
Understanding the Microbiome Using QIIME
40
Understanding the Microbiome Using QIIME Hu Huang ([email protected]) Biomedical Informatics & Computational Biology University of Minnesota IIHG 2014 Bioinforma=cs Short course
Transcript of Understanding the Microbiome Using QIIME

Understanding the Microbiome Using QIIME
Hu Huang ([email protected])
Biomedical Informatics & Computational Biology
University of Minnesota
IIHG 2014 Bioinforma=cs Short course

Outline
! Introduction to QIIME
! QIIME workflow
! SourceTracker
! MWAS package

What is QIIME?
! QIIME (“chime”, Quantitative Insights Into Microbial Ecology)
! An open-source pipeline written in Python
! Wraps the popular algorithms rather than re-implements them
! Comparison and analysis of microbial communities
! Supports a variety of sequencing platforms

What is QIIME?
Hamady et al. Error-correcting barcodes for pyrosequencing hundreds of samples in multiplex. Nature Methods, 2008

What is QIIME?
! Third-party packages (dependencies) integrated in
QIIME
! Latest version: 1.8.0
! GreenGenes version: v13_8
! USEARCH:
! v5.2.236 : USEARCH
! v6.1: USEARCH61
! Other op=onal packages
! Cytoscape – visualiza=on
! SourceTracker
! R 3.0 – supervised learning
1. Python-‐2.7.0 17. cd-‐hit
2. QIIME-‐1.8.0 18. rdp-‐classifier-‐2.2
3. Setuptools 19. blast
4. MySQL-‐python 20. muscle
5. SQLAIchemy 21. infernal
6. PyCogent-‐1.5.3 22. cytoscapeSource
7. PyNAST-‐1.2.2 23. Clearcut.source
8. NumPy-‐1.7.1 24. Muther
9. Matplotlib-‐1.3.1 25. uclustq
10. Mpi4py 26. R 3.0.2
11. Lxml 27. AmpliconNoise
12. Sphinx 28. ViennaRNA
13. RAxML 29. pprospector
14. FastTree 30. microbiomeu=l
15. cdbfasta 31. Biom-‐format-‐1.3.1
16. Qcli-‐0.1.0 32. Emperor-‐0.9.3 ... ...

Why use QIIME? ! Integrated most popular functions and packages ! Constantly evolving – well maintained and updating regularly
! The code is tested properly
! Support multiple sequencing platforms (454, Illumina...)

QIIME Installa3on
! New version (v1.8.0) provides multiple easy options ! Virtual Machine version based on Ubuntu - QIIME Virtual Box
! All dependencies are pre-installed ! Based on Ubuntu system ! http://qiime.org/install/virtual_box.html
! Mac OS X version – MacQIIME ! Automated installation steps ! Jeff Werner Lab (http://www.wernerlab.org/software/macqiime)
! Linux systems – QIIME-deploy ! Ubuntu, CentOS and RedHat ! Either v1.8.0 or v1.8.0dev ! GitHub (https://github.com/qiime/qiime-deploy)
! Installing using pip ! Manually installing QIIME and dependencies
! http://qiime.org/install/install.html?highlight=usearch#manually-installing-qiime

QIIME Installa3on
! Installing QIIME and dependencies using pip on Mac
1. Install Homebrew (http://brew.sh/)
2. Run command: brew install gfortran
3. Run command: sudo easy_install pip
4. Run command: sudo pip install numpy==1.7.1
5. Run command: pip install qiime
6. Dependencies: Pyqi and other others if needed
! Notes: in steps 4 and 5, there is an Apple bug with Xcode 5.1, must run as: sudo ARCHFLAGS=-Wno-error=unused-command-line-argument-hard-error-in-future pip install numpy==1.7.1

QIIME Installa3on
! Configuring QIIME and dependencies
1. Dependencies: Pyqi and others if needed hap://qiime.org/install/install.html?highlight=usearch#manually-‐installing-‐qiime
2. Necessary Data files (hap://qiime.org/home_sta=c/dataFiles.html)
1) GreenGenes core set sequence file 2) GreenGenes alignment landmask file 3) Marker gene reference OTUs, taxonomies and trees 4) GreenGenes version: latest version is v13_8, but could also use v13_5 GreenGenes v13_5: hap://greengenes.secondgenome.com/downloads/database/13_5
3. Set up qiime_config file (http://qiime.org/install/qiime_config.html)
1) Customize QIIME environment
2) Could only change the necessary values

QIIME Installa3on
! print_qiime_cofig.py

QIIME Workflow
DeLong, Ed. Microbial Metagenomics, Metatranscriptomics, and Metaproteomics. Vol. 531. p.378, Academic Press, 2013

QIIME supported files
! Sequence files ! .fastq ! .fasta/.fna and/or .qual
! Mapping file (.txt) ! links sequences with sample IDs ! contains all metadata ! Tab-delimited text file ! Format:

QIIME supported files
! OTU table ! Classical format - (sample X OTU matrix)
! Human-friendly – readable ! May use a lot of storage space
OTU identifiers
Sample identifiers
OTU taxonomic information

QIIME supported files
! OTU table ! BIOM format - (sample X observation contingency matrix)
! Space efficient ! Include more metadata ! Human-unfriendly (hard to read)

QIIME workflow
! Preprocessing ! Sequence file format conversion
! .fastq to .fasta + .qual
convert_fastaqual_fastq.py -c fastq_to_fastaqual -f
seqs.fastq -o fastaqual/
! .fasta + .qual to .fastq
convert_fastaqual_fastq.py -f seqs.fasta -q seqs.qual
-o fastqfiles/

QIIME workflow: Preprocessing
! Quality control
quality_scores_plot.py
-q seqs.qual
-o quality_histogram/
! Truncate bad read locations
truncate_fasta_qual_files.py -f seqs.fna -q seqs.qual -b 100
-o filtered100/

QIIME workflow: Preprocessing
! Phred Quality Score: Q = - 10 log10 P
! P : base-calling error probability (system error rate)
! Commonly used threshold: Q = 25 (or P = 0.32 % or reads accuracy = 99.68%)
Q = 20 (or P = 0.20% or reads accuracy = 98 %)
! (99%) ^ 10 = 90.43%; (99%) ^ 20 = 81.79%; (99%) ^ 50 = 60.50%; (99%) ^100 = 36.60%
!

QIIME workflow: Preprocessing
! Multiplexed sequence structure
! Demultiplexing requires a valid mapping file
validate_mapping_file.py -m mapfile.txt -o mapping_output
Adapter 1 Barcode Linker Primer Desired sequence Reverse Primer Adapter 2
This area is all we need.

QIIME workflow: Preprocessing
! Demultiplexing, removing primers/barcodes
split_libraries.py -m mapfile.txt -f seqs.fasta -b 10 -l 50 -o slout/
split_libraries_fastq.py -i seqs.fastq -b seqs_barcodes.fastq --barcode_type 10 -o slout_r3_q20/ -m mapfile.txt -q 20 –r 3
! Other useful commands ! Count sequences count_seqs.py -i seqs.fna
! Reverse complement sequences adjust_seq_orientation.py -i seqs.fna
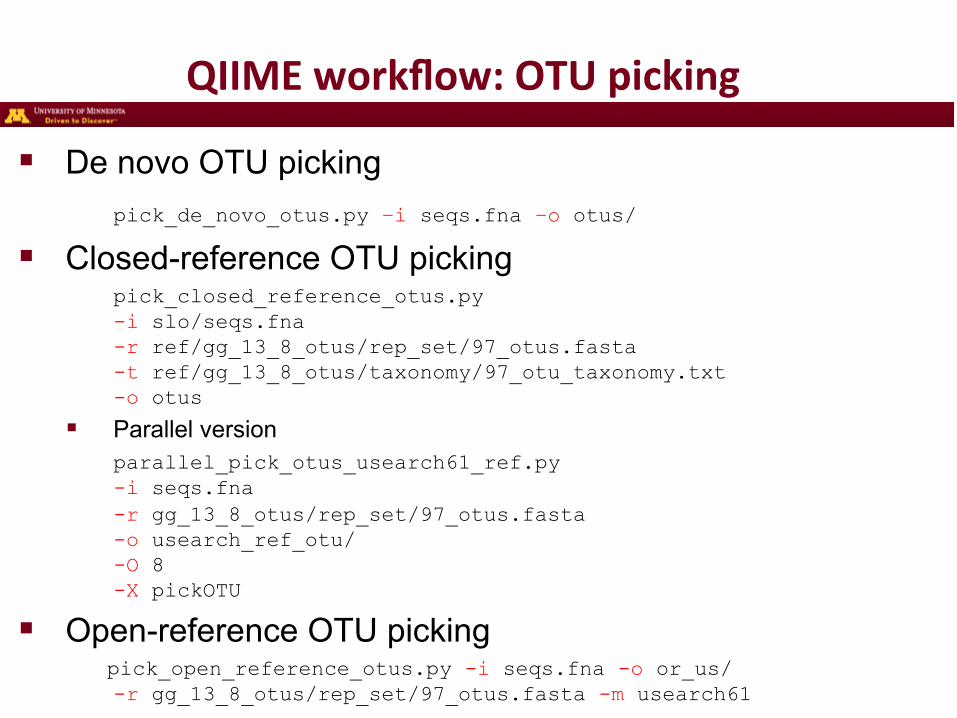
QIIME workflow: OTU picking
! De novo OTU picking pick_de_novo_otus.py –i seqs.fna –o otus/
! Closed-reference OTU picking pick_closed_reference_otus.py -i slo/seqs.fna -r ref/gg_13_8_otus/rep_set/97_otus.fasta -t ref/gg_13_8_otus/taxonomy/97_otu_taxonomy.txt -o otus
! Parallel version parallel_pick_otus_usearch61_ref.py -i seqs.fna -r gg_13_8_otus/rep_set/97_otus.fasta -o usearch_ref_otu/ -O 8 -X pickOTU
! Open-reference OTU picking pick_open_reference_otus.py -i seqs.fna -o or_us/ -r gg_13_8_otus/rep_set/97_otus.fasta -m usearch61

QIIME workflow: BIOM table
! pick_OTU Output
! Make OTU BIOM table make_otu_table.py -i seqs_otus.txt –t /gg_13_8_otus/taxonomy/97_otu_taxonomy.txt –o seqs_otus.biom
biom add-metadata -i seqs_otus.biom -o biom-taxa.biom --observation-metadata-fp /panfs/roc/groups/8/knightsd/public/gg_13_8_otus/taxonomy/97_otu_taxonomy.txt --observation-header "OTU_ID,taxonomy" --sc-separated taxonomy
One cluster
Cluster Center (OTU ID)
Sample ID

QIIME workflow: BIOM table
! OTU BIOM table
! Summarize BIOM table biom summarize-table -i seqs_otus.biom -o biom_summary.txt
! Convert BIOM table to classical OTU table biom convert -i seqs_otus.biom -o otu_table.txt -b --header-key taxonomy

QIIME workflow: Summarize Taxa
! Taxonomic levels (L1~L7) L1: Kingdom level, e.g. k__Bacteria L2: Phylum level, e.g. k__Bacteria;p__Acidobacteria
L3: Class level, e.g. k__Bacteria;p__Acidobacteria;c__Chloracidobacteria L4: Oder level, e.g. k__Bacteria;p__Acidobacteria;c__Solibacteres;o__Solibacterales L5: Family level,
e.g. k__Bacteria;p__Actinobacteria;c__Actinobacteria (class);o__Acidimicrobiales;f__CL500-29 L6: Genus level,
e.g. k__Bacteria;p__Actinobacteria;c__Actinobacteria (class);o__Actinomycetales; f__Actinosynnemataceae;g__Lentzea
L7: Species level, e.g. k__Bacteria;p__Actinobacteria;c__Actinobacteria; o__Bifidobacteriales;f__Bifidobacteriaceae; g__Bifidobacterium;s__breve
! Taxa plots – pie, bar, area charts summarize_taxa_through_plots.py -i seqs_otus.biom -o taxa_summary -m mapfile.txt -p summarize_param.txt -c SAMPTYPE

QIIME workflow: Diversity Analysis
! Alpha diversity ! Distance op=ons:
PD_whole_tree, observed_species, Chao1, Shannon!alpha_rarefaction.py -i seqs_otus.biom -m mapfile.txt -o alpha_div/ -p alpha_params.txt -t /panfs/roc/groups/8/knightsd/public/gg_13_8_otus/trees/97_otus.tree
! Beta diversity– PCoA plots in 3D !beta_diveristy_through_plots.py -i seqs_otus.biom -m mapfiles.txt -o beta_div/ -t /panfs/roc/groups/8/knightsd/public/gg_13_8_otus/trees/97_otus.tree -e 2000

Microbial Source Tracking
! Community-‐wide microbial source tracking
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.
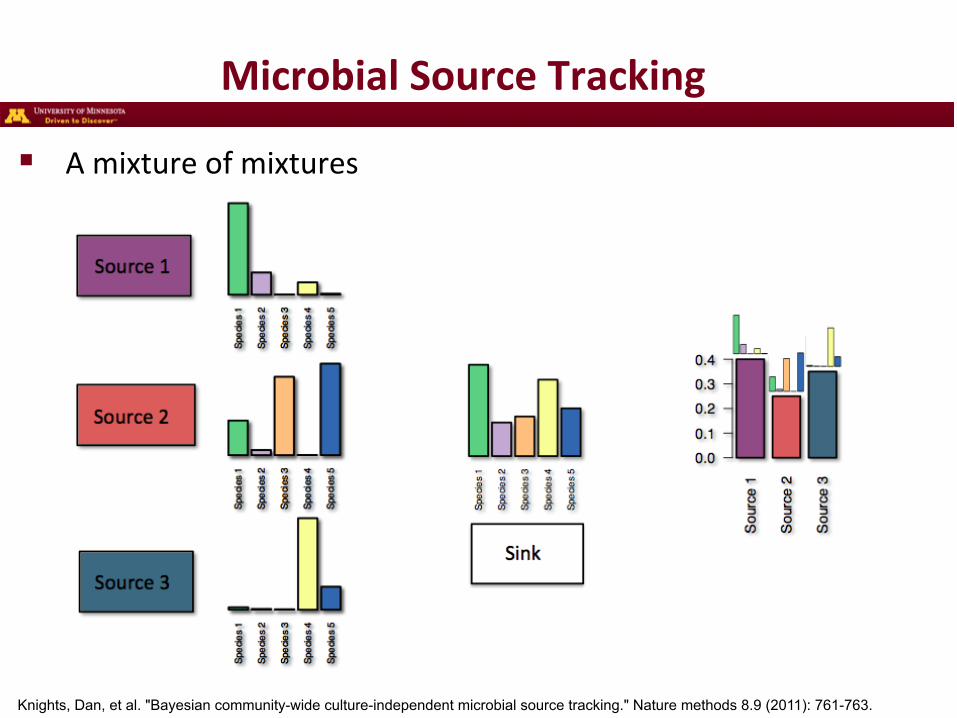
Microbial Source Tracking
! A mixture of mixtures
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.

Microbial Source Tracking
! Community-‐wide microbial source tracking
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.

Microbial Source Tracking
! A mixture of mixtures
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.

Microbial Source Tracking – Previous work
! Linear regression ! Minimize
! Naive Bayes ! Assumes independence of features ! Not a mixture model
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.

Microbial Source Tracking – SourceTacker
! Probabilis=c Topic models ! Idea: each document is some mix of topics ! Each word in the document belongs to a topic
! Latent Dirichlet Alloca=on (LDA) with some known priors ! Use Gibbs Sampling (Markov Chain Monte Carlo)
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.
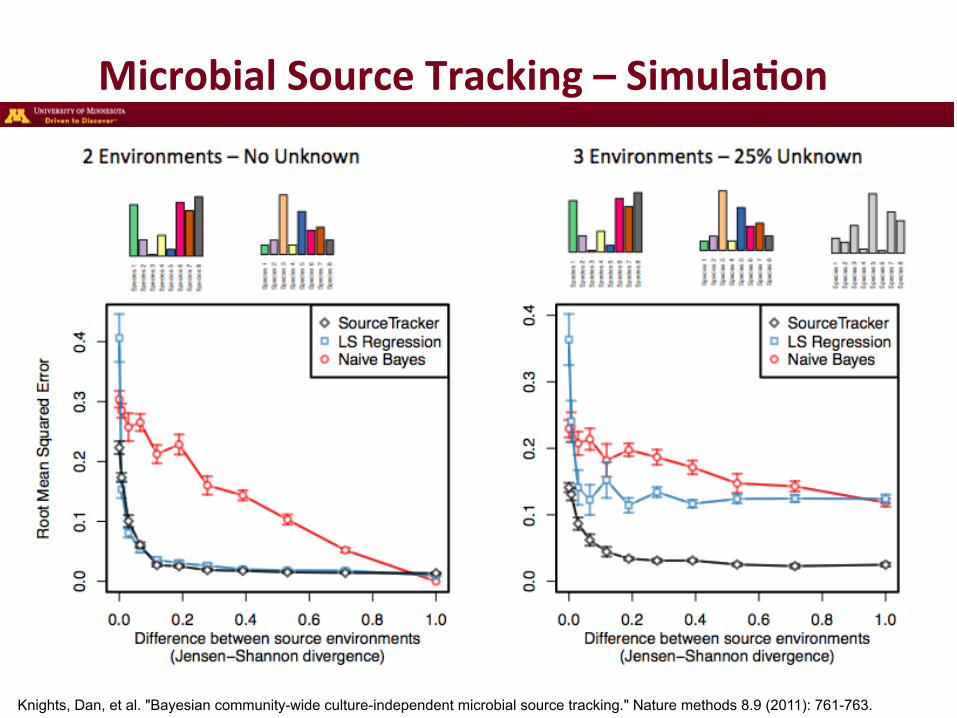
Microbial Source Tracking – Simula3on
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.
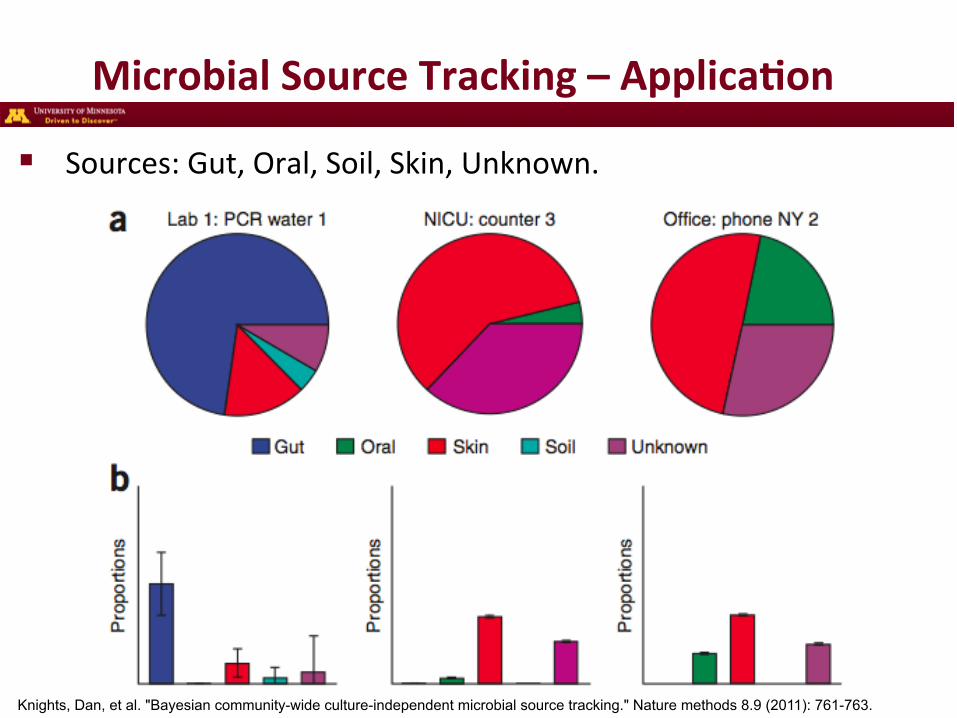
Microbial Source Tracking – Applica3on
Knights, Dan, et al. "Bayesian community-wide culture-independent microbial source tracking." Nature methods 8.9 (2011): 761-763.
! Sources: Gut, Oral, Soil, Skin, Unknown.

Microbial Source Tracking – SourceTacker
! Configura=on
! QIIME comes with $SOURCETRACKER_PATH environment variable
! Commands
echo $SOURCETRACKER_PATH
# show help information Rscript $SOURCETRACKER_PATH/sourcetracker_for_qiime.r -h # run sourcetracker Rscript $SOURCETRACKER_PATH/sourcetracker_for_qiime.r -i otus.txt -m map.txt -o st_output -r 100 -n 10

Online Resources
! QIIME Documents
! hap://qiime.org/tutorials/index.html
! Knights Lab Wiki
! haps://sites.google.com/site/knightslabwiki/

Microbiome-‐wise Associate Study (MWAS) package
! Extend the func=ons already integrated in QIIME ! All func=ons are implemented in R ! Will be released soon!

Microbiome-‐wise Associate Study (MWAS) package
! Machine learning techniques ! QIIME only has Random Forest classifier supervised_learning.py -i otu_table.biom -m map.txt -c Treatment -o ml_output
! New features: ! Feature selec=on ! Support vector machines (SVM)
! Radial basis kernel (RBF) ! Linear kernel ! Sparse UniFrac kernel
! Mul=nomial logis=c regression

Microbiome-‐wise Associate Study (MWAS) package
! Statistical testing ! Available in QIIME: adonis, ANOSIM, BEST, Moran’s
I, MRPP, PERMANOVA, PERMDISP, and db-RDA
! New features: ! Effect size ! Power calcula=on ! This feature is also available independently
as a web applica=on ! Web server URL will be released soon!

MWAS package

MWAS package
! Visualization ! Available in QIIME: PCoA plots, heatmaps, OTU-sample
network, area/pie/bar charts
! New features: ! Customized heatmap
-‐ showing mul=dimensional factors
! Super-‐node PCoA plots -‐ reflec=ng taxonomic composi=on on PCoA plots
! Gradient PCoA plots with color gradient
! ROC curve from the classifier training

Acknowledgement
Dr. Dan Knights (PI)
Pajau (PJ) Vangay
Dr. Tonya Ward


















