through natural pathways reflecting total elimination of the initial material and its degradation byproducts with no residual side effects.
Bioerodable The same as bioresorbable, but the degradation is from the surface.
Bioabsorbable Polymer dissolution in body fluids without any polymer chain cleavage or molecular mass decrease, as happens for water-soluble implants.
From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor &
Francis Editorial Group, London, UK, 2014 [In Press].
Aliphatic polyesters are bulk-eroding polymers, meaning that erosion happens
throughout the polymer bulk and not exclusively from the surface inwards, as happens with
surface-eroding polymers such as polyanhydrides (273). Both degradation mechanisms are
illustrated in Figure 1.8.. For less hydrophobic polymers, water is rapidly transported into the
matrix and the polymer degradation rate is similar everywhere within the matrix, leading to
erosion throughout the polymer matrix (i.e., bulk erosion). In contrast, in hydrophobic

General introduction
47
polymers, water is excluded from the bulk of the matrix leading to erosion only at the surface.
In reality, many polymers erode by a combination of surface and bulk erosion (274). Erosion
depends on the degradation, dissolution and diffusion processes. For PLGA, the dissolution of
water-soluble oligomers (up to nonamers) is often assumed to be the rate-limiting process of
erosion (275). The in vivo degradation of PLGA (and also other polyesters) happens by a
chemical hydrolysis reaction of ester bonds in its backbone resulting in the production of the
original monomers (LA and GA in the case of PLGA) (276). The reaction can be catalyzed by
acids or bases, acids being more relevant. The source of the catalyst can be external (non-
autocatalytic reaction) or internal (autocatalytic reaction) when it happens by the carboxylic
acid end groups of the polymer chain (275). The degradation rate of PLGA is related to the
monomer ratio used in production: GA is slightly more hydrophilic than LA, leading to higher
hydrolysis rates with its higher content (277). From the several monomer ratio combinations
available in the market (50:50, 65:35, 75:25 and 80:20), PLGA 50:50 is a fairly hydrophilic
polymer and has the fastest biodegradation rate of the PLGA polymers (278). For instance,
PLGA 50:50 of 14 KDa has demonstrated to be almost completely degraded in water within
30 days (279).
Figure 1.8. Mechanisms of bulk and surface erosion of polymers. From Peppas N. and Narasimah B.
(2014) J Control Release; pii: S0168-3659(14)00450-7.
Concerning other aliphatic polyester degradation products, hydrolytic degradation of
PHB results in the formation of D-(−)-3-hydroxybutyric acid, a normal blood constituent in
common with acetoacetate and acetone, constitutes one of the three ketone bodies produced
endogenously by the ketogenesis process (280). The PPF breaks down into nontoxic waste

Chapter 1
48
products of fumaric acid and propylene glycol (235). PCL degradation is a homogenous
process that happens in two stages: (i) the non-enzymatic hydrolytic cleavage of ester groups,
and (ii) intracellular degradation for more crystalline polymers and low molecular weight
(less than 3000 KDa). Acetyl coenzyme A and 6-hydroxyl caproic acid are hydrolysis
intermediates that are eliminated through the Krebs cycle (231).
Biodegradation is closely related with the cargo release process. Upon immersion in
aqueous environments, the polymer starts to absorb water immediately causing pore
formation. When the pore network becomes big enough, the cargo release starts (268).
Several mechanistic mathematical models for prediction of drug release from PLGA bulk
polymer have been proposed (275,281). These models are often based on studies with PLGA
microspheres (MSs) and specifically address interactions between three main influent
phenomena: polymer degradation, erosion and cargo transport. Models could be further
improved by considering the effects of autocatalysis on degradation kinetics and oligomer
transport. Deeper insights on the mechanistic coupling between drug diffusion rate and
polymer erosion are also important to characterize those degradation mechanistic models.
There are only four possible ways for molecules to be released from PLGA-based
DDSs: (i) transport through water-filled pores by diffusion (the most common release process
due to the cargo physicochemical characteristics such as a protein or a peptide that are too
large and hydrophilic to be transported through the polymer phase); (ii) transport through
water-filled pores by convention or osmotic pumping; (iii) transport through the polymer; and
(iv) dissolution or erosion of the polymer (which does not require drug transport) (268,282).
Cargo release from aliphatic polyester particulate delivery system is often described to
happen in three phases (Figure 1.9.): an initial phase with high release rate (burst release
phase); an intermediated slow release phase of the molecules entrapped in the polymeric
matrix (lag phase); and a second increased release rate phase (final release phase). The first
phase of the release happens due to the release of adsorbed molecules to the particle surface
and reflects diffusion/controlled dissolution of drug molecules accessible to the solid/liquid
interface. This phenomenon seems to be accentuated by the high hydrophobicity of the
polymer and hydrophilicity of the entrapped molecule (283). The second phase results from
the entrapped drug release that is closely related to the bulk degradation of the polymer (201).
Some studies point only two degradation phases: a first phase that is dependent on particle
size with no polymer mass loss and a second phase that is independent from particle size
during which mass loss occurs (281).

General introduction
49
Figure 1.9. Typical cargo release curve from aliphatic polyester-based particulate vaccine delivery
systems. An exemplificative curve of the release behavior of a macromolecule from an aliphatic polyester-
based particulate delivery system is represented. The release from aliphatic polyester particulate delivery
systems normally happens in three phases. It starts with a “burst release phase” (1.) that can function as
the vaccine priming dose. The following “lag phase” (2.) and the final “sustained final phase” (3.) can
provide controlled release of the antigens and adjuvants and mimic vaccine boosting doses. From Silva
JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor & Francis
Editorial Group, London, UK, 2014 [In Press].
Selection of the appropriate polymer material to achieve the expected cargo release
time scale is normally a primordial consideration in formulation design. The choice of PLGA
polymer for antigen delivery depends on the goal of the immunization strategy. Generally, the
burst release is often considered to avoid when the particulate delivery systems are used for
general drug delivery. However, for vaccine delivery the burst effect can mimic the priming
doses required for current vaccination programs. The release profile is also considered
advantageous because the dose of antigen necessary to stimulate the secondary response is
much lower than that required for evoking a primary response (284). For instance, MSs
prepared with PLGA polymers with different degradation rates (different LA:GA ratios)
yielded particles with different dimensions with fast or slow antigen release profiles with the
aim of developing a single-injection vaccines (167).

Chapter 1
50
4.3. Inherent adjuvanticity
Synthetic polymers are normally referred to have only a limited capacity to elicit
antibody formation or to induce cellular immune responses. Even that the negligible adjuvant
properties are pointed out as one of the disadvantages of aliphatic polyester for vaccine
delivery systems, these materials can also exhibit significant immunomodulation activity in a
complex process dependent on inter-related physicochemical factors such as hydrodynamic
size, morphology, composition and surface properties (285). This immunomodulation
capacity can be considered as an intrinsic adjuvant effect, and is mainly related to their
particulate and physicochemical characteristics and passive targeting of the immune system
(52). These factors are enumerated in Table 1.9.. Aliphatic polyesters can be derivatized or
particles can be decorated in a way that the adjuvant effects on the immune system become
more prominent. Several strategies will be mentioned later on section 6..
Table 1.9. Inherent adjuvanticity of aliphatic polyester particulate delivery systems for vaccines.
• Ability to protect the immunogens from rapid degradation and clearance;
• efficient internalization by APCs; • extracellular reservoirs of immunogens due to sustained release
occurring for lengthy periods (depot effect); • intracellular reservoirs of immunogens in APCs after particle
uptake so that intracellularly released antigens can be presented by the APCs to specific T cells over extended periods of time;
• capacity to directly migrate to lymph nodes after peripheral administration (for particles < 200 nm);
• codelivery of several immunogens, being either several antigens or antigens and adjuvant molecules;
• capacity to induce presentation of antigens through both MHC class I and MHC class II complexes.
From Silva J.M. et al. (2013) J Control Release; 168:179–199.
It is well known that non-functionalized polymeric particles can be rapidly removed
from the blood stream by macrophages of the mononuclear phagocytic system (MPS), also
known as the reticuloendothelial system (RES). This process is mediated by the adsorption of
opsonins (mainly complement proteins such as C3, C4, and C5 and immunoglobulins) to
particle surface and recognition of these molecules by the macrophage phagocytic receptors
(286). It seems that hydrophobic surfaces and chemical groups such as -NH3, -OH or -COOH
can influence the activation of complement but it is not clear if aliphatic polyester particles are
able to induce complement activation or other direct effects on the immune system (287).
There are some evidences that several polymer-related parameters, such as chain length,
architecture (e.g., linear, branched, star-shaped, diblock and triblock copolymers, dendrimers),
functional groups, hydrophobicity/hydrophilicity, and solution properties (monomer versus

General introduction
51
micellar forms) may affect complement activation differently and through different initiation
pathways (288,289). A study on PLGA MPs and NPs demonstrated potent inflammatory
stimulus after the uptake process with NF-κB translocation to cell nucleus and further pro-
inflammatory cytokines production, mainly by MP-stimulated cells (290). On the other hand,
it has been demonstrated that empty PLGA MPs with or without polycationic coatings did not
induced significant maturation and did not affect the capacity of DCs to respond to a
maturation-inducing cocktail afterwards (291). Further studies are needed to clarify the role of
these systems on the activation of the immune system.
5. The influence of physicochemical factors on NP performance
Modulation of immune responses by particulate delivery systems is deeply connected
to their capacity to target the immune system, not only concerning specific tissues and cellular
types but also at a subcellular level. Depending on the internalization mechanism and
intracellular traffic, different immunomodulatory effects may be considered (1,39).
Understanding the interdependent roles that physicochemical characteristics of nanocarriers
play on the cellular uptake and intracellular traffic and fate is extremely important. Many
factors, such as size, charge, geometry, adhesion energy, surface tension of membrane and
bending rigidity ratio between nanocarrier and membrane, can have an impact on their
performance. These parameters affect the so-called “wrapping time,” which describes how a
membrane enclosed and internalize a particle (292,293).
5.1. Influence of size
Size is one of most important factors affecting the uptake of particulate vaccines by
APCs and also on determining their intracellular fate. The cellular uptake can be considered
as a result of the competition between thermodynamic driving force for wrapping and the
receptor diffusion kinetics. The thermodynamic driving force refers to the amount of free
energy required to drive the NPs into the cell while the receptor diffusion kinetics refer to the
kinetics of recruitment of receptors to the binding site. These are fundamental factors that
dictate the amount of NPs that are taken up by cells, as well as the time needed for that (294).
Very small NPs might not produce enough free energy to be wrapped by cell
membrane and bigger NPs might require a too long wrapping time due to the slower receptor
diffusion kinetics (295). As so, it is almost utopical to think about an optimal size for particle
uptake by APCs. There are evidences pointing out that these cells have evolved to effectively
process any antigen with similar dimensions to pathogens, ranging from viruses (20 - 100 nm)

Chapter 1
52
to bacteria and even cells (in the micrometer range) (185). DCs and macrophages have been
shown to optimally phagocyte particles up to 10 and 30 μm in diameter, respectively (296).
Particle size also have an impact on the endocytic mechanism and on the intracellular
fate (accumulation in specific organelles such as lysosomes, endoplasmic reticulum (ER) or
Golgi apparatus, escape to cytosol, exocytosis, etc) and, when dealing with APCs, the type of
immune response generated (297). Each endocytic pathway seems to be defined by a specific
size range of the engulfed soluble or particulate material. In general, virus-sized particles (20
– 200 nm) are usually taken up via classic receptor-mediated endocytosis (clathrin-mediated)
(particles < 200 nm) or endocytosis through caveolae (particles within 50 – 100 nm) and tend
to generate a virus-like immune response with activation of CTLs and Th1 cells (298,299).
Endocytosis of larger sized particles (> 0.5 µm) occurs mainly via macropinocytosis.
Phagocytosis happens only in phagocytic cells, such as APCs, and seems to be more related
with the presence of opsonins at NP surface than with size itself (286). Phagocytosis
preferentially generates a bacteria-like immune response with activation of Th2 cells and
production of antibodies (300).
It has been suggested that NPs smaller than 50 nm tend to aggregate during cellular
uptake. Moreover, the higher surface curvature associated with smaller NPs restricts the
binding and the mutual adsorption between NPs and cells. For NPs larger than 50 nm, the
slower receptor diffusion rate led to much fewer NPs internalized by cells (301). On the other
hand, Gao et al. (2005) demonstrated that, in a size range from tens to hundreds of
nanometers, the fastest wrapping time and receptor-ligand interaction was verified for NPs
with ~55 nm leading to higher intracellular accumulation. For bigger NPs (> 50 nm), the
wrapping time was slower because of the slower receptor diffusion kinetics since more
receptors were taken up during the receptor-ligand binding process and were not available for
binding (295).
Particulate size cannot be ignored when considering the route of administration of a
particulate vaccine. The more traditional routes of vaccine administration are the
subcutaneous (s.c.) and intramuscular (i.m.) injections, the last is often preferred since it has
been recognized that adjuvanted vaccines given via s.c. route induce an unacceptable rate of
injection site reaction (302). Nevertheless, while the two routes are convenient and suitable
for almost any type of material and formulation, neither the muscular tissue nor the subcutis is
particularly rich in APCs. Thus, introducing antigens at sites with a higher density of APCs,
such as the epidermis, lymph nodes, or the afferent lymphatics, with appropriate formulations
and injection devices may induce more efficacious immune responses and protection (303).

General introduction
53
The epidermis is particularly rich in specialized DCs called Langerhans cells (LCs),
which extend dendritic processes to sample a large area of the skin and eventually detect
antigens from pathogens that invade the epidermis (303). After acquisition of the antigen in
the periphery, LCs migrate to regional lymph nodes where they can present antigen to naïve
and memory T cells. As so, a vaccine injected into the epidermis will certainly reach the
lymph nodes for stimulation of immune responses. However, the intradermal (i.d) route is
underexplored for vaccine administration, likely due to the relative difficulty in efficient
intradermal administration, and due to presence of alum in many vaccines, which is
contraindicated for i.d. application due to the risk of long-lasting local adverse reactions
(303). Moreover, the skin of small rodents and humans differ not only in their thickness, but
also in the compactness and connection to adjacent tissues, which might compromise the
translation of pre-clinical studies approaching the impact of different routes of administration
in the immune response.
The majority of lymph node resident DCs have an immature phenotype and can
respond to activator signals and mature in situ (304,305). When targeting the lymph nodes,
additional size aspects must be taken into account. Initial lymph vessels have diameters
around 10 - 60 µm and the sinusoid in the spleen varies from 150 to 200 nm (306). Only
particles of 20 - 200 nm can efficiently enter the lymphatic system and bigger particles only
enter the lymphatic system if they are associated with DCs, which will take approximately 24
hours to arrive in the lymph nodes (307).
Mucosal surfaces, such as gastrointestinal, nasal and vaginal tracts are very often the
portal of entry of the majority of infectious microorganism. Mucosal vaccines have the ability
to confer a protective local immunity to mucosal barriers, in addition to a systemic immune
response. As so, there has been an increasing demand for the development of novel vaccines
that leads to the induction of immune response in systemic circulation as well as at mucosal
surfaces against infectious diseases (308,309). The mucus consists of a physically crosslinked,
viscoelastic hydrogel, with mesh sizes in the order of ~100 nm, which penetration is largely
restricted for particles greater in diameter than a few hundred nanometres (310). It has been
demonstrated that PLGA MPs are suitable for injection into all the aforementioned sites (s.c.,
i.m., i.d., intra-lymph node (i.ln.) administration) but that the type and extent of immune
response varies with the site of administration (303).

Chapter 1
54
5.2. Influence of chemical constitution and hydrophilicity/hydrophobicity
Studies using particulate delivery systems with distinct chemical natures have
demonstrated that hydrophilicity/hydrophobicity can have an impact on the extent of cellular
uptake and intracellular trafficking and fate (311). Zhao F. et al. (2011) reviewed the major
experimental findings of the effects of physicochemical and surface properties of NPs on the
cellular uptake pathway. Nevertheless, the proposed mechanisms of cellular uptake for NPs
studied in different laboratories are sometimes inconsistent or even totally conflicting (312).
Non-functionalized PLGA NPs seem to interact with DCs through β2integrins. The
involvement of these adhesion molecules might explain the higher adhesion of PLGA to DCs
comparing with other biomaterials and also the capacity of PLGA NPs to induce DC
maturation even in the absence of further stimuli (313). A systematic understanding of the
uptake and trafficking processes of particulate delivery systems within cells hence becomes
most important.
5.3. Influence of charge
Surface charge has a profound effect on the internalization capability of particulate
delivery systems. Positively charged particles seem to allow higher extent of internalization,
apparently as a result of the ionic interactions established between positively charged particles
and the negative charge of cell membrane (314–316). Normally, fast and efficient uptake is
associated with positively charged copolymers due to non-specific adsorptive endocytosis,
whereas slow uptake is associated with negatively charged copolymers. Subsequent
intracellular trafficking indicates that some of positively charged NPs could escape from
lysosomes after being internalized and exhibit perinuclear localization, whereas the negatively
charged and neutral NPs prefer to colocalize within lysosomes (317,318). Taking into account
the authors’ experience, polymeric particle charge mainly depends on the chemical nature of
the polymer, the chemical nature of the stabilizing agent and the pH of the dispersant.
The pKa of LA is 3.1 and that of GA is 3.8. As so, PLGA particles are normally
negatively charged at the physiological pH as carboxylic groups on the polymer chain are
ionized at pH 7.4. Notwithstanding, it has been described that PLGA particles reverse their
surface charge at more acidic pH, as that in the endo-lysosomal compartment (86,319). This
reversal of surface charge can explain the capacity of PLGA particles to rapidly escape from
endosomes and lysosomes due to interaction with the membranes of these organelles (316).
This property is a great advantage for a vaccine delivery system where a cellular immune
response is aimed, as it can contribute to the accumulation of antigens in the cytosol and their

General introduction
55
cross-presentation through the MHC class I pathway. Surface charge of aliphatic polyester-
based particulate delivery systems can be easily changed by several techniques such as the use
of PEG-containing copolymers or recurrence to cationic polymer coatings such as chitosan or
poly(ethylene imine) (PEI) (320–324).
5.4. Influence of shape
Particle shape is another parameter of great impact in the context of cellular uptake
and intracellular trafficking. Venkataraman S. et al. (2011) provides a comprehensive
summary on the current understandings of the influence of nanostructures with different
shapes on important biological processes in drug delivery (325). Emerging studies have
clearly shown that non-spherical particles possess drug loading capacities and biological
behaviors that frequently deviate from their historically well-studied spherical counterparts
(326). Spherical-shaped NPs have demonstrated higher cellular uptake comparing to their
sheet-shaped or nanorod-shaped counterparts (294,327). These studies show that the selection
of the NP shape can influence the accumulation and concentration rates of the NPs in a cell.
The optimization of such parameters will lead to improvement in the design of smart
nanoparticulate carriers for various biomedical applications including vaccination.
6. Targeting NPs to the immune system
Generally, two main approaches can be used to target particulate vaccines to the
immune system: passive targeting and active targeting. Passive targeting relies on relatively
simple characteristics such as size, shape, surface charge and hydrophobicity/hydrophilicity of
the particulate vaccine delivery system that influence the rate and extent of internalization by
APCs (185). On the other hand, active targeting is based on surface functionalization of
particles with ligands directed against surface receptors of cells that can function as a gateway
to the immune system (328).
6.1. Passive targeting
Passive targeting to the immune system is an inherent property of particulate vaccine
delivery systems since their particulate nature induces per se the internalization by APCs
(185). Factors that influence the rate and extend of internalization by APCs, such as size,
geometry, surface charge, adhesion energy, surface tension of membrane and bending rigidity
ratio between particle and membrane, hydrophobicity and hydrophilicity, are relatively easy
to manipulate on aliphatic polyester-based particulate delivery systems (245). For instance,

Chapter 1
56
Reddy et al. (2006) presented an interesting study where polypropylene sulphide NPs were
targeted to lymphatic DCs by virtue of their small size, without using any targeting ligand
(329). These parameters have been discussed in more detail in section 5..
A classic example of the passive target potential of nanocarries is the accumulation of
NPs in the tumor microenvironment over time due to the enhanced permeability and retention
(EPR) effect (330). This phenomenon can be explained by the poorly developed leaky
vasculature containing inter-endotelial gaps and the failure to drain extravasated materials due
to an impaired lymphatic system, inherent characteristic of the majority of tumors. The EPR
effect is mostly exploited for anticancer nanomedicines as it is expected to increase the
therapeutic efficacy of chemotherapeutics due to tumor accumulation (331). In an
immunotherapeutic approach, the EPR effect can be seen as a potential way of targeting
immune cells in the tumor microenvironment which are known to play important roles in the
tumor and metastasis development (332).
6.2. Active targeting
Functionalization of particulate-delivery systems can take advantage of different classes
of ligands such as peptides, antibodies, proteins, polysaccharides, glycolipids, glycoproteins,
and lectins (333). Targeting the immune system with functionalizing particulate vaccine
delivery systems can be based on the introduction of ligands for specific cell surface receptors
of defined cell types of the immune system and enhance cellular specific internalization (334).
Also, the recognition of some specific ligands by their receptors on APCs can also contribute
to the enhancement of the immunogenicity of the particulate vaccine, providing an intrinsic
“danger signal” that induces the activation of innate and adaptive immune mechanisms
(321,335). A schematic representation of a functionalized particle designed as a vaccine
delivery system is presented on Figure 1.10..

General introduction
57
Figure 1.10. Schematic representation of a functionalized particle intended to function as a vaccine
delivery system. NPs can be functionalized with antibodies directed to receptors expressed on cells of the
immune systems (e.g. anti-DEC205 to target DCs). Carbohydrates are frequently used for active targeting
of NPs. Simple carbohydrate residues, such as mannose or fucose, are used to target C-type lectins
expressed on DCs. More complex carbohydrates can be also used, such as hyaluronic acid which targets
CD44 receptors on several tumor cell types. NPs can be equipped with an immunogenic coating in order
to increase their recognition by the immune system (e.g. coating with immunogenic polymers such as
chitosan or pluronic block copolymers). The cargo to be delivered by NPs can be either adsorbed at NP
surface or/and entrapped in the polymeric matrix.
An important issue for vaccine design is the consideration of the ligands carried by a
number of pathogens that can interact with receptors on cells of the immune system. As
mentioned in section 3., APCs possess a broad spectrum of cell surface receptors (the PRRs)
that recognize conservative elements exclusively expressed on pathogens (the PAMPs) and
are involved in the initiation, promotion and execution of immune responses (33,144,336).
Recognition of PAMPs by PRRs induces activation of APCs by triggering intracellular
signaling pathways that lead to the induction of inflammatory cytokines, chemokines, IFNs
and upregulation of costimulatory molecules (184). Several PRRs have been targeted by
PLGA NPs functionalized with ligands or antibodies specific for these receptors (Table
1.10.).

Chapter 1
58
Table 1.10. Pattern Recognition Receptors (PRRs) targeted by functionalized PLGA NPs.
PRRs Ligand Antigen Targeted cells
Study design Reference
Toll-like receptors (TLRs)
TLR4 TLR4 ligand: MPLA Ovalbumin (OVA) DCs Preclinical
(mice) (337)
TLR4 TLR4 ligand: 7-acyl lipid A
Tyrosinase-related protein 2 (TRP2) DCs Preclinical
(mice) (167)
TLR4 TLR4 ligand: LPS OVA DCs In vitro (168)
TLR3 + TLR9 TLR3 ligand:
Poly(I:C)
TLR9 ligand: CpG OVA DCs Preclinical
(mice) (165)
TLR4 + TLR7 TLR4 ligand:
MPLA
TLR7 ligand: R837 OVA DCs Preclinical
(mice) (163)
TLR4 or TLR9 TLR4 ligand:
MPLA
TLR9 ligand: CpG
Tetanus toxoid (TT) or MUC1 lipopeptide DCs In vitro (249)
C-Type lectins
Mannose receptor (MR) mannan OVA DCs Preclinical
(mice) (338)
Mannose receptor (MR) mannose OVA Macrophages
In vitro
Preclinical (mice)
(339)
DC-SIGN humanized
targeting antibody hD1
TT DCs In vitro (340)
α-l-fucose
LTA (Lotus tetragonolobus from Winged or Asparagus pea)
hepatitis B surface antigen (HBsAg) M cells Preclinical
(mice) (341)
DEC-205 Anti-DEC-205 mAb OVA
DCs
T cells
In vitro
Preclinical (mice)
(342)
Siglecs
Siglec-7 Anti-siglec-7 polyclonal antibody -
Fibroblast transfected
with Siglec 7 In vitro (343)
Claudins
Claudin 4 C-terminal 30 amino acids of CPE (CPE30)
influenza hemagglutinin (HA) M cells Preclinical
(mice) (344)
Integrins
β1α5Integrin RGD OVA M cells In vitro
Preclinical (mice)
(254)
β1α5Integrin RGD RGDp
OVA M cells In vitro Preclinical
(339)

General introduction
59
LDV LDVp
(mice)
Other targets
STAT3 STAT3 inhibitor:
JSI-124 (cucurbitacin I)
- DCs In vitro (345)
STAT3 + TLR4
STAT3 inhibitor: JSI-124
(cucurbitacin I) TLR4 ligand: 7-acyl lipid A
- DCs In vitro
Preclinical (mice)
(346)
From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor &
Francis Editorial Group, London, UK, 2014 [In Press].
Active targeting is often performed by the functionalization of particulate vaccine
delivery systems with antibodies (Abs). The advantage of this approach is the high specificity
and affinity of Abs for their counterpart ligands. However, their size impedes tissue
penetration and the Fc-part of the Ab may induce nonspecific uptake, Fc-R triggering or
evoke immunogenic reactions against it. These problems may be overcome by using the
smaller single-chain Fv antibody (scFv), humanized Abs or ligands for the receptors, either of
natural or synthetic origins and even modified to have increased affinity (347,348).
As mentioned before, the family of TLRs is the largest and most extensively studied
class of PRRs. They are mainly expressed on APCs but also on several T cell subsets, such as
conventional αβT cells, Treg cells, and γδT cells as well as NKT cells, and serve central roles
in the induction of innate and adaptive immune responses (147). Mammalian TLRs are
classified into several groups based on the type of PAMPs they recognize and cellular
localization (see Fig. 1.4.). As many studies have demonstrated direct costimulatory effects of
TLR agonists in both CD4+ and CD8+ T cells and also B cells in enhancing their proliferation,
activation and effector functions, the use of TLR agonists as vaccine adjuvants is an attractive
target of research (349,350). Moreover, synthetic molecules mimicking original PAMPs are
also recognized by TLRs, which increases their potential for clinical investigation (154).
Nevertheless, as TLRs do not have a direct effect on the uptake of antigens it might be
advantageous to associate the DC activation potential of TLR ligands with other factors that
can have a direct action on the uptake of antigens by APCs.
Other PRRs expressed by APCs that are often targeted by particulate vaccine delivery
systems are the C-type lectin receptors (CLRs). CLRs are a family of transmembrane
receptors characterized by the presence of carbohydrate-binding domains. CLRs are involved
in the recognition and internalization of many glycosylated self and pathogenic antigens.

Chapter 1
60
CLRs are highly specific receptors that facilitate antigen internalization and processing.
Depending on the CLR targeted, either CD4+ or CD8+ T cell responses can be specifically
induced (347,351). For instance, the mannose receptor (MR) recognizes sugars terminated in
D-mannose, L-fucose or N-acetyl glucosamine present on a range of bacteria, viruses, and
yeast (352). Antigens targeted to the MR seem to be directed to distinct early endosomes that
remain stable over extended periods within APCs, ending by being presented exclusively by
the cross-presentation pathway to CD8+ T cells through MHC class I (353). For instance,
PLGA NPs decorated with mannan, a natural polymannose isolated from the cell wall of
Saccharomyces cerevisae, has demonstrated a strong binding affinity and increased CD4+ and
CD8+ T cell responses compared to non-functionalized NPs (338). Another C-type lectin
receptor that has been extensively employed for functionalization of vaccine NPs to APCs is
the DEC-205 (354). Ovalbumin-containing PLGA NPs decorated with different densities of
anti-DEC-205 mAb induced stronger DC and T cell activation with the increasing of the
density of ligands on the surface (342).
The Siglec (sialic acid-binding immunoglobulin-like lectins), another family of PRRs,
is differentially expressed on leukocytes and other immune cells, including APCs (355,356).
Siglecs bind specific sialic acid-containing carbohydrates (sialoglycans) and are believed to
function as endocytic receptors in innate immune recognition of sialylated pathogens,
including both bacteria (e.g. N. meningitidis) and viruses (e.g. HIV) (357). Synthetic glycan
ligands can be used to target these receptors (358).
DC-specific intracellular adhesion molecule-3 grabbing non-integrin (DC-SIGN) is
one of the most DC-restricted C-type lectin receptors in humans that can also mediate antigen
presentation. DC-SIGN specifically binds high-mannose glycans and Lewis-type antigens
which are present on pathogens such as HIV and Mycobacterium and its targeting has
demonstrated to improved antigen presentation of NPs and induced stronger antigen-
dependent T cell responses (340,359).
M cells are specialized cells present in mucosal-associated lymphoid tissue (MALT)
that have a crucial role on mucosal antigen sampling (360). Targeting of these M cells is a
way of getting direct access to APCs (361). Integrins are a family of receptors that can be
targeted on M cells. This family is composed of twenty-four αβ heterodimeric members that
not only mediate the attachment of cells to the extracellular matrix but also take part in
specialized cell-cell interactions (362). β1-integrins at the surface of M cells mediate the
uptake of bacteria, alone or in complex with α5-integrins. Garinot et al. (2007) demonstrated
in vitro higher internalization of PEGylated PLGA NPs grafted with the β1 intregrin peptide

General introduction
61
ligand RGD (Arg-Gly-Asp) and in vivo studies demonstrated that those NPs concentrated in
M cells and induced an antigen-specific IgG response on mice (254). As RGD peptide is
susceptible to degradation, several RGD analogs as well as mannose were grafted on the PEG
chain of PCL–PEG copolymer and incorporated in PLGA-based NPs and demonstrated to
increase the transport of NPs across an in vitro model of human M cells, enhanced NP
internalization by macrophages in vitro, and induced higher production of IgG antibodies than
the i.m. injection of free ovalbumin or intraduodenal administration of either non-targeted or
RGD-NPs (339). Claudins are a family of integral membrane proteins that are found on tight
junctions that are highly implicated on paracellular transport (363). Claudin 4 is one of such
receptors and is highly expressed on M cells in the colon and nasopharynx surface epithelium
that has also been successfully targeted with functionalized PLGA NPs (344).
7. Aliphatic polyester-based NPs applied to cancer vaccines, prophylactic
vaccines and other immunotherapeutic strategies
Aliphatic polyesters-based particulate delivery systems have been tested as carriers for
antigens and adjuvants for prophylactic and therapeutic vaccination strategies, as well as for
the delivery of immnunomodulatory agents. Some examples are presented on Table 1.11..
Vaccines for infectious diseases currently under development aim to mimic the
immune response stimulated by the natural infectious agent in order to induce a protective and
specific immunity against the disease-causing pathogen. However, even that killed or live
attenuated microorganisms and bacterial toxoids are highly effective in the induction of
effector and memory immune responses, related safety issues have driven contemporary
vaccine to be preferentially based on defined and purified recombinant subunit proteins (47).
The comprehension of the mechanisms of microbial evasion and of the host immune system
complex mucosal network has led to the development of several strategies focused on the
induction of protective local immune responses through the administration of mucosal
vaccines based on specific adjuvants or delivery systems (254,339,364). The major advantage
of those mucosal vaccines is their ability to simultaneously induce systemic and local
protective immune responses, through the production of antigen-specific antibodies and the
induction of cellular immune responses (365–367).
As represented on Figure 1.11.A, for the majority of the extracellular pathogens,
protective immunity depends on the presence of antibodies that are able to: (i) bind toxins and
physically block their interaction with host cells; (ii) have a bactericidal effect mediated by
the activation of the classical pathway of complement activation; or (iii) opsonise bacteria that

Chapter 1
62
are then ingested by macrophages (368). In the case of intracellular pathogens (Fig. 1.11.B),
the generalizations are not so easy to apply. It is consensual that a cellular immune response,
where CTLs derived from CD8+ T cells play the most preeminent effector role not only in the
protection against tumors (as represented in Figure 1.4.) but are also essential in the protection
against intracellular pathogens (369). Even though, it would be simplistic to consider that a
cellular immune response should be the only concern of a vaccine against an intracellular
pathogen since it seems that a synergy of different responses might be needed to provide the
adequate protection (370). The role of antibodies in the protection against intracellular
pathogens is not negligible. Antibodies have been shown to provide protection against
intracellular pathogens by exerting a significant immunoregulatory effect on T-cell immunity
and, in some cases, through the interaction with the pathogen before it enters the target cells,
during cell-to-cell spread of the pathogen or even inside the cell upon membrane crossing
(371,372).
The great difficulty in developing such a vaccine is the identification of a single
component (or a limited pool of components) that provides high levels of protection when
used as an immunogen. Instead of a single gene product, such as a toxin with a major role in
the pathogenesis, intracellular pathogens are often equipped with a battery of virulence factors
with high complexity and limited visibility to the immune system, which are generally not
good targets for antibody neutralization (373–375). For many pathogens it is necessary to use
a mixture of subunits to elicit a protective response that is comparable to that elicited by a live
attenuated vaccine (368). The selection of the best candidate antigens requires the application
of distinct methods such as, for example, the identification of antigens deeply involved in
pathogenesis, bacterial survival or conserved sites of vulnerability between strains (376).
Specific features of pathogens such as size, shape and surface molecule organization
seem to be key factors for their recognition and internalization by DCs (292,293). Particulate
vaccines are able to provide an improved interaction with DCs due to their high surface area,
which can provide electrostatic or receptor-interactions (185). From all the classes of
particulate vaccine delivery systems, NPs, especially those made of biodegradable and
biocompatible polymers, have attracted special attention (292,293).

General introduction
63
Figure 1.11 Desired immune responses against extracellular and intracellular pathogens. A. Once
recognized and internalized by DCs, extracellular pathogens are digested in endo-lysosomal organelles
and resultant peptides are loaded on MHC class II molecules. MHC-antigen complexes are presented on

Chapter 1
64
DC surface to CD4+ T cells. The recognition of the antigens happens through the TCR on T cells. The
costimulatory environment resultant from pathogen infection and DC maturation causes T cell activation,
differentiation and proliferation. Upon extracellular pathogen infection, the cytokine priming by DCs
favors a Th2 profile, which results in the activation of B cells and secretion of antibodies against the
pathogen. These antibodies can act through direct pathogen neutralization, complement activation and/or
signaling the pathogen to phagocytic cells through opsonization. The recruitment of cells of the innate
immune system, such as NK cells, granulocytes and macrophages, plays also an important role in the
immune response against extracellular pathogens. Memory Th1 and Th2 cells are also induced and
rapidly reacquire their lineage-specific effector functions upon antigen reencounter. B. Intracellular
pathogens seem to need a much more complex immune response with the involvement of cellular and
humoral branches of the immune system. Antigens from intracellular pathogens are presented through
MHC class I complexes to CD8+ T cells inducing the differentiation of CTLs, which are the main effector
cells in pathogen-infected cell lysis. CD8+ T cells can also develop a memory phenotype that will allow a
prompt response in the case of a second infection. The presentation of pathogen antigens through MHC
class II to CD4+ T cells is crucial for the enhancement of CTL functions through the auxiliary action of Th
cells. Th1 cells enhance CTL functions through the secretion of cytokines (such as IL-2, IL-12 and IFN-γ)
and Th2 cells stimulate the secretion of antibodies by B cells. Players from the innate immune system,
such as NK cells, granulocytes and macrophages, are also recruited in the destruction of intracellular
pathogens.
Aliphatic polyesters-based particulate delivery systems have been tested as carriers for
antigens and adjuvants for prophylactic vaccination strategies (Table 1.11.A). In recent
studies, our group demonstrated that polymeric MSs and NSs were able to influence
Streptococcus equi (S. equi) enzymatic extract presentation by the immune system, inducing a
mucosal and balanced Th1/Th2 immune response crucial for protection against S. equi
infection (255,364,377). Those studies focused mainly on mucosal immunization, which is
thought to be more appropriate for a respiratory infection, such as equine strangles, while
emphasizing the mucosal adjuvant properties of PCL NSs containing S. equi protein extract.
Although some state that aliphatic polyesters homopolymers are quite limited in terms
of conjugation possibilities, the use of these polymers in the formulation of particulate
delivery systems broaden the possibilities of conjugation strategies of different classes of
molecules. For instance, Florindo et al. (2009) tested PLA NSs as vaccine delivery systems
for the delivery of the S. equi M-like protein (SeM) concomitantly with other molecules with
adjuvant properties, such as spermine, oleic acid, alginate and glycol-chitosan (171). Guo and
Gemeinhart (2008) also demonstrated that chitosan can easily be adsorbed to PLGA NPs,
yielding NPs with highly positive zeta potential, which are useful to increase the loading of
negatively charged biomolecules, such as proteins, peptides and nucleic acids (378). Roth-

General introduction
65
Walter et al. (2005) functionalized PLGA MSs with Aleuria aurantia lectin taking advantage
of the available surface carboxylic groups of the polymer for covalent coupling with the
protein (379).
Aliphatic polyester NPs are under investigation for several immunotherapeutic
applications, including the reversion of allergies, asthma and autoimmune diseases. This set
of diseases is normally characterized by IgE-mediated reactions initiated by the degranulation
of mast cells and basophils when their surface-bound IgE molecules are cross-linked through
the binding of the allergen. The reversion of these conditions depends on the shifting of the
immune response against the allergen from a predominantly Th2-type response (IgE and IL-4)
towards a Th1-like immune response (IgG and IFN-γ) (380). Despite the proven clinical
efficacy of standard allergen-specific immunotherapy, this laborious and time-consuming
procedure has reduced patient compliance. Alternative approaches aiming at enhanced
efficacy, shorter treatment duration and lower number of interventions, while simultaneously
providing a high-safety profile, are under intense investigation (381). Examples of aliphatic
polyester particulate delivery systems under investigation to immunotherapy are presented in
Table 1.11.C. These nanomaterials are particularly attractive for allergy immunotherapy due
to their capacity to provide controlled release of allergens overtime and coencaptulate them
with Th1-inducing immunoadjuvants (379,380).
Table 1.11. Examples of aliphatic polyester particulate delivery systems applied to prophylactic
vaccination, cancer therapeutic vaccines and immunotherapy. A. Aliphatic polyester particulate delivery systems for prophylactic vaccines Carrier Aim Study design Outcome Ref.
PLA NSsa
Application: horse protection from the infectious disease strangles. Aim: evaluate the humoral and cellular immune responses induced by i.m. vaccination with Streptococcus equi enzymatic extract or purified Streptococcus equi M-like protein (SeM) entrapped in PLA NSs and the effect of including molecules with adjuvant properties such as spermine (SP), oleic acid (OA), alginate (ALG) and glycol-chitosan (GCS).
Design: Preclinical (mice), immune response evaluation after i.m.b immunizations. Groups:
1. PLA-PVASeM NSs 2. PLA-GCSSeM NSs 3. PLA-ALGSeM NSs 4. PLA-SPSeM NSs 5. PLA-OASeM NSs 6. PLA-PVASeM+CpG NSs 7. SeM+CpG sol.c 8. SeM sol.
• The humoral immune response induced by NSs was markedly higher than that elicited by soluble antigens, isolated or co-admixed with CpG;
• the IgG and IgG subtypes and cytokine titres indicated that NSs with GCS developed a more balanced Th1/Th2 response for both purified SeM and S. equi enzymatic extract proteins, although those induced by the pure antigen-entrapped particles were higher than the S. equi tested vaccines composed by total antigens entrapped in polymeric NSs.
(171)
PLGA NPsd
Application: protection against lethal avian H5N1 influenza virus. Aim: understand the critical parameters of innate immunity that program antibody responses
Design: Preclinical (mice), immune response evaluation after s.c.e immunizations. Groups:
1. NP(HA) + NP(MPL+R837) 2. NP(HA) + NP(R837)
• Synergistic enhancement in the antigen-specific antibody responses after primary and secondary immunization of mice with NP(MPL+R837) plus NP(HA).
(163)
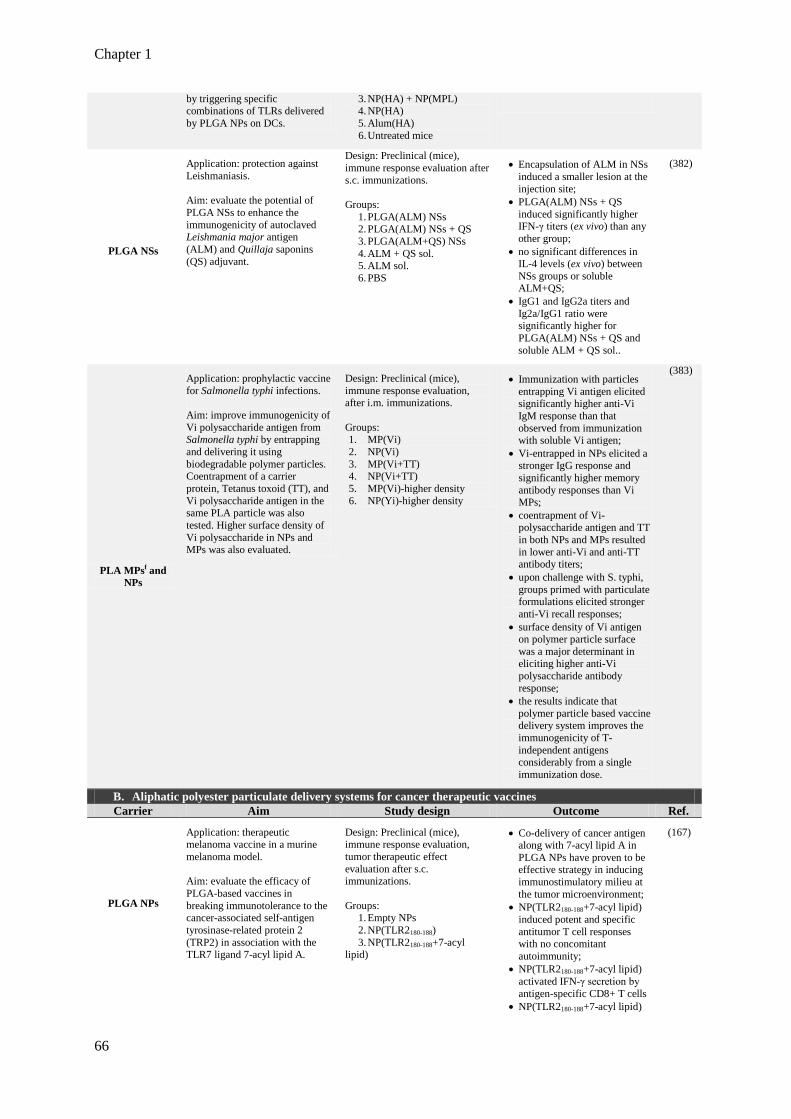
Chapter 1
66
by triggering specific combinations of TLRs delivered by PLGA NPs on DCs.
3. NP(HA) + NP(MPL) 4. NP(HA) 5. Alum(HA) 6. Untreated mice
PLGA NSs
Application: protection against Leishmaniasis. Aim: evaluate the potential of PLGA NSs to enhance the immunogenicity of autoclaved Leishmania major antigen (ALM) and Quillaja saponins (QS) adjuvant.
Design: Preclinical (mice), immune response evaluation after s.c. immunizations. Groups:
1. PLGA(ALM) NSs 2. PLGA(ALM) NSs + QS 3. PLGA(ALM+QS) NSs 4. ALM + QS sol. 5. ALM sol. 6. PBS
• Encapsulation of ALM in NSs induced a smaller lesion at the injection site;
• PLGA(ALM) NSs + QS induced significantly higher IFN-γ titers (ex vivo) than any other group;
• no significant differences in IL-4 levels (ex vivo) between NSs groups or soluble ALM+QS;
• IgG1 and IgG2a titers and Ig2a/IgG1 ratio were significantly higher for PLGA(ALM) NSs + QS and soluble ALM + QS sol..
(382)
PLA MPsf and NPs
Application: prophylactic vaccine for Salmonella typhi infections. Aim: improve immunogenicity of Vi polysaccharide antigen from Salmonella typhi by entrapping and delivering it using biodegradable polymer particles. Coentrapment of a carrier protein, Tetanus toxoid (TT), and Vi polysaccharide antigen in the same PLA particle was also tested. Higher surface density of Vi polysaccharide in NPs and MPs was also evaluated.
Design: Preclinical (mice), immune response evaluation, after i.m. immunizations. Groups: 1. MP(Vi) 2. NP(Vi) 3. MP(Vi+TT) 4. NP(Vi+TT) 5. MP(Vi)-higher density 6. NP(Yi)-higher density
• Immunization with particles entrapping Vi antigen elicited significantly higher anti-Vi IgM response than that observed from immunization with soluble Vi antigen;
• Vi-entrapped in NPs elicited a stronger IgG response and significantly higher memory antibody responses than Vi MPs;
• coentrapment of Vi-polysaccharide antigen and TT in both NPs and MPs resulted in lower anti-Vi and anti-TT antibody titers;
• upon challenge with S. typhi, groups primed with particulate formulations elicited stronger anti-Vi recall responses;
• surface density of Vi antigen on polymer particle surface was a major determinant in eliciting higher anti-Vi polysaccharide antibody response;
• the results indicate that polymer particle based vaccine delivery system improves the immunogenicity of T-independent antigens considerably from a single immunization dose.
(383)
B. Aliphatic polyester particulate delivery systems for cancer therapeutic vaccines Carrier Aim Study design Outcome Ref.
PLGA NPs
Application: therapeutic melanoma vaccine in a murine melanoma model. Aim: evaluate the efficacy of PLGA-based vaccines in breaking immunotolerance to the cancer-associated self-antigen tyrosinase-related protein 2 (TRP2) in association with the TLR7 ligand 7-acyl lipid A.
Design: Preclinical (mice), immune response evaluation, tumor therapeutic effect evaluation after s.c. immunizations. Groups:
1. Empty NPs 2. NP(TLR2180-188) 3. NP(TLR2180-188+7-acyl
lipid)
• Co-delivery of cancer antigen along with 7-acyl lipid A in PLGA NPs have proven to be effective strategy in inducing immunostimulatory milieu at the tumor microenvironment;
• NP(TLR2180-188+7-acyl lipid) induced potent and specific antitumor T cell responses with no concomitant autoimmunity;
• NP(TLR2180-188+7-acyl lipid) activated IFN-γ secretion by antigen-specific CD8+ T cells
• NP(TLR2180-188+7-acyl lipid)
(167)

General introduction
67
induced of therapeutic antitumor effect;
• the results validated the potential of PLGA-based cancer vaccines to break self-tolerance against cancer antigen.
PLGA NPs
Application: develop a novel delivery approach for tumor antigenic peptides in order to elicit enhanced immune responses using PLGA NPs encapsulating tumor antigenic peptides. Aim: explore a novel strategy for effective peptide delivery that enhances the cytoplasmic delivery of peptides into DCs, eliciting a more robust tumor antigen-specific CTL response both in vitro and in vivo. The following peptides were tested: ovalbumin (OVA) peptide 256–264, and mouse six-transmembrane epithelial antigen of the prostate (mSTEAP): 326–335.
Design: Preclinical (mice), immune response evaluation, tumor therapeutic effect evaluation after s.c. immunizations. Groups:
1. empty NPs 2. OVA emulsified in IFA 3. mSTEAP emulsified in IFA 4. NP(OVA) 5. NP(mSTEAP)
• CTL activity in the mice immunized with NPs encapsulating peptides was significantly higher than in those immunized with peptides emulsified in IFA;
• peptide dose encapsulated in NPs was 63 times less than that emulsified in IFA, but it induced a more robust CTL response;
• the lystic capacity of the CTLs induced by NPs encapsulating mSTEAP peptide was 1.82–2.22-fold higher than of the CTLs induced by mSTEAP peptide emulsified in IFA.
(384)
C. Aliphatic polyester particulate delivery systems of immunotherapeutic agents
PLGA MSsg
Application: counteract the Th2-dominated type I allergy. Aim: construct a novel allergen delivery system with the ability for mucosal targeting and evaluate their capacity to induce allergen-specific immune. PLGA MSs were loaded with birch pollen allergens (BP), and functionalized with the α- l-fucose specific Aleuria aurantia lectin (AAL).
Design: preclinical (mice), immune response evaluation, after oral immunizations.
Groups:
1. Empty MSs 2. MS(BP) 3. ALL-MS(BP) 4. BP 5. mSTEAP emulsified in IFA
• AAL-MS(BP) induced a significant production of allergen-specific IgG2a comparing to MS(BS) but not IgG1;
• BS alone induced IgG2a and IgG1, a typical antibody subclass during Th2-type responses in mice.
(379)
PLGA MPs
Application: subcutaneous allergen-specific immunotherapy (SIT) to counteract the Th2-dominated type I allergy Aim: evaluate the capacity of a PLGA MS-based vaccine to be used in SIT and trigger IgG2a antibodies. PLGA MSs were loaded with the major allergen of bee venom, phospholipase A2 (PLA2). The effect of coencapsulation or admixed CpG with or without the cationic stabilizer protamine was also evaluated.
Design: preclinical (mice), immune response evaluation in bee venom pre-sensitized mice followed by oral immunizations with the vaccine.
Groups:
1. MS(PLA2) 2. PLA2 sol. + Alum 3. MS(PLA2+CpG)
• CpG lowered the dose-threshold for induction of allergen-specific immune responses; • only CpG-containing
preparations induced IgG2a antibodies, suggesting that a potent allergy vaccine based on PLGA MPs should also contain CpG or an equivalent Th1-triggering adjuvant; • coencapsulated protamine
exerted its effect through stabilization and improved encapsulation of CpG.
(380)
PLGA/1,2-dioleoyl-3-
(trimethylammonium) propane
(DOTAP)/ligand asialofetuin (AF)
NPs
Application: immunotherapy for hepatocellular carcinoma Aim: development of a non-viral vector for IL-12 gene transfer in hepatocellular carcinoma by employing lipopolymeric Asialoglycoprotein receptor-targeted cationic (PLGA/DOTAP/AF) NPs for plasmid IL-12 (pIL-12) encapsulation or adsorption.
Design: preclinical (mice), antitumor activity and cytokine levels evaluation after intratumoral administration of pIl-12-encapsulated or adsorbed NPs.
Groups:
1. No treatment 2. PBS 3. Naked pIL-12 4. Empty NPs
• Suppression of tumor growth strictly dependent on the presence of the ligand AF; • injection of naked pIL-12
had no effect on tumor growth; • empty NPs by themselves
have no significatively effect on tumor suppression or progression; • AF-NP(pIL-12) and Plain
NP(pIL-12) inhibited significatively the growth of
(385)

Chapter 1
68
5. NP(pIL-12) (encapsulated) 6. NP-pIL-12 (adorbed) 7. AF-NP(pIL-12)
(encapsulated) 8. AF-NP-pIL-12 (adorbed)
tumors with their complete disappearance; • AF-NP-pIl-12 resulted in a
modest antitumor activity, with a slight growth inhibition and very low levels of IL-12 detected.
a nanospheres; b intramuscular; c solution; d nanoparticles; e subcutaneous; f microparticles; g microspheres.
From Silva JM. et al. (2014) Encyclopedia of Biomedical Polymers and Polymeric Biomaterials, Taylor &
Francis Editorial Group, London, UK, 2014 [In Press].
8. Translational aspects and regulatory requirements
Polymeric NPs have demonstrated promising results as particulate vaccine delivery
systems and several advantages over the currently available strategies. The field of particulate
vaccine delivery systems is rapidly evolving and those based on aliphatic polyester polymers
are closer to make a major contribution to improve vaccinology in the 21st century. The
expectation is that the particulate vaccine delivery systems along with the recent developed
techniques in detecting the most appropriate bacterial targets will be better at providing potent
antigen-specific humoral and cellular immune responses and will allow next generation
vaccines to be devised against a range of infectious diseases. With the development of many
new vaccines, children immunization programs have to deliver a large number of
inoculations, a burden for them and for their parents (386) Combining antigens is one of the
solutions despite their technical, clinical, and regulatory difficulties. Nanoparticulate delivery
systems can have an important role in combining several antigens in one vaccine. An aging
population requires prevention to several infectious diseases, such as influenza or
pneumococcal, which require regular booster doses. Polymeric NPs with a prolonged antigen
and adjuvant release might also solve the problem of the need of regular booster injections for
elderly. Nevertheless, several difficulties still need to be overcome. These difficulties are both
related to the vaccine development process - need for validated methods to clinically evaluate
new vaccine strategies; selection of the most relevant antigens and epitopes; determination of
the optimal target population, doses and schedules - as well as with the particulate delivery
systems - determination of whole body and cellular pharmacokinetics; stability; drug release
rates and translation to clinic.
Current translational requirements for nanoparticulate-based systems in
immunomodulation against microbial pathogens have to deal with aspects related to: (i)
conventional approaches for translational research of antimicrobial medicinal products (387–
389); (ii) aspects related to immunomodulation looking at risk-benefit specific assessment and
the toxicological relevance of activating a number of immunomodulatory pathways (390);

General introduction
69
(iii) the importance of immunomodulatory effects of a number of particulate systems, either
activating specific cascades, or potential deleterious effects through non-intended
accumulation of synthetic polymers (391) or potential for generating a stronger immune
response with non-intended systemic impact (392).
Improved understanding of the role of the immune system in controlling tumor growth
and recent developments in the field of nanoparticulate cancer vaccines have created a degree
of knowledge that promises to lead to favorable results with this approach in the near future.
Nevertheless, several questions remain unanswered and difficulties need to be overcome.
Methods to clinically evaluate cancer vaccine responses need to be validated. More
information about the most relevant tumor antigens and epitopes, the optimal patient
population and optimal doses and schedules need to be collected.
Regulatory issues regarding this area deal with two sets of issues: the materials
involved and the cancer vaccine approach in itself. From the materials side, certainly a
number of polymeric and macromolecular carriers have made the way to clinical practice and
are being used for parenteral administration, which might facilitate the access to regulatory
appraisal. But in any specific case a full assessment will have to be considered and the
nonclinical requirements to clinical translation will vary according also to disease
pathophysiology and its impact on overall clinical benefit-risk assessment, together with
materials’ properties and administration routes (2,393–395). A number of activities
particularly dealing with vaccines have a growing place in EMA’s agenda. Even though,
current priorities go further than specific anti-infectious vaccines and even consider DNA
vaccines, meanwhile they lack a strong committement on the Cancer Therapeutic Vaccines
area (396). Current regulatory compliance for clinical development of vaccines applicable
also to cancer vaccines in Europe has long time dealt both with “Note for guidance on the
clinical evaluation of vaccines” (397) and also “Guideline on adjuvants in vaccines for human
use” (398) as well as other relevant documents for vaccines in general. Meanwhile, FDA as
issued in October 2011 a relevant guidance document prepared by the Office of Cellular,
Tissue and Gene Therapies in FDA’s Center for Biologics Evaluation and Research. The
guidance on “Clinical Considerations for Therapeutic Cancer Vaccines” aims at providing
guidance to “sponsors who wish to submit an Investigational New Drug application (IND) for
a therapeutic cancer vaccine with recommendations on critical clinical considerations for
investigational studies of these products” (399). This will have a major impact in present and
future plans to move Cancer Therapeutic Vaccines.

Chapter 1
70
Tumor vaccines might have the capacity to combat already established malignant
disease, but it must be kept in mind that the success of immunological strategies will correlate
with the stage of the disease. The ideal scenario for the application of a therapeutic cancer
vaccine might be minimal residual disease with low numbers of aberrant or cancer stem cells,
where the actively induced immune mechanisms would have a fair chance to prevent tumor
relapse through reduction of circulating tumor cells and micrometastases. In addition,
prophylactic TAA-based vaccines may be conceivable in cases of hereditary predisposition.
Notwithstanding these remaining issues, the development of nanoparticulate cancer
vaccines has received great impetus and efforts are being applied to the development of novel
vaccine adjuvants and nanoparticulate delivery systems, moving from the understanding of
the inherent cellular mechanisms to the evaluation of therapeutic potential in clinical practice.

Chapter 2


73
Chapter 2 - Development of Functionalized Nanoparticles for Vaccine
Delivery to Dendritic Cells: a Mechanistic Approach
Joana M. Silva1,2, Gaëlle Vandermeulen2, Vanessa G. Oliveira3, Sandra N. Pinto4, Catarina
Rodrigues1, Ana Salgado1, Carlos A. Afonso1, Ana S. Viana5, Christine Jérôme6, Liana C.
Silva1, Luís Graça3, Véronique Préat2 & Helena F. Florindo1
1Instituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia, Universidade de Lisboa,
1649-003 Lisbon, Portugal. 2Louvain Drug Research Institute, Advanced Drug Delivery & Biomaterials, Université Catholique de Louvain,
1200 Brussels, Belgium. 3Instituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, 1649-025 Lisbon,
Portugal. 4Centro de Química-Física Molecular & Institute of Nanoscience and Nanotechnology, Instituto Superior
Técnico, Universidade de Lisboa, 1049-001 Lisbon, Portugal. 5Centro de Química e Bioquímica, Faculdade de Ciências da Universidade de Lisboa, 1749-016 Lisbon,
Portugal. 6Center for Education and Research on Macromolecules, Université de Liège, 4000 Liège, Belgium.
Accepted for publication in Nanomedicines, Special Issue Nanotechnology for Vaccine
Development (November 2014)


Nanoparticle delivery to dendritic cells
75
Abstract
Aim: Produce biodegradable nanoparticles to target antigen-presenting cells and evaluate
their potential to be used as a vaccine delivery system. Materials & methods: Untargeted
PEGylated PLGA-based nanoparticles and mannose-grafted nanoparticles were formulated
and physicochemically characterized. Immortalized and primary antigen-presenting cells were
used to study nanoparticle internalization patterns. The endocytic pathways and intracellular
trafficking followed by nanoparticles were also investigated. Results & discussion:
Nanoparticles displayed mannose residues available for binding at the nanoparticle surface.
Different nanoparticle internalization patterns by immortalized and primary antigen
presenting cells were verified. Macropinocytosis, clathrin-mediated endocytosis, caveolin-
and lipid raft-dependent endocytosis are involved in nanoparticles internalization.
Nanoparticles demonstrate both endo-lysosomal and cytosolic localizations and a tendency to
accumulate nearby the endoplasmic reticulum. Conclusion & future perspective: The
developed nanoparticles might drive antigens to be presented through MHC class I and II
molecules to both CD8+ and CD4+ T cells, favoring a complete and coordinated immune
response.
Keywords
Nanoparticles, PLGA, immunotherapy, vaccine delivery systems, mannose receptor
targeting, dendritic cells.


Nanoparticle delivery to dendritic cells
77
1. Introduction
Increasing evidence of the active role of the immune system in non-infectious diseases
such as cancer, autoimmune diseases and allergies, justifies the need for the development of
new strategies for vaccine delivery and immunotherapy (400). Biodegradable nanoparticles
(NPs) are promising delivery systems have been shown to enhance the uptake of antigens and
adjuvants by antigen-presenting cells (APCs), improving immune responses (171,172). These
carriers provide a suitable platform to target the immune system and co-deliver antigens and
adjuvants simultaneously to the same APC, enabling its coordinated activation (3). In
addition, NPs have a large surface area that promote electrostatic and/or receptor-interacting
properties in a greater extent compared to soluble antigens (185). NPs can also protect from
immediate degradation a variety of molecules commonly used as key subunit vaccine
components, such as proteins, peptides, nucleic acids and oligonucleotides (186,187).
Furthermore, biodegradable polymeric NPs, such as those based on aliphatic polyesters, are
able to modulate the release profiles of the entrapped agents through the manipulation of
polymers’ physicochemical properties (191).
NP functionalization further allows the active targeting of APCs improving the
vaccine outcome. The mannose receptor (MR/CD206), a C-type lectin receptor, is involved in
the internalization of pathogens through the recognition of carbohydrate-binding domains. It
recognizes sugars terminating in D-mannose, L-fucose or N-acetyl glucosamine present on
pathogens (351). As so, NP functionalization with mannose can be used to improve APC
targeting and provide direct intracellular access to immunotherapeutic molecules.
The cellular internalization mechanisms and intracellular trafficking pathways
followed by NPs determine the way in which antigens are processed and the nature of
downstream events (1). Cell-surface and intracellular interactions seem to be influenced by
NP physicochemical characteristics. For instance, each endocytic pathway is potentially
adapted to a range of particle sizes and normally relates to a specific type of immune response
(300). Also, extracellular antigens, like those delivered by NPs, are normally directed to
lysosomes where they are processed and loaded onto MHC class II molecules for subsequent
presentation to CD4+ T cells. However, an alternative antigen presentation pathway (cross-
presentation) has also been described, whereby NPs are able to escape from endo-lysosomal
compartments to the cytosol and provide access to MHC class I molecules, insuring the
activation of CD8+ T cells (193). How exactly the NP-related factors contribute to the

Chapter 2
78
determination of these cellular mechanisms is insufficiently understood and must be analyzed
in order to better predict the in vivo performance of these nanosystems.
Herein, we report the development of aliphatic polyester-based NPs functionalized to
target APCs and be used as a vaccine delivery system. NPs were physicochemically
characterized and NP-cell interaction profiles were analyzed. Moreover, we studied the
intracellular trafficking profile that could predict presentation of antigens through both MHC
class I and II, supporting the potential induction of a complete and coordinated immune
response.
2. Materials and Methods
2.1. Materials
Poly(D,L-lactic-co-glycolide) (PLGA) Resomer® RG 502 (lactide:glycolide 50:50)
(average Mw = 7,000 – 17,000 g/mol), sodium cholate hydrate, sucrose, dichloromethane
(DCM), dimethilsulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,3-(4,5-dimethylthiazol-2-
yl)-2,5-diphenyltetrazolium bromide (MTT), paraformaldehyde, concanavalin A (Con A)
(lectin from Canavalia ensiformis (Jack bean), Type IV) grafted to fluorescein isothiocyanate
(FITC) (Con A-FITC), lipopolysaccharide (LPS) from Escherichia coli, bovine serum
albumin (BSA), rottlerin and cytochalasin D were purchased from Sigma-Aldrich (St. Louis,
MO, USA). Chlorpromazine hydrochloride, genistein, nystatin and dynasore were purchased
from Enzo Life Sciences (Farmingdale, NY, USA).
Poly(D,L-lactic-co-glycolide-b-ethylene glycol) (PEG-b-PLGA) (average Mw(PEG) =
4,600 g/mol and Mw(PLGA) = 16,500 g/mol) and poly(ε-caprolactone-b-ethylene glycol)
(PEG-b-PCL) (average Mw(PEG) = 6,000 g/mol and Mw(PCL) = 12,000 g/mol) were
synthesized by ring opening polymerization (402,403). Fluorescein derivative- (F-PLGA) and
rhodamine 6G derivative-grafted poly(D,L-lactic-co-glycolide) (R-PLGA) polymers were
synthesized as described by Freichels et al. (2011) (404). Rhodamine 6G derivative (9-(2-
((carboxymethoxy)carbonyl)phenyl)-3,6-bis(ethylamino)-2,7-dimethylxanthylium bromide)
was synthesized as described elsewhere (405). Mannose-grafted poly(ε-caprolactone-b-
ethylene glycol) (man-PEG-b-PCL) (average Mw(PCL) = 17,000 g/mol and Mw(PEG) =
2,000 g/mol) was synthesized by the Huisgen 1,3-dipolar cycloaddition, where each PCL
chain was grafted with three PEG chains, with 30 % of the PEG chains containing one
mannose residue (406).
Ovalbumin (OVA) Alexa Fluor® 647, wheat germ agglutinin (WGA) Alexa Fluor®
594, goat anti-rabbit IgG Alexa Fluor 488® antibody, Image-iTTM FX Signal Enhancer and

Nanoparticle delivery to dendritic cells
79
AlamarBlue® reagent were purchased from Life Technologies (Carlsbad, CA, USA). The
fluorescence-conjugated rat anti-mouse monoclonal antibodies for flow cytometry analysis
PE-CD11c, FITC-CD206, FITC-Rat IgG2a к isotype control, APC-CD80 and APC-Armenian
Hamster IgG isotype control were purchased from Biolegend (San Diego, CA, USA). Anti-
EEA1, anti-Rab7a, anti-LAMP1 and anti-calnexin immunofluorescence rabbit anti-mouse
antibodies were acquired from Novus Biologicals (Littleton, CO, USA).
2.2. Preparation of NPs
NPs were prepared by the double emulsion-solvent evaporation method, as previously
reported with modifications (254). Two different kinds of NPs were formulated: non-
functionalized NPs (hereafter NPs), constituted by a blend of PLGA, PEG-b-PLGA and PEG-
b-PCL in a 70:15:15 ratio; and functionalized (mannose-grafted) NPs (hereafter man-NPs)
using a blend of PLGA, PEG-b-PLGA and man-PEG-b-PCL (70:15:15). Polymers were
dissolved in DCM (NPs) or DCM:DMSO 70:30 (man-NPs). Fluorescent NPs were formulated
by replacing one tenth of the PLGA mass by F-PLGA (NP-F) or R-PLGA (NP-R). Fifty
microliters of phosphate buffer containing Ca2+ and Mg2+ (DPBS) were added to the polymer
solution and mixtures were emulsified using an ultrasonic processor for 15 seconds at 70 W.
A second emulsion was performed with 1 % (w/v) sodium cholate aqueous solution under the
same conditions. The double emulsion was then poured drip into a 0.3 % (w/v) sodium
cholate aqueous solution, and stirred at 37 °C for 1 h. The NP suspension was washed twice
with ultrapure water by centrifugation at 22000 × g for 45 min, 4 °C (Beckman Coulter
Avanti® J-E Centrifuge JA-20) and finally resuspended in DPBS.
2.3. Physicochemical characterization of NPs
NP size was determined by Dynamic Light Scattering (DLS) using a Malvern Nano
ZS (Malvern Instruments, UK). Cumulative analysis was used to determine the Z-average
size. The zeta potential of NPs was measured using the same device by Laser Doppler
Velocimetry (LDV) in combination with Phase Analysis Light Scattering (PALS). NPs were
diluted in DPBS or phosphate-citrate buffer 200 mM pH 6.0 or 5.0. Electrophoretic mobility
was determined at 25 °C using the Helmholtz-von Smoluchowski model. For stability studies,
NPs were resuspended in DPBS, phosphate-citrate buffer 200 mM pH 6.0 or 5.0 or in cell
culture medium (CCM), i.e., RPMI 1640 + GlutamaxTM supplemented with 10 % (v/v) FBS,
104 Unit/ml/10,000 μg/ml penicillin - streptomycin (PEST), 50 µM β-mercaptoethanol, 10

Chapter 2
80
mM HEPES and 1 mM sodium pyruvate, kept at 4, 25 or 37 °C protected from light, and size
and zeta potential were analyzed as described above at the defined time points.
To determine NP morphology using atomic force microscopy (AFM), NPs were
diluted in purified water at a concentration of 5 mg/ml. Samples were prepared by placing a
drop of the colloidal suspension onto freshly cleaved mica for 15 min, and drying with pure
N2. Samples were than analyzed by AFM in tapping mode in air at room temperature, using a
Nanoscope IIIa Multimode AFM (Digital Instruments, Veeco), and etched silicon tips (ca.
300 kHz), at a scan rate of ca. 1.6 Hz.
To determine surfactant washing efficiency, the supernatants from NP centrifugations
were collected for sodium cholate quantification by high-performance liquid chromatography
method (HPLC) using a Beckman System Gold: UV-vis detector (Beckman 166), Beckman
126 solvent module and Midas autosampler. Samples (20 µl) were injected onto a Shodex
PROTEIN KW-803 series column (8.0 mm ID x 300 mm, 5 µm particle size, 300 Å pore size)
and eluted with 50 mM sodium phosphate buffer (pH 7.0) plus 0.3 M NaCl at 1 ml/min for 20
min at room temperature. Elution was monitored at 220 nm by spectrophotometric analysis.
The amount of sodium cholate in each sample was calculated using a linear standard curve
generated in the range of 0.5 – 5.0 mg/ml, r2 = 0.9979. The surfactant washing efficiency was
calculated as the percentage of sodium cholate quantified in the supernatants comparing to the
total amount used for NP formulation.
2.4. Detection of mannose on functionalized NPs by Lectin Recognition
Assay
Con A was used to detect mannose residues at man-NP surface. man-NPs (1 mg/ml)
were incubated for 1 h with Con A (0.7 mg/ml) in DPBS supplemented with 3 % (w/v) BSA.
Non-targeted NPs were used as a control. NPs were then washed twice with supplemented
DPBS by centrifugation at 22000 × g for 45 min, 4 °C (Beckman Coulter Avanti® J-E
Centrifuge JA-20). Finally, NPs were resuspended in DPBS and RFU were measured using
the Tecan Infinite® Pro 200 Multimode plate reader at 494 nm excitation and 518 nm
emission wavelengths. NPs were also loaded in 8-well Ibidi® µ-Slide microscopy chambers
(Martinsried, Germany) in order to confirm the presence of aggregates induced by Con A
binding. Images were obtained by confocal microscopy using a Leica TCS SP5 (Leica
Mycrosystems CMS GmbH, Mannheim, Germany) inverted microscope (DMI6000) with a
63× water (1.2 numerical aperture) apochromatic objective. Images were processed using the
WCIF ImageJ Software (University Health Network, Toronto, Canada).

Nanoparticle delivery to dendritic cells
81
2.5. Cell line culture conditions
Murine immature dendritic cells (DCs) (JAW SII cell line, ATCC#CRL-11904) were
propagated in αMEM with 10 % (v/v) FBS, 5 ng/ml murine GM-CSF and 4 mM L-glutamine.
Murine macrophage-like cells (J744 cell line), kindly given by Professor Marie-Paule
Mingeot-Leclercq, Université Catholique de Louvain, Belgium, were propagated in RPMI
medium with 10 % (v/v) FBS and 104 U/ml/10 mg/ml PEST.
2.6. Generation of murine bone-marrow derived DCs
Immature bone marrow-derived DCs (BMDCs) were generated as described by Inaba
et al. (1992) (407), with some modifications. Fresh bone marrows flushed from femurs and
tibias from C57BL/6 female mice (Jackson Laboratories) were depleted of erythrocytes by
ACK lysis buffer and cultured for 7 days in RPMI 1640 + GlutamaxTM with 10 % (v/v) FBS,
104 Unit/ml/10,000 μg/ml PEST, 50 µM β-mercaptoethanol, 10 mM HEPES, 1 mM sodium
pyruvate and 20 ng/ml murine GM-CSF. On days 3 and 6, plates were gently swirled, and
medium replaced. On day 7, loosely and non-adherent cells were harvested and cryopreserved
at - 80 °C. DC purity was tested by flow cytometry analysis of PE-CD11c monoclonal
antibody-surface stained cells. Cell cultures had approximately 75 % of CD11c+ DCs.
2.7. In vitro cell viability in the presence of NPs
Cell viability of JAW SII DCs, murine macrophage-like J744.1 cells and BMDCs in
the presence of NPs was evaluated by using the MTT or AlamarBlue® assays (364,408).
Briefly, 4 × 104 cells were seeded in 96-well plates, incubated overnight and treated with
different concentrations of NPs for 48 h. Cells were then washed and the MTT reagent was
added and incubated for 3 h. Formazan crystals were dissolved in DMSO. Absorbance was
quantified at 560 nm using the MultiSkan Ex plate reader (Thermo Fisher Scientific,
Waltham, MA). Alternatively, AlamarBlue® reagent was added at 10 % (v/v) and incubated
for 3 h. Fluorescence measurements were made at 530 nm/590 nm using a FLUOstar Omega
microplate reader (BMG Labtech, Ortenberg, Germany). Triton 0.5 % (v/v) and cell culture
medium were used as positive and negative controls, respectively.
2.8. Nanoparticle uptake by flow cytometry analysis
JAW SII DCs or murine macrophage-like J744.1 cells (5 × 105 /ml/well) were seeded
in 12-well plates and incubated overnight. Cells were then incubated with NP-F (100 and 250
μg/ml) for 3, 8, 24 and 42 h. BMDCs (1.3 × 105 /200 µl/well) were seeded in 96-well flat

Chapter 2
82
bottom plate and incubated overnight. NP-F (100 and 250 mg/ml) were added and incubated
for the indicated times. Cells were then washed with DPBS and resuspended in flow
cytometry buffer. BMDCs were also stained with anti-mouse fluorescently labeled
monoclonal antibodies (PE-CD11c, FITC-CD206, APC-CD80) for 20 min, harvested by
centrifugation and resuspended in the same buffer. The individual fluorescence of 10,000
cells was collected for each sample using a FACScan (BD Biosciences) or a LSR Fortessa
cytometer (BD Biosciences) and analyzed with the CellQuest software (BD Biosciences) or
FlowJo software version 7.6.5 for Microsoft (TreeStar, San Carlos, CA).
2.9. Confocal microscopy imaging analysis
For qualitative uptake studies, BMDCs (2 × 105 /200 µl/well) were seeded in 8-well
Ibidi® µ-Slide microscopy chambers and NP-F were added at the indicated concentrations for
18 h. Cells were then washed with DPBS buffer at 4 °C and stained with WGA-Alexa Fluor®
594 for plasma membrane and Golgi apparatus. Images were obtained by confocal
microscopy as mentioned and processed using the WCIF ImageJ Software. Three-
dimensional (3D) projection images were obtained from 0.4 µm Z-stacks and processed using
the Leica Application Suite-Advanced Fluorescence (LAS-AF) software.
To study intracellular trafficking, BMDCs (2 × 105 /200 µl/well) were seeded in 8-well
Ibidi® μ-Slide microscopy chambers and incubated with NP-R at 500 µg/ml for 3 or 18 h.
Cells were then washed with DPBS buffer at 4 °C, fixed with 4 % (w/v) paraformaldehyde
and permeabilized with 0.2 % (v/v) triton X-100 for 10 min. Nonspecific fluorescence was
prevented by blocking samples with Image-iTTM FX Signal Enhancer for 30 min. Anti-EEA1,
anti-Rab7a, anti-LAMP1 and anti-calnexin rabbit anti-mouse antibodies were added at 1:100
for 2 h. Cells were washed with DPBS and the secondary goat anti-rabbit IgG Alexa Fluor
488® antibody was added at 1:400 for 2 h. Final nucleus contrast was achieved with Hoescht®
332 at 1 µg/ml for 10 min. Images were obtained by confocal microscopy and processed as
mentioned.
For colocalization analysis, images were processed using the WCIF ImageJ Software,
colocalization plug-in. Raw images from each channel were used. Background was
automatically subtracted and thresholds for red and green channels were calculated. A
colocalization finder was run and Pearson’s correlation coefficient (Rr) as well as Mander’s
coefficients (M1 and M2) were calculated. The percentages of colocalization for each channel
were given by the number of voxels for each channel which have both green and red channel
intensities above the threshold.

Nanoparticle delivery to dendritic cells
83
2.10. Study of endocytic pathways involved in NP internalization
BMDCs were seeded in 8-well Ibidi® μ-Slide microscopy chambers for confocal
microscopy (2 × 105 /200 µl/well) or in 96-well flat bottom plate (1.5 × 105 /150 µl/well) for
flow cytometry and challenged for 30 min with one of the following molecules: cytochalasin
D (10 µM), rottlerin (10 µM), chlorpromazine (10 µg/ml), dynasore (80 µM), genistein (100
and 200 µM), nystatin (50 and 100 µM) and sucrose (0.45 M). NPs were then added (500
µg/ml) for 18 h. To check energy-dependency of NP internalization, NPs were added to
BMDCs at 500 µg/ml for 3, 18 or 24 h at 4 or 37 °C. For the experiments performed at 4 °C,
BMDCs were kept at that temperature for 30 min before the addition of NPs. Analyses were
performed by confocal microscopy or flow cytometry as described. OVA Alexa Fluor® 647
(10 µg/ml), used as clathrin-mediated endocytosis (CME) control, was incubated for 30 min
after BMDC treatment with the inhibitors.
2.11. Statistics
All results are expressed as mean ± standard deviation (SD). T-test, one-way ANOVA
and Tukey's post test or two-way ANOVA and Bonferroni’s post test were performed to
demonstrate statistical differences (P < 0.05), using the software GraphPad Prism version
5.00 (GraphPad Software.Inc, CA, USA).
3. Results
3.1. Physicochemical characterization of NPs
Biodegradable polymeric NPs were produced by the double emulsion-solvent
evaporation method using a blend of three aliphatic polyester copolymers: (i) PLGA; (ii)
PLGA pegylated counterpart (PEG-b-PLGA) and (iii) pegylated poly-ε-caprolactone (PEG-b-
PCL) (254).
NPs were physicochemically characterized by the determination of size and
polydispersity Index (PdI) (Table 2.1.). NPs were in a size range between 130 ± 13 nm and
143 ± 22 nm, with a narrow particle size distribution (PdI ranging from 0.09 ± 0.04 to 0.16 ±
0.05). Sodium cholate was quantified by HPLC in the supernatants obtained from the
washings of NPs and demonstrated to be efficiently removed. The surfactant washing
efficiency of NPs was 100.1 ± 6.1 %. The average recovered amount of sodium cholate in
each of the three supernatants was 98.2 ± 7.0 % in the first, 1.6 ± 0.7 % in the second and 0.2
± 0.1 % in the third (data not shown).

Chapter 2
84
In order to potentiate NP interaction with APCs, a targeted delivery system to
MR/CD206 on APCs was also developed. man-NPs were produced by replacing the polymer
PEG-b-PCL by its mannose-grafted equivalent man-PEG-b-PCL. The presence of mannose
did not affect NP size distribution (Table 2.1.).
Table 2.1. Nanoparticle size and polydispersity index (PdI) (mean ± SD; N ≥ 5, n = 3).
Nanoparticles Sizea (nm) PdIb
NP 135 ± 20 0.11 ± 0.07
NP-F 143 ± 22 0.09 ± 0.04
NP-R 130 ± 13 0.16 ± 0.05
man-NP 135 ± 19 0.18 ± 0.06
man-NP-F 132 ± 05 0.18 ± 0.09 a Z-average hydrodynamic diameter. b PdI, polydispersity index.
The zeta potential was measured in three pH values mimicking intracellular
compartments: pH 7.4 (extracellular and cytosolic environments), pH 6.0 (early endosomes)
and pH 5.0 (lysosomes). At pH 7.4, NPs presented a zeta potential around -3 mV confirming
the presence of PEG chains shielding the negative charges of PLGA at NP surface (Fig. 2.1.).
At pH 6.0 and 5.0, zeta potential of NPs and man-NPs became closer to neutrality or even
slightly positive.
Figure 2.1. Zeta potential of NPs and man-NPs at three pH values mimicking intracellular compartments:
DPBS at pH 7.4 (mimicking cytosol) and in 200 mM phosphate/citrate buffer at pH 6.0 (mimicking early
endosomes) or pH 5.0 (mimicking lysosomes). Mean ± SD; N = 3, n = 3. Statistics: one-way ANOVA and
Tukey’s post test. Relative to zeta potential at pH 7.4: ** p < 0.01. NS, statistically non-significant. Two-
way ANOVA and Bonferroni’s post test were used to compare NPs and man-NPs.

Nanoparticle delivery to dendritic cells
85
AFM was used to further characterize the size, shape and surface morphology of NPs
and man-NPs. Both types of NPs presented smooth surface and spherical topography, as can
be seen in 3D images (Fig. 2.2.A and 2.2.C). Section analyses of the diameters demonstrate a
homogenous size distribution but smaller diameters (~15 nm less) compared to the
hydrodynamic diameters obtained by DLS. This can be explained by the fact that the PEG
chains were not in a hydrated state, hence not being extended as they were during the DLS
measurements.
Figure 2.2. Analysis of NP morphology and size by atomic force microscopy. 3D images of NPs (A) and
man-NPs (C), as well as section analysis of NPs (B) and man-NPs (D) where mean diameters are
presented. Mean diameters ± SD were calculated from fifty individual NPs from section analysis of five
different areas imaged for each type of NP.
3.2. Detection of mannose residues at man-NP surface by lectin-recognition
assay
To investigate if mannose residues were exposed at man-NP surface, a lectin (Con A)
recognition assay using DLS, confocal microscopy and fluorescence intensity measurements
was developed. Con A would induce man-NP aggregation if mannose residues were
accessible for binding at NP surface (Fig. 2.3.A) (409). DLS analysis demonstrated that Z-
average hydrodynamic diameter of man-NPs increased from 107 ± 0.4 nm to 572 ± 36 nm,
and PdI increased from 0.126 ± 0.01 to 0.613 ± 0.12 after their incubation with Con A (Fig.

Chapter 2
86
2.3.B). This augmentation in size and PdI was not observed for non-functionalized NPs.
Moreover, confocal microscopy images after treatment with Con A clearly showed bigger
aggregates for man-NPs when compared to NPs (Fig. 2.3.C) and their fluorescence intensity
was significantly higher (p < 0.05) (Fig.2.3.D). These observations demonstrate that mannose
residues were exposed at man-NP surface and were accessible for binding.
Figure 2.3. Detection of mannose residues at man-NP surface using a lectin recognition assay. A.
Schematic representation of man-NP aggregation in the presence of Con A due to the binding of mannose
residues to one of the four binding sites in Con A tetramers. B. NP size distribution represented as
frequency curves of Intensity (%) in function of diameter determined by DLS for man-NPs (upper graph)
and NPs (lower graph), before and after the incubation with Con A. C. Confocal microscopy images of the
NP-Con A-FITC aggregates (scale bars = 20 µm). D. Fluorescence given in arbitrary units (a.u.) of NP-
Con A-FITC aggregates. Mean ± SD; N = 3, n = 3. Statistical analysis: one-way ANOVA and Tukey’s post
test.
3.3. Effect of pH, temperature and medium composition on NP stability
In order to predict the stability and degradation profiles of NPs at different conditions,
NPs were resuspended in DPBS (pH 7.4), 200 mM phosphate/citrate buffer (pH 6.0 or pH
5.0) or CCM, incubated at three temperatures (4, 25 and 37 °C) and their size and zeta
potential were followed for 45 days. NPs did not show any tendency to aggregate, even in the
presence of serum, a constituent of CCM (Fig. 2.4.). The zeta potential decreased with time,

Nanoparticle delivery to dendritic cells
87
which is consistent with the degradation of the aliphatic polyesters and exposure of an
increasing number of carboxylic groups. The decrease of the zeta potential was more
accentuated at 25 and 37 °C and at pH 7.4 and it was similar in DPBS or CCM.
Figure 2.4. The effect of temperature (4 °C, 25 °C and 37 °C) in DPBS (A), the effect of pH (DPBS at pH
7.4 or phosphate/citrate buffer 200 mM at pH 6.0 or 5.0) at 37 °C (B) and the effect of fluid constitution
(DPBS or cell culture medium, CCM) at 37 °C on NP size are represented as the overlay of frequency
curves of Intensity (%) as a function of the diameter over time determined by DLS (histograms). The
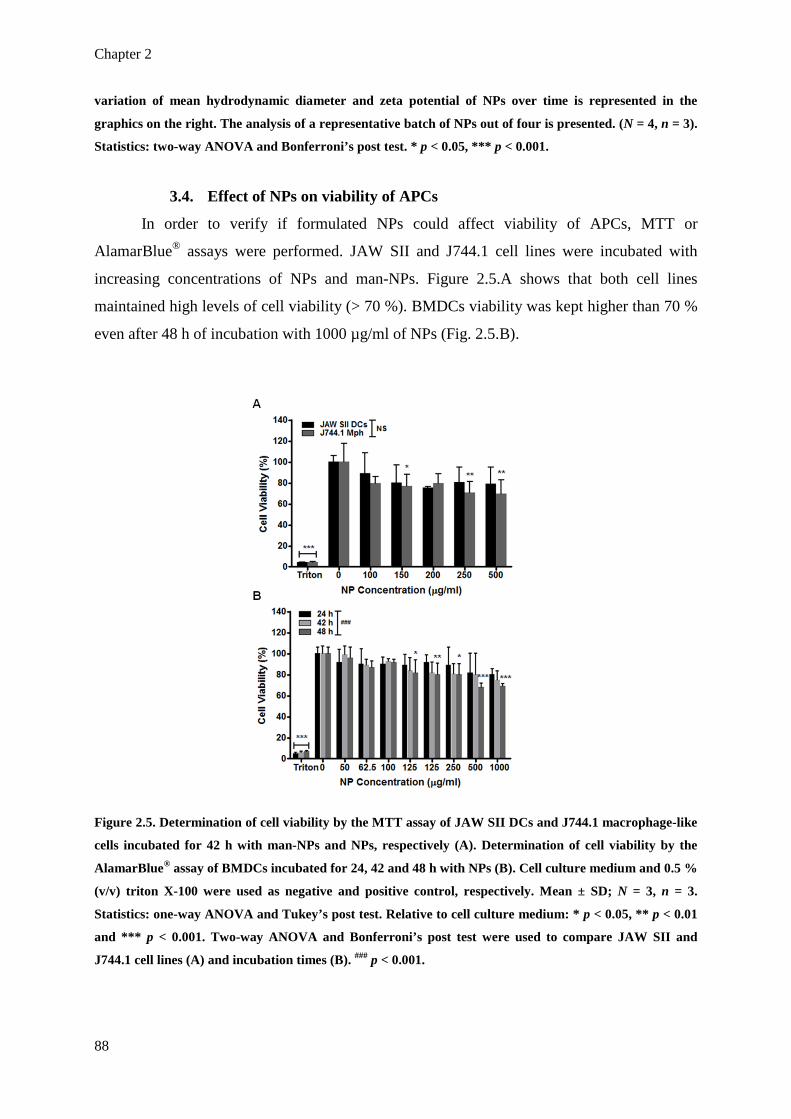
Chapter 2
88
variation of mean hydrodynamic diameter and zeta potential of NPs over time is represented in the
graphics on the right. The analysis of a representative batch of NPs out of four is presented. (N = 4, n = 3).
Statistics: two-way ANOVA and Bonferroni’s post test. * p < 0.05, *** p < 0.001.
3.4. Effect of NPs on viability of APCs
In order to verify if formulated NPs could affect viability of APCs, MTT or
AlamarBlue® assays were performed. JAW SII and J744.1 cell lines were incubated with
increasing concentrations of NPs and man-NPs. Figure 2.5.A shows that both cell lines
maintained high levels of cell viability (> 70 %). BMDCs viability was kept higher than 70 %
even after 48 h of incubation with 1000 µg/ml of NPs (Fig. 2.5.B).
Figure 2.5. Determination of cell viability by the MTT assay of JAW SII DCs and J744.1 macrophage-like
cells incubated for 42 h with man-NPs and NPs, respectively (A). Determination of cell viability by the
AlamarBlue® assay of BMDCs incubated for 24, 42 and 48 h with NPs (B). Cell culture medium and 0.5 %
(v/v) triton X-100 were used as negative and positive control, respectively. Mean ± SD; N = 3, n = 3.
Statistics: one-way ANOVA and Tukey’s post test. Relative to cell culture medium: * p < 0.05, ** p < 0.01
and *** p < 0.001. Two-way ANOVA and Bonferroni’s post test were used to compare JAW SII and
J744.1 cell lines (A) and incubation times (B). ### p < 0.001.

Nanoparticle delivery to dendritic cells
89
3.5. Uptake of NPs by APCs
To verify if the produced NPs could be internalized by APCs, flow cytometry analyses
were performed. NPs were internalized by JAW SII and J744.1 cell lines as early as after 3 h
of incubation. Internalization levels increased with the concentration and incubation time
(Fig. 2.6.A and 2.6.B). After 24 and 42 h, NP internalization levels were significantly superior
to those obtained after only 3 h (p < 0.001). The uptake levels were statistically different for
both NP concentrations tested (unpaired t-test analysis).
BMDCs were used to confirm the uptake patterns of NPs in primary cells.
Surprisingly, a totally different pattern of internalization was observed. Similar levels of
positive cells (around 60 %) were obtained after 3, 18 or 24h of incubation (Fig. 2.6.C).
Differences observed between NP concentration and times of incubation were not statistically
significant. It was possible to exclude the possibility of NP-F adsorption onto plasma
membranes since NP-F have a clear intracellular localization shown by 3D confocal
microscopy images (Fig. 2.7.). Moreover, regardless the concentration used, high levels of
NP-F can be seen inside BMDCs, corroborating the results from flow cytometry analysis (Fig.
2.4.C).
Figure 2.6. Internalization of NPs by JAW SII murine immature DCs (A), J744.1 murine macrophage-like
cells (B) and BMDCs (C), expressed by percentage of positive cells in the population sorted by a flow
cytometer. Mean ± SD; N = 3, n = 3. One-way ANOVA and Tukey’s post test were used to compare
uptake at any time point to 3 h of incubation in the respective NP concentration: NS, statistically non-
significant; * p < 0.05, ** p < 0.01 and *** p < 0.001. Two-way ANOVA and Bonferroni’s post test were
used to compare uptake at 100 µg/ml vs. 250 µg/ml: # p < 0.05, ## p < 0.01 and ### p < 0.001.

Chapter 2
90
Figure 2.7. Confocal microscopy images obtained after 18 h of incubation of BMDCs with three different
concentrations of NPs (250, 500 and 1000 µg/ml). Plasma membrane was stained with WGA-Alexa Fluor®
594. Representative images of three independent experiments are shown. Scale bars = 15 µm.
3.6. Endocytic pathways involved in NP internalization
There was a clear inhibition of NP internalization at 4 °C suggesting energy-
dependency of the process (Figure 2.8.A). To study the involvement of CME in the
internalization of NPs by BMDCs, chlorpromazine, dynasore and a hypertonic sucrose
solution were used. Chlorpromazine and hypertonic sucrose solution are known to prevent the
recycling of clathrin and consequently disable the formation of clathrin-coated pits (410,411).
Dynasore is an inhibitor of dynamin and consequently prevents clathrin-coated pits and
caveoli from pinching off from cell membrane (412). BMDCs were incubated with NPs for
18 h after treatment with one of those molecules for 30 min. Cells were then analyzed by flow
cytometry and confocal microscopy. OVA-Alexa Fluor® 647 was used as a control since it is

Nanoparticle delivery to dendritic cells
91
known to be internalized by CME (413). Confocal microscopy images demonstrate an
apparent total inhibition of NP internalization by dynasore, a very significant inhibition by
chlorpromazine and a partial inhibition by sucrose (Fig. 2.8.B). When analyzed by flow
cytometry, the internalization of NPs was reduced by 40 to 60 % (Fig. 2.8.C). Therefore,
CME seems to be partially involved in the internalization of NPs by BMDCs. Further
evidence of the partial involvement of CME in NP internalization was obtained by analyzing
the colocalization of NPs and OVA-Alexa Fluor® 647 after their coincubation on BMDCs.
Colocalization analysis was based on the approach of Costes et al. (2004) using the WCIF
ImageJ software and a colocalization plug-in (414). After 30 min of simultaneous incubation
of NPs and OVA, their colocalization was near 20 % (Fig. 2.8.D). M1, M2 and Rr were in
accordance. After 18 h of incubation of NPs followed by 30 min of incubation with OVA-
Alexa Fluor® 647, the colocalization of NPs with OVA increased to ~40 %, while
colocalization of OVA with NPs increased to ~60 %. MNPs increased from 0.27 ± 0.08 to 0.52
± 0.17 while MOVA increased from 0.39 ±0.12 to 0.73 ± 0.17, suggesting an increase of
dependence of both channels with time. Rr increased from 0.32 ± 0.11 to 0.55 ± 0.04,
suggesting a prominent correlation of both channels with time.
Since macropinocytosis is a constitutive process in DCs it is likely to be involved in
NP internalization (415). Cytochalasin D, a known actin disrupter, and rottlerin which
interfers with cytoskeletal rearrangements through the inhibition of the delta protein kinase C
(PKCδ), were used. Confocal microscopy images (Fig. 2.8.E) as well as flow cytometry
analyses (Fig. 2.8.F) demonstrate that both molecules extensively inhibited the internalization
of NPs by BMDCs (p < 0.001). The vehicle used for these molecules (DMSO) had no effect
on NP internalization at the concentration used. As expected, macropinocytosis was involved
in the internalization of the nanocarriers by BMDCs.
Genistein, a specific inhibitor of tyrosine kinases, and nystatin, which complexes with
cholesterol, inhibit the caveolae- and lipid raft-dependent endocytosis (410,416). Figure 2.8.G
demonstrates that both molecules reduced the internalization of NPs by BMDCs in 60 %.
Genistein and nystatin concentrations were doubled to 200 and 100 µM, respectively, and the
reduction of NP internalization was similar, demonstrating that the maximum inhibition was
reached and caveolin- and lipid raft-dependent endocytosis is only partially involved in the
internalization of NPs by BMDCs.

Chapter 2
92
Figure 2.8. Effect of endocytic inhibitors on the internalization of NPs by BMDCs. A. Energy-dependency
of NP internalization by BMDCs. Clathrin-mediated endocytosis (CME) involvement was analyzed by
confocal microscopy (B) and flow cytometry (C) after treatment with dynasore, chlorpromazine and

Nanoparticle delivery to dendritic cells
93
hypertonic sucrose solution. For colocalization analysis of NPs and OVA in BMDCs (D), NP-R (green)
were added to BMDCs at 500 µg/ml and incubated for 30 min or 18 h. OVA-Alexa Fluor® 647 (red) were
simultaneously added at 10 µg/ml and incubated for 30 min. Hoescht® 342 was used for nucleus contrast
(blue). Colocalized pixels from red and green channels, obtained through WCIF ImageJ software, appear
in white (right). This software was also used to quantify the percentage of colocalization of the NPs with
OVA (%ColNP) and OVA with NPs (%ColOVA), the Mander’s coefficients for both entities (MNPs and
MOVA) and the Pearson’s correlation coefficient (Rr). Mean ± SD from at least 25 cells from six
independent images is represented. Macropinocytosis involvement analyzed by confocal microscopy (E)
and flow cytometry (F) after treatment with rottlerin and cytochalasin D. G. Caveolin and lipid raft
involvement analyzed by flow cytometry after treatment with genistein and nystatin. DMSO was tested as
the vehicle control. Scale bars = 25 µm. Results from flow cytometry analysis are expressed as normalized
median fluorescence intensity relative to untreated samples (mean ± SD; N = 3, n = 3). Statistics: one-way
ANOVA and Tukey’s post test. *** p < 0.001. Two-way ANOVA and Bonferroni’s post test were used to
compare uptake at 4 °C and 37 °C at different time points (A).
3.7. Intracellular trafficking followed by NPs in BMDCs
To evaluate the potential of NPs to deliver antigens to MHC class I and II complexes,
the intracellular trafficking of NPs was followed in BMDCs. NP-R were added to BMDCs
and immunofluorescence antibodies were used to visualize organelles: anti-calnexin for
endoplasmic reticulum (ER), anti-EEA1 for early endosomes (EE), anti-rab7a for EE and late
endosomes (LE) and anti-LAMP1 for LE and lysosomes (Ly). Figure 2.9.A shows
representative images of three independent experiments of the incubation of BMDCs with
NP-R (red) for 30 min, 1, 2, 3 and 18 h, and staining of each organelle (green). Red-green
merged images present the colocalized pixels (in white). Figure 2.9.B represents the
percentage of colocalization of NPs with each organelle. After 30 min, NPs presented a
colocalization of 35.4 ± 15.4 %, 46.3 ± 14.6 %, 55.9 ± 13.2 % and 61.9 ± 13.2 % with EEA1,
Rab7a, LAMP1 and calnexin, respectively. After 3 h, NPs colocalization with organelles
increased to 51.6 ± 10.0 %, 62.7 ± 9.8 %, 66.3 ± 13.3 % and 77.1 ± 8.8 % for EEA1, Rab7a,
LAMP1 and calnexin, respectively. The percentage of colocalization of NPs with EEA1,
Rab7a and LAMP1 after 18 h decreased to 46.1 ± 13.8 %, 56.9 ± 10.2 % and 47.2 ± 22.1 %,
respectively. Only the colocalization with calnexin increased (79.1 ± 8.2 %). This suggests
that NPs rapidly reach the endo-lysosomal pathway since after only 30 min considerable
amounts of NPs colocalized with those organelles (between 35.4 ± 15.4 % for EEA1 and 61.9
± 13.2 % for calnexin). The levels of colocalization increased for all the organelles during the
first 3 h, decreasing then at 18 h, with the exception of calnexin, which remained very high (~
80 %) and kept almost constant from 3 to 18 h of NP incubation. The percentage of

Chapter 2
94
colocalization of the organelles with NPs varied from 32.6 ± 13.0 % to 77.1 ± 8.8 %
suggesting no saturation of the organelles with NPs (Fig. 2.10.A). M1 was in accordance to
what was obtained for the percentage of colocalization of the red with the green channels
(Fig. 2.9.C). Once again, the highest colocalization was obtained between NPs and calnexin.
M2 varied in accordance with the M1, suggesting the dependence of the staining of NPs and
organelles (Fig. 2.10.B). Rr values were higher than 0.5 at all time points, implying high
correlation levels between NP and organelle stains (Fig. 2.9.D).
Figure 2.9. Intracellular trafficking and colocalization analysis of NPs in BMDCs. A. Confocal microscopy
images obtained after 30 min, 1, 2, 3 and 18 h of BMDCs incubation with NP-R (red). Cells were
incubated with rabbit anti-mouse anti-EEA1 for early endosomes (EE), anti-Rab7a for EE and late
endosomes (EE + LE), anti-LAMP1 for LE and lysosomes (LE + Lys) and anti-calnexin for the
endoplasmic reticulum (ER) and then labeled with goat anti-rabbit IgG Alexa Fluor® 488 (green).

Nanoparticle delivery to dendritic cells
95
Hoescht® 342 was used for nuclei contrast (blue). Scale bars = 15 µm. Colocalized pixels from red and
green channels, obtained through WCIF ImageJ software, appear in white below the respective image.
This software was also used to quantify the percentage of colocalization of NPs with each organelle-
labeling protein (B), the Mander’s coefficient values for the red channel (M1) (C) and the Pearson’s
correlation coefficient (Rr) (D). Mean ± SD from at least 25 cells from six independent images is
represented.
Figure 2.10. Percentage of colocalization of organelles (green pixels) with NPs (red pixels) (A). Mander’s
coefficient of green channel (M2) (B). Mean ± SD from at least 25 cells from six independent images is
represented.
3.8. In vitro targeting of APCs with mannose-grafted NPs
To verify if man-NPs could be actively targeted to APCs, flow cytometry analyses
were performed to compare the internalization levels of NPs and man-NPs. For JAW SII DC
line, a significantly higher uptake was obtained for man-NPs (15.6 ± 0.9 % and 43.8 ± 1.6 %
for 100 and 250 µg/ml of man-NPs comparing to 7.5 ± 0.4 % and 17 ± 7.6 % for 100 and 250
µg/ml of non-functionalized NPs) (Fig. 2.11.A). Increasing the amount of man-PEG-b-PCL in
the formulation to 30 % w/w (man(30%)-NPs) resulted in higher internalization levels (42.7 ±
0.1 % and 66.7 ± 1.1 %, for 100 and 250 µg/ml, respectively) (p < 0.001). Similar
observations were made on the macrophage-like cell line after 3 h of incubation with 250
µg/ml of non-targeted NPs, man(15%)-NP and man(30%)-NPs (10.1 ± 1.0 %, 14.0 ± 1.2 %
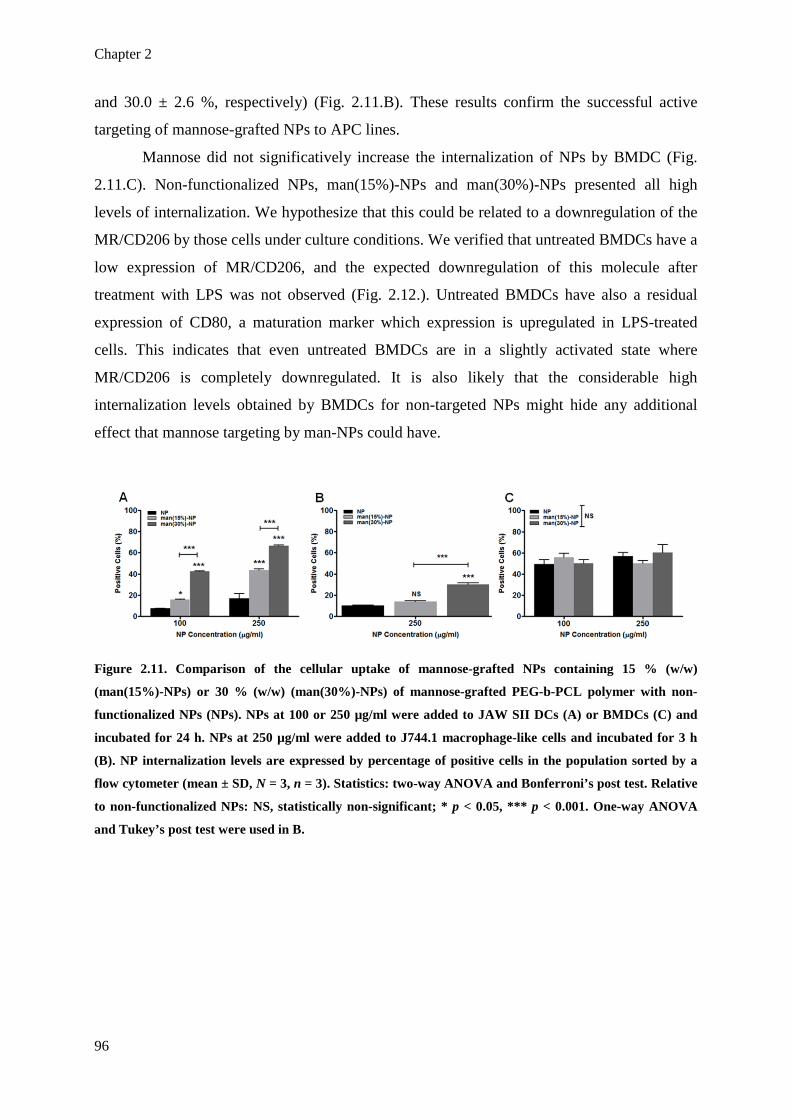
Chapter 2
96
and 30.0 ± 2.6 %, respectively) (Fig. 2.11.B). These results confirm the successful active
targeting of mannose-grafted NPs to APC lines.
Mannose did not significatively increase the internalization of NPs by BMDC (Fig.
2.11.C). Non-functionalized NPs, man(15%)-NPs and man(30%)-NPs presented all high
levels of internalization. We hypothesize that this could be related to a downregulation of the
MR/CD206 by those cells under culture conditions. We verified that untreated BMDCs have a
low expression of MR/CD206, and the expected downregulation of this molecule after
treatment with LPS was not observed (Fig. 2.12.). Untreated BMDCs have also a residual
expression of CD80, a maturation marker which expression is upregulated in LPS-treated
cells. This indicates that even untreated BMDCs are in a slightly activated state where
MR/CD206 is completely downregulated. It is also likely that the considerable high
internalization levels obtained by BMDCs for non-targeted NPs might hide any additional
effect that mannose targeting by man-NPs could have.
Figure 2.11. Comparison of the cellular uptake of mannose-grafted NPs containing 15 % (w/w)
(man(15%)-NPs) or 30 % (w/w) (man(30%)-NPs) of mannose-grafted PEG-b-PCL polymer with non-
functionalized NPs (NPs). NPs at 100 or 250 µg/ml were added to JAW SII DCs (A) or BMDCs (C) and
incubated for 24 h. NPs at 250 µg/ml were added to J744.1 macrophage-like cells and incubated for 3 h
(B). NP internalization levels are expressed by percentage of positive cells in the population sorted by a
flow cytometer (mean ± SD, N = 3, n = 3). Statistics: two-way ANOVA and Bonferroni’s post test. Relative
to non-functionalized NPs: NS, statistically non-significant; * p < 0.05, *** p < 0.001. One-way ANOVA
and Tukey’s post test were used in B.

Nanoparticle delivery to dendritic cells
97
Figure 2.12. Surface expression of the mannose receptor (MR/CD206) and the costimulatory molecule
CD80 by BMDCs before and after treatment with LPS. Black lines: untreated BMDCs. Colored lines:
BMDCs treated with 100 ng/ml LPS from E.coli for 24 h. Tinted curves: isotype antibody control.
Numbers correspond to median fluorescence intensity levels. One representative experiment out of three
is represented.
4. Discussion
The aim of this study was to develop a nanoparticulate delivery system to target
APCs, deliver antigens to be presented by MHC class I (in the endoplasmic reticulum) and II
molecules (in the lysosomes) and potentiate the encounter of immunoadjuvants with their
intracellular receptors. We also intended to increase the knowledge on NP-cell interaction, NP
functionalization impact, endocytic pathways and intracellular trafficking.
A reproducible formulation method has been developed using biodegradable and
biocompatible aliphatic polyester copolymers to produce ~130 nm NPs with narrow particle
size distribution, smooth surface and spherical morphology. PLGA, the main constituent of
NPs, is approved for parenteral use in humans (186,193). PEG-b-PLGA and PEG-b-PCL,
were used in order to provide a PEG source in NP formulation, to chemically functionalize
the polymers with ligands and increase NP stability due to PEG repulsive properties (254).
PEG-b-PCL was also used due to its easiness of functionalization (231). The ratio of the
polymers in the formulation has been previously optimized in terms of shielding of negative
charges at physiologic pH without hindering ligand accessibility (254). In order to potentiate
NP interaction with APCs, functionalization of NPs with mannose was developed to target the
MR/CD206 on APCs, which is involved in the recognition and internalization of pathogens
(351).
The uptake efficiency of particulate delivery systems by APCs depends on several
particle properties, such as size, shape, surface charge, hydrophobicity/hydrophilicity and

Chapter 2
98
receptor interactions (417). The developed NPs should be able to travel through the lymphatic
system after peripheral injection and arrive directly to the lymph nodes, where high number of
DCs can be found. Only NPs up to 200 nm can be delivered into the lymphatic circulation
(185,418). Bigger particles will need transportation by DCs. NP charge can also influence
their internalization and intracellular trafficking (314). Zeta potential measurements showing
a tendency to revert NP surface charge from negative values to neutral or even positive
surface charge, possibly due to protonation of the exposed acidic groups on PLGA chains, can
explain the capacity of PLGA NPs to escape from endo-lysosomes due to their association
with the membranes of those organelles (86,319). This property constitutes an advantage for a
vaccine delivery system where a cellular immune response is required, as is the case of anti-
viral or cancer vaccines. Escape from endo-lysosomes to cytosol is known to be one of the
mechanisms of cross-presentation of exogenous antigens to CD8+ T cells, potentiating the
activation of cytotoxic T lymphocytes (CTLs) (1). The maintenance of NP size over time
along with the decrease of zeta potential is in accordance with a bulk eroding profile,
characteristic of the aliphatic polyesters (419).
To verify if the produced NPs were internalized by APCs, JAW SII and J744.1 cell
lines were incubated with fluorescent NPs containing fluorescein derivative or rhodamine
derivative-grafted PLGA. The use of fluorophore-grafted polymers in our formulation
constitute a great advantage over the use of soluble probes entrapped in NPs, since it
eliminates the risk of misinterpretation due to probe leakage. Internalization levels increased
with NP concentration and incubation time. The hydrophilic surface of PEGylated NPs could
confer a “stealth-like” effect and delay the NP recognition and uptake by APCs (420). The
internalization levels obtained for both cell lines were not statistically different, reinforcing
the previous observation that DCs and macrophages have equivalent internalization capacities
(35). BMDCs were used to confirm the uptake patterns of NPs in physiologic-closer
conditions. Surprisingly, a totally different pattern of NP internalization was observed.
BMDCs internalized NPs more promptly in a time- and concentration-independent manner.
To the best of our knowledge this is the first time that profiles of NP internalization by APC
of different natures are compared. These results suggest that immortalized cell lines of APCs
might have lost their avidness for particulate entities. APC lines have been pointed out as an
effective alternative to the use of primary cells in order to avoid several experimental issues
related to their use, such as culture heterogeneity, contamination with other cell types,
variability due to donor characteristics and need of animals (421). However, our study shows
that careful attention must be paid to the interpretation of the results obtained with cell lines

Nanoparticle delivery to dendritic cells
99
since they might not always be correlated with what happens in vivo, contrary to the
observations made before by others, where BMDC and immortalized DC lines were
compared in other contexts (421,422).
The endocytic pathways followed by NPs are determinant of its intracellular
trafficking and final fate and can determine the type of induced immune response. The
formulated NPs presented mean diameters that fall into the range usually taken up via CME
(< 150 nm) and tend to generate a virus-like immune response with activation of CTLs and T
helper (Th) 1 cells (300). Our study demonstrates that CME is partially involved in NP
internalization by BMDCs. Since macropinocytosis is a constitutive process in DCs, this
mechanism could be involved in the internalization of NPs, even though it normally happens
to particles bigger than 0.5 µm (300,415). Macropinocytosis drives antigens to be presented
preferentially by MHC class II molecules and induces mainly a humoral immune response,
but it can also drive antigens for presentation by MHC class I (415). As expected,
macropinocytosis was highly involved in the internalization of the nanocarrier by BMDCs.
The total inhibition of NP internalization by dynasore, which inhibits both CME and caveolin-
dependent endocytosis, suggested that this last mechanism could also participate in the
internalization of NPs, albeit the size range normally internalized by this pathway (50 – 80
nm) is smaller than our NP mean size. Surprisingly, caveolin- and lipid raft inhibitors
genistein and nystatin reduced NP internalization to 40 %.
As several endocytic mechanisms seem to be involved in the internalization of our
NPs by BMDCs, we hypothesized that these NPs could also have distinct intracellular
pathways. Our results suggest that NPs rapidly reach the endo-lysosomal pathway since after
only 30 min of incubation considerable amounts of NPs colocalized with the organelles.
Then, NPs might travel through endo-lysosomal vesicles but might also escape to the cytosol,
as reported previously (86,319). This property of PLGA-based NPs can be explained by the
reversion of surface charge at lysosomal pH (Fig. 2.1.). The colocalization with ER protein
marker calnexin was very high (~ 80 %) and kept almost at a constant level from throughout
the study. Extracellular antigens are usually digested in endo-lysosomes, and processed
peptides are loaded onto MHC class II molecules. By contrast, intracellular antigens are
degraded by the proteasome, released into the cytosol and subsequently translocated into the
ER by the transporter associated with antigen processing (TAP) to be loaded into the peptide-
binding site of MHC class I molecules. This assembly typically happens in the ER (1). Our
observations suggest that NPs have the potential to function as an efficient vaccine delivery

Chapter 2
100
system that might provide access to MHC class I and II pathways of antigen presentation,
promoting a complete and coordinated immune response.
Based on the results obtained, a hypothetical schematic representation of the
intracellular trafficking of the developed NPs is presented in Figure 2.13.. Firstly,
considerable colocalization levels (50 to 70 %) with endo-lysosomal vesicles suggest the high
potential of this carrier to induce the presentation of antigens through MHC class II molecules
to CD4+ T cells. Secondly, high levels of colocalization with the ER protein marker calnexin
suggest a considerable potential for cross-presentation of antigens through the MHC class I
molecules to CD8+ T cells. Thirdly, colocalization with endosomes suggests that NPs are able
to deliver immunoadjuvants to the compartment where some have their receptors, such as the
ligands of the Toll-like receptors 3 and 9. Additionally, the decrease in the colocalization of
NPs with EE, LE and Lys from 3 to 18 h suggests that NPs might escape from these vesicles
to the cytosol. This phenomenon might translate in the accumulation of NPs in the cytosol and
prolonged release of the entrapped antigens over time, demonstrating potential to be used as a
single-dose vaccine.
The most common method of generating ligand-targeted NPs involves coupling
targeting moieties directly to preformed NPs as a second step of the NP preparation (423).
However, this approach results in variations in coupling yields and side reactions that can
cause chemical and conformational changes to the ligand, resulting in highly heterogeneous
NP populations with inconsistent outcomes (424). The use of the mannose-grafted polymer
man-PEG-b-PCL in our formulation obviates this problem. Even if it is established that long
PEG polymers, including PEG with 2000 Da, fold into a mushroom like globular structure
that can hide a large fraction of the conjugated ligand (425), we have verified that mannose
residues are available for binding at man-NP surface. Surprisingly, the effect of active
targeting of man-NP to APCs was verified in immortalized APC lines but not in BMDCs. We
hypothesized that this could be related to the downregulation of the MR/CD206 by those cells
under culture conditions. It is also likely that the considerable high internalization levels
obtained by BMDCs for non-targeted NPs hide any additional effect that mannose targeting
by man-NPs may have. In fact, the benefit of the functionalization of NPs for active targeting
must be verified in vivo.

Nanoparticle delivery to dendritic cells
101
Figure 2.13. Schematic representation of the hypothetical intracellular trafficking of the developed NPs in
DCs. NPs are internalized by DCs and contained within early endosomes (EE), where NPs start to degrade
and release some of their cargo. Immunoadjuvants with receptors located in the endosomal membrane
might get access to them. As endosomes start to mature, their pH drops and NPs become neutral or
positively charged, being able to interact with endo-lysosomal membranes and escape to cytosol. Here,
NPs accumulate nearby the endoplasmic reticulum (ER), where the loading of antigens on MHC class I
molecules happens. MHC class I-peptide complexes are transported to the cell surface in exocytic vesicles
to be presented to CD8+ T cells. Some NPs continue to degrade in the lysosomes, releasing their cargo.
Here, peptides can be loaded on MHC class II molecules. MHC class II-peptide complexes are then
transferred to cell surface to be presented to CD4+ T cells.
5. Conclusions & future perspective
We aimed at developing biodegradable NPs to target APCs and evaluate their
potential to be used as a vaccine delivery system by exploring their profile of cellular
interaction. We were able to produce spherical NPs with ~130 nm functionalized with
mannose residues, which were available for binding at NP surface. NP internalization profiles
were different for immortalized APCs and primary BMDCs, the last revealing a more prompt
NP internalization, independent of mannose functionalization. These results highlight the
importance of choosing physiologic-closer models to better predict NP behavior in vivo.
Multiple endocytic pathways were involved in NP internalization, namely macropinocytosis,

Chapter 2
102
CME and caveolin- and lipid raft- dependent endocytosis. This fact demonstrates the
versatility of the present system since different endocytic pathways might lead to distinct
intracellular trafficking pathways and fates. NPs demonstrated both an endo-lysosomal and
cytosolic localizations and a tendency to accumulate nearby the ER. This system may provide
intracellular access to immunoadjuvants and drive antigens to be presented by both MHC
class I and II molecules to CD8+ and CD4+ T cells leading to a complete and coordinated
immune response. Our study highlights the need to fully understand the mechanisms by which
the vaccine delivery nanosystems interact with the immune system.
Vaccine delivery nanosystems can (i) be loaded with both antigen(s) and adjuvant(s),
(ii) be taken up by APCs, (iii) be presented to MHC class I and II, and (iv) be administered by
different routes (193). Hence, we foresee a great applicability of this vaccine delivery
nanosystem on a wide range of vaccine formulations, from prophylactic vaccines to viral
infectious diseases and therapeutic cancer vaccines.
The selection of the polymer for a potential clinical application will require either the
use of well accepted polymers already approved by EMA/FDA for parenteral use (e.g. PLGA)
or the development of smarter polymers which will need to be tested before their approval as
vaccine excipient. Scaling up process will also be required and cost/benefit should be
assessed. If extensive studies have been performed in preclinical models, in particular in
rodent models, further studies with vaccine delivery nanosystems must be performed both in
primate and humans before their marketing authorization is granted and their clinical use
increases.
6. Executive Summary
• Aliphatic polyester-based NPs were developed and functionalized to target APCs.
NPs were physicochemically characterized and NP-cell interaction profiles were analyzed.
• Spherical NPs were produced with ~130 nm and slightly negative zeta potential that
reverse to neutral/positive values in acidic pH.
• NPs did not affect cell viability of the APC lines JAW SII and J744.1 and primary
BMDCS.
• NP internalization profiles were different for immortalized APCs and primary
BMDCs, the last revealing a more prompt NP internalization, independent from mannose
functionalization of NPs.

Nanoparticle delivery to dendritic cells
103
• Mannose residues are exposed and available for binding at mannose-grafted NP
surface.
• NPs are extensively internalized by macropinocytosis but clathrin-mediated,
caveolin- and lipid raft- dependent endocytosis are also partially involved.
• NPs demonstrated endo-lysosomal and cytosolic localizations and a tendency to
accumulate nearby the endoplasmic reticulum.
• The developed NPs are promising vaccine delivery systems that can provide direct
access to endo-lysosomal vesicles and cytosol, potentially driving antigens to be presented by
both MHC class I and II molecules to CD8+ and CD4+ T cells.
Acknowledgments
The authors which to thank M. Leclercq from Unit of Cellular and Molecular
Pharmacology, Catholic University of Louvain, Brussels, Belgium, for supplying J744.1
murine macrophage-like cell line. We also thank Professor M. Prieto from Instituto Superior
Tecnico (Lisbon, Portugal) for accessibility to confocal microscope facilities.
Financial & competing interests disclosure
This work was supported by Fundação para a Ciência e a Tecnologia, Ministério da
Educação e Ciência, Portugal (PhD Grant SFRH/BD/64295/2009 to JMS, PTDC/SAU-
FAR/119389/2010, Compromisso para a Ciência 2008 to LCS, and Pest-
OE/SAU/UI4013/2011) and by Fonds de la Recherche Scientifique Médicale, Belgium. GV is
a postdoctoral researcher of the Fonds de la Recherche Scientifique-FNRS. The authors have
no other relevant affiliations or financial involvement with any organization or entity with a
financial interest or financial conflict with the subject matter or materials discussed in the
manuscript.
Ethical conduct of research
Animal experimental procedures were reviewed and approved by the Direcção-Geral
de Alimentação e Veterinária, Portugal.


Chapter 3


107
Chapter 3 - Combination of peptides and adjuvants in mannose-
functionalized nanoparticles for melanoma immunotherapy
Joana M. Silvaa,b, Eva Zupancica, Gaëlle Vandermeulenb, Vanessa G. Oliveirac, Ana Salgadoa,
Mafalda Videiraa, Manuela Gaspara, Luís Graçac, Véronique Préatb & Helena F. Florindoa
aInstituto de Investigação do Medicamento (iMed.ULisboa), Faculdade de Farmácia, Universidade de
Lisboa, 1649-003 Lisbon, Portugal. bLouvain Drug Research Institute, Advanced Drug Delivery & Biomaterials, Université Catholique de
Louvain, 1200 Brussels, Belgium. cInstituto de Medicina Molecular, Faculdade de Medicina da Universidade de Lisboa, 1649-025 Lisbon,
Portugal. dCenter for Education and Research on Macromolecules, Université de Liège, 4000 Liège, Belgium.
Submitted for publication


Nanoparticles for melanoma immunotherapy
109
Abstract
We hypothesized that the coentrapment of multiple melanoma-associated antigens
along with Toll-like receptor ligands in mannose-functionalized aliphatic polyester-based
nanoparticles could be an effective antitumor strategy. High entrapment efficiencies of
antigens and immunoadjuvants in 150 nm nanoparticles were obtained. In OT II mice, the
coentrapment of ovalbumin and the Toll-like receptor ligands Poly(I:C) and CpG was crucial
to induce high levels of IFN-γ and IL-2, as well as high IgG2c/IgG1 ratios. Mannose-
functionalization of nanoparticles potentiated the Th1 immune response. The nanoparticulate
vaccines decreased the growth rate of murine B16F10 melanoma tumors in both therapeutic
and prophylatic settings. The combination of MHC class I- and class II-restricted melanoma
antigens in different mannose-functionalyzed nanoparticles containing Toll like receptor
ligands induced the highest tumor growth delay. Overall, we demonstrate that the
combination of active targeting of nanoparticles with their multifunctional properties in terms
of antigen and adjuvant delivery has high cancer immunotherapeutic potential.
Graphical abstract
Keywords
Cancer vaccine, nanoparticles, PLGA, mannose receptor targeting, TLR, antigen
presenting cells, melanoma


Nanoparticles for melanoma immunotherapy
111
1. Introduction
There are increasing evidences that the immune system is able to mount responses
against tumors that can be enhanced using a number of strategies (4). Cancer immunotherapy
must envision the activation of cell-mediated immunity, particularly CD8+ cytotoxic T cells.
Activated CD8+ T cells are able to directly kill malignant cells and also acquire a long-lasting
memory phenotype, important in the prevention of cancer relapses (183). CD4+ T helper (Th)
1 cells are also essential to obtain an effective antitumor response by enhancing clonal
expansion of cytotoxic T lymphocytes (CTLs) at the tumor site and promoting the generation
and maintenance of a memory phenotype (426). To activate both CD8+ and CD4+ T cells
against tumor, tumor-associated antigens (TAAs) must be presented through major
histocompatibility complex (MHC) class I and II molecules in an immunostimulatory context
(36).
Several vaccination strategies have been tested as whole tumor cells, cell lysates,
proteins or specific peptide fragments, leading to the generation of tumor-specific T cell
responses (427). Nevertheless, the therapeutic benefit of these strategies remains unclear and
further well designed studies are needed to clarify the potential of each approach. Improved
cancer vaccination strategies depend on the targeted delivery of TAAs to antigen presenting
cells (APCs) - dendritic cells (DCs) by excellence but also macrophages and B cells - which
present antigens to T cells (38). Notwithstanding, their capacity to properly stimulate T cells
depends on the presentation of antigens in an immunostimulatory environment, which can be
achieved by using molecules with known adjuvant potential, such as the Toll-like receptor
(TLR) ligands. The stimulation of TLRs in APCs activates intracellular signaling pathways
that culminate in the induction of inflammatory cytokines, chemokines, interferons and
upregulation of costimulatory molecules that provide the proper environment for T cell
stimulation (184). Some studies have demonstrated that the combined administration of TLR
ligands and antigens enhances specific immune responses (170,428). However, additional
strategies are needed in order to (i) prevent their prompt in vivo degradation, (ii)
simultaneously target antigens and adjuvants to an APC and (iii) promote their sustained and
modulated release in those cells. This can be accomplished by the use of biodegradable
nanoparticles (NPs), which have been shown to improve immune responses by enhancing the
uptake of antigens and adjuvants by APCs (171,172). NPs are able to target the immune
system and co-deliver antigens and adjuvants simultaneously to the same APC, enabling its
coordinated activation (3). The large surface area of NPs potentiates electrostatic and/or
receptor-interactions in a greater extent compared to soluble antigens (185). By providing

Chapter 3
112
direct intracellular access, the engagement of intracellular receptors is facilitated, which
improves the efficacy of vaccine adjuvants (190). Also, NPs avoid immediate degradation of
the key vaccine components i.e. proteins, peptides, nucleic acids and oligonucleotides
(186,187). Furthermore, biodegradable polymeric NPs can provide different release profiles
to the entrapped molecules according to polymer’s physicochemical properties (191). Finally,
NP functionalization further allows the active targeting of APCs potentiating the delivery of
their cargo at the intracellular level and thus improving the vaccine outcome (192). All these
advantageous properties confer intrinsic adjuvanticity to aliphatic polyester-based NPs and
make them promising vaccine delivery systems (193).
We hypothesized that the coentrapment of several melanoma-associated antigens
(MAAs) along with TLR ligands in mannose-functionalized aliphatic polyester-based NPs
could be an effective antitumor strategy. The developed nanoparticulate vaccines were able to
induce antitumor immune responses in a murine melanoma model, when used in therapeutic
and prophylactic approaches. We investigated multiple factors that have an impact on the
therapeutic effect of a cancer vaccine, as the importance of using an actively targeted delivery
system, the synergy between TLR ligands and the relevance of combining multiple TAAs,
including MHC class I- and class II-restricted peptides.
2. Materials and methods
2.1. Materials
Poly(D,L-lactic-co-glycolide) (PLGA) Resomer® RG 502 (lactide:glycolide 50:50)
(average Mw = 7,000 – 17,000 g/mol), sodium cholate hydrate, dichloromethane (DCM),
Tris, fluorescamine, lipopolysaccharide (LPS) from Escherichia coli and bovine serum
albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO,USA). Poly(D,L-lactic-
co-glycolide-b-ethylene glycol) (PEG-b-PLGA) (average Mw(PEG) = 4,600 g/mol and
Mw(PLGA) = 16,500 g/mol) and poly(ε-caprolactone-b-ethylene glycol) (PEG-b-PCL)
(average Mw(PEG) = 6,000 g/mol and Mw(PCL) = 12,000 g/mol) were synthesized by ring
opening polymerization followed by a “clip” and “click” reaction of α-methoxy-ω-alkyne
poly(ethylene glycol) (PEG) to N-hydroxy-succinimidyl activated esters (402,403). Mannose-
grafted poly(ε-caprolactone-b-ethylene glycol) (man-PEG-b-PCL) (average Mw(PCL) =
17,000 g/mol and Mw(PEG) = 2,000 g/mol) was synthesized by the Huisgen 1,3-dipolar
cycloaddition, where each PCL long chain was grafted with three PEG chains, with 30 % of
PEG chains containing one mannose residue (406).

Nanoparticles for melanoma immunotherapy
113
Dimethilsulfoxide (DMSO) and hydrochloric acid were purchased from Merck
(Darmstadt, Germany). The MicroBCATM Protein assay kit and the Oligreen® ssDNA
quantitation kit were supplied by Thermo Fisher Scientific Inc. (Rockford, IL USA) and Life
Technologies (Carlsbad, CA, USA), respectively.
Melan-A:26-35(L27) (hereafter Melan-A:26 or M), ELAGIGILTV, gp100:209-
217(2M) (hereafter gp100:209 or G209), IMDQVPFSV and gp100:44-59 (hereafter gp100:44
or G44), WNRQLYPEWTEAQRLD, were synthesized by JPT Peptide Technologies GmbH
(Berlin, Germany). CpG ODN 1826 VaccigradeTM (hereafter CpG or C),
TCCATGACGTTCCTGACGTT, and Poly(I:C) (HMW) VaccigradeTM (hereafter Poly(I:C)
or P) were purchased from InvivoGen (San Diego, CA, USA).
Dulbecco’s Phosphate Buffered Saline (DPBS) (1×), RPMI + Glutamax® medium, heat
inactivated fetal bovine serum (FBS), trypsin EDTA 0.05 %, penicilin/streptomycin (PEST)
10,000 Unit/ml/10,000 µg/ml, sodium pyruvate 100 mM, HEPES 1 M, 2-mercaptoethanol 50
mM, ACK lysing buffer and AlamarBlue® reagent were purchase from Life Technologies
(Carlsbad, CA, USA).
Rat anti-mouse PE-conjugated anti-CD4 for flow cytometry was purchased from
Miltenyi Biotec (Koln Germany). Rat anti-mouse APC-conjugated anti-CD8α and PerCP-
Cy5.5-conjugated anti-CD3 monoclonal antibodies for flow cytometry were purchased from
Biolegend (San Diego, CA, USA). Horseradish peroxidase (HRP) conjugate goat anti-mouse
IgG (1:2000), IgG1 (1:6000) and IgG2c (1:10000) for ELISA were purchased from AbD
Serotech (Raleigh, NC, USA). TMB One Component HRP substrate was acquired from Tebu-
bio (Le Perray-en-Yvelines, France). DuoSet® ELISA Development kits for IFN-γ, IL-2, IL-4,
IL-5, IL-6, IL-10 and Granzyme B (Grz B) were purchased from R&D Systems Europe
(Abingdon, UK). Anti-mouse ELISA sets for IL-2, IL-4, IL-6 and IL-10 were also acquired
from Southern biotech (Birmingham, AL, USA).
2.2. Mice
Female transgenic OT-II mice were bred in-house at Instituto Gulbenkian de Ciência
(Oeiras, Portugal), under conventional, non-specific pathogen-free conditions.
Male C57Bl6 mice 8-10 weeks old were purchased from Charles River (Wilmington,
MA, USA.).
Procedures were reviewed and ethically approved by the local ethics committee and
Portuguese competent authority for animal protection, Direcção Geral de Alimentação e
Veterinária, Lisbon, Portugal.

Chapter 3
114
2.3. Preparation of NPs
NPs were aseptically prepared at room temperature by the double emulsion solvent
evaporation method, as previously reported with modifications (254). Two different types of
NPs were formulated: non-functionalized NPs (hereafter NPs), constituted by a blend of
PLGA, PEG-b-PLGA and PEG-b-PCL in a 70:15:15 ratio; and functionalized (mannose-
grafted) NPs (hereafter man-NPs) using PLGA, PEG-b-PLGA and man-PEG-b-PCL
(70:15:15). Polymers were dissolved in DCM or DCM:DMSO 70:30 to prepare NPs or man-
NPs, respectively. Aqueous solutions of antigens (Melan-A:26, gp100:209 or gp100:44) at 5
mg/ml and adjuvants CpG (5 mg/ml) and Poly(I:C) (2.5 mg/ml), were added to the polymer
solutions (50 mg/ml) and mixtures were emulsified using an ultrasonic processor for 15 s at
70 W. Ovalbumin (OVA) (10 mg/ml) was also used as a model antigen. A second emulsion
was performed with 1 % (w/v) sodium cholate aqueous solution under the same conditions.
The double emulsion was then poured drip into a 0.3 % (w/v) sodium cholate aqueous
solution, and stirred at 37 °C for 1 h. The NP suspension was then washed twice with
ultrapure water by centrifugation at 22000 × g for 45 min, 4 °C (Beckman Coulter Avanti® J-
E Centrifuge JA-20) and finally resuspended in DPBS.
2.4. Physicochemical characterization of NPs
NP size was determined by Dynamic Light Scattering (DLS) in DPBS using a Malvern
Nano ZS (Malvern Instruments, UK). Cumulant analysis was used to determine the Z-average
size. The zeta potential of NPs was measured using the same device by Laser Doppler
Velocimetry (LDV) in combination with Phase Analysis Light Scattering (PALS). NPs were
diluted in DPBS and electrophoretic mobility was determined at 25 °C using the Helmholtz-
von Smoluchowski model.
2.5. Determination of antigen and adjuvant loadings
The supernatants of NPs were collected by centrifugations to quantify indirectly the
entrapped agents. The entrapment efficiency [EE% (w/w), Eq. (1)] and the loading capacity
[LC µg/mg, Eq. (2)] of Melan-A:26, gp100:44 and OVA were determined by the
MicroBCATM assay according to the manufacturer’s instructions (364). For the quantification
of gp100:209, the fluorescent dye fluorescamine was used (429). Relative Fluorescence Units
(RFU) were measured using a FluorocountTM fluorometer (Packard, NY, USA), at 360 nm
excitation and 460 nm emission wavelengths. Both adjuvants CpG and Poly(I:C) were

Nanoparticles for melanoma immunotherapy
115
quantified by the Oligreen® ssDNA quantitation kit (430). RFU were measured using the
fluorometer at 485 nm excitation and 530 nm emission wavelengths.
The loading of OVA was also quantified by high-performance liquid chromatography
method (HPLC), using a Beckman System Gold: UV-vis detector (Beckman 166), Beckman
126 solvent module and Midas autosampler. Samples (20 µl) were injected onto a Shodex
PROTEIN KW-803 series column (8.0 mm ID x 300 mm, 5 µm particle size, 300 Å pore size)
and eluted with 50 mM sodium phosphate buffer (pH 7.0) + 0.3 M NaCl at 1 ml/min for 20
min at room temperature. OVA elution was monitored at 220 nm by spectrophotometric
analysis. The amount of OVA in each sample was calculated using a standard curve generated
with known concentrations of the protein (0.5 – 10.0 μg/ml, r2 = 0.99).
(1)
(2)
2.6. Integrity of the NP-entrapped OVA
OVA standard solution (250 µg/ml) and NP suspensions (20 mg/ml) were mixed with
SDS-containing loading buffer and digested at 95 °C for 20 min. Samples were then loaded
onto a SDS PAGE system composed by a stacking gel at 4 % (w/v) (2.5 cm depth) and a
resolving gel at 12 % (w/v) (5.5 cm depth), both prepared with an acrylamide:bisacrylamide
solution 29:1 at 30 % (w/v) (Bio-Rad, UK). Electrophoresis was performed in a PowerpackTM
Basic Power Supply (Bio-Rad, UK) in a SDS-containing discontinuous buffer system,
initially at 30 V for approximately 30 min and then at 190 V for approximately 1 h. The
resolving gel was then stained with a Coomassie blue solution.
2.7. Cell line culture conditions
Murine melanoma B16F10 cell line (ATCC#CRL-6475) were propagated in RPMI-
1640 + GlutamaxTM supplemented with 10 % (v/v) FBS, 10,000 Unit/ml/10,000 μg/ml PEST,
50 µM β-mercaptoethanol, 10 mM HEPES and 1 mM sodium pyruvate (hereafter complete
medium).

Chapter 3
116
2.8 Characterization of the immune response induced by the nanoparticulate
vaccines in OT II mice
Female OT II mice were subcutaneously immunized on day 1 and boosted on day 15
and 29, with 100 µl of one of the following formulations (n = 5): 100 µg OVA entrapped in
NP (NP[OVA]) or 100 µg OVA admixed with 20 µg CpG and 40 µg Poly(I:C) in PBS
(OVA+P+C sol.) or entrapped in NPs (NP[OVA+P+C]) or man-NPs (man-NP[OVA+P+C]).
Blood samples were collected on weeks 1, 3, 5, 7, 9 and 12 after the first immunization. Sera
were separated from clotted blood by centrifugation (18,000 × g, 20 min at 4 °C, Allegra 64R,
Beckman, USA) and stored at - 20 °C till quantification of immunoglobulins by ELISA.
Twelve weeks after the first immunization mice were sacrificed and spleens were aseptically
isolated.
2.9. Therapeutic efficacy of the nanoparticulate vaccines
Male C57Bl6 6-8 weeks old mice were randomized into one of the following treatment
groups (n = 6): (1) PBS, (2) Melan-A:26 (M) + Poly(I:C) (P) in solution, (3) M + gp100:209
(G209) + P + CpG (C) in solution, (4) NP[M+P], (5) NP[M+P+C], (6) man-NP[M+P+C], (7)
man-NP[M+G209+P+C] and (8) man-NP[M+P+C] + man-NP[G209+P+C]. Mice were
subcutaneously challenged in the right flank with 1 × 105 B16F10 cells suspended in PBS
with 2 % (v/v) FBS (day 0). One week after the tumor challenge, mice were immunized 3
times with subcutaneous injections of 100 µl of the vaccine in the left groin with 1-week
interval (days 7, 14 and 21). When two different types of NPs were administered [group (8)]
50 µl of each NP suspension were administered in adjacent spots. Each mice received 100 µg
of antigen (50 µg of each when two antigens were used), 40 µg of Poly(I:C) and 20 µg of
CpG, entrapped in 4 mg of NPs or man-NPs. Mice were monitored every two days for pain or
distress, tumor volume and body weight. Tumor growth was evaluated by measuring two
perpendicular tumor dimensions using a caliper. Tumor volumes (V, mm3) were calculated
using the following formula: V = ½ × (L × W2), where L (length) is the longest dimension
and W (width) is the perpendicular dimension to the length. Mice were sacrificed on day 26.
Spleens and tumor tissues were excised and weighted.
2.10. Protection efficacy of the nanoparticulate vaccines against tumor
challenge
Male C57Bl6 6-8 weeks old mice were randomized into the same immunization groups
referred in section 2.9.. An additional group (9) was assayed where mice received Melan-

Nanoparticles for melanoma immunotherapy
117
A:26, Poly(I:C) and CpG entrapped in man-NPs mixed with man-NPs entrapping gp100:44,
Poly(I:C) and CpG (man-[M+P+C] + man-NP[G44+P+C]). Mice received 3 subcutaneous
injections of 100 µl of vaccine with 2-week intervals (days -42, -28 and -14) in the left groin.
When two different types of NPs were administered (groups (8) and (9)), 50 µl of each were
administered in adjacent spots. Each mice received 100 µg of antigen (50 µg of each when
two antigens were used), 40 µg of Poly(I:C) and 20 µg of CpG, entrapped in 4 mg of NPs or
man-NPs. On day 0, mice were subcutaneously challenged in the right flank with 1 × 105
B16F10 cells suspended in PBS with 2 % (v/v) FBS. Mice were monitored every two days for
pain or distress, tumor volume and body weight. Mice were sacrificed on day 19. Spleens and
tumor tissues were excised and weighed. Tumor growth was evaluated as mentioned before.
2.11. Cytokine and Granzyme B quantification
Spleens were aseptically removed from sacrificed mice and placed in sterile PBS. A
crude suspension of splenocytes was prepared by filtration through 70 µm strainers in
complete medium. The cell suspension was spun at 200 × g for 10 min, the cell pellet
resuspended and erythrocytes were lyzed using ACK lysing buffer. The centrifugation was
repeated and the cell pellet was resuspended in fresh complete medium. Cells were then
seeded in sterile 96-well tissue culture plates at the amount of 1 × 106 cells in 200 µl of media
per well with or without 20 µg/ml of antigen. Plates were incubated at 37 °C in a humidified
incubator at 5 % CO2. After 72 h of re-stimulation, splenocyte culture supernatants were
collected and cytokine production was assessed using DuoSet® ELISA Development kits
(R&D) for IFN-γ, IL-2, IL-4, IL-5, IL-6 and IL-10 or anti-mouse ELISA sets (Southern
biotech) for IL-2, IL-4, IL-6 and IL-10, according to the manufacturer’s instructions. The
concentrations, expressed in pg/ml, were determined by reference to cytokine standard curves.
To assess Granzyme B (Grz B) production, splenocytes (effector cells, E) from mice
in the protection study were seeded in sterile 96-well tissue culture plates at the amount of 1 ×
106 cells in 100 µl of complete media. Murine melanoma B16F10 cells (target cells, T) were
added in the following day in a 100:1 ratio in 100 µl of complete media. Breast cancer MDA-
MB-231 cells (ATCC-HTB-26) were used as unspecific target cell control in the same E:T
ratio. After 4 h of incubation, supernatants were collected and analyzed for Grz B production
using a murine Granzyme B DuoSet® ELISA Development kits (R&D). Relative ratios of the
Grz B production by each group were determined by comparing the production in the
presence or absence of target cells.

Chapter 3
118
2.12. Splenocyte proliferation assay
Splenocytes harvested from mice in the protection study were seeded in sterile 96-well
tissue culture plates at the amount of 5 × 105 in 200 µl of complete media per well in the
presence of 20 µg/ml of antigen (Melan-A:26, gp100:209 or gp100:44), 1 μg/ml of LPS,
Triton X-100 0.025 % (v/v), or left untreated for 48 h. AlamarBlue® reagent was added at 10
% (v/v) of the total volume and incubated for 3 h. Fluorescence measurements were made at
530 nm/590 nm using a FLUOstar Omega microplate reader (BMG Labtech, Ortenberg,
Germany). Stimulation Index (SI) was calculated using the formula SI = (RFUstimulated –
RFUmedium) - (RFUunstimulated – RFUmedium), where RFUstimulated is the mean RFU of treated
splenocytes, RFUunstimulated is the mean RFU of untreated splenocytes and RFUmedium is the
mean RFU of the wells containing only medium.
2.13. Analysis of spleen T lymphocyte subsets
Splenocytes from mice in the protection assay were seeded in sterile 96-well tissue
culture plates at the amount of 1 × 106 cells in 200 µl of complete media per well and
stimulated with 20 µg/ml of antigen (Melan-A:26, gp100:209 or gp100:44) for 72 h.
Splenocytes were then stained with anti-mouse fluorescently labeled monoclonal antibodies
for flow cytometry (PerCPCy5.5-CD3, PE-CD4 and APC-CD8α) for 20 min after Fc blocking
with rat anti-mouse CD16/CD32 antibody for 15 min. Splenocytes were harvested by
centrifugation and analysed by flow cytometry. The individual fluorescence of 50,000 cells
was collected for each sample using a LSR Fortessa cytometer (BD Biosciences) and
analyzed with FlowJo software version 7.6.5 for Microsoft (TreeStar, San Carlos, CA).
2.14. Statistics
All results are expressed as mean ± standard deviation (SD). T-test, one-way ANOVA
and Tukey's post test or two-way ANOVA and Bonferroni’s post test were performed to
demonstrate statistical differences (p < 0.05), using the software GraphPad Prism version 5.00
(GraphPad Software.Inc, CA, USA).

Nanoparticles for melanoma immunotherapy
119
3. Results
3.1. NP physicochemical characterization
3.1.1. NP size and zeta potential
The double emulsion-solvent evaporation method was used to produce biodegradable
polymeric NPs from a blend of three aliphatic polyester copolymers: (i) PLGA; (ii) pegylated
PLGA (PEG-b-PLGA) and (iii) pegylated poly-ε-caprolactone (PEG-b-PCL) or mannose
grafted polymer (man-PEG-b-PCL) (254). PLGA, the main constituent of these NPs (70 %
w/w), is a biodegradable polymer approved for parenteral use in humans and is the most
studied aliphatic polyester for biomedical applications (186,193). PEG-b-PLGA and PEG-b-
PCL were introduced in the formulation to functionalize the polymers with ligands and
increase NP stability due to PEG repulsive properties (254). The ratio of the three different
polymers in the formulation has been previously optimized in our laboratory in terms of
shielding of PLGA negative charges at physiologic pH without hindering ligand accessibility
(254).
To physicochemically characterize the produced NPs, size (Z-average hydrodynamic
diameters), polydispersity index (PdI) and zeta potential were determined (Table 3.1.). The
formulation method was very reproducible. NPs displayed a size range between 145 ± 28 nm
and 190 ± 27 nm, and a narrow particle size distribution (PdI ranging from 0.11 ± 0.02 to 0.28
± 0.02), indicating a homogenous NP population. At pH 7.4, NPs presented a zeta potential
ranging from -0.8 ± 0.8 to -6.2 ± 0.8 mV (Table 3.1.).
Table 3.1. Nanoparticle size, polydispersity index (PdI) and zeta potential (mean ± SD; N ≥ 5, n = 3).
Formulations Sizea (nm) PdIb Zeta Potential (mV)
NP[OVA] 161 ± 26 0.17 ± 0.05 -4.1 ± 1.0 NP[OVA+P] 162 ± 09 0.14 ± 0.06 -6.0 ± 14.7 NP[OVA+C] 148 ± 23 0.15 ± 0.06 -4.1 ± 1.8 NP[OVA+P+C] 145 ± 28 0.13 ± 0.03 -3.5 ± 1.9 man-NP[OVA+P+C] 185 ± 17 0.22 ± 0.02 -5.5 ± 2.8 NP[M] 159 ± 11 0.11 ± 0.02 -0.8 ± 0.8 NP[G209] 159 ± 21 0.13 ± 0.03 -1.4 ± 0.4 NP[M+P] 158 ± 17 0.13 ± 0.02 -4.4 ± 0.1 NP[M+C] 167 ± 06 0.11 ± 0.02 -1.4 ± 0.2 NP[M+P+C] 162 ± 17 0.12 ± 0.02 -4.1 ± 0.8 man-NP[M+P+C] 169 ± 02 0.18 ± 0.07 -4.4 ± 1.5 man-NP[G209+P+C] 157 ± 19 0.28 ± 0.02 -3.7 ± 0.5 man-NP[G44+P+C] 147 ± 25 0.25 ± 0.06 -6.2 ± 0.8 man-NP[M+G209+P+C] 190 ± 27 0.28 ± 0.02 -3.7 ± 1.2
a Z-average hydrodynamic diameter. b Polydispersity index.

Chapter 3
120
3.1.2. Loading of antigens and adjuvants
NPs were able to coentrap different molecules such as OVA, the MAAs Melan-A,
gp100:209 and gp100:44, the oligonucleotide CpG and the dsRNA analog Poly(I:C) (Table
3.2.). The amount of antigens and adjuvants used in the formulation of NPs was adjusted
according to the doses needed for the in vivo proof of concept studies.
To formulate NPs and man-NPs containing OVA, 50 µg of the protein per mg of
polymer were used. A high EE % of OVA in NPs and man-NPs was obtained (between 71.0 ±
24.1 and 79.2 ± 14.0 % (w/w)). It was possible to load between 25.7 ± 10.2 to 39.6 ± 7.0 µg
of OVA per mg of NPs and man-NPs. These results were also confirmed by HPLC (data not
shown). The presence of CpG and/or Poly(I:C) had no significant impact on the loading
capacity of these formulations.
To formulate non-functionalized NPs containing MAAs, 25 μg of either Melan-A:26
or gp100:209 per mg of polymer were used. The EE % obtained ranged from 63.0 ± 20.6 to
71.2 ± 10.2 % (w/w) and the LC 15.8 ± 5.2 and 19.7 ± 3.3 µg of peptide per mg of NPs. To
formulate man-NP[M+P+C], 50 µg of Melan-A:26 per mg of polymer were used. To
formulate either man-NP[G209+P+C] or man-NP[G44+P+C], 25 µg of peptide per mg of
polymer were used. Finally, 25 μg of each peptide per mg of polymer were used to formulate
man-NP[M+G209+P+C]. When, 25 µg of peptide per mg of polymer were used, the EE %
ranged from 80.1 ± 5.2 to 83.6 ± 10.6 % (w/w) with a LC of 20 μg of peptide per mg of man-
NPs. When 50 μg of peptide per mg of polymer were used, the loading increased to values
between 30.0 ± 0.9 and 36.2 ± 1.1 µg of peptide per mg of man-NPs, with a slight decreased
in the EE % (60.0 ± 1.8 to 72.4 ± 2.2 % (w/w)).
The amounts of the immunoadjuvants used in the formulation of all NPs were 20 µg of
Poly(I:C) and 10 μg of CpG per mg of polymer. It was possible to load approximately 11 μg
of Poly(I:C) and 7 μg of CpG, when used individually. When both immunoadjuvants were
coentrapped, the total LC ranged from 15.8 ± 2.1 to 22.5 ± 0.8 µg/mg, and the EE % varied
between 52.7 ± 7.0 and 75.0 ± 2.6 % (w/w).

Nanoparticles for melanoma immunotherapy
121
Table 3.2. Entrapment Efficiency (EE %) and Loading Capacity (LC µg/mg) of antigens and
immunoadjuvants in NPs (mean ± SD; N ≥ 5, n = 3).
Formulations EE % Antigen LCa Antigen EE % Adjuvant LC Adjuvant
NP[OVA] 71.0 ± 24.1 25.7 ± 10.2 - - NP[OVA+P] 79.2 ± 14.0 39.6 ± 7.0 52.0 ± 10.5 10.4 ± 2.1
NP[OVA+C] 74.0 ± 12.5 28.7 ± 11.7 78.0 ± 7.0 7.8 ± 0.7
NP[OVA+P+C] 74.0 ± 21.4 30.2 ± 11.1 52.7 ± 7.0b 15.8 ± 2.1b
man-NP[OVA+P+C] 76.1 ± 25.8 33.7 ± 15.0 53.7 ± 4.7b 16.1 ± 1.4b
NP[M] 71.2 ± 10.2 17.8 ± 2.6 - - NP[G209] 66.0 ± 9.1 16.5 ± 2.3 - - NP[M+P] 67.9 ± 26.0 16.1 ± 7.8 57.9 ± 3.9 11.6 ± 0.8 NP[M+C] 63.0 ± 20.6 15.8 ± 5.2 66.1 ± 22.8 6.6 ± 2.3 NP[M+P+C] 65.6 ± 10.9 19.7 ± 3.3 75.0 ± 2.6b 22.5 ± 0.8b
man-NP[M+P+C] 60.0 ± 1.8 30.0 ± 0.9 54.3 ± 8.3b 16.3 ± 2.5b
man-NP[G209+P+C] 83.6 ± 10.6 20.9 ± 2.7 49.0 ± 4.3b 14.7 ± 1.3b
man-NP[G44+P+C] 80.1 ± 5.2 20.0 ± 1.3 49.0 ± 10.7b 13.9 ± 3.2b
man-NP[M+G209+P+C] 72.4 ± 2.2c 36.2 ± 1.1c 47.0 ± 6.0b 14.1 ± 1.8b
a Loading Capacity corresponds to µg of entrapped molecule per mg of polymer. b Total values for CpG and Poly(I:C). c Total values for Melan-A:26 and gp100:209.
3.1.3. Ovalbumin integrity after NP formulation
In order to verify if OVA is degraded during NP formulation, a SDS PAGE analysis
was performed. OVA-entrapping NPs, in the presence or absence of the adjuvants CpG and
Poly(I:C), were applied to the gel. Plain NPs as well as an OVA standard solution and
commercial molecular weight standards were used as controls. OVA maintained its molecular
weight (45 KDa) after the formulation procedure since the bands obtained for all samples of
NPs containing OVA were very similar to those obtained for OVA standard and no other
bands with different molecular weights were detected (Fig. 3.1.). OVA integrity is maintained
independently from the presence of the adjuvants CpG and Poly(I:C) entrapped in the same
NPs.
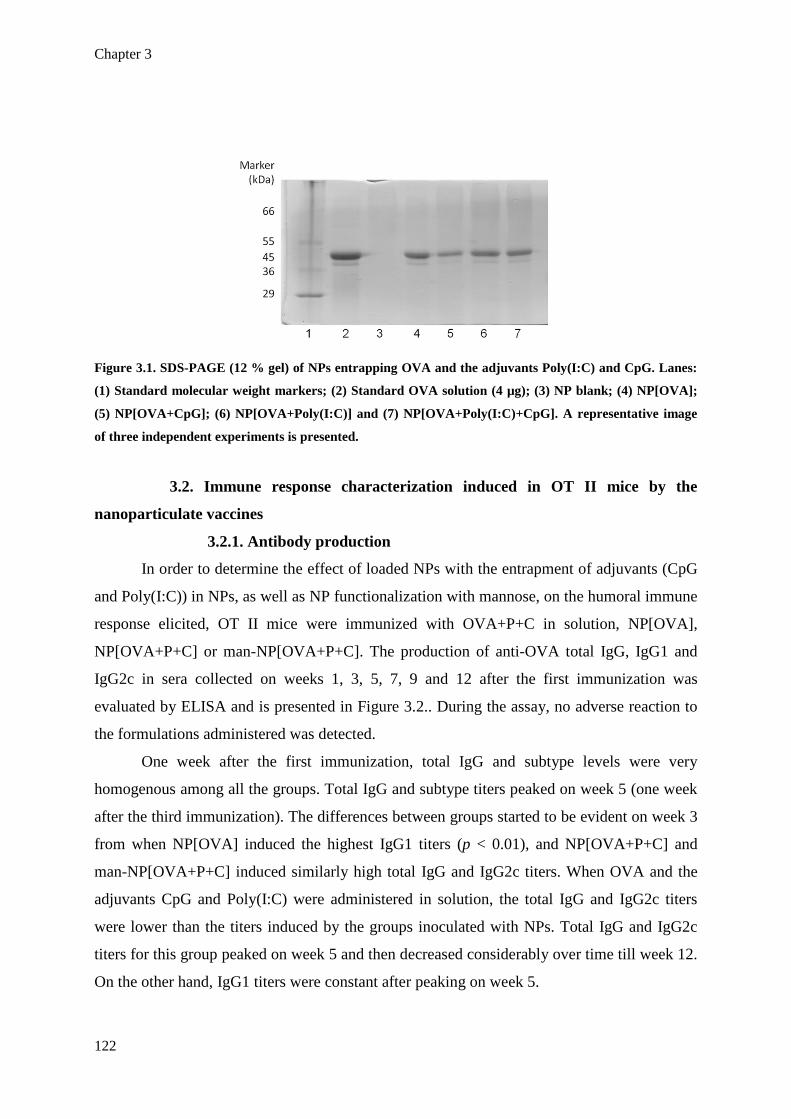
Chapter 3
122
Figure 3.1. SDS-PAGE (12 % gel) of NPs entrapping OVA and the adjuvants Poly(I:C) and CpG. Lanes:
(1) Standard molecular weight markers; (2) Standard OVA solution (4 µg); (3) NP blank; (4) NP[OVA];
(5) NP[OVA+CpG]; (6) NP[OVA+Poly(I:C)] and (7) NP[OVA+Poly(I:C)+CpG]. A representative image
of three independent experiments is presented.
3.2. Immune response characterization induced in OT II mice by the
nanoparticulate vaccines
3.2.1. Antibody production
In order to determine the effect of loaded NPs with the entrapment of adjuvants (CpG
and Poly(I:C)) in NPs, as well as NP functionalization with mannose, on the humoral immune
response elicited, OT II mice were immunized with OVA+P+C in solution, NP[OVA],
NP[OVA+P+C] or man-NP[OVA+P+C]. The production of anti-OVA total IgG, IgG1 and
IgG2c in sera collected on weeks 1, 3, 5, 7, 9 and 12 after the first immunization was
evaluated by ELISA and is presented in Figure 3.2.. During the assay, no adverse reaction to
the formulations administered was detected.
One week after the first immunization, total IgG and subtype levels were very
homogenous among all the groups. Total IgG and subtype titers peaked on week 5 (one week
after the third immunization). The differences between groups started to be evident on week 3
from when NP[OVA] induced the highest IgG1 titers (p < 0.01), and NP[OVA+P+C] and
man-NP[OVA+P+C] induced similarly high total IgG and IgG2c titers. When OVA and the
adjuvants CpG and Poly(I:C) were administered in solution, the total IgG and IgG2c titers
were lower than the titers induced by the groups inoculated with NPs. Total IgG and IgG2c
titers for this group peaked on week 5 and then decreased considerably over time till week 12.
On the other hand, IgG1 titers were constant after peaking on week 5.

Nanoparticles for melanoma immunotherapy
123
Figure 3.2. Anti-OVA specific immunoglobulin sera titers from OT II mice immunized with OVA- and
adjuvant-loaded NPs. OT II mice were immunized with OVA and the adjuvants CpG and Poly(I:C) in
solution, NP[OVA], NP[OVA+P+C] or man-NP[OVA+P+C] on weeks 0, 2 and 4. The production of anti-
OVA total IgG (A), IgG1 (B) and IgG2c (C) in sera collected on weeks 1,3,5,7,9 and 12 after the first
immunization were evaluated by ELISA. Sera from naїve OT II mice were used as control. The titres
reported are the reciprocal of serum dilution that gave an optical density 5 % higher than the control at
the same dilution. D. Th1/Th2 indexes in OT II mice immunized with OVA and adjuvant-loaded NPs,
calculated as a ratio of the obtained mean IgG2c and IgG1 titers for each group at each time point. The
grid line at Th1/Th2 = 1 separates the Th1-biased indexes (> 1) from the Th2-biased ones (< 1). The bars
correspond to mean ± SD; n = 3. Statistics: two-way ANOVA and Bonferroni’s post test. Relative to levels
determined on naїve mice: * p < 0.05, ** p < 0.01 and *** p < 0.001.
To assess the Th1/Th2 polarity of the immune response induced by the different
immunization strategies tested, a Th1/Th2 (IgG2c/IgG1) index was calculated. A Th1/Th2
index < 1 stands for a Th2 polarization while a Th1/Th2 index > 1 stands for a Th1
polarization. Groups immunized with NP[OVA+P+C] and man-NP[OVA+P+C] induced very
high Th1/Th2 indexes, mainly on weeks 3 and 5. The indexes were constant (above 10) from
week 7 to week 12 for both groups (Fig. 3.2.D). The Th1/Th2 index for the NP[OVA] group

Chapter 3
124
was constant (around 5) till week 5 and then decreased to levels below 1 on week 9. The
groups that received OVA and the adjuvants in solution also induced a temporary Th1-biased
immune response (from week 3 to week 7) that reversed to a Th2 profile on week 9.
3.2.2. Cytokine secretion
Cytokine levels were determined to complement the information obtained from the
IgG subtype production in order to understand the overall profile of the immune response
induced by the different vaccine formulations. Mice were sacrificed on week 12 after
immunizations, their spleens were aseptically removed and splenocytes were put in culture
and restimulated with 20 µg/ml of OVA. After 72 h of restimulation, cytokines were
quantified in culture supernatants by ELISA. Cytokine levels are represented in Figure 3.3..
NP[OVA+P+C] and man-NP[OVA+P+C] induced the highest levels of the Th1 cytokine IL-2
(p < 0.01). Surprisingly, only man-NP[OVA+P+C] induced significant superior levels of IFN-
γ (p < 0.001) while NP[OVA+P+C] induced levels of IFN-γ comparable to the other groups.
NP[OVA] and OVA+P+C sol. induced similar low levels of both cytokines. Concerning the
secretion of the Th2 cytokines IL-4, IL-6 and IL-10, all groups induced similar low levels of
IL-4 and no detectable levels of IL-10 (data not shown). OVA and the adjuvants in solution
induced a significantly higher level of IL-6 (p < 0.05) comparing to the NP-based groups.
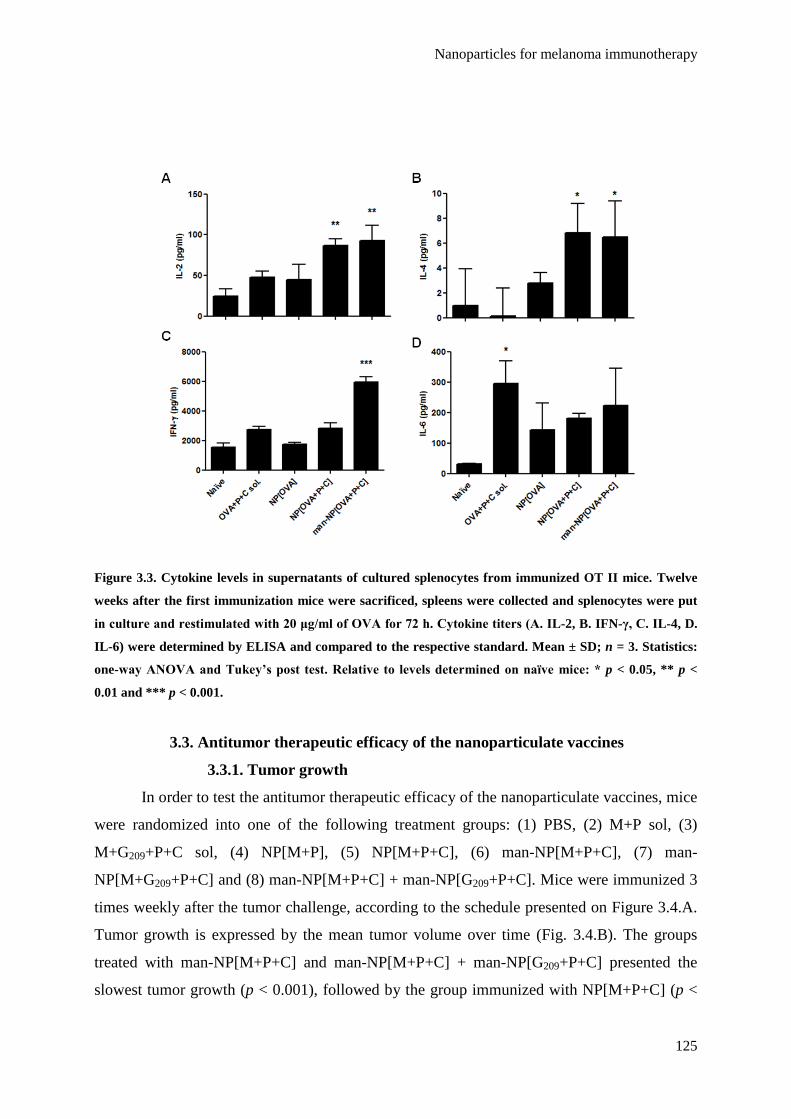
Nanoparticles for melanoma immunotherapy
125
Figure 3.3. Cytokine levels in supernatants of cultured splenocytes from immunized OT II mice. Twelve
weeks after the first immunization mice were sacrificed, spleens were collected and splenocytes were put
in culture and restimulated with 20 μg/ml of OVA for 72 h. Cytokine titers (A. IL-2, B. IFN-γ, C. IL-4, D.
IL-6) were determined by ELISA and compared to the respective standard. Mean ± SD; n = 3. Statistics:
one-way ANOVA and Tukey’s post test. Relative to levels determined on naїve mice: * p < 0.05, ** p <
0.01 and *** p < 0.001.
3.3. Antitumor therapeutic efficacy of the nanoparticulate vaccines
3.3.1. Tumor growth
In order to test the antitumor therapeutic efficacy of the nanoparticulate vaccines, mice
were randomized into one of the following treatment groups: (1) PBS, (2) M+P sol, (3)
M+G209+P+C sol, (4) NP[M+P], (5) NP[M+P+C], (6) man-NP[M+P+C], (7) man-
NP[M+G209+P+C] and (8) man-NP[M+P+C] + man-NP[G209+P+C]. Mice were immunized 3
times weekly after the tumor challenge, according to the schedule presented on Figure 3.4.A.
Tumor growth is expressed by the mean tumor volume over time (Fig. 3.4.B). The groups
treated with man-NP[M+P+C] and man-NP[M+P+C] + man-NP[G209+P+C] presented the
slowest tumor growth (p < 0.001), followed by the group immunized with NP[M+P+C] (p <

Chapter 3
126
0.001) and then NP[M+P] (p < 0.01). The groups immunized with antigens and adjuvants
solutions and the saline control had similar tumor growth. Surprisingly, the group immunized
with man-NP[M+G209+P+C] had a tumor growth rate similar to these groups too.
The mean mass of tumors per 100 g of final body weight and the mean mass of
spleens per 100 g of initial body weight are represented on Figures 3.4.C and 3.4.D,
respectively. These data are in accordance with the obtained mean tumor growth curves, with
the group immunized with man-NP[M+P+C] + man-NP[G209+P+C] presenting the highest
spleen mass per 100 g of initial body weight and the lowest mean tumor mass per 100 g of
final body weight.
Figure 3.4. Antitumor therapeutic efficacy of the nanoparticulate vaccines. A. Schedule of the therapeutic
assay. B. Mean tumor growth curves given by mean tumor volume over time, determined by V = ½ × (L ×
W2), where L (length) is the longest dimension and W (width) is the perpendicular dimension to the
length. C. Mean final tumor weight per 100 g of final body weight. D. Mean final spleen weight per 100 g
of initial body weight. Mean ± SD, N = 6, n = 3. Statistics: two-way ANOVA and Bonferroni’s post test (B)
or one-way ANOVA and Tukey’s post test (C & D); * p < 0.05, ** p < 0.01 and *** p < 0.001.

Nanoparticles for melanoma immunotherapy
127
3.3.2. Cytokine secretion
In order to understand the overall profile of immune response induced by the different
vaccines, the Th1 cytokines IFN-γ and IL-2 and the Th2 cytokines IL-4, IL-5, IL-6 and IL-10
were determined by ELISA after 72 h of splenocyte culture in the presence of Melan-A:26
(Fig. 3.5.A) or gp100:209 (Fig. 3.5.B). The groups immunized with man-NP[M+P+C] + man-
NP[G209+P+C], man-NP[M+P+C] and NP[M+P+C] had the highest levels of the Th1
cytokines IFN-γ and IL-2 (p < 0.001) after splenocyte restimulation with Melan-A:26.
Stimulation with gp100:209 induced high secretion of IFN-γ and IL-2 by the groups that
received this antigen delivered in NPs (man-NP[M+P+C] + man-NP[G209+P+C] and man-
NP[M+G209+P+C]) but not by the group that received it in solution. On the other hand, the
highest levels of the Th2 cytokines IL-4, IL-5 and IL-6 were secreted by splenocytes from the
groups immunized with the antigens and adjuvants in solution and the saline control, either
after restimulation with Melan-A:26 or gp100:209. All groups produced undetectable levels
of IL-10 (data not shown).
Figure 3.5. Cytokine levels in the supernatants of cultured splenocytes from mice in the therapeutic study.
Splenocytes were restimulated with 20 μg/ml of Melan-A:26 (A) or gp100:209 (B) for 72 h, and cytokines
were measured in culture supernatants by ELISA. Mean ± SD, n = 3. Statistics: one-way ANOVA and
Tukey’s post test. Relative to the saline control group: * p < 0.05, ** p < 0.01 and *** p < 0.001.

Chapter 3
128
3.4. Protection efficacy of the nanoparticulate vaccine against tumor
challenge
3.4.1. Tumor growth
The antitumor protection capacity of the nanoparticulate vaccines was also evaluated
in a prophylactic setting. Mice were randomized into the same groups tested in the therapeutic
assay. An additional group was included in order to test the combination of MHC class I
(Melan-A:26) and class II (gp100:44) antigens, where mice received man-NP[M+P+C] +
man-NP[G44+P+C]. Mice were immunized 3 times every two weeks before the tumor
challenge (Fig. 3.6.A). Tumor growth curves demonstrate that the additional group tested in
this assay (man-NP[M+P+C] + man-NP[G44+P+C]) induced the slowest tumor growth rate (p
< 0.001) (Fig. 3.6.B). This group presented the lowest mean tumor mass per 100 g of final
body weight (Fig. 3.6.D) and the highest mean spleen mass per 100 g if initial body weight
(Fig. 3.6.E). The remaining groups produced similar results to those obtained in the
therapeutic setting in terms of mean tumor growth curves and mean tumor and spleen
weights. Photographs of the excised tumors representative of each group can be seen in
Figure 3.6.C.
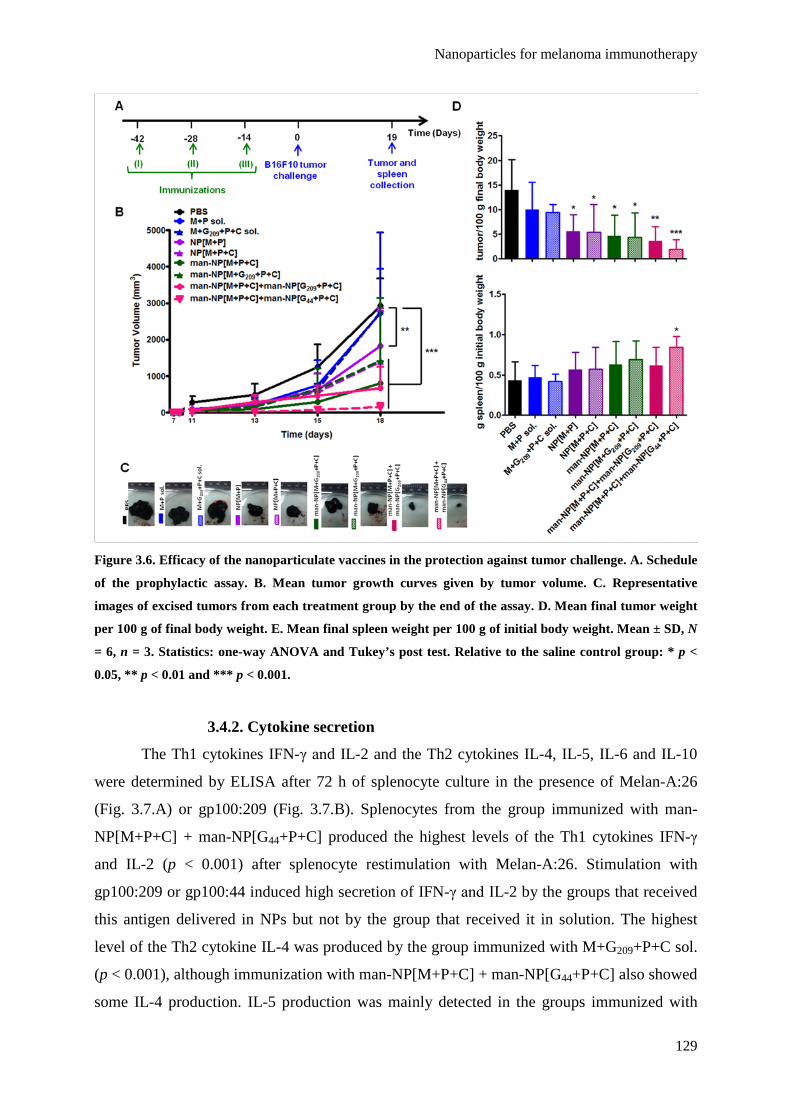
Nanoparticles for melanoma immunotherapy
129
Figure 3.6. Efficacy of the nanoparticulate vaccines in the protection against tumor challenge. A. Schedule
of the prophylactic assay. B. Mean tumor growth curves given by tumor volume. C. Representative
images of excised tumors from each treatment group by the end of the assay. D. Mean final tumor weight
per 100 g of final body weight. E. Mean final spleen weight per 100 g of initial body weight. Mean ± SD, N
= 6, n = 3. Statistics: one-way ANOVA and Tukey’s post test. Relative to the saline control group: * p <
0.05, ** p < 0.01 and *** p < 0.001.
3.4.2. Cytokine secretion
The Th1 cytokines IFN-γ and IL-2 and the Th2 cytokines IL-4, IL-5, IL-6 and IL-10
were determined by ELISA after 72 h of splenocyte culture in the presence of Melan-A:26
(Fig. 3.7.A) or gp100:209 (Fig. 3.7.B). Splenocytes from the group immunized with man-
NP[M+P+C] + man-NP[G44+P+C] produced the highest levels of the Th1 cytokines IFN-γ
and IL-2 (p < 0.001) after splenocyte restimulation with Melan-A:26. Stimulation with
gp100:209 or gp100:44 induced high secretion of IFN-γ and IL-2 by the groups that received
this antigen delivered in NPs but not by the group that received it in solution. The highest
level of the Th2 cytokine IL-4 was produced by the group immunized with M+G209+P+C sol.
(p < 0.001), although immunization with man-NP[M+P+C] + man-NP[G44+P+C] also showed
some IL-4 production. IL-5 production was mainly detected in the groups immunized with
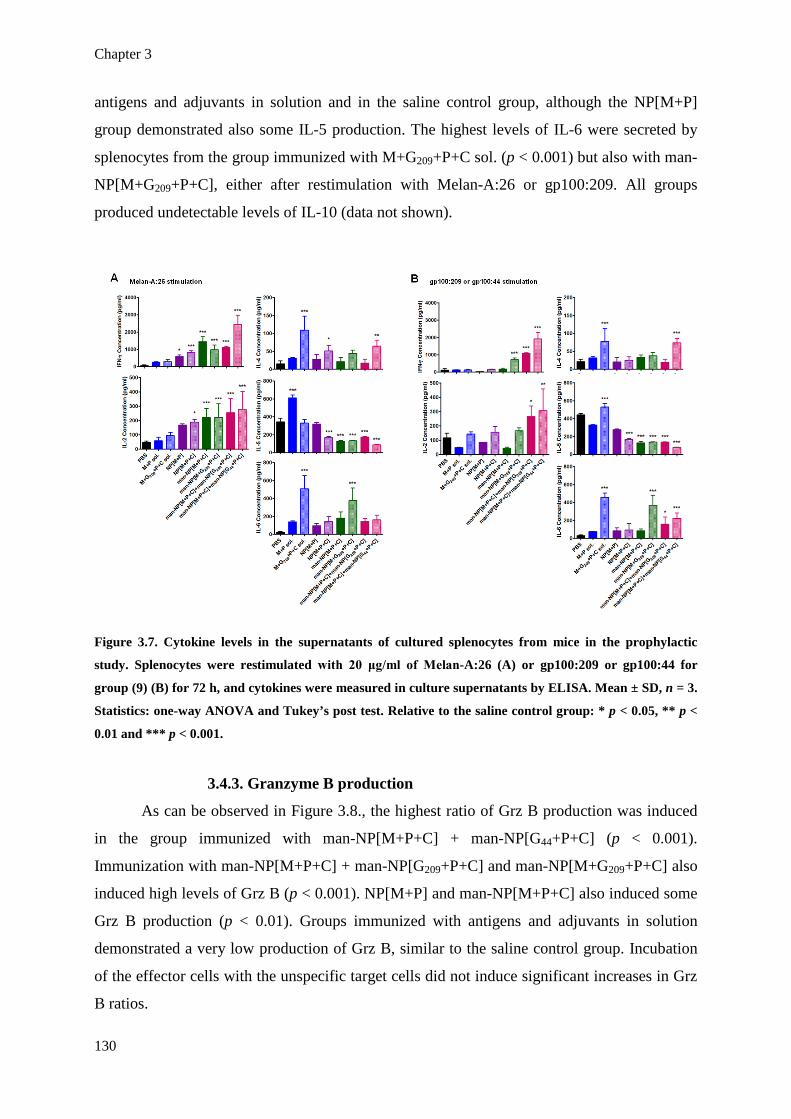
Chapter 3
130
antigens and adjuvants in solution and in the saline control group, although the NP[M+P]
group demonstrated also some IL-5 production. The highest levels of IL-6 were secreted by
splenocytes from the group immunized with M+G209+P+C sol. (p < 0.001) but also with man-
NP[M+G209+P+C], either after restimulation with Melan-A:26 or gp100:209. All groups
produced undetectable levels of IL-10 (data not shown).
Figure 3.7. Cytokine levels in the supernatants of cultured splenocytes from mice in the prophylactic
study. Splenocytes were restimulated with 20 μg/ml of Melan-A:26 (A) or gp100:209 or gp100:44 for
group (9) (B) for 72 h, and cytokines were measured in culture supernatants by ELISA. Mean ± SD, n = 3.
Statistics: one-way ANOVA and Tukey’s post test. Relative to the saline control group: * p < 0.05, ** p <
0.01 and *** p < 0.001.
3.4.3. Granzyme B production
As can be observed in Figure 3.8., the highest ratio of Grz B production was induced
in the group immunized with man-NP[M+P+C] + man-NP[G44+P+C] (p < 0.001).
Immunization with man-NP[M+P+C] + man-NP[G209+P+C] and man-NP[M+G209+P+C] also
induced high levels of Grz B (p < 0.001). NP[M+P] and man-NP[M+P+C] also induced some
Grz B production (p < 0.01). Groups immunized with antigens and adjuvants in solution
demonstrated a very low production of Grz B, similar to the saline control group. Incubation
of the effector cells with the unspecific target cells did not induce significant increases in Grz
B ratios.

Nanoparticles for melanoma immunotherapy
131
Figure 3.8. Relative granzyme B (Grz B) production by splenocytes collected from mice in the protection
study. Splenocytes (effector cells, E) were cultured with B16F10 cells (target cells, T) or breast cancer
MDA-MB-231 cells as unspecific target control (unsp T). After 4 h of incubation, Grz B levels were
measured in culture supernatants by ELISA. Relative ratios of the Grz B production were determined by
comparing the production in the presence or absence of target cells. Mean ± SD, n = 3. Statistics: one-way
ANOVA and Tukey’s post test. Relative to levels of the saline control group: * p < 0.05, ** p < 0.01 and
*** p < 0.001.
3.4.4. Splenocyte proliferation assay
Splenocyte proliferation capacity from immunized mice in the protection study is
shown in Figure 3.9.. Stimulation of splenocytes with Melan-A:26 induced proliferation in all
groups (p < 0.001) except in the saline control. The highest SI was verified in the groups
immunized with man-NP[M+P+C] + man-NP[G44+P+C] and man-NP[M+P+C] + man-
NP[G209+P+C]. Stimulation with gp100:209 caused high proliferation (p < 0.001) of the
splenocytes from the man-NP[M+P+C] + man-NP[G209+P+C] group. Similarly, stimulation
with gp100:44 caused high proliferation (p < 0.001) of the splenocytes of the group
immunized with man-NP[M+P+C] + man-NP[G44+P+C]. In both cases, the SI of the
remaining groups was not significantly augmented and any stimulation was seen in the saline
control group. The mitogen LPS induced high proliferation (p < 0.001) of the same groups as
well as the group immunized with NP[M+P+C]. Some proliferation was observed for the rest
of the groups, including the saline control, but not statistically significant.

Chapter 3
132
Figure 3.9. Splenocyte proliferation assay from mice in the protection study. Splenocytes were
restimulated with 20 µg/ml of each antigen (Melan-A:26, gp100:209 or gp100:44), 1 μg/ml of the mitogen
LPS from E. coli, Triton X-100 0.025 % (v/v) as a negative control, or left untreated for 48 h. Splenocyte
proliferation was measured using the AlamarBlue® reagent. Stimulation Index (SI) was calculated as the
ratio of the proliferation in each sample to the proliferation of the untreated splenocytes for each
immunization group. Mean ± SD, n = 3. Statistics: two-way ANOVA and Bonferroni’s post test; * p < 0.05,
** p < 0.01 and *** p < 0.001.
3.4.5. Spleen T lymphocytes subset analysis
Splenocytes from mice in the protection study were harvested and restimulated with
the peptides Melan-A:26, gp100:209 and gp100:44. After 72 h, the percentages of CD3+CD4+
and CD3+CD8+ T cells were quantified by flow cytometry. Figure 3.10. shows representative
histograms of the percentages of CD3+ versus CD4+ and CD3+ versus CD8+ T cells for each
group in the protection assay, after restimulation with the antigens. Mean percentages of each
T cell subset are also presented in the graphics on the right. It is possible to observe that the
groups immunized with man-NP[M+P+C] + man-NP[G44+P+C] and man-NP[M+P+C] +
man-NP[G209+P+C] had a superior (p < 0.001) percentage of CD3+CD4+ and CD3+CD8+ T
cells. The groups immunized with NP[M+P+C] and man-NP[M+P+C] also showed slightly
higher levels (p < 0.05) of CD3+CD4+ T cells but not CD3+CD8+ T cells. After restimulation
with gp100:209 (Fig. 3.10.C) or gp100:44 (Fig. 3.10.D), the groups immunized with man-
NP[M+P+C] + man-NP[G209+P+C] and man-NP[M+G209+P+C] demonstrated superior
percentage of CD3+CD4+ T cells (p < 0.001), comparing to the saline control group cells.
Surprisingly, any group demonstrated to induce higher percentages of CD3+CD8+ T cells.
Similarly, the group immunized with man-NP[M+P+C] + man-NP[G44+P+C] showed
superior levels of CD3+CD4+ T cells (p <0.001) but not of CD3+CD8+ T cells (Fig. 3.10.E &
3.10.F).

Nanoparticles for melanoma immunotherapy
133
Figure 3.10. Representative
flow cytometry histograms
of the percentages of CD3+
versus CD4+ and CD3+
versus CD8+ T cells for
each group in the
protection study, after
restimulation with Melan-
A:26 (A), gp100:209 (C) or
gp100:44 (D). Mean
percentages of each T cell
subset after restimulation
Melan-A:26 (B), gp100:44
(E) or gp100:209 (F) are
presented. Mean ± SD, n =
3. Statistics: one-way
ANOVA and Tukey’s post
test. Relative to the saline
control group: * p < 0.05,
** p < 0.01 and *** p <
0.001.
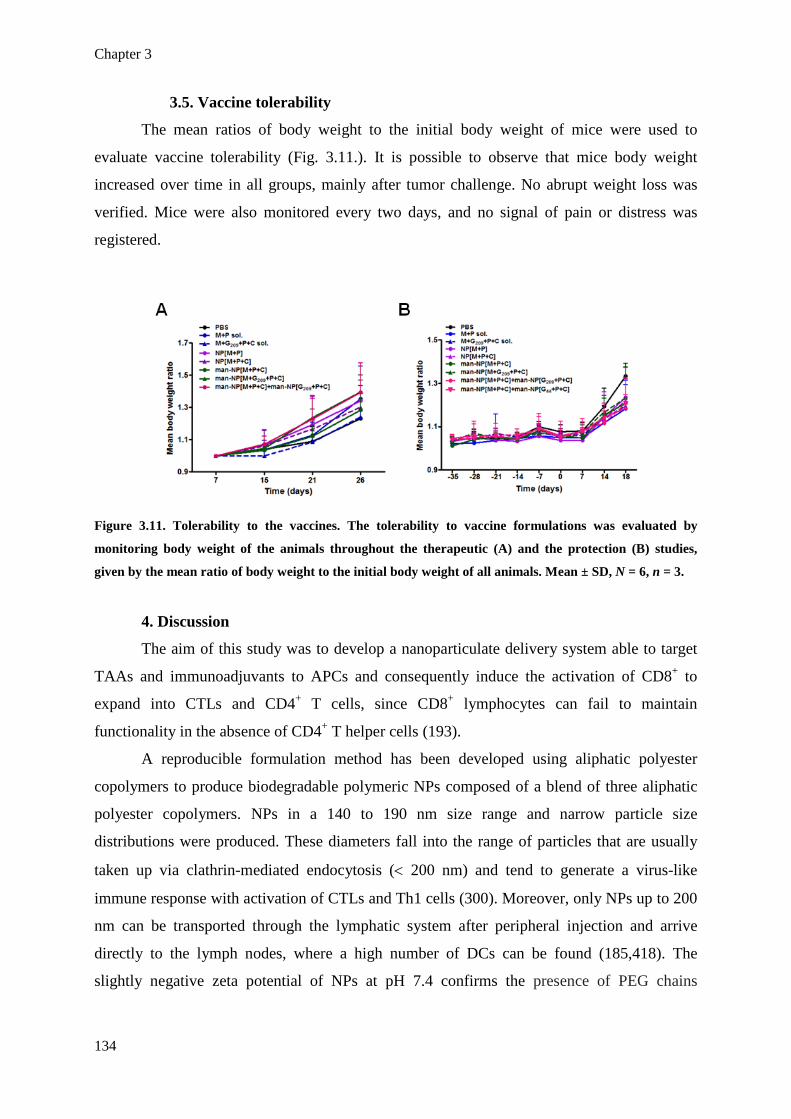
Chapter 3
134
3.5. Vaccine tolerability
The mean ratios of body weight to the initial body weight of mice were used to
evaluate vaccine tolerability (Fig. 3.11.). It is possible to observe that mice body weight
increased over time in all groups, mainly after tumor challenge. No abrupt weight loss was
verified. Mice were also monitored every two days, and no signal of pain or distress was
registered.
Figure 3.11. Tolerability to the vaccines. The tolerability to vaccine formulations was evaluated by
monitoring body weight of the animals throughout the therapeutic (A) and the protection (B) studies,
given by the mean ratio of body weight to the initial body weight of all animals. Mean ± SD, N = 6, n = 3.
4. Discussion
The aim of this study was to develop a nanoparticulate delivery system able to target
TAAs and immunoadjuvants to APCs and consequently induce the activation of CD8+ to
expand into CTLs and CD4+ T cells, since CD8+ lymphocytes can fail to maintain
functionality in the absence of CD4+ T helper cells (193).
A reproducible formulation method has been developed using aliphatic polyester
copolymers to produce biodegradable polymeric NPs composed of a blend of three aliphatic
polyester copolymers. NPs in a 140 to 190 nm size range and narrow particle size
distributions were produced. These diameters fall into the range of particles that are usually
taken up via clathrin-mediated endocytosis (< 200 nm) and tend to generate a virus-like
immune response with activation of CTLs and Th1 cells (300). Moreover, only NPs up to 200
nm can be transported through the lymphatic system after peripheral injection and arrive
directly to the lymph nodes, where a high number of DCs can be found (185,418). The
slightly negative zeta potential of NPs at pH 7.4 confirms the presence of PEG chains

Nanoparticles for melanoma immunotherapy
135
shielding the highly negative charges of PLGA at NP surface. It also indicates that sodium
cholate, which is an anionic tensioactive, might be efficiently removed from NP surface.
NPs demonstrated great versatility in terms of their capacity to coentrap different
molecules including a 45 KDa protein (OVA), very small peptides (~ 1 KDa) (Melan-A:26,
gp100:209 and gp100:44) and even the oligonucleotide CpG and the dsRNA analog
Poly(I:C). EE % around 70 % and LC ranging from 20 to 40 µg of antigen and from 7 to 11
µg of immunoadjuvant per mg of polymer indicate that these NPs are adequate for mice
immunization, since few milligrams of NPs were necessary to deliver the amount of peptide
and adjuvant normally used in pre-clinical studies (431). Biological responses do not to
correlate with doses and very small amounts of antigens and adjuvants seem to be necessary
to obtain effective immune responses (72). The MHC class I-restricted MAAs - Melan-A:26
and gp100:209 - have been used as they represent two highly immunogenic epitopes that are
frequently recognized by tumor-infiltrating T lymphocytes (TILs) (16). A MHC class II-
restricted melanoma antigen (gp100:44) has also been used to test the impact of co-
presentation of MHC class I and class II peptides in the antitumor immune response. Two
TLR ligands, Poly(I:C) and CpG, were specifically chosen because they have been described
as potent Th1-inducing immunoadjuvants (148). Their receptors, TLR3 and TLR9
respectively, are both localized in the endosomal membrane where NPs can easily get access
(184). Moreover, the dissimilar distribution pattern among human leukocytes and the
induction of parallel signaling pathways by these TLR ligands might allow a synergistic
action in the promotion of cytokine release and broader activation of APCs (432). To the best
of our knowledge, neither gp100:209 or gp100:44 have been encapsulated in any particulate
delivery system. On the other hand, Melan-A:26 has already been incorporated in virosomes
and liposomes, but considerably lower EE % values have been reported (429,433).
In order to potentiate NP interaction with APCs, a mannose-functionalized delivery
system was developed to target the mannose receptor (MR/CD206) on APCs, which is
involved in the recognition and internalization of pathogens (351). Several studies suggest
that mannose-grafted nanocarriers can have an increased uptake by APCs, resulting in
enhanced humoral and cellular antitumor responses (339,434).
The immune response profile induced by OVA-containing NPs was characterized in
OT II mice which are transgenic mice in which all CD4+ T cells express transgenic αβ-TCRs
that are specific for the OVA323–339 determinant in the context of H-2-I-Ab (435). This mice
model provides CD4+ T cell homogeneity that allows the characterization of immune
response profiles (Th1/Th2) without unspecific immune response bias (436). In this study, it

Chapter 3
136
was possible to verify by the immunoglobulins and cytokines produced that the administration
of OVA and the immunoadjuvants in solution induced a Th2-biased immune response. OVA-
containing NPs in the absence of the adjuvants induced a weak Th1-biased immune response
that reverted to Th2 three weeks after the last immunization. As so, it seems that antigens
delivered by NPs are not sufficient to induce a long lasting Th1 profile in the absence of
immunoadjuvants. When OVA and the immunoadjuvants were coentrapped in NPs or man-
NPs, a strong and long lasting Th1-biased immune response was induced. This demonstrates
that both the use of NPs and the coentrapment of Th1-immunoadjuvants along with the
antigen are essential to induce a Th1-biased immune response. Poly(I:C) and CpG are known
by their Th1-inducing properties (148). However, it is likely that these molecules are not able
to exert their effect when administered in solution, probably due to degradation and/or low
uptake by APCs.
In order to characterize the antitumor therapeutic efficacy of the developed NPs, the
MAAs were entrapped in NPs and used in combination with the immunoadjuvants Poly(I:C)
and CpG and administered to B16F10-bearing mice. Tumor growth rates and tumor and
spleen weights suggest that the combination of man-NPs coentrapping different MHC class I-
restricted MAAs along with two TLR ligands (man-NP(M+P+C] + man-NP[G209+P+C])
induced the most effective antitumor immune response. Using only one MAA along with two
TLR ligands-containing man-NPs (man-NP[M+P+C]) was equally very effective in tumor
growth reduction. However, when two MAAs were combined in the same man-NP, a
detrimental effect was obtained, since mean tumor volume over time was similar between the
group treated with man-NP[M+G209+P+C] and the groups that received antigens and
adjuvants in solution. The presence of both antigens in the same NP, and consequently, in the
same APC, could induce competition to the MHC class I complexes (437). However, when
the antigens are carried by different NPs, the chances that they are processed by different
APCs increase, and a stronger antitumor immune response was induced. Treatment with
NP[M+P+C] was inferior to man-NP[M+P+C], which demonstrate that mannose-
functionalization increased NP antitumor efficacy. NP[M+P+C] was slightly superior to
NP[M+P], suggesting a synergistic effect between both TLR ligands when they are
administered concomitantly, as previously reported (166,438). The groups immunized with
antigens and adjuvants in solution and the saline control had similar higher tumor growth
rates, which demonstrates the inefficacy of these molecules when administered in solution.
The nanoparticulate vaccines were also evaluated in a prophylactic setting. Here, an
additional group was tested, where man-NPs containing a MHC class I or a MHC class II-

Nanoparticles for melanoma immunotherapy
137
restricted MAA with both TLR ligands were combined (man-NP[M+P+C] + man-
NP[G44+P+C]). In recent years it has became evident that CD4+ T cells also play a critical
role in the development of effective antitumor immunity. Indeed, CD8+ lymphocytes can fail
to maintain functionality in large part because of the absence of CD4+ T cell help (426,439).
CD4+ T helper cells can enhance antitumor CTL responses by enhancing clonal expansion at
the tumor site, promoting the generation and maintenance of the memory CTL phenotype,
preventing activation-induced cell death (AICD) and functioning as APCs for CD8+ T cells
(26). We hypothesized that the combination of MHC class I and class II-restricted peptides
could induce both CD8+ and CD4+ activation and lead to a superior antitumor immune
response. In fact, the group immunized with the combination of man-NPs containing MHC
class I- and MHC class II-restricted peptides demonstrated the most significant tumor growth
rate reduction and the best results in terms of tumor and spleen weights, Th1-cytokine
production, Grz B secretion, splenocyte proliferation and percentage of CD4+ and CD8+ T
cells. The remaining groups tested in the protection assay demonstrated similar results to what
was obtained in the therapeutic setting.
5. Conclusions
We report the successful development of a cancer immunotherapeutic strategy by
using biodegradable polymeric NPs to target TAAs and TLR ligands to APCs and induce an
antitumor immune response. We were able to produce 140 – 190 nm NPs, either plain or
functionalized with mannose residues, that could successfully coentrap various antigens and
immunoadjuvants. Several factors that could affect the performance of the vaccine were
evaluated, such as (i) the importance of using a nanoparticulate delivery system; (ii) the
impact of mannose-functionalization of NPs for active targeting of APCs; (iii) the potential
synergy between two TLR ligands; (iv) the impact of the combination of two peptide antigens
either in separated or in the same NP; and (v) the effect of using a combination of NPs
containing MHC class I- and class II-restricted peptides.
All together, the in vivo results obtained lead us to the following conclusions:
• The coentrapment of antigens and immunoadjuvants in NPs is essential to induce a
long lasting Th1 immune response;
• Mannose functionalization of NPs improves NP performance in terms of the
desired Th1/antitumor immune response;
• The immunoadjuvants CpG and Poly(I:C) exert a synergistic effect when
coentrapped in NPs;

Chapter 3
138
• Immunization with two MHC class I-restricted MAAs in separated man-NPs was
slightly superior comparing to when only one antigen was used;
• The use of two MHC class I-restricted MAAs (Melan-A:26 and gp100:209)
coentrapped in the same man-NPs has a detrimental effect on the antitumor immune response,
possibly due to a competition of both peptides to the MHC class I molecules;
• The combination of man-NPs containing either a MHC class I- or class II-restricted
MAAs along with both immunoadjuvants elicited the most potent antitumor immune
response, suggesting the importance of the activation of both CD4+ and CD8+ T cells in the
efficacy of the antitumor immune response.
Akgwledgements
This work was supported by Fundação para a Ciência e a Tecnologia, Ministério da
Educação e Ciência, Portugal (PhD Grant SFRH/BD/64295/2009 to JMS, PTDC/SAU-
FAR/119389/2010 and Pest-OE/SAU/UI4013/2011) and by Fonds de la Recherche
Scientifique Médicale, Belgium. GV is a postdoctoral researcher of the Fonds de la Recherche
Scientifique-FNRS. The authors have no other relevant affiliations or financial involvement
with any organization or entity with a financial interest or financial conflict with the subject
matter or materials discussed in the manuscript.

Nanoparticles for melanoma immunotherapy
139
Appendix
1. Induction of DC maturation by NPs
In order to properly stimulate T cells to start an effective immune response, DC
maturation must be induced upon the internalization of the antigens to be presented. Given
that, it is important to evaluate the capacity of the antigen- and immunoadjuvant-loaded NPs
to induce the maturation of DCs. To access the effect of different NP formulations containing
the model antigen OVA and the TLR ligands Poly(I:C) and CpG on the maturation induction
of DCs, the surface expression of the costimulatory molecules CD40 and CD86 was
quantified in the CD11c+ population of BMDCs after 24 h of incubation with the different NP
formulations.
1.1. Method
BMDCs were seeded in 96-flat bottom well plates (2 × 105/200 µl) and let to become
adherent overnight. Cells were treated in triplicate with one of the following treatments: OVA
2.5 µg/ml, LPS 100 ng/ml, Poly(I:C) 5 µg/ml, NP[blank], NP[OVA], NP[OVA+P],
NP[OVA+P+C] and man-NP[OVA+P+C] or left untreated. NPs were added at a
concentration of 500 µg/ml. NPs carried 12.5 µg/mg of OVA and 2.5 µg/mg of Poly(I:C) or
CpG. After 24 h of incubation, BMDCs were washed with FACS buffer and stained with anti-
mouse fluorescently labeled monoclonal antibodies for flow cytometry (PE-CD11c, FITC-
CD86 and APC-CD40) for 20 min after Fc blocking with rat anti-mouse CD16/CD32
antibody for 15 min. BMDCs were harvested by centrifugation and analysed by flow
cytometry. The individual fluorescence of 10,000 cells was collected for each sample using a
LSR Fortessa cytometer (BD Biosciences) and analyzed with FlowJo software version 7.6.5
for Microsoft (TreeStar, San Carlos, CA).
1.2. Results
To access the effect of different NP formulations containing the model antigen OVA
and the TLR ligands Poly(I:C) and CpG on the maturation induction of DCs, the surface
expression of the costimulatory molecules CD40 and CD86 was quantified in the CD11c+
population of BMDCs after 24 h of incubation with different NP formulations. Untreated and
LPS-treated BMDCs were used as controls. OVA and Poly(I:C) were also tested in solution.
The surface expression of CD40 and CD86 by CD11c+ BMDCs upon different treatments is

Chapter 3
140
represented in the histograms presented in Figure 3.12.A along with the histograms of
untreated cells and isotype control. Figure 3.12.B presents the expression of the costimulatory
molecules by median fluorescence intensities in the graphics. Untreated BMDCs presented
basal levels of CD40 and CD86 expression. These costimulatory molecules were highly
upregulated by treatment with LPS (p < 0.001), with CD40 demonstrating a much higher
upregulation than CD86. OVA and Poly(I:C) added to BMDCs in solution had slightly
different effects on the expression of CD40 and CD80. OVA significantly increased the
expression of CD40 (p < 0.01) but not CD86, while Poly(I:C) had the opposite effect. The
great majority of NP formulations induced a significant increase (p < 0.001) in CD40 and
CD86 expression. Comparing to OVA solutions, OVA entrapped in NPs induced significant
upregulation of CD40 (p < 0.05) but mainly of CD86 (p < 0.001). Surprisingly, the addition of
the TLR ligands Poly(I:C) or CpG to the NP formulations did not significantly increased the
expression of both molecules. Plain NPs also demonstrate to significantly increased the
expression of both costimulatory molecules (p < 0.001) comparing to untreated cells and were
not significantly different from the antigen and adjuvant-loaded NPs. man-NP[OVA+P+C]
induced very different effects on the expression of CD40 and CD86. While this formulation
induced lower upregulation of CD40 comparing to the other NP formulations tested, it
induced a much higher CD86 expression (p < 0.001) comparing to any other NP formulation
or even LPS.

Nanoparticles for melanoma immunotherapy
141
Figure 3.12. Expression of the costimulatory molecules CD40 and CD86 by BMDCs upon incubation with
NPs. BMDCs were incubated for 24 h with OVA 2.5 µg/ml, LPS 100 ng/ml, Poly(I:C) 5 µg/ml, NP[blank],
NP[OVA], NP[OVA+P], NP[OVA+P+C] and man-NP[OVA+P+C)] or left untreated. After staining with
anti-mouse fluorescently labeled monoclonal antibodies for flow cytometry (PE-CD11c, FITC-CD86 and
APC-CD40) 10,000 cells were analyzed in a flow cytometer. A. Upregulation of CD40 and CD86 presented
in histograms. Each histogram presents the isotype control (blue line), untreated cells (red line) and
treated cells (orange or green line for CD40 or CD86, respectively). B. Median fluorescence intensity
translating the expression of CD40 or CD86 for each condition tested. Mean ± SD, N = 3, n = 3. Statistics:
one-way ANOVA and Tukey’s post test. * p < 0.05, ** p < 0.01, *** p < 0.001, NS, statistically non-
significant.
2. Discussion & conclusions
For T cells to be effectively activated by DCs, the presentation of the antigen by DCs
to T cells needs to happen in an immunostimulatory environment. This means that, along with
the recognition of the MHC-antigen complex presented by DCs, T cells need to receive other
stimuli from DCs. This stimuli results from the maturation of DCs which induces the
upregulation of cell surface molecules, also known as costimulatory molecules, such as

Chapter 3
142
CD40, the receptor for CD40L expressed by naïve T cells, and also CD80 and CD86 (B7-1
and B7-2 in humans, respectively), which are the ligands for the immunoglobulin superfamily
member CD28, also expressed by naïve T cells. Also, T cells need cytokines and chemokines,
such as IL-12 and type I IFNs, which are produced by DCs and directly affect T cells in a
paracrine fashion (41,42).
The analysis of the expression of CD40 and CD86 by BMDCs treated with different
NP formulations revealed that NPs are able to induce the upregulation of those costimulatory
factors. The slightly different effects that were observed in the expression of CD40 and CD86
might be explained by the fact that the induction of the expression of these costimulatory
molecules happens in different moments of the DC maturation process. CD86 is known to be
an affector costimulatory molecule, since its upregulation on DCs is induced in an early stage
upon stimulation. CD40 expression happens later on as it is known to be an effector
costimulatory molecule (440). This can explain the fact that NP formulations, mainly man-
NP[OVA+P+C], had a stronger effect on the induction of CD86 than of CD40. We
hypothesized that a longer time of incubation would be necessary to see a higher effect of
these NPs on CD40 expression. Even though, these results suggest that mannose
functionalization can significantly increase the induction of the maturation process of DCs,
which may happen due to an increase of the uptake of the mannose-functionalized NPs, but
more likely due to the direct effect of mannose on triggering the C-type lectin receptor
MR/CD206 and consequently induce the activation of DCs.
NPs loaded with OVA demonstrated to be more effective in the induction of the
expression of CD40 (p < 0.05) and CD86 (p < 0.001) than OVA solution, which demonstrates
the importance of using NPs as vaccine delivery systems. Surprisingly, the coentrapment of
the TLR ligands Poly(I:C) and CpG did not significantly increase the effect observed for
NP[OVA]. We hypothesized that the 24 h of incubation used in this experiment were not
enough to observe the effect of antigen and adjuvant-loaded NP maximal effect. We alert to
the fact that the full potentiating effect of the immune response by of loaded NPs is probably
more notorious in vivo due to the more efficient targeting and internalization of the antigens
and adjuvants by DCs.
Another surprising result observed in this experiment is the fact that plain NPs (i.e.
NPs deprived of any payload) induced also the upregulation of both costimulatory molecules
in a level not significantly different from the antigen/adjuvant-loaded NPs. There are several
possible explanations for this observation. One hypothesis is the fact that the formulated NPs
have intrinsic immunogenicity. There is a great debate around the possibility of aliphatic

Nanoparticles for melanoma immunotherapy
143
polyester-based particles being able to induce complement activation or other direct effects on
the immune system (287). A study on PLGA MPs and NPs demonstrated potent inflammatory
stimulus after the uptake process with NF-κB translocation to cell nucleus and further pro-
inflammatory cytokine production (290). On the other hand, other study has demonstrated that
empty PLGA MPs did not induced significant maturation and did not affect the capacity of
DCs to respond to a maturation-inducing cocktail afterwards (291). We do not exclude the
possibility of some contamination with pirogens or endotoxins of some of the bulk material
used to formulate the NPs since it was not possible to get pirogen/endotoxin-free grade for
some of the reagents used. There is also the possibility that plain NPs increased the
immunogenicity of serum proteins present in the cell culture medium due to adsorption of
those proteins to the NPs during the incubation period and consequent increase of their
internalization. We also hypothesized that BMDCs are more susceptible in vitro to the effects
of external factors since we also verified that untreated cells also have a basal expression of
the costimulatory factors and they downregulate the expression the MR/CD206.
In conclusion, it was possible to observe that NPs are able to induce the upregulation
of the costimulatory factors CD40 and CD86, mainly when NPs were functionalized with
mannose. However, some of the results obtained, such as the low effect of TLR ligand-loaded
NPs and the high stimulation caused by empty NPs, were not expected. We conclude that the
full potentiating effect of the immune response by loaded NPs would be more notorious in
vivo due to the more efficient targeting and internalization of the antigens and adjuvants by
DCs. The real potential of the NP formulations might not be possible to observe in vitro due
to several limitations imposed by the nature of these experiments, such as the limited time of
incubation which might impose limited antigen and immunoadjuvant release from NPs; the
unrealistic environment provided to DCs, which might cause some basal level of maturation;
and the absence of the in vivo complex environment, which might further enhance the
importance of using antigen- and immunoadjuvant-loaded NPs over those molecules in
solution.


Chapter 4


Integrative discussion
147
Chapter 4 - Integrative discussion
Despite the fact that more than a century has passed from the first observations of the
active role of the immune system in combating cancer, very few immunotherapeutic strategies
have been approved to clinical use. Some of the clinical trials of the first cancer vaccines have
provided evidence of clinical benefit thus encouraging the development of other vaccines. The
identification and characterization of the antigens to be targeted, the definition of the desired
immune response to be elicited by a vaccine, and the choice of the appropriate vaccine
delivery system, are among the main difficulties drawing vaccine development back. An
optimistic perspective would see that there is an enormous amount of information and
techniques available for guiding vaccine development in the context of cancer
immunotherapy. Several TAAs have been identified and can be produced in large scale by
recombinant techniques, either as fusion proteins in bacterial or other systems, or as soluble
molecules in eukaryotic systems. Synthetic peptides mimicking immunodominant tumor
antigens to be presented by either class I or class II MHC molecules can also be produced.
Cytokines have been described and there are much more knowledge of how the immune
system functions, in particular how cellular immune responses are generated and how
peptides can be presented. DCs have also been discovered and have shown to play a central
role in generating immune responses in patients with cancer, establishing a bridge between
the innate and the adaptive branches of the immune system. Several methods have also been
applied for the generation of optimal immune responses, such as DNA vaccines, gene therapy,
hybrid cell vaccination, tumor cell vaccination and cell-free vaccines. In addition, there are
several methods of assessing cellular immunity: measuring CTLs, T-cell proliferation,
intracellular and extracellular cytokine production by cells in culture using ELISA or
multiplex and flow cytometry, polyfunctional T-cell assays, ELISPOT and MHC class I/II
tetramers.
Vaccination has had a significant impact in global heath as they permitted the control
of many infectious diseases and the eradication of smallpox (44,45). Although similar in
concept to the more conventional vaccines directed against infectious diseases, cancer
vaccines differ in several critical areas. Cancer vaccines are envisioned to be administered
after the onset or detection of disease thus they would be considered therapeutic rather than
prophylactic vaccines. As many tumor antigens are intracellular proteins rather than cell
surface molecules, cancer vaccines should be designed to elicit cell-mediated immunity rather

Chapter 4
148
then humoral responses. Lastly, the target of most cancer vaccines might be self-molecules
instead of foreign antigens, which raises the concern of whether vaccination will be
sufficiently robust enough to initiate an immune response rather than immunological self-
tolerance (7).
Several vaccine strategies have been evaluated in clinical trials for cancer therapy,
including administration of peptides and proteins mixed with adjuvants, injection of
recombinant viruses or other recombinant microorganisms, delivery of killed tumor cells, or
infusion of protein- or peptide-activated DCs to patients (8).
Since the first clinical trial of a melanoma antigen gene-1 (MAGE-1)-derived peptide-
based vaccine was reported in 1995 (441), several peptide and protein-based vaccines have
reached the clinical trials. The earlier generations of peptide vaccines were composed by one
to several HLA class I-restricted epitope peptides of a single HLA type. Currently, various
types of next-generation peptide vaccines are under development: (i) multivalent long peptide
vaccines; (ii) multi-peptide vaccines consisting of CTL- and helper-epitopes; (iii) peptide
cocktail vaccines; (iv) hybrid peptide vaccines; (v) personalized peptide vaccines; and (vi)
peptide-pulsed DC vaccines (442). Viral vectors, either from non-lytic or oncolytic nature,
expressing TAAs and immunostimulatory molecules, such as cytokines, GM-CSF or
costimulatory molecules, are also being tested in clinical trials. However, the therapeutic
benefit of delivering transgenes using viruses needs to become clearer and easier to define
(443). Tumor cell vaccines have an advantage in that the antigen does not need to be defined
or identified prior to vaccination. However, the mechanisms of action and the relevant target
of the vaccine may be difficult to elucidate. In addition, there may be significant regulatory
and manufacturing issues that will need to be considered before commercialization. Of
particular interest is Melacine, an allogeneic melanoma cell line lysate vaccine combined with
a potent adjuvant. Despite data from phase II clinical trials not indicating a benefit in either
early stage or advanced stage melanoma, these studies conducted to the critical finding that
HLA expression can influence the efficacy of a cancer vaccine (444,445).
Ex vivo loaded DC-based vaccines have been intensively studied over the past decade.
At the time of writing this thesis, 300 clinical trials with DC-based immunotherapies were
registered, not only for cancer therapy but also for other immune-related diseases, such as
HIV infection and type 1 diabetes (446). FDA and EMA have recently approved Sipuleucel-T
as the first DC-based vaccine (Provenge®, Dendreon Corp.) for the therapy of asymptomatic
metastatic castrate-resistant prostate cancer (9). The requirement of leukapheresis and cell
culture processing are pointed out as drawbacks of this strategy, due to its high cost and

Integrative discussion
149
limited number of vaccinations that can be used. The combination of nanotechnological and
biochemical advances has been pointing out NP-based vaccines as being one possible strategy
to overcome these drawbacks. This strategy focus on loading and activating DCs in vivo, by
targeting antigens and immunoadjuvant molecules to these cells without the need for
isolatation (447).
Since the lecture of Richard Feynman (1959), which is considered the beginning of the
“nanotechnology era”, the outcome of nanomedicines has been modest. The concepts of
antibody-conjugates, liposomes, NPs, and polymer-conjugates stem from the 1970s. These
strategies were seen as competing technologies to the only one that would eventually stand up
as the “magic bullet” for all drug-targeting applications. However, the advantages and
disadvantages of each one have emerged. Antibodies have great potential for selective
targeting but high risk of immunogenicity. Liposomes are able to transport high quantities of
drug, but drug release from the liposomes might be difficult to control and they are prone to
be captured by the RES. Similarly, NPs are also difficult to avoid capture by RES after
intravenous injection. The combination of several technologies in one delivery system is more
likely to lead to its desired performance. Since 1990, several nanomedicines have been
approved for clinical use by regulatory authorities, mostly for cancer therapy, such as
liposomes (e.g. DaunoXome®), polymer-coated liposomes (Doxil®/Caelyx®), polymeric drugs
(Copaxone®), antibodies (Herceptin®, Avastin™) and antibody conjugates (Mylotarg®),
polymer-protein conjugates (Oncaspar®, Neulasta®), and, lately, NPs containing paclitaxel
(Abraxane™) (2). The application of nanomedicines for cancer vaccines and immunotherapy,
or even for vaccine delivery in general, is giving the first steps, and the present research work
was developed aiming to contribute to the acceleration of the process.
The main goal of the research work presented in this thesis was to develop a
melanoma therapeutic vaccine by using biodegradable polymeric NPs to deliver MAAs and
immunoadjuvant molecules and induce an antitumor immune response. As described in
chapter 2, the aim of the study started to be the development of a nanoparticulate delivery
system to target APCs, deliver antigens to be presented by MHC class I (in the endoplasmic
reticulum) and II molecules (in the lysosomes) and potentiate the encounter of
immunoadjuvants with their intracellular receptors. Increase the knowledge on NP-cell
interaction, NP functionalization impact, endocytic pathways and intracellular trafficking was
also under focus. Spherical NPs with ~130 nm were produced in a reproducible way. It was
possible to produce also NPs functionalized with mannose residues, which were available for
binding at NP surface. The study provided insight on the different NP internalization profiles

Chapter 4
150
by immortalized APCs and primary BMDCs as well as on the multiple endocytic pathways
that were involved in NP internalization. NPs demonstrated both an endo-lysosomal and
cytosolic localizations and a tendency to accumulate nearby the ER. This system may provide
intracellular access to immunoadjuvants and drive antigens to be presented by both MHC
class I and II molecules to CD8+ and CD4+ T cells, and consequently it has potential to induce
a complete and coordinated immune response. This preliminary research provided sufficient
information to go a step further and test the developed NPs as a cancer vaccine delivery
system in a preclinical context.
In chapter 3, we report the results of the preclinical evaluation of the developed cancer
immunotherapy strategy by using biodegradable polymeric NPs to target TAAs and TLR
ligands to APCs and induce an antitumor immune response. We were able to produce 140 –
190 nm NPs, either plain or functionalized with mannose residues, that could successfully
coentrap various antigens and immunoadjuvants. Several factors that could affect the
performance of the vaccine and have an impact on the antitumor immune response were
evaluated, such as (i) the importance of using a NPs for vaccine delivery; (ii) the impact of
mannose-functionalization of NPs for active targeting of APCs; (iii) the potential synergy
between two TLR ligands; (iv) the impact of the combination of two peptide antigens either in
separated or in the same NP; and (v) the effect of using a combination of NPs containing
MHC class I- and class II-restricted peptides.
As mentioned in the section “Aim and outline of the thesis”, throughout the
development of this work, several questions were raised. The answers to these questions were
addressed throughout this manuscript and summarily presented here:
1. Is it possible to produce biodegradable polymeric NPs with the desired
physicochemical characteristics in a reproducible way? Is was possible to produce
biodegradable polymeric NPs in a reproducible way with the desired physicochemical
characteristics for cancer vaccine delivery, such as a mean diameter around 150 nm, zeta
potential close to neutrality at pH 7.4 and spherical morphology. A mean diameter aroud 150
nm was desired primarly because it is known that NPs with diameters up to 150 nm seem to
be mainly taken up via CME and tend to generate a virus-like immune response with
activation of CTLs and T helper (Th) 1 cells (300). Moreover, NPs up to 200 nm are able to
travel through the lymphatic system after peripheral injection and arrive directly to the lymph
nodes, where a high number of DCs can be found. NPs with zeta potential close to neutrality
at physiologic pH were desired because highly negative NPs might be repulsed from the
negatively charged plasma membranes, highly positively charged NPs are generally very

Integrative discussion
151
toxic due to high penetration in plasma membranes and neutral NPs are more prone to
aggregate. The slightly negatively zeta potential verified for the produced NPs is likely to
contribute to the high internalization levels obtained, the low tocxicity verified and the
absence of aggregation of NPs over time.
2. Can biodegradable polymeric NPs entrap MAAs and immunoadjuvant
molecules? The produced NPs were able to entrap several MAAs (Melan-A:26-35(27L),
gp100:209-217(2M) and gp100:44-59) and immunoadjuvant molecules (Poly(I:C) and CpG)
in appropriate quantities for in vivo use (20 a 40 µg of antigen and 7 a 11 µg of adjuvant per
mg of polymer). The obtained loadings ensured the immunization of mice with the standard
doses mostly referred in literature (100 µg, 40 µg of Poly(I:C) and 20 µg of CpG) delivered in
4 mg of NPs, which demonstrated to be easily administered subcutaneously and well
tolerated.
3. Can biodegradable polymeric NPs target and be internalized by APCs? The
produced NPs demonstrated to be internalized by APCs of different natures. To the best of
our knowledge this was the first time that the profile of NP internalization by immortalized
and primary APCs was compared. The observation that immortalized APCs (DCs and
macrophages) and primary DCs have distinct patterns of NP internalization might contribute
to the awareness of the scientific community for the impact of the chosen models to the
outcome of the experiment. Mannose functionalization of NPs revealed to increase the in vivo
efficacy of the nanoparticulate vaccines. However, in vitro it increased internalization by
immortalized cell lines but not by primary DCs. Once again, this observation reinforces the
difficulty of translation of the observations made in vitro to the preclinical and clinical reality.
4. Which endocytic mechanisms are involved in the internalization of the NPs?
Several endocytic mechanisms demonstrated to be involved in the internalization of the NPs
by DCs, namely macropinocytosis, clathrin-mediated endocytosis, caveolin- and lipid raft-
dependent endocytosis. Successful navigation of the endocytic pathways is usually a
prerequisite to the achievement of the intracellular targets of nanomedicines, with
simultaneous reduced access to sites of toxicity (448). When targeting APCs for the activation
of the immune system, considerations about size, endocytic pathways and final immune
response must come together. Each endocytic pathway seems to be defined by a specific size
range of the engulfed soluble or particulate material and apparently drives the activation of a
specific branch of the immune system (298,299). The fact that multiple endocytic pathways
seem to be involved in the internalization of the produced NPs might be related with the fact,
even that NPs present low PdI and are a monodispersed population in terms of diameter, there

Chapter 4
152
are still some variability of the NP size. It is possible that the narrow size distribution of NP
population is heterogeneous enough to activate distinct endocytic pathways. On the other end,
it might also be utopical to think that only one endocytic pathway is activated by one single
particulate entity. It is well know that macropinocytosis is a constitutive process in DCs (415).
As so, it has a great probability of being involved in NP internalization by these cells. It is
also likely, that specific physicochemical features of NPs, such as size, shape and charge, also
contribute to the activation of other(s) endocytic pathways. Despite the high number of
nanoparticulate systems under research, there is still a lack of knowledge on the kinetics of
these systems at the cellular level. There is a need to use endocytosis-relevant clinical
biomarkers to better select those patients most likely to benefit from nanomedicine therapy
since it is becoming evident that endocytic pathways are disregulated in many diseases and
this has the potential to significantly impact nanomedicine performance in terms of safety and
efficacy (448).
5. Once internalized by APCs, which are the intracellular traffic and final fate of the
NPs? The knowledge of the endocytic pathways involved in NP internalization provides
information that can help to predict if NPs can achieve the desired final intracellular target.
However, only the observation of their intracellular traffic can confirm that. The produced
NPs demonstrated both an endo-lysosomal and cytosolic localizations and a tendency to
accumulate nearby the ER overtime. This pattern of intracellular traffic allowed the prediction
that this nanosystem could provide intracellular access to the immunoadjuvants CpG and
Poly(I:C), which receptors are located at the endosomal membrane, and also drive antigens to
be presented by both MHC class I (in the ER) and II (in lysosomes) molecules to CD8+ and
CD4+ T cells leading to a complete and coordinated immune response. The knowledge of the
intracellular traffic and final fate of particulate delivery systems constitutes an important and
underrated tool to evaluate the potential of these systems and support rational decisions
concerning the choice of the best systems to each application.
6. Which is the profile of the immune response induced by NPs entrapping antigens
and immunoadjuvants? The transgenic mice model OT II allowed the characterization of the
profile of the immune responses induced by NPs and the impact of coentrapping the
immunoadjuvants CpG and Poly(I:C) with the antigen as well as the effect of mannose
functionalization on the final outcome. The fact that this mice model provides CD4+ T cell
homogeneity avoids unspecific immune response bias in the characterization of immune
response profiles in terms Th1/Th2 balance (436). This study supported three valuable
conclusions that confirm the observations made by others. Firstly, NPs improved the vaccine

Integrative discussion
153
outcome of the antigen and adjuvants administered in solution, as verified before (3,169,449).
Secondly, the immunoadjuvants CpG and Poly(I:C) coentrapped with the antigen in NPs
reverted a Th2-biased immune response to a long lasting Th1 profile. It has been verified
before that NPs coentrapping TLR ligands along with the antigens induced better immune
responses than the NPs containing only the antigen (165–167,169,170,449). Thirdly, mannose
functionalization of NPs further improved the vaccine outcome, as also verified before
(339,434). This study might seem redundant since the information collected only confirms
others’ observations. However, when developing a new nanoparticulate delivery system for
biomedical applications, assumptions should be avoided since there are several factors that
can affect the performance of these systems, from physicochemical properties to the models
and conditions used to test them.
7. Can NPs induce an antitumor immune response? Different NP-based
immunizations were tested in a melanoma murine model, either in a therapeutic (after tumor
challenge) or prophylactic settings (before tumor challenge). All the NP-based vaccines
reduced the tumor growth rate when compared to the antigens and adjuvants administered in
solution. NP formulations also induced higher levels of granzyme B production by
splenocytes, higher splenocyte proliferation, higher CD3+CD4+ and CD3+CD8+ splenocyte
frequencies and higher production of IFN-γ and IL-2 by restimulated splenocytes. Given that,
it is possible to conclude that NP-based vaccines are able to induce an antitumor immune
response. It is important to keep in mind that cancer vaccine research aims to develop
therapeutic strategies for cancer rather than prophylactic vaccines. However, the high tumor
growth rate of mice tumor models might not allow the development of the antitumor immune
response to its greater extension since this process might take longer than the tumor
development. Moreover, the melanoma model used (B16F10) is highly aggressive and
metastatic. This cell line is obtained from ten repeated passages of lung colonies obtained
from C57Bl6 mice after their intravenous injection with the murine epithelial melanoma
B16F0 cell line (ATCC) (450). These limitant aspects of the animal models available for the
preclinical development justify the application of the NP formulations in a prophylactic
setting, in an attempt to increase the time for the immune response development before the
invasiveness of the tumor reaches a level that compromises the immune system of the animals
under test.
8. Which is the impact of the coentrapment of different MAAs and immunoadjuvant
molecules in the antitumor response induced by the NPs? To the best of our knowledge this is
the first time that several antigens are coentrapped in the same nanoparticulate delivery

Chapter 4
154
system and tested as a cancer vaccine. This research provides valuable information on the
impact of combining several antigens in the same NP or in separated NPs. It was possible to
observe that the coentrapment of Melan-A:26-35(27L) and gp100:209-217(2M) in the same
NP (man-NP[M+G209+P+C]) had a detrimental effect in the antitumor immune response. On
the other hand, when the peptides were administered in separated NPs (man-NP(M+P+C] +
man-NP[G209+P+C]), there was an improvement of the antitumor response relatively to the
use of only one peptide. The presence of both antigens in the same NP, and consequently, in
the same APC, could induce competition to the MHC class I complexes (437). However,
when the antigens are carried by different NPs, the chances that they are processed by
different APCs increase, and a stronger antitumor immune response was induced. This
information might contribute to the successful design of effective particulate vaccines in the
future. The present research also took insight on the impact of combining MHC class I- and
class II-restricted peptides in the immunization regimens. Even that the induction of CTLs is
the primary goal in antitumor immunization, it is well established that CD8+ lymphocytes can
fail to maintain functionality in the absence of CD4+ T helper cells (193). As so, it would
make sense to combine both types of antigens in an attempt to activate both CD8+ and CD4+
T cells. However, there are very few studies combining MHC class I- and class II-restricted
peptides for antitumor immunization, probably due to the late identification of MHC class II-
restricted tumor peptides (451). Surprisingly, Phan G. (2003) and coworkers verified that the
addition of the class II peptide appeared to decrease immunologic response of circulating
PBMC to the class I-restricted peptide (452). The authors hypothesized that this unexpected
result could have involved increased CD4+CD25+ Treg activity, a possible increased
apoptosis of activated CD8+ T cells, the trafficking of sensitized CD8+ reactive cells out of the
peripheral blood or even bias in the sequential, nonrandomized nature of patient enrollment. It
has also been demonstrated that the peptide Melan-A:26-35(27L) could elicit peptide-specific
CTLs in the absence of a Th peptide due to its increased immunogenicity resultant from the
single aminoacid modification (453). However, it remained to be proven if the presence of a
Th peptide would not further improve the immune response obtained. The present research
demonstrates that mice immunized with a combination of NPs containing either the MHC
class I-restricted peptide Melan-A:26-35(27L) either the MHC class II-restricted peptide
gp100:44-59 (man-NP[M+P+C] + man-NP[G44+P+C]) had a very significant reduction of the
tumor growth rate and improved the overall immune response comparing to mice that
received either only one MHC class I-restricted peptide (man-NP[M+P+C]) either two MHC
class I-restricted peptides entrapped in separated NPs (man-NP(M+P+C] + man-

Integrative discussion
155
NP[G209+P+C]). These results reinforce the fact that activation of CD4+ T might be an
advantageous strategy for the induction of potent antitumor immune responses.


Chapter 5


General conclusions & future perspectives
159
Chapter 5 - General conclusions & future perspectives
The research work described in this thesis highlights the potential of NPs, in particular
those constituted by biodegradable aliphatic polyesters, for use as cancer vaccines to target
tumor antigens and adjuvants to DCs for priming antigen-specific T cell responses. Improved
understanding of the role of the immune system in controlling tumor growth and recent
developments in the field of nanoparticulate cancer vaccines have created a degree of
knowledge that promises to lead to favorable results with this approach in a near future.
However, there are still more limitations in the knowledge of the pathways of cancer immune
evasion rather than in the ability to develop new vaccines and immunotherapies capable of
addressing them. Notably, it is clear that preclinical model systems can be poorly predictive
of clinical challenges, with promising preclinical results often not translating into patient
survival benefits. This may be due, at least in part, to the genetic diversity of the outbred
human population, presenting highly heterogeneous malignancies, but may also reflect
qualitative differences between slow-growing clinical disease and the rapidly growing
transplantable tumors that are often used in murine models.
Tumor vaccines might have the capacity to combat already established malignant
disease, but it must be kept in mind that the success of immunological strategies will correlate
with the stage of the disease. The ideal scenario for the application of a therapeutic cancer
vaccine might be minimal residual disease with low numbers of aberrant or cancer stem cells,
where the actively induced immune mechanisms would have a fair chance to prevent tumor
relapse through reduction of circulating tumor cells and micrometastases. The determination
of the stage of disease has been prioritized over immune stage of the host. The evaluation of
individual host immunity should be considered in the selection of the most appropriate
treatment protocols.
To shift favorably the balance between anticancer effector cells and tumor-induced
immune suppression, cancer vaccines might be more effective in combination with other
therapeutic strategies (454). In theory, the combined use of different approaches to cancer
therapy may reduce the probability of resistance to emerge. Also, it might improve drug
tolerability when different approaches are used sequentially (455). The concomitant
administration of cytotoxic agents and cancer vaccines has been previously avoided due to
potential immune suppression by chemotherapeutics. However, evidences started to point out
that common chemotherapy agents positively influence adaptive and innate antitumor

Chapter 5
160
immune responses and also provide the tumor burden reduction needed for the cancer vaccine
to overcome tumor suppression (456,457). More recently, the use of metronomic
chemotherapy (low-dose chemotherapeutics at short intervals) has demonstrated to promote
tumor eradication not only exerting an antiangiogenic activity but also by stimulating
anticancer immunity and selectively eliminating immunosuppressive cells, while toxicity is
maintained to a mild and short-lived level (458). Cancer vaccines could also beneficiate from
their combination with the new targeted therapies (vemurafenib, dabrafenib and trametinib) or
the immune checkpoint inhibitor of CTLA-4 ipilimumab. There are evidences that targeted
therapies decrease production of the immunosuppressive factor IL-10 and enhance expression
of tumor-specific antigens creating a favorable environment for the action of immunotherapy
(459).
When analysing the main barriers for cancer vaccine efficacy presented on Table 1.2.,
the developed strategy might be able to revert some of them. The developed NP-based
vaccine might contribute to overcome the fact that MHC class I molecules are down-regulated
in most tumor cells and consequently they do not constitute good antigen sources for
presentation to DCs. Tumor cells can even be able to present directly their antigens to T cells
but will rather induce tolarization of T cells due to the lack of effective costimulation (75). T
cell ignorance to tumor antigens due to its “self” nature might be also reverted by the
developed strategy as it potentiate the proper activation of DCs and consequently an
immunostimulatory environment for DC-T cell contact, which might overcome the
“ignorance” threshold of T cells. Helpless CTLs are not long lasting and die early in the
absence of CD4+ helper T cells (26). The fact that NPs have great versatility in terms of the
molecules that can be loaded into their matrix allows the combination of MHC class I- and
class II-restricted peptides in the same immunization regimen, which was translated in a very
high tumor growth rate reduction.
Notwithstanding all these promising advantages of the developed antitumor vaccine,
there are still bigger challenges related to T-cell or tumor cell biology that need to be
considered. For instance, if tumor cells “edit” their immunologic print, they can avoid
immunosurveillance and recognition by activated T cells. As so, it might be necessary to use
personalized vaccination to select the antigens with higher CTL precursor frequency and
adequate the vaccine composition to the patient. Also, the sustained release over time that
polymeric NPs can provide to the entrapped molecules is pointed out as one of their
advantages for vaccine delivery. However, it is also known that the chronic presence of tumor
antigens can induce T cell exhaustion by activation of the programmed death-1 (PD-1)

General conclusions & future perspectives
161
molecule on T cells that ultimately leads to T cell death (79). As so, further studies should be
conducted in order to analyze the in vivo kinetics and biodynamics of the antigens and
adjuvants entrapped in biodegradable NPs. The tumor microenvironment in solid tumors is an
immunoregulatory network composed by malignant cells surrounded by a tumor-conditioned
stroma that contains extracellular matrix and a variety of nonmalignant populations, including
myeloid cells, lymphocytes, fibroblasts, and endothelial cells (71). It is likely that successful
antitumor immunotherapeutic strategies encompass the targeting of the immune cells within
the tumor microenvironment to revert their state of anergy and tumor tolerance and effectively
destroy tumor cells. These strategies might include the combination of NP functionalization
with tumor-specific targeting ligands or antibodies along with the delivery of
immunostimulatory molecules.
In spite of the great advances in vaccinology over the last century, a number of
problems still remain. There are a considerable number of infectious diseases for which an
effective vaccine has not been developed such as HIV, malaria and tuberculosis. Also, the
currently approved vaccines have weaknesses in several aspects: (i) inadequacy for specific
populations such as newborns, elderly and immunocompromised individuals; (ii) long-term
maintenance of both effector and central memory; (iii) adjuvants capable of selectively
stimulating distinct cell types and induce the immune response with the desired profile; and
(iv) alternative immunization routes with feasibility for massive vaccination of populations,
namely in subdeveloped countries, such as mucosal immunization. Recent developments in
the field of particulate delivery systems has alerted to their great potential as
immunomodulation tools not only for cancer therapy but also for vaccination against
infectious diseases and other diseases where the immune system plays a prominent role, such
allergy and autoimmune diseases. The long history of use of aliphatic polyesters for
biomedical applications has proven their safety and flexibility for modification and
functionalization. Particulate vaccine delivery systems based on these polymers have
demonstrated promising results and several advantages over the currently available strategies.
Nevertheless, several questions remain unanswered and difficulties need to be overcome.
These difficulties are both related to the vaccine development process - need for validated
methods to clinically evaluate new vaccine strategies, selection of most relevant antigens and
epitopes, determination of the optimal target population, doses and schedules - as well as with
the particulate delivery systems – determination of whole body and cellular pharmacokinetics,
stability, drug release rates and translation to clinic.

Chapter 5
162
The present research work reports the successful development of biodegradable
polymeric NPs with great potential to be used as a vaccine delivery system. It was possible to
prove this concept in a cancer immunotherapy strategy by using the developed biodegradable
polymeric NPs to target TAAs and TLR ligands to APCs and induce an antitumor immune
response. The present work contributes with valuable information on several factors that can
affect the performance of a vaccine, such as (i) the fact that coentrapment of antigens and
immunoadjuvants in NPs demonstrated to be essential to induce a long lasting Th1 immune
response; (ii) the fact that mannose functionalization of NPs improved NP performance in
terms of the desired Th1/antitumor immune response; (iii) the fact that the immunoadjuvants
CpG and Poly(I:C) demonstrated to exert a synergistic effect when coentrapped in NPs; (iv)
the fact that immunization with two MHC class I-restricted MAAs in separated man-NPs was
slightly superior comparing to when only one antigen was used; (v) the fact that the use of
two MHC class I-restricted MAAs coentrapped in the same man-NPs demonstrated to have a
detrimental effect on the antitumor immune response; and (vi) the fact that the combination of
man-NPs containing either a MHC class I- or class II-restricted MAAs along with both
immunoadjuvants demonstrated the most potent antitumor immune response, suggesting the
importance of the activation of both CD4+ and CD8+ T cells in the efficacy of the antitumor
immune response. This information will certainly contribute to the improvement of the
development of novel immunization approaches, not only for cancer therapy but also for other
diseases where the immune system plays a central role. The field of particulate vaccine
delivery systems is rapidly evolving moment by moment and those based on aliphatic
polyester polymers are closer to make a major contribution to improved vaccinology, cancer
therapy and immunotherapy in the 21st century.

References
163
References
1. Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462(7272):449–60.
2. Duncan R, Gaspar R. Nanomedicine (s) under the Microscope. Mol Pharm. 2011;8(6):2101–41.
3. Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, et al. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine. 2008;26(13):1626–37.
4. Begley J, Ribas A. Targeted therapies to improve tumor immunotherapy. Clin Cancer Res. 2008;14(14):4385–91.
5. Dimberu PM, Leonhardt RM. Cancer immunotherapy takes a multi-faceted approach to kick the immune system into gear. Yale J Biol Med. 2011;84(4):371–80.
6. Speiser DE, Romero P. Molecularly defined vaccines for cancer immunotherapy, and protective T cell immunity. Semin Immunol. 2010;22(3):144–54.
7. Henderson RA, Mossman S, Nairn N, Cheever MA. Cancer vaccines and immunotherapies: emerging perspectives. Vaccine. 2005;23(17-18):2359–62.
8. Schlom J. Therapeutic Cancer Vaccines: Current Status and Moving Forward. JNCI J Natl Cancer Inst. 2012;104(8):599–613.
9. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
10. Markovic SN, Erickson LA, Rao RD, McWilliams RR, Kottschade LA, Creagan ET, et al. Malignant melanoma in the 21st century, part 1: epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin Proc. 2007;82(3):364–80.
11. NCCN. NCCN Guidelines for Patients - Melanoma, Version 1.2014. National Comprehensive Cancer Network; 2014.
12. Melanoma Skin Cancer [Internet]. American Cancer Society. 2014. Obtido de: http://www.cancer.org/cancer/skincancer-melanoma/detailedguide/melanoma-skin-cancer-key-statistics
13. Markovic SN, Erickson LA, Rao RD, Weenig RH, Pockaj BA, Bardia A, et al. Malignant melanoma in the 21st century, part 2: staging, prognosis, and treatment. Mayo Clin Proc. 2007;82(4):490–13.
14. Yang AS, Chapman PB. The history and future of chemotherapy for melanoma. Hematol Oncol Clin North Am. 2009;23(3):583–97.
15. Olszanski AJ. Current and future roles of targeted therapy and immunotherapy in advanced melanoma. J Manag Care Pharm JMCP. 2014;20(4):346–56.
16. Benlalam H, Labarrière N, Linard B, Derré L, Diez E, Pandolfino MC, et al. Comprehensive analysis of the frequency of recognition of melanoma associated antigen (MAA) by CD8

164
melanoma infiltrating lymphocytes (TIL): implications for immunotherapy. Eur J Immunol. 2001;31(7):2007–15.
17. Barnetson R, Halliday G. Regression in skin tumours: a common phenomenon. Australas J Dermatol. 1997;38(Suppl 1):S63–65.
18. Rao UN, Lee SJ, Luo W, Mihm MC, Kirkwood JM. Presence of tumor-infiltrating lymphocytes and a dominant nodule within primary melanoma are prognostic factors for relapse-free survival of patients with thick (t4) primary melanoma: pathologic analysis of the e1690 and e1694 intergroup trials. Am J Clin Pathol. 2010;133(4):646–53.
19. Yaziji H, Gown AM. Immunohistochemical markers of melanocytic tumors. Int J Surg Pathol. 2003;11(1):11–5.
20. Nicholas C, Lesinski GB. Immunomodulatory cytokines as therapeutic agents for melanoma. Immunotherapy. 2011;3(5):673–90.
21. Vujanovic L, Butterfield LH. Melanoma cancer vaccines and anti-tumor T cell responses. J Cell Biochem. 2007;102(2):301–10.
22. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15.
23. Weiss SA, Chandra S, Pavlick AC. Update on vaccines for high-risk melanoma. Curr Treat Options Oncol. 2014;15(2):269–80.
24. Huang Y, Shah S, Qiao L. Tumor resistance to CD8+ T cell-based therapeutic vaccination. Arch Immunol Ther Exp Warsz. 2007;55(4):205–17.
25. Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood. 2010;115(9):1678–9.
26. Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti tumor immune responses. Immunol Rev. 2008;222(1):129–44.
27. Walsh KP, Mills KHG. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. Novembro de 2013;34(11):521–30.
28. Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med. 1999;190(5):617–27.
29. Becker Y. Molecular immunological approaches to biotherapy of human cancers-a review, hypothesis and implications. Anticancer Res. 2006;26(2A):1113–34.
30. Gilboa E. The promise of cancer vaccines. Nat Rev Cancer. 2004;4(5):401–11.
31. Rossi M, Young JW. Human dendritic cells: potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J Immunol. 2005;175(3):1373–81.
32. Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113(15):3418–27.
33. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–20.

References
165
34. Gajewski TF, Fuertes MB, Woo S-R. Innate immune sensing of cancer: clues from an identified role for type I IFNs. Cancer Immunol Immunother. 2012;61(8):1343–7.
35. Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307(5715):1630–4.
36. Kadowaki N. Dendritic cells: a conductor of T cell differentiation. Allergol Int. 2007;56(3):193–9.
37. Martín-Fontecha A, Sebastiani S, Höpken UE, Uguccioni M, Lipp M, Lanzavecchia A, et al. Regulation of Dendritic Cell Migration to the Draining Lymph Node. J Exp Med. 2003;198(4):615–21.
38. Palucka AK, Ueno H, Fay J, Banchereau J. Dendritic cells: a critical player in cancer therapy? J Immunother. 2008;31(9):793–805.
39. Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–96.
40. Raghavan M, Del Cid N, Rizvi SM, Peters LR. MHC class I assembly: out and about. Trends Immunol. 2008;29(9):436–43.
41. Hwang I, Ki DL. Receptor-mediated T cell absorption of antigen presenting cell-derived molecules. Front Biosci. 2011;16:411–21.
42. Takeuchi S, Furue M. Dendritic cells: ontogeny. Allergol Int. 2007;56(3):215–23.
43. Muranski P, Restifo NP. Adoptive immunotherapy of cancer using CD4+ T cells. Curr Opin Immunol. 2009;21(2):200–8.
44. Verardi PH, Titong A, Hagen CJ. A vaccinia virus renaissance: new vaccine and immunotherapeutic uses after smallpox eradication. Hum Vaccin Immunother. 2012;8(7):961–70.
45. Ravanfar P, Satyaprakash A, Creed R, Mendoza N. Existing antiviral vaccines. Dermatol Ther. 2009;22(2):110–28.
46. Riedel S. Edward Jenner and the history of smallpox and vaccination. Proc Bayl Univ Med Cent. 2005;18(1):21–5.
47. Plotkin SA. History of Vaccine Development. Plotkin SA, editor. New York, NY: Springer New York; 2011. 364 p.
48. Waldmann TA. Immunotherapy: past, present and future. Nat Med. 2003;9(3):269–77.
49. Lundin JI, Checkoway H. Endotoxin and cancer. Env Health Perspecs. 2009;117(9):1344–50.
50. Vacchelli E, Martins I, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial watch: Peptide vaccines in cancer therapy. Oncoimmunology. 2012;1(9):1557–76.
51. Malyala P, O’Hagan DT, Singh M. Enhancing the therapeutic efficacy of CpG oligonucleotides using biodegradable microparticles. Adv Drug Deliv Rev. 2009;61(3):218–25.
52. Krishnamachari Y, Geary SM, Lemke CD, Salem AK. Nanoparticle Delivery Systems in Cancer Vaccines. Pharma Res. 2011;28(2):215–36.

166
53. Matera L. The choice of the antigen in the dendritic cell-based vaccine therapy for prostate cancer. Cancer Treat Rev. 2010;36(2):131–41.
54. Van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde BJ, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. J Immunol. 2007;178(5):2617–21.
55. Novellino L, Castelli C, Parmiani G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother. 2005;54(3):187–207.
56. Zhao L, Liu Z, Fan D. Overview of mimotopes and related strategies in tumor vaccine development. Expert Rev Vaccines. 2008;7(10):1547–55.
57. Parmiani G, Castelli C, Dalerba P, Mortarini R, Rivoltini L, Marincola FM, et al. Cancer immunotherapy with peptide-based vaccines: what have we achieved? Where are we going? J Natl Cancer Inst. 2002;94(11):805–18.
58. Higgins JP, Bernstein MB, Hodge JW. Enhancing immune responses to tumor-associated antigens. Cancer Biol Ther. 2009;8(15):1440–9.
59. Pejawar-Gaddy S, Rajawat Y, Hilioti Z, Xue J, Gaddy DF, Finn OJ, et al. Generation of a tumor vaccine candidate based on conjugation of a MUC1 peptide to polyionic papillomavirus virus-like particles. Cancer Immunol Immunother. 2010;59(11):1685–96.
60. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84.
61. Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13(20):5991–4.
62. Liao Z-L, Tang X-D, Lü M-H, Wu Y-Y, Cao Y-L, Fang D-C, et al. Antitumor effect of new multiple antigen peptide based on HLA-A0201-restricted CTL epitopes of human telomerase reverse transcriptase (hTERT). Cancer Sci. 2012;103(11):1920–8.
63. Parmiani G, Russo V, Marrari A, Cutolo G, Casati C, Pilla L, et al. Universal and stemness-related tumor antigens: potential use in cancer immunotherapy. Clin Cancer Res. 2007;13(19):5675–9.
64. Anichini A, Molla A, Mortarini R, Tragni G, Bersani I, Di Nicola M, et al. An expanded peripheral T cell population to a cytotoxic T lymphocyte (CTL)-defined, melanocyte-specific antigen in metastatic melanoma patients impacts on generation of peptide-specific CTLs but does not overcome tumor escape from immune surveillance in metastatic lesions. J Exp Med. 1999;190(5):651–67.
65. Maeda Y, Hida N, Niiya F, Katagiri K, Harada M, Yamana H, et al. Detection of peptide-specific CTL-precursors in peripheral blood lymphocytes of cancer patients. Br J Cancer. 2002;87(7):796–804.
66. Noguchi M, Sasada T, Itoh K. Personalized peptide vaccination: a new approach for advanced cancer as therapeutic cancer vaccine. Cancer Immunol Immunother. 2012;62(5):919–29.
67. Sensi M, Anichini A. Unique tumor antigens: evidence for immune control of genome integrity and immunogenic targets for T cell-mediated patient-specific immunotherapy. Clin Cancer Res. 2006;12(17):5023–32.

References
167
68. Itoh K, Yamada A. Personalized peptide vaccines: a new therapeutic modality for cancer. Cancer Sci. 2006;97(10):970–6.
69. Poland GA, Kennedy RB, Ovsyannikova IG. Vaccinomics and Personalized Vaccinology: Is Science Leading Us Toward a New Path of Directed Vaccine Development and Discovery? Rall GF, editor. PLoS Pathog. 2011;7(12):e1002344.
70. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. 2013;25(2):214–21.
71. Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45.
72. Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy–revisited. Nat Rev Drug Discov. 2011;10(8):591–600.
73. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang K-M, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. 2012;42(12):3174–9.
74. Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest. 2013;123(3):1044–56.
75. Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol. 2004;22(6):1136–51.
76. Ray S, Chhabra A, Mehrotra S, Chakraborty NG, Ribas A, Economou J, et al. Obstacles to and opportunities for more effective peptide-based therapeutic immunization in human melanoma. Clin Dermatol. 2009;27(6):603–13.
77. Prado-Garcia H, Romero-Garcia S, Aguilar-Cazares D, Meneses-Flores M, Lopez-Gonzalez JS. Tumor-induced CD8+ T-cell dysfunction in lung cancer patients. Clin Dev Immunol. 2012;2012:741741.
78. Mehrotra S, Chhabra A, Chattopadhyay S, Dorsky DI, Chakraborty NG, Mukherji B. Rescuing melanoma epitope-specific cytolytic T lymphocytes from activation-induced cell death, by SP600125, an inhibitor of JNK: implications in cancer immunotherapy. J Immunol. 2004;173(10):6017–24.
79. Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia-specific T cells results in T-cell exhaustion and disease progression. Blood. 2009;114(8):1528–36.
80. Zielinski C, Knapp S, Mascaux C, Hirsch F. Rationale for targeting the immune system through checkpoint molecule blockade in the treatment of non-small-cell lung cancer. Ann Oncol. 2013;24(5):1170–9.
81. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105–15.
82. Morita R, Hirohashi Y, Sato N. Depletion of Tregs in vivo: a promising approach to enhance antitumor immunity without autoimmunity. Immunotherapy. 2012;4(11):1103–5.

168
83. Whiteside TL. Disarming suppressor cells to improve immunotherapy. Cancer Immunol Immunother. 2012;61(2):283–8.
84. Zarour HM, Kirkwood JM, Kierstead LS, Herr W, Brusic V, Slingluff CL Jr, et al. Melan-A/MART-1(51-73) represents an immunogenic HLA-DR4-restricted epitope recognized by melanoma-reactive CD4(+) T cells. Proc Natl Acad Sci U A. 2000;97(1):400–5.
85. Kerkar SP, Sanchez-Perez L, Yang S, Borman ZA, Muranski P, Ji Y, et al. Genetic engineering of murine CD8+ and CD4+ T cells for preclinical adoptive immunotherapy studies. J Immunother. 2011;34(4):343–52.
86. Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, et al. Enhanced and prolonged cross presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88.
87. Bukur J, Jasinski S, Seliger B. The role of classical and non-classical HLA class I antigens in human tumors. Semin Cancer Biol. 2012;22(4):350–8.
88. Bubeník J. MHC class I down-regulation: tumour escape from immune surveillance? (review). Int J Oncol. 2004;25(2):487–91.
89. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Ann Rev Immunol. 2007;25:267–96.
90. Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U A. 2012;109(41):16618–23.
91. Nawrocki S, Mackiewicz A. Genetically modified tumour vaccines--where we are today. Cancer Treat Rev. 1999;25(1):29–46.
92. Taddei ML, Giannoni E, Comito G, Chiarugi P. Microenvironment and tumor cell plasticity: An easy way out. Cancer Lett. 2013;341(1):80–96.
93. Rivoltini L, Carrabba M, Huber V, Castelli C, Novellino L, Dalerba P, et al. Immunity to cancer: attack and escape in T lymphocyte–tumor cell interaction. Immunol Rev. 2002;188(1):97–113.
94. Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, et al. Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol. 1995;154(8):3961.
95. Labarriere N, Pandolfino MC, Raingeard D, Le Guiner S, Diez E, Le Drean E, et al. Frequency and relative fraction of tumor antigen specific T cells among lymphocytes from melanoma invaded lymph nodes. Int J Cancer. 1998;78(2):209–15.
96. Benlalam H, Linard B, Guilloux Y, Moreau-Aubry A, Derré L, Diez E, et al. Identification of five new HLA-B* 3501-restricted epitopes derived from common melanoma-associated antigens, spontaneously recognized by tumor-infiltrating lymphocytes. J Immunol. 2003;171(11):6283–9.
97. Rivoltini L, Squarcina P, Loftus DJ, Castelli C, Tarsini P, Mazzocchi A, et al. A superagonist variant of peptide MART1/Melan A27–35 elicits anti-melanoma CD8+ T cells with enhanced functional characteristics: implication for more effective immunotherapy. Cancer Res. 1999;59(2):301–6.

References
169
98. Valmori D, Fonteneau JF, Lizana CM, Gervois N, Liénard D, Rimoldi D, et al. Enhanced generation of specific tumor-reactive CTL in vitro by selected Melan-A/MART-1 immunodominant peptide analogues. J Immunol. 1998;160(4):1750–8.
99. Blanchet JS, Valmori D, Dufau I, Ayyoub M, Nguyen C, Guillaume P, et al. A new generation of Melan-A/MART-1 peptides that fulfill both increased immunogenicity and high resistance to biodegradation: implication for molecular anti-melanoma immunotherapy. J Immunol. 2001;167(10):5852–61.
100. Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med. 1998;4(3):321–7.
101. Borbulevych OY, Baxter TK, Yu Z, Restifo NP, Baker BM. Increased immunogenicity of an anchor-modified tumor-associated antigen is due to the enhanced stability of the peptide/MHC complex: implications for vaccine design. J Immunol. 2005;174(8):4812–20.
102. Smith KA, Meisenburg BL, Tam VL, Pagarigan RR, Wong R, Joea DK, et al. Lymph Node–Targeted Immunotherapy Mediates Potent Immunity Resulting in Regression of Isolated or Metastatic Human Papillomavirus–Transformed Tumors. Clin Cancer Res. 2009;15(19):6167–76.
103. Walker EB, Haley D, Miller W, Floyd K, Wisner KP, Sanjuan N, et al. gp100(209–2M) Peptide Immunization of Human Lymphocyte Antigen-A2+ Stage I-III Melanoma Patients Induces Significant Increase in Antigen-Specific Effector and Long-Term Memory CD8+ T Cells. Clin Cancer Res. 2004;10(2):668–80.
104. Nawrath M, Pavlovic J, Moelling K. Synergistic effect of a combined DNA and peptide vaccine against gp100 in a malignant melanoma mouse model. J Mol Med. 2001;79(2):133–42.
105. Ginsberg BA, Gallardo HF, Rasalan TS, Adamow M, Mu Z, Tandon S, et al. Immunologic response to xenogeneic gp100 DNA in melanoma patients: comparison of particle-mediated epidermal delivery with intramuscular injection. Clin Cancer Res. 2010;16(15):4057–65.
106. Tagawa ST, Lee P, Snively J, Boswell W, Ounpraseuth S, Lee S, et al. Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with Stage IV melanoma. Cancer. 2003;98(1):144–54.
107. Bioley G, Jandus C, Tuyaerts S, Rimoldi D, Kwok WW, Speiser DE, et al. Melan-A/MART-1-Specific CD4 T Cells in Melanoma Patients: Identification of New Epitopes and Ex Vivo Visualization of Specific T Cells by MHC Class II Tetramers. J Immunol. 2006;177(10):6769–79.
108. Reynolds SR, Celis E, Sette A, Oratz R, Shapiro RL, Johnston D, et al. HLA-independent heterogeneity of CD8+ T cell responses to MAGE-3, Melan-A/MART-1, gp100, tyrosinase, MC1R, and TRP-2 in vaccine-treated melanoma patients. J Immunol. 1998;161(12):6970–6.
109. Cobb BA, Kasper DL. Coming of age: carbohydrates and immunity. Eur J Immunol. 2005;35(2):352–6.
110. De Libero G, Mori L. How the immune system detects lipid antigens. Prog Lipid Res. 2010;49(2):120–7.
111. Facciotti F, Cavallari M, Angénieux C, Garcia-Alles LF, Signorino-Gelo F, Angman L, et al. Fine tuning by human CD1e of lipid-specific immune responses. Proc Natl Acad Sci USA. 2011;108(34):14228–33.

170
112. De Libero G, Collmann A, Mori L. The cellular and biochemical rules of lipid antigen presentation. Eur J Immunol. 2009;39(10):2648–56.
113. Godfrey DI, Pellicci DG, Patel O, Kjer-Nielsen L, McCluskey J, Rossjohn J. Antigen recognition by CD1d-restricted NKT T cell receptors. Semin Immunol. 2010;22(2):61–7.
114. Schiefner A, Wilson IA. Presentation of lipid antigens by CD1 glycoproteins. Curr Pharm Des. 2009;15(28):3311–7.
115. Fujii S-I, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198(2):267–79.
116. Hakomori S. Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 1996;56(23):5309–18.
117. Heimburg-Molinaro J, Lum M, Vijay G, Jain M, Almogren A, Rittenhouse-Olson K. Cancer vaccines and carbohydrate epitopes. Vaccine. 2011;29(48):8802–26.
118. Pashov A, Monzavi-Karbassi B, Kieber-Emmons T. Glycan mediated immune responses to tumor cells. Hum Vaccin. 2011;7 Suppl:156–65.
119. Monzavi-Karbassi B, Pashov A, Jousheghany F, Artaud C, Kieber-Emmons T. Evaluating strategies to enhance the anti-tumor immune response to a carbohydrate mimetic peptide vaccine. Int J Mol Med. 2006;17(6):1045–52.
120. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15(17):5323–37.
121. Vlad AM, Kettel JC, Alajez NM, Carlos CA, Finn OJ. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol. 2004;82:249–93.
122. Gaidzik N, Westerlind U, Kunz H. The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem Soc Rev. 2013;42(10):4421–42.
123. Kaiser A, Gaidzik N, Westerlind U, Kowalczyk D, Hobel A, Schmitt E, et al. A synthetic vaccine consisting of a tumor-associated sialyl-T(N)-MUC1 tandem-repeat glycopeptide and tetanus toxoid: induction of a strong and highly selective immune response. Angew Chem Int Ed Engl. 2009;48(41):7551–5.
124. Cai H, Huang Z-H, Shi L, Sun Z-Y, Zhao Y-F, Kunz H, et al. Variation of the Glycosylation Pattern in MUC1 Glycopeptide BSA Vaccines and Its Influence on the Immune Response. Angew Chem Int Ed Engl. 2012;51(7):1719–23.
125. Mazorra Z, Mesa C, Fernández A, Fernández LE. Immunization with a GM3 ganglioside nanoparticulated vaccine confers an effector CD8(+) T cells-mediated protection against melanoma B16 challenge. Cancer Immunol Immunother. 2008;57(12):1771–80.
126. O’Hagan DT, De Gregorio E. The path to a successful vaccine adjuvant--’the long and winding road’. Drug Discov Today. 2009;14(11-12):541–51.
127. Kool M, Fierens K, Lambrecht BN. Alum adjuvant: some of the tricks of the oldest adjuvant. J Med Microbiol. 2012;61(Pt 7):927–34.

References
171
128. Mbow ML, De Gregorio E, Valiante NM, Rappuoli R. New adjuvants for human vaccines. Curr Opin Immunol. 2010;22(3):411–6.
129. Wilschut J. Influenza vaccines: the virosome concept. Immunol Lett. 2009;122(2):118–21.
130. Morel S, Didierlaurent A, Bourguignon P, Delhaye S, Baras B, Jacob V, et al. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine. 2011;29(13):2461–73.
131. Didierlaurent AM, Morel S, Lockman L, Giannini SL, Bisteau M, Carlsen H, et al. AS04, an aluminum salt- and TLR4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J Immunol. 2009;183(10):6186–97.
132. Vandepapelière P, Horsmans Y, Moris P, Van Mechelen M, Janssens M, Koutsoukos M, et al. Vaccine adjuvant systems containing monophosphoryl lipid A and QS21 induce strong and persistent humoral and T cell responses against hepatitis B surface antigen in healthy adult volunteers. Vaccine. 2008;26(10):1375–86.
133. Aucouturier J, Dupuis L, Ganne V. Adjuvants designed for veterinary and human vaccines. Vaccine. 2001;19(17-19):2666–72.
134. Thompson BS, Chilton PM, Ward JR, Evans JT, Mitchell TC. The low-toxicity versions of LPS, MPL adjuvant and RC529, are efficient adjuvants for CD4+ T cells. J Leukoc Biol. 2005;78(6):1273–80.
135. Schellack C, Prinz K, Egyed A, Fritz JH, Wittmann B, Ginzler M, et al. IC31, a novel adjuvant signaling via TLR9, induces potent cellular and humoral immune responses. Vaccine. 2006;24(26):5461–72.
136. Halperin SA, McNeil S, Langley JM, Smith B, MacKinnon-Cameron D, McCall-Sani R, et al. Safety and immunogenicity of different two-dose regimens of an investigational hepatitis B vaccine (hepatitis B surface antigen co-administered with an immunostimulatory phosphorothioate oligodeoxyribonucleotide) in healthy young adults. Vaccine. 2012;30(36):54458.
137. Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117(5):1184–94.
138. Yuzhalin AE, Kutikhin AG. Interleukin-12: clinical usage and molecular markers of cancer susceptibility. Growth Factors. 2012;30(3):176–91.
139. Pichichero ME. Improving vaccine delivery using novel adjuvant systems. Hum Vaccin. 2008;4(4):262–70.
140. Chuang C-M, Monie A, Hung C-F, Wu T-C. Treatment with imiquimod enhances antitumor immunity induced by therapeutic HPV DNA vaccination. J Biomed Sci. 2010;17:32.
141. Wu JJ, Huang DB, Tyring SK. Resiquimod: a new immune response modifier with potential as a vaccine adjuvant for Th1 immune responses. Antivir Res. 2004;64(2):79–83.
142. Nicodemus CF, Berek JS. TLR3 agonists as immunotherapeutic agents. Immunotherapy. 2010;2(2):137–40.
143. Mizel SB, Bates JT. Flagellin as an adjuvant: cellular mechanisms and potential. J Immunol. 2010;185(10):5677–82.

172
144. Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420(1):1–16.
145. Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunolo Rev. 2009;227(1):248–63.
146. Wesch D, Peters C, Oberg H-H, Pietschmann K, Kabelitz D. Modulation of γδ T cell responses by TLR ligands. Cell Mol Life Sci. 2011;68(14):2357–70.
147. Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann Rev Immunol. 2003;21(1):335–76.
148. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EEM, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201(9):1435–46.
149. Sariol CA, Martínez MI, Rivera F, Rodríguez IV, Pantoja P, Abel K, et al. Decreased dengue replication and an increased anti-viral humoral response with the use of combined Toll-like receptor 3 and 7/8 agonists in macaques. PloS One. 2011;6(4):e19323.
150. Duthie MS, Windish HP, Fox CB, Reed SG. Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev. 2011;239(1):178–96.
151. Liu G, Zhang L, Zhao Y. Modulation of immune responses through direct activation of Toll like receptors to T cells. Clin Exp Immunol. 2010;160(2):168–75.
152. Avalos AM, Latz E, Mousseau B, Christensen SR, Shlomchik MJ, Lund F, et al. Differential cytokine production and bystander activation of autoreactive B cells in response to CpG-A and CpG-B oligonucleotides. J Immunol. 2009;183(10):6262–8.
153. Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol. 2004;34(1):251–62.
154. Agrawal S, Kandimalla E. Synthetic agonists of Toll-like receptors 7, 8 and 9. Bioch Soc Trans. 2007;35(6):1461–7.
155. Barata T, Teo I, Lalwani S, Simanek E, Zloh M, Shaunak S. Computational design principles for bioactive dendrimer based constructs as antagonists of the TLR4-MD-2-LPS complex. Biomaterials. 2011;32(33):8702–11.
156. Barata TS, Teo I, Brocchini S, Zloh M, Shaunak S. Partially glycosylated dendrimers block MD-2 and prevent TLR4-MD-2-LPS complex mediated cytokine responses. PLoS Comput Biol. 2011;7(6):e1002095.
157. Goldinger SM, Dummer R, Baumgaertner P, Mihic-Probst D, Schwarz K, Hammann-Haenni A, et al. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8+ T-cell responses in melanoma patients. Eur J Immunol. 2012;42(11):3049–61.
158. Link BK, Ballas ZK, Weisdorf D, Wooldridge JE, Bossler AD, Shannon M, et al. Oligodeoxynucleotide CpG 7909 delivered as intravenous infusion demonstrates immunologic modulation in patients with previously treated non-Hodgkin lymphoma. J Immunother. 2006;29(5):558–68.
159. Carpentier A, Laigle-Donadey F, Zohar S, Capelle L, Behin A, Tibi A, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006;8(1):60–6.

References
173
160. Manegold C, Gravenor D, Woytowitz D, Mezger J, Hirsh V, Albert G, et al. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(24):3979–86.
161. Engel AL, Holt GE, Lu H. The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Exp Rev Clin Pharmacol. 2011;4(2):275–89.
162. Warger T, Osterloh P, Rechtsteiner G, Fassbender M, Heib V, Schmid B, et al. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood. 2006;108(2):544–50.
163. Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470(7335):543–7.
164. Tross D, Petrenko L, Klaschik S, Zhu Q, Klinman DM. Global changes in gene expression and synergistic interactions induced by TLR9 and TLR3. Mol Immunol. 2009;46(13):2557–64.
165. Lee YR, Lee YH, Im SA, Yang IH, Ahn GW, Kim K, et al. Biodegradable nanoparticles containing TLR3 or TLR9 agonists together with antigen enhance MHC-restricted presentation of the antigen. Arch Pharm Res. 2010;33(11):1859–66.
166. Stone GW, Barzee S, Snarsky V, Santucci C, Tran B, Langer R, et al. Nanoparticle-delivered multimeric soluble CD40L DNA combined with Toll-Like Receptor agonists as a treatment for melanoma. PLoS One. 2009;4(10):e7334.
167. Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, et al. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26(39):5046–57.
168. Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27(23):3013–21.
169. Diwan M, Tafaghodi M, Samuel J. Enhancement of immune responses by co-delivery of a CpG oligodeoxynucleotide and tetanus toxoid in biodegradable nanospheres. J Control Release. 2002;85(1-3):247–62.
170. Slütter B, Jiskoot W. Dual role of CpG as immune modulator and physical crosslinker in ovalbumin loaded N-trimethyl chitosan (TMC) nanoparticles for nasal vaccination. J Control Release. 2010;148(1):117–21.
171. Florindo HF, Pandit S, Gonçalves L, Videira M, Alpar O, Almeida AJ. Antibody and cytokine-associated immune responses to S. equi antigens entrapped in PLA nanospheres. Biomaterials. 2009;30(28):5161–9.
172. Clawson C, Huang CT, Futalan D, Martin Seible D, Saenz R, Larsson M, et al. Delivery of a peptide via poly (d, l-lactic-co-glycolic) acid nanoparticles enhances its dendritic cell-stimulatory capacity. Nanomed. 2010;6(5):651–61.
173. Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55(3):237–45.

174
174. Gao Q, Qiu S-J, Fan J, Zhou J, Wang X-Y, Xiao Y-S, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol. 2007;25(18):2586–93.
175. Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, et al. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J Immunol. 2011;186(4):1963–9.
176. Oberg H-H, Juricke M, Kabelitz D, Wesch D. Regulation of T cell activation by TLR ligands. Eur J Cell Biol. 2011;90(6-7):582–92.
177. Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell–mediated CD8 tolerance. Nat Immunol. 2004;5(5):508–15.
178. Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197(4):403–11.
179. Chen Q, Davidson TS, Huter EN, Shevach EM. Engagement of TLR2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J Immunol. 2009;183(7):4458–66.
180. Olivier A, Sainz-Perez A, Dong H, Sparwasser T, Majlessi L, Leclerc C. The adjuvant effect of TLR agonists on CD4(+) effector T cells is under the indirect control of regulatory T cells. Eur J Immunol. 2011;41(8):2303–13.
181. Tanchot C, Terme M, Pere H, Tran T, Benhamouda N, Strioga M, et al. Tumor-Infiltrating Regulatory T Cells: Phenotype, Role, Mechanism of Expansion In Situ and Clinical Significance. Cancer Microenviron. 2012;6(2):147–57.
182. Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, et al. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. 2011;17(7):1765–75.
183. Arens R, Schoenberger SP. Plasticity in programming of effector and memory CD8+ T cell formation. Immunolo Rev. 2010;235(1):190–205.
184. Kawai T, Akira S. TLR signaling. 2007;19(1):24–32.
185. Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10(11):787–96.
186. Danhier F, Ansorena E, Silva, JM. J, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: An overview of biomedical applications. J Control Release. 2012;161(2):505–22.
187. Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55(3):329–47.
188. Damm J, Wiegand F, Harden LM, Gerstberger R, Rummel C, Roth J. Fever, sickness behavior, and expression of inflammatory genes in the hypothalamus after systemic and localized subcutaneous stimulation of rats with the Toll-like receptor 7 agonist imiquimod. Neuroscience. 2012;201:166–83.

References
175
189. Tacken PJ, Zeelenberg IS, Cruz LJ, van Hout-Kuijer MA, van de Glind G, Fokkink RG, et al. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood. 2011;118(26):6836–44.
190. Nguyen DN, Mahon KP, Chikh G, Kim P, Chung H, Vicari AP, et al. Lipid-derived nanoparticles for immunostimulatory RNA adjuvant delivery. Proc Natl Acad Sci USA. 2012;109(14):E797–803.
191. Jain RA, Rhodes CT, Railkar AM, Malick AW, Shah NH. Controlled release of drugs from injectable in situ formed biodegradable PLGA microspheres: effect of various formulation variables. Eur J Pharm Biopharm. 2000;50(2):257–62.
192. Fernandez-Megia E, Novoa-Carballal R, Quiñoá E, Riguera R. Conjugation of bioactive ligands to PEG-grafted chitosan at the distal end of PEG. Biomacromolecules. 2007;8(3):833–42.
193. Silva JM, Videira M, Gaspar R, Préat V, Florindo HF. Immune system targeting by biodegradable nanoparticles for cancer vaccines. J Control Release. 2013;168(2):179–99.
194. Pulapura S, Kohn J. Trends in the development of bioresorbable polymers for medical applications. J Biomater Appl. 1992;6(3):216–50.
195. Beck LR, Cowsar DR, Lewis DH, Cosgrove RJ Jr, Riddle CT, Lowry SL, et al. A new long-acting injectable microcapsule system for the administration of progesterone. Fertil Steril. 1979;31(5):545–51.
196. Benita S, Benoit JP, Puisieux F, Thies C. Characterization of drug-loaded poly(d,l-lactide) microspheres. J Pharm Sci. 1984;73(12):1721–4.
197. Pitt CG, Marks TA, Schindler A. Biodegradable drug delivery systems based on aliphatic polyesters: application to contraceptives and narcotic antagonists. NIDA Res Monogr. 1981;28:232–53.
198. Juni K, Ogata J, Nakano M, Ichihara T, Mori K, Akagi M. Preparation and evaluation in vitro and in vivo of polylactic acid microspheres containing doxorubicin. Chem Pharm Bull. 1985;33(1):313–8.
199. Wakiyama N, Juni K, Nakano M. Preparation and evaluation in vitro of polylactic acid microspheres containing local anesthetics. Chem Pharm Bull. 1981;29(11):3363–8.
200. Guiziou B, Armstrong DJ, Elliott PN, Ford JL, Rostron C. Investigation of in-vitro release characteristics of NSAID-loaded polylactic acid microspheres. J Microencapsul. 1996;13(6):701–8.
201. Corrigan OI, Li X. Quantifying drug release from PLGA nanoparticulates. Eur J Pharm Sci. 2009;37(3-4):477–85.
202. Okada H, Doken Y, Ogawa Y, Toguchi H. Preparation of three-month depot injectable microspheres of leuprorelin acetate using biodegradable polymers. Pharma Res. 1994;11(8):1143–7.
203. Isurugi K, Niijima T, Akaza H, Aso Y, Koiso K, Fuse M, et al. Treatment of prostatic cancer with TAP-144-SR, a depot preparation of LH-RH agonist (leuprolide). J Chemother. 1989;1(4 Suppl):1237–41.

176
204. Nicholson RI, Walker KJ, Maynard PV. Anti-tumour potential of a new luteinizing hormone releasing hormone analogue, ICI 118630. Eur J Cancer. 1980;Suppl 1:295–9.
205. O’Hagan DT, Rahman D, McGee JP, Jeffery H, Davies MC, Williams P, et al. Biodegradable microparticles as controlled release antigen delivery systems. Immunology. 1991;73(2):239–42.
206. Lo RY. The development of subunit and synthetic vaccines using recombinant DNA technology. Biotechnol Adv. 1987;5(2):235–56.
207. Hudson D, Margaritis A. Biopolymer nanoparticle production for controlled release of biopharmaceuticals. Crit Rev Biotechnol. 2013;34(2):161–79.
208. Ali M, Brocchini S. Synthetic approaches to uniform polymers. Adv Drug Deliv Rev. 2006;58(15):1671–87.
209. Ulery BD, Nair LS, Laurencin CT. Biomedical applications of biodegradable polymers. J Polym Sci Polym Phys. 2011;49(12):832–64.
210. Seyednejad H, Ghassemi AH, van Nostrum CF, Vermonden T, Hennink WE. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J Control Release. 2011;152(1):168–76.
211. Athanasiou KA, Niederauer GG, Agrawal C. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials. 1996;17(2):93–102.
212. Hyon SH, Jamshidi K, Ikada Y. Synthesis of polylactides with different molecular weights. Biomaterials. 1997;18(22):1503–8.
213. Albertsson A-C, Varma IK. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules. 2003;4(6):1466–86.
214. Thomas CM. Stereocontrolled ring-opening polymerization of cyclic esters: synthesis of new polyester microstructures. Chem Soc Rev. 2010;39(1):165–73.
215. Kobayashi S, Uyama H, Kimura S. Enzymatic polymerization. Chem Rev. 2001;101(12):3793–818.
216. Dordick JS. Enzymatic and chemoenzymatic approaches to polymer synthesis. Trends Biotechnol. 1992;10(8):287–90.
217. Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem Rev. 2004;104(12):6147–76.
218. Platel R, Hodgson L, Williams C. Biocompatible Initiators for Lactide Polymerization. Polym Rev. 2008;48(1):11–63.
219. Kricheldorf HR. Syntheses and application of polylactides. Chemosphere. 2001;43(1):49–54.
220. Labet M, Thielemans W. Synthesis of polycaprolactone: a review. Chem Soc Rev. 2009;38(12):3484–504.
221. Huatan H, Collett JH, Attwood D, Booth C. Preparation and characterization of poly(epsilon-caprolactone) polymer blends for the delivery of proteins. Biomaterials. 1995;16(17):1297–303.

References
177
222. Abe H, Doi Y. Structural effects on enzymatic degradabilities for poly[(R)-3-hydroxybutyric acid] and its copolymers. Int J Biol Macromol. 1999;25(1-3):185–92.
223. Qian H, Wohl AR, Crow JT, Macosko CW, Hoye TR. A Strategy for Control of «Random» Copolymerization of Lactide and Glycolide: Application to Synthesis of PEG-b-PLGA Block Polymers Having Narrow Dispersity. Macromolecules. 2011;44(18):7132–40.
224. Peter SJ, Yaszemski MJ, Suggs LJ, Payne RG, Langer R, Hayes WC, et al. Characterization of partially saturated poly(propylene fumarate) for orthopaedic application. J Biomater SCi Polym Ed. 1997;8(11):893–904.
225. Liu J, Chen P, Li J, Jiang S-H, Jiang Z-Q, Gu Q. Synthesis of poly(ethylene adipate-co-l-lactic acid) copolymers via ring opening polymerization. Polym Bull. 2011;66(2):187–97.
226. Lee CW, Urakawa R, Kimura Y. Copolymerization of γ-valerolactone and β-butyrolactone. Eur Polym J. 1998;34(1):117–22.
227. Freichels H, Pourcelle V, Auzély-Velty R, Marchand-Brynaert J, Jérôme C. Synthesis of poly(lactide-co-glycolide-co-ε-caprolactone)-graft-mannosylated poly(ethylene oxide) copolymers by combination of «clip» and «click» chemistries. Biomacromolecules. 2012;13(3):760–8.
228. Lasprilla AJR, Martinez GAR, Lunelli BH, Jardini AL, Filho RM. Poly-lactic acid synthesis for application in biomedical devices - a review. Biotechnol Adv. 2012;30(1):321–8.
229. Shao J, Sun J, Bian X, Cui Y, Li G, Chen X. Investigation of poly(lactide) stereocomplexes: 3-armed poly(L-lactide) blended with linear and 3-armed enantiomers. J Phys Chem B. 2012;116(33):9983–91.
230. Gunatillake P, Mayadunne R, Adhikari R. Recent developments in biodegradable synthetic polymers. Biotechnol Annu Rev. 2006;12:301–47.
231. Woodruff MA, Hutmacher DW. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog Polym Sci. 2010;35(10):1217–56.
232. Francis L, Meng D, Knowles J, Keshavarz T, Boccaccini AR, Roy I. Controlled Delivery of Gentamicin Using Poly(3-hydroxybutyrate) Microspheres. Int J Mol Sci. 2011;12(7):4294–314.
233. Errico C, Bartoli C, Chiellini F, Chiellini E. Poly(hydroxyalkanoates)-based polymeric nanoparticles for drug delivery. J Biomed Biotechnol. 2009;2009:571702.
234. Wróbel-Kwiatkowska M, Skórkowska-Telichowska K, Dymińska L, Maczka M, Hanuza J, Szopa J. Biochemical, mechanical, and spectroscopic analyses of genetically engineered flax fibers producing bioplastic (poly-beta-hydroxybutyrate). Biotechnol Prog. 2009;25(5):1489–98.
235. Dreifke MB, Ebraheim NA, Jayasuriya AC. Investigation of potential injectable polymeric biomaterials for bone regeneration. J Biomed Mater Res A. 2013;101(8):2436–47.
236. Mistry AS, Mikos AG, Jansen JA. Degradation and biocompatibility of a poly(propylene fumarate)-based/alumoxane nanocomposite for bone tissue engineering. J Biomed Mater Res A. 2007;83(4):940–53.
237. Malberg S, Hoglund A, Albertsson A-C. Macromolecular design of aliphatic polyesters with maintained mechanical properties and a rapid, customized degradation profile. Biomacromolecules. 2011;12(6):2382–8.

178
238. Ryner M, Albertsson A-C. Resorbable and highly elastic block copolymers from 1,5-dioxepan-2-one and L-lactide with controlled tensile properties and hydrophilicity. Biomacromolecules. 2002;3(3):601–8.
239. Pang X, Zhuang X, Tang Z, Chen X. Polylactic acid (PLA): Research, development and industrialization. Biotechnol. 2010;5(11):1125–36.
240. Köse GT, Kenar H, Hasirci N, Hasirci V. Macroporous poly(3-hydroxybutyrate-co-3-hydroxyvalerate) matrices for bone tissue engineering. Biomaterials. 2003;24(11):1949–58.
241. Jack KS, Velayudhan S, Luckman P, Trau M, Grøndahl L, Cooper-White J. The fabrication and characterization of biodegradable HA/PHBV nanoparticle-polymer composite scaffolds. Acta Biomater. 2009;5(7):2657–67.
242. Khang G, Kim SW, Cho JC, Rhee JM, Yoon SC, Lee HB. Preparation and characterization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) microspheres for the sustained release of 5-fluorouracil. Biomed Mater Eng. 2001;11(2):89–103.
243. Mendes JBE, Riekes MK, de Oliveira VM, Michel MD, Stulzer HK, Khalil NM, et al. PHBV/PCL microparticles for controlled release of resveratrol: physicochemical characterization, antioxidant potential, and effect on hemolysis of human erythrocytes. ScientificWorldJournal. 2012;2012:542937.
244. Lü JM, Wang X, Marin-Muller C, Wang H, Lin PH, Yao Q, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Exp Rev Mol Diagn. 2009;9(4):325–41.
245. Jain RA. The manufacturing techniques of various drug loaded biodegradable poly (lactide-co-glycolide)(PLGA) devices. Biomaterials. 2000;21(23):2475–90.
246. Soderquist RG, Sloane EM, Loram LC, Harrison JA, Dengler EC, Johnson SM, et al. Release of plasmid DNA-encoding IL-10 from PLGA microparticles facilitates long-term reversal of neuropathic pain following a single intrathecal administration. Pharm Res. 2010;27(5):841–54.
247. Cun D, Jensen DK, Maltesen MJ, Bunker M, Whiteside P, Scurr D, et al. High loading efficiency and sustained release of siRNA encapsulated in PLGA nanoparticles: Quality by design optimization and characterization. Eur J Pharm Biopharm. 2010;77(1):26–35.
248. Solbrig C, Saucier-Sawyer J, Cody V, Saltzman W, Hanlon D. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol Pharm. 2007;4(1):47–57.
249. Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. Biodegradable nanoparticle mediated antigen delivery to human cord blood derived dendritic cells for induction of primary T cell responses. J Drug Target. 2003;11(8-10):495–507.
250. Prasad S, Cody V, Saucier-Sawyer JK, Saltzman WM, Sasaki CT, Edelson RL, et al. Polymer nanoparticles containing tumor lysates as antigen delivery vehicles for dendritic cell–based antitumor immunotherapy. Nanomed. 2011;7(1):1–10.
251. Thomas CM, Rawat A, Hope-Weeks LJ, Ahsan F. Aerosolized PLA and PLGA Nanoparticles Enhance Humoral, Mucosal and Cytokine Responses to Hepatitis B Vaccine. Mol Pharm. 2010;27:905–19.
252. Tian J, Yu J. Cell biology of antigen processing in vitro and in vivo. Fish Shellfish Immunol. 2010;30(1):109–17.

References
179
253. Jiang W, Gupta RK, Deshpande MC, Schwendeman SP. Biodegradable poly (lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv Drug Deliv Rev. 2005;57(3):391–410.
254. Garinot M, Fiévez V, Pourcelle V, Stoffelbach F, Des Rieux A, Plapied L, et al. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J Control Release. 2007;120(3):195–204.
255. Florindo HF, Pandit S, Gonçalves LMD, Alpar HO, Almeida AJ. New approach on the development of a mucosal vaccine against strangles: Systemic and mucosal immune responses in a mouse model. Vaccine. 2009;27(8):1230–41.
256. Diwan M, Elamanchili P, Cao M, Samuel J. Dose sparing of CpG oligodeoxynucleotide vaccine adjuvants by nanoparticle delivery. Curr Drug Deliv. 2004;1(4):405–12.
257. Bates FS, Hillmyer MA, Lodge TP, Bates CM, Delaney KT, Fredrickson GH. Multiblock Polymers: Panacea or Pandora’s Box? Science. 2012;336(6080):434–40.
258. Kissel T, Li Y, Unger F. ABA-triblock copolymers from biodegradable polyester A-blocks and hydrophilic poly(ethylene oxide) B-blocks as a candidate for in situ forming hydrogel delivery systems for proteins. Adv Drug Deliv Rev. 2002;54(1):99–134.
259. Xiong X-B, Falamarzian A, Garg SM, Lavasanifar A. Engineering of amphiphilic block copolymers for polymeric micellar drug and gene delivery. J Control Release. 2011;155(2):248–61.
260. Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92(7):1343–55.
261. Yang Y, Kuang Y, Liu Y, Li W, Jiang Z, Xiao L, et al. Immunogenicity of multiple-epitope antigen gene of HCV carried by novel biodegradable polymers. Comp Immunol Microbiol Infect Dis. 2011;34(1):65–72.
262. Blencowe A, Tan JF, Goh TK, Qiao GG. Core cross-linked star polymers via controlled radical polymerisation. Polymer. 2009;50(1):5–32.
263. Cameron DJA, Shaver MP. Aliphatic polyester polymer stars: synthesis, properties and applications in biomedicine and nanotechnology. Chem Soc Rev. 2011;40(3):1761–76.
264. Jansen J, Tibbe MP, Mihov G, Feijen J, Grijpma DW. Photo-crosslinked networks prepared from fumaric acid monoethyl ester-functionalized poly(D,L-lactic acid) oligomers and N-vinyl-2-pyrrolidone for the controlled and sustained release of proteins. Acta Biomater. 2012;8(10):3652–9.
265. Zotzmann J, Alteheld A, Behl M, Lendlein A. Amorphous phase-segregated copoly(ether)esterurethane thermoset networks with oligo(propylene glycol) and oligo[(rac-lactide)-co-glycolide] segments: synthesis and characterization. J Mater Sci Mater Med. 2009;20(9):1815–24.
266. Nagahama K, Saito T, Ouchi T, Ohya Y. Biodegradable Nano-aggregates of Star-Shaped 8-arm PEG-PLLA Block Co-polymers for Encapsulation of Water-Soluble Macromolecules. J Biomater SCi Polym Ed. 2010;[Epub ahead of print].
267. Ferreira SA, Gama FM, Vilanova M. Polymeric nanogels as vaccine delivery systems. Nanomed. 2013;9(2):159–73.

180
268. Fredenberg S, Wahlgren M, Reslow M, Axelsson A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems--a review. Int J Pharm. 2011;415(1-2):34–52.
269. Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1-2):113–42.
270. Chye Joachim Loo S, Ooi CP, Hong Elyna Wee S, Chiang Freddy Boey Y. Effect of isothermal annealing on the hydrolytic degradation rate of poly (lactide-co-glycolide)(PLGA). Biomaterials. 2005;26(16):2827–33.
271. Vert M, Li SM, Spenlehauer G, Guerin P. Bioresorbability and biocompatibility of aliphatic polyesters. J Mater Sci Mater Med. 1992;3(6):432–46.
272. Weir NA, Buchanan FJ, Orr JF, Dickson GR. Degradation of poly-L-lactide. Part 1: in vitro and in vivo physiological temperature degradation. Proc Inst Mech Eng H. 2004;218(5):307–19.
273. Göpferich A, Tessmar J. Polyanhydride degradation and erosion. Adv Drug Deliv Rev. 2002;54(7):911–31.
274. Peppas NA, Narasimhan B. Mathematical models in drug delivery: How modeling has shaped the way we design new drug delivery systems. J Control Release Off J Control Release Soc. 2014;pii: S0168-3659(14)00450-7.
275. Ford Versypt AN, Pack DW, Braatz RD. Mathematical modeling of drug delivery from autocatalytically degradable PLGA microspheres - A review. J Control Release. 2013;165(1):29–37.
276. Li S. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. J Biomed Mater Res. 1999;48(3):342–53.
277. Houchin ML, Topp EM. Chemical degradation of peptides and proteins in PLGA: a review of reactions and mechanisms. J Pharm Sci. 2008;97(7):2395–404.
278. Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly (D, L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125(3):193–209.
279. Waeckerle-Men Y, Groettrup M. PLGA microspheres for improved antigen delivery to dendritic cells as cellular vaccines. Adv Drug Deliv Rev. 2005;57(3):475–82.
280. Laeger T, Metges CC, Kuhla B. Role of beta-hydroxybutyric acid in the central regulation of energy balance. Appetite. 2010;54(3):450–5.
281. Dunne M, Corrigan I, Ramtoola Z. Influence of particle size and dissolution conditions on the degradation properties of polylactide-co-glycolide particles. Biomaterials. 2000;21(16):1659–68.
282. Batycky RP, Hanes J, Langer R, Edwards DA. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J Pharm Sci. 1997;86(12):1464–77.
283. Benoit MA, Baras B, Gillard J. Preparation and characterization of protein-loaded poly (epsilon-caprolactone) microparticles for oral vaccine delivery. Int J Pharm. 1999;184(1):73–84.

References
181
284. Gao H, Wang Y-N, Fan Y-G, Ma J-B. Conjugates of poly(DL-lactide-co-glycolide) on amino cyclodextrins and their nanoparticles as protein delivery system. J Biomed Mater Res A. 2007;80(1):111–22.
285. Storni T, Kundig T, Senti G, Johansen P. Immunity in response to particulate antigen-delivery systems. Adv Drug Delive Rev. 2005;57(3):333–55.
286. Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102.
287. Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol Immunol. 2007;44(1-3):82–94.
288. Moghimi SM, Andersen AJ, Ahmadvand D, Wibroe PP, Andresen TL, Hunter AC. Material properties in complement activation. Adv Drug Deliv Rev. 2011;63(12):1000–7.
289. Mosqueira VC, Legrand P, Gulik A, Bourdon O, Gref R, Labarre D, et al. Relationship between complement activation, cellular uptake and surface physicochemical aspects of novel PEG-modified nanocapsules. Biomaterials. 2001;22(22):2967–79.
290. Nicolete R, Santos DF dos, Faccioli LH. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int Immunopharmacol. 2011;11(10):1557–63.
291. Fischer S, Uetz-von Allmen E, Waeckerle-Men Y, Groettrup M, Merkle HP, Gander B. The preservation of phenotype and functionality of dendritic cells upon phagocytosis of polyelectrolyte-coated PLGA microparticles. Biomaterials. 2007;28(6):994–1004.
292. Yi X, Shi X, Gao H. Cellular Uptake of Elastic Nanoparticles. Phys Rev Lett. 2011;107(9):98101.
293. Tzlil S, Deserno M, Gelbart WM, Ben-Shaul A. A statistical-thermodynamic model of viral budding. Biophys J. 2004;86(4):2037–48.
294. Chithrani BD, Chan WCW. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7(6):1542–50.
295. Gao H, Shi W, Freund LB. Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci USA. 2005;102(27):9469–74.
296. Sapin A, Garcion E, Clavreul A, Lagarce F, Benoit JP, Menei P. Development of new polymer-based particulate systems for anti-glioma vaccination. Int J Pharm. 2006;309(1-2):1–5.
297. Mottram PL, Leong D, Crimeen-Irwin B, Gloster S, Xiang SD, Meanger J, et al. Type 1 and 2 immunity following vaccination is influenced by nanoparticle size: formulation of a model vaccine for respiratory syncytial virus. Mol Pharm. 2007;4(1):73–84.
298. Ehrlich M, Boll W, van Oijen A, Hariharan R, Chandran K, Nibert ML, et al. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118(5):591–605.
299. Wang Z, Tiruppathi C, Minshall RD, Malik AB. Size and dynamics of caveolae studied using nanoparticles in living endothelial cells. ACS Nano. 2009;3(12):4110–6.
300. Xiang SD, Scholzen A, Minigo G, David C, Apostolopoulos V, Mottram PL, et al. Pathogen recognition and development of particulate vaccines: Does size matter? Methods. 2006;40(1):1–9.

182
301. Gan Q, Dai D, Yuan Y, Qian J, Sha S, Shi J, et al. Effect of size on the cellular endocytosis and controlled release of mesoporous silica nanoparticles for intracellular delivery. Biomed Microdevices. 2011;14(2):259–70.
302. Cook IF. Evidence based route of administration of vaccines. Hum Vaccin. 2008;4(1):67–73.
303. Johansen P, Mohanan D, Martínez-Gómez JM, Kündig TM, Gander B. Lympho-geographical concepts in vaccine delivery. J Control Release. 2010;148(1):56–62.
304. Villadangos JA, Heath WR. Life cycle, migration and antigen presenting functions of spleen and lymph node dendritic cells: limitations of the Langerhans cells paradigm. Semin Immunol. 2005;17(4):262–72.
305. Wilson NS, El-Sukkari D, Belz GT, Smith CM, Steptoe RJ, Heath WR, et al. Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood. 2003;102(6):2187–94.
306. Cho K, Wang X, Nie S, Chen ZG, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–6.
307. Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38(5):1404–13.
308. Garg K, Mangal S, Khambete H, Sharma K, Tyagi K. Mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymers. Recent Pat Drug Deliv Formul. 2010;4(2):114–28.
309. Foster N, Hirst BH. Exploiting receptor biology for oral vaccination with biodegradable particulates. Adv Drug Deliv Rev. 2005;57(3):431–50.
310. Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61(2):75–85.
311. Chiu YL, Ho YC, Chen YM, Peng SF, Ke CJ, Chen KJ, et al. The characteristics, cellular uptake and intracellular trafficking of nanoparticles made of hydrophobically-modified chitosan. J Control Release. 2010;146(1):152–9.
312. Zhao F, Zhao Y, Liu Y, Chang X, Chen C, Zhao Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7(10):1322–37.
313. Rogers TH, Babensee JE. The role of integrins in the recognition and response of dendritic cells to biomaterials. Biomaterials. 2010;32:1270–9.
314. Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298(2):315–22.
315. Vasir JK, Labhasetwar V. Quantification of the force of nanoparticle-cell membrane interactions and its influence on intracellular trafficking of nanoparticles. Biomaterials. 2008;29(31):4244–52.
316. Thurn KT, Brown EM, Wu A, Vogt S, Lai B, Maser J, et al. Nanoparticles for applications in cellular imaging. Nanoscale Res Lett. 2007;2(9):430–41.
317. Liu J, Bauer H, Callahan J, Kopecková P, Pan H, Kopecek J. Endocytic uptake of a large array of HPMA copolymers: Elucidation into the dependence on the physicochemical characteristics. J Control Release. 2010;143(1):71–9.

References
183
318. Yue ZG, Wei W, Lv PP, Yue H, Wang LY, Su ZG, et al. Surface Charge Affects Cellular Uptake and Intracellular Trafficking of Chitosan-based Nanoparticles. Biomacromolecules. 2011;12(7):2440–6.
319. Panyam M J, Zhou WENZ, Prabha S, Sahoo SK, Labhase V. Rapid endo-lysosomal escape of poly (DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16(10):1217–26.
320. Danhier F, Lecouturier N, Vroman B, Jér me C, Marchand-Brynaert J, Feron O, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133(1):11–7.
321. Van den Berg JH, Oosterhuis K, Hennink WE, Storm G, van der Aa LJ, Engbersen JF, et al. Shielding the cationic charge of nanoparticle-formulated dermal DNA vaccines is essential for antigen expression and immunogenicity. J Control Release. 2010;141(2):234–40.
322. Florindo HF, Pandit S, Gonçalves LMD, Alpar HO, Almeida AJ. Surface modified polymeric nanoparticles for immunisation against equine strangles. Int J Pharm. 2010;390(1):25–31.
323. Tahara K, Sakai T, Yamamoto H, Takeuchi H, Hirashima N, Kawashima Y. Improved cellular uptake of chitosan-modified PLGA nanospheres by A549 cells. Int J Pharm. 2009;382(1-2):198–204.
324. Sirsi SR, Schray RC, Wheatley MA, Lutz GJ. Formulation of polylactide-co-glycolic acid nanospheres for encapsulation and sustained release of poly(ethylene imine)-poly(ethylene glycol) copolymers complexed to oligonucleotides. J Nanobiotechnology. 2009;7(1):1.
325. Venkataraman S, Hedrick JL, Ong ZY, Chuan Y, Ee PL, Hammond PT, et al. The effects of polymeric nanostructure shape on drug delivery. Adv Drug Deliv Rev. 2011;63(14-15):1228–46.
326. Arnida, Janát-Amsbury MM, Ray A, Peterson CM, Ghandehari H. Geometry and surface characteristics of gold nanoparticles influence their biodistribution and uptake by macrophages. Eur J Pharm Biopharm. 2011;77(3):417–23.
327. Heng BC, Zhao X, Tan EC, Khamis N, Assodani A, Xiong S, et al. Evaluation of the cytotoxic and inflammatory potential of differentially shaped zinc oxide nanoparticles. Arch Toxicol. 2011;85(12):1517–28.
328. Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–15.
329. Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J Control Release Off J Control Release Soc. 1 de Maio de 2006;112(1):26–34.
330. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–92.
331. Upreti M, Jyoti A, Sethi P. Tumor microenvironment and nanotherapeutics. Transl Cancer Res. 2013;2(4):309–19.
332. Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis. 2014;7:9–18.

184
333. Kelly C, Jefferies C, Cryan SA. Targeted Liposomal Drug Delivery to Monocytes and Macrophages. J Drug Deliv. 2011;2011:727241.
334. Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20(1):89–95.
335. Fernandez-Megia E, Novoa-Carballal R, Quiñoá E, Riguera R. Conjugation of bioactive ligands to PEG-grafted chitosan at the distal end of PEG. Biomacromolecules. 2007;8(3):833–42.
336. Kadowaki N, Ho S, Antonenko S, de Waal Malefyt R, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194(6):863–70.
337. Hamdy S, Elamanchili P, Alshamsan A, Molavi O, Satou T, Samuel J. Enhanced antigen specific primary CD4+ and CD8+ responses by codelivery of ovalbumin and toll like receptor ligand monophosphoryl lipid A in poly (D, L lactic co glycolic acid) nanoparticles. J Biomed Mater Res A. 2007;81(3):652–62.
338. Hamdy S, Haddadi A, Shayeganpour A, Samuel J, Lavasanifar A. Activation of Antigen-Specific T Cell-Responses by Mannan-Decorated PLGA Nanoparticles. Pharm Res. 2011;28(9):2288–301.
339. Fievez V, Plapied L, Des Rieux A, Pourcelle V, Freichels H, Wascotte V, et al. Targeting nanoparticles to M cells with non-peptidic ligands for oral vaccination. Eur J Pharm Biopharm. 2009;73(1):16–24.
340. Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, et al. Targeted PLGA nano-but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release. 2010;144(2):118–26.
341. Mishra N, Tiwari S, Vaidya B, Agrawal GP, Vyas SP. Lectin anchored PLGA nanoparticles for oral mucosal immunization against hepatitis B. J Drug Target. 2010;19(1):67–78.
342. Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, Fahmy TM. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials. 2011;32(11):3094–105.
343. Scott CJ, Marouf WM, Quinn DJ, Buick RJ, Orr SJ, Donnelly RF, et al. Immunocolloidal targeting of the endocytotic siglec-7 receptor using peripheral attachment of siglec-7 antibodies to poly (lactide-co-glycolide) nanoparticles. Pharma Res. 2008;25(1):135–46.
344. Rajapaksa TE, Stover-Hamer M, Fernandez X, Eckelhoefer HA, Lo DD. Claudin 4-targeted protein incorporated into PLGA nanoparticles can mediate M cell targeted delivery. J Control Release. 2010;142(2):196–205.
345. Molavi O, Mahmud A, Hamdy S, Hung RW, Lai R, Samuel J, et al. Development of a Poly (d, l-lactic-co-glycolic acid) Nanoparticle Formulation of STAT3 Inhibitor JSI-124: Implication for Cancer Immunotherapy. Mol Pharm. 2010;7(2):364–74.
346. Molavi O, Ma Z, Hamdy S, Lavasanifar A, Samuel J. Immunomodulatory and anticancer effects of intra-tumoral co-delivery of synthetic lipid A adjuvant and STAT3 inhibitor, JSI-124. Immunopharmacol Immunotoxicol. 2009;31(2):214–21.
347. Unger WWJ, van Kooyk Y. «Dressed for success» C-type lectin receptors for the delivery of glyco-vaccines to dendritic cells. Curr Opin Immunol. 2010;23(1):131–7.

References
185
348. Flacher V, Tripp CH, Stoitzner P, Haid B, Ebner S, Del Frari B, et al. Epidermal Langerhans cells rapidly capture and present antigens from C-type lectin-targeting antibodies deposited in the dermis. J Invest Dermatol. 2009;130(3):755–62.
349. Liu G, Zhang L, Zhao Y. Modulation of immune responses through direct activation of Toll like receptors to T cells. Clin Exp Immunol. 2010;160(2):168–75.
350. Bekeredjian Ding I, Jego G. Toll like receptors–sentries in the B cell response. Immunology. 2009;128(3):311–23.
351. Van Kooyk Y. C-type lectins on dendritic cells: key modulators for the induction of immune responses. Biochem Soc Trans. 2008;36(Pt 6):1478–81.
352. Gazi U, Martinez-Pomares L. Influence of the mannose receptor in host immune responses. Immunobiology. 2009;214(7):554–61.
353. Burgdorf S, Kautz A, Böhnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science. 2007;316(5824):612–6.
354. Tel J, Benitez Ribas D, Hoosemans S, Cambi A, Adema GJ, Figdor CG, et al. DEC 205 mediates antigen uptake and presentation by both resting and activated human plasmacytoid dendritic cells. Eur J Immunol. 2011;41(4):1014–23.
355. O’Reilly MK, Paulson JC. Siglecs as targets for therapy in immune-cell-mediated disease. Trends Pharmacol Sci. 2009;30(5):240–8.
356. Jandus C, Simon HU, von Gunten S. Targeting Siglecs--A novel pharmacological strategy for immuno-and glycotherapy. Biochem Pharmacol. 2011;82(4):323–32.
357. Von Gunten S, Bochner BS. Basic and clinical immunology of Siglecs. Ann N Acad Sci. 2008;1143:61–82.
358. Collins BE, Paulson JC. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr Opin Chem Biol. 2004;8(6):617–25.
359. Joshi MD, Unger WWJ, Litjens M, van Bloois L, Kalay H, van Kooyk Y, et al. DC-SIGN mediated antigen-targeting using glycan-modified liposomes: Formulation considerations. Int J Pharm. 2011;416(2):426–32.
360. Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6(2):148–58.
361. Des Rieux A, Fievez V, Garinot M, Schneider YJ, Préat V. Nanoparticles as potential oral delivery systems of proteins and vaccines: a mechanistic approach. J Control Release. 2006;116(1):1–27.
362. Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. 2010;339(1):269–80.
363. Peter Y, Goodenough D. Claudins. Curr Biol. 2004;14(8):R293–4.
364. Florindo H, Pandit S, Lacerda L, Gonçalves L, Alpar H, Almeida A. The enhancement of the immune response against S. equi antigens through the intranasal administration of poly--caprolactone-based nanoparticles. Biomaterials. 2009;30(5):879–91.
365. Watts PJ, Smith A. Re-formulating drugs and vaccines for intranasal delivery: maximum benefits for minimum risks? Drug Discov Today. 2011;16(1-2):4–7.

186
366. Krauland AH, Guggi D, Bernkop-Schnürch A. Thiolated chitosan microparticles: a vehicle for nasal peptide drug delivery. Int J Pharm. 2006;307(2):270–7.
367. Alpar HO, Eyles JE, Williamson ED, Somavarapu S. Intranasal vaccination against plague, tetanus and diphtheria. Adv Drug Deliv Rev. 2001;51(1-3):173–201.
368. Titball RW. Vaccines against intracellular bacterial pathogens. Drug Discov Today. 2008;13(13-14):596–600.
369. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. 2013;14(5):509–13.
370. Igietseme JU, Eko FO, He Q, Black CM. Antibody regulation of Tcell immunity: implications for vaccine strategies against intracellular pathogens. Exp Rev Vaccines. 2004;3(1):23–34.
371. Casadevall A, Pirofski L. Antibody-mediated regulation of cellular immunity and the inflammatory response. Trends Immunol. 2003;24(9):474–8.
372. McEwan WA, Mallery DL, Rhodes DA, Trowsdale J, James LC. Intracellular antibody-mediated immunity and the role of TRIM21. BioEssays. 2011;33(11):803–9.
373. Manish M, Rahi A, Kaur M, Bhatnagar R, Singh S. A Single-Dose PLGA Encapsulated Protective Antigen Domain 4 Nanoformulation Protects Mice against Bacillus anthracis Spore Challenge. PloS One. 2013;8(4):e61885.
374. Camejo A, Carvalho F, Reis O, Leitão E, Sousa S, Cabanes D. The arsenal of virulence factors deployed by Listeria monocytogenes to promote its cell infection cycle. Virulence. 2011;2(5):379–94.
375. Barat S, Willer Y, Rizos K, Claudi B, Mazé A, Schemmer AK, et al. Immunity to intracellular Salmonella depends on surface-associated antigens. PLoS Pathog. 2012;8(10):e1002966.
376. Zepp F. Principles of vaccine design—Lessons from nature. Vaccine. 2010;28:C14–C24.
377. Florindo H, Pandit S, Goncalves L, Alpar H, Almeida A. S. equi antigens adsorbed onto surface modified poly-epsilon-caprolactone microspheres induce humoral and cellular specific immune responses. Vaccine. 2008;26(33):4168–77.
378. Guo C, Gemeinhart RA. Understanding the adsorption mechanism of chitosan onto poly(lactide-co-glycolide) particles. Eur J Pharm Biopharm. 2008;70(2):597–604.
379. Roth-Walter F, Schöll I, Untersmayr E, Ellinger A, Boltz-Nitulescu G, Scheiner O, et al. Mucosal targeting of allergen-loaded microspheres by Aleuria aurantia lectin. Vaccine. 2005;23(21):2703–10.
380. Gómez JMM, Fischer S, Csaba N, Kündig TM, Merkle HP, Gander B, et al. A Protective Allergy Vaccine Based on CpG- and Protamine-Containing PLGA Microparticles. Pharm Res. 2007;24(10):1927–35.
381. Weiss R, Scheiblhofer S, Machado Y, Thalhamer J. New approaches to transcutaneous immunotherapy: targeting dendritic cells with novel allergen conjugates. Curr Opin Allergy Clin Immunol. 2013;13(6):669–76.
382. Tafaghodi M, Eskandari M, Kharazizadeh M, Khamesipour A, Jaafari MR. Immunization against leishmaniasis by PLGA nanospheres loaded with an experimental autoclaved Leishmania major (ALM) and Quillaja saponins. Trop Biomed. 2010;27(3):639–50.

References
187
383. Anish C, Goswami DG, Kanchan V, Mathew S, Panda AK. The immunogenic characteristics associated with multivalent display of Vi polysaccharide antigen using biodegradable polymer particles. Biomaterials. 2012;33(28):6843–57.
384. Ma W, Chen, Kaushal, McElroy, Zhang, Ozkan, et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomedicine. 2012;7:1475–87.
385. Díez S, Navarro G, de ILarduya CT. In vivo targeted gene delivery by cationic nanoparticles for treatment of hepatocellular carcinoma. J Gene Med. 2009;11(1):38–45.
386. Loucq C. Vaccines today, vaccines tomorrow: a perspective. Clin Expl Vaccine Res. 2013;2(1):4.
387. Lewis K. Platforms for antibiotic discovery. Nat Rev Drug Discov. 2013;12(5):371–87.
388. FDA. FDA Guidance documents for anti-microbial clinical development [Internet]. 2013. Obtido de: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064980.htm.
389. European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP),. Guideline on the evaluation of medicinal products indicated for treatment of bacterial infections. [Internet]. Obtido de: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003417.pdf.
390. Sathish JG, Sethu S, Bielsky M-C, de Haan L, French NS, Govindappa K, et al. Challenges and approaches for the development of safer immunomodulatory biologics. Nat Rev Drug Discov. 2013;12(4):306–24.
391. McNeil SE. Nanoparticle therapeutics: a personal perspective. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(3):264–71.
392. Zolnik BS, González-Fernández A, Sadrieh N, Dobrovolskaia MA. Nanoparticles and the immune system. Endocrinology. 2010;151(2):458–65.
393. Gaspar R. Regulatory issues surrounding nanomedicines: setting the scene for the next generation of nanopharmaceuticals. Nanomedicine Lond. 2007;2(2):143–7.
394. Gaspar R, Duncan R. Polymeric carriers: Preclinical safety and the regulatory implications for design and development of polymer therapeutics. Adv Drug Deliv Rev. 2009;61(13):1220–31.
395. Bowman D, Maynard A, Gaspar R. Chapter 14: Therapeutic products: regulating drugs and medical devices. International Handbook on Regulating Nanotechnologies. Northampton, MA: Edward Elgar Pub; 2010. p. 291– 320.
396. European Medicines Agency C. Vaccines Working Party – «Workplan for 2013» [Internet]. 2013. Obtido de: http://www.ema.europa.eu/docs/en_GB/document_library/Work_programme/2010/02/WC500073590.pdf
397. European Medicines Agency C. Note For Guidance on the Clinical Evaluation of Vaccines [Internet]. (EMEA/CHMP/VWP/164653/2005; 2005. Obtido de: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003875.pdf

188
398. European Medicines Agency C. Guideline on Adjuvants in Vaccines for Human Use [Internet]. EMEA/CHMP/VEG/134716/2004; 2005. Obtido de: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003809.pdf
399. Food and Drugs Administration O of C, Tissue and Gene Therapies. Clinical Considerations for Therapeutic Cancer Vaccines [Internet]. FDA’s Center for Biologics Evaluation and Research; 2013. Obtido de: (http://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/vaccines/ucm278673.pdf )
400. França RFO, da Silva CC, De Paula SO. Recent advances in molecular medicine techniques for the diagnosis, prevention, and control of infectious diseases. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2013;32(6):723–8.
401. Macho Fernandez E, Chang J, Fontaine J, Bialecki E, Rodriguez F, Werkmeister E, et al. Activation of invariant Natural Killer T lymphocytes in response to the α-galactosylceramide analogue KRN7000 encapsulated in PLGA-based nanoparticles and microparticles. Int J Pharm. 2012;423(1):45–54.
402. Freichels H, Pourcelle V, Le Duff CS, Marchand Brynaert J, Jérôme C. «Clip» and «Click» Chemistries Combination: Toward Easy PEGylation of Degradable Aliphatic Polyesters. Macromol Rapid Commun. 2011;32(7):616–21.
403. Vangeyte P, Leyh B, Heinrich M, Grandjean J, Bourgaux C, Jérôme R. Self-assembly of poly(ethylene oxide)-b-poly(epsilon-caprolactone) copolymers in aqueous solution. Langmuir. 2004;20(20):8442–51.
404. Freichels H, Danhier F, Préat V, Lecomte P, Jérôme C. Fluorescent labeling of degradable poly (lactide-co-glycolide) for cellular nanoparticles tracking in living cells. Int J Artif Organs. 2011;34(2):152–60.
405. Afonso CA, Santhakumar V, Lough A, Batey RA. An Expedient Synthesis of Cationic Rhodamine Fluorescent Probes Suitable for Conjugation to Amino Acids and Peptides. Synthesis. 2003;(17):2647–54.
406. Rieger J, Freichels H, Imberty A, Putaux JL, Delair T, Je ro me C, et al. Polyester nanoparticles presenting mannose residues: toward the development of new vaccine delivery systems combining biodegradability and targeting properties. Biomacromolecules. 2009;10(3):651–7.
407. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–702.
408. Ashany D, Savir A, Bhardwaj N, Elkon KB. Dendritic cells are resistant to apoptosis through the Fas (CD95/APO-1) pathway. J Immunol. 1999;163(10):5303–11.
409. Wang X, Ramström O, Yan M. Dynamic light scattering as an efficient tool to study glyconanoparticle-lectin interactions. Analyst. 2011;136(20):4174–8.
410. Haspot F, Lavault A, Sinzger C, Laib Sampaio K, Stierhof Y-D, Pilet P, et al. Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner. PloS One. 2012;7(4):e34795.
411. Bhattacharyya S, Warfield KL, Ruthel G, Bavari S, Aman MJ, Hope TJ. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology. 2010;401(1):18–28.

References
189
412. Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation. Vaccine. 2010;28(50):7897–906.
413. Autenrieth SE, Autenrieth IB. Variable antigen uptake due to different expression of the macrophage mannose receptor by dendritic cells in various inbred mouse strains. Immunology. 2009;127(4):523–9.
414. Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86(6):3993–4003.
415. Norbury CC. Drinking a lot is good for dendritic cells. Immunology. 2006;117(4):443–51.
416. Dos Santos T, Varela J, Lynch I, Salvati A, Dawson KA. Effects of Transport Inhibitors on the Cellular Uptake of Carboxylated Polystyrene Nanoparticles in Different Cell Lines. Schnur JM, editor. PLoS ONE. 2011;6(9):e24438.
417. Ahsan F, Rivas IP, Khan MA, Torres Suarez AI. Targeting to macrophages: role of physicochemical properties of particulate carriers--liposomes and microspheres--on the phagocytosis by macrophages. J Control Release. 2002;79(1-3):29–40.
418. Platt CD, Ma JK, Chalouni C, Ebersold M, Bou-Reslan H, Carano RAD, et al. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc Natl Acad Sci U A. 2010;107(9):4287–92.
419. Mollica F, Biondi M, Muzzi S, Ungaro F, Quaglia F, Rotonda MI, et al. Mathematical modelling of the evolution of protein distribution within single PLGA microspheres: prediction of local concentration profiles and release kinetics. J Mater Sci Mater Med. 2007;19(4):1587–93.
420. Shan X, Yuan Y, Liu C, Tao X, Sheng Y, Xu F. Influence of PEG chain on the complement activation suppression and longevity in vivo prolongation of the PCL biomedical nanoparticles. Biomed Microdevices. 2009;11(6):1187–94.
421. Jiang X, Shen C, Rey-Ladino J, Yu H, Brunham RC. Characterization of Murine Dendritic Cell Line JAWS II and Primary Bone Marrow-Derived Dendritic Cells in Chlamydia muridarum Antigen Presentation and Induction of Protective Immunity. Infec Immun. 2008;76(6):2392–401.
422. Mendoza L, Bubeník J, Símová J, Jandlová T, Vonka V, Mikysková R. Prophylactic, therapeutic and anti-metastatic effects of BMDC and DC lines in mice carrying HPV 16-associated tumours. Int J Oncol. 2003;23(1):243–7.
423. Horak D, Babic M, Jendelová P, Herynek V, Trchová M, Pientka Z, et al. D-mannose-modified iron oxide nanoparticles for stem cell labeling. Bioconjug Chem. 2007;18(3):635–44.
424. Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. ACS Nano. 2013;7(4):2935–47.
425. Tirosh O, Barenholz Y, Katzhendler J, Priev A. Hydration of polyethylene glycol-grafted liposomes. Biophys J. 1998;74(3):1371–9.
426. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300(5617):337–9.

190
427. Vujanovic L, Butterfield LH. Melanoma cancer vaccines and anti tumor T cell responses. J Cell Biochem. 2007;102(2):301–10.
428. Hamdy S, Elamanchili P, Alshamsan A, Molavi O, Satou T, Samuel J. Enhanced antigen specific primary CD4+ and CD8+ responses by codelivery of ovalbumin and toll like receptor ligand monophosphoryl lipid A in poly (D, L lactic co glycolic acid) nanoparticles. J Biomed Mater Res A. 2007;81(3):652–62.
429. Adamina M, Bolli M, Albo F, Cavazza A, Zajac P, Padovan E, et al. Encapsulation into sterically stabilised liposomes enhances the immunogenicity of melanoma-associated Melan-A/MART-1 epitopes. Br J Cancer. 2004;90(1):263–9.
430. Hafner AM, Corthesy B, Textor M, Merkle HP. Tuning the immune response of dendritic cells to surface-assembled poly (I: C) on microspheres through synergistic interactions between phagocytic and TLR3 signaling. Biomaterials. 2011;32(10):2651–61.
431. Kalariya M, Ganta S, Amiji M. Multi-Compartmental Vaccine Delivery System for Enhanced Immune Response to gp100 Peptide Antigen in Melanoma Immunotherapy. Pharm Res. 2012;29(12):3393–403.
432. Schreibelt G, Tel J, Sliepen KH, Benitez-Ribas D, Figdor CG, Adema GJ, et al. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol Immunother. 2010;59(10):1573–82.
433. Schumacher R, Amacker M, Neuhaus D, Rosenthal R, Groeper C, Heberer M, et al. Efficient induction of tumoricidal cytotoxic T lymphocytes by HLA-A0201 restricted, melanoma associated, L (27) Melan-A/MART-1 (26-35) peptide encapsulated into virosomes in vitro. Vaccine. 2005;23(48-9):5572.
434. He LZ, Crocker A, Lee J, Mendoza-Ramirez J, Wang XT, Vitale LA, et al. Antigenic targeting of the human mannose receptor induces tumor immunity. J Immunol. 2007;178(10):6259–67.
435. Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40.
436. Leung S, Smith D, Myc A, Morry J, Baker JR. OT-II TCR transgenic mice fail to produce anti-ovalbumin antibodies upon vaccination. Cell Immunol. 2013;282(2):79–84.
437. Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol. 1999;17:51–88.
438. Lee Y-R, Lee Y-H, Im S-A, Yang I-H, Ahn GW, Kim K, et al. Biodegradable nanoparticles containing TLR3 or TLR9 agonists together with antigen enhance MHC-restricted presentation of the antigen. Arch Pharm Res. 2010;33(11):1859–66.
439. Bourgeois C, Veiga Fernandes H, Joret AM, Rocha B, Tanchot C. CD8 lethargy in the absence of CD4 help. Eur J Immunol. 2002;32(8):2199–207.
440. Crow MK. Modification of accessory molecule signaling. Springer Semin Immunopathol. 2006;27(4):409–24.
441. Hu X, Chakraborty NG, Sporn JR, Kurtzman SH, Ergin MT, Mukherji B. Enhancement of cytolytic T lymphocyte precursor frequency in melanoma patients following immunization with the MAGE-1 peptide loaded antigen presenting cell-based vaccine. Cancer Res. 1996;56(11):2479–83.

References
191
442. Yamada A, Sasada T, Noguchi M, Itoh K. Next-generation peptide vaccines for advanced cancer. Cancer Sci. 2013;104(1):15–21.
443. Cawood R, Hills T, Wong SL, Alamoudi AA, Beadle S, Fisher KD, et al. Recombinant viral vaccines for cancer. Trends Mol Med. 2012;18(9):564–74.
444. Sondak VK, Liu P-Y, Tuthill RJ, Kempf RA, Unger JM, Sosman JA, et al. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: overall results of a randomized trial of the Southwest Oncology Group. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20(8):2058–66.
445. Sosman JA, Unger JM, Liu P-Y, Flaherty LE, Park MS, Kempf RA, et al. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: impact of HLA class I antigen expression on outcome. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20(8):2067–75.
446. NIH. www.clinicaltrials.gov. National Institutes of Health; 2014.
447. Paulis LE, Mandal S, Kreutz M, Figdor CG. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr Opin Immunol. 2013;25(3):389–95.
448. Duncan R, Richardson SCW. Endocytosis and Intracellular Trafficking as Gateways for Nanomedicine Delivery: Opportunities and Challenges. Mol Pharm. 2012;9(9):2380–402.
449. Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27(23):3013–21.
450. Nakamura K, Yoshikawa N, Yamaguchi Y, Kagota S, Shinozuka K, Kunitomo M. Characterization of mouse melanoma cell lines by their mortal malignancy using an experimental metastatic model. Life Sci. 2002;70(7):791–8.
451. Greten TF, Jaffee EM. Cancer vaccines. J Clin Oncol Off J Am Soc Clin Oncol. 1999;17(3):1047–60.
452. Phan GQ, Touloukian CE, Yang JC, Restifo NP, Sherry RM, Hwu P, et al. Immunization of patients with metastatic melanoma using both class I- and class II-restricted peptides from melanoma-associated antigens. J Immunother Hagerstown Md 1997. 2003;26(4):349–56.
453. Men Y, Miconnet I, Valmori D, Rimoldi D, Cerottini JC, Romero P. Assessment of immunogenicity of human Melan-A peptide analogues in HLA-A*0201/Kb transgenic mice. J Immunol Baltim Md 1950. 1999;162(6):3566–73.
454. Elliott RL. Combination cancer immunotherapy «Expanding Paul Ehrlich’s Magic Bullet Concept». Surg Oncol. 2012;21(1):53–5.
455. Azijli K, Stelloo E, Peters GJ, van Den Eertwegh AJM. New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer Res. 2014;34(4):1493–505.
456. Kohlmeyer J, Cron M, Landsberg J, Bald T, Renn M, Mikus S, et al. Complete Regression of Advanced Primary and Metastatic Mouse Melanomas following Combination Chemoimmunotherapy. Cancer Res. 2009;69(15):6265–74.
457. John J, Ismail M, Riley C, Askham J, Morgan R, Melcher A, et al. Differential effects of Paclitaxel on dendritic cell function. BMC Immunol. 2010;11:14.

192
458. Sheng Sow H, Mattarollo SR. Combining low-dose or metronomic chemotherapy with anticancer vaccines: A therapeutic opportunity for lymphomas. OncoImmunology. 2013;2(12):e27058.
459. Ashworth MT, Daud AI. Combinatorial approach to treatment of melanoma. Hematol Oncol Clin North Am. 2014;28(3):601–12.

Curriculum vitae
193
Curriculum vitae
Academic degrees
Master in Pharmaceutical Sciences, Faculty of Pharmacy University of Lisbon,
Portugal, April 2009 (Final Classification: 16/20).
Current scientific/professional position
• PhD student (supervisors: Prof. Helena Florindo, Prof. Mafalda Videira, Prof.
Véronique Préat), Faculty of Pharmacy University of Lisbon, January 2010/2014. Expected
conclusion date: 3rd October 2014.
Domain of specializations: Pharmacy - Pharmaceutical Technology. Specialization on
cancer therapeutic vaccines: development of a dendritic cell-targeted peptide vaccine through
the coencapsulation of tumor antigens and adjuvants using biodegradable polymeric
nanoparticles with different functionalizations. Intracellular traffic and endocytic mechanisms
of nanocarriers and their interaction with biological membranes. Immunological mechanisms
induced by different nanoparticulate vaccines and in vivo therapeutic potential.
Host Labs:
InTraCell_ADD: Intracellular Trafficking Modulation for Advance Drug Delivery
Group
Instituto de Investigação do Medicamento (iMed-ULisboa)
Faculdade de Farmácia da Universidade de Lisboa
Av. Prof. Gama-Pinto 1649-003 Lisbon
Portugal
&
Advanced Drug Delivery and Biomaterials
Louvain Drug Research Institute (LDRI)
Université Catholique de Louvain
Avenue Mounier, Van Helmont Tower
73, bte B1.73.12, 1200 Woluwe-Saint-Lambert
Brussels, Belgium.
• Postdoctoral fellow at the McKetta Department of Biomedical Engineering,
Cockrell School of Engineering, The University of Texas at Austin, USA, under the
supervision of prof. Nicholas A. Peppas (20th October 2014 – (…)).

194
Scientific papers in peer-review international journals
1. Silva JM, Vandermeulen G, Pinto SN, Rodrigues C, Salgado A, Afonso CA, Viana
A, Jérôme C, Silva LC, Préat V and Florindo HF. Development of Functionalized
Nanoparticles for Vaccine Delivery to Dendritic Cells: a Mechanistic Approach,
Nanomedicines – Nanotechnology for Vaccine Development Special Issue, 2014 (accepted).
(IF 5.26)
2. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, Design of a novel
nanoparticulate platform to deliver tumor associated antigens, Procedia in Vaccinology, 2014,
DOI: 10.1016/j.provac.2014.07.011.
3. Silva JM, Videira M, Gaspar R, Préat V, Florindo HF, Immune System Targeting by
Nanoparticles for Cancer Vaccines. J Control Release, 2013, 168(2):179-99. DOI:
10.1016/j.jconrel.2013.03.010. (IF 7.63)
4. Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V, PLGA-based
nanoparticles: an overview of biomedical applications, J Control Release, 2012, 161(2):505-
22. DOI: 10.1016/j.jconrel.2012.01.043. Invited Review. (IF 7.63)
Scientific papers in preparation:
1. Silva JM, Zupancic E, Vandermeulen G, Oliveira VG, Salgado A, Videira M,
Gaspar M, Graca L, Préat V and Florindo HF, Combination of peptides and adjuvants in
mannose-functionalized NPs for melanoma immunotherapy, J Control Release, 2014
(submitted). (IF 7.63)
2. Peres C, Matos A, Conniot J, Zupancic E, Sainz V, Silva JM and Florindo HF,
Poly(lactic acid) nanomedicines: a potential vaccine delivery system, Wiley Interdisciplin
Rev Nanomed Nanobiotechnol – (in final preparation). (IF 5.68)
3.
Book chapters
1. Silva JM, Zupancic E, Peres C, Silva LC, Gaspar R, Préat V and Florindo HF,
Immunomodulation against microbial pathogens through dendritic cells, In: Microbial
pathogens and strategies for combating them: science, technology and education (A. Méndez-
Vilas, Ed., Formatex Research Center, Badajoz, Spain, 2013, pp. 1686-1705. ISBN (13)
Volume 3: 978-84-942134-1-0. - Invited Book Chapter.
2. Silva JM, Préat V, Florindo HF, Role of the Aliphatic Polyesters as Particulate
Vaccine Delivery Systems, In: Encyclopedia of Biomedical Polymers and Polymeric

Curriculum vitae
195
Biomaterials, Taylor & Francis Editorial Group, London, UK, 2014 [In Press] - Invited Book
Chapter.
Grants and prizes
1. II Simpósio Nacional de Nanociência e Nanotecnologia Biomédica, Universidade
Lusófona, 11th October 2013, Lisbon, Portugal (Best panel communication prize).
2. First International Symposium on Profiling – ISPROF, 2nd - 4rd September 2013,
Costa de Caparica, Portugal (Best oral communication prize).
3. 5th iMed.UL Post-Graduate Students Meeting, 18th July 2013, Faculty of Pharmacy
University of Lisbon, Portugal (Best panel communication prize).
4. 38th Federation of European Biochemical Societies (FEBS) Congress “Mechanisms
in Biology”, 6th - 11th July 2013, St. Petersburg, Russia (Congress Bursary awarded).
5. 3rd iMed.UL Post-Graduate Students Meeting, 12th December 2011, Faculty of
Pharmacy University of Lisbon, Portugal (Best oral communication prize).
6. 3rd European Science Foundation Summer School - Nanomedicine 2011, 24th June
2011, Lutherstadt Wittenberg, Germany (Travel grant awarded from the conference).
Teaching and supervision of scientific work
1. Teaching theoric and practical classes of Nanomedicines of the of the 5th year of the
Master in Pharmaceutical Sciences of the Faculty of Pharmacy of the University of Lisbon,
Department of Pharmaceutical Technology and Galenic Pharmacy (November 2013).
2. Teaching of laboratorial classes of Pharmaceutical Technology II of the 4rd year of
the Master in Pharmaceutical Sciences of the Faculty of Pharmacy of the University of
Lisbon, Department of Pharmaceutical Technology and Galenic Pharmacy (September 2013 -
February 2014).
3. Supervision of the undergraduate research project of Tiago Cairrão and Isabel
Marcos from the 5th year of the Master in Pharmaceutical Sciences of the Faculty of
Pharmacy of the University of Lisbon, Department of Pharmaceutical Technology and
Galenic Pharmacy (September 2012 - February 2013).
4. Supervision of the undergraduate extracurricular research internship of Ana Pragana
at Louvain Drug Research Institute, Pharmaceutics & Drug Delivery Group, Université
Catholique de Louvain, Brussels, Belgium (March-June 2011).
5. Supervision of the undergraduate research project of Ana Pragana and Diogo Ferreira
from the 4rd year of the Master in Pharmaceutical Sciences of the Faculty of Pharmacy of the

196
University of Lisbon, Department of Pharmaceutical Technology and Galenic Pharmacy
(July-August 2010).
Communications
Oral Communications:
A. Invited Oral Communications
1. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate using polymeric nanoparticles, VI Congresso Científico
AEFFUP - Química Terapêutica e Novas Tecnologias, 2nd November 2011, Oporto, Portugal.
B. Selected Oral Communications
1. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Zupancic E, Graça L, Préat V,
Florindo H, Multifunctional Cancer Vaccine Using Ligand-Modified Nanoparticles, 7th
Vaccine & ISV Congress, 27th - 29th October 2013, Barcelona, Spain.
2. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Videira M, Silva L, Graça L, Préat
V, Florindo H, Development of dendritic cell-targeted peptide vaccine against melanoma by
multifunctional nanoparticles, 1st International Symposium on Profiling – ISPROF, 2nd - 4rd
September 2013, Costa de Caparica, Portugal.
3. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate using polymeric nanoparticles, III Congress of the Portuguese
Society of Pharmaceutical Sciences and IX Spanish-Portuguese Conference on Controlled
Drug Delivery, 14th October 2011, Oporto, Portugal.
4. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate: coencapsulation of melanoma antigenic peptides and Toll-like
ligands as adjuvants into polymeric nanoparticles, 3rd European Science Foundation Summer
School - Nanomedicine 2011, 24th June 2011, Lutherstadt Wittenberg, Germany.
5. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate: coencapsulation of a melanoma peptide antigen and CpG
oligonucleotide as an adjuvant into polymeric nanoparticles, 15th Forum of Pharmaceutical
Sciences, Belgium Society of Pharmaceutical Sciences 13th May 2011, Spa, Belgium.

Curriculum vitae
197
C. Other Oral Communications
1. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate using polymeric nanoparticles, 3rd iMed.UL Post-Graduate
Students Meeting, 12th December 2011, Faculty of Pharmacy University of Lisbon, Portugal.
2. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate using polymeric nanoparticles, Nanomedicines and Drug
Delivery Systems Group - iMed.UL, 28th July 2011, Faculty of Pharmacy University of
Lisbon, Portugal.
Panel Communications:
1. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Zupancic E, Videira M, Gaspar M,
Graça L, Préat V, Florindo H1, Ligand-modified nanoparticles as a multifunctional cancer
vaccine, 6th iMed.UL Post-Graduate Students Meeting, 2th July 2014, Faculty of Pharmacy
University of Lisbon, Portugal.
2. Silva JM, Zupancic E, Peres C, Sainz V, Videira M, Graca L, Moreira JN, Preat V &
Florindo HF, Development of multifunctional aliphatic polyester-based biodegradable
nanoparticles for vaccine delivery and immunotherapy, Showcasing Portuguese Pharma
Science Advances in Discovery, Development and Manufacturing, Hovione & MIT Portugal
Symposium, 30th June 2014, Lisbon, Portugal.
3. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Zupancic E, Graca L, Préat V and
Florindo HF, Dendritic Cell-Targeted Multifunctional Cancer Vaccine Using Ligand-
Modified Nanoparticles, 9th World Meeting on Pharmaceutics, Biopharmaceutics and
Pharmaceutical Technology (PBP 2014), 31st March – 3rd April 2014, Lisbon, Portugal.
4. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, A new nanoparticulate
platform for tumor associated antigen delivery, 9th World Meeting on Pharmaceutics,
Biopharmaceutics and Pharmaceutical Technology (PBP 2014), 31st March – 3rd April 2014,
Lisbon, Portugal.
5. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, Novel
immunotherapeutic approach against breast cancer, Informa’s annual Vaccines: Formulation
Development, Manufacturing and Novel Production Technologies, December 2013, Brussels,
Belgium.
6. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, Design of a new
nanoparticulate platform to deliver tumor associated antigens, 7th Vaccine & ISV Congress,
27th - 29th October 2013, Barcelona, Spain.

198
7. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Zupancic E, Graca L, Préat V and
Florindo HF, Ligand-Modified Nanoparticles as a Multifunctional Approach for a Cancer
Vaccine, II Simpósio Nacional de Nanociência e Nanotecnologia Biomédica, Universidade
Lusófona, 11th October 2013, Lisbon, Portugal.
8. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, Novel nanoparticulate
platform for delivery of tumour associated antigens, II Simpósio Nacional de Nanociência e
Nanotecnologia Biomédica, Universidade Lusófona, 11th October 2013, Lisbon, Portugal.
9. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, Design of a new
nanoparticulate platform to deliver tumor associated antigens, 13th International Conference
Progress in Vaccination Against Cancer (PIVAC13), October 2013, Amsterdam, Netherlands.
10. Silva JM, Vandermeulen G, Oliveira G, Pinto S, Videira M, Graca L, Préat V and
Florindo HF, Development of Dendritic Cell-Targeted Peptide Vaccine Against Melanoma by
Ligand-Modified Nanoparticles, 40th Annual Meeting & Exposition of the Controlled Release
Society, 21st - 24rd July 2013, Honolulu, Hawaii, USA.
11. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Zupancic E, Videira M, Graca L,
Préat V, Florindo H, Dendritic Cell-Targeted Multifunctional Cancer Vaccine Using Ligand-
Modified Nanoparticles, 5th iMed.UL Post-Graduate Students Meeting, 18th July 2013,
Faculty of Pharmacy University of Lisbon, Portugal.
12. Zupancic E, Silva JM, Videira M, Moreira JN, Florindo HF, Design of a new
immunotherapeutic approach against breast cancer, 5th iMed.UL Post-Graduate Students
Meeting, 18th July 2013, Faculty of Pharmacy University of Lisbon, Portugal.
13. Silva JM, Vandermeulen G, Oliveira V, Pinto S, Graca L, Préat V and Florindo HF,
Dendritic Cell-Targeted Multifunctional Vaccine Against Melanoma Using Ligand-Modified
Nanoparticles, 38th Federation of European Biochemical Societies (FEBS) Congress
“Mechanisms in Biology”, 6th - 11th July 2013, St. Petersburg, Russia.
14. Silva JM, Pinto S, Vandermeulen G, Videira M, Florindo H, Préat V, Dendritic cell-
targeting EMBO 1st International Workshop on Molecular Medicine of Sphingolipids,
October 2012, Ramot, Israel.
15. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate using polymeric nanoparticles, XXVI Scientific Meeting GTRV
– Group Thematique de Recherche sur la Vectorisation, 5th December 2011, Université
Catholique de Louvain, Brussels, Belgium.
16. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate: coencapsulation of melanoma antigenic peptides and Toll-like

Curriculum vitae
199
ligands as adjuvants into polymeric nanoparticles, 38th Annual Meeting & Exposition of the
Controlled Release Society – Innovative and Low-cost Technologies for Healthcare and
Consumer Products, 2nd August 2011, Maryland, USA.
17. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate: coencapsulation of melanoma antigenic peptides and Toll-like
ligands as adjuvants into polymeric nanoparticles, Louvain Drug Research Institute
Inauguration, March 12th 2011, Université Catholique de Louvain, Brussels, Belgium.
18. Silva JM, Vandermeulen G, Videira M, Florindo H, Préat V, Design of a therapeutic
melanoma vaccine candidate: coencapsulation of a melanoma peptide antigen and a CpG
oligonucleotides as an adjuvant into polymeric nanoparticles using polymeric nanoparticles,
2nd iMed.UL Post-Graduate Students Meeting, 3rd December 2010, Faculty of Pharmacy
University of Lisbon, Portugal.





















