
Type I Alveolar Epithelial Cells Mount Innate Immune ... · The Journal of Immunology Type I...
11
of August 25, 2018. This information is current as Pneumococcal Pneumonia Innate Immune Responses during Type I Alveolar Epithelial Cells Mount Mizgerd Ramirez, Matthew R. Jones, Lee J. Quinton and Joseph P. Kazuko Yamamoto, Joseph D. Ferrari, Yuxia Cao, Maria I. ol.1200634 http://www.jimmunol.org/content/early/2012/07/27/jimmun published online 27 July 2012 J Immunol average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2012 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on August 25, 2018 http://www.jimmunol.org/ Downloaded from by guest on August 25, 2018 http://www.jimmunol.org/ Downloaded from
Transcript of Type I Alveolar Epithelial Cells Mount Innate Immune ... · The Journal of Immunology Type I...

of August 25, 2018.This information is current as
Pneumococcal PneumoniaInnate Immune Responses during Type I Alveolar Epithelial Cells Mount
MizgerdRamirez, Matthew R. Jones, Lee J. Quinton and Joseph P. Kazuko Yamamoto, Joseph D. Ferrari, Yuxia Cao, Maria I.
ol.1200634http://www.jimmunol.org/content/early/2012/07/27/jimmun
published online 27 July 2012J Immunol
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2012 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Type I Alveolar Epithelial Cells Mount Innate ImmuneResponses during Pneumococcal Pneumonia
Kazuko Yamamoto,*,† Joseph D. Ferrari,* Yuxia Cao,* Maria I. Ramirez,*
Matthew R. Jones,* Lee J. Quinton,* and Joseph P. Mizgerd*
Pneumonia results from bacteria in the alveoli. The alveolar epithelium consists of type II cells, which secrete surfactant and
associated proteins, and type I cells, which constitute 95% of the surface area and meet anatomic and structural needs. Other
than constitutively expressed surfactant proteins, it is unknown whether alveolar epithelial cells have distinct roles in innate im-
munity. Because innate immunity gene induction depends on NF-kB RelA (also known as p65) during pneumonia, we generated
a murine model of RelA mutated throughout the alveolar epithelium. In response to LPS, only 2 of 84 cytokine transcripts (CCL20
and CXCL5) were blunted in lungs of mutants, suggesting that a very limited subset of immune mediators is selectively elaborated
by the alveolar epithelium. Lung CCL20 induction required epithelial RelA regardless of stimulus, whereas lung CXCL5 expres-
sion depended on RelA after instillation of LPS but not pneumococcus. RelA knockdown in vitro suggested that CXCL5 induction
required RelA in type II cells but not type I cells. Sorted cell populations from mouse lungs revealed that CXCL5 was induced
during pneumonia in type I cells, which did not require RelA. TLR2 and STING were also induced in type I cells, with RelA
essential for TLR2 but not STING. To our knowledge, these data are the first direct demonstration that type I cells, which
constitute the majority of the alveolar surface, mount innate immune responses during bacterial infection. These are also, to our
knowledge, the first evidence for entirely RelA-independent pathways of innate immunity gene induction in any cell during
pneumonia. The Journal of Immunology, 2012, 189: 000–000.
The epithelium represents the first line of defense againstpathogens in the lung. Epithelial cells provide a mechan-ical barrier to prevent infection, and they can produce
chemokines and cytokines that recruit and activate phagocytic cellsto eradicate organisms and infected cells (1, 2). The alveolar ep-ithelium consists of two main populations: alveolar type I (AT1)and type II (AT2) epithelial cells. AT2 cells synthesize and secretepulmonary surfactant, express chemokines and cytokines, andparticipate in the innate immune response of the lung (3). Despitebeing the predominant cell in the alveolar space by number, AT2cells cover only 5% of the surface. The remaining 95% is coveredby large attenuated AT1 cells (4–6). Although constituting somuch of the surface area of the lung, very little is known about anypotential contribution of AT1 cells to pulmonary innate immunity.Proinflammatory cytokines orchestrate innate immunity and are
mediated by multiple transcription factors, including NF-kB. Of
the five NF-kB proteins, only p50 and RelA (also known as p65)are readily detectable in lung nuclear fractions during acute pul-monary inflammation (7–9). p50 limits the expression of inflam-matory cytokines and prevents lung injury during pneumonia (10,11). In contrast, RelA drives inflammatory responses by promot-ing the expression of many cytokines, and the deletion of RelAfrom all cells severely compromises antibacterial host defense (12,13). Mice with a surfactant protein C (SPC)-driven dominant-negative (dn)IkBa inhibitor of NF-kB have increased bacterialburdens during pneumococcal pneumonia (12), suggesting thatNF-kB in AT2 cells contributes to host defense. These mice alsohave decreased neutrophil recruitment and inflammatory cyto-kines after LPS inhalation (14), indicating that NF-kB in AT2cells participates in acute inflammatory responses. However, thednIkBa protein is not specific to distinct NF-kB proteins, and theefficacy of NF-kB inhibition by this approach is based on dy-namic stoichiometry, which has not been analyzed in these lungcells. Importantly, neither these nor other studies to date have ex-amined roles of AT1 cells. The goal of the present study was toevaluate unique roles of alveolar epithelial cells in innate immu-nity mediator expression elicited by bacterial stimuli in the lungsand to assess their dependence on NF-kB RelA.
Materials and MethodsMice
Relaflx/flx mice (15) (provided by Dr. Roland Schmid, Technical Universityof Munich, Munich, Germany) were bred with SPC-reverse tetracyclinetransactivator (rtTA)tg/2/(tetO)7CMV-Cretg/tg mice (16) (provided byDr. Jeffrey A. Whitsett, Cincinnati Children’s Hospital Medical Center,Cincinnati, OH) to generate colonies of wild-type (WT) control mice(SPC-rtTA2/2/(tetO)7CMV-Cretg/tg/Relaflx/flx) or RelAD/D mice (SPC-rtTAtg/2/(tetO)7CMV-Cretg/tg/Relaflx/flx) in which the Rela gene is selectively mutatedin alveolar epithelial cells. Results obtained from RelAD/D mice werecompared with sex-matched littermate controls. Doxycycline was providedin the chow (625 mg/kg, S-5086; Bio-Serv) to all mice throughout ges-tation and nursing to induce Cre-recombinase–mediated Rela mutation in
*Pulmonary Center, Boston University School of Medicine, Boston, MA 02118; and†Department of Molecular Microbiology and Immunology, Nagasaki UniversityGraduate School of Biomedical Sciences, Nagasaki 852-8588, Japan
Received for publication February 23, 2012. Accepted for publication June 26, 2012.
This work was supported by National Institutes of Health Grants HL068153 (toJ.P.M.), HL079392 (to J.P.M.), HL104053 (to M.R.J.), HL092956 (to L.J.Q.), andHL083034 (to M.I.R.). K.Y. was supported by an American Lung Association SeniorResearch Fellowship.
The sequences presented in this article have been submitted to the National Centerfor Biotechnology Information Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE38398 and GSE38399).
Address correspondence and reprint requests to Dr. Joseph P. Mizgerd, PulmonaryCenter, Boston University School of Medicine, 72 E. Concord Street, Boston, MA02118. E-mail address: [email protected]
Abbreviations used in this article: AT1, alveolar epithelial type I; AT2, alveolarepithelial type II; dn, dominant-negative; PRR, pattern recognition receptor; qRT-PCR, quantitative RT-PCR; rtTA, reverse tetracycline transactivator; siRNA, smallinterfering RNA; SPC, surfactant protein C; STING, stimulator of IFN gene; WT,wild-type.
Copyright� 2012 by The American Association of Immunologists, Inc. 0022-1767/12/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1200634
Published July 27, 2012, doi:10.4049/jimmunol.1200634 by guest on A
ugust 25, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
the alveolar epithelium of the rtTA-transgenics, including both AT1 cellsand AT2 cells (17). Mice were not exposed to the doxycycline diet afterweaning from their mothers at 3 wk age, preventing effects of confoundingby doxycycline during experiments. At the time of experimentation, micewere 7–11 wk age. Experiments with nontransgenic mice were performedusing C57BL/6 mice. All experimental protocols were approved by theBoston University Institutional Animal Care and Use Committee.
Pneumonia
Mice were anesthetized by i.p. injection of ketamine (50 mg/kg)/xylazine(5 mg/kg). An angiocatheter was placed down the left bronchus, and micereceived intratracheal instillations of 50 ml saline containing 106 CFUStreptococcus pneumoniae serotype 3 (6303; American Type CultureCollection) or 50 mg LPS (Escherichia coli O111:B4; Sigma-Aldrich) intothe left lung lobe. These stimuli were chosen because they are relevant tohuman health, are each well characterized in murine models of pulmonaryinflammation, and are to our knowledge the only two bacterial stimuli thathave been studied in the context of epithelial NF-kB roles (12, 14, 18).Mice were euthanized by an aerosolized isoflurane overdose 15 h afterS. pneumoniae instillation or 6 h after LPS instillation.
Bacterial clearance
Lungs were homogenized in sterile distilled H2O containing proteaseinhibitors (19). Homogenates were serially diluted, plated on 5% sheepblood agar plates using the drop plate method (20), and incubated over-night at 37˚C. Colonies were counted to quantify CFU/lung.
Lung histology
Alveolar neutrophils were quantified by morphometric analyses of H&E-stained sections of left (instilled) lung lobes, as described previously (17,21, 22).
RelA interference in murine lung epithelial cell lines
The murine AT1-like cell line E10 [(23), obtained from Dr. Alvin Malkinson(University of Colorado, Aurora, CO)], was cultured in CMRL-1066medium containing 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin,and 100 mg/ml streptomycin. The murine AT2 cell-derived MLE15 cell
line [(24), obtained from Dr. Jeffrey A. Whitsett (Cincinnati Children’sHospital Medical Center, Cincinnati, OH)] was cultured in a 1:1 mixture ofDMEM and F12 medium supplemented with 2% FBS, 100 U/ml penicillin,100 mg/ml streptomycin, 5 mg/ml insulin, 10 mg/ml transferrin, 30 nMsodium selenite, 10 nM hydrocortisone, 10 nM b-estradiol, and 10 mMHEPES. Cells were studied at ∼80% confluence. Cells were transfectedby using 0.1 mM RelA-targeting small interfering RNA (siRNA; sense,59-GCUCAAGAUCUGCCGAGUAUU-39, antisense, 59-PUACUCGGC-AGAUCUUGAGCUU-39) or control nontargeting siRNA (D-001810-01-20),which did not complement known sequences in the mouse transcriptome(both from Dharmacon). After 24 h, cells were washed once with PBS andwere stimulated after another 24 h culture with fresh media. Cells werestimulated for 6 h with 10 ng/ml recombinant murine TNF-a, 10 mg/mlLPS, or 5 mg/ml BSA (all from Sigma-Aldrich). The cell pellets werecollected after washing with PBS and resuspended in 1 ml TRIzol (Invitrogen)for RNA isolation.
Immunoblot assay
Western blots were performed to determine siRNA-mediated knockdownof RelA in E10 and in MLE15 cells, as previously described (12). Abs forRelA (product no. 3034) and pan-actin (product no. 4968S) were purchasedfrom Cell Signaling Technology.
RNA purification and analyses
Total RNA was isolated from mouse lung, alveolar epithelial cell lines,and isolated cells with an RNeasy Mini kit (Qiagen) or TRIzol reagent(Invitrogen) and an RNase-free DNase set (Qiagen). Quantitative RT-PCR(qRT-PCR) was performed using the TaqMan RNA-to-CT one-step kit andthe StepOnePlus real-time PCR systems (both from Applied Biosystems).The primers and TaqMan probes for mouse CXCL5, TNF-a, IL-6, IL-1a,IL-1b, CXCL1, CXCL2, TLR2, and TLR4 were previously published(12, 25, 26). The primers and TaqMan probe set were designed for mouseCCL20, G-CSF, and Ly6G using the CLC DNAWorkbench software (CLCbio) with the following sequences: CCL20, forward, 59-CCTCAGCCTAA-GAGTCAAGAAGA-39, reverse, 59-ACAAGTCCACTGGGACACAAA-39,probe, 59-ACACAGCCCAAGGAGGAAATGATCACAGC-39; G-CSF,forward, 59-TTCCCCTGGTCACTGTCAGC-39, reverse, 59-CACAG-CTTGTAGGTGGCACAC-39, probe, 59-ACCATCCCTGCCTCTGCC-
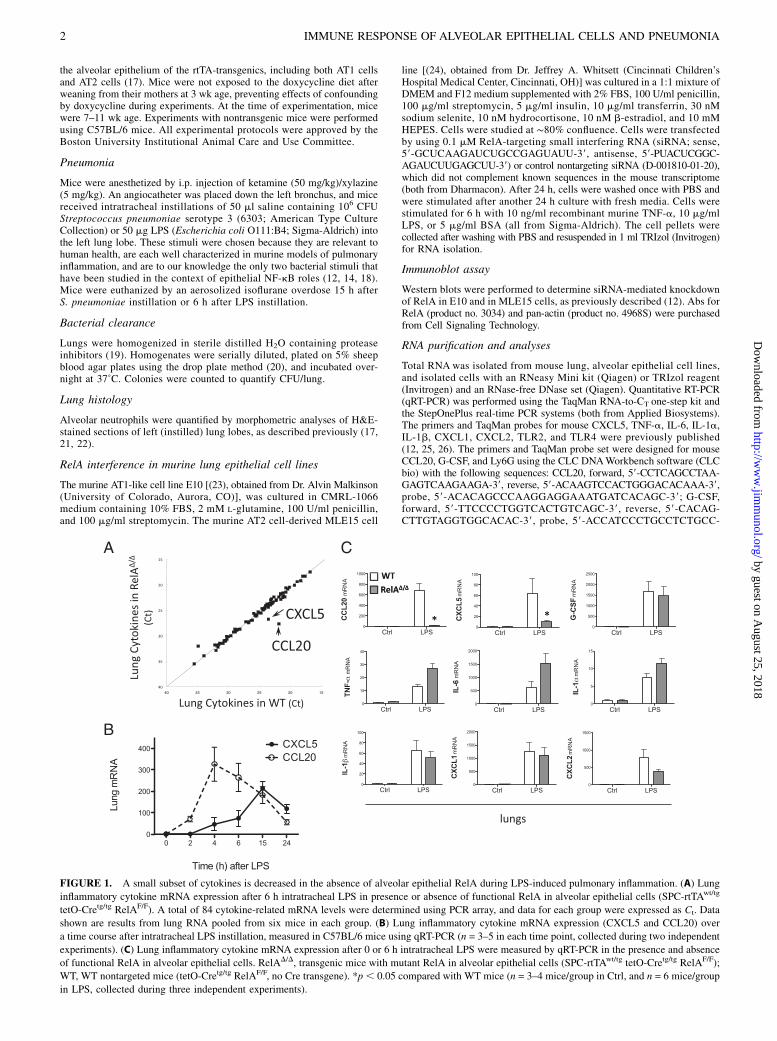
FIGURE 1. A small subset of cytokines is decreased in the absence of alveolar epithelial RelA during LPS-induced pulmonary inflammation. (A) Lung
inflammatory cytokine mRNA expression after 6 h intratracheal LPS in presence or absence of functional RelA in alveolar epithelial cells (SPC-rtTAwt/tg
tetO-Cretg/tg RelAF/F). A total of 84 cytokine-related mRNA levels were determined using PCR array, and data for each group were expressed as Ct. Data
shown are results from lung RNA pooled from six mice in each group. (B) Lung inflammatory cytokine mRNA expression (CXCL5 and CCL20) over
a time course after intratracheal LPS instillation, measured in C57BL/6 mice using qRT-PCR (n = 3–5 in each time point, collected during two independent
experiments). (C) Lung inflammatory cytokine mRNA expression after 0 or 6 h intratracheal LPS were measured by qRT-PCR in the presence and absence
of functional RelA in alveolar epithelial cells. RelAD/D, transgenic mice with mutant RelA in alveolar epithelial cells (SPC-rtTAwt/tg tetO-Cretg/tg RelAF/F);
WT, WT nontargeted mice (tetO-Cretg/tg RelAF/F, no Cre transgene). *p, 0.05 compared with WT mice (n = 3–4 mice/group in Ctrl, and n = 6 mice/group
in LPS, collected during three independent experiments).
2 IMMUNE RESPONSE OF ALVEOLAR EPITHELIAL CELLS AND PNEUMONIA
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
CCGAAG-39; Ly6G, forward, 59-TCCTGTGTGCTCATCCTTCTT-39,reverse, 59-TCCAGAGCAACGCAAAATCCAT-39, probe, 59-TTCCTGCA-ACACAACTACCTGCCCCTT-39. TaqMan gene expression assays wereused for podoplanin (T1a) (Mm00494716_m1), caveolin-1 (Mm00483057_m1), Nkx2-1 (Mm00447558_ml), Sftpc (Mm00488144_m1), Sftpb(Mm00455681_ml), CC10 (Mm00442046_m1), PECAM-1 (Mm01242584_m1), p75 (Mm01309638_m1), and stimulator of IFN gene (STING;Mm01158117_m1; all from Applied Biosystems) mRNA analyses. Foreach sample, values were normalized to the content of 18S rRNA (27, 28).RT2 Profiler mouse inflammatory cytokines and receptors PCR array(PAMM-011; SABiosciences) was used for the multianalyte qRT-PCRanalysis of mouse inflammatory cytokines. Results of the PCR arrayswere deposited at National Center for Biotechnology Information GeneExpression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), retrievable withaccession numbers GSE38398 and GSE38399. Efficacy of Rela gene rear-rangement was determined by examining sizes of amplification productsusing electrophoresis. For Rela RT-PCR, isolated RNA (1 mg) was reverse-transcribed using SuperScript VILO cDNA synthesis kit (Applied Biosystems),and PCR reactions were performed with GoTaq DNA polymerase (Promega).Primers sequences for Rela were: forward, 59-ACAATAACCCCTTTCA-CGTTCCTA-39; reverse, 59-CCCAAGTCTTCATCAGCATCAA-39. Full-length or rearranged Rela products were indicated by amplification productsof ∼939 or 490 bp, respectively.
ELISA
ELISA kits (R&D Systems) for murine CXCL5 and CCL20 levels wereused to measure their respective concentrations in lung homogenate or insupernatants of murine lung cell lines.
Isolation of AT1 cells from lung digests
After euthanasia, lungs were perfused with 10 ml HBSS via the rightventricle through the pulmonary artery. One milliliter porcine elastase(4.5U; Roche Diagnostics) was instilled through a tracheal cannula fol-lowed by 0.5 ml low melting agarose solution warmed to 45˚C. Lungswere immediately covered with ice for 2 min to gel the agarose and in-cubated in 2 ml elastase for 45 min at 37˚C. After this incubation, lunglobes were gently separated from the bronchi and minced in RPMI 1640medium (Life Technologies) containing 50% FBS and 100 U/ml DNase I(Qiagen). Cells in suspension were subsequently filtered through 100-, 70-,and 40-mm nylon mesh. Cell suspensions from these lung digests weresubjected to FACS. The following Abs and isotype controls were used:PE-conjugated hamster anti-mouse podoplanin (T1a), PE-conjugated ham-ster IgG (both hamster products from BioLegend), FITC-conjugated ratanti-mouse CD45, FITC-conjugated rat IgG2b, rat anti-mouse CD16/CD32,and 7-aminoactinomycin D (all nonhamster products from BD Pharmin-gen). Cell isolation was performed using a MoFlo cell sorter. Collectedcells were stored in TRIzol (Invitrogen).
Immunofluorescence staining of sorted cells
Cells were stained for AT1 and AT2 markers using immunofluorescence.Cytocentrifuged cell preparations sorted from lungs of C57BL/6 micewere fixed with 4% paraformaldehyde, blocked, and permeabilized.Slides were labeled with caveolin-1 or pro-SPC primary Abs followedby Alexa Fluor 488-conjugated donkey anti-rabbit IgG secondary Abs.Immunofluorescence images were taken using the AxioVision system(Zeiss).
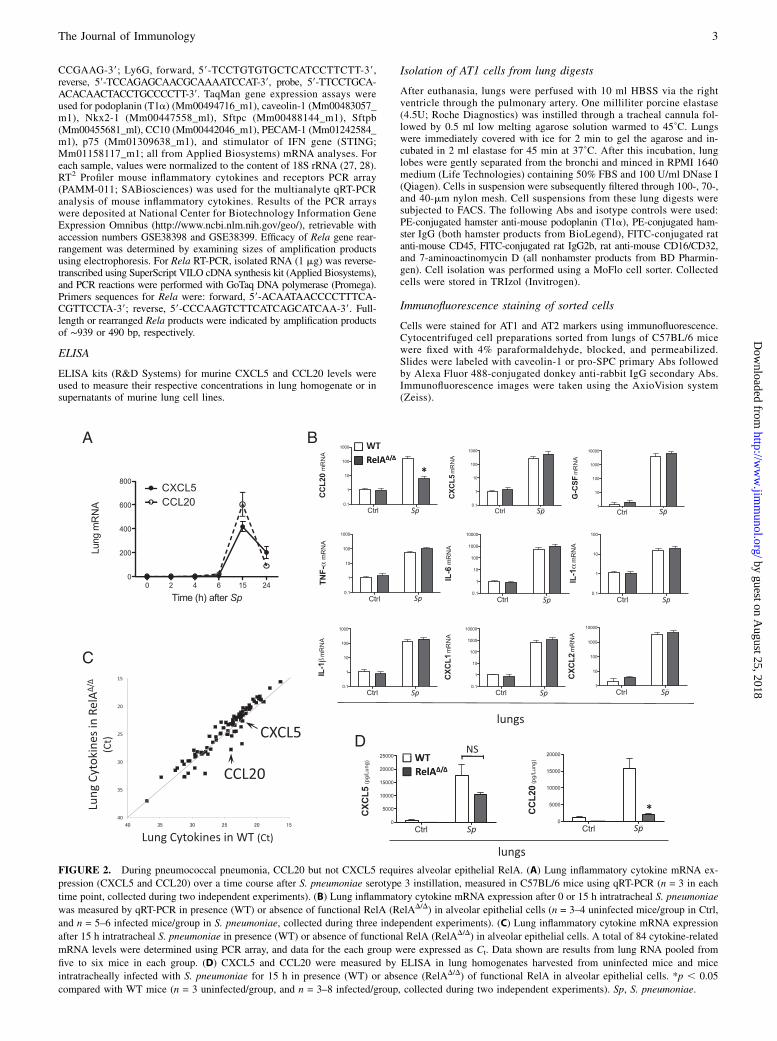
FIGURE 2. During pneumococcal pneumonia, CCL20 but not CXCL5 requires alveolar epithelial RelA. (A) Lung inflammatory cytokine mRNA ex-
pression (CXCL5 and CCL20) over a time course after S. pneumoniae serotype 3 instillation, measured in C57BL/6 mice using qRT-PCR (n = 3 in each
time point, collected during two independent experiments). (B) Lung inflammatory cytokine mRNA expression after 0 or 15 h intratracheal S. pneumoniae
was measured by qRT-PCR in presence (WT) or absence of functional RelA (RelAD/D) in alveolar epithelial cells (n = 3–4 uninfected mice/group in Ctrl,
and n = 5–6 infected mice/group in S. pneumoniae, collected during three independent experiments). (C) Lung inflammatory cytokine mRNA expression
after 15 h intratracheal S. pneumoniae in presence (WT) or absence of functional RelA (RelAD/D) in alveolar epithelial cells. A total of 84 cytokine-related
mRNA levels were determined using PCR array, and data for the each group were expressed as Ct. Data shown are results from lung RNA pooled from
five to six mice in each group. (D) CXCL5 and CCL20 were measured by ELISA in lung homogenates harvested from uninfected mice and mice
intratracheally infected with S. pneumoniae for 15 h in presence (WT) or absence (RelAD/D) of functional RelA in alveolar epithelial cells. *p , 0.05
compared with WT mice (n = 3 uninfected/group, and n = 3–8 infected/group, collected during two independent experiments). Sp, S. pneumoniae.
The Journal of Immunology 3
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Statistical analyses
Statistical analyses were performed using GraphPad Prism software. Datawere presented as means 6 SE. Real-time RT-PCR data were expressed asfold induction and therefore used geometric means 6 geometric SE usedas descriptive statistics. Statistical comparisons among groups for contin-uous variables were performed using a Student t test or a two-way ANOVAfollowed by Bonferroni post hoc analysis. A p value,0.05 was consideredsignificant.
ResultsPhenotype of the alveolar epithelial RelA mutant mouse model
We generated a mouse model devoid of functional RelA in alveolarepithelial cells. Breeding was designed so that all offspring shouldbe homozygous for tetO-Cre and floxed RelA, with half beingnegative and half positive for SPC-rtTA. When no doxycyclinechow was included in the breeding cages, that ratio was observed.However, the inclusion of doxycycline chow during gestation andnursing resulted in a skewing from Hardy–Weinberg equilibrium(29, 30), with only 10% instead of 50% of the offspring beingpositive for the rtTA transgene and hence mutant for RelA inalveolar epithelial cells when genotyped at weaning (3 wk age).Furthermore, some but not all adult mice that survived RelAmutation via such a strategy displayed emphysematous-like en-larged alveolar air spaces. These results could indicate that RelAin epithelial cells is essential for preventing lethality during em-bryonic and neonatal periods and for lung development postna-tally. However, these phenotypes more likely represent an artifactof the strategy, as evidenced by similar findings using the SPC-rtTA/tetO-Cre system in the absence of floxed genes (31, 32), withvariable penetrance that depends on genetic background. The in-terpretation that these phenotypes resulted from the rtTA-basedstrategy rather than the RelA deletion is also supported by the factthat we observe neither phenotype in mice lacking RelA in all cells(12, 13). Although these phenotypes precluded extensive analysesof integrated responses in these mice, we were able to use them fora limited set of studies to interrogate cell-specific gene expressionduring acute pulmonary inflammation.
Alveolar epithelial cell RelA is required for induction ofselected cytokines
The expression of lung inflammatory cytokines was evaluated usingPCR array. To our surprise, during LPS-induced pulmonary in-flammation, only 2 of 84 cytokines in the PCR array were de-creased in the lungs of mice with RelA mutated in alveolarepithelial cells, CCL20 and CXCL5 (Fig. 1A). One gene, C-reactiveprotein, appeared to be increased in mutant mice in this array,but this was not followed up, as the mRNA levels were very loweven in mutants (Fig. 1A). qRT-PCR confirmed that CCL20 andCXCL5 were significantly induced by LPS in the lungs, withCXCL5 peaking at 6–15 h and CCL20 peaking at 4–6 h (Fig. 1B).Induction of each was almost entirely abrogated by the muta-tion of RelA in alveolar epithelial cells (Fig. 1C). Seven othercytokines represented on the array (CXCL1, CXCL2, IL-1a,IL-1b, TNF-a, IL-6, and G-CSF) were measured by qRT-PCR,which confirmed that they were induced by LPS but were notsignificantly affected by the mutation of RelA in alveolar epi-thelial cells (Fig. 1C). That most cytokines examined were com-parable in WT and mutant lungs suggests that the strategy totarget alveolar epithelial RelA did not have global or widespreadimmunity consequences, but rather had very selective effects on asmall subset of mediators. The strategy in these mice was designedto selectively target alveolar epithelial cells (16), suggesting AT2or AT1 cells as potentially critical sources of the cytokines CCL20and CXCL5 during LPS-induced pulmonary inflammation.
To determine whether cytokines showed similar dependence onRelA in alveolar epithelial cells in response to pneumococcus, qRT-PCR was performed on lung mRNA extracts from mice withpneumococcal pneumonia. Whereas neither CCL20 nor CXCL5was induced by 6 h, both were strongly induced by 15 h afterinfection (Fig. 2A), consistent with prior publications demon-strating innate immunity gene induction responses to pneumo-cocci to involve slower kinetics compared with Gram-negativebacterial products (7, 12, 26, 33). As with LPS-induced inflam-mation, many genes induced in the lungs did not require RelAspecifically in alveolar epithelial cells (Fig. 2B). Also similar toLPS-induced inflammation, CCL20 induction did depend on RelAin alveolar epithelial cells, being .90% diminished by the mu-tation of RelA in alveolar epithelial cells (Fig. 2B). Interestingly,CXCL5 was induced despite RelA mutation in the alveolar epi-thelial cells 15 h after pneumococcal infection (Fig. 2B), whichsharply contrasted with the results from lungs 6 h after LPS in-stillation. A PCR array suggested similar cytokine expression
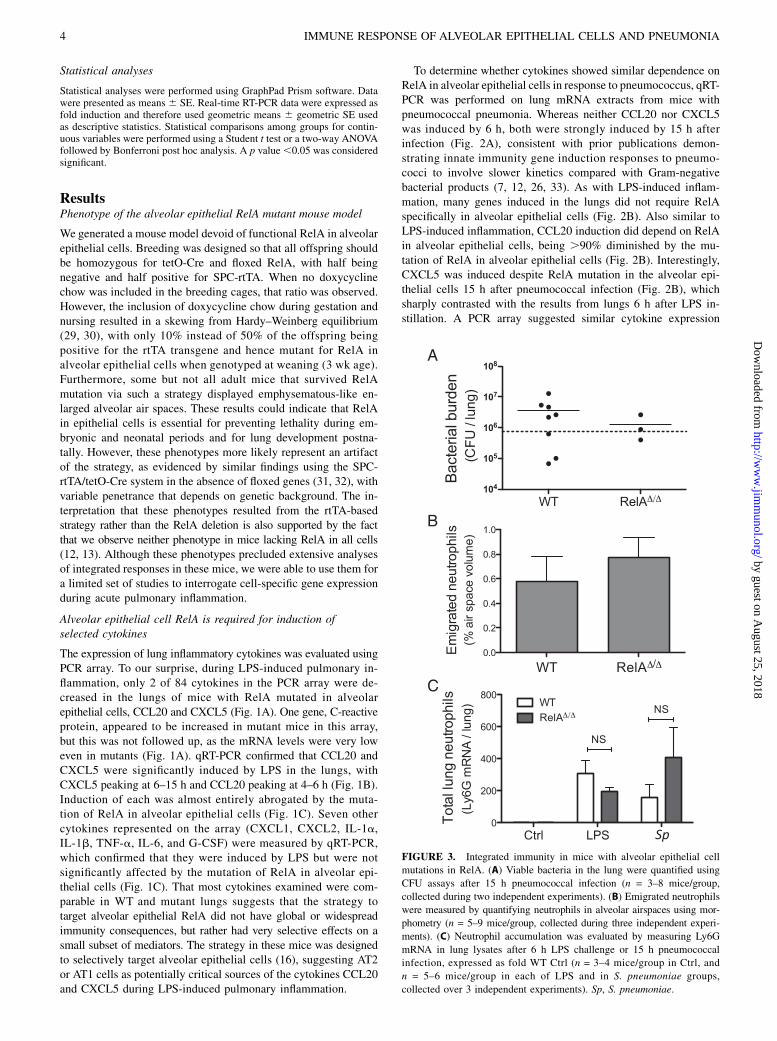
FIGURE 3. Integrated immunity in mice with alveolar epithelial cell
mutations in RelA. (A) Viable bacteria in the lung were quantified using
CFU assays after 15 h pneumococcal infection (n = 3–8 mice/group,
collected during two independent experiments). (B) Emigrated neutrophils
were measured by quantifying neutrophils in alveolar airspaces using mor-
phometry (n = 5–9 mice/group, collected during three independent experi-
ments). (C) Neutrophil accumulation was evaluated by measuring Ly6G
mRNA in lung lysates after 6 h LPS challenge or 15 h pneumococcal
infection, expressed as fold WT Ctrl (n = 3–4 mice/group in Ctrl, and
n = 5–6 mice/group in each of LPS and in S. pneumoniae groups,
collected over 3 independent experiments). Sp, S. pneumoniae.
4 IMMUNE RESPONSE OF ALVEOLAR EPITHELIAL CELLS AND PNEUMONIA
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
patterns in the lungs of mutant mice compared with WT afterpneumococcus infection (Fig. 2C). CXCL5 was unaffected by RelAmutation, CCL20 was diminished, and two additional transcripts(for CXCR3 and IL-10Rb) were potentially blunted by epithelialRelA mutation during pneumococcal pneumonia. Although theseresults suggest additional new lines of inquiry, we chose to main-tain focus on the epithelial-derived chemokines and pursue thesuggestion that CCL20 is consistently dependent on epithelialRelA whereas CXCL5 dependency appears to differ after LPS orpneumococcus. Importantly, the disparate roles for alveolar epi-thelial RelA in pneumococcal induction of CXCL5 and CCL20were similarly observed when examined at the protein level usingELISA (Fig. 2D). These results suggest a heretofore unappre-ciated stimulus specificity relating to the role of alveolar epithelialRelA in CXCL5 induction, which could result from distinct cellsources (e.g., AT2 versus AT1 cells) and/or distinct molecular reg-ulation (RelA-dependent versus -independent pathways) within thesame cells.
Integrated immune responses
Limitations of the animal model, discussed above, precluded ex-tensive analyses of integrated immune responses, but exploratorystudies were performed in an effort to reveal roles of epithelialRelA. Bacterial burdens were measured 24 h after pneumococcalinfection. Neither WT nor mutant mice showed effective hostdefense during this infection, and there were no significant dif-ferences between genotypes (Fig. 3A). In this transgenic model,there were no significant effects of epithelial RelA mutation onneutrophil recruitment in the lungs, measured by quantifying neu-trophils in the alveolar air spaces using morphometry after LPSinstillation (Fig. 3B) and by quantifying neutrophil-specific Ly6G
mRNA in the lung using qRT-PCR after either LPS or pneumo-coccus instillation (Fig. 3C). Thus, these data do not reveal rolesfor epithelial RelA in integrated innate immune responses. How-ever, the limitations of this transgenic mouse model system, par-ticularly the perinatal lethality phenotype, restrict the numbers andtypes of experiments that can be done using this model. A betterunderstanding of the functional significance of epithelial RelA inintegrated pulmonary immunity will require alternative and improvedmodels for cell-specific targeting of this transcription factor.
Distinct cytokine expression patterns in alveolar epithelial celllines in vitro
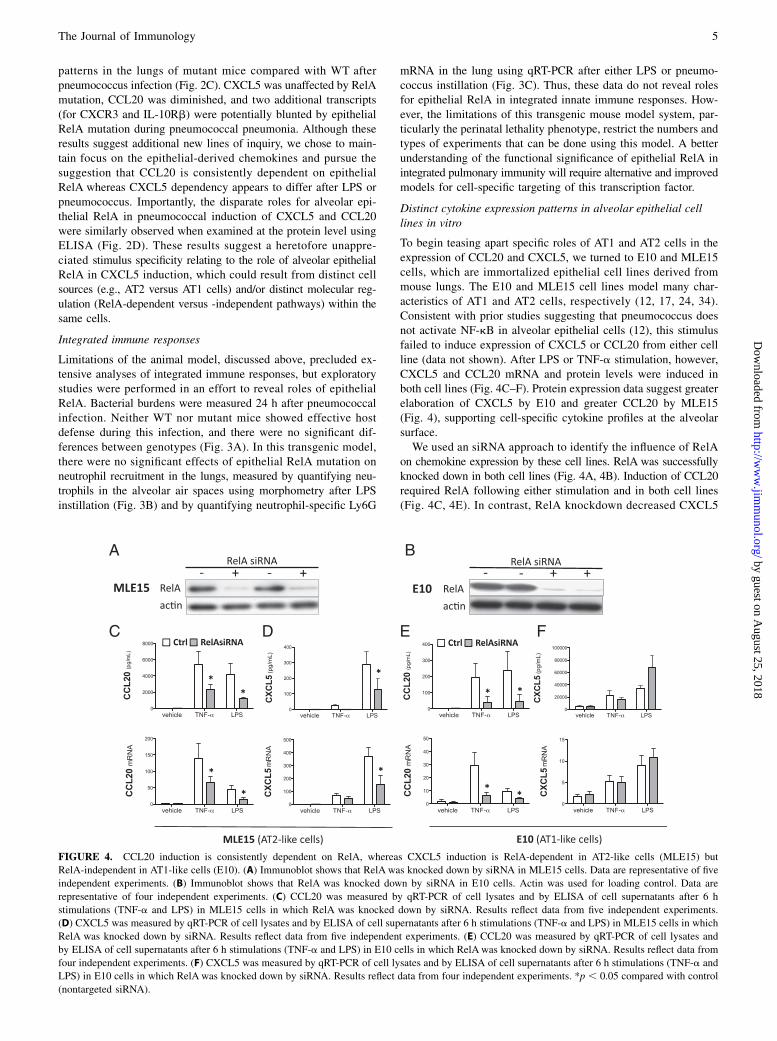
To begin teasing apart specific roles of AT1 and AT2 cells in theexpression of CCL20 and CXCL5, we turned to E10 and MLE15cells, which are immortalized epithelial cell lines derived frommouse lungs. The E10 and MLE15 cell lines model many char-acteristics of AT1 and AT2 cells, respectively (12, 17, 24, 34).Consistent with prior studies suggesting that pneumococcus doesnot activate NF-kB in alveolar epithelial cells (12), this stimulusfailed to induce expression of CXCL5 or CCL20 from either cellline (data not shown). After LPS or TNF-a stimulation, however,CXCL5 and CCL20 mRNA and protein levels were induced inboth cell lines (Fig. 4C–F). Protein expression data suggest greaterelaboration of CXCL5 by E10 and greater CCL20 by MLE15(Fig. 4), supporting cell-specific cytokine profiles at the alveolarsurface.We used an siRNA approach to identify the influence of RelA
on chemokine expression by these cell lines. RelA was successfullyknocked down in both cell lines (Fig. 4A, 4B). Induction of CCL20required RelA following either stimulation and in both cell lines(Fig. 4C, 4E). In contrast, RelA knockdown decreased CXCL5
FIGURE 4. CCL20 induction is consistently dependent on RelA, whereas CXCL5 induction is RelA-dependent in AT2-like cells (MLE15) but
RelA-independent in AT1-like cells (E10). (A) Immunoblot shows that RelA was knocked down by siRNA in MLE15 cells. Data are representative of five
independent experiments. (B) Immunoblot shows that RelA was knocked down by siRNA in E10 cells. Actin was used for loading control. Data are
representative of four independent experiments. (C) CCL20 was measured by qRT-PCR of cell lysates and by ELISA of cell supernatants after 6 h
stimulations (TNF-a and LPS) in MLE15 cells in which RelA was knocked down by siRNA. Results reflect data from five independent experiments.
(D) CXCL5 was measured by qRT-PCR of cell lysates and by ELISA of cell supernatants after 6 h stimulations (TNF-a and LPS) in MLE15 cells in which
RelA was knocked down by siRNA. Results reflect data from five independent experiments. (E) CCL20 was measured by qRT-PCR of cell lysates and
by ELISA of cell supernatants after 6 h stimulations (TNF-a and LPS) in E10 cells in which RelA was knocked down by siRNA. Results reflect data from
four independent experiments. (F) CXCL5 was measured by qRT-PCR of cell lysates and by ELISA of cell supernatants after 6 h stimulations (TNF-a and
LPS) in E10 cells in which RelA was knocked down by siRNA. Results reflect data from four independent experiments. *p , 0.05 compared with control
(nontargeted siRNA).
The Journal of Immunology 5
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
induction only in MLE15 cells (Fig. 4D, 4F). These data suggestthe unexpected hypothesis that AT1 cells may be important sourcesof CXCL5, and they may be able to produce this chemokine ina RelA-independent manner.
In vivo responses of primary AT1 cells during pneumonia
To elucidate distinct cytokine expression patterns from differentepithelial cell types in vivo, AT1 cells were isolated from the lungusing Ab against T1a, also known as podoplanin or RT140, asurface protein expressed by AT1 cells (35–39). In lung single-cell suspensions, the T1a+ cell population was clearly separatedfrom T1a2 cells (Fig. 5A). T1a+ cells express high podoplaninand caveolin-1, but not markers of other epithelial cells (SPC,SPB, Nkx2-1, or CC10), suggesting successful separation of AT1cells from AT2 cells and Clara cells (Fig. 5B). Immunofluores-cence of sorted cells confirmed that caveolin-1 staining, char-acteristic of AT1 cells, was observed in the T1a+ population butnot the T1a2 population, whereas pro-SPC staining, characteristicof AT2 cells, showed the opposite pattern (Fig. 5C). Completepurity was not achieved, as a fraction of cells in the T1a+ pop-
ulation did not stain for caveolin-1 (Fig. 5C). Potential T1a+ cellsin addition to AT1 cells may include lymphatic endothelial cells,basal cells, and pleural cells (40, 41). PECAM-1 and p75, char-acteristic of lymphatic endothelial cells and basal cells, respec-tively, were less prominent in the T1a+ population compared withthe nonselected cells (Fig. 5B). Muc16, characteristic of pleuralcells but not other cells from the lung (42), was higher in the T1a+
population (data not shown), suggesting cells from the visceralpleura as the other cells included with AT1 cells in these prepa-rations. We interpret these T1a+ cell pools as greatly enrichedfor AT1 cells and devoid of their most closely related and ap-posed AT2 cells, although we recognize that they are not pureAT1 cell populations.During LPS-induced pulmonary inflammation, both CXCL5
and CCL20 were induced in cells other than AT1 cells and notin the T1a+ cells (Fig. 5D, 5E). During pneumococcal pneu-monia, as after LPS, CCL20 was only induced in cells other thanAT1 cells (Fig. 5F). In contrast, CXCL5 was strongly induced inthe T1a+ AT1 cells during pneumococcal pneumonia (Fig. 5G).These results indicate that CXCL5 is induced in AT1 cells during
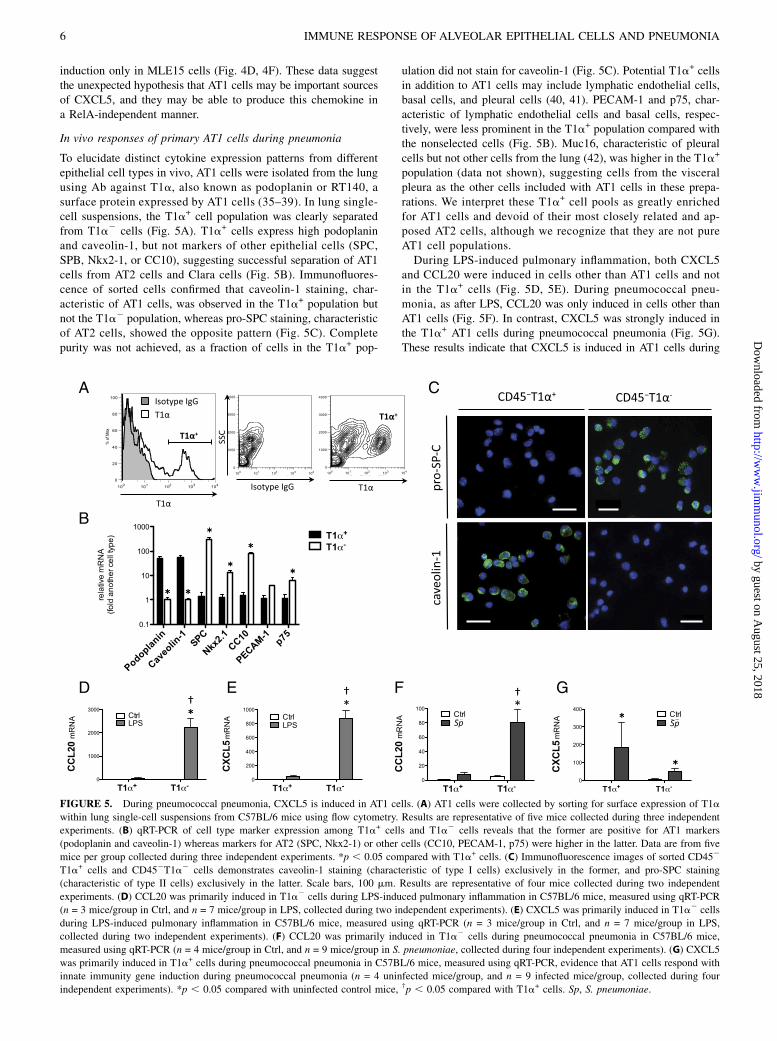
FIGURE 5. During pneumococcal pneumonia, CXCL5 is induced in AT1 cells. (A) AT1 cells were collected by sorting for surface expression of T1a
within lung single-cell suspensions from C57BL/6 mice using flow cytometry. Results are representative of five mice collected during three independent
experiments. (B) qRT-PCR of cell type marker expression among T1a+ cells and T1a2 cells reveals that the former are positive for AT1 markers
(podoplanin and caveolin-1) whereas markers for AT2 (SPC, Nkx2-1) or other cells (CC10, PECAM-1, p75) were higher in the latter. Data are from five
mice per group collected during three independent experiments. *p , 0.05 compared with T1a+ cells. (C) Immunofluorescence images of sorted CD452
T1a+ cells and CD452T1a2 cells demonstrates caveolin-1 staining (characteristic of type I cells) exclusively in the former, and pro-SPC staining
(characteristic of type II cells) exclusively in the latter. Scale bars, 100 mm. Results are representative of four mice collected during two independent
experiments. (D) CCL20 was primarily induced in T1a2 cells during LPS-induced pulmonary inflammation in C57BL/6 mice, measured using qRT-PCR
(n = 3 mice/group in Ctrl, and n = 7 mice/group in LPS, collected during two independent experiments). (E) CXCL5 was primarily induced in T1a2 cells
during LPS-induced pulmonary inflammation in C57BL/6 mice, measured using qRT-PCR (n = 3 mice/group in Ctrl, and n = 7 mice/group in LPS,
collected during two independent experiments). (F) CCL20 was primarily induced in T1a2 cells during pneumococcal pneumonia in C57BL/6 mice,
measured using qRT-PCR (n = 4 mice/group in Ctrl, and n = 9 mice/group in S. pneumoniae, collected during four independent experiments). (G) CXCL5
was primarily induced in T1a+ cells during pneumococcal pneumonia in C57BL/6 mice, measured using qRT-PCR, evidence that AT1 cells respond with
innate immunity gene induction during pneumococcal pneumonia (n = 4 uninfected mice/group, and n = 9 infected mice/group, collected during four
independent experiments). *p , 0.05 compared with uninfected control mice, †p , 0.05 compared with T1a+ cells. Sp, S. pneumoniae.
6 IMMUNE RESPONSE OF ALVEOLAR EPITHELIAL CELLS AND PNEUMONIA
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
pneumococcal pneumonia, which differs from LPS-induced in-flammation and which is to our knowledge the first direct evidenceof an innate immune response from type I alveolar epithelial cells.
AT1 cells induce CXCL5 independent of RelA duringpneumococcal pneumonia
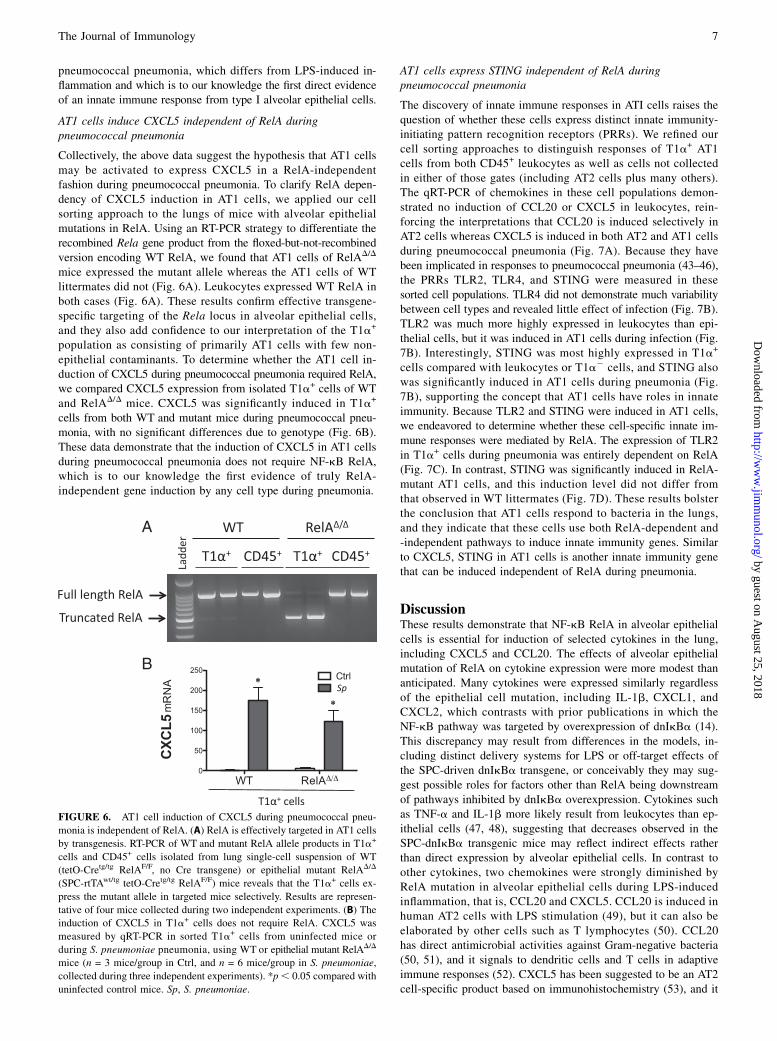
Collectively, the above data suggest the hypothesis that AT1 cellsmay be activated to express CXCL5 in a RelA-independentfashion during pneumococcal pneumonia. To clarify RelA depen-dency of CXCL5 induction in AT1 cells, we applied our cellsorting approach to the lungs of mice with alveolar epithelialmutations in RelA. Using an RT-PCR strategy to differentiate therecombined Rela gene product from the floxed-but-not-recombinedversion encoding WT RelA, we found that AT1 cells of RelAD/D
mice expressed the mutant allele whereas the AT1 cells of WTlittermates did not (Fig. 6A). Leukocytes expressed WT RelA inboth cases (Fig. 6A). These results confirm effective transgene-specific targeting of the Rela locus in alveolar epithelial cells,and they also add confidence to our interpretation of the T1a+
population as consisting of primarily AT1 cells with few non-epithelial contaminants. To determine whether the AT1 cell in-duction of CXCL5 during pneumococcal pneumonia required RelA,we compared CXCL5 expression from isolated T1a+ cells of WTand RelAD/D mice. CXCL5 was significantly induced in T1a+
cells from both WT and mutant mice during pneumococcal pneu-monia, with no significant differences due to genotype (Fig. 6B).These data demonstrate that the induction of CXCL5 in AT1 cellsduring pneumococcal pneumonia does not require NF-kB RelA,which is to our knowledge the first evidence of truly RelA-independent gene induction by any cell type during pneumonia.
AT1 cells express STING independent of RelA duringpneumococcal pneumonia
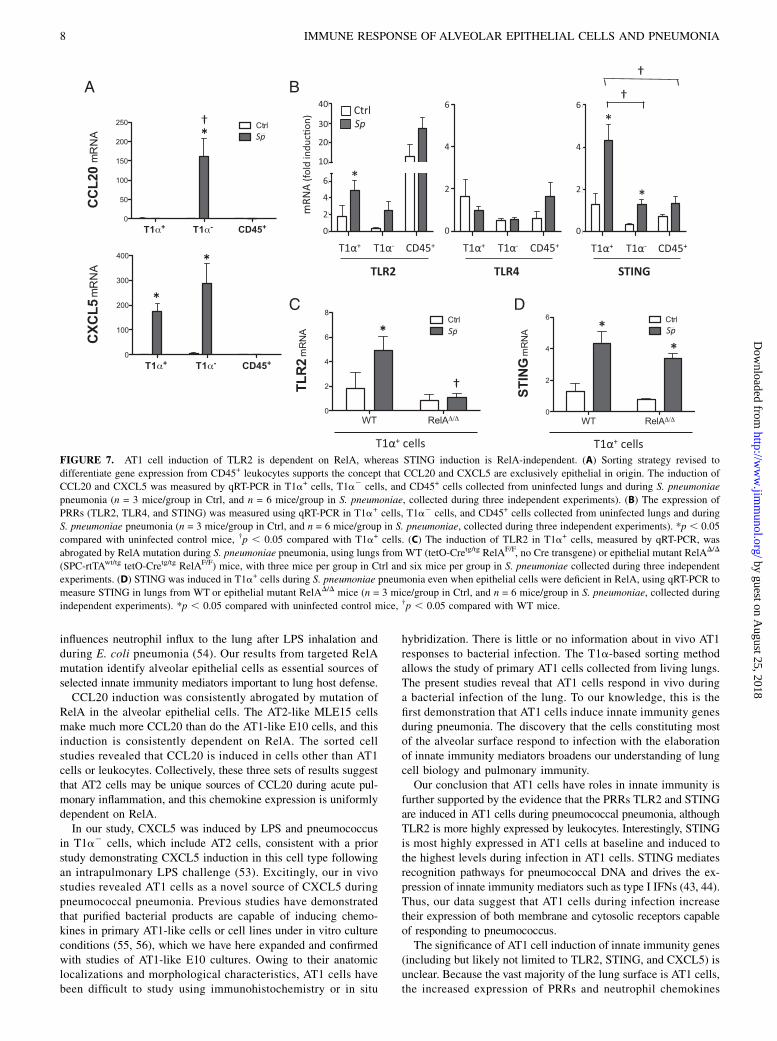
The discovery of innate immune responses in ATI cells raises thequestion of whether these cells express distinct innate immunity-initiating pattern recognition receptors (PRRs). We refined ourcell sorting approaches to distinguish responses of T1a+ AT1cells from both CD45+ leukocytes as well as cells not collectedin either of those gates (including AT2 cells plus many others).The qRT-PCR of chemokines in these cell populations demon-strated no induction of CCL20 or CXCL5 in leukocytes, rein-forcing the interpretations that CCL20 is induced selectively inAT2 cells whereas CXCL5 is induced in both AT2 and AT1 cellsduring pneumococcal pneumonia (Fig. 7A). Because they havebeen implicated in responses to pneumococcal pneumonia (43–46),the PRRs TLR2, TLR4, and STING were measured in thesesorted cell populations. TLR4 did not demonstrate much variabilitybetween cell types and revealed little effect of infection (Fig. 7B).TLR2 was much more highly expressed in leukocytes than epi-thelial cells, but it was induced in AT1 cells during infection (Fig.7B). Interestingly, STING was most highly expressed in T1a+
cells compared with leukocytes or T1a2 cells, and STING alsowas significantly induced in AT1 cells during pneumonia (Fig.7B), supporting the concept that AT1 cells have roles in innateimmunity. Because TLR2 and STING were induced in AT1 cells,we endeavored to determine whether these cell-specific innate im-mune responses were mediated by RelA. The expression of TLR2in T1a+ cells during pneumonia was entirely dependent on RelA(Fig. 7C). In contrast, STING was significantly induced in RelA-mutant AT1 cells, and this induction level did not differ fromthat observed in WT littermates (Fig. 7D). These results bolsterthe conclusion that AT1 cells respond to bacteria in the lungs,and they indicate that these cells use both RelA-dependent and-independent pathways to induce innate immunity genes. Similarto CXCL5, STING in AT1 cells is another innate immunity genethat can be induced independent of RelA during pneumonia.
DiscussionThese results demonstrate that NF-kB RelA in alveolar epithelialcells is essential for induction of selected cytokines in the lung,including CXCL5 and CCL20. The effects of alveolar epithelialmutation of RelA on cytokine expression were more modest thananticipated. Many cytokines were expressed similarly regardlessof the epithelial cell mutation, including IL-1b, CXCL1, andCXCL2, which contrasts with prior publications in which theNF-kB pathway was targeted by overexpression of dnIkBa (14).This discrepancy may result from differences in the models, in-cluding distinct delivery systems for LPS or off-target effects ofthe SPC-driven dnIkBa transgene, or conceivably they may sug-gest possible roles for factors other than RelA being downstreamof pathways inhibited by dnIkBa overexpression. Cytokines suchas TNF-a and IL-1b more likely result from leukocytes than ep-ithelial cells (47, 48), suggesting that decreases observed in theSPC-dnIkBa transgenic mice may reflect indirect effects ratherthan direct expression by alveolar epithelial cells. In contrast toother cytokines, two chemokines were strongly diminished byRelA mutation in alveolar epithelial cells during LPS-inducedinflammation, that is, CCL20 and CXCL5. CCL20 is induced inhuman AT2 cells with LPS stimulation (49), but it can also beelaborated by other cells such as T lymphocytes (50). CCL20has direct antimicrobial activities against Gram-negative bacteria(50, 51), and it signals to dendritic cells and T cells in adaptiveimmune responses (52). CXCL5 has been suggested to be an AT2cell-specific product based on immunohistochemistry (53), and it
FIGURE 6. AT1 cell induction of CXCL5 during pneumococcal pneu-
monia is independent of RelA. (A) RelA is effectively targeted in AT1 cells
by transgenesis. RT-PCR of WT and mutant RelA allele products in T1a+
cells and CD45+ cells isolated from lung single-cell suspension of WT
(tetO-Cretg/tg RelAF/F, no Cre transgene) or epithelial mutant RelAD/D
(SPC-rtTAwt/tg tetO-Cretg/tg RelAF/F) mice reveals that the T1a+ cells ex-
press the mutant allele in targeted mice selectively. Results are represen-
tative of four mice collected during two independent experiments. (B) The
induction of CXCL5 in T1a+ cells does not require RelA. CXCL5 was
measured by qRT-PCR in sorted T1a+ cells from uninfected mice or
during S. pneumoniae pneumonia, using WT or epithelial mutant RelAD/D
mice (n = 3 mice/group in Ctrl, and n = 6 mice/group in S. pneumoniae,
collected during three independent experiments). *p, 0.05 compared with
uninfected control mice. Sp, S. pneumoniae.
The Journal of Immunology 7
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
influences neutrophil influx to the lung after LPS inhalation andduring E. coli pneumonia (54). Our results from targeted RelAmutation identify alveolar epithelial cells as essential sources ofselected innate immunity mediators important to lung host defense.CCL20 induction was consistently abrogated by mutation of
RelA in the alveolar epithelial cells. The AT2-like MLE15 cellsmake much more CCL20 than do the AT1-like E10 cells, and thisinduction is consistently dependent on RelA. The sorted cellstudies revealed that CCL20 is induced in cells other than AT1cells or leukocytes. Collectively, these three sets of results suggestthat AT2 cells may be unique sources of CCL20 during acute pul-monary inflammation, and this chemokine expression is uniformlydependent on RelA.In our study, CXCL5 was induced by LPS and pneumococcus
in T1a2 cells, which include AT2 cells, consistent with a priorstudy demonstrating CXCL5 induction in this cell type followingan intrapulmonary LPS challenge (53). Excitingly, our in vivostudies revealed AT1 cells as a novel source of CXCL5 duringpneumococcal pneumonia. Previous studies have demonstratedthat purified bacterial products are capable of inducing chemo-kines in primary AT1-like cells or cell lines under in vitro cultureconditions (55, 56), which we have here expanded and confirmedwith studies of AT1-like E10 cultures. Owing to their anatomiclocalizations and morphological characteristics, AT1 cells havebeen difficult to study using immunohistochemistry or in situ
hybridization. There is little or no information about in vivo AT1responses to bacterial infection. The T1a-based sorting methodallows the study of primary AT1 cells collected from living lungs.The present studies reveal that AT1 cells respond in vivo duringa bacterial infection of the lung. To our knowledge, this is thefirst demonstration that AT1 cells induce innate immunity genesduring pneumonia. The discovery that the cells constituting mostof the alveolar surface respond to infection with the elaborationof innate immunity mediators broadens our understanding of lungcell biology and pulmonary immunity.Our conclusion that AT1 cells have roles in innate immunity is
further supported by the evidence that the PRRs TLR2 and STINGare induced in AT1 cells during pneumococcal pneumonia, althoughTLR2 is more highly expressed by leukocytes. Interestingly, STINGis most highly expressed in AT1 cells at baseline and induced tothe highest levels during infection in AT1 cells. STING mediatesrecognition pathways for pneumococcal DNA and drives the ex-pression of innate immunity mediators such as type I IFNs (43, 44).Thus, our data suggest that AT1 cells during infection increasetheir expression of both membrane and cytosolic receptors capableof responding to pneumococcus.The significance of AT1 cell induction of innate immunity genes
(including but likely not limited to TLR2, STING, and CXCL5) isunclear. Because the vast majority of the lung surface is AT1 cells,the increased expression of PRRs and neutrophil chemokines
FIGURE 7. AT1 cell induction of TLR2 is dependent on RelA, whereas STING induction is RelA-independent. (A) Sorting strategy revised to
differentiate gene expression from CD45+ leukocytes supports the concept that CCL20 and CXCL5 are exclusively epithelial in origin. The induction of
CCL20 and CXCL5 was measured by qRT-PCR in T1a+ cells, T1a2 cells, and CD45+ cells collected from uninfected lungs and during S. pneumoniae
pneumonia (n = 3 mice/group in Ctrl, and n = 6 mice/group in S. pneumoniae, collected during three independent experiments). (B) The expression of
PRRs (TLR2, TLR4, and STING) was measured using qRT-PCR in T1a+ cells, T1a2 cells, and CD45+ cells collected from uninfected lungs and during
S. pneumoniae pneumonia (n = 3 mice/group in Ctrl, and n = 6 mice/group in S. pneumoniae, collected during three independent experiments). *p , 0.05
compared with uninfected control mice, †p , 0.05 compared with T1a+ cells. (C) The induction of TLR2 in T1a+ cells, measured by qRT-PCR, was
abrogated by RelA mutation during S. pneumoniae pneumonia, using lungs from WT (tetO-Cretg/tg RelAF/F, no Cre transgene) or epithelial mutant RelAD/D
(SPC-rtTAwt/tg tetO-Cretg/tg RelAF/F) mice, with three mice per group in Ctrl and six mice per group in S. pneumoniae collected during three independent
experiments. (D) STING was induced in T1a+ cells during S. pneumoniae pneumonia even when epithelial cells were deficient in RelA, using qRT-PCR to
measure STING in lungs from WT or epithelial mutant RelAD/D mice (n = 3 mice/group in Ctrl, and n = 6 mice/group in S. pneumoniae, collected during
independent experiments). *p , 0.05 compared with uninfected control mice, †p , 0.05 compared with WT mice.
8 IMMUNE RESPONSE OF ALVEOLAR EPITHELIAL CELLS AND PNEUMONIA
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

may limit the spread of microbes throughout alveoli or acrossepithelial barriers. The fact that we did not see defects in in-flammation and host defense is certainly not evidence for a lackof a role; these particular challenges and the limitations of thistransgenic mouse model (discussed above) may have been in-adequate to reveal functionally significant effects of AT1-derivedgenes. It is tempting to speculate that AT1 responses may be es-pecially important in settings where microbes subvert macrophageresponses, because alveolar macrophages are typically sentinel cellsalerting to lung infection (2, 25). The functional significance ofinnate immunity gene induction by AT1 cells now becomes animportant focus for future research.Not only do the results indicate that CXCL5, TLR2, and STING
are induced in AT1 cells during infection, they also illuminatedistinct transcriptional regulation in these cells. TLR2 inductionin AT1 cells was entirely abrogated by RelA deletion, revealingthat AT1 cells can use RelA to induce the expression of innateimmunity mediators, similar to prior observations with other cells(12, 13, 25). In contrast, the induction of CXCL5 and STINGwas unaffected by the mutation of RelA. This was unexpectedand, to our knowledge, represents the first observations of trulyNF-kB RelA-independent innate immunity gene induction in pneu-monic lungs. These results highlight novel pathways for innateimmunity gene expression yet to be discovered.Most significantly, these results demonstrate that AT1 cells are
underappreciated players in lung immunity and antibacterial hostdefense. These cells constitute the vast majority of the surface withwhich bacteria can interact during lung infection. Which productsthey elaborate, by what recognition and signaling and transcrip-tional pathways, and what effects they have on integrated immuneand physiological processes now become critical questions. Animproved understanding of the mechanisms and the significanceof AT1 cell responses to infection may suggest new suscepti-bility determinants and therapeutic approaches for acute bacterialpneumonia.
AcknowledgmentsWe are grateful to Dr. Laertis Oikonomou, Dr. Jesus Paez-Cortez, Anne C.
Hinds, and Meenakshi Lakshminarayanan (Pulmonary Center, Boston Uni-
versity School of Medicine) for help with immunofluorescence staining.
DisclosuresThe authors have no financial conflicts of interest.
References1. Parker, D., and A. Prince. 2011. Innate immunity in the respiratory epithelium.
Am. J. Respir. Cell Mol. Biol. 45: 189–201.2. Mizgerd, J. P. 2008. Acute lower respiratory tract infection. N. Engl. J. Med. 358:
716–727.3. Mason, R. J. 2006. Biology of alveolar type II cells. Respirology 11(Suppl.):
S12–S15.4. Crapo, J. D., B. E. Barry, P. Gehr, M. Bachofen, and E. R. Weibel. 1982. Cell
number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis.126: 332–337.
5. McElroy, M. C., and M. Kasper. 2004. The use of alveolar epithelial type Icell-selective markers to investigate lung injury and repair. Eur. Respir. J. 24:664–673.
6. Stone, K. C., R. R. Mercer, P. Gehr, B. Stockstill, and J. D. Crapo. 1992. Al-lometric relationships of cell numbers and size in the mammalian lung. Am.J. Respir. Cell Mol. Biol. 6: 235–243.
7. Jones, M. R., B. T. Simms, M. M. Lupa, M. S. Kogan, and J. P. Mizgerd. 2005.Lung NF-kB activation and neutrophil recruitment require IL-1 and TNF re-ceptor signaling during pneumococcal pneumonia. J. Immunol. 175: 7530–7535.
8. Blackwell, T. S., L. H. Lancaster, T. R. Blackwell, A. Venkatakrishnan, andJ. W. Christman. 1999. Differential NF-kB activation after intratracheal endo-toxin. Am. J. Physiol. 277: L823–L830.
9. Mizgerd, J. P., M. L. Scott, M. R. Spieker, and C. M. Doerschuk. 2002. Functionsof IkB proteins in inflammatory responses to Escherichia coli LPS in mouselungs. Am. J. Respir. Cell Mol. Biol. 27: 575–582.
10. Saccani, A., T. Schioppa, C. Porta, S. K. Biswas, M. Nebuloni, L. Vago,B. Bottazzi, M. P. Colombo, A. Mantovani, and A. Sica. 2006. p50 nuclearfactor-kB overexpression in tumor-associated macrophages inhibits M1 in-flammatory responses and antitumor resistance. Cancer Res. 66: 11432–11440.
11. Mizgerd, J. P., M. M. Lupa, M. S. Kogan, H. B. Warren, L. Kobzik, andG. P. Topulos. 2003. Nuclear factor-kB p50 limits inflammation and preventslung injury during Escherichia coli pneumonia. Am. J. Respir. Crit. Care Med.168: 810–817.
12. Quinton, L. J., M. R. Jones, B. T. Simms, M. S. Kogan, B. E. Robson,S. J. Skerrett, and J. P. Mizgerd. 2007. Functions and regulation of NF-kB RelAduring pneumococcal pneumonia. J. Immunol. 178: 1896–1903.
13. Alcamo, E., J. P. Mizgerd, B. H. Horwitz, R. Bronson, A. A. Beg, M. Scott,C. M. Doerschuk, R. O. Hynes, and D. Baltimore. 2001. Targeted mutation ofTNF receptor I rescues the RelA-deficient mouse and reveals a critical role forNF-kB in leukocyte recruitment. J. Immunol. 167: 1592–1600.
14. Skerrett, S. J., H. D. Liggitt, A. M. Hajjar, R. K. Ernst, S. I. Miller, andC. B. Wilson. 2004. Respiratory epithelial cells regulate lung inflammation inresponse to inhaled endotoxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L143–L152.
15. Algul, H., M. Treiber, M. Lesina, H. Nakhai, D. Saur, F. Geisler, A. Pfeifer,S. Paxian, and R. M. Schmid. 2007. Pancreas-specific RelA/p65 truncationincreases susceptibility of acini to inflammation-associated cell death followingcerulein pancreatitis. J. Clin. Invest. 117: 1490–1501.
16. Perl, A. K., S. E. Wert, A. Nagy, C. G. Lobe, and J. A. Whitsett. 2002. Earlyrestriction of peripheral and proximal cell lineages during formation of the lung.Proc. Natl. Acad. Sci. USA 99: 10482–10487.
17. Quinton, L. J., M. R. Jones, B. E. Robson, B. T. Simms, J. A. Whitsett, andJ. P. Mizgerd. 2008. Alveolar epithelial STAT3, IL-6 family cytokines, and hostdefense during Escherichia coli pneumonia. Am. J. Respir. Cell Mol. Biol. 38:699–706.
18. Poynter, M. E., C. G. Irvin, and Y. M. Janssen-Heininger. 2003. A prominent rolefor airway epithelial NF-kB activation in lipopolysaccharide-induced airwayinflammation. J. Immunol. 170: 6257–6265.
19. Greenberger, M. J., R. M. Strieter, S. L. Kunkel, J. M. Danforth, R. E. Goodman,and T. J. Standiford. 1995. Neutralization of IL-10 increases survival in a murinemodel of Klebsiella pneumonia. J. Immunol. 155: 722–729.
20. Herigstad, B., M. Hamilton, and J. Heersink. 2001. How to optimize the dropplate method for enumerating bacteria. J. Microbiol. Methods 44: 121–129.
21. Mizgerd, J. P., H. Kubo, G. J. Kutkoski, S. D. Bhagwan, K. Scharffetter-Kochanek, A. L. Beaudet, and C. M. Doerschuk. 1997. Neutrophil emigration inthe skin, lungs, and peritoneum: different requirements for CD11/CD18 revealedby CD18-deficient mice. J. Exp. Med. 186: 1357–1364.
22. Weibel, E. R., C. C. Hsia, and M. Ochs. 2007. How much is there really? Whystereology is essential in lung morphometry. J. Appl. Physiol. 102: 459–467.
23. Smith, G. J., R. K. Kumar, C. P. Hristoforidis, and A. W. Lykke. 1985. Ex-pression of a type 2 pneumocyte-specific antigen by a cell strain from normaladult mouse lung. Cell Biol. Int. Rep. 9: 1115–1122.
24. Wikenheiser, K. A., D. K. Vorbroker, W. R. Rice, J. C. Clark, C. J. Bachurski,H. K. Oie, and J. A. Whitsett. 1993. Production of immortalized distal respira-tory epithelial cell lines from surfactant protein C/simian virus 40 large tumorantigen transgenic mice. Proc. Natl. Acad. Sci. USA 90: 11029–11033.
25. Pittet, L. A., L. J. Quinton, K. Yamamoto, B. E. Robson, J. D. Ferrari, H. Algul,R. M. Schmid, and J. P. Mizgerd. 2011. Earliest innate immune responses requiremacrophage RelA during pneumococcal pneumonia. Am. J. Respir. Cell Mol.Biol. 45: 573–581.
26. Quinton, L. J., M. R. Jones, B. E. Robson, and J. P. Mizgerd. 2009. Mechanismsof the hepatic acute-phase response during bacterial pneumonia. Infect. Immun.77: 2417–2426.
27. Jones, M. R., L. J. Quinton, B. T. Simms, M. M. Lupa, M. S. Kogan, andJ. P. Mizgerd. 2006. Roles of interleukin-6 in activation of STAT proteins andrecruitment of neutrophils during Escherichia coli pneumonia. J. Infect. Dis.193: 360–369.
28. Schmittgen, T. D., B. A. Zakrajsek, A. G. Mills, V. Gorn, M. J. Singer, andM. W. Reed. 2000. Quantitative reverse transcription-polymerase chain reactionto study mRNA decay: comparison of endpoint and real-time methods. Anal.Biochem. 285: 194–204.
29. Hardy, G. H. 1908. Mendelian proportions in a mixed population. Science 28:49–50.
30. Stern, C. 1943. The Hardy-Weinberg law. Science 97: 137–138.31. Sisson, T. H., J. M. Hansen, M. Shah, K. E. Hanson, M. Du, T. Ling,
R. H. Simon, and P. J. Christensen. 2006. Expression of the reverse tetracycline-transactivator gene causes emphysema-like changes in mice. Am. J. Respir. CellMol. Biol. 34: 552–560.
32. Morimoto, M., and R. Kopan. 2009. rtTA toxicity limits the usefulness of theSP-C-rtTA transgenic mouse. Dev. Biol. 325: 171–178.
33. Mizgerd, J. P., M. M. Lupa, J. Hjoberg, J. C. Vallone, H. B. Warren, J. P. Butler,and E. S. Silverman. 2004. Roles for early response cytokines during Escher-ichia coli pneumonia revealed by mice with combined deficiencies of all sig-naling receptors for TNF and IL-1. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L1302–L1310.
34. Cao, Y., T. Vo, G. Millien, J. B. Tagne, D. Kotton, R. J. Mason, M. C. Williams,and M. I. Ramirez. 2010. Epigenetic mechanisms modulate thyroid transcriptionfactor 1-mediated transcription of the surfactant protein B gene. J. Biol. Chem.285: 2152–2164.
35. Dobbs, L. G., M. C. Williams, and R. Gonzalez. 1988. Monoclonal antibodiesspecific to apical surfaces of rat alveolar type I cells bind to surfaces of cultured,but not freshly isolated, type II cells. Biochim. Biophys. Acta 970: 146–156.
The Journal of Immunology 9
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

36. Rishi, A. K., M. Joyce-Brady, J. Fisher, L. G. Dobbs, J. Floros, J. VanderSpek,J. S. Brody, and M. C. Williams. 1995. Cloning, characterization, and de-velopment expression of a rat lung alveolar type I cell gene in embryonicendodermal and neural derivatives. Dev. Biol. 167: 294–306.
37. Gonzalez, R. F., and L. G. Dobbs. 1998. Purification and analysis of RTI40,a type I alveolar epithelial cell apical membrane protein. Biochim. Biophys. Acta1429: 208–216.
38. Vanderbilt, J. N., and L. G. Dobbs. 1998. Characterization of the gene andpromoter for RTI40, a differentiation marker of type I alveolar epithelial cells.Am. J. Respir. Cell Mol. Biol. 19: 662–671.
39. Dahlin, K., E. M. Mager, L. Allen, Z. Tigue, L. Goodglick, M. Wadehra, andL. Dobbs. 2004. Identification of genes differentially expressed in rat alveolartype I cells. Am. J. Respir. Cell Mol. Biol. 31: 309–316.
40. Vanderbilt, J. N., L. Allen, R. F. Gonzalez, Z. Tigue, J. Edmondson, D. Ansaldi,A. M. Gillespie, and L. G. Dobbs. 2008. Directed expression of transgenes toalveolar type I cells in the mouse. Am. J. Respir. Cell Mol. Biol. 39: 253–262.
41. Rock, J. R., M. W. Onaitis, E. L. Rawlins, Y. Lu, C. P. Clark, Y. Xue, S. H. Randell,and B. L. Hogan. 2009. Basal cells as stem cells of the mouse trachea andhuman airway epithelium. Proc. Natl. Acad. Sci. USA 106: 12771–12775.
42. Wang, Y., D. J. Cheon, Z. Lu, S. L. Cunningham, C. M. Chen, R. Z. Luo,D. Xing, S. Orsulic, R. C. Bast, Jr., and R. R. Behringer. 2008. MUC16 ex-pression during embryogenesis, in adult tissues, and ovarian cancer in the mouse.Differentiation 76: 1081–1092.
43. Parker, D., F. J. Martin, G. Soong, B. S. Harfenist, J. L. Aguilar, A. J. Ratner,K. A. Fitzgerald, C. Schindler, and A. Prince. 2011. Streptococcus pneumoniaeDNA initiates type I interferon signaling in the respiratory tract. mBio 2:e00016–e00011.
44. Koppe, U., K. Hogner, J. M. Doehn, H. C. Muller, M. Witzenrath, B. Gutbier,S. Bauer, T. Pribyl, S. Hammerschmidt, J. Lohmeyer, et al. 2012. Streptococcuspneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type IIFN production in macrophages, which regulates RANTES production in mac-rophages, cocultured alveolar epithelial cells, and mouse lungs. J. Immunol. 188:811–817.
45. Malley, R., P. Henneke, S. C. Morse, M. J. Cieslewicz, M. Lipsitch, C. M. Thompson,E. Kurt-Jones, J. C. Paton, M. R. Wessels, and D. T. Golenbock. 2003. Recog-nition of pneumolysin by Toll-like receptor 4 confers resistance to pneumo-coccal infection. Proc. Natl. Acad. Sci. USA 100: 1966–1971.
46. Dessing, M. C., R. A. Hirst, A. F. de Vos, and T. van der Poll. 2009. Role ofToll-like receptors 2 and 4 in pulmonary inflammation and injury induced bypneumolysin in mice. PLoS ONE 4: e7993.
47. Hajjar, A. M., H. Harowicz, H. D. Liggitt, P. J. Fink, C. B. Wilson, andS. J. Skerrett. 2005. An essential role for non-bone marrow-derived cells incontrol of Pseudomonas aeruginosa pneumonia. Am. J. Respir. Cell Mol. Biol.33: 470–475.
48. Noulin, N., V. F. Quesniaux, S. Schnyder-Candrian, B. Schnyder, I. Maillet,T. Robert, B. B. Vargaftig, B. Ryffel, and I. Couillin. 2005. Both hemopoieticand resident cells are required for MyD88-dependent pulmonary inflammatoryresponse to inhaled endotoxin. J. Immunol. 175: 6861–6869.
49. Thorley, A. J., P. Goldstraw, A. Young, and T. D. Tetley. 2005. Primary humanalveolar type II epithelial cell CCL20 (macrophage inflammatory protein-3alpha)-induced dendritic cell migration. Am. J. Respir. Cell Mol. Biol. 32: 262–267.
50. Yang, D., Q. Chen, D. M. Hoover, P. Staley, K. D. Tucker, J. Lubkowski, andJ. J. Oppenheim. 2003. Many chemokines including CCL20/MIP-3a displayantimicrobial activity. J. Leukoc. Biol. 74: 448–455.
51. Starner, T. D., C. K. Barker, H. P. Jia, Y. Kang, and P. B. McCray, Jr. 2003.CCL20 is an inducible product of human airway epithelia with innate immuneproperties. Am. J. Respir. Cell Mol. Biol. 29: 627–633.
52. Stick, S. M., and P. G. Holt. 2003. The airway epithelium as immune modulator:the LARC ascending. Am. J. Respir. Cell Mol. Biol. 28: 641–644.
53. Jeyaseelan, S., R. Manzer, S. K. Young, M. Yamamoto, S. Akira, R. J. Mason,and G. S. Worthen. 2005. Induction of CXCL5 during inflammation in therodent lung involves activation of alveolar epithelium. Am. J. Respir. CellMol. Biol. 32: 531–539.
54. Mei, J., Y. Liu, N. Dai, M. Favara, T. Greene, S. Jeyaseelan, M. Poncz, J. S. Lee,and G. S. Worthen. 2010. CXCL5 regulates chemokine scavenging and pul-monary host defense to bacterial infection. Immunity 33: 106–117.
55. Gentry, M., J. Taormina, R. B. Pyles, L. Yeager, M. Kirtley, V. L. Popov,G. Klimpel, and T. Eaves-Pyles. 2007. Role of primary human alveolar epithelialcells in host defense against Francisella tularensis infection. Infect. Immun. 75:3969–3978.
56. Thorley, A. J., D. Grandolfo, E. Lim, P. Goldstraw, A. Young, and T. D. Tetley.2011. Innate immune responses to bacterial ligands in the peripheral humanlung: role of alveolar epithelial TLR expression and signalling. PLoS ONE 6:e21827.
10 IMMUNE RESPONSE OF ALVEOLAR EPITHELIAL CELLS AND PNEUMONIA
by guest on August 25, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from


















