
The Immunogenic Effect of Local Radiation Therapy … · Luis Alberto de la Maza Borja ... was to...
164
The Immunogenic Effect of Local Radiation Therapy in a Murine Model of Mesothelioma by Luis Alberto de la Maza Borja A thesis submitted in conformity with the requirements for the degree of Master of Science Institute of Medical Science University of Toronto © Copyright by Luis Alberto de la Maza Borja 2015
Transcript of The Immunogenic Effect of Local Radiation Therapy … · Luis Alberto de la Maza Borja ... was to...

The Immunogenic Effect of Local Radiation Therapy in a Murine Model of Mesothelioma
by
Luis Alberto de la Maza Borja
A thesis submitted in conformity with the requirements for the degree of Master of Science
Institute of Medical Science University of Toronto
© Copyright by Luis Alberto de la Maza Borja 2015

ii
The Immunogenic Effect of Local Radiation Therapy in a Murine
Model of Mesothelioma
Luis A. de la Maza Borja
Master of Science
Institute of Medical Science
University of Toronto
2015
Abstract
Malignant pleural mesothelioma (MPM) is a rare cancer that arises from the mesothelial surfaces
of the pleural cavity. It is associated with asbestos inhalation, with 70% of cases being associated
with documented asbestos exposure. MPM has a poor prognosis and the outlook has not been
improved by newer therapeutic interventions. A new approach developed in our lab, focusing on
Surgery for Mesothelioma After Radiation Therapy (SMART) showed promising results in a
phase I/II clinical trial. We believe that radiation is important to achieving activation of the
immune system and may contribute to the benefits observed in patients. The goal of this project
was to develop a mouse model to analyze the immunogenic effect of Local Radiation Therapy
(LRT) and its impact on immune cell recruitment and activation in the context of MPM. Results
from these studies may have clinical implication for the treatment of MPM, where combination
of LRT and other treatments such as immunotherapy may prove useful.

iii
Acknowledgments
I would like to thank my supervisor Dr. Marc de Perrot, the members of my lab, Licun Wu,
Yidan Zhao and Hannah Yun. Many thanks to my friend and bro Matthew Wu, together we
learned, grew and overcome difficulties in the lab and in our life projects. Also I would like to
thank the members of my PAC committee, Dr Pam Ohashi, Dr. Mark Cattral, Dr. Andrea
McCart and Dr. John Cho. Without the help and support of Carolyn, Ingrid, Corrina and Marlene
the meetings with my advisors would not have been possible. The support of my family many
kilometers away was instrumental in the completion of this work, specially my parents who
always believed in me and were curious about my work, Ana Maria G Borja Cano and Carlos A.
de la Maza Gonzalez. My brothers and sister who inspired me to keep working and supported me
through difficult times, Laura Patricia, Carlos Alejandro and Jose Eduardo. My uncle and aunt
Magda and Gerardo de la Maza Gonzales, without their support this would not have been
possible. Most importantly, my best friend, colleague and wife, Marisol Davila Foyo, who spent
many hours deliberating about my work, preparing assignments and listening to my
presentations, big thanks to you my love.

iv
Contributions
This work and my Master’s degree was supported partly by CONACyT and the MARF grant
(Mesothelioma Applied Research Foundation) and by the Princess Margaret Hospital
Foundation. Matthew Wu performed immunofluorescence staining of tumor samples.

v
Table of Contents
Contents
Contributions .................................................................................................................................. iv
Table of Contents ............................................................................................................................ v
List of Tables ................................................................................................................................. ix
List of Figures ................................................................................................................................. x
List of Abbreviations ..................................................................................................................... xi
Chapter 1 ......................................................................................................................................... 1
1 General Background ................................................................................................................... 1
1.1 Malignant Pleural Mesothelioma ........................................................................................ 1
1.1.1 Historical Background ............................................................................................ 1
1.1.2 Epidemiology .......................................................................................................... 2
1.1.3 Etiology ................................................................................................................... 4
1.1.4 Pathogenesis ............................................................................................................ 6
1.1.5 Clinical Presentation ............................................................................................... 8
1.1.6 Diagnosis ................................................................................................................. 9
1.1.7 Current Management ............................................................................................ 11
1.1.8 Prospective treatment Options for MPM .............................................................. 18
1.2 Tumor Immunity ............................................................................................................... 26
1.2.1 The immune response ........................................................................................... 26
1.2.2 Adaptive immunity ............................................................................................... 27
1.2.3 The T cell immune response ................................................................................. 28
1.2.4 Tumor Escape ....................................................................................................... 30
1.3 Radiation Therapy of tumors and the Immune System .................................................... 32
1.3.1 Brief history .......................................................................................................... 32

vi
1.3.2 Radiation and its interaction with matter .............................................................. 32
1.3.3 Factors affecting the cellular response to radiation .............................................. 34
1.3.4 Cellular Sensing and Response to Radiation ........................................................ 34
1.3.5 Cell death response ............................................................................................... 36
1.3.6 Immunogenic Cell Death ...................................................................................... 37
1.3.7 Tumor microenvironment ..................................................................................... 39
1.4 The immune response to surgery ...................................................................................... 42
1.4.1 The Surgical Stress response ................................................................................ 42
1.4.2 Post-surgical cytokine cascades ............................................................................ 44
1.4.3 Postoperative tumor progression ........................................................................... 46
1.5 Summary ........................................................................................................................... 46
2 Hypothesis and Aims ............................................................................................................... 49
3 Materials and Methods ............................................................................................................. 51
3.1 Murine Cell lines ............................................................................................................... 51
3.2 Mice .................................................................................................................................. 52
3.3 In vivo Tumor Growth experiments ................................................................................. 52
3.4 Local Radiation Therapy ................................................................................................... 52
3.5 Surgical Resection of Subcutaneous Tumors ................................................................... 53
3.6 Combination therapy with LRT and Surgery .................................................................... 55
3.7 In vivo depletion of CD4+ and CD8+ specific T cells ..................................................... 55
3.8 Anti-CTLA-4 therapy ....................................................................................................... 56
3.9 Blood Collection ............................................................................................................... 56
3.10 Tumor Digestion ............................................................................................................... 56
3.11 Isolation of Lymphocytes from Spleens and Lymph Nodes ............................................. 57
3.12 Flow Cytometry ................................................................................................................ 57
3.13 Immunofluorescence ......................................................................................................... 58

vii
3.14 Immunohistochemistry ..................................................................................................... 58
3.15 Ovalbumin ELISA ............................................................................................................ 59
3.16 Statistical Analysis ............................................................................................................ 60
4 Results ...................................................................................................................................... 61
4.1 Development of a Mouse Model of Malignant Mesothelioma ......................................... 61
4.1.1 Local Radiation Therapy, Right Flank Model ...................................................... 61
4.1.2 Combination therapy with LRT and Surgery ........................................................ 65
4.2 T cells infiltrate tumors after LRT .................................................................................... 68
4.2.1 Tumor infiltrating CD8+ T Cells in Untreated and Radiated Mouse Tumor
Tissue .................................................................................................................... 68
4.2.2 A Large Proportion of Tumor Infiltrating Lymphocytes are OVA-specific ......... 71
4.2.3 Expression of 4-1BB and PD-1 by Tumor Infiltrating Cells ................................ 73
4.2.4 Depletion of CD4+ T cells and CD8+ T cells partially abrogates the effect of
LRT on tumor growth ........................................................................................... 74
4.3 Immunological Protective Memory After LRT and Surgery ............................................ 77
4.3.1 Role of T cell on the protection against rechallenge ............................................. 79
4.4 CTLA-4 blockade improves the beneficial effect of LRT on tumors ............................... 82
5 Discussion ................................................................................................................................ 84
5.1 Development of a mouse model of MPM treated with LRT followed by surgery ........... 85
5.2 T cell tumor infiltration ..................................................................................................... 88
5.2.1 Upregulation of the activation marker 4-1BB and decrease in the exhaustion
marker PD-1 .......................................................................................................... 90
5.2.2 Depletion of lymphocytes partially abrogated the effect of radiation .................. 91
5.3 Protective memory response after LRT and Surgery ........................................................ 92
5.3.1 Depletion and rechallenge ..................................................................................... 93
5.4 CTLA-4 blockade synergized with LRT .......................................................................... 94
6 Conclusions .............................................................................................................................. 96

viii
7 Limitations ............................................................................................................................. 100
Future Directions ........................................................................................................................ 102
8 References .............................................................................................................................. 105

ix
List of Tables
Table 1 Worldwide Trends in the Epidemiologic Features of Malignant Mesothelioma
Table 2. Diagnosis of Mesothelioma

x
List of Figures
Figure 1. A possible mechanism for asbestos induced oncogenesis.. ............................................. 7
Figure 2. SMART Study schema. RT, radiotherapy. .................................................................... 17
Figure 3 Partial resection of a subcutaneous tumor with blunt dissection.. .................................. 54
Figure 4. Schematic of radiation treatment in tumor bearing mice. ............................................. 62
Figure 5 Increasing doses of Local Radiation Therapy and its effect on tumor growth.. ............. 64
Figure 6 Schematic of LRT and Surgery in AB12 tumor bearing mice.. ..................................... 66
Figure 7. The effect of combination therapy with LRT and Surgery.. ......................................... 67
Figure 8. CD3+CD8+ cell tumor infiltration after LRT compared to untreated tumors.. ............ 70
Figure 9. CD8+ lymphocytes infiltrating AE17-OVA tumor are OVA specific.. ........................ 72
Figure 10. 4-1BB and PD-1 expression tetramer specific CD8+ T cells after LRT. .................... 74
Figure 11. LRT and CD4+ CD8+ T cell depletion. ...................................................................... 76
Figure 12. AE17 OVA Rechallenge 90 days after treatment ........................................................ 78
Figure 13. CD4+ and CD8+ T cells role during rechallenge.. ...................................................... 81
Figure 14. Combination therapy with LRT and CTLA-4 shows a synergistic effect on tumor
growth. .......................................................................................................................................... 83

xi
List of Abbreviations
Ab Antibody
APC Antigen Presenting Cell
ATP Adenosine Triphosphate
BSA Bovine Serum Albumin
CRT Calreticulin
CTCF Corrected Total Cell Fluorescence
CTLA-4 Cytotoxic T-Lymphocyte-Associated Antigen 4
DAMP Danger Associated Molecular Pattern
DC Dendritic Cell
EGF Epidermal Growth Factor
ELISA Enzyme-Linked Immunosorbent Assay
EPD Extended Pleurectomy-Decortication
EPP Extrapleural Pneumonectomy
ERK Extracellular Signal-Related Kinase
FBS Fetal Bovine Serum
HMGB-1 High Mobility Group Box 1
ICAM Intracellular Adhesion Molecule
ICD Immunogenic Cell Death
IL Interleukin
LFA Lymphocyte Function Associated Antigen
LRT Local Radiation Therapy
mAb Monoclonal Antibody
Mac-1 Macrophage Receptor 1
MHC Major Histocompatibility

xii
MPM Malignant Pleural Mesothelioma
NF-κB Nuclear Factor Kappa Light Chain Enhancer of Activated B Cells
NK Natural Killer
NLRP3 NOD-like Receptor Family Pyrin Domain Containing 3
PD-1 Programmed Death-1
PD-L1 Programmed Death Ligand 1
PAMP Pathogen Associated Molecular Pattern
PRR Pathogen Recognition Receptor
RAGE Receptor for Advanced Glycation End Products
ROS Reactive Oxygen Species
SMART Surgery for Mesothelioma After Radiation Therapy
SV40 Simian Virus 40
TCR T Cell Receptor
TLR Toll-Like Receptor
TNF Tumor Necrosis Factor
VCAM Vascular Cell Adhesion Molecule
VLA Very Late Antigen
WT-1 Wilms Tumor Protein

1
Chapter 1
1 General Background
1.1 Malignant Pleural Mesothelioma
Mesothelioma is a rare and insidious malignancy with dismal prognosis. It arises from the
mesothelial surfaces of the pleural cavity, peritoneum, tunica vaginalis or pericardium. Pleural
mesothelioma is the most common type, accounting for about 70% of all malignant
mesothelioma cases (Yang, Testa, and Carbone 2008). The main risk factor associated with
development of MPM is chronic inhalational exposure to asbestos (Fuhrer and Lazarus 2011).
MPM is characterized by insidious growth and clinical presentation at an advanced stage of
disease. Currently, even with aggressive multimodality interventions including invasive surgery,
prognosis remains poor (Raja, Murthy, and Mason 2011).
1.1.1 Historical Background
The story of the discovery of mesothelioma and its causation by asbestos is long and complex.
The earliest mention of a possible tumor of the chest wall, likely mesothelioma, was by Joseph
Lieutaud in 1767. There were other mentions of patients with pleural tumors after this, but it was
only in 1870 that Wagner published the first pathological description of a primary malignancy of

2
the pleura, which he called endothelioma. By 1920, Du Bray and Rosson suggested that the term
endothelioma was not appropriate and proposed the term mesothelioma of the pleura (Hsu 2006).
In 1943, in Germany Dr. H.W. Wedler reported the first case of diffuse mesothelioma in a
patient with asbestosis, but the link between asbestos and MPM was not discovered until 1960 in
South Africa. In 1960, Wagner et al. reported a MPM epidemic among asbestos miners in the
North West of Cape Province. This was the first convincing link between asbestos exposure and
MPM (Wagner, Sleggs, and Marchand 1960).
1.1.2 Epidemiology
The worldwide incidence of Malignant Pleural Mesothelioma (MPM) is approximately 0.9 case
per 100,000 persons (Driece et al. 2010). The rate is variable between countries partly because of
asbestos exposure. In North America the incidence is about 2000-3000 new cases per year
(Yang, Testa, and Carbone 2008). In the United States (Bruce W S Robinson and Lake 2005) it
is possible that the disease has already reached its peak. Peak incidence is predicted to occur
between 2015 and 2020 in Canada (Bruce W S Robinson and Lake 2005; Cree et al. 2009),
Europe (Peto et al. 1999; Marinaccio et al. 2005; Hodgson et al. 2005) and Australia (Bruce W S
Robinson and Lake 2005; B. M. Robinson 2012; Clements et al. 2007). Moreover, In Japan (B.
M. Robinson 2012) and other non-western countries where asbestos regulation occurred later, the
peak in mesothelioma incidence will be delayed as well (Table 1).
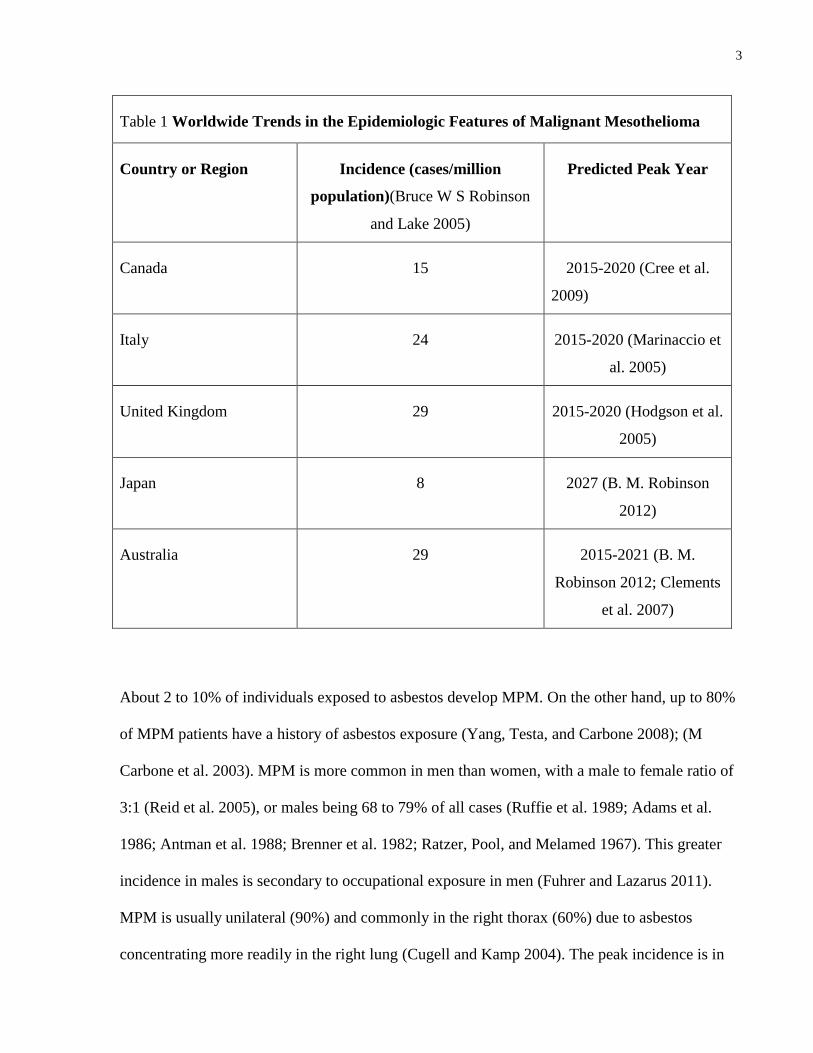
3
Table 1 Worldwide Trends in the Epidemiologic Features of Malignant Mesothelioma
Country or Region Incidence (cases/million
population)(Bruce W S Robinson
and Lake 2005)
Predicted Peak Year
Canada 15 2015-2020 (Cree et al.
2009)
Italy 24 2015-2020 (Marinaccio et
al. 2005)
United Kingdom 29 2015-2020 (Hodgson et al.
2005)
Japan 8 2027 (B. M. Robinson
2012)
Australia 29 2015-2021 (B. M.
Robinson 2012; Clements
et al. 2007)
About 2 to 10% of individuals exposed to asbestos develop MPM. On the other hand, up to 80%
of MPM patients have a history of asbestos exposure (Yang, Testa, and Carbone 2008); (M
Carbone et al. 2003). MPM is more common in men than women, with a male to female ratio of
3:1 (Reid et al. 2005), or males being 68 to 79% of all cases (Ruffie et al. 1989; Adams et al.
1986; Antman et al. 1988; Brenner et al. 1982; Ratzer, Pool, and Melamed 1967). This greater
incidence in males is secondary to occupational exposure in men (Fuhrer and Lazarus 2011).
MPM is usually unilateral (90%) and commonly in the right thorax (60%) due to asbestos
concentrating more readily in the right lung (Cugell and Kamp 2004). The peak incidence is in

4
between the 6th and 8th decade of life and in up to 80% of patients a past asbestos exposure can
be identified (Ismail-Khan et al. 2006).
1.1.3 Etiology
Chronic asbestos exposures is the most important risk factor associated with subsequent
development of pleural mesothelioma (Ismail-Khan et al. 2006; Raja, Murthy, and Mason 2011;
Suzuki, Yuen, and Ashley 2005; Yang, Testa, and Carbone 2008). The word asbestos comes
from the ancient Greek ἄσβεστος, meaning "inextinguishable" (Yang, Testa, and Carbone 2008).
There are six minerals defined by the United States Environmental Protection Agency as
“asbestos”. The two major forms of asbestos are the serpentine and amphibole. Serpentine or
white asbestos are feathery fibers and the only member of this class is chrysotile. Amphibole
asbestos is defined by long and thin fibers and include crocidolite (blue asbestos) (Leigh and
Robinson 2002; Hodgson and Darnton 2000; Bruce W S Robinson and Lake 2005). The
association of amphibole asbestos exposure and MPM is well accepted but whether chrysotile
fibers carries less risk of mesothelioma is still debated (Suzuki, Yuen, and Ashley 2005;
Yarborough 2006; Powers and Carbone 2002; Hodgson and Darnton 2000).
SV40 (Simian Virus 40), a DNA virus, has been implicated as a cofactor in the causation of
MPM (M Carbone et al. 1994). This virus blocks tumor-suppressor genes and is a potent
oncogenic virus in human and rodent cells; SV40 DNA has been found in MPM and atypical
mesothelial proliferations (Shivapurkar et al. 2002). SV40 is believed to have contaminated the
Salk polio vaccine that was used from 1955 to 1963 in the US. However, epidemiologic data is
not consistent with an etiologic effect of exposure to SV40-contaminated polio virus and the

5
development of cancer (Bocchetta and Carbone 2005; Fisher, Weber, and Carbone; Heinonen et
al. 1973; Innis 1968; Strickler et al. 1998). Subsequently, SV40 was considered harmless to
humans for many years. Finally, during the past decade interest in the association of SV40 and
human cancer has resurfaced and now the experimental data strongly associates SV40 with
human tumors, and more specifically with MPM (M Carbone et al. 2003; Bocchetta et al. 2000;
Jasani et al. 2001; Gazdar, Butel, and Carbone 2002).
Genetics may also play a role in MPM as demonstrated in some Cappadocian villages in Turkey
where an MPM epidemic caused up to 50% of all deaths (Michele Carbone et al. 2007). In these
villages, MPM was prevalent in some families but not others, even though all houses contained
similar levels of erionite. Erionite is a different type of mineral fiber and is one of the most
potent inducers of MPM in animal studies (Hill, Edwards, and Carthew 1990). MPM appeared to
be inherited in an autosomal dominant pattern according to pedigree studies. The result of
mineralogical studies and pedigree analysis suggested that MPM in Cappadocia is caused by
erionite exposure in a genetically predisposed population (gene and environment) (Yang, Testa,
and Carbone 2008). Recent work done by Dr. Carbone has led to the identification of BAP1 as
the gene mutated and associated with high rates of mesothelioma in 2 clusters of families in
United States and possibly in Cappadocia (Michele Carbone et al. 2013).
Radiation has also been linked with MPM development in animal studies (Sanders and Jackson
1972). Furthermore, multiple case reports have documented MPM in patients who received
radiation therapy for other malignancies decades before, with an average interval of 21 years
(Yang, Testa, and Carbone 2008; Amin, Mason, and Rowe 2001; Travis et al. 2005).

6
1.1.4 Pathogenesis
The development of MPM and asbestos pathogenicity are still not fully understood (Michele
Carbone, Kratzke, and Testa 2002). It is thought that asbestos is inhaled and fibers reach the
alveoli. Fibers are not easily removed and eventually translocate to the pleura via the lymphatics
or by direct extension (Powers and Carbone 2002). Asbestos fibers in the pleural space cause
persistent inflammation and secretion of cytokines, recruitment of macrophages and neutrophils
(Choe et al. 1997). Pleural mesothelial cells (PMC) secretes monocyte chemoattractant protein-1
(MCP-1) in response to asbestos. MCP-1 attracts monocytes that in turn differentiate into
macrophages and causes accumulation of macrophages within the pleural space (Tanaka et al.
2000). Upon differentiation into macrophages these cells phagocytose, and together with pleural
mesothelial cells release tumor necrosis factor alpha (TNF-α) and IL-1beta (IL-1β) (Y. Zhang et
al. 1993; Wang et al. 2004). Asbestos, also induces the expression of the TNF- α receptor 1
(TNF-R1) on pleural mesothelial cells. Activation of the TNF- α receptor in PMC consequently
activates NF-κB signaling that promotes survival and division of these cells (Yang et al. 2006).
Concurrently, the chronic state of inflammation upregulates other growth factors, including
platelet derived growth factor (PDGF), insulin-like growth factor (IGF), fibroblast growth factor
(FGF), and tumor growth factor (TGF). These factors appear to stimulate mesothelioma cell
proliferation and angiogenesis (Fuhrer and Lazarus 2011; Liu and Klominek 2004). The damage
and repair cycle contributes to DNA damage and subsequent transformation of PMC into
cancerous cells.
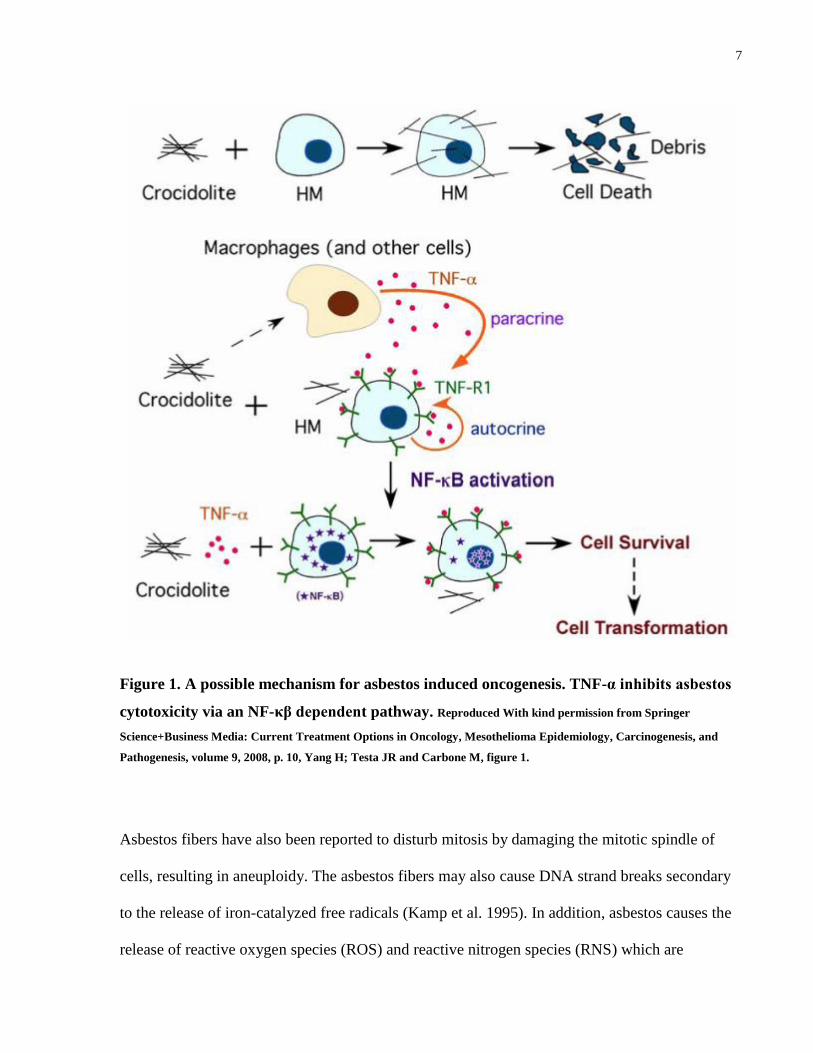
7
Figure 1. A possible mechanism for asbestos induced oncogenesis. TNF-α inhibits asbestos
cytotoxicity via an NF-κβ dependent pathway. Reproduced With kind permission from Springer
Science+Business Media: Current Treatment Options in Oncology, Mesothelioma Epidemiology, Carcinogenesis, and
Pathogenesis, volume 9, 2008, p. 10, Yang H; Testa JR and Carbone M, figure 1.
Asbestos fibers have also been reported to disturb mitosis by damaging the mitotic spindle of
cells, resulting in aneuploidy. The asbestos fibers may also cause DNA strand breaks secondary
to the release of iron-catalyzed free radicals (Kamp et al. 1995). In addition, asbestos causes the
release of reactive oxygen species (ROS) and reactive nitrogen species (RNS) which are

8
genotoxic and lead to a broad spectrum of mutations (Xu et al. 2002). Another mechanism by
which asbestos affects the pleura is induced phosphorylation of mitogen-activated protein (MAP)
kinases and extracellular signal-regulated kinases (ERK) 1 and 2. The phosphorylation of these
proteins increases expression of oncogenes such as activator protein (AP)-1 and subsequent
increased mitosis of PMC (Ramos-Nino, Timblin, and Mossman 2002).
In summary, asbestos promotes a chronic inflammatory environment, repeated cell injury and
DNA damage together with expression of cytokines and chemokines promoting proliferation,
survival and the activation of oncogenes. This may lead to PMC accumulating DNA damage and
subsequently development of MPM.
1.1.5 Clinical Presentation
Patients with MPM typically present with progressive dyspnea, chest wall pain and pleural
effusion. Dyspnea develops secondary to large pleural effusions and chest wall pain is a
consequence of chest wall infiltration. Mesothelioma is suspected in any patient with
unexplained pleural effusion and chest wall pain (Bruce W S Robinson, Musk, and Lake 2005).
Other symptoms include cough, fatigue and a mass on the chest wall. Constitutional symptoms
like weight loss, fever, night sweats and cachexia may be present in late stages of MPM.
Presentation of these symptoms at diagnosis of MPM is associated with poor prognosis (Bruce
W S Robinson, Musk, and Lake 2005).
On physical exam, the most common findings are those of pleural effusion, that is, dullness to
percussion on the affected side, decreased breath sounds and a lack of egophony. Clubbing

9
occurs in in less than 1 percent of cases (Raja, Murthy, and Mason 2011; Fuhrer and Lazarus
2011).
There are no specific laboratory findings diagnostic of MPM but eosinophilia, thrombocytosis,
and anemia of chronic diseases may be present (Ruffie et al. 1989).
1.1.6 Diagnosis
Physical exam and chest X-ray show pleural effusion in most cases (80% to 95%). Imaging
studies and biopsy are needed for definitive diagnosis. Computed chest tomography (CT) with
contrast is sensitive and is the one of the most frequent technique used to evaluate patients with
MPM. CT is sensitive to detect pleural effusion, pleural masses and lymph nodes in the hilum
and mediastinum. Magnetic resonance imaging (MRI) of the chest with contrast can provide
more information on chest wall and diaphragm invasion that may be important when considering
curative surgery. Positron emission tomography (PET) offers additional information when
assessing patients, occasionally demonstrating metastasis to contralateral thorax or extrathoracic
lymph nodes (Marom et al. 2002).
Pleural fluid cytology has a low sensitivity for diagnosis of MPM, yielding a positive diagnosis
in 33 to 84% of patients (Whitaker 2000). Blind core biopsy only modestly increases the
sensitivity. A CT-guided core needle biopsy is 87% sensitive depending on the bulkiness of the
disease and video-assisted thoracoscopy (VATS) directed pleural biopsy may increase diagnostic
accuracy to 95% to 98%. However, this procedure increases the possibility of seeding tumor
cells along the surgical incision. Some authors report tumor growth in the chest wall after VATS

10
in up to 20% of patients (Ismail-Khan et al. 2006; Bruce W S Robinson, Musk, and Lake 2005;
Nguyen et al. 1999).
Immuno-histochemical staining is usually needed for the definitive diagnosis of MPM. The most
common diagnostic problem is distinguishing mesothelioma from adenocarcinoma (Tang et al.
2001). MPM is typically positive for calretinin (88%), vimentin (50%), Wilm’s tumor 1 (WT1)
and epithelial membrane antigen (EMA, 85%) (Bruce W S Robinson, Musk, and Lake 2005).
Adenocarcinoma usually lacks these markers and is positive for carcinoembryonic antigen
(84%), CD15 (77%) and Ber-EP-4 (82%) (Martensson 1990). Histological diagnosis is also
useful to determine the subtype. Epithelial mesothelioma which has the best prognosis is the
most common and is present in 60 to 70% of cases. Sarcomatoid, characterized by spindle-
shaped cells is more aggressive and is present in 10-15% of cases. Biphasic, a combination of
epithelial and sarcomatoid, is seen in 15-30% of patients (Tischoff et al. 2011).
Despite many diagnostic options, it is common that a definitive diagnosis of MPM is delayed due
to low clinical suspicion for the disease.
Table 2. Diagnosis of Mesothelioma
1. Imaging
- Chest Radiography
o Unilateral Pleural effusion
o Localised mass
o Lung encasement by tumour rind
o Diffuse lobular masses
o Plaques
- Computed tomography
o Fluid only:74%
o Localised or diffuse pleural mass: 92%
o Thickening of interlobular fissure: 86%

11
o Chest wall invasion 18%
o Signs of asbestos exposure:20%
- Magnetic resonance imaging
o Can be helpful in planning of radiotherapy to localised disease
o Good for assessing tumour extent and chest wall invasion
- Positron emission tomography
o Useful for assessing tumour likelihood, and extent
o Can be helpful in staging
2. Cytopathology
o Pleural or ascetic fluid is often blood-stained
o Malignant cells seen in fluid: 33-84%
o Fine needle aspiration sampling of masses is useful
o Characteristic pattern of staining (eg, EMA positive, CEA negative)
3. Histopathology
o Closed biopsy: 30-50% positive
o Direct thoracoscopic biopsy:98%
o Immunohistochemistry (EMA, WT1, calretinin positive and CEA, CD15, Ber-EP4 negative)
4. Blood tests
o Non-specific features of malignancy (anemia, thrombocytosis, raised ESR)
o Abnormal liver function tests
o Serum mesothelin-related protein
5. Pulmonary function tests
o Restrictive pattern with increased maximum expiratory flow rates
o Volume changes vary according to amount of pleural fluid
o Changes in FVC are an accurate guide to disease progression or regression
Adapted from The Lancet, Vol. 366, Robinson BW; Musk AW; Lake RA, Malignant Mesothelioma, P 397-408, Copyright 2015,
with permission from Elsevier.
1.1.7 Current Management
Management of MPM continues to be very challenging. The limited number of patients, the
difficulty in objectively assessing responses and the lack of good quality randomised control
trials are challenges in studying effective therapies (Mossman et al. 2013).

12
Currently, the management of MPM is a multidisciplinary treatment approach in most centres,
and is based on the extent of disease, patient’s overall health condition and comorbidities and
patient’s desire for an aggressive treatment. Current guidelines recommend surgery for patients
with clinical stages I-III as part of the multimodal approach. For patients with stage IV disease,
sarcomatoid histology and medically inoperable stage I-III, chemotherapy is the treatment of
choice (Ettinger et al. 2012). Because most patients present with advanced disease, the mainstay
of therapy is chemotherapy (Raja, Murthy, and Mason 2011).
1.1.7.1 Surgery
Surgery alone is unable to improve survival and needs to be combined within a multimodality
approach. The goal of surgery is macroscopic complete resection (MCR). There currently are
two major surgical approaches that can achieve MCR: extrapleural pneumonectomy (EPP) and
pleurectomy/decortication (P/D). EPP is a well standardized surgical technique and consists of
en bloc resection of the parietal and visceral pleura with the ipsilateral lung, pericardium and
diaphragm. P/D is not standardized everywhere, in some centres P/D is defined as macroscopic
tumor removal with pleurectomy of the parietal pleura and decortication of the visceral pleura,
while others include resection of pericardium and diaphragm. This latter approach is now
nominated “extended” P/D (EPD) (Opitz 2014).
The optimal surgical approach is not clear and the role of EPP has been the subject of debate
after the publication of the MARS I trial, a multicentre randomised feasibility study. The MARS
I trial, in contrast to other phase II studies, concluded that EPP offers no benefit and possibly
harms patients compared to P/D. However, the study included only 16 patients in the EPP arm

13
and was designed as a feasibility study (Opitz 2014; Treasure et al. 2011). Recently, the initial
analysis of the IASLC reported a survival advantage in patients undergoing EPP compared to
P/D (Valerie W. Rusch et al. 2012). In conclusion, the role of EPP versus P/D remains highly
controversial and there is no clear indication as to which operation is more advantageous.
Furthermore, during the IMIG meeting in Boston in 2012, the role of surgery including P/D and
EPP in the treatment of MPM was reviewed and the attendees of the meeting agreed on the
following points:
A) Surgical MCR and control of micrometastatic disease play a vital role in the
multimodality treatment of MPM;
B) Surgical cytoreduction is indicated when MCR is deemed achievable;
C) The type of surgery (EPP or P/D) depends on clinical factors and on individual surgical
judgement and expertise;
D) All patients with MPM should initially be evaluated in a multidisciplinary setting;
E) Clinical staging should be performed before therapy;
F) The histologic subtype should be identified by tissue biopsy before initiation of therapy
(V. Rusch et al. 2013).

14
1.1.7.2 Chemotherapy
Patients that are not a good candidate for surgery are treated with chemotherapy. The
combination an antifolate drug such as pemetrexed and cisplatin achieves the best overall
survival and quality of life, and is currently the first-line chemotherapy regimen. Cisplatin is a
platinum-based drug that causes apoptosis through the cross-linking of DNA (Tanida et al.
2012). Pemetrexed, chemically similar to folic acid, works by inhibiting three enzymes involved
in purine and pyrimidines synthesis: thymidylate synthase, dihydrofolate reductase and
glycinamide ribonucleotide formyltransferase (Mita et al. 2006). The combination is the most
common regimen used for non-surgical candidates but also in the neo-adjuvant or adjuvant
setting. In a phase III study by Vogelzang et al (Vogelzang et al. 2003), the median survival for
patients who received pemetrexed and cisplatin was significantly longer than in patients
receiving only cisplatin (12.1 vs 9.3 months). The time to progression was also longer in the
combination group (5.7 vs. 3.9 months), as was the objective response rate (41 vs. 17 percent).
These differences were more striking after supplementation with folic acid and vitamin B12
during therapy and this is currently the standard of care. Second line chemotherapeutic agents
that have shown moderate increase in survival times are gemcitabine (Garland 2011; Castagneto
et al. 2005; Nowak et al. 2002) and vinorelbine (Garland 2011; Stebbing et al. 2009; Muers et al.
2008) alone or in combination with cisplatin.
1.1.7.3 Radiotherapy
Radiation has been typically used for gross tumor control as palliative intent or as part of
multimodality treatment for adjuvant local control in the postoperative setting. Adjuvant

15
radiotherapy seems to reduce local recurrence rates in some series, but its role in the
management of MPM remains unclear (Baldini 2004; V W Rusch et al. 2001).
There are some particular challenges with radiation therapy in MPM, such as the large target
volume and the presence of vital structures in the field. It is particularly challenging when the
lung is in place, as is the case when radiation follows P/D. Therefore, the main application of
radiotherapy is postoperatively following EPP since the lung is removed (Kotova, Wong, and
Cameron 2015). However, some groups have used radiation after P/D and report acceptable
toxicity profile (Minatel et al. 2014; Rosenzweig et al. 2012).
Various fractionation modalities have been used, but the most successful and most accepted are
3D conformal (3D-CRT) and intensity modulated radiotherapy (IMRT). The combination of EPP
and IMRT has been very successful at controlling local disease, but ultimately most patients will
present with metastatic disease (Ahamad et al. 2003). In the palliative setting, radiotherapy is
used for pain control and prevention or relief of obstructive symptoms (Stahel et al. 2010).
Finally, newer approaches such as the role of preoperative radiation was evaluated in the
feasibility study, Surgery for Mesothelioma After Radiation Therapy (SMART) trial. In this
study, high dose hypofractionated radiation was given to the hemi thorax 1 week prior to EPP.
Initial results showed that the protocol is feasible and reported promising survival data (Cho et
al. 2014).

16
1.1.7.3.1 SMART Trial
The “SMART” approach for resectable malignant pleural mesothelioma, was developed by Dr.
de Perrot and Dr. Cho at the University of Toronto. SMART was conceived after they observed
successful local control with hemithoracic radiation after EPP without direct effect in controlling
distant failures. The most common site of distant failure were the abdominal peritoneal cavity
and contralateral lung. They hypothesized that a mechanism of failure could be inadvertent
tumor spillage at the time of EPP. In this context, neoadjuvant radiotherapy was developed with
the presumption that its tumoricidal/tumorostatic effect on tumor cells could prevent the ability
of clonogens to proliferate in distant places if intraoperative spillage occurred. To limit the risk
of toxicity of the lung, they proposed the protocol with a short accelerated course of high-dose
hypofractionated hemithoracic radiation followed by EPP.
They conducted a seamless phase I/II feasibility study on surgically resectable MPM. The study
aim was to evaluate the feasibility of SMART. The primary endpoint was that the proportion of
patients treated with grade 5 (GS5) treatment-related mortality should not exceed 8%. Secondary
aims included morbidity, local and distant recurrence, disease free survival and overall survival
rates. Radiation dose to the target volume was 25 Gy in five daily fractions with a boost of 5Gy
to the tumor and tract sites. IMRT technique was used. All patients underwent EPP within 1
week of completion of IMRT. Cases with lymph node involvement were offered adjuvant
chemotherapy with cisplatin and an antifolate agent such as pemetrexed after EPP.
Initial results were published with 25 patients completing IMRT and EPP. The study showed that
the protocol is feasible without elevated perioperative morbidity and mortality. There was one
patient who developed grade 5 toxicity. After a median follow up of 23 months overall survival
reached 58% at 3 years. For the epithelial subtype alone the study reported the very promising

17
result of 84% survival at 3 years. While these results are encouraging, further study with a larger
number of patients is still in progress.
The authors postulated that the short course of radiation induces a tumoricidal/tumorostatic effect
that prevents implantation of tumor cells to distant sites after EPP. Furthermore, based on
growing evidence (Y. Lee et al. 2009; Levy et al. 2013; Kalbasi et al. 2013), they suggested that
hypofractionated radiation, not only has a direct cytotoxic effect, but also activates the immune
system against the tumor. Thus, the protocol of radiation followed by EPP may have an
important beneficial impact on the immune system by activating cytotoxic T cells and by
removing the immunosuppressive environment created by the tumor (Cho et al. 2014).
Figure 2. SMART Study schema. RT, radiotherapy.
Reproduced With kind permission from Wolters Kluwer Health, Inc. Journal of Thoracic Oncology, A Feasibility Study
Evaluating Surgery for Mesothelioma After Radiation Therapy: The “SMART” Approach for Resectable Malignant Pleural
Mesothelioma, volume 9, issue 3, p. 397-402, 2014, Cho, B.C., Jon Feld, Natasha Leighl, et al., figure 1.

18
1.1.8 Prospective treatment Options for MPM
Based on the understanding of the biology of mesothelioma there are many trials evaluating
newer therapeutic approaches. Many of these trials involve novel drugs affecting the molecular
signalling of tumor cells or balancing the immune system toward an antitumor profile. Some of
the targeted mechanisms include: Tyrosine kinase inhibitors, antibody conjugated toxins,
immune checkpoint inhibitors, gene therapy and tumor vaccines (Kotova, Wong, and Cameron
2015).
1.1.8.1 Tyrosine Kinase inhibitors
Several studies of MPM tumors have shown overexpression of protein targets, specifically
epidermal growth factor receptor (EGFR) (Mezzapelle et al. 2013; Okuda et al. 2008) and
vascular endothelial growth factor (VEFG) (Ohta et al. 1999; Demirag et al. 2005). EGFR a
tyrosine kinase receptor, through its activation promotes cellular proliferation and angiogenesis
and interferes with apoptosis. In-vitro inhibition of EGFR signaling in MPM cells leads to
decrease cell proliferation (Jänne et al. 2002). Although the effect was very promising in vitro,
several clinical trials evaluating drugs targeting the intracellular tyrosine kinase such as gefitinib
and erlotinib have failed to demonstrate any benefit in survival (Govindan et al. 2005; Garland et
al. 2007). However, in non-small cell lung cancer patients, treatment with chemotherapy and anti
EGFR monoclonal antibodies (cetuximab), targeting the extracellular domain of EGFR,
significantly improved overall survival compared to chemotherapy alone (Pirker et al. 2009). The
phase II Mesomab trial (NCT00551252 ) is evaluating cetuximab in combination with
chemotherapy in MPM patients.

19
VEGF is a powerful endothelial cell-specific mitogen associated with tumor neovascularisation
and cell proliferation. Expression of VEGF and its receptor VEGFR-1, -2 and -3 have been
demonstrated in human mesothelioma cells lines (König et al. 2000; Masood et al. 2003). VEGF
overexpression in MPM tumor samples is associated with poor prognosis (Demirag et al. 2005).
In-vitro studies showed that inhibition of VEGF greatly reduces MPM cell viability (Masood et
al. 2003). Hence, inhibition of this pathway has received great attention as a potential anti-
neoplastic therapy. Results of numerous clinical trials evaluating VEGF inhibition in MPM have
failed to show any significant difference in survival (Jahan et al. 2012; Dowell et al. 2012; Papa
et al. 2013; Ceresoli et al. 2013). However, an ongoing clinical trial (NCT00651456) presented at
the 2015 American Society of Clinical Oncology Annual Meeting presented promising results in
patients treated with bevacizumab. The group showed significant longer survival in patients with
MPM treated with Pemetrexed, Cisplatin and Bevacizumab compared to patients treated with
Pemetrexed and Cisplatin only (Zalcman et al. 2015).
1.1.8.2 Antibody conjugated toxins
Mesothelin is a protein present on normal mesothelium and overexpressed on epithelial cancer
like mesothelioma, ovarian cancer, lung adenocarcinoma and pancreatic adenocarcinoma (Chang
et al. 1992). Its restricted expression on normal tissues and overexpression on neoplastic cells
make mesothelin a good target for antibody based therapy. Amatuximab (MORAb-009) is a
chimeric high-affinity monoclonal IgG1/k antibody targeting mesothelin. In-vitro, after binding
mesothelin the antibody is internalized and elicits antibody-dependent cellular cytotoxicity
(ADCC). Preclinical studies with xenografts, combination treatment with amatuximab and
chemotherapy was more effective than either amatuximab or chemotherapy alone (Hassan et al.

20
2007). A phase I study demonstrated amatuximab is well tolerated (Hassan et al. 2010). In a
phase II clinical trial, amatuximab was combined with pemetrexed and cisplatin in patients with
unresectable MPM. The study did not meet the primary endpoint of 3-month improvement in
progression free survival over historical controls. However, the authors highlighted the median
OS of 14.8 months in this particular population with 87% of patients with stage III/IV disease.
Together with the fact that a third of the patients were alive at the time of analysis, the authors
suggested that Amatuximab potentially had an antitumor activity (Hassan et al. 2014).
CRS-207 is a live-attenuated double deleted Listeria monocytogenes strain that was engineered
to express human mesothelin. CRS-207 induced mesothelin-specific T-cell responses that
correlated with regression of murine tumors in preclinical studies. A phase I clinical trial
determined that it was well tolerated and demonstrated an induction of tumor antigen-specific T
cells responses in patients with advanced cancer. A CD8 T cell specific for mesothelin response
was induced in six out of 10 patients but did not correlate with clinical response (Le et al. 2012).
SS1P is an immunotoxin consisting of the variable fragment of an anti-mesothelin antibody
linked to a truncated form of Pseudomonas exotoxin A (PE38). When SS1P binds its target,
PE38 is internalized and kill cells by ADP ribosylation and inactivation of elongation factor 2
(EF-2) and apoptosis (Pastan et al. 2007). A phase I trial showed that SS1P was well tolerated
and had modest clinical activity. However, many patients developed neutralizing antibodies that
prevented additional therapy (Kreitman et al. 2009). A second study showed better results after
combining SS1P with pentostatin and cyclophosphamide, to induce an immunosuppressive state
and abrogate the production of neutralizing antibodies (Hassan et al. 2013).
Interleukin-4 (IL-4) receptors are present on several human tumors including mesothelioma,
making it a potential clinical target. A recombinant IL4 toxin called cpIL-4-PE was designed

21
with fragments of IL-4 fused with a variant form of the exotoxin PE38 (Puri et al. 1996). This
IL-4 toxin binds mesothelioma cells and inhibit protein synthesis in vitro. Pre-clinical studies in
mice are promising and show reduced tumor volumes and prolonged survival in mice treated
with the toxin compared to controls (Beseth et al. 2004).
1.1.8.3 Mesothelin-specific Chimeric Antigen Receptor T cells
Tumor antigen specific T cells have been engineered by the introduction of chimeric antigen
receptors (CARs) that have antibody-based external receptor structures and cytosolic domains
that encode signal transduction modules of the T cell receptor. This concept was first developed
by Dr. Eshhar and his group created the first functional CAR T cells by 1989 (Gross et al. 1989).
The recognition element is derived from an antibody variable region and this retargets T cells in
an MHC unrestricted manner and are not patient specific, thus the same chimeric antigen
receptor can be used for multiple patients. Clinical trials have documented safety but poor in
vivo persistence and expression of the transgene (Kershaw et al. 2006; Till et al. 2008). Second
and third generation CAR T cells have been engineered to overcome these shortcomings and
incorporate costimulatory signaling domains into the signaling module (Till et al. 2012; Heiblig
et al. 2015).
The application of CAR T cells to treat solid malignancies has been limited in part due to the
potential of CAR-based therapies to cause on-target off-tumor toxicity through recognition of
normal cells that express the target antigens (Lamers et al. 2013). To circumvent this toxicity
several groups have incorporated safety genes such as inducible caspase 9 transgene with
variable results (Di Stasi et al. 2011).

22
Mesothelin is overexpressed in the majority of MPM but it is also expressed at low level on
normal peritoneal, pleural and pericardial mesothelial surfaces. In a preclinical model Carpenito
et al., (Carpenito et al. 2009; Zhao et al. 2010) evaluated mRNA electroporated mesothelin CAR
T cells (CARTmeso cells) and observed potent antitumor effects against established tumor
xenografts. mRNA electroporated T cells transiently express the mesothelin-targeting CAR, thus
reducing off-tumor toxicity. The same group reported the use of CARTmeso cells is feasible and
safe in humans and reported two cases from an ongoing trial (Gregory L Beatty et al. 2014).
Clinical and laboratory evidence of antitumor activity was demonstrated in both patients. Their
data support the feasibility of CAR T cells as a novel strategy for the treatment of patients with
solid malignancies such as MPM.
1.1.8.4 Gene Therapy
1.1.8.4.1 Suicide Gene Therapy
In this approach, tumor cells are transduced with genes that encode for proteins that metabolize
prodrugs into toxic metabolites. These metabolites accumulate in tumor cells and leads to its
death or “suicide”. A common gene is thymidine kinase gene (HSVtk) from the herpes simplex
virus-1. Thymidine kinase metabolizes the nontoxic antiviral ganciclovir into GCV-
monophosphate which is then catalyzed by cell kinases into ganciclovir triphosphate. This
metabolite is a competitive inhibitor of DNA polymerase and disrupts DNA synthesis (Markham
and Faulds 1994; Vachani et al. 2010). A phase 1 clinical trial of adenovirus gene therapy
(Ad.HSVtk/GCV) in patients with MPM was done to evaluate safety, immunologic responses,
transgene expression and clinical responses. A single intrapleural dose of the vector was given

23
followed by GCV I.V. twice daily for 14 days. The vector was well tolerated and deemed safe.
23/30 patients had demonstrated gene transfer. Post treatment antibody responses against the
tumors were seen and proliferative T cell responses were generated in serum and pleural fluid.
Clinical responses were seen and 2 patients showed long term survival (7 and 10 years) (Sterman
et al. 2005). In another phase1 clinical trial, ovarian carcinoma cells transfected with the HSVtk
gene (PA1-STK) were infused intrapleurally followed by GCV. Some of the cells were followed
with 99Tc and it was shown that the labeled cells adhered preferentially to intrapleural
mesothelioma deposits. The transfected cells were thought to have exerted a bystander effect on
mesothelial cells–the killing of neighboring cells not transduced with the vector. Patients showed
minimal side effects and the authors concluded this therapy is feasible in humans (Harrison et al.
2000).
1.1.8.4.2 Cytokine Gene Therapy
This therapy is based on the administration of viral vectors encoding cytokine genes that may
lead to high expression of immunostimulatory cytokines. Some of the cytokines may have a
cytotoxic effect or activate the immune system. There are several trials that evaluated
administration of IL-2 with good results in phase I and II trials (Astoul et al. 1998; Tan et al.).
Recently gene therapy has centered on IFN which plays a central role in the activation of the
immune system and may have a direct anti-tumor cytotoxic effect (Sterman et al. 2011; Odaka et
al. 2001; Sterman et al. 2007).

24
1.1.8.5 Tumor Vaccines
Dendritic cell vaccines are used to improve effective tumor antigen presentation and successfully
activate the immune system. Dendritic cells are the most potent antigen presenting cells.
Preclinical data has shown encouraging results and may be a valuable strategy in cancers like
mesothelioma (Palucka and Banchereau 2012; Hegmans et al. 2010). Besides autologous tumor
lysates, calretinin, mesothelin and WT-1 have been used as antigens. In the case of WT-1,
measurable CD4 and CD8 T cells responses were elicited, but there were no clinical response
(Krug et al. 2010). With autologous tumor antigens in a phase I clinical trial, the vaccine was
well tolerated and there were several partial responses (Hegmans et al. 2010). Currently, there
are several phase I-II trials running using WT-1, Mesothelin and 5T4 as vaccine antigens
(Kotova, Wong, and Cameron 2015).
1.1.8.6 Immune Checkpoint Inhibitors
Antibodies that modulates the immune system by binding to checkpoint molecules and shifting
the immunes system towards an anti-cancer response are starting to be used in MPM. Immune
checkpoints are pathways that regulate and modulate immune responses and are crucial for
maintaining self-tolerance. Some of these checkpoints are triggered by ligand-receptor
interactions leading to the potential for a therapeutic target, by blocking these interactions
(Brahmer and Pardoll 2013).
Cytotoxic T lymphocyte antigen-4 (CTLA-4), also known as CD152 is one of the receptors most
actively studied in the context of cancer immunotherapy (Postow, Harding, and Wolchok 2012).

25
Briefly, CTLA-4 receptor raises the threshold for T lymphocyte activation by sequestering the
co-stimulatory signals provided by CD80 and CD86 present on antigen-presenting cells. By
blocking the CTLA-4 receptor with antibodies, the negative regulatory effect of the receptor can
be reversed and a therapeutic response against tumor cells can be induced. We have shown in our
lab in a mouse model of mesothelioma that blockade of CTLA-4 demonstrates an anticancer
effect and is correlated with inhibition of cancer cell repopulation when combined with
chemotherapy (L. Wu et al. 2012). A phase II clinical trial evaluated tremelimumab, an anti-
CTLA-4 antibody. The study enrolled chemotherapy-resistant advanced mesothelioma patients
and showed clinical responses in 2/29 patients, stabilization in 9/29 and overall survival at one
year was 48% (Calabrò et al. 2013). Currently there are two fully human antibodies, ipilimumab
and tremelimumab, both block CTLA-4 interaction with B7 ligands. The antibodies have
undergone extensive study in melanoma (Hodi et al. 2010; Sanford 2012; Ascierto 2013; Larkin
et al. 2015) and ipilimumab was approved to treat patients with advanced melanoma in the US,
Canada and the European Union. Tremelimumab was approved recently by the FDA to treat
MPM.
The other immune checkpoint studied for cancer immunotherapy is the programmed death 1
(PD-1) pathway. The pathway PD1 and programmed death ligand 1 (PD-L1) limits the activity
of T cells in peripheral tissues during inflammation. PD-1 is found on the surface of T cells and
PD-L1 is expressed on the surface of tumor cells (Zou and Chen 2008). Expression of PD-L1
was first demonstrated in murine mesothelioma (Currie et al. 2009) and later on human
mesothelioma specimens. Expression of PD-L1 was seen mostly on sarcomatoid and biphasic
subtype of MPM and was associated with poor prognosis in 2 recent publications (Mansfield et
al. 2014; Cedrés et al. 2015). Studies in our lab demonstrated no effect of anti PD-1 mAb alone
on tumor growth in a subcutaneous murine mesothelioma model. However, we have observed

26
dramatic tumor shrinkage when combined with local radiotherapy (unpublished data). Clinical
trials in melanoma and lung cancer have shown durable clinical activity with anti PD-1
immunotherapy (Topalian et al. 2012; Lipson et al. 2013; Hamid et al. 2013). Currently, there are
no clinical trials evaluating the role of anti PD-1 in MPM, but it remains a promising option.
1.2 Tumor Immunity
1.2.1 The immune response
The immune system consists of two integrated systems, the innate and the adaptive system. They
each consist of both cellular and non-cellular components. The innate system provides us with a
rapid immune response against great variety of pathogens by recognizing evolutionary conserved
molecules (O. Krysko et al. 2013). Adaptive immunity involves an orchestrated response
involving cellular and humoral components. The adaptive immune system requires more time
but is more specific. Furthermore, the adaptive immune system generates an antigen specific
response against pathogen molecules not previously encountered by the host and in many cases
results in immunological memory (Alberts et al. 2002). This project is focused on the adaptive
system as it provides a specific response as well as immune memory. In cancer a successful
adaptive response can specifically target cancerous cells and prevent the recurrence of disease
(D. S. Chen and Mellman 2013).

27
1.2.2 Adaptive immunity
Adaptive immunity is predominantly comprised of B and T cell lymphocytes. There are two
main branches of the adaptive immune response: the humoral immune response mediated by B
cells, and the cell-mediated immune response directed by T cells. The humoral immune response
is characterized by the production of antibodies. Antibodies bind to their targets and mediate the
pathogen clearance or inactivation. Cellular mediated responses include direct killing of target
cells such as virally infected or cancerous cells by T cells or NK cells, as wells as activation of
other cells through production of cytokines by T cells (Norvell 2013).
1.2.2.1 T cells
T cells, derive their name from their maturation in the thymus and are broadly classified into
cytotoxic, helper and regulatory T cells. In cancer, T cells are the principal mediators of
antitumor immunity. CD8+ T cells recognize pathogen peptides loaded on the major
histocompatibility complex (MHC) class I molecules by antigen presenting cells. Naïve CD8 T
cells can differentiate into cytotoxic T cells (CTL) that recognize and kill virally infected or
cancerous cells. CTLs kill their targets through perforin mediated cytotoxicity and Fas/FasL
interaction (Nagata and Golstein 1995; Henkart 1994; Seki et al. 2002). There are numerous
studies that have examined and found a positive correlation between the presence and number of
CD8+ tumor-infiltrating T cells (TILs) and good prognosis. This positive association has been
reported in colorectal cancer, ovarian and breast cancer among others (Bachmayr-Heyda et al.
2013; Matkowski et al. 2009; Nosho et al. 2010). Our group, reported that high levels of CD8+
TILs between cycles of chemotherapy is associated with longer progression-free survival as well

28
as delayed recurrence in MPM in a murine model (Licun Wu et al. 2011). Furthermore, our
group reported the association in MPM patients of high levels of CD8+ tumor infiltrating
lymphocytes with better survival in patients undergoing EPP (Anraku et al. 2008). The
importance of CD8 T cells in immune therapy is highlighted by the attempts to activate and
increase their number in order to improve survival in patients (Dudley et al. 2002; Fourcade et al.
2010).
1.2.3 The T cell immune response
For an anticancer immune response to effectively kill cancer cells, a series of events must occur
to ultimately activate and expand T cells.
In the first step immature dendritic cells (DC) uptake tumor antigens present in the extracellular
environment primarily through pinocytosis. The up-taken antigens are then cleaved and
processed for loading onto the MHC molecules. If DCs receive specific signals or sense
“danger” they upregulate costimulatory molecules and the chemokine receptor CCR7. CCR7
recognizes the chemokines produce by lymphoid tissue CCL19 and CCL21 and leads the DC to
the T-cell zones of the local lymph nodes. The role of DC in the lymph nodes is to present
antigens to specific T lymphocytes. The signals involved in DC maturation may include
pathogen-associated molecular patterns (PAMPs) or damage-associated molecular pattern
molecules (DAMPs) and proinflammatory cytokines and factors released by dying tumor cells.
In the second step, DCs present the antigens on MHC I and MHC II molecules to naïve T cells
resulting in the priming and activation of effector T cell responses against tumor specific
antigens. To activate T cells three stimulatory events must occur. The first signal involves the

29
specific peptide-MHC complex and T cell receptor (TCR) interaction which is stabilized by
either CD4 or CD8 molecules on T cells. The second signal involves the costimulatory
interaction of B7.1 and B7.2 on DCs binding to CD28 on T cells. Additional costimulatory
interactions include OX40L-OX40, CD70-CD27, CD40-CD40L, CD137L-CD137, ICOSL-ICOS
(Driessens, Kline, and Gajewski 2009). The last signal involves cytokines secreted by APCs that
differentiate T cells into one of many T cell subsets that include but are not limited to CD8
cytotoxic, Th1, Th2, Th17, and T regulatory cells. The encounter with an specific antigen in the
presence of a co-stimulatory signal triggers T cell proliferation and, induces the synthesis of IL-2
and the α chain of the IL-2 receptor (CD25). Association of the α chain with the β and γ chains
forms a high affinity IL-2 receptor, allowing T cells to respond to very low concentrations of IL-
2. IL-2 then stimulates T cell proliferation, differentiation and survival. After 4-5 days of rapid
proliferation induced by IL-2, activated T cells differentiate into effector T cells that can
synthesize all the molecules required for their cytotoxic functions.
In the next step the activated effector T cells traffics to the tumor where it recognizes and binds
to cancer cells through interaction of the TCR and the antigen MHC complex. This interaction
results in immune attack without the need for co-stimulation. T cells will then kill the target
cancer cells. Killing of the cancer cell releases additional tumor associated antigens and this in
turn can increase the intensity of the response against the tumor. The antitumor immune response
will be defined at this point, with a balance between the ratio of effector T cells versus regulatory
T cells. Experimental evidence has shown that tumor rejection requires T cells that have
functionally differentiated to become CD4+ Th1 and CD8+ cytotoxic T cells (CTL) (Nishimura
et al. 2000).

30
1.2.4 Tumor Escape
The idea of immune surveillance for eradicating nascent transformed cells before they are
clinically detected was first proposed by Ehrlich in the early 20th century (Ehrlich 1909). Fifty
years later, experimental evidence that tumors could be repressed by the immune system came
from tumor transplantation models. This led to the formal hypothesis of “cancer
immunosurveillance” by Burnet and Thomas (Burnet 1957). Both speculated that lymphocytes
acted as sentinels in recognizing and eliminating continuously arising, nascent transformed cells
(Burnet 1970). However, subsequent experiments based on experimental immunosuppression
(Kaplan 1971; Stutman 1975) or using nude mice (O Stutman 1974; Osias Stutman 1979) failed
to prove the immunosurveillance hypothesis at that time and led to its abandonment. It was until
the 1990s when experimental animal models using knockout mice validated the existence of
cancer immune surveillance in both chemically induced and spontaneous tumors. At the same
time the central roles of T cells, NK, NKT, Interferons and perforin were clarified in cancer
immune surveillance (Dighe et al. 1994; Russell and Ley 2002; van den Broek et al. 1996;
Shankaran et al. 2001).
Finally, the current concept of cancer immunoediting leading from immune surveillance to
immune escape was proposed. Three essential phases were proposed by the Schreiber group
(Dunn et al. 2002): elimination; equilibrium; and escape. Briefly, in the elimination phase,
transformed cells can be eliminated by immune effector cells such as NK and T cells. Tumor
cells are recognized initially by NK, NKT or T cells which are then stimulated to produce IFN γ
which will lead to accumulation and activation of immune cells and eventually tumor cell lysis.
In the equilibrium phase, the host immune system and tumor cells that have survived the
elimination process enter into a dynamic equilibrium. In this phase, immune cells exert potent

31
selection pressure on the tumor cells that is enough to contain, but not fully eliminate. During
this period of selection new variants with different mutations arise and provide them with
increased resistance to immune attack. Lastly, in the escape process, tumor cells that have
acquired resistance to immunologic detection or elimination begin to expand in an uncontrolled
manner that may eventually lead to clinical disease. Eventually, during tumor progression,
tumor-derived soluble factors can induce several mechanism for escape from immune attack in
the tumor microenvironment (R. Kim, Emi, and Tanabe 2007).
As described previously, there is significant complexity for mounting an antitumor immune
response, among other factors, the priming and effector phases are separated by time and space.
Priming occurs in lymph nodes, the effector functions must operate within the tumor mass. There
are several obstacles that the system must overcome. During priming, the lack of “danger”
signals from innate immune cells, poor recruitment of DCs for cross-presentation, and
inadequate expression of costimulatory ligands on tumor cells or APCs can hinder the immune
response. Furthermore, during the effector phase there may be inadequate recruitment of
lymphocytes due to abnormal blood vessels and cytokines, activation of inhibitory receptors on T
cells such as CTLA-4 and PD-1, extrinsic suppressive cells (TREGs, myeloid-derived
suppressive cells-MDSC), metabolic inhibitors (IDO, arginase) and inhibitory cytokines (IL-10,
TGF-β) (Gajewski et al. 2011). All of the previous can limit the impact of the T cell response on
the tumor and eventually lead to progression of clinical disease and if left unchecked may result
in the death of the host.

32
1.3 Radiation Therapy of tumors and the Immune System
1.3.1 Brief history
In November of 1895, Wilhelm Conrad Rӧntgen described the x-ray and very soon after,
potential applications were recognized in different fields. Shortly after in January 1896, Emil
Grubbé treated a woman who suffered of an open inoperable carcinoma of the breast. The same
year, Despeignes was the first to publish his results using x-rays to treat gastric carcinoma
(Buschke 1958). Radiation therapy increased in popularity and advances have continued to the
present.
1.3.2 Radiation and its interaction with matter
When radiation interacts with matter, there is an absorption of energy from radiation and this
may lead to excitation or ionization. Excitation occurs when an electron in an atom is moved to a
higher energy level. Ionization occurs when there is enough energy to eject the electron from the
atom. In the latter case, radiation is called ionizing. Gamma and x-rays are example of ionization
radiation. Radiation can be classified as directly or indirectly ionizing. Charged particles, such as
electrons or protons, are examples of directly ionizing. Provided they have enough energy they
can disrupt the atoms of the structure they pass through and produce chemical and biologic
changes. On the other hand x-rays are indirectly ionizing; they do not disrupt the structure of the
atoms by themselves but they are absorbed in the material and give up their energy to produce
fast moving charged particles (electrons) that can in turn produce damage (Hall and Giaccia
2006).

33
Gamma and x-rays do not differ in nature or in properties, and can be considered from two
different standpoints, as electromagnetic waves or as a stream of photons or packets of energy.
Like radio waves and visible light, gamma and X-rays are forms of electromagnetic radiation. X-
rays occupy the short wavelength end of the electromagnetic spectrum. Also, x-rays can be
considered as packets of energy called photons. When x-ray photons are absorbed by matter at
high energies as when used in radiotherapy the Compton Process will dominate. In this process
the photon will interact with a “free” electron and part of the energy of the photon is given to the
electron as kinetic energy. The photon will continue deflected from its original path and with less
energy. The result is a photon of reduced energy and a fast electron. Electrons can then ionize
other atoms, break chemical bonds and initiate the change of events that will result in biological
damage (Hall and Giaccia 2006).
The main biologic effects of radiation results from damage to DNA. Radiation interacts with
atoms in the cells, mainly water and produces free radicals (hydroxyl radical - OH▪) that can
diffuse and reach DNA. It is estimated that 2/3 of the x-ray damage to DNA in cells is caused by
the hydroxyl radical. Estimations suggest that 1-2 Gy of x-ray radiation can result in 105
ionization events per cells, resulting in 1000-2000 single-stranded DNA breaks (SSB) and 40
double-stranded breaks (DSB) (Lewanski and Gullick 2001). SSB are of little biologic
consequence, as they are repaired readily by the cell mechanisms. However, DSBs are the most
important lesion and may result in cell killing (Radford 1985).

34
1.3.3 Factors affecting the cellular response to radiation
The final outcome in a cell after being exposed to radiation will depend mainly on the stage of
the cell cycle but also on the presence of free radical scavengers and biomolecules (oxygen).
Cells are most sensitive at or just before mitosis. Resistance is greatest in the latter part of the S
phase, as DNA damage can be repaired more rapidly at this stage (Pateras et al. 2015). Cells that
divide frequently like tumor cells or lymphocytes are more radiosensitive than those that divide
rarely such as nerve and muscle cells. In the case of a tumor, where cell division is not
synchronized, not all cells will be successfully eliminated.
The presence or absence of oxygen will influence the biologic effect of x-rays. Oxygen at the
time of radiation fixes ionization damage and makes it more difficult to repair. If cells are in
hypoxic condition they will become more resistant to radiation (Palcic and Skarsgard 1984). A
clinical study used hyperbaric chamber trying to improve the effect of radiation. They reported a
slightly improved tumor control compared to radiotherapy on normoxic conditions, but the
treatment proved to be costly and difficult to administer (Overgaard and Horsman 1996).
1.3.4 Cellular Sensing and Response to Radiation
Ionizing radiation damage have effects on transcription, DNA synthesis, cell cycle regulation and
may trigger apoptosis or cell death (Valerie and Povirk 2003). Radiation induced DNA damage,
such as DSB, is a lethal DNA lesion. However, cells are equipped with sensors that recognize
these lesions immediately after formation and a signaling cascade is started that ultimately may
result in cell cycle arrest, giving the cell time for repair if the damage is not extensive, either by

35
homologous recombination or non-homologous end-joining pathways (Valerie and Povirk 2003).
The most important sensors are ataxia-telangiectaxia mutated (ATM) and the MRN complex
(Mre11, Rad50 and Nbs1) (Lavin 2007; J.-H. Lee and Paull 2004). ATM is activated through a
functional MRN complex, which recognizes and migrates to DSBs after ionizing radiation
(Lavin 2007). The most important proteins activated by ATM are surveillance proteins, such as
p53 (Canman et al. 1998), CHK2 (Matsuoka et al. 2000) and DNA-protein kinase (B. P. C. Chen
et al. 2007). CHK2 phosphorylates in turn p53 (Shieh et al. 2000) leading to a release of p53
from MDM2. The dissociation of MDM2-p53, results in stabilization of p53. ATM also directly
phosphorylate p53 increasing its activity (Canman et al. 1998). As a results p53 translocate into
the nucleus and binds target genes, such as p21. The upregulation of p21 ultimately results in cell
cycle arrest at the G1/S checkpoint through inhibition of the Cdk2-cyclin E-PCNA complex
(Kaina, Roos, and Christmann 2010). Additionally, CHK1 via phosphorylation of Cdc25a and
Cdc25c leads to dephosphorylation of CDK2 and Cdk1-CyclinB, inducing G1/S and G2/M arrest
(Sanchez et al. 1997; Peng et al. 1997; Mailand et al. 2000).
The main apoptosis pathways activated by DNA damage involve the Fas/Caspase 8 signaling
and the apoptosome formation, both can be dependent or independent of p53/p73 (Pietsch et al.
2008). In all cases, signals converge on Caspase 3 to induce apoptosis. The upregulation of
p53/p73 in turn upregulates the fas receptor/caspase-8 apoptotic pathway (Bennett et al. 1998).
In 50% or more of all human cancers p53 is mutated (Soussi and Lozano 2005). In cells where
p53 is inactive, DNA damage activates the endogenous or mitochondrial apoptotic pathway. In
this pathway the decline of Bcl-2 leads to leakiness of the mitochondria, cytochrome c release
and activation of the apoptosome (Apaf-1, ATP, procaspase-9, and cytochrome c). Caspase 9
then cleaves caspase-3 and other downstream caspases leading to activation of caspase-activated

36
DNase (CAD). CAD ultimately cleaves DNA into the typical nucleosomal fragments (Haupt et
al. 2003).
Other factors modulate the effect of DNA damage, such as, p53 (Batista et al. 2007), Jun
kinase/p38 kinase (Hamdi et al. 2005), caspase-2(Robertson et al. 2004), NF-κB (Karin and Ben-
Neriah 2000) and Akt (Gottlieb et al. 2002). These various pathways will converge and compete.
Some transduce cell death signals, with others transducing survival signals. The summation of all
these signals are carefully regulated by the cell context and the level of DNA damage, with the
net effect of either survival, death by apoptosis or necrotic cell death in the case of high non
tolerable levels of DNA lesions (Kaina, Roos, and Christmann 2010).
1.3.5 Cell death response
Apoptosis is one type of programed cell death and is the prevalent form of cell death under
normal conditions and daily tissue regeneration. Cells undergoing apoptosis are rapidly cleared
by macrophages and dendritic cells and induce immune tolerance (Voll et al. 1997; Albert et al.
1998). Apoptosis is the main response to radiotherapy in the hematopoietic system and the
intrinsic death pathway is the major signaling mechanism (Heylmann et al. 2014; Eriksson and
Stigbrand 2010). Significantly less apoptosis is observed in cells of epithelial origin and in this
kind of cells, radiotherapy can trigger necroptosis or necrosis(Mantel et al. 2010; Vandenabeele
et al. 2010). Necroptosis is characterized by the production of ROS, lipid peroxidation, swelling
of organelles, rupture of the plasma membrane, and release of intracellular contents
(Vandenabeele et al. 2010). Also, secondary necrosis can occur when apoptotic dying cells failed
to be phagocytosed in time. This can occur when a large number of cells undergo apoptosis and

37
overwhelms the phagocytic cells, as in the context of tumor radiotherapy (Silva 2010). Mitotic
catastrophe occurs as a result of improper entry into mitosis resulting in aberrant cell division. It
is one of the main mechanism of cell death secondary to radiation induced DNA damage in cells
with defective cell cycle checkpoints and impaired DNA repair mechanisms (p53 mutations)
(Eriksson and Stigbrand 2010). Cells that entered into mitotic catastrophe may survive for days,
transit into senescence or die by delayed apoptosis, or necroptosis (Lauber et al. 2012). Mitotic
catastrophe is a delayed type of cell death, days after treatment initiation, and may explain the
slow clinical regression of solid tumors. Senescence is a condition of permanent cell cycle arrest.
Senescence secondary to radiation is observed in cells where cell cycle checkpoints are still
intact (Eriksson and Stigbrand 2010). Cell death by necrosis or necroptosis is not
immunologically silent and can trigger a potent inflammatory immune response. Necrosis
releases cellular debris and pro-inflammatory molecules into the extracellular space including
high-mobility group box 1 proteins (HMGB-1), heat-shock proteins (HSP), calreticulin and ATP
(Rock and Kono 2008; Kono and Rock 2008). These mediators can orchestrate the recruitment
of immune cells, according to the danger hypothesis proposed by Matzinger (Matzinger 1998),
and eventually this lead to activation of innate and adaptive immune system (Tesniere et al.
2008; Jonathan, Bernhard, and McKenna 1999; Illidge 1998). Therefore, the type of death,
timing of clearance, and factors released during cell death will elicit a response from the host that
will shape the state of the immune system after irradiation.
1.3.6 Immunogenic Cell Death
Cell death and necrosis in a radiated tumor may alter the activation state of the immune system.
The danger hypothesis states that dying cells release signals that aid the immune system to

38
recognize forms of non-physiological cell death. This form of cell death releasing specific
signals that stimulate adaptive immune responses has been named “immunogenic cell death”
(ICD) (Tesniere et al. 2008; Obeid et al. 2007; Golden et al. 2012; Golden and Apetoh 2015;
Kepp et al. 2011). In necrotic cell death, these signals involve HSP, HMGB-1, Calreticulin and
ATP among others. These endogenous molecules that deliver danger signals in response to stress
and trigger ICD are known as damage associated molecular patterns (DAMPs). Although the
details of ICD continue to be elucidated and other molecules cannot be excluded, 3 necessary
components have been characterized, the exposure of the endoplasmic reticulum (ER) chaperon
calreticulin (CRT) on the outer surface of the plasma membrane, the release of the non-histone
chromatin-binding HMGB1 and ATP release (Ma et al. 2010; Golden and Apetoh 2015; Obeid et
al. 2007). Calreticulin is an “eat me” signal and enables phagocytes and dendritic cells to
efficiently engulf dead cells. Calreticulin is recognized by CD91 (LRP1) positive cells
(macrophages and DCs) unless they simultaneously express the “do-not-eat me” signal CD47
(Garg et al. 2012; D. V Krysko et al. 2012; Kroemer et al. 2013). ATP is a “find me signal” and
promotes recruitment of APC by binding to the receptors, purinergic receptor P2Y, G-protein
coupled, 2 (P2Y2) and purinergic receptor P2X ligand-gated ion channel 7 (P2X7) (D. V Krysko
et al. 2012; Bezu et al. 2015). Stimulation of P2Y2 is required for monocyte attraction and
activation of the P2X7 receptors on dendritic cells activates the NALP3 inflammasome, a
multimeric danger-sensing platform that promotes and drives the secretion of IL-1β (Garg et al.
2012). This cytokine is required for the polarization of IFN-γ producing CD8+ T cells
(Ghiringhelli et al. 2009). HMGB-1 exerts its immunostimulatory effects through TLR4 and
advanced glycosylation end product-specific receptor (RAGE). Binding of HMGB-1 to these
receptors on innate immune cells such as neutrophils, macrophages and monocytes stimulates the
production of pro-inflammatory cytokines (TNF-α, IL-1, IL-6 and IL-8) (Rovere-Querini et al.

39
2004; G. Chen et al. 2004). In summary, danger signals released from necrotic cells as a result of
radiation play a role in the activation of immune responses that can tip the balance against the
tumor.
1.3.7 Tumor microenvironment
1.3.7.1 Cytokine expression
Radiotherapy has a significant effect on the modulation of immune responses and this effect is
due in part to the balance between pro-inflammatory and anti-inflammatory cytokines and
chemokines. Radiation leads to an increase in IFN-γ production (A. A. Lugade et al. 2008). IFN-
γ has pleiotropic effects in the tumor microenvironment, including the upregulation of MHC
class I and II expression (Weber and Rosenberg 1988; Dighe et al. 1994), activation of
macrophages (Boehm et al. 1997; Xie, Whisnant, and Nathan 1993), inhibition of the production
of immunosuppressive molecules (Hirte and Clark 1991), and enhancement of the secretion of
antiangiogenic chemokines (Sgadari, Angiolillo, and Tosato 1996; Arenberg et al. 1996). IFN-γ
production by infiltrating T cells enhances expression of VCAM-1 and ICAM-1 to further
enhance T cell infiltration (A. A. Lugade et al. 2008; Caldenhoven et al. 1994). Furthermore,
IFN-γ activates STAT1 which drives the expression of multiple chemokines including CXCL9
(MIG), CXCL10 (IP-10). These chemokines are potent signals for T cell activation through the
receptor CXCR3 (Burnette and Weichselbaum 2013). However, IFN-γ has also the potential to
down regulate antigen presentation by tumor cells (G L Beatty and Paterson 2000), either
through down regulation of tumor antigen protein expression of by less efficient processing of
tumor antigens (S. Morel et al. 2000). TNF-α is another cytokine upregulated after radiation (van

40
Valen et al. 1997; Rübe et al. 2004; Fedorocko, Egyed, and Vacek 2002) and mediates its effects
in concert with IL-6 and IL-1 (Desai et al. 2013). TNF- α is a cytokine with dual effects, at low
concentrations promotes tumor angiogenesis, tumor cell survival and metastasis, but at high
levels prevents tumor growth and induce the expression of adhesion molecules and increase
vascular permeability, together with IFN-γ (S. Kim et al. 2009; Lumniczky and Sáfrány 2015).
The main negative regulator of inflammations in a radiated tumor include TGF-β (Roedel et al.
2002) and IL-10. TGF-β is upregulated and activated from its latent form after radiation
(Barcellos-Hoff et al. 1994). TGF-β is another pleiotropic cytokine and play a role in
extracellular remodeling accompanied by cancer cell migration and invasion (De Wever and
Mareel 2003). TGF-β secreted locally in the tumor microenvironment after radiation modulates
the local inflammatory response, suppresses DCs and CTL functions and promotes the
infiltration of CD4 Regulatory T cells (Barcellos-Hoff et al. 1994). IL-10 another anti-
inflammatory cytokine, secreted by cancer cells and apoptotic lymphocytes and monocytes also
influence the outcome of the immune response (Gao et al. 1998; Lumniczky and Sáfrány 2015).
The understanding of the intricate signalling and effects of cytokines in the tumor
microenvironment after radiation therapy will be essential for instituting better interventions
such as immunotherapeutic interventions.
1.3.7.2 Changes in the tumor microenvironment
Tumor cells undergo a process of immune-editing, and develop several mechanisms by which
neoplastic cells can escape immune recognition and elimination. As a result the tumor
microenvironment is an immunosuppressive environment. Radiation causes important changes in
the tumour cells that have implications for its interaction with the immune system.

41
One escape mechanism of tumor cells is the reduction of MHC I molecules on the surface
leading to inadequate antigen presentation (Rabinovich, Gabrilovich, and Sotomayor 2007).
However, after irradiation MHC Class I are up-regulated in a variety of tumors, both in vitro and
in vivo (Chiriva-Internati et al.; Ciernik et al. 1999; Reits et al. 2006). Furthermore, the cellular
damage induced by radiation increases the peptide concentration and repertoire displayed in
radiated cells (Reits et al. 2006). It has been shown by Reits et al. (Reits et al. 2006) that tumor
cells with upregulated MHCI molecules are eliminated more efficiently by tumor-specific
cytotoxic T lymphocytes (CTLs). Additionally, upregulation of the death receptor CD95, in a
p53 dependent way, was reported on multiples tumor models (Sheard, Uldrijan, and Vojtesek
2003; I.-C. Park et al.). CD95 signaling is also enhanced and leads to an increase in tumor
immunogenicity, since it can improve the cytotoxic effect of CD95 ligand-expressing CD8+ T
lymphocytes (Luce et al. 2009). Both, MHC class I upregulation and CD95 may enhance tumor
cell recognition and killing after radiation. T cells require co-stimulation to become fully
activated, Seo et al.(Seo et al. 1999) demonstrated that CD80 was upregulated on B cell
lymphoma and the same was demonstrated on myeloid leukemia cells after irradiation (A. Morel
et al. 1998). Thus, radiation can enhance co-stimulation and in this way it may prevent T cell
anergy. Adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) (Hallahan,
Kuchibhotla, and Wyble 1996; Garnett et al. 2004) and vascular cell adhesion molecule-1
(VCAM-1) (A. A. Lugade et al. 2008; A. a Lugade et al. 2005) have been shown to be up-
regulated on tumour cells or in the tumour vasculature after irradiation. This increased
expression of adhesion molecules enhance the migration of lymphocytes into the tumor and
increase the interaction between immune cells (Carlos 2001). Radiation also greatly increase the
level of tumour-associated antigens (TAAs) released in the tumor microenvironment (Garnett et
al. 2004; Hareyama et al. 1991). The increased availability of released TAAs for uptake by

42
circulating DCs can result in tumor-specific immune attack. Zhang et al. (B. Zhang et al. 2007)
demonstrated that irradiation tumor with low expression of antigen, caused a significant release
of antigen and that was enough to cause tumor cell killing by cytotoxic T lymphocytes. In
summary, radiation causes increased surface expression of MHC class I and CD95 on tumor
cells, increased expression of adhesion molecules in vessels, expression of costimulatory
molecules and increase levels of TAAs. All these effects increase tumor cell recognition by
effector cells, promote lymphocyte infiltration, prevent T cell anergy, and render tumor cells
more susceptible to CTL killing.
1.4 The immune response to surgery
1.4.1 The Surgical Stress response
Surgery or trauma lead to the “surgical stress” response. This response is a combination of
metabolic, immunologic and hematologic changes occurring after injury or trauma. The severity
of the response is proportional to the magnitude of injury and reflects increased demand of organ
function (Kehlet 1997). It is generally believed that the final outcome of the surgical stress
response is postoperative immunosuppression. This is particularly important in patients with
cancer since immunosuppression may promote tumor proliferation and metastasis (Dąbrowska
and Słotwiński 2014).
Following surgical trauma, the nervous system activates the stress response by sending impulses
to the hypothalamus via afferent nerves from the injury site. This in turn stimulates the
hypothalamic-pituitary-adrenal (HPA) axis and sympathetic nervous system. The final outcome

43
is the elevation of cortisol, glucagon, catecholamines, aldosterone, vasopressin and a host of
inflammatory cytokines in an effort to provide the body with energy, retain fluid and salt and
maintain cardiovascular homeostasis. However, this state can result in outcomes such as
hyperglycemia, cardiovascular instability and immunosuppression (Desborough 2000).
The hypersecretion of cortisol and catecholamines, due to surgical stress, has both anti-
inflammatory and immunosuppressant effects (Ogawa et al. 2000). Cortisol shift the Th1/Th2
balance by interfering with the production of Type 1 cytokines and increasing the synthesis of
Type 2 cytokines, by acting directly on CD4+ T cells and indirectly by inhibiting IL-12
production by monocytes (Marik and Flemmer 2012). Also, cortisol induces downregulation of
IL-12 receptors in T cells and NK cells (Ilia J. Elenkov 2004). IL-12 is involved in the
differentiation of naïve T cells into Th1 cells, and stimulates the production of IFN-γ by T cells
while inhibiting IL-4 synthesis. Catecholamines, through the stimulation of β2-adrenergic
receptors on macrophages/monocytes, contribute to the shift in Th1/Th2 balance by inhibiting
IL-12 synthesis and enhancing the production of IL-10 (Mizuno et al. 2005). Prostaglandin E2
(PGE2) which is produced by macrophages after tissue injury further increases Th2 cytokine
production while reducing Th2 cytokines(Lorne et al. 2008). Cortisol and PGE2 act
synergistically and cause immunosuppression after trauma. Furthermore, cortisol impedes
aggregation of macrophages and neutrophils at site of injury, and decreases phagocytosis. In
addition, it induces apoptosis, in T lymphocytes and promotes Th2 cell dominance (Jameson et
al. 1997). Regulatory T cells might also be involved in the immune suppression after surgery, as
demonstrated by MacConmara (MacConmara et al. 2006), who demonstrated significant increase
in Tregs by day 7 after trauma, these regulatory T cells were a significant source of Th2
cytokines in this cohort of patients. Furthermore, Ochoa et al. (Bryk et al. 2010; Zhu, Herrera,
and Ochoa 2010) demonstrated that arginase 1 (ARG1) is induced in myeloid-derived suppressor

44
cells (MDSCs) after surgery and trauma. Th2 cytokines, catecholamines and PGE2 induce the
expression of MDSC and act synergistically to increase the expression of ARG1 in these cells
(Marik and Flemmer 2012). This leads to a state of arginine deficiency, which is required for
lymphocyte proliferation, causing further immune depression. Moreover, the combination of
surgery, neuroendocrine response and analgesics, especially opioids, depress natural killer cell
activity (Beilin et al. 2003; Melamed et al. 2003). NK cells are an important component of the
innate immune system and play a crucial role in identifying and lysing tumor cells.
In summary, the activation of the surgical stress response causes elevation of cortisol,
catecholamines and PGE2 that result in a switch in the Th1/Th2 balance, promoting release of
Th2 cytokines. Th2 cytokines in turn induce the expression of ARG1, which depletes cellular
arginine and together with an increase in Tregs result in further impairment of T cell proliferative
responses.
1.4.2 Post-surgical cytokine cascades
The changes after surgery are related to the extent of surgical trauma and neuroendocrine stress
response (Pirttikangas et al. 1995). There is a balance between the release of pro and anti-
inflammatory cytokines. Multiple regulator mechanisms exist to maintain homeostasis and avoid
an unbalanced inflammatory state. Following surgical trauma there is an early proinflammatory
immune response, also called systemic inflammatory response syndrome (SIRS) and a late
adaptive response or compensatory anti-inflammatory response syndrome (CARS) (Kimura et al.
2010). After tissue damage, the initial proinflammatory response is mediated primarily by the
cells of innate immune system, whereby phagocytic and endothelial cells release IL-1β and TNF-

45
α. These cytokines are necessary mediators that direct the inflammatory response to sites of
infection and injury, and play an essential role in promoting wound healing and maintaining
homeostasis (Lin, Calvano, and Lowry 2000). IL-1 and TNF-α leads to cleavage of Iκβ and the
subsequent activation of NF-κβ triggering the synthesis of other proinflammatory cytokines. IL-6
and IFN-γ are the primary proinflammatory mediators induced by NF-κβ. IL-6 exerts both pro
and anti-inflammatory effects and has been shown to correlate with the duration of surgery and
the extent of injury. (Menger and Vollmar 2004). IL-6 induces the production of acute phase
reactants such as C-reactive protein and procalcitonin. Also, IL-6 plays a major role in the
proliferation of polymorphonuclear (PMN) progenitors in the bone marrow and later in the
function of mature PMNs (Botha et al. 1995). Lastly, IL-6 act as an anti-inflammatory cytokine
and helps controlling local or systemic acute inflammatory responses (Xing et al. 1998). IL-6
exerts anti-inflammatory properties during injury by attenuating TNF-α and IL-1 activity,
furthermore, it promotes the release of sTNFRs and IL-1ra (Lin, Calvano, and Lowry 2000).
The early proinflammatory response following surgery is a result of a predominance of the Th1
cytokines (IL-2, IL-12, and IFN-γ). However, the increased surgical stress release of
glucocorticoids, catecholamines and acute phase reactants often results in a shift towards the
anti-inflammatory Th2 predominance (cytokines IL-4, IL-5, IL-6, IL-10, and IL-13) later in the
postsurgical period with consequential depressed cellular immunity (Menger and Vollmar 2004;
Lin, Calvano, and Lowry 2000).

46
1.4.3 Postoperative tumor progression
Surgery is the most successful therapy when treating patients with solid tumors (Demicheli et al.
2008). However, occasionally radical surgery accelerates growth and dissemination of residual
malignant cells (Kal, Struikmans, and Barten-van Rijbroek 2008). In the wound bed, cells that
normally divide infrequently, are induced to proliferate rapidly, epithelial cells and stromal cells
migrate and new blood vessels are recruited. Thus, a wound response would appear to provide a
highly favourable milieu for cancer progression (Hofer et al. 1999). Furthermore, enhanced
tumor progression following surgery is thought to be largely mediate by NK cell suppression
(Colacchio, Yeager, and Hildebrandt 1994; Shamgar Ben-Eliyahu, Page, and Schleifer 2007; S
Ben-Eliyahu et al. 1999). Animal studies indicate that the majority of anesthetics have a
profound suppressive effect on NK cells and some of them are associated with increased
metastases (Welden et al. 2009). A balanced Th1/Th2 ratio is important for anticancer immunity
(Melamed et al. 2003). The characteristic change in balance towards an Th2 type immune
response after surgery is associated with depressed cellular immunity and tumor surveillance
(Wada et al. 2007). Finally, surgical manipulation may result in shedding of tumor cells and
release of growth and angiogenic factors (VEGF) (Hormbrey et al. 2003), thus promoting
metastasis and tumor growth. In summary, a favourable milieu for proliferation, the anti-
inflammatory response following surgery with the dominant Th2 response, and NK cell
suppression contribute to tumor progression and metastasis after surgical trauma.
1.5 Summary

47
MPM is a cancer of the pleura associated with the inhalation of asbestos fibers. Due to a long
incubation time between exposure and onset of disease, the incidence of disease in North
America has increased by 65 percent over the past two decades worldwide and is projected to
continue to increase until at least the year 2020. Moreover, the continued use in the developing
world suggests that MPM will continue to rise worldwide for the foreseeable future. Median
survival of MPM patients without treatment is 4 to 9 months. Non-resectable MPM is treated
with a pemetrexed and platinum based chemotherapy regimen that increases median survival
from 9 to 12 months in epithelial disease. Conventional therapies to MPM offer little
improvement in survival, thus newer therapeutics are needed.
The group of Dr. De Perrot and Dr. Cho has developed a new approach consisting in
hypofractionated radiation followed by Extrapleural Pneumonectomy which differs from
previous approaches in which radiation is given after surgical resection. A feasibility study of
Surgery for Mesothelioma After Radiation Therapy (SMART) has demonstrated that this
procedure results in a significant increase in 3-year survival in patients with epithelial disease
from 53% to 84% compared to previous treatment modalities. The authors discussed, based on
accumulating evidence, that high-dose hypofractionated radiation is able to stimulate the immune
system in addition to mediating direct tumor killing. Cytotoxic T lymphocytes circulate through
the body and are able to recognize and eradicate tumor cells. However, a strong inhibitory tumor
microenvironment renders T cells unable to inhibit tumor growth. Radiation has been shown to
reverse this immunosuppressive environment and promote a strong inflammatory response which
may shift the balance towards an anti-tumor response. In the SMART approach the pro
inflammatory effect of radiation together with the removal of the immunosuppressive tumor may
have contributed to the beneficial effects seen in patients. This pro inflammatory effect of
radiation may open the door to newer therapeutic approaches against cancer such as combination

48
with immunotherapy. In this project, we investigated the effect of local radiation therapy in
combination with surgery and immunotherapy in a mouse model of mesothelioma. We analyzed
the immune response after radiation and the role of CD8+ T cells. Finally we assessed the role of
lymphocytes in the generation of a protective immunological memory in our mouse model. The
following chapter will outline the hypothesis and aims of this study.

49
2 Hypothesis and Aims
Radiation therapy (RT) is an important modality in the treatment of mesothelioma and many
other tumors. Until recently RT was used based on the ability to eradicate cancer cells by means
of its cytotoxic effect [1]. In regards of its interaction with the immune system, it was considered
an immune attenuator. If we consider the effect of radiation to any particular cell in isolation, it
will be almost always detrimental. However, if we account for the interaction between tumor cell
death, enhanced antigen expression on tumor cells, and inflammatory signals from the irradiated
tissue which affects lymphocytes and dendritic cells (DC), we could have a beneficial effect on
the immune system.
The goal of this study was to investigate the effect of local radiation therapy of a tumor in
combination with surgery and immunotherapy in the context of mesothelioma in a mouse model.
The goal of this translational research projects was to use the findings of this study to understand
and improve the treatment approach of MPM.
The objectives of this study were threefold: 1) to develop a mouse model of MPM to study the
effect of local radiation therapy and surgery; 2) to evaluate CD8+ T cell immune response on
tumors after radiation therapy; 3) to investigate the effect of radiotherapy combined with
immunotherapy, specifically the antibody anti CTLA-4.
For the first model a subcutaneous tumor is established in the right flank of the mouse. Tumor
growth is monitored and treated when the tumor reaches a threshold area. The tumor is irradiated

50
and/or removed surgically. Being a subcutaneous model it is easy to follow tumor growth and
evaluate the effect of different treatments.
I propose combination therapy with hypofractionated local radiation and surgery will have better
tumor control than either therapy alone. For the second objective, I postulate activation of the
immune system will play a role and there will be more CD8+ T cell tumor infiltration in the
group receiving radiation therapy either alone or in combination. CD8+ T cell infiltration will
correlate with tumor control and will provide treated animals with immunological protective
memory. This immunological memory will protect mice when rechallenged with the same tumor
and depletion of lymphocytes will make them susceptible to rechallenge.
For the last objective, we will examine immunotherapy in the mouse model to further support
the role of immune activation after local hypofractionated radiation. Tumor bearing mice will be
treated with anti CTLA-4 antibody alone or in combination with radiation, surgery or both. It is
hypothesized that anti CTLA-4 will have a synergistic effect when combined with radiation
therapy. The hypothesis that radiation induces an immunogenic cell death and promotes
recruitment and function of T cells within the tumor microenvironment supports the idea that a
tumor can be converted into and in situ vaccine. This effect of radiation might be relevant and
provide a synergistic effect when combined with immune checkpoint inhibitors such as anti
CTLA-4 antibody.

51
3 Materials and Methods
3.1 Murine Cell lines
AB12 and AE17 malignant pleural mesothelioma cell lines were both derived from an asbestos-
induced tumor in a BALB/c and C57BL/6 mouse, respectively. AB12 was kindly donated by Dr.
Jay Kolls, University of Pittsburgh, Pittsburgh, PA, in 2008. AE17 was obtained from the
European Collection of Cell Cultures. AE17-OVA was developed by stably transfecting the
parental cell line (AE17) with secretory ovalbumin (sOVA).(Jackaman et al. 2003) The cell line
was kindly provided by Dr. Steven Albelda, University of Pennsylvania, Philadelphia, PA, and
Dr. Delia Nelson, University of Western Australia, Crawley, WA, Australia.
AB12 and AE17 were grown in RPMI 1640 culture media (Life Technologies Inc., Burlington
ON, CAN) supplemented with 10% heat inactivated fetal bovine serum (Life Technologies Inc.,
Burlington ON, CAN), 2 mM L-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin
and non-essential amino acids. The transfected cell line AE17-OVA was maintained in the same
medium supplemented with 400 μg/L neomycin analog G418 (geneticin; Invitrogen). Cells were
plated in tissue-culture coated flasks (BD Biosciences Canada, Mississauga, ON).Additionally,
cultures were grown in a 37oC and 5% CO2 environment and passaged when 70% confluent. For
passage, cells were trypsinized using 0.25% trypsin and split 1 in 10.

52
3.2 Mice
Eight to twelve week old C57BL/6 and BALB/c wild type mice were purchased from the
Jackson Laboratory (Maine, USA). All mice were housed at the Toronto Medical Discovery
Tower’s Animal Resource Centre under pathogen-free conditions in accordance with
institutional and national animal care and ethics protocols.
3.3 In vivo Tumor Growth experiments
Mice were injected subcutaneously in the right flank with 1x106 AB12 cells, AE17 or AE17-
OVA cells in 100μL of PBS at day 0. For rechallenge experiments cells were injected s.c. in the
left flank. After removing fur and cleaning the skin, injections were made with a syringe and 25-
27G needles. Tumor growth and weight was monitored 3 days a week. Tumor dimensions were
measured using microcallipers. Mice were regularly checked and sacrificed when tumor
dimension reached 150mm2 or showed signs of ulceration as per institutional ethics protocols.
3.4 Local Radiation Therapy
For all experiments involving local radiation therapy, eight to twelve week old BALB/c mice or
C57BL/6 mice were injected subcutaneously in the right flank with 1x106 AB12 cells or AE17-
OVA cells in 100μL of PBS, respectively at day 0. On radiation day, tumors were either sham-
irradiated or irradiated with a total dose of 15 Gy over 3 days. Radiation was given using the X-
Rad 225Cx small animal image-guided irradiator (Precision X-Ray, Branford CT, USA). The

53
irradiator has a 225kVp x-ray tube (Varian Associates, Palo Alto, CA) and a flat-panel silicon
detector mounted on a 360o rotation C-arm gantry.(Moretti 2010) The automated stage is
movable on the x, y and z axis. It is all housed in a self-shielded cabinet and is remotely
controlled by a computer (Dell Precision 690, Intel Xeon CPU running Windows XP). The mean
targeting displacement error is approximately ≤0.1mm in the x-y-z planes. Radiation was given
to mice under isoflurane anesthesia and positioned on the irradiation stage. To initially visualize
the animal and the tumor fluoroscopic mode was used. To target precisely the tumor a scout
cone-beam computed tomography was done at a 40kVp tube potential and 0.5mA current. The
tomography was then reconstructed at a 0.4mm voxel size (Moretti 2010). The beam source was
collimated to either a 1.5 cm or 2 cm diameter circular field. To confirm the area to be irradiated
the tumor was then visualized under fluoroscopic imaging with the collimator in place,
immediately prior to delivery of treatment. Radiation was delivered at a tube potential of 225kVp
and a 13 mA current for a dose rate of 3.02 Gy/min. The daily dose was given from 2 angles,
half from above and half from below. Total dose was given in divided fractions over 3 days
according to treatment protocols. After radiation mice were placed back in their cages and
housing facilities.
3.5 Surgical Resection of Subcutaneous Tumors
Mice with flank tumors under general anesthesia with isoflurane were shaved and cleaned with
isopropanol. Tear gel was applied on both eyes and a heating lamp was used to prevent
hypothermia. Skin around the tumor was infiltrated with Marcaine (bupivacaine 0.25%) prior to
incision. Two different approaches were used depending on the experiment. We named the first

54
approach, “Blunt Surgery”, and the second and more aggressive approach, the “Radical
Surgery”. For both approaches, a 1 cm incision was made adjacent to the tumor. Blunt Surgery
was achieved using careful blunt dissection. All macroscopic tumor was removed but no skin or
surrounding healthy-looking tissue (Figure 3)
Figure 3 Partial resection of a subcutaneous tumor with blunt dissection. Under general
anesthesia the animal was positioned in the lateral decubitus position with the tumor exposed and
removal of the tumor was performed. A) After infiltration around the tumor with bupivacaine a 1
cm incision was made along the tumor. B) With blunt dissection the tumor was removed. C)
Complete macroscopic resection of the tumor was done but no skin or surrounding tissue. D) The
wound was closed with 6-0 prolene.
A
C
B
D

55
Radical Surgery was performed removing skin on top of the tumor and a 0.5 cm margin of
healthy skin around the tumor. All macroscopic tumor was removed including vascular supply
and 0.5 cm margin of healthy-looking subcutaneous tissue around the tumor. Sterile prolene 6-0
or 5-0 sutures were used to close wounds. Marcaine was administered immediately after closing
the wound along the suture for postoperative analgesia and the mice were observed until
complete recovery. Mice were then monitored 6hrs, 24hrs and 48 hrs after surgery and
meloxicam 1mg/kg SC was given for postoperative analgesia in case of mild or moderate pain.
3.6 Combination therapy with LRT and Surgery
For the experiments involving combination therapy with LRT and Surgery tumors were
inoculated in the right flank. After 7 to 10 days when all tumors were larger than 6 mm2, mice
were treated with LRT for 3 consecutive days as described before and depending of the
experiment, 24hrs, 5 days or 7 days after, surgery was performed.
3.7 In vivo depletion of CD4+ and CD8+ specific T cells
Anti-CD4 MAb from rat GK 1.5 hybridoma or anti-CD8 Mab from 2.43 rat hybridoma (Bio X
Cell, West Lebanon, NH, USA) were diluted to a final concentration of 1 mg/mL with PBS or
2mg/ml for double depletion. Intraperitoneal injections for 3 consecutive days with 0.2 ml (0.2
mg) of purified Mab were performed. For double depletion the total volume injected was 0.2 ml,
consisting of 0.1 ml of each Mab at a concentration of 2mg/mL. By day 6, peripheral lymphoid

56
organs were depleted and one control was sacrificed to assess satisfactory depletion (>95%). The
depleted condition was maintained with 0.2 mg injections of MAb every 3 days.
3.8 Anti-CTLA-4 therapy
Mouse monoclonal antibody (9D9) to CTLA-4 (Bio X Cell, West Lebanon, NH, USA) was
diluted to a final concentration of 1mg/mL with PBS and kept at 4°C until further use.
Intraperitoneal injections were made with 0.2 mL (0.2 mg) of the purified antibody every 3 days
for the length of the specified treatment.
3.9 Blood Collection
Blood was collected by tail venipuncture or heart puncture using heparinized capillary blood
collection tubes (Fischer Scientific Co., Toronto, ON). Red blood cells were lysed with ACK
lysing buffer (Life Technologies Inc. Burlington, ON). Finally, remaining cells were washed
twice with phosphate buffered saline, pH 7.4 (Life Technologies Inc., Burlington ON, CAN).
3.10 Tumor Digestion
Tumors were removed and placed in 15 mL conical tubes filled RPMI 1640 culture media and
stored on ice until further use. To prepare for dissociation the tissue was chopped to small pieces
(approx. 2 mm) and transferred to 15 mL conical tubes containing digestion media consisting in

57
RPMI 1640, DNAse (Roche 10104159001) and Liberase TM (Roche Diagnostics, Laval,
Quebec, Canada). Tubes were placed in a shaking water bath for 30 minutes and when the pieces
were soft and malleable the solution was filtered and mashed through a 70 μm cell strainer. Cells
were then washed with PBS and remaining cells were counted and viability was assessed.
3.11 Isolation of Lymphocytes from Spleens and Lymph Nodes
Spleens and inguinal and axillary lymph nodes were isolated in phosphate buffered saline (PBS)
and filtered through a 70μm cell strainer. Red blood cells were lysed with ACK lysing buffer
(150mM NH4Cl, 10mM KHCO3, 0.1mM NA2EDTA) for 10 minutes at room temperature and
washed twice with PBS after lysis. Remaining cells were counted using trypan blue exclusion
and a hemocytometer.
3.12 Flow Cytometry
Cells were resuspended in FACS buffer (PBS, 2% FBS, EDTA 5mM) and stained for 30 minutes
at 4 C with α-CD16/CD32 Fc block (BD, Pharmingen; San Diego, CA, USA), and a combination
of the following mouse specific antibodies: CD3, CD4, CD8, CD44, CD45, CD69, CD137 (4-
1BB), TIM3, PD-1, ICOS. Cells stained with tetramer were incubated for 30 min with the Class I
H-2Kb SIINFEKL tetramer prior to surface staining. All samples were then washed twice with
FACS buffer and analyzed immediately. FACS analysis was conducted using a BD LSR II flow
cytometer (BD Biosciences, Mississauga ON, CAN). Analysis was performed using FlowJo
V10 (FlowJo LLC, Ashland, USA) software.

58
3.13 Immunofluorescence
Frozen tissue samples on slides were fixed with cold acetone for 10 minutes. Paraffin embedded
samples were deparaffinised with Xylene, 100% ethanol, 95% ethanol, and 70% ethanol
respectively and antigen retrieval was performed by immersing samples in 100°C citrate buffer
for 20 minutes. Samples were blocked with 5% BSA in Tris-buffered saline for one hour before
the addition of primary Ab. After incubating overnight at 4°C, sections were washed in
TBS+0.2% Tween 20. Slides were subsequently incubated for 1 hour at room temperature with
the appropriate fluorescently labelled secondary Ab. Slides were further washed with
TBS+0.2% Tween 20 before adding mounting media with DAPI nuclear stain. Cover slips were
placed on top and sealed with nail polish.
Fluorescently labelled cells or tissues were visualized with the WaveFX (Quorum Technologies
Inc, Guelph ON, CAN) confocal microscope system. Pictures were analyzed using ImageJ
V1.47 (National Institute of Health, USA). Corrected total cell fluorescence (CTCF) was
calculated by the formula CTCF = Integrated Density – (Area of cell x mean fluorescence of
background reading).
3.14 Immunohistochemistry
Tumors were snap frozen in liquid nitrogen and kept at –80oC on dry ice until staining. Frozen
samples were sliced into 5µm thick sections using a microtome. Frozen sections were fixed with
1% paraformaldehyde for 1 hour before staining. Sections were blocked with serum (5% BSA in
Tris-buffered saline) for one hour before the addition of primary anti-CD3, or CD8 antibody at a

59
1:100 dilution. After incubating overnight at 4oC, sections were washed in TBS+0.2% Tween
20. Slides were subsequently incubated for 1 hour at room temperature with anti-rabbit-HRP
secondary Ab kit (Vector Laboratories Inc., Burlington ON, CAN). After washing, the HRP
substrate DAB (3, 3-diaminobenzidine) (Vector Laboratories Inc., Burlington ON, CAN) was
added to each slide at 100 µL per section. Sections were then counterstained with hematoxylin,
dehydrated, and mounted with mounting media (Fischer Scientific, Ottawa ON, CAN).
Immunostained slides were imaged at 200x magnification with the Aperio ImageScope digital
scanner and visualized with Aperio ImageScope Viewer version 12.1 (Vista, CA, USA).
Scanning was provided by the Advanced Optical Microscopy Facility (Toronto ON, CAN)
3.15 Ovalbumin ELISA
AE17-OVA and AE17 cell culture supernatants were collected 3 days after seeding cells. Cells
were then trypsinized and washed twice with PBS. For cell lysates, cells were collected by
centrifugation, 5 min at 1000xG. Cells were then subjected to ultrasonication for 4 cycles on ice.
Cell lysates were collected by centrifugation at 1500xG for 10 minutes at 4 °C to remove cellular
debris. Cell lysate or media was placed in wells coated with a biotin-conjugated antibody
specific to OVA from an ELISA kit (Biomatik corporation, Cambridge, ON). Samples were left
for 2 hours to bind anti-Ova antibodies. Avidin conjugated to Horseradish Peroxidase (HRP) was
then added to each well and incubated for one hour. Finally TMB substrate solution was added
and those wells containing OVA, biotin-conjugated antibody and enzyme-conjugated Avidin
exhibited a change in color. Reaction was terminated by the addition of sulphuric acid.

60
Concentrations were determined by four-parameter logistic test using a standard curve. Samples
were measured in duplicate.
3.16 Statistical Analysis
Statistical analysis was performed with GraphPad Prism 5 (GraphPad Inc, La Jolla CA, USA).
More than 2 groups were compared using one-way ANOVA analysis. Student’s T-test was used
to analyze two groups. A P-value of less than 0.05 was considered statistically significant.
Results have been presented as Mean ± Standard Error of the Mean (SEM). * indicates p<0.05,
** indicates p<0.01, and *** indicates p<0.001 in all results figures.

61
4 Results
4.1 Development of a Mouse Model of Malignant Mesothelioma
To develop a mouse model of mesothelioma, BALB/c mice and C57BL/6 mice were inoculated
with the cell lines AB12 and AE17-OVA respectively.
4.1.1 Local Radiation Therapy, Right Flank Model
To determine the radiation sensitivity of tumors in vivo, tumor irradiation with 4 different doses
was conducted. Initially experiments were done using BALB/c mice and the cell line AB12
following a previous model used by our group. The tumor was inoculated subcutaneously in the
right flank with 1x106 AB12 cells. The right flank was chosen because it is accessible for
consistent caliper measurements and readily accessible for surgical resection of the tumors.
Tumors were locally irradiated with targeted x-rays starting 7 days after tumor inoculation
(Error! Reference source not found.). On the first day of treatment, mice were randomized into
the following groups: 1) no treatment, 2) 15Gy over 3 days (5Gy x 3), 3) 22.5Gy over 3 days
(7.5Gy x 3), 4) 30Gy over 3 days (10Gy x 3) and 5) 22.5Gy in a single dose. Tumor growth was
followed every 3 days with caliper measurement before and after treatment until they reached a
humane endpoint, at that point mice were sacrificed. Tumor size is expressed as tumor area in
squared millimeters using the longest length and the perpendicular width (Length x width).

62
Figure 4. Schematic of radiation treatment in tumor bearing mice. Tumor was inoculated on
day 0. In the treated group, Local Radiation Therapy (LRT) started on day 7. Depending on the
group, radiation was give on 3 consecutive days or on a single dose. Tumor was monitored every
3 days with caliper measurements.
Untreated mice showed the most rapid tumor growth and greatest tumor area. After 22 days
tumor growth decelerated (results not shown) and mice were in distress, at that time point mice
were sacrificed. The tumor growth in untreated mice could be described as a sigmoidal curve.
Radiation with 30Gy (10x3) and 22.5G in one fraction exhibited an excellent response to
radiation compared to no treatment. Tumor growth stopped for 10 days after treatment started
and tumor area was significantly lower on day 16, 19, and 22 compared to no treatment (
Figure 5). However, irradiated mice showed signs of distress and lost 10 to 15% of total body
weight during the first 2 weeks after treatment (data not shown). Radiation with 15Gy (5x3) and

63
22.5Gy (7.5x3) displayed a good response, with tumor growth deceleration for 7 to 10 days after
treatment. Tumor area was significantly lower compared to untreated mice at days 16, 19 and 22.
There was no significant difference among LRT treatment groups.
Overall, tumor growth slowed down in mice treated with local radiation therapy (LRT) compared
to untreated controls for at least 6 days in all cases. The difference was significant for all groups
receiving 15Gy and 30Gy at in 3 fractions. After approximately 7 to 10 days post-radiation,
tumor growth accelerated in all groups. This result confirms that the tumor model is sensitive to
LRT. For the following experiments 15Gy was used, consistent with others in our group and
because it was the lowest radiation dose with good effect in tumor growth without accompanied
weight loss or mouse distress.

64
A
B
Figure 5 Increasing doses of Local Radiation Therapy and its effect on tumor growth. A)
Tumor area is represented as the mean of 5 mice. All 4 LRT doses showed an excellent response,
with tumor growth stable for at least 7 days B) Tumor is significantly smaller in the 4 LRT
groups compared to untreated mice on days 16, 19 and 22. Values shown are the mean ± SEM of
5 mice per time-point. ** < 0.05, n.s not significant comparing treated groups to untreated.

65
4.1.2 Combination therapy with LRT and Surgery
To evaluate the different treatment modalities in the BALB/c mouse model and to assess whether
or not LRT combined with surgery had a benefit, the different treatment modalities were
compared. In this experiment blunt surgery was used to remove tumors. Mice were randomized
the first day of LRT into the following groups: 1) No treatment, 2) Local Radiation Therapy
15Gy (5x3), 3) Surgery, 4) LRT and Surgery. In the LRT and Surgery group, blunt surgery was
performed 5 days after completion of LRT (Figure 6). For statistical analysis the day of surgical
removal of the tumor is day 0 for the groups treated with blunt surgery, otherwise day 0 is the
day of the inoculation of the tumor.
Mice treated with surgery alone had tumor recurrence as early as 48 hours after surgery. Tumor
growth rate after tumor resection was more rapid than untreated tumors and tumor size was
significantly larger than untreated mice at days 17 and 24. In the group treated with LRT alone
tumor growth slowed down significantly compared to untreated mice. Tumor area was
significantly smaller at day 24 and 28. Finally, in the combination group, tumor growth slowed
down after LRT and prior to surgical removal. Tumor growth after surgery was not observed
until at least 7 days later. Tumor size was significantly smaller compared to untreated tumors on
days 17 and 28 (n=5, p=0.035; n=5, p=0.0415) (Figure 7).
Overall, tumor growth after blunt surgery alone was accelerated compared to untreated mice.
Conversely, tumor growth slowed down in the mice treated with LRT alone or LRT and blunt
surgery. This suggest blunt surgery alone may have negative effects in the treatment of tumors
and is not a good therapeutic approach in this model. However, when LRT was given prior to
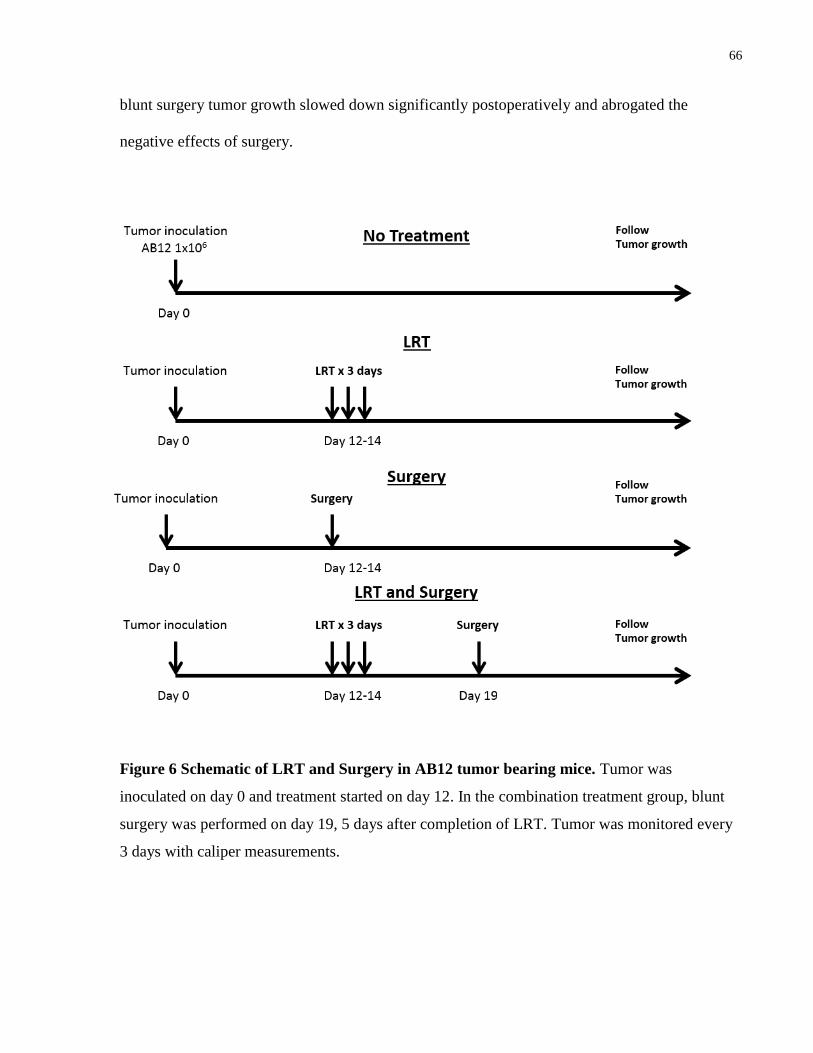
66
blunt surgery tumor growth slowed down significantly postoperatively and abrogated the
negative effects of surgery.
Figure 6 Schematic of LRT and Surgery in AB12 tumor bearing mice. Tumor was
inoculated on day 0 and treatment started on day 12. In the combination treatment group, blunt
surgery was performed on day 19, 5 days after completion of LRT. Tumor was monitored every
3 days with caliper measurements.

67
Figure 7. The effect of combination therapy with LRT and Surgery. LRT alone or in
combination with blunt surgery slows down tumor growth. However, blunt surgery alone accelerates
tumor growth compared to no treatment. A) Untreated group B) Blunt surgery alone day 12. C) LRT
alone day 12 D) Combination group, LRT at day 12 and blunt surgery 5 days after. E) Groups means
showing a significant negative effect of surgery alone and a significant beneficial effect on the groups
treated with LRT F) Significant difference at days 17 and 24 in the surgery group compared to no
treatment and at days 24 and 28 in the LRT treated groups compared to untreated. In E) and F) the day of
surgical removal of the tumor is day 0 for the groups treated with blunt surgery, otherwise day 0 is the
day of the inoculation of the tumor. Values shown in E) and F) are the mean ± SEM of 5 mice per time-
point. ** < 0.05 compared to untreated, §<0.05 compared to surgery.

68
4.2 T cells infiltrate tumors after LRT
4.2.1 Tumor infiltrating CD8+ T Cells in Untreated and Radiated Mouse
Tumor Tissue
To evaluate if the immune system was involved in the effect of LRT on tumor growth we
analyzed CD8+ T cell tumor infiltration after LRT. CD8+ T Cells are responsible for mediating
antigen-specific tumor cell killing. Staining for CD8+ T Cells was performed on untreated and
radiated tumors to study the effect of radiation in terms of CD8+ T Cell recruitment. The
recruitment of the CD8+ T Cell immune response was measured by immunofluorescent staining
of tumor tissue for CD3+CD8+ cells. Tumor samples from 15Gy radiated and untreated mice
were taken 2, 7, and 12 days after LRT for the pathology analysis and on day 7 for FACS
analysis. Tumors in the treated group were visibly smaller compared to untreated tumors at days
7 and 12 after LRT. To confirm results, analysis was done with flow cytometry of the treated or
untreated tumors, stained with conjugated antibodies specific for CD45, CD3 and CD8. For
FACS analysis 3 tumor samples were pooled and analyzed as a single sample.
The number of CD3+CD8 T cells on the slides was quantified at each time point by averaging
the cell counts of 5 random fields (Figure 8a). The count of double positive cells 2 days after
LRT was not significantly different (18.40 ± 4.739 vs. 15.20 ± 2.818 cells, p= 0.5775). However,
7 days after LRT the count was significantly higher in the LRT group compared to untreated
tumor (32.20 ± 8.225 vs 10.60 ± 3.076 cells, p=0.0398). Similarly, 12 days after LRT, treated
tumors showed higher counts compared to untreated (55.60 ± 13.07 vs. 1.600 ± 0.5099 cells,
p=0.0033) (Figure 8b). Peak staining occurred 12 days after initiating radiation. It was also
noted that the number of double positive cells decreased in the untreated tumor over time (days 7

69
and 12) and as the tumor grew bigger. This was in contrast to what was observed in the treated
tumor. We confirmed these results with FACS, where we observed 5.19% CD45+ CD3+CD8+
cells infiltrating the treated tumor compared to only 1.03% in the untreated tumor 7 days after
the first day of radiation (Figure 8c).
In summary, treated tumors showed a significant increase in the number of infiltrating
CD3+CD8+ cells on days 7 and 12 after LRT. This suggest LRT recruits CD3+CD8+ T cells
into the tumor.

70
C
Figure 8. CD3+CD8+ cell tumor infiltration after LRT compared to untreated tumors.
Immunofluorescent staining of tumor 2, 7 and 12 days after LRT, compared to untreated tumor
controls. A) Images show DAPI (405nm), CD3 (488 nm) and CD8 (633nm) merged staining. B)
Average cell count of 5 random x200 magnified fields. *<0.05, **<0.005 C) FACS analysis of
the treated and untreated tumor 7 days after the first day of radiation. Doublets and dead cells
were excluded before gating on CD45/CD3. FACS confirms the increase number of CD3+CD8
cell infiltrating the tumor after LRT compared to untreated control. The graph represent 4 tumors
pooled into a single tube.

71
4.2.2 A Large Proportion of Tumor Infiltrating Lymphocytes are OVA-
specific
To examine whether or not the increased number of tumor infiltrating CD8 T cells in the treated
tumor were tumor-specific, tumor infiltrating cells were stained using H-2Kb tetramers
containing the OVA protein-derived peptide SIINFEKL. For this experiment C57BL/6 mice and
the cell line AE17-OVA were used. Ten days after LRT treated and untreated tumors were
excised, and analyzed with FACS.
After gating for live cells, CD3 and CD8 double positive cells were identified. Out of the
CD3+CD8+ double positive population, the proportion of CD44+ and SIINFEKL tetramer cells
were identified (Figure 9). Radiated tumors showed greater proportion of tetramer specific
CD8+ T cells compared to untreated tumors. This difference was close to significance comparing
4 tumors in each group (30.60 ± 6.785 n=4 vs 14.99 ± 2.554 n=4, p=0.07).
The result of this experiment gives further evidence that LRT promotes recruitment of
lymphocytes into the tumor. About 30% of the recruited lymphocytes are specific for the OVA
derived peptide SIINFEKL in the treated tumor compared to only 14.99% in the untreated group.
Together with the previous experiment this suggests radiation of a tumor with 15Gy in 3
fractions in our model stimulates recruitment of CD8+ T cells to tumor tissue. A high proportion
of the CD8+ T cells are specific against our tumor antigen OVA.

72
A B
Figure 9. CD8+ lymphocytes infiltrating AE17-OVA tumor are OVA specific. A)
Representative flow cytometry graph gated on CD3+, CD8+. There is greater proportion (50.8%)
of CD44+ Tetramer+ double positive cells in the radiated group compared to untreated tumor
(21.4%). B) Graph comparing proportion of tumor specific CD8+ T cells in radiated and
untreated tumor.

73
4.2.3 Expression of 4-1BB and PD-1 by Tumor Infiltrating Cells
To assess the phenotype of tumor infiltrating lymphocytes following LRT we assessed the
expression of the inhibitory receptor, PD-1 and the activation marker 4-1BB on tumor specific
CD8 T cells. C57BL/6 mice and the cell line AE17-OVA were used. LRT treated tumor and
untreated controls were analyzed 3 and 10 days after completion of LRT. Flow cytometric
analysis was utilized to quantify the frequency of CD8+ Tetramer+ 4-1BB+ or CD8+ Tetramer+
PD-1+.
The early activation marker 4-1BB was significantly upregulated in the treated tumors 3 days
following radiation compared to untreated controls (8.932±2.521 n=5 vs 1.840 ±0.280 n=5,
p=0.0233) but 9 days later there was no difference (4.938±3.393 n=4 vs 4.1418±1.078 n=4,
p=0.0831) (Figure 10). In the case of PD-1, significantly fewer lymphocytes expressed the
marker at day 3 and 9 after radiation (16.13±5.453 n=4 vs 38.63±5.138 n=4, p=0.0239; and
30.45±12.39 n=4 vs 67.7±6.495 n=4, p=0.0374) (Figure 10).
In summary, radiation enhances the expression of 4-1BB on tumor specific infiltrating
lymphocytes and downregulates the expression of PD-1. This suggest that LRT may reverse T
cell exhaustion and restore T cell activation temporarily.
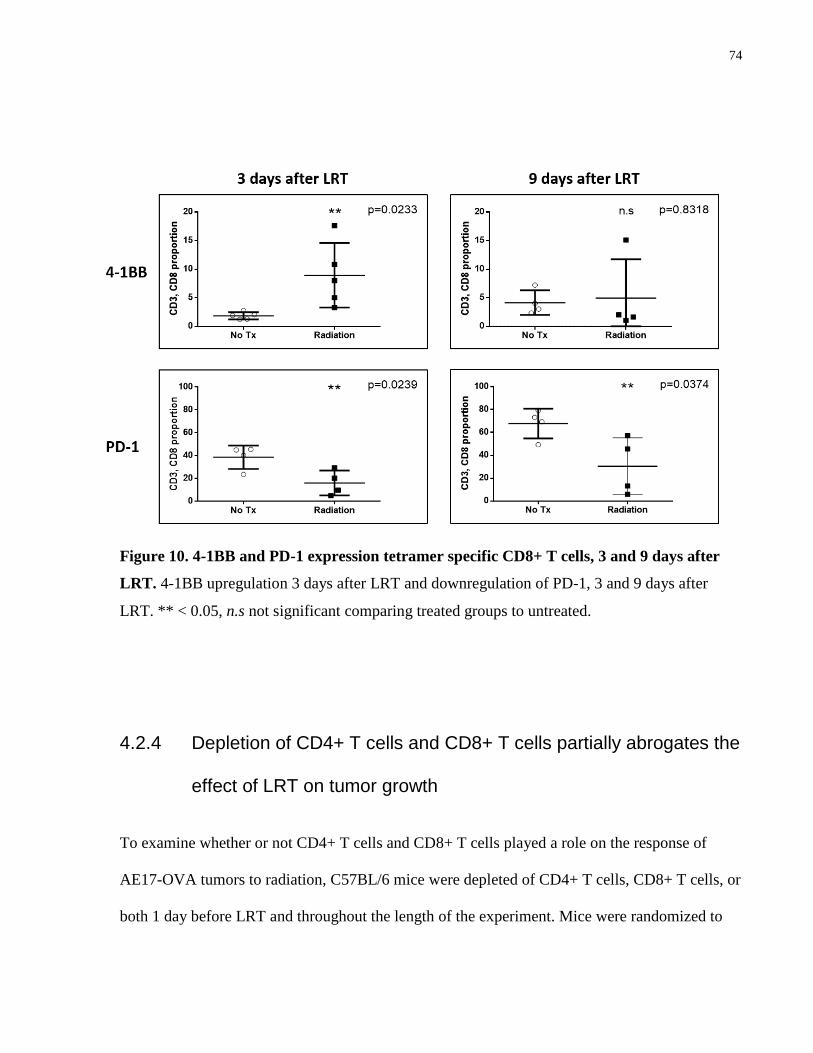
74
Figure 10. 4-1BB and PD-1 expression tetramer specific CD8+ T cells, 3 and 9 days after
LRT. 4-1BB upregulation 3 days after LRT and downregulation of PD-1, 3 and 9 days after
LRT. ** < 0.05, n.s not significant comparing treated groups to untreated.
4.2.4 Depletion of CD4+ T cells and CD8+ T cells partially abrogates the
effect of LRT on tumor growth
To examine whether or not CD4+ T cells and CD8+ T cells played a role on the response of
AE17-OVA tumors to radiation, C57BL/6 mice were depleted of CD4+ T cells, CD8+ T cells, or
both 1 day before LRT and throughout the length of the experiment. Mice were randomized to

75
the following groups: 1) No treatment, 2) LRT only, 3) LRT and CD4 depletion, 4) LRT and
CD8 depletion, 5) LRT and double depletion.
All groups treated with LRT showed significantly delayed tumor growth compared to the
untreated group at days 17, 20 and 24. Double depleted animals treated with LRT showed
significant greater tumor size than animals treated with LRT at days 24 and 27. In comparing
LRT only group and LRT and CD8 depleted groups there was a trend towards larger tumor area
in the CD8 depleted group at day 24 and 27. There was also a trend for smaller tumor size in
CD4 depleted animals treated with LRT compared to LRT only at days 24 and 27. Finally, CD8
depleted mice treated with LRT showed significant greater tumor size than CD4 depleted mice at
day 24 and 27 (Figure 11).
In summary, tumor size was significantly larger in double depleted animals treated with LRT
compared to mice treated with LRT only. This suggest T cells play a role in the beneficial effect
of LRT on tumors. Although not significant, there is a trend toward larger tumors in the LRT and
CD8 depletion group, and, toward smaller tumors in the LRT and CD4 depletion group
compared to the LRT only group. This suggests that the beneficial effect of LRT is partially
mediated by CD8+ T cell activity. On the other hand, CD4+ T cells may play a detrimental role
on radiated tumors, as shown by the trend towards smaller tumors when mice were depleted.
This effect is probably related to CD4+ regulatory cells activity within the tumor.

76
A
B
/
Figure 11. LRT and CD4+ CD8+ T cell depletion. Tumor growth in mice treated with LRT
and depletion of CD4+, CD8+ or both. Values shown are the mean tumor area in mm2 of 5 mice
per time point and are expressed as mean±SEM. ** < 0.05, compared to untreated; §<0.05
compared to LRT; ¶ compared to LRT + depletion of CD4+.

77
4.3 Immunological Protective Memory After LRT and Surgery
In this experiment, the goal was to investigate the role of LRT before surgical resection of
tumors in generating effective immunological memory response. C57BL/6 mice were inoculated
with AE17-OVA cells and after 9 days were randomized into the following treatment groups 1)
Surgery, 2)LRT and Surgery 24 hours later, 3)LRT and Surgery 7 days later. Radical surgery
was performed in this experiment to maximize the number of cured mice in order to rechallenge
them in the opposite flank 90 days after the initial treatment. Each group consisted of 10 mice.
All 10 mice in the surgery group were tumor free 90 days after treatment, and 9 mice in both
groups treated with LRT and surgery were tumor free. One mouse in the two groups treated with
LRT and surgery was lost during surgery due to tumor infiltration of the chest wall. After
rechallenge in the opposite flank, tumor size was significantly smaller in the group treated with
LRT and radical surgery after 7 days, compared to the other 2 groups (Surgery only and LRT and
Surgery 24hrs later). In the group treated with LRT and surgery after 7 days, and rechallenged
after 90 days, 3 out of 9 mice completely rejected the tumor. There was no rejections in the other
two groups (Figure 12).
This suggest that LRT 7 days before surgical removal of tumors contributes to generating a
protective immunological memory response. This protection was not generated in the group
treated only with radical surgery or when surgery was done one day after radiation.

78
A
B
C
Figure 12. AE17 OVA Rechallenge 90 days after treatment. A) Mice treated with radical
surgery only, LRT and surgery after 24 hrs or LRT and surgery after 7 day were rechallenged in
the opposite flank 90 days after treatment. B) Mice treated with LRT and radical surgery after 7
days grew significantly smaller tumors compared to the surgery and to LRT and surgery after 24
hrs. Values shown are the mean tumor area in mm2 of 10 mice per time point in the surgery
group and 9 mice in the other groups and are expressed as mean±SEM. ** < 0.05, compared to
LRT and surgery after 7 days.

79
4.3.1 Role of T cell on the protection against rechallenge
To investigate whether or not T cells play a role in the protection against AE17-OVA
rechallenge after LRT and surgery, C57BL/6 mice that rejected tumors after rechallenge in the
previous experiment were depleted of lymphocytes and then rechallenged again.
Animals treated and cured with LRT and surgery were rechallenged and completely rejected the
tumor. These mice were pooled from multiple previous experiments after varying periods of time
ranging from two to 14 months after rejection of the tumor and then prior to the depletion
experiment all mice were rechallenged with AE17-OVA in the left flank to assess if they still had
protective memory response against the tumor. Twenty one mice were collected and 20 rejected
the tumor for the second time (data not shown). Lastly, these 20 mice were rechallenged once
more after randomization into the following groups: 1) CD4 depletion n=6, 2) CD8 depletion
n=7, 3) Double depletion n=7. Depletion of lymphocytes started one week before AE17-OVA
cells inoculation (Figure 13a).
Double depleted mice displayed the fastest tumor growth rate, similar to mice challenged for the
first time. Tumor size was significantly larger than the other two groups starting at day 9 and at
every time point until mice were sacrificed. All CD8 depleted mice completely rejected the
tumor. CD4 depleted mice rejected 1 of 6 tumors and growth rate of the remaining 5 was
significantly slower than double depleted animal but also significantly faster than CD8 depleted
mice. Tumor size in CD4 depleted mice was significantly larger than tumors in CD8 depleted
mice at days 12, 15, 19 and 22, but significantly smaller than tumor size in double depleted mice
at every time point after day 9(Figure 13b and c).

80
These results suggest that both CD4+ and CD8+ T cells are required to fully protect against
rechallenge. CD4+ T cells are critically required for an effective memory response as 5/6
depleted animals failed to reject a tumor when rechallenged. However, tumor growth was
significantly delayed compared to double depleted mice suggesting CD8 lymphocytes can
protect the animal but need intact CD4 function to be fully protective. All CD8 depleted mice
completely rejected the tumors, suggesting that CD4 T cells are sufficient to mount an effective
response against the tumor, probably by recruiting other immune cells than CD8 T cells.

81
Figure 13. CD4+ and CD8+ T cells role during rechallenge. A) Mice that had previously
rejected a tumor after rechallenge were depleted of CD4+ T cells, CD8+ T cells or both and
rechallenged one more time. B) Tumor size was significantly larger in double depleted mice
compared to CD4 or CD8 single depleted mice. Tumor size was also significantly larger in CD4
depleted mice compared to CD8 depleted mice but smaller than double depleted mice. C) AE-
17-OVA tumor growth of double depleted mice is faster compared to CD4 or CD8 single
depleted mice. Values shown are the mean tumor area in mm2 of 6 mice per time point in the
CD4 group and 7 mice in the other groups and are expressed as mean ± SEM. ** < 0.05,
compared to CD8, §<0.005, compared to CD4.
A
B C

82
4.4 CTLA-4 blockade improves the beneficial effect of LRT on tumors
In this experiment, the goal was to determine the role of anti-CTLA-4 combined with LRT in
improving AE17-OVA tumor growth control. CTLA-4 is a negative regulator of T cell activation
and blockade of its function induces effective antitumor immunity. Therefore, the combination of
LRT and anti-CTLA4 may show a synergistic effect. Tumor bearing C57BL/6 mice were
randomized and received 1) No treatment, 2) anti-CTLA4, 3) LRT, 4) LRT and anti-CTLA-4.
Each group consisted of 4 mice. Anti-CTLA-4 and LRT started on day 9 in all groups, and the
anti-CTLA-4 dose was repeated every 3 days for a total of 3 doses.
The group treated only with anti-CTLA4 was not significantly different than the untreated group.
Tumor size of mice treated with anti-CTLA-4 was significantly larger at day 16 and 18
compared to LRT, and at day 16, 18 and 21 compared to the combination group. Mice treated
with the combination group had significantly smaller tumors at day 18 and 21 compared to LRT
only. In a similar experiment where 5 mice were treated with LRT and anti CTLA-4 antibody we
observed consistent results and with longer follow up 2 out of 5 mice completely rejected the
tumor (results not shown) (Figure 14) .
Overall, blockade of CTLA-4 during radiation treatment showed a synergistic beneficial effect
on tumor growth. However, blockade of CTLA-4 with no LRT did not significantly affect tumor
growth.

83
Figure 14. Combination therapy with LRT and CTLA-4 shows a synergistic effect on
tumor growth. A) Schematic of the experiment. B) Tumor growth significantly slows down in
the combination group compared to the antiCTLA-4 group and to the LRT group. C)
Combination therapy is significantly better at controlling tumor size at day 18 and 21. Values
shown are the mean tumor area in mm2 of 4 mice per time point in all groups and are expressed
as mean ± SEM. ** < 0.05, compared to LRT, §<0.005, compared to anti-CTLA-4.

84
5 Discussion
Radiation therapy is an important treatment modality for mesothelioma and cancer in general.
Until recently, the beneficial effect was based on the ability of radiation to eradicate cancer cells
by means of its cytotoxic effect (Demaria, Bhardwaj, et al. 2005). However, the effects of
radiation may extend beyond the elimination of radiosensitive tumor cells. Although radiation
therapy is a localized therapy, it can have a major impact on the immune system and have a
systemic effect. Considerable evidence indicates that radiation therapy has the potential to
enhance tumor immunogenicity by promoting cross priming and eliciting antitumor T-cell
responses (Demaria et al. 2004). The generation of inflammation and modification of the tumor
microenvironment after RT results in an immunogenic milieu which could provide a unique
opportunity to combine radiation and immunotherapy to obtain a synergistic effect on the tumor
killing. Furthermore, recent studies have shown that local radiation can eradicate the targeted
tumor, but also distant metastasis away from the radiation field through specific activation of the
immune system against the targeted tumor cells, this is called the abscopal effect (B. Park, Yee,
and Lee 2014). The abscopal mechanism remains unexplained, although a variety of underlying
biologic events can be hypothesized, including a possible role for the immune system.
The aims of this project were to evaluate the effect of Local Radiation Therapy (LRT) combined
with surgery in a mouse model of mesothelioma and to investigate the involvement of the
immune system. As mentioned before, radiation may have a systemic impact on the immune
system by activating immune cells, and, by surgically removing the tumor, the
immunosuppressive factors produced by the tumor tissue may add to the immunogenic effect of

85
LRT. Lastly, the addition of immunotherapeutic molecules may shift the balance toward a strong
immune response against the tumor. Thus, in this study 1) a mouse model of LRT followed by
surgery was developed, 2) CD8+ T cells were analyzed within the tumor after LRT, and 3) the
effect of immunotherapy together with LRT was examined in this model.
5.1 Development of a mouse model of MPM treated with LRT
followed by surgery
The first part of the project involved the development of the mouse model. A previous mouse
model used by our group was examined initially. This model consisted of BALB/c mice
inoculated subcutaneously in the right flank or leg with 2x106 AB12 cells. The number of
inoculated cells was lowered to 1x106 AB12 cells to slow tumor growth. This change resulted in
slower tumor growth and implantation success was 100% (data not shown). The next step was to
evaluate if the tumor was sensitive to radiation and to find the best radiation dose and regimen.
Multiple studies have shown evidence that LRT at typical doses consistently elicits some
activation of the innate and adaptive immune system (McBride et al. 2004; A. a Lugade et al.
2005; Keisari et al. 2014; Demaria et al. 2004). On the other hand, the proportion of cells
undergoing immunogenic cell death and the recruitment of inhibitory cells versus DCs is
variable depending on the dose and fractionation (Demaria and Formenti 2012; Klug et al. 2013).
Also, some authors speculate that antigens released after radiation-induced-cell death need to be
pulsed to overcome a critical threshold to achieve antitumor immune responses (Frey et al.
2014). Several recent studies have shown that hypofractionated doses are more effective at
activating the immune system and eliciting an abscopal effect with in-vivo models and in clinical

86
reports compared to standard radiation (Dewan et al. 2009; Camphausen et al. 2003; Golden et
al. 2013; Postow et al. 2012; Victor et al. 2015; Filatenkov et al. 2015). Similar benefits have
been demonstrated by others with single ablative dose regimens (Reits et al. 2006; Demaria,
Kawashima, et al. 2005; Shiraishi et al. 2008; Matsumura et al. 2008). Overall multiple
preclinical and clinical publications support the immunogenic effect of radiation but there is no
consensus about the best radiation regimen to be used for the best antitumor immune response.
Finding the optimal regimen may affect the ability of radiation to make an in-situ vaccine out of
a tumor.
All doses evaluated in our model provided good tumor control but the highest doses resulted in
side effects as demonstrated by weight loss and animal distress. Although the literature is not
decisive, we favor in our group the use of hypofractionated radiation as the better approach for
activating the immune system since it is clinically relevant. Furthermore, the 15Gy dose
fractionated over 3 days mimics more closely the SMART protocol and is consistent with other
projects in our group.
Lastly, the combination therapy with LRT and Surgery was evaluated in the mouse model. A
significant increase in tumor growth rate and a detrimental effect on survival was observed in the
group treated with blunt surgery alone compared to untreated and the LRT groups. The increase
in tumor growth rate can be explained by the cytokines and growth factors released at the site of
the excised tumor (Kal, Struikmans, and Barten-van Rijbroek 2008; Tsuchiya et al. 2003).
Among the many molecules present at the surgical wound, transforming growth factor beta
(TGF-β)(Hofer et al. 1998), epidermal growth factor (EGF) (Tagliabue et al. 2003), vascular
endothelial growth factor (VEGF) (Hormbrey et al. 2003) and basic fibroblast growth factor,
importantly promote tumor growth in addition to promoting healing on the surgical wound.

87
These factors promote neoangiogenesis, wound healing and cancer cell proliferation (Demicheli
et al. 2008). Furthermore, dysfunction of the immune system may play a role in the progression
of tumors after surgical stress (Menges et al. 2012). The postoperative inflammatory state
induces inhibition of cell-mediated immunity (Cardinale et al.), the release of glucocorticoids, a
reduced rate of T cell proliferation, and lymphocytopenia (Dhabhar et al. 1996; Ogawa et al.
2000), and activation of the sympathetic nervous system results in catecholamine mediated
inhibition of NKT cells (I J Elenkov et al. 2000; Rosenne et al. 2014). Thus, cytokines and
growth factors promoting tumor growth and factors depressing the immune response may
explain the increased rate of tumor growth after blunt surgery.
The LRT group had a significant decrease in tumor growth compared to blunt surgery and
untreated groups. Tumor growth slowed down significantly, but it only lasted for 7 to 10 days
before tumor growth accelerated once more. As discussed previously there is growing evidence
that support the immunogenic effect of radiation. However, the demand faced by the immune
system by a rapidly growing tumor is challenging. Clinically detectable tumors double in size in
a period of time measured in weeks or months, but tumors treated with radiation can progress
faster with a doubling time in the order of days. It has been reported that accelerated growth of
some tumor cells surviving radiation proceeds at a rate 20 times faster than before treatment
(Withers 1993; Yom 2015; Abe et al. 1991). As a result, without debulking and inhibition of
tumor cell repopulation, the immune system may not be capable of eliminating or controlling the
tumor.
In our model, in the combined therapy group with LRT and surgery, tumor growth was
significantly delayed compared to the untreated and blunt surgery group. In the combination
group, tumor growth was delayed before and after blunt surgery and one of five mice was cured.

88
Hence, the combination therapy abrogates the negative effect observed in the group treated with
surgery alone and may have the advantage of removing the immunosuppressive tumor bulk, thus
promoting the benefit of LRT activating the immune system. Adding immunotherapy to further
activate the immune response against the rapidly proliferating tumor cells might be beneficial to
balance the immune response towards a cytotoxic immune response.
5.2 T cell tumor infiltration
When tumors were analyzed for CD8 T cell infiltration, we observed in the radiated tumor 2
days after treatment an important decrease in infiltrating T cells. This was most likely secondary
to the cytotoxic effect of radiotherapy as T cells are extremely sensitive to radiation (Manda et
al. 2012; Heylmann et al. 2014). However, although radiation may initially kill lymphocytes, it
does not have systemic cytotoxic effects and lymphocytes from the systemic pool re-infiltrate the
tumor not long after. As we observed in our experiment, at day 7, treated tumors had been
repopulated and at day 12 we observed a dramatic increase in infiltrating T cells. This contrasts
with the progressive decrease in infiltrating lymphocytes in the entire tissue section of untreated
tumor. We further demonstrated that this rise in tumor infiltrating lymphocytes after LRT was
antigen specific as we found an increase in OVA-tetramer CD8+ T cells in the radiated tumor.
Lymphocyte trafficking and infiltration into solid tumors secondary to radiation has been
examined in different murine models. Lugade et. al., treated B16-OVA tumors and this led to
increased priming of tumor specific T cells in the draining lymph node and increased infiltration
of CD8+ T cells in the tumor (A. a Lugade et al. 2005). Also, OT1 CD8+ T cells specific for
OVA that were activated ex-vivo and adoptively transferred were directed to the radiated tumor.

89
In a different model, Takeshima et. al. reported increased number of tumor specific T cells
infiltrating the tumor and draining lymph nodes following radiation, but a major reduction
despite the radiation in mice whose lymph nodes were surgically ablated or genetically defective
(Takeshima et al. 2010). Thus, radiation of a tumor promotes tumor infiltration by lymphocytes.
Radiation creates a pro inflammatory environment where danger signals and cytokines are
abundant creating a concentration gradient that directs the migration of lymphocytes towards the
tumor (A. a Lugade et al. 2005). One of the cytokines that play an important role is IFN-γ (B.
Park, Yee, and Lee 2014). Mice deficient in IFN-γ are unable to upregulate expression of
VCAM-1 and MHC class 1 in the tumor vasculature after radiotherapy, important for T cell
infiltration and tumor cell target recognition (A. A. Lugade et al. 2008). ICAM-1 is also
upregulated after radiation in human colon cancer and gastric adenocarcinoma cells (Chiriva-
Internati et al.). ICAM-1 facilitates lymphocyte infiltration. Secretion of chemokines such as
CCL2 (Draghiciu et al. 2014) and CXCL16, which bind CXCR6 on Th1 and CD8 T cells, are
increased after radiation in mouse and human breast cancer cells, and this leads to increased
migration of CD8 T cells to the tumor (Matsumura et al. 2008). This evidence suggest that
radiation of a tumor may direct lymphocytes to the tumor by upregulation of adhesion molecules
and an increase in the secretion of chemokines.
Finally, decreasing number of infiltrating T cells in the untreated tumor was observed over time.
Initially the tumor was infiltrated by a similar number of T cells probably by the initial immune
response after tumor cell injection. However, the immunosuppressive environment of the tumor
may have led to anergy and apoptosis of tumor infiltrating T cells in the untreated tumors. The
immunosuppressive tumor environment involves infiltrating T regulatory cells, M2
macrophages, PD-L1 and TGF-β expression. Also the downregulation of MHC and loss of tumor

90
associated antigen expression, allow the tumor to evade specific immune recognition. Radiation
may also reverse this immunosuppressive state by the release of proinflammatory molecules.
5.2.1 Upregulation of the activation marker 4-1BB and decrease in the
exhaustion marker PD-1
Tumor specific infiltrating lymphocytes were further analyzed on days 3 and 7 after local
radiation therapy and compared with untreated tumors as controls. At day 3 there was a
significant upregulation of the activation marker 4-1BB and downregulation of PD-1. At day 7,
4-1BB returned to similar levels as the untreated tumor but persistent lower expression of PD-1
was observed. 4-1BB is mainly present in activated T cells but not resting T cells and has
important effects on T cell proliferation and function. Signaling through 4-1BB in lymphocytes
can stimulate in a CD28 independent way, production of IL-2, a critical step for activation of T
cells but also for prevention of an anergic state (Saoulli et al. 1998). 4-1BB prevents apoptosis
and increase survival of tumor infiltration lymphocytes via upregulation of the antiapoptotic
pathways involving expression of Bcl-x and Bfl-1 (H.-W. Lee et al. 2002). In addition, 4-1BB
also enhances cytotoxic T lymphocyte cytolytic activity (Hernandez-Chacon et al. 2011). PD-1
on the other hand identifies T cells with an exhausted phenotype that cannot maintain appropriate
cytokine production (Fourcade et al. 2010). PD-1 in physiological conditions plays a role in
limiting damage to normal tissue during inflammatory response (Topalian, Drake, and Pardoll
2012). One of its ligands, PD-L1 is common in tumors including mesothelioma and has been
shown to correlate with poor prognosis (Cedrés et al. 2015). Furthermore, intratumoral
infiltration of PD-1 positive T cells was also correlated with progression of malignant tumors in

91
humans (Richendollar et al. 2011; Thompson et al. 2007). Also, PD-1 upregulation on CD8 T
cells is associated with T cell dysfunction, low proliferation and low activation (Keir et al. 2008).
In our experiment, the high expression of PD-1 and low expression of 4-1BB suggest
lymphocytes in untreated tumors may have been initially recognized tumor antigens but were no
longer in an activated state. The decreased PD-1 expression in tumor specific infiltrating
lymphocytes at days 3 and 9, together with the initial decrease in CD8+ T cells 2 days after
radiation, suggest radiation eliminated exhausted lymphocytes and new CD8+ T cells
repopulated the tumor. Again, the pro-inflammatory environment after irradiation may have been
responsible for the increased expression of the activation marker 4-1BB. Overall, radiation
therapy led to increased tumor-specific lymphocyte infiltration and these lymphocytes expressed
higher activation markers and downregulated the exhaustion marker PD-1. These lymphocytes
may have been responsible for at least part of the beneficial effect of radiation.
5.2.2 Depletion of lymphocytes partially abrogated the effect of radiation
Double depletion of CD4+ and CD8+ T cells during hypofractionated radiation significantly
abolished the effect of radiation. Depletion of CD8+ T cells partially abolished the therapeutic
effect of radiation as shown by the trend towards increased tumor growth rate compared to
radiation alone. Depletion of CD4+ T caused a trend towards the opposite direction, insinuating
an improved effect of radiation. These findings suggest both CD4+ and CD8+ T cells play a
major role in the response to radiation. While the direct cytotoxic effect of radiation was
preserved in all depleted groups, based on our results, CD4+ and CD8+ T cells together appear
to account for at least a third of the response to radiation. While depletion of CD8+ T cell did not
significantly reduced the effect of radiation, we noted a strong trend towards faster tumor growth

92
compared to radiation only. Also, the difference between the CD4 depleted and CD8 depleted
group was significant, suggesting CD8+ T cells might be responsible for large part of the lost
therapeutic effect in the double depleted mice. Regulatory T cells (CD4+CD25+FoxP3+) were
also depleted in the CD4 depleted group and that might explain the trend towards better tumor
control after radiation as reported by others including our group (Licun Wu et al. 2011; Son et al.
2015). In this CD4 depleted group, CD8+ T cells might have been activated by DCs exposed to
danger signals in the strong inflammatory tumor environment after radiation without the help of
CD4 T cells. Strengthening this idea, other groups have shown that this activation of CD8 T cells
without help of CD4 T helper leads to impaired CD8 memory responses (Novy et al. 2007; Sun,
Williams, and Bevan 2004). Our observations are in line with the literature indicating CD8 T
cells are required for the therapeutic effects of ablative radiation (Y. Lee et al. 2009; Gupta et al.
2012).
5.3 Protective memory response after LRT and Surgery
LRT was shown to result in the induction of protective immune memory response against the
tumor as demonstrated in the rechallenge experiments 90 days after LRT followed by radical
surgery. The group treated with radical surgery and the group treated with LRT followed by
radical surgery after 24 hrs displayed an accelerated tumor growth rate compared to LRT
followed by radical surgery after 7 days. In addition we did not observe rejections in these two
groups. By contrast, 3 out 10 mice that were treated with hypofractionated radiation 7 days
before radical surgery rejected the tumor, and the other 7 mice had a significantly reduced tumor

93
growth rate compared to the other 2 groups. The cell line AE17-OVA was weakly immunogenic,
as demonstrated by the fact that radical surgery does not provide protective memory.
Overall LRT before surgery contributed to generating a partially protective immunological
memory, but only when surgery was performed after 7 days and not when it was performed after
24 hrs. This might be as 7 days are required to reach the peak of the adaptive T cell immune
response in mice (Busch et al. 1998). Others have shown that, 5 days after RT, there is an
increase in DC loaded with tumor peptides in the draining lymph node and in the tumor (Y. Lee
et al. 2009). Together, these findings suggest that radiation might activate DCs within the tumor,
and then promotes maturation and migration of DCs to the draining lymph node to present
antigens and activate T cells which then will migrate to the tumor. This is in agreement with our
CD8+ T cell infiltration experiment where we observed important repopulation of T cells within
the tumor at 7 and 12 days but not at 2 days.
5.3.1 Depletion and rechallenge
Mice previously protected against AE17-OVA failed to reject the tumor against rechallenge
when CD4+ and CD8+ T cells were depleted and when CD4+ alone was depleted. CD4+
depletion preserved some protection against the tumor as 1 out of 6 mice rejected the tumor and
the tumor growth rate curve was slower.
These results suggest that both T cell subsets are required for an optimal protection. However,
for memory response, CD4 T cells are the key players. As mentioned in the depletion experiment
during LRT, there are some situations where CD8 T cells can be activated by mature DCs
without the help of CD4 T helper cells during the primary response (Novy et al. 2007;

94
Andreasson et al. 2010). However, these CD8 T cells will not mount a strong memory response
if rechallenged. Not only are CD4 T cells important during the primary encounter to create an
adequate memory CD8 T cell response, but they are crucial during the memory response to
expand and activate the pool of memory CD8 T cells. Despite the fact that all CD8 depleted
animals completely rejected the tumor, CD8 T cells are important during the memory response,
but they need CD4 T cell help. Perhaps, in the CD8 depleted mice, thanks to redundancy in the
immune system and after the very efficient tumor vaccine created in situ by LRT, CD4 T cells
orchestrated the memory immune response and activated other effector cells such as NKT cells
and macrophages and that was enough to reject the tumor.
5.4 CTLA-4 blockade synergized with LRT
Finally, we observed improvement of the therapeutic effects of hypofractionated radiation when
the immune checkpoint CTLA-4 was blocked. We observed a significant and dramatic
synergistic effect in the group treated with the combination therapy but no effect in the group
treated with the antibody alone. The observation that in the absence of an active immune
response, the beneficial effect of anti-CTLA-4 is minimal has been reported before by our group
and others, suggesting that combination with other therapeutic modalities is important (Licun
Wu et al. 2015; Davila, Kennedy, and Celis 2003; Grosso and Jure-Kunkel 2013; Vanpouille-
Box et al. 2015). It is believed anti-CLTA-4 therapy is not effective as a single agent because of
the dominant immunosuppressive tumor environment. Radiation and its capacity to create
inflammation and immunogenic cell death might be an excellent therapeutic adjunct to CTLA-4
blockade. This idea together with the dramatic effect of CTLA-4 in combination with LRT in

95
preclinical models is recognized and has emerged as a promising strategy in the clinical setting.
Since 2011, ipilimumab (Bristol-Meyers Squibb) an anti-CTLA-4 drug was approved for patients
with metastatic or unresectable melanoma and there are some encouraging reports. In MPM,
Tremelimumab therapy (AstraZeneca) has been explored in patients with unresectable disease
and results were published recently (Calabrò et al. 2013). Furthermore, a large multicenter
randomized trial with Tremelimumab was initiated in patients with unresectable and
chemotherapy resistant mesothelioma. However, currently there are no trials in MPM using
CTLA-4 blockade in combination with LRT.

96
6 Conclusions
The goal of this project was to develop a mouse model to recreate the effects seen with SMART
treatment in the clinic and asses the immunogenic effect of local hypofractionated radiation in a
model of malignant mesothelioma. It was suggested in the SMART trial that the benefits
observed in patients might have been related to activation of the immune system. The purpose of
the model was to investigate this suggestion and to find ways of improving it.
First, tumor sensitivity to radiation together with a safe dose and radiation regimen was assessed.
Several recent studies have shown that hypofractionated doses might be more effective at
activating the immune system and eliciting an abscopal effect compared to standard fractionated
radiation. Single dose and fractionated regimens were compared in the first part of the project.
All doses provided good and similar tumor control but the highest doses resulted in unacceptable
side effects in the animals. To be consistent with other projects in our group and to mimic more
closely the SMART protocol, 15 Gy over 3 days was chosen as the dose and regimen for the rest
of the project.
Afterwards, a subcutaneous mouse model was developed where we combined local radiation
therapy and surgery and we obtained similar results as seen in the clinic. LRT caused significant
tumor growth deceleration for a brief period of time. When the tumor was surgically removed,
the beneficial effect of local radiation therapy was preserved for a period of time. This was in
contrast to mice treated with blunt surgery only, where a significant increase in tumor growth
rate and detrimental effect on survival was observed after surgical removal of the tumor. The
beneficial effect provided by local radiation therapy before surgical removal of a tumor might be

97
secondary to immune activation. Furthermore, combination therapy abrogated the negative effect
observed in the group treated with surgery alone and may have the advantage of removing the
immunosuppressive tumor bulk, thus promoting the benefit of LRT activating the immune
system. Alternatively, the tumorostatic effect of radiation may have prevented or delayed
successful implantation and proliferation of tumor cells after surgery.
Furthermore, we analyzed tumors to elucidate if there was evidence of an involvement of the
immune system. We found an increase in tumor infiltrating T cells in the radiated tumor after 7
days. Conversely, in untreated tumors there was a distinct lack of tumor infiltrating T cells, likely
secondary to the strong immunosuppressive environment of the tumor. The tumor environment
may lead to anergy and apoptosis of tumor infiltrating T cells in the untreated tumors. A large
number of the lymphocytes in the radiated tumor were specific against the tumor and exhibited
an activated profile whereas untreated tumors upregulated exhaustion markers on T cells. These
findings suggest a strong activity of the immune system in our model after hypofractionated
radiation.
Radiation creates a pro inflammatory environment with abundant danger signals and cytokines,
providing a concentration gradient that attracts lymphocytes into the tumor. Radiation also
causes upregulation of adhesion molecules present on the tumor blood vessels and increased
secretion of chemokines. Together these effects after radiation of a tumor directs lymphocytes to
the tumor and favor its activity against tumor cells.
Depletion experiments supported the role of the immune system on the therapeutic response of
radiation therapy and the memory protection provided by radiation and surgery. Animals treated
with hypofractionated radiation and surgery were partially protected to tumor rechallenge,
compared to mice treated only with surgery which were not protected. This protection against

98
tumor rechallenged was lost after depleting CD4+ and CD8+ lymphocytes on mice previously
protected.
Lastly, we observed an improvement of the therapeutic effect of hypofractionated radiation
when the immune checkpoint CTLA-4 was blocked. A dramatic synergistic effect was observed
in the group treated with radiation and CTLA-4 blockade while single therapy with anti CTLA-4
showed no difference when compared with untreated mice. This lack of effect of anti CTLA-4
therapy might be secondary to the dominant immunosuppressive tumor environment. However,
when tumors are treated with radiation the transient inflammatory milieu might be a trigger to
initiate an immune response against the tumors. This synergistic effect of radiation and blockade
of the immune checkpoint inhibitor anti-CTLA-4 supported the potential benefit of
immunomodulation of the immune system in combination with hypofractionated radiation.
Malignant Pleural Mesothelioma remains a major public health concern and at present only few
patients are candidate to the potentially curative surgery within a multimodality treatment
including chemotherapy and radiotherapy. Also, there is no second line treatment that
significantly prolongs survival of patients. Thus new alternatives are needed.
The goal of this project was to translate findings into clinical practice. In vivo date suggests that
radiation is effective in creating an inflammatory tumor environment and recruiting immune cells
into the tumor. Radiation therapy is emerging as an optimal partner for immunotherapy such as
anti CTLA-4 because of its ability to induce immune activation. Radiation immunogenicity and
combination with immunotherapy warrants further investigation and supports the potential
benefit of immunomodulating therapy in combination with hypofractionated radiation.

99
Future development of clinical trials directed at the combination of radiation, surgery and
immunotherapy will potentially open new avenues in the treatment of mesothelioma.

100
7 Limitations
Although the project started with the weakly immunogenic cell line AB12, most of the
experiments were performed with the cell line AE17-OVA. Although this cell line is not highly
immunogenic, as mice treated with radical surgery did not reject the tumor after rechallenge, the
artificially transfected OVA antigen does increase its immunogenicity. It may be worth,
repeating these experiments with the AB12 mouse model.
A limitation with animal models and cell lines is the difference in the tumor microenvironment
and subsequent immune editing as compared to spontaneous tumor. In spontaneous tumors, it
takes long periods of inflammation to develop a tumor whereas injection of cells takes a few
days. This may lead to important differences in the tumor environment and to the immune
response against the tumor.
Another limitation of our model is the subcutaneous location of the tumor. The connective tissue
and blood supply found in the subcutaneous space is certainly different than the pleural space.
This might affect the response to radiation and the effect on immune cell migration.
In regards to tumor specific T cells, significant differences were not found, most likely as a result
of the number of animals analyzed and the single time point used. It may be beneficial to
increase the number of animals and repeat the analysis on the same time points used for
immunofluorescent staining at 2, 7 and 12 days. Other difficulties arose while analyzing tumor
infiltrating lymphocytes as tumors can differ in size dramatically. Untreated tumors are up to 10
times larger than radiated tumors. While disrupting tumors into a single cell suspension,

101
lymphocytes might be artificially concentrated and the number overestimated in untreated
tumors.
Finally the cytotoxic activity of infiltrating lymphocytes was not investigated. The fact that there
is an increase number of T cells may not be relevant if they are unable to lyse tumor cells. Future
studies will need to look at the functional status of cytotoxic T cells and their capacity to secrete
relevant cytokines to achieve their role.

102
Future Directions
There is no consensus about the best radiation regimen to be used for the best antitumor immune
response. It would be interesting to compare the ability of single-dose, hypofractionated and
fractionated radiation and its ability to activate the immune response. Finding the optimal
regimen for combination therapy remains to be done.
An intrapleural model would be a more clinically relevant model of MPM. As it was discussed in
the limitations there are several weaknesses in our subcutaneous model. Injection of tumor cells
in the thoracic cavity under direct visualisation can be done through a small incision under
anesthesia. Tumor growth would be more difficult to monitor, but it is possible to perform CT or
PET scan to have an approximate evaluation of the tumor growth. For radiotherapy, the XRAD
225cx would allow us to provide hypofractionated hemithoracic radiation to a mouse intrapleural
tumor without compromising other vital organs. Finally, pneumonectomy could be performed
with a similar technique to what is done in lung transplant model by others in the Thoracic
Surgery Research Laboratories.
Also to resemble more closely to what is seen in human MPM, future studies could involve
mouse MPM developed from asbestos exposure. Spontaneously developed tumors may be less
immunogenic than cell lines due to slow selection of cancer cell clones that escape the immune
system, potentially better modelling clinical cases of MPM. Spontaneous development is
possible as asbestos tumor development in mice has been extensively demonstrated. However,
long incubation period and low number of mice that develop tumors after asbestos exposure still
poses a potential challenge.

103
To assess the systemic effect of immune activation after hypofractionated radiation a metastatic
cell line can be used. AB12 and AE17 are poorly metastatic in vivo and systemic protection
against the tumor cannot be assessed properly. To simulate metastasis in a different mouse model
in our group a tumor is injected in two sites in the same animal at the same time point. Only one
of the two sites is treated with radiation and the systemic effect is evaluated in the untreated
tumor. However, this model does not resemble closely the biology of spontaneous metastatic
tumors. To overcome this difficulty we could utilize a spontaneous metastatic cell line that have
been reported in other preclinical tumor models such as breast cancer. The limitation with such a
cell line is that there is no metastatic mesothelioma cell line reported in the literature.
Other alternative to support our findings on the beneficial effect of combination therapy with
radiation and surgery, could be a different animal model. Other authors have reported
intrapleural mesothelioma in rats. Similar results in different species could provide stronger
evidence for the beneficial immunogenic effect of radiation therapy.
Lastly, as mentioned previously beside mesothelin and WT-1 there are only a few known
specific malignant pleura mesothelioma associated tumor antigens. Finding other mesothelioma
associated tumor antigens might be useful to implement immunotherapeutic approaches in MPM
patients.
In regards to infiltrating lymphocytes into the tumors, we have not analyzed the functional status
of T cells. CD8 T cells may be present in the tumor in an anergic state and unable to lyse tumor
cells. An important future experiment involves the analysis of T cells infiltrating the tumor. Flow
cytometry analysis and PCR analysis to investigate the capacity of T cells to secrete cytokines
such as IFN γ and to produce granzyme B and perforin to lyse tumor cells is the next step in this
project.

104
The SMART approach in patients involved radiation of mesothelioma before extrapleural
pneumonectomy. Previously, adjuvant radiation was done after extrapleural pneumonectomy.
The rationale on SMART beneficial effect was that the irradiated tumors released danger signals
and tumor associated antigens before tumor removal causing activation and priming of the
immune system. In this project a mouse model was stablished and we demonstrated longer
survival in mice treated with the SMART approach and increased infiltration of lymphocytes
within the tumor. To further understand if the increase in survival in patients treated with
SMART is immune mediated, tumors infiltrating lymphocytes could be measured and analyzed
for activation and exhaustion markers after the completion of treatment. Tumor of SMART
treated patients could be compared with patients treated with extrapleural pneumonectomy
without radiotherapy.
Finally, the following and ultimate goal of this project would be to translate this findings into the
clinic. A clinical trial comparing patients treated with SMART vs SMART combined with
immunotherapy could be considered. Similar trials involving immune checkpoint inhibitors are
undergoing in other tumors in patients. However, to the best or our knowledge there are no
undergoing clinical trial involving hypofractionated radiation and immune checkpoint inhibitors
in MPM patients.

105
8 References
Abe, Y, M Urano, L A Kenton, J Kahn, and C G Willet. 1991. “The Accelerated Repopulation of
a Murine Fibrosarcoma, FSA-II, during the Fractionated Irradiation and the Linear-
Quadratic Model.” International Journal of Radiation Oncology, Biology, Physics 21 (6):
1529–34. http://www.ncbi.nlm.nih.gov/pubmed/1938563.
Adams, V I, K K Unni, J R Muhm, J R Jett, D M Ilstrup, and P E Bernatz. 1986. “Diffuse
Malignant Mesothelioma of Pleura. Diagnosis and Survival in 92 Cases.” Cancer 58 (7):
1540–51. http://www.ncbi.nlm.nih.gov/pubmed/3742473.
Ahamad, Anesa, Craig W Stevens, W Roy Smythe, Ara A Vaporciyan, Ritsuko Komaki, Jason F
Kelly, Zhongxing Liao, George Starkschall, and Kenneth M Forster. 2003. “Intensity-
Modulated Radiation Therapy: A Novel Approach to the Management of Malignant Pleural
Mesothelioma.” International Journal of Radiation Oncology, Biology, Physics 55 (3):
768–75. http://www.ncbi.nlm.nih.gov/pubmed/12573764.
Albert, M L, S F Pearce, L M Francisco, B Sauter, P Roy, R L Silverstein, and N Bhardwaj.
1998. “Immature Dendritic Cells Phagocytose Apoptotic Cells via alphavbeta5 and CD36,
and Cross-Present Antigens to Cytotoxic T Lymphocytes.” The Journal of Experimental
Medicine 188 (7): 1359–68.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2212488&tool=pmcentrez&ren
dertype=abstract.
Alberts, Bruce, Alexander Johnson, Julian Lewis, Martin Raff, Keith Roberts, and Peter Walter.
2002. “Lymphocytes and the Cellular Basis of Adaptive Immunity.” Garland Science.
http://www.ncbi.nlm.nih.gov/books/NBK26921/.
Amin, A M, C Mason, and P Rowe. 2001. “Diffuse Malignant Mesothelioma of the Peritoneum
Following Abdominal Radiotherapy.” European Journal of Surgical Oncology : The
Journal of the European Society of Surgical Oncology and the British Association of
Surgical Oncology 27 (2): 214–15. doi:10.1053/ejso.2000.1024.

106
Andreasson, Kalle, Mathilda Eriksson, Karin Tegerstedt, Torbjörn Ramqvist, and Tina Dalianis.
2010. “CD4+ and CD8+ T Cells Can Act Separately in Tumour Rejection after
Immunization with Murine Pneumotropic Virus Chimeric Her2/neu Virus-like Particles.”
PLoS ONE 5 (7). doi:10.1371/journal.pone.0011580.
Anraku, Masaki, Kristopher S Cunningham, Zhihong Yun, Ming-Sound Tsao, Li Zhang, Shaf
Keshavjee, Michael R Johnston, and Marc de Perrot. 2008. “Impact of Tumor-Infiltrating T
Cells on Survival in Patients with Malignant Pleural Mesothelioma.” The Journal of
Thoracic and Cardiovascular Surgery 135 (4): 823–29. doi:10.1016/j.jtcvs.2007.10.026.
Antman, K, R Shemin, L Ryan, K Klegar, R Osteen, T Herman, G Lederman, and J Corson.
1988. “Malignant Mesothelioma: Prognostic Variables in a Registry of 180 Patients, the
Dana-Farber Cancer Institute and Brigham and Women’s Hospital Experience over Two
Decades, 1965-1985.” Journal of Clinical Oncology : Official Journal of the American
Society of Clinical Oncology 6 (1): 147–53. http://www.ncbi.nlm.nih.gov/pubmed/3335886.
Arenberg, D A, S L Kunkel, P J Polverini, S B Morris, M D Burdick, M C Glass, D T Taub, M D
Iannettoni, R I Whyte, and R M Strieter. 1996. “Interferon-Gamma-Inducible Protein 10
(IP-10) Is an Angiostatic Factor That Inhibits Human Non-Small Cell Lung Cancer
(NSCLC) Tumorigenesis and Spontaneous Metastases.” The Journal of Experimental
Medicine 184 (3): 981–92.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2192788&tool=pmcentrez&ren
dertype=abstract.
Ascierto, Paolo A. 2013. “Ipilimumab in the Treatment of Metastatic Melanoma: A Summary of
Recent Studies.” Tumori 99 (6): 302e – 5e. doi:10.1700/1390.15474.
Astoul, P, D Picat-Joossen, J R Viallat, and C Boutin. 1998. “Intrapleural Administration of
Interleukin-2 for the Treatment of Patients with Malignant Pleural Mesothelioma: A Phase
II Study.” Cancer 83 (10): 2099–2104. http://www.ncbi.nlm.nih.gov/pubmed/9827714.
Bachmayr-Heyda, Anna, Stefanie Aust, Georg Heinze, Stephan Polterauer, Christoph Grimm,
Elena Ioana Braicu, Jalid Sehouli, et al. 2013. “Prognostic Impact of Tumor Infiltrating

107
CD8+ T Cells in Association with Cell Proliferation in Ovarian Cancer Patients--a Study of
the OVCAD Consortium.” BMC Cancer 13 (January): 422. doi:10.1186/1471-2407-13-422.
Baldini, Elizabeth H. 2004. “External Beam Radiation Therapy for the Treatment of Pleural
Mesothelioma.” Thoracic Surgery Clinics 14 (4): 543–48. doi:10.1016/S1547-
4127(04)00108-2.
Barcellos-Hoff, M H, R Derynck, M L Tsang, and J A Weatherbee. 1994. “Transforming Growth
Factor-Beta Activation in Irradiated Murine Mammary Gland.” The Journal of Clinical
Investigation 93 (2): 892–99. doi:10.1172/JCI117045.
Batista, Luís F Z, Wynand P Roos, Markus Christmann, Carlos F M Menck, and Bernd Kaina.
2007. “Differential Sensitivity of Malignant Glioma Cells to Methylating and
Chloroethylating Anticancer Drugs: p53 Determines the Switch by Regulating Xpc, ddb2,
and DNA Double-Strand Breaks.” Cancer Research 67 (24): 11886–95. doi:10.1158/0008-
5472.CAN-07-2964.
Beatty, G L, and Y Paterson. 2000. “IFN-Gamma Can Promote Tumor Evasion of the Immune
System in Vivo by down-Regulating Cellular Levels of an Endogenous Tumor Antigen.”
Journal of Immunology (Baltimore, Md. : 1950) 165 (10): 5502–8.
http://www.ncbi.nlm.nih.gov/pubmed/11067903.
Beatty, Gregory L, Andrew R Haas, Marcela V Maus, Drew A Torigian, Michael C Soulen,
Gabriela Plesa, Anne Chew, et al. 2014. “Mesothelin-Specific Chimeric Antigen Receptor
mRNA-Engineered T Cells Induce Anti-Tumor Activity in Solid Malignancies.” Cancer
Immunology Research 2 (2): 112–20. doi:10.1158/2326-6066.CIR-13-0170.
Beilin, Benzion, Yehuda Shavit, Evelyn Trabekin, Boris Mordashev, Eduard Mayburd,
Alexander Zeidel, and Hanna Bessler. 2003. “The Effects of Postoperative Pain
Management on Immune Response to Surgery.” Anesthesia and Analgesia 97 (3): 822–27.
http://www.ncbi.nlm.nih.gov/pubmed/12933409.
Ben-Eliyahu, S, G G Page, R Yirmiya, and G Shakhar. 1999. “Evidence That Stress and Surgical
Interventions Promote Tumor Development by Suppressing Natural Killer Cell Activity.”

108
International Journal of Cancer. Journal International Du Cancer 80 (6): 880–88.
http://www.ncbi.nlm.nih.gov/pubmed/10074922.
Ben-Eliyahu, Shamgar, Gayle G Page, and Steven J Schleifer. 2007. “Stress, NK Cells, and
Cancer: Still a Promissory Note.” Brain, Behavior, and Immunity 21 (7): 881–87.
doi:10.1016/j.bbi.2007.06.008.
Bennett, M, K Macdonald, S W Chan, J P Luzio, R Simari, and P Weissberg. 1998. “Cell
Surface Trafficking of Fas: A Rapid Mechanism of p53-Mediated Apoptosis.” Science (New
York, N.Y.) 282 (5387): 290–93. http://www.ncbi.nlm.nih.gov/pubmed/9765154.
Beseth, Bryce D, Robert B Cameron, Pamela Leland, Liang You, Frederick Varricchio, Robert J
Kreitman, Richard A Maki, David M Jablons, Syed R Husain, and Raj K Puri. 2004.
“Interleukin-4 Receptor Cytotoxin as Therapy for Human Malignant Pleural Mesothelioma
Xenografts.” The Annals of Thoracic Surgery 78 (2): 436–43; discussion 436–43.
doi:10.1016/j.athoracsur.2004.03.010.
Bezu, Lucillia, Ligia C Gomes-de-Silva, Heleen Dewitte, Karine Breckpot, Jitka Fucikova,
Radek Spisek, Lorenzo Galluzzi, Oliver Kepp, and Guido Kroemer. 2015. “Combinatorial
Strategies for the Induction of Immunogenic Cell Death.” Frontiers in Immunology 6
(January): 187. doi:10.3389/fimmu.2015.00187.
Bocchetta, M, and Michele Carbone. 2005. “SV40-Mediated Oncogenesis.” In Malignant
Mesothelioma: Pathogenesis, Diagnosis, and Translational Therapies, edited by H Pass,
Nicholas J. Vogelzang, and Michele Carbone, 1st ed., 34–59. New York, NY: Springer.
Bocchetta, M, I Di Resta, A Powers, R Fresco, A Tosolini, J R Testa, H I Pass, P Rizzo, and M
Carbone. 2000. “Human Mesothelial Cells Are Unusually Susceptible to Simian Virus 40-
Mediated Transformation and Asbestos Cocarcinogenicity.” Proceedings of the National
Academy of Sciences of the United States of America 97 (18): 10214–19.
doi:10.1073/pnas.170207097.
Boehm, U, T Klamp, M Groot, and J C Howard. 1997. “Cellular Responses to Interferon-
Gamma.” Annual Review of Immunology 15 (January): 749–95.
doi:10.1146/annurev.immunol.15.1.749.

109
Botha, A J, F A Moore, E E Moore, A Sauaia, A Banerjee, and V M Peterson. 1995. “Early
Neutrophil Sequestration after Injury: A Pathogenic Mechanism for Multiple Organ
Failure.” The Journal of Trauma 39 (3): 411–17.
http://www.ncbi.nlm.nih.gov/pubmed/7473901.
Brahmer, Julie R, and Drew M Pardoll. 2013. “Immune Checkpoint Inhibitors: Making
Immunotherapy a Reality for the Treatment of Lung Cancer.” Cancer Immunology
Research 1 (2): 85–91. doi:10.1158/2326-6066.CIR-13-0078.
Brenner, J, P P Sordillo, G B Magill, and R B Golbey. 1982. “Malignant Mesothelioma of the
Pleura: Review of 123 Patients.” Cancer 49 (11): 2431–35.
http://www.ncbi.nlm.nih.gov/pubmed/7074556.
Bryk, Jodie A, Petar J Popovic, Mazen S Zenati, Veronica Munera, John P Pribis, and Juan B
Ochoa. 2010. “Nature of Myeloid Cells Expressing Arginase 1 in Peripheral Blood after
Trauma.” The Journal of Trauma 68 (4): 843–52. doi:10.1097/TA.0b013e3181b026e4.
Burnet. 1970. “The Concept of Immunological Surveillance.” Progress in Experimental Tumor
Research 13 (January): 1–27. http://www.ncbi.nlm.nih.gov/pubmed/4921480.
Burnet, M. 1957. “Cancer: A Biological Approach. III. Viruses Associated with Neoplastic
Conditions. IV. Practical Applications.” British Medical Journal 1 (5023): 841–47.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1973618&tool=pmcentrez&ren
dertype=abstract.
Burnette, Byron, and Ralph R. Weichselbaum. 2013. “Radiation as an Immune Modulator.”
Seminars in Radiation Oncology 23 (4). Elsevier: 273–80.
doi:10.1016/j.semradonc.2013.05.009.
Busch, D H, I M Pilip, S Vijh, and E G Pamer. 1998. “Coordinate Regulation of Complex T Cell
Populations Responding to Bacterial Infection.” Immunity 8 (3): 353–62.
http://www.ncbi.nlm.nih.gov/pubmed/9529152.
Buschke, F. 1958. “Progress in Radiation Therapy: Introduction.” Progress in Radiation Therapy
1 (January): 1–12. http://www.ncbi.nlm.nih.gov/pubmed/13601954.

110
Calabrò, Luana, Aldo Morra, Ester Fonsatti, Ornella Cutaia, Giovanni Amato, Diana Giannarelli,
Anna Maria Di Giacomo, et al. 2013. “Tremelimumab for Patients with Chemotherapy-
Resistant Advanced Malignant Mesothelioma: An Open-Label, Single-Arm, Phase 2 Trial.”
The Lancet Oncology 14 (11): 1104–11. doi:10.1016/S1470-2045(13)70381-4.
Caldenhoven, E, P Coffer, J Yuan, A Van de Stolpe, F Horn, W Kruijer, and P T Van der Saag.
1994. “Stimulation of the Human Intercellular Adhesion Molecule-1 Promoter by
Interleukin-6 and Interferon-Gamma Involves Binding of Distinct Factors to a Palindromic
Response Element.” The Journal of Biological Chemistry 269 (33): 21146–54.
http://www.ncbi.nlm.nih.gov/pubmed/7914891.
Camphausen, Kevin, Marsha A Moses, Cynthia Ménard, Mary Sproull, Wolf-Dietrich Beecken,
Judah Folkman, and Michael S O’Reilly. 2003. “Radiation Abscopal Antitumor Effect Is
Mediated through p53.” Cancer Research 63 (8): 1990–93.
http://www.ncbi.nlm.nih.gov/pubmed/12702593.
Canman, C E, D S Lim, K A Cimprich, Y Taya, K Tamai, K Sakaguchi, E Appella, M B Kastan,
and J D Siliciano. 1998. “Activation of the ATM Kinase by Ionizing Radiation and
Phosphorylation of p53.” Science (New York, N.Y.) 281 (5383): 1677–79.
http://www.ncbi.nlm.nih.gov/pubmed/9733515.
Carbone, M, H I Pass, L Miele, and M Bocchetta. 2003. “New Developments about the
Association of SV40 with Human Mesothelioma.” Oncogene 22 (33): 5173–80.
doi:10.1038/sj.onc.1206552.
Carbone, M, H I Pass, P Rizzo, M Marinetti, M Di Muzio, D J Mew, A S Levine, and A
Procopio. 1994. “Simian Virus 40-like DNA Sequences in Human Pleural Mesothelioma.”
Oncogene 9 (6): 1781–90. http://www.ncbi.nlm.nih.gov/pubmed/8183577.
Carbone, Michele, Salih Emri, A Umran Dogan, Ian Steele, Murat Tuncer, Harvey I Pass, and Y
Izzettin Baris. 2007. “A Mesothelioma Epidemic in Cappadocia: Scientific Developments
and Unexpected Social Outcomes.” Nature Reviews. Cancer 7 (2): 147–54.
doi:10.1038/nrc2068.

111
Carbone, Michele, Robert A Kratzke, and Joseph R Testa. 2002. “The Pathogenesis of
Mesothelioma.” Seminars in Oncology 29 (1): 2–17.
http://www.ncbi.nlm.nih.gov/pubmed/11836664.
Carbone, Michele, Haining Yang, Harvey I Pass, Thomas Krausz, Joseph R Testa, and Giovanni
Gaudino. 2013. “BAP1 and Cancer.” Nature Reviews. Cancer 13 (3): 153–59.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3792854&tool=pmcentrez&ren
dertype=abstract.
Cardinale, F, I Chinellato, S Caimmi, D G Peroni, F Franceschini, M Miraglia Del Giudice, and
R Bernardini. “Perioperative Period: Immunological Modifications.” International Journal
of Immunopathology and Pharmacology 24 (3 Suppl): S3–12.
http://www.ncbi.nlm.nih.gov/pubmed/22014920.
Carlos, T M. 2001. “Leukocyte Recruitment at Sites of Tumor: Dissonant Orchestration.”
Journal of Leukocyte Biology 70 (2): 171–84.
http://www.ncbi.nlm.nih.gov/pubmed/11493608.
Carpenito, Carmine, Michael C Milone, Raffit Hassan, Jacqueline C Simonet, Mehdi Lakhal,
Megan M Suhoski, Angel Varela-Rohena, et al. 2009. “Control of Large, Established
Tumor Xenografts with Genetically Retargeted Human T Cells Containing CD28 and
CD137 Domains.” Proceedings of the National Academy of Sciences of the United States of
America 106 (9): 3360–65. doi:10.1073/pnas.0813101106.
Castagneto, Bruno, Silvia Zai, Diego Dongiovanni, Alberto Muzio, Sergio Bretti, Gianmauro
Numico, Mario Botta, and G Sinaccio. 2005. “Cisplatin and Gemcitabine in Malignant
Pleural Mesothelioma: A Phase II Study.” American Journal of Clinical Oncology 28 (3):
223–26. http://www.ncbi.nlm.nih.gov/pubmed/15923792.
Cedrés, Susana, Santiago Ponce-Aix, Jon Zugazagoitia, Irene Sansano, Ana Enguita, Alejandro
Navarro-Mendivil, Alex Martinez-Marti, Pablo Martinez, and Enriqueta Felip. 2015.
“Analysis of Expression of Programmed Cell Death 1 Ligand 1 (PD-L1) in Malignant
Pleural Mesothelioma (MPM).” PloS One 10 (3): e0121071.
doi:10.1371/journal.pone.0121071.

112
Ceresoli, G L, P A Zucali, M Mencoboni, M Botta, F Grossi, D Cortinovis, N Zilembo, et al.
2013. “Phase II Study of Pemetrexed and Carboplatin plus Bevacizumab as First-Line
Therapy in Malignant Pleural Mesothelioma.” British Journal of Cancer 109 (3): 552–58.
doi:10.1038/bjc.2013.368.
Chang, K, L H Pai, J K Batra, I Pastan, and M C Willingham. 1992. “Characterization of the
Antigen (CAK1) Recognized by Monoclonal Antibody K1 Present on Ovarian Cancers and
Normal Mesothelium.” Cancer Research 52 (1): 181–86.
http://www.ncbi.nlm.nih.gov/pubmed/1727378.
Chen, Benjamin P C, Naoya Uematsu, Junya Kobayashi, Yaniv Lerenthal, Andrea Krempler,
Hirohiko Yajima, Markus Löbrich, Yosef Shiloh, and David J Chen. 2007. “Ataxia
Telangiectasia Mutated (ATM) Is Essential for DNA-PKcs Phosphorylations at the Thr-
2609 Cluster upon DNA Double Strand Break.” The Journal of Biological Chemistry 282
(9): 6582–87. doi:10.1074/jbc.M611605200.
Chen, Daniel S., and Ira Mellman. 2013. “Oncology Meets Immunology: The Cancer-Immunity
Cycle.” Immunity 39 (1): 1–10. doi:10.1016/j.immuni.2013.07.012.
Chen, Guoqian, Mary F Ward, Andrew E Sama, and Haichao Wang. 2004. “Extracellular
HMGB1 as a Proinflammatory Cytokine.” Journal of Interferon & Cytokine Research : The
Official Journal of the International Society for Interferon and Cytokine Research 24 (6):
329–33. doi:10.1089/107999004323142187.
Chiriva-Internati, Maurizio, Fabio Grizzi, Justin Pinkston, K John Morrow, Nicholas D’Cunha,
Eldo E Frezza, Pier Carlo Muzzio, W Martin Kast, and Everardo Cobos. “Gamma-
Radiation Upregulates MHC Class I/II and ICAM-I Molecules in Multiple Myeloma Cell
Lines and Primary Tumors.” In Vitro Cellular & Developmental Biology. Animal 42 (3-4):
89–95. doi:10.1290/0508054.1.
Cho, B C John, Ron Feld, Natasha Leighl, Isabelle Opitz, Masaki Anraku, Ming-Sound Tsao,
David M Hwang, Andrew Hope, and Marc de Perrot. 2014. “A Feasibility Study Evaluating
Surgery for Mesothelioma After Radiation Therapy: The ‘SMART’ Approach for
Resectable Malignant Pleural Mesothelioma.” Journal of Thoracic Oncology : Official

113
Publication of the International Association for the Study of Lung Cancer 9 (3): 397–402.
doi:10.1097/JTO.0000000000000078.
Choe, N., S. Tanaka, W. Xia, D. R. Hemenway, V. L. Roggli, and E. Kagan. 1997. “Pleural
Macrophage Recruitment and Activation in Asbestos-Induced Pleural Injury.”
Environmental Health Perspectives 105 Suppl (September): 1257–60.
doi:10.1289/ehp.97105s51257.
Ciernik, I F, P Romero, J A Berzofsky, and D P Carbone. 1999. “Ionizing Radiation Enhances
Immunogenicity of Cells Expressing a Tumor-Specific T-Cell Epitope.” International
Journal of Radiation Oncology, Biology, Physics 45 (3): 735–41.
http://www.ncbi.nlm.nih.gov/pubmed/10524430.
Clements, Mark, Geoffrey Berry, Jill Shi, Sandra Ware, Deborah Yates, and Anthony Johnson.
2007. “Projected Mesothelioma Incidence in Men in New South Wales.” Occupational and
Environmental Medicine 64 (11): 747–52. doi:10.1136/oem.2006.031823.
Colacchio, T A, M P Yeager, and L W Hildebrandt. 1994. “Perioperative Immunomodulation in
Cancer Surgery.” American Journal of Surgery 167 (1): 174–79.
http://www.ncbi.nlm.nih.gov/pubmed/8311130.
Cree, M, M Lalji, B Jiang, K Carriere, J Beach, and A Kamruzzaman. 2009. “Explaining
Alberta’s Rising Mesothelioma Rates - Chronic Diseases in Canada - Volume 29, No. 4,
2009 - Public Health Agency Canada.” Chronic Diseases in Canada. Government of
Canada, Public Health Agency of Canada. 29 (4): 144–52. http://www.phac-
aspc.gc.ca/publicat/hpcdp-pspmc/29-4/ar_01-eng.php.
Cugell, David W, and David W Kamp. 2004. “Asbestos and the Pleura: A Review.” Chest 125
(3): 1103–17. http://www.ncbi.nlm.nih.gov/pubmed/15006974.
Currie, Andrew J, Amy Prosser, Alison McDonnell, Amanda L Cleaver, Bruce W S Robinson,
Gordon J Freeman, and Robbert G van der Most. 2009. “Dual Control of Antitumor CD8 T
Cells through the Programmed Death-1/programmed Death-Ligand 1 Pathway and
Immunosuppressive CD4 T Cells: Regulation and Counterregulation.” Journal of
Immunology (Baltimore, Md. : 1950) 183 (12): 7898–7908. doi:10.4049/jimmunol.0901060.

114
Dąbrowska, Aleksandra M., and Robert Słotwiński. 2014. “The Immune Response to Surgery
and Infection.” Central European Journal of Immunology 4 (4): 532–37.
doi:10.5114/ceji.2014.47741.
Davila, Eduardo, Richard Kennedy, and Esteban Celis. 2003. “Generation of Antitumor
Immunity by Cytotoxic T Lymphocyte Epitope Peptide Vaccination, CpG-
Oligodeoxynucleotide Adjuvant, and CTLA-4 Blockade.” Cancer Research 63 (12): 3281–
88. http://www.ncbi.nlm.nih.gov/pubmed/12810660.
De Wever, Olivier, and Marc Mareel. 2003. “Role of Tissue Stroma in Cancer Cell Invasion.”
The Journal of Pathology 200 (4): 429–47. doi:10.1002/path.1398.
Demaria, Sandra, Nina Bhardwaj, William H McBride, and Silvia C Formenti. 2005.
“Combining Radiotherapy and Immunotherapy: A Revived Partnership.” International
Journal of Radiation Oncology, Biology, Physics 63 (3): 655–66.
doi:10.1016/j.ijrobp.2005.06.032.
Demaria, Sandra, and Silvia C. Formenti. 2012. “Radiation as an Immunological Adjuvant:
Current Evidence on Dose and Fractionation.” Frontiers in Oncology 2 (October): 1–7.
doi:10.3389/fonc.2012.00153.
Demaria, Sandra, Noriko Kawashima, Anne Marie Yang, Mary Louise Devitt, James S. Babb,
James P. Allison, and Silvia C. Formenti. 2005. “Immune-Mediated Inhibition of
Metastases after Treatment with Local Radiation and CTLA-4 Blockade in a Mouse Model
of Breast Cancer.” Clinical Cancer Research 11 (2 I): 728–34. doi:11/2/728 [pii].
Demaria, Sandra, Bruce Ng, Mary Louise Devitt, James S. Babb, Noriko Kawashima, Leonard
Liebes, and Silvia C. Formenti. 2004. “Ionizing Radiation Inhibition of Distant Untreated
Tumors (abscopal Effect) Is Immune Mediated.” International Journal of Radiation
Oncology Biology Physics 58 (3): 862–70. doi:10.1016/j.ijrobp.2003.09.012.
Demicheli, R, M W Retsky, W J M Hrushesky, M Baum, and I D Gukas. 2008. “The Effects of
Surgery on Tumor Growth: A Century of Investigations.” Annals of Oncology : Official
Journal of the European Society for Medical Oncology / ESMO 19 (11): 1821–28.
doi:10.1093/annonc/mdn386.

115
Demirag, Funda, Ebru Unsal, Aydin Yilmaz, and Atalay Caglar. 2005. “Prognostic Significance
of Vascular Endothelial Growth Factor, Tumor Necrosis, and Mitotic Activity Index in
Malignant Pleural Mesothelioma.” Chest 128 (5): 3382–87. doi:10.1378/chest.128.5.3382.
Desai, Sejal, Amit Kumar, S Laskar, and B N Pandey. 2013. “Cytokine Profile of Conditioned
Medium from Human Tumor Cell Lines after Acute and Fractionated Doses of Gamma
Radiation and Its Effect on Survival of Bystander Tumor Cells.” Cytokine 61 (1): 54–62.
doi:10.1016/j.cyto.2012.08.022.
Desborough, J P. 2000. “The Stress Response to Trauma and Surgery.” British Journal of
Anaesthesia 85 (1): 109–17. http://www.ncbi.nlm.nih.gov/pubmed/10927999.
Dewan, M. Zahidunnabi, Ashley E. Galloway, Noriko Kawashima, J. Keith Dewyngaert, James
S. Babb, Silvia C. Formenti, and Sandra Demaria. 2009. “Fractionated but Not Single-Dose
Radiotherapy Induces an Immune-Mediated Abscopal Effect When Combined with Anti-
CTLA-4 Antibody.” Clinical Cancer Research 15 (17): 5379–88. doi:10.1158/1078-
0432.CCR-09-0265.
Dhabhar, F S, A H Miller, B S McEwen, and R L Spencer. 1996. “Stress-Induced Changes in
Blood Leukocyte Distribution. Role of Adrenal Steroid Hormones.” Journal of Immunology
(Baltimore, Md. : 1950) 157 (4): 1638–44. http://www.ncbi.nlm.nih.gov/pubmed/8759750.
Di Stasi, Antonio, Siok-Keen Tey, Gianpietro Dotti, Yuriko Fujita, Alana Kennedy-Nasser,
Caridad Martinez, Karin Straathof, et al. 2011. “Inducible Apoptosis as a Safety Switch for
Adoptive Cell Therapy.” The New England Journal of Medicine 365 (18): 1673–83.
doi:10.1056/NEJMoa1106152.
Dighe, A S, E Richards, L J Old, and R D Schreiber. 1994. “Enhanced in Vivo Growth and
Resistance to Rejection of Tumor Cells Expressing Dominant Negative IFN Gamma
Receptors.” Immunity 1 (6): 447–56. http://www.ncbi.nlm.nih.gov/pubmed/7895156.
Dowell, Jonathan E, Frank R Dunphy, Robert N Taub, David E Gerber, Likheng Ngov,
Jingsheng Yan, Yang Xie, and Hedy Lee Kindler. 2012. “A Multicenter Phase II Study of
Cisplatin, Pemetrexed, and Bevacizumab in Patients with Advanced Malignant

116
Mesothelioma.” Lung Cancer (Amsterdam, Netherlands) 77 (3): 567–71.
doi:10.1016/j.lungcan.2012.05.111.
Draghiciu, Oana, Mateusz Walczak, Baukje Nynke Hoogeboom, Kees L M C Franken, Kees J M
Melief, Hans W Nijman, and Toos Daemen. 2014. “Therapeutic Immunization and Local
Low-Dose Tumor Irradiation, a Reinforcing Combination.” International Journal of
Cancer. Journal International Du Cancer 134 (4): 859–72. doi:10.1002/ijc.28418.
Driece, Hermen a L, Sabine Siesling, Paul H J J Swuste, and Alex Burdorf. 2010. “Assessment
of Cancer Risks due to Environmental Exposure to Asbestos.” Journal of Exposure Science
& Environmental Epidemiology 20 (5). Nature Publishing Group: 478–85.
doi:10.1038/jes.2009.56.
Driessens, Gregory, Justin Kline, and Thomas F. Gajewski. 2009. “Costimulatory and
Coinhibitory Receptors in Anti-Tumor Immunity.” Immunological Reviews 229 (1): 126–
44. doi:10.1111/j.1600-065X.2009.00771.x.
Dudley, Mark E, John R Wunderlich, Paul F Robbins, James C Yang, Patrick Hwu, Douglas J
Schwartzentruber, Suzanne L Topalian, et al. 2002. “Cancer Regression and Autoimmunity
in Patients after Clonal Repopulation with Antitumor Lymphocytes.” Science (New York,
N.Y.) 298 (5594): 850–54. doi:10.1126/science.1076514.
Dunn, Gavin P, Allen T Bruce, Hiroaki Ikeda, Lloyd J Old, and Robert D Schreiber. 2002.
“Cancer Immunoediting: From Immunosurveillance to Tumor Escape.” Nature Immunology
3 (11): 991–98. doi:10.1038/ni1102-991.
Ehrlich, Paul. 1909. “Über Den Jetzigen Stand Der Karzinomforschung.” Ned. Tijdschr.
Geneeskd. 5: 273–90.
Elenkov, I J, R L Wilder, G P Chrousos, and E S Vizi. 2000. “The Sympathetic Nerve--an
Integrative Interface between Two Supersystems: The Brain and the Immune System.”
Pharmacological Reviews 52 (4): 595–638.
http://www.ncbi.nlm.nih.gov/pubmed/11121511.

117
Elenkov, Ilia J. 2004. “Glucocorticoids and the Th1/Th2 Balance.” Annals of the New York
Academy of Sciences 1024 (1): 138–46. doi:10.1196/annals.1321.010.
Eriksson, David, and Torgny Stigbrand. 2010. “Radiation-Induced Cell Death Mechanisms.”
Tumour Biology : The Journal of the International Society for Oncodevelopmental Biology
and Medicine 31 (4): 363–72. doi:10.1007/s13277-010-0042-8.
Ettinger, D S, W Akerley, H Borghaei, a Chang, R T Cheney, L R Chirieac, T a D’Amico, et al.
2012. “Malignant Pleural Mesothelioma. Clinical Practice Guidelines in Oncology.” J Natl
Compr Canc Netw 10 (1): 26–41. doi:10/1/26 [pii].
Fedorocko, P, A Egyed, and A Vacek. 2002. “Irradiation Induces Increased Production of
Haemopoietic and Proinflammatory Cytokines in the Mouse Lung.” International Journal
of Radiation Biology 78 (4): 305–13. doi:10.1080/09553000110104614.
Filatenkov, Alexander, Jeanette Baker, Antonia M S Mueller, Justin Kenkel, G-One Ahn,
Suparna Dutt, Nigel Zhang, et al. 2015. “Ablative Tumor Radiation Can Change the Tumor
Immune Cell Microenvironment to Induce Durable Complete Remissions.” Clinical Cancer
Research : An Official Journal of the American Association for Cancer Research, April.
doi:10.1158/1078-0432.CCR-14-2824.
Fisher, S G, L Weber, and M Carbone. “Cancer Risk Associated with Simian Virus 40
Contaminated Polio Vaccine.” Anticancer Research 19 (3B): 2173–80.
http://www.ncbi.nlm.nih.gov/pubmed/10472327.
Fourcade, Julien, Zhaojun Sun, Mourad Benallaoua, Philippe Guillaume, Immanuel F Luescher,
Cindy Sander, John M Kirkwood, Vijay Kuchroo, and Hassane M Zarour. 2010.
“Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen-Specific
CD8+ T Cell Dysfunction in Melanoma Patients.” The Journal of Experimental Medicine
207 (10): 2175–86. doi:10.1084/jem.20100637.
Frey, Benjamin, Yvonne Rubner, Lorenz Kulzer, Nina Werthmöller, Eva-Maria Weiss, Rainer
Fietkau, and Udo S Gaipl. 2014. “Antitumor Immune Responses Induced by Ionizing
Irradiation and Further Immune Stimulation.” Cancer Immunology, Immunotherapy : CII 63
(1): 29–36. doi:10.1007/s00262-013-1474-y.

118
Fuhrer, Gregory, and Angeline a. Lazarus. 2011. “Mesothelioma.” Disease-a-Month 57 (1): 40–
54. doi:10.1016/j.disamonth.2010.12.004.
Gajewski, Thomas F, Mercedes Fuertes, Robbert Spaapen, Yan Zheng, and Justin Kline. 2011.
“Molecular Profiling to Identify Relevant Immune Resistance Mechanisms in the Tumor
Microenvironment.” Current Opinion in Immunology 23 (2): 286–92.
doi:10.1016/j.coi.2010.11.013.
Gao, Y, J M Herndon, H Zhang, T S Griffith, and T A Ferguson. 1998. “Antiinflammatory
Effects of CD95 Ligand (FasL)-Induced Apoptosis.” The Journal of Experimental Medicine
188 (5): 887–96.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2213381&tool=pmcentrez&ren
dertype=abstract.
Garg, Abhishek D, Dmitri V Krysko, Tom Verfaillie, Agnieszka Kaczmarek, Gabriela B
Ferreira, Thierry Marysael, Noemi Rubio, et al. 2012. “A Novel Pathway Combining
Calreticulin Exposure and ATP Secretion in Immunogenic Cancer Cell Death.” The EMBO
Journal 31 (5): 1062–79. doi:10.1038/emboj.2011.497.
Garland, Linda L, Cathryn Rankin, David R Gandara, Saul E Rivkin, Katherine M Scott,
Raymond B Nagle, Andres J P Klein-Szanto, Joseph R Testa, Deborah A Altomare, and
Ernest C Borden. 2007. “Phase II Study of Erlotinib in Patients with Malignant Pleural
Mesothelioma: A Southwest Oncology Group Study.” Journal of Clinical Oncology :
Official Journal of the American Society of Clinical Oncology 25 (17): 2406–13.
doi:10.1200/JCO.2006.09.7634.
Garland, Linda L. 2011. “Chemotherapy for Malignant Pleural Mesothelioma.” Current
Treatment Options in Oncology 12 (2): 181–88. doi:10.1007/s11864-011-0152-6.
Garnett, Charlie T, Claudia Palena, Mala Chakarborty, Kwong-yok Tsang, Jeffrey Schlom, and
James W Hodge. 2004. “Sublethal Irradiation of Human Tumor Cells Modulates Phenotype
Resulting in Enhanced Killing by Cytotoxic T Lymphocytes.” Cancer Research 64: 7985–
94.

119
Gazdar, Adi F, Janet S Butel, and Michele Carbone. 2002. “SV40 and Human Tumours: Myth,
Association or Causality?” Nature Reviews. Cancer 2 (12): 957–64. doi:10.1038/nrc947.
Ghiringhelli, François, Lionel Apetoh, Antoine Tesniere, Laetitia Aymeric, Yuting Ma, Carla
Ortiz, Karim Vermaelen, et al. 2009. “Activation of the NLRP3 Inflammasome in Dendritic
Cells Induces IL-1beta-Dependent Adaptive Immunity against Tumors.” Nature Medicine
15 (10): 1170–78. doi:10.1038/nm.2028.
Golden, Encouse B, and Lionel Apetoh. 2015. “Radiotherapy and Immunogenic Cell Death.”
Seminars in Radiation Oncology 25 (1): 11–17. doi:10.1016/j.semradonc.2014.07.005.
Golden, Encouse B, Sandra Demaria, Peter B Schiff, Abraham Chachoua, and Silvia C Formenti.
2013. “An Abscopal Response to Radiation and Ipilimumab in a Patient with Metastatic
Non-Small Cell Lung Cancer.” Cancer Immunology Research 1 (6): 365–72.
doi:10.1158/2326-6066.CIR-13-0115.
Golden, Encouse B., Ilenia Pellicciotta, Sandra Demaria, Mary H. Barcellos-Hoff, and Silvia C.
Formenti. 2012. “The Convergence of Radiation and Immunogenic Cell Death Signaling
Pathways.” Frontiers in Oncology 2 (August): 1–13. doi:10.3389/fonc.2012.00088.
Gottlieb, Tanya M, Juan Fernando Martinez Leal, Rony Seger, Yoichi Taya, and Moshe Oren.
2002. “Cross-Talk between Akt, p53 and Mdm2: Possible Implications for the Regulation
of Apoptosis.” Oncogene 21 (8): 1299–1303. doi:10.1038/sj.onc.1205181.
Govindan, Ramaswamy, Robert A Kratzke, James E Herndon, Gloria A Niehans, Robin
Vollmer, Dorothy Watson, Mark R Green, and Hedy L Kindler. 2005. “Gefitinib in Patients
with Malignant Mesothelioma: A Phase II Study by the Cancer and Leukemia Group B.”
Clinical Cancer Research : An Official Journal of the American Association for Cancer
Research 11 (6): 2300–2304. doi:10.1158/1078-0432.CCR-04-1940.
Gross, G, G Gorochov, T Waks, and Z Eshhar. 1989. “Generation of Effector T Cells Expressing
Chimeric T Cell Receptor with Antibody Type-Specificity.” Transplantation Proceedings
21 (1 Pt 1): 127–30. http://www.ncbi.nlm.nih.gov/pubmed/2784887.

120
Grosso, Joseph F., and Maria N. Jure-Kunkel. 2013. “CTLA-4 Blockade in Tumor Models: An
Overview of Preclinical and Translational Research.” Cancer Immunity 13 (January): 5.
Gupta, Anurag, Hans Christian Probst, Van Vuong, Alexandro Landshammer, Sabine Muth,
Hideo Yagita, Reto Schwendener, Martin Pruschy, Alexander Knuth, and Maries van den
Broek. 2012. “Radiotherapy Promotes Tumor-Specific Effector CD8+ T Cells via Dendritic
Cell Activation.” Journal of Immunology (Baltimore, Md. : 1950) 189 (2): 558–66.
doi:10.4049/jimmunol.1200563.
Hall, Eric J., and Amato J. Giaccia. 2006. “Physics and Chemistry of Radiation Absorption.” In
Radiobiology for the Radiologist, 6th ed., 5–15. Lippincott Williams & Wilkins.
Hallahan, D, J Kuchibhotla, and C Wyble. 1996. “Cell Adhesion Molecules Mediate Radiation-
Induced Leukocyte Adhesion to the Vascular Endothelium.” Cancer Research 56 (22):
5150–55. http://www.ncbi.nlm.nih.gov/pubmed/8912850.
Hamdi, Mohamed, Jaap Kool, Paulien Cornelissen-Steijger, Francoise Carlotti, Herman E
Popeijus, Corina van der Burgt, Josephine M Janssen, et al. 2005. “DNA Damage in
Transcribed Genes Induces Apoptosis via the JNK Pathway and the JNK-Phosphatase
MKP-1.” Oncogene 24 (48): 7135–44. doi:10.1038/sj.onc.1208875.
Hamid, Omid, Caroline Robert, Adil Daud, F Stephen Hodi, Wen-Jen Hwu, Richard Kefford,
Jedd D Wolchok, et al. 2013. “Safety and Tumor Responses with Lambrolizumab (anti-PD-
1) in Melanoma.” The New England Journal of Medicine 369 (2): 134–44.
doi:10.1056/NEJMoa1305133.
Hareyama, M, K Imai, K Kubo, H Takahashi, H Koshiba, Y Hinoda, M Shidou, A Oouchi, A
Yachi, and K Morita. 1991. “Effect of Radiation on the Expression of Carcinoembryonic
Antigen of Human Gastric Adenocarcinoma Cells.” Cancer 67 (9): 2269–74.
http://www.ncbi.nlm.nih.gov/pubmed/1901513.
Harrison, L H, P O Schwarzenberger, P S Byrne, A J Marrogi, J K Kolls, and K E McCarthy.
2000. “Gene-Modified PA1-STK Cells Home to Tumor Sites in Patients with Malignant
Pleural Mesothelioma.” The Annals of Thoracic Surgery 70 (2): 407–11.
http://www.ncbi.nlm.nih.gov/pubmed/10969653.

121
Hassan, Raffit, Steven J Cohen, Martin Phillips, Ira Pastan, Elad Sharon, Ronan J Kelly, Charles
Schweizer, Susan Weil, and Daniel Laheru. 2010. “Phase I Clinical Trial of the Chimeric
Anti-Mesothelin Monoclonal Antibody MORAb-009 in Patients with Mesothelin-
Expressing Cancers.” Clinical Cancer Research : An Official Journal of the American
Association for Cancer Research 16 (24): 6132–38. doi:10.1158/1078-0432.CCR-10-2275.
Hassan, Raffit, Wolfgang Ebel, Eric L Routhier, Rina Patel, J Bradford Kline, Jingli Zhang,
Qimin Chao, et al. 2007. “Preclinical Evaluation of MORAb-009, a Chimeric Antibody
Targeting Tumor-Associated Mesothelin.” Cancer Immunity 7 (January): 20.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2935758&tool=pmcentrez&ren
dertype=abstract.
Hassan, Raffit, Hedy L Kindler, Thierry Jahan, Lyudmila Bazhenova, Martin Reck, Anish
Thomas, Ira Pastan, et al. 2014. “Phase II Clinical Trial of Amatuximab, a Chimeric
Antimesothelin Antibody with Pemetrexed and Cisplatin in Advanced Unresectable Pleural
Mesothelioma.” Clinical Cancer Research : An Official Journal of the American
Association for Cancer Research 20 (23): 5927–36. doi:10.1158/1078-0432.CCR-14-0804.
Hassan, Raffit, Andrew C Miller, Elad Sharon, Anish Thomas, James C Reynolds, Alexander
Ling, Robert J Kreitman, et al. 2013. “Major Cancer Regressions in Mesothelioma after
Treatment with an Anti-Mesothelin Immunotoxin and Immune Suppression.” Science
Translational Medicine 5 (208): 208ra147. doi:10.1126/scitranslmed.3006941.
Haupt, Susan, Michael Berger, Zehavit Goldberg, and Ygal Haupt. 2003. “Apoptosis - the p53
Network.” Journal of Cell Science 116 (Pt 20): 4077–85. doi:10.1242/jcs.00739.
Hegmans, Joost P, Joris D Veltman, Margaretha E Lambers, I Jolanda M de Vries, Carl G
Figdor, Rudi W Hendriks, Henk C Hoogsteden, Bart N Lambrecht, and Joachim G Aerts.
2010. “Consolidative Dendritic Cell-Based Immunotherapy Elicits Cytotoxicity against
Malignant Mesothelioma.” American Journal of Respiratory and Critical Care Medicine
181 (12): 1383–90. doi:10.1164/rccm.200909-1465OC.

122
Heiblig, Maël, Mohamed Elhamri, Mauricette Michallet, and Xavier Thomas. 2015. “Adoptive
Immunotherapy for Acute Leukemia: New Insights in Chimeric Antigen Receptors.” World
Journal of Stem Cells 7 (7): 1022–38. doi:10.4252/wjsc.v7.i7.1022.
Heinonen, O P, S Shapiro, R R Monson, S C Hartz, L Rosenberg, and D Slone. 1973.
“Immunization during Pregnancy against Poliomyelitis and Influenza in Relation to
Childhood Malignancy.” International Journal of Epidemiology 2 (3): 229–35.
http://www.ncbi.nlm.nih.gov/pubmed/4359832.
Henkart, P A. 1994. “Lymphocyte-Mediated Cytotoxicity: Two Pathways and Multiple Effector
Molecules.” Immunity 1 (5): 343–46. http://www.ncbi.nlm.nih.gov/pubmed/7882166.
Hernandez-Chacon, Jessica Ann, Yufeng Li, Richard C Wu, Chantale Bernatchez, Yijun Wang,
Jeffrey S Weber, Patrick Hwu, and Laszlo G Radvanyi. 2011. “Costimulation through the
CD137/4-1BB Pathway Protects Human Melanoma Tumor-Infiltrating Lymphocytes from
Activation-Induced Cell Death and Enhances Antitumor Effector Function.” Journal of
Immunotherapy (Hagerstown, Md. : 1997) 34 (3): 236–50.
doi:10.1097/CJI.0b013e318209e7ec.
Heylmann, Daniel, Franz Rödel, Thomas Kindler, and Bernd Kaina. 2014. “Radiation Sensitivity
of Human and Murine Peripheral Blood Lymphocytes, Stem and Progenitor Cells.”
Biochimica et Biophysica Acta 1846 (1): 121–29. doi:10.1016/j.bbcan.2014.04.009.
Hill, R J, R E Edwards, and P Carthew. 1990. “Early Changes in the Pleural Mesothelium
Following Intrapleural Inoculation of the Mineral Fibre Erionite and the Subsequent
Development of Mesotheliomas.” Journal of Experimental Pathology (Oxford, England) 71
(1): 105–18.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1998670&tool=pmcentrez&ren
dertype=abstract.
Hirte, Hal, and David A. Clark. 1991. “Generation of Lymphokine-Activated Killer Cells in
Human Ovarian Carcinoma Ascitic Fluid: Identification of Transforming Growth Factor-Β
as a Suppressive Factor.” Cancer Immunology Immunotherapy 32 (5): 296–302.
doi:10.1007/BF01789047.

123
Hodgson, J T, and A Darnton. 2000. “The Quantitative Risks of Mesothelioma and Lung Cancer
in Relation to Asbestos Exposure.” The Annals of Occupational Hygiene 44 (8): 565–601.
http://www.ncbi.nlm.nih.gov/pubmed/11108782.
Hodgson, J T, D M McElvenny, A J Darnton, M J Price, and J Peto. 2005. “The Expected
Burden of Mesothelioma Mortality in Great Britain from 2002 to 2050.” British Journal of
Cancer 92 (3): 587–93. doi:10.1038/sj.bjc.6602307.
Hodi, F Stephen, Steven J O’Day, David F McDermott, Robert W Weber, Jeffrey A Sosman,
John B Haanen, Rene Gonzalez, et al. 2010. “Improved Survival with Ipilimumab in
Patients with Metastatic Melanoma.” The New England Journal of Medicine 363 (8): 711–
23. doi:10.1056/NEJMoa1003466.
Hofer, S O, G Molema, R A Hermens, H J Wanebo, J S Reichner, and H J Hoekstra. 1999. “The
Effect of Surgical Wounding on Tumour Development.” European Journal of Surgical
Oncology : The Journal of the European Society of Surgical Oncology and the British
Association of Surgical Oncology 25 (3): 231–43. doi:10.1053/ejso.1998.0634.
Hofer, S O, D Shrayer, J S Reichner, H J Hoekstra, and H J Wanebo. 1998. “Wound-Induced
Tumor Progression: A Probable Role in Recurrence after Tumor Resection.” Archives of
Surgery (Chicago, Ill. : 1960) 133 (4): 383–89.
http://www.ncbi.nlm.nih.gov/pubmed/9565118.
Hormbrey, Emma, Cheng Han, Anthony Roberts, Duncan Angus McGrouther, and Adrian L
Harris. 2003. “The Relationship of Human Wound Vascular Endothelial Growth Factor
(VEGF) after Breast Cancer Surgery to Circulating VEGF and Angiogenesis.” Clinical
Cancer Research : An Official Journal of the American Association for Cancer Research 9
(12): 4332–39. http://www.ncbi.nlm.nih.gov/pubmed/14555503.
Hsu, Elisabeth. 2006. “The History of.” In Tropical Medicine, edited by Harvey I. Pass, Nicholas
J. Vogelzang, and Michele Carbone, 1st ed., 3–20. New York, NY: Springer.
doi:10.1016/j.trstmh.2005.09.020.
Illidge, T M. 1998. “Radiation-Induced Apoptosis.” Clinical Oncology (Royal College of
Radiologists (Great Britain)) 10 (1): 3–13. http://www.ncbi.nlm.nih.gov/pubmed/9543608.

124
Innis, M D. 1968. “Oncogenesis and Poliomyelitis Vaccine.” Nature 219 (5157): 972–73.
http://www.ncbi.nlm.nih.gov/pubmed/4299754.
Ismail-Khan, Roohi, Lary a Robinson, Charles C Williams, Christopher R Garrett, Gerold
Bepler, and George R Simon. 2006. “Malignant Pleural Mesothelioma: A Comprehensive
Review.” Cancer Control : Journal of the Moffitt Cancer Center 13 (4): 255–63.
Jackaman, Connie, Christine S Bundell, Beverley F Kinnear, Alison M Smith, Pierre Filion,
Deborah van Hagen, Bruce W S Robinson, and Delia J Nelson. 2003. “IL-2 Intratumoral
Immunotherapy Enhances CD8+ T Cells That Mediate Destruction of Tumor Cells and
Tumor-Associated Vasculature: A Novel Mechanism for IL-2.” Journal of Immunology
(Baltimore, Md. : 1950) 171 (10): 5051–63. doi:10.4049/jimmunol.171.10.5051.
Jahan, Thierry, Lin Gu, Robert Kratzke, Arkadiusz Dudek, Gregory A Otterson, Xiaofei Wang,
Mark Green, Everett E Vokes, and Hedy Lee Kindler. 2012. “Vatalanib in Malignant
Mesothelioma: A Phase II Trial by the Cancer and Leukemia Group B (CALGB 30107).”
Lung Cancer (Amsterdam, Netherlands) 76 (3): 393–96.
doi:10.1016/j.lungcan.2011.11.014.
Jameson, P, J P Desborough, A E Bryant, and G M Hall. 1997. “The Effect of Cortisol
Suppression on Interleukin-6 and White Blood Cell Responses to Surgery.” Acta
Anaesthesiologica Scandinavica 41 (2): 304–8.
http://www.ncbi.nlm.nih.gov/pubmed/9062617.
Jänne, Pasi A, Michele L Taffaro, Ravi Salgia, and Bruce E Johnson. 2002. “Inhibition of
Epidermal Growth Factor Receptor Signaling in Malignant Pleural Mesothelioma.” Cancer
Research 62 (18): 5242–47. http://www.ncbi.nlm.nih.gov/pubmed/12234991.
Jasani, B, A Cristaudo, S A Emri, A F Gazdar, A Gibbs, B Krynska, C Miller, et al. 2001.
“Association of SV40 with Human Tumours.” Seminars in Cancer Biology 11 (1): 49–61.
doi:10.1006/scbi.2000.0346.
Jonathan, E C, E J Bernhard, and W G McKenna. 1999. “How Does Radiation Kill Cells?”
Current Opinion in Chemical Biology 3 (1): 77–83.
http://www.ncbi.nlm.nih.gov/pubmed/10021401.

125
Kaina, Bernd, Wynand Roos, and Markus Christmann. 2010. “DNA Damage Response and the
Balance Between Cell Survival and Cell Death.” In Checkpoint Controls and Targets in
Cancer Therapy, edited by Zahid H. Siddik, First, 95–108. Springer Science & Business
Media.
Kal, Henk B, Henk Struikmans, and Angelique D Barten-van Rijbroek. 2008. “Surgical Stress
and Accelerated Tumor Growth.” Anticancer Research 28 (2A): 1129–32.
http://www.ncbi.nlm.nih.gov/pubmed/18507064.
Kalbasi, Anusha, Carl H. June, Naomi Haas, and Neha Vapiwala. 2013. “Radiation and
Immunotherapy: A Synergistic Combination.” Journal of Clinical Investigation.
doi:10.1172/JCI69219.
Kamp, D W, V A Israbian, S E Preusen, C X Zhang, and S A Weitzman. 1995. “Asbestos
Causes DNA Strand Breaks in Cultured Pulmonary Epithelial Cells: Role of Iron-Catalyzed
Free Radicals.” The American Journal of Physiology 268 (3 Pt 1): L471–80.
http://www.ncbi.nlm.nih.gov/pubmed/7900829.
Kaplan, H S. 1971. “Role of Immunologic Disturbance in Human Oncogenesis: Some Facts and
Fancies.” British Journal of Cancer 25 (4): 620–34.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2008860&tool=pmcentrez&ren
dertype=abstract.
Karin, M, and Y Ben-Neriah. 2000. “Phosphorylation Meets Ubiquitination: The Control of NF-
[kappa]B Activity.” Annual Review of Immunology 18 (January): 621–63.
doi:10.1146/annurev.immunol.18.1.621.
Kehlet, H. 1997. “Multimodal Approach to Control Postoperative Pathophysiology and
Rehabilitation.” British Journal of Anaesthesia 78 (5): 606–17.
http://www.ncbi.nlm.nih.gov/pubmed/9175983.
Keir, Mary E, Manish J Butte, Gordon J Freeman, and Arlene H Sharpe. 2008. “PD-1 and Its
Ligands in Tolerance and Immunity.” Annual Review of Immunology 26 (January): 677–
704. doi:10.1146/annurev.immunol.26.021607.090331.

126
Keisari, Yona, Ilan Hochman, Hila Confino, Rafi Korenstein, and Itzhak Kelson. 2014.
“Activation of Local and Systemic Anti-Tumor Immune Responses by Ablation of Solid
Tumors with Intratumoral Electrochemical or Alpha Radiation Treatments.” Cancer
Immunology, Immunotherapy 63 (1): 1–9. doi:10.1007/s00262-013-1462-2.
Kepp, Oliver, Lorenzo Galluzzi, Isabelle Martins, Frederic Schlemmer, Sandy Adjemian,
Mickael Michaud, Abdul Qader Sukkurwala, Laurie Menger, Laurence Zitvogel, and Guido
Kroemer. 2011. “Molecular Determinants of Immunogenic Cell Death Elicited by
Anticancer Chemotherapy.” Cancer Metastasis Reviews 30 (1): 61–69. doi:10.1007/s10555-
011-9273-4.
Kershaw, Michael H, Jennifer A Westwood, Linda L Parker, Gang Wang, Zelig Eshhar, Sharon
A Mavroukakis, Donald E White, et al. 2006. “A Phase I Study on Adoptive
Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer.” Clinical Cancer
Research : An Official Journal of the American Association for Cancer Research 12 (20 Pt
1): 6106–15. doi:10.1158/1078-0432.CCR-06-1183.
Kim, Ryungsa, Manabu Emi, and Kazuaki Tanabe. 2007. “Cancer Immunoediting from Immune
Surveillance to Immune Escape.” Immunology 121 (1): 1–14. doi:10.1111/j.1365-
2567.2007.02587.x.
Kim, Sunhwa, Hiroyuki Takahashi, Wan-Wan Lin, Pascal Descargues, Sergei Grivennikov,
Youngjun Kim, Jun-Li Luo, and Michael Karin. 2009. “Carcinoma-Produced Factors
Activate Myeloid Cells through TLR2 to Stimulate Metastasis.” Nature 457 (7225): 102–6.
doi:10.1038/nature07623.
Kimura, Fumio, Hiroaki Shimizu, Hiroyuki Yoshidome, Masayuki Ohtsuka, and Masaru
Miyazaki. 2010. “Immunosuppression Following Surgical and Traumatic Injury.” Surgery
Today 40 (9): 793–808. doi:10.1007/s00595-010-4323-z.
Klug, Felix, Hridayesh Prakash, Peter E Huber, Tobias Seibel, Noemi Bender, Niels Halama,
Christina Pfirschke, et al. 2013. “Low-Dose Irradiation Programs Macrophage
Differentiation to an iNOS+/M1 Phenotype That Orchestrates Effective T Cell
Immunotherapy.” Cancer Cell 24 (5): 589–602. doi:10.1016/j.ccr.2013.09.014.

127
König, J, E Tolnay, T Wiethege, and K Müller. 2000. “Co-Expression of Vascular Endothelial
Growth Factor and Its Receptor Flt-1 in Malignant Pleural Mesothelioma.” Respiration;
International Review of Thoracic Diseases 67 (1): 36–40. doi:29460.
Kono, Hajime, and Kenneth L Rock. 2008. “How Dying Cells Alert the Immune System to
Danger.” Nature Reviews. Immunology 8 (4): 279–89. doi:10.1038/nri2215.
Kotova, Svetlana, Raymond M Wong, and Robert B Cameron. 2015. “New and Emerging
Therapeutic Options for Malignant Pleural Mesothelioma : Review of Early Clinical Trials.”
Cancer Management and Research 7: 51–63.
Kreitman, Robert J, Raffit Hassan, David J Fitzgerald, and Ira Pastan. 2009. “Phase I Trial of
Continuous Infusion Anti-Mesothelin Recombinant Immunotoxin SS1P.” Clinical Cancer
Research : An Official Journal of the American Association for Cancer Research 15 (16):
5274–79. doi:10.1158/1078-0432.CCR-09-0062.
Kroemer, Guido, Lorenzo Galluzzi, Oliver Kepp, and Laurence Zitvogel. 2013. “Immunogenic
Cell Death in Cancer Therapy.” Annual Review of Immunology 31 (January): 51–72.
doi:10.1146/annurev-immunol-032712-100008.
Krug, Lee M, Tao Dao, Andrew B Brown, Peter Maslak, William Travis, Sara Bekele, Tatyana
Korontsvit, et al. 2010. “WT1 Peptide Vaccinations Induce CD4 and CD8 T Cell Immune
Responses in Patients with Mesothelioma and Non-Small Cell Lung Cancer.” Cancer
Immunology, Immunotherapy : CII 59 (10): 1467–79. doi:10.1007/s00262-010-0871-8.
Krysko, O, T Løve Aaes, C Bachert, P Vandenabeele, and D V Krysko. 2013. “Many Faces of
DAMPs in Cancer Therapy.” Cell Death & Disease 4 (January): e631.
doi:10.1038/cddis.2013.156.
Krysko, Dmitri V, Abhishek D Garg, Agnieszka Kaczmarek, Olga Krysko, Patrizia Agostinis,
and Peter Vandenabeele. 2012. “Immunogenic Cell Death and DAMPs in Cancer Therapy.”
Nature Reviews. Cancer 12 (12): 860–75. doi:10.1038/nrc3380.
Lamers, Cor Hj, Stefan Sleijfer, Sabine van Steenbergen, Pascal van Elzakker, Brigitte van
Krimpen, Corrien Groot, Arnold Vulto, et al. 2013. “Treatment of Metastatic Renal Cell

128
Carcinoma with CAIX CAR-Engineered T Cells: Clinical Evaluation and Management of
on-Target Toxicity.” Molecular Therapy : The Journal of the American Society of Gene
Therapy 21 (4): 904–12. doi:10.1038/mt.2013.17.
Larkin, James, Vanna Chiarion-Sileni, Rene Gonzalez, Jean Jacques Grob, C Lance Cowey,
Christopher D Lao, Dirk Schadendorf, et al. 2015. “Combined Nivolumab and Ipilimumab
or Monotherapy in Untreated Melanoma.” The New England Journal of Medicine 373 (1):
23–34. doi:10.1056/NEJMoa1504030.
Lauber, Kirsten, Anne Ernst, Michael Orth, Martin Herrmann, and Claus Belka. 2012. “Dying
Cell Clearance and Its Impact on the Outcome of Tumor Radiotherapy.” Frontiers in
Oncology 2 (January): 116. doi:10.3389/fonc.2012.00116.
Lavin, M F. 2007. “ATM and the Mre11 Complex Combine to Recognize and Signal DNA
Double-Strand Breaks.” Oncogene 26 (56): 7749–58. doi:10.1038/sj.onc.1210880.
Le, Dung T, Dirk G Brockstedt, Ran Nir-Paz, Johannes Hampl, Shruti Mathur, John Nemunaitis,
Daniel H Sterman, et al. 2012. “A Live-Attenuated Listeria Vaccine (ANZ-100) and a Live-
Attenuated Listeria Vaccine Expressing Mesothelin (CRS-207) for Advanced Cancers:
Phase I Studies of Safety and Immune Induction.” Clinical Cancer Research : An Official
Journal of the American Association for Cancer Research 18 (3): 858–68.
doi:10.1158/1078-0432.CCR-11-2121.
Lee, Hyeon-Woo, Su-Jung Park, Beom K Choi, Hyun Hwa Kim, Kyung-Ok Nam, and Byoung S
Kwon. 2002. “4-1BB Promotes the Survival of CD8+ T Lymphocytes by Increasing
Expression of Bcl-xL and Bfl-1.” Journal of Immunology (Baltimore, Md. : 1950) 169 (9):
4882–88. http://www.ncbi.nlm.nih.gov/pubmed/12391199.
Lee, Ji-Hoon, and Tanya T Paull. 2004. “Direct Activation of the ATM Protein Kinase by the
Mre11/Rad50/Nbs1 Complex.” Science (New York, N.Y.) 304 (5667): 93–96.
doi:10.1126/science.1091496.
Lee, Youjin, Sogyong L. Auh, Yugang Wang, Byron Burnette, Yang Wang, Yuru Meng,
Michael Beckett, et al. 2009. “Therapeutic Effects of Ablative Radiation on Local Tumor

129
Require CD8 + T Cells: Changing Strategies for Cancer Treatment.” Blood 114 (3): 589–
95. doi:10.1182/blood-2009-02-206870.
Leigh, J., and Bruce Robinson. 2002. “The History of Mesothelioma in Australia 1945-2001.” In
Mesothelioma, edited by Bruce W.S. Robinson and A. Philippe Chahinian, 55–110.
London: Martin Dunitz Ltd.
Levy, Antonin, Cyrus Chargari, Morgane Cheminant, Noémie Simon, Céline Bourgier, and Eric
Deutsch. 2013. “Radiation Therapy and Immunotherapy: Implications for a Combined
Cancer Treatment.” Critical Reviews in Oncology/Hematology 85 (3). Elsevier Ireland Ltd:
278–87. doi:10.1016/j.critrevonc.2012.09.001.
Lewanski, C R, and W J Gullick. 2001. “Radiotherapy and Cellular Signalling.” The Lancet.
Oncology 2 (6): 366–70. doi:10.1016/S1470-2045(00)00391-0.
Lin, E, S E Calvano, and S F Lowry. 2000. “Inflammatory Cytokines and Cell Response in
Surgery.” Surgery 127 (2): 117–26. doi:10.1067/msy.2000.101584.
Lipson, Evan J, William H Sharfman, Charles G Drake, Ira Wollner, Janis M Taube, Robert A
Anders, Haiying Xu, et al. 2013. “Durable Cancer Regression off-Treatment and Effective
Reinduction Therapy with an Anti-PD-1 Antibody.” Clinical Cancer Research : An Official
Journal of the American Association for Cancer Research 19 (2): 462–68.
doi:10.1158/1078-0432.CCR-12-2625.
Liu, Zhiwen, and Julius Klominek. 2004. “Chemotaxis and Chemokinesis of Malignant
Mesothelioma Cells to Multiple Growth Factors.” Anticancer Research 24 (3 A): 1625–30.
Lorne, Emmanuel, Jaroslaw W Zmijewski, Xia Zhao, Gang Liu, Yuko Tsuruta, Young-Jun Park,
Hervé Dupont, and Edward Abraham. 2008. “Role of Extracellular Superoxide in
Neutrophil Activation: Interactions between Xanthine Oxidase and TLR4 Induce
Proinflammatory Cytokine Production.” American Journal of Physiology. Cell Physiology
294 (4): C985–93. doi:10.1152/ajpcell.00454.2007.
Luce, Audrey, Aurélie Courtin, Céline Levalois, Sandrine Altmeyer-Morel, Paul-Henri Romeo,
Sylvie Chevillard, and Jérôme Lebeau. 2009. “Death Receptor Pathways Mediate Targeted

130
and Non-Targeted Effects of Ionizing Radiations in Breast Cancer Cells.” Carcinogenesis
30 (3): 432–39. doi:10.1093/carcin/bgp008.
Lugade, Amit a, James P Moran, Scott a Gerber, Robert C Rose, John G Frelinger, and Edith M
Lord. 2005. “Local Radiation Therapy of B16 Melanoma Tumors Increases the Generation
of Tumor Antigen-Specific Effector Cells That Traffic to the Tumor.” Journal of
Immunology (Baltimore, Md. : 1950) 174 (12): 7516–23.
doi:10.4049/jimmunol.174.12.7516.
Lugade, Amit A, Elizabeth W Sorensen, Scott A Gerber, James P Moran, John G Frelinger, and
Edith M Lord. 2008. “Radiation-Induced IFN-Gamma Production within the Tumor
Microenvironment Influences Antitumor Immunity.” Journal of Immunology (Baltimore,
Md. : 1950) 180 (5): 3132–39. http://www.ncbi.nlm.nih.gov/pubmed/18292536.
Lumniczky, Katalin, and Géza Sáfrány. 2015. “The Impact of Radiation Therapy on the
Antitumor Immunity: Local Effects and Systemic Consequences.” Cancer Letters 356 (1):
114–25. doi:10.1016/j.canlet.2013.08.024.
Ma, Yuting, Oliver Kepp, François Ghiringhelli, Lionel Apetoh, Laetitia Aymeric, Clara Locher,
Antoine Tesniere, et al. 2010. “Chemotherapy and Radiotherapy: Cryptic Anticancer
Vaccines.” Seminars in Immunology 22 (3): 113–24. doi:10.1016/j.smim.2010.03.001.
MacConmara, Malcolm P, Adrian A Maung, Satoshi Fujimi, Ann M McKenna, Adam Delisle,
Peter H Lapchak, Selwyn Rogers, James A Lederer, and John A Mannick. 2006. “Increased
CD4+ CD25+ T Regulatory Cell Activity in Trauma Patients Depresses Protective Th1
Immunity.” Annals of Surgery 244 (4): 514–23. doi:10.1097/01.sla.0000239031.06906.1f.
Mailand, N, J Falck, C Lukas, R G Syljuâsen, M Welcker, J Bartek, and J Lukas. 2000. “Rapid
Destruction of Human Cdc25A in Response to DNA Damage.” Science (New York, N.Y.)
288 (5470): 1425–29. http://www.ncbi.nlm.nih.gov/pubmed/10827953.
Manda, Katrin, Annegret Glasow, Daniel Paape, and Guido Hildebrandt. 2012. “Effects of
Ionizing Radiation on the Immune System with Special Emphasis on the Interaction of
Dendritic and T Cells.” Frontiers in Oncology 2 (January): 102.
doi:10.3389/fonc.2012.00102.

131
Mansfield, Aaron Scott, Anja C Roden, Tobias Peikert, Yuri M Sheinin, Susan M Harrington,
Christopher J Krco, Haidong Dong, and Eugene D Kwon. 2014. “B7-H1 Expression in
Malignant Pleural Mesothelioma Is Associated with Sarcomatoid Histology and Poor
Prognosis.” Journal of Thoracic Oncology : Official Publication of the International
Association for the Study of Lung Cancer 9 (7): 1036–40.
doi:10.1097/JTO.0000000000000177.
Mantel, Frederick, Benjamin Frey, Stefan Haslinger, Petra Schildkopf, Renate Sieber, Oliver J
Ott, Barbara Lödermann, et al. 2010. “Combination of Ionising Irradiation and
Hyperthermia Activates Programmed Apoptotic and Necrotic Cell Death Pathways in
Human Colorectal Carcinoma Cells.” Strahlentherapie Und Onkologie : Organ Der
Deutschen Röntgengesellschaft ... [et Al] 186 (11): 587–99. doi:10.1007/s00066-010-2154-
x.
Marik, Paul E., and Mark Flemmer. 2012. “The Immune Response to Surgery and Trauma.”
Journal of Trauma and Acute Care Surgery 73 (4): 801–8.
doi:10.1097/TA.0b013e318265cf87.
Marinaccio, Alessandro, Fabio Montanaro, Marina Mastrantonio, Raffaella Uccelli, Pierluigi
Altavista, Massimo Nesti, Adele Seniori Costantini, and Giuseppe Gorini. 2005.
“Predictions of Mortality from Pleural Mesothelioma in Italy: A Model Based on Asbestos
Consumption Figures Supports Results from Age-Period-Cohort Models.” International
Journal of Cancer. Journal International Du Cancer 115 (1): 142–47.
doi:10.1002/ijc.20820.
Markham, A, and D Faulds. 1994. “Ganciclovir. An Update of Its Therapeutic Use in
Cytomegalovirus Infection.” Drugs 48 (3): 455–84.
http://www.ncbi.nlm.nih.gov/pubmed/7527763.
Marom, Edith M, Jeremy J Erasmus, Harvey I Pass, and Edward F Patz. 2002. “The Role of
Imaging in Malignant Pleural Mesothelioma.” Seminars in Oncology 29 (1): 26–35.
http://www.ncbi.nlm.nih.gov/pubmed/11836666.

132
Martensson, G. 1990. “Diagnosing Malignant Pleural Mesothelioma.” The European Respiratory
Journal : Official Journal of the European Society for Clinical Respiratory Physiology 3
(9): 985–86.
Masood, Rizwan, Ajay Kundra, SuTao Zhu, Guangbin Xia, Pierluigi Scalia, D Lynne Smith, and
Parkash S Gill. 2003. “Malignant Mesothelioma Growth Inhibition by Agents That Target
the VEGF and VEGF-C Autocrine Loops.” International Journal of Cancer. Journal
International Du Cancer 104 (5): 603–10. doi:10.1002/ijc.10996.
Matkowski, Rafal, Iwona Gisterek, Agnieszka Halon, Aleksandra Lacko, Krzysztof Szewczyk,
Urszula Staszek, Marek Pudelko, et al. 2009. “The Prognostic Role of Tumor-Infiltrating
CD4 and CD8 T Lymphocytes in Breast Cancer.” Anticancer Research 29 (7): 2445–51.
http://www.ncbi.nlm.nih.gov/pubmed/19596912.
Matsumura, Satoko, Baomei Wang, Noriko Kawashima, Steve Braunstein, Michelle Badura,
Thomas O Cameron, James S Babb, et al. 2008. “Radiation-Induced CXCL16 Release by
Breast Cancer Cells Attracts Effector T Cells.” Journal of Immunology (Baltimore, Md. :
1950) 181 (5): 3099–3107.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2587101&tool=pmcentrez&ren
dertype=abstract.
Matsuoka, S, G Rotman, A Ogawa, Y Shiloh, K Tamai, and S J Elledge. 2000. “Ataxia
Telangiectasia-Mutated Phosphorylates Chk2 in Vivo and in Vitro.” Proceedings of the
National Academy of Sciences of the United States of America 97 (19): 10389–94.
doi:10.1073/pnas.190030497.
Matzinger, P. 1998. “An Innate Sense of Danger.” Seminars in Immunology 10 (5): 399–415.
doi:10.1006/smim.1998.0143.
McBride, William H, Chi-Shiun Chiang, Jennifer L Olson, Chun-Chieh Wang, Ji-Hong Hong,
Frank Pajonk, Graeme J Dougherty, Keisuke S Iwamoto, Milena Pervan, and Yu-Pei Liao.
2004. “A Sense of Danger from Radiation.” Radiation Research 162 (1): 1–19.
http://www.ncbi.nlm.nih.gov/pubmed/15222781.

133
Melamed, Rivka, Shahar Bar-Yosef, Guy Shakhar, Keren Shakhar, and Shamgar Ben-Eliyahu.
2003. “Suppression of Natural Killer Cell Activity and Promotion of Tumor Metastasis by
Ketamine, Thiopental, and Halothane, but Not by Propofol: Mediating Mechanisms and
Prophylactic Measures.” Anesthesia and Analgesia 97 (5): 1331–39.
http://www.ncbi.nlm.nih.gov/pubmed/14570648.
Menger, Michael D, and Brigitte Vollmar. 2004. “Surgical Trauma: Hyperinflammation versus
Immunosuppression?” Langenbeck’s Archives of Surgery / Deutsche Gesellschaft Für
Chirurgie 389 (6): 475–84. doi:10.1007/s00423-004-0472-0.
Menges, P, W Kessler, C Kloecker, M Feuerherd, S Gaubert, S Diedrich, J van der Linde, et al.
2012. “Surgical Trauma and Postoperative Immune Dysfunction.” European Surgical
Research. Europäische Chirurgische Forschung. Recherches Chirurgicales Européennes
48 (4): 180–86. doi:10.1159/000338196.
Mezzapelle, R, U Miglio, O Rena, A Paganotti, S Allegrini, J Antona, F Molinari, et al. 2013.
“Mutation Analysis of the EGFR Gene and Downstream Signalling Pathway in Histologic
Samples of Malignant Pleural Mesothelioma.” British Journal of Cancer 108 (8): 1743–49.
doi:10.1038/bjc.2013.130.
Minatel, Emilio, Marco Trovo, Jerry Polesel, Tania Baresic, Alessandra Bearz, Giovanni
Franchin, Carlo Gobitti, et al. 2014. “Radical Pleurectomy/decortication Followed by High
Dose of Radiation Therapy for Malignant Pleural Mesothelioma. Final Results with Long-
Term Follow-Up.” Lung Cancer (Amsterdam, Netherlands) 83 (1): 78–82.
doi:10.1016/j.lungcan.2013.10.013.
Mita, Alain C, Christopher J Sweeney, Sharyn D Baker, Andrew Goetz, Lisa A Hammond,
Amita Patnaik, Anthony W Tolcher, et al. 2006. “Phase I and Pharmacokinetic Study of
Pemetrexed Administered Every 3 Weeks to Advanced Cancer Patients with Normal and
Impaired Renal Function.” Journal of Clinical Oncology : Official Journal of the American
Society of Clinical Oncology 24 (4): 552–62. doi:10.1200/JCO.2004.00.9720.
Mizuno, Kenji, Hideo Kohka Takahashi, Hiromi Iwagaki, Goutaro Katsuno, Huda A S M
Kamurul, Satoru Ohtani, Shuji Mori, Tadashi Yoshino, Masahiro Nishibori, and Noriaki

134
Tanaka. 2005. “Beta2-Adrenergic Receptor Stimulation Inhibits LPS-Induced IL-18 and IL-
12 Production in Monocytes.” Immunology Letters 101 (2): 168–72.
doi:10.1016/j.imlet.2005.05.008.
Morel, A, N Fernandez, A de La Coste, H Haddada, M Viguier, B S Polla, B Antoine, and A
Kahn. 1998. “Gamma-Ray Irradiation Induces B7.1 Costimulatory Molecule Neoexpression
in Various Murine Tumor Cells.” Cancer Immunology, Immunotherapy : CII 46 (5): 277–
82. http://www.ncbi.nlm.nih.gov/pubmed/9690456.
Morel, S, F Lévy, O Burlet-Schiltz, F Brasseur, M Probst-Kepper, A L Peitrequin, B Monsarrat,
et al. 2000. “Processing of Some Antigens by the Standard Proteasome but Not by the
Immunoproteasome Results in Poor Presentation by Dendritic Cells.” Immunity 12 (1):
107–17. http://www.ncbi.nlm.nih.gov/pubmed/10661410.
Moretti, Amanda. 2010. “Development of a Model to Study the Abscopal Effect : Combining
Image-Guided Radiation Therapy and Immunotherapy in Cancer Treatment by
Development of a Model to Study the Abscopal Effect : Combining Image-Guided
Radiation Therapy and Immunotherapy.” University of Toronto.
Mossman, Brooke T, Arti Shukla, Nicholas H Heintz, Claire F Verschraegen, Anish Thomas,
and Raffit Hassan. 2013. “New Insights into Understanding the Mechanisms, Pathogenesis,
and Management of Malignant Mesotheliomas.” The American Journal of Pathology 182
(4): 1065–77. doi:10.1016/j.ajpath.2012.12.028.
Muers, Martin F, Richard J Stephens, Patricia Fisher, Liz Darlison, Christopher M B Higgs,
Erica Lowry, Andrew G Nicholson, et al. 2008. “Active Symptom Control with or without
Chemotherapy in the Treatment of Patients with Malignant Pleural Mesothelioma (MS01):
A Multicentre Randomised Trial.” Lancet (London, England) 371 (9625): 1685–94.
doi:10.1016/S0140-6736(08)60727-8.
Nagata, S, and P Golstein. 1995. “The Fas Death Factor.” Science (New York, N.Y.) 267 (5203):
1449–56. http://www.ncbi.nlm.nih.gov/pubmed/7533326.

135
Nguyen, G K, M R Akin, R R Villanueva, and J Slatnik. 1999. “Cytopathology of Malignant
Mesothelioma of the Pleura in Fine-Needle Aspiration Biopsy.” Diagnostic Cytopathology
21 (4): 253–59. http://www.ncbi.nlm.nih.gov/pubmed/10495318.
Nishimura, T, M Nakui, M Sato, K Iwakabe, H Kitamura, M Sekimoto, A Ohta, T Koda, and S
Nishimura. 2000. “The Critical Role of Th1-Dominant Immunity in Tumor Immunology.”
Cancer Chemotherapy and Pharmacology 46 Suppl (January): S52–61.
http://www.ncbi.nlm.nih.gov/pubmed/10950149.
Norvell, Amanda. 2013. “Components of the Immune System.” In Cancer Immunotherapy, 11–
24. New Jersey: Elsevier. doi:10.1016/B978-0-12-394296-8.00002-6.
Nosho, Katsuhiko, Yoshifumi Baba, Noriko Tanaka, Kaori Shima, Marika Hayashi, Jeffrey A
Meyerhardt, Edward Giovannucci, Glenn Dranoff, Charles S Fuchs, and Shuji Ogino. 2010.
“Tumour-Infiltrating T-Cell Subsets, Molecular Changes in Colorectal Cancer, and
Prognosis: Cohort Study and Literature Review.” The Journal of Pathology 222 (4): 350–
66. doi:10.1002/path.2774.
Novy, Patricia, Michael Quigley, Xiaopei Huang, and Yiping Yang. 2007. “CD4 T Cells Are
Required for CD8 T Cell Survival during Both Primary and Memory Recall Responses.”
Journal of Immunology (Baltimore, Md. : 1950) 179 (12): 8243–51.
http://www.ncbi.nlm.nih.gov/pubmed/18056368.
Nowak, A K, M J Byrne, R Williamson, G Ryan, A Segal, D Fielding, P Mitchell, A W Musk,
and B W S Robinson. 2002. “A Multicentre Phase II Study of Cisplatin and Gemcitabine
for Malignant Mesothelioma.” British Journal of Cancer 87 (5): 491–96.
doi:10.1038/sj.bjc.6600505.
Obeid, Michel, Antoine Tesniere, François Ghiringhelli, Gian Maria Fimia, Lionel Apetoh, Jean-
Luc Perfettini, Maria Castedo, et al. 2007. “Calreticulin Exposure Dictates the
Immunogenicity of Cancer Cell Death.” Nature Medicine 13 (1): 54–61.
doi:10.1038/nm1523.
Odaka, M, D H Sterman, R Wiewrodt, Y Zhang, M Kiefer, K M Amin, G P Gao, et al. 2001.
“Eradication of Intraperitoneal and Distant Tumor by Adenovirus-Mediated Interferon-Beta

136
Gene Therapy Is Attributable to Induction of Systemic Immunity.” Cancer Research 61
(16): 6201–12. http://www.ncbi.nlm.nih.gov/pubmed/11507073.
Ogawa, K, M Hirai, T Katsube, M Murayama, K Hamaguchi, T Shimakawa, Y Naritake, T
Hosokawa, and T Kajiwara. 2000. “Suppression of Cellular Immunity by Surgical Stress.”
Surgery 127 (3): 329–36. doi:10.1067/msy.2000.103498.
Ohta, Y, V Shridhar, R K Bright, G P Kalemkerian, W Du, M Carbone, Y Watanabe, and H I
Pass. 1999. “VEGF and VEGF Type C Play an Important Role in Angiogenesis and
Lymphangiogenesis in Human Malignant Mesothelioma Tumours.” British Journal of
Cancer 81 (1): 54–61. doi:10.1038/sj.bjc.6690650.
Okuda, K, H Sasaki, O Kawano, H Yukiue, T Yokoyama, M Yano, and Y Fujii. 2008.
“Epidermal Growth Factor Receptor Gene Mutation, Amplification and Protein Expression
in Malignant Pleural Mesothelioma.” Journal of Cancer Research and Clinical Oncology
134 (10): 1105–11. doi:10.1007/s00432-008-0384-4.
Opitz, Isabelle. 2014. “Management of Malignant Pleural Mesothelioma-The European
Experience.” Journal of Thoracic Disease 6 (SUPPL.2): 238–52. doi:10.3978/j.issn.2072-
1439.2014.05.03.
Overgaard, J, and MR Horsman. 1996. “Modification of Hypoxia-Induced Radioresistance in
Tumors by the Use of Oxygen and Sensitizers.” Seminars in Radiation Oncology 6 (1): 10–
21. doi:10.1053/SRAO0060010.
Palcic, B, and L D Skarsgard. 1984. “Reduced Oxygen Enhancement Ratio at Low Doses of
Ionizing Radiation.” Radiation Research 100 (2): 328–39.
http://www.ncbi.nlm.nih.gov/pubmed/6494444.
Palucka, Karolina, and Jacques Banchereau. 2012. “Cancer Immunotherapy via Dendritic Cells.”
Nature Reviews Cancer 12 (4): 265–77. doi:10.1038/nrc3258.
Papa, Sophie, Sanjay Popat, Riyaz Shah, A Toby Prevost, Rohit Lal, Blair McLennan, Paul
Cane, et al. 2013. “Phase 2 Study of Sorafenib in Malignant Mesothelioma Previously
Treated with Platinum-Containing Chemotherapy.” Journal of Thoracic Oncology : Official

137
Publication of the International Association for the Study of Lung Cancer 8 (6): 783–87.
doi:10.1097/JTO.0b013e31828c2b26.
Park, Bonggoo, Cassian Yee, and Kyung Mi Lee. 2014. “The Effect of Radiation on the Immune
Response to Cancers.” International Journal of Molecular Sciences 15 (1): 927–43.
doi:10.3390/ijms15010927.
Park, In-Chul, Sang-Hyeok Woo, Myung-Jin Park, Hyung-Chahn Lee, Su-Jae Lee, Young-Joon
Hong, Seung-Hoon Lee, Seok-Il Hong, and Chang-Hun Rhee. “Ionizing Radiation and
Nitric Oxide Donor Sensitize Fas-Induced Apoptosis via up-Regulation of Fas in Human
Cervical Cancer Cells.” Oncology Reports 10 (3): 629–33.
http://www.ncbi.nlm.nih.gov/pubmed/12684635.
Pastan, Ira, Raffit Hassan, David J FitzGerald, and Robert J Kreitman. 2007. “Immunotoxin
Treatment of Cancer.” Annual Review of Medicine 58 (January): 221–37.
doi:10.1146/annurev.med.58.070605.115320.
Pateras, Ioannis S., Sophia Havaki, Xenia Nikitopoulou, Konstantinos Vougas, Paul Townsend,
Michalis I. Panayiotidis, Alexandros G. Georgakilas, and Vassilis G. Gorgoulis. 2015. “The
DNA Damage Response and Immune Signaling Alliance: Is It Good or Bad? Nature
Decides When and Where.” Pharmacology & Therapeutics, July.
doi:10.1016/j.pharmthera.2015.06.011.
Peng, C Y, P R Graves, R S Thoma, Z Wu, A S Shaw, and H Piwnica-Worms. 1997. “Mitotic
and G2 Checkpoint Control: Regulation of 14-3-3 Protein Binding by Phosphorylation of
Cdc25C on Serine-216.” Science (New York, N.Y.) 277 (5331): 1501–5.
http://www.ncbi.nlm.nih.gov/pubmed/9278512.
Peto, J, a Decarli, C La Vecchia, F Levi, and E Negri. 1999. “The European Mesothelioma
Epidemic.” British Journal of Cancer 79 (3-4): 666–72. doi:10.1038/sj.bjc.6690105.
Pietsch, E C, S M Sykes, S B McMahon, and M E Murphy. 2008. “The p53 Family and
Programmed Cell Death.” Oncogene 27 (50): 6507–21. doi:10.1038/onc.2008.315.

138
Pirker, Robert, Jose R Pereira, Aleksandra Szczesna, Joachim von Pawel, Maciej Krzakowski,
Rodryg Ramlau, Ihor Vynnychenko, et al. 2009. “Cetuximab plus Chemotherapy in Patients
with Advanced Non-Small-Cell Lung Cancer (FLEX): An Open-Label Randomised Phase
III Trial.” Lancet (London, England) 373 (9674): 1525–31. doi:10.1016/S0140-
6736(09)60569-9.
Pirttikangas, C O, M Salo, M Mansikka, J Grönroos, K Pulkki, and O Peltola. 1995. “The
Influence of Anaesthetic Technique upon the Immune Response to Hysterectomy. A
Comparison of Propofol Infusion and Isoflurane.” Anaesthesia 50 (12): 1056–61.
http://www.ncbi.nlm.nih.gov/pubmed/8546287.
Postow, Michael A, Margaret K Callahan, Christopher A Barker, Yoshiya Yamada, Jianda Yuan,
Shigehisa Kitano, Zhenyu Mu, et al. 2012. “Immunologic Correlates of the Abscopal Effect
in a Patient with Melanoma.” The New England Journal of Medicine 366 (10): 925–31.
doi:10.1056/NEJMoa1112824.
Postow, Michael A, James Harding, and Jedd D Wolchok. 2012. “Targeting Immune
Checkpoints: Releasing the Restraints on Anti-Tumor Immunity for Patients with
Melanoma.” Cancer Journal (Sudbury, Mass.) 18 (2): 153–59.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3751414&tool=pmcentrez&ren
dertype=abstract.
Powers, Amy, and Michele Carbone. 2002. “The Role of Environmental Carcinogens, Viruses
and Genetic Predisposition in the Pathogenesis of Mesothelioma.” Cancer Biology and
Therapy 1 (4): 348–53. doi:10.4161/cbt.1.4.4.
Puri, R K, D S Hoon, P Leland, P Snoy, R W Rand, I Pastan, and R J Kreitman. 1996.
“Preclinical Development of a Recombinant Toxin Containing Circularly Permuted
Interleukin 4 and Truncated Pseudomonas Exotoxin for Therapy of Malignant
Astrocytoma.” Cancer Research 56 (24): 5631–37.
http://www.ncbi.nlm.nih.gov/pubmed/8971168.

139
Rabinovich, Gabriel A, Dmitry Gabrilovich, and Eduardo M Sotomayor. 2007.
“Immunosuppressive Strategies That Are Mediated by Tumor Cells.” Annual Review of
Immunology 25 (January): 267–96. doi:10.1146/annurev.immunol.25.022106.141609.
Radford, I R. 1985. “The Level of Induced DNA Double-Strand Breakage Correlates with Cell
Killing after X-Irradiation.” International Journal of Radiation Biology and Related Studies
in Physics, Chemistry, and Medicine 48 (1): 45–54.
http://www.ncbi.nlm.nih.gov/pubmed/3874180.
Raja, Siva, Sudish C. Murthy, and David P. Mason. 2011. “Malignant Pleural Mesothelioma.”
Current Oncology Reports 13 (4): 259–64. doi:10.1007/s11912-011-0177-9.
Ramos-Nino, Maria E., Cynthia R. Timblin, and Brooke T. Mossman. 2002. “Mesothelial Cell
Transformation Requires Increased AP-1 Binding Activity and ERK-Dependent Fra-1
Expression.” Cancer Research 62 (21): 6065–69.
Ratzer, E R, J L Pool, and M R Melamed. 1967. “Pleural Mesotheliomas. Clinical Experiences
with Thirty-Seven Patients.” The American Journal of Roentgenology, Radium Therapy,
and Nuclear Medicine 99 (4): 863–80. http://www.ncbi.nlm.nih.gov/pubmed/6021390.
Reid, A, N de Klerk, G Ambrosini, N Olsen, S C Pang, and A W Musk. 2005. “The Additional
Risk of Malignant Mesothelioma in Former Workers and Residents of Wittenoom with
Benign Pleural Disease or Asbestosis.” Occupational and Environmental Medicine 62 (10):
665–69. doi:10.1136/oem.2004.018531.
Reits, Eric a, James W Hodge, Carla a Herberts, Tom a Groothuis, Mala Chakraborty, Elizabeth
K Wansley, Kevin Camphausen, et al. 2006. “Radiation Modulates the Peptide Repertoire,
Enhances MHC Class I Expression, and Induces Successful Antitumor Immunotherapy.”
The Journal of Experimental Medicine 203 (5): 1259–71. doi:10.1084/jem.20052494.
Richendollar, Bill G, Brad Pohlman, Paul Elson, and Eric D Hsi. 2011. “Follicular Programmed
Death 1-Positive Lymphocytes in the Tumor Microenvironment Are an Independent
Prognostic Factor in Follicular Lymphoma.” Human Pathology 42 (4): 552–57.
doi:10.1016/j.humpath.2010.08.015.

140
Robertson, John D, Vladimir Gogvadze, Andrey Kropotov, Helin Vakifahmetoglu, Boris
Zhivotovsky, and Sten Orrenius. 2004. “Processed Caspase-2 Can Induce Mitochondria-
Mediated Apoptosis Independently of Its Enzymatic Activity.” EMBO Reports 5 (6): 643–
48. doi:10.1038/sj.embor.7400153.
Robinson, Benjamin M. 2012. “Malignant Pleural Mesothelioma: An Epidemiological
Perspective.” Annals of Cardiothoracic Surgery 1 (4): 491–96. doi:10.3978/j.issn.2225-
319X.2012.11.04.
Robinson, Bruce W S, and Richard A Lake. 2005. “Advances in Malignant Mesothelioma.” The
New England Journal of Medicine 353 (15): 1591–1603. doi:10.1056/NEJMra050152.
Robinson, Bruce W S, Arthur W Musk, and Richard A Lake. 2005. “Malignant Mesothelioma.”
Lancet (London, England) 366 (9483): 397–408. doi:10.1016/S0140-6736(05)67025-0.
Rock, Kenneth L, and Hajime Kono. 2008. “The Inflammatory Response to Cell Death.” Annual
Review of Pathology 3 (January): 99–126.
doi:10.1146/annurev.pathmechdis.3.121806.151456.
Roedel, F, N Kley, H U Beuscher, G Hildebrandt, L Keilholz, P Kern, R Voll, M Herrmann, and
R Sauer. 2002. “Anti-Inflammatory Effect of Low-Dose X-Irradiation and the Involvement
of a TGF-beta1-Induced down-Regulation of Leukocyte/endothelial Cell Adhesion.”
International Journal of Radiation Biology 78 (8): 711–19.
doi:10.1080/09553000210137671.
Rosenne, Ella, Liat Sorski, Lee Shaashua, Elad Neeman, Pini Matzner, Ben Levi, and Shamgar
Ben-Eliyahu. 2014. “In Vivo Suppression of NK Cell Cytotoxicity by Stress and Surgery:
Glucocorticoids Have a Minor Role Compared to Catecholamines and Prostaglandins.”
Brain, Behavior, and Immunity 37 (March): 207–19. doi:10.1016/j.bbi.2013.12.007.
Rosenzweig, Kenneth E, Marjorie G Zauderer, Benjamin Laser, Lee M Krug, Ellen Yorke,
Camelia S Sima, Andreas Rimner, Raja Flores, and Valerie Rusch. 2012. “Pleural Intensity-
Modulated Radiotherapy for Malignant Pleural Mesothelioma.” International Journal of
Radiation Oncology, Biology, Physics 83 (4): 1278–83. doi:10.1016/j.ijrobp.2011.09.027.

141
Rovere-Querini, Patrizia, Annalisa Capobianco, Paola Scaffidi, Barbara Valentinis, Federica
Catalanotti, Marta Giazzon, Ingrid E Dumitriu, et al. 2004. “HMGB1 Is an Endogenous
Immune Adjuvant Released by Necrotic Cells.” EMBO Reports 5 (8): 825–30.
doi:10.1038/sj.embor.7400205.
Rübe, Claudia E, Falk Wilfert, Jan Palm, Jochem König, Susanne Burdak-Rothkamm, Li Liu,
Andreas Schuck, Normann Willich, and Christian Rübe. 2004. “Irradiation Induces a
Biphasic Expression of pro-Inflammatory Cytokines in the Lung.” Strahlentherapie Und
Onkologie : Organ Der Deutschen Röntgengesellschaft ... [et Al] 180 (7): 442–48.
doi:10.1007/s00066-004-1265-7.
Ruffie, P, R Feld, S Minkin, Y Cormier, A Boutan-Laroze, R Ginsberg, J Ayoub, F A Shepherd,
W K Evans, and A Figueredo. 1989. “Diffuse Malignant Mesothelioma of the Pleura in
Ontario and Quebec: A Retrospective Study of 332 Patients.” Journal of Clinical
Oncology : Official Journal of the American Society of Clinical Oncology 7 (8): 1157–68.
http://www.ncbi.nlm.nih.gov/pubmed/2666592.
Rusch, V W, K Rosenzweig, E Venkatraman, L Leon, A Raben, L Harrison, M S Bains, R J
Downey, and R J Ginsberg. 2001. “A Phase II Trial of Surgical Resection and Adjuvant
High-Dose Hemithoracic Radiation for Malignant Pleural Mesothelioma.” The Journal of
Thoracic and Cardiovascular Surgery 122 (4): 788–95. doi:10.1067/mtc.2001.116560.
Rusch, Valerie, Elizabeth H. Baldini, Raphael Bueno, Marc De Perrot, Raja Flores, Seiki
Hasegawa, Walter Klepetko, et al. 2013. “The Role of Surgical Cytoreduction in the
Treatment of Malignant Pleural Mesothelioma: Meeting Summary of the International
Mesothelioma Interest Group Congress, September 11-14, 2012, Boston, Mass.” Journal of
Thoracic and Cardiovascular Surgery 145 (4): 909–10. doi:10.1016/j.jtcvs.2013.01.039.
Rusch, Valerie W., Dorothy Giroux, Catherine Kennedy, Enrico Ruffini, Ayten K. Cangir, David
Rice, Harvey Pass, et al. 2012. “Initial Analysis of the International Association For the
Study of Lung Cancer Mesothelioma Database.” Journal of Thoracic Oncology 7 (11):
1631–39. doi:10.1097/JTO.0b013e31826915f1.

142
Russell, John H, and Timothy J Ley. 2002. “Lymphocyte-Mediated Cytotoxicity.” Annual
Review of Immunology 20 (January): 323–70.
doi:10.1146/annurev.immunol.20.100201.131730.
Sanchez, Y, C Wong, R S Thoma, R Richman, Z Wu, H Piwnica-Worms, and S J Elledge. 1997.
“Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to
Cdk Regulation through Cdc25.” Science (New York, N.Y.) 277 (5331): 1497–1501.
http://www.ncbi.nlm.nih.gov/pubmed/9278511.
Sanders, C L, and T A Jackson. 1972. “Induction of Mesotheliomas and Sarcomas from ‘Hot
Spots’ of 239 PuO 2 Activity.” Health Physics 22 (6): 755–59.
http://www.ncbi.nlm.nih.gov/pubmed/5084007.
Sanford, Mark. 2012. “Ipilimumab: In Previously Treated Patients with Advanced Melanoma.”
BioDrugs : Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy 26 (3):
185–93. doi:10.2165/11208200-000000000-00000.
Saoulli, K, S Y Lee, J L Cannons, W C Yeh, A Santana, M D Goldstein, N Bangia, et al. 1998.
“CD28-Independent, TRAF2-Dependent Costimulation of Resting T Cells by 4-1BB
Ligand.” The Journal of Experimental Medicine 187 (11): 1849–62.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2212301&tool=pmcentrez&ren
dertype=abstract.
Seki, Naoko, Alan D Brooks, Clive R D Carter, Timothy C Back, Erin M Parsoneault, Mark J
Smyth, Robert H Wiltrout, and Thomas J Sayers. 2002. “Tumor-Specific CTL Kill Murine
Renal Cancer Cells Using Both Perforin and Fas Ligand-Mediated Lysis in Vitro, but Cause
Tumor Regression in Vivo in the Absence of Perforin.” Journal of Immunology (Baltimore,
Md. : 1950) 168 (7): 3484–92. http://www.ncbi.nlm.nih.gov/pubmed/11907109.
Seo, A, F Ishikawa, H Nakano, H Nakazaki, K Kobayashi, and T Kakiuchi. 1999. “Enhancement
of B7-1 (CD80) Expression on B-Lymphoma Cells by Irradiation.” Immunology 96 (4):
642–48.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2326785&tool=pmcentrez&ren
dertype=abstract.

143
Sgadari, C, A L Angiolillo, and G Tosato. 1996. “Inhibition of Angiogenesis by Interleukin-12 Is
Mediated by the Interferon-Inducible Protein 10.” Blood 87 (9): 3877–82.
http://www.ncbi.nlm.nih.gov/pubmed/8611715.
Shankaran, V, H Ikeda, A T Bruce, J M White, P E Swanson, L J Old, and R D Schreiber. 2001.
“IFNgamma and Lymphocytes Prevent Primary Tumour Development and Shape Tumour
Immunogenicity.” Nature 410 (6832): 1107–11. doi:10.1038/35074122.
Sheard, Michael A, Stjepan Uldrijan, and Borivoj Vojtesek. 2003. “Role of p53 in Regulating
Constitutive and X-Radiation-Inducible CD95 Expression and Function in Carcinoma
Cells.” Cancer Research 63 (21): 7176–84.
http://www.ncbi.nlm.nih.gov/pubmed/14612511.
Shieh, S Y, J Ahn, K Tamai, Y Taya, and C Prives. 2000. “The Human Homologs of Checkpoint
Kinases Chk1 and Cds1 (Chk2) Phosphorylate p53 at Multiple DNA Damage-Inducible
Sites.” Genes & Development 14 (3): 289–300.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=316358&tool=pmcentrez&rend
ertype=abstract.
Shiraishi, Kenshiro, Yoshiro Ishiwata, Keiichi Nakagawa, Shoji Yokochi, Chiho Taruki, Teruo
Akuta, Kuni Ohtomo, Kouji Matsushima, Takuya Tamatani, and Shiro Kanegasaki. 2008.
“Enhancement of Antitumor Radiation Efficacy and Consistent Induction of the Abscopal
Effect in Mice by ECI301, an Active Variant of Macrophage Inflammatory Protein-1alpha.”
Clinical Cancer Research : An Official Journal of the American Association for Cancer
Research 14 (4): 1159–66. doi:10.1158/1078-0432.CCR-07-4485.
Shivapurkar, Narayan, Kenichi Harada, Jyotsna Reddy, Richard H Scheuermann, Yin Xu, Robert
W McKenna, Sara Milchgrub, Steven H Kroft, Ziding Feng, and Adi F Gazdar. 2002.
“Presence of Simian Virus 40 DNA Sequences in Human Lymphomas.” Lancet 359 (9309):
851–52. doi:10.1016/S0140-6736(02)07921-7.
Silva, Manuel T. 2010. “Secondary Necrosis: The Natural Outcome of the Complete Apoptotic
Program.” FEBS Letters 584 (22): 4491–99. doi:10.1016/j.febslet.2010.10.046.

144
Son, Cheol-Hun, Jae-Ho Bae, Dong-Yeok Shin, Hong-Rae Lee, Wol-Soon Jo, Kwangmo Yang,
and You-Soo Park. 2015. “Combination Effect of Regulatory T-Cell Depletion and Ionizing
Radiation in Mouse Models of Lung and Colon Cancer.” International Journal of Radiation
Oncology, Biology, Physics 92 (2): 390–98. doi:10.1016/j.ijrobp.2015.01.011.
Soussi, T, and G Lozano. 2005. “p53 Mutation Heterogeneity in Cancer.” Biochemical and
Biophysical Research Communications 331 (3): 834–42. doi:10.1016/j.bbrc.2005.03.190.
Stahel, R. a., W. Weder, Y. Lievens, and E. Felip. 2010. “Malignant Pleural Mesothelioma:
ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up.” Annals of
Oncology 21 (SUPPL. 5): 2009–11. doi:10.1093/annonc/mdq173.
Stebbing, Justin, Thomas Powles, Kirsty McPherson, Jonathan Shamash, Paula Wells, Michael T
Sheaff, Sarah Slater, Robin M Rudd, Dean Fennell, and Jeremy P C Steele. 2009. “The
Efficacy and Safety of Weekly Vinorelbine in Relapsed Malignant Pleural Mesothelioma.”
Lung Cancer (Amsterdam, Netherlands) 63 (1): 94–97. doi:10.1016/j.lungcan.2008.04.001.
Sterman, Daniel H, Andrew Haas, Edmund Moon, Adriana Recio, Daniel Schwed, Anil Vachani,
Sharyn I Katz, et al. 2011. “A Trial of Intrapleural Adenoviral-Mediated Interferon-α2b
Gene Transfer for Malignant Pleural Mesothelioma.” American Journal of Respiratory and
Critical Care Medicine 184 (12): 1395–99. doi:10.1164/rccm.201103-0554CR.
Sterman, Daniel H, Adri Recio, Richard G Carroll, Colin T Gillespie, Andrew Haas, Anil
Vachani, Veena Kapoor, et al. 2007. “A Phase I Clinical Trial of Single-Dose Intrapleural
IFN-Beta Gene Transfer for Malignant Pleural Mesothelioma and Metastatic Pleural
Effusions: High Rate of Antitumor Immune Responses.” Clinical Cancer Research : An
Official Journal of the American Association for Cancer Research 13 (15 Pt 1): 4456–66.
doi:10.1158/1078-0432.CCR-07-0403.
Sterman, Daniel H, Adriana Recio, Anil Vachani, Jing Sun, Lumei Cheung, Peter DeLong,
Kunjlata M Amin, et al. 2005. “Long-Term Follow-up of Patients with Malignant Pleural
Mesothelioma Receiving High-Dose Adenovirus Herpes Simplex Thymidine
Kinase/ganciclovir Suicide Gene Therapy.” Clinical Cancer Research : An Official Journal

145
of the American Association for Cancer Research 11 (20): 7444–53. doi:10.1158/1078-
0432.CCR-05-0405.
Strickler, H D, P S Rosenberg, S S Devesa, J Hertel, J F Fraumeni, and J J Goedert. 1998.
“Contamination of Poliovirus Vaccines with Simian Virus 40 (1955-1963) and Subsequent
Cancer Rates.” JAMA 279 (4): 292–95. http://www.ncbi.nlm.nih.gov/pubmed/9450713.
Stutman. 1975. “Immunodepression and Malignancy.” Advances in Cancer Research 22
(January): 261–422. http://www.ncbi.nlm.nih.gov/pubmed/766581.
Stutman, O. 1974. “Tumor Development after 3-Methylcholanthrene in Immunologically
Deficient Athymic-Nude Mice.” Science (New York, N.Y.) 183 (4124): 534–36.
http://www.ncbi.nlm.nih.gov/pubmed/4588620.
Stutman, Osias. 1979. “Chemical Carcinogenesis in Nude Mice: Comparison between Nude
Mice from Homozygous Matings and Heterozygous Matings and Effect of Age and
Carcinogen Dose.” Journal of the National Cancer Institute 62 (2): 353–58.
http://www.ncbi.nlm.nih.gov/pubmed/283266.
Sun, Joseph C, Matthew A Williams, and Michael J Bevan. 2004. “CD4+ T Cells Are Required
for the Maintenance, Not Programming, of Memory CD8+ T Cells after Acute Infection.”
Nature Immunology 5 (9): 927–33. doi:10.1038/ni1105.
Suzuki, Yasunosuke, Steven R Yuen, and Richard Ashley. 2005. “Short, Thin Asbestos Fibers
Contribute to the Development of Human Malignant Mesothelioma: Pathological
Evidence.” International Journal of Hygiene and Environmental Health 208 (3): 201–10.
doi:10.1016/j.ijheh.2005.01.015.
Tagliabue, Elda, Roberto Agresti, Maria Luisa Carcangiu, Cristina Ghirelli, Daniele Morelli,
Manuela Campiglio, Maritza Martel, et al. 2003. “Role of HER2 in Wound-Induced Breast
Carcinoma Proliferation.” Lancet (London, England) 362 (9383): 527–33.
doi:10.1016/S0140-6736(03)14112-8.
Takeshima, Tsuguhide, Kenji Chamoto, Daiko Wakita, Takayuki Ohkuri, Yuji Togashi, Hiroki
Shirato, Hidemitsu Kitamura, and Takashi Nishimura. 2010. “Local Radiation Therapy

146
Inhibits Tumor Growth through the Generation of Tumor-Specific CTL: Its Potentiation by
Combination with TH1 Cell Therapy.” Cancer Research 70 (7): 2697–2706.
doi:10.1158/0008-5472.CAN-09-2982.
Tan, Y, M Xu, W Wang, F Zhang, D Li, X Xu, J Gu, and R M Hoffman. “IL-2 Gene Therapy of
Advanced Lung Cancer Patients.” Anticancer Research 16 (4A): 1993–98.
http://www.ncbi.nlm.nih.gov/pubmed/8712732.
Tanaka, S, N Choe, a Iwagaki, D R Hemenway, and E Kagan. 2000. “Asbestos Exposure
Induces MCP-1 Secretion by Pleural Mesothelial Cells.” Experimental Lung Research 26
(4): 241–55.
Tang, P, S K Vatsia, S Teichberg, and E Kahn. 2001. “Pulmonary Adenocarcinoma Simulating
Malignant Mesothelioma.” Archives of Pathology & Laboratory Medicine 125 (12): 1598–
1600. doi:10.1043/0003-9985(2001)125<1598:PASMM>2.0.CO;2.
Tanida, Satoshi, Tsutomu Mizoshita, Keiji Ozeki, Hironobu Tsukamoto, Takeshi Kamiya,
Hiromi Kataoka, Daitoku Sakamuro, and Takashi Joh. 2012. “Mechanisms of Cisplatin-
Induced Apoptosis and of Cisplatin Sensitivity: Potential of BIN1 to Act as a Potent
Predictor of Cisplatin Sensitivity in Gastric Cancer Treatment.” International Journal of
Surgical Oncology 2012 (January): 862879. doi:10.1155/2012/862879.
Tesniere, A, T Panaretakis, O Kepp, L Apetoh, F Ghiringhelli, L Zitvogel, and G Kroemer. 2008.
“Molecular Characteristics of Immunogenic Cancer Cell Death.” Cell Death and
Differentiation 15 (1): 3–12. doi:10.1038/sj.cdd.4402269.
Thompson, R Houston, Haidong Dong, Christine M Lohse, Bradley C Leibovich, Michael L
Blute, John C Cheville, and Eugene D Kwon. 2007. “PD-1 Is Expressed by Tumor-
Infiltrating Immune Cells and Is Associated with Poor Outcome for Patients with Renal Cell
Carcinoma.” Clinical Cancer Research : An Official Journal of the American Association
for Cancer Research 13 (6): 1757–61. doi:10.1158/1078-0432.CCR-06-2599.
Till, Brian G, Michael C Jensen, Jinjuan Wang, Eric Y Chen, Brent L Wood, Harvey A
Greisman, Xiaojun Qian, et al. 2008. “Adoptive Immunotherapy for Indolent Non-Hodgkin

147
Lymphoma and Mantle Cell Lymphoma Using Genetically Modified Autologous CD20-
Specific T Cells.” Blood 112 (6): 2261–71. doi:10.1182/blood-2007-12-128843.
Till, Brian G, Michael C Jensen, Jinjuan Wang, Xiaojun Qian, Ajay K Gopal, David G Maloney,
Catherine G Lindgren, et al. 2012. “CD20-Specific Adoptive Immunotherapy for
Lymphoma Using a Chimeric Antigen Receptor with Both CD28 and 4-1BB Domains: Pilot
Clinical Trial Results.” Blood 119 (17): 3940–50. doi:10.1182/blood-2011-10-387969.
Tischoff, Iris, Matthias Neid, Volker Neumann, and Andrea Tannapfel. 2011. “Pathohistological
Diagnosis and Differential Diagnosis.” Recent Results in Cancer Research. Fortschritte Der
Krebsforschung. Progrès Dans Les Recherches Sur Le Cancer 189 (January): 57–78.
doi:10.1007/978-3-642-10862-4_5.
Topalian, Suzanne L, Charles G Drake, and Drew M Pardoll. 2012. “Targeting the PD-1/B7-
H1(PD-L1) Pathway to Activate Anti-Tumor Immunity.” Current Opinion in Immunology
24 (2): 207–12. doi:10.1016/j.coi.2011.12.009.
Topalian, Suzanne L, F Stephen Hodi, Julie R Brahmer, Scott N Gettinger, David C Smith,
David F McDermott, John D Powderly, et al. 2012. “Safety, Activity, and Immune
Correlates of Anti-PD-1 Antibody in Cancer.” The New England Journal of Medicine 366
(26): 2443–54. doi:10.1056/NEJMoa1200690.
Travis, Lois B, Sophie D Fosså, Sara J Schonfeld, Mary L McMaster, Charles F Lynch, Hans
Storm, Per Hall, et al. 2005. “Second Cancers among 40,576 Testicular Cancer Patients:
Focus on Long-Term Survivors.” Journal of the National Cancer Institute 97 (18): 1354–
65. doi:10.1093/jnci/dji278.
Treasure, Tom, Loic Lang-Lazdunski, David Waller, Judith M. Bliss, Carol Tan, James Entwisle,
Michael Snee, et al. 2011. “Extra-Pleural Pneumonectomy versus No Extra-Pleural
Pneumonectomy for Patients with Malignant Pleural Mesothelioma: Clinical Outcomes of
the Mesothelioma and Radical Surgery (MARS) Randomised Feasibility Study.” The
Lancet Oncology 12 (8). Elsevier Ltd: 763–72. doi:10.1016/S1470-2045(11)70149-8.

148
Tsuchiya, Yasunori, Shigeaki Sawada, Isaku Yoshioka, Yasukata Ohashi, Mitsuhiro Matsuo,
Yuko Harimaya, Kazuhiro Tsukada, and Ikuo Saiki. 2003. “Increased Surgical Stress
Promotes Tumor Metastasis.” Surgery 133 (5): 547–55. doi:10.1067/msy.2003.141.
Vachani, Anil, Edmund Moon, Elliot Wakeam, and Steven M. Albelda. 2010. “Gene Therapy for
Mesothelioma and Lung Cancer.” American Journal of Respiratory Cell and Molecular
Biology 42 (4): 385–93. doi:10.1165/rcmb.2010-0026RT.
Valerie, Kristoffer, and Lawrence F Povirk. 2003. “Regulation and Mechanisms of Mammalian
Double-Strand Break Repair.” Oncogene 22 (37): 5792–5812. doi:10.1038/sj.onc.1206679.
Van den Broek, M E, D Kägi, F Ossendorp, R Toes, S Vamvakas, W K Lutz, C J Melief, R M
Zinkernagel, and H Hengartner. 1996. “Decreased Tumor Surveillance in Perforin-Deficient
Mice.” The Journal of Experimental Medicine 184 (5): 1781–90.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2192859&tool=pmcentrez&ren
dertype=abstract.
Van Valen, F, V Kentrup-Lardong, B Truckenbrod, C Rübe, W Winkelmann, and W W Jürgens.
1997. “Regulation of the Release of Tumour Necrosis Factor (TNF)alpha and Soluble TNF
Receptor by Gamma Irradiation and Interferon Gamma in Ewing’s Sarcoma/peripheral
Primitive Neuroectodermal Tumour Cells.” Journal of Cancer Research and Clinical
Oncology 123 (5): 245–52. http://www.ncbi.nlm.nih.gov/pubmed/9201246.
Vandenabeele, Peter, Lorenzo Galluzzi, Tom Vanden Berghe, and Guido Kroemer. 2010.
“Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion.” Nature Reviews.
Molecular Cell Biology 11 (10): 700–714. doi:10.1038/nrm2970.
Vanpouille-Box, Claire, Karsten A Pilones, Erik Wennerberg, Silvia C Formenti, and Sandra
Demaria. 2015. “In Situ Vaccination by Radiotherapy to Improve Responses to Anti-
CTLA-4 Treatment.” Vaccine, July. doi:10.1016/j.vaccine.2015.05.105.
Victor, Christina Twyman-Saint, Andrew J. Rech, Amit Maity, Ramesh Rengan, Kristen E.
Pauken, Erietta Stelekati, Joseph L. Benci, et al. 2015. “Radiation and Dual Checkpoint
Blockade Activate Non-Redundant Immune Mechanisms in Cancer.” Nature 520 (7547):
373–77. doi:10.1038/nature14292.

149
Vogelzang, Nicholas J., James J. Rusthoven, James Symanowski, Claude Denham, E. Kaukel,
Pierre Ruffie, Ulrich Gatzemeier, et al. 2003. “Phase III Study of Pemetrexed in
Combination with Cisplatin versus Cisplatin Alone in Patients with Malignant Pleural
Mesothelioma.” Journal of Clinical Oncology 21 (14): 2636–44.
doi:10.1200/JCO.2003.11.136.
Voll, R E, M Herrmann, E A Roth, C Stach, J R Kalden, and I Girkontaite. 1997.
“Immunosuppressive Effects of Apoptotic Cells.” Nature 390 (6658): 350–51.
doi:10.1038/37022.
Wada, Hiroki, Shuhji Seki, Tetsuya Takahashi, Nobuaki Kawarabayashi, Hideyuki Higuchi,
Yoshiko Habu, Shinya Sugahara, and Tomiei Kazama. 2007. “Combined Spinal and
General Anesthesia Attenuates Liver Metastasis by Preserving TH1/TH2 Cytokine
Balance.” Anesthesiology 106 (3): 499–506.
http://www.ncbi.nlm.nih.gov/pubmed/17325508.
Wagner, J C, C a Sleggs, and P Marchand. 1960. “Diffuse Pleural Mesothelioma and Asbestos
Exposure in the North Western Cape Province.” British Journal of Industrial Medicine 17:
260–71.
Wang, Yaohe, Steven P Faux, Gunnel Hallden, David H Kirn, Cathy E Houghton, Nicholas R
Lemoine, and Graham Patrick. 2004. “Interleukin-1beta and Tumour Necrosis Factor-Alpha
Promote the Transformation of Human Immortalised Mesothelial Cells by Erionite.”
International Journal of Oncology 25 (1): 173–78.
http://www.ncbi.nlm.nih.gov/pubmed/15202003.
Weber, J S, and S A Rosenberg. 1988. “Modulation of Murine Tumor Major Histocompatibility
Antigens by Cytokines in Vivo and in Vitro.” Cancer Research 48 (20): 5818–24.
http://www.ncbi.nlm.nih.gov/pubmed/3139284.
Welden, Brett, Gerald Gates, Resti Mallari, and Normalynn Garrett. 2009. “Effects of
Anesthetics and Analgesics on Natural Killer Cell Activity.” AANA Journal 77 (4): 287–92.
http://www.ncbi.nlm.nih.gov/pubmed/19731847.

150
Whitaker, D. 2000. “The Cytology of Malignant Mesothelioma.” Cytopathology : Official
Journal of the British Society for Clinical Cytology 11 (3): 139–51.
http://www.ncbi.nlm.nih.gov/pubmed/10877273.
Withers, HR. 1993. “Treatment-Induced Accelerated Human Tumor Growth.” Seminars in
Radiation Oncology 3 (2): 135–43. doi:10.1054/SRAO00300135.
Wu, L., Z. Yun, T. Tagawa, K. Rey-McIntyre, and M. de Perrot. 2012. “CTLA-4 Blockade
Expands Infiltrating T Cells and Inhibits Cancer Cell Repopulation during the Intervals of
Chemotherapy in Murine Mesothelioma.” Molecular Cancer Therapeutics 11 (8): 1809–19.
doi:10.1158/1535-7163.MCT-11-1014.
Wu, Licun, Matthew Onn Wu, Luis De Maza, Zhihong Yun, Julie Yu, John Cho, and Marc De
Perrot. 2015. “Targeting the Inhibitory Receptor CTLA-4 on T Cells Increased Apscopal
Effects in Murine Mesothelioma Model.”
Wu, Licun, Zhihong Yun, Tetsuzo Tagawa, Katrina Rey-McIntyre, Masaki Anraku, and Marc de
Perrot. 2011. “Tumor Cell Repopulation between Cycles of Chemotherapy Is Inhibited by
Regulatory T-Cell Depletion in a Murine Mesothelioma Model.” Journal of Thoracic
Oncology : Official Publication of the International Association for the Study of Lung
Cancer 6 (9): 1578–86. doi:10.1097/JTO.0b013e3182208ee0.
Xie, Q W, R Whisnant, and C Nathan. 1993. “Promoter of the Mouse Gene Encoding Calcium-
Independent Nitric Oxide Synthase Confers Inducibility by Interferon Gamma and Bacterial
Lipopolysaccharide.” The Journal of Experimental Medicine 177 (6): 1779–84.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2191051&tool=pmcentrez&ren
dertype=abstract.
Xing, Z, J Gauldie, G Cox, H Baumann, M Jordana, X F Lei, and M K Achong. 1998. “IL-6 Is
an Antiinflammatory Cytokine Required for Controlling Local or Systemic Acute
Inflammatory Responses.” Journal of Clinical Investigation 101 (2): 311–20.
doi:10.1172/JCI1368.
Xu, An, Hongning Zhou, Dennis Zengliang Yu, and Tom K. Hei. 2002. “Mechanisms of the
Genotoxicity of Crocidolite Asbestos in Mammalian Cells: Implication from Mutation

151
Patterns Induced by Reactive Oxygen Species.” Environmental Health Perspectives 110
(10): 1003–8. doi:10.1289/ehp.021101003.
Yang, Haining, Maurizio Bocchetta, Barbara Kroczynska, Amira G Elmishad, Yuanbin Chen,
Zemin Liu, Concetta Bubici, et al. 2006. “TNF-Alpha Inhibits Asbestos-Induced
Cytotoxicity via a NF-kappaB-Dependent Pathway, a Possible Mechanism for Asbestos-
Induced Oncogenesis.” Proceedings of the National Academy of Sciences of the United
States of America 103 (27): 10397–402. doi:10.1073/pnas.0604008103.
Yang, Haining, Joseph R. Testa, and Michele Carbone. 2008. “Mesothelioma Epidemiology,
Carcinogenesis, and Pathogenesis.” Current Treatment Options in Oncology 9 (2-3): 147–
57. doi:10.1007/s11864-008-0067-z.
Yarborough, Charles M. 2006. “Chrysotile as a Cause of Mesothelioma: An Assessment Based
on Epidemiology.” Critical Reviews in Toxicology 36 (2): 165–87.
http://www.ncbi.nlm.nih.gov/pubmed/16736942.
Yom, Sue S. 2015. “Accelerated Repopulation as a Cause of Radiation Treatment Failure in
Non-Small Cell Lung Cancer: Review of Current Data and Future Clinical Strategies.”
Seminars in Radiation Oncology 25 (2): 93–99. doi:10.1016/j.semradonc.2014.12.002.
Zalcman, Gerard, Julien Mazieres, Jacques Margery, Laurent Greillier, Clarisse Audigier-
Valette, Denis Moro-Sibilot, Oliver Molinier, et al. 2015. “Bevacizumab 15mg/kg plus
Cisplatin-Pemetrexed (CP) Triplet versus CP Doublet in Malignant Pleural Mesothelioma
(MPM): Results of the IFCT-GFPC-0701 MAPS Randomized Phase 3 Trial.” Journal of
Clinical Investigations (ASCO Meeting Abstracts) 33 (15_suppl).
http://hwmaint.meeting.ascopubs.org/cgi/content/abstract/33/15_suppl/7500.
Zhang, Bin, Natalie A Bowerman, Joseph K Salama, Hank Schmidt, Michael T Spiotto, Andrea
Schietinger, Ping Yu, et al. 2007. “Induced Sensitization of Tumor Stroma Leads to
Eradication of Established Cancer by T Cells.” The Journal of Experimental Medicine 204
(1): 49–55. doi:10.1084/jem.20062056.
Zhang, Y, T C Lee, B Guillemin, M C Yu, and W N Rom. 1993. “Enhanced IL-1 Beta and
Tumor Necrosis Factor-Alpha Release and Messenger RNA Expression in Macrophages

152
from Idiopathic Pulmonary Fibrosis or after Asbestos Exposure.” Journal of Immunology
(Baltimore, Md. : 1950) 150 (9): 4188–96. http://www.ncbi.nlm.nih.gov/pubmed/8473757.
Zhao, Yangbing, Edmund Moon, Carmine Carpenito, Chrystal M Paulos, Xiaojun Liu, Andrea L
Brennan, Anne Chew, et al. 2010. “Multiple Injections of Electroporated Autologous T
Cells Expressing a Chimeric Antigen Receptor Mediate Regression of Human Disseminated
Tumor.” Cancer Research 70 (22): 9053–61. doi:10.1158/0008-5472.CAN-10-2880.
Zhu, Xinmei, Gabriel Herrera, and Juan B Ochoa. 2010. “Immunosupression and Infection after
Major Surgery: A Nutritional Deficiency.” Critical Care Clinics 26 (3): 491–500, ix.
doi:10.1016/j.ccc.2010.04.004.
Zou, Weiping, and Lieping Chen. 2008. “Inhibitory B7-Family Molecules in the Tumour
Microenvironment.” Nature Reviews. Immunology 8 (6): 467–77. doi:10.1038/nri2326.


















