
The Possible Role of Volcanic Aquifers in Prebiologic Genesis of Organic Compounds and RNA
27
THE POSSIBLE ROLE OF VOLCANIC AQUIFERS IN PREBIOLOGIC GENESIS OF ORGANIC COMPOUNDS AND RNA JOHN WASHINGTON USEPA, National Exposure Research Laboratory 960 College Station Road, Athens, GA 30605-2700, e-mail: [email protected] (Received 7 July 1999) Abstract. In a volcanic aquifer, a wide range of physical and chemical conditions are not merely possible, but to be expected: relatively oxidizing and reducing environments both are present; hot and moderate temperatures can be expected; distillation and reflux conditions are probable to allow concentration of reactants, stimulation of reaction and fractionation of isotopes; apatite, hydroxides, clays and sulfide minerals are present to act as chromatographic media for separating compounds, to serve as catalytic surfaces and to provide potential energy sources; supersaturated precipitation of optically active crystals is reasonable, allowing for chromatographic separation of racemic mix- tures by the resulting fixed chiral phase; and saturated and unsaturated conditions both are present for promotion of constructive reactions and inhibiting destructive hydrolysis reactions. Because the multitude of physical-chemical environments makes the setting robust with respect to circumventing commonly identified problems in origin-of-life theories, even if objections to details proposed herein are identified, the setting is favorable for devising alternatives. This paper describes a theory for the genesis of organic compounds, including RNA, in the mixing zone of juvenile and meteoric waters above a leaky semi-confined aquifer. Starting with basic reactants for best-guess conditions on Archean Earth, parallel sequences of specific reactions are pro- posed that culminate with RNA oligonucleotides, key molecules in contemporary life. All proposed reactions, or close analogues, are experimentally confirmed and all are set in plausible Archean conditions. Calculations indicate that the proposed reactions would yield C isotopic compositions that are consistent with observed biologic C. 1. Introduction There are at least three general theories regarding the genesis of life on Earth, each conventionally thought mutually exclusive from the others: 1) the warm primor- dial soup of Miller (1953, 1987, 1974) generating a self-replicatory molecule; 2) the chemoautotrophic proto-metabolism of Wachtershauser (1988a, 1988b, 1990, 1992) leading to a self-replicatory molecule; and 3) the generation of a proto- membrane (Russell and Hall, 1997) that subsequently evolved metabolism and self-replicatory capabilities. Each of these theories, and their variants, is attended by several significant obstacles to a complete and specific description of the re- actions leading from probable early-Earth simple molecules to a self-replicating modern molecule. Generally, obstacles to complete theories of biogenesis fall into one of two cat- egories: 1) conditions necessary for hypothesized reactants to exist are thought not Origins of Life and Evolution of the Biosphere 30: 53–79, 2000. © 2000 Kluwer Academic Publishers. Printed in the Netherlands.
-
Upload
john-washington -
Category
Documents
-
view
212 -
download
0
Transcript of The Possible Role of Volcanic Aquifers in Prebiologic Genesis of Organic Compounds and RNA

THE POSSIBLE ROLE OF VOLCANIC AQUIFERS IN PREBIOLOGICGENESIS OF ORGANIC COMPOUNDS AND RNA
JOHN WASHINGTONUSEPA, National Exposure Research Laboratory 960 College Station Road, Athens, GA
30605-2700, e-mail: [email protected]
(Received 7 July 1999)
Abstract. In a volcanic aquifer, a wide range of physical and chemical conditions are not merelypossible, but to be expected: relatively oxidizing and reducing environments both are present; hotand moderate temperatures can be expected; distillation and reflux conditions are probable to allowconcentration of reactants, stimulation of reaction and fractionation of isotopes; apatite, hydroxides,clays and sulfide minerals are present to act as chromatographic media for separating compounds,to serve as catalytic surfaces and to provide potential energy sources; supersaturated precipitationof optically active crystals is reasonable, allowing for chromatographic separation of racemic mix-tures by the resulting fixed chiral phase; and saturated and unsaturated conditions both are presentfor promotion of constructive reactions and inhibiting destructive hydrolysis reactions. Because themultitude of physical-chemical environments makes the setting robust with respect to circumventingcommonly identified problems in origin-of-life theories, even if objections to details proposed hereinare identified, the setting is favorable for devising alternatives.
This paper describes a theory for the genesis of organic compounds, including RNA, in themixing zone of juvenile and meteoric waters above a leaky semi-confined aquifer. Starting with basicreactants for best-guess conditions on Archean Earth, parallel sequences of specific reactions are pro-posed that culminate with RNA oligonucleotides, key molecules in contemporary life. All proposedreactions, or close analogues, are experimentally confirmed and all are set in plausible Archeanconditions. Calculations indicate that the proposed reactions would yield C isotopic compositionsthat are consistent with observed biologic C.
1. Introduction
There are at least three general theories regarding the genesis of life on Earth, eachconventionally thought mutually exclusive from the others: 1) the warm primor-dial soup of Miller (1953, 1987, 1974) generating a self-replicatory molecule; 2)the chemoautotrophic proto-metabolism of Wachtershauser (1988a, 1988b, 1990,1992) leading to a self-replicatory molecule; and 3) the generation of a proto-membrane (Russell and Hall, 1997) that subsequently evolved metabolism andself-replicatory capabilities. Each of these theories, and their variants, is attendedby several significant obstacles to a complete and specific description of the re-actions leading from probable early-Earth simple molecules to a self-replicatingmodern molecule.
Generally, obstacles to complete theories of biogenesis fall into one of two cat-egories: 1) conditions necessary for hypothesized reactants to exist are thought not
Origins of Life and Evolution of the Biosphere30: 53–79, 2000.© 2000Kluwer Academic Publishers. Printed in the Netherlands.

54 JOHN WASHINGTON
plausible for early-Earth, e.g., cyanide in a mildly oxidizing atmosphere; 2) requis-ite conditions for synthesis of one necessary organic compound, e.g., sugar, are in-compatible with the synthesis or stability of another necessary organic compound,e.g., nitrogenous bases.
Regarding obstacle 1, the majority of evidence suggests that life started at leastas early as the early Archean, 3.5 to 3.9 billion years before present (bybp). Forexample: microfossils have been identified in 3.4 bybp South African sediments(Knoll and Barghoorn, 1977); fossilized bacteria have been studied in 3.5 bybprock from the Warrawoona Group in Australia (Awramik, 1981; 1982; Awramiketal., 1983; Schopf and Packer, 1987; Schopf and Walter, 1983); and stromatoliteshave been identified in 3.5 bybp rock of the Pilbara group of Australia (Walter,1983). Still further back, isotopic data have been used to imply life was present atthe time the 3.8-bybp rocks of Isua, Greenland were formed (Schidlowskiet al.,1979; Hayeset al., 1983; Schidlowskiet al., 1983). Interpretations regarding lifevestiges in the Isua metasediments have been received with skepticism, however,because of concerns regarding effects of metamorphism on isotopic ratios. Theseconcerns appear to have been addressed by analysis of the C-isotope ratios in singleapatite crystals (Mojzsiset al., 1996) and in carbonaceous microparticles (Rosing,1999) in rocks of western Greenland. Based on these studies, it is reasonable toconsider best guesses of early-Archean conditions as those from which life hasarisen, specifically 3.8 bybp.
Regarding obstacle 2, Goethe (1831), the early mineralogist and philosopher,described a scheme for genesis of life in which compounds are formed in parallelreaction sequences, then moved to different environments, and mixed to completecreation (see Endnote). In this paper, I expand on Goethe’s idea to hypothesizeparallel reaction sequences taking place in a groundwater system – starting withsimple molecules in plausible concentrations and settings for the early-ArcheanEarth, and culminating in the genesis of self-replicatory naked genes, specificallythe RNA oligonucleotides proposed by Gilbert (1986). By considering separatereaction sequences, in which reactants and products are carried, separated by clayschromatographically, and mixed in groundwater, obstacles to other theories areovercome. In this paper, these major obstacles are identified initalics and followedby a description of the manner in which they are addressed by this hypothesis.
2. Summary of Hypothesis
The idea is depicted in Figure 1 which shows the proposed chemical-reactionscheme as well as the flow regime that might allow the reaction scheme to progress.RNA is composed of three basic components, ribose sugar, phosphatic backbone,and nitrogenous bases. For these components, the theory calls for: 1) formaldehydeto form in the Archean atmosphere, fall with meteoric water to the ground where itconcentrates by distillation and polymerizes to form sugars that separate chroma-

ORGANIC SYNTHESIS IN AQUIFERS 55

56 JOHN WASHINGTON
Figure 1. Cartoon of the process. See text for explanation. References for the depicted composi-tions and processes: 1) (Pintoet al., 1980); 2) (Kasting, 1993); 3) (Walker, 1964); 4) (Dong andDasgupta, 1986); 5) (Yamagataet al., 1991); 6) (Hsu, 1989); 7) (Dixon, 1989); 8) (Borchardt, 1989);9) (Farberov and Speranskaya, 1955); 10) (Gabel and Ponnamperuma, 1967); 11) (Morrison andBoyd, 1959); 12) (Jacques, 1993; Jacqueset al., 1981); 13) (Dhanesar and Gisch, 1988); 14) (Ferrisand Hagan, 1984); 15) (Loweet al., 1963); 16) (Ferriset al., 1972); 17) (Oro and Kimball, 1962); 18and 19) (Ferris and Hagan, 1984); 20) (Ferriset al., 1972); 21) (Ferriset al., 1978); 22) (Oro, 1960;Oro and Kimball, 1961; Oro and Kimball, 1962); 23–25) (Ferris and Hagan, 1984); 26) (Shapiro andKlein, 1966); 27) (Fulleret al., 1972; Ponnamperumaet al., 1963); 28) (Beck and Orgel, 1967); 29)(Ferris and Ertem, 1992; Ferriset al., 1996).
tographically; 2) phosphates to outgas from volcanos to the atmosphere, condenseas high-energy polyphosphates by fast quenching, fall with meteoric water to theground, and become immobilized by mineral precipitation; 3) nitrogenous basesto form from chemically reducing, primordial hydrothermal fluids, and purify andconcentrate by chromatography and adsorption; and 4) the sugar, phosphates andnitrogenous bases mix in a clay lens, the sugars and phosphates flowing at the uppersurface of the lens and the bases upwelling from below, located in the dischargezone of a watershed.
3. Assumed and Inferred Early-Archean Conditions
3.1. ASSUMED ATMOSPHERIC AND METEORIC-WATER COMPOSITION
For this paper the Archean atmosphere is assumed to be that described by Kasting(1990, 1993). For some of the more relevant gases: PCO+CO2 = 0.2–10 bar; PN2 =0.2 bar; PH2 = 10−3 bar; PO2 = 10−13 bar; PHCO = 10−12; CO/CO2 = 4× 10−4; andrelative humidity = 77%. Using a photochemical production path for formaldehyde(H2CO) (Pintoet al., 1980), Kasting modeled PH2CO = 10−10 bar. For his ‘basecase’, Kasting (1990) used PCO2 = 0.2 bar and calculated a surface temperature of278◦K.
Using the interpolation software of Bethke (1998), which operates on the Law-rence-Livermore database, for 5◦C:
CO2(g) + H2O = H+ + HCO−3 (1)
has a Keq = 10−7.71. For PCO2 = 0.2, and assuming [H+] ≈ [HCO−3 ], the pH of rainwould be about 4.2. Using Henry’s Law Constants for 5◦C of KH2
H = 103.1 (bar-L)/mol and KO2
H = 102.7 (bar-L)/mol for H2 and O2, respectively (Bethke, 1998),rainfall [H2] = 10−6 M and [O2] = 10−15.7 M.
Rain water in equilibrium with the assumed temperature and formaldehyde con-centration of the Archean atmosphere would have 10−6 M formaldehyde, almost all

ORGANIC SYNTHESIS IN AQUIFERS 57
of it hydrated to methylene glycol (Dong and Dasgupta, 1986); this concentrationcompares conservatively with measurements of our modern, relatively oxidizingenvironment of up to 50-times this value (Gaffneyet al., 1987).
3.2. ASSUMED STATE OF THE MANTLE-DERIVED FLUIDS
The oxidation state of magma-exsolved volatiles commonly is assumed to reflectthat of the upper mantle from which they migrate (e.g., Holland, 1984). There isdisagreement regarding the oxidation state of the Archean mantle, some arguingfor an oxidation state buffered by the iron-wustite (IW) system (Kastinget al.,1993), others arguing for a higher oxidation state, similar to modern conditions,near the quartz-fayalite-magnetite (QFM) buffer (Holland, 1984). Figure 2 depictsthe relative positions of these buffers along with the N and S fluid couples fora conservatively low, assumed fH2O of 5 atm (e.g., Holland, 1984) and illustratesthat the difference between these buffers can affect the fNH3/fN2 ratio by orders ofmagnitude. Observations of modern hydrothermal systems show that N2 tends todominate over reduced N species such as NH3 (Ellis, 1979) suggesting that QFMis insufficiently reducing to cause NH3 to prevail over N2 even at low temperat-ures where it is thermodynamically stable (Figure 2). Consequently, for this paper,Archean mantle redox is assumed to have been controlled by IW.
Giggenbach (1997) notes that the degree to which hydrothermal fluids reactwith solid couples, such as IW, is a function of the rise velocity of the fluid andwhether the fluid is ‘dry’ or an aqueous solution; presumably, also, it is a functionof the fluid to rock ratio. Hence, it is possible that the IW buffer could draw theN pool into the NH3 region for the conditions depicted in Figure 2. More likely,however, is that fH2O could have been higher than the 5 atm assumed in Figure 2.Each 5 atm increase in fH2O shifts the equilibrium N2-NH3 couple upwards one logfO2 unit, effectively closing the gap between N2-NH3 and IW in the 800–1000◦Crange at fH2O∼20 atm, a reasonable value.
Base synthesis has been documented from solutions as dilute as about 2 mmolN/mol H2O (Ferriset al., 1978), but the yield was unquantified. Quantified pro-cesses of base formation described below depend upon higher concentrations of N,say > 18 mmol/mol, being present in the upwelling juvenile fluids (Oro, 1960; Oroand Kimball, 1961, 1962). While such concentrations seem reasonable, they are farfrom certain. Pineau and Javoy (1994) report magma-exsolved gas, isolated frommid-ocean ridge basalt (MORB) gas vesicles, as having about [N] = 2.5 mmol/molCO2+H2O (the dominant species). Giggenbach (1997) reports [N] in three, present-day vapor discharges believed to be of mantle origin as ranging from 0.6 to 2.2mmol/mol as N; these values are in the range used by Ferris and coworkers (1978),but only about 10% of the concentrations used by Oro and coworkers.
Oro et al. (1990) summarize several studies in which the terrestrial deficit ofnoble gases relative to solar concentrations was used to infer the nearly completeloss of the primordial atmosphere and that the present atmosphere accumulated

58 JOHN WASHINGTON
Figure 2.At temperatures of about 1000◦C and more, fluids tend to equilibrate with solid bufferssuch as IW; however, at temperatures below 1000◦C, fluid reactions with solids tend to slow sothat fluid redox can to be controlled internally, often by SO2-H2S in modern systems (Giggenbach,1997). For the 5 atm-fH2O depicted in Figure 2, fluids cooling from 1000◦C in equilibrium withIW will have N dominantly as N2 and S dominantly as H2S. Since the S is vastly reduced underIW conditions, the S couple is less effective at oxidizing reduced solutes than it is at reducingoxidized compounds so the SO2-H2S should not exercise an overwhelming effect on maintaining Nin its oxidized form. N2-NH3 and SO2-H2S from Holland (1984). QFM and IW from Chou (1987).IW(ex.) is extrapolated from experimental range.
from outgassing of internal volatiles plus subsequent accretion of minor comets andother volatile-rich bodies. Such a scenario would be consistent with higher N con-centrations in outgassing fluids during the Archean than at present. Holland (1984)tabulated the masses of the present-day atmospheric, hydrospheric, biospheric andcrustal reservoirs of N and water; assuming these reservoirs accumulated by out-gassing, the average [N] over geologic time is about 4 mmol/mol. Assuming thepresent-day average [N] of about 2 mmol/mol has been about constant for the last3.5 bybp, the average [N] for the period 4.5 to 3.5 bybp was about 11 mmol/mol,close to 18 mmol/mol. Some recent modeling, however, would argue against higherN concentrations in volcanic emissions at 3.8 bybp; Tolstikhin and Marty (1998)concluded that about 97% of Earth’s present N reservoir had been outgassed by 4.3bybp, about 0.5× 109 y earlier than this proposed genesis of life. This observation,along with the observation that a large fraction of the N might have been presentas N2, imply that if juvenile water was the source of N for the primordial bases as

ORGANIC SYNTHESIS IN AQUIFERS 59
TABLE I
Major-chemical and normative-mineral compositionof mid-oceanic ridge basalt
Normative
Oxide Weight % Mineral Weight %
SiO2 50.67 Anorthite 30.6
Al22O3 15.45 Diopside 21.5
CaO 11.72 Albite 21.0
FeO(1) 9.67 Hypersthene 16.9
MgO 9.05 Magnetite 3.7
Na2O 2.51 Olivine 3.5
TiO2 1.28 Ilmenite 2.4
P2O5 0.20 Orthoclase 1.1
K2O 0.15 Apatite 0.3
Sum 100.70 Sum 101.0
1) Sum of ferrous and ferric as ferrous.
suggested in this paper, the bases were produced much earlier than the 3.8 bybprocks when magmas likely were exceptionally N rich.
3.3. INFERRED CONDITIONS IN ARCHEAN SOILS AND GROUNDWATER
SYSTEMS
For this description, I assume a gabbro bedrock of MORB elemental compositionand normative primary-mineral composition (Table I).
Nesbitt and Wilson (1992) studied modern chemical weathering of basalts indetail and many of the general reactions they report can be expected to have oc-curred in the assumed Archean environment. Regarding primary-mineral weather-ing, Nesbitt and Wilson (1992) found the order of susceptibility to be olivines >glass& plagioclases& clinopyroxenes > Fe-Ti oxides.
Waters collected from settings of little weathering generally are in equilib-rium with smectites, the dominant, first-generation, authigenic minerals producedin weathering of basaltic primary minerals (Nesbitt and Wilson, 1992). Becauseof acidity consumption in dissolution of minerals, waters that have reacted withfresh basalt tend to have high pHs, commonly ranging from 8 to 9.5 in both coolgroundwaters (Nesbitt and Wilson, 1992) and hydrothermal groundwaters (Ellis,1979). Because of these high pHs, and the high assumed Archean PCO2, carbonateminerals can be expected to have precipitated early in the weathering process as

60 JOHN WASHINGTON
well. For example, the olivine forsterite and the plagioclase anorthite might weatherto Mg-beidellite and dolomite according to:
Mg2SiO4 + 0.8694Ca[Al2Si2O8] + 2.015H+ + 1.739CO2(g) =
Mg0.165A2.33Si3.67O10(OH)2 + 0.8694CaMg(CO3)2 +
1.007Mg2+ + 0.2612H2O (2)
At 5 ◦C, log K = 35.12 (Bethke, 1998), so this reaction would have proceededstrongly to the right. Depending on conditions, smectites other than beidellite alsowould form. At the high pHs of these incipient weathering environments, Fe-bearing primary minerals might have given rise to ferrous hydroxides accordingto:
Fe2+ + 2H2O = Fe(OH)2 + 2H+ (3)
where log K = –13.97 at 5◦C (Bethke, 1998).As primary minerals are depleted, precipitation of smectites shifts to dissolution
with the consequent precipitation of kaolin minerals (Nesbitt and Wilson, 1992).Waters collected from these settings tends toward equilibrium with kaolin phasesand, as the reactions proceed, pH tends downward (Nesbitt and Wilson, 1992).Weathering beidellite to kaolinite is an example:
Mg0.165Al2.33Si3.67O10(OH)2 + 1.165H2O + 0.33H+ =
0.165Mg2+ + 1.165Al2Si2O5(OH)4 + 1.34SiO2(aq) (4)
where log K = –2.47 at 5◦C (Bethke, 1998). In detail, several researchers havefound that in volcanic settings the kaolin mineral halloysite generally dominatesover kaolinite (Dixon, 1989). Subject to lower pHs of this moderately weatheredsetting, ferrous hydroxides, if present, would have dissolved, probably reprecipit-ating as siderite (FeCO3). With further leaching carbonate dissolution would haveensued, poising pH at moderate values for as long the phases persisted. As weath-ering progresses further, kaolins dissolve, often equilibrating with gibbsite (Nesbittand Wilson, 1992):
Al2Si2O5(OH)4 + H2O = 2Al(OH)3 + 2SiO2(aq) (5)
At 5 ◦C, Keq = –9.12 (Bethke, 1998) so this reaction would proceed as aSi ap-proached 10−4.6 M.
The progress of these reactions would have buffered the pH of soil and ground-waters to higher values than that of the pH = 4.2 for precipitation. With PCO2
buffered by the atmosphere in the vadose zone, water equilibrating with gibbsite, amineral that would have approached equilibrium with water more closely than theclays according to:
Al(OH)3 + 3CO2(g) = Al3+ + 3HCO−3 (6)

ORGANIC SYNTHESIS IN AQUIFERS 61
would have yielded water having pH≈ 4.9, using Keq = 10−13.9 (Bethke, 1998) andstoichiometric ratios of reaction products. In groundwaters with no buffering effectfrom the atmosphere, waters in equilibrium with gibbsite would have had pHs ofabout 5.6 assuming stoichiometric ratios among reactants and products.
The authigenic mineral deposits, consisting of clay minerals and hydroxides,would have had significant permanent cation exchange capacities as well as pH-dependent anion exchange capacities for the inferred pH conditions. Aluminosilic-ate clays have mostly permanent cation exchange capacities (CECs); smectites,for example, have about 110–130 meq/100 g (Borchardt, 1989) and kaolins arereported to have CECs of about 1–5 meq/100 g (Dixon, 1989). In contrast, gibbsitehas a pH-dependent exchange capacity with a zero-point of charge (ZPC)≈ 8.0 to9.2 (Hsu, 1989). For pHs of 4.9 to 5.6, these hydroxides would have had positivelycharged surfaces and served to scavenge anions from solution, notably includingH2PO−4 and negatively charged polyphosphates.
On steep, igneous slopes, under likely aggressive early-Archean physical andchemical conditions, there would have been significant downslope wasting yieldinghuge lahar, colluvial, alluvial and deltaic lenses (Figure 1). The deposits wouldhave had thickening accumulations as one progressed downslope. On steep slopesand with no plant roots for stabilization, the surface might well have been dom-inated by wasting coarse fragments, and resultantly blanketed in thick talus fields.As primary minerals weathered in these deposits, authigenic hydroxides and clayswould have precipitated and winnowed their way downward to finer pores wherethey would cement, commonly giving rise to the fragipans that often occur incolluvial aprons at mountain-base slopes (Smeck and Ciolkosz, 1989).
Such a scenario might have resulted in relatively coarse-grained materials withhigh hydraulic conductivities over Al-hydroxide- and carbonate-cemented fines ofrelatively lower hydraulic conductivities, in turn overlying basalt fractured by cool-ing and consequently relatively conductive. For vesicular basalts, the high porosityalso might have caused it to be even more conductive. Such a lens of largely sec-ondary minerals, consequently, would act hydraulically as an aquitard, curbing butnot stopping the mixing of the meteoric water dominantly in the talus/colluviumand juvenile waters in the fractured/vesicular basalt.
For this given stratigraphy, on the slopes of a large shield volcano, with up-welling hydrothermal fluids, groundwater flow patterns could have been relativelycomplex. With precipitation recharge infiltrating the coarser, relatively conductivesediments of the upper reaches of the slope, an unconfined, water-table aquifer withdownward gradients might be expected (Figure 1). For these conditions, shallowflow would have followed in the downslope direction the low-permeability beddingof the authigenic hydroxides and clays, with some of the water leaking downwardthrough the aquitard to the relatively permeable, fractured/disaggregated basaltwhich would act as a semi-confined aquifer. Depending on several factors, thewater-table aquifer in the upper reaches of the watershed also could have beenperched sporadically, seasonally or permanently in some locations.

62 JOHN WASHINGTON
At some point in the midslope area, the hydraulic head of the semi-confined(shallow gabbro) aquifer would have declined to a potential less than the headof the juvenile hydrothermal fluids (Figure 1). In this location, the juvenile fluidswould upwell and mix with the meteoric waters that had managed to flow throughthe lens of low-permeability authigenic minerals.
Finally, near the toe of the slope, the topographic elevation, and consequentlythe hydraulic head of the water-table aquifer, would have decreased to a value lessthan the hydraulic head of the confined system (Figure 1). Here the mixed meteoric-juvenile waters would upwell, co-mingle further with shallow meteoric waters andissue forth as springs or shallow marine vents.
The presence of two or more, largely separate, groundwater-flow systems is animportant element of this hypothesis; so it is worth noting possibilities other thanthat described above for such flow patterns. Roweet al. (1995) and Sanfordetal. (1995) characterized the geochemistry and hydrogeology of the Poas Volcano,Costa Rica. They describe several warm-water springs that are chemically similarto the crater lake, yet are located 1 to 5 km from the crater. Based on the absenceof dilution and on modeling with3H, the authors concluded that there is littlemixing of water along the flow path from point of recharge to point of dischargefor these springs. Yet, these springs are located among others that apparently dis-charge meteoric water. The authors describe several factors that could contributeto, or single-handedly cause, largely separate flows of deep- and shallow-sourcesprings in volcanic settings: 1) old, weathered, permeable lava deposits are buriedby younger, less permeable deposits; 2) deep waters, often having higher salinity,can be denser than meteoric waters and consequently flow largely separately; and3) deep waters are unusually aggressive, and dissolve rock relatively quickly, en-hancing permeability along their flow paths. Finally, it is worth noting that even ina homogeneous, isotropic system, two sources of water to a system can be arrangedspatially and in flow rates so that their flows remain largely separate to the point ofdischarge.
Because of variations in topographic surface, thickness of the clay lens, vari-ations in hydraulic conductivity associated with deposits from different lava flows,seasonal or secular precipitation changes, changes in temperature or salinity ofjuvenile fluids, and changes in rates of upwelling, these watersheds could havehad a wide array of hydrologic and chemical settings. To the extent that there wasseasonality in precipitation or evaporation, or there were large recharge events, orsporadicity in migration of hydrothermal fluids, the groundwater-flow patterns andthe vadose/groundwater interface would have varied, both spatially and in time.Springs, streams, rivers and mud flats would have been present, and these featurescould have been ephemeral depending upon above-identified factors. Because ofchanges in recharge or flow patterns, previously wet locations could be left dry,baking in radiation filtering through the thin Archean atmosphere – and vice versa.
Because hydrothermal fluids likely were quite enriched in solutes, their flowpathslikely were ubiquitously enriched in sulfide minerals.

ORGANIC SYNTHESIS IN AQUIFERS 63
4. Features of Formose Formation
The formation of sugars from CH2O often is called the formose reaction. Multiplemethods now have been documented to generate ribose from CH2O or formoseintermediates. Gabel and Ponnamperuma (1967) produced monosaccharides fromaqueous solutions of CH2O by refluxing in the presence of corundum, kaolinite andillite, of which related minerals were described above as likely secondary mineralsfor the hypothetical setting. Among the three minerals, corundum yielded highermonosaccharide production than the clays. Corundum is the dehydrated analogueof gibbsite. Krishnamurthyet al. (1998) produced large yields of ribose from mil-limolar concentrations of formose intermediates in the presence of layered hydrox-ide minerals having positive surface charge. Zubay (1998) reports that Pb(OH)2
catalyzed the formose reaction in the presence of Mg(OH)2, the yield of pentosesbeing about 30% of the product, but initial CH2O concentrations were high.
A commonly cited impediment to prebiotic ribose generation by these methods isthat their syntheses have been documented to proceed only at 10−3 M and higherof CH2O or formose intermediates, at least 103-times the concentration of likelyArchean rainfall. In fact, starting with CH2O as opposed to intermediates, Reidand Orgel (1967) failed to identify sugar production from 10−3 M solutions.
A reasonable method of concentrating the CH2O from likely precipitation con-centrations to formose-reaction range is by distillation, especially considering thatdistillation temperatures and conditions would be similar to those required for therefluxing that Gabel and Ponnamperuma (1967) employed in their synthesis. Dis-tillation processes have been shown to occur in modern geothermal systems; forexample, Pineau and Javoy (1994) have shown the distribution of CO2 and H2O inthe vesicles of MORB basalts to be controlled by Rayleigh distillation.
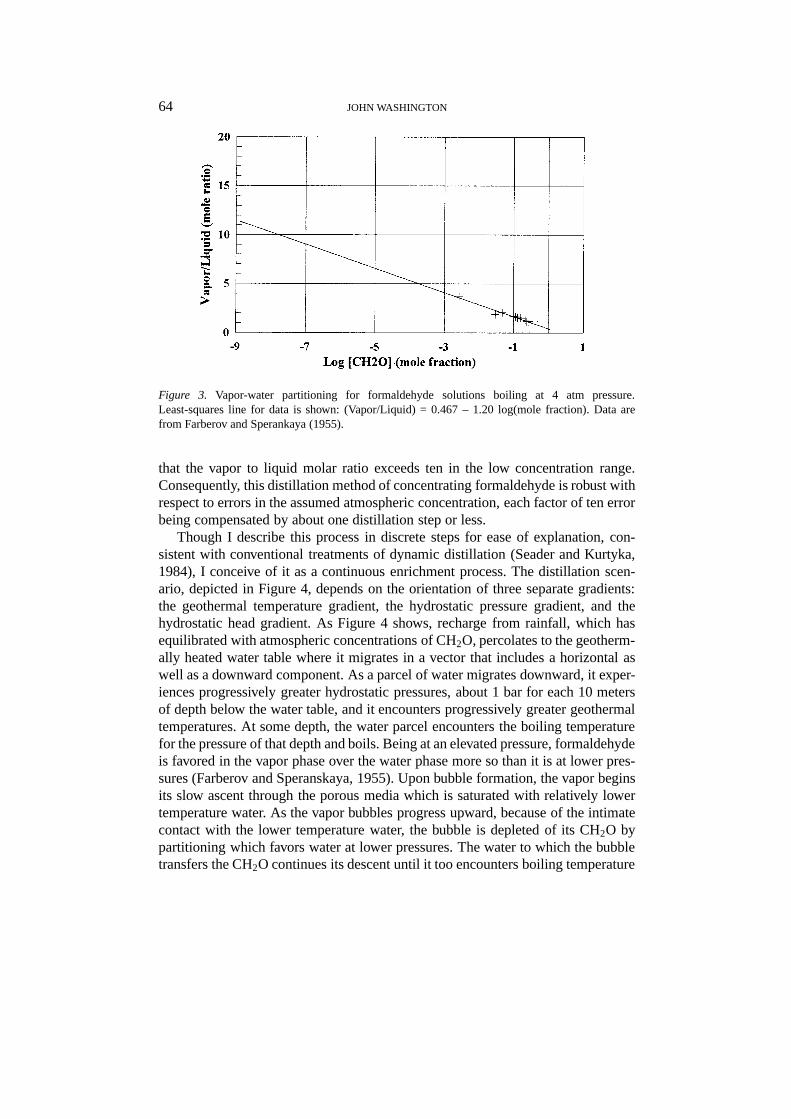
According to Farberov and Speranskaya (1955), whose work was cited liberallyin Walker (1964), at atmospheric pressure, the vapor over boiling aqueous solutionsof CH2O has just slightly lower concentrations of CH2O than does the solution,measured as mole %. Furthermore, as dilute CH2O-H2O solutions slowly enrichvia boiling under atmospheric pressure, the efficiency of concentration decreasesfurther as the vapor and liquid concentrations approach azeotropic equipartioningwhich occurs over the interval 4 to 25 mole % (Farberov and Speranskaya, 1955).Consequently, distillation of CH2O at nearly one-atmosphere pressures is an inef-ficient process. However, the vapor phase is favored progressively with increasingpressures, especially over the increment of 2 to 4 bar. Figure 3 shows that thevapor-partitioning data of Farberov and Speranskaya (1955) for distillation at 4 barplots linearly when the vapor/liquid molar ratio is plotted against the log of thesolution concentration. Using the rough approximation of a linear extrapolationto the concentration range of likely Archean rainfall, depending on efficiency ofexchange and other factors, dynamic, ideal-binary distillation (Seader and Kurtyka,1984) in roughly 4 steps could yield the 10−3 M concentration needed for the for-mose reaction to proceed (Table II). Referring again to Figure 3, it also is notable
64 JOHN WASHINGTON
Figure 3. Vapor-water partitioning for formaldehyde solutions boiling at 4 atm pressure.Least-squares line for data is shown: (Vapor/Liquid) = 0.467 – 1.20 log(mole fraction). Data arefrom Farberov and Sperankaya (1955).
that the vapor to liquid molar ratio exceeds ten in the low concentration range.Consequently, this distillation method of concentrating formaldehyde is robust withrespect to errors in the assumed atmospheric concentration, each factor of ten errorbeing compensated by about one distillation step or less.
Though I describe this process in discrete steps for ease of explanation, con-sistent with conventional treatments of dynamic distillation (Seader and Kurtyka,1984), I conceive of it as a continuous enrichment process. The distillation scen-ario, depicted in Figure 4, depends on the orientation of three separate gradients:the geothermal temperature gradient, the hydrostatic pressure gradient, and thehydrostatic head gradient. As Figure 4 shows, recharge from rainfall, which hasequilibrated with atmospheric concentrations of CH2O, percolates to the geotherm-ally heated water table where it migrates in a vector that includes a horizontal aswell as a downward component. As a parcel of water migrates downward, it exper-iences progressively greater hydrostatic pressures, about 1 bar for each 10 metersof depth below the water table, and it encounters progressively greater geothermaltemperatures. At some depth, the water parcel encounters the boiling temperaturefor the pressure of that depth and boils. Being at an elevated pressure, formaldehydeis favored in the vapor phase over the water phase more so than it is at lower pres-sures (Farberov and Speranskaya, 1955). Upon bubble formation, the vapor beginsits slow ascent through the porous media which is saturated with relatively lowertemperature water. As the vapor bubbles progress upward, because of the intimatecontact with the lower temperature water, the bubble is depleted of its CH2O bypartitioning which favors water at lower pressures. The water to which the bubbletransfers the CH2O continues its descent until it too encounters boiling temperature

ORGANIC SYNTHESIS IN AQUIFERS 65
TABLE II
Estimated Rayleigh distillation of formaldehyde at 4 atm. where 10% of the liquid isvaporized and condensed, approximated using the fractionation factor for H2CO3 – CO2
Distillation Incoming Incoming KD Condensate Condensate
Step [H2C(OH)2] δ13C (1) [H2C(OH)2] δ13C
(M) (h PDB (M) (h PDB)
1 1.00× 10−6 –13.10 9.75 9.75× 10−6 –13.96
2 9.75× 10−6 –13.86 8.56 8.35× 10−5 –14.63
3 8.35× 10−5 –14.63 7.45 6.22× 10−4 –15.39
4 6.22× 10−4 –15.39 6.40 3.98× 10−3 –16.15
5 3.98× 10−3 –16.15 5.43 2.16× 10−2 –16.91
6 2.16× 10−2 –16.91 4.55 9.85× 10−2 –17.67
7 9.85× 10−2 –17.67 3.67 3.71× 10−1 –18.43
8 3.71× 10−1 –18.43 3.07 1.14× 100 –19.19
9 1.14× 100 –19.19 2.49 2.84× 100 –19.95
10 2.84× 100 –19.95 2.02 5.72× 100 –20.71
11 5.72× 100 –20.71 1.65 9.44× 100 –21.46
12 9.44× 100 –21.46 1.39 1.31× 101 –22.22
13 1.31× 101 –22.22 1.22 1.60× 101 –22.98
14 1.60× 101 –22.98 1.12 1.78× 101 –23.73
(1) Vapor/liquid partitioning ratio (mole fractions) using a regression against the ex-perimental data of Farberov and Speranskaya (1955). See Figure 3 for regressionequation.
and the cycle is repeated. Since the groundwater has a horizontal component ofmovement as well as a vertical, with each cycle, the progressively enriched watersare transferred downgradient and away from the heat source.
There are several other appealing qualities to this scenario:
1) As summarized above and described below in more detail, the proposed sourceof the nitrogenous bases for the RNA is from upwelling juvenile waters. As such,setting of the formose reaction in a groundwater recharge zone, where the ground-water-flow direction is downward, or setting it over a clay lens, circumvents aconventional stumbling block in theories of the origin of life, the reaction of CH2Owith cyanide or similar compounds to form glyconitrile or similar products at theexpense of completion of the formose reaction(Shapiro, 1988). Either a downward-flow direction or a poorly permeable aquiclude can serve as satisfactory agents formaintaining the required isolation of the CH2O from the reduced N forms.
2) Several steps in thesynthesis of nitrogenous bases generally are more efficientunder alkaline conditions. Conversely, ribose is known to isomerize to other sugars

66JO
HN
WA
SH
ING
TO
N
Figure 4. Depiction of the dynamic distillation of CH2O. As the meteoric-source water migrates along the hydrostatic head gradient downward andlaterally in the recharge zone, it encounters progressively greater geothermal temperatures as well as greater hydrostatic pressures. Boiling temperaturesare encountered at depths that are a function of these latter two gradients. At elevated pressures vapors are enriched in formaldehyde, relative to boilingaqueous solutions. As bubbles ascend, formaldehyde redissolves in water, to be carried downward and laterally again, resulting in enrichment of thewater.

ORGANIC SYNTHESIS IN AQUIFERS 67
relatively rapidly under alkaline conditions(Sochaet al., 1981). For the setting de-scribed, however, expected pH for the shallow groundwater in which minerals areequilibrating with atmospheric CO2, is in the range of 4.9 to 5.6. In mildly acidicconditions, depending upon temperature, ribose decomposition can be slowed tothe time scale of years (Larralde and Miller, 1994).
3) This distillation is proposed for a setting rich in smectite clays. Ethylene glycolconcentrates strongly in the interlayer of smectites (Brown, 1961). Formaldehydein solutions of the concentrations described here hydrates almost entirely to methyl-ene glycol, so smectite clays could aid concentration of CH2O solutions.
4) Farberov and Speranskaya (1955) also noted thatincreased efficiency of CH2Opartitioning to the gas phase with increasing pressure was counterbalanced atpressures exceeding 4 bar by degradation of the CH2Ovia the Cannizzaro-Tischenkoreaction to formic acid and methanol:
2CH2O + H2O = HCOOH + CH3OH (7)
A 10-meter water column equates to approximately 1 bar of pressure so, assumingArchean atmospheric pressure was at least 1 bar,this observation seems to imposea depth limit on origin of life at ‘deep-sea vents’ at about 30 meters or less, unlessthe theory does not require sugar formation from CH2O. This reaction also appearsto constrain Archean PCO2 to less than about 4 bar if ribose is to be produced bythe formose reaction.
These formose reactions probably produce racemic mixes of several species ofsugars, of which ribose is only a fraction. Below, the possible isolation of D-ribosewill be discussed.
5. Polyphosphate Polymerization
Phosphorus binds adjacent nucleotides as 3′,5′-phosphodiester bonds on riboses(Usher and McHale, 1976). Such bonds have not been documented as forming fromorthophosphate but only from relatively high-energy polyphosphate forms such aspyrophosphate (Chyba and McDonald, 1995). Polyphosphates are uncommon innature, but Yamagataet al. (1991) have shown experimentally, and identified inmodern volcanic emissions, the presence of pyrophosphate and tripolyphosphate,the compounds metastabilized by rapid quenching following explosive eruption.
Upon volcanic eruption, the polyphosphates could migrate to soil and ground-water by dry deposition or rain-out. Once in soil and groundwater, the polyphos-phatic anions would be subject to scavenging by, and concentration on, Al-hydroxi-des which would have positively charged surfaces at the pHs expected in the sys-tem. This idea is bolstered by the observation that P has been found to concentratein gibbsite and other authigenic Al phases in modern basaltic soils (Williams and

68 JOHN WASHINGTON
Walker, 1969). A large fraction of the Al-scavenged P undoubtedly would havebeen occluded and unavailable for further reaction; however, in modern soils, sig-nificant fractions of P associated with Al(OH)3 are not occluded (Williams andWalker, 1969).
In areas of significant P accumulation, however, probably a more importantfate for P was precipitation as ferrous pyrophosphates. Lindsay (1979) providesa solubility product for Fe2P2O7 of log K= –17.60. Assuming pH∼ 4.9, PCO2
∼ 0.2 atm and aFe2+ to be controlled by equilibrium with siderite, as describedabove, then Lindsay’s (1979) values for the solubility of ferrous pyrophosphateand siderite, and the dissociation of anions of pyrophosphoric acid, can be used tocalculate an equilibrium activity for H2P2O2−
7 of about 10−6.1 M. This activity is inthe range reported for orthophosphate in modern soils (Hetrick and Schwab, 1992),suggesting the feasibility of their existence despite scavenging by Al hydroxides.
An alternative possible source of polyphosphates in the proposed environmentis the heating of hydroxyapatite to 100◦C in the presence of urea and ammoniumchloride (Osterberget al., 1973).
6. Purine and Pyrimidine Production
RNA bases include two purines, adenine and guanine, and two pyrimidines, cytosineand uracil. This scenario calls for N-bearing, chemically reducing, primordial hy-drothermal fluids to upwell from depth to a confined-aquifer setting where theycan mix with CH2O-rich meteoric waters leaking downward from an unconfinedaquifer and react to form the nuclear bases (Figure 1).Reduced-N compounds arenot stable in the presence of the mildly oxidizing Archean atmosphere assumedhere (Kasting, 1993); however, since these N-bearing fluids are proposed to beupwelling under a low-permeability clay lens, they would be largely isolated fromthese oxidizing conditions during synthesis.
6.1. ADENINE
Adenine (C5H5N5) was synthesized under reasonable early-Earth conditions byOro and co-workers in the early 1960s (Oro, 1960; Oro and Kimball, 1961, 1962)by refluxing aqueous solutions containing 1 to 15 M HCN and NH3. Such heatedconditions are consistent with the hydrothermal origin of the juvenile waters pro-posed as the source of the bases in this theory.
As noted above, 1+ M HCN has not been shown to have been present in Archeanjuvenile fluids; however, Ferriset al. (1978) have produced adenine under moreconservative conditions of 0.1 M HCN, pH≈9 and room temperature. The yieldsin this procedure were not quantified, but they probably were not high. It is possiblethat more aggressive conditions or a longer reaction time might have given higheryields. Also, dilute HCN solutions might have been concentrated by distillation as

ORGANIC SYNTHESIS IN AQUIFERS 69
described above for CH2O or by chromatographic isolations as described in Section8.
6.2. GUANINE
Guanine (C5H5N5O) has been synthesized under prebiotic conditions by thecondensation of 4-aminoimidazole-5-carbonitrile (C3H6N4O; AICN) with urea(H2NCONH2) (Ferris and Hagan, 1984). Ferris and Hagan (1984) also describesynthesis of guanine from 4-aminoimidazole-5-carboxamide (C3H6N4O; AICA)and potassium cyanate (KCNO). AICN has been formed from reaction of diamino-maleonitrile ((CN)2C2(NH2)2) and formamide (HCONH2) (Ferris et al., 1972),both of which are produced in condensation of HCN solutions (Ferris, 1992; Ferriset al., 1972; Oro and Kimball, 1962). Urea is produced by hydrolysis of HCN oli-gomers (Ferris, 1992; Ferriset al., 1978). Oro and Kimball (1962) found that AICAand other imidazole derivatives also are produced by condensation of solutionsof HCN. Potassium cyanate can be produced by hydrolysis of cyanogen (C2N2)(Ferris and Hagan, 1984), which can be produced from HCN by several methods.
6.3. CYTOSINE
Cyanoacetaldehyde (OHCCH2CN) has been reacted with guanidine ((NH2)2CNH)to form 2,4-diaminopyrimidine (C4H6N4) which, in turn, hydrolyzes to form cytosine(C4H5N3O) (Ferris and Hagan, 1984). Cyanoacetaldehyde is produced by hydro-lysis of cyanoacetylene (HC3N) which can be produced in a reduced carbon-nitrogenenvironment by spark discharge (Ferris and Hagan, 1984). Guanidine has beenproduced by oligomerization of HCN (Loweet al., 1963). Sanchezet al. (1966)also produced cytosine by condensation of cyanoacetylene with urea.Synthesis ofcytosine by the method of Sanchez was performed by heating dry samples. Dry-heatconditions would be reasonable in ground-water discharge areas during dry timeswhen the water table would drop to let previously wet materials dry in the vadosezone.
6.4. URACIL
Uracil (C4H4N2O2) also is produced by reacting cyanoacetaldehyde with guanidine(Ferris and Hagan, 1984).
More generally, uracil (C4H4N2O2) is efficiently formed from cytosine by deam-inization; Shapiro and Klein (1966) found that cytosine deaminates to form uracilat pHs as high as 5.0, and that these conditions do not deaminate the adenineand guanine nucleosides. Interestingly, for Kasting’s ‘base-case’ conditions as-sumed here of PCO2 = 0.2 bar, atmospheric water equilibrating with gibbsite un-der open-system (vadose) conditions would have a pH≈ 4.9 so this deaminationcould proceed. Conversely, for closed-system (groundwater) conditions, pH≈ 5.6,presumably inhibiting this reaction.

70 JOHN WASHINGTON
7. Isotope Isolation
The most compelling evidence for life at 3.8 bybp is the identification of isotopic-ally light, reduced C in rocks from Isua, Greenland, on the order ofδ13C≈ –19hrelative to Pee Dee Belemnite (PDB, Rosing, 1999). This value falls in the rangecommon for organic matter in Recent sediment ofδ13C ≈ –10 to –30h, and isrelatively close to the average coalδ13C of –25h and the average petroleumδ13Cof –28h relative to PDB (Faure, 1977).Because of this longstanding isotopic-fractionation pattern, hypothetical processes leading to the earliest life should beexpected to yield light-C composition.
In RNA, C is present in the nucleic acids and ribose. For the hypothesis de-scribed in this paper, nucleic acids are produced from juvenile waters upwellingfrom extruding mantle material. In their study of mantle-source C in vesicles ofmodern MORB, Pineau and Javoy (1994) found that the reduced-C fraction hadδ13C≈ –16h relative to PDB, a value close to the –19h of Rosing (1999). It isreasonable to assume similar fractionations might have occurred during formationof reduced C in juvenile fluids during the Archean.
For the hypothesis described in this paper, ribose is produced by polymerizationof atmospheric-source CH2O which is produced photolytically from reduction ofCO2 (Pintoet al., 1980). Assumptions implicit in inferring life at 3.8 bybp basedon δ13C values for reduced C seems to be that theδ13C for the atmosphere wasroughly similar to the present and that biogenic processes active at that time hadeffects on fractionation similar to that of modern life. As such, a reasonable firstapproximation ofδ13C for the Archean atmosphere is that it was about the sameas at present,δ13C ≈ –7h (Faure, 1977). This assumption is consistent with: 1)detailed isotopic studies of the global C budget which document no gross, long-term changes inδ13C for organic C or carbonate C over the last 2.5 bybp (DesMaraiset al., 1992); and 2) the observation that the dominant Earthly pool for C,carbonate rocks, also haveδ13C≈ –7h to 0h.
The concentrations of CH2O produced by atmospheric photolysis hypothetic-ally were controlled kinetically, not by equilibrium (Pintoet al., 1980). Isotopicfractionation of such trace atmospheric gases usually is controlled by kinetic iso-topic effects (Conny and Currie, 1996) that generally favor faster reaction of thelighter isotope; the relative rates being an exponential function of differences in thepopulations of vibrational energy levels between the initial reactant and the trans-ition state for each isotope (Bigeleisen, 1949). The differences in the populations ofvibrational energy levels between initial reactants and the transition state often areapproximated by the differences in zero-point energies (ZPE, Conny and Currie,1996).
The value of the kinetic isotopic fraction factor (k12/k13) for the photolyticreduction of CO2 to CH2O has not been reported, but the value has been repor-ted for the photolytic oxidation of CH4 to CH2O as being k12/k13(CH4+OH6CH2O) =1.0054 (Cantrell, 1990). For the production of CH2O by both reduction of CO via

ORGANIC SYNTHESIS IN AQUIFERS 71
HCO· and oxidation of CH4 via CH3·, the initial reactants and the transition statesdiffer by a single H+. According to the Schrodinger Equation, ZPE is inverselyproportional to the mass of the molecule (Atkins, 1982), so the differences inZPE between CO and HCO·, and CH4 and CH3· probably are not grossly dis-similar. Based on this reasoning, the kinetic fraction factor for the reduction ofCO2 to CH2O is a reasonable first-order approximation of the kinetic fractionationfactor for the oxidation of to CH4 to CH2O. Assuming thatδ13C ≈ –7h for theArchean atmospheric reservoir and that k12/13 = 1.0054 is roughly applicable tothe photolytic production of CH2O from CO2, δ13CCH2O≈ –12.3h in the Archeanatmosphere.
Dissolution of CH2O in rainwater and distillation of CH2O solutions both in-volve phase changes. I have not found fractionation factors for reactions exchan-ging CH2O between air and water. However, because of its similar compositionand because it hydrates upon dissolution, the fractionation factor for CO2-H2CO3
(αH2CO3−CO2) is a reasonable first-order approximation of the fractionation factorfor CH2O-H2C(OH)2 (αH2C(OH)2−CH2O). Also, sinceαH2CO3−CO2 is closer to unitythan most fractionation factors, its use in estimating isotopic fractionation for othercompounds is conservative. Deineset al. (1974) reported the variation ofαH2CO3−CO2
with temperature as:
1000 lnαH2CO3−CO2 = 0.0063(106/T2) - 0.91 (8)
where T is temperature in degrees kelvin. Using equation 8, for T = 278◦K andδ13CCH2O ≈ –12.3h, and assuming the atmospheric reservoir of CH2O is largerelative to the amount dissolved in rainwater,δ13CH2C(OH)2≈ –13.1h.
Concentration of light C during distillation in the groundwater system will beaffected by depletion of the light isotope from the source water as distillation pro-ceeds so Rayleigh-distillation effects must be accounted for (Faure, 1977). Theresults of several Rayleigh distillation steps on [H2C(OH)2] and δ13CH2C(OH)2 aresummarized in Table II for distillations wherein 10% of the each parcel of ground-water is boiled and recondensed. The concentrations are based on the least-squaresregression of Farberov and Speranskaya (1955) data (Figure 3); theδ13CH2C(OH)2
values are based onαH2CO3−CO2 calculated for 373◦K using equation 8 and theRayleigh distillation equation (Faure, 1977). As Table II shows, as few as 8 distil-lation steps will produce waters havingδ13CH2C(OH)2 ≈ –19h; a number of stepsthat is remarkably close to the 4 estimated necessary for the formose reaction toproceed. Such waters could be diluted to lower, but still effective, concentrationswith little effect on the isotopic composition. Also, note that ifαH2C(OH)2−CH2O is alittle larger than theαH2CO3−CO2 value used here to estimateδ13CH2C(OH)2, then forthe assumed conditions,δ13C necessarily would be as negative as the documentedvalues for Archean reduced C.
The calculations described above show that reactions in the ribose-producingsequence up through distillation can account for isotopic compositions of ribose-source C being consistent with biologic residue. However, additional isotope frac-

72 JOHN WASHINGTON
tionation seems likely to have occurred by: 1) kinetic isotope effects in the formosereactions; and 2) chromatographic separation during transport through clays (Co-plen and Hanshaw, 1973). Also, Table II shows that isotopic fractionation becomeseven more pronounced as distillation proceeds beyond 8 steps. Hence, it appearsquite reasonable that fluids mixing nucleic acids of aboutδ13C≈ –16h, or evenheavier C, with ribose formed by the above-described process could yield RNA inthe rangeδ13C≈ –19h.
It is interesting to note that the largest kinetic isotope effect in modern bio-logical systems comes about during the fixation of C from CO2 by ribulose-1,5-bisphosphate carboxylase (RuBP, Schidlowski, 1982) in the Calvin Cycle; andRuBP is synthesized by DNA (Zubay, 1993). Since it is commonly argued thatDNA did not develop for some time after life evolved, some as yet unidentifiedbiological process should have been active during the period intervening betweenthe above-hypothesized, inefficient process and the advent of DNA.
8. Nucleotide Nucleation
Nucleosides are assembled from the ribose and bases, nucleotides are formed fromnucleosides and phosphates, and oligonucleotides are chains of nucleotides. Thehypothetical formation of the oligonucleotide components was described above;however, the locations for formation of these components are separate, so the com-ponents must be transported to a common location. Moreover,for ribose and thebases, they must be separated from complex witches’ brews of sugar enantiomersand similar compounds, many of which probably would have hindered nucleotideand oligonucleotide formation (Joyce, 1989). In particular, Joyce et al. (1994)found that template-directed oligomerization reactions are inhibited in racemicmixtures by molecules of opposite-handedness. Finally, these components shouldmix in an environment similar to experimental conditions where their reaction hasbeen confirmed to proceed.
8.1. ISOLATION OF NUCLEOTIDE REACTANTS FROM COMPLEX SOLUTIONS
Upon formation, the ribose and nuclear bases would need to be transported to acommon location in order to be assembled into nucleosides and nucleotides. Thiswould take place by groundwater advection from their locations of formation, nearthe top of the watershed in the unconfined aquifer for ribose and up from theconfined aquifer for the bases. In both cases, the solutes would migrate along theirflowpaths through primary minerals, hydroxides and clays. Groundwater migra-tion is well documented to induce chromatographic separation of solutes (Appelo,1996). This separation has been shown to be especially effective during migrationthrough clays (Coplen and Hanshaw, 1973; Hanshaw and Coplen, 1973). Alsonucleic acids are separated in modern HPLC columns composed of hydroxyapatite

ORGANIC SYNTHESIS IN AQUIFERS 73
(Supelco, 1998), a mineral common in volcanic settings (Deeret al., 1982); re-member the stable C analyses suggesting the presence of life in the Isua rocks wereperformed on single apatite crystals (Mojzsiset al., 1996). With all this in mind,it seems probable that complex solutions would have been subject to significantchromatographic separations as they passed through vast, mineralogically complexwatersheds on the flanks of volcanos.
8.2. CHIRAL SEPARATION OFD-RIBOSE FROM OTHER SUGARS
There have been no detailed analyses of all the species formed in the alumina-,clay-, hydroxide-, or Pb-catalyzed formose; however, assuming the product mix issimilar to the alkali-catalyzed formose reaction,ribose was just a fraction of thereaction products that appear to include tens of species (Shapiro, 1988). Also, theformose reaction produces racemic mixes of all the chiral products. It is from thiscomplex mixture that the correct enantiomer of ribose would have to be extracted,as it turns out, D-ribose.
Aqueous pentoses and hexoses are subject to isomerization, bond fission andother degradative reactions; ribose, for example, isomerizes to arabinose and ribu-lose (Shapiro, 1988). Arabinose degrades to erythrose, thence to mesotartaric acid(Morrison and Boyd, 1959). Mesotartaric acid is optically inactive. Other sug-ars, however, give rise to chiral tartaric acid. For example, xylose degrades tothreose thence to chiral tartaric acid (Morrison and Boyd, 1959). It is with saltsof tartaric acid that Pasteur first discovered the phenomenon of enantioselectivecrystallization from racemic solutions (Jacques, 1993). For several of these salts,the enantiomers differ in solubility (Jacqueset al., 1981). Since tartaric acid isa probable decomposition product of pentose solutions and several salts of tar-taric acid have been shown to precipitate optically active crystals, it is reasonablethat such crystals could be expected downgradient of the formose-reaction zone,supersaturation being achieved by cooling from distillation temperatures or byevaporation.
Such optically active crystals, deposited in an aquifer, could induce enantiose-lective chromatography of racemic ribose, essentially acting as a fixed-chiral-phaseliquid chromatograph similar to that described by Dhanesar and others (1988;Wainer and Doyle, 1983). Alternatively, the optically active crystals might dissolvein dilute solutions and sorb to clays to form chiral clays (e.g., Fraileet al., 1998)that could act as the fixed chiral phase. Note that modern biological moleculesinclude D-ribose and L-enzymes; chiral chromatography allows for this possibil-ity since these molecules probably would have different locations of synthesis sodiffering ‘retention times’ could allow for them to be isolated at similar locations.
8.3. FORMATION OF NUCLEOSIDES, NUCLEOTIDES AND OLIGONUCLEOTIDES
Nucleosides have been produced with reasonable yield from ribose and purines indry heat (Fuller et al., 1972)and from solutions of ribose and purines that have

74 JOHN WASHINGTON
been subjected to UV radiation (Ponnamperumaet al., 1963a). The environmentsof both of these syntheses are reasonable possibilities. The dry heat could occur inbaking muds under low groundwater conditions and the UV radiation could occurin a warm pool or seep of a discharge zone.
Attempts to form pyrimidine nucleosides under plausible prebiologic condi-tions have not been as successful as for the purines, yields being < 0.1% (Joyce,1989). However, the pyrimidine nucleosides could have been concentrated, andthe purine nucleosides separated from by-product impurities, by further chroma-tographic separations. Such an effect would be possible at a midslope ephemeralseep or wetland where the nucleosides are formed during dry times and then, duringthe next wet season, transported back into the groundwater system via downwardhydraulic gradients. Distillation also might affect concentration of the pyrimidinenucleosides.
Nucleotides have been produced from nucleosides, again under dry-heat condi-tions as opposed to the saturated environments normally proposed for origin of life.Perhaps the most relevant synthesis of activated nucleotides for our setting is alsoone of the simplest, by dry heating of nucleosides with solid calcium pyrophos-phates (Becket al., 1967). This reaction seems likely because the pyrophosphatesalt of Ca used in these experiments is very similar to the ferrous pyrophosphatehypothesized above to be present.
Other possibilities include, Lohrmann and Orgel (1971) produced nucleotidesfrom nucleosides, urea and phosphate by heating to 65–100◦C in the presenceof hydroxyapatite. This same reaction also can proceed without heating if cyano-gen (C2N2) or cyanacetylene are added to the mix (Lohrmann and Orgel, 1968).Lohrmann (1975) produced nucleoside polyphosphates from nucleoside mono-phosphates and trimetaphosphate.
In addition, Ponnamperuma and others (1963b) reported the synthesis of aden-osine triphosphate (ATP) by UV irradiation of adenine, ribose and ethyl metaphos-phate. This synthesis has not been confirmed independently. Also, the presenceof ethyl metaphosphate under prebiologic conditions needs to be addressed (per-sonal communication from James Ferris). If these limitations are tackled, however,such a synthesis might be especially relevant because: 1) ATP is the ‘universalenergy intermediary in contemporary terrestrial organisms’ (Ponnamperumaet al.,1963b); 2) adenine is the easiest of the four RNA bases to produce and, likely, waspresent in the highest concentrations for the conditions described herein; and 3) fora groundwater discharge zone, exposure of concentrated adenosine-polyphosphatesolutions to UV radiation is easy to imagine as occurring on tidal flats or similarsettings as described above.
These proposed methods of nucleotide synthesis also have the appeal of anothermetabolic analogue in modern life. In modern life, nucleotides are formed almostentirely by two independent pathways, the de-novo synthesis and by reconstruc-tion from purine bases through the addition of a ribose-phosphate moiety (Zubay,1993). A problem in using either of these pathways as an analogue model for

ORGANIC SYNTHESIS IN AQUIFERS 75
prebiologic synthesis of nucleotides is that for both pathways an essential precursoris phosphoribosylpyrophosphate (PRPP) and PRPP has only been synthesized bio-logically (Zubay, 1993). However, a third pathway for the synthesis of nucleotides,the Salvage Pathway, also proceeds in minor amounts in modern life. The SalvagePathway occurs by phosphorylation of nucleosides and it does not require PRPP asa precursor (Zubay, 1993); the Salvage Pathway is the same general synthesis routeas that proposed in this scheme for the generation of nucleotides: base, nucleoside,nucleotide.
Oligonucleotides have been formed from nucleotides by various schemes. Mostsyntheses have been on mineral surfaces and most of these have achieved oligomersof only about 5 to 10 monomers long (Dinget al., 1996; Ferris and Ertem, 1992).It is generally believed, however, that 30-mers to 60-mers would be required forautoreplication. Recently, Ferriset al. (1996) succeeded in synthesizing oligomersup to 55 monomers long, with the principal products being 20–40mers.This syn-thesis was done in an alternating unsaturated-saturated setting on montmorillonitesurfaces with successive feedings of monomer solutions, a scenario that is hard toimagine near a deep-sea vent. As noted above, montmorillonite can be expectedin our proposed environment so such a scenario can be easily imagined at an eph-emeral spring or on a mud flat discharge zone subjected to repeated wettings anddryings. The wet-dry cycle could be caused by seasons, sporadic recharge eventsor simply tidal fluctuations.
Worth noting is the fact that RNA is subject to hydrolytic degradation in bulkaqueous environments such as deep-sea vents (Pace, 1991), a problem avoided bysetting RNA synthesis in or near the vadose zone as we have done. Also notableis the fact that these successful oligomerizations have been conducted in pH≈8 solutions (Ertem and Ferris, 1998) and this pH is in the range expected forgroundwaters equilibrated with montmorillonites (Ellis, 1979; Nesbitt and Wilson,1992).
Ertem and Ferris (1998) identified montmorillonite’s catalytic activity for oli-gomerization as being located in the clay interlayer. With this in mind, it is in-triguing to remember that halloysite clay is common in volcanic soils. Unlike mostclays, halloysite exists as a tube having an open interior diameter of about 200Å (van Olphen, 1977) or about 10× the ∼ 20 Å diameter of DNA (Morrisonand Boyd, 1959). With the cation-enriched diffuse layer just off its surface, hal-loysite might provide an ideal environment for the sorption (Akimet al., 1998)and oligomerization of the RNA having the negatively-charged, phosphorated outersurface.
In completing the story, several additional points are noteworthy: 1) the expec-ted relative locations for this hypothetical groundwater system of the Al-hydroxides,with which Gabel and Ponnamperuma had the most success in the formose reac-tion, and the montmorillonites, with which Ferriset al. found their greatest successin long-chain oligomerization, are ideal for the reaction series to proceed; 2) withlarger molecules being synthesized in and near a clay-lens groundwater system,

76 JOHN WASHINGTON
as proposed herein, rates of water movement can be on the order of centimetersper year in some areas, and highly ordered molecule assembly might well be pro-moted in settings where molecular motion is dominated by diffusion rather thanby mass flow; 3) ion distribution in clay lenses often is modeled with Donnan-equilibrium concepts that were first applied to equilibrium across cell membranes,so this proposed setting might promote primitive-metabolism type reactions; 4)excepting atmospheric synthesis of formaldehyde, this proposed genesis can beatmosphere-independent, shielded from threat of late meteor bombardment; 5) re-cent RNA analysis and modeling (Galtieret al., 1999) suggests that the earliestorganisms were not from the hot settings one would expect at deep-sea vents, butfrom environments of closer to 40◦C, a temperature that suits this hypothesis.
Given the concept of forming oligonucleotides with Earth systems providing en-vironments rich in requisite high-energy compounds and clays functioning as crudemembranes, such a setting might be thought of as a primitive “geometabolism”.In this light, Miller’s warm primordial soup (1974), Wachterhauser’s chemoauto-trophs (Wachterhauser, 1990) and Russell and Hall’s protomembrane (1997) arenot necessarily at odds but potentially pieces of the same puzzle.
Endnote: ‘The feeble force that was life’s starting point, like the compellingstrength that from it sprang and took and gave, ordained to shape its own design,assimilating first like elements, and then unlike, that force is now divested of allrights and privileges... by mixing many hundred substances – the mixture is whatmatters – carefully and seal it tight with clay in a retort, then re-distill it properly,our secret labors will be finished. It works! The moving mass grows clearer, andmy conviction the more certain: what’s been extolled as Nature’s mystery can beinvestigated, if but Reason dare, and what she used to let be just organic we canproduce by crystallizing.’ the character Wagner in Faust Part II, Act II by Goethe.
Acknowledgments
I thank Arthur Rose, James Ferris, Martin Schoonen, Robert Swank, Wayne Gar-rison and Paul Chen for their reviews of drafts of this paper, their thoughtfulideas and helpful criticism that resulted in substantial improvements. The researchdescribed herein was developed by the author, an employee of the U.S. Environ-mental Protection Agency, on his own time. It was conducted independently ofEPA employment and has not been subjected to the Agency’s peer and adminis-trative review. Therefore, the conclusions and opinions drawn are solely those ofthe author and should not be construed to reflect the views of the Agency.

ORGANIC SYNTHESIS IN AQUIFERS 77
References
Akim, L. G., Bailey, G.W. and Shevchenko, S. M.: 1998, ‘A Computational Chemistry Approachto Study the Interactions of Humic Substances with Mineral Surfaces’, in G> Davies and E. A.Ghabbour (eds),Humic Substances: Structures, Properties and Uses, pp. 133–145, The RoyalSociety of Chemistry.
Appelo, C. A. J.: 1996, ‘Multicomponent Ion Exchange and Chromatography in Natural Systems’,in P. C. Lichtner, C. I. Steefel, E. H. Oelkers, E. H. (eds),Reactive Transport in Porous Madia,Vol. 34, pp. 193–228, Mineralogical Society of America.
Atkins, P. W.: 1982,Physical Chemistry, W. H. Freeman and Co.Awramik, S. M.: 1981, ‘The Pre-Phanerozoic Biosphere – Three Billion Years of Crises and Op-
portunities’, in M. Nitecki (ed),Biotic Crises in Ecological and Evolutionary Time, pp. 83–102,Academic Press.
Awramik, S. M.: 1982, ‘The Pre-Phanerozoic Fossil Record’, in H. D. Holland and M. Schidlowski(ed),Mineral Deposits and the Evolution of the Biosphere, pp. 67–81, Springer-Verlag.
Awramik, S. M., Schopf, J. W. and Walter, M. R.: 1983,Precambrian Res.20, 357–374.Beck, A. R. L. and Orgel, L. E.: 1967,Science157, 952.Bethke, C. M.: 1998, ‘The Geochemist’s Workbench’, University of Illinois.Bigeleisen, J.: 1949,J. Chem. Phys.17, 675–678.Borchardt, G.: 1989, ‘Smectites’, in J. B. Dixon and S. B. Weed (eds),Minerals in Soil Environments,
pp. 675–727, Soil Science Society of America.Brown, G.: 1961,The X-Ray Identification and Crystal Structures of Clay Minerals, Mineralogical
Society.Cantrell, C. A.: 1990,J. Geophys. Res.95, 455–462.Chyba, C. F. and McDonald, G. D.: 1995,Ann. Rev. Earth Planet. Sci.23, 215–249.Conny, J. M. and Currie, L. A.: 1996,Atmospheric Environment30, 621–638.Coplen, T. B. and Hanshaw, B. B.: 1973,Geochim. Cosmochim. Acta37.Deer, W. A., Howie, R. A. and Zussman, J.: 1982,An Introduction to the Rock Forming Minerals,
Halsted Press (U.S.A. distributor).Deines, P., Langmuir, D. and Harmon, R. S.: 1974,Geochim. Cosmochim. Acta38, 1147–1164.Des Marais D. J., Strauss, H., Summons, R. E. and Hayes, J. M.: 1992,Nature359, 605–609.Dhanesar, S. C. and Gisch, D. J.: 1988,J. Chromatog.461, 407.Ding, P. Z., Kawamura, K. and Ferris, J. P.: 1996,Orig. Life Evol. Biosphere26, 151–171.Dixon, J. B.: 1989, ‘Kaolin and Serpentine Group Minerals’, in J. B. Dixon and S. B., Weed (eds),
Minerals in Soil Environments, pp. 467–526, Soil Science Society of America.Dong, S. and Dasgupta, P. K.: 1986,Environ. Sci. Technol.20, 637–640.Ellis, A. J.: 1979, ‘Explored Geothermal Systems’, in H. L. Barnes (ed),Geochemistry of Hydro-
thermal Ore Deposits, pp. 632–683, John Wiley and Sons.Ertem, G. and Ferris, J. P.: 1998,Orig. Life Evol. Biosphere28, 485–499.Farberov, M. I. and Speranskaya, V. A.: 1955,J. App. Chem. of the USSR28, 205–208.Faure, F.: 1977,Principles of Isotope Geology, John Wiley and Sons.Ferris, J. P.: 1992,Orig. Life Evol. Biosphere22, 109–134.Ferris, J. P., Donner, D. B. and Lotz, W.: 1972,J. Am. Chem. Soc.94, 6968–6974.Ferris, J. P. and Ertem, G.: 1992,Science257, 1387–1389.Ferris, J. P. and Hagan, W. J., Jr.: 1984,Tetrahedron40, 1093–1120.Ferris, J. P., Hill, A. R. J., Liu, R. and Orgel, L. E.: 1996,Nature381, 59–61.Ferris, J. P., Joshi, P. C., Edelson, E. H. and Lawless, J. G.: 1978,J. Mol. Evol.11, 293–311.Fraile, J. M., Garcia, J. I., Mayoral, J. A. and Tarnai, T.: 1998,Tetrahedron: Asymmetry9, 3997–4008.Fuller, W. D., Sanchez, R. A. and Orgel, L. E.: 1972,J. Mol. Biol.67, 25–33.Gabel, N. W. and Ponnamperuma, C.: 1967,Nature216, 453–455.Gaffney, J. S., Streit, G. E., Spall, W. D. and Hall, J. H.: 1987,Environ. Sci. Technol.21(6), 519–524.

78 JOHN WASHINGTON
Galtier, N., Tourasse, N. and Gouy, M.: 1999,Science283, 220–221.Giggenbach, W. F.: 1997, ‘The Origin and Evolution of Fluids in Magmatic-Hydrothermal Systems’,
in H. L. Barnes (ed),Geochemistry of Hydrothermal Ore Deposits, pp. 737–796, John Wiley &Sons, Inc.
Gilbert, W.: 1986,Nature319(20), 618.Goethe, J. W.: 1831,Faust Part II. Princeton University Press.Hanshaw, B. B. and Coplen, T. B.: 1973,Geochim. Cosmochim. Acta37, 2311–2327.Hayes, J. M., Kaplan, I. R. and Wedeking, K. W.: 1983, ‘Precambrian Organic Geochemistry, Pre-
servation of the Record’, in J. W. Schopf (ed),The Earth’s Earliest Biosphere: Its Origin andEvolution, Princeton University Press.
Hetrick, J. A. and Schwab, A. P.: 1992,Soil Science Society of America Journal56, 755–761.Holland, H. D.: 1984,The Chemical Evolution of the Atmosphere and Oceans, Princeton University
Press.Hsu, P. H.: 1989, ‘Aluminum Hydroxides and Oxyhydroxides’, in J. B. Dixon and S. B. Weed (ed),
Minerals in Soil Environments, Soil Science Society of America.Jacques, J.: 1993,The Molecule and its Double, McGraw-Hill, Inc.Jacques, J., Collet, A. and Wilen, S. H.: 1981,Enantiomers, Racemates, and Resolutions, John Wiley
and Sons.Joyce, G. F.: 1989,Nature338, 217–224.Joyce, G. F., Visser, G. M., van Boeckel, C. A. A., van Boom, J. H., Orgel, L. E. and Westrenen v.:
1994,Nature310, 602–604.Kasting, J. F.: 1990,Orig. Life Evol. Biosphere20, 199–231.Kasting, J. F.: 1993,Science259, 920–926.Kasting, J. F., Eggler, D. H. and Raeburn, S. P.: 1993,J. Geol.101, 245–257.Knoll, A. H. and Barghoorn, E. S.: 1977,Science198, 396–398.Krishnamurthy, R., Pitsch S. and Arrhenius G.: 1998,Orig. Life Evol. Biosphere29, 139–152.Larralde, R. and Miller S. L.: 1994, ‘Kinetics of Decomposition of Ribose’,207th National Meeting
of the American Chemical Society.Lindsay, W. L.: 1979,Chemical Equilibria in Soils, John Wiley and Sons.Lohrmann, R.: 1975,Journal of Molecular Evolution6, 237–252.Lohrmann, R. and Orgel L. E.: 1968,Science161, 64–66.Lohrmann, R. and Orgel L. E.: 1971,Science171, 490–494.Lowe, C. U., Rees M. W. and Markham R.: 1963,Nature199, 219–222.Miller, S. L.: 1953,Science117, 528–529.Miller, S. L.: 1987, Which Organic Compounds Could Have Occurred on the Prebiotic Earth?Cold
Spring Harbor Symp. Quant. Biol., 17–27.Miller, S. L. and Orgel, L. E.: 1974,The Origins of Life on the Earth, Prentice Hall.Mojzsis, S. J., Arrhenius, G., McKeegan, K. D., Harrison, T. M., Nutman, A. P. and Friend, C. R. L.:
1996,Nature384, 55–59.Morrison, R. T. and Boyd, R. N.: 1959,Organic Chemistry, Allyn and Bacon, Inc.Nesbitt, H. W. and Wilson, R. E.: 1992,Am. J. Sci.292, 440–777.Oro, J.: 1960,Biochem. Biophys. Res. Commun.2, 407–412.Oro, J. and Kimball, A. P.: 1961,Arch. Biochem. Biophys.34, 217–227.Oro, J. and Kimball, A. P.: 1962,Arch. Biochem. Biophys.96, 293–313.Oro, J., Miller, S. L. and Lazcano, A.: 1990,Annu. Rev. Earth Planet. Sci.18, 317–356.Osterberg, R., Orgel, L. E. and Lohrmann, R.: 1973,J. Mol. Evol.2, 231.Pace, N. R.: 1991,Cell 65, 531–533.Pineau, F. and Javoy, M.: 1994,Earth Planet. Letts.123, 179–198.Pinto, J. P., Gladstone, G. R. and Yung, Y. L.: 1980,Science210, 183–185.Ponnamperuma, C., Mariner, R. and Sagan, C.: 1963a,Nature198, 1199–1200.Ponnamperuma, C., Sagan, C. and Mariner, R.: 1963b,Nature199, 223–226.

ORGANIC SYNTHESIS IN AQUIFERS 79
Reid, C. and Orgel, L. E.: 1967,Nature216, 455.Rosing, M. T.: 1999,Science, 674–676.Rowe, G. L., Jr., Brantley, S. L., Fernandez, J. F. and Borgia A.: 1995,Journal of Volcanology and
Geothermal Research64, 233–267.Russell, M. J. and Hall, A. J.: 1997,Journal of the Geol. Soc., London154, 377–402.Sanchez, R. A., Ferris, J. P., Orgel, L. E.: 1966,Science154, 784–786.Sanford, W. E., Konikow, L. F., Rowe, G. and Brantley, S.: 1995,Journal of Volcanology and
Geothermal Research64, 233–267.Schidlowski, M.: 1982, Stable Isotopes and the Evolution of Life: An Overview’, in H.-L. Schmidt,
H. Forstel and K. Heinzinger (eds),Stable Isotopes, pp. 95–101. Elsevier Scientific PublishingCompany.
Schidlowski, M., Appel, P. W. U., Eichmann, R. and Junge, C. E.: 1979,Geochim. Cosmochim. Acta43, 189–199.
Schidlowski, M., Hayes, J. M. and Kaplan, I. R.: 1983, ‘Isotopic Inferences of Ancient Bio-chemistries: Carbon, Sulfur, Hydrogen and Nitrogen’, in J. W. Schopf (ed),The Earth’s EarliestBiosphere: Its Origin and Evolution, pp. 149–186. Princeton University Press.
Schopf, J. W. and Packer, B. M.: 1987,Science237, 70–73.Schopf, J. W. and Walter, M. R.: 1983, Archean Microfossils: ‘New Evidence of Ancient Microbes’,
in J. W. Schopf (ed),The Earth’s Earliest Biosphere: Its Origin and Evolution, pp. 214–239,Princeton University Press.
Seader, J. D. and Kurtyka, Z. M.: 1984, ‘Distillation’, in R. H. Perry, D. W. Green and J. O. Maloney(eds),Perry’s Chemical Engineers’ Handbook, pp. 97, McGraw-Hill, Inc.
Shapiro, R.: 1988,Orig. Life Evol. Biosphere18, 71–85.Shapiro, R. and Klein, R. S.: 1966,Biochemistry5, 2358–2362.Smeck, N. E. and Ciolkosz, E. J.: 1989, Fragipans: Their Occurrence, Classification, and Genesis,
pp. 153. Soil Science Society of America.Socha, R. F., Weiss, A. H. and Sakharov, M. M.: 1981,Journal of Catalysis67, 207–217.Supelco: 1998,Supelco Chromatography Products, Supelco.Tolstikhin, I. N. and Marty, B.: 1998,Chem. Geol.147(1–2), 27–52.Usher, D. A. and McHale, A. H.: 1976,Proceedings of the National Academy of Science73, 1149–
1153.Van Olphen, H.: 1977,An Introduction to Clay Colloid Chemistry, John Wiley and Sons.Wachtershauser, G.: 1988a,Microbiol. Rev.52, 452–484.Wachtershauser, G.: 1988b,Syst. Appl. Microbiol.10, 207–210.Wachtershauser, G.: 1990,Orig. Life Evol. Biosph.20, 173–176.Wachtershauser, G.: 1992,Prog. Biophys. Mol. Biol.58, 85.Wainer, I. W. and Doyle, T. D.: 1983,Journal of Chromatography259, 465.Walker, J. F.: 1964,Formaldehyde, Reinhold Publishing Company.Walter, M. R.: 1983, ‘Archean Stromatolites: Evidence of the Earth’s Earliest Benthos’, in J. W.
Schopf (ed),The Earth’s Earliest Biosphere: Its Origin and Evolution, pp. 187–212, PrincetonUniversity Press.
Williams, J. D. H. and Walker, T. W.: 1969,Soil Science107, 22–30.Yamagata, Y., Watanabe, H., Saitoh, M. and Namba, T.: 1991,Nature352, 516–519.Zubay, G.: 1993,Biochemistry, Wm. C. Brown Publishers.Zubay, G.: 1998,Orig. Life Evol. Biosphere28, 13–26.


















