
Unilateral periventricular leukomalacia in association with pyruvate dehydrogenase deficiency
3
Unilateral periventricular leukomalacia in association with pyruvate dehydrogenase deficiency RUCHI SHARMA 1 | MARK J SHARRARD 2 | DANIEL J CONNOLLY 3 | SANTOSH R MORDEKAR 1 1 Department of Paediatric Neurology. 2 Department of Paediatric Metabolic Medicine. 3 Department of Paediatric Radiology, Sheffield Children's Hospital, Sheffield, UK. Correspondence to Dr Santosh R Mordekar, Ryegate Children's Centre, Tapton Crescent, Sheffield S10 5DD, UK. E-mail: [email protected] This article is commented on by Brown on pages 395–396 of this issue. PUBLICATION DATA Accepted for publication 10th July 2011. Published online 7th September 2011. ABBREVIATIONS PDH Pyruvate dehydrogenase PVL Periventricular leukomalacia Pyruvate dehydrogenase (PDH) deficiency is a major cause of primary lactic acidosis and neurological dysfunction in infancy and early childhood. A deficiency of PDH E1 alpha, a subunit of the PDH complex, is a prominent cause of congenital lactic acidosis. We describe a female infant born at term and delivered by emergency Caesarean section because of fetal distress. There was no parental consanguinity. She presented at 5 months of age with failure to thrive, microcephaly, hypertonia, and developmental impairment. Her plasma and cerebrospinal fluid lactate were raised. She had raised plasma pyruvate with a normal lactate–pyruvate ratio. Magnetic resonance imaging of the brain showed a focal dilatation of the right lateral ventricle with unilateral periventricular leukomalacia (PVL) with subependymal cyst. Skin fibroblast culture assay revealed PDH deficiency, confirmed by mutation analysis of the E1 alpha subunit. At 18 months of age, she has hypertonia and global impairment and is making slow progress. Denver II assessment showed delay in gross motor, fine motor, adaptive, personal, social, and language categories. She has been treated with dichloroacetate and a ketogenic diet since the age of 10 and 13 months respectively, without any side effects. To our knowledge, unilateral PVL as a neuroradiological feature has not been described in children with PDH deficiency. PDH deficiency should be considered as a differential diagnosis if PVL is unilateral and if the perinatal history is not typical of PVL. Defects in the pyruvate dehydrogenase (PDH) complex are an important cause of primary lactic acidosis, a frequent manifes- tation of metabolic disease in children. Clinical symptoms can vary considerably in individuals with PDH deficiency and may be due to a defect in the E1, E2, E3, X-lipoate, or PDH phos- phatase component of the complex. The great majority of PDH deficiencies result from mutations in the X-linked PDH (E1) alpha subunit gene (PDHA1). Although males are more frequently affected, the defect also occurs in females, as is evident in our case. 1 Various clinical presentations have been described, includ- ing congenital lactic acidosis, callosal agenesis, 1 infantile spasms, 2 and other types of early-onset epilepsy, Leigh encephalopathy, recurrent ataxia, and chronic neuropathy. 3 Our female infant has a focal dilatation of the right lateral ventricle with unilateral periventricular leukomalacia (PVL) in association with PDH deficiency, which has not been previ- ously described. Consent has been obtained the family for publication of the case report. CASE REPORT A term female infant was born as a first child of her healthy, non-consanguineous parents. The mother had hypertension during pregnancy from 24 weeks, which required medication. The fetus was noted to be small for date in utero. She was born with a birthweight of 2.214kg (<0.4th centile) by emergency Caesarean section for fetal distress. Apgar scores were nine at 1 minute and nine at 5 minutes. She did not require special care baby unit admission. At 5 months of age, she was referred to hospital for failure to thrive and concerns about motor development. Her weight was 5.07kg (<0.4th centile), height 81cm (between 0.4th and 2nd centile), and head circumference 37.8cm (<0.4th centile). On examination she had a left eye intermittent convergent squint, upper limb hypertonia, and global developmental impairment. Investigations included normal toxoplasmosis, rubella, cytomegalovirus, and herpes screening, skull radio- graph, karyotype, and a coeliac screen. Urinary organic acid analysis showed increased lactate excretion together with increased fumarate, glutarate, and 2-ketoglutarate. Her fasting and postfeed lactate was high (2.9mmol ⁄ L prefeed; 5.5mmol ⁄ L post feed). Her plasma pyruvate levels were 216lmol ⁄ L (typical range 45–90lmol ⁄ L) giving her a lactate– pyruvate ratio of 14. Cerebrospinal fluid lactate was 3.3 mmol ⁄ L. This strongly suggested the possibility of a defect in energy metabolism. Magnetic resonance imaging ª The Authors. Developmental Medicine & Child Neurology ª 2011 Mac Keith Press DOI: 10.1111/j.1469-8749.2011.04108.x 469 DEVELOPMENTAL MEDICINE & CHILD NEUROLOGY CASE REPORT
-
Upload
ruchi-sharma -
Category
Documents
-
view
216 -
download
3
Transcript of Unilateral periventricular leukomalacia in association with pyruvate dehydrogenase deficiency

Unilateral periventricular leukomalacia in association with pyruvatedehydrogenase deficiency
RUCHI SHARMA1 | MARK J SHARRARD2 | DANIEL J CONNOLLY3 | SANTOSH R MORDEKAR1
1 Department of Paediatric Neurology. 2 Department of Paediatric Metabolic Medicine. 3 Department of Paediatric Radiology, Sheffield Children's Hospital, Sheffield, UK.
Correspondence to Dr Santosh R Mordekar, Ryegate Children's Centre, Tapton Crescent, Sheffield S10 5DD, UK. E-mail: [email protected]
This article is commented on by Brown on pages 395–396 of this issue.
PUBLICATION DATA
Accepted for publication 10th July 2011.Published online 7th September 2011.
ABBREVIATIONSPDH Pyruvate dehydrogenasePVL Periventricular leukomalacia
Pyruvate dehydrogenase (PDH) deficiency is a major cause of primary lactic acidosis and
neurological dysfunction in infancy and early childhood. A deficiency of PDH E1 alpha, a subunit of
the PDH complex, is a prominent cause of congenital lactic acidosis. We describe a female infant
born at term and delivered by emergency Caesarean section because of fetal distress. There was no
parental consanguinity. She presented at 5 months of age with failure to thrive, microcephaly,
hypertonia, and developmental impairment. Her plasma and cerebrospinal fluid lactate were raised.
She had raised plasma pyruvate with a normal lactate–pyruvate ratio. Magnetic resonance imaging
of the brain showed a focal dilatation of the right lateral ventricle with unilateral periventricular
leukomalacia (PVL) with subependymal cyst. Skin fibroblast culture assay revealed PDH deficiency,
confirmed by mutation analysis of the E1 alpha subunit. At 18 months of age, she has hypertonia
and global impairment and is making slow progress. Denver II assessment showed delay in gross
motor, fine motor, adaptive, personal, social, and language categories. She has been treated with
dichloroacetate and a ketogenic diet since the age of 10 and 13 months respectively, without any
side effects. To our knowledge, unilateral PVL as a neuroradiological feature has not been described
in children with PDH deficiency. PDH deficiency should be considered as a differential diagnosis if
PVL is unilateral and if the perinatal history is not typical of PVL.
Defects in the pyruvate dehydrogenase (PDH) complex are animportant cause of primary lactic acidosis, a frequent manifes-tation of metabolic disease in children. Clinical symptoms canvary considerably in individuals with PDH deficiency and maybe due to a defect in the E1, E2, E3, X-lipoate, or PDH phos-phatase component of the complex. The great majority ofPDH deficiencies result from mutations in the X-linked PDH(E1) alpha subunit gene (PDHA1). Although males are morefrequently affected, the defect also occurs in females, as isevident in our case.1
Various clinical presentations have been described, includ-ing congenital lactic acidosis, callosal agenesis,1 infantilespasms,2 and other types of early-onset epilepsy, Leighencephalopathy, recurrent ataxia, and chronic neuropathy.3
Our female infant has a focal dilatation of the right lateralventricle with unilateral periventricular leukomalacia (PVL) inassociation with PDH deficiency, which has not been previ-ously described. Consent has been obtained the family forpublication of the case report.
CASE REPORTA term female infant was born as a first child of her healthy,non-consanguineous parents. The mother had hypertension
during pregnancy from 24 weeks, which required medication.The fetus was noted to be small for date in utero. She was bornwith a birthweight of 2.214kg (<0.4th centile) by emergencyCaesarean section for fetal distress. Apgar scores were nine at1 minute and nine at 5 minutes. She did not require specialcare baby unit admission.
At 5 months of age, she was referred to hospital for failureto thrive and concerns about motor development. Her weightwas 5.07kg (<0.4th centile), height 81cm (between 0.4th and2nd centile), and head circumference 37.8cm (<0.4th centile).
On examination she had a left eye intermittent convergentsquint, upper limb hypertonia, and global developmentalimpairment. Investigations included normal toxoplasmosis,rubella, cytomegalovirus, and herpes screening, skull radio-graph, karyotype, and a coeliac screen. Urinary organic acidanalysis showed increased lactate excretion together withincreased fumarate, glutarate, and 2-ketoglutarate. Her fastingand postfeed lactate was high (2.9mmol ⁄ L prefeed;5.5mmol ⁄ L post feed). Her plasma pyruvate levels were216lmol ⁄ L (typical range 45–90lmol ⁄ L) giving her a lactate–pyruvate ratio of 14. Cerebrospinal fluid lactate was3.3 mmol ⁄ L. This strongly suggested the possibility of adefect in energy metabolism. Magnetic resonance imaging
ª The Authors. Developmental Medicine & Child Neurology ª 2011 Mac Keith Press DOI: 10.1111/j.1469-8749.2011.04108.x 469
DEVELOPMENTAL MEDICINE & CHILD NEUROLOGY CASE REPORT
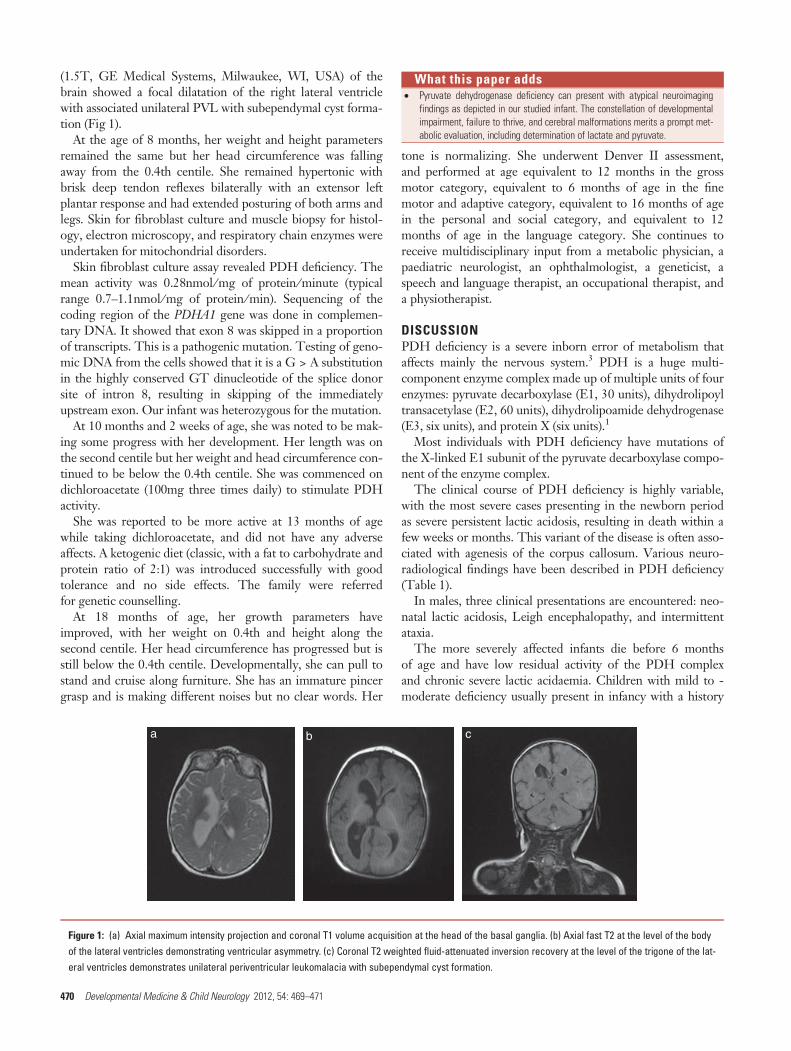
(1.5T, GE Medical Systems, Milwaukee, WI, USA) of thebrain showed a focal dilatation of the right lateral ventriclewith associated unilateral PVL with subependymal cyst forma-tion (Fig 1).
At the age of 8 months, her weight and height parametersremained the same but her head circumference was fallingaway from the 0.4th centile. She remained hypertonic withbrisk deep tendon reflexes bilaterally with an extensor leftplantar response and had extended posturing of both arms andlegs. Skin for fibroblast culture and muscle biopsy for histol-ogy, electron microscopy, and respiratory chain enzymes wereundertaken for mitochondrial disorders.
Skin fibroblast culture assay revealed PDH deficiency. Themean activity was 0.28nmol ⁄ mg of protein ⁄ minute (typicalrange 0.7–1.1nmol ⁄ mg of protein ⁄ min). Sequencing of thecoding region of the PDHA1 gene was done in complemen-tary DNA. It showed that exon 8 was skipped in a proportionof transcripts. This is a pathogenic mutation. Testing of geno-mic DNA from the cells showed that it is a G > A substitutionin the highly conserved GT dinucleotide of the splice donorsite of intron 8, resulting in skipping of the immediatelyupstream exon. Our infant was heterozygous for the mutation.
At 10 months and 2 weeks of age, she was noted to be mak-ing some progress with her development. Her length was onthe second centile but her weight and head circumference con-tinued to be below the 0.4th centile. She was commenced ondichloroacetate (100mg three times daily) to stimulate PDHactivity.
She was reported to be more active at 13 months of agewhile taking dichloroacetate, and did not have any adverseaffects. A ketogenic diet (classic, with a fat to carbohydrate andprotein ratio of 2:1) was introduced successfully with goodtolerance and no side effects. The family were referredfor genetic counselling.
At 18 months of age, her growth parameters haveimproved, with her weight on 0.4th and height along thesecond centile. Her head circumference has progressed but isstill below the 0.4th centile. Developmentally, she can pull tostand and cruise along furniture. She has an immature pincergrasp and is making different noises but no clear words. Her
tone is normalizing. She underwent Denver II assessment,and performed at age equivalent to 12 months in the grossmotor category, equivalent to 6 months of age in the finemotor and adaptive category, equivalent to 16 months of agein the personal and social category, and equivalent to 12months of age in the language category. She continues toreceive multidisciplinary input from a metabolic physician, apaediatric neurologist, an ophthalmologist, a geneticist, aspeech and language therapist, an occupational therapist, anda physiotherapist.
DISCUSSIONPDH deficiency is a severe inborn error of metabolism thataffects mainly the nervous system.3 PDH is a huge multi-component enzyme complex made up of multiple units of fourenzymes: pyruvate decarboxylase (E1, 30 units), dihydrolipoyltransacetylase (E2, 60 units), dihydrolipoamide dehydrogenase(E3, six units), and protein X (six units).1
Most individuals with PDH deficiency have mutations ofthe X-linked E1 subunit of the pyruvate decarboxylase compo-nent of the enzyme complex.
The clinical course of PDH deficiency is highly variable,with the most severe cases presenting in the newborn periodas severe persistent lactic acidosis, resulting in death within afew weeks or months. This variant of the disease is often asso-ciated with agenesis of the corpus callosum. Various neuro-radiological findings have been described in PDH deficiency(Table 1).
In males, three clinical presentations are encountered: neo-natal lactic acidosis, Leigh encephalopathy, and intermittentataxia.
The more severely affected infants die before 6 monthsof age and have low residual activity of the PDH complexand chronic severe lactic acidaemia. Children with mild to -moderate deficiency usually present in infancy with a history
a b c
Figure 1: (a) Axial maximum intensity projection and coronal T1 volume acquisition at the head of the basal ganglia. (b) Axial fast T2 at the level of the bodyof the lateral ventricles demonstrating ventricular asymmetry. (c) Coronal T2 weighted fluid-attenuated inversion recovery at the level of the trigone of the lat-eral ventricles demonstrates unilateral periventricular leukomalacia with subependymal cyst formation.
What this paper adds• Pyruvate dehydrogenase deficiency can present with atypical neuroimaging
findings as depicted in our studied infant. The constellation of developmentalimpairment, failure to thrive, and cerebral malformations merits a prompt met-abolic evaluation, including determination of lactate and pyruvate.
470 Developmental Medicine & Child Neurology 2012, 54: 469–471

of developmental impairment, hypotonia, failure to thrive,and seizures. The course of the diseases is often punctuatedby bouts of severe lactic acidosis, often precipitated byintercurrent illnesses. Presentation with relapsing intermit-tent ataxia is also described in males.4
Females with PDHE1 alpha deficiency tend to have a morehomogeneous clinical presentation, although with variableseverity. This includes dysmorphic features (narrow head withfrontal bossing, wide nasal bridge with upturned nose, longphiltrum, and flared nostrils), microcephaly, moderate tosevere intellectual disability, and a spastic diplegia or quadri-plegia resembling a non-progressive encephalopathy. Seizuresare reported in almost all affected females.4 These usuallypresent within the first 6 months of life and are often presentas epileptic spasms or severe myoclonic seizures. Our infanthas not been affected by seizures to date.
Plasma lactate levels in PDH deficiency are usually persis-tently elevated. The lactate pyruvate ratio is characteristicallynormal.1 Urinary organic acid analysis in patients with E1alpha defects is unremarkable apart from the presence ofexcess lactate and some 2-hydroxybutyrate. Our infant wasunusual in that she had tricarboxylic acid cycle intermediates(fumarate and 2-ketoglutarate) in the urine. These may be
seen in neonates but not usually at this age. If present at thisage, they are usually suggestive of respiratory chain disorderrather than PDH. Our child’s respiratory chain enzyme activi-ties were within normal limits.
In children with mitochondrial encephalopathies, the mostcommon magnetic resonance imaging features are bilateralsymmetric abnormalities in the basal ganglia and brainstem.The presence of white matter abnormality has been describedonly in a few cases.5
PVL and parenchymal venous infarction complicating ger-minal matrix ⁄ intraventricular haemorrhage have long beenrecognized as the two significant white matter diseases respon-sible for the majority of cases of cerebral palsy in survivors ofpreterm birth. However, subependymal cysts are usually notassociated with PVL in survivors of preterm birth. The pres-ence of PVL with cysts should warrant further neurometabolic investigations, especially if the perinatal history isnot typical of PVL.
To our knowledge, unilateral ventricular dilatation andPVL with subependymal cyst has not been reported before inindividuals with PDH deficiency. PDH deficiency should beconsidered as a differential diagnosis if PVL is unilateral and ifthe perinatal history is not typical of PVL.
REFERENCES
1. Robinson BH. Lactic acidemia: disorders of pyruvate carbox-
ylase and pyruvate dehydrogenase. In: Scriver CR, Beaudet
AL, Sly WS, editors. The Metabolic and Molecular Basis of
Inherited Disease, Vol. 2, 8th edn. London: McGraw-Hill
Professional, 2011; 2278–82.
2. Wada N, Matsuishi T, Nonaka M, Yoshino M. Pyruvate
dehydrogenase E1 alpha subunit deficiency in a female
patient: evidence of antenatal origin of brain damage and pos-
sible etiology of infantile spasms. Brain Dev 2004; 26: 57–60.
3. Barnerias C, Saudubray JM, Touati G, et al. Pyruvate dehy-
drogenase complex deficiency: four neurological phenotypes
with differing pathogenesis. Dev Med Child Neurol 2010; 52:
e1–9.
4. De Meirleir LJ, Vancoster R, Lissens W. Disorders of pyru-
vate metabolism and the tricarboxylic acid cycle. In: Saudu-
bray J-M, van den Berghe G, Walter JH, editors. Inborn
Metabolic Diseases, 5th edn. New York: Springer, 2011:
167–8.
5. Moroni I, Bugiani M, Castelli G, Lamantea E, Uziel G.
Cerebral white matter involvement in children with
mitochondrial encephalopathies. Neuropediatrics 2002; 33:
79–85.
6. Michotte A, De Meirleir L, Lissens W, et al. Neuropatho
logical findings of a patient with pyruvate dehydrogenase E1
alpha deficiency presenting as a cerebral lactic acidosis. Acta
Neuropathol 1993; 85: 674–8.
7. Raissaki M, Grafakou O, Giannopoulos A, et al. Develop-
ment of subdural effusions in association with PDH defi-
ciency. Eur Radiol 2005; 15: 2205–20.
Table 1: Neuroradiological findings described in pyruvate dehydrogenase deficiency.
White matter abnormalities Brainstem Cortical Cerebellar Other
Periventricular pseudocysts2 Palladi and caudate nucleiabnormalities
Abnormalities of corpus callosumincluding agenesis, hypoplasiaand partial dysgenesis
Vascular proliferationon cerebellum
Subduraleffusions7
Periventricular white matterdemyelination
Abnormalities of thalamus,putamen, and dentate nuclei
Cortical atrophy, absent pyramids – –
Subcortical white matterinvolvement
– Polymicrogyria – –
Enlargement of lateral ventriclesand brain atrophy
– Heterotopic nodule of grey matterin subependymal regions7
– –
Progressive vacuolization ofaffected white matter5
– Decreased cortical sulcation andgyration
– –
Large subependymal cysts – – – –
Case Report 471


















