
The spreading depression theory - Semantic Scholar · 2017-10-19 · noxious stimuli. Finally,...
12
Brain (1994), 117, 199-210 REVIEW ARTICLE Pathophysiology of the migraine aura The spreading depression theory Martin Lauritzen Laboratory of Clinical Neurophysiology, Rigshospitalet and Medical Physiological Institute, Panum Institute, University of Copenhagen, Denmark Correspondence to: Martin Lauritzen, Medical Physiological Institute, Panum Institute, Blegdamsvej 3, DK-22OO Copenhagen, Denmark Summary The characteristic form and development of sensory disturbances during migraine auras suggests that the underlying mechanism is a disturbance of the cerebral cortex, probably the cortical spreading depression (CSD) of Leio. The demonstration of unique changes of brain blood flow during attacks of migraine with aura, which have been replicated in animal experiments during CSD, constitutes another important line of support for the 'spreading depression' theory, which may be a key to an understanding of the migraine attack. Cortical spreading depression is a short-lasting depolarization wave that moves across the cortex at a rate of 3—5 mm/min. A brief phase of excitation heralds the reaction which is immediately followed by prolonged nerve cell depression synchronously with a dramatic failure of brain ion homeostasis, efflux of excitatory amino acids from nerve cells and enhanced energy metabolism. Recent experimental work has shown that CSD in the neocortex of a variety of species including man is dependent on activation of a single receptor, the N-methyl-D-aspartate receptor, one of the three subtypes of glutamate receptors. The combined experimental and clinical studies point to fruitful areas in which to look for migraine treatments of the future and provide a framework within which important aspects of the migraine attack can be modelled. Key words: migraine; cerebral blood flow; spreading cortical depression; glutamate; nitric oxide The theories The migraine aura may be defined as any neurological disturbance that appears shortly before or during the development of a migraine headache. Seemingly similar migraine auras may have different features, suggesting involvement of different brain regions. The headache is most often throbbing and unilateral, on the side of the head relevant for the focal symptoms (Wolff, 1963; Olesen et al., 1990). To explain this sequence of events Wolff (1963) suggested that the focal symptoms were due to transient constriction of a cerebral artery and the headache to a sterile inflammatory reaction around the walls of dilated cephalic vessels. Several clinical arguments were in favour of the vascular concept. First, the pulsating quality of the pain in migraine and the existence of many other headache syndromes secondary to vascular disease such as stroke, subarachnoid haemorrhage, arterial hypertension and temporal arteritis indicated a vascular mechanism. Secondly, vascular headaches were effectively treated with ergotamine which constricts cerebral arteries without decreasing cerebral blood flow (Tfelt-Hansen et al., 1991). Thirdly, extensive work on the localization of intracranial pain sources pointed towards the blood vessels and associated structures at the ventral surface of the brain as most pain- sensitive, while the brain parenchyma itself was insensitive to noxious stimuli. Finally, inhalation of gas mixtures containing 10% carbon dioxide and 90% oxygen prevented headache and aborted the aura, supposedly due to vasodilatation and increased oxygen supply to the region suffering from vasospasm. The functional anatomical basis of vascular headaches is most likely the trigeminovascular system and there is little doubt that the source of pain in migraine resides within the blood vessels (Moskowitz, 1992; Goadsby and Edvinsson, 1993). The vascular theory has much merit with respect to the headache, but cannot explain all the phenomena which are associated with the migraine aura. In particular, the stereotyped recurrence of an unprovoked, transient disturbance of brain function is reminiscent of epilepsy more than of any known © Oxford University Press 1994 at Aston University on January 15, 2014 http://brain.oxfordjournals.org/ Downloaded from
Transcript of The spreading depression theory - Semantic Scholar · 2017-10-19 · noxious stimuli. Finally,...

Brain (1994), 117, 199-210
REVIEW ARTICLE
Pathophysiology of the migraine auraThe spreading depression theory
Martin Lauritzen
Laboratory of Clinical Neurophysiology, Rigshospitalet andMedical Physiological Institute, Panum Institute, Universityof Copenhagen, Denmark
Correspondence to: Martin Lauritzen, MedicalPhysiological Institute, Panum Institute, Blegdamsvej 3,DK-22OO Copenhagen, Denmark
SummaryThe characteristic form and development of sensory disturbancesduring migraine auras suggests that the underlying mechanismis a disturbance of the cerebral cortex, probably the corticalspreading depression (CSD) of Leio. The demonstration ofunique changes of brain blood flow during attacks of migrainewith aura, which have been replicated in animal experimentsduring CSD, constitutes another important line of support forthe 'spreading depression' theory, which may be a key to anunderstanding of the migraine attack. Cortical spreadingdepression is a short-lasting depolarization wave that movesacross the cortex at a rate of 3—5 mm/min. A brief phase ofexcitation heralds the reaction which is immediately followed
by prolonged nerve cell depression synchronously with adramatic failure of brain ion homeostasis, efflux of excitatoryamino acids from nerve cells and enhanced energy metabolism.Recent experimental work has shown that CSD in the neocortexof a variety of species including man is dependent on activationof a single receptor, the N-methyl-D-aspartate receptor, oneof the three subtypes of glutamate receptors. The combinedexperimental and clinical studies point to fruitful areas in whichto look for migraine treatments of the future and provide aframework within which important aspects of the migraine attackcan be modelled.
Key words: migraine; cerebral blood flow; spreading cortical depression; glutamate; nitric oxide
The theoriesThe migraine aura may be defined as any neurologicaldisturbance that appears shortly before or during thedevelopment of a migraine headache. Seemingly similarmigraine auras may have different features, suggestinginvolvement of different brain regions. The headache is mostoften throbbing and unilateral, on the side of the head relevantfor the focal symptoms (Wolff, 1963; Olesen et al., 1990).
To explain this sequence of events Wolff (1963) suggestedthat the focal symptoms were due to transient constriction ofa cerebral artery and the headache to a sterile inflammatoryreaction around the walls of dilated cephalic vessels.
Several clinical arguments were in favour of the vascularconcept. First, the pulsating quality of the pain in migraine andthe existence of many other headache syndromes secondary tovascular disease such as stroke, subarachnoid haemorrhage,arterial hypertension and temporal arteritis indicated a vascularmechanism. Secondly, vascular headaches were effectivelytreated with ergotamine which constricts cerebral arteries
without decreasing cerebral blood flow (Tfelt-Hansen et al.,1991). Thirdly, extensive work on the localization of intracranialpain sources pointed towards the blood vessels and associatedstructures at the ventral surface of the brain as most pain-sensitive, while the brain parenchyma itself was insensitive tonoxious stimuli. Finally, inhalation of gas mixtures containing10% carbon dioxide and 90% oxygen prevented headache andaborted the aura, supposedly due to vasodilatation and increasedoxygen supply to the region suffering from vasospasm. Thefunctional anatomical basis of vascular headaches is most likelythe trigeminovascular system and there is little doubt that thesource of pain in migraine resides within the blood vessels(Moskowitz, 1992; Goadsby and Edvinsson, 1993).
The vascular theory has much merit with respect to theheadache, but cannot explain all the phenomena which areassociated with the migraine aura. In particular, the stereotypedrecurrence of an unprovoked, transient disturbance of brainfunction is reminiscent of epilepsy more than of any known
© Oxford University Press 1994
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

200 M. Lauritzen
disorder of the peripheral vasculature (Gowers, 1907). The focalsymptoms in migraine develop in a characteristic 'creeping'fashion, commonly used to differentiate migraine from epilepsy.Several investigators have described their own migraine aura.Lashley's scintillation-scotomas developed symmetrically in thevisual fields, suggesting a cortical localization of the symptoms(Lashley, 1941). The disturbance started at the visual field centreand propagated to the peripheral (temporal) parts within~ 10-15 min, while function returned to normal within another10-15 min. The aura symptoms indicated a wave of intenseexcitation in the primary visual cortex that moved at the speedof 3 mm/min, followed by a longer period of inhibition(Lashley, 1941) (Fig. 1). Similar calculations can be madewith respect to somatosensory symptoms developing along thesensory homunculus (Lord, 1986). The very orderlydevelopment of the aura makes a vascular origin a remotepossibility, while a primary disturbance of cortical nerve cellfunction, probably cortical spreading depression (CSD) is amore attractive explanation (Leao and Morison, 1945; Milner,1958).
Cortical spreading depression in animals, including monkeys,gives rise to specific changes of behaviour which mimicimportant features of the migraine aura (BureS et al., 1974,1984). The rat is the most popular animal for studies ofbehaviour in which single unilateral waves of CSD inducecontralateral sensory neglect and motor impairment of theforepaw lasting for 15—30 min, i.e. much shorter than the bloodflow reduction (BureS et al., 1984; Lauritzen, 1987a). Rats donot experience hippocampal or CSD as aversive (Koroleva andBures, 1993). The analogy can hardly be taken further due tothe obvious differences between rat and human brains, but itis important that the behavioural changes caused by CSD inanimals are consistent with the transient neurological deficitsrecorded during the migraine aura.
This review summarizes clinical and experimental data whichsuggest that the development of a CSD in migraine patientstriggers the aura, the changes of cerebral blood flow andpossibly migraine headaches. The key to this theory of migraineis an understanding of the mechanisms of brain blood flowchanges during attacks of migraine as summarized in thefollowing.
The 'spreading oligaemia': arterial or arteriolarvasospasm?Methods for the measurement of regional cerebral blood flow(rCBF) in man with a high spatial resolution are well-established(Sveinsdottir et al., 1977; Celsis et al., 1981; Lauritzen, 1987a).Studies during acute migraine attacks, provoked by angiographyor occurring spontaneously, have revealed unique changes ofrCBF which were not observed in > 1,000 patients with othercategories of neurological disorders examined with the sametypes of equipment (Olesen et al., 1981; Lauritzen, 1987a;Olesen, 1991). The migraine symptoms which developed duringthe provoked attacks were similar, but not identical to thesymptoms which developed during spontaneous attacks.
V3
Fig. 1 Successive maps of a scintillation-scotoma to showcharacteristic distribution of the fortification figures. In each casethe asterisk indicates the fixation point. Knowledge of theretinotopic organization of the visual cortex allowed Lashley(1941) to calculate the speed of propagation of theexcitation—depression wave as ~3 mm/min. Reproduced withpermission from K. M. Lashley (published in Archives ofNeurology and Psychiatry ; 46: 331-9; © American MedicalAssociation 1941)
Therefore, the pathophysiology of induced and spontaneousattacks may be different. On the other hand, the disparities ofsymptoms between the two types of attacks were felt to be minorand it was therefore assumed that the changes of rCBF duringprovoked and spontaneous attacks of migraine were similar.The data from the studies in Copenhagen including both patientswith induced and spontaneous attacks of migraine aresummarized in the following, while the results of earlier studieshave been reviewed in detail in other recent publications (Olesenetal., 1981, 1990; Lauritzen, \9%la,b; Olesen, 1991).
At the very beginning of migraine attacks rCBF decreasesin the posterior part of the brain. Subsequently, the low flowregion spreads into the parietal and temporal lobes at rate of2 - 3 mm/min for the next 30-60 min to various extent inindividual patients, the so-called 'spreading oligaemia' (Olesenet al., 1981). The spread of reduced rCBF does not match theterritories of supply of large arteries, but follows the corticalsurface. Arterial vasospasm is therefore not the mechanism ofreduced flow in migraine, which appears to be due to arteriolarvasoconstriction. The rCBF remains constant in hypo- as wellas normoperfused brain regions despite variations of the meanarterial blood pressure, i.e. autoregulation is preserved. Thisis another strong argument against arterial vasospasm as themechanism of reduced flow, and an equally strong argumentfor cortical arterioles as the site of increased resistance.Vasospasm of a large artery would cause compensatorydilatation of the arterioles if severe enough to reduce rCBF.In this situation, increased blood pressure would lead to anincrease of rCBF in the oligaemic region because ofautoregulatory dilatation of the cortical arterioles, but inmigraine rCBF remained constant. Therefore, the reduced rCBFin migraine is probably due to arteriolar, not arterialvasoconstriction.
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

Migraine and spreading depression 201
Vascular reactivity to mental tasks and changes of arterialCO2 is impaired in the hypoperfused parts of the brain, butnormal in neighbouring non-invaded regions. Thus, there is noevidence of vascular dysfunction before the blood flowdecreases. The effects of hypercapnic hyperoxia on migrainesymptoms (Wolff, 1963) are not explained by dilatation ofresistance vessels, since rCBF in hypoperfused regions isinsensitive to changes of PaCO2 during migraine attacks. TherCBF changes are consistent with a process propagating at arate of 2 —3 mm/min, while constricting pial and corticalarterioles, i.e. the vessels which are imbedded in and stronglyinfluenced by changes of the local neuronal microenvironment(Lauritzen, 1987a).
The 'spreading oligaemia': relation tosymptomsDuring migraine attacks provoked by angiography, the aurasymptoms usually appeared at some time point during the earlyphase of spread of the oligaemia, lasted for 15 — 30 min andthen ceased while the hypoperfusion continued to spread whilethe patients developed migraine headaches (Olesen et al., 1981,1990; Lauritzen, 1987a).
The studies of spontaneous attacks confirmed that corticalblood flow was reduced during the initial part of the headacheperiod after the aura symptoms had altogether vanished.Eventually, after ~ 2 - 6 h, patchy regions of increased flowsometimes developed in cortical regions which were previouslyhypoperfused (Lauritzen, 1987a; Andersen et al., 1988).Interestingly, the side of the headache usually corresponded tothe side of the vascular changes (Olesen ex al., 1990) suggestingthat the process which triggered the changes of rCBF alsostimulated the vascular nociceptors. The average rCBFreduction amounts to - 2 0 - 2 5 % , with only small areas ofischaemia in the apparently homogeneously perfused oligaemicregion, suggesting that ischaemia is not of primary importancein the development of focal migraine symptoms. In conclusion,the time base of the focal symptoms and the reduced rCBF areclearly different. This is a strong argument against a simpleversion of the 'vascular' theory of migraine. The focalneurological symptoms develop while the 'spreading oligaemia'expands, but the flow reduction is usually moderate and doesnot explain the focal symptoms. On the other hand, the datado not exclude that a critically low rCBF, under certainconditions, may contribute to a persistent neurological deficit.The headache in turn is associated with decreased, normal orincreased rCBF and slightly dilated cerebral arteries, which mayconstrict in response to anti-migraine medication (Olesen et al.,1990; Friberg et al., 1991).
Compton scatter and rCBF during migraineattacksIt has been claimed that brain ischaemia is the primary causeof neurological deficits during classic migraine attacks, and that
rCBF in the hypoperfused regions is overestimated due toCompton scattered radiation from 133Xe in the neighbouringnormoperfused regions (Skyhej Olsen et al., 1987; SkyhajOlsen and Lassen, 1989). Dissipation of energy from l33Xe inorganic tissue occurs particularly from Compton scatter, causingchange of the direction and energy of the emitted photon. TheCompton scatter has almost the same energy as the primaryradiation. Therefore, it is not possible to discriminate accuratelythe primary radiation from Compton scatter, and the radiationfrom every tissue element is influenced by its surroundings.This source of error is specially important in regions with lowisotope distribution (low flow) since scatter from adjacentnormally perfused tissue influences the radiation and accord-ingly the steepness of the clearance curves, leading to anoverestimation of rCBF. The controversy of the importance ofCompton scatter for rCBF measurements with 133Xe has beena major issue in the ongoing discussion of migraine patho-physiology and will be discussed here in some detail.
Skyhoj Olsen et al. (1987, 1989) reanalysed the rCBF datafrom all the migraine patients who had been investigated withthe 133Xe intracarotid technique. The purpose was to estimatethe degree of rCBF reduction when taking Compton scatterinto consideration. They selected three rCBF values eachrepresenting a value of one of the 254 channels: the highestrCBF value 'closest to' the non-affected region, the lowest rCBFvalue 'furthest away' from the non-affected region and the rCBFin the 'centre' of the low-flow region. The correlations betweeneach of the three sets of rCBF values and the mean rCBF ofthe non-affected region were then calculated. The correlationcoefficients were significantly different from zero and apparentlygrowing with increasing distance from the non-affected region.This led the authors to conclude that the lowest value of rCBFin the low-flow region represented the 'true value' of rCBF,as the values closer to the non-affected region were moreinfluenced by Compton scatter as indicated by a highercorrelation coefficient.
There are serious problems with this type of analysis, inparticular with respect to the method of data selection (Kronborget al., 1990). The rCBF value of the region 'closest to' the non-affected region was selected as the highest value in one singlechannel of the low-flow region and represents neither a typicalrCBF value nor the median or mean value of a group or rowof detectors aligned in the border zone between 'non-affected'and 'affected' regions. Similarly, the lowest rCBF of the low-flow region was selected as the lowest value in one singlechannel of the low-flow region 'furthest away' from the 'non-affected' region. Clearly, the selection procedure was the basisfor the variations of correlation coefficients observed, whichcould not be used as arguments for uniformity of rCBF in thelow-flow region. In addition, there were geometrical problemsrelated to defining areas such as 'closest to', 'centre of and'furthest away' from the boundary of a 'non-affected' regionof a three-dimensional structure, the brain, with equipment thatmeasures rCBF along two axes only. First, the distance fromthe low-flow region to the non-affected region was not the samein all patients, since the size of the low-flow region varied
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

202 M. Lauritzen
between 9 and 84 cm2. Secondly, it is also important that thelow-flow region only exceptionally conformed to any well-defined geometrical figure and that the form of the low-flowregion varied between measurements. Thus, the metaspatialscale 'closest to', 'centre' and 'furthest away' had to be regardedwith the highest degree of scepticism. Thirdly, the strongpositive correlation between rCBF in the low-flow regions andrCBF in the 'non-affected' regions were partly due to individualrCBF levels for the patients. The correlation coefficientsdecreased considerably when this was taken into account andhuge variations of the correlation pattern were observed betweenpatients. This made the interpretation of the calculatedpopulation correlations very difficult (Kronborg et al., 1990).Fourthly, an examination of the original data of six of the 11patients revealed no anterior-to-posterior gradient of rCBFvalues as expected from the papers by Skyhaj Olsen et al.(1987, 1989). Therefore, it was doubted that this kind ofstatistical analysis had any prospect of contributing to ourunderstanding of the role of Compton scatter when using 133Xefor measurements of rCBF (Kronborg et al., 1990).
In a subsequent paper, a mathematical model was suggestedto explain the impact of Compton scattered radiation for themeasured rCBF (Skyhaj Olsen and Lassen, 1989). The mainconclusion of the paper was that the 'spreading oligaemia'resulted from Compton scatter radiating from a normallyperfused region to a region of increasingly reduced rCBF, i.e.the spread of the 'spreading oligaemia' was apparent, not real.The basis for the choice of equation was, however, difficultto define and very different conclusions could be reached bysmall alterations of the (arbitrary) constants used in the equations(Dalgaard et al., 1991). Alternative mathematical models gavedifferent results suggesting that this approach was of very limitedutility for the interpretation of the migraine data (Dalgaardet al., 1991). Taken together, there is no doubt that Comptonscatter leads to an overestimation of rCBF in low-flow regions,in migraine patients and in patients with other neurologicaldiseases. We are, however, ignorant of the magnitude of thecorrection factor and it is not possible at the moment to makeany categorical conclusion with respect to the importance ofischaemia for migraine. However, it should be clear thatischaemia during migraine attacks is the exception, not the rule,and in most migraine patients the benign nature of the diseaseis reflected in a mild blood flow reduction (Kronborg et al.,1990; Dalgaard et al., 1991).
At this point I will return to discuss the physiological basisof the 'spreading oligaemia' which has a number of importantfeatures in common with Leao's CSD.
Cortical spreading depression: basic featuresThe literature concerning CSD is overwhelming, but a numberof reviews cover all but the most recent studies (BureS et al.,1974; Somjen, 1979; Nicholson and Kraig, 1981; Martins-Ferreira, 1984; Do Carmo, 1992; Somjen et al., 1992;Lehmenkuhler et al., 1993). The reaction was first identified
in rabbit cerebral cortex (Leao, 1944a,*). The basic observationwas that the EEG following mild noxious stimuli would becomecompletely extinguished for a minute or so and that thedepression would propagate very slowly across a wide corticalregion.
Cortical spreading depression has been induced in most greymatter regions studied so far, e.g. in the cortex, thehippocampus and the cerebellum of a variety of species (BureSet al., 1974). It has been observed in human cortical tissue invitro (Avoli et al., 1991), and in human hippocampus andstriatum in vivo (Sramka et al., 1977/8). Thus, human corticaltissues do support the development of CSD, but a recordingof CSD from the human neocortex in vivo is still missing (Gloor,1986).
Successful elicitation of CSD in experiments depends on thesusceptibility of the tissue and the trigger factor involved.Hypoglycaemia and hypoxia lower the threshold (BureS et al.,1974). Common methods of triggering CSD include localelectrical or mechanical stimulation or injections of highconcentrations of KC1. Potassium plays a central role for CSDand it is reasonable to assume that any disturbance of K+
homeostasis would predispose the brain region to CSD(Grafstein, 1963). Brain K+ clearance systems are heavilydependent on the capacity of glial cells (Nicholson and Kraig,1981). In humans the lowest glial —neuronal cell ratio is in theprimary visual cortex (Bailey and von Bonin, 1951). Therefore,one would expect human CSD to be initiated occipitally. Asis well known, visual auras are indeed very frequent in migraine(Olesen et al., 1990).
Neurons and glial cells depolarize during CSD, giving riseto an intense, but transient spike activity (seconds) when thereaction enters the tissue (Sugaya et al., 1975). Neuronal silenceimmediately follows, lasting for a few minutes, but evokedpotentials usually take a longer time to recover, 15-30 min(BureS et al., 1974). This sequence of brief excitation fol-lowed by a short-lasting depression is supposed to be theneurophysiological basis of the sensory symptoms duringmigraine auras (Leao and Morison, 1945; Milner, 1958;Gardner-Medwin, 1981; Lord, 1986; Lauritzen, 1987a,*).
The depolarization is associated with dramatic changes in thedistribution of ions between the intra- and extracellularcompartments: K+ and hydrogen ions leaves the cells, whileNa+ , Ca2+ and Cl" enters together with water, as the size ofthe extracellular space decreases to approximately half of thecontrol values (Fig. 2) (Nicholson and Kraig, 1981; Hansen,1985). A return to normal of most ion concentrations and ofthe size of the extracellular space occur spontaneously after 3 0 -60 s, whereas Ca2+ and pH usually take a few more minutesto recover. There is, at the moment, no satisfactory explanationof the spreading mechanism of CSD, but the spread probablyinvolves the diffusion of one or more chemical mediators, mostlikely K+ and glutamate, into the extracellular compartment(Nicholson, 1993). It has been suggested that a calcium wavein glial cells underlies CSD, but this still remains to be proven(Leibowitz, 1992). A simplistic scheme of the mechanism of
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from
Migraine and spreading depression
(mM)
203
log [carl . M
-1
-2
-3
- 4
- 7
- « L
J 008
20 nV
unitact.
1 rin
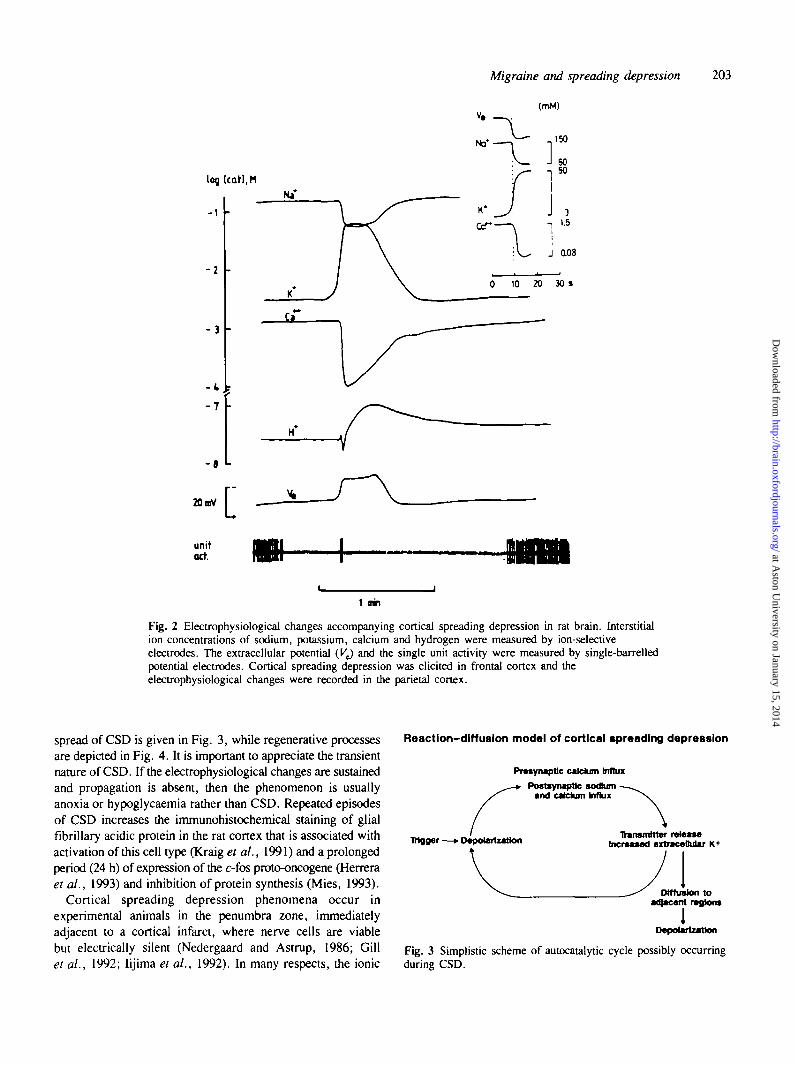
Fig. 2 Electrophysiological changes accompanying cortical spreading depression in rat brain. Interstitialion concentrations of sodium, potassium, calcium and hydrogen were measured by ion-selectiveelectrodes. The extracellular potential {VJ and the single unit activity were measured by single-barrelledpotential electrodes. Cortical spreading depression was elicited in frontal cortex and theelectrophysiological changes were recorded in the parietal cortex.
spread of CSD is given in Fig. 3, while regenerative processesare depicted in Fig. 4. It is important to appreciate the transientnature of CSD. If the electrophysiological changes are sustainedand propagation is absent, then the phenomenon is usuallyanoxia or hypoglycaemia rather than CSD. Repeated episodesof CSD increases the immunohistochemical staining of glialfibrillary acidic protein in the rat cortex that is associated withactivation of this cell type (Kraig et al., 1991) and a prolongedperiod (24 h) of expression of the c-fos proto-oncogene (Herreraet al., 1993) and inhibition of protein synthesis (Mies, 1993).
Cortical spreading depression phenomena occur inexperimental animals in the penumbra zone, immediatelyadjacent to a cortical infarct, where nerve cells are viablebut electrically silent (Nedergaard and Astrup, 1986; Gillet al., 1992; Iijima et al., 1992). In many respects, the ionic
Reaction-diffusion model of cortical spreading depression
Presynapttc calcium Influx
Postsynapttc socftumand calcium Influx
retoaseIncreased extracellular K+Trigger—• Depolarization
Diffusion toadjacent regions
1Depolarization
Fig. 3 Simplistic scheme of autocatalytic cycle possibly occurringduring CSD.
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

204 M. Lauritzen
Regenerative processes In cortical spreading depression
Depolarization
Transmitterrelease
Transmitterreabsofptlon
IncreasedextraceUar K+
Q MrhtpotBiinlhm
UUTrvnl rTl#qiraOK+dspersai
Increasedhtracelular Na+
Increased H»+l K+
ATPase acthrlty
Increased energymetabotsm
Fig. 4 Simplistic scheme of regenerative processes assumed tooccur during CSD.
disequilibria during CSD resemble transient ischaemia, but thereis usually no shortage of energy supply during CSD (Lauritzenet al., 1990). These dramatic changes of neuronal function andion homeostasis are associated with profound changes of thelocal circulation.
Brain blood flow during CSD and migraine:precise communicationCortical blood flow may decrease before CSD or during theonset of depolarization, but the vasoconstriction is variable andusually brief (Lauritzen, 1987a). Propagation of CSD is totallyindependent of the vascular reaction: cortical, hippocampal,cerebellar and retinal nervous tissue support CSD perfectly wellin vitro (BureS et al., 1974; Martins-Ferreira, 1984; Do Carmo,1992). Therefore, it is questionable whether or not CSD setsup a cycle of potassium-induced vasoconstriction of importancefor its propagation (Young and Van Vliet, 1992).
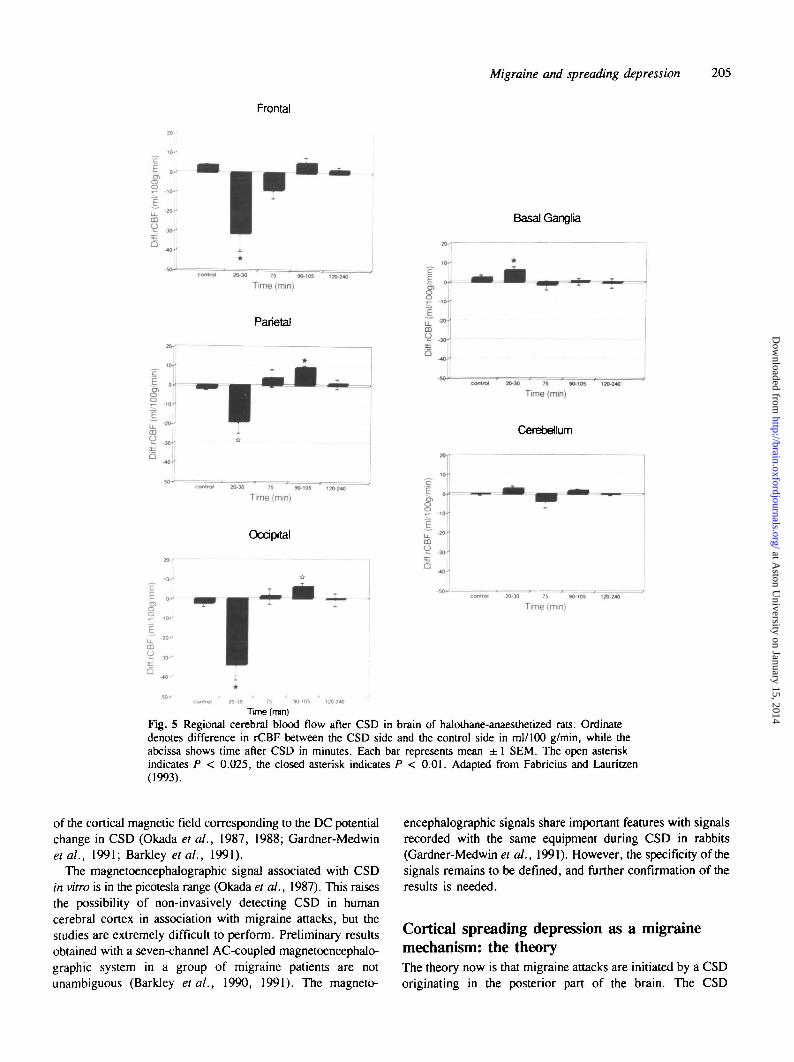
During return to normal of ionic changes, rCBF increasesby ~ 100% in anaesthetized animals (Leao, 1944ft; Lauritzen,1987a), simultaneously with the release of lactate to the braininterstitial fluid (Scheller et al., 1992). The flow rise lasts for1 - 2 min, being rapidly succeeded by a 20-30% reduction dueto cortical arteriolar vasoconstriction, while rCBF in non-invaded regions remains constant (Mies and Paschen, 1984;Lauritzen, 1987a; Kocher, 1990; Shibata et al., 1990;Duckrow, 1991; Piper et al., 1991; LaCombe et al., 1992).The hypoperfusion in rats persists for — 1 h. Blood pressureautoregulation of rCBF is preserved in the entire brain. Incontrast, the responsiveness to changes of PaCC^ (Lauritzen,1987a; Piper et al., 1991; Lacombe et al., 1992), basalforebrain stimulation (Lacombe, 1992) and vasoactivesubstances applied directly to the pial arterioles are markedlyimpaired in the CSD cortex (Wahl et al., 1987). At 90 minafter CSD, cortical rCBF increases transiently (Fig. 5)(Fabricius and Lauritzen, 1992).
Thus, CSD in rats may be associated with a brief initial flow
rise succeeded by a moderate but prolonged decrease of rCBFdue to pial arteriolar vasoconstriction. The reduced rCBF isassociated with preserved autoregulation and decreased vascularreactivity to chemical stimuli including CCv A brief, smallhyperperfusion follows. This pattern of rCBF changes comparesfavourably with the rCBF changes associated with attacks ofmigraine (Lauritzen, 1987a; Olesen, 1991).
Energy metabolism in migraine and spreadingdepressionOur knowledge of the changes of energy rich phosphatecompounds and oxygen and glucose metabolism duringmigraine attacks is minimal. This is due to the inherentdifficulties in investigating short-lasting paroxysmal disorderslike migraine, in which the disturbed metabolic function mayhave disappeared at the time the patient is referred to, or appearsat the clinic. A PET study has shown normal cerebral oxygenmetabolism in the hypoperfused regions of a patient withmigraine aura at the time of neurological deficits (Herold et al.,1985). The data suggest that ischaemia is not the cause of thefocal deficits in migraine. Following the first minutes of CSDin rats, the cerebral metabolic rate of glucose remains normaldespite the low cerebral blood flow (Lauritzen, 1987a). Byanalogy, the behavioural changes at this time point after CSDare also not caused by impaired glucose metabolism.
Magnetic resonance spectroscopy applied to migraine patientsat 3 -48 h after the beginning of the attack showed a decreaseof the phosphocreatine (PC) concentration, an increase ofinorganic phosphate, while ATP and intracellular pH remainedconstant (Welch et al., 1989). The absence of pH changes wastaken as argument against the 'vascular' theory since pH wasfound to be reduced in the affected region in almost all patientswith a stroke or a transient ischaemic attack (Welch et al.,1989). Intracellular pH during and following CSD is unknown,but PC decreases by - 3 8 % while ATP remains constant atthe CSD wave front (Lauritzen et al., 1990), concomitantly withan increase of cortical oxygen consumption which is matchedby a rise in blood flow (Mayevsky and Weiss, 1991). Thus,there are similar changes of PC and ATP during CSD andmigraine attacks, suggesting an increased turnover rate of thesecompounds, but no evidence of ischaemia.
The magnetoencephalographic experienceMagnetoencephalography is a technique by which the brain'selectrical activity is recorded by sensors of magnetic fields andnot by electrodes as the conventional EEG (Cohen and Cuffin,1983). Magnetoencephalography measures slow changes ofbrain electrical activity accurately, which is not possible by EEG(Okada et al., 1988). Cerebral spreading depression depolarizesthe cortex for ~ 1 min and changes the brains DC potential,the steady potential around which the EEG oscillates (Leao,1951). The major purpose of the ongoing magnetoencephalo-graphic research in migraine has been to record a slow change
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from
Migraine and spreading depression 205
Frontal
. - -10-'
1co
QV Basal Ganglia
30-30 75 90-105 120-240
Time (min)
Parietal
control 20-30 75 90-105 120-240
Time (min)
Cerebellum
20-30 75 90-105 120-240
Time (min)
Occiprtal
Icontrol 20-30 75 90-105 120-240
Time (mm)
Tune (mm)Fig. 5 Regional cerebral blood flow after CSD in brain of halothane-anaesthetized rats. Ordinatedenotes difference in rCBF between the CSD side and the control side in ml/100 g/min, while theabcissa shows time after CSD in minutes. Each bar represents mean ± 1 SEM. The open asteriskindicates P < 0.025, the closed asterisk indicates P < 0.01. Adapted from Fabricius and Lauritzen(1993).
of the cortical magnetic field corresponding to the DC potentialchange in CSD (Okada el al., 1987, 1988; Gardner-Medwinetal., 1991; Barkley et al., 1991).
The magnetoencephalographic signal associated with CSDin vitro is in the picotesla range (Okada et al., 1987). This raisesthe possibility of non-invasively detecting CSD in humancerebral cortex in association with migraine attacks, but thestudies are extremely difficult to perform. Preliminary resultsobtained with a seven-channel AC-coupled magnetoencephalo-graphic system in a group of migraine patients are notunambiguous (Barkley etal., 1990, 1991). The magneto-
encephalographic signals share important features with signalsrecorded with the same equipment during CSD in rabbits(Gardner-Medwin et al., 1991). However, the specificity of thesignals remains to be defined, and further confirmation of theresults is needed.
Cortical spreading depression as a migrainemechanism: the theoryThe theory now is that migraine attacks are initiated by a CSDoriginating in the posterior part of the brain. The CSD
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

206 M. Lauritzen
Fig. 6 Hypothesis of development of a migraine attack based onaspects of CSD and migraine, summarized in the text. Thefigures represent lateral views of the human brain at differenttime intervals after the start of the attack, spaced by —30 min.The dotted area represents the region of reduced rCBF, thestriped area represents the region of neuronal depolarizationduring the first minutes of CSD and the arrows show thedirection of progression of CSD. 1, Initially during a migraineattack a CSD is elicited at the occipital pole, spreading anteriorlyat the lateral, mesial, and ventral sides of the brain. At the CSDwave front, transient ionic and metabolic disequilibria triggerperturbed neuronal function, rCBF changes and neurologicalsymptoms. 2, Following CSD, cortical rCBF decreases by20-30% for 2 - 6 h. 3, rCBF in regions not invaded by CSDremains normal until encountered by CSD. 4, The region ofreduced rCBF expands as the CSD moves anteriorly. 5,Somatosensory symptoms from the extremities appear when theCSD invades the primary sensory cortex at the post-central gyrus.6, CSD usually stops on reaching the central sulcus, but in manypatients it does not even propagate this far. The ventral spread ofCSD causes activation of pain-sensitive fibers and headache. 7,Full-scale attack. The CSD has stopped and is now detectable asa persistent reduction of cortical rCBF. At this tune the patientsuffers from headache, but has no focal deficits. Reproduced withpermission from Elsivier Publications Cambridge (published inTrends in Neuroscience 1987; 10: 8-13).
progresses anteriorly with a constant speed of 2 - 3 mm/min,triggering the migraine aura at the wave front where the nervecells depolarize, and the long-lasting blood flow changes in itswake. The changes of rCBF in migraine are according to thisviewpoint, not the primary event, but secondary to CSD (Fig. 6)(Lauritzen, 1987*).
The generation of headache from intracranial sources requiresactivation of pain-sensitive fibres that are located at the ventralsurface of the brain. Recent data suggest that CSD activatesnociceptive fibres in rats (Moskowitz etal., 1993), butconscious rats do not experience CSD as an aversive stimulus
(Koroleva and Bures, 1993). The latency period between onsetof aura and headache may reflect the time it takes for the CSDto propagate from the occipital cortex to the pain triggering zone(Moskowitz, 1984), but the mechanism by which CSD mayinflict pain is still far from understood.
The aura and headache may represent separate effects of CSDon different brain structures-the cortex and the nociceptivevascular afferents. This may explain the variations of migrainesymptoms observed in the same patient and also in differentpatients. As is well known, headaches are observed quitefrequently following epileptic attacks (Schon and Blau, 1987),which are associated with marked changes of the neuronalmicroenvironment similar to, but less profound than those foundduring CSD (Somjen, 1979; Avoli etal., 1991)
Some research perspectives in experimentalmigraine researchIt appears that CSD displays sufficiently important similaritiesto the migraine attack that it can be considered as a diseasemodel. The recent development of drugs directed againstglutamate receptors have given us important clues to synapticevents which are clearly important for CSD.
Van Harreveld (1959) was the first to point out that glutamatetriggered CSD. A few years later it was shown that an agonistof a glutamate subtype receptor, iV-methyl-D-aspartate(NMDA), was 100 times more potent than glutamate in elicitingCSD (Curtis and Watkins, 1961). Indeed, the brain cortexreleases excitatory amino acids, including glutamate andaspartate, to the interstitial fluid during CSD, but the increaseis brief, lasting for only ~ 1 min (Fig. 7) (Van Harreveld andKooiman, 1965; Fabricius etal., 1993).
Cortical spreading depression is blocked by variouscompetitive and non-competitive NMDA antagonists (VanHarreveld, 1984; Gorelova etal., 1987; Hernandez-Caceresetal., 1987; Mody etal., 1987; Lauritzen etal., 1988;Marrannes etal., 1988; Avoli etal., 1991; Lauritzen andHansen, 1992) but not by antagonists of non-NMDA glutamatesubtype receptors (Lauritzen and Hansen, 1992). NMDAreceptor activation in turn triggers the synthesis of nitric oxide,which is both an important vasodilator and a neurotransmitter(Garthwaite et al., 1988). This is of interest since nitroglycerinprovokes headache in man (Iversen et al., 1989).
The fact that hypercapnic hyperoxia aborted the migraine auraand prevented the headache could not be explained byvasodilatation as discussed in a preceding section, but CSD inrats is reliably inhibited by hypercapnic hyperoxia (Gardner-Medwin, 1981) (Fig. 8). This result strengthens the case thatCSD underlies the migraine aura (Gardner-Medwin, 1981). Themechanism of action of hypercapnia on CSD and the auraremains uncertain, but hypercapnia stimulates nitric oxidesynthesis in the brain (Iadecola, 1992; Wang etal., 1992;Pelligrino etal., 1993; Fabricius and Lauritzen, 1994).Interestingly, amylnitrite, a well-known donor of nitric oxide,curtails the migraine aura (Wolff, 1963).
Magnetic resonance spectroscopy has revealed low brain
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

Migraine and spreading depression 207
Microdialysis, rat neocortexCortical Spreading Depression
GLLTTAMATE
Microdialysis, rat neocortexCortical Spreading Depression
• A S i w t i i a t J i 5 \ 5 S 4 I I 5TIME
-*- GLYCINE (™>
Fig. 7 Maximal concentration in dialysate of glutamate (upperpanel), and glycine (lower panel) during single episodes ofcortical spreading depression of rat brain. Graphs represent meanvalues ±1 SD of eight experiments at different time pointsduring CSD. The CSD was elicited by needle stab in the frontalcortex while the dialysate was from the parietal cortex. Time zerocorresponds to maximum of negative DC potential recorded witha single-barelled micro-electrode placed adjacent to themicrodialysis probe. Amino acid concentrations were determinedby HPLC (from Fabricius et al., 1993).
magnesium in migraine patients (Swanson, 1988; Ramadanet al., 1989), a condition that strongly facilitates the elicitationof CSD in turtle, rat and human brain tissue in vitro (Modyet al., 1987; Lauritzen et al., 1988; Avoli et al., 1991). Plasmalevels of glutamate and aspartate are elevated in migrainepatients suggesting impairment of the amino acid reuptakemechanisms (Ferrari et al., 1990). Glutamate metabolism inmigraine patients appears to be a fruitful area for future migraineresearch.
Drugs used for prophylactic migraine treatment includingmethysergide, propanolol, pizotifen, clonidine or flunarizineare ineffective as blockers of CSD (Hansen et al., 1984;Marranes et al., 1986). Probably these substances influence thesequence of events which cause pain (Moskowitz, 1992) rather
3
M.so
10
nU
20 ntf
KCI f
_JL_JL__JL_V
Fig. 8 Extracellular [K+]c in parietal cortex (upper trace) and DCpotential change in the frontal cortex (lower trace) in rats exposedto CSD and cerebral ischaemia. After the fourth CSD MK801, aNMDA-antagonist, at 10 mg/kg was given intravenously(indicated by arrow). The drug blocked CSD completely, but didnot delay or curtail the [K+]e changes during anoxicdepolarization induced by intravenous injection of KCI (fromLauritzen and Hansen, 1992).
than the CSD itself. Sumatriptan, die newly developed drugfor treatment of migraine headaches, decreases the cortical inputto the trigeminal nucleus caudalis of the brainstem withoutaffecting the CSD itself (Moskowitz et al., 1993). Ergotamineon the other hand increases the threshold for CSD in rats(Marranes et al., 1986).
Our understanding of the mechanisms of migraine is stillminimal. Future clinical studies will need to focus further onmetabolic and electrophysiological variables in the humanneocortex during migraine attacks, including measurements ofthe skull DC potential (Lehmenkuhler et al., 1991). With thesequalifications in mind, it is on the other hand clear that migrainehas advanced to a disease category in which some of themechanisms can be defined in terms of basic physiology.
AcknowledgementsThis study was supported by the Friis Foundation, the DanishMedical Research Council, the NOVO Foundation, theFoundation for Experimental Research in Neurology, theDanish Migraine Society, Lykfelds Legat, Fonden af 1870, andthe Cool-Sorption Foundation.
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

208 M. Lauritzen
ReferencesAndersen AR, Friberg L, Olsen TS, Olesen J. Delayed hyperemiafollowing hypoperfusion in classic migraine. Single photon emissioncomputed tomographic demonstration [see comments]. Arch Neurol1988; 45: 154-9. Comment in: Arch Neurol 1989; 46: 60S. Commentin: Arch Neurol 1989; 46: 605-6.
Avoli M, Drapeau C, Louvel J, Pumain R, Olivier A, Villemure J-G. Epileptiform activity induced by low extracellular magnesium inthe human cortex maintained in vitro. Ann Neurol 1991; 30: 589-96.
Bailey P, von Bonin G. The isocortex of man. Urbana: Universityof Illinois Press, 1951.
Barkley GL, Tepley N, Nagel-Leiby S, Moran JE, Simkins RT, WelchKMA. Magnetoencephalographic studies of migraine. Headache 1990;30: 428-34.
Barkley GL, Moran JE, Takanashi Y, Tepley N. Techniques for DCmagnetoencephalography [review]. J Clin Neurophysiol 1991; 8:189-99.
BureS J, BureSovd O, Kfivinek J. The mechanism and applicationsof Leao's spreading depression of electroencephalographic activity.Prague: Academia, 1974.
BureS J, Buresova' O, Kfivanek J. The meaning and significance ofLeSo's spreading depression [review]. An Acad Bras Cienc 1984; 56:385-400.
Celsis P, Goldman T, Henriksen L, Lassen NA. A method forcalculating regional cerebral blood flow from emission computedtomography of inert gas concentrations. J Comput Assist Tomogr 1981;5: 641-5 .
Cohen D, Cuffin BN (1983) Demonstration of useful differencesbetween magnetoencephalogram and electroencephalogram.Electroencephalogr Clin Neurophysiol 1983; 56: 38-51 .
Curtis DR, Watkins JC. Analogues of glutamic acid and y-amino-n-butyric acids having potent actions on mammalian neurones. Nature
Dalgaard P, Kronborg D, Lauritzen M. Migraine with aura, cerebralischemia, spreading depression, and compton scatter [letter, comment].Headache 1991; 31:49-53. Comment on: Headache 1989; 29: 15-20.Comment on: Headache 1990; 30: 269-72.
Do Carmo RJ, editor. Spreading depression. Berlin: Springer Verlag,1992.
Duckrow RB. Regional cerebral blood flow during spreading corticaldepression in conscious rats. J Cereb Blood Flow Metab 1991; 11:150-4.
Fabricius M, Lauritzen M. Transient hyperemia succeeds oligemia inthe wake of cortical spreading depression. Brain Res 1992; 602:350-3 .
Fabricius M, Lauritzen M. Examination of the role of nitric oxide forthe hypercapnic rise of cerebral blood flow in rats. Am J Physiol 1994.In press.
Fabricius M, Jensen LH, Lauritzen M. Microdialysis of interstitialamino acids during spreading depression and anoxic depolarization inrat neocortex. Brain Res 1993; 612: 61 -9 .
Ferrari MD, Odink J, Bos KD, Malessy MJA, Bruyn GW.
Neuroexcitatory plasma amino acids are elevated in migraine.Neurology 1990; 40: 1582-6.
Friberg L, Olesen J, Iversen HK, Sperling B. Migraine pain associatedwith middle cerebral artery dilatation: reversal by sumatriptan [seecomment]. Lancet 1991; 338: 13-17. Comment in: Lancet 1991; 338:1084-5.
Gardner-Medwin AR. Possible roles of vertebrate neuroglia inpotassium dynamics, spreading depression and migraine. J Exp Biol1981; 95: 111-27.
Gardner-Medwin AR, Tepley N, Barkley GL, Moran J, Nagel-LeibyS, Simkins RT etal.. Magnetic fields associated with spreadingdepression in anesthetised rabbits. Brain Res 1991; 540: 153-8.
Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derivedrelaxing factor release on activation of NMDA receptors suggests roleas intercellular messenger in the brain. Nature 1988; 336: 385-8.
Gill R, Andim* P, Hillered L, Persson L, Hagberg H. The effect ofMK-801 on cortical spreading depression in the penumbral zonefollowing focal ischaemia in the rat. J Cereb Blood Flow Metab 1992;12: 371-9.
Gloor P. Regional cerebral upflow in migraine. Trends Neurosci 1986;6: 21.
Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine:studies characterizing cerebrovascular and neuropeptide changes seenin humans and cats. Ann Neurol 1993; 33: 48-56.
Gorelova NA, Koroleva VI, Amemori T, Pavlik V, Bures J. Ketamineblockade of cortical spreading depression in rats. ElectroecephalogrClin Neurophysiol 1987; 66: 440-7.
Cowers WR. The border-land of epilepsy, faints, vagal attacks, vertigo,and their treatment. London, Churchill, 1907.
Grafstein B. Neuronal release of potassium during spreadingdepression. In: Brazier MAB, editor. Brain function, Vol. 1, Corticalexcitability and steady potentials. Berkeley: University of CaliforniaPress, 1963; 87-124.
Hansen AJ. Effect of anoxia on ion distribution in the brain [review].Physiol Rev 1985; 65: 101-48.
Hansen AJ, Lauritzen M, Tfelt-Hansen P. Spreading cortical depressionand antimigraine drugs. In: Amery WK, Van Nueten JM, WauquierA, editors. The pharmacological basis of migraine therapy. London:Pitman, 1984; 161-70.
Hernandez-Caceres J, Macias-Gonzales R, Brozek G, Bures J.Systemic ketamine blocks cortical spreading depression, but does notdelay the onset of terminal anoxic depolarization in rats. Brain Res1987; 437: 360-4.
Herold S, Gibbs JM, Jones AKP, Brooks DJ, Frachiowiak RSJ, LeggNJ. Oxygen metabolism in migraine. J Cereb Blood Flow Metab 1985;5: S445-6.
Herrera DG, Maysinger D, Gadient R, Boeckh C, Otten U, CuelloAC. Spreading depression induces c-fos-like immunoreactivity andNGF mRNA in the rat cerebral cortex. Brain Res 1993; 602: 99-103.
Iadecola C. Does nitric oxide mediate the increases in cerebral bloodflow dieted by hypercapnia? Proc Natl Acad Sci USA 1992; 89:3913-16.
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

Migraine and spreading depression 209
Iijima T, Mies G, Hossmann K-A. Repeated negative DC deflectionsin rat cortex following middle cerebral artery occlusion are abolishedby MK-801: effect on volume of ischemic injury. J Cereb Blood FlowMetab 1992; 12: 727-33.
Iversen HK, Olesen J, Tfelt-Hansen P. Intravenous nitroglycerin asan experimental model of vascular headache. Basic characteristics. Pain1989; 38: 17-24.
Kocher M. Metabolic and hemodynamic activation of postischemicrat brain by cortical spreading depression. J Cereb Blood Flow Metab1990; 10: 564-71.
Koroleva VI, Bures J. Rats do not experience cortical or hippocampalspreading depression as aversive. Neurosci Lett 1993; 149: 153-6.
Kraig RP, Dong LM, Thisted R, Jaeger CB. Spreading depressionincreases immunohistochemical staining of glial fibrillary acidic protein.J Neurosci 1991; 11: 2187-98.
Kronborg D, Dalgaard P, Lauritzen M. Ischemia may be the primarycause of neurological deficits in classic migraine [letter,comment]. ArchNeurol 1990; 47: 124-5. Comment on Arch Neurol 1987; 44:156-61.
LaCombe P, Sercombe R, Correze JL, Springhetti V, Seylaz J.Spreading depression induces prolonged reduction of cortical bloodflow reactivity in the rat. Exp Neurol 1992; 117: 278-86.
Lashley KS. Patterns of cerebral integration indicated by the scotomasof migraine. Arch Neurol Psychiatry 1941; 46: 331-9.
Lauritzen M. Cerebral blood flow in migraine and cortical spreadingdepression [review]. Acta Neurol Scand Suppl 1987a; 76; Suppl 113:1-40.
Lauritzen M. Cortical spreading depression as a putative migrainemechanism. Trends Neurosci 1987£>; 10: 8 — 13.
Lauritzen M, Hansen AJ. The effect of glutamate receptor blockadeon anoxic depolarization and cortical spreading depression. J CerebBlood Flow Metab 1992; 12: 223-9.
Lauritzen M, Rice ME, Okada YC, Nicholson C. Quisqualate, kainate,and NMDA can initiate spreading depression in the turtle cerebellum.Brain Res 1988; 475: 317-27.
Lauritzen M, Hansen AJ, Kronborg D, Wieloch T. Cortical spreadingdepression is associated with arachidonic acid accumulation andpreservation of energy charge. J Cereb Blood Flow Metab 1990; 10:115-22.
Leao AAP. Spreading depression of activity in the cerebral cortex.J Neurophysiol 1944a; 7: 359-90.
Le3o AAP. Pial circulation and spreading depression of activity in thecerebral cortex. J Neurophysiol 1944b; 7: 391-6.
Leao AAP. The slow voltage variation of cortical spreading depressionof activity. Electroencephalogr Clin Neurophysiol 1951; 3: 315-21.
Leao AAP, Morison RS. Propagation of spreading cortical depression.J Neurophysiol 1945; 8: 33-45.
Lehmenkuhler A, Richter F, Scheller D, Speckmann E-J. NoninvasiveDC recordings from the skull and the skin during cortical spreadingdepression: a model of detection of migraine. In: Olesen J, editor.Migraine and other headaches: the vascular mechanisms. New York:Raven Press, 1991; 167-70.
Lehmenkuhler A, Grotemeyer K-H, Tegtmeier F, editors. Migraine:basic mechanisms and treatment. Urban and Schwarzenberg, 1993.
Leibowitz DH. The glial spike theory. I. On an active role of neurogliain spreading depression and migraine. Proc R Soc Lond Biol, 1992;250: 287-95.
Lord GDA. Clinical characteristics of the migrainous aura. In: AmeryWK, Wauquier A, editors. The prelude to the migraine attack. London,Bailliere Tindall, 1986: 87-98
Marrannes R, Wauquier A, Reid K, De Prins E. Effects of drugs oncortical spreading depression. In: Amery WK, Wauquier A, editors.The prelude to the migraine attack. London: Bailliere Tindall, 1986;158-73
Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for arole of the N-methyl-D-aspartate (NMDA) receptor in corticalspreading depression in the rat. Brain Res 1988; 457: 226—40.
Martins-Ferreira H, editor. Current views on Leao's spreadingdepression. An Acad Bras Cienc 1984; 56: 369-531.
Mayevsky A, Weiss HR. Cerebral blood flow and oxygen consumptionin cortical spreading depression. J Cereb Blood Flow Metab 1991;11: 29-836.
Mies G. Inhibition of protein synthesis during repetitive corticalspreading depression. J Neurochem 1993; 60: 360-3.
Mies G, Paschen W. Regional changes of blood flow, glucose andATP content determined on brain sections during a single passage ofspreading depression in rat brain cortex. Exp Neurol 1984; 84:249-58.
Milner PM. Note on a possible correspondence between the scotomatasof migraine and spreading depression of Leao. Electroencephalogr ClinNeurophysiol 1958; 10: 705.
Mody I, Lambert JDC, Heinemann U. Low extracellular magnesiuminduces epileptiform activity and spreading depression in rathippocampal slices. J Neurophysiol 1987; 57: 869-88.
Moskowitz MA (1984) The neurobiology of vascular head pain. AnnNeurol 1984; 16: 157-68.
Moskowitz MA. Neurogenic versus vascular mechanisms ofsumatriptan and ergot alkaloids in migraine [review]. Trends PharmacolSci 1992; 13: 307-11.
Moskowitz MA, Nozaki K, Kraig RP. Neocortical spreadingdepression provokes the expression of C-fos protein-likeimmunoreactivity within trigeminal nucleus caudalis viatrigeminovascular mechanisms. J Neurosci 1993; 13: 1167 — 77.
Nedergaard M, Astrup J. Infarct rim: effect of hyperglycemia on directcurrent potential and (l4Q2-deoxyglucose phosphorylation. J CerebBlood Flow Metab 1986; 6: 607-15.
Nicholson C. Volume transmission and the propagation of spreadingdepression. In: Lehmenkuhler A, Grotemeyer K-H, Tegtmeier F,editors. Migraine: basic mechanisms and treatment. Munich: Urbanand Schwarzenberg, 1993; 293-308.
Nicholson C, Kraig RP. The behavior of extracellular ions duringspreading depression. In: Zeuthen T, editor. The application of ion-selective microelectrodes. Amsterdam: Elsevier, 1981: 217—38.
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from

210 M. Lauritzen
Okada Y, Lauritzen M, Nicholson C. MEG source models andphysiology. Phys Med Biol 1987; 32: 43 -51 .
Okada YC, Lauritzen M, Nicholson C. Magnetic field associated withspreading depression: a model for the detection of migraine. BrainRes 1988; 442: 185-90.
Olesen J. Cerebral and extracranial circulatory disturbances in migraine:pathophysiological implications. Cerebrovasc Brain Metab Rev 1991;3: 1-28.
Olesen J, Larsen B, Lauritzen M. Focal hyperemia followed byspreading oligemia and impaired activation of rCBF in classic migraine.Ann Neurol 1981; 9: 344-52.
Olesen J, Friberg L, Olsen TS, Iversen HK, Lassen NA, AndersenAR et al. Timing and topography of cerebral blood flow, aura, andheadache during migraine attacks. Ann Neurol 1990; 28: 791-8.
Pelligrino DA, Koenig HM, Albrecht RF. Nitric oxide synthesis andregional cerebral blood flow responses to hypercapnia and hypoxiain the rat. J Cereb Blood Flow Metab 1993; 13: 80-7 .
Piper RD, Lambert GA, Duckworth JW. Cortical blood flow changesduring spreading depression in cats. Am J Physiol 1991; 261:H96-102.
Ramadan NM, Halvorson H, Vande-Linde A, Levine SR, HelpemJA, Welch KMA. Low brain magnesium in migraine. Headache 1989;29: 590-3 .
Scheller D, Kolb J, Tegtmeier F, Lehmenkuhler A. Extracellularchanges of inorganic phosphate are different during spreadingdepression and global cerebral ischemia of rats. Neurosci Lett 1992;141: 269-72.
Schon F, Blau JN. Post-epileptic headache and migraine. J NeurolNeurosurg Psychiatry 1987; 50: 1148-52.
Shibata M, Leffler CW, Busija DW. Cerebral hemodynamics duringcortical spreading depression in rabbits. Brain Res 1990; 530: 267-74.
Skyhoj Olsen T, Lassen NA. Blood flow and vascular reactivity duringattacks of classic migraine-limitations of the Xe-133 intraarterialtechnique. Headache 1989; 29: 15-20.
Skyhej Olsen T, Friberg L, Lassen NA. Ischemia may be the primarycause of the neurologic deficits in classic migraine [see comments].Archs Neurol 1987; 44: 156-61. Comment in: Arch Neurol 1990;47: 124-7.
Somjen GG. Extracellular potassium in the mammalian central nervoussystem [review]. Annu Rev Physiol 1979; 41: 159-77.
Somjen GG, Aitken PG, Cz£h GL, Herreras O, Jing J, Young JN.Mechanism of spreading depression: a review of recent findings and
a hypothesis [review]. Can J Physiol Pharmacol 1992; 70 Suppl:S248-54.
Sramka M, Broiek G, BureS J, N&lvornfk P. Functional ablation byspreading depression: possible use in human stereotactic neurosurgery.Appl Neurophysiol 1977/8; 40: 48 -61 .
Sugaya E, Yakato M, Noda Y. Neuronal and glial activity duringspreading depression in cerebral cortex of cat. J Neurophysiol 1975;38: 822-41.
Sveinsdottir E, Larsen B, Rommer P, Lassen NA. A multidetectorscintillation camera with 254 channels. J Nucl Med 1977; 18: 168-74.
Swanson DR. Migraine and magnesium: eleven neglected connections[review]. Perspect Biol Med 1988; 31: 526-57.
Tfelt-Hansen P, Sperling B, Andersen AR. The effect of ergotamineon human cerebral blood flow and cerebral arteries. In: Olesen J, editor.Migraine and other headaches: the vascular mechanisms. New York:Raven Press, 1991: 339-43.
Van Harreveld A. Compounds in brain extracts causing spreadingdepression of cerebral cortical activity and contraction of crustaceanmuscle. J Neurochem 1959; 3: 300-15.
Van Harreveld A. The nature of the chick's magnesium-sensitive retinalspreading depression. J Neurobiol 1984; 15: 333—43.
Van Harreveld A, Kooiman M. Amino acid release from the cerebralcortex during spreading depression and asphyxiation. J Neurochem1965; 12: 431-9.
Wahl M, Lauritzen M, Schilling L. Change of cerebrovascularreactivity after cortical spreading depression in cats and rats. BrainRes 1987; 411: 72-80.
Wang Q, Paulson OB, Lassen NA. Effect of nitric oxide blockadeby A^nitro-L-arginine on cerebral blood flow response to changes incarbon dioxide tension. J Cereb Blood Flow Metab 1992; 12: 947-53.
Welch KMA, Levine SR, d'Andrea GD, Schultz LR, Helpern JA.Preliminary observations on brain energy metabolism in migrainestudied by in vivo phosphorus 31 NMR spectroscopy. Neurology 1989;39: 538-41.
Wolff HG. Headache and other head pain. 2nd ed. New York: OxfordUniversity Press, 1963.
Young DB, Van Vliet BN. Migraine with aura: a vicious cycleperpetuated by potassium-induced vasoconstriction [review]. Headache1992; 32: 24-34.
Received June 15, 1993. Revised September 14, 1993.Accepted November 7, 1993
at Aston U
niversity on January 15, 2014http://brain.oxfordjournals.org/
Dow
nloaded from


















