
Two Family B DNA Polymerases from Aeropyrum pernix, an Aerobic ...
9
JOURNAL OF BACTERIOLOGY, 0021-9193/99/$04.0010 Oct. 1999, p. 5984–5992 Vol. 181, No. 19 Copyright © 1999, American Society for Microbiology. All Rights Reserved. Two Family B DNA Polymerases from Aeropyrum pernix, an Aerobic Hyperthermophilic Crenarchaeote ISAAC K. O. CANN, 1 SONOKO ISHINO, 1 NORIMICHI NOMURA, 2 YOSHIHIKO SAKO, 2 AND YOSHIZUMI ISHINO 1 * Department of Molecular Biology, Biomolecular Engineering Research Institute, Suita, Osaka 565-0874, 1 and Laboratory of Marine Microbiology, Division of Applied Bioscience, Graduate School of Agriculture, Kyoto University, Kyoto 606-8502, 2 Japan Received 16 April 1999/Accepted 29 July 1999 DNA polymerase activities in fractionated cell extract of Aeropyrum pernix, a hyperthermophilic crenarchae- ote, were investigated. Aphidicolin-sensitive (fraction I) and aphidicolin-resistant (fraction II) activities were detected. The activity in fraction I was more heat stable than that in fraction II. Two different genes (polA and polB) encoding family B DNA polymerases were cloned from the organism by PCR using degenerated primers based on the two conserved motifs (motif A and B). The deduced amino acid sequences from their entire coding regions contained all of the motifs identified in family B DNA polymerases for 3*35* exonuclease and polymerase activities. The product of polA gene (Pol I) was aphidicolin resistant and heat stable up to 80°C. In contrast, the product of polB gene (Pol II) was aphidicolin sensitive and stable at 95°C. These properties of Pol I and Pol II are similar to those of fractions II and I, respectively, and moreover, those of Pol I and Pol II of Pyrodictium occultum. The deduced amino acid sequence of A. pernix Pol I exhibited the highest identities to archaeal family B DNA polymerase homologs found only in the crenarchaeotes (group I), while Pol II exhibited identities to homologs found in both euryarchaeotes and crenarchaeotes (group II). These results provide further evidence that the subdomain Crenarchaeota has two family B DNA polymerases. Furthermore, at least two DNA polymerases work in the crenarchaeal cells, as found in euryarchaeotes, which contain one family B DNA polymerase and one heterodimeric DNA polymerase of a novel family. DNA polymerases are indispensable components of the mo- lecular machinery responsible for replicating and repairing the genome of every organism. Studies of members of the domains Bacteria and Eucarya show that for these fundamental pro- cesses of life, multiple DNA polymerases are required (18). However, in the third domain of life, Archaea, the mechanisms involved in DNA replication and repair remain cryptic. Two major subdomains, Euryarchaeota and Crenarchaeota, are now known to exist in Archaea (1). In the complete genome sequences of Methanococcus jannaschii and Pyrococcus horiko- shii in Euryarchaeota, one DNA polymerase gene each was predicted from the similarity of the deduced amino acid se- quences to family B DNA polymerases, and no more pol-like genes were found (4, 16). The fact that these euryarchaeotes may survive with just a single DNA polymerase was very per- plexing (8, 11, 19). Recently, however, a novel two-subunit DNA polymerase whose sequence has no similarity to that of any DNA polymerase family was identified in Pyrococcus fu- riosus (28), in addition to a family B DNA polymerase isolated earlier from this organism (30). Through a protein homology search in the complete genome sequences of M. jannaschii, Archaeoglobus fulgidus, Methanobacterium thermoautotrophi- cum, and P. horikoshii, homologs of this novel DNA polymer- ase were found (7, 15). Based on the conservation of this DNA polymerase in these euryarchaeotes, members of Euryarcha- eota seem to possess at least two DNA polymerases, one of family B and a second, heterodimeric DNA polymerase. On the other hand, the demonstration of more than one DNA polymerase in cell extracts of the organisms in Crenar- chaeota has often been attributed to proteolytic degradation or contamination (20). The evidence, at the gene level, suggesting that multiple DNA polymerases occur in this subdomain orig- inates from research with Sulfolobus solfataricus P2 and Pyro- dictium occultum PL-19. Two genes which seemed to encode family B DNA polymerases were cloned from S. solfataricus (23); then two family B DNA polymerase genes were cloned from P. occultum, and the gene products were proved to be functional DNA polymerases (29). Edgell et al. predicted from the deduced amino acid sequence that the sulfolobales have three family B DNA polymerase genes, and they proposed that the DNA polymerases encoded by these genes be designated B1, B2, and B3 (9). From these reports, it is possible that the organisms in Crenarchaeota have multiple family B DNA poly- merases. However, there is no report on the expression of any of the three DNA polymerase genes of S. solfataricus P2, even though a B1 ortholog of S. solfataricus P2 has been cloned from S. solfataricus MT4, expressed, and characterized in detail (21, 22). For an understanding of archaeal DNA replication, it is necessary to know how many DNA polymerases function in the cells. To obtain further experimental evidence that Crenarcha- eota may contain multiple DNA polymerases in general, we designed an experiment to investigate the existence of multiple DNA polymerases in Aeropyrum pernix, a hyperthermophilic member of this subdomain, both in vivo and at the gene level. In this report, we demonstrate the presence of two family B DNA polymerases in this organism. In addition, we discuss the biochemical properties of the two groups of family B DNA polymerases found in Archaea, one of which exists only in Crenarchaeota whereas the other exists in Euryarchaeota as well. * Corresponding author. Mailing address: Department of Molecular Biology, Biomolecular Engineering Research Institute, 6-2-3 Furuedai, Suita, Osaka 565-0874, Japan. Phone: 81-6-872-8208. Fax: 81-6-872- 8219. E-mail: [email protected]. 5984 on March 16, 2018 by guest http://jb.asm.org/ Downloaded from
Transcript of Two Family B DNA Polymerases from Aeropyrum pernix, an Aerobic ...

JOURNAL OF BACTERIOLOGY,0021-9193/99/$04.0010
Oct. 1999, p. 5984–5992 Vol. 181, No. 19
Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Two Family B DNA Polymerases from Aeropyrum pernix, anAerobic Hyperthermophilic Crenarchaeote
ISAAC K. O. CANN,1 SONOKO ISHINO,1 NORIMICHI NOMURA,2 YOSHIHIKO SAKO,2
AND YOSHIZUMI ISHINO1*
Department of Molecular Biology, Biomolecular Engineering Research Institute, Suita, Osaka 565-0874,1 andLaboratory of Marine Microbiology, Division of Applied Bioscience, Graduate School of Agriculture, Kyoto University,
Kyoto 606-8502,2 Japan
Received 16 April 1999/Accepted 29 July 1999
DNA polymerase activities in fractionated cell extract of Aeropyrum pernix, a hyperthermophilic crenarchae-ote, were investigated. Aphidicolin-sensitive (fraction I) and aphidicolin-resistant (fraction II) activities weredetected. The activity in fraction I was more heat stable than that in fraction II. Two different genes (polA andpolB) encoding family B DNA polymerases were cloned from the organism by PCR using degenerated primersbased on the two conserved motifs (motif A and B). The deduced amino acid sequences from their entire codingregions contained all of the motifs identified in family B DNA polymerases for 3*35* exonuclease andpolymerase activities. The product of polA gene (Pol I) was aphidicolin resistant and heat stable up to 80°C.In contrast, the product of polB gene (Pol II) was aphidicolin sensitive and stable at 95°C. These properties ofPol I and Pol II are similar to those of fractions II and I, respectively, and moreover, those of Pol I and PolII of Pyrodictium occultum. The deduced amino acid sequence of A. pernix Pol I exhibited the highest identitiesto archaeal family B DNA polymerase homologs found only in the crenarchaeotes (group I), while Pol IIexhibited identities to homologs found in both euryarchaeotes and crenarchaeotes (group II). These resultsprovide further evidence that the subdomain Crenarchaeota has two family B DNA polymerases. Furthermore,at least two DNA polymerases work in the crenarchaeal cells, as found in euryarchaeotes, which contain onefamily B DNA polymerase and one heterodimeric DNA polymerase of a novel family.
DNA polymerases are indispensable components of the mo-lecular machinery responsible for replicating and repairing thegenome of every organism. Studies of members of the domainsBacteria and Eucarya show that for these fundamental pro-cesses of life, multiple DNA polymerases are required (18).However, in the third domain of life, Archaea, the mechanismsinvolved in DNA replication and repair remain cryptic.
Two major subdomains, Euryarchaeota and Crenarchaeota,are now known to exist in Archaea (1). In the complete genomesequences of Methanococcus jannaschii and Pyrococcus horiko-shii in Euryarchaeota, one DNA polymerase gene each waspredicted from the similarity of the deduced amino acid se-quences to family B DNA polymerases, and no more pol-likegenes were found (4, 16). The fact that these euryarchaeotesmay survive with just a single DNA polymerase was very per-plexing (8, 11, 19). Recently, however, a novel two-subunitDNA polymerase whose sequence has no similarity to that ofany DNA polymerase family was identified in Pyrococcus fu-riosus (28), in addition to a family B DNA polymerase isolatedearlier from this organism (30). Through a protein homologysearch in the complete genome sequences of M. jannaschii,Archaeoglobus fulgidus, Methanobacterium thermoautotrophi-cum, and P. horikoshii, homologs of this novel DNA polymer-ase were found (7, 15). Based on the conservation of this DNApolymerase in these euryarchaeotes, members of Euryarcha-eota seem to possess at least two DNA polymerases, one offamily B and a second, heterodimeric DNA polymerase.
On the other hand, the demonstration of more than one
DNA polymerase in cell extracts of the organisms in Crenar-chaeota has often been attributed to proteolytic degradation orcontamination (20). The evidence, at the gene level, suggestingthat multiple DNA polymerases occur in this subdomain orig-inates from research with Sulfolobus solfataricus P2 and Pyro-dictium occultum PL-19. Two genes which seemed to encodefamily B DNA polymerases were cloned from S. solfataricus(23); then two family B DNA polymerase genes were clonedfrom P. occultum, and the gene products were proved to befunctional DNA polymerases (29). Edgell et al. predicted fromthe deduced amino acid sequence that the sulfolobales havethree family B DNA polymerase genes, and they proposed thatthe DNA polymerases encoded by these genes be designatedB1, B2, and B3 (9). From these reports, it is possible that theorganisms in Crenarchaeota have multiple family B DNA poly-merases. However, there is no report on the expression of anyof the three DNA polymerase genes of S. solfataricus P2, eventhough a B1 ortholog of S. solfataricus P2 has been cloned fromS. solfataricus MT4, expressed, and characterized in detail (21,22).
For an understanding of archaeal DNA replication, it isnecessary to know how many DNA polymerases function in thecells. To obtain further experimental evidence that Crenarcha-eota may contain multiple DNA polymerases in general, wedesigned an experiment to investigate the existence of multipleDNA polymerases in Aeropyrum pernix, a hyperthermophilicmember of this subdomain, both in vivo and at the gene level.In this report, we demonstrate the presence of two family BDNA polymerases in this organism. In addition, we discuss thebiochemical properties of the two groups of family B DNApolymerases found in Archaea, one of which exists only inCrenarchaeota whereas the other exists in Euryarchaeota aswell.
* Corresponding author. Mailing address: Department of MolecularBiology, Biomolecular Engineering Research Institute, 6-2-3 Furuedai,Suita, Osaka 565-0874, Japan. Phone: 81-6-872-8208. Fax: 81-6-872-8219. E-mail: [email protected].
5984
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

MATERIALS AND METHODS
Preparation of cell extracts for enzyme assay. A. pernix K1 was cultivated asdescribed earlier (26). Ten grams of cells was suspended in buffer A (50 mMTris-HCl [pH 8.0], 0.1 mM EDTA, 10% glycerol, 0.5 mM dithiothreitol) to avolume of 40 ml and disrupted by three passages through a French pressure cell(SLM Aminco, Rochester, N.Y.). Cell debris was removed by centrifugation at48,000 3 g for 30 min at 4°C. The supernatant was dialyzed against buffer Aovernight, and 2.5 ml of the dialysate was applied to an anion-exchange column(HiTrap Q; Pharmacia Biotech, Uppsala, Sweden) fitted to a high-pressureliquid chromatograph (AKTA Explorer 10S; Pharmacia Biotech). After beingloaded with the lysate, the column was washed with 4 column volumes of equil-ibration buffer (buffer A), followed by elution with a linear NaCl gradient (0 to100%) developed with 50 ml of buffer B (buffer A containing 1 M NaCl) at a flowrate of 2 ml/min. The effluents were monitored by absorbance at 280 nm, andfractions of volume 1.5 ml were collected. The contents of each fraction wasanalyzed for DNA polymerase activity, and the positive fractions were pooled forrechromatography.
DNA polymerase assay. The DNA polymerase assay was carried out as de-scribed previously (15). In brief, the assay mixture contained, in a volume of 30ml, 20 mM Tris-HCl (pH 8.8), 2 mM MgCl2, 2 mM b-mercaptoethanol, 0.2 mg ofactivated calf thymus DNA per ml, 40 mM deoxynucleoside triphosphates con-taining 60 nM [methyl-3H]TTP (Amersham, Buckinghamshire, United King-dom), and 3 ml of each fraction. Activated DNA was prepared as outlined byRichardson (24), and the amount of radioactivity incorporated into DNA strandswas counted by a scintillation counter.
Activity gel analysis. In situ analysis of DNA polymerase activity to identify theactive polypeptides in the peak fractions of the A. pernix cell extracts or recom-binant DNA polymerases I and II [Pol I and Pol II] after purification were doneas described earlier (12, 31). (The family B DNA polymerase in euryarchaeotesis designated Pol I [the first DNA polymerase discovered in this subdomain] todistinguish it from the euryarchaeal heterodimeric DNA polymerase referred toas Pol II.) The conditions were the same as those described in previous reports(14, 28).
PCR and DNA sequencing. Genomic DNA from A. pernix K1 was prepared asdescribed by Sako and others (26). Degenerate primers were designed based ontwo conserved motifs A (SLYPSII) and C (VIYGDTD), which are found inpolymerase regions I and II of archaeal family B DNA polymerases (29). Theprimers were used to amplify, via PCR, an approximately 400-bp fragment fromgenomic DNA of A. pernix. PCR was performed in a thermal cycler; the thermalprofiles involved 30 cycles of denaturation at 94°C for 30 s, annealing at 45°C for50 s, and extension at 72°C for 50 s. Approximately 20 ng of genomic DNA servedas the template in the reaction. All PCR reagents were purchased from acommercial source (TaKaRa Shuzo, Kyoto, Japan), and each was used as de-scribed by the manufacturer. The PCR fragment obtained was ligated into a TAcloning vector (pT7 Blue; Novagen, Milwaukee, Wis.), and the product was usedin transforming E. coli JM109 cells. The nucleotide sequences of cloned DNAfragments were determined by a capillary sequencer (ABI Prism 310 geneticanalyzer; Applied Biosystems, Foster City, Calif.), and the BLAST search pro-gram (19a) was used to identify DNA fragments coding for polypeptides withsimilarities to known archaeal family B DNA polymerases.
Genomic Southern hybridization. Aliquots of A. pernix genomic DNA (1.2 mg)were independently digested with BamHI, HindIII, PstI, XbaI, and ScaI. Theproducts of digestion were resolved by electrophoresis on 0.7% agarose gel andtransferred onto a nylon membrane (Hybond-N1; Amersham). Two differentDNA fragments obtained by PCR (ApeI and ApeII; approximately 400 bp each,which translated into amino acid sequences showing high similarity to archaealfamily B DNA polymerases) were used as probes for Southern hybridization asdescribed elsewhere (29). A chemiluminescent labeling and detection system(Gene Images; Amersham) was applied in the procedure to visualize signals.
Genomic walking library construction. The genomic walking library of A.pernix was constructed by using the Universal Genome Walker kit (ClontechLaboratories, Palo Alto, Calif.) as described by the manufacturer. Briefly, thegenomic DNA of A. pernix was digested to completion with EcoRV, PvuII, DraI,ScaI, and StuI. Thus, the DNA fragments generated were all blunt ended. Theproduct of each restriction digest was ligated at 16°C to a blunt-ended adapter,which is a double-stranded deoxyoligonucleotide with a known sequence, in avolume of 8 ml. Ligation mixtures were heated at 70°C for 5 min to end thereaction, and the volume was increased to 80 ml. One microliter of each of thefive libraries served as a template in the genome walking PCR.
Genomic walking to sequence the entire pol genes. To obtain the completenucleotide sequence of the gene which ApeI constituted part of, two primers,ApeI-F1 (59 CTGAAGTCTCTCAGCATGC-39) and ApeI-R1 (59-TATGAAAGTGTATATTAACGCGAG-39) were designed for PCR to obtain nucleotidesequence upstream and downstream of ApeI, respectively. The primers werecombined individually with the adapter primer (AP1; 59 CCTGTAGTCTATTCCAACCCTC-39), supplied by the manufacturer, for the PCR amplifications. Fivedifferent reactions, each containing an aliquot of a different library as the tem-plate, were carried out for ApeI-F1 and likewise ApeI-R1. In the case of ApeII,three primers were used together with the adapter primer for PCR to obtain theentire gene from which it originated. Two of the three primers (ApeII-F1 [59-ACCTCTCAAGTATTTTCTTGA-39] and ApeII-F2 [59-ACAGCCTCCCTATAC
TCCC-39]) were used to obtain nucleotide sequence upstream of ApeII. Toobtain nucleotide sequence at the downstream region, ApeII-R1 (59-TTCAGGAAGAGCCCTCCCGGCTTCTTCAA-39) was used. The complete structuralgene (polA) for Pol I was amplified by using ApeI-N (59-GTTACCATGGCTGGTCCTGCTAAGCCTAAG-39, NcoI tagged) and ApeI-C (59-GAATCCATATGTGGTTATCACGAGTCGAAA-39, NdeI tagged) as forward and reverseprimers, respectively (restriction enzyme recognition sequences are underlined).The entire gene (polB) coding for Pol II was amplified with ApeII-N (59-ATACATATGAGGGGGTCAACCCCCGTTATC-39, NdeI tagged) as the forwardprimer and ApeII-C (59 GATGCGGCCGCATAAGGTACTTCATCCTCCTACACACCC-39, NotI tagged) as the reverse primer. In all of these PCR ampli-fications, the thermal profiles involved 30 cycles of denaturation at 95°C for 30 s,annealing temperature of 58°C for 1 min, and extension at 72°C for 2 min 30 s.The PCR fragments were ligated into a TA cloning vector (pT7 Blue; Novagen),and DNA inserts in isolated recombinant plasmids were sequenced in bothorientations as described above. To ensure accuracy of PCR, TaKaRa LA Taq(TaKaRa Shuzo) was used for amplifications, and several clones were sequencedindependently to correct possible errors.
Cloning and expression of A. pernix polA and polB. The plasmid containingpolA in pT7 Blue was digested with NcoI and NdeI to release polA, while the oneharboring polB in pT7 Blue was digested with NdeI and NotI to obtain polB. ThepolA was inserted into an NcoI/NdeI-digested pET15b (Novagen), and the re-combinant plasmid, pAPP1, was used for transformation of Escherichia coliBL21(DE3) cells. The gene was expressed by incubating transformed cells inLuriani-Bertani broth supplemented with ampicillin (100 mg/ml) at 37°C for 18 h.The polB was expressed in a similar fashion except that the expression plasmid,pAPP2, was made by insertion of the gene into the NdeI-NotI digested pET21a(Novagen).
Purification of A. pernix Pol I and Pol II. Pol I and Pol II were prepared fromE. coli BL21(DE3)/pAPP1 and BL21(DE3)/pAPP2, respectively. One-liter cul-tures of cells harboring the pol genes were harvested by centrifugation at 7,000 3g for 10 min. The cell pellets were suspended in 30 ml of buffer A and lysed bytwo passages through a French pressure cell. The lysates were each centrifugedat 48,000 3 g for 30 min, and the supernatant from each preparation was heatedat 80°C for 15 min followed by recentrifugation. The supernatant containing PolI was applied to a gel filtration column (Superdex 200; Pharmacia) equilibratedwith buffer A, followed by elution with the same buffer. Fractions were examinedfor DNA polymerase activity, and the proteins eliciting activity were identified bythe in situ method as described above. In the case of Pol II, after gel filtration,the fractions containing DNA polymerase activity were pooled, dialyzed againstbuffer A, and applied to an anion-exchange column (HiTrap Q; Pharmacia), andthe chromatography was developed with a 0 to 1 M NaCl gradient.
Primer extension analysis. The primer extension abilities of the purified Pol Iand Pol II were demonstrated by using a M13 single-stranded DNA primed witha 59-labeled oligonucleotide as the substrate (28). To anneal the primer to thetemplate, 1.0 mg of M13 single-stranded DNA and 1.0 pmol of 32P-labeledprimer (45 bases in length) in a buffer containing 20 mM Tris-HCl (pH 6.3) and1.5 mM MgCl2 were heated at 95°C for 3 min to ensure denaturation. Themixture was gently cooled to room temperature. Primer extension was carriedout at 68°C. Each reaction mixture at this temperature received 0.05 U of eitherA. pernix Pol I or Pol II, and the reaction was initiated by adding deoxynucleosidetriphosphates (dNTPs) to a concentration of 250 mM. The total volume of themixture was 20 ml, and aliquots (5 ml) were taken at 1, 2, and 3 min afterinitiation of the reaction. Each aliquot was dispensed into an Eppendorf tubecontaining 3 ml of stop solution (98% deionized formamide, 1 mM EDTA, 0.1%xylene cyanol, 0.1% bromphenol blue), and 2.5 ml of each was analyzed bypolyacrylamide gel electrophoresis (PAGE) in the presence of 8 M urea. Thesame amount of Thermus aquaticus (Taq) DNA polymerase (TaKaRa Taq) wasused to compare the primer extension abilities. One unit was defined as theamount of enzyme catalyzing the incorporation of 10 nmol of dTMP per 30 minat 70°C into DNA, using the conditions described earlier (15). The amount ofTaq DNA polymerase used in this experiment corresponds to 2.5 U according tothe manufacturer’s specification.
Computer analysis. With the exception of A. pernix Pol I and Pol II, all DNApolymerases were retrieved from GenBank. The proteins and their accessionnumbers are as follows: P. occultum Pol I (B1; D38573) and Pol II (B3; D38574),S. solfataricus B1 (U92875) and B3 (X71597), Sulfurisphaera ohwakuensis B1(AB008894), Sulfolobus acidocaldarius B1 (U33846), Cenarchaeum symbiosumB1 (AF028831), Thermococcus litoralis Pol I (M47198), P. furiosus Pol I(D12983), and A. fulgidus Pol I (AE001070). Amino acid sequence comparisonswere carried out at a web site (7a).
Nucleotide sequence accession numbers. The nucleotide sequences of A. per-nix Pol I and Pol II have been submitted to the DDBJ and assigned accession no.AB017500 and AB017501, respectively.
RESULTS
Detection of DNA polymerase activities in cell extracts of A.pernix. Cell extracts of A. pernix were fractionated by an anion-exchange chromatography, and aliquots from the fractionswere analyzed for DNA polymerase activity. Two peaks of
VOL. 181, 1999 TWO FAMILY B DNA POLYMERASES FROM A. PERNIX 5985
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
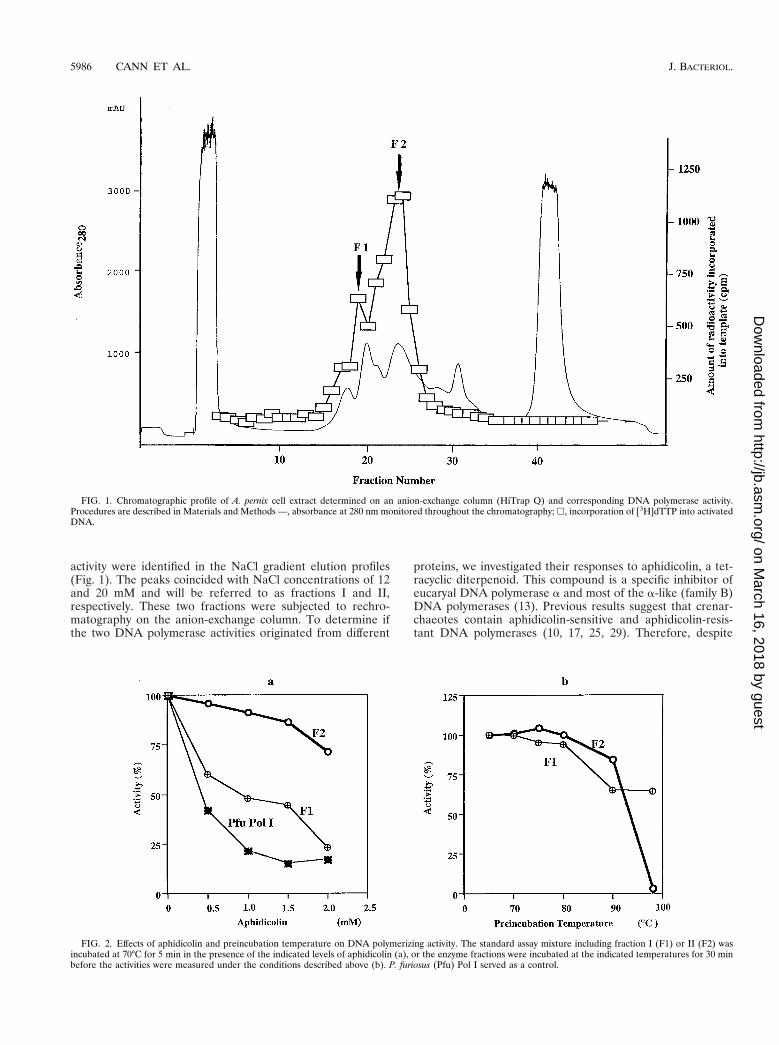
activity were identified in the NaCl gradient elution profiles(Fig. 1). The peaks coincided with NaCl concentrations of 12and 20 mM and will be referred to as fractions I and II,respectively. These two fractions were subjected to rechro-matography on the anion-exchange column. To determine ifthe two DNA polymerase activities originated from different
proteins, we investigated their responses to aphidicolin, a tet-racyclic diterpenoid. This compound is a specific inhibitor ofeucaryal DNA polymerase a and most of the a-like (family B)DNA polymerases (13). Previous results suggest that crenar-chaeotes contain aphidicolin-sensitive and aphidicolin-resis-tant DNA polymerases (10, 17, 25, 29). Therefore, despite
FIG. 1. Chromatographic profile of A. pernix cell extract determined on an anion-exchange column (HiTrap Q) and corresponding DNA polymerase activity.Procedures are described in Materials and Methods —, absorbance at 280 nm monitored throughout the chromatography; h, incorporation of [3H]dTTP into activatedDNA.
FIG. 2. Effects of aphidicolin and preincubation temperature on DNA polymerizing activity. The standard assay mixture including fraction I (F1) or II (F2) wasincubated at 70°C for 5 min in the presence of the indicated levels of aphidicolin (a), or the enzyme fractions were incubated at the indicated temperatures for 30 minbefore the activities were measured under the conditions described above (b). P. furiosus (Pfu) Pol I served as a control.
5986 CANN ET AL. J. BACTERIOL.
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
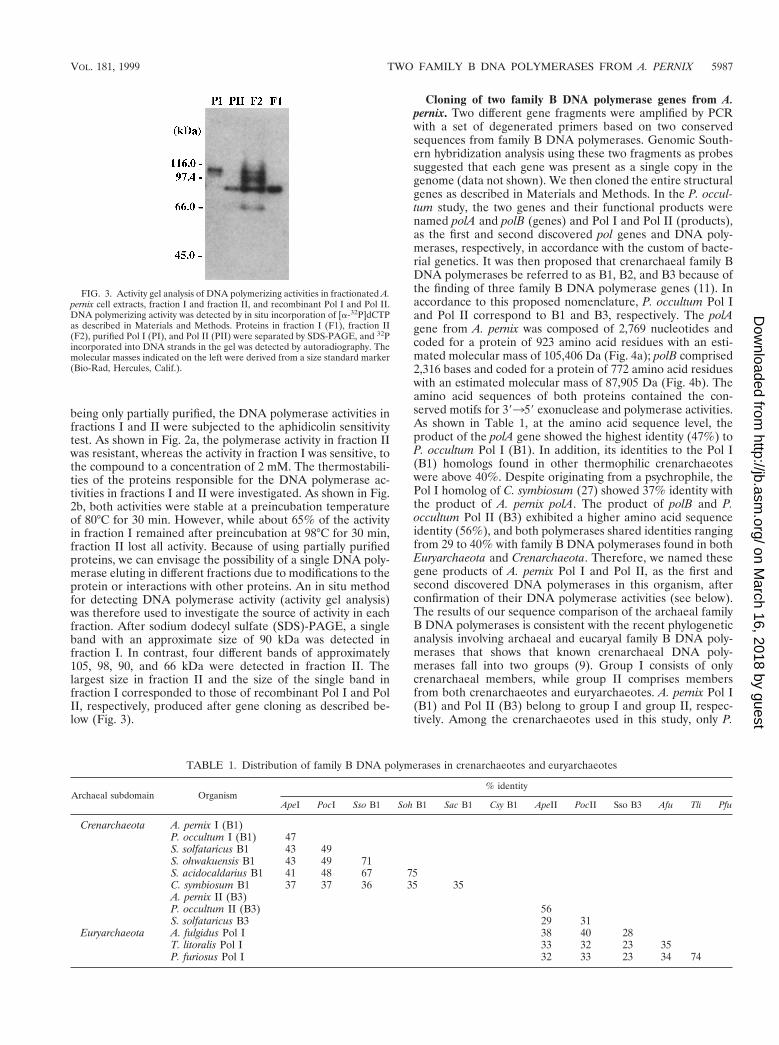
being only partially purified, the DNA polymerase activities infractions I and II were subjected to the aphidicolin sensitivitytest. As shown in Fig. 2a, the polymerase activity in fraction IIwas resistant, whereas the activity in fraction I was sensitive, tothe compound to a concentration of 2 mM. The thermostabili-ties of the proteins responsible for the DNA polymerase ac-tivities in fractions I and II were investigated. As shown in Fig.2b, both activities were stable at a preincubation temperatureof 80°C for 30 min. However, while about 65% of the activityin fraction I remained after preincubation at 98°C for 30 min,fraction II lost all activity. Because of using partially purifiedproteins, we can envisage the possibility of a single DNA poly-merase eluting in different fractions due to modifications to theprotein or interactions with other proteins. An in situ methodfor detecting DNA polymerase activity (activity gel analysis)was therefore used to investigate the source of activity in eachfraction. After sodium dodecyl sulfate (SDS)-PAGE, a singleband with an approximate size of 90 kDa was detected infraction I. In contrast, four different bands of approximately105, 98, 90, and 66 kDa were detected in fraction II. Thelargest size in fraction II and the size of the single band infraction I corresponded to those of recombinant Pol I and PolII, respectively, produced after gene cloning as described be-low (Fig. 3).
Cloning of two family B DNA polymerase genes from A.pernix. Two different gene fragments were amplified by PCRwith a set of degenerated primers based on two conservedsequences from family B DNA polymerases. Genomic South-ern hybridization analysis using these two fragments as probessuggested that each gene was present as a single copy in thegenome (data not shown). We then cloned the entire structuralgenes as described in Materials and Methods. In the P. occul-tum study, the two genes and their functional products werenamed polA and polB (genes) and Pol I and Pol II (products),as the first and second discovered pol genes and DNA poly-merases, respectively, in accordance with the custom of bacte-rial genetics. It was then proposed that crenarchaeal family BDNA polymerases be referred to as B1, B2, and B3 because ofthe finding of three family B DNA polymerase genes (11). Inaccordance to this proposed nomenclature, P. occultum Pol Iand Pol II correspond to B1 and B3, respectively. The polAgene from A. pernix was composed of 2,769 nucleotides andcoded for a protein of 923 amino acid residues with an esti-mated molecular mass of 105,406 Da (Fig. 4a); polB comprised2,316 bases and coded for a protein of 772 amino acid residueswith an estimated molecular mass of 87,905 Da (Fig. 4b). Theamino acid sequences of both proteins contained the con-served motifs for 39359 exonuclease and polymerase activities.As shown in Table 1, at the amino acid sequence level, theproduct of the polA gene showed the highest identity (47%) toP. occultum Pol I (B1). In addition, its identities to the Pol I(B1) homologs found in other thermophilic crenarchaeoteswere above 40%. Despite originating from a psychrophile, thePol I homolog of C. symbiosum (27) showed 37% identity withthe product of A. pernix polA. The product of polB and P.occultum Pol II (B3) exhibited a higher amino acid sequenceidentity (56%), and both polymerases shared identities rangingfrom 29 to 40% with family B DNA polymerases found in bothEuryarchaeota and Crenarchaeota. Therefore, we named thesegene products of A. pernix Pol I and Pol II, as the first andsecond discovered DNA polymerases in this organism, afterconfirmation of their DNA polymerase activities (see below).The results of our sequence comparison of the archaeal familyB DNA polymerases is consistent with the recent phylogeneticanalysis involving archaeal and eucaryal family B DNA poly-merases that shows that known crenarchaeal DNA poly-merases fall into two groups (9). Group I consists of onlycrenarchaeal members, while group II comprises membersfrom both crenarchaeotes and euryarchaeotes. A. pernix Pol I(B1) and Pol II (B3) belong to group I and group II, respec-tively. Among the crenarchaeotes used in this study, only P.
FIG. 3. Activity gel analysis of DNA polymerizing activities in fractionated A.pernix cell extracts, fraction I and fraction II, and recombinant Pol I and Pol II.DNA polymerizing activity was detected by in situ incorporation of [a-32P]dCTPas described in Materials and Methods. Proteins in fraction I (F1), fraction II(F2), purified Pol I (PI), and Pol II (PII) were separated by SDS-PAGE, and 32Pincorporated into DNA strands in the gel was detected by autoradiography. Themolecular masses indicated on the left were derived from a size standard marker(Bio-Rad, Hercules, Calif.).
TABLE 1. Distribution of family B DNA polymerases in crenarchaeotes and euryarchaeotes
Archaeal subdomain Organism% identity
ApeI PocI Sso B1 Soh B1 Sac B1 Csy B1 ApeII PocII Sso B3 Afu Tli Pfu
Crenarchaeota A. pernix I (B1)P. occultum I (B1) 47S. solfataricus B1 43 49S. ohwakuensis B1 43 49 71S. acidocaldarius B1 41 48 67 75C. symbiosum B1 37 37 36 35 35A. pernix II (B3)P. occultum II (B3) 56S. solfataricus B3 29 31
Euryarchaeota A. fulgidus Pol I 38 40 28T. litoralis Pol I 33 32 23 35P. furiosus Pol I 32 33 23 34 74
VOL. 181, 1999 TWO FAMILY B DNA POLYMERASES FROM A. PERNIX 5987
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

FIG. 4. Nucleotide and deduced amino acid sequences of A. pernix polA (a) and polB (b) genes. The regions used in designing primers for genome walking PCRare indicated by black arrows; grey arrows indicated the primers used in amplifying the structural gene for each polymerase. The regions including the exonucleasemotifs (3) and the polymerase motifs (32) are underlined by solid and broken lines, respectively; boldface letters represent putative box A sequences.
5988
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

FIG. 4—Continued.
5989
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from
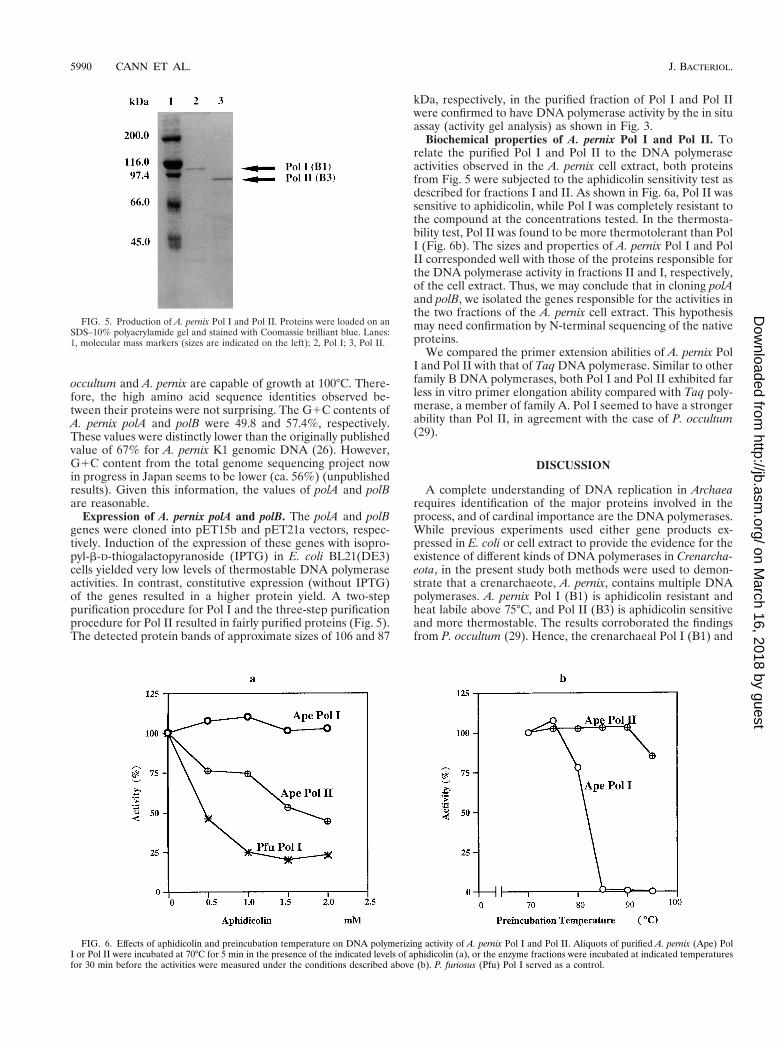
occultum and A. pernix are capable of growth at 100°C. There-fore, the high amino acid sequence identities observed be-tween their proteins were not surprising. The G1C contents ofA. pernix polA and polB were 49.8 and 57.4%, respectively.These values were distinctly lower than the originally publishedvalue of 67% for A. pernix K1 genomic DNA (26). However,G1C content from the total genome sequencing project nowin progress in Japan seems to be lower (ca. 56%) (unpublishedresults). Given this information, the values of polA and polBare reasonable.
Expression of A. pernix polA and polB. The polA and polBgenes were cloned into pET15b and pET21a vectors, respec-tively. Induction of the expression of these genes with isopro-pyl-b-D-thiogalactopyranoside (IPTG) in E. coli BL21(DE3)cells yielded very low levels of thermostable DNA polymeraseactivities. In contrast, constitutive expression (without IPTG)of the genes resulted in a higher protein yield. A two-steppurification procedure for Pol I and the three-step purificationprocedure for Pol II resulted in fairly purified proteins (Fig. 5).The detected protein bands of approximate sizes of 106 and 87
kDa, respectively, in the purified fraction of Pol I and Pol IIwere confirmed to have DNA polymerase activity by the in situassay (activity gel analysis) as shown in Fig. 3.
Biochemical properties of A. pernix Pol I and Pol II. Torelate the purified Pol I and Pol II to the DNA polymeraseactivities observed in the A. pernix cell extract, both proteinsfrom Fig. 5 were subjected to the aphidicolin sensitivity test asdescribed for fractions I and II. As shown in Fig. 6a, Pol II wassensitive to aphidicolin, while Pol I was completely resistant tothe compound at the concentrations tested. In the thermosta-bility test, Pol II was found to be more thermotolerant than PolI (Fig. 6b). The sizes and properties of A. pernix Pol I and PolII corresponded well with those of the proteins responsible forthe DNA polymerase activity in fractions II and I, respectively,of the cell extract. Thus, we may conclude that in cloning polAand polB, we isolated the genes responsible for the activities inthe two fractions of the A. pernix cell extract. This hypothesismay need confirmation by N-terminal sequencing of the nativeproteins.
We compared the primer extension abilities of A. pernix PolI and Pol II with that of Taq DNA polymerase. Similar to otherfamily B DNA polymerases, both Pol I and Pol II exhibited farless in vitro primer elongation ability compared with Taq poly-merase, a member of family A. Pol I seemed to have a strongerability than Pol II, in agreement with the case of P. occultum(29).
DISCUSSION
A complete understanding of DNA replication in Archaearequires identification of the major proteins involved in theprocess, and of cardinal importance are the DNA polymerases.While previous experiments used either gene products ex-pressed in E. coli or cell extract to provide the evidence for theexistence of different kinds of DNA polymerases in Crenarcha-eota, in the present study both methods were used to demon-strate that a crenarchaeote, A. pernix, contains multiple DNApolymerases. A. pernix Pol I (B1) is aphidicolin resistant andheat labile above 75°C, and Pol II (B3) is aphidicolin sensitiveand more thermostable. The results corroborated the findingsfrom P. occultum (29). Hence, the crenarchaeal Pol I (B1) and
FIG. 5. Production of A. pernix Pol I and Pol II. Proteins were loaded on anSDS–10% polyacrylamide gel and stained with Coomassie brilliant blue. Lanes:1, molecular mass markers (sizes are indicated on the left); 2, Pol I; 3, Pol II.
FIG. 6. Effects of aphidicolin and preincubation temperature on DNA polymerizing activity of A. pernix Pol I and Pol II. Aliquots of purified A. pernix (Ape) PolI or Pol II were incubated at 70°C for 5 min in the presence of the indicated levels of aphidicolin (a), or the enzyme fractions were incubated at indicated temperaturesfor 30 min before the activities were measured under the conditions described above (b). P. furiosus (Pfu) Pol I served as a control.
5990 CANN ET AL. J. BACTERIOL.
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

Pol II (B3) homologs may represent the aphidicolin-resistantand aphidicolin-sensitive DNA polymerases detected previ-ously in other crenarchaeal cells.
In the in situ DNA polymerase activity analysis with thefractionated cell extract (fraction II), three smaller bands cor-responding to 98-, 90-, and 65-kDa proteins were detected inthe autoradiogram in addition to the band corresponding toPol I (105 kDa). These proteins may represent the degradationproducts of Pol I, or they may be entirely different DNApolymerases. The band of 90 kDa seems to be from unsepa-rated Pol II. Some contamination of Pol II in fraction II can bepredicted from the observations that to compare with purifiedPol I (Fig. 6), the activity from fraction II was less aphidicolinresistant and more heat stable (Fig. 2). The properties ofpurified Pol I and fraction II may be comparable when some ofaphidicolin-sensitive and heat-stable activity is subtracted fromthe total activity of fraction II. It should be noted here that PolII is very stable, and it always gave a band signal much strongerthan that of the same amount of Pol I in the activity gelanalysis. Edgell and coworkers (9) have suggested the exis-tence of three family B DNA polymerase in S. solfataricus P2from their work and previous literature (25). Therefore, itwould be noteworthy to detect a third DNA polymerase fromA. pernix. The size of the expected third DNA polymerase in S.
solfataricus is around 74 kDa from the nucleotide sequence ofthe third gene (23). The sizes of the bands observed in ourexperiment are somewhat different from this size. From theassay profile in the anion-exchange chromatography shown inFig. 1, another activity may exist in fraction 21. However, insitu activity gel analysis using fractions around fraction II (e.g.,fraction 23) gave basically the same band signals as in Fig. 3(data not shown). The existence of a third DNA polymerasecould not be confirmed due to insufficient evidence. Furtherexperiments are necessary to answer this interesting question.
We have attempted to isolate from A. pernix and othercrenarchaeotes the genes that code for the heterodimericDNA polymerase found thus far in the euryarchaeotes (7),with consistently negative results. It is our hypothesis that theheterodimeric DNA polymerase occurs only in Euryarchaeota.We propose here that in the archaeal domain, the crenarchae-otes at least have two family B DNA polymerases, while theeuryarchaeotes have one (Table 1). Instead, the euryarchae-otes have, in addition to their family B DNA polymerase, aheterodimeric DNA polymerase. It will be interesting to knowthe biological roles of each DNA polymerase in Archaea, es-pecially why the forms of DNA polymerases in the cells are sodifferent between Euryarchaeota and Crenarchaeota.
Even though their amino acid sequences are typically that offamily B DNA polymerases, comparison of the structure-func-tion relationship of DNA polymerases Pol I and Pol II inCrenarchaeota will be of further interest, and the results arelikely to contribute to our understanding of the basic mecha-nism of DNA synthesis in these organisms. In vitro primerextension abilities of Pol I and Pol II by themselves wereobviously lower than that of Taq DNA polymerase (Fig. 7).The low processivity of the archaeal family B DNA poly-merases is known (20). In Eucarya, proliferating cell nuclearantigen (PCNA) functions as an accessory factor to enhancethe processivity of DNA polymerase d (and ε), the replicativeDNA polymerase, severalfold (2). The PCNA homologs inArchaea may stimulate the processivity of these DNA poly-merases in vivo as observed in eucaryal cells. We confirmed thefunctional interaction between P. furiosus Pol I (B3) and itsPCNA homologs in vitro (5). Furthermore, we have found twoputative homologs of PCNA from A. pernix and cloned them(6). There is only one PCNA homolog each in the euryarchaealgenomes. It is possible that each DNA polymerase in A. pernixhas a specific PCNA as the partner for elongation reaction, andthe next step is to investigate the effect of each homolog on PolI and Pol II. The results will help to clarify the role of eachDNA polymerase in the process of DNA replication in Cren-archaeota.
ADDENDUM IN PROOF
The complete genome sequence of Aeropyrum pernix hasbeen published (Y. Kawarabayasi et al., DNA Res. 6:83–101,1999).
REFERENCES1. Barns, S. M., J. D. Delwiche, J. D. Palmer, and N. R. Pace. 1996. Perspectives
on archaeal diversity, thermophily and monophyly from environmentalrRNA sequences. Proc. Natl. Acad. Sci. USA 93:9188–9193.
2. Bauer, G. A., and P. M. J. Burgers. 1988. The yeast analog of mammaliancyclin/proliferating-cell nuclear antigen interacts with mammalian DNApolymerase delta. Proc. Natl. Acad. Sci. USA 85:7506–7510.
3. Bernad, A., L. Blanco, J. M. Lazaro, G. Martin, and M. Salas. 1989. Aconserved 39359 exonuclease active site in prokaryotic and eukaryotic DNApolymerases. Cell 59:219–228.
4. Bult, C. J., O. White, G. J. Olsen, L. Zhou, R. D. Fleischmann, G. G. Sutton,J. A. Blake, L. M. FitzGerald, R. A. Clayton, J. D. Gocayne, A. R. Kerlavage,B. A. Dougherty, J.-F. Tomb, M. D. Adams, C. I. Reich, R. Overbeek, E. F.Kirkness, K. G. Weinstock, J. M. Merrick, A. Glodek, J. L. Scott, N. S. M.
FIG. 7. Chain elongation ability of A. pernix Pol I and Pol II. A 32P-labeledprimer annealed to M13 DNA served as the template. Aliquots (5 ml) of reactionmixture were sampled at 1, 2, and 3 min after initiation of reaction, and theirproducts were resolved on an 8% polyacrylamide gel containing 8 M urea. TaqDNA polymerase was used as a control.
VOL. 181, 1999 TWO FAMILY B DNA POLYMERASES FROM A. PERNIX 5991
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from

Geoghagen, J. F. Weidman, J. L. Fuhrmann, E. A. Presley, D. Nguyen, T. R.Utterback, J. M. Kelley, J. D. Peterson, P. W. Sadow, M. C. Hanna, M. D.Cotton, M. A. Hurst, K. M. Roberts, B. P. Kaine, M. Borodovsky, H.-P.Klenk, C. M. Fraser, H. O. Smith, C. R. Woese, and J. C. Venter. 1996.Complete genome sequence of the methanogenic archaeon, Methanococcusjannaschii. Science 273:1058–1073.
5. Cann, I. K. O., S. Ishino, I. Hayashi, K. Komori, H. Toh, K. Morikawa, andY. Ishino. Unpublished data.
6. Cann, I. K. O., S. Ishino, Y. Kawarabayashi, H. Kikuchi, Y. Sako, and Y.Ishino. Unpublished data.
7. Cann, I. K. O., K. Komori, H. Toh, S. Kanai, and Y. Ishino. 1998. Aheterodimeric DNA polymerase: evidence that members of euryarchaeotapossess a distinct DNA polymerase. Proc. Natl. Acad. Sci. USA 95:14250–14255.
7a.CLUSTALW. 5 February 1998, revision date. [Online.] http://www.genome-.ad.jp/SIT/CLUSTALW.html. [15 March 1999, last date accessed.]
8. Edgell, D. R., and W. F. Doolittle. 1996. Archaebacterial genomics: thecomplete genome sequence of Methanococcus jannaschii. Bioessays 19:1–4.
9. Edgell, D. R., H.-P. Klenk, and W. F. Doolittle. 1997. Gene duplications inevolution of archaeal family B DNA polymerases. J. Bacteriol. 179:2632–2640.
10. Elie, C., A. M. De Rocondo, and P. Forterre. 1989. Thermostable DNApolymerase from the archaebacterium Sulfolobus acidocaldarius. Eur. J. Bio-chem. 178:619–626.
11. Gray, M. W. 1996. The third form of life. Nature 383:299.12. Hamatake, R. K., H. Hasegawa, A. B. Clark, K. Bebenek, T. A. Kunkel, and
A. Sugino. 1990. Purification and characterization of DNA polymerase IIfrom the yeast Saccharomyces cerevisiae. J. Biol. Chem. 265:4072–4083.
13. Huberman, J. A. 1981. New view of the biochemistry of eukaryotic DNAreplication revealed by aphidicolin, an unusual inhibitor of DNA polymerasea. Cell 23:647–648.
14. Imamura, M., T. Uemori, I. Kato, and Y. Ishino. 1995. A non-a-like DNApolymerase from the hyperthermophilic archaeon Pyrococcus furiosus. Biol.Pharm. Bull. 18:1647–1652.
15. Ishino, Y., K. Komori, I. K. O. Cann, and Y. Koga. 1998. A novel DNApolymerase family found in Archaea. J. Bacteriol. 180:2232–2236.
16. Kawarabayasi, Y., M. Sawada, H. Horikawa, Y. Haikawa, Y. Hino, S.Yamamoto, M. Sekine, S. Baba, H. Kosugi, A. Hosoyama, Y. Nagai, M.Sakai, K. Ogura, R. Otsuka, H. Nakazawa, M. Takamiya, Y. Ohfuku, T.Funahashi, T. Tanaka, Y. Kudoh, J. Yamazaki, N. Kushida, A. Oguchi, K.Aoki, T. Yoshizawa, Y. Nakamura, F. T. Robb, K. Horikoshi, Y. Masuchi, H.Shizuya, H. Kikuchi. 1998. Complete sequence and gene organization of thegenome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshiiOT3. DNA Res. 5:55–76.
17. Klimczak, L. J., F. Grummt, and K. J. Burger. 1985. Purification and char-acterization of DNA polymerase from the archaeabacterium Sulfolobus aci-docaldarius. Nucleic Acids Res. 13:5269–5281.
18. Kornberg, A., and T. A. Baker. 1992. DNA replication, 2nd ed. W. H.Freeman and Co., New York, N.Y.
19. Morell, V. 1996. Life’s last domain. Science 273:1043–1045.19a.National Center for Biotechnology Information. 13 April 1999, revision date.
BLAST. [Online.] http://www.ncbi.nlm.nih.gov/. National Center for Bio-technology Information, National Institutes of Health, Bethesda, Md. [1March 1999, last date accessed.]
20. Perler, F. B., S. Kumar, and H. Kong. 1996. Thermostable DNA poly-merases. Adv. Protein Chem. 48:377–435.
21. Pisani, F., C. De Martino, and M. Rossi. 1992. A DNA polymerase from thearchaeon Sulfolobus solfataricus shows sequence similarity to family B DNApolymerases. Nucleic Acids Res. 20:2711–2716.
22. Pisani, F., and M. Rossi. 1994. Evidence that an archaeal a-like DNApolymerase has a molecular organization of its associated catalytic activities.J. Biol. Chem. 269:7887–7892.
23. Prangishvili, D., and H.-P. Klenk. 1994. The gene for a 74 kDa DNApolymerase from the archaeon Sulfolobus solfataricus. Syst. Appl. Microbiol.16:665–671.
24. Richardson, C. C. 1966. DNA polymerase from Escherichia coli, p. 263–276.In G. L. Cantoni and D. R. Davies (ed.), Procedures in nucleic acid research.Harper & Row, New York, N.Y.
25. Rossi, M., R. Rella, M. Pensa, S. Bartolucci, M. De Rosa, A. Gambacorta,C. A. Raia, and N. Dell’Aversano Orabona. 1986. Structure and properties ofa thermophilic and thermostable DNA polymerase isolated from Sulfolobussolfataricus. Syst. Appl. Microbiol. 7:337–341.
26. Sako, Y., N. Nomura, A. Uchida, Y. Ishida, H. Morii, Y. Koga, T. Hoaki, andT. Maruyama. 1996. Aeropyrum pernix gen. nov., sp. nov., a novel aerobichyperthermophilic archaeon growing at temperatures up to 100°C. Int. J.Syst. Bacteriol. 46:1070–1077.
27. Schleper, C., R. V. Swanson, E. J. Mathur, and E. F. Delong. 1997. Charac-terization of a DNA polymerase from the uncultivated psychrophilic ar-chaeon Cenarchaeum symbiosum. J. Bacteriol. 179:7803–7811.
28. Uemori, T., Y. Sato, I. Kato, H. Doi, and Y. Ishino. 1997. A novel DNApolymerase in the hyperthermophilic archaeon, Pyrococcus furiosus: genecloning, expression, and characterization. Genes Cells 2:499–512.
29. Uemori, T., Y. Ishino, H. Doi, and I. Kato. 1995. The hyperthermophilicarchaeon Pyrodictium occultum has two a-like DNA polymerases. J. Bacte-riol. 177:2164–2177.
30. Uemori, T., Y. Ishino, H. Toh, K. Asada, and I. Kato. 1993. Organization andnucleotide sequence of the DNA polymerase gene from the archaeon Pyro-coccus furiosus. Nucleic Acids Res. 21:259–265.
31. Wernette, C. M., and L. S. Kaguni. 1986. A mitochondrial DNA polymerasefrom embryos of Drosophila melanogaster. J. Biol. Chem. 261:14764–14770.
32. Wong, S. W., A. F. Wahl, P.-M. Yuan, N. Arai, B. E. Pearson, K.-L. Arai, D.Korn, M. W. Hunkapiller, and T. S.-F. Wang. 1988. Human DNA polymer-ase a gene expression is cell proliferation dependent and its primary struc-ture is similar to both prokaryotic and eukaryotic replicative DNA poly-merases. EMBO J. 7:37–47.
5992 CANN ET AL. J. BACTERIOL.
on March 16, 2018 by guest
http://jb.asm.org/
Dow
nloaded from


















