
THE JOURNAL OF BIOL~ICAL CHEMISTRY Vol. 269, No. 38, … · Cloning and Expression in EXPERIMENTAL...
9
THE JOURNAL OF BIOL~ICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc. Vol. 269, No. 38, Issue of September 23, pp. 23808-23816, 1994 Printed in U.S.A. Cloning and Expression of the Mammalian Multifunctional Protein CAD in Escherichia coZi CHARACTERIZATION OF THE RECOMBINANT PROTEIN AND A DELETION MUTANT LACKING THE MAJOR INTERDOMAIN LINKER* (Received for publication, February 18, 1994) Hedeel I. Guy and David R. Evans$ From the Department of Biochemistry, Wayne State University School of Medicine, Detroit, Michigan 48201 The multifunctional protein CAD catalyzes the first three steps in de nouo pyrimidine biosynthesis in mam- malian cells. Glutamine-dependent carbamyl-phosphate synthetase (CPSase), aspartate transcarbamylase, and dihydroorotase activities are carried by a 243-kDa polypeptide chain that is organized into discrete func- tional domains connected by interdomain linkers. One of the connecting chain segments, the DA linker bridg- ing the dihydroorotase and aspartate transcarbamylase domains, is unusually long (109 residues) and conserved in length in all eukaryotic species. A plasmid(pCK- CAD10) that encodes the entire 243-kDapolypeptide was constructed and expressed in Escherichia coli. The re- combinant protein was purified to homogeneity by ion exchange and gel filtration chromatography. The puri- fied protein had kinetic parameters that were close to those obtained for native CAD. Moreover, the CPSase activity was allosterically regulated. Gel filtration showed that the recombinant protein had the same mo- lecular mass as native CAD. Thus, this complex mamma- lian protein is expressed and folds correctly in bacterial cells and, despite its extreme protease sensitivity,can be isolated intact. A deletion mutant that lacked the DA linker was then constructed. The kinetic parameters of the mutant protein were, for the most part, unaltered, showing that the DA linker is not essential for the proper folding or optimal functioning of the individual domains. However, a significant decrease in the thermal stability of the CPSase domain suggested that the linker helps to stabilize the complex. Moreover, the channeling of carbamyl phosphate, determined by measuring the extent to which the exogenously added intermediate could dilute the endogenous carbamylphosphate pool, was appreciably reduced when the DA linker was re- moved.Thus, although the domains function autono- mously, some of the linkers are important for interdo- main interactions in CAD. CAD is a large multifunctional protein that initiates de novo pyrimidine biosynthesis in mammalian cells (1-3). The 243- kDapolypeptide is organized into functional domains (4-9) that have glutamine-dependent carbamyl-phosphate synthe- tase (CPSase’; EC 6.3.5.51, aspartate transcarbamylase Grant GM47399. The costs of publication of this article were defrayed in * This work was supported by National Institutes of Health Research part by the payment of page charges. This article must therefore be herebv marked “advertisement” in accordance with 18 U.S.C. Section ~~~~ ~ ~ 1734 solely to indicate this fact. t To whom correspondence should be addressed. Tel.: 313-577-1016; Fak 313-577-1510. tase; ATCase, aspartate transcarbamylase; DHOase, dihydroorotase; The abbreviations used are: CPSase, carbamyl-phosphate synthe- (ATCase;EC 2.1.3.21, and dihydroorotase (DHOase; EC 3.5.2.3) activities. The structural organization of the molecule renders it hypersensitive to proteases that cleave at 16 sites within chain segments that connect the domains and subdomains (9). The CAD polypeptide associates to form hexamers and other large oligomers (3, 10). The carbamyl-phosphate synthetase activity is allosterically regulated, UTP inhibits and 5-phospho-a-~-ribosyl pyrophos- phate activates (11-13). Moreover, cAMP-dependent protein ki- nase-mediated phosphorylation (14, 15) activates CPSase and abolishes UTP inhibition. In vivo, CAD is themajor regulatory locus of the pyrimidine biosynthetic pathway (16). The inter- mediates carbamyl phosphate and carbamyl aspartate have been shown to be partially channeled from one active site to the next within the CAD complex (17-19). CAD is encoded by a complex gene >25 kb in length that is interrupted by at least 37 intervening sequences (20). Early attempts to construct a full-length cDNA have resulted in trun- cated molecules that lack the coding sequenceat the 5‘-end of the molecule (20,21).We have sequenced pCAD142, the largest cDNA clone with a 6.5-kb insert (22-241, and have constructed and sequenced an overlapping clone containing the 5“coding sequences (25). The availability of these two partial cDNA clones made it possible to construct a full-length cDNA. One such cDNA molecule was expressed in mammalian cells (261, but attempts to express it in bacterial systems resulted in ex- tensive proteolytic cleavages. The compactly folded functional domains are covalently con- nected by segments of the polypeptide chain of variable lengths. One of these linkers, the DAlinker, which bridges the aspartate transcarbamylase and dihydroorotase domains, is very large (12 kDa) and very hydrophilic and has an unusual amino acid composition, rich in proline, hydroxyamino acids, and charged residues (22).The length, but not the sequence, of the DAlinker is conserved in other eukaryotic multifunctional proteins (27- 29). We suggested that this linker may function to bring the carbamyl-phosphate synthetase and aspartate transcarbamyl- ase domains into proximity to allow channeling of carbamyl phosphate (22). We report here the construction of a plasmid (pCK-CAD10) encoding the entire CAD polypeptide. When transformed into Escherichia coli, the fully functional protein is expressed and can be isolated intact. The purified recombinant protein has properties that are indistinguishable from those of CAD iso- lated from mammalian cells. A deletion mutant lacking the entire DA linker has also been expressed. While the catalytic activities of the CAD domains are not altered, the deletion reduces the stability of the complex and effectively eliminates carbamyl phosphate channeling. GLNase, glutaminase; kb, kilobase(s); DTT, dithiothreitol. 23808
Transcript of THE JOURNAL OF BIOL~ICAL CHEMISTRY Vol. 269, No. 38, … · Cloning and Expression in EXPERIMENTAL...

THE JOURNAL OF BIOL~ICAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 269, No. 38, Issue of September 23, pp. 23808-23816, 1994 Printed in U.S.A.
Cloning and Expression of the Mammalian Multifunctional Protein CAD in Escherichia coZi CHARACTERIZATION OF THE RECOMBINANT PROTEIN AND A DELETION MUTANT LACKING THE MAJOR INTERDOMAIN LINKER*
(Received for publication, February 18, 1994)
Hedeel I. Guy and David R. Evans$ From the Department of Biochemistry, Wayne State University School of Medicine, Detroit, Michigan 48201
The multifunctional protein CAD catalyzes the first three steps in de nouo pyrimidine biosynthesis in mam- malian cells. Glutamine-dependent carbamyl-phosphate synthetase (CPSase), aspartate transcarbamylase, and dihydroorotase activities are carried by a 243-kDa polypeptide chain that is organized into discrete func- tional domains connected by interdomain linkers. One of the connecting chain segments, the DA linker bridg- ing the dihydroorotase and aspartate transcarbamylase domains, is unusually long (109 residues) and conserved in length in all eukaryotic species. A plasmid (pCK- CAD10) that encodes the entire 243-kDa polypeptide was constructed and expressed in Escherichia coli. The re- combinant protein was purified to homogeneity by ion exchange and gel filtration chromatography. The puri- fied protein had kinetic parameters that were close to those obtained for native CAD. Moreover, the CPSase activity was allosterically regulated. Gel filtration showed that the recombinant protein had the same mo- lecular mass as native CAD. Thus, this complex mamma- lian protein is expressed and folds correctly in bacterial cells and, despite its extreme protease sensitivity, can be isolated intact. A deletion mutant that lacked the DA linker was then constructed. The kinetic parameters of the mutant protein were, for the most part, unaltered, showing that the DA linker is not essential for the proper folding or optimal functioning of the individual domains. However, a significant decrease in the thermal stability of the CPSase domain suggested that the linker helps to stabilize the complex. Moreover, the channeling of carbamyl phosphate, determined by measuring the extent to which the exogenously added intermediate could dilute the endogenous carbamyl phosphate pool, was appreciably reduced when the DA linker was re- moved. Thus, although the domains function autono- mously, some of the linkers are important for interdo- main interactions in CAD.
CAD is a large multifunctional protein that initiates de novo pyrimidine biosynthesis in mammalian cells (1-3). The 243- kDa polypeptide is organized into functional domains (4-9) that have glutamine-dependent carbamyl-phosphate synthe- tase (CPSase’; EC 6.3.5.51, aspartate transcarbamylase
Grant GM47399. The costs of publication of this article were defrayed in * This work was supported by National Institutes of Health Research
part by the payment of page charges. This article must therefore be herebv marked “advertisement” in accordance with 18 U.S.C. Section ~~~~ ~ ~
1734 solely to indicate this fact. t To whom correspondence should be addressed. Tel.: 313-577-1016;
Fak 313-577-1510.
tase; ATCase, aspartate transcarbamylase; DHOase, dihydroorotase; The abbreviations used are: CPSase, carbamyl-phosphate synthe-
(ATCase; EC 2.1.3.21, and dihydroorotase (DHOase; EC 3.5.2.3) activities. The structural organization of the molecule renders it hypersensitive to proteases that cleave at 16 sites within chain segments that connect the domains and subdomains (9). The CAD polypeptide associates to form hexamers and other large oligomers (3, 10).
The carbamyl-phosphate synthetase activity is allosterically regulated, UTP inhibits and 5-phospho-a-~-ribosyl pyrophos- phate activates (11-13). Moreover, cAMP-dependent protein ki- nase-mediated phosphorylation (14, 15) activates CPSase and abolishes UTP inhibition. In vivo, CAD is the major regulatory locus of the pyrimidine biosynthetic pathway (16). The inter- mediates carbamyl phosphate and carbamyl aspartate have been shown to be partially channeled from one active site to the next within the CAD complex (17-19).
CAD is encoded by a complex gene >25 kb in length that is interrupted by at least 37 intervening sequences (20). Early attempts to construct a full-length cDNA have resulted in trun- cated molecules that lack the coding sequence at the 5‘-end of the molecule (20,21). We have sequenced pCAD142, the largest cDNA clone with a 6.5-kb insert (22-241, and have constructed and sequenced an overlapping clone containing the 5“coding sequences (25). The availability of these two partial cDNA clones made it possible to construct a full-length cDNA. One such cDNA molecule was expressed in mammalian cells (261, but attempts to express it in bacterial systems resulted in ex- tensive proteolytic cleavages.
The compactly folded functional domains are covalently con- nected by segments of the polypeptide chain of variable lengths. One of these linkers, the DAlinker, which bridges the aspartate transcarbamylase and dihydroorotase domains, is very large (12 kDa) and very hydrophilic and has an unusual amino acid composition, rich in proline, hydroxyamino acids, and charged residues (22). The length, but not the sequence, of the DAlinker is conserved in other eukaryotic multifunctional proteins (27- 29). We suggested that this linker may function to bring the carbamyl-phosphate synthetase and aspartate transcarbamyl- ase domains into proximity to allow channeling of carbamyl phosphate (22).
We report here the construction of a plasmid (pCK-CAD10) encoding the entire CAD polypeptide. When transformed into Escherichia coli, the fully functional protein is expressed and can be isolated intact. The purified recombinant protein has properties that are indistinguishable from those of CAD iso- lated from mammalian cells. A deletion mutant lacking the entire DA linker has also been expressed. While the catalytic activities of the CAD domains are not altered, the deletion reduces the stability of the complex and effectively eliminates carbamyl phosphate channeling.
GLNase, glutaminase; kb, kilobase(s); DTT, dithiothreitol.
23808

Cloning and Expression in
EXPERIMENTAL PROCEDURES Plasmids and Strains-The 9.1-kb plasmid pCAD142 (21) was a gift
of George Stark (Cleveland Clinic, Cleveland, OH) and consists of pBR322 with a 6.5-kb cDNAinsert containing 2167 amino acids of CAD. The 7.1-kb plasmid pHG-GLN52 (30) consists of a 1.1-kb insert encod- ing the CAD GLNase domain in a plasmid derived from pEK81(31). The insert is located immediately downstream of the pyrBI promoter, which controls the expression ofE. coli ATCase. When transformed into the E. colipyrBI mutant EK1104 and grown in limiting uracil, the mammalian GLNase domain is expressed (30). The E. coli host strain EK1104 (F-, ara, thi, A-lac, ApyrBI, pyrF", rpsL) lacks the E. coli pyrB and pyrI genes and has a leaky pyrF mutation, so it produces only low levels of orotidylate phosphoribosyltransferase. The E. coli mutant strains used in this study each lack one of the pyrimidine biosynthetic activities. L132 (32) is defective in carA, which encodes the CPSase amidotrans- ferase subunit; C600 (32) is defective in carB, which encodes the large CPSase synthetase subunit; EK1104 and TB2 (31) are defective in pyrBI, which encodes the aspartate transcarbamylase subunits; and X7014a (E. coli Genetic Stock Center strain 5358) is defective inpyrC, which encodes dihydroorotase.
Cell Growth and Recombinant DNA Methods-Cells harboring the recombinant plasmids were grown routinely from a single colony in 50 pg/ml ampicillin. For induction of recombinant proteins under the con- trol of the pyrBI promoter, the transformed EK1104 or L673 cells were grown in minimal medium consisting of 6 giliter Na,HPO,, 3 giliter KH,PO,, 1 giliter NH,Cl, 0.5 g/liter NaC1,5 giliter casamino acids (Difco 0230), 4 giliter glucose, 0.5 mgiliter ZnS0,.7H,O, 0.5 mgiliter FeS0,.7H,O, 0.1 m~ CaCl,, 1 m~ MgS0,.7H,O, 5 mgiliter thiamine, and 10 mgiliter tryptophan supplemented with 12 pg/ml uracil and 100-200 mg/liter ampicillin. Under these conditions, there is sufficient uracil to sustain growth for 16-20 h, after which time growth is arrested or slowed, and the recombinant protein is expressed. Growth was moni- tored spectrophotometrically at 600 nm, and the cells were typically allowed to grow for 22 h aRer inoculation. The cells were harvested by centrifugation at 2,700 x g for 30 min at 4 "C in a Sorvall R3C centri- fuge. Plasmids were isolated either using a commercial rapid miniprep procedure (BIO 101, Inc.) or by CsCl gradient centrifugation (32). Transformation and preparation of competent E. coli cells were carried out by the procedure of Hanahan (33). DNAfragments were gel-purified following electrophoresis on 0.8% agarose by extraction onto glass beads (Geneclean kit) or by the low melting point agarose method (32). Re- striction digests, exonuclease I11 digestions, ligations, and other DNA methods were carried out using standard protocols (32). The method of Kunkel et al. (Ref. 35; see also Ref. 34) was used to produce site-directed mutants.
Protein Methods-Native CAD was isolated from an overproducing strain of Syrian hamster cells (BK-128) as described previously (3, 5). Both the native and recombinant proteins were stored in 0.02 M Tris- HCl, pH 7.4, 0.05 M KCl, 4 m~ aspartate, 4 mM glutamine, 1 m~ DTT, 0.01 m~ EDTA, 5% glycerol, and 30% Me,SO (storage buffer A) at -70 "C. Protein concentrations were determined by the dye-binding method of Bradford (36) using bovine serum albumin as a standard. SDS gel electrophoresis was carried out on 10% polyacrylamide gels using the procedure of Laemmli (37). The gels were stained with Coo- massie Brilliant Blue or electroblotted onto nitrocellulose for immuno- blotting (7, 38, 39). CAD antibodies were prepared as described previously (7).
Enzyme Assays-Glutamine-dependent carbamyl-phosphate synthe- tase and aspartate transcarbamylase activities were assayed using the radiometric procedures described previously (6, 17, 27, 41). The dihy- droorotase activity was measured colorimetrically in the reverse direc- tion by assaying the formation of carbamyl aspartate from dihydrooro- tate (6, 17, 41). Saturation curves were fitted to the Michaelis-Menten equation, the Hill equation, or a modified Michaelis-Menten equation that incorporates substrate inhibition (7), and the kinetic parameters were derived by nonlinear least-squares analysis using the program Minsq (Micromath).
Measurement of Carbamyl Phosphate Channeling-Carbamyl phos- phate channeling was measured using an approach similar to that used for the yeast (42) and CAD (18) complexes, in which the dilution of endogenously synthesized carbamyl phosphate is measured as a func- tion of added carbamyl phosphate. The assays were conducted at 37 "C and contained saturating 5 m~ ['4C]sodium bicarbonate (0.6 mCi/ mmol), 3.5 m~ glutamine, 1 m~ aspartate, 0.1 M KCl, 0.1 M Tris-HC1, pH 7.4, 7.5% Me,SO (v/v), 2.5% glycerol (v/v), 9.8 pg of CAD or an equiva- lent amount of partially purified protein, and a variable concentration of carbamyl phosphate in a total volume of 1 ml. Saturating concentra-
E. coli of Mammalian CAD 23809
tions of sodium bicarbonate and glutamine were used, but the concen- tration of aspartate at 1 mM was only 0.1 of the K,,,, and the reaction times were kept to 5 min to slow down the ATCase reaction. Attenuation of the ATCase activity was necessary to reduce the amount of exogenous carbamyl phosphate consumed during the incubation period, a factor that was particularly important when low concentrations of the unla- beled intermediate were used. Under these conditions, we observed no carbamyl phosphate inhibition of the CPSase activity at product con- centrations up to 5 mM, which complicated the calculation of dilution curves in previous studies (18). The concentration of the radiolabeled carbamyl phosphate plus carbamyl aspartate at the end of the 5-min incubation period was measured by the addition of 0.6 ml of 2 M am- monium chloride and heating at 90 "C for 15 min, followed by the addition of 0.4 ml of 14% perchloric acid. This procedure (43) converts the carbamyl phosphate to acid-stable urea. The reaction was found to be 90% complete over a 20-fold range of carbamyl phosphate concen- tration, so a correction of 10% was applied to the measured carbamyl phosphate concentration. The concentration of carbamyl aspartate was measured by adding 1 ml of 40% trichloroacetic acid, which converts the acid-labile carbamyl phosphate to carbonic acid, which was quantita- tively removed by heating at 90 "C for 15 min, followed by the addition of 0.1 g of powdered dry ice to purge the residual [14Clcarbon dioxide. The samples were allowed to stand overnight prior to counting. The concentration of carbamyl phosphate was determined by the difference. A second type of experiment was camed out under identical conditions, except that the sodium bicarbonate was unlabeled and exogenous ["Clcarbamyl phosphate (25 pCi/mmol) was used. In this case, the K,,, and V,, for ATCase under the conditions used for channeling could be calculated, and the total amount of radiolabeled plus unlabeled car- bamyl phosphate consumed during the 5-min assay period could be measured. The theoretical dilution curves assuming no channeling were calculated by the method outlined by Christopherson and Jones (18).
RESULTS
Construction of Recombinants Because of the large size of the insert and the consequent
rarity of unique restriction sites, the full-length CAD cDNA was cloned in a stepwise fashion starting with the plasmid pHG-GLN52 (30). This recombinant, which was derived from pEK81 (311, expresses the CAD GLNase domain under the control of the E. coli pyrBI promoter (30).
A SmaUHincII fragment of pCAD142 (CAD nucleotides 1095-3696) was inserted into the SmaI site of pHG-GLN52 (Fig. 1). The resulting plasmid, pHG-17, which encodes the GLNase domain, and most of the CPSase domain, was used to make the full-length CAD cDNA molecule.' The purified 3.7-kb XbaI fragment of pCAD142 (nucleotides 3126-6863 in the CAD coding sequence) was inserted into the XbaI site (nucleotide 3162) of the intermediate plasmid pHG-17 to form pCK- CAD10.
Expression of Recombinant Protein When the recombinant plasmid pCK-CAD10 was trans-
formed into a series of E. coli mutants defective in one of the first three activities in the pyrimidine biosynthetic pathway (Table I), complementation was observed for each mutant, in-
~
Two other recombinants were first constructed. The plasmid pHG- CAD1 was made by inserting a fragment of pCAD142 (XhoIIEglII, nucleotides 3211-6863) into the XhoIIEcoRI sites of pHG-17 after the 5'-extensions of the mismatched sites were trimmed with mung bean nuclease. The recombinant complemented E. coli mutants defective in each of the three CAD activities. Immunoblots of induced pHG-CAD1 cell extracts showed a small amount of immunoreactive protein of the
ciably higher molecular mass. Attempts to dissociate the high molecular expected molecular mass and more intensely staining species of appre-
mass form of CAD, which was assumed to be an aggregate, using re- ducing agents and a variety of detergents were unsuccessful. The plas- mid pHG-CAD2 was constructed by inserting a HindIIIIXhoI fragment of pHG-17 (nucleotides -250 to 3211) encoding the E. coli pyrBI regu- latory region and the first half of the CAD coding sequence into pCAD142. The pHG-CAD2 transformants produced a protein of the
but in relatively small amounts. expected molecular mass that complemented all of the E. coli mutants,

23810 Cloning and Expression in E. coli of Mammalian CALI
I Smal
I Xbal(3162) I &Ill
HincII fragment of pCAD142 encoding most of the CAD CPSase domain FIG. 1. Construction of recombinant plasmids. A 2.6-kb SmaIl
was inserted into the SmaI site of pHG-GLN52, a plasmid (30) that expresses the CAD GLNase domain, to form the intermediate recombi- nant pHG-17. To construct the plasmid pCK-CAD10, the 3.7-kb XbaI fragment of pCAD142 was inserted into the XbaI site of pHG-17 after the 5”phosphate groups had been removed with calf intestinal phos- phatase (CIP). Expression of CAD was under the control of the pyrBI promoter (reg (cross-hatched region)).
TABLE I Mutant E. coli strains corndemented bv DCK-CAD10
Strain Genotype Phenotype
L132 carA Lacks GLNase (mows on high NH,)
.I
C600 carB Lacks CPSase (uracil
TB2
X7014a
auxotroph)
auxotroph)
auxotroph)
PY rB Lacks ATCase (uracil
PY rC Lacks DHOase (uracil
dicating that sequences encoding each of the domains are pre- sent in the plasmid and that the sequence is in-frame through the entire 6.5-kb coding region. Moreover, enough of the protein is produced to sustain the growth of the cells.
SDS gel electrophoresis and immunoblots of cell extracts showed that the protein was produced in relatively small amounts, -0.5% of the total protein in the cell extract. How- ever, it was soluble and had the expected polypeptide molecular mass. No low molecular mass species were detected by immu- noblotting, indicating that the protein is not cleaved by endog- enous proteases. The relative CPSase, ATCase, and DHOase activities measured in cell extracts (Table 11) were close to those of CAD isolated from BHK-128 cells.
TABLE I1 Enzymatic activities of native and recombinant CAD
Enzyme Recombinant CAD (partially purified)”
activity
CPSase 20 1 200 1 ATCase 633 32 7500 38 DHOase 277 14 2200 11
Native CAD (purified)
nmol/min/mg Ratio nmol/min/mg Ratio
“The recombinant protein was partially purified by one cycle of DEAE chromatography as described under “Results.”
Purification of Recombinant CAD A 4-liter culture of pCK-CALllO/EK1104 was grown for 22 h
in minimal medium supplemented with 12 pg/ml uracil. The cells were harvested; resuspended to a final volume of 10 ml in 0.05 M Tris-HC1, pH 7.4, 1 mM DTT, 5% glycerol (suspension buffer); and sonicated on ice for 3 min. The sonicate was cen- trifuged at 29,000 x g a t 4 “C for 30 min, and the supernatant was applied to a DEAE-Sephacryl column equilibrated with suspension buffer. CAD was eluted with a linear 0-0.4 M so- dium chloride gradient. Fractions having ATCase activity were pooled, dialyzed overnight against suspension buffer, and re- applied to an identical DEAE-Sephacryl column. The column was eluted with 2 column volumes of suspension buffer and then eluted with a 0-3 M NaCl gradient. The active fractions (Fig. 2) were pooled, chromatographed on a Bio-Gel A-5m gel filtration column equilibrated with storage buffer A, and eluted with the same buffer. Fractions containing the nearly homoge- neous protein (Fig. 2) were pooled and stored at -70 “C. This procedure, summarized in Table 111, resulted in 140 pg of pro- tein that was -95% pure.
Characterization of Recombinant CAD Size and Subunit Structure-The recombinant protein,
which comigrated with native CAD on SDS gel electrophoresis, had a molecular weight of 240,000 f 25,000. Similarly, the elution profile of purified recombinant CAD on a calibrated Bio-Gel A-5m column was superimposable (Fig. 3 A ) . This result indicates that the recombinant protein, like the protein puri- fied from mammalian cells, consists primarily of hexamers.
Kinetic Parameters of Constituent Enzymes-Analysis of sub- strate saturation curves of glutamine-dependent carbamyl- phosphate synthetase, aspartate transcarbamylase, and dihy- droorotase gave the kinetic parameters summarized in Table IV.
Regulation of Carbamyl-phosphate Synthetase-ATP satura- tion curves of the carbamyl-phosphate synthetase activity showed that the protein was regulated by allosteric effectors. At 2 mM ATP, the CPSase activity was activated 282% by 5-phos- pho-a-D-ribosyl pyrophosphate and inhibited 90% by UTP. Sim- ilar results were obtained with CAD purified from BHK-128 cells.
Construction of CAD Deletion Mutant To assess the function of the linker bridging the dihydrooro-
tase and aspartate transcarbamylase domains, exonuclease di- gestions were carried out to produce a family of clones in which the linker was partially or completely eliminated. The CAD recombinant was cleaved at a unique SauI site (nucleotide 5438) near the amino-terminal end of the linker and then di- gested with exonuclease 111. Timed aliquots were taken, and the partially digested plasmid was gel-purified, religated, and transformed into the E. coli pyrBI mutant TB2. The mutants that complemented the host defect were selected for further characterization. This selection method ensures that the mu- tants recovered remained in-frame downstream of the deletion and that the ATCase domain was functional. The size of the

Cloning and Expression in E. coli of Mammalian CAD 23811
DEAE
Peak Fractions
BIOGEL A5m
c. CAD -
Peak Fractions
- 210K + 116K * 97 K - 66 K - 43 K + 29K
FIG. 2. Purification of recombinant CAD. AQ-liter culture of pCK- CAD10-transformed EK1104 cells was harvested after growing for 22 h, and the cells were resuspended in 10 ml of suspension buffer. The suspension was sonicated, centrifuged at 29,000 x g for 30 min, and then chromatographed twice on a 1.3 x 17-cm DEAE-Sephacryl column (see “Experimental Procedures”). The column fractions from the second chromatographic step were analyzed by SDS gel electrophoresis (left panel ), pooled, and then applied to a 2.7 x 36.5-cm Bio-Gel A-5m column equilibrated in storage buffer A. Also shown is the SDS gel of the peak fractions from the gel filtration column (right panel) .
deletion was determined approximately by restriction site map- ping. Some mutants had a significant portion of the DHOase and CPSase domains deleted, in which case, the corresponding activities were lost (Fig. 4). However, in those mutants, such as A100, in which the deletion was confined to the linker region, all of the catalytic activities were preserved (data not shown).
Once we had established that an appreciable part of the DA linker could be removed without significantly altering the ex- pression and folding of the protein or the function of the indi- vidual domains, a plasmid was constructed in which the entire linker was removed. Site-directed mutagenesis was used to introduce a second unique site, SstII, at the carboxyl-terminal end of the DA linker (nucleotide 5767) of pCK-CAD10. The deletion was carried out (Fig. 4) by first digesting the plasmid with SauI and then adding a single thymidine by reaction with the Klenow fragment and dTTP. The plasmid was then cut with SstII, and the extensions on both ends were removed with mung bean nuclease to allow blunt end ligation. Following religation, the deletion mutant was transformed into E. coli EK1104. The resulting plasmid was verified by digestion with BalI, a site that was created at the new junction only if all the steps described above worked properly.
Characterization of the Deletion Mutant Size and Subunit Structure-As expected, deletion of the
12-kDa DA linker did not significantly alter the mobility of the polypeptide on SDS gel electrophoresis since this region of the molecule represents 4 % of the mass of the protein. While it is difficult t o determine accurately the molecular mass of the CAD oligomer because of its large size, removal of the DAlinker increased the elution volume of the complex on Sephacryl S-500 column chromatography (Fig. 3B). Using intact CAD and E. coli ATCase as molecular mass markers, we estimated that when the linker is removed, the oligomer behaves as species with a molecular mass of -1.1 x 10‘ Da.
Kinetics of the Deletion Mutant-The plasmid pCK-CAD10 and the deletion mutant were transformed into E. coli strains L673, EK1104, andX7014a, which lackE. coli CPSase,ATCase, and DHOase, respectively. The substrate saturation curves for each of the catalytic activities of partially purified recombinant
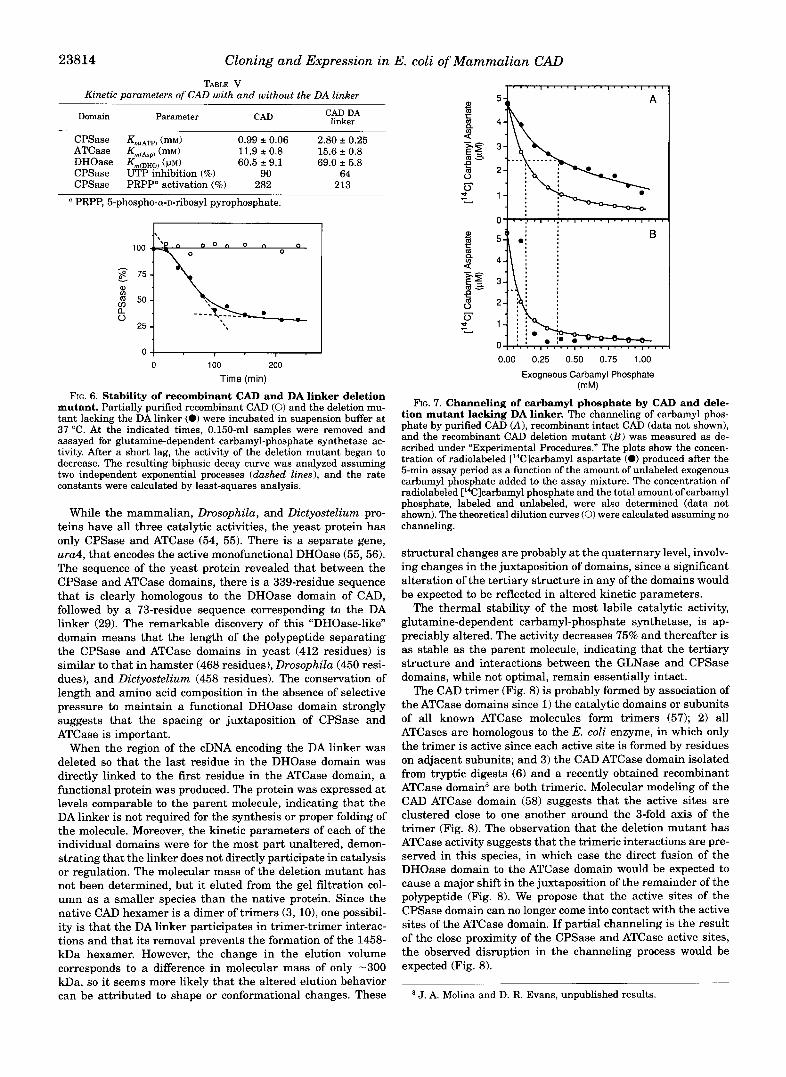
CAD and the deletion mutant were found to be similar (Fig. 5 and Table V). For glutamine-dependent carbamyl-phosphate synthetase, the K,,, for ATP increased %fold in the deletion mutant. The aspartate saturation curves for ATCase and the dihydroorotate saturation curve for DHOase were not appre- ciably altered by the removal of the DA linker. Similarly, dele- tion of the linker had little effect on UTP inhibition or 5-phos- pho-a-D-ribosyl pyrophosphate activation (Table V).
Thermal Stability of the Recombinant CAD a n d the Deletion Mutant
The thermal stability of the protein was assessed by incu- bating recombinant CAD and the deletion mutant at 37 “C and measuring the loss of glutamine-dependent carbamyl-phos- phate synthetase activity, the most labile catalytic activity, as a function of time. Under these conditions (Fig. 6), the recombi- nant protein was found to be relatively stable over the 4-h incubation period. In contrast, the activity of the deletion mu- tant decreased rapidly. After an initial 20-min lag phase, the activity decayed exponetially, with an inactivation constant of 1.8 x s-’, to 25% of the initial value. Thereafter, the CPSase activity of the deletion mutant was as stable as that of the native molecule.
Channeling of Carbamyl Phosphate We had previously suggested that the linker may be essential
for channeling of the unstable intermediate carbamyl phos- phate between the active site of CPSase, where it is produced, to the active site of ATCase, where it is used to make carbamyl aspartate (22). To test this idea, carbamyl phosphate channel- ing was measured in native and recombinant CAD and in the deletion mutant by determining the extent to which exogenous carbamyl phosphate can dilute carbamyl phosphate produced endogenously by the CPSase domain. For CAD purified from hamster cells and recombinant CAD (Fig. 7A), the amount of radiolabeled carbamyl aspartate produced from endogenously synthesized carbamyl phosphate decreased as increasing con- centrations of unlabeled carbamyl phosphate were added to the incubation mixture. There was an equivalent increase in the concentration of radiolabeled carbamyl phosphate in the assay mixture, indicating that the rate of carbamyl phosphate syn- thesis is not altered by the addition of unlabeled carbamyl phosphate (data not shown). The concentration of exogenous carbamyl phosphate required to give a 50% dilution of the radiolabeled intermediate was 370 VM.
The decrease in radiolabeled carbamyl aspartate with in- creasing exogenous carbamyl phosphate was much more pre- cipitous (Fig. 7B) when the same experiment was conducted with the deletion mutant. In this case, the addition of only 80 p~ produced a 50% dilution of the endogenous carbamyl phos- phate, indicating that channeling is severely curtailed or abol- ished in the mutant.
DISCUSSION
A full-length CAD cDNA has been constructed from two over- lapping cDNA clones, pHG-GLN52 and pCAD142. When trans- formed into E. coli mutants, the cDNA could complement de- fects in the first three steps of the pathway, indicating that the functional protein is expressed. A 243-kDa polypeptide could be seen by SDS gel electrophoresis, and immunoblotting showed that the protein had not been cleaved into smaller fragments. The ratio of carbamyl-phosphate synthetase, aspartate trans- carbamylase, and dihydroorotase was 1:38:11, close to the value obtained for the protein purified from hamster cells. The recombinant protein was purified to near homogeneity by ion exchange and gel filtration chromatography. The steady-state kinetic parameters and regulatory properties of the purified
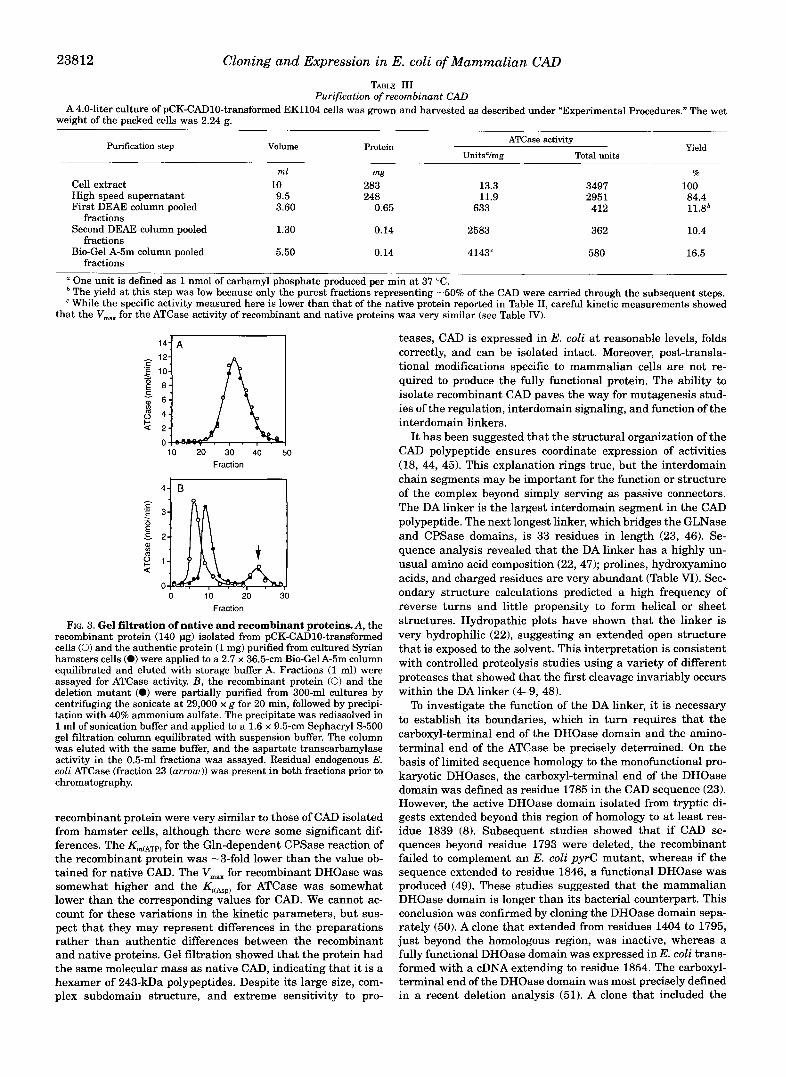
Cloning and Expression in E. coli of Mammalian CAD TABLE I11
Purification of recombinant CAD A4.0-liter culture of pCK-CAD10-transformed EK1104 cells was grown and harvested as described under "Experimental Procedures." The wet
weight of the packed cells was 2.24 E.
Purification step Volume ATCase activity
UnitsVmg Total units Protein Yield
ml mg %
Cell extract 10 283 13.3 3497 100 High speed supernatant First DEAE column pooled
Second DEAE column pooled 1.30 0.14 2583 362 10.4
Bio-Gel A-5m column pooled 5.50 0.14 4143" 580 16.5
9.5 248 11.9 295 1 3.60 412 0.65 633
84.4 11.8*
fractions
fractions
fractions One unit is defined as 1 nmol of carbamyl phosphate produced per min at 37 "C. The yield at this step was low because only the purest fractions representing -50% of the CAD were carried through the subsequent steps. While the specific activity measured here is lower than that of the native protein reported in Table 11, careful kinetic measurements showed
that the V,, for the ATCase activity of recombinant and native proteins was very similar (see Table IV).
E 6 4 2 0 10
Fraction
. 50
Fraction
FIG. 3. Gel filtration of native and recombinant proteins.A, the recombinant protein (140 pg) isolated from pCK-CAD10-transformed cells (0) and the authentic protein (1 mg) purified from cultured Syrian hamsters cells (0) were applied to a 2.7 x 36.5-cm Bio-Gel A-5m column equilibrated and eluted with storage buffer A. Fractions (1 ml) were assayed for ATCase activity. B, the recombinant protein (0) and the deletion mutant (0) were partially purified from 300-ml cultures by centrifuging the sonicate at 29,000 x g for 20 min, followed by precipi- tation with 40% ammonium sulfate. The precipitate was redissolved in 1 ml of sonication buffer and applied to a 1.6 x 9.5-cm Sephacryl S-500 gel filtration column equilibrated with suspension buffer. The column was eluted with the same buffer, and the aspartate transcarbamylase activity in the 0.5-ml fractions was assayed. Residual endogenous E. coli ATCase (fraction 23 (arrow)) was present in both fractions prior to chromatography.
recombinant protein were very similar to those of CAD isolated from hamster cells, although there were some significant dif- ferences. The Km(ATp) for the Gln-dependent CPSase reaction of the recombinant protein was -3-fold lower than the value ob- tained for native CAD. The V,, for recombinant DHOase was somewhat higher and the Kj(ABp) for ATCase was somewhat lower than the corresponding values for CAD. We cannot ac- count for these variations in the kinetic parameters, but sus- pect that they may represent differences in the preparations rather than authentic differences between the recombinant and native proteins. Gel filtration showed that the protein had the same molecular mass as native CAD, indicating that it is a hexamer of 243-kDa polypeptides. Despite its large size, com- plex subdomain structure, and extreme sensitivity to pro-
teases, CAD is expressed in E. coli at reasonable levels, folds correctly, and can be isolated intact. Moreover, post-transla- tional modifications specific to mammalian cells are not re- quired to produce the fully functional protein. The ability to isolate recombinant CAD paves the way for mutagenesis stud- ies of the regulation, interdomain signaling, and function of the interdomain linkers.
I t has been suggested that the structural organization of the CAD polypeptide ensures coordinate expression of activities (18, 44, 45). This explanation rings true, but the interdomain chain segments may be important for the function or structure of the complex beyond simply serving as passive connectors. The DA linker is the largest interdomain segment in the CAD polypeptide. The next longest linker, which bridges the GLNase and CPSase domains, is 33 residues in length (23, 46). Se- quence analysis revealed that the DA linker has a highly un- usual amino acid composition (22,47); prolines, hydroxyamino acids, and charged residues are very abundant (Table VI). Sec- ondary structure calculations predicted a high frequency of reverse turns and little propensity to form helical or sheet structures. Hydropathic plots have shown that the linker is very hydrophilic (22), suggesting an extended open structure that is exposed to the solvent. This interpretation is consistent with controlled proteolysis studies using a variety of different proteases that showed that the first cleavage invariably occurs within the DA linker (4-9, 48).
To investigate the function of the DA linker, it is necessary to establish its boundaries, which in turn requires that the carboxyl-terminal end of the DHOase domain and the amino- terminal end of the ATCase be precisely determined. On the basis of limited sequence homology to the monofunctional pro- karyotic DHOases, the carboxyl-terminal end of the DHOase domain was defined as residue 1785 in the CAD sequence (23). However, the active DHOase domain isolated from tryptic di- gests extended beyond this region of homology to at least res- idue 1839 (8). Subsequent studies showed that if CAD se- quences beyond residue 1793 were deleted, the recombinant failed to complement an E. coli pyrC mutant, whereas if the sequence extended to residue 1846, a functional DHOase was produced (49). These studies suggested that the mammalian DHOase domain is longer than its bacterial counterpart. This conclusion was confirmed by cloning the DHOase domain sepa- rately (50). A clone that extended from residues 1404 to 1795, just beyond the homologous region, was inactive, whereas a fully functional DHOase domain was expressed in E. coli trans- formed with a cDNA extending to residue 1854. The carboxyl- terminal end of the DHOase domain was most precisely defined in a recent deletion analysis (51). A clone that included the

Cloning and Expression in E. coli of Mammalian CAD 23813 TAEILE IV
Steady-state kinetic parameters of native and recombinant CAD Enzyme Substrate Km vm,
M pmol I min I mg
Recombinant CAD Gln CPSase ATCase" DHOase
Native CAD Gln CPSase ATCase" DHOase
ATP Aspartate Dihydroorotate
ATP Aspartate Dihvdroorotate
(0.672 -c 0.056) x (9.50 2 2.13) x (69.2 f 6.0) x
(2.14 f 0.16) x (6.31 f 1.4) x (60.4 2 8.9) x
0.132 f 0.003 27.5 f 3.93 3.50 f 0.05
0.194 f 0.006 26.5 f 3.11
1.88 2 0.10
The ATCase data were fit to a modified Michaelis-Menten equation that takes into consideration substrate inhibition (17): u = V,, [S]/(K, + [SI + K,4S12). The K, for CAD purified from hamster cells is 42.0 k 11.5 mM, while that for the recombinant protein is 20.9 f 4.9 mM.
GLN CPS-A CPS-B DHO DAInk ATC I I I I
ADA Ink - - - - - - - - - - - - " FIG. 4. Construction of CAD DA linker deletion mutants. The
diagram shows the CAD domain structure and the construction of the deletion mutants (the numbers correspond to amino acid residues). The DHOase domain (DHO) ends at residue 1813, which is 24 residues (lightly shaded area) beyond the region defined by sequence homology (darkly shaded area). The ATCase domain (ATC) begins at residue 1922 (darkly shaded area), which is 5 residues beyond the region defined by homology (lightly shaded area). A series of deletion mutants were iso- lated by cutting the plasmid pHG-CAD1 (see Footnote 2) at a naturally occurring S a d site (5438 base pairs) at the amino-terminal end of the DA linker (DA Ink), digesting with exonuclease 111, and selecting for in-frame deletions as described under "Experimental Procedures." The dashed lines represent the approximate locations of the deletions. To excise the entire DA linker, the plasmid pCK-CAD10 was cut at the S a d site, and then to preserve the reading frame, a complementary deoxythymidine was added to the 3"extension by reaction with dlTP and the Klenow fragment. The remaining 2 bases in the extension were removed with mung bean nuclease. At the carboxyl-terminal end of the DA linker (5767 base pairs), an unique SstII restriction site was intro- duced by site-directed mutagenesis. Following restriction, the 5'-exten- sion was removed with mung bean nuclease. The ends of the linearized plasmid were then joined by blunt end ligation with T4 DNA ligase. GLN, GLNase domain; CPS-A and CPS-B, CPSase domains A and B.
DHOase coding sequences up to residue 1806 was inactive, whereas a second clone that extended to residue 1813 was fully active. The amino-terminal end of the ATCase domain was defined (22) by homology to E. coli aspartate transcarbamylase as residue 1918. However, Bacillus subtillis ATCase has a ho- mologous sequence, but is 5 residues shorter (52), beginning at a point corresponding to residue 1923 in the CAD sequence. The amino-terminal end of the ATCase domain of the multi- functional yeast protein was also defined at this point (53). Thus, the CAD DA linker consists of 109 amino acids and ex- tends from residue 1814, the first residue following the small- est active DHOase, to residue 1922, the residue immediately preceding the start of the smallest active ATCase.
It is interesting to compare the DA linker of the mammalian protein with multifunctional pyrimidine biosynthetic com- plexes from other eukaryotes (Table VI). While the DA linker is approximately the same size in all of these organisms, ranging from 78 to 109 residues, the sequence is very poorly conserved.
""""""_
2.0
c 'E 1.5
E . - c 1.0
I 2 0.5 0
m
0 L ATP (rnM)
0 Aspartate (rnM)
10 20 30 40
0 0 100 200 300 400
FIG. 5. Kinetics of deletion mutant. The plasmid pCK-CAD10 and the DA linker deletion mutant were transformed into cell lines L673, EK1104, and X7014a, which lack E. coli CPSase, ATCase, and DHOase, respectively. The ATP saturation curve for glutamine-dependent CPSase, the aspartate saturation curve for ATCase, and the dihydro- orotate saturation curve for DHOase were measured in cell extracts of transformants lacking the corresponding E. coli background activity. The highest point of the curves for the deletion mutant (0) was nor- malized to the corresponding highest value obtained for the intact pro- tein (0). The CPSase data were fitted to the Hill equation, the ATCase data to a modified version of the Michaelis-Menten equation that in- corporates substrate inhibition, and the DHOase data to the unmodified Michaelis-Menten equation by nonlinear least-squares analysis. While precise values for V,, could not be determined because the proteins were not purified to homogeneity, the level of expression of these two proteins was similar in each cell line, and the normalization factors for the CPSase,ATCase, and DHOase curves were similar (1.1,1.3, and 1.3, respectively), indicating that the maximum velocity is not substantially different in the intact protein and the deletion mutant.
There are, however, certain common characteristics. All are very hydrophilic, with a composition ranging from 43% hydro- philic residues in CAD to 65% in the Dictyostelium protein. Although the unusually high proline content of the CAD DA linker does not occur universally, all four proteins have a large number of hydroxyamino acids and charged residues.
23814 Cloning and Expression in E. coli of Mammalian CAD
TABLE V Kinetic parameters of CAD with and without the DA linker
CAD DA linker Domain Parameter CAD
CPSase Km,ATp) (mM) 0.99 2 0.06 2.80 f 0.25 ATCase (mM) 11.9 -c 0.8 15.6 f 0.8 DHOase Km(DHOl ( p ~ ) CPSase UTP inhibition (%)
60.5 2 9.1 69.0 2 5.8 90 64
CPSase PRPP" activation (%) 282 213
PRPP, 5-phospho-a-D-ribosyl pyrophosphate.
0 100 200 Time (min)
FIG. 6. Stability of recombinant CAD and DA linker deletion mutant. Partially purified recombinant CAD (0) and the deletion mu- tant lacking the DA linker (0) were incubated in suspension buffer at 37 "C. At the indicated times, 0.150-ml samples were removed and
tivity. After a short lag, the activity of the deletion mutant began to assayed for glutamine-dependent carbamyl-phosphate synthetase ac-
two independent exponential processes (dashed lines), and the rate decrease. The resulting biphasic decay curve was analyzed assuming
constants were calculated by least-squares analysis.
While the mammalian, Drosophila, and Dictyostelium pro- teins have all three catalytic activities, the yeast protein has only CPSase and ATCase (54, 55). There is a separate gene, ura4, that encodes the active monofunctional DHOase (55,56). The sequence of the yeast protein revealed that between the CPSase and ATCase domains, there is a 339-residue sequence that is clearly homologous to the DHOase domain of CAD, followed by a 73-residue sequence corresponding to the DA linker (29). The remarkable discovery of this "DHOase-like" domain means that the length of the polypeptide separating the CPSase and ATCase domains in yeast (412 residues) is similar to that in hamster (468 residues), Drosophila (450 resi- dues), and Dictyostelium (458 residues). The conservation of length and amino acid composition in the absence of selective pressure to maintain a functional DHOase domain strongly suggests that the spacing or juxtaposition of CPSase and ATCase is important.
When the region of the cDNA encoding the DA linker was deleted so that the last residue in the DHOase domain was directly linked to the first residue in the ATCase domain, a functional protein was produced. The protein was expressed at levels comparable to the parent molecule, indicating that the DA linker is not required for the synthesis or proper folding of the molecule. Moreover, the kinetic parameters of each of the individual domains were for the most part unaltered, demon- strating that the linker does not directly participate in catalysis or regulation. The molecular mass of the deletion mutant has not been determined, but it eluted from the gel filtration col- umn as a smaller species than the native protein. Since the native CAD hexamer is a dimer of trimers (3, lo) , one possibil- ity is that the DA linker participates in trimer-trimer interac- tions and that its removal prevents the formation of the 1458- kDa hexamer. However, the change in the elution volume corresponds to a difference in molecular mass of only -300 kDa, so it seems more likely that the altered elution behavior can be attributed to shape or conformational changes. These
0.00 0.25 0.50 0.75 1.00
Exogneous Carbamyl Phosphate (mM)
FIG. 7. Channeling of carbamyl phosphate by CAD and dele- tion mutant lacking DA linker. The channeling of carbamyl phos- phate by purified CAD (A), recombinant intact CAD (data not shown), and the recombinant CAD deletion mutant ( B ) was measured as de- scribed under "Experimental Procedures." The plots show the concen- tration of radiolabeled [14Clcarbamyl aspartate (0) produced after the 5-min assay period as a function of the amount of unlabeled exogenous carbamyl phosphate added to the assay mixture. The concentration of radiolabeled ['4Clcarbamyl phosphate and the total amount of carbamyl phosphate, labeled and unlabeled, were also determined (data not shown). The theoretical dilution curves (0) were calculated assuming no channeling.
structural changes are probably at the quaternary level, involv- ing changes in the juxtaposition of domains, since a significant alteration of the tertiary structure in any of the domains would be expected to be reflected in altered kinetic parameters.
The thermal stability of the most labile catalytic activity, glutamine-dependent carbamyl-phosphate synthetase, is ap- preciably altered. The activity decreases 75% and thereafter is as stable as the parent molecule, indicating that the tertiary structure and interactions between the GLNase and CPSase domains, while not optimal, remain essentially intact.
The CAD trimer (Fig. 8 ) is probably formed by association of the ATCase domains since 1) the catalytic domains or subunits of all known ATCase molecules form trimers (57); 2) all ATCases are homologous to the E. coli enzyme, in which only the trimer is active since each active site is formed by residues on adjacent subunits; and 3) the CAD ATCase domain isolated from tryptic digests (6) and a recently obtained recombinant ATCase domain3 are both trimeric. Molecular modeling of the CAD ATCase domain (58) suggests that the active sites are clustered close to one another around the 3-fold axis of the trimer (Fig. 8). The observation that the deletion mutant has ATCase activity suggests that the trimeric interactions are pre- served in this species, in which case the direct fusion of the DHOase domain to the ATCase domain would be expected to cause a major shift in the juxtaposition of the remainder of the polypeptide (Fig. 8). We propose that the active sites of the CPSase domain can no longer come into contact with the active sites of the ATCase domain. If partial channeling is the result of the close proximity of the CPSase and ATCase active sites, the observed disruption in the channeling process would be expected (Fig. 8).
J. A. Molina and D. R. Evans, unpublished results.

Cloning and Expression in E. coli of Mammalian CAD 23815 TABLE VI
Comparison of eukaryotic DA linkers from hamster (CAD), Drosophila, Dictyostelium, and Yeast ~~
Sequence similarityb Protein" Length in Proline Hydroxy Charged Hydrophilic Hydro hobic
CAD DRO DIC YST residues residues residues residues' residues' resiluesc
CAD 15.0% 9.6% 11.3% 104 25 (24.0%) 14 (13.5%) 24 (23.1%) 45 (43.3%) 25 (24.0%) DRO 16 11.3% 5.9% 86 14 (16.2%) 16 (18.6%) 22 (25.6%) 45 (52.3%) 21 (24.4%) DIC 10 12 10.0% 94 6 (6.4%) 27 (28.7%) 18 (19.1%) 61 (64.8%) 24 (25.5%) YST 11 6 10 73 6 (8.2%) 11 (15.1%) 17 (23.3%) 34 (46.6%) 26 (35.0%)
~~ ~
Multifunctional pyrimidine complexes are from hamster (CAD), Drosophila (DRO), Dictyostelium (DIC), and yeast (YST). Partial sequencing (53) of human CAD showed that the human and hamster sequences in the DA linker region are very similar.
* Entries represent percent identities, where indicated, and number of amino acid identities based on the aligned sequences (60). The overall sequence length is 107 residues. The alignment was achieved by introducing 3,21, 13, and 34 deletions into the sequences of CAD, DRO, DIC, and YST, respectively.
and hydrophobic residues include Met, Ala, Val, Leu, Ile, Phe, and Trp. e Charged residues include Asp, Glu, Lys, k g , and His; hydrophilic residues include hydroxy residues, charged residues, and Asn, Gln, and Cys;
Intact CAD
A DHO
CAD without the DA Linker
DHO A
ASP
Channeling of CP 7 ASP No Channeling of CP
m . ATCase
-\GLN CPSase FIG. 8. Schematic representation of CAD monomer and trimer. A , shown is the CAD monomer illustrating all of the reactions catalyzed
by the complex. Bicarbonate is activated by 1 mol of ATP to form carboxyl phosphate (CBX), which reacts with ammonia generated by glutamine hydrolysis on the GLNase domain, forming carbamate (CBM). Carbamate reacts with a second mole ofATP to form carbamyl phosphate (CP) . In intact CAD, the active site of the CPSase B domain (shaded) is postulated to be in close proximity to the active site of the ATCase domain (shaded 1. Carbamyl phosphate is partially channeled to the ATCase domain, where carbamyl aspartate (CAsp) is formed. Carbamyl aspartate is then converted to dihydroorotate (DHO) by the DHOase domain. When the DA linker (wavy line) is deleted, the CPSase B and ATCase domains are no longer in close contact, carbamyl phosphate is not sequestered, and channeling is abolished. B, the CAD trimer is postulated to be held together by strong noncovalent interactions between the ATCase domains (shaded area) with the active sites (hatched region) clustered on the lower side of the trimer. Note that the GLNase domain and the CPSase A and B subdomains are represented as a single sphere for simplicity. We propose that the interactions between the ATCase domains are preserved when the DA linker is removed, but there are major dislocations of the remainder of the polypeptide chain, so the active sites (hatched region) of the CPSase domain are no longer close to those of the ATCase domain.
The channeling of carbamyl phosphate by CAD is not abso- lute. Carbamyl phosphate can freely enter the complex, and some of the endogenously synthesized intermediate is released into the bulk phase. However, the dilution of the endogenous pool of carbamyl phosphate occurs more slowly than predicted from the intrinsic kinetic parameters of the system, indicating that the intermediate is partially sequestered. In contrast, de- letion of the DA linker allows equilibration of endogenous and exogenous carbamyl phosphate, suggesting that the interme- diate is released directly into the solvent phase.
The question then becomes, is the magnitude of channeling in the native molecule sufficient to maintain the selective pres- sure necessary to conserve the linker throughout the course of evolution? While in vitro studies have shown that channeling is weak, it is possible that it may be more effective in vivo and that the leakage of carbamyl phosphate from the CAD complex may be more significant in the purified protein than in situ. Recent arguments challenge the physiological significance of channeling in vivo (45). A mutant that has an unlinked mar- ginally active ATCase has been found to grow almost as well in
media lacking uridine as the parental strain, suggesting that channeling is not essential for the normal functioning of the pathway. However, it is possible that the individual domains associate in the cell to form a noncovalently linked functional complex in which the ATCase and CPSase domains are associ- ated. This type of complex cannot be formed in the deletion mutant since it is likely that the fusion of the DHOase and ATCase domains introduces constraints that preclude contact between the CPSase and ATCase domains. Moreover, the sug- gestion that channeling is not needed in cultured cells growing under near optimal conditions does not necessarily mean that the process does not contribute to the overall economy of the cell. Channeling cannot increase the overall rate of de novo pyrimidine biosynthesis, which at steady state is set by the CPSase activity of the complex, but it could result in more rapid changes at steady state, thereby decreasing the response time to allosteric effectors (40, 59).
While the physiological significance of the limited channel- ing of carbamyl phosphate by CAD remains a moot point, its abolition in the deletion mutant is interesting from a mecha-

23816 Cloning and Expression in
nistic standpoint. The simplest explanation for our results is that the active sites of the CPSase B and ATCase domains of CAD are in proximity. Moreover, this study shows that chain segments connecting the domains can be an important part of the complex that allows interdomain interactions to occur and that stabilizes the optimal conformation of multidomain pro- teins.
Acknowledgments-We thank Dr. Carol Lusty (Public Health Re- search Institute of the City of New York, New York) and Dr. Evan Kantrowitz (Boston College, Chestnut Hill, MA) for generous gifts of plasmids and strains.
REFERENCES
2. Mori, M., Ishida, H., and Tatibana, M. (1975) Biochemistry 14,2622-2630 1. Shoaf, W. T., and Jones, M. E. (1973) Biochemistry 12, 4039-4051
3. Coleman, P. F., Suttle, D. P., and Stark, G. R. (1977) J. Biol. Chem. 262,
4. Davidson, J. N., Rumsby, P. C., and Tamaren, J. (1981) J. Biol. Chem. 266,
5. Mally, M. I., Grayson, D. R., and Evans, D. R. (1981) Proc. Natl. Acad. Sci.
6. Grayson, D. R., and Evans, D. R. (1983) J. B i d . Chem. 268,4123-4129 7. Grayson, D. R., Lee, L., and Evans, D. R. (1985) J. B i d . Chem. 260, 15840-
8. Kelly, R. E., Mally, M. I., and Evans, D. R. (1986) J. B i d . Chem. 261, 6073-
9. Kim, H., Kelly, R. E., and Evans, D. R. (1992) J. Eiol. Chem. 267,7177-7184 10. Lee, L., Kelly, R. E., Pastra-Landis, S. C., and Evans, D. R. (1985) Proc. Natl.
11. Tatibana, M., and Ito, K. (1967)Eiochem. Biophys. Res. Commun. 26,221-227 12. Levine, R. L., Hoogenraad, N. J., and Kretchmer, N. (1971) Biochemistry 10,
13. Tatibana, M., and Shigesada, K. (1972) J. Biochem. (Tokyo) 72,549-560 14. Carrey, E. A,, Campbell, D. G., and Hardie, D. G. (1985) EMBO J. 4,3735-3742 15. Carrey, E. A,, and Hardie, D. G. (1988) Eur. J. Biochem. 171,583-588 16. Chen, J.-J., and Jones, M. E. (1979) J. Biol. Chem. 254, 2697-2704 17. Mally, M. I., Grayson, D. R., and Evans, D. R. (1980) J. B i d . Chem. 266,
18. Christopherson, R. I., and Jones, M. E. (198O)J. Biol. Chem. 266,11381-11395 19. Otsuki, T., Mori, M., and Tatibana, M. (1982) J. Biochem. (Tokyo) 92, 1431-
20. Padgett, R. A,, Wahl, G. M., and Stark, G. R. (1982) Mol. Cell. B i d . 2,293-301 21. Shigesada, K., Stark, G. R., Maley, J. A,, Niswander, L.A., and Davidson, J. N.
22. Simmer, J. P., Kelly, R. E., Scully, J. L., Grayson, D. R., Rinker, A. G., Jr., (1985) Mol. Cell. Biol. 5, 1735-1741
Bersh. S. T., and Evans, D. R. (1989) Proc. Natl. Acad. Sci. U. S. A. 86,
6379-6385
52206225
U. S. A. 78,66474651
15849
6083
Acad. Sci. U. S. A. 82, 6802-6806
3694-3699
11372-11380
1437
43822386 23. Simmer, J. P., Kelly, R. E., Rinker, A. G., Jr., Zimmermann, B. H., Scully, J. L.,
Kim, H., Bergh, S . T., and Evans, D. R. (1990) Proc. Natl. Acad. Sci. U. S. A. 87.174-178 ~~
24. Simmer, J. P., Kelly R. E., Rinker, A. G., Jr., Scully, J. L., and Evans, D. R. , - ~
(1990) J. B i d . Chem. 265,10395-10402
E. coli of Mammalian CAD 25. Bein, K, Simmer, J. P., and Evans, D. R. (1991)J. B i d . Chem. 266,3791-3799 26. Musmanno, L. A,, Jamison, R. S., Barnett, R. S., Buford, E., and Davidson, J.
27. Freund, J. N., and Jarry, B. P. (1987) J. Mol. Biol. 193, 1-13 28. Faure, M., Camonis, J. H., and Jacquet, M. (1989) Eur. J. Biochem. 179,
29. Souciet, J. L., Nagy, M., Le Gouar, M., Lacroute, F., and Potier, S. (1989) Gene
30. Guy, H. I., and Evans, D. R. (1994) J. Biol. Chem. 269, 7702-7708 31. Nowlan, S. F., and Kantrowitz, E. R. (1985) J. B i d . Chem. 260, 14712-14716 32. Maniatis, T., Fritsch, E. F., and Sambrook, J. (1982) Molecular Cloning: A
Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
33. Hanahan, D. (1985) in DNA Cloning-A Practical Approach (Glover, D., ed) pp. 109-135, IRL Press, New York
34. Zoller, M. J., and Smith, M. (1987) Methods Ezymol. 164,329350 35. Kunkel, T. A,, Roberts, J. D., and Zakour, R. A. (1987) Methods Ezymol. 164,
36. Bradford, M. M. (1976) Anal. Biochem. 72,248-254 37. Laemmli, U. K. (1970) Nature 227,680-685 38. Towbin, H., Staehlin, T., and Gordon, J. (1982) Proc. Natl. Acad. Sei. U. S. A.
39. Hawkes, R., Niday, E., and Gordon, J. (1982)Anal. Biochem. 119,142-147 40. Easterby, J. S. (1989) Biochem. J. 264,605-607 41. Prescott, L. M., and Jones, M. E. (1969)Anal. Biochem. 32,408-419 42. Lue, P. F., and Kaplan, J. G. (1970) Biochim. Biophys. Acta 220,365-372 43. Carrey, E. A. (1986) Biochem. J. 236,327-335 44. Stark, G. R. (1977) *ends Biochem. Sci. 2,64-66 45. Davidson, J. N., Chen, K. C., Jamison, R. S., Musmanno, L. A,, and Kern, C. B.
46. Maley, J. M., andDavidson, J. N. (1988)Biochem. Biophys. Res. Commun. 164,
47. Williams, N. K., Simpson, R. J., Moritz, R. L., Peide, Y., CroRs, L., Minasian, 1047-1053
E., Leach, S. J., Wake, R. G., and Christopherson, R. I. (1990) Gene (Amst. 1 94,283-288
48. Rumsby, P. C., Campbell, P. C., Niswander, L. A,, and Davidson, J. N. (1984) Biochem. J. 2 1 7 , 4 3 5 4 0
49. Musmanno, L. A., Maley, J. A,, and Davidson, J. N. (1991) Gene (Amst.) 99, 211-216
50. Zimmermann, B. H., and Evans, D. R. (1993) Biochemistry 32, 1519-1527 51. Williams, N. K., Peide, Y., Seymour, K. K., Ralston, G. B., and Christopherson,
N. (1992) Somatic Cell Mol. Genet. 18,309418
345-348
(Amst. ) 79, 59-70
367-382
76,43504354
(1993) B~WSSUYS 15, 157-164
52. Lerner, C. G., and Switzer, R. L. (1986) J. Biol. Chem. 261, 1115611165 63. Davidson. .J. N.. Rao. G. N.. Niswander. L..Andreano. C.. Tamer. C.. and Chen.
R. I., (1993) Protein Eng. 6 ,333340
.~ ~ " . ~ ~ , .. ~
K.-C (1990) DNA'Cell h l . 9,667-676
Chem. 264, 836G3374 54. Nagy, M., Le Gouar, M., Potier, S., Souciet, J. L., and Herve, G. (1989) J. B i d .
55. Lacroute, E (1964) C. R. Acad. Sci. Paris Ser. ZZZ 269, 1357-1359 56. Lacroute, F. (1968) J. Bacteriol. 96,824-832 57. Bergh, S. T., and Evans, D. R. (1993) Proc. Natl. Acad. Sei. U. S. A. 90,9818-
58. Scully, J. L., and Evans, D. R. (1991) Proteins Struct. Funct. Genet. 9,191-206 59. Davis, R. H. (1967) in OrganizationalEiosynthesis (Vogel, H. J., Lampen, J. 0..
60. Simmer, J. P. (1990) The DNA Sequence of the Coding Region of the Multi- and Bryson, V., eds) pp. 303322, Academic Press, New York
functional Protein CAD, Ph.D. dissertation, Wayne State University
9822


















