Understanding rheumatic fever
8
REVIEW ARTICLE Understanding rheumatic fever Pedro Ming Azevedo • Rosa Rodrigues Pereira • Luiza Guilherme Received: 21 April 2011 / Accepted: 10 September 2011 / Published online: 28 September 2011 Ó Springer-Verlag 2011 Abstract Through a comprehensive review of the recent findings on rheumatic fever, we intend to propose a new physiopathologic model for this disease. A Medline search was performed for all articles containing the terms rheu- matic fever or rheumatic heart disease in title or abstract from 1970 to 2011. Best evidence qualitative technique was used to select the most relevant. The scientific interest on rheumatic fever has notably diminished throughout the twentieth century as evidenced by the comparison of the proportion of articles in which RF was a subject in 1950 (0.26%) and today (0.03%) [Pubmed]. However, RF remains a major medical and social problem in the devel- oping world and in the so-called hotspots, where it still causes around 500.000 deaths each year, not too different from the pre-antibiotic era. The role of genetic factors in RF susceptibility is discussed. Familiar aggregation, simi- larity of disease patterns between siblings, identical twin, and HLA correlation studies are evidence for a genetic influence on RF susceptibility. The suspect-involved genes fall mainly into those capable of immunologic mediation. Molecular mimicry explains the triggering of RF, but an intense and sustained inflammation is needed to cause sequels. Also, RF patients vary greatly in terms of symp- toms. It is likely that a genetic background directing immune response towards a predominantly Th1 or Th2 pattern contributes to these features. The recent findings on rheumatic fever provide important insight on its physio- pathology that helps understanding this prototype post- infectious autoimmune disease giving insights on other autoimmune conditions. Keywords Rheumatic fever Á Genetics Á Immunology Á Physiopathology Introduction Incidence of rheumatic fever has dramatically declined in the developed world, where it remains around 0.2–1.9 per 100.000 habitants [1], mainly due to sporadic outbreaks. The scientific interest on this disease has proportionally diminished as evidenced by the comparison of the propor- tion of articles in which RF was a subject in 1950 (0.26%) and today (0.03%) [Pubmed]. However, RF remains a major medical and social problem in the developing world and in the so-called hotspots, where the incidence still is from 20 to 51 per 100.000 habitants, causing around 500.000 deaths each year [2], not too different from the pre-antibiotic era. Also RF is the prototype of post-infectious autoimmune disease. It is likely that understanding RF will help to decipher other autoimmune diseases. Rheumatic fever (RF) is an inflammatory disease that affects susceptible children and teenagers (aged 3–19 years) [3]. It is mediated by humoral and cellular autoimmune responses that occur as delayed sequelae of Streptococcus pyogenes infection [4]. Bacterial factors are important determinants of disease acquisition, but only 0.3–3% of individual infected by a Streptococcus cepa known to be rheumatogenic will develop disease [3, 5]. Environmental factors such as poor quality of life and limited access to medical facilities partially explain susceptibility, but indi- vidual factors are known to play a role. A small proportion of patients develop the full picture of symptoms (American P. M. Azevedo (&) Á R. R. Pereira Á L. Guilherme Universidade de Sao Paulo, Sao Paulo, Brazil e-mail: [email protected] 123 Rheumatol Int (2012) 32:1113–1120 DOI 10.1007/s00296-011-2152-z
Transcript of Understanding rheumatic fever

REVIEW ARTICLE
Understanding rheumatic fever
Pedro Ming Azevedo • Rosa Rodrigues Pereira •
Luiza Guilherme
Received: 21 April 2011 / Accepted: 10 September 2011 / Published online: 28 September 2011
� Springer-Verlag 2011
Abstract Through a comprehensive review of the recent
findings on rheumatic fever, we intend to propose a new
physiopathologic model for this disease. A Medline search
was performed for all articles containing the terms rheu-
matic fever or rheumatic heart disease in title or abstract
from 1970 to 2011. Best evidence qualitative technique
was used to select the most relevant. The scientific interest
on rheumatic fever has notably diminished throughout the
twentieth century as evidenced by the comparison of the
proportion of articles in which RF was a subject in 1950
(0.26%) and today (0.03%) [Pubmed]. However, RF
remains a major medical and social problem in the devel-
oping world and in the so-called hotspots, where it still
causes around 500.000 deaths each year, not too different
from the pre-antibiotic era. The role of genetic factors in
RF susceptibility is discussed. Familiar aggregation, simi-
larity of disease patterns between siblings, identical twin,
and HLA correlation studies are evidence for a genetic
influence on RF susceptibility. The suspect-involved genes
fall mainly into those capable of immunologic mediation.
Molecular mimicry explains the triggering of RF, but an
intense and sustained inflammation is needed to cause
sequels. Also, RF patients vary greatly in terms of symp-
toms. It is likely that a genetic background directing
immune response towards a predominantly Th1 or Th2
pattern contributes to these features. The recent findings on
rheumatic fever provide important insight on its physio-
pathology that helps understanding this prototype post-
infectious autoimmune disease giving insights on other
autoimmune conditions.
Keywords Rheumatic fever � Genetics � Immunology �Physiopathology
Introduction
Incidence of rheumatic fever has dramatically declined in
the developed world, where it remains around 0.2–1.9 per
100.000 habitants [1], mainly due to sporadic outbreaks.
The scientific interest on this disease has proportionally
diminished as evidenced by the comparison of the propor-
tion of articles in which RF was a subject in 1950 (0.26%)
and today (0.03%) [Pubmed]. However, RF remains a major
medical and social problem in the developing world and in
the so-called hotspots, where the incidence still is from 20
to 51 per 100.000 habitants, causing around 500.000 deaths
each year [2], not too different from the pre-antibiotic era.
Also RF is the prototype of post-infectious autoimmune
disease. It is likely that understanding RF will help to
decipher other autoimmune diseases.
Rheumatic fever (RF) is an inflammatory disease that
affects susceptible children and teenagers (aged 3–19 years)
[3]. It is mediated by humoral and cellular autoimmune
responses that occur as delayed sequelae of Streptococcus
pyogenes infection [4]. Bacterial factors are important
determinants of disease acquisition, but only 0.3–3% of
individual infected by a Streptococcus cepa known to be
rheumatogenic will develop disease [3, 5]. Environmental
factors such as poor quality of life and limited access to
medical facilities partially explain susceptibility, but indi-
vidual factors are known to play a role. A small proportion of
patients develop the full picture of symptoms (American
P. M. Azevedo (&) � R. R. Pereira � L. Guilherme
Universidade de Sao Paulo, Sao Paulo, Brazil
e-mail: [email protected]
123
Rheumatol Int (2012) 32:1113–1120
DOI 10.1007/s00296-011-2152-z

Heart Association) [6], and only one- to two-thirds will have
carditis [7, 8]. Again, individual factor is determinant of
disease pattern and is poorly understood. Familiar aggrega-
tion [9, 10], similarity of disease patterns between siblings
and identical twin [11] and HLA correlation studies [8] are
evidence for a genetic influence on RF susceptibility and
manifestation. The suspect-involved genes fall mainly into
those capable of immunologic mediation. It is likely that a
genetic background directing immune response towards a
predominantly Th1 or Th2 pattern contributes to these fea-
tures. Molecular mimicry explains the triggering of RF, but
an intense and sustained inflammation is needed to cause
sequels. The purpose of the present article is to review the
recent findings on RF and to expose how they help to build a
physiopathological model for this disease.
Methods
A Medline search was performed for all articles containing
the terms rheumatic fever or rheumatic heart disease in title
or abstract from 1970 to 2011. ‘Best evidence’ qualitative
technique was used to select the most relevant.
Results
Genetic contribution to susceptibility
Different HLA class II antigens associations have been
observed in several populations. The HLA-DR7 was the
most frequently associated with the disease [12]. The fact
that several HLA class II antigens are associated with the
development of RF/rheumatic heart disease (RHD) in
diverse countries is consistent with the possibility that
different strains of group A streptococci are implicated in
the development of RF/RHD in different countries. The
variable association may also be due to the important role
that HLA class II antigens play in antigen presentation to
the T-cell receptors (TCR) [4]. Accordingly, Guilherme
and colleagues studied the T-cell reactivity in the periph-
eral blood of 74 Brazilian RHD patients and found that on
those with severe RHD the immunodominant M5 (81–96)
epitope was preferentially presented to T cells in the con-
text of DR7 and DR53 molecules [13].
Since evidence of genetic influence on RF came into
light, many genes suspected to play a role in susceptibility
have been studied (Table 1). The great majority of them
Table 1 Gene polymorphisms
associated with RF and/or its
manifestations
MBL mannose-binding lectin,
TLR2 toll-like receptor-2, FCN2ficolin-2, TNFa tumor necrosis
factor-a, TGFb transforming
growth factor-b, CTLA-4cytotoxic T-lymphocyte
antigen-4, IL1RN interleukin-1
receptor antagonist gene, IL10interleukin-10, ACEangiotensin-converting enzyme,
AoR aortic regurgitation, MVLmultivalvular lesion, OCDobsessive compulsive disorder,
NS not significant, NC not
calculated
Gene polymorphisms Assoc. OR P Ref.
Innate immunity
MBL AA RHD 1.99 B0.02 [14]
MBL YA/YA & YA/XA RHD 2.48 & 2.42 0.035 & 0.001 [15]
MBL defectives alleles AoR 3.5 0.0022 [16]
TLR2 Arg753Gln & Arg753Arg RF 97.1 & 0.01 \10-3 & \10-3 [17]
FCN2 -986/-602/-4 G/G/A &
A/G/A
RHD 1.6 & 0.3125 0.021 & 0.008 [18]
Adaptive immunity
IL1RN A1 & A1A1 SRHD 0.11 & 0.092 0.031 & 0.017 [37]
IL1RN A1A1 RHD 2.2 \0.05 [39]
IL-10 -1082 AA & GG RHD/
MVL
3.1 & 5.2/5.2 & NS \0.05 & \0.05/\0.05 &
NS
[39]
FCc RIIA RR & RIIIB NA2 RF 4.98 & NS 0.0022 & NS [40]
TNFa G-308A & G-238A RF 1.4 & 1.9 0.026 & 0.015 [41]
TNFa G-308A RF/RHD 3.4/3.3 \0.0032/\0.0055 [42]
TNFa G-308A & G-238G RHD/
MVL
10.8 & 14.1/8.65 &
NS
\10-3 & \10-3/\10-3 &
NS
[43]
TNFa -308A & -238A OCD NC & NC \0.0005 & 0.0099 [44]
TNFa -308AA RHD/
MVL
5.7/10.6 \10-3/\0.05 [39]
TGF-b1 C-509T & T869C RHD 1.49 & NC \10-3 & 0.04 [49]
TGF-b1 C-509T & T869T &
T869TT
RHD 1.78 & 1.89 & 3.37 0.04 & 0.02 & 0.02 [45]
CTLA-4 ?49GG RHD 3.1 0.016 [28]
Others
ACE II RHD NC \0.003 [46]
ACE II RHD 2.12 0.02 [48]
1114 Rheumatol Int (2012) 32:1113–1120
123

are somehow involved with the regulation of the immune
system.
The innate immune response provides immediate
defense against infection, recruits immune cells to sites of
infection and activates the adaptive immune system
through antigen presentation, production of cytokines and
activation of the complement cascade. It is expected that
variations within genes codifying proteins involved in
innate immune response may influence the predisposition
to RF or it sequels. Of note, mannose-binding lectin (MBL)
is an innate pattern-recognition collectin, an acute-phase
protein known to play a key role in pathogen clearance
[14]. It binds to several pathogen’s surface sugars,
including N-acetylglucosamine (GlcNAc), the major im-
munoepitope of group A streptococcal cell wall carbohy-
drates that has immunological similarity with human heart
valve’s laminin [15, 16]. The toll-like receptors (TLRs)
family is a key player in host immunity by mediating
inflammatory reactions against a wide range of pathogens.
TLR-2 is reported to interact with different bacterial struc-
tures, including lipoproteins, peptidoglycan and lipoteichoic
acid, some of them present in Streptococcus’ structures [17].
Ficolins (FCN) are pattern-recognition proteins involved in
innate immunity, which upon binding to their specific
pathogen-associated molecular patterns on the microbial
surfaces trigger the immune response either by binding to
collectin cellular receptors or by initiating the complement
lectin pathway. Ficolin-2 was shown to bind to lipoteichoic
acid, a cell wall constituent in all Gram-positive bacteria
such as Streptococcus [18]. Polymorphisms of the genes
codifying the above protein were associated with RF and/or
its manifestations (Table 1).
As further discussed later on, the adaptive immune
system plays a crucial role on the maintenance and
expansion of inflammation that leads to tissue damage seen
in RF. To date were associated with RF and/or manifes-
tations the genes for the interleukin-1 (IL-1) receptor
antagonist (IL1RN), tumor necrosis factor-a (TNFa),
transforming growth factor-b (TGFb) and cytotoxic
T-lymphocyte antigen-4 (CTLA-4) (Table 1). Interleukin-1
a and interleukin-1 b are produced by a wide variety of
cells only upon stimulation and promote the expression of
adhesion molecules and directly activate a number of cells
involved in immune response. An enhanced production of
IL-1 by peripheral blood mononuclear cells (PBMC) has
been implicated as the initial event of the cytokine dys-
crasias seen in RF [19]. Both IL-1a and IL-1b bind to the
same cellular receptor (IL-1R), which has a soluble
antagonist (IL1-RA). It acts by competitively linking to
IL-1 receptor without initiating the intracellular cascade
that signalizes the inflammation. The ratio IL-1ra/IL-1 is
important to determine the intensity and duration of the
inflammatory response [20]. The allelic variation of the
gene codifying IL1-RA (IL1RN) influences the leukocyte
IL-1RA production and immune response [21]. Most of the
studies associate IL-1RN’s allele 2 (A2) with an exacer-
bated immune response and the IL-1RN’s allele 1 (A1)
with a somehow milder immunological state [22]. Tumor
necrosis factor is one of the most important proinflamma-
tory cytokines. In RF and RHD, increased plasma levels of
TNF-a have been demonstrated [23–25]. TNF-a (also IL-1
and IL-2) production in the valvular lesions of RF patients
was correlated with Aschoff nodule progression [26]. It is
possible that TNF-a gene polymorphism interferes with
inflammatory response by influencing the levels of the
cytokine, but linkage disequilibrium with MHC class II
(both located on chromosome 6) is an alternative. Trans-
forming growth factor-beta (TGF-b) is a protein that con-
trols proliferation, cellular differentiation, and other
functions in most cells, including many within the immune
system and reparatory mechanisms. It was postulated that
TGF-b could be responsible for the increased valvular
fibrosis and calcification in the pathogenesis of RHD [27].
Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) is
a negative regulator of T-cell activation and proliferation
during the immune response. CTLA-4 gene polymorphism
has been shown to affect the inhibitory function of CTLA-4
[28]. Fc-gamma RIIA and Fc-gamma RIIIB are low-
affinity receptors for the FC region of immunoglobulin
gamma, present in many immune cells. Interleukin-10 (IL-
10) has pleiotropic effects in immunoregulation and
inflammation. It downregulates the expression of Th1
cytokines, MHC class II antigens, and costimulatory mol-
ecules on macrophages. It also enhances B-cell survival,
proliferation, and antibody production.
Triggering autoimmunity
The mechanism through which the group A Streptococcus
pyogenes pharyngeal infection triggers the autoimmunity
responsible to RF’s sequels is molecular mimicry. Several
streptococcal and human protein cross-reactive antibodies
found in the sera of RF patients and immunized rabbits and
mice have been described [29]. Antibodies against strep-
tococci’ wall GlcNAc display cross-reactivity against
laminin, a protein present in extracellular matrix that sur-
rounds heart cells and in the valves. Cardiac myosin and
vimentin are other major target antigens. Cunningham and
colleagues identified a 5 aminoacid residues’ epitope of the
Streptococcus N-terminal M5 and M6 proteins that cross-
reacts with cardiac myosin [30]. Antibodies from RF
patients were able to recognize several cardiac myosin
epitopes [31]. Sydenham’s chorea (SC) is mediated by
antibodies able to bind neuronal cells. Antibodies that
cross-react with lysoganglioside GM1 from neuronal cells
and streptococcal GlcNAc are capable of mediating signal
Rheumatol Int (2012) 32:1113–1120 1115
123

transduction, triggering dopamine release from neuronal
cells [32]. Cunningham’s group showed that in RF patients’
cross-reactive antibodies upregulate the adhesion molecule
VCAM-1 after binding to the endothelial surface leading to
cellular infiltration, inflammation, and valve scarring [33,
34]. These data establish that the antimyosin and antilami-
nin cross-reactive antibodies are implicated in the physio-
pathology of RF and bridge the early humoral immune
response to the later cellular infiltration and tissue damage.
Role of cellular immune response
As above described, humoral antibody–mediated autoim-
munity has an important role in initiating cardiac inflam-
mation and attracting cells that will perpetuate and expand
inflammation, now via cellular immune response. T lym-
phocytes CD4? are the major effectors of heart lesions in
RHD [35–37]. Molecular mimicry for T cells is manly
mediated by the recognition of bacteria and/or self-antigens
through HLA class II antigen presentation by antigen-
presenting cells (APCs), such as macrophages, dendritic
cells, and B lymphocytes. Pathogen epitopes that present
structural or sequential similarity to self-epitopes might
activate by the molecular mimicry mechanism autoreactive
T lymphocytes that have escaped immune tolerance. These
autoreactive T cells can also activate B cells that will
produce pathogen- and self-antigen-specific antibodies,
amplifying the process.
Several authors described peripheral and intracardiac
T cells capable of cross-recognizing a number of strepto-
coccus and self-epitopes, mainly derived from the immu-
nodominant bacterial M5 protein and cardiac myosin [31,
38–40], laminin, and tropomyosin [31, 40]. These M5
epitopes are also preferentially recognized by peripheral
T lymphocytes from RHD patients when compared to
normal individuals [13]. A high proportion (63%) of the
T-cell clones present in valvular tissue were reactive
against the LMM region of myosin in one study [13].
Guilherme and colleagues assessed the peripheral and int-
ralesional T-cell repertoire of RHD patients and found
evidence that some antigen-driven T-cell population
migrates from periphery to the heart where they are
expanded in an oligoclonal manner [41]. Several intrale-
sional T-cell clones present the same amino acid sequences
in the CDR3 region of the TCR conferring compatibility to
recognize numerous heart tissue proteins and LMM region
cardiac myosin peptides, indicating degeneracy in antigen
recognition [38, 42, 43]. Together, these data provide
support for the idea that antigen-specific peripheral T-cell
populations migrate to the heart tissue, expand locally, and
become capable of recognizing new self-antigens that are
distinct from the pathogen-inducing epitope, by an epitope-
spreading mechanism [44] (Fig. 1).
Intense and long-lasting immune response
One- to two-thirds of RF patients will have rheumatic
carditis [7, 8], and only a portion of them will suffer from a
severe and persistent form of this manifestation. It is not
rare that the initial heart inflammation subsides, leaving no
sequels. The factors determining the heart evolution are not
completely understood, but it is possible that some RF
patients will go through the epitope-spreading phenomena
and develop an intense and long-lasting cellular immune
response while others will not go much further on the initial
humoral-mediated inflammation. Accordingly, RF mild
carditis, as Sydenham chorea and arthritis, is believed to be
mediated by humoral immunity (Th-2-type immune
response) [3, 29, 32], while severe carditis is cellular
immunity–mediated (Th-1-type immune response) [35–37].
Cytokines in rheumatic fever
Antigen-activated CD4? T cells polarize to Th1 or Th2 or
Th17 subsets, depending on the cytokine secreted. Th1
cells are involved in the cellular immune response and
produce IL-1, IL-2, IFNc, and TNFa. Th2 cells mediate
humoral and allergic immune responses and produce IL-4,
IL-5, and IL-13. Th17 has more recently been described as
a type of proinflammatory response mediated by IL-17.
Streptococcus pyogenes throat infection triggers an
inflammatory reaction that involves several proinflamma-
tory cytokines, such as IL-1, IL-6, and TNFa.
Several evidence suggest that FR patients have a more
severe and long-lasting immune reaction. Acute RF (ARF)
patients’ lymphocytes have exacerbated in vitro cellular
reactivity as compared to patients with acute post-strepto-
coccal glomerulonephritis peaking at 1–6 months and
Fig. 1 Epitope spreading
1116 Rheumatol Int (2012) 32:1113–1120
123

lasting for at least 2 years after onset [45]. Upon stimula-
tion, acute ARF patients’ peripheral blood mononuclear
cells (PBMC) produce larger amounts of IL-1 and IL-2
than PBMC from normal controls, streptococcal pharyn-
gitis (SP), or chronic rheumatic heart disease (CRHD)
patients, and the IL-2 overproduction persisted after
48 weeks. ARF and acute rheumatic heart disease (ARHD)
patients’ PBMC also expressed higher proportions of IL-2
receptors (CD25). The IL-1 is considered to be an amplifier
of IL-2 and IL-2r production, and thus, the author specu-
lated that IL-1 is the major responsible factor for the per-
sistent inflammatory process [19]. In other study, the
plasma concentration of IL-1 and IL-2 was higher in ARF
than those in SP, CRHD, or normal controls, and the pro-
duction of IL-2 in ARF and CRHD directly correlated with
increased percentages of CD4? (T helper) and CD25?
cells in the peripheral blood [25].
In the heart tissue (myocardium and valves) of acute and
chronic RHD patients were identified by immunohisto-
chemistry a large number of mononuclear cells that were
able to secrete inflammatory cytokines (TNFa and IFNc)
and the regulatory cytokine IL-10. The analysis of the
cytokine profile of these cells suggested that the regression
of myocardium inflammation does not depend solely on the
regulatory function of IL-10. The differential cytokine
polarization in the atrium versus valvular tissue was
speculatively attributed by the authors to immigrant auto-
reactive T cells, local chemokines produced by inflamma-
tory cells, and adhesion molecules. The predominant IFN-cand TNF-a expression in the heart suggested that Th1-type
cytokines could mediate chronicity of RHD. The
production of IL-4 (antiinflammatory cytokine) seemed to
be crucial for a protective role by the fact that in the valves
there are only few cells producing this cytokine and the
lack of this cytokine probably leads to the worsening of
valvular lesions [46]. Fraser and colleagues divided the
progression of the Aschoff nodules into 3 chronologic
stages: macrophages only, accumulation of first T lym-
phocytes, and finally B lymphocytes. They concluded that
TNF-a and IL-1 secretion in macrophages is required for
T and B lymphocytes activation and aggregation, sug-
gesting that the macrophages arrive at the scene of rheu-
matic injury prior to lymphocytes. Il-2 is usually expressed
later and was found only in the lymphoid aggregates [26].
The previously mentioned associations between genes
polymorphism RF and/or its manifestations suggest that
genetic variation of cytokine genes influences their pro-
duction, the cytokine profile, and finally RF outcome. Our
group found a protective association (OR = 0.092,
P = 0.017) of the A1A1 IL1RN genotype, classically
connected to a milder immune response, with the devel-
opment of severe carditis in 84 RF Brazilian patients [47].
This is interesting because severe carditis is Th-1-mediated
and IL1-RA production is a major signal to inflammation
end in Th-1 immune response [48], diminishing the pos-
sibility of chronification and epitope-spreading. Also,
Morris and colleagues suggested an enhanced production
of IL-1 by peripheral blood mononuclear cells (PBMC) as
the initial event of the cytokine dyscrasias seen in RF [19],
and Guilherme et al. pointed the importance of the local
Th1-type cytokines produced by infiltrating inflammatory
cells in mediating chronicity of RHD [46].
Fig. 2 Rheumatic fever
physiopathology
Rheumatol Int (2012) 32:1113–1120 1117
123

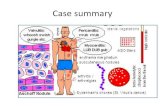
Physiopathological model of disease
Under the light of the mentioned findings, a model of RF
physiopathology can be proposed (Fig. 2), where a pharynx
infection by a Streptococcus pyogenes bearing epitopes
similar to human structures (molecular mimicry) is initially
fought by the innate immunity that triggers local adaptive
immunity that will produce exacerbated quantities of pro-
inflammatory cytokines and antibodies, some of them
capable of cross-recognize human and bacterial epitopes.
The cytokines act systemically, activating and expanding
multiple lymphocytes clones. At the joints, the antibodies
precipitate in form of immunocomplex and activate com-
plement-inducing arthritis. Few antibodies recognize basal
ganglia neurons and induce dopamine secretion leading to
Sydenham’s chorea and probably obsessive compulsive
disorders in some patients. Autoantibodies against heart
structures, mainly derived from myosin, laminin, and
tropomyosin, promote adhesion molecules as VCAM-1
expression recruiting initially monocytes and macrophages.
These mononuclear cells produce a gamma of cytokines
that acts locally with proinflammatory and antiinflamma-
tory properties. The balance between these cytokines can
be influenced by host genetic background and will deter-
mine whether peripheral lymphocytes will be attracted to
the heart, where epitope spreading phenomena may occur,
leading to persistent and intense inflammation and heart
damage. If antiinflammatory cytokines overcome inflam-
mation end, no heart damage occurs.
Conclusion
Rheumatic fever is a prototype of post-infectious autoim-
mune disease and offers important clues to understand
other immune conditions. The accumulation of information
about RF now permits a construction of a physiopatho-
logical model that hopefully will be soon traduced into
befits to millions that still are affected by this disease.
Acknowledgments The work was supported only by academical
grants. RMRP receives funds by CNPQ #300559/2009-7.
Conflicts of interest We have no financial or other relationships
that could lead to a conflict of interest.
References
1. Hochberg MC (ed) (2003) Rheumatic fever. In: Rheumatology,
vol 2, 3rd edn. Elsevier Health Sciences, New York,
pp 1131–1141
2. Carapetis JR, Steer AC, Mulholland EK, Weber M (2005) The
global burden of group A streptococcal diseases. Lancet Infect
Dis 5(11):685–694
3. Ayoub EM (2001) Rheumatic fever. In: Rich RR (ed) Clinical
immunology principles & practice, vol 2, 2nd edn. Mosby, St
Louis, pp 1–7
4. Guilherme L, Kalil J (2004) Rheumatic fever: from sore throat to
autoimmune heart lesions. Int Arch Allergy Immunol
134(1):56–64. doi:10.1159/000077915
5. (2004) Rheumatic fever and rheumatic heart disease: report of a
WHO expert consultation. World Health Organ Technical Report
Series 923:1–122
6. Dajani AS, Ayoub E, Bierman FZ et al (1992) Guidelines for the
diagnosis of rheumatic fever. Jones Criteria, 1992 update. Special
writing Group of the Committee on rheumatic fever, endocarditis,
and Kawasaki disease of the council on cardiovascular disease in
the young of the American Heart Association. JAMA 268(15):
2069–2073
7. Carapetis JR, Currie BJ, Mathews JD (2000) Cumulative incidence
of rheumatic fever in an endemic region: a guide to the suscepti-
bility of the population? Epidemiol Infect 124(2):239–244
8. Guilherme L, Ramasawmy R, Kalil J (2007) Rheumatic fever and
rheumatic heart disease: genetics and pathogenesis. Scand J
Immunol 66(2–3):199–207
9. Gray F, Quinn R, WQuinn J (1952) A long term survey of
rheumatic and non-rheumatic families; with particular reference
to environment and heredity. Am J Med 13(4):400–412
10. Wilson MG, Schweitzer M (1954) Pattern of hereditary suscep-
tibility in rheumatic fever. Circulation 10(5):699–704
11. Spagnuolo M, Taranta A (1968) Rheumatic fever in siblings. Sim-
ilarity of its clinical manifestations. N Engl J Med 278(4):183–188
12. Guilherme L, Kalil J (2010) Rheumatic fever and rheumatic heart
disease: cellular mechanisms leading autoimmune reactivity and
disease. J Clin Immunol 30(1):17–23. doi:10.1007/s10875-009-
9332-6
13. Guilherme L, Oshiro SE, Fae KC, Cunha-Neto E, Renesto G,
Goldberg AC, Tanaka AC, Pomerantzeff PM, Kiss MH, Silva C,
Guzman F, Patarroyo ME, Southwood S, Sette A, Kalil J (2001)
T-cell reactivity against streptococcal antigens in the periphery
mirrors reactivity of heart-infiltrating T lymphocytes in rheumatic
heart disease patients. Infect Immun 69(9):5345–5351
14. Messias Reason IJ, Schafranski MD, Jensenius JC, Steffensen R
(2006) The association between mannose-binding lectin gene
polymorphism and rheumatic heart disease. Hum Immunol
67(12):991–998. doi:10.1016/j.humimm.2006.08.296
15. Schafranski MD, Pereira Ferrari L, Scherner D, Torres R,
Jensenius JC, de Messias-Reason IJ (2008) High-producing
MBL2 genotypes increase the risk of acute and chronic carditis in
patients with history of rheumatic fever. Mol Immunol
45(14):3827–3831. doi:10.1016/j.molimm.2008.05.013
16. Ramasawmy R, Spina GS, Fae KC, Pereira AC, Nisihara R,
Messias Reason IJ, Grinberg M, Tarasoutchi F, Kalil J, Guil-
herme L (2008) Association of mannose-binding lectin gene
polymorphism but not of mannose-binding serine protease 2 with
chronic severe aortic regurgitation of rheumatic etiology. Clin
Vaccine Immunol 15(6):932–936. doi:10.1128/CVI.00324-07
17. Berdeli A, Celik HA, Ozyurek R, Dogrusoz B, Aydin HH (2005)
TLR-2 gene Arg753Gln polymorphism is strongly associated
with acute rheumatic fever in children. J Mol Med 83(7):
535–541. doi:10.1007/s00109-005-0677-x
18. Messias-Reason IJ, Schafranski MD, Kremsner PG, Kun JF (2009)
Ficolin 2 (FCN2) functional polymorphisms and the risk of rheu-
matic fever and rheumatic heart disease. Clin Exp Immunol
157(3):395–399. doi:10.1111/j.1365-2249.2009.03975.x
19. Morris K, Mohan C, Wahi PL, Anand IS, Ganguly NK (1993)
Enhancement of IL-1, IL-2 production and IL-2 receptor gener-
ation in patients with acute rheumatic fever and active rheumatic
heart disease; a prospective study. Clin Exp Immunol
91(3):429–436
1118 Rheumatol Int (2012) 32:1113–1120
123

20. Hirsch E, Irikura V, Paul S, Hirsh D (1996) Functions of inter-
leukin 1 receptor antagonist in gene knockout and overproducing
mice. Proc Natl Acad Sci 93:11008–11013
21. Danis VA, Millington M, Hyland VJ, Grennan D (1995) Cytokine
production by normal human monocytes: inter-subject variation
and relationship to an IL-1 receptor antagonist (IL-1Ra) gene
polymorphism. Clin Exp Immunol 99(2):303–310
22. Witkin SS, Gerber S, Ledger WJ (2002) Influence of interleukin-
1 receptor antagonist gene polymorphism on disease. Clin Infect
Dis 34(2):204–209
23. Yegin O, Coskun M, Ertug H (1997) Cytokines in acute rheu-
matic fever. Eur J Pediatr 156(1):25–29
24. Samsonov MY, Tilz GP, Pisklakov VP, Reibnegger G, Nassonov
EL, Nassonova VA, Wachter H, Fuchs D (1995) Serum-soluble
receptors for tumor necrosis factor-alpha and interleukin-2, and
neopterin in acute rheumatic fever. Clin Immunol Immunopathol
74(1):31–34. doi:S0090122985710057
25. Narin N, Kutukculer N, Ozyurek R, Bakiler AR, Parlar A,
Arcasoy M (1995) Lymphocyte subsets and plasma IL-1 alpha,
IL-2, and TNF-alpha concentrations in acute rheumatic fever and
chronic rheumatic heart disease. Clin Immunol Immunopathol
77(2):172–176
26. Fraser WJ, Haffejee Z, Jankelow D, Wadee A, Cooper K (1997)
Rheumatic Aschoff nodules revisited. II: cytokine expression
corroborates recently proposed sequential stages. Histopathology
31(5):460–464
27. Chou HT, Chen CH, Tsai CH, Tsai FJ (2004) Association
between transforming growth factor-beta1 gene C-509T and
T869C polymorphisms and rheumatic heart disease. Am Heart
J 148(1):181–186. doi:10.1016/j.ahj.2004.03.032
28. Duzgun N, Duman T, Haydardedeoglu FE, Tutkak H (2009)
Cytotoxic T lymphocyte-associated antigen-4 polymorphism in
patients with rheumatic heart disease. Tissue Antigens 74(6):
539–542. doi:10.1111/j.1399-0039.2009.01347.x
29. Cunningham MW (2000) Pathogenesis of group A streptococcal
infections. Clin Microbiol Rev 13(3):470–511
30. Cunningham MW, McCormack JM, Fenderson PG, Ho MK,
Beachey EH, Dale JB (1989) Human and murine antibodies
cross-reactive with streptococcal M protein and myosin recognize
the sequence GLN-LYS-SER-LYS-GLN in M protein. J Immu-
nol 143(8):2677–2683
31. Cunningham MW (2004) T cell mimicry in inflammatory heart
disease. Mol Immunol 40(14–15):1121–1127. doi:10.1016/
j.molimm.2003.11.023
32. Kirvan CA, Swedo SE, Heuser JS, Cunningham MW (2003)
Mimicry and autoantibody-mediated neuronal cell signaling in
Sydenham chorea. Nat Med 9(7):914–920. doi:10.1038/nm892
33. Roberts S, Kosanke S, Terrence Dunn S, Jankelow D, Duran CM,
Cunningham MW (2001) Pathogenic mechanisms in rheumatic
carditis: focus on valvular endothelium. J Infect Dis 183(3):
507–511. doi:10.1086/318076
34. Galvin JE, Hemric ME, Ward K, Cunningham MW (2000)
Cytotoxic mAb from rheumatic carditis recognizes heart valves
and laminin. J Clin Invest 106(2):217–224. doi:10.1172/JCI7132
35. Guilherme L, Weidebach W, Kiss MH, Snitcowsky R, Kalil J
(1991) Association of human leukocyte class II antigens with
rheumatic fever or rheumatic heart disease in a Brazilian popu-
lation. Circulation 83(6):1995–1998
36. Kemeny E, Grieve T, Marcus R, Sareli P, Zabriskie JB (1989)
Identification of mononuclear cells and T cell subsets in rheu-
matic valvulitis. Clin Immunol Immunopathol 52(2):225–237
37. Raizada V, Williams RC Jr, Chopra P, Gopinath N, Prakash K,
Sharma KB, Cherian KM, Panday S, Arora R, Nigam M,
Zabriskie JB, Husby G (1983) Tissue distribution of lymphocytes
in rheumatic heart valves as defined by monoclonal anti-T cell
antibodies. Am J Med 74(1):90–96
38. Fae KC, da Silva DD, Oshiro SE, Tanaka AC, Pomerantzeff PM,
Douay C, Charron D, Toubert A, Cunningham MW, Kalil J,
Guilherme L (2006) Mimicry in recognition of cardiac myosin
peptides by heart-intralesional T cell clones from rheumatic heart
disease. J Immunol 176(9):5662–5670. doi:176/9/5662
39. Yoshinaga M, Figueroa F, Wahid MR, Marcus RH, Suh E,
Zabriskie JB (1995) Antigenic specificity of lymphocytes isolated
from valvular specimens of rheumatic fever patients. J Autoim-
mun 8(4):601–613. doi:0896-8411(95)90011-X
40. Ellis NM, Li Y, Hildebrand W, Fischetti VA, Cunningham MW
(2005) T cell mimicry and epitope specificity of cross-reactive T
cell clones from rheumatic heart disease. J Immunol 175(8):
5448–5456. doi:175/8/5448
41. Guilherme L, Dulphy N, Douay C, Coelho V, Cunha-Neto E,
Oshiro SE, Assis RV, Tanaka AC, Pomerantzeff PM, Charron D,
Toubert A, Kalil J (2000) Molecular evidence for antigen-driven
immune responses in cardiac lesions of rheumatic heart disease
patients. Int Immunol 12(7):1063–1074
42. Wucherpfennig KW, Allen PM, Celada F, Cohen IR, De Boer R,
Garcia KC, Goldstein B, Greenspan R, Hafler D, Hodgkin P,
Huseby ES, Krakauer DC, Nemazee D, Perelson AS, Pinilla C,
Strong RK, Sercarz EE (2007) Polyspecificity of T cell and B cell
receptor recognition. Semin Immunol 19(4):216–224. doi:
10.1016/j.smim.2007.02.012
43. Fae K, Kalil J, Toubert A, Guilherme L (2004) Heart infiltrating
T cell clones from a rheumatic heart disease patient display a
common TCR usage and a degenerate antigen recognition pattern.
Mol Immunol 40(14–15):1129–1135. doi:10.1016/j.molimm.
2003.11.007
44. Lehmann PV, Forsthuber T, Miller A, Sercarz EE (1992)
Spreading of T-cell autoimmunity to cryptic determinants of an
autoantigen. Nature 358(6382):155–157. doi:10.1038/358155a0
45. Read SE, Reid HF, Fischetti VA, Poon-King T, Ramkissoon R,
McDowell M, Zabriskie JB (1986) Serial studies on the cellular
immune response to streptococcal antigens in acute and conva-
lescent rheumatic fever patients in Trinidad. J Clin Immunol
6(6):433–441
46. Guilherme L, Cury P, Demarchi LM, Coelho V, Abel L, Lopez
AP, Oshiro SE, Aliotti S, Cunha-Neto E, Pomerantzeff PM, Ta-
naka AC, Kalil J (2004) Rheumatic heart disease: proinflamma-
tory cytokines play a role in the progression and maintenance of
valvular lesions. Am J Pathol 165(5):1583–1591. doi:165/5/158347. Azevedo PM, Bauer R, Caparbo Vde F, Silva CA, Bonfa E,
Pereira RM (2010) Interleukin-1 receptor antagonist gene
(IL1RN) polymorphism possibly associated to severity of rheu-
matic carditis in a Brazilian cohort. Cytokine 49(1):109–113. doi:
10.1016/j.cyto.2009.09.003
48. Granowitz EV, Santos AA, Poutsiaka DD, Cannon JG, Wilmore
DW, Wolff SM, Dinarello CA (1991) Production of interleukin-
1-receptor antagonist during experimental endotoxaemia. Lancet
338(8780):1423–1424. doi:0140-6736(91)92725-H
49. Kamal H, Hussein G, Hassoba H, Mosaad N, Gad A, Ismail M
(2010) Transforming growth factor-beta1 gene C-509T and
T869C polymorphisms as possible risk factors in rheumatic heart
disease in Egypt. Acta Cardiol 65(2):177–183
50. Settin A, Abdel-Hady H, El-Baz R, Saber I (2007) Gene poly-
morphisms of TNF-alpha(-308), IL-10 (-1082), IL-6 (-174), and
IL-1Ra (VNTR) related to susceptibility and severity of rheu-
matic heart disease. Pediatr Cardiol 28(5):363–371
51. Berdeli A, Celik HA, Ozyurek R, Aydin HH (2004) Involvement
of immunoglobulin FcgammaRIIA and FcgammaRIIIB gene
polymorphisms in susceptibility to rheumatic fever. Clin Bio-
chem 37(10):925–929. doi:10.1016/j.clinbiochem.2004.06.007
52. Ramasawmy R, Fae KC, Spina G, Victora GD, Tanaka AC,
Palacios SA, Hounie AG, Miguel EC, Oshiro SE, Goldberg AC,
Kalil J, Guilherme L (2007) Association of polymorphisms
Rheumatol Int (2012) 32:1113–1120 1119
123

within the promoter region of the tumor necrosis factor-alpha
with clinical outcomes of rheumatic fever. Mol Immunol
44(8):1873–1878. doi:10.1016/j.molimm.2006.10.001
53. Sallakci N, Akcurin G, Koksoy S, Kardelen F, Uguz A, Coskun
M, Ertug H, Yegin O (2005) TNF-alpha G-308A polymorphism
is associated with rheumatic fever and correlates with increased
TNF-alpha production. J Autoimmun 25(2):150–154. doi:
10.1016/j.jaut.2005.05.005
54. Hernandez-Pacheco G, Flores-Dominguez C, Rodriguez-Perez
JM, Perez-Hernandez N, Fragoso JM, Saul A, Alvarez-Leon E,
Granados J, Reyes PA, Vargas-Alarcon G (2003) Tumor necrosis
factor-alpha promoter polymorphisms in Mexican patients with
rheumatic heart disease. J Autoimmun 21(1):59–63. doi:S089
6841103000799
55. Hounie AG, Cappi C, Cordeiro Q, Sampaio AS, Moraes I,
Rosario MC, Palacios SA, Goldberg AC, Vallada HP, Machado-
Lima A, Nakano E, Kalil J, Pauls D, Pereira CA, Guilherme L,
Miguel EC (2008) TNF-alpha polymorphisms are associated with
obsessive-compulsive disorder. Neurosci Lett 442(2):86–90. doi:
10.1016/j.neulet.2008.07.022
56. Davutoglu V, Nacak M (2005) Influence of angiotensin-con-
verting enzyme gene insertion/deletion polymorphism on rheu-
matic valve involvement, valve severity and subsequent valve
calcification. J Heart Valve Dis 14(3):277–281
57. Chou HT, Tsai CH, Tsai FJ (2004) Association between angio-
tensin I-converting enzyme gene insertion/deletion polymor-
phism and risk of rheumatic heart disease. Jpn Heart J
45(6):949–957. doi:JST.JSTAGE/jhj/45.949
1120 Rheumatol Int (2012) 32:1113–1120
123