
TLR signaling and effector functions are intact in XLA neutrophils
7
TLR signaling and effector functions are intact in XLA neutrophils Thomas U. Marron a , Kaileen Rohr b , Monica Martinez-Gallo a , Joyce Yu a , c , Charlotte Cunningham-Rundles a , d , ⁎ a Immunology Institute, Mount Sinai School of Medicine, New York, NY, USA b Mount Sinai School of Medicine, Graduate School of Biological Sciences, New York, NY, USA c Department of Pediatrics, Weill Cornell Medical Center, New York, NY, USA d Department of Medicine and Pediatrics, Mount Sinai School of Medicine, New York, NY, USA Received 13 May 2010; accepted with revision 21 June 2010 Available online 14 July 2010 KEYWORDS Bruton's tyrosine kinase; BTK; X-linked agammaglobulinemia; XLA; Toll-like receptors; TLR; Neutrophils; Respiratory burst Abstract Toll-like receptors (TLRs) are essential components of the innate immune system, and their ligands are important activators of neutrophils. Bruton's tyrosine kinase (Btk) has been reported to mediate signaling through toll-like receptors (TLRs) in many cell types, however, the role of Btk in TLR activation of neutrophils remains unclear. Impaired TLR-induced neutrophil function was found in mice with loss of Btk and in humans with TLR-signaling defects, but the integrity of TLR pathways in X-linked agammaglobulinemia (XLA) neutrophils has not been assessed. In this study LPS (TLR4) or an imidazoquinoline compound (TLR7/8) activated XLA neutrophil shedding of surface CD62L, and phosphorylated MAP kinases p38, JNK and ERK. TLR activation also induced normal respiratory burst and retarded apoptosis for XLA neutrophils, comparable to normal controls. These data demonstrate that the loss of Btk in XLA neutrophils does not impair functional responses to TLR signals. © 2010 Elsevier Inc. All rights reserved. Introduction X-linked agammaglobulinemia (XLA) is a primary immuno- deficiency disease resulting from mutations in a cytoplas- mic tyrosine kinase, Bruton's tyrosine kinase (Btk). The defect is characterized by a block in B cell maturation leading to a loss of peripheral blood B cells and severely reduced serum immunoglobulins [1,2]. Prior to receiving replacement immune globulin (Ig), patients with XLA experience invasive pyogenic infections, resulting in significant morbidity and mortality [3–5]. Interestingly, although Btk expression is abundant in neutrophils as well Abbreviations: XLA, X-linked agammaglobulinemia; Btk, Bruton's tyrosine kinase; Ig, immunoglobulin; TLR, Toll-like receptor; LPS, lipopolysaccharide; MFI, mean fluorescence intensity; DHR, dihydrorhodamine 123; SOD, superoxide dismutase; PMA, phorbol 12-myristate 13-acetate; GM-CSF, granulocyte macrophage colony- stimulating factor; HBSS, Hanks balanced salt solution; ssRNA, single- stranded RNA; PI, propidium iodide. ⁎ Corresponding author. Departments of Medicine and Pediatrics, The Immunology, Institute, Mount Sinai School of Medicine, 1425 Madison Avenue, New York, NY 10029, USA. Fax: +1 212 987 5593. E-mail address: [email protected] (C. Cunningham-Rundles). 1521-6616/$ – see front matter © 2010 Elsevier Inc. All rights reserved. doi:10.1016/j.clim.2010.06.011 available at www.sciencedirect.com Clinical Immunology www.elsevier.com/locate/yclim Clinical Immunology (2010) 137, 74–80
-
Upload
thomas-u-marron -
Category
Documents
-
view
215 -
download
0
Transcript of TLR signaling and effector functions are intact in XLA neutrophils

ava i l ab l e a t www.sc i enced i r ec t . com
C l i n i ca l Immuno logy
www.e l sev i e r . com/ l oca te /yc l im
Clinical Immunology (2010) 137, 74–80
TLR signaling and effector functions are intact inXLA neutrophilsThomas U. Marron a, Kaileen Rohr b, Monica Martinez-Gallo a,Joyce Yu a,c, Charlotte Cunningham-Rundles a,d,⁎
a Immunology Institute, Mount Sinai School of Medicine, New York, NY, USAb Mount Sinai School of Medicine, Graduate School of Biological Sciences, New York, NY, USAc Department of Pediatrics, Weill Cornell Medical Center, New York, NY, USAd Department of Medicine and Pediatrics, Mount Sinai School of Medicine, New York, NY, USA
Received 13 May 2010; accepted with revision 21 June 2010Available online 14 July 2010
Abbreviations: XLA, X-linked agammtyrosine kinase; Ig, immunoglobulin;lipopolysaccharide; MFI, mean fludihydrorhodamine 123; SOD, superox12-myristate 13-acetate; GM-CSF, grastimulating factor; HBSS, Hanks balancstranded RNA; PI, propidium iodide.⁎ Corresponding author. Department
The Immunology, Institute, Mount SinMadison Avenue, New York, NY 10029,
E-mail address: Charlotte.Cunningh(C. Cunningham-Rundles).
1521-6616/$ – see front matter © 201doi:10.1016/j.clim.2010.06.011
KEYWORDSBruton's tyrosine kinase;BTK;X-linkedagammaglobulinemia;XLA;Toll-like receptors;TLR;Neutrophils;Respiratory burst
Abstract Toll-like receptors (TLRs) are essential components of the innate immune system,and their ligands are important activators of neutrophils. Bruton's tyrosine kinase (Btk) has beenreported to mediate signaling through toll-like receptors (TLRs) in many cell types, however, therole of Btk in TLR activation of neutrophils remains unclear. Impaired TLR-induced neutrophilfunction was found in mice with loss of Btk and in humans with TLR-signaling defects, but theintegrity of TLR pathways in X-linked agammaglobulinemia (XLA) neutrophils has not beenassessed. In this study LPS (TLR4) or an imidazoquinoline compound (TLR7/8) activated XLAneutrophil shedding of surface CD62L, and phosphorylated MAP kinases p38, JNK and ERK. TLRactivation also induced normal respiratory burst and retarded apoptosis for XLA neutrophils,comparable to normal controls. These data demonstrate that the loss of Btk in XLA neutrophilsdoes not impair functional responses to TLR signals.
© 2010 Elsevier Inc. All rights reserved.aglobulinemia; Btk, Bruton'sTLR, Toll-like receptor; LPS,orescence intensity; DHR,ide dismutase; PMA, phorbolnulocyte macrophage colony-ed salt solution; ssRNA, single-
s of Medicine and Pediatrics,ai School of Medicine, 1425USA. Fax: +1 212 987 [email protected]
0 Elsevier Inc. All rights reser
vedIntroduction
X-linked agammaglobulinemia (XLA) is a primary immuno-deficiency disease resulting from mutations in a cytoplas-mic tyrosine kinase, Bruton's tyrosine kinase (Btk). Thedefect is characterized by a block in B cell maturationleading to a loss of peripheral blood B cells and severelyreduced serum immunoglobulins [1,2]. Prior to receivingreplacement immune globulin (Ig), patients with XLAexperience invasive pyogenic infections, resulting insignificant morbidity and mortality [3–5]. Interestingly,although Btk expression is abundant in neutrophils as well
.

Figure 1 Btk expression in XLA cells: Leukocytes of 8 XLApatients examined by western blot, demonstrated no Btkexpression in contrast to cells of 3 normal controls.
75TLR signaling and effector functions are intact in XLA neutrophils
as many other cells of hematopoietic lineage, patientsmaintained on sufficient Ig therapy are generally healthy[6,7], suggesting that Btk is either dispensable outside theB-cell compartment, and/or that compensatory kinasesmaintain normal functions in other cells [8]. However, anumber of recent studies have demonstrated that Btk playsan important, or possibly essential, role in signaling throughseveral Toll-like receptors (TLRs) which recognize con-served pathogen-associated molecular patterns such aslipopolysaccharides (LPS), bacterial DNA, or single-strand-ed viral RNA (ssRNA) [9–19]. These observations havesuggested that in addition to the loss of humoral immunity,subjects with XLA might have additional innate immunedefects.
The first suggestion that Btk might be important in TLRsignaling came from work in Xid mice in which a pointmutation in the pleckstrin homology domain of the Btk generesults in loss of Btk function. LPS induced production of TNF-α and IL-1β was found impaired in these mice and Xidmacrophages and neutrophils had reduced generation ofreactive oxygen intermediates and defective LPS clearance[16,20,21]. Further work in Btk deficient mice and thehuman monocyte line THP1 showed that TLR2, 4, 7, 8 and 9ligands phosphorylated Btk, and that Btk could be co-immunoprecipitated with myeloid differentiation primaryresponse gene (88) (MyD88), toll-interleukin 1 receptordomain containing adaptor protein (TIRAP, also known asthe MyD88 adaptor-like protein, or Mal), and Interleukin-1receptor-associated kinase 1 (IRAK1), key components of theTLR signaling complex [9,12,22,23].
TLRs have selected patterns of distribution on immunecells, with the final effector functions differing depending onthe cell type. Neutrophils, the most abundant immune celland first responders to infection, express most of the TLRs[24]. When neutrophil TLRs bind their ligands, signalingpathways are activated which triggers the shedding ofsurface L-selectin, upregulation of surface integrins, primingof respiratory burst, increased cytokine production andphagocytosis, and slowed progression to apoptosis [24].
Taking advantage of TLR induced changes in adhesionmolecules, Bernuth et al. showed the impaired shedding ofL-selectin was characteristic of subjects with mutations inNF-κB essential modulator (NEMO) or IL-1R associatedkinase (IRAK-4); this test was suggested for clinicalscreening for these defects. Neutrophils of these subjectshad previously been shown to have decreased LPS-inducedNADPH oxidase activation, impaired superoxide production[25] and defective neutrophil migration and phagocytosis[26]. However, if Btk is integral to TLR signaling inneutrophils, subjects with XLA should show similar neutro-phil defects. XLA patients occasionally are found to beneutropenic, typically coinciding with the severe infectionthat suggests the presence of an immunodeficiency andpotentially, neutrophil dysfunction [4,27], however, neu-tropenia is not a characteristic of XLA patients on sufficientIg replacement. To date no study has directly assessed TLRinduction of pro-inflammatory signaling pathways andeffector functions in primary XLA neutrophils. The presentstudy assesses the activation and downstream activity ofTLR4 and TLR7/8 effector functions to examine whether thedistinct signaling pathways activated by extracellular andendosomal TLRs are intact.
Materials and methods
Patients and controls
This study included 8 XLA patients of age 12 to 43 years. Allpatients were profoundly hypogammaglobulinemic, hadfewer than 0.1% B cells in peripheral blood. Mutationalanalyses to confirm the diagnosis of XLA were kindlyperformed by Dr. M. E. Conley. Lack of Btk expression bythese patients was confirmed by western blot as describedbelow (Fig. 1). All subjects were well at the time of the studyand on replacement Ig in standard doses; blood samples werecollected before monthly infusions. Control subjects werehealthy adult volunteers. These studies were done using anInstitutional Review Board approved protocol and informedconsent.
Neutrophil isolation
Whole blood was mixed 1:1 with 3% dextran (MP Biomedicals,Solon, OH) in PBS and erythrocytes allowed to sediment for30 min. The leukocyte-rich supernatant was then applied toa Ficoll-Hypaque (GE Healthcare, Uppsala, Sweden) densitygradient centrifugation. PBMCs were removed, and remain-ing erythrocytes in the pelleted mixture were lysed byhypotonic NaCl treatment. Purified neutrophils were resus-pended in Hanks' Balanced Salt Solution (HBSS, Gibco,Carlsbad, CA) supplemented with 10% Fetal Calf Serum, 1%penicillin/streptomycin.
TLR induced CD62L shedding
The assay for neutrophil CD62L shedding was performed asdescribed by von Bernuth et al. Briefly, 100 μL of heparinizedwhole blood was incubated for 1 h at 37 °C alone or in thepresence of the of a water-soluble R848, imidazoquinolinecompound TLR7/8 ligand CL097 (0.1 μg/mL–5.0μg/mL)(Invivogen, San Diego, CA), LPS (1 ng/mL–1 μg/mL) (Invivo-gen) or as a control, phorbol 12-myristate 13-acetate (PMA)(1 ng/mL–1 μg/mL) (Sigma, St. Louis, MO). In the control,CL097, and PMA treated cells 10 μg/mL polymixin B (Sigma)was added before treatment with TLR ligand. Erythrocyteswere then lysed in (1.3 M NH4Cl, 100 mM KHCO3, 1 mMEDTA). Remaining leukocytes were incubated for 15 min onice in PBS+2% fetal calf serum with FITC-conjugatedmonoclonal anti-CD62L BD Biosciences, San Diego, CA) or
76 T.U. Marron et al.
isotype control, washed three times in PBS/FCS; CD62Lsurface expression was analyzed via flow cytometry on aFACSCalibur (BD Biosciences). Granulocytes were gatedaccording to forward-scatter/sideways-scatter and themean fluorescence intensity (MFI) of CD62L surface expres-sion determined.
Western blot
Neutrophils in complete HBSS media were plated at 5×106
cells/mL and allowed to rest for 6 h at 37 °C. After 1 h,neutrophils were incubated for an optimized time of 5 minwith the optimized concentrations of 2.5 μg/mL CL097 or100 ng/mL LPS, and subsequently centrifuged at 4 °C andlysed with lysis buffer (1% Triton X-100, 20 mM Tris [pH 7.4],40 mM NaCl, 5 mM EDTA, 50 mM NaF, 30 mM Na4P2O7)supplemented with protease and phosphatase inhibitors(Thermo Scientific, Rockford, IL, USA). Protein was quantifiedwith the Micro BCA Protein Assay Kit (Thermo Scientific), andsubsequently fractionated on 12% SDS-PAGE gels (ThermoScientific), transferred to nitrocellulose membrane (What-
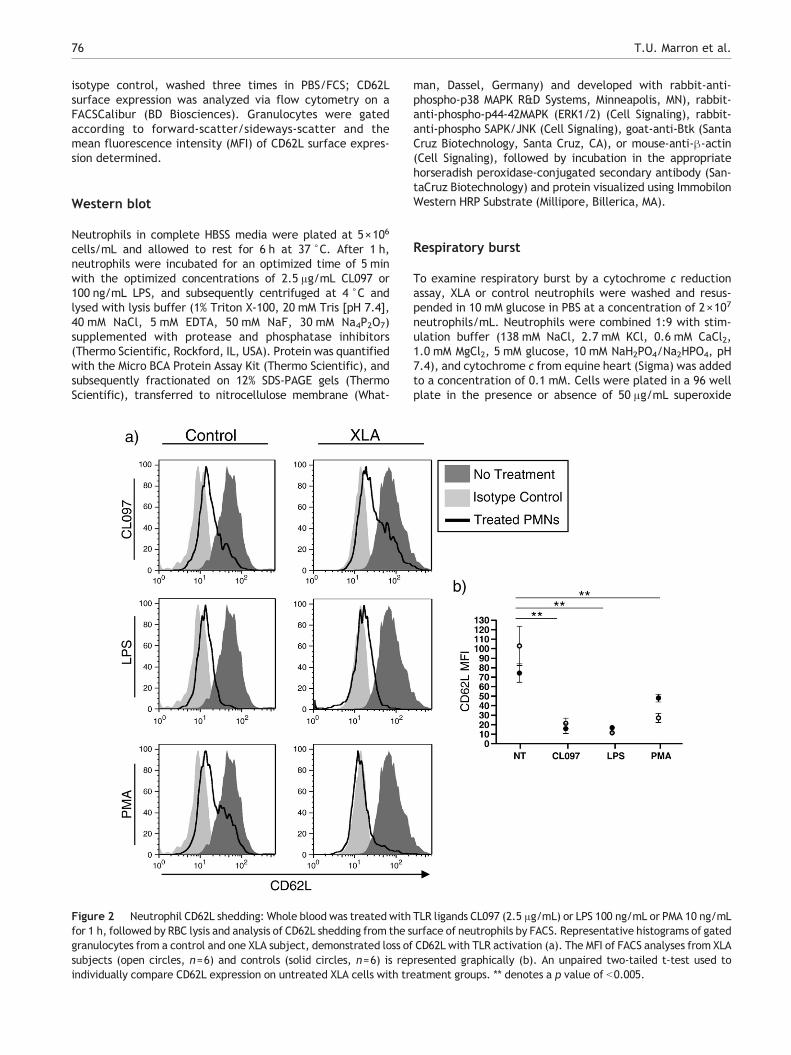
Figure 2 Neutrophil CD62L shedding: Whole blood was treated withfor 1 h, followed by RBC lysis and analysis of CD62L shedding from the sgranulocytes from a control and one XLA subject, demonstrated loss ofsubjects (open circles, n=6) and controls (solid circles, n=6) is repindividually compare CD62L expression on untreated XLA cells with tr
man, Dassel, Germany) and developed with rabbit-anti-phospho-p38 MAPK R&D Systems, Minneapolis, MN), rabbit-anti-phospho-p44-42MAPK (ERK1/2) (Cell Signaling), rabbit-anti-phospho SAPK/JNK (Cell Signaling), goat-anti-Btk (SantaCruz Biotechnology, Santa Cruz, CA), or mouse-anti-β-actin(Cell Signaling), followed by incubation in the appropriatehorseradish peroxidase-conjugated secondary antibody (San-taCruz Biotechnology) and protein visualized using ImmobilonWestern HRP Substrate (Millipore, Billerica, MA).
Respiratory burst
To examine respiratory burst by a cytochrome c reductionassay, XLA or control neutrophils were washed and resus-pended in 10 mM glucose in PBS at a concentration of 2×107
neutrophils/mL. Neutrophils were combined 1:9 with stim-ulation buffer (138 mM NaCl, 2.7 mM KCl, 0.6 mM CaCl2,1.0 mM MgCl2, 5 mM glucose, 10 mM NaH2PO4/Na2HPO4, pH7.4), and cytochrome c from equine heart (Sigma) was addedto a concentration of 0.1 mM. Cells were plated in a 96 wellplate in the presence or absence of 50 μg/mL superoxide
TLR ligands CL097 (2.5 μg/mL) or LPS 100 ng/mL or PMA 10 ng/mLurface of neutrophils by FACS. Representative histograms of gatedCD62L with TLR activation (a). The MFI of FACS analyses from XLAresented graphically (b). An unpaired two-tailed t-test used toeatment groups. ** denotes a p value of b0.005.

77TLR signaling and effector functions are intact in XLA neutrophils
dismutase (SOD, MP Biomedicals). Neutrophils were stimu-lated with 2.5 μg/mL CL097, 100 ng/mL LPS or 10 ng/mL PMAfor 20 min and absorbance of reduced cytochrome c read at550nM absorbance. SOD-inhibitable cytochrome c reductionwas assessed by subtracting absorbance in samples pre-treated with SOD, which corrects for non-superoxidemediated reduction. Results are given relative to thebaseline respiratory activity.
Production of superoxide radicals was also quantifiedusing the dihydrorhodamine (DHR) assay. For this, neutro-phils in suspension in HBSS complete media were incubatedwith 5 μg/mL DHR (EMD Darmstadt, Germany) and allowed torest at 37 °C for 30 min. Samples were treated with TLRligands as above for 90 min. Neutrophils were subsequentlywashed in PBS and intracellular rhodamine visualized onFACScalibur. Data are presented as the mean fluorescence ofrhodamine in activated samples, divided by the meanfluorescence of untreated cells.
TLR induced cell survival
Effective TLR activation prolongs the life span of neutrophils,delaying apoptosis [28]. To examine this effect in Btkdeficient cells, neutrophils were plated in HBSS completemedia at 2×106 cells/mL and treated with 2.5 μg/mL CL097,100 ng/mL LPS or 20 ng/mL GM-CSF as a positive control,(eBioscience, San Diego, CA). After 48 h, cells were washedwith PBS, stained for Annexin 5 and propidium iodide (PI) (BDPharmingen, San Diego, CA) and assessed by flow cytometry.
Statistical analysis
Statistical analysis was performed using PRISM 4.0 (Graph-Pad). An unpaired two-tailed t-test was used for one-to-onecomparisons of individual treatment groups to the correlat-ing no-treatment controls, or comparing within a treatmentgroup for differences between control and XLA patientcellular responses.
Figure 3 MAPK signaling: Neutrophils isolated from blood of contro(2.5 μg/mL) or LPS (100 ng/mL), and phosphorylation of MAP kinasesresults for two controls and four XLA subject are shown in comparis
Results
Shedding of CD62
As shedding of leukocyte surface CD62L occurs uponsuccessful TLR activation, we treated blood from XLApatients and normal controls, with optimized concentrationsof LPS, CL097 or as a control, PMA to assess whether thisearly marker of TLR signaling was intact. Neutrophils of XLAsubjects exhibited the same loss of neutrophil surface CD62Las control neutrophils when exposed to these ligands,suggesting that TLR4 and TLR7/8 signaling activation isretained (Fig. 2a, b).
MAP kinase activation
TLRs activation also leads to phosphorylation of the MAPkinase families p38, ERK, and JNK in neutrophils, all of whichare involved in mediating effector functions; thus weassessed whether these MAP kinases required Btk to attainan activated phosphorylated state. LPS and CL097 treatmentstimulated an increase in phosphorylation of p38, ERK, andJNK in both control (n=6) and XLA neutrophils (n=6). Figure 3ows representative data for one XLA subject and a control.
TLR induced neutrophil oxidative burst
As TLR signaling pathways in neutrophils appeared intact,TLR effector functions were then assessed. TLR activation ofneutrophils normally leads to priming the cells for therelease of reactive oxygen species needed to kill phagocy-tosed pathogens [25,29]. To examine priming of respiratoryburst, isolated XLA or control neutrophils were incubatedwith cytochrome c and stimulated with LPS, CL097 or, as acontrol, PMA. The level of cytochrome c reduction by thesuperoxide radicals produced following neutrophil activationwas then assessed. As shown in Figure 4a induction of
ls (n=6) or XLA subjects (n=6) were treated for 5 min with CL097p38, JNK and ERK was assessed by western blot. Representativeon to β-actin.
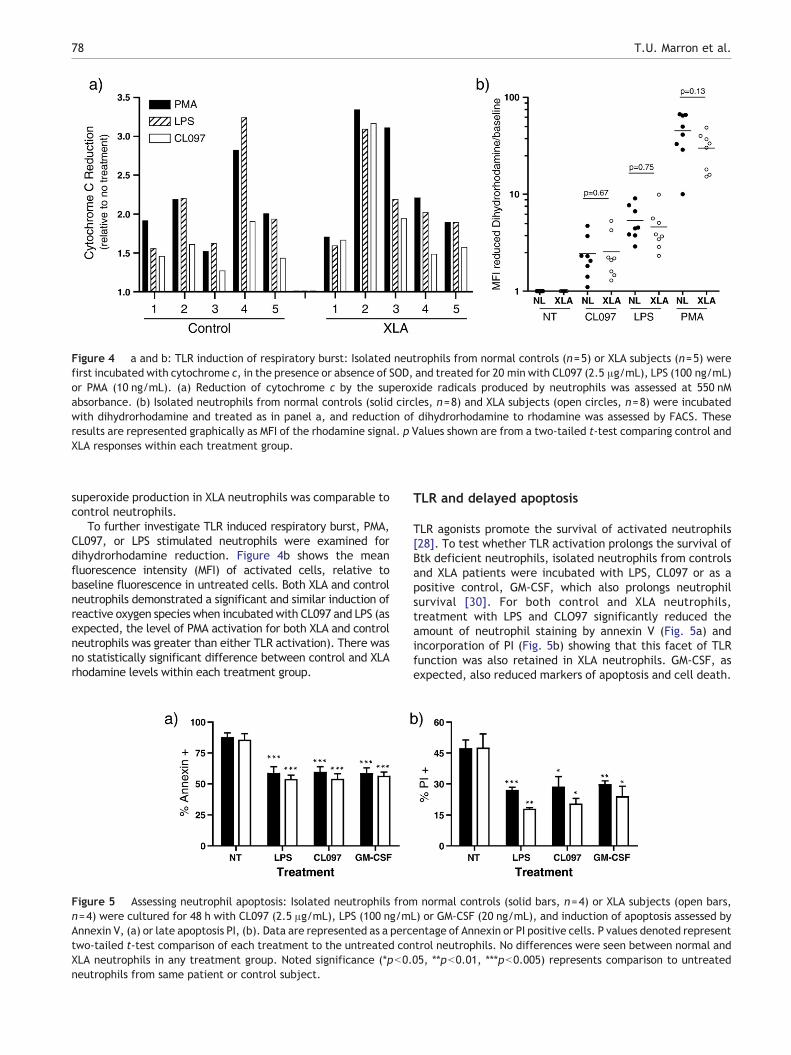
Figure 4 a and b: TLR induction of respiratory burst: Isolated neutrophils from normal controls (n=5) or XLA subjects (n=5) werefirst incubated with cytochrome c, in the presence or absence of SOD, and treated for 20 min with CL097 (2.5 μg/mL), LPS (100 ng/mL)or PMA (10 ng/mL). (a) Reduction of cytochrome c by the superoxide radicals produced by neutrophils was assessed at 550 nMabsorbance. (b) Isolated neutrophils from normal controls (solid circles, n=8) and XLA subjects (open circles, n=8) were incubatedwith dihydrorhodamine and treated as in panel a, and reduction of dihydrorhodamine to rhodamine was assessed by FACS. Theseresults are represented graphically as MFI of the rhodamine signal. p Values shown are from a two-tailed t-test comparing control andXLA responses within each treatment group.
78 T.U. Marron et al.
superoxide production in XLA neutrophils was comparable tocontrol neutrophils.
To further investigate TLR induced respiratory burst, PMA,CL097, or LPS stimulated neutrophils were examined fordihydrorhodamine reduction. Figure 4b shows the meanfluorescence intensity (MFI) of activated cells, relative tobaseline fluorescence in untreated cells. Both XLA and controlneutrophils demonstrated a significant and similar induction ofreactive oxygen species when incubated with CL097 and LPS (asexpected, the level of PMA activation for both XLA and controlneutrophils was greater than either TLR activation). There wasno statistically significant difference between control and XLArhodamine levels within each treatment group.
Figure 5 Assessing neutrophil apoptosis: Isolated neutrophils fromn=4) were cultured for 48 h with CL097 (2.5 μg/mL), LPS (100 ng/mAnnexin V, (a) or late apoptosis PI, (b). Data are represented as a perctwo-tailed t-test comparison of each treatment to the untreated conXLA neutrophils in any treatment group. Noted significance (*pb0.neutrophils from same patient or control subject.
TLR and delayed apoptosis
TLR agonists promote the survival of activated neutrophils[28]. To test whether TLR activation prolongs the survival ofBtk deficient neutrophils, isolated neutrophils from controlsand XLA patients were incubated with LPS, CL097 or as apositive control, GM-CSF, which also prolongs neutrophilsurvival [30]. For both control and XLA neutrophils,treatment with LPS and CLO97 significantly reduced theamount of neutrophil staining by annexin V (Fig. 5a) andincorporation of PI (Fig. 5b) showing that this facet of TLRfunction was also retained in XLA neutrophils. GM-CSF, asexpected, also reduced markers of apoptosis and cell death.
normal controls (solid bars, n=4) or XLA subjects (open bars,L) or GM-CSF (20 ng/mL), and induction of apoptosis assessed byentage of Annexin or PI positive cells. P values denoted representtrol neutrophils. No differences were seen between normal and05, **pb0.01, ***pb0.005) represents comparison to untreated

79TLR signaling and effector functions are intact in XLA neutrophils
Discussion
Based predominantly on mouse models and cell lines, anumber of previous studies have concluded that Btk is a keycomponent of selected cell-specific TLR signaling pathways[9–11,14,17,31]. LPS stimulated Xid or Btk−/− murinemacrophages displayed impaired production of TNF-α andIL-1β [17,20], IL-10[17], impaired AP-1 and NF-κB activation[10,12,17], as well as diminished production of reactiveoxygen intermediates [16]. On the other hand, studies onhuman XLA dendritic cells, monocytes and macrophages haveprovided contradictory results, showing both reduced[18,22,32] or normal TLR cytokine secretion responses[33,34].
TLR pathways are crucial to activation of a variety ofinflammatory cells including neutrophils, and it is notsurprising that neutrophil function might be impaired inpatients with mutations in IRAK4 or NEMO, key signalingintermediates in the NF-κB pathway activated by TLRligation [25,26]. The original observation that an individualwith IRAK4 deficiency had defective CD62L shedding [35] ledto the suggestion of a clinical test to screen for TLR defects[36]. While L-selectin shedding was clearly abnormal insubjects with IRAK-4 and NEMO defects, the neutrophils ofXLA subjects examined here demonstrated normal loss ofCD62L upon TLR activation, suggesting that this marker ofneutrophil activation is Btk-independent. To further assessTLR-induced signaling, activation of p38, ERK, and JNK wasassessed, as TLR activation of these pathways regulatesimportant neutrophil functions such as adhesion, migration,activation of NF-κB signaling, and downstream cytokinerelease [26,37,38]. However, there was no difference inactivation of these kinases in XLA neutrophils compared withcontrols. Turning to an essential downstream effectorfunction, respiratory burst, we found that TLR activatedXLA neutrophils were also comparable to controls. Inaddition, XLA neutrophils treated with TLR agonists demon-strated delayed apoptosis, further confirming the fidelity ofTLR activation and signaling pathways in the absence of Btk.Other TLR-induced effector functions, such as phagocytosisor chemokine production, have not been investigated in XLAneutrophils, so further studies are warranted.
The results of these studies appear to be in concert withthe clinical phenotype of XLA subjects, as patients main-tained on adequate Ig-replacement therapy do not haveinfections suggestive of innate immune defects[25,36,39,40]. While XLA patients may be neutropenic duringacute infections [27], which might suggest acceleratedapoptosis or increased susceptibility to stress, XLA neutro-phils displayed normally enhanced survival on culture withTLR ligands. However, it remains possible that sufficientreplacement Ig could ameliorate the outcomes of any Btkrelated TLR defects. Possibly in concert with this, Btk−/−mice challenged with LPS had defective neutrophil mediatedclearance, which was reversed by infusions with murine IgMnatural antibody [21].
A possible explanation for why Btk deficiency does nothave significant clinical effects outside the B cell compart-ment is that other members of the Tec kinase family mayable to compensate for Btk deficiency. The Xid and Btk−/−
mice have a much less severe phenotype than XLA, and onegroup has demonstrated this is due to compensatory activity
of the homologous Tec kinase [41]. Human neutrophils areknown to also express Tec as well as Bmx, another Tec kinasefamily member, and Btk, Tec and Bmx are often activated inconcert upon neutrophil stimulation. It is possible that thesekinases are able to compensate for the loss of Btk in XLAneutrophils [42]. We conclude that as for mast cells [31], Btkappears dispensable for essential TLR induced functions inneutrophils.
Acknowledgments
This work was supported by the National Institutes of Health,AI 101093, AI-467320, AI-48693, NIAID Contract 03-22, TheJeffrey Modell Foundation, and the David S GottesmanImmunology Chair. Baxter Healthcare also supports a studyon the demographics of immune deficiency in New York State.We thank Dr. M. E. Conley for genetic anaylses of the patients.
References
[1] M.E. Conley, B cells in patients with X-linked agammaglobulin-emia, J. Immunol. 134 (1985) 3070–3074.
[2] D. Vetrie, I. Vorechovsky, P. Sideras, J. Holland, A. Davies, F.Flinter, L. Hammarstrom, C. Kinnon, R. Levinsky, M. Bobrow,The gene involved in X-linked agammaglobulinaemia is amember of the src family of protein-tyrosine kinases.[seecomment][erratum appears in Nature 1993 Jul 22;364(6435):362], Nature 361 (1993) 226–233.
[3] M.E. Conley, Genetics of hypogammaglobulinemia: what do wereally know? Curr. Opin. Immunol. 21 (2009) 466–471.
[4] J.E. Farrar, J. Rohrer, M.E. Conley, Neutropenia in X-linkedagammaglobulinemia, Clin. Immunol. Immunopathol. 81 (1996)271–276.
[5] J.A. Winkelstein, M.C. Marino, H.M. Lederman, S.M. Jones, K.Sullivan, A.W. Burks, M.E. Conley, C. Cunningham-Rundles, H.D.Ochs, X-linked agammaglobulinemia: report on a United Statesregistry of 201 patients, Medicine 85 (2006) 193–202.
[6] V.Howard, J.M.Greene, S. Pahwa, J.A.Winkelstein, J.M. Boyle,M.Kocak, M.E. Conley, The health status and quality of life of adultswith X-linked agammaglobulinemia, Clin. Immunol. 118 (2006)201–208.
[7] J.A. Winkelstein, M.E. Conley, C. James, V. Howard, J. Boyle,Adults with X-linked agammaglobulinemia: impact of diseaseon daily lives, quality of life, educational and socioeconomicstatus, knowledge of inheritance, and reproductive attitudes,Medicine 87 (2008) 253–258.
[8] Y. Qiu, H.J. Kung, Signaling network of the Btk family kinases,Oncogene 19 (2000) 5651–5661.
[9] S.L. Doyle, C.A. Jefferies, C. Feighery, L.A. O'Neill, Signaling byToll-like receptors 8 and 9 requires Bruton's tyrosine kinase,J. Biol. Chem. 282 (2007) 36953–36960.
[10] S.L. Doyle, C.A. Jefferies, L.A. O'Neill, Bruton's tyrosine kinaseis involved in p65-mediated transactivation and phosphoryla-tion of p65 on serine 536 during NFkappaB activation bylipopolysaccharide, J. Biol. Chem. 280 (2005) 23496–23501.
[11] M. Hasan, G. Lopez-Herrera, K.E. Blomberg, J.M. Lindvall, A.Berglof, C.I. Smith, L. Vargas, Defective Toll-like receptor 9-mediated cytokine production in B cells from Bruton's tyrosinekinase-deficient mice, Immunology 123 (2008) 239–249.
[12] C.A. Jefferies, S. Doyle, C. Brunner, A. Dunne, E. Brint, C.Wietek, E. Walch, T. Wirth, L.A. O'Neill, Bruton's tyrosinekinase is a Toll/interleukin-1 receptor domain-binding proteinthat participates in nuclear factor kappaB activation by Toll-like receptor 4, J. Biol. Chem. 278 (2003) 26258–26264.

80 T.U. Marron et al.
[13] H. Kumar, T. Kawai, S. Akira, Pathogen recognition in theinnate immune response, Biochem. J. 420 (2009) 1–16.
[14] K.G. Lee, S. Xu, E.T. Wong, V. Tergaonkar, K.P. Lam, Bruton'styrosine kinase separately regulates NFkappaB p65RelA activa-tion and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells, J. Biol. Chem. 283 (2008) 11189–11198.
[15] M. Liljeroos, R. Vuolteenaho, S. Morath, T. Hartung, M.Hallman, M. Ojaniemi, Bruton's tyrosine kinase together withPI 3-kinase are part of Toll-like receptor 2 multiproteincomplex and mediate LTA induced Toll-like receptor 2responses in macrophages, Cell. Signal. 19 (2007) 625–633.
[16] A. Mangla, A. Khare, V. Vineeth, N.N. Panday, A. Mukhopadhyay,B. Ravindran, V. Bal, A.George, S. Rath, Pleiotropic consequencesof Bruton tyrosine kinase deficiency in myeloid lineages lead topoor inflammatory responses, Blood 104 (2004) 1191–1197.
[17] N.W. Schmidt, V.T. Thieu, B.A. Mann, A.N. Ahyi, M.H. Kaplan,Bruton's tyrosine kinase is required for TLR-induced IL-10production, J. Immunol. 177 (2006) 7203–7210.
[18] K. Sochorova, R. Horvath, D. Rozkova, J. Litzman, J.Bartunkova, A. Sediva, R. Spisek, Impaired Toll-like receptor8-mediated IL-6 and TNF-alpha production in antigen-present-ing cells from patients with X-linked agammaglobulinemia,Blood 109 (2007) 2553–2556.
[19] H. Taneichi, H. Kanegane, M.M. Sira, T. Futatani, K. Agematsu, M.Sako, H. Kaneko, N. Kondo, T. Kaisho, T. Miyawaki, Toll-likereceptor signaling is impaired in dendritic cells from patients withX-linkedagammaglobulinemia, Clin. Immunol. 126 (2008) 148–154.
[20] S. Mukhopadhyay, M. Mohanty, A. Mangla, A. George, V. Bal, S.Rath, B. Ravindran, Macrophage effector functions controlled byBruton's tyrosine kinase aremore crucial than the cytokine balanceof T cell responses for microfilarial clearance, J. Immunol. 168(2002) 2914–2921.
[21] R.R. Reid, A.P. Prodeus, W. Khan, T. Hsu, F.S. Rosen, M.C.Carroll, Endotoxin shock in antibody-deficient mice: unravelingthe role of natural antibody and complement in the clearanceof lipopolysaccharide, J. Immunol. 159 (1997) 970–975.
[22] N.J. Horwood, T.H. Page, J.P.McDaid, C.D. Palmer, J. Campbell,T. Mahon, F.M. Brennan, D. Webster, B.M. Foxwell, Bruton'styrosine kinase is required for TLR2 and TLR4-induced TNF, butnot IL-6, production, J. Immunol. 176 (2006) 3635–3641.
[23] N. Kadowaki, S. Ho, S. Antonenko, R.W. Malefyt, R.A.Kastelein, F. Bazan, Y.J. Liu, Subsets of human dendritic cellprecursors express different toll-like receptors and respond todifferent microbial antigens, J. Exp. Med. 194 (2001) 863–869.
[24] F. Hayashi, T.K. Means, A.D. Luster, Toll-like receptors stimulatehuman neutrophil function, Blood 102 (2003) 2660–2669.
[25] A. Singh, K.A. Zarember, D.B. Kuhns, J.I. Gallin, Impairedprimingand activation of the neutrophil NADPH oxidase in patients withIRAK4 or NEMO deficiency, J. Immunol. 182 (2009) 6410–6417.
[26] G. Bouma, R. Doffinger, S.Y. Patel, E. Peskett, J.C. Sinclair, G.Barcenas-Morales, L. Cerron-Gutierrez, D.S. Kumararatne, E.G.Davies, A.J. Thrasher, S.O. Burns, Impaired neutrophil migra-tion and phagocytosis in IRAK-4 deficiency, Br. J. Haematol.147 (2009) 153–156.
[27] C. Kozlowski, D.I. Evans, Neutropenia associated with X-linkedagammaglobulinaemia, J. Clin. Pathol. 44 (1991) 388–390.
[28] A. Lee, M.K. Whyte, C. Haslett, Inhibition of apoptosis andprolongation of neutrophil functional longevity by inflamma-tory mediators, J. Leukoc. Biol. 54 (1993) 283–288.
[29] C. Elbim, G. Lizard, Flow cytometric investigation of neutrophiloxidative burst and apoptosis in physiological and pathologicalsituations, Cytom. A 75 (2009) 475–481.
[30] J.B. Klein, M.J. Rane, J.A. Scherzer, P.Y. Coxon, R. Kettritz,J.M. Mathiesen, A. Buridi, K.R. McLeish, Granulocyte-macro-phage colony-stimulating factor delays neutrophil constitutiveapoptosis through phosphoinositide 3-kinase and extracellularsignal-regulated kinase pathways, J. Immunol. 164 (2000)4286–4291.
[31] C.N. Zorn, S. Keck, R.W. Hendriks, M. Leitges, M.A. Freuden-berg, M. Huber, Bruton's tyrosine kinase is dispensable for theToll-like receptor-mediated activation of mast cells, Cell.Signal. 21 (2009) 79–86.
[32] N.J. Horwood, T.Mahon, J.P.McDaid, J. Campbell, H.Mano, F.M.Brennan, D. Webster, B.M. Foxwell, Bruton's tyrosine kinase isrequired for lipopolysaccharide-induced tumor necrosis factoralpha production, J. Exp. Med. 197 (2003) 1603–1611.
[33] H. Jyonouchi, L. Geng, G.A. Toruner, K. Vinekar, D. Feng, P.Fitzgerald-Bocarsly, Monozygous twins with a microdeletionsyndrome involving BTK, DDP1, and two other genes; evidenceof intact dendritic cell development and TLR responses, Eur. J.Pediatr. 167 (2008) 317–321.
[34] R. Perez de Diego, E. Lopez-Granados, M. Pozo, C. Rodriguez, P.Sabina, A. Ferreira, G. Fontan, M.C. Garcia-Rodriguez, S.Alemany, Bruton's tyrosine kinase is not essential for LPS-inducedactivation of human monocytes, J. Allergy Clin. Immunol. 117(2006) 1462–1469.
[35] D.B. Kuhns, D.A. Long Priel, J.I. Gallin, Loss of L-selectin(CD62L) on human neutrophils following exudation in vivo, Cell.Immunol. 164 (1995) 306–310.
[36] H. von Bernuth, C. Picard, Z. Jin, R. Pankla, H. Xiao, C.L. Ku, M.Chrabieh, I.B. Mustapha, P. Ghandil, Y. Camcioglu, J. Vascon-celos, N. Sirvent, M. Guedes, A.B. Vitor, M.J. Herrero-Mata, J.I.Arostegui, C. Rodrigo, L. Alsina, E. Ruiz-Ortiz, M. Juan, C.Fortuny, J. Yague, J. Anton, M. Pascal, H.H. Chang, L. Janniere,Y. Rose, B.Z. Garty, H. Chapel, A. Issekutz, L. Marodi, C.Rodriguez-Gallego, J. Banchereau, L. Abel, X. Li, D. Chaussabel,A. Puel, J.L. Casanova, Pyogenic bacterial infections in humanswith MyD88 deficiency, Science 321 (2008) 691–696.
[37] K. Aomatsu, T. Kato, H. Fujita, F. Hato, N. Oshitani, N. Kamata,T. Tamura, T. Arakawa, S. Kitagawa, Toll-like receptor agonistsstimulate human neutrophil migration via activation ofmitogen-activated protein kinases, Immunology 123 (2008)171–180.
[38] I. Sabroe, S.K. Dower, M.K. Whyte, The role of Toll-likereceptors in the regulation of neutrophil migration, activa-tion, and apoptosis, Clin. Infect. Dis. 41 (Suppl 7) (2005)S421–S426.
[39] T.W. Kim, K. Staschke, K. Bulek, J. Yao, K. Peters, K.H. Oh, Y.Vandenburg, H. Xiao, W. Qian, T. Hamilton, B. Min, G. Sen, R.Gilmour, X. Li, A critical role for IRAK4 kinase activity in Toll-likereceptor-mediated innate immunity, J. Exp. Med. 204 (2007)1025–1036.
[40] J.S. Orange, A. Jain, Z.K. Ballas, L.C. Schneider, R.S. Geha, F.A.Bonilla, The presentation and natural history of immunodefi-ciency caused by nuclear factor kappaB essential modulatormutation, J. Allergy Clin. Immunol. 113 (2004) 725–733.
[41] W. Ellmeier, S. Jung, M.J. Sunshine, F. Hatam, Y. Xu, D.Baltimore, H. Mano, D.R. Littman, Severe B cell deficiency inmice lacking the tec kinase family members Tec and Btk, J.Exp. Med. 192 (2000) 1611–1624.
[42] G. Lachance, S. Levasseur, P.H. Naccache, Chemotactic factor-induced recruitment and activation of Tec family kinases inhuman neutrophils. Implication of phosphatidynositol 3-kinases, J. Biol. Chem. 277 (2002) 21537–21541.


















