
Collagen Kinase Receptors as Potential Therapeutic Targets ...


TRP CHANNELS AS THERAPEUTIC
TARGETSFrom Basic Science to
Clinical Use
Edited by
ARPAD SZALLASI MD, PHDDepartment of Pathology, Monmouth Medical Center,
Long Branch, NJ, USA
AMSTERDAM • BOSTON • HEIDELBERG • LONDON
NEW YORK • OXFORD • PARIS • SAN DIEGO
SAN FRANCISCO • SINGAPORE • SYDNEY • TOKYOAcademic Press is an imprint of Elsevier

TRP Channels as Therapeutic Targets 279 © 2015 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/B978-0-12-420024-1.00016-3
C H A P T E R
16Activating, Inhibiting, and
Highjacking TRP Channels for Relief from Itch
Lindsey M. Snyder,1,3,4 Huizhen Huang,1,3,5 Sarah E. Ross1,2,3,4,*
1Department of Neurobiology, University of Pittsburgh, Pittsburgh, PA, USA2Department of Anesthesiology, University of Pittsburgh, Pittsburgh, PA, USA
3Pittsburgh Center for Pain Research, Pittsburgh, PA, USA4Center for Neuroscience Research at the University of Pittsburgh, Pittsburgh PA, USA
5Tsinghua University School of Medicine, Beijing, China
*Corresponding author:
O U T L I N E
Introduction 279
Which Sensory Neurons Signal Itch? 280
Activating TRP Channels to Block Itch 282
Inhibiting TRP Channels to Block Itch 284
Highjacking TRP Channels to Deliver Relief from Itch 286
An Unexpected Role for TprV3: Mutations in TRPV3 Result in Itchy Lesions Found in Olmsted Syndrome 287
Conclusions 289
References 289
INTRODUCTION
Itch is an unpleasant sensation that produces the desire to scratch [1]. Moreover, scratching relieves the itch, at least temporarily. This behavior—scratching in response to an aversive stimulus—is highly conserved across species. All mammals and birds show itch behavior. Fish will rub themselves against a rough surface to remove an irritant. Even fruit flies display

280 16. TRP CHANNELS AND ITCH
site-directed grooming behavior in response to an irritating substance [2]. The conservation of this behavioral response across the animal kingdom underscores the idea that itch has an important protective function: itch triggers a behavior that removes harmful agents from the body’s surface in the short term, and because itch feels unpleasant, an organism learns to avoid itch-inducing situations in the long term.
However, there are many pathological conditions for which itch is no longer protective. In such cases, itch becomes a chronic condition that significantly decreases the quality of life. Numerous diseases are associated with severe itch, including atopic dermatitis, liver disease, postherpetic itch, small fiber neuropathies, and even some cancers [3]. Pathological itch from these and other diseases can be just as debilitating as pain, and there is a great unmet need for new treatment options [4].
In many ways, itch is like pain—both are aversive sensations that evolved to protect us, and both are initially detected by similar (though likely distinct) subsets of primary afferents [5]. Indeed, the transformation from an aversive somatosensory stimulus in the periphery to the sensory percept of either pain or itch in the brain appears to involve parallel neural path-ways comprised of peripheral neurons that detect pruritogens, the superficial dorsal horn of the spinal cord, the parabrachial nucleus, the dorsal raphe, the thalamus, and several re-gions of the cortex. Thus, functional magnetic resonance imaging performed on the brain of a person experiencing either pain or itch might be almost indistinguishable [6]. Indeed, we do not yet understand how itch and pain are differentially encoded in the nervous system. Nevertheless, we know that they must be because itch and pain feel qualitatively different and elicit distinct behavioral responses.
WHICH SENSORY NEURONS SIGNAL ITCH?
There is great interest in determining which sensory neurons mediate itch and how these afferents are different than those that are involved in signaling pain. Identifying such fibers has important therapeutic implications because this knowledge could potentially allow the development of selective therapies for pruritus. Several years ago, microneurography exper-iments in humans led to the long-awaited discovery of itch-specific sensory neurons—these fibers responded to histamine, and their activity corresponded to the sensation of itch [7]. However, there was no molecular marker that allowed us to identify these cells. (Histamine responsiveness on its own is insufficient to identify an itch fiber because histamine activates numerous sensory neurons, likely including those that are involved in signaling pain). Thus, although there was good evidence for the existence of itch-selective sensory afferents, their identity remained elusive.
In the last few years, several groups have reported the discovery of markers that define itch-mediating sensory afferents, including gastrin releasing peptide (GRP), Mas-related G-protein receptor A3 (MrgprA3), and Naturietic Polypeptide b (Nppb) [8–11]. But these reports are somewhat contradictory, and so the identity of itch fibers remains controversial. Both GRP and Nppb are attractive candidates as potential markers of itch afferents because both of these peptides cause itch when injected intrathecally [9,10]. Thus, it stands to reason that primary afferents that release these peptides might be involved in itch. However, the idea that GRP is a marker of itch neurons has come under scrutiny of late. The analysis of GRP expression in dorsal root ganglia (DRG) using antibodies originally suggested that

WHICH SENSORY NEURONS SIGNAL ITCH? 281
GRP is expressed in subsets of primary sensory afferents [9]. Yet, it has been difficult to corroborate these findings with methods that detect GRP mRNA, and the inability to detect GRP message in DRG sensory neurons has now called the specificity of the GRP antibody into question [12], though this subject remains controversial [13]. Rather, GRP mRNA is found in abundance in spinal interneurons, and so it seems likely that these spinal inter-neurons (rather than DRG neurons) are the main source of endogenous GRP in the spinal cord. Nppb, on the other hand, remains a potential marker for itch neurons. Nppb mRNA is clearly expressed in subsets of primary afferents, and Nppb is required for itch sensation [10]. What remains to be determined is the degree to which Nppb is a bona fide marker of itch neurons—in other words, one that marks all itch afferents and only itch afferents.
The strongest evidence for a marker that defines a specific population of sensory afferents that are tuned for itch comes from the analysis of a small population of DRG neurons that coexpress the MrgprA3 and MrgprC11. These related GPCRs were originally discovered in a screen for genes that are selectively expressed in peptidergic afferents [14]. Subsequently it was found that these two proteins are receptors for pruritogens. Specifically, MrgprA3 is a receptor for chloroquine, whereas MrgprC11 is activated by Bam8-22 and SLIGRL [15,16]. Because MrgprA3 and MrgprC11 are activated by pruritogens, it was logical to infer that the neurons that express these receptors are itch afferents. However, direct evidence that MrgprA3/C11-expressing neurons mediate itch was only possible on the genetic targeting of this population. The selective expression of cre recombinase in these cells made it possible to vi-sualize this population clearly and enabled loss- and gain-of-function (LOF and GOF) studies to rigorously test the role of these afferents in itch. Importantly, the ablation of the MrgprA3 population with diphtheria toxin significantly reduced (although did not completely elimi-nate) itch, and selective activation of the MrgprA3 population resulted in scratching behavior [11]. The human counterpart to these receptors is thought to be MrgprX1, which also re-sponds to a broad array of pruritogens when ectopically expressed, but whether this gene is a marker for itch-mediating neurons in humans is unknown [15,17].
The genetic labeling of the MrgprA3-expressing population of sensory neurons in a mouse was an important advance because it allowed us to see itch fibers for the first time [11]. Intriguingly, this population of neurons coexpresses CGRP and IB4, two mostly nonoverlap-ping markers that are thought to define distinct subsets of C fibers, the so-called peptidergic and nonpeptidergic classes, respectively. Furthermore, the central terminals of the MrgprA3 population ramify within a very narrow band of lamina II within the superficial spinal cord (newly termed II middle) that is between the peptidergic and nonpeptidergic layers. Hence the identification of MrgprA3-expressing itch afferents has challenged the conventional clas-sification scheme for sensory neurons by revealing the existence of yet a third subtype that mediates itch and shows intermediate properties with respect to neurochemical expression as well as the laminar distribution of its central terminals. Importantly, the peripheral targeting of these neurons is entirely consistent with a role for these cells in itch: these neurons exclu-sively target structures in which we feel itch, such as the skin, but not any other regions of the body such as muscle or internal organs. Furthermore, within the skin, these neurons show an extremely superficial pattern of innervation, with terminations in the stratum granulosum. The loss- and gain-of function studies along with the specialized distribution of MrgprA3-expressing cells provide very compelling evidence that MrgprA3-expressing neurons medi-ate itch. Nonetheless, it should be noted that these fibers likely represent just one of several subpopulations of itch afferents [13,18].

282 16. TRP CHANNELS AND ITCH
ACTIVATING TRP CHANNELS TO BLOCK ITCH
Although we are still sorting out identity of all of the subtypes of primary sensory afferents that mediate itch, there is good evidence that these fibers belong to a larger population of sen-sory neurons that express TRPV1. This idea implies that, although TRPV1-targeted therapies may not be specific, they should nevertheless be effective at blocking itch. In mice, chemical ablation of TRPV1-expressing sensory neurons caused an almost complete loss of itch be-havior [19]. Similarly in humans, repeated topical application of capsaicin is commonly used as a treatment for itch [20]. For instance, in a double-blind trial, low-concentration capsaicin treatment (0.025%, four times daily) caused a significant decrease in pruritic psoriasis [21]. This treatment seems paradoxical, and in some ways it is, for the initial response to capsaicin is intense burning, stinging, and itch. However, when TRPV1-expressing sensory nerve fi-bers are exposed to repeated applications of TRPV1 agonists, TRPV1-expressing sensory neu-rons show retraction of peripheral processes and impaired signaling (Figure 16.1), an effect that has been termed defunctionalization [22]. More recently, the use of a high- concentration capsaicin treatment (8% capsaicin patch) has been developed [23]. The advantage to this approach is that a single application can cause the defunctionalization of TRPV1-expressing sensory neurons. In anecdotal reports, an 8% capsaicin patch was successfully used to treat notalgia paraesthetica [24]. In addition, the fact that capsaicin patches significantly reduce postherpetic neuralgia strongly suggests that such treatment will also be effective for pos-therpetic itch [25].
However, the treatment of intractable itch with capsaicin, though it can be effective, is a long way from a perfect solution. The treatment itself (not surprisingly) can be quite painful—indeed, concentrated capsaicin is applied in the presence of a local anesthetic. Furthermore, on the retraction of TRPV1-expressing fibers, there is a multimodal loss in sensitivity, including pain, itch, and warm/hot temperatures, and so the treatment is not particularly specific for itch. For these reasons, capsaicin therapy for pruritus only makes sense if the itch is localized to a specific region of the skin. Finally, the effects of capsai-cin treatment are temporary, lasting up to 12 weeks, whereas the peripheral terminals from TRPV1-expressing sensory neurons regenerate and reinnervate the skin. Despite these short-comings, low-dose capsaicin treatment is commonly used for the treatment of itch [20], and the newer high-concentration formulas have the potential to be broadly effective for numer-ous types of localized, neuropathic itch.
Activating TRPV1 is not the only strategy to combat itch—the other is via TRPM8. Many over-the-counter itch remedies contain menthol as an active ingredient. Now research from our own lab may have uncovered the underlying mechanism for how menthol inhibits itch [26]. Menthol is known to activate TRPM8, which is expressed on sensory afferents that con-vey cool [27]. Our work suggests that menthol acts as a counterstimulus that inhibits itch via the activation of a specific population of inhibitory neurons in the dorsal spinal cord, which we have termed B5-I neurons. In particular, we show that B5-I neurons, which function to in-hibit itch, get direct input from TRPM8 afferents. In addition, we find that, whereas menthol inhibits itch in wild-type mice, this counterstimulus has no effect in mice that are lacking B5-I neurons. These data suggest that B5-I neurons are the cellular basis for the inhibition of itch by menthol. In this regard, menthol and scratching may be two types of counterstimulation that work via analogous spinal circuits. But activation of TRPM8 as a strategy to inhibit itch
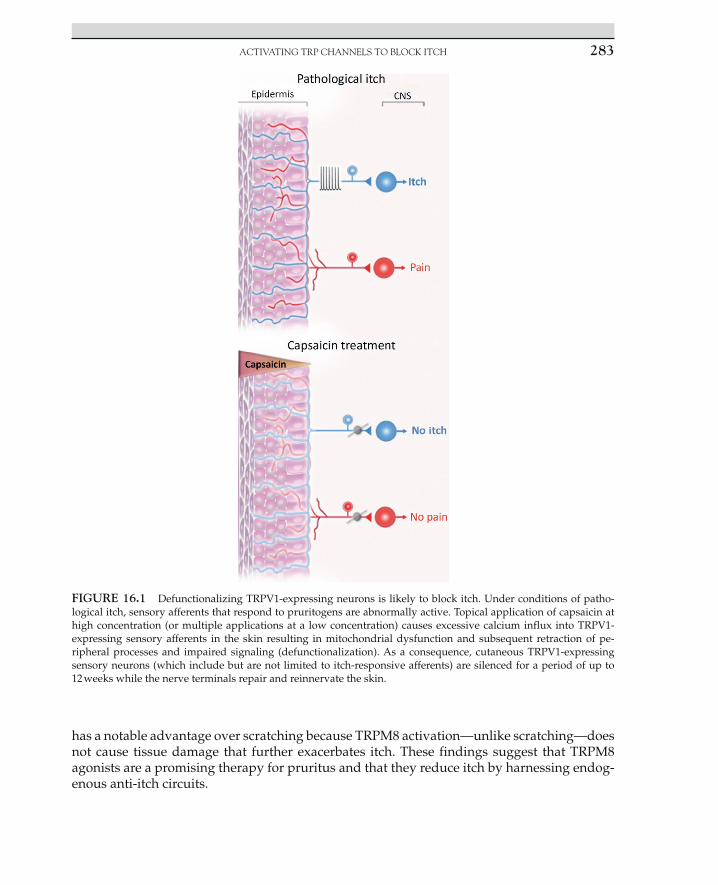
ACTIVATING TRP CHANNELS TO BLOCK ITCH 283
has a notable advantage over scratching because TRPM8 activation—unlike scratching—does not cause tissue damage that further exacerbates itch. These findings suggest that TRPM8 agonists are a promising therapy for pruritus and that they reduce itch by harnessing endog-enous anti-itch circuits.
FIGURE 16.1 Defunctionalizing TRPV1-expressing neurons is likely to block itch. Under conditions of patho-logical itch, sensory afferents that respond to pruritogens are abnormally active. Topical application of capsaicin at high concentration (or multiple applications at a low concentration) causes excessive calcium influx into TRPV1-expressing sensory afferents in the skin resulting in mitochondrial dysfunction and subsequent retraction of pe-ripheral processes and impaired signaling (defunctionalization). As a consequence, cutaneous TRPV1-expressing sensory neurons (which include but are not limited to itch-responsive afferents) are silenced for a period of up to 12 weeks while the nerve terminals repair and reinnervate the skin.

284 16. TRP CHANNELS AND ITCH
INHIBITING TRP CHANNELS TO BLOCK ITCH
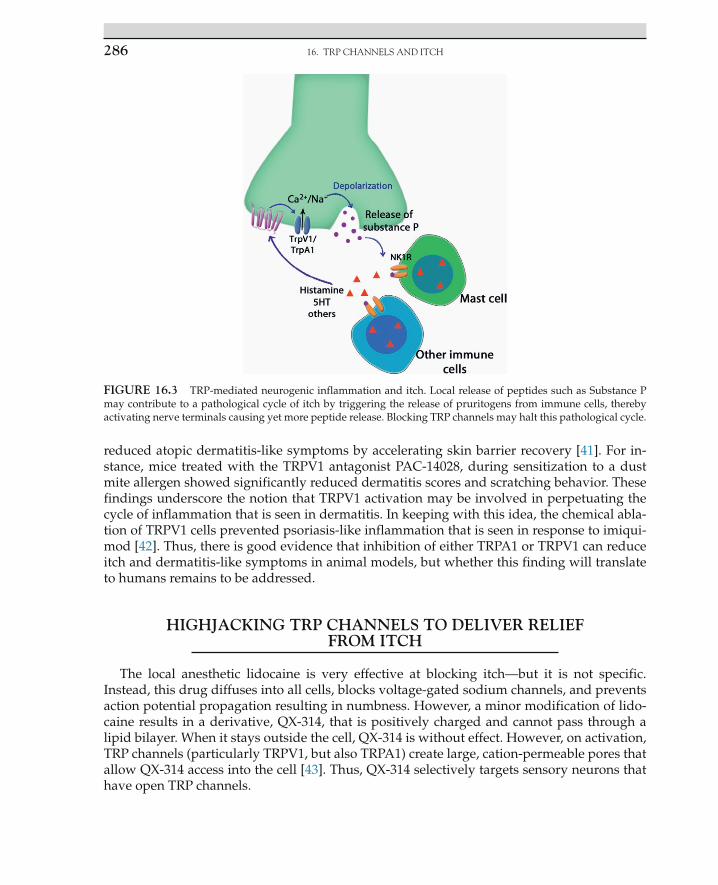
Numerous chemical agents cause itch, and recently we have begun to get a clearer picture of how they do so. Activated dermal mast cells release histamine and serotonin, which bind to receptors on sensory neurons and cause itch. Foreign materials from plants and insects often have protease activity that can cause itch [28]. For instance, spicules from the tropical plant cowhage contain a protease that activates PAR2, and proteolytic cleavage products including BAM8-22 and SLIGRL cause itch by activating MrgprC11 [15,16,28]. Often itch is part of an immune response that involves the release of numerous cytokines, such as interleukin 31 and TSLP, which act on cytokine receptors [29,30]. What these itch receptors have in common is that they are all metabotropic rather than ionotropic receptors—in other words, they are not sufficient to trigger an action potential by themselves. Instead, these metabotropic receptors need to be coupled through signaling pathways to a channel that allows current influx to support the generation of an action potential.
Notably, in every case that has been examined in detail, it has been found that the channels that are activated downstream of itch receptors are TRP channels. In particular, H1 recep-tor for histamine is coupled to TRPV1 via phospholipase A2 [19,31]. Analogously, MrgprA3, MrgprC11, and TSPLR appear to be coupled to TRPA1, whereas IL-31R may be able to couple to either TRP channel [29,30,32]. These studies emphasize the important idea that TRPV1 and TRPA1 are more than just pain sensors. Rather, it is now emerging that these channels are inte-grators of diverse noxious stimuli, including those that induce sensations of itch (Figure 16.2).
Whether antagonists for TRPV1 and TRPA1 can block itch in humans is currently unknown. Several TRPV1 antagonists have failed in clinical trials for the treatment of pain because, at least when delivered systemically, these antagonists caused dangerous increases in body tem-perature in some patients [35]. This hyperthermia is likely due an underappreciated role for TRPV1 in sensing and regulating temperature. But the disappointing outcome of this clinical trial does not rule out the possibility that TRPV1 (and possibly TRPA1) antagonists could be used safely for the treatment of localized itch. In particular, the topical application of a TRPV1 antagonist to a delimited area of affected skin would be unlikely to trigger hyperthermia that
FIGURE 16.2 Pruritogens activate metabotropic receptors that require TRPV1 and/or TRPA1 for signaling. Many pruritogens signal through G-protein coupled receptors (e.g., histamine, SLIGRL, Bam8-22) or cytokine recep-tors (e.g., IL-31, TSLP), which cannot, by themselves, trigger an action potential. In many instances, these receptors appear to couple to either TRPV1 or TRPA1, which allow current influx and provide the depolarization potential. (A notable exception is hydrogen peroxide-mediated itch, which may be due to its ability to activate TRPA1 directly [33,34]). Thus, TRP antagonists may block itch that is caused by numerous, diverse pruritogens.

INHIBITING TRP CHANNELS TO BLOCK ITCH 285
is seen on systemic treatment with the drug. Thus, TRPV1 and TRPA1 antagonists remain an attractive candidate for the treatment of localized itch.
Indeed, there is now good evidence for the idea that blocking TRPs—particularly TRPA1—may be an effective way to treat some types of pruritus based on the work from two groups who have used several different animal models of chronic itch. In particular, allergic contact dermatitis was modeled by repeated exposure of the mice to urishiol (allergen found in poi-son ivy, poison oak, and sumac) or the chemical allergen oxazolone, which caused itchy skin accompanied by edema formation and increases in skin thickness [36]. Dry skin-induced itch was modeled through the repeated application of acetone/ether followed by water (AEW model) to disrupt barrier integrity of the skin, resulting in spontaneous itch [37]. Importantly, irrespective of which model was used, in all these experiments, TRPA1 knockout mice con-sistently showed a significant decrease in scratching behavior as well as reduced severity of dermatitis relative to wild-type controls. Moreover, acute inhibition of TRPA1 with TRPA1 antagonists also reduced scratching behavior in these animal models of chronic itch. These findings raise the possibility that TRPA1 antagonists may reduce chronic pruritus in humans, just as they do in rodent models.
Intriguingly, not only did TRPA1-deficient mice display lower dermatitis pathology rela-tive to controls, but these mice also lacked changes in gene and protein expression observed in wild-type mice as a result of these models of dermatitis. Specifically, although mice lack-ing TRPA1 showed a normal systemic immune response, they did not show an induction of pro-inflammatory cytokines and peptides that is seen in a normal cutaneous inflamma-tory response [36]. These findings suggest that TRPA1 activation in primary afferent neurons not only regulates the transmission of itch sensation but also may play a role in regulating the expression of genes involved in the neurogenic inflammatory response in chronic itch conditions.
Normally scratching relieves itch sensation, but in chronic conditions the itch sensation is unrelenting and only temporarily relieved by scratching. Excessive scratching causes damage of the skin and release of inflammatory mediators that further sensitizes cutaneous afferents in the skin involved in transmitting itch and hence amplify the itch sensation. Interestingly, the sensitization to allergens caused an increase in substance P gene expression and protein levels in the skin that was reduced in TRPA1!/! mice [36]. Substance P is a peptide released by local immune cells and cutaneous afferent nerve terminals in the skin and has been found to be up-regulated in the skin of atopic dermatitis patients and correlated with disease severity [38]. Substance P binds to its receptor, the neurokinin-1 receptor (NK1R), which is expressed on mast cells in the skin. This leads to release of inflammatory mediators that further sensitize afferent nerve terminals [5]. There is also some evidence the NK1R is expressed on cutaneous afferent nerve terminals [39,40]. An increase in substance P can intensify the itch signal by ac-tivating and sensitizing afferent terminals through either mechanism. This, in turn, increases the desire to scratch, thus perpetuating the cycle (Figure 16.3). Interestingly, an NK1R antag-onist during treatment was found to significantly reduce dermatitis scores and scratching behavior [36]. This discovery suggests that TRPA1 channel activation may be a critical com-ponent of SP-NK1R-induced enhancement of primary afferent responses in chronic pruritus.
Although these studies highlight the key role of TRPA1 in chronic itch, there is also some evidence for the involvement of TRPV1. In particular, in two models of allergic contact der-matitis (oxazolone and exposure to dust mite extract), it was found that antagonism of TRPV1
286 16. TRP CHANNELS AND ITCH
reduced atopic dermatitis-like symptoms by accelerating skin barrier recovery [41]. For in-stance, mice treated with the TRPV1 antagonist PAC-14028, during sensitization to a dust mite allergen showed significantly reduced dermatitis scores and scratching behavior. These findings underscore the notion that TRPV1 activation may be involved in perpetuating the cycle of inflammation that is seen in dermatitis. In keeping with this idea, the chemical abla-tion of TRPV1 cells prevented psoriasis-like inflammation that is seen in response to imiqui-mod [42]. Thus, there is good evidence that inhibition of either TRPA1 or TRPV1 can reduce itch and dermatitis-like symptoms in animal models, but whether this finding will translate to humans remains to be addressed.
HIGHJACKING TRP CHANNELS TO DELIVER RELIEF FROM ITCH
The local anesthetic lidocaine is very effective at blocking itch—but it is not specific. Instead, this drug diffuses into all cells, blocks voltage-gated sodium channels, and prevents action potential propagation resulting in numbness. However, a minor modification of lido-caine results in a derivative, QX-314, that is positively charged and cannot pass through a lipid bilayer. When it stays outside the cell, QX-314 is without effect. However, on activation, TRP channels (particularly TRPV1, but also TRPA1) create large, cation-permeable pores that allow QX-314 access into the cell [43]. Thus, QX-314 selectively targets sensory neurons that have open TRP channels.
FIGURE 16.3 TRP-mediated neurogenic inflammation and itch. Local release of peptides such as Substance P may contribute to a pathological cycle of itch by triggering the release of pruritogens from immune cells, thereby activating nerve terminals causing yet more peptide release. Blocking TRP channels may halt this pathological cycle.

AN UNEXPECTED ROLE FOR TPRV3 287
Importantly, active itch-sensing neurons likely belong to this class. Evidence for the idea that QX-314 can inhibit itch came from a recent study that used QX-314 as a tool to reveal the existence of two distinct populations of itch fibers [44]. In this clever experiment, Robertson et al. took advantage of the fact that metabotropic itch receptors couple to TRPV1 and/or TRPA1 for function. Thus, coadministration of histamine and QX-314 resulted in the opening and then silencing of TRPV1 channels on histamine-responsive sensory neurons. Because this population had been silenced, histamine could no longer cause itch when it was reapplied 30 minutes later. However, chloroquine—a pruritogen that is thought to act on a distinct population—was still able to cause itch in these mice. Conversely, the coadministration of chloroquine and QX-314 rendered chloroquine-responsive cells insensitive to a second chloro-quine challenge, but these mice retained sensitivity to histamine. These experiments were used as evidence of the existence of two separate populations of itch-sensing neurons. However, the implications of this study are far greater, for this study showed that both histamine- dependent and histamine-independent itch can be silenced with QX-314. Moreover, by specifically silencing sensory neurons that are overactive under conditions of pathological pruritus, it may be possible to selectively target itch (Figure 16.4).
AN UNEXPECTED ROLE FOR TPRV3: MUTATIONS IN TRPV3 RESULT IN ITCHY LESIONS FOUND IN OLMSTED SYNDROME
Several years ago, a spontaneous dominant mutation occurred in the DS mouse strain. These mutant mice never grew hair and hence were termed DS-Nh for no hair. In addition, Nh mice developed dermatitis and showed abnormal scratching behavior indicative of itch [45–49]. A few years later, a dominant mutation in an inbred rat strain likewise resulted in the absence of fur and signs of dermatitis (named Ht for Hypotrochotic, having abnormal hair) [50,51]. Nh mice and Ht rats showed many features that were similar to atopic dermatitis in humans, including elevated levels of IgE and IL-4, increased numbers of mast and other im-mune cells, and the presence of Staphylococcus aureus in the skin. Through positional cloning, it was subsequently discovered that the hairless and dermatitis phenotypes in both the Nh mice and Ht rats were caused by an amino acid substitution in TRPV3 at Gly573 [52]. The amino acid substitutions at this site turned out to be a gain-of-function mutation, which sensitizes TRPV3 such that it is abnormally active [53]. TRPV3 is highly expressed in keratinocytes [54],
FIGURE 16.4 Highjacking TRP channels to deliver relief from itch. QX-314 is a cell impermeant sodium channel blocker that can only gain access to the cell upon entry through a large diameter channel such as a TRP channel. Thus, QX-314 has the potential to selectively inactivate neurons that are pathologically activated in chronic itch.

288 16. TRP CHANNELS AND ITCH
where it plays an important role in keratinocyte function that supports skin barrier formation and hair growth [54–58]. These findings suggest that abnormally elevated TRPV3 activity in keratinocytes results in disruptions of the epidermal barrier, immune infiltration, and patho-logical itch, as well as the loss of fur (Figure 16.5).
Now humans expressing variants in the TRPV3 gene have been discovered, and such variants cause a rare condition called Olmsted syndrome involving itchy skin lesions, par-ticularly on the palms, soles, and face, as well as hair loss. The first described cases were also dominant, gain-of-function variants at Gly573, just as seen in rodents. However, subse-quently other variants in TRPV3 have also been found to cause Olmsted syndrome [59–62]. Intriguingly, some involve variants in TRPV3 that are recessive in nature and are thought to result in loss of TRPV3 function [63,64]. Patients with recessive variants in TRPV3 also have thickened, lesioned skin on the soles of their feet that is associated with severe itch and some loss of eyelashes. These findings raise the possibility that gain- and loss-of-function variants in the TRPV3 channel result in overlapping phenotypes, suggesting that either too much or too little activity of this channel can result in skin barrier dysfunction that is accompanied by immune infiltration and itch at the site of lesions, as well as alopecia.
What is not currently known is whether abnormal TRPV3 activity underlies other, more common types of dermatological itch. There is some precedent for this idea because TRPV3 mRNA is significantly up-regulated in the area of lesional skin of patients with atopic der-matitis [65]. Furthermore, the activation of TRPV3 in keratinocytes results in the release of numerous inflammatory mediators that could potentially trigger itch [66–69]. Recently, sev-eral small-molecule TRPV3 antagonists have been identified, but whether they inhibit itch remains to be seen.
FIGURE 16.5 Mutations in TRPV3 causing itch due to Olmsted syndrome (OS). Two dominant mutations (G573C and G573S) in TRPV3 were first discovered in rodents, where they caused atopic-dermatitis like symptoms, skin barrier dysfunction, abnormal hair growth, and itch. Since then, dominant gain-of-function (GOF) mutations at this site have been described in humans where they cause Olmsted syndrome, a rare skin disease that involves the thickening of the skin, barrier dysfunction, and itchy lesions as well as alopecia. Most recently, recessive mutations that likely result in loss of function (LOF) have also been found to cause this syndrome, suggesting that appropriate TRPV3 function—neither too much, nor too little—is essential for normal keratinocyte function to support skin and hair growth. Whether abnormal TRPV3 activity underlies more common forms of itch is unknown.

CONCLUSIONS
The knowledge that TRPV1 and TRPA1 are key molecular integrators of itch has raised the possibility that these channels may be a good therapeutic target for the treatment of pruritus. There are many possible ways that TRPs could be targeted—from activating, to inhibiting, and even highjacking these channels to silence itch-mediating primary afferents. Moreover, there are numerous new TRP agonists and antagonists that, though originally developed as analgesics, should be considered for the treatment of itch. Even TRP compounds that have failed when given systemically have the potential to be effective and safe for the treatment of localized pruritus.
References [1] Ross SE. Pain and itch: insights into the neural circuits of aversive somatosensation in health and disease. Curr
Opin Neurobiol 2011;21(6):880–7. [2] Phillis RW, Bramlage AT, Wotus C, Whittaker A, Gramates LS, Seppala D, et al. Isolation of mutations affecting
neural circuitry required for grooming behavior in Drosophila melanogaster. Genetics 1993;133(3):581–92. [3] Weisshaar E, Dalgard F. Epidemiology of itch: adding to the burden of skin morbidity. Acta Derm Venereol
2009;89(4):339–50. [4] Bathe A, Weisshaar E, Matterne U. Chronic pruritus–more than a symptom: a qualitative investigation into
patients’ subjective illness perceptions. J Adv Nurs 2013;69(2):316–26. [5] Ikoma A, Steinhoff M, Stander S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nat Rev Neurosci
2006;7(7):535–47. [6] Pfab F, Valet M, Napadow V, Tolle TR, Behrendt H, Ring J, et al. Itch and the brain. Chem Immunol Allergy
2012;98:253–65. [7] Schmelz M, Schmidt R, Bickel A, Handwerker HO, Torebjork HE. Specific C-receptors for itch in human skin.
J Neurosci 1997;17(20):8003–8. [8] Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF. Cellular basis of itch sensation. Science 2009;325(5947):1531–4. [9] Sun YG, Chen ZF. A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature
2007;448(7154):700–3. [10] Mishra SK, Hoon MA. The cells and circuitry for itch responses in mice. Science 2013;340(6135):968–71. [11] Han L, Ma C, Liu Q, Weng HJ, Cui Y, Tang Z, et al. A subpopulation of nociceptors specifically linked to itch.
Nat Neurosci 2013;16(2):174–82. [12] Fleming MS, Ramos D, Han SB, Zhao J, Son YJ, Luo W. The majority of dorsal spinal cord gastrin releasing pep-
tide is synthesized locally whereas neuromedin B is highly expressed in pain- and itch-sensing somatosensory neurons. Mol Pain 2012;8:52.
[13] Liu XY, Wan L, Huo FQ, Barry DM, Li H, Zhao ZQ, et al. B-type natriuretic peptide is neither itch-specific nor functions upstream of the GRP-GRPR signaling pathway. Mol Pain 2014;10(1):4.
[14] Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ. A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 2001;106(5):619–32.
[15] Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell 2009;139(7):1353–65.
[16] Liu Q, Weng HJ, Patel KN, Tang Z, Bai H, Steinhoff M, et al. The distinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. Sci Signal 2011;4(181):ra45.
[17] Sikand P, Dong X, LaMotte RH. BAM8-22 peptide produces itch and nociceptive sensations in humans inde-pendent of histamine release. J Neurosci 2011;31(20):7563–7.
[18] Liu Q, Sikand P, Ma C, Tang Z, Han L, Li Z, et al. Mechanisms of itch evoked by beta-alanine. J Neurosci 2012;32(42):14532–7.
[19] Imamachi N, Park GH, Lee H, Anderson DJ, Simon MI, Basbaum AI, et al. TRPV1-expressing primary af-ferents generate behavioral responses to pruritogens via multiple mechanisms. Proc Natl Acad Sci USA 2009;106(27):11330–5.
REFERENCES 289

290 16. TRP CHANNELS AND ITCH
[20] Papoiu AD, Yosipovitch G. Topical capsaicin. The fire of a ‘hot’ medicine is reignited. Expert Opin Pharmacother 2010;11(8):1359–71.
[21] Ellis CN, Berberian B, Sulica VI, Dodd WA, Jarratt MT, Katz HI, et al. A double-blind evaluation of topical cap-saicin in pruritic psoriasis. J Am Acad Dermatol 1993;29(3):438–42.
[22] Anand P, Bley K. Topical capsaicin for pain management: therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8% patch. Br J Anaesth 2011;107(4):490–502.
[23] Capsaicin patch (Qutenza) for postherpetic neuralgia. Med Lett Drugs Ther 2011;53(1365):42–3. [24] Metz M, Krause K, Maurer M, Magerl M. Treatment of notalgia paraesthetica with an 8% capsaicin patch. Br J
Dermatol 2011;165(6):1359–61. [25] Wallace M, Pappagallo M. Qutenza(R): a capsaicin 8% patch for the management of postherpetic neuralgia.
Expert Rev Neurother 2011;11(1):15–27. [26] Kardon AP, Polgar E, Hachisuka J, Snyder LM, Cameron D, Savage S, et al. Dynorphin acts as a neuromodula-
tor to inhibit itch in the dorsal horn of the spinal cord. Neuron 2014;82(3):573–86. [27] Knowlton WM, McKemy DD. TRPM8: from cold to cancer, peppermint to pain. Curr Pharm Biotechnol
2011;12(1):68–77. [28] Reddy VB, Iuga AO, Shimada SG, LaMotte RH, Lerner EA. Cowhage-evoked itch is mediated by a novel cyste-
ine protease: a ligand of protease-activated receptors. J Neurosci 2008;28(17):4331–5. [29] Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31
receptor mediates T helper cell-dependent itch: involvement of TRPV1 and TRPA1. J Allergy Clin Immunol 2014;133(2):448–60, e7.
[30] Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic derma-titis cytokine TSLP activates neurons to induce itch. Cell 2013;155(2):285–95.
[31] Shim WS, Tak MH, Lee MH, Kim M, Koo JY, Lee CH, et al. TRPV1 mediates histamine-induced itching via the activation of phospholipase A2 and 12-lipoxygenase. J Neurosci 2007;27(9):2331–7.
[32] Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, et al. TRPA1 is required for histamine- independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci 2011;14(5):595–602.
[33] Liu T, Ji RR. Oxidative stress induces itch via activation of transient receptor potential subtype ankyrin 1 in mice. Neurosci Bull 2012;28(2):145–54.
[34] Sawada Y, Hosokawa H, Matsumura K, Kobayashi S. Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur J Neurosci 2008;27(5):1131–42.
[35] Brederson JD, Kym PR, Szallasi A. Targeting TRP channels for pain relief. Eur J Pharmacol 2013; 716(1–3):61–76.
[36] Liu B, Escalera J, Balakrishna S, Fan L, Caceres AI, Robinson E, et al. TRPA1 controls inflammation and prurito-gen responses in allergic contact dermatitis. Faseb J 2013;27(9):3549–63.
[37] Wilson SR, Nelson AM, Batia L, Morita T, Estandian D, Owens DM, et al. The ion channel TRPA1 is required for chronic itch. J Neurosci 2013;33(22):9283–94.
[38] Toyoda M, Nakamura M, Makino T, Hino T, Kagoura M, Morohashi M. Nerve growth factor and substance P are useful plasma markers of disease activity in atopic dermatitis. Br J Dermatol 2002;147(1):71–9.
[39] Carlton SM, Zhou S, Coggeshall RE. Localization and activation of substance P receptors in unmyelinated axons of rat glabrous skin. Brain Res 1996;734(1–2):103–8.
[40] von Banchet GS, Schaible HG. Localization of the neurokinin 1 receptor on a subset of substance P-positive and isolectin B4-negative dorsal root ganglion neurons of the rat. Neurosci Lett 1999;274(3):175–8.
[41] Yun JW, Seo JA, Jang WH, Koh HJ, Bae IH, Park YH, et al. Antipruritic effects of TRPV1 antagonist in murine atopic dermatitis and itching models. J Invest Dermatol 2011;131(7):1576–9.
[42] Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 2014;510:157–61.
[43] Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 2007;449(7162):607–10.
[44] Roberson DP, Gudes S, Sprague JM, Patoski HA, Robson VK, Blasl F, et al. Activity-dependent silencing reveals functionally distinct itch-generating sensory neurons. Nat Neurosci 2013;16(7):910–18.
[45] Haraguchi M, Hino M, Tanaka H, Maru M. Naturally occurring dermatitis associated with Staphylococcus aureus in DS-Nh mice. Exp Anim 1997;46(3):225–9.

[46] Hikita I, Yoshioka T, Mizoguchi T, Tsukahara K, Tsuru K, Nagai H, et al. Characterization of dermatitis arising spontaneously in DS-Nh mice maintained under conventional conditions: another possible model for atopic dermatitis. J Dermatol Sci 2002;30(2):142–53.
[47] Yoshioka T, Hikita I, Asakawa M, Hirasawa T, Deguchi M, Matsutani T, et al. Spontaneous scratching behaviour in DS-Nh mice as a possible model for pruritus in atopic dermatitis. Immunology 2006;118(3):293–301.
[48] Watanabe A, Takeuchi M, Nagata M, Nakamura K, Hirasawa T, Nakao H, et al. Spontaneous development of dermatitis in DS-Nh mice under specific pathogen-free conditions. Exp Anim 2003;52(1):77–80.
[49] Watanabe A, Takeuchi M, Nagata M, Nakamura K, Nakao H, Yamashita H, et al. Role of the Nh (Non-hair) muta-tion in the development of dermatitis and hyperproduction of IgE in DS-Nh mice. Exp Anim 2003;52(5):419–23.
[50] Asakawa M, Yoshioka T, Hikita I, Matsutani T, Hirasawa T, Arimura A, et al. WBN/Kob-Ht rats spontaneously develop dermatitis under conventional conditions: another possible model for atopic dermatitis. Exp Anim 2005;54(5):461–5.
[51] Tani S, Noguchi M, Hosoda Y, Sugibayasi K, Morimoto Y. Characteristics of spontaneous erythema appeared in hairless rats. Exp Anim 1998;47(4):253–6.
[52] Asakawa M, Yoshioka T, Matsutani T, Hikita I, Suzuki M, Oshima I, et al. Association of a mutation in TRPV3 with defective hair growth in rodents. J Invest Dermatol 2006;126(12):2664–72.
[53] Xiao R, Tian J, Tang J, Zhu MX. The TRPV3 mutation associated with the hairless phenotype in rodents is con-stitutively active. Cell Calcium 2008;43(4):334–43.
[54] Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, Hergarden AC, et al. A heat-sensitive TRP channel expressed in keratinocytes. Science 2002;296(5575):2046–9.
[55] Imura K, Yoshioka T, Hikita I, Tsukahara K, Hirasawa T, Higashino K, et al. Influence of TRPV3 mutation on hair growth cycle in mice. Biochem Biophys Res Commun 2007;363(3):479–83.
[56] Cheng X, Jin J, Hu L, Shen D, Dong XP, Samie MA, et al. TRP channel regulates EGFR signaling in hair morpho-genesis and skin barrier formation. Cell 2010;141(2):331–43.
[57] Yoshioka T, Imura K, Asakawa M, Suzuki M, Oshima I, Hirasawa T, et al. Impact of the Gly573Ser substitution in TRPV3 on the development of allergic and pruritic dermatitis in mice. J Invest Dermatol 2009;129(3):714–22.
[58] Borbiro I, Lisztes E, Toth BI, Czifra G, Olah A, Szollosi AG, et al. Activation of transient receptor potential vanilloid-3 inhibits human hair growth. J Invest Dermatol 2011;131(8):1605–14.
[59] Duchatelet S, Pruvost S, de Veer S, Fraitag S, Nitschke P, Bole-Feysot C, et al. A new TRPV3 missense mutation in a patient with Olmsted syndrome and erythromelalgia. JAMA Dermatol 2014;150(3):303–6.
[60] Danso-Abeam D, Zhang J, Dooley J, Staats KA, Van Eyck L, Van Brussel T, et al. Olmsted syndrome: exploration of the immunological phenotype. Orphanet J Rare Dis 2013;8:79.
[61] Lai-Cheong JE, Sethuraman G, Ramam M, Stone K, Simpson MA, McGrath JA. Recurrent heterozygous mis-sense mutation, p.Gly573Ser, in the TRPV3 gene in an Indian boy with sporadic Olmsted syndrome. Br J Dermatol 2012;167(2):440–2.
[62] Lin Z, Chen Q, Lee M, Cao X, Zhang J, Ma D, et al. Exome sequencing reveals mutations in TRPV3 as a cause of Olmsted syndrome. Am J Hum Genet 2012;90(3):558–64.
[63] Duchatelet S, Guibbal L, de Veer S, Fraitag S, Nitschke P, Zarhrate M, et al. Olmsted syndrome with erythrome-lalgia caused by recessive TRPV3 mutations. Br J Dermatol 2014;171:675–8.
[64] Eytan O, Fuchs-Telem D, Mevorach B, Indelman M, Bergman R, Sarig O, et al. Olmsted syndrome caused by a homozygous recessive mutation in TRPV3. J Invest Dermatol 2014;134:1752–4.
[65] Bianchi P, Ribet V, Casas C, Lejeune O, Schmitt AM, Redoules D. Analysis of gene expression in atopic derma-titis using a microabrasive method. J Invest Dermatol 2012;132(2):469–72.
[66] Mandadi S, Sokabe T, Shibasaki K, Katanosaka K, Mizuno A, Moqrich A, et al. TRPV3 in keratinocytes trans-mits temperature information to sensory neurons via ATP. Pflugers Arch 2009;458(6):1093–102.
[67] Xu H, Delling M, Jun JC, Clapham DE. Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat Neurosci 2006;9(5):628–35.
[68] Huang SM, Lee H, Chung MK, Park U, Yu YY, Bradshaw HB, et al. Overexpressed transient receptor poten-tial vanilloid 3 ion channels in skin keratinocytes modulate pain sensitivity via prostaglandin E2. J Neurosci 2008;28(51):13727–37.
[69] Phelps CB, Wang RR, Choo SS, Gaudet R. Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J Biol Chem 2010;285(1):731–40.
REFERENCES 291


















