
The Golgi-associated protein p115 is required for the...
123
Molecular Mechanisms of the Unconventional Secretion of Macrophage Migration Inhibitory Factor (MIF) Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades einer Doktorin der Naturwissenschaften genehmigte Dissertation vorgelegt von Diplom-Biologin Melanie Merk aus Freiburg im Breisgau Berichter: Universitätsprofessor Dr. Jürgen Bernhagen Universitätsprofessor Dr. Werner Baumgartner Tag der mündlichen Prüfung: 19. Dezember 2008 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.
Transcript of The Golgi-associated protein p115 is required for the...

Molecular Mechanisms of the
Unconventional Secretion of Macrophage
Migration Inhibitory Factor (MIF)
Von der Fakultät für Mathematik, Informatik und Naturwissenschaften
der RWTH Aachen University zur Erlangung des akademischen Grades
einer Doktorin der Naturwissenschaften genehmigte Dissertation
vorgelegt von
Diplom-Biologin
Melanie Merk
aus
Freiburg im Breisgau
Berichter: Universitätsprofessor Dr. Jürgen Bernhagen
Universitätsprofessor Dr. Werner Baumgartner
Tag der mündlichen Prüfung: 19. Dezember 2008
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek
online verfügbar.

II
TABLE OF CONTENTS
A. Abbreviations ..................................................................................... VI
B. Acknowledgments .............................................................................. IX
C. Publications........................................................................................ XI
1 INTRODUCTION ................................................................ 1
1.1 Macrophage migration inhibitory factor.................................... 1
1.1.1 MIF – An Overview ...................................................................... 1
1.1.2 MIF in Inflammatory Disease ...................................................... 2
1.1.2.1 Endotoxemia and Sepsis ........................................................... 3
1.1.2.2 Rheumatoid Arthritis ............................................................... 4
1.1.2.3 Atherosclerosis .......................................................................... 4
1.1.2.4 Asthma ...................................................................................... 5
1.1.3 Molecular Mechanisms of MIF Action ......................................... 6
1.1.3.1 ERK 1/2 Activation ................................................................... 6
1.1.3.2 MIF Inhibits JAB1 Activity ...................................................... 7
1.1.3.3 MIF in Apoptosis ....................................................................... 7
1.1.3.4 MIF Upregulates TLR4 Expression ......................................... 8
1.1.3.5 MIF Receptors ........................................................................... 8
1.1.4 Structure, Enzymatic Activities and Inhibitors .......................... 9
1.1.4.1 Structure ................................................................................... 9
1.1.4.2 Enzymatic Activities ............................................................... 10
1.1.4.3 Small Molecule Inhibitors ...................................................... 11
1.1.5 The Structural Homolog D-Dopachrome Tautomerase ............ 12
1.2 Classical Secretion in Eukaryotes.............................................. 14
1.2.1 ER/Golgi Transport and the Golgi Apparatus ........................... 15
1.2.1.1 ER to Golgi Transport ............................................................. 15
1.2.1.2 The Golgi Apparatus ............................................................... 16
1.2.2 The Tethering Protein p115 ....................................................... 17
1.3 Non-Classical Secretion ................................................................ 19
1.3.1 Vesicle-Independent Translocation ........................................... 20
1.3.1.1 Membrane Blebbing/Exovesicles ............................................ 20
1.3.1.2 Plasma Membrane-Resident Transporters ............................ 20
1.3.2 Vesicle-dependent secretion ....................................................... 21
1.3.2.1 Lysosomal Secretion ............................................................... 21
1.3.2.2 Exosomes/Multivesicular Bodies ............................................ 22
1.3.3 Role of ABC transporters in unconventional secretion ............. 23
1.3.4 Unconventional Secretion of MIF .............................................. 24
2 MATERIAL AND METHODS .......................................... 25
2.1 Cell lines, Bacteria and Plasmids ............................................... 25
2.1.1 Cell lines ..................................................................................... 25

III
2.1.2 Bacteria ....................................................................................... 25
2.1.3 Plasmids ...................................................................................... 25
2.2 Equipment, Consumables and Chemicals ................................ 26
2.2.1 Equipment .................................................................................. 26
2.2.2 Consumables ............................................................................... 26
2.2.3 Chemicals .................................................................................... 27
2.2.4 Multi-Component Systems ......................................................... 28
2.3 Primary and Secondary Antibodies ........................................... 29
2.3.1 Primary Antibodies .................................................................... 29
2.3.2 Secondary Antibodies ................................................................. 29
2.4 RNAi-targeting Sequences ........................................................... 30
2.5 Quantitative PCR Primers ........................................................... 30
2.6 Media, Buffer and Solutions ........................................................ 30
2.6.1 Media ........................................................................................... 30
2.6.2 Buffers and Solutions ................................................................. 31
2.6.2.1 General Buffers ....................................................................... 31
2.6.2.2 Flow Cytometry ....................................................................... 31
2.6.2.3 Human MIF ELISA ................................................................ 32
2.6.2.4 Murine MIF ELISA ................................................................. 32
2.6.2.5 Murine DDT ELISA ................................................................ 33
2.6.2.6 Ni2+-Column Purification ........................................................ 33
2.6.2.7 MIF Purification ..................................................................... 34
2.6.2.8 DDT Purification ..................................................................... 34
2.6.2.9 SDS-PAGE and Western Blot ................................................. 35
2.6.2.10 Coomassie Staining ............................................................. 35
2.6.2.11 Immunofluorescent Staining .............................................. 35
2.7 Molecular Biology Techniques .................................................... 36
2.7.1 Measurement of DNA/RNA Concentration ............................... 36
2.7.2 Polymerase Chain Reaction (PCR) ............................................ 36
2.7.3 Enzymatic DNA Digestion ......................................................... 37
2.7.4 Agarose Gel Electrophoresis ...................................................... 37
2.7.5 Isolation and Purification of DNA from Agarose Gels .............. 37
2.7.6 DNA Ligation .............................................................................. 37
2.7.7 Heat shock Transformation of E. coli ........................................ 37
2.7.8 Plasmid Isolation ........................................................................ 38
2.7.9 Isolation of RNA and cDNA Synthesis ...................................... 38
2.7.10 Quantitative PCR ....................................................................... 38
2.7.11 Design and Production of short hairpin RNA ........................... 38
2.7.12 Preparation of Adenoviruses ...................................................... 39
2.8 Cell Culture Techniques ............................................................... 39
2.8.1 Cultivation and Treatment ........................................................ 39
2.8.2 Isolation of Macrophages from Blood......................................... 40

IV
2.8.3 Transient Transfection ............................................................... 40
2.8.4 Preparation of Cell Lysates ........................................................ 41
2.8.5 Immunofluorescent staining for microscopy ............................. 41
2.8.6 Fluorescence-based Quantification of FGF Export ................... 41
2.8.7 Chlamydia trachomatis (L1) Infection ...................................... 42
2.9 Proteinchemistry and Immunology Techniques .................... 42
2.9.1 Determination of Protein Concentration ................................... 42
2.9.2 SDS-PAGE .................................................................................. 42
2.9.3 Coomassie Staining .................................................................... 43
2.9.4 Western Blot ............................................................................... 43
2.9.5 Sandwich ELISA ........................................................................ 44
2.9.5.1 Human MIF ELISA ................................................................ 44
2.9.5.2 Murine MIF ELISA ................................................................. 44
2.9.5.3 Murine DDT ELISA ................................................................ 44
2.9.5.4 TNF-α, IL-1β and IL-6 ELISA ................................................ 45
2.9.6 Protein Purification .................................................................... 45
2.9.6.1 Overexpression of Proteins ..................................................... 45
2.9.6.2 Purification of Histidine-tagged Proteins .............................. 45
2.9.6.3 Purification of MIF ................................................................. 46
2.9.6.4 Purification of DDT ................................................................. 46
2.9.7 In vitro MIF/p115 Binding Studies ............................................ 46
2.10 In vivo Mouse Experiments .......................................................... 47
2.10.1 LPS Shock ................................................................................... 47
2.10.1.1 Serum Collection ................................................................. 47
2.10.2 Isolation of Bone Marrow-Derived Macrophages ...................... 48
3 RESULTS ........................................................................... 49
3.1 p115 Mediates the Secretion of MIF........................................... 49
3.1.1 p115 is a Novel Interaction Partner of MIF .............................. 49
3.1.2 The MIF/p115 Interaction is Direct and Specific ...................... 50
3.1.3 MIF Release from Human Monocytes ....................................... 51
3.1.4 Macrophage Activation Leads to a Redistribution of MIF ....... 53
3.1.5 p115 is Required for MIF Release in Monocytes ....................... 54
3.1.5.1 RNAi-Mediated Depletion of p115 ......................................... 54
3.1.5.2 Depletion of p115 Inhibits the Release of MIF ...................... 55
3.1.6 FGF-2 and FGF-4 Export in p115-Depleted HeLa cells ........... 57
3.1.7 Role of p115 in MIF Secretion in Macrophages ........................ 58
3.1.7.1 MIF and p115 are Co-Secreted in Activated Macrophages ... 58
3.1.7.2 Adenovirus-Mediated Depletion of p115 ................................ 62
3.1.7.3 p115 is Required for the Secretion of MIF in Macrophages .. 64
3.1.8 p115 Mediates the Release of MIF from Infected Cells ............ 67
3.1.9 Small Molecule Inhibitor Blocks MIF Secretion ....................... 70
3.1.9.1 Immunorecognition of 4-IPP-Modified MIF ........................... 70
3.1.9.2 4-IPP Targets the MIF/p115 Interaction ............................... 71
3.1.9.3 Effect of 4-IPP in LPS-Shock Model ....................................... 74

V
3.2 D-Dopachrome Tautomerase (DDT) ........................................... 76
3.2.1 Purification of D-Dopachrome Tautomerase ............................. 76
3.2.1.1 Purification of Histidine-tagged DDT .................................... 76
3.2.1.2 Purification of Native DDT Protein ....................................... 77
3.2.2 Establishment of a DDT-ELISA ................................................ 79
3.2.3 DDT is released from macrophages ........................................... 81
3.2.4 Effect of mif-gene deletion on DDT production ......................... 82
4 DISCUSSION ..................................................................... 84
4.1 Role of p115 in MIF Secretion ..................................................... 84
4.1.1 MIF is Released from a Pre-Formed Pool .................................. 85
4.1.2 MIF and p115 Associate at the Perinuclear Site ...................... 85
4.1.3 MIF and p115 Interact Directly ................................................. 86
4.1.4 p115 Is Required for the Stimulated Release of MIF ............... 87
4.1.4.1 p115 Overexpression does not Affect MIF Secretion ............. 89
4.1.4.2 MIF and p115 are Co-Secreted ............................................... 89
4.1.5 4-IPP Inhibits the Release of MIF and p115 ............................. 90
4.2 The Structural Homologue DDT ................................................. 92
5 SUMMARY AND OUTLOOK ........................................... 94
6 REFERENCES ................................................................... 97

VI
A. Abbreviations
Amino acids are abbreviated in the three letter code.
4-IPP 4-iodo-6-phenylpyrimidine
4-OT 4-oxalocrotonate tautomerase
ABC ATP binding cassette
ATP Adenosine triphosphate
CC Coiled-coiled
CD74 major histocompatibility complex, class II
invariant chain (Ii)
CHMI 5-carboxymethyl-2-hydroxymuconate
isomerase
CLP Cegal ligation and puncture
COP Coat protein complex
CSN5 Subunit 5 of the COP9 signalosome
Ct Chlamydia trachomatis
CXXC Cys-Xaa-Xaa-Cys
DDT D-Dopachrome tautomerase
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
DTT Dithiothreitol
E. coli Escherichia coli
EDTA Ethylenediaminetetraacetic acid
EGTA Ethylene glycol tetraacetic acid
ELISA Enzyme-linked immunosorbent assay
ER Endoplasmatic reticulum
ERK Extracellular signal-regulated kinase
FCS Fetal calf serum
FGF Fibroblast growth factor
GAPDH Glyceraldehyde 3-phosphate dehydrogenase
GFP Green fluorescent protein
HASPB Hydrophilic acylated surface protein B

VII
HSP Heat shock protein
HSPG Heparan sulfate proteoglycan
IB Immunoblot
IFN Interferon
IgG Immune globulin G
IL Interleukin
IP Immunoprecipitation
IPTG Isopropyl β-D-thiogalactopyranoside
ISO-1 (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-
isoxazole acetic acid methyl ester
JAB1 c-JUN activation domain-binding protein 1
JNK c-JUN N-terminal kinase
Kd Dissociation constant
kd kilo-Dalton (1 kDa = 1.6605 • 10-21 g)
LPS Lipopolysaccharide
MDR Multi-drug resistance
MIF Macrophage migration inhibitory factor
MVB Multivesicular body
NAPQI N-acetyl-p-benzoquinone imine
NMR Nuclear magnetic resonance
OD Optical density
PAGE Polyacrylamide gel electrophoresis
PBS Phosphate-buffered saline
PCR Polymerase chain reaction
PDB Protein data bank
qPCR quantitative PCR
PI(4,5)P2 Phosphatidylinositol 4,5 bisphosphate
RA Rheumatoid arthritis
RNA Ribonucleic acid
RNAi RNA interference
RT Room temperature

VIII
SDS Sodium dodecyl sulfate
siRNA short interfering RNA
SNARE soluble NSF attachment receptor
SOD1 Cu, Zn superoxide dismutase
shRNA short hairpin RNA
TBS Tris-buffered saline
tER transitional endoplasmatic reticulum
TH T helper
TLR Toll-like receptor
TMB 3,3’,5,5’-Tetramethylbenzidine
TNF Tumor necrosis factor
TPOR Thiol protein oxidoreductase
Tris 2-amino-2-hydroxymethylpropane-1,3-diol
VTC Vesicular tubular cluster

IX
B. Acknowledgments
In the course of my dissertation, I was very fortunate to have the guidance
of Prof. Jürgen Bernhagen of the Department of Biochemistry and
Molecular Cell Biology at the RWTH Aachen University Hospital and Prof.
Rick Bucala of the Department of Internal Medicine at the Yale University
School of Medicine.
I want to thank Prof. Jürgen Bernhagen for providing the topic of my
thesis and making it possible for me to spend major parts of my thesis
abroad at Yale in the collaborative lab of Prof. Bucala. His scientific advice
and support were invaluable for the successful completion of my thesis.
I thank Prof. Rick Bucala for his continuous willingness to always discuss
the progress of my project and to help me not to lose track of the bigger
picture.
I thank Prof. Baumgartner for reviewing my thesis and accepting the Co-
Referat; and Prof. Rink for his participation in my committee.
I thank Prof. Walter Nickel and Antje Ebert of the Biochemistry Center in
Heidelberg for their help with the FGF studies, and Prof. Paula Kavathas
and Dr. Seung-Joon Lee from Yale University for their support with the C.
trachomatis experiments.
My thanks to all my colleagues in the Bernhagen and Bucala labs for their
support; especially to Lin Leng and Hongqi Lue for their unceasing advice
in lab techniques, Juan Fan for her patience while training me in mouse
experiments, and my bench-mates Birgitt Lennartz, Jason Griffith, Utako
Kaneyuki, Tiffany Sun and Tarah Connolly who were always helpful with
discussions.

X
I thank my fiancé Swen Zierow, for his help with the purification of
proteins, and his emotional support throughout my studies. I’m fortunate
to have you in my life.
I thank my family for their constant encouragement throughout my
academic studies and my life.
Lastly, I thank the Studienstiftung des deutschen Volkes for their generous
financial support of my thesis work.

XI
C. Publications
Parts of my thesis were published in international peer-reviewed journals:
1) D. Kamir, S. Zierow, L. Leng, Y. Cho, Y. Diaz, J. Griffith, C. McDonald,
M. Merk, R.A. Mitchell, J. Trent, Y. Chen, Y.K. Kwong, H. Xiong, J.
Vermeire, M. Cappello, D. McMahon-Pratt, J. Walker, J. Bernhagen, E.
Lolis, and R. Bucala. 2008. A leishmania ortholog of macrophage
migration inhibitory factor modulates host macrophage responses. J
Immunol 180:8250-8261.
2) M. Merk, J. Baugh, S. Zierow, L. Leng, U. Pal, S. Lee, A. Ebert, Y.
Mizue, J. Trent, R. Mitchell, W. Nickel, P. Kavathas, J. Bernhagen, and R.
Bucala. 2008. The Golgi-associated Protein p115 Mediates the Secretion of
Macrophage Migration Inhibitory Factor (MIF). Mol Biol Cell (in revision)

Introduction – 1
1 Introduction
1.1 Macrophage migration inhibitory factor
1.1.1 MIF – An Overview
While developing an assay to measure the migration of peritoneal cells in
capillary tubes, macrophage migration inhibitory factor (MIF) was
discovered in 1962 (1). The first function contributed to MIF was its
inhibitory action on the random migration of macrophages (2), and in the
following years, MIF was shown to activate macrophages leading to their
increased cell surface adhesion and phagocytosis (3). The molecular
mechanisms underlying MIF’s functions nevertheless remained unknown.
In 1989, a human MIF cDNA was first isolated and cloned, and the
expressed polypeptide had the expected molecular weight of 12.5 kDa (4).
Bernhagen et al. first cloned and purified murine MIF as a protein
released from anterior pituitary cells in response to bacterial
lipopolysaccharide (LPS) (5, 6). Soon macrophages were also described as a
major source of MIF; they were observed to release MIF after stimulation
with pro-inflammatory stimuli such as LPS, tumor necrosis factor (TNF) α
and interferon (IFN) γ, and even after stimulation with low concentrations
of anti-inflammatory glucocorticoids (7, 8). In 1999, a MIF knockout mouse
was constructed, showing that the deletion of the mif gene reduces
inflammatory responses (9). The discovery of the MIF cell surface receptor
CD74 (10), and later of the chemokine receptors CXCR2 and CXCR4 (11),
helped to improve the understanding of the molecular mechanism of MIF,
and reports about an association of high expression MIF alleles and
severity of inflammatory diseases further emphasized its role in disease
pathology (12-14).

Introduction – 2
1.1.2 MIF in Inflammatory Disease
Inflammation is the characteristic immune response of tissue to harmful
stimuli. It is a protective attempt to remove the injurious stimulus and
start the healing process. Causes for inflammation can be of physical
(mechanical, thermal, radiation), chemical (toxins, acids, allergens) or
biological (bacteria, viruses, parasites, fungi) nature.
There are two types of inflammation: acute and chronic. Acute
inflammation displays the five classic signs of inflammation: swelling,
redness, pain, heat, and loss of function. In contrast, chronic inflammation
does not show these symptoms, but is characterized by concurrent
inflammation, tissue destruction, and attempts at repair (15). MIF is an
important mediator in both acute and chronic inflammation. It was shown
to be a key mediator in the pathogenesis of endotoxemia/sepsis, arthritis,
glomerulonephritis, atherosclerosis, inflammatory bowel disease and
several other inflammatory and immune conditions (16).
MIF is a ubiquitously expressed protein that is most notably present in
tissue that is in direct contact with the host’s environment including
endothelial cells, and fibroblasts, the gastrointestinal tract, pituitary
glands, kidney, liver, lung, pancreas, skin and eyes. Immune cells are the
major source of circulating MIF, with monocytes/macrophages as the main
contributor, followed by T cells and dendritic cell. B cells, neutrophils,
eosinophils, basophils, and mast cells also release MIF (17).
The release of MIF is a common occurrence in inflammatory diseases, for
example the level of MIF is 5 – 10 times higher in serum of patients with
rheumatoid arthritis compared to a healthy control group (18). Table 1 lists
known stimuli causing the release of MIF.
Table 1: Stimuli responsible for MIF release. Table adapted from (17).
Stimulus Cell type
Endotoxin Monocytes/macrophages,
neutrophils, dendritic cells
Exotoxin Macrophages, splenocytes
Peptidoglycan PBMCs
Heat killed E. coli PBMCs

Introduction – 3
Heat killed S. aureus PBMCs
M. tuberculosis Macrophages
Influenza virus Pulmonary epithelial cells
TNF-α, INFγ Macrophages
C5a Eosinophils, neutrophils
IL-5 Eosinophils
IL-9 Mast cells
Nitric oxide Fetal membranes
Anti-CD3 antibodies T cells
Low-dose glucocorticoids Monocytes/macrophages, T cells
1.1.2.1 Endotoxemia and Sepsis
MIF is rapidly released by specialized pathway from pre-formed pools in
immune and pituitary cells after stimulation with LPS, a virulence factor
in the cell wall of gram-negative bacteria (5, 8, 19).
In a murine model of endotoxemia, MIF was rapidly released after
intraperitoneal injection of LPS, and peaked 8 – 20 h after stimulation.
The concurrent administration of recombinant MIF protein and LPS
decreased the survival rate compared to the control group (5). Moreover,
neutralizing MIF via antibodies, the deletion of the mif gene, or the
administration of small molecule MIF inhibitor prior to LPS challenge
improved the chances for survival significantly (5, 9, 20). These results are
in accord with models of E. coli injection in the peritoneum or cecal
ligation and puncture (CLP). Peritonitis leads to an increased MIF level in
the serum and treatment with anti-MIF antibodies has a protective effect
(21).
Of note, if mice with sublethal peritonitis had a secondary infection with
S. typhimurium, P. aeruginosa, or L. monocytogenes 48 h after the first
infection, the administration of recombinant MIF had a protective effect,
whereas applying anti-MIF antibody further enhanced the negative effect
of bacterial superinfection. This indicates that MIF has an important
regulatory role in the inflammatory response, with likely a positive
function in immune suppressed animals by reenabling the organism to
react adequately to a secondary infection (22).

Introduction – 4
1.1.2.2 Rheumatoid Arthritis
MIF is closely associated with the development of rheumatoid arthritis
(RA) (16). RA is the most common inflammatory disease of the joints, it is
estimated that 1% of adults in the U.S. and Europe suffer from RA. Key
pathologic characteristics of RA are a marked expansion of the synovium
by infiltrating leukocytes, the activation of apoptosis of resident cells, and
the participation of cytokines such as TNF-α and IL-1β. MIF is a key
regulator for the migration of leukocytes into the joint in RA (23).
In 1997, the role of MIF in inflammatory arthritis was reported for the
first time (24). In a experimental model of collagen type-II induced
arthritis the treatment with neutralizing anti-MIF antibodies before
immunization with collagen led to a delayed onset of RA, lowered the
frequency of arthritis, and reduced the levels of IgG2a. Treatment with
neutralizing anti-MIF antibody also showed protective effects in the model
of adjuvant-induced arthritis and antigen-induced arthritis (25, 26).
Recent studies employing mif-/- mice confirmed the importance of MIF in
arthritis. The severity of the disease, both in collagen-induced arthritis
and antigen-induced arthritis, was less pronounced in mice with a deletion
of the mif gene (27, 28). It was shown that both Th1 and Th2 cells
expressed high quantities of MIF, and that MIF stimulates the expression
of MMP (matrix metalloproteinase) in FLS (29).
In RA patients, MIF has been detected in synovial fluid, macrophages,
endothelial cells, fibroblast-like synoviocytes (FLS), and CD3+ synovial
lymphoid aggregates (30, 31), and MIF production is associated with
disease activity.
1.1.2.3 Atherosclerosis
Atherosclerosis is characterized by the growth of atherosclerotic plaques in
the vascular wall over the period of many years, leading to hardening and
constriction of arteries. Inflammation and accumulation of large numbers
of white blood cells (monocyte-derived macrophages, macrophage-derived

Introduction – 5
foam cells, to a lesser degree Th1 cells) is now regarded as a crucial force in
the initiation and progression of atherosclerotic lesion formation (32). Both
cytokines (TNF-α, GM-CSF, IL-12) and chemokines (CXCL8, MCP1) are
pivotal for the recruitment of monocytes and the activation of intimal
macrophages/foam cells.
Recent studies indicate an important role for MIF in atherogenesis,
atheroma formation and vascular disease. In 2002, the strong
overexpression of MIF in human atheroma lesions was reported for the
first time. It was also reported that the increased expression of MIF is
accompanied by an increase in disease progression. MIF protein was
upregulated in HUVEC cells when the cells were incubated with oxidized
LDL. Furthermore, stimulation of macrophages with oxidized LDL led to
an enhanced secretion of MIF (33). Importantly, through interaction with
the chemokine receptors CXCR2 and CXCR4, MIF is instrumental in
inflammatory leukocyte recruitment in atherosclerosis, targeting
monocytes and neutrophils through CXCR2 and T cells through CXCR4
(11). MIF is also causally linked to the development of atherosclerosis
lesions. Mice lacking the low-density lipoprotein receptor (ldlr) are prone
to develop atheroma. In comparison, mice lacking both ldlr and mif exhibit
a reduction in atheroma lesions (34). Furthermore, it was reported that
the neutralization of MIF in apoE-/- (apolipoprotein E) mice results in a
marked reduction in inflammatory responses associated with
atherosclerosis (35). It is also proposed that MIF serves as a link between
rheumatoid arthritis and atherosclerosis (36).
1.1.2.4 Asthma
Asthma is a chronic, inflammatory disease of the respiratory system. In
response to allergens such as tobacco smoke or air pollution, the airways
constrict, become inflamed and produce excessive amounts of mucus.
Studies in both mice and humans have supported a role of T helper 2 (Th2)
cells in asthma which mediate allergic inflammation by producing

Introduction – 6
cytokines leading to the production of IgE, and the differentiation,
proliferation and activation of eosinophils (37).
Rossi and colleagues showed that human eosinophils stimulated with C5a
or IL-5 release high amounts of MIF in vitro. Furthermore the
bronchoalveolar lavage fluid obtained from asthmatic patients contains
significantly elevated levels of MIF as compared to nonatopic normal
volunteers (38). In 2005, genetic studies observing the prevalence of
different MIF polymorphisms in asthma patients were performed. The
MIF gene has a functional promoter polymorphism consisting of a
tetranucleotide sequence, CATT, which is repeated five to eight times. A
high number of repeats results in an increase in MIF promoter activity
(39). While there was no association between particular alleles and
asthma incidence, a significant association between a mild form of asthma
and low-expression, 5-CATT MIF allele, was observed. Furthermore the
study demonstrated that mif-deficient mice show less pulmonary
inflammation and lower airway hyperresponsiveness than wildtype mice.
This was in accord with the finding that mif-/- mice have lower titers of
IgE and TH2 cytokines (13).
1.1.3 Molecular Mechanisms of MIF Action
1.1.3.1 ERK 1/2 Activation
Different stimuli lead either to a transient (rapid, within minutes) or
sustained (prolonged, for hours) activation of extracellular signal-
regulated (ERK) mitogen-activated protein kinases. MIF promotes both
the transient and the sustained ERK 1/2 signaling pathway (40, 41).
The sustained phase of ERK activation was shown to occur through
autocrine and paracrine stimulation and depends on protein kinase A (41,
42). It has been established that the sustained ERK 1/2 activation is
associated with an activation of cytosolic phospholipase A2, overriding of
glucocorticoids and a promotion of cell proliferation.

Introduction – 7
Lue et al demonstrated that the stimulation of fibroblasts with MIF leads
rapidly to a transient activation of the ERK 1/2 signaling pathway. They
also established that transient ERK phosphorylation by MIF is dependent
on a Src-kinase and leads to the phosphorylation and activation of the
transcription factor Elk.
1.1.3.2 MIF Inhibits JAB1 Activity
In a yeast two-hybrid screen JAB1 (c-JUN-activation domain-binding
protein-1) was identified as an intracellular binding partner of MIF (43).
JAB1, also known as CSN5 (subunit 5 of the COP9 signalosome),
regulates the degradation of p27Kip1 and activation of JNK (c-JUN N-
terminal kinase) and is a co-activator of AP-1, a transcription factor
implicated in cell growth, transformation and cell death. MIF antagonizes
these effects via the inhibition of the JAB1-dependent AP-1 transcription.
Furthermore, MIF stabilizes the cell cycle inhibitor p27Kip1 by inhibiting
the JAB1-mediated degradation of p27Kip1.
Recently, it was reported that JAB1 traps MIF inside cells in order to
prevent MIF secretion. The depletion of JAB1 protein led to an increased
secretion of MIF (44), whereas the overexpression of JAB1 led to an
inhibition of sustained ERK activation (40). Although there is no evidence
for an active participation of JAB1 in the secretory pathway of MIF, JAB1
nevertheless seems to have the ability to block MIF secretion and its
autocrine and paracrine functions.
1.1.3.3 MIF in Apoptosis
p53 has a central role in the regulation of the cell cycle. In a screen to
identify proteins which by-pass p53-dependent cell cycle control, MIF was
identified as a negative regulator of p53 activity (45). MIF sustains
macrophage survival by suppressing activation-induced, p53-dependent
apoptosis (46). Mitchell and colleagues reported that LPS-challenge in mif-
deficient mice results in decreased macrophage viability, decreased

Introduction – 8
proinflammatory function, and increased apoptosis when compared to
wild-type macrophages. In 2008, it was reported that MIF and p53
interact physically, and that cysteine-81 in MIF is of critical importance
for the physical and functional interaction (47). It is important to note that
in comparable studies a direct, physical interaction between MIF and p53
could not be detected (personal communication, J. Bernhagen).
1.1.3.4 MIF Upregulates TLR4 Expression
MIF has been implicated in the host response to LPS challenge in
numerous studies. Toll like receptor (TLR) 4 forms a receptor complex
with LPS binding protein and CD14, with TLR4 being the essential
signaling component when LPS is bound to the receptor complex. It was
demonstrated that mif-/- macrophages have a reduced expression level of
TLR4, but not of LBP or CD14. The downregulation of TLR4 in mif-
deficient mice leads to a hyporesponsiveness towards LPS resulting in a
reduced production of TNF-α, IL-1β, IL-6 and IL-12. (48).
1.1.3.5 MIF Receptors
Cytokines bind to their cell surface receptors and trigger an intracellular
signaling cascade. CD74 was discovered as the cell surface receptor of MIF
in 2003 (10). Surface plasmon resonance experiments between MIF and
soluble CD7473-232 revealed an equilibrium Kd of ~9 nM. Leng et al
reported that the MIF-dependent phosphorylation of ERK 1/2, the
production of prostaglandin E2 and cell proliferation requires the cell
surface expression of CD74. CD74 does not possess any obvious signal
transduction domains; therefore, the molecular mechanism of signaling
remained unclear for some time. Recent data showed that MIF’s binding
to CD74 leads to the phosphorylation of CD74 at the intracytoplasmic
domain in a CD44-dependent manner (49). The co-receptor CD44 was
demonstrated to be the missing link of MIF/CD74-mediated sustained
ERK activation via Src-kinases.

Introduction – 9
In 2007, the chemokine receptors CXCR2 and CXCR4 were discovered as
non-cognate receptors for MIF in MIF-mediated migratory and
atherogenic functions (11). MIF competed with cognate ligands for CXCR2
and CXCR4 binding, and directly binds to CXCR2 and CXCR4 with a Kd of
1.4 and 20 nM, respectively. Monocyte arrest induced by MIF involved
both CXCR2 and CD74. Furthermore, it was described that CD74 and
CXCR2 co-localize, suggesting a MIF signaling pathway via a
CD74/CXCR2 complex.
1.1.4 Structure, Enzymatic Activities and Inhibitors
1.1.4.1 Structure
The MIF protein consists of 114 amino acids. In 1996, the structure of MIF
was solved independently by crystallography (50, 51) and NMR (52). The
secondary structure of the MIF monomer consists of two α-helices and six
β-strands, four of these strands form a β-sheet (Fig. 1).
The structural analysis by crystallography revealed MIF to be a trimer,
stabilized by hydrogen bonds and a hydrophobic core. The three
dimensional structure of MIF possesses a strong homology to D-dopa-
chrome tautomerase (DDT) and the bacterial enzymes 5-carbodymethyl-2-
hydroxymuconate isomerase (CHMI) and 4-oxalocrotonate tautomerase (4-
OT) (53). These proteins are members of the MIF/tautomerase super-
family.
Although crystallographic analysis revealed MIF to be a trimer, other
studies, such as NMR and size exclusion chromatography, support the
idea that MIF forms a monomer or dimer (52, 54). The question of the
physiological state of MIF is still not answered to satisfaction.
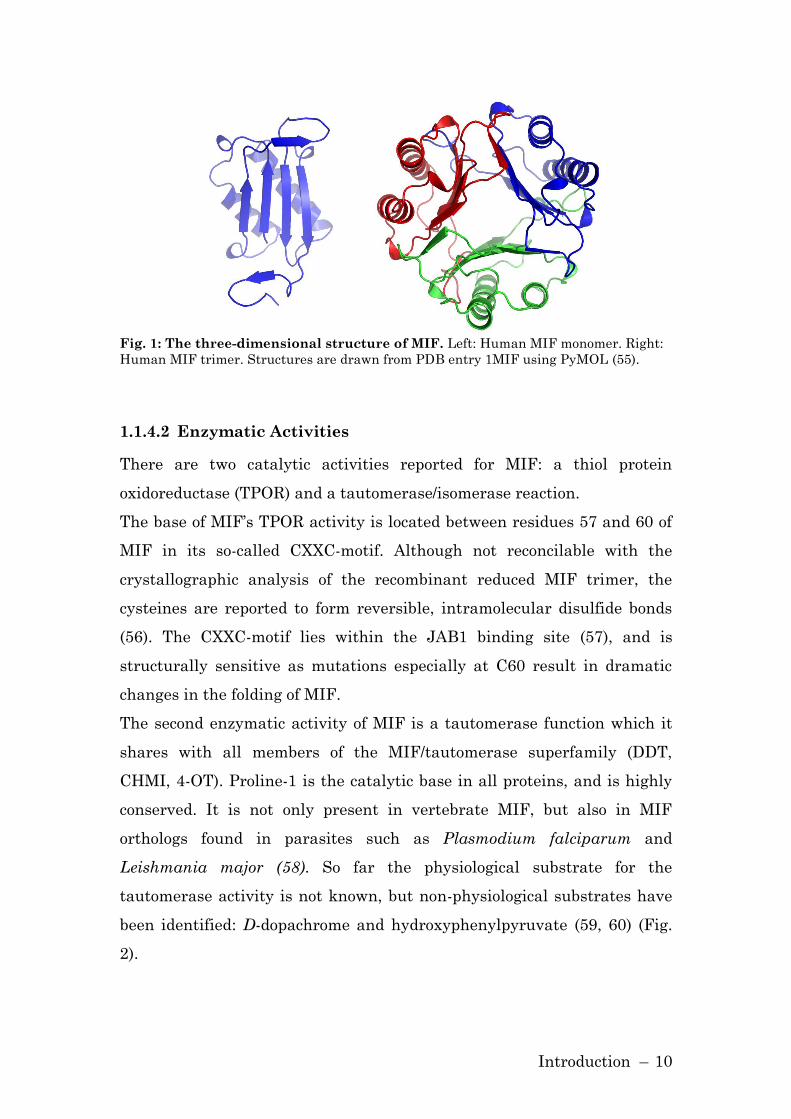
Introduction – 10
Fig. 1: The three-dimensional structure of MIF. Left: Human MIF monomer. Right:
Human MIF trimer. Structures are drawn from PDB entry 1MIF using PyMOL (55).
1.1.4.2 Enzymatic Activities
There are two catalytic activities reported for MIF: a thiol protein
oxidoreductase (TPOR) and a tautomerase/isomerase reaction.
The base of MIF’s TPOR activity is located between residues 57 and 60 of
MIF in its so-called CXXC-motif. Although not reconcilable with the
crystallographic analysis of the recombinant reduced MIF trimer, the
cysteines are reported to form reversible, intramolecular disulfide bonds
(56). The CXXC-motif lies within the JAB1 binding site (57), and is
structurally sensitive as mutations especially at C60 result in dramatic
changes in the folding of MIF.
The second enzymatic activity of MIF is a tautomerase function which it
shares with all members of the MIF/tautomerase superfamily (DDT,
CHMI, 4-OT). Proline-1 is the catalytic base in all proteins, and is highly
conserved. It is not only present in vertebrate MIF, but also in MIF
orthologs found in parasites such as Plasmodium falciparum and
Leishmania major (58). So far the physiological substrate for the
tautomerase activity is not known, but non-physiological substrates have
been identified: D-dopachrome and hydroxyphenylpyruvate (59, 60) (Fig.
2).

Introduction – 11
Fig. 2: The tautomerase reaction of MIF. A) MIF catalyzes D-Dopachrome to 5,6-
dihydroxyindole-2-carboxylic acid. B) Keto-enol isomerization of both p-hydroxyphenyl-
pyruvate and phenylpyruvate
1.1.4.3 Small Molecule Inhibitors
The active site of MIF around proline-1 was reported to be of importance
for the pro-inflammatory actions of MIF. Mutational analysis revealed
proline-1 to be involved in the counter-regulation of glucocorticoids,
neutrophil priming and the upregulation of metalloproteinase-1 and 3 in
rheumatoid arthritis synovial fibroblast (61-63). Prototypic small molecule
inhibitors of MIF have been described that bind to the protein’s amino-
terminal tautomerase region. Prominent inhibitors are N-acetyl-p-
benzoquinone imine (NAPQI), (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-
isoxazole acetic acid methyl ester (ISO-1), and 4-iodo-6-phenylpyrimidine
(4-IPP) (61, 64, 65) (Fig. 3).
ISO-1 binds reversibly to MIF and does not only inhibit the tautomerase
activity of MIF, but also inhibits MIF’s ability to counter-regulate
glucocorticoids (61), and protects mice from endotoxic shock (20). NAPQI
binds covalently to MIF and the modification of MIF with NAPQI results
in an inhibition of its tautomerase activity, decreased cell binding ability
and a decreased immunorecognition (64). The inhibitor 4-IPP was found
via computational virtual screening and also binds covalently to the amino
acid proline-1. In contrast to NAPQI, which only bound to one third of MIF
molecules, almost all MIF molecules were modified by 4-IPP. This is

Introduction – 12
reflected in the IC50 determined by D-dopachrome tautomerase assay; ISO-
1 has an IC50 of ~50 µM, NAPQI of ~40 µM, and 4-IPP of ~5 µM.
Functionally, 4-IPP was reported to inhibit MIF-dependent cell motility
and growth in vitro (65).
Fig. 3: Small molecule MIF inhibitors. Left: NAPQI, Middle: ISO-1, Right: 4-IPP
1.1.5 The Structural Homolog D-Dopachrome Tautomerase
D-dopachrome and p-hydroxyphenylpyruvate serve as substrate for the
tautomerase activity of both MIF and D-dopachrome tautomerase (60, 66).
But in contrast to MIF, which converts D-dopachrome to 5,6-
dihydroxyindole-2-carboxylic acid, DDT decarboxylases D-dopachrome to
5,6-dihydroxyindole. The enzymatic activity of DDT was first described in
melanocytes (67); higher activities later were reported in human liver and
blood (68).
Murine DDT protein shares only 28% identity and 45% homology with
murine MIF. Of note, proline-1, the catalytic base of the tautomerase
activity in MIF is conserved in DDT. Furthermore, the active side residues
lysine-32 and isoleucine-64 also are present in DDT (Fig. 4). Interestingly,
DDT possesses no CXXC-motif which is associated with the TPOR activity
in MIF (56).

Introduction – 13
Fig. 4: Sequence alignment of mouse MIF with mouse D-dopachrome
tautomerase. The secondary structure of mouse MIF is shown (Pdb: 1MFI). Elements of
the secondary structures are indicated by α for α-helix and β for a β-strand of β-sheet, η
for a short α-helix and T for turns. The sequence identity of mouse DDT and mouse MIF
is 28%. Invariant residues among DDT and MIF are marked by black background; if
residues are similar they are marked by a box.
Human DDT protein was crystallized in 1999. The overall folding and
subunit topology are almost identical to human MIF, with two αβα motifs
related by a pseudo 2-fold symmetry and similar trimeric β-sheet packing
(Fig. 5) (69).
Fig. 5: Human D-dopachrome tautomerase. Left: Human DDT monomer. Right:
Human DDT trimer. Structures are drawn from PDB entry 1DPT with PyMOL (55).
To date, little is known about the biological functions and mechanisms of
action of DDT. It is highly expressed in the liver, and also found in
kidneys, brain, spleen, lung and the heart (70). In 2003, it was reported
that DDT activity is increased in UVB-induced blister fluid.
Correspondingly, the activity of MIF was also found to be increased (71). A

Introduction – 14
more recent study suggests a role for DDT in cancer. Coleman et al
reported about the individual and cooperative action of MIF and DDT in
angiogenesis showing that the depletion of MIF and/or DDT reduces the
basal secretion level of CXCL8 and VEGF from non-small cell lung
carcinoma cells. The transcriptional regulation of CXCL8 by MIF and DDT
appears to involve JNK, c-JUN and AP-1. Furthermore, they showed that
the MIF receptor CD74 is necessary both for MIF and DDT induced JNK
activation (72).
1.2 Classical Secretion in Eukaryotes
Over the last two decades, considerable progress was achieved in
improving the understanding of protein export. The process requires
multiple components including the endoplasmatic reticulum (ER), the
Golgi, and secretory vesicles.
Proteins targeted for the conventional export into the extracellular milieu
possess a signal peptide. This sequence directs the ribosome to the rough
ER membrane and initiates the transport of the growing peptide chain
across it into the lumen of the ER (73, 74).
The ER is one of the largest intracellular compartments in cells and
consists of a large network of membrane cisternae (75). Proteins are
translated into the lumen of the ER, where they either fold into their
native structure by themselves or with the aid of chaperones. The proteins
are screened for their proper folding (76), before being transported to the
Golgi in cargo vesicles (77). Misfolded proteins are degraded by the 26S
proteasome (78).
The Golgi, a highly dynamic organelle, is polarized with a cis-side facing
the ER, and a trans-side, communicating with the plasma membrane. On
their way through the Golgi, proteins are further processed mainly by
“trimming” of N-glycosylations obtained in the ER (79). Over the trans-
Golgi network, cargo proteins exit the Golgi and are transported to the

Introduction – 15
plasma membrane in vesicles. Proteins are released into the extracellular
space via a fusion of the carrier vesicles with the plasma membrane (80)
(Fig. 6).
Fig. 6: Secretory pathway. Proteins destined for export are transported in COPII-
coated vesicles from the ER to the cis-Golgi; they are leaving the Golgi at the trans-side
and are transported in vesicles to the plasma membrane. Resident ER proteins are
returned from the Golgi to the ER in COPI-coated vesicles.
1.2.1 ER/Golgi Transport and the Golgi Apparatus
1.2.1.1 ER to Golgi Transport
Proteins leave the ER not uniformly across the ER, but at distinct exit
sides, also called transitional ER (tER) sides. At these sites, no ribosomes
are docked to the ER, and an increase in budding activity is evident by
electron microscopy (81, 82). At the tER, COPII-coated vesicles bud off
carrying cargo to the Golgi.

Introduction – 16
These vesicles uncoat and fuse to form tubular clusters, termed vesicular-
tubular clusters (VTCs) (83). VTCs become part of the cis-Golgi network,
and ER-resident proteins are recycled in COPI-coated vesicles in
retrograde vesicle transport (84).
The ability of transport vesicles to fuse and form tubes is conferred by
tethering factors which ensure specific binding interaction and SNAREs
(soluble NSF attachment receptor) which facilitate the fusion of
membranes (85). Tethering proteins act like ropes between a vesicle and
target membrane. In addition to multi-component tethers such as the
exocyst, which facilitates the binding of vesicles to the plasma membrane
(86) or conserved oligomeric Golgi complex which is of importance in the
intra-Golgi transport (87), there are also filamentous tethering proteins
such as p115.
SNAREs are subdivided into v- and t-SNAREs referring to their presence
on either vesicles or target membranes. When v- and t-SNAREs are
pairing, they form a SNAREpin and fusion occurs.
It was reported that the tethering protein p115 which shares some
sequence homology with SNAREs, is required (at least in vitro) for the
initial interaction of SNAREs (88).
After cargo proteins reach the Golgi from the ER by tethering and fusion
of their carrier vesicles, proteins traverse through the Golgi.
1.2.1.2 The Golgi Apparatus
Over the last 30 years, one of the most controversial discussions in protein
export concerned the question how cargo proteins cross the Golgi and are
modified by resident Golgi proteins.
Two models are mainly discussed: On the one hand, there is the idea that
cargo arrives at the cis-side of the Golgi and then traverse through the
Golgi to the trans-side, the resident Golgi proteins stationary in their
tubular structure. On the other hand, there is the hypothesis that as
vesicles arrive from the ER, they form cis-tubular structures and then

Introduction – 17
progress to mature into trans-tubular structures. This implies that cargo
proteins remain in their distinct structures and resident Golgi proteins are
shuttled between the cisternae (89). In recent years, more and more
evidence supports this second hypothesis of cisternal maturation.
Recently, live imaging in yeast has provided direct evidence in support of
this hypothesis (90, 91).
1.2.2 The Tethering Protein p115
The transport between ER and Golgi requires the tethering and fusion of
vesicles. p115, belonging to the family of golgin proteins, is one of the best
studied tethering proteins involved in this process (92).
Golgins were originally identified as a group of proteins found in the sera
of patients with a variety of autoimmune diseases (93). Members of the
golgin family possess a coiled-coiled region forming a rod-like structure
(94). Localization studies showed that p115 predominantly localizes to the
cis-Golgi, and it was also detected on COPII-coated vesicles (95, 96).
p115 has a globular head, a extended coiled-coiled (CC) region and a short
acidic tail (Fig. 9). It forms a parallel homodimer by dimerization in the
CC region (97). Distinct domains interact with different proteins: the
acidic carboxy-terminal tail interacts with the golgin proteins GM130 (98)
and giantin (95), the CC1 domain of p115, a region with SNARE homology,
interacts with SNARE family members such as syntaxin 5 (88, 99), the
guanosine triphosphatase Rab1 mediates the recruitment of p115 by
interacting with the globular head (96).
p115, located at the cis-Golgi, tethers COPI-coated vesicles due to its
interaction with giantin and GM130. Furthermore, p115, located to
COPII-coated vesicles, tethers these vesicles in a SNARE-dependent
process to form VTCs. These VTCs then dock with the cis-Golgi facilitated
by an interaction between p115 and GM130. All tethering functions of

Introduction – 18
p115 are mediated through the interaction of p115 with Rab1 (Fig. 7)
(100).
Fig. 7: Tethering functions of p115. A) COPII-coated vesicles are tethered by p115.
Rab1 recruits p115 on these vesicles by interaction with the globular head. Tethered
vesicles fuse in a SNARE-dependent manner and form VTCs. B) Docking of VTCs to the
cis-Golgi is facilitated by the interaction of p115 and GM130. The acidic carboxy-terminal
tail of p115 binds to a basic amino-terminal region of GM130. C) Role of p115 in the
tethering of COPI-coated vesicles. Giantin, located on the COPI-coated vesicles interacts
with GM130 and p115 on the cis-side of the Golgi. Figure taken from (100).
In various studies it was demonstrated that p115 is essential for the
integrity of the Golgi. Both a depletion of p115 below detectable levels and
the injection of antibodies targeting p115 led to a fragmentation of the
Golgi (92, 101-103). It was furthermore reported that the fragmentation of
the Golgi by targeting p115 led to a decreased protein transport from the
ER to the cell surface (92).

Introduction – 19
1.3 Non-Classical Secretion
In contrast to the classical secretion of proteins where proteins are
exported via the ER/Golgi route, the pathway of non-classical secretion is
not uniform. Unconventionally secreted proteins do not possess a
hydrophobic signal peptide and therefore are not translocated into the
lumen of the ER. In contrast to classically secreted proteins, proteins
lacking a signal peptide are translated on free ribosomes. Evidence for this
heterogeneous group of proteins that is secreted from cells without
entering the ER/Golgi pathway came from the observation that leaderless
proteins are not inhibited in their secretion by brefeldin A (104-107).
Brefeldin A is an anti-fungal drug that interferes with the anterograde
protein transport from the ER to the Golgi, and causes a rapid
fragmentation of the Golgi (108, 109).
Distinct mechanisms were suggested to describe non-conventional protein
secretion including vesicular transport via either lysosomes and exosomes,
and vesicle-independent export via plasma membrane shedding and direct
translocation mediated by transporters in the plasma membrane (Fig.
8)(110).
Fig. 8: Potential export routes of non-conventionally secreted proteins. 1: Export
by secretory lysosomes; 2: Export mediated by plasma membrane-resident transporters;
3: Export through the release of exosomes derived from multivesicular bodies; 4: Export
mediated by plasma membrane shedding of microvesicles. Figure taken from (110).

Introduction – 20
1.3.1 Vesicle-Independent Translocation
1.3.1.1 Membrane Blebbing/Exovesicles
The plasma membrane is highly dynamic, and its reorganization is
controlled by small Rho GTPases. Specific Rho GTPases destabilize the
actin filaments resulting in the formation of rounded cell protrusions,
called membrane blebs (111). There are multiple, distinct types of
membrane blebs including necrotic and apoptotic blebs.
The export of the Leishmania protein HASPB (hydrophilic acylated
surface protein B) belongs to the group of unconventionally secreted
proteins employing the process of membrane blebbing (112). HASPB is
only produced in the infectious stages of the parasite life cycle, and is then
synthesized on free ribosomes. The amino-terminus of the protein becomes
both myristolylated and palmitoylated which are necessary modifications
for a membrane anchorage of the protein (113). Denny et al observed that
myristylation and palmitoylation of HASPB at its amino-terminal SH-4
domain leads to its redistribution to the plasma membrane. Both the
deletion of the complete amino-terminus and the mutation of the
myristylation site led to a redistribution of HASPB in the cytoplasm.
Mutation of the palmitoylation site led to an accumulation of HASPB at
the Golgi. In the secretion model proposed based on these observations,
HASPB is first located to the Golgi and subsequently transported to the
plasma membrane, probably using the conventional vesicular transport
system. Once anchored to the plasma membrane, the SH4-domain of
HASPB induces membrane blebbing in a Rho-dependent manner.
1.3.1.2 Plasma Membrane-Resident Transporters
Secretion facilitated by transporters residing in the plasma membrane is
another possible way to export proteins independent of the ER/Golgi
pathways. Fibroblast growth factors are heparan-binding growth factors
that are activators of tumor-induced angiogenesis. Fibroblast growth
factor (FGF) 2 is believed to be exported by direct translocation.

Introduction – 21
In an in vitro system employing inside-out vesicles it was demonstrated
that FGF-2 is translocated across the plasma membrane and that the
membrane-derived vesicles contain the molecular machinery necessary for
an efficient export of FGF-2. The process is independent of ATP or a
membrane potential; other proteins, among these MIF, were not able to
translocate into the vesicles (114). Before FGF-2 is able to translocate
across the plasma membrane, it needs to be targeted to the plasma
membrane. This likely occurs through the interaction with
phosphatidylinositol 4,5 bisphosphate (PI(4,5)P2) (115). It was
demonstrated that the downregulation of PI(4,5)P2 leads to a decreased
export of FGF-2. So far, the actual transporter of FGF-2 in the plasma
membrane has not been elucidated, but it is believed that heparan sulfate
proteoglycans (HSPGs) probably serve as an extracellular trap to ensure
the transport of FGF-2 into the extracellular space (116).
1.3.2 Vesicle-dependent secretion
1.3.2.1 Lysosomal Secretion
Secretory lysosomes share many features with conventional lysosomes.
But in contrast to conventional lysosomes which are multi-vesicular in
structure, secretory lysosomes have a more diverse structure. Both
secretory and conventional lysosomes contain classical markers such as
LAMP-1 and Hsc70, but secretory lysosomes additionally contain a specific
cell-type specific set of components (117).
Among the proteins reported to be secreted via lysosomes are interleukin-
1 (IL-1) β, heat shock protein (HSP) 70 and HMGB1 (118-120).
Andrei et al demonstrated that IL-1β is co-fractioning with endolysosomes
and that an inhibition of endolysosomal proteases leads to an increased
IL-1β secretion. Via immunolocalization they revealed that IL-1β-positive
vesicles also stain for the lysosomal marker LAMP-1 (118). Upon
stimulation of cells, it is believed that these vesicles undergo fusion with

Introduction – 22
the plasma membrane and release IL-1β into the extracellular milieu
(121). Similarly HSP70, a protein released from prostate carcinoma cells
after heat shock, appears in the extracellular space at the same time that
LAMP-1 appears on the cell surface, suggesting that lysosomes facilitate
the unconventional secretion of HSP70.
1.3.2.2 Exosomes/Multivesicular Bodies
Exosomes represent another vesicle-based mechanism for export of
proteins independent of the ER/Golgi pathway. Exosomes are a distinct set
of membrane vesicles homogenous in form and size (122, 123). Exosomes
were first identified as a mechanism for externalization of obsolete
membrane proteins during reticulocyte maturation (124).
The characterization of exosomes is based on morphology and biochemical
criteria. The diameter of exosomes is between 50 and 90 nm, and they are
released when multivesicular bodies (MBVs) fuse with the plasma
membrane. Many cell types are known to release exosomes including
hematopoietic cells, reticulocytes, B- and T-lymphocytes, dendritic cells
and tumor cells (125).
It was recently reported that the unconventional secretion of Cu, Zn
superoxide dismutase (SOD1) is facilitated by exosomes (126). SOD1 was
detected on isolated exosomes, but not on membrane blebs. In this study,
the authors furthermore found p115 both on exosomes and membrane
blebs.
Another role for exosomes in the process of non-classical protein secretion
is the export of the heat shock protein 90α (HSP90α) (127). In human
keratinocytes, transforming growth factor α stimulates the rapid secretion
of HSP90α via an exosomal secretion pathway. The inhibitor dimethyl
amiloride inhibited the secretion of HSP90α in a concentration-dependent
manner by blocking exosome-mediated protein secretion, while Brefeldin
A did not show any inhibitory effects.

Introduction – 23
A further complication in the study of unconventional protein secretion
comes from the fact that for some unconventionally secreted proteins
several potential secretory pathways were described for one protein.
Potential secretory pathways that have been described for interleukin-1
(IL-1β) for instance, include cell surface blebbing and the formation of
microvesicles that lyse in the extracellular space (128), and the fusion of
endolysosomal vesicles with the cell surface plasma membrane (118).
1.3.3 Role of ABC transporters in unconventional secretion
The ATP binding cassette (ABC) transporter superfamily is one of the
largest protein families known, with members in almost all organisms
from bacteria to mammals and plants. These transmembrane proteins
actively transport specific substrates across extra- and intracellular
membranes. In eukaryotes, these proteins mainly transfer molecules to
the outside of the cell. Classified in seven groups, 48 ABC transporter
proteins are known in humans (129). Multiple drug resistance (MDR)-1
was the first ABC transporter characterized in humans and it is also the
best studied. MDR transporters are important in pharmacology, for
example, they pump tumor suppressor drugs out of cancer cells (130).
ABCA1 is a member of the subfamily ABCA and has been implicated as a
regulator in cellular cholesterol and phospholipid homeostasis.
Additionally, ABCA1 is reported to be involved in the unconventional
secretion of several proteins; among these are IL-1β, HSP70, and Annexin-
1 (119, 131, 132). It was demonstrated that the pharmacological inhibitor
of ABCA1, glyburide, inhibits the release of these proteins. In the case of
Annexin-1 the results obtained by using pharmacological inhibition were
confirmed in siRNA experiments targeting ABCA1. To date, no study was
undertaken in regard to Annexin-1 and vesicles. However, it remains
unclear whether ABC transporters are directly or indirectly involved in
the unconventional secretion pathway of these proteins.

Introduction – 24
1.3.4 Unconventional Secretion of MIF
As mentioned previously, MIF belongs to the heterogonous group of
proteins that do not possess a signal peptide and are secreted by a
specialized, unconventional pathway. MIF from bovine liver cell lysates or
monocytes/macrophages does not become post-translationally N-
glycosylated although two potential N-glycosylation sites are present in
the MIF cDNA sequence (6, 105). Only after the experimental fusion of
the leader-sequence of IL-2 to MIF, N-glycosylations of MIF were
observed, indicating that native MIF is excluded from the classical
secretory organelles ER and Golgi. Furthermore, brefeldin A, an inhibitor
of the ER/Golgi pathway did not affect the stimulated release of MIF from
monocytes/macrophages and neutrophils, whereas glyburide and
probenicide, inhibitors of the ABCA1 transporter, blocked the MIF
secretion (105, 133). Of note, there is evidence that MIF is localized to
vesicles in certain cell types (pituitary cells, β-cells of the pancreatic islets,
and epididymal cells) (134-136). Interestingly, other unconventionally
secreted proteins that are inhibited in their secretion by glyburide (IL-1β,
HSP70) are also associated with vesicles.

Material and Methods – 25
2 Material and Methods
2.1 Cell lines, Bacteria and Plasmids
2.1.1 Cell lines
Description Cell type Morphology
HeLa Human cervix carcinoma
cells
Epithelial, adherent
HEK 293 Human embryonal kidney
cells
Epithelial, adherent
RAW 264.7 Murine ascites leukemia
cells
Macrophage/monocyte,
adherent
THP-1 Human peripheral blood
leukemia cells
Monocyte, suspension
2.1.2 Bacteria
Strain Genotype Origin
E. coli
DH5α
F- mcrA Δ(mrr-hsdRMS-mcrBC)φ80lacZ
ΔM15 ΔlacX74 recA1 araD139 Δ(araleu)
7697 galU galK rpsL (StrR) endA1 nupG
Invitrogen,
Carlsbad, CA
E. coli
BL21 (DE3)
F- ompT hsdSB (rB-mB-) gal dcm rne131
(DE3)
Invitorgen,
Carlsbad, CA
2.1.3 Plasmids
Plasmid Insert Resistance/
Promotor
Origin
pET22b p115 Amp/ T7 this work
pET22b DDT Amp/ T7 this work
pET11b MIF Amp/ T7 Bucala +
Bernhagen
lab
pENTR/U6 shRNA targeting p115 or
lacZ
Kan/U6 this work
pAd/Block-iT
Dest
shRNA targeting p115 or
lacZ
Amp/U6 this work
pAd/CMV/V5-
DEST
p115 Amp/CMV this work
pAd/CMV/V5-
DEST
lacZ Amp/CMV this work

Material and Methods – 26
2.2 Equipment, Consumables and Chemicals
2.2.1 Equipment
Equipment Manufacturer
Äkta FPLC system GE Healthcare, Piscataway, NJ
Amaxa nucleofector Amaxa, Gaithersburg, MD
Amaxa nucleofector Amaxa, Cologne, Germany
Analytical balance Acculab, Columbia, MD
Beckman Alegra6 centrifuge Beckman, Fullerton, CA
Centrifuge 5417 R Eppendorf Eppendorf, Westbury, NY
Centrifuge 5417 R Eppendorf Eppendorf, Wesseling-Berzdorf,
Germany
CO2 incubator Heracell Thermo Scientific, Waltham, MA
CO2 incubator Heracell Heraeus Instruments, Hanau,
Germany
Electrophoresis Power Supply BioRad, Hercules, CA
Heat block thermo mixer comfort Eppendorf, Westbury, NY
HERA-Safe sterile bench Heraeus Instruments, Hanau,
Germany
HERA-Safe sterile bench Thermo Scientific, Waltham, MA
iCycler BioRad, Hercules, CA
HiTrap chelating HP column GE Healthcare, Piscataway, NJ
Light microscope Carl Zeiss, Thornwood, NY
Light microscope Olympus CK40 Olympus Co GmbH, Hamburg,
Germany
LSM510 confocal microscope Carl Zeiss, Thornwood, NY
NuPAGE/XCell II Blot Module Invitrogen, Carlsbad, CA
NuPAGE/XCell SureLock Invitrogen, Carlsbad, CA
ND-1000 Spectrophotometer NanoDrop, Wilmington, DE
PTC-100 PCR BioRad, Hercules, CA
Q Sepharose Anion exchange
chromatography column
GE Healthcare, Piscataway, NJ
Superdex 75 size exclusion column GE Healthcare, Piscataway, NJ
Sorvall Centrifuge Thermo Scientific, Waltham, MA
SpectraMax Plus Molecular Devices, Sunnyvale, CA
2.2.2 Consumables
Consumables Manufacturer
BioMax MS Film Kodak, Rochester, NY
BioMax MR Film Kodak, Rochester, NY
Blotting paper Whatman, Clifton, NJ
C8 Sep-Pak Waters, Milford, MA
Cell culture equipment BD Falcon, Bedford, MA

Material and Methods – 27
Cell culture equipment Greiner, Frickenhausen, Germany
Centricon Amicon Bioseparations
Culture Slides BD Falcon, Bedford, MA
Eppendorf tubes Eppendorf, Westbury, NY
Eppendorf tubes Eppendorf GmbH, Wesseling-
Berzdorf, Germany
ImmunoModule ELISA plate Nunc, Rochester, NY
ImmunoModule ELISA plate Nunc, Roskilde, Germany
NuPAGE 4-12% Bis-Tris-Gels Invitrogen, Carlsbad, CA
Polypropylene tubes BD Falcon, Bedford, MA
PVDF membrane Millipore, Bedford, MS
Thin wall PCR tubes BioRad, Hercules, CA
2.2.3 Chemicals
Chemicals Manufacturer
Acetonitrile J.T.Baker, Phillipsburg, NJ
Agarose Sigma, St. Louis, MO
Ampicillin Sigma, St. Louis, MO
Bacto Agar BD, Franklin Lakes, NJ
BSA American Bioanalytical, Natick, MA
Bradford Reagent BioRad, Hercules, CA
Dimethylsulfoxide Sigma, St. Louis, MO
DMEM Invitrogen, Carlsbad, CA
DMEM Invitrogen, Eggenstein, Germany
DNA ladder Invitrogen, Carlsbad, CA
Dry milk powder BioRad, Hercules, CA
DTT Sigma, St. Louis, MO
EDTA American Bioanalytical, Natick, MA
FBS Invitrogen, Carlsbad, CA
FBS Invitrogen, Eggenstein, Germany
Formaldehyde American Bioanalytical, Natick, MA
Glycerol Sigma, St. Louis, MO
Goat serum Sigma, St. Louis, MO
human Serum Cambrex, Walkersville, MD
Imidazole Sigma, St. Louis, MO
iQ Sybr Green BioRad, Hercules, CA
Kanamycin Sigma, St. Louis, MO
Lipofectamine 2000 Invitrogen, Carlsbad, CA
Lipofectamine 2000 Invitrogen, Eggenstein, Germany
Lipopolysaccharide 0111:B4 Sigma, St. Louis, MO
Lipopolysaccharide 0111:B4 Sigma, Munich, Germany
Luria Broth medium Invitrogen, Carlsbad, CA
Na3VO4 Sigma, St. Louis, MO
NaCl J.T.Baker, Phillipsburg, NJ
NaF Sigma, St. Louis, MO

Material and Methods – 28
β-Mercaptoethanol Sigma, St. Louis, MO
Methanol Fisher scientific, Pittsburgh, PA
Nonidet P-40 Roche, Indianapolis, IN
NuPAGE LDS sample buffer Invitrogen, Carlsbad, CA
NuPAGE SDS running buffer Invitrogen, Carlsbad, CA
NuPAGE transfer buffer Invitrogen, Carlsbad, CA
Phosphate buffered saline Invitrogen, Carlsbad, CA
Penicillin/Streptomycin Invitrogen, Carlsbad, CA
PIC Complete Roche, Indianapolis, IN
Protein Free Blocking buffer Thermo Scientific, Waltham, MA
Protein G Sepharose Sigma, St. Louis, MO
RPMI 1640 Invitrogen, Carlsbad, CA
RPMI 1640 Invitrogen, Eggenstein, Germany
RT² SYBR Green SuperArray, Frederick, MD
SeaBlue 2Plus Invitrogen, Carlsbad, CA
SOC medium Invitrogen, Carlsbad, CA
Sodiumdodecylsulfate Sigma, St. Louis, MO
Superblock Pierce, Rockford, IL
Streptavidin-HRP Promega, Madison, WI
Stripping buffer Thermo Scientific, Waltham, MA
TAE buffer Invitrogen, Carlsbad, CA
TMB substrate system Dako, Carpinteria, CA
Tris American Bioanalytical, Natick, MA
Triton-X-100 Sigma, St. Louis, MO
Trypsin/EDTA Invitrogen, Carlsbad, CA
Tween-20 Sigma, St. Louis, MO
2.2.4 Multi-Component Systems
Kits Manufacturer
AccuScript cDNA synthesis Stratagene, La Jolla, CA
Biotin protein labeling Roche, Indianapolis, IN
ELISA (TNF-α, IL-1β, IL-6) ebioscience, San Diego, CA
EZ-Link NHS-SS-Biotin Pierce, Rockford, IL
GeneClean Qbiogene, Carlsbad California
QIAprep MiniPrep Qiagen, Valencia, CA
QIAprep MiniPrep Qiagen, Hilden, Germany
RNeasy Mini Kit Qiagen, Valencia, CA
Qiashredder Qiagen, Valencia, CA
SlowFade Antifade Invitrogen, Carlsbad, CA
Super Signal West Dura (ECL) Pierce, Rockford, IL

Material and Methods – 29
2.3 Primary and Secondary Antibodies
2.3.1 Primary Antibodies
Antigen Application* Species/Type** Manufacturer
ß-Actin WB Mouse/M Sigma, St. Louis, MO
Ct-LPS FACS Mouse/M DAKO, Carpinteria, CA
DDT WB/ELISA Rabbit/P this work
GAPDH WB Goat/P Santa Cruz
Biotechnology, Santa
Cruz, CA
MIF (3H2F) ELISA Mouse/M Bucala lab
MIF (XIV14.3) ELISA Mouse/M Bucala lab
MIF (N20) WB/ELISA Goat/P Santa Cruz
Biotechnology, Santa
Cruz, CA
MIF (IIID9) IFA Mouse/M Bucala lab
MIF
(MAB289)
ELISA Mouse/M R&D Systems GmbH,
Wiesbaden, Germany
p115 WB/IFA Rabbit/P Calbiochem, San Diego,
CA *WB: Western blot, IP: immunoprecipitation, IFA: Immunofluorescent assay, FACS: fluorescent –
activated cell sorting, ELISA: Enzyme-Linked Immunosorbent Assay
**P: IgG polyclonal, M: monoclonal (IgG1)
2.3.2 Secondary Antibodies
Description Application* Species Manufacturer
Anti-goat HRP WB/ELISA Donkey Santa Cruz Biotechnology,
Santa Cruz, CA
Anti-mouse HRP WB Horse Cell signaling, Danvers, MA
Anti-mouse
Biotin
ELISA Goat R&D Systems GmbH,
Wiesbaden, Germany
Anti-mouse
Alexa568
IFA Rabbit Invitrogen, Carlsbad, CA
Anti-rabbit HRP WB Goat Cell signaling, Danvers, MA
Anti-rabbit
Alexa488
IFA Goat Invitrogen, Carlsbad, CA
*WB: Western blot, IFA: Immunofluorescent assay, ELISA: Enzyme-Linked Immunosorbent Assay

Material and Methods – 30
2.4 RNAi-targeting Sequences
Description Sequence
RNAi GM130 5'-ATGAGAACATGGAGATCACC-3'
RNAi p115-2 5'-GCAGCTGGATTCATCTAATAG-3'
RNAi p115-1 5'-GCAGTTGGTCCAAGGCTTAT-3'
RNAi lacZ provided by Invitrogen, Carlsbad, CA
RNAi PIP-K Validated siRNA, Qiagen, Hilden, Germany
2.5 Quantitative PCR Primers
Description Sequence
MIF-forward 5'-CGGACAGGGTCTACATCAA-3'
MIF-reverse 5'-CTTAGGCGAAGGTGGAGTT-3'
β-Actin-forward 5'-GGATGCAGAAGGAGATCACTG-3'
β-Actin-reverse 5'-CGATCCACACGGAGTACTTG-3'
For quantification of p115 mRNA levels, p115 and β-Actin primers were
designed and synthesized from SuperArray.
2.6 Media, Buffer and Solutions
2.6.1 Media
All media contained 1 U/ml penicillin/streptomycin
Complete growth medium for HEK293,
HeLa and RAW264.7
Concentration,
percent
DMEM 90
FBS 10
Complete growth medium for THP-1 Concentration,
percent
RPMI 1640 90
FBS 10
Complete growth medium for primary human
macrophages
Concentration,
percent
RPMI 80
human AB serum 20

Material and Methods – 31
Complete growth medium for bone marrow
derived macrophages
Concentration,
percent
DMEM 50
FBS 20
L929 supernatant 30
2.6.2 Buffers and Solutions
All buffers were prepared with distilled water, if not stated otherwise.
2.6.2.1 General Buffers
TBS, pH 7.4 Concentration
Tris-HCl, pH 7.4 20 mM
NaCl 150 mM
TBS-T, pH 7.4 Concentration
TBS (buffer)
Tween-20 0.05%
RIPA Concentration
Tris-HCl, pH 7.4 50 mM
NaCl 150 mM
EDTA 2 mM
Nonident P-40 1%
Sodium deoxycholate 0.5%
SDS 0.1%
Protease inhibitors complex 1x
2.6.2.2 Flow Cytometry
Staining buffer Concentration
PBS (buffer)
FBS 1%
Sodium azide 0.1%
Fixing buffer Concentration
PBS (buffer)
Formaldehyde 3.7%

Material and Methods – 32
2.6.2.3 Human MIF ELISA
Washing buffer Concentration
PBS (buffer)
Tween-20 0.05%
Blocking buffer Concentration
PBS (buffer)
Sucrose 1%
BSA 1%
Reaction buffer Concentration
PBS (buffer)
EDTA 1mM
BSA 0.1%
Tween-20 0.05%
Primary antibody Concentration
PBS (buffer)
Anti-MIF antibody clone 3H2F 10 µg/ml
HRP-coupled secondary
antibody
Concentration
Reaction buffer (buffer)
Peroxidase-conjugated anti-MIF
antibody
1x
2.6.2.4 Murine MIF ELISA
Washing and reaction buffer are the same as for human MIF ELISA. For
blocking, Protein Free T20 Blocking buffer is used.
Primary antibody Concentration
PBS (buffer)
anti-MIF antibody clone XIV.14.3 15 µg/ml
Secondary antibody Concentration
reaction buffer (buffer)
Anti-MIF antibody N20 0.6 µg/ml

Material and Methods – 33
Tertiary antibody Concentration
reaction buffer (buffer)
Anti-goat antibody HRP conjugated 2 µg/ml
2.6.2.5 Murine DDT ELISA
Washing and blocking buffer are the same as for human MIF ELISA.
Primary antibody Concentration
PBS (buffer)
Anti-DDT antibody 15 µg/ml
Reaction buffer Concentration
TBS (buffer)
BSA 0.1%
Tween-20 0.05%
Secondary antibody Concentration
reaction buffer (buffer)
anti-DDT antibody (biotinylated) 0.6 µg/ml
Tertiary antibody Concentration
reaction buffer (buffer)
Streptavidin-HRP conjugate 2 µg/ml
2.6.2.6 Ni2+-Column Purification
Binding buffer Concentration
Tris-HCl, pH 8 50 mM
NaCl 200 mM
Glycerol 10%
Imidazole 20 mM
β-mercaptoethanol 10 mM

Material and Methods – 34
Elution buffer Concentration
Tris-HCl, pH 8 50 mM
NaCl 200 mM
Glycerol 10%
Imidazole 500 mM
β-mercaptoethanol 10 mM
2.6.2.7 MIF Purification
Binding buffer Concentration
Tris-HCl, pH 7.4 50 mM
NaCl 150 mM
Elution buffer Concentration
Tris-HCl, pH 7.4 50 mM
NaCl 2000 mM
Denaturation buffer Concentration
TBS (buffer)
Urea 8 M
Renaturation buffer Concentration
TBS (buffer)
DTT 5 mM
2.6.2.8 DDT Purification
Binding buffer Concentration
Tris-HCl, pH 7.4 50 mM
NaCl 50 mM
Elution buffer Concentration
Tris-HCl, pH 7.4 50 mM
NaCl 1000 mM

Material and Methods – 35
2.6.2.9 SDS-PAGE and Western Blot
Running buffer Concentration
MES running buffer 20x (NuPAGE) 1x For separation of low molecular weight proteins
Running buffer Concentration
MOPS running buffer 20x
(NuPAGE)
1x
For separation of high molecular weight proteins
Transfer buffer Concentration
Transfer buffer 20x (NuPAGE) 1x
Methanol 10%
Blocking buffer Concentration
TBS-T (buffer)
Dry milk powder 5%
Washing buffer Concentration
TBS-T (buffer)
2.6.2.10 Coomassie Staining
Staining solution Concentration
Coomassie Brilliant Blue R-250 0.25%
Methanol 40%
Glacial acidic acid 10%
Destaining solution Concentration
Methanol 40%
Glacial acidic acid 10%
2.6.2.11 Immunofluorescent Staining
Fixing solution Concentration
PBS (buffer)
Formaldehyde 3.7%
Triton-X-100 0.1%

Material and Methods – 36
Blocking solution Concentration
PBS (buffer)
Goat serum 5%
2.7 Molecular Biology Techniques
2.7.1 Measurement of DNA/RNA Concentration
The concentration and purity of DNA/RNA was assessed by photometric
analysis. The absorption of the nucleotides was measured at OD260 and
OD280. One OD260 equals 50 µg/ml DNA and 40 µg/ml RNA. The ratio
between OD260 vs. OD280 assesses the purity of the sample with respect to
protein contamination. A quotient of 1.8 to 2.0 indicates an
uncontaminated sample.
2.7.2 Polymerase Chain Reaction (PCR)
DNA fragments were amplified using native Pfu DNA polymerase
(Stratagene). In contrast to the Taq polymerase, Pfu polymerase has a
proofreading activity, resulting in fewer mutations in the process of
amplification.
The reaction mixture was prepared according to the manufacturer’s
recommendations. The profile for amplification included:
Step Number of cycles Temperature Duration
1 1 95 ºC 45 sec
2 25 – 30 95 ºC
Tm – 5 ºC
72 ºC
45 sec
45 sec
1 min per kb of PCR target
3 1 72 ºC 10 min

Material and Methods – 37
2.7.3 Enzymatic DNA Digestion
For the hydrolytic restriction of DNA, vector/insert DNA was incubated
with the desired restrictionendonucleases according to the manufacturer’s
instructions.
2.7.4 Agarose Gel Electrophoresis
In order to identify and/or purify DNA after PCR or DNA restriction, DNA
was separated according to its mass in 1% agarose gels. The running
buffer contained ethidium bromide for visualization of DNA under UV
light.
2.7.5 Isolation and Purification of DNA from Agarose Gels
DNA from PCR reactions or restriction reactions were run on an agarose
gel and the desired bands cut from the gel with a razor blade. DNA was
eluted using GENECLEAN® III Kit according to their instructions.
2.7.6 DNA Ligation
For the ligation reactions, T4 DNA ligase was used. 50 – 100 ng vector
DNA were incubated with the insert at a molar ratio of 1:3 in the
recommended buffers with 5 U T4 Ligase at 4 ºC over night.
2.7.7 Heat shock Transformation of E. coli
After thawing an aliquot of competent cells on ice, bacteria were incubated
with plasmid DNA for 30 min on ice prior to transformation. Heatshock
transformation occurred via incubation for 30 sec at 42 ºC. Subsequently,
bacteria were kept on ice for two minutes and then incubated at 37 ºC for 1
h in SOC medium. Bacteria were then plated on LB-agar plates containing
the appropriate antibiotics.

Material and Methods – 38
2.7.8 Plasmid Isolation
Plasmids were isolated with the Plasmid Mini Kit from Qiagen according
to the manufacturer’s recommendations.
2.7.9 Isolation of RNA and cDNA Synthesis
For the isolation of RNA the RNeasy kit was used according to the
manufacturer’s instruction. RNA was converted to cDNA by reverse
transcriptase. Oligo-dT primers were used for priming polyadenylated
RNA. For an efficient cDNA synthesis, RNA together with the primers
were denatured at 65 ºC for 5 min, followed by a 5 min cooling period at
room temperature to allow the primers to anneal to the RNA. The
synthesis reaction was carried out at 42 ºC for 1 h. After heat inactivation
of the reverse transcriptase, cDNA was stored at –20 ºC.
2.7.10 Quantitative PCR
Quantitative PCR (qPCR) is a modification of the polymerase chain
reaction, and is used to analyze gene expression. After the synthesis of
cDNA, primers specifically amplify the gene of interest in the presence of
SYBR green which only emits fluorescence after intercalating into double
stranded DNA.
The qPCR was performed on the iCycler from BioRad. The amplification
profile included denaturation at 95 °C for 5 min, followed by one step
annealing and elongation for 1 min at 60 °C for a total of 40 cycles. For
verification of specific product amplification a melting curve was
performed.
2.7.11 Design and Production of short hairpin RNA
The Block-it RNAi designer from Invitrogen was used to design short
hairpin RNA molecules (shRNA) specific to human p115. Two constructs
were designed: p115-1 from position 2625 to 2645 (5'-

Material and Methods – 39
GCAGCTGGATTCATCTAATAG-3') and p115-2 from position 1601 to 1620
(5'-GCAGTTGGTCCAAGGCTTAT-3') in the human p115 mRNA sequence
(NM_003715). Both constructs share no significant sequence homology
with other genes as analyzed by BLAST search. As negative control,
shRNA constructs against lacZ (provided from Invitrogen) were generated.
Oligonucleotides were cloned into the RNA expression vector pENTR/U6
and confirmed by sequencing.
2.7.12 Preparation of Adenoviruses
shRNA constructs p115-2 and lacZ in the pENTR/U6 vector were
recombined with the adenoviral vector pAd/BLOCK-iT-DEST using
Gateway LR Clonase according to the manufacturer’s protocol. The
resulting p115/pAd plasmid was transfected into HEK293 and amplified
following the standard procedure described in the manual. The viral titer
was determined using AdenoX Rapid Titer Kit (Clontech, Palo Alto, CA).
2.8 Cell Culture Techniques
2.8.1 Cultivation and Treatment
All cell culture work was performed in BL2-approved laminar flow hoods.
For cultivation, cells were kept in a humidified atmosphere at 37 ºC with
5% CO2. All media and solution were sterile and endotoxin free. Adherent
cells were passaged at a confluence of 70 – 80%. Cells were washed twice
with PBS and then detached from culture flask using Trypsin/EDTA. Cells
were re-plated in fresh medium at a confluence of 10%. Suspension cell
were passaged every third day.
For differentiation of THP-1 monocytes to macrophages, cells were treated
with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) for 24 h.
Subsequently, cells were cultured for additional 48 h (137).

Material and Methods – 40
For MIF secretion assays, cells were incubated in RPMI + l% FBS medium
for 3 h prior to stimulation to quiescent cells. MIF secretion was
stimulated with lipopolysaccharide (LPS). THP-1 monocytes and primary
human macrophages were stimulated with 10 µg/ml LPS, PMA-
differentiated macrophages were stimulated with 0.1 µg/ml LPS.
2.8.2 Isolation of Macrophages from Blood
Human PBMCs were isolated by Ficoll-Hypaque gradient centrifugation.
The cells were resuspended in RPMI 1640 medium supplemented with
20% human AB serum and plated at 2.5x106 cells per well in 24-well cell
culture plates. After 2 h of culture, the adherent cells were washed
extensively with PBS and cultured for 1 week to allow differentiation into
monocyte-derived macrophages (MDMs).
2.8.3 Transient Transfection
Depending on the cell type, different methods were employed for
transfection. HeLa and HEK293 cells were transfected with Lipofectamine
2000, a cationic, lipid-based transfection reagent. THP-1 monocytes were
transfected by Amaxa electroporation utilizing program V-001 and the
nucleofector solution V.
Adenovirus-mediated transfection was performed to efficiently transfer
plasmids into differentiated THP-1 macrophages and primary human
macrophages. PMA-differentiated cells were incubated with a multiplicity
of infection (MOI) of 50 over night; primary macrophages were incubated
with 50 MOI of adenovirus for 2 h.
Depending on the cell type, transfection method and construct, cells were
cultured between 48 and 96 h after transfection.

Material and Methods – 41
2.8.4 Preparation of Cell Lysates
The preparation of cell lysates for Western blot analysis included the
following steps: washing of cells with ice cold PBS, lysis of cells by
incubation in RIPA buffer containing protease and phosphatase inhibitors
for 20 min at 4 ºC, removal of genomic DNA and cell debris by
centrifugation for 10 min at 14,000 rpm at 4 ºC.
2.8.5 Immunofluorescent staining for microscopy
Cells were plated at a confluency of 80% on culture slides. For fixation and
permeablization, cells were incubated with fixation solution for 20 min at
37 ºC. Cells were thoroughly washed with PBS, and then incubated with
blocking buffer for 30 min at RT. Cells were incubated with primary
antibody over night at 4 ºC, washed three times with PBS and then
incubated with secondary antibody for 1 h at RT. To reduce fading during
microscopy, the cells were mounted with Antifade. Primary and secondary
antibodies were diluted in blocking solution according to the
recommendations of the manufacturer.
2.8.6 Fluorescence-based Quantification of FGF Export
HeLa cells were genetically modified to express FGF-2-GFP and FGF-4-
GFP fusion proteins in a doxycycline-dependent manner (138). To quantify
the secretion of FGF fusion proteins, cells were transfected with
knockdown constructs targeting either p115, PIP-K (115), or a control
sequence. After 96 h, the expression of the fusion protein was induced by
doxycycline for 24 h. Cells were prepared for flow cytometry to detect cell
surface associated FGF-2-GFP and FGF-4-GFP, respectively. Primary
antibodies were decorated with APC-conjugated secondary antibodies and
GFP fluorescence (intracellular and cell surface) and APC fluorescence
(cell surface) were determined employing FACSCalibur flow cytometer

Material and Methods – 42
(BD Bioscience). Untransduced HeLa cells were used to determine
autofluorescence signals.
2.8.7 Chlamydia trachomatis (L1) Infection
THP-1 cells were infected with C. trachomatis at an MOI of 10 for 1 h at
RT by slow rotation in RPMI medium. Cells were washed with medium
and resuspended in RMPI containing 10% FBS without antibiotics and
cultivated at 1x106 cells per ml in 12 well plates. The infection rate was
analyzed by flow cytometry. For FACS analysis, cells were washed in
staining buffer and then fixed in 3.7% formaldehyde in PBS for 20 min at
RT. Subsequently cells were permeabilized with BD Perm/Wash by
incubating for 20 min on ice. Cells were stained with mouse anti-Ct LPS
FITC antibody.
2.9 Proteinchemistry and Immunology Techniques
2.9.1 Determination of Protein Concentration
The measurement of protein concentration was performed according to
Bradford (139). After diluting the Bradford reagent 1:5 in water, samples
were diluted 1:100 in Bradford solution. After incubation for 5 min at RT,
the absorbance was measured at 595 nm. The concentration of the
samples was calculated by interpolation from a BSA standard curve.
2.9.2 SDS-PAGE
Proteins were separated according to their size in the NuPAGE system
using Bis-Tris 4 – 12% polyacrylamide gradient gels. Samples were boiled
for 5 min in LDS sample loading buffer and separated at 180 V in
NuPAGE/XCell SureLock chambers. Depending on the size of the protein
analyzed, either MES running buffer for small molecular weight proteins

Material and Methods – 43
(3 – 50 kd) or MOPS running buffer for large molecular weight proteins
(50 – 200 kd) was used.
2.9.3 Coomassie Staining
Coomassie brilliant blue is a triphenylmethane dye binding to the basic
amino acids of proteins. To stain proteins after a SDS/PAGE, the gel is
incubated with the Coomassie staining solution for 30 min at RT. For
visualization of proteins, the gel was washed with destaining solution
until only the dye bound to the proteins remains. The destaining solution
was changed repeatedly.
2.9.4 Western Blot
Proteins separated in a SDS-PAGE were transferred to a methanol-
activated PVDF membrane using the XCell II Blot Module. The unspecific
binding sites on the membrane were blocked by incubating the membrane
for 2 h at RT in blocking buffer. The primary antibody was diluted 1:1000
or according the manufacturer’s recommendations in blocking buffer and
the membrane was incubated with primary antibody solution either for 2
h at RT or over night at 4 ºC. Before the incubation with the HRP-coupled
secondary antibody, the membrane was washed 3 times. The secondary
antibody was diluted in blocking solution at 1:5000 and incubated with the
membrane for 1 h. After washing the membrane, proteins were visualized
by detecting the chemiluminescence emitted after addition of the ECL
substrate. If the membrane was re-probed, the membrane was incubated
in stripping buffer for at least 30 min at RT. After a thorough washing the
membrane, immunoblotting procedure was repeated. For densitometric
analysis, the Scion Image program from NIH was used.

Material and Methods – 44
2.9.5 Sandwich ELISA
2.9.5.1 Human MIF ELISA
The human MIF ELISA is a one-step solid phase assay performed as
described previously (13). The samples were mixed with anti-MIF
antibody-HRP construct and added to an ELISA plate pre-coated with
anti-MIF antibody 3H2F. During the incubation, the MIF antigen binds
simultaneously to the capture antibody on one side and the HRP-coupled
anti-MIF antibody on the other side. After incubation and washing to
remove unbound enzyme, the amount of bound enzyme in each well was
detected by adding the substrate TMB. The reaction was stopped by
adding an acidic stop solution, and the resulting color read at 450 nm in a
microtiter plate spectrophotometer. The concentration of MIF in the
samples was calculated by interpolation from the MIF standard curve.
2.9.5.2 Murine MIF ELISA
MIF concentration was measured as described previously (21). Briefly, 96-
well microtiter plates were coated with the anti-MIF monoclonal antibody,
washed, and blocked. 50 µl aliquots of each sample were added to the plate
and incubated overnight at 4 °C. The plate was washed and incubated
with polyclonal anti-MIF antibody (N-20). This was followed by the
addition of anti-goat IgG antibody conjugated to HRP, and the antibody
complexes were quantified after the addition of the substrate TMB.
2.9.5.3 Murine DDT ELISA
96-well microtiter plates were coated with 15 µg/ml polyclonal anti-DDT
antibody over night at 4 ºC. Subsequently the plate was washed and
blocked with 1% BSA, 1% Sucrose in PBS for 2 h at RT. 50 µl aliquots of
each sample were added to the plate and incubated for 2 h at RT. After
washing, biotinylated anti-DDT antibody was added. This was followed by
washing and the addition of a streptavidin-HRP conjugate, which was

Material and Methods – 45
incubated for 1 h at RT. The amount of bound enzyme in each well was
detected by adding the substrate TMB. The DDT concentrations were
calculated by extrapolation from a sigmoidal quadratic standard curve
using mouse DDT.
2.9.5.4 TNF-α, IL-1β and IL-6 ELISA
To measure the concentration of TNF-α, IL-1β and IL-6 the commercially
available ELISA kits from ebioscience were used according to the
manufacturer’s instructions.
2.9.6 Protein Purification
2.9.6.1 Overexpression of Proteins
The DNA sequence of the protein of interest was cloned into the bacterial
expression vectors (pET from Novagen). After transforming the constructs
into competent E. coli BL21 (DE3), cells were cultured over night. The
following day, LB medium was inoculated with the over night culture to
obtain an OD600 of 0.1. Bacteria were grown until an OD600 of 0.6, and the
expression of the protein was induced by the addition of IPTG to a final
concentration of 1 mM. The expression lasted for 4 h; afterwards the
bacteria were pelleted by centrifugation for 10 min at 3,000 rpm at 4 ºC.
The supernatant was discarded and pellets stored at –20 ºC.
2.9.6.2 Purification of Histidine-tagged Proteins
After overexpression of histidine-tagged p115 or DDT, bacteria were
resuspended in binding buffer and lysed by French Press. The crude
bacterial extract was purified using a HiTrap chelating HP column with a
linear gradient of 20 – 400 mM imidazole in 50 mM Tris, pH 8.0 and 200
mM NaCl. The peak fractions, as determined by SDS-PAGE, were
subjected to gel filtration chromatography. Protein purity was verified by
SDS-PAGE/Coomassie staining and Western blotting.

Material and Methods – 46
2.9.6.3 Purification of MIF
MIF was purified as described previously (6). Briefly, after overexpression,
pelleted bacterial were resuspended in 50 mM Tris-HC1 pH 7.5, 150 mM
NaCl and lysed using a French Press. The lysates was sterile filtered and
then subjected to a MonoQ anion exchange chromatography. The MIF
containing fractions were applied to a C8-SepPak reverse phase column.
Weakly bound material was eluted with 20% acetonitrile. MIF then was
eluted with six column volumes of 60% acetonitrile, frozen at –80 ºC, and
lyophilized. For renaturation, MIF was dissolved in 20 mM sodium
phosphate buffer containing 8 M urea and 5 mM DTT and dialyzed
against 20 mM sodium phosphate buffer containing 5 mM DTT, followed
by 20 mM sodium phosphate buffer alone.
2.9.6.4 Purification of DDT
Native DDT was overexpressed in E. coli BL21 (DE3). The bacteria pellet
was resuspended in 50 mM Tris-HCl, pH 7.4, 50 mM NaC1 and lysed
using a French Press. The lysate was sterile-filtered and subjected to a Q
Sepharose anion exchange chromatography. The column was equilibrated
with Tris-buffered saline. DDT was eluted with a salt gradient (50 mM
Tris-HCl, pH 7.4, 1 M NaC1) and 7.5 ml fractions were collected. The
DDT-containing fractions were combined, concentrated and subjected to a
Superdex 75 size exclusion chromatography. One fraction contained pure
DDT protein. One liter of E. coli culture resulted in 10 mg DDT protein.
The protein was stored at –20 ºC. LPS content of the purified protein was
not analyzed.
2.9.7 In vitro MIF/p115 Binding Studies
96-well protein-absorbent plates were coated with 1 µM p115 per well.
After coating over night, the plate was washed with TBS-T and blocked
with Superblock. After pre-incubating MIF at concentrations ranging from

Material and Methods – 47
0 – 4 µM with 150 nM biotinylated MIF for 2 h, the samples were added to
the plate and incubated over night at 4°C. For detection of bound
biotinylated MIF, a streptavidin-alkaline phosphatase conjugate was
added after washing the plate. Using p-nitrophenylphosphate as a
substrate for the alkaline phosphatase, the amount of captured protein
was measured at 405 nm. For a reverse set-up of the assay, 1 µM MIF was
coated and 150 nM biotinylated p115 incubated with unlabeled p115 at
concentrations from 0 – 4 µM. Values are plotted as percent absorbance at
405 nm relative to wells containing biotinylated MIF or p115 alone.
2.10 In vivo Mouse Experiments
2.10.1 LPS Shock
LPS shock experiments were performed with 6 – 8 week old female
BALB/c mice.
4-IPP was diluted in DMSO to a concentration of 1 M. For the
administration of 4-IPP, the stock solution was diluted in corn oil to a final
concentration of 10 mg/ml. 100 µl of 4-IPP or vehicle control was injected
intraperitoneally 2 h prior to LPS injection. 17.5 mg/kg LPS (0111:B4) was
intraperitoneally injected for survival experiments (5). When serum
cytokines levels were monitored, shock was induced with 15 mg/kg LPS.
2.10.1.1 Serum Collection
Blood was collected from mice by cardiac puncture. The mice were
anaesthetized with isoflurane. After the mouse was under a deep surgical
anesthesia, blood samples were taken from the heart by a thoracotomy
through the diaphragm. Blood was withdrawn slowly to prevent the heart
from collapsing.
Blood samples were incubated at 37 ºC for 30 min and then at 4 ºC over
night. The next day, samples were centrifuged for 10 min at 1,600 rpm,
and serum supernatant was transferred to a fresh tube.

Material and Methods – 48
2.10.2 Isolation of Bone Marrow-Derived Macrophages
Bone marrow-derived macrophages were isolated from C57BL/6 mice. To
obtain the tibia and femur for isolation of bone marrow, mice were
sacrificed and legs dissected. After the removal of tissue from the bone,
bone marrow was expelled by a jet of medium from both ends of the bone.
4x106 cells were plated per petri dish in 10 ml of conditioned medium.
After 4 days of cultivation, the medium was exchanged and cells were
cultured for at least another 24 h. For experiments the cells were scraped
off and plated at a concentration of 0.5x106/ml.

Results – 49
3 Results
3.1 p115 Mediates the Secretion of MIF
3.1.1 p115 is a Novel Interaction Partner of MIF
In a yeast two-hybrid screen in the laboratory of Prof. Bucala, p115 was
identified as a novel interaction partner of MIF. The interaction was
affirmed by co-immunoprecipitation in a mammalian cell system.
For the yeast two-hybrid screen, MIF cDNA served as “bait” and a human
pituitary cDNA library was the prey (5). After co-transformation of bait
and prey plasmids into Saccharomyces cereviciae cdc25H, 90 colonies were
identified in a primary screen, but secondary and tertiary screening ruled
out 75. The remaining 15 colonies carried proteins binding specifically to
MIF. By DNA sequencing, one of the positive clones contained the carboxy
terminal 780 bases of p115. p115 is a large protein of 962 amino acids, and
consists of a globular head, four conserved coiled-coiled (CC) domains and
a short acidic tail (97) with MIF binding at the CC region and/or the acidic
tail (Fig. 9).
Fig. 9: Schematic structure of p115. Top: Full length p115 with its globular head
(blue), four coiled-coiled domains (black), and an acidic tail (red). Bottom: Carboxy-
terminus of p115 (residues 702-962) that interacts with MIF.
The intracellular interaction between MIF and p115 was further
investigated by co-immunoprecipitation in mammalian cells. In the
prostate carcinoma cell line LNcap, an endogenous interaction between
MIF and p115 was observed by Western blot analysis for p115 after
immunoprecipitation with anti-MIF, but not an isotype control antibody
(Fig. 10).
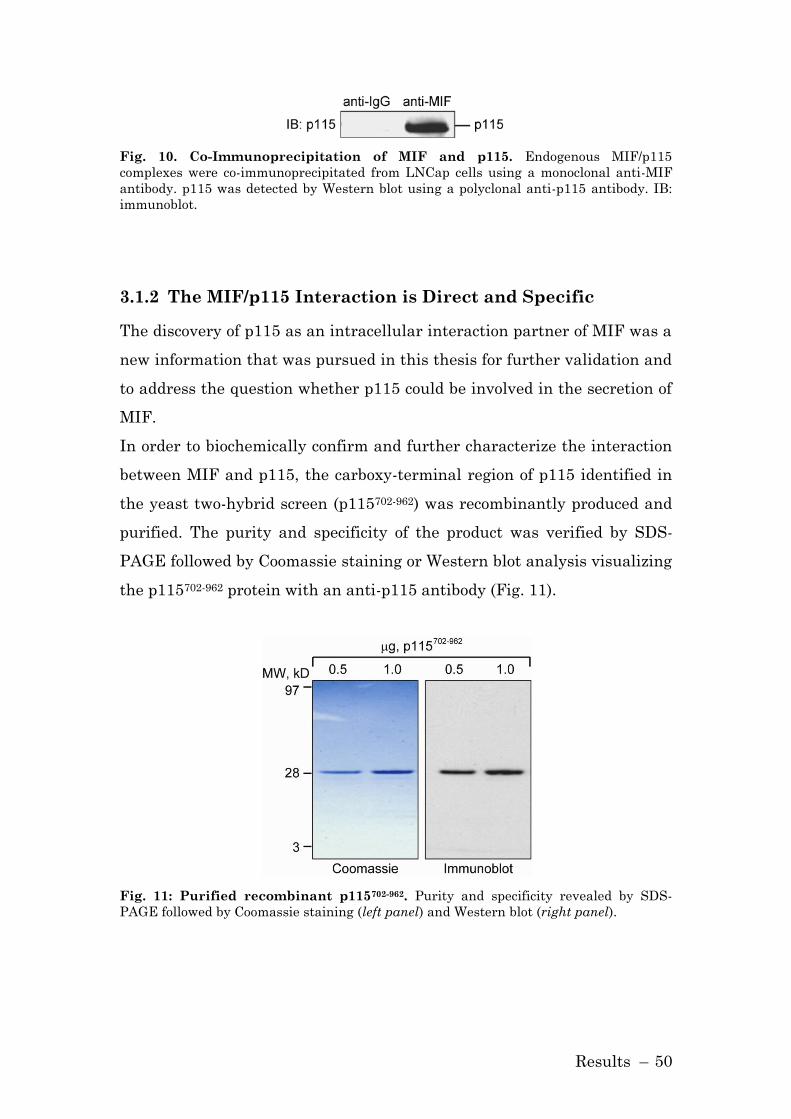
Results – 50
Fig. 10. Co-Immunoprecipitation of MIF and p115. Endogenous MIF/p115
complexes were co-immunoprecipitated from LNCap cells using a monoclonal anti-MIF
antibody. p115 was detected by Western blot using a polyclonal anti-p115 antibody. IB:
immunoblot.
3.1.2 The MIF/p115 Interaction is Direct and Specific
The discovery of p115 as an intracellular interaction partner of MIF was a
new information that was pursued in this thesis for further validation and
to address the question whether p115 could be involved in the secretion of
MIF.
In order to biochemically confirm and further characterize the interaction
between MIF and p115, the carboxy-terminal region of p115 identified in
the yeast two-hybrid screen (p115702-962) was recombinantly produced and
purified. The purity and specificity of the product was verified by SDS-
PAGE followed by Coomassie staining or Western blot analysis visualizing
the p115702-962 protein with an anti-p115 antibody (Fig. 11).
Fig. 11: Purified recombinant p115702-962. Purity and specificity revealed by SDS-
PAGE followed by Coomassie staining (left panel) and Western blot (right panel).

Results – 51
A specific interaction between MIF and p115 was observed in an in vitro
competition binding assay employing recombinant, immobilized p115702-962
and biotinylated MIF. As shown in Fig. 12, increasing concentrations of
MIF, but not the addition of equimolar amounts of control protein
(lysozyme), inhibited the interaction of biotinylated MIF with p115.
Protein-protein interaction was verified by performing the reverse assay,
i.e. measuring the binding of biotinylated p115702-962 to immobilized MIF.
Fig. 12: MIF binding to p115 demonstrated by a competitive binding assay. Left
panel: MIF binds to immobilized p115702-962 in a concentration-dependent manner. Right
panel: p115702-962 binds to immobilized MIF in a concentration-dependent manner.
Lysozyme served as a negative control. Results are expressed as mean±S.D. of duplicate
measurements and are representative for three independent experiments.
3.1.3 MIF Release from Human Monocytes
Although MIF does not possess a leader sequence, it is released from cells
in both a regulated and a specific manner. Human THP-1 monocytes were
stimulated with 10 µg/ml LPS and an increase in the concentration of MIF
in the supernatant was observed (Fig. 13, A). MIF release was detectable 2
h after stimulation and reached its plateau after 4 h.
The observed increase in the supernatant content of MIF was not the
result of cell death, as the cell viability was verified by lactate
dehyrogenase (LDH) assay. Supernatants from both LPS-stimulated and
solvent control treated cells showed 3 – 5 mU/ml LDH activity. In
contrast, cells treated with 0.1% Triton-X-100, a detergent, showed 100
mU/ml LDH activity.

Results – 52
The increase in MIF protein concentration in the supernatant after LPS
stimulation was not accompanied by an increase in the level of MIF or
p115 mRNA in the monitored period of 6 h (Fig. 13, B). These data
indicate that the initial release of MIF from THP-1 monocytes occurs from
pre-formed pools and does not require the transcription of MIF mRNA.
The results are in accord with previous reports that were based on the
immunostaining and in situ hybridization of murine tissues after LPS
administration in vivo (19, 140).
Fig. 13: Cell stimulation increases MIF secretion without affecting MIF or p115
mRNA levels. A) Time course of MIF secretion from LPS-stimulated, THP-1 monocytes.
THP-1 cells (1×106/ml) were treated with LPS (10 µg/ml) or PBS (control) and
supernatants were collected at the indicated time for measurement of MIF by specific
ELISA. Results are expressed as mean±S.E. of duplicate measurements from two
independent experiments (n=4). P values were calculated by unpaired Student’s t-test for
all time points. *p<0.01 for stimulated vs. unstimulated cells. B) MIF and p115 mRNA
levels in LPS-stimulated THP-1 monocytes. RNA was isolated at indicated time points
and analyzed by quantitative PCR. Results are expressed as mean±S.E. of three
independent experiments.

Results – 53
3.1.4 Macrophage Activation Leads to a Redistribution of
MIF
The cellular distribution of MIF in macrophages in response to LPS
stimulation was studied. In unstimulated macrophages, MIF appears
present throughout the cytoplasm and p115 is localized predominantly in
the perinuclear area in a pattern that is consistent with the Golgi (Fig. 14,
A), which is in agreement with prior reports (95, 98). Upon activation of
primary macrophages with LPS, MIF redistributes from the cytoplasm to
a plasma membrane-proximal area, and a portion of p115 now appears
dispersed in the cytoplasm (Fig. 14, B).

Results – 54
Fig. 14: Immunolocalization of p115 and MIF in macrophages. A) Macrophages
were plated on a four-chamber culture slide (2.5×105 cells/ chamber). The cells were
cultivated overnight, fixed with formaldehyde, and permeabilized with Triton-X-100.
MIF (red) and p115 (green) were visualized by immunostaining with specific antibodies
(1:200). Yellow indicates an overlapping signal after merging. B) Macrophages were
stimulated with LPS (10 µg/ml) or PBS (unstimulated) for 4 h, fixed with formaldehyde,
and permeabilized with Triton-X-100. MIF and p115 were visualized by immunostaining
with specific antibodies. Images are representative for n=40 cells. Scale bar=10 µm.
3.1.5 p115 is Required for MIF Release in Monocytes
3.1.5.1 RNAi-Mediated Depletion of p115
p115 plays an essential role in vesicle transfer from the endoplasmatic
reticulum to the Golgi complex and in Golgi re-assembly after mitosis (92,
141). MIF secretion is not inhibited by brefeldin A, a pharmacological

Results – 55
inhibitor of the Golgi-retrograde transport, MIF does not enter the Golgi,
and it is not glycosylated (105). It was nevertheless of interest to examine
whether p115 was necessary for the release of MIF in response to
activating stimuli. RNA interference (RNAi) constructs were generated for
the gene-specific silencing of p115 and their efficacy tested in human
HeLa and THP-1 cells. As shown in Fig. 15, two RNAi constructs specific
for p115 (p115-1, p115-2) reduced the concentration of p115 protein by 50
– 70% in the two cell types tested.
Fig. 15: RNAi-mediated depletion of p115. HeLa or THP-1 cells were transfected with
RNAi constructs specific for p115 (p115-1, p115-2) or a control plasmid (mock) and
analyzed for cellular p115 content by Western blotting 48 h after transfection.
3.1.5.2 Depletion of p115 Inhibits the Release of MIF
The objective was to address, whether a p115 knockdown reduces the
amount of MIF produced by LPS-stimulated THP-1 monocytes.
Transfection of the p115-1 or p115-2 RNAi sequences resulted in a 30 –
40% decrease in MIF release in response to LPS, while basal levels of MIF
production were unaffected (Fig. 16). Of note, the LPS-stimulated
secretion of the two conventionally-secreted cytokines, TNF- and IL-6,
which both bear hydrophobic signal peptides, was not affected by a
reduction in p115 protein. These data point to a potential specificity of
p115 in mediating MIF secretion, and also indicate that the cellular
depletion of p115 by the RNAi approach used herein does not appreciably
affect the functioning of the ER/Golgi pathway.

Results – 56
Fig. 16: p115 depletion inhibits MIF secretion. THP-1 monocytes (1x106/ml) were
transfected with p115 RNAi or mock RNAi plasmids and stimulated with LPS for 4 h.
Supernatants were collected and analyzed for MIF, TNF and IL-6 by specific ELISA.
Results are expressed as the mean±S.E. of four independent experiments. Statistical
significance was determined for each p115 RNAi construct against mock RNAi by
unpaired Student’s t-test, *p<0.05, **p<0.01.
GM130 is a protein known to interact with the carboxy-terminal acidic tail
of p115 (95). With the purpose of studying the role of GM130 in MIF
secretion, GM130 was depleted in THP-1 cells by siRNA. The depletion of
GM130 protein in THP-1 cells had no effect on LPS-stimulated MIF
release (Fig. 17).
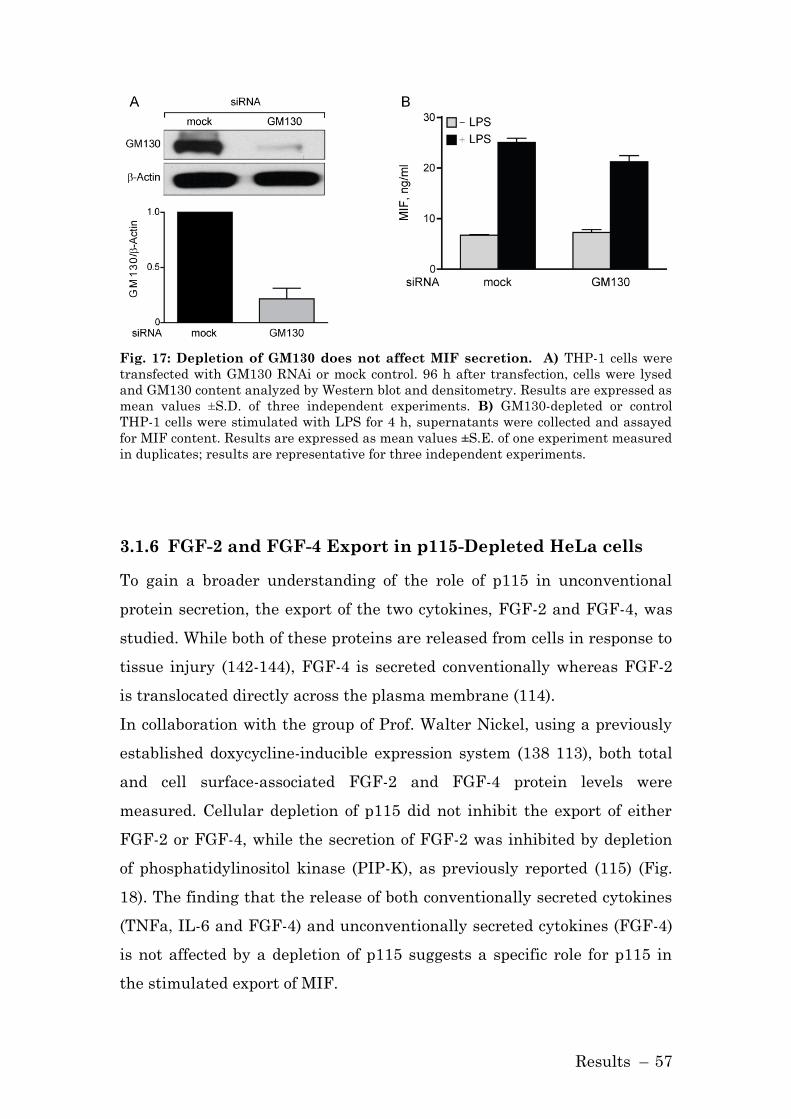
Results – 57
Fig. 17: Depletion of GM130 does not affect MIF secretion. A) THP-1 cells were
transfected with GM130 RNAi or mock control. 96 h after transfection, cells were lysed
and GM130 content analyzed by Western blot and densitometry. Results are expressed as
mean values ±S.D. of three independent experiments. B) GM130-depleted or control
THP-1 cells were stimulated with LPS for 4 h, supernatants were collected and assayed
for MIF content. Results are expressed as mean values ±S.E. of one experiment measured
in duplicates; results are representative for three independent experiments.
3.1.6 FGF-2 and FGF-4 Export in p115-Depleted HeLa cells
To gain a broader understanding of the role of p115 in unconventional
protein secretion, the export of the two cytokines, FGF-2 and FGF-4, was
studied. While both of these proteins are released from cells in response to
tissue injury (142-144), FGF-4 is secreted conventionally whereas FGF-2
is translocated directly across the plasma membrane (114).
In collaboration with the group of Prof. Walter Nickel, using a previously
established doxycycline-inducible expression system (138 113), both total
and cell surface-associated FGF-2 and FGF-4 protein levels were
measured. Cellular depletion of p115 did not inhibit the export of either
FGF-2 or FGF-4, while the secretion of FGF-2 was inhibited by depletion
of phosphatidylinositol kinase (PIP-K), as previously reported (115) (Fig.
18). The finding that the release of both conventionally secreted cytokines
(TNFa, IL-6 and FGF-4) and unconventionally secreted cytokines (FGF-4)
is not affected by a depletion of p115 suggests a specific role for p115 in
the stimulated export of MIF.

Results – 58
Fig. 18: FGF export does not depend on p115. HeLa cells were transfected with
mock, p115-2 or PIP-K RNAi constructs, FGF-GFP fusion protein production was induced
by doxycycline after 96 h. FGF-GFP fusion proteins were measured after an additional 24
h as total or cell surface fluorescence. Data are shown relative to the background level of
GFP. Results are expressed as mean±S.D. of three independent experiments.
3.1.7 Role of p115 in MIF Secretion in Macrophages
3.1.7.1 MIF and p115 are Co-Secreted in Activated Macrophages
In order to determine whether p115 is also of importance for stimulated
MIF release in macrophages, THP-1 monocytes were differentiated into
macrophages. Human THP-1 monocytes acquire many of the
characteristics of mature, primary macrophages upon differentiation with
phorbol 12-myristate 13- acetate (PMA), such as an increase in the
expression of the LPS co-receptor CD14 and the induction of cytokine
expression (137). Differentiated THP-1 macrophages thus respond to low
doses of LPS and show a plateau in the MIF secretion response at an LPS
concentration of 0.1 µg/ml. In addition to a lower threshold dose for
stimulating secretion, differentiated THP-1 also secrete ~5 fold more MIF
protein than undifferentiated THP-1 cells (Fig. 19).
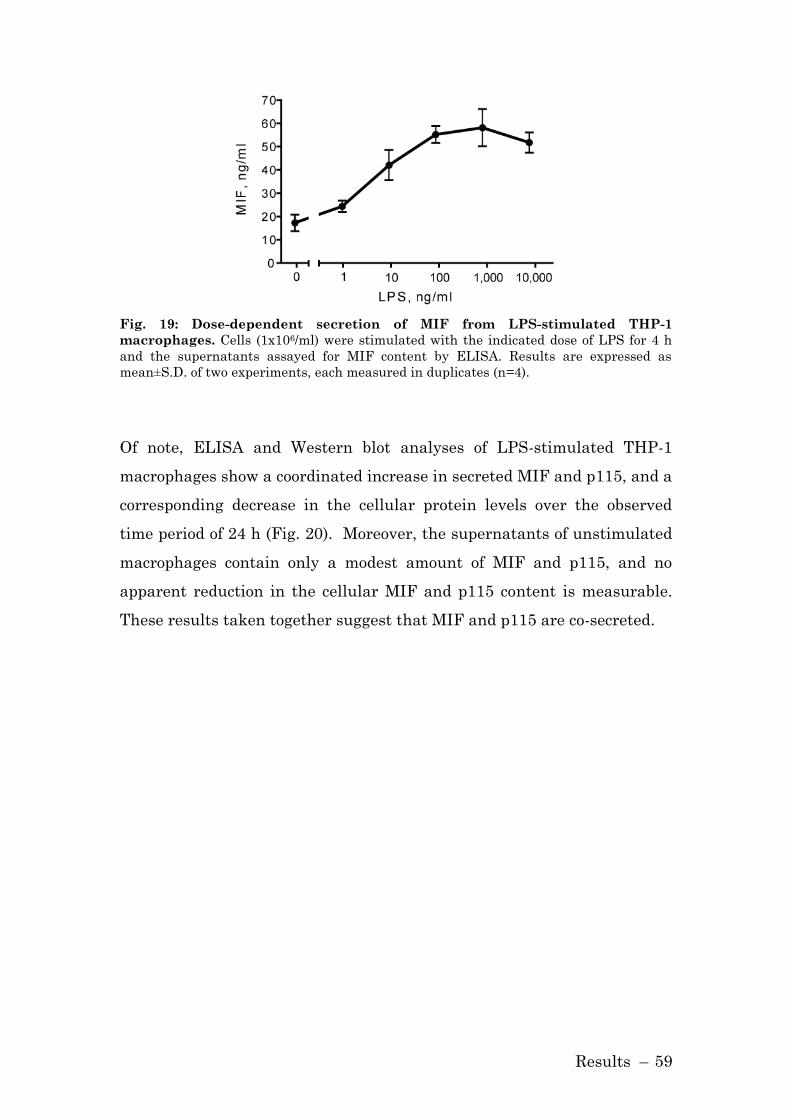
Results – 59
Fig. 19: Dose-dependent secretion of MIF from LPS-stimulated THP-1
macrophages. Cells (1x106/ml) were stimulated with the indicated dose of LPS for 4 h
and the supernatants assayed for MIF content by ELISA. Results are expressed as
mean±S.D. of two experiments, each measured in duplicates (n=4).
Of note, ELISA and Western blot analyses of LPS-stimulated THP-1
macrophages show a coordinated increase in secreted MIF and p115, and a
corresponding decrease in the cellular protein levels over the observed
time period of 24 h (Fig. 20). Moreover, the supernatants of unstimulated
macrophages contain only a modest amount of MIF and p115, and no
apparent reduction in the cellular MIF and p115 content is measurable.
These results taken together suggest that MIF and p115 are co-secreted.

Results – 60
Fig. 20: Co-secretion of MIF and p115 from macrophages. A) Differentiated THP-1
macrophages were stimulated with 0.1 µg/ml LPS or PBS (control) and the supernatants
were collected at the indicated time points. MIF concentration was measured by ELISA;
p115 release was determined by immunoblotting and subsequent densitometric analysis.
B) Cell lysates of THP-1 macrophages stimulated with LPS or control (PBS) were
prepared at the indicated time points. Intracellular MIF concentration was measured by
ELISA, and intracellular p115 level was determined by Western blot and densitometric
analysis. The ELISA results are expressed as mean values ±S.D. of two independent
experiments measured in duplicate. Densitometric results are expressed as mean values
±S.D. of at least two independent experiments. Statistical significance was determined
by Student’s t-test comparing LPS-stimulated vs. control time point, *p<0.05, **p<0.01.

Results – 61
Of note, the LPS stimulation of bone-marrow derived macrophages also
led to the export of both MIF and p115 protein (Fig. 21), affirming the
results obtained from THP-1 macrophages.
Fig. 21: Co-Secretion of MIF and p115 in primary macrophages. Bone-marrow
derived primary macrophages (0.5×106/ml) were stimulated with 1 µg/ml LPS or control
(PBS) and the supernatants collected at the indicated time points. MIF concentration was
measured by ELISA and p115 content was determined by immunoblotting.
Further evidence that MIF and p115 are co-secreted came from the
observation that glyburide, an pharmacological inhibitor known to inhibit
the secretion of MIF (105, 133), also shows an inhibitory effect on the
secretion of p115 (Fig. 22).
Fig. 22: The ABC-transporter inhibitor glyburide inhibits the secretion of MIF
and p115. THP-1 macrophages were pre-treated with glyburide (2.5 µM) or solvent
control (DMSO) for 1 h. Subsequently, MIF secretion was stimulated with LPS, and
supernatant were collected 4 h later. MIF concentration in supernatants was measured
by ELISA, and p115 concentration by Western blot analysis. MIF-ELISA: n=3
measurements, p<0.05.

Results – 62
3.1.7.2 Adenovirus-Mediated Depletion of p115
In order to deliver p115 siRNA to differentiated THP-1 macrophages, an
adenovirus construct was utilized. Adenoviruses do not infect monocytes
because of their low expression levels of integrins 3 and 5, which
mediate adenovirus uptake. Upon differentiation to macrophages, the
expression level of the integrins is up-regulated and adenoviruses
interact with the cells (145). The transfection of adenovirus encoding p115
siRNA, but not a control adenovirus led to a reduced intracellular p115
protein level in THP-1 macrophages (Fig. 23, A).
It has been reported that a depletion of p115 below detectable levels
compromises the structure of the Golgi and causes its fragmentation (102,
103). By immunofluorescent microscopy it was confirmed that the
morphology of the Golgi was not affected by the RNAi approach used
herein (Fig. 23, B).
Adenovirus-mediated delivery of p115 siRNA was also a successful
approach to reduce the p115 protein level in human peripheral blood
monocyte-derived macrophages (Fig. 23, C).

Results – 63
Fig. 23: Depletion of p115 by adenovirus-encoded siRNA in macrophages. A)
Differentiated THP-1 macrophages (1x106/ml) were transfected with an MOI of 50 of
adenovirus encoding siRNA against p115 or mock control siRNA. 96 h after transfection,
the cells were lysed and the protein content analyzed by Western blot and densitometry.
The results are expressed as mean values ±S.D. of three independent experiments. B)
THP-1 macrophages were differentiated on culture slides and p115 depleted by
adenovirus transfection. 96 h after transfection, cells were fixed and permeabilized and
stained for p115 (green) and the Golgi marker GM130 (red). Scale bar=10µm. C)
Adenovirus-mediated depletion of p115 reduces p115 protein level in primary human
macrophages. Human peripheral blood monocyte-derived macrophages (1x106/ml) were
transfected with 100 MOI of adenovirus carrying siRNA against p115 or mock control.
The cells were analyzed 48 h later for p115 content by Western blot. Knockdown of p115
was evaluated by densitometry. The results are expressed as mean values ±S.D. of three
independent experiments.

Results – 64
3.1.7.3 p115 is Required for the Secretion of MIF in Macrophages
After p115 was depleted in macrophages via adenoviruses carrying siRNA
targeting p115, the release of cytokines was stimulated with LPS. MIF
secretion in p115 depleted macrophages was reduced in response to LPS
stimulation in comparison to control cells over the 24 h experimental
period (Fig. 24, A). By contrast, the release of TNF-, IL-6 and IL-1 was
not affected by adenovirus encoded p115 siRNA, confirming that partial
p115 reduction does not interfere with the integrity of the Golgi
apparatus, while having a specific role in the release of MIF. Adenovirus-
mediated delivery of p115 siRNA also reduced p115 protein production in
human peripheral blood monocyte-derived macrophages (Fig. 23, C), and
reduced MIF release after stimulation with LPS by almost 50% (Fig. 24,
C).

Results – 65
Fig. 24: LPS-stimulated MIF secretion in macrophages. A) Time course of LPS-
stimulated MIF secretion in control or p115 siRNA treated THP-1 macrophages.
Supernatants were collected at the indicated time points and MIF content was measured
by ELISA. Supernatants also were assayed for TNF-, IL-6 and IL-1 by ELISA. The
results are expressed as mean values ±S.E. of triplicate measurements of two
independent experiments (n=6). Statistical significance was determined by comparing the
curves obtained in p115 knockdown vs. mock control by two-way ANOVA analysis. C)
LPS-stimulated MIF secretion is reduced in p115 siRNA, but not in mock siRNA treated
PBMCs. MIF secretion was stimulated with 10 µg/ml LPS for 4 h and the supernatants
analyzed for MIF content by ELISA. The results are expressed as mean values ±S.D. of
two independent experiments each measured in duplicates (n=4). Statistical significance
was determined by an unpaired Student’s t-test, *p<0.001.

Results – 66
The inhibition of MIF secretion also appeared to be dose-dependent with
respect to the level of p115 depletion (Fig. 25). When compared to controls,
a ~50% depletion of p115 was associated with a ~20% inhibition of MIF
release, while a ~75% depletion of p115 was associated with a ~40%
inhibition of MIF release.
Fig. 25: Dose-dependent effect of p115 depletion on MIF secretion. Differentiated
THP-1 macrophages were transfected with 50 MOI of adenovirus encoding siRNA against
p115 or mock control siRNA. 48 h after transfection (50% depletion of p115 compared to
mock treated cells) and 96 h after transfection (75% depletion of p115 compared to mock
treated cells), the cells were stimulated for 4 h with 0.1 µg/ml LPS. The supernatants
were analyzed for MIF release by ELISA, and data are shown relative to mock treated
cells. Results are representative for two independent experiments measured in
duplicates. Statistical significance was determined by Student’s t-test, *p<0.05, **p<0.01.
In p115-depletion studies, it was shown that the efficient release of MIF
from monocytes/macrophages in response to LPS depends on the presence
of p115. To determine whether an excess of p115 protein leads to an
enhanced secretion of MIF, p115 was overexpressed in THP-1
macrophages utilizing an adenovirus construct carrying a p115 expression
construct. The overexpression of p115 was confirmed by Western blot, and
the effect of p115 overexpression on LPS stimulated MIF secretion was
evaluated by ELISA (Fig. 26). The overexpression of p115 had no effect on
the LPS-stimulated release of MIF or the conventionally secreted cytokine
TNF-.
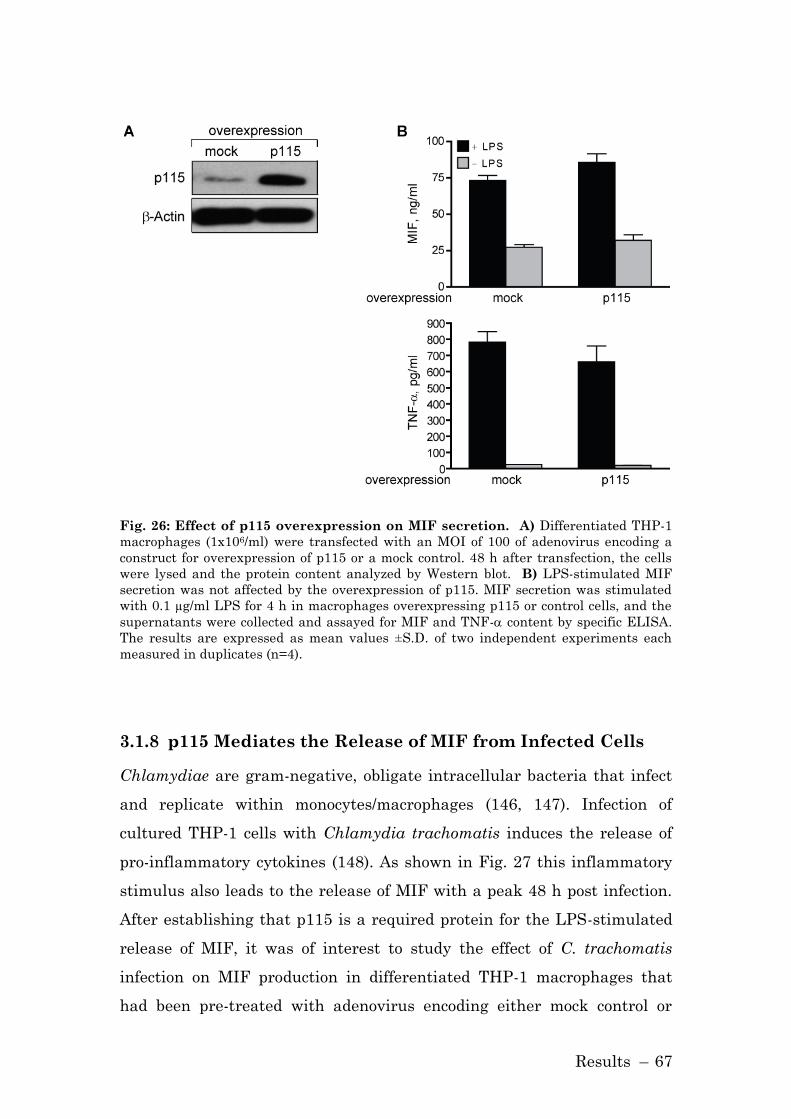
Results – 67
Fig. 26: Effect of p115 overexpression on MIF secretion. A) Differentiated THP-1
macrophages (1x106/ml) were transfected with an MOI of 100 of adenovirus encoding a
construct for overexpression of p115 or a mock control. 48 h after transfection, the cells
were lysed and the protein content analyzed by Western blot. B) LPS-stimulated MIF
secretion was not affected by the overexpression of p115. MIF secretion was stimulated
with 0.1 µg/ml LPS for 4 h in macrophages overexpressing p115 or control cells, and the
supernatants were collected and assayed for MIF and TNF- content by specific ELISA.
The results are expressed as mean values ±S.D. of two independent experiments each
measured in duplicates (n=4).
3.1.8 p115 Mediates the Release of MIF from Infected Cells
Chlamydiae are gram-negative, obligate intracellular bacteria that infect
and replicate within monocytes/macrophages (146, 147). Infection of
cultured THP-1 cells with Chlamydia trachomatis induces the release of
pro-inflammatory cytokines (148). As shown in Fig. 27 this inflammatory
stimulus also leads to the release of MIF with a peak 48 h post infection.
After establishing that p115 is a required protein for the LPS-stimulated
release of MIF, it was of interest to study the effect of C. trachomatis
infection on MIF production in differentiated THP-1 macrophages that
had been pre-treated with adenovirus encoding either mock control or
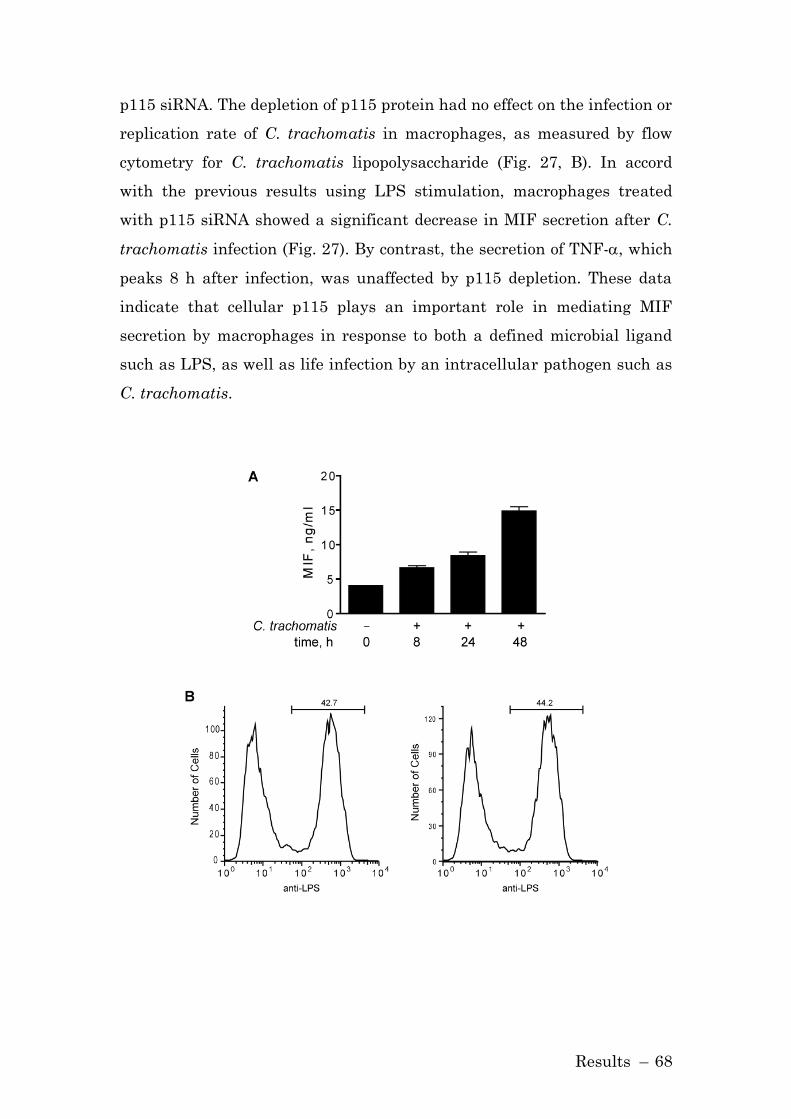
Results – 68
p115 siRNA. The depletion of p115 protein had no effect on the infection or
replication rate of C. trachomatis in macrophages, as measured by flow
cytometry for C. trachomatis lipopolysaccharide (Fig. 27, B). In accord
with the previous results using LPS stimulation, macrophages treated
with p115 siRNA showed a significant decrease in MIF secretion after C.
trachomatis infection (Fig. 27). By contrast, the secretion of TNF-, which
peaks 8 h after infection, was unaffected by p115 depletion. These data
indicate that cellular p115 plays an important role in mediating MIF
secretion by macrophages in response to both a defined microbial ligand
such as LPS, as well as life infection by an intracellular pathogen such as
C. trachomatis.

Results – 69
Fig. 27: p115 knockdown inhibits MIF secretion in Chlamydia trachomatis
infected human macrophages. A) THP-1 cells (1x106/ml) were infected with 10 MOI
of C. trachomatis. Supernatants were collected at the indicated time points and analyzed
for MIF release by ELISA. B) p115-depleted or mock control THP-1 macrophages were
analyzed for C. trachomatis infection and replication by flow cytometry. 24 h post
infection, cells were washed, permeabilized, and stained for C. trachomatis using an anti-
LPS antibody. The results are representative for at least three independent experiments.
Statistical significance of infection of p115 depleted vs. mock treated cells was analyzed
by unpaired Student’s t-test. Mock siRNA: 38±5%, p115 siRNA: 39±8%. C) THP-1
macrophages were transfected with 50 MOI of adenovirus encoding a p115 or control
(mock) siRNA construct 96 h prior to infection with C. trachomatis. Supernatants were
collected at the indicated time points and the levels of MIF and TNF- were analyzed by
ELISA. Results are expressed as mean±S.E. of one experiment measured in triplicates.
Data are representative of three independent experiments. Statistical significance was
analyzed by unpaired Student’s t-test, *p<0.01.

Results – 70
3.1.9 Small Molecule Inhibitor Blocks MIF Secretion
Due to the unique structure of MIF and its tautomerase activity, several
prototypic small molecule inhibitors targeting MIF function have been
designed (61, 64). These inhibitors (ISO-1 and NAPQI) bind to MIF’s
amino-terminal proline thus inhibiting the tautomerase activity. Mitchell
and colleagues recently described the suicide substrate, 4-iodo-6-
phenylpyrimidine (4-IPP), and showed by crystallography that 4-IPP
covalently binds to the amino-terminus of MIF (65).
3.1.9.1 Immunorecognition of 4-IPP-Modified MIF
In a prior report by Senter et al. it was observed that the covalent
modification of the amino-terminus of MIF alters its immunorecognition in
a two-antibody sandwich ELISA. Indeed, MIF that had been incubated
with 4-IPP showed increased immunorecognition by a MIF-specific
monoclonal antibody (clone MAB289) when compared to native MIF. By
contrast, when analyzing the 4-IPP/MIF complex with a different anti-
MIF mAb (clone 3H2F), a diminished recognition compared to native MIF
was observed (Fig. 28).
Fig. 28: 4-IPP modifies the immunorecognition of MIF in a two antibody
sandwich ELISA. Quantification of 4-IPP modified MIF (MIF-4-IPP) or unmodified MIF
(MIF) by two distinct ELISAs. A) MIF-4-IPP shows decreased immunorecognition by the
monoclonal anti-MIF antibody 3H2F. B) MIF-4-IPP displays enhanced detection by the
monoclonal anti-MIF antibody MAB289.

Results – 71
By contrast, the immunodetection of MIF by Western blot, which detects
denatured, linearized proteins, was not affected by 4-IPP modification
(Fig. 29).
Fig. 29: Immunorecognition of 4-IPP modified MIF by Western blot. MIF was
incubated with either vehicle control (DMSO), 0.1x molar excess of 4-IPP, or 10x molar
excess of 4-IPP over night. Samples were run on a SDS-PAGE and transferred to a PVDF
membrane. Unmodified and 4-IPP modified MIF were quantified and detected by
Coomassie staining, Ponceau S staining, and Western blot (polyclonal anti-MIF
antibody).
3.1.9.2 4-IPP Targets the MIF/p115 Interaction
Small molecule MIF inhibitors are able to inhibit MIF functions. It was
reported that ISO-1 effectively protects mice from LPS shock and West
Nile virus infection (20, 149). In these models, ISO-1 exhibited similar
protective properties as neutralizing anti-MIF antibody or genetic mif
deletion (21, 149).
To analyze whether the prototypic inhibitor 4-IPP inhibits the release of
MIF after LPS stimulation in THP-1 macrophages, Western blot analysis
was employed to quantify released MIF. Fig. 30, A shows that 4-IPP
effectively blocked the release of both MIF and p115 after LPS stimulation
in macrophages. Analysis of the cytosolic concentration of MIF and p115
in response to LPS treatment showed that the level of both proteins in the
cytoplasm was decreased by ~50% after stimulation with LPS, while in
macrophages pre-treated with 4-IPP the intracellular concentration of
MIF and p115 remained unchanged.
4-IPP also reduced the secretion of MIF from monocytes infected with
Chlamydia trachomatis (Fig. 30, B).

Results – 72
Fig. 30: 4-IPP inhibits MIF release from macrophages. A) THP-1 macrophages
(1×106/ml) were pre-treated with 1 µM of 4-IPP for 1 h prior to stimulation with 0.1 µg/ml
LPS, and MIF and p115 was measured in supernatants and cell lysates by Western
blotting. Western blots were quantified by densitometry relative to β-actin. Mean±S.E.
of at least two independent experiments are stated below the blots. Statistical
significance for the comparison of LPS + 4-IPP vs. LPS alone was analyzed by an
unpaired Student’s t-test, *p<0.05. 4-IPP did not compromise cell viability, supernatants
showed LDH activity of >5 mU/ml LDH. B) THP-1 monocytes were infected with an MOI
of 10 of C. trachomatis and treated after infection with 1 µM of 4-IPP or control over the
time of infection. Supernatants were collected at the indicated time points and analyzed
for MIF content by Western blot. Results are representative for three experiments.
In contrast, 4-IPP did not affect the LPS-stimulated release of TNF-α or
IL-1β (Fig. 31, A); this affirms that 4-IPP has no overall effect on the
secretion mechanism of cells. Furthermore, the influence of the reversible
MIF inhibitor ISO-1 on MIF release from stimulated THP-1 macrophages
was investigated, but no effect was observed (Fig. 31, B).

Results – 73
Fig. 31: 4-IPP does not target release of other cytokines, ISO-1 does not
influence MIF release. A) Supernatants of LPS-stimulated THP-1 macrophages that
had been treated with 4-IPP or control assayed for the release of TNF-α and IL-1β. The
results are expressed as mean±S.D. of one experiment measured in duplicates. Data are
representative for two independent experiments. B) THP-1 macrophages pre-treated
with ISO-1 were stimulated with 0.1 µg/ml LPS. MIF release at the indicated time points
was analyzed by ELISA. The results are expressed as mean±S.D. of one experiment
measured in duplicates. Data are representative for two independent experiments.
The observation that 4-IPP alters the immunorecognition of MIF by anti-
MIF monoclonal antibodies (Fig. 28) suggests that 4-IPP may impart a
significant conformational affect on MIF that may influence its binding to
p115. To test this hypothesis, the impact of 4-IPP on the competitive
binding of MIF to p115 was examined in vitro. As shown in Fig. 32, 4-IPP,
but not the small molecule MIF inhibitors ISO-1 or NAPQI (61, 64),
increase MIF binding to p115. The enhanced binding of MIF to p115 in the
presence of 4-IPP thus may play a role in the inhibitory action of 4-IPP on
MIF release from cells, perhaps by interfering with additional protein
interactions necessary for secretion.

Results – 74
Fig. 32: 4-IPP enhances the binding of MIF to p115. Competitive binding to
immobilized p115 of biotinylated MIF vs. increasing concentrations of MIF pre-incubated
with the small molecule MIF inhibitors 4-IPP, NAPQI, ISO-1, or solvent control. Results
are expressed as mean±S.E. of one experiment measured in duplicate. The data are
representative of three independent experiments.
3.1.9.3 Effect of 4-IPP in LPS-Shock Model
The small molecular MIF inhibitor ISO-1 has a protective effect in the
LPS-shock model (20). Therefore, it was examined, whether the prototypic
inhibitor, 4-IPP, also protects mice from LPS shock. 4-IPP had no
significant effect on the survival following LPS challenge (Fig. 33).
Fig. 33: 4-IPP does not protect mice in a model of endotoxemia. Female BALB/c
mice (6-8 weeks) were intraperitoneally injected with 1 mg 4-IPP or vehicle control
(DMSO and corn oil). 2h later, LPS was injected (17.5 mg/kg). Mice were observed for 6
days. Data points are from one experiment (N=10 mice per group, p=NS), representative
for three independent experiments.

Results – 75
In addition to studying the effect of 4-IPP on the survival of mice in the
LPS-shock model, it was of interest to analyze whether the administration
of 4-IPP modulates the release of MIF and other pro-inflammatory
cytokines. In contrast to the in vitro experiments, 4-IPP did not
significantly inhibit the release of MIF into the serum, although a modest
reduction of MIF levels was observed. Cytokines downstream of MIF
(TNF-α, IL-6) were also not targeted in their release by 4-IPP in the
examined model (Fig. 34).
Fig. 34: 4-IPP does not affect the release of MIF after LPS injection. Female
BALB/c mice (6-8 weeks) were intraperitoneally injected with 1 mg 4-IPP or vehicle
control (DMSO and corn oil). 2h later, a sublethal dose of LPS was injected (15 mg/kg). At
the indicated time points, serum was collected and assayed for cytokine content by
ELISA. (N=5 mice per group per time point), the experiment was performed three times
independently.

Results – 76
3.2 D-Dopachrome Tautomerase (DDT)
Like MIF, D-dopachrome tautomerase (DDT), catalyzes enzymatic
reactions of the non-physiological substrate D-dopachrome in its amino-
terminal tautomerase active site.
Since the “rediscovery” of MIF in the early 90s, the knowledge about MIF
grew steadily, but little is known about DDT. Crystallographic analysis of
DDT revealed that although MIF and DDT share only limited sequence
homology, the three-dimensional structures of the two proteins show
remarkable similarity (69). The gene structure of MIF and DDT also show
similarities: The two proteins are closely linked on chromosome 22 (in
humans) or chromosome 10 (in mice), respectively; moreover, the two
proteins have an identical exon structure (150). Nevertheless, studies
addressing the biological functions of the structural homolog of MIF are
limited (72), and the molecular action of DDT remains unknown.
3.2.1 Purification of D-Dopachrome Tautomerase
3.2.1.1 Purification of Histidine-tagged DDT
In order to expedite the process of obtaining pure DDT protein, mouse
DDT cDNA was cloned into the pET22b vector. The resulting fusion
protein possesses a short carboxy-terminal histidine tag (6x His), and was
overexpressed in E. coli BL21 (DE3). Utilizing the carboxy-terminal
histidine tag, DDT was purified over a Ni2+ column. The purity of the
protein was determined by SDS-PAGE and Coomassie staining (Fig. 35).

Results – 77
Fig. 35: Purification of Histidine-tagged mouse DDT. Purified recombinant mouse
DDT revealed by SDS-PAGE followed by Coomassie staining.
The purified protein was sent to Pocono Rabbit Farm & Laboratory
(PRF&L) for the production of a polyclonal anti-DDT antibody. A rabbit
was periodically injected with 100 µg of DDT protein in Complete Freund’s
adjuvant in the popliteal lymph nodes, serum was collected after day 28,
42 and 70.
3.2.1.2 Purification of Native DDT Protein
From the MIF protein it is known that the amino-terminus is of
importance not only for the tautomerase activity, but also for various
biological actions (ERK 1/2 activation, glucocorticoids overriding ability,
CD74 receptor binding). Similarly, the carboxy-terminus was shown to be
crucial for the trimer formation (51), thus it was of importance to purify
recombinant, native DDT protein, to successfully perform characterization
studies.
Two stop codons were cloned after the DDT cDNA in pET22b to prevent
the expression of the carboxy-terminal histidine-tag and obtain native
DDT protein; the insertion was confirmed by DNA sequencing. After the
overexpression in E. coli BL21 (DE3), DDT was sequentially purified
utilizing anion exchange chromatography, followed by gel filtration. Fig.
36 shows the purification steps performed for obtaining pure protein.
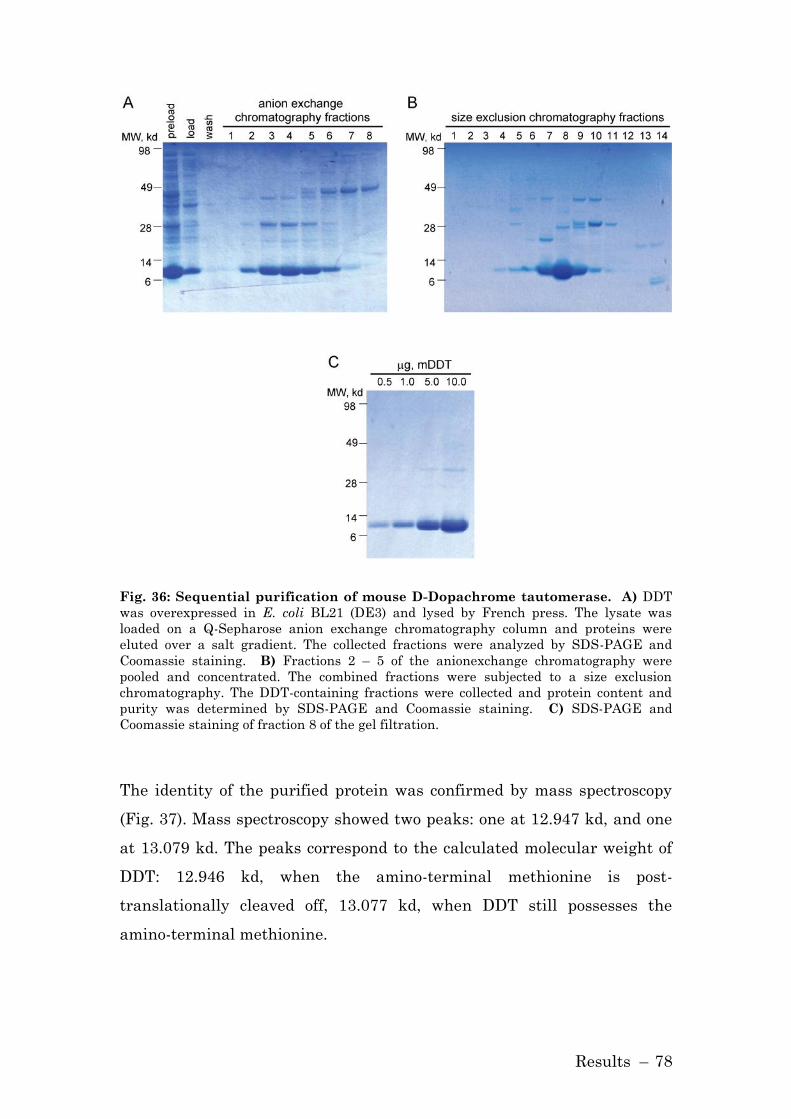
Results – 78
Fig. 36: Sequential purification of mouse D-Dopachrome tautomerase. A) DDT
was overexpressed in E. coli BL21 (DE3) and lysed by French press. The lysate was
loaded on a Q-Sepharose anion exchange chromatography column and proteins were
eluted over a salt gradient. The collected fractions were analyzed by SDS-PAGE and
Coomassie staining. B) Fractions 2 – 5 of the anionexchange chromatography were
pooled and concentrated. The combined fractions were subjected to a size exclusion
chromatography. The DDT-containing fractions were collected and protein content and
purity was determined by SDS-PAGE and Coomassie staining. C) SDS-PAGE and
Coomassie staining of fraction 8 of the gel filtration.
The identity of the purified protein was confirmed by mass spectroscopy
(Fig. 37). Mass spectroscopy showed two peaks: one at 12.947 kd, and one
at 13.079 kd. The peaks correspond to the calculated molecular weight of
DDT: 12.946 kd, when the amino-terminal methionine is post-
translationally cleaved off, 13.077 kd, when DDT still possesses the
amino-terminal methionine.

Results – 79
Fig. 37: Mass spectroscopy of D-Dopachrome tautomerase. Electrospray ionization
mass spectrometry of murine DDT showing a molecular mass (m/z) that lies within
0.02% accuracy of the predicted m/z (13.077 kd and 12.946 kd).
3.2.2 Establishment of a DDT-ELISA
MIF and DDT share 28% sequence identity and the overall topology and
trimeric formations of DDT are similar to those of MIF. Therefore, the
newly produced polyclonal anti-DDT antibody was analyzed for cross
reactivity with both human and murine MIF protein in Western Blot
analysis. Fig. 38 demonstrates that the antibody detects DDT protein,
while exhibiting no cross reactivity for murine or human MIF.

Results – 80
Fig. 38: Polyclonal anti-DDT antibody shows no cross reactivity for MIF in
Western blot. 100, 10 and 1 ng protein (DDT, mMIF or hMIF) were separated on a
SDS-PAGE gel and transferred on PVDF membrane. The membrane was probed with the
IgG fraction of the serum obtained from PRF&L.
In order to analyze and quantify, if DDT is released from cells, an indirect
ELISA for DDT was established. The produced and purified anti-DDT
antibody served both as a capture and detection antibody; the antibody
was biotinylated when used as a detection antibody. The developed ELISA
detects DDT protein in a range of 100 – 5000 pg/ml, and shows no
measureable cross reactivity with MIF (Fig. 39).
Fig. 39: Specific detection of DDT protein in a newly established ELISA. A)
ELISA plate was coated with polyclonal anti-DDT antibody (15 µg/ml). After incubation
with blocking solution, either DDT or MIF protein was added in a range of 100 – 5000
pg/ml. For the detection of proteins, first biotinylated anti-DDT, then a streptavidin-HRP
conjugate was added to the plate. Protein concentration was detected by colorimetric
measurement of TMB conversion. B) DDT ELISA does not significantly cross react with
mouse MIF. A microtiter plate was coated with anti-DDT antibody and either DDT alone
(100 – 5000 pg/ml) or DDT (100 – 5000 pg/ml) plus 50 ng/ml MIF was added. Protein was
detected by colorimetric measurement at 450 nm.

Results – 81
3.2.3 DDT is released from macrophages
To date, little is known about the biological function of DDT. Both MIF
and DDT are leaderless proteins, and it was of interest to analyze whether
DDT like MIF is released from cells in response to a pro-inflammatory
stimulus. Fig. 40 illustrates that not only MIF, but also DDT is released
from bone marrow-derived macrophages in response to LPS.
Fig. 40: Release of DDT and MIF in response to LPS. Bone marrow-derived
macrophages were stimulated with LPS (1 µg/ml) ore PBS (control). At the indicated time
points, supernatants were collected and assayed for DDT and MIF by specific ELISA. The
results are representative for three independent experiments.
The pattern of release of DDT and MIF after stimulation with LPS is very
similar. Both proteins peak at 16 h, exhibiting a bell-shaped curve often
observed for released cytokines.

Results – 82
3.2.4 Effect of mif-gene deletion on DDT production
Both MIF and DDT contribute to the secretion of CXCL8 and VEGF (72)
and both proteins are released in response to LPS stimulation of
macrophages. Given these overlapping functions of the two proteins it was
considered that a loss of the mif-gene might be compensated by higher
expression levels of DDT. Therefore, the intracellular DDT concentration
in wildtype and mif-/- macrophages was measured (Fig. 41, A), but no
difference was observed. These results were also reflected when comparing
DDT concentration in serum of wildtype vs. mif-/- mice (Fig. 41, B). Last, it
was analyzed whether the deletion of the mif-gene had an effect on the
stimulated release of DDT, but again no difference between wildtype and
mif-/- macrophages was measurable (Fig. 41, C).
Fig. 41: Comparison of DDT production. A) Bone-marrow derived macrophages of
wildtype (mif+/+) and MIF knockout (mif-/-) were lysed and DDT concentration was
measured by ELISA, total protein concentration was analyzed using Bradford, (N=8 mice
per group). B) Serum samples of mif+/+ and mif-/- were collected, and DDT concentration
was analyzed by Bradford (N=6 mice per group). C) Bone-marrow derived macrophages
of wildtype (mif+/+) and MIF knockout (mif-/-) were stimulated with LPS (1 µg/ml) or
control (PBS). At the indicated time points, supernatants were collected and assayed for
DDT and MIF by specific ELISA. The results are representative for three independent
experiments.

Results – 83
The functional role of DDT is still unclear. In general DDT might either
have similar functions as MIF, thus supporting MIF in its pro-
inflammatory, anti-apoptotic role or it might be a counter-regulator of
MIF, attenuating the effects of MIF.

Discussion – 84
4 Discussion
4.1 Role of p115 in MIF Secretion
Macrophage migration inhibitory factor plays an important upstream role
in the regulation of diverse cellular responses (8, 151, 152) and its role in
human pathology has been emphasized by the finding that high
expression MIF alleles are associated with the incidence or the severity of
inflammatory and oncologic diseases (12-14, 153).
MIF is constitutively expressed in a large number of tissues,
predominantly in those that are in contact with the host’s environment.
(19, 154, 155). The release of MIF, especially from immune cells, occurs in
many inflammatory diseases, but the precise mechanism underlying the
secretion of MIF, which lacks an amino-terminal signal sequence, has
remained enigmatic (4, 5). MIF has been reported to be associated with
vesicles (134-136), and its release from stimulated monocytes is reduced
by glyburide and probenicide, which are pharmacological inhibitors mainly
targeting the ABCA1 transporter (105, 133).
In many inflammatory diseases, the amount of released MIF positively
correlates with disease severity, e.g. in sepsis, high MIF levels in the
serum are in direct relation with a fatal outcome (156-159). Moreover, the
administration MIF-neutralizing agents such as anti-MIF antibodies or
the MIF inhibitor ISO-1, were shown to be protective in diverse disease
models (20, 149). Together, these data clearly demonstrate the importance
of understanding the molecular mechanisms underlying MIF secretion.
To this end, a yeast two-hybrid screen was performed to identify
intracellular proteins that interact with MIF. In this screen, p115 was
found as a new binding partner of MIF. The specificity of the screen was
confirmed by the finding that 93% of the specifically interacting clones
were MIF itself, which associates into a non-covalently associated
homotrimer (50, 51). However, a cDNA clone encoding the carboxyl
terminal 260 amino acids of the Golgi-associated protein p115 also was

Discussion – 85
identified. Co-immunoprecipitation studies confirmed the interaction
between MIF and p115.
4.1.1 MIF is Released from a Pre-Formed Pool
Lipopolysaccharide (LPS) was one of the first stimuli reported to cause the
secretion of MIF from cells (5, 8). In order to determine the pathway by
which MIF is released from immune cells, it was indispensable to
establish a reliable system to monitor the stimulated release of MIF.
Monocytes stimulated with LPS effectively release MIF, while the mRNA
level of both MIF and p115 remain unchanged. These findings are in
accord with previous observations in murine tissue (19, 140) and clearly
show that MIF is secreted from pre-formed stores and is not synthesized
de novo. In contrast, other cytokines, such as TNF-α and IL-1β, show a
dramatic increase in their mRNA expression level 1 – 3 h post LPS
stimulation (160, 161). A possible explanation for the difference in mRNA
expression profiles may be that TNF-α and IL-1β operate exclusively as
cytokines, whereas MIF has cytokine functions, but in addition also has an
important intracellular role. For instance, it was reported that MIF
contributes to cell proliferation via direct interaction with the cell cycle
regulators p53 and JAB1 (43, 45, 47).
4.1.2 MIF and p115 Associate at the Perinuclear Site
Previous studies have demonstrated that p115 is localized at the Golgi
(95). Immunofluorescent microscopy analysis in macrophages showed that
MIF is localized throughout the cytoplasm, and that MIF and p115
associate most notably in the area of the Golgi. The comparison of MIF
and p115 distribution in unstimulated vs. LPS-stimulated macrophages
furthermore showed that upon stimulation MIF is found predominantly in
a plasma membrane proximal area, whereas unstimulated cells stain for
MIF predominantly in the perinuclear ring. p115 is localized at the Golgi,

Discussion – 86
and upon stimulation a portion of p115 is dispersed in the cytoplasm. This
suggests that the interaction between MIF and p115 occurs early in the
secretory pathway and that MIF and p115 are co-secreted in vesicles. p115
may be essential in the transport of MIF from the perinuclear ring to the
plasma membrane and then out of the cell. This hypothesis is supported
by data showing a co-secretion of MIF and p115 in LPS stimulated cells.
It was reported that MIF is detected in vesicles in pituitary cells, cells of
the testis, and β-cells of the pancreas (134-136). It is of interest to further
characterize the location of MIF and p115 in immune cells. Electron
microscopy images labelled for MIF and p115 might help to understand
whether MIF and p115 localize together at vesicles or if p115 just
momentarily associated with MIF, presumably to help transfer MIF into
vesicles.
4.1.3 MIF and p115 Interact Directly
The interaction between MIF and p115 was discovered in a yeast two-
hybrid screen and affirmed in several co-immunoprecipitation studies in
mammalian cell systems. Furthermore, the interaction of the two proteins
was confirmed biochemically in a competition binding assay utilizing
recombinant MIF and p115702-962. The assay demonstrated that the two
proteins bind directly and specifically without necessarily requiring
additional, intermediary proteins for the interaction. The percent of
competition measured was between 50 and 60%. In contrast, the percent of
competition reached between MIF and its receptor CD74 is near 100%
(58).
A likely explanation for the levelling of the competition curves at 50 – 60%
is that there is some non-specific background binding to the plate which
cannot be fully blocked by the Superblock reagent. Alternatively, the
biotinylated species (both biotin-p115 and biotin-MIF) may have higher
non-specific binding than the non-modified, native protein. It also is

Discussion – 87
possible that there exist two conformational species of the immobilized
protein on the plate, and that only one can be competed for.
4.1.4 p115 Is Required for the Stimulated Release of MIF
p115 is a Golgi-associated protein known to be predominantly localized to
the cis-Golgi and the vesicular tubular clusters (VTC). It was
demonstrated that the cellular depletion of p115 causes a decrease in MIF
export from stimulated cells.
Previous reports describe that the depletion of p115 below detectable
levels and the microinjection of anti-p115 antibodies causes a
fragmentation of the Golgi leading to a delay in protein export (101-103,
162). In contrast, the partial depletion of p115 by the RNAi technique used
in this study neither affected the structural morphology of the Golgi nor
reduced the export of ER/Golgi-dependent proteins such as TNF-α, IL-6
and FGF-4. The release of the unconventionally secreted protein IL-1β,
which occurs via vesicles (118), and FGF-2, which translocates across the
plasma membrane (114) also was not affected by a depletion of p115. That
p115 is necessary for the release of MIF but not other cytokines, whether
conventionally or unconventionally secreted, suggests a specific and
unique role in the export of MIF (Fig. 42).

Discussion – 88
Fig. 42: Scheme of p115-dependent MIF secretion. Left: p115 is localized at the cis-
side of the Golgi where it mediates the tethering of vesicles arriving at the Golgi from the
ER, contributing to the conventional secretion of proteins. MIF and p115 also interact in
the area of the Golgi. Right: p115 is partially depleted, but still mediates the tethering of
ER-derived vesicles at the Golgi. MIF is not longer effectively released from cells.
The role of p115 in the transfer of vesicles from the ER to the Golgi has
been established for some time (97). In this process, the carboxy-terminus
of p115 interacts with the golgin protein GM130 to tether COPII-coated
vesicles to the cis-side of the Golgi. While the depletion of p115 inhibits
the stimulated release of MIF, the depletion of GM130 has no measureable
effect on the release of MIF. This observation indicates that the role of
p115 in MIF secretion is different from its role in the conventional
secretion of proteins. To confirm this hypothesis one can analyze the role
of Rab1, a small GTPase, in the release of MIF, since p115 is a Rab1
effector protein. The active form of Rab1 binds to the amino-terminal head
of p115 (96). Rab1 exists in two isoforms, Rab1A and Rab1B; both have
been suggested to regulate anterograde transport of cargo between the ER
and the Golgi apparatus (163). Therefore, an efficient knockdown of Rab1
for a comprehensive study of its role in MIF secretion is difficult to
accomplish. Another approach to study the effect of Rab1 on MIF release
is the microinjection of guanine nucleotide dissociation inhibitor (GDI). It

Discussion – 89
was demonstrated that the administration of GDI effectively blocks the
function of Rab1 by blocking the transfer of stomatitis virus glycoprotein
(VSV-G) from the ER to the cell surface (164). The disadvantage of this
approach is that in contrast to the previous studies the conventional
secretion pathway is compromised; therefore any effect seen after the
microinjection of GDI could be either contributed to a direct effect on the
MIF secretion or due to the compromising of the cell system, which then
secondarily leads to a reduced secretion rate of MIF.
4.1.4.1 p115 Overexpression does not Affect MIF Secretion
For the intracellular binding partner of MIF, JAB1, it was demonstrated
that both its depletion and its overexpression lead to the modulation of the
MIF signaling pathway (40, 44).
While the depletion of p115 consistently decreased MIF release in various
experimental approaches, the overexpression of p115 had no affect on
either the stimulated release of MIF or TNF-α. These observations
indicate that p115 may not be the only limiting factor in the process of
MIF release, but that other proteins are likely required. To address this
hypothesis it is of interest to screen for MIF interaction partners in the
mammalian cell system. Thus far in efforts to identify new MIF binding
partner only yeast two-hybrid screens were performed. Other approaches,
like tandem affinity purification (TAP) in the mammalian cell system
(165) might lead to the discovery of new MIF interaction partners or even
a multi-protein complex, including MIF and p115, thus possibly
identifying other proteins involved in the pathway of MIF export.
4.1.4.2 MIF and p115 are Co-Secreted
Macrophages stimulated with LPS export both MIF and p115 in a pattern
that suggests the coupled release of the two proteins. This observation is

Discussion – 90
supported by reports linking both MIF (134-136) and p115 (126) to
vesicles. The co-secretion of the two proteins affirms the previous
observations that p115 mediates the release of MIF.
So far it is unknown whether MIF and p115 locate to exosomes in
macrophages. To address this question, supernatants of stimulated
macrophages have to be examined. One potential approach is the isolation
of vesicles by gradient centrifugation and analyzing the vesicles for MIF
and p115. Another method might be the further examination of the two
proteins in the supernatant after cross-linking or co-immunoprecipitation.
A positive outcome of these experiments would support the idea that MIF
and p115 are released together, possible in exosomes.
It is also conceivable that p115 mediates a generalized, non-classical
secretory response by activated monocytes/macrophages that results in the
release of a p115 macromolecular complex containing numerous proteins,
of which MIF is one. Further elucidation of both the signals and the
protein-protein interactions underlying this export pathway may be
informative and enhance our understanding of the acute secretory
response of activated monocytes/macrophages.
4.1.5 4-IPP Inhibits the Release of MIF and p115
4-IPP is the first pharmacological inhibitor for MIF found to block the
secretion of MIF in macrophages in vitro. This was demonstrated via LPS
stimulation and C. trachomatis infection. Furthermore 4-IPP treatment
prevents the export of p115 after LPS stimulation, thus affirming the
hypothesis that MIF and p115 are released in a complex. These data
identify 4-IPP as a potentially selective inhibitor of the p115/MIF
secretory pathway. 4-IPP covalently modifies the MIF amino-terminus,
and alters the protein’s conformational integrity so as to increase its
binding interaction with p115. One possibility might be that MIF’s
enhanced binding to p115 in the presence of 4-IPP disrupts its normal

Discussion – 91
trafficking pathway and secondarily reduces MIF secretion. 4-IPP thus
provides proof-of-concept for the possibility of reducing MIF-dependent
responses in the earliest phase of cell activation by interfering with MIF’s
cytoplasmic release.
The administration of 4-IPP did not improve the survival in a murine
model of LPS shock. Furthermore, 4-IPP did not reduce the serum level of
MIF and other cytokines downstream of MIF in response to LPS shock.
Numerous possibilities might explain why 4-IPP blocks MIF secretion in
vitro, while it does not inhibit MIF release in vivo. 4-IPP is a highly
lipophilic reagent; due to its hydrophobic properties, 4-IPP easily crosses
the plasma membrane, but the solubility in hydrophilic solvents in
concentrations necessary for an in vivo administration is unsatisfactory.
Both MIF inhibitors, 4-IPP and ISO-1, are first dissolved in DMSO, but
while ISO-1 is diluted in water for the intraperitoneal injection, 4-IPP has
to be diluted in corn oil to avoid precipitation. Mitchell and colleagues are
in the process of modifying 4-IPP to improve its solubility in water, and it
is of interest whether a decreased hydrophobicity provides protection in
the LPS shock model. Variables, such as the time point and dose of 4-IPP
administration also contribute to the outcome of the experiment. In this
study, 4-IPP was injected 1.5 h prior to LPS, likewise to the protocol for
the administration of neutralizing antibody and ISO-1 (5, 20). In contrast
to neutralizing antibody or ISO-1, it is not known whether 4-IPP
interferes with MIF’s ability to interact with its receptor CD74, thus a
different time point of administration might be favourable. Once the
solubility of the compound is improved, various concentrations and time
points of administration should to be tested. Furthermore, in the LPS-
shock model, MIF is not only released by specific secretion, but also due to
cell death, and this latter source of MIF cannot be blocked by 4-IPP.
Nevertheless, inhibitors such as 4-IPP may be attractive in clinical
application in those settings, such as severe inflammation, in which rapid
intervention is desired to prevent the initiation of a tissue-damaging,
cytokine cascade (166).

Discussion – 92
4.2 The Structural Homologue DDT
Most cytokines, such as IL-1, belong to large superfamilies. MIF has no
cytokine family, and its protein family comprises only a few members: D-
dopachrome tautomerase in eukaryotes and the two bacterial proteins 4-
oxalocrotonate tautomerase (4-OT) and 5-carboxymethyl-2-hydroxy-
muconate isomerase (CHMI) (167).
The members of the MIF/tautomerase superfamily all possess a
tautomerase active site that has its catalytic base in the amino-terminal
proline-1. To date, the physiological function for this enzymatic activity is
unknown, but in the case of MIF, several biological functions are linked to
this region (61).
The three-dimensional structures of human MIF and DDT are highly
conserved; the protomeric polypeptides of the two proteins consist of six β-
strands and two α-helixes. Four of the β-strands form a β-sheet; the
remaining two β-strands are important for the formation of the trimer by
forming hydrogen bonds to the adjacent protomers (50, 69) (Fig. 1 and Fig.
5). However, while the knowledge about MIF’s prominent role as regulator
of the innate and adaptive immunity has grown tremendously over the
last decades, nothing is known about the physiological role of DDT.
In order to establish the basic tools for a better understanding of the
biological functions of DDT, the native, recombinant protein was purified
and the first specific polyclonal antibody targeting murine DDT was
produced. This antibody does not show any cross reactivity to MIF.
Utilizing this new antibody, an indirect ELISA for DDT was successfully
developed and used to analyze the release of DDT after stimulation of
macrophages with LPS. Both DDT and MIF are secreted from cells in
response to LPS. Additionally it was shown that both proteins are released
constitutively at a low level. However, herein the molecular cascade
triggered by DDT after its release in response to a pro-inflammatory
stimulus was not addressed. Detailed studies have to be conducted to
elucidate whether DDT exerts similar biological activities than MIF, or
whether it acts as a cross-regulator of MIF. Both scenarios are well

Discussion – 93
documented in immunology. For instance, the chemokines CXCL9,
CXCL10, and CXCL11 all bind to the same receptor CXCR3 eliciting an
increase in intracellular Ca2++ level and activating phosphoinositide-3 and
MAPK kinase (168).
Some arguments can be made supporting the hypothesis that MIF and
DDT share some biological abilities. First, the mRNA and protein levels of
DDT are unchanged in mif knockout cells compared to wildtype control
cells. Second, MIF and DDT are released together after stimulation with
LPS from macrophages. Third, the depletion of either MIF or DDT by
siRNA results in similar effects (transcriptional regulation of IL-8), both
involving the receptor CD74 (72).
With the help of the purified, native DDT protein and anti-DDT antibody,
future experiments can be performed to further address these questions.
Additionally, on a molecular level, the details of the release of DDT have
not been addressed. Established protocols from this work might aid in the
determination whether the release of DDT also depends on p115, and
considering the structural conservation of the active sites, whether the
secretion can also be inhibited by the small molecule inhibitor 4-IPP.
Anti-MIF therapies have been shown to be beneficial in many
inflammatory diseases. However, these approaches might not be complete
without considering DDT’s possible role in disease development, therefore
studying the molecular mechanisms underlying DDT might improve
established anti-MIF treatments of inflammatory disease.

Summary and Outlook – 94
5 Summary and Outlook
Macrophage migration inhibitory factor is a pro-inflammatory cytokine
that plays a pivotal role in the pathogenesis of inflammatory disease such
as septic shock and rheumatoid arthritis. The release of MIF from
inflammatory cells is a common feature in these diseases, and a
correlation between the severity of disease and MIF production is
observed. MIF is a leaderless protein that is secreted from cells by a
specialized, non-classical export pathway, possibly involving the
transporter ABCA1. The release of MIF nevertheless is tightly regulated
with respect to its production in response to different inflammatory,
proliferative, and activating stimuli. In recent studies it was demonstrated
that the administration of neutralizing anti-MIF antibodies and small
molecule MIF inhibitors is advantageous in inflammatory diseases. These
approaches have in common that they all target MIF only after it is
released from cells. A more effective approach in treating MIF-related
diseases may be to target MIF before it is released from cells. The
unconventional secretion pathway of MIF might permit to specifically
block the release of MIF without compromising the general secretion
mechanism of the cell. To date, little is known about the molecular
mechanisms involved in the release of MIF.
My thesis demonstrates that MIF associates with the novel MIF
interaction partner p115, a Golgi-associated protein that takes part in the
ER/Golgi secretion pathway. By confocal microscopy, I was able to
demonstrate that MIF and p115 are associating in the cytoplasm;
following activation, macrophages redistribute MIF to the plasma
membrane together with a portion of p115. This finding agrees with
results obtained from Western blot and ELISA analysis, demonstrating
that upon LPS stimulation both MIF and p115 are co-secreted,
accompanied by a corresponding decrease in intracellular protein
concentration. Accordingly, I demonstrated that the depletion of p115 from
monocytes/macrophages by RNAi results in an inhibition of MIF release

Summary and Outlook – 95
after LPS stimulation. Intriguingly, the secretion of other pro-
inflammatory cytokines, both conventionally released (TNF-α, IL-6) and
unconventionally released (IL-1β) was not affected by the partial depletion
of p115. Similarly, the depletion of p115 in HeLa cells did not affect the
conventional (FGF-4) or unconventional (FGF-2) secretion. This
observation leads to the assumption that it is possible to specifically target
the secretion of MIF without compromising the normal and necessary
secretory mechanism of cells. It is important to note that the effect of p115
depletion on MIF secretion was not only observed after LPS stimulation,
but also after bacterial infection with Chlamydia trachomatis. Notably,
the prototypic small molecule MIF inhibitor, 4-iodo-6-phenylpyrimidine,
inhibits MIF secretion by targeting the interaction between MIF and p115.
My data reveal p115 to be a critical intermediary component in the
regulated secretion of MIF from monocytes/macrophages.
After the discovery of GRASP65 in the unconventional secretion of acyl-
CoA binding protein in Dictyostelium (169), p115 is the second protein of
the Golgi complex attributed with a dual function in secretion. On one
hand p115 is involved in the classical secretion pathway tethering vesicles
arriving from the ER to the Golgi, and on the other hand it is involved in
the unconventional secretion of MIF. The discovery that 4-IPP specifically
blocks the secretion of MIF by interfering with MIF’s interaction with
p115 gives hope in the search of a potent approach in treating MIF-related
diseases by blocking its release from cells. It seems feasible to perform a
screening of MIF inhibitors that specifically target MIF’s ability to
interact with p115 and block its release from cells.
In addition, in my thesis I report on the initial characterization of the
structural homolog of MIF, D-dopachrome tautomerase (DDT). After the
successful purification of recombinant, native murine DDT, a specific anti-
DDT antibody was produced and utilized for the establishment of a DDT-
specific ELISA. It was shown that DDT is released from macrophages in
response to LPS in a pattern similar to that of MIF. Furthermore, it was

Summary and Outlook – 96
shown that the deletion of the mif gene has no effect on the production of
DDT. The precise biological function of DDT remains unclear and the
detailed characterization of the secretion pathway of DDT was beyond the
scope of this thesis, but the comparable release patterns of MIF and DDT
suggest related functions of the two proteins.

References – 97
6 References
1. George, M., and J.H. Vaughan. 1962. In vitro cell migration as a
model for delayed hypersensitivity. Proc Soc Exp Biol Med 111:514-
521.
2. David, J.R. 1966. Delayed hypersensitivity in vitro: its mediation by
cell-free substances formed by lymphoid cell-antigen interaction.
Proc Natl Acad Sci U S A 56:72-77.
3. Nathan, C.F., H.G. Remold, and J.R. David. 1973. Characterization
of a lymphocyte factor which alters macrophage functions. J Exp
Med 137:275-290.
4. Weiser, W.Y., P.A. Temple, J.S. Witek-Giannotti, H.G. Remold, S.C.
Clark, and J.R. David. 1989. Molecular cloning of a cDNA encoding
a human macrophage migration inhibitory factor. Proc Natl Acad
Sci U S A 86:7522-7526.
5. Bernhagen, J., T. Calandra, R.A. Mitchell, S.B. Martin, K.J. Tracey,
W. Voelter, K.R. Manogue, A. Cerami, and R. Bucala. 1993. MIF is a
pituitary-derived cytokine that potentiates lethal endotoxaemia.
Nature 365:756-759.
6. Bernhagen, J., R.A. Mitchell, T. Calandra, W. Voelter, A. Cerami,
and R. Bucala. 1994. Purification, bioactivity, and secondary
structure analysis of mouse and human macrophage migration
inhibitory factor (MIF). Biochemistry 33:14144-14155.
7. Calandra, T., J. Bernhagen, C.N. Metz, L.A. Spiegel, M. Bacher, T.
Donnelly, A. Cerami, and R. Bucala. 1995. MIF as a glucocorticoid-
induced modulator of cytokine production. Nature 377:68-71.
8. Calandra, T., J. Bernhagen, R.A. Mitchell, and R. Bucala. 1994. The
macrophage is an important and previously unrecognized source of
macrophage migration inhibitory factor. J Exp Med 179:1895-1902.
9. Bozza, M., A.R. Satoskar, G. Lin, B. Lu, A.A. Humbles, C. Gerard,
and J.R. David. 1999. Targeted disruption of migration inhibitory
factor gene reveals its critical role in sepsis. J Exp Med 189:341-346.
10. Leng, L., C.N. Metz, Y. Fang, J. Xu, S. Donnelly, J. Baugh, T.
Delohery, Y. Chen, R.A. Mitchell, and R. Bucala. 2003. MIF signal
transduction initiated by binding to CD74. J Exp Med 197:1467-
1476.
11. Bernhagen, J., R. Krohn, H. Lue, J.L. Gregory, A. Zernecke, R.R.
Koenen, M. Dewor, I. Georgiev, A. Schober, L. Leng, T. Kooistra, G.
Fingerle-Rowson, P. Ghezzi, R. Kleemann, S.R. McColl, R. Bucala,
M.J. Hickey, and C. Weber. 2007. MIF is a noncognate ligand of
CXC chemokine receptors in inflammatory and atherogenic cell
recruitment. Nat Med 13:587-596.
12. Meyer-Siegler, K.L., P.L. Vera, K.A. Iczkowski, C. Bifulco, A. Lee,
P.K. Gregersen, L. Leng, and R. Bucala. 2007. Macrophage
migration inhibitory factor (MIF) gene polymorphisms are

References – 98
associated with increased prostate cancer incidence. Genes Immun
8:646-652.
13. Mizue, Y., S. Ghani, L. Leng, C. McDonald, P. Kong, J. Baugh, S.J.
Lane, J. Craft, J. Nishihira, S.C. Donnelly, Z. Zhu, and R. Bucala.
2005. Role for macrophage migration inhibitory factor in asthma.
Proc Natl Acad Sci U S A 102:14410-14415.
14. Radstake, T.R., F.C. Sweep, P. Welsing, B. Franke, S.H. Vermeulen,
A. Geurts-Moespot, T. Calandra, R. Donn, and P.L. van Riel. 2005.
Correlation of rheumatoid arthritis severity with the genetic
functional variants and circulating levels of macrophage migration
inhibitory factor. Arthritis Rheum 52:3020-3029.
15. Coussens, L.M., and Z. Werb. 2002. Inflammation and cancer.
Nature 420:860-867.
16. Javeed, A., Y. Zhao, and Y. Zhao. 2008. Macrophage-migration
inhibitory factor: role in inflammatory diseases and graft rejection.
Inflamm Res 57:45-50.
17. Kaech, C., T. Roger, and T. Calandra. 2007. MIF in Innate
Immunity and Infectious Diseases. In MIF Most Interesting Factor.
R. Bucala, editor World Scientific Publishing Co. Pte. Ltd.,
Hckensack, NJ. 107-132.
18. Morand, E.F., M. Leech, H. Weedon, C. Metz, R. Bucala, and M.D.
Smith. 2002. Macrophage migration inhibitory factor in rheumatoid
arthritis: clinical correlations. Rheumatology (Oxford) 41:558-562.
19. Bacher, M., A. Meinhardt, H.Y. Lan, W. Mu, C.N. Metz, J.A.
Chesney, T. Calandra, D. Gemsa, T. Donnelly, R.C. Atkins, and R.
Bucala. 1997. Migration inhibitory factor expression in
experimentally induced endotoxemia. Am J Pathol 150:235-246.
20. Al-Abed, Y., D. Dabideen, B. Aljabari, A. Valster, D. Messmer, M.
Ochani, M. Tanovic, K. Ochani, M. Bacher, F. Nicoletti, C. Metz,
V.A. Pavlov, E.J. Miller, and K.J. Tracey. 2005. ISO-1 binding to the
tautomerase active site of MIF inhibits its pro-inflammatory
activity and increases survival in severe sepsis. J Biol Chem
280:36541-36544.
21. Calandra, T., B. Echtenacher, D.L. Roy, J. Pugin, C.N. Metz, L.
Hultner, D. Heumann, D. Mannel, R. Bucala, and M.P. Glauser.
2000. Protection from septic shock by neutralization of macrophage
migration inhibitory factor. Nat Med 6:164-170.
22. Pollak, N., T. Sterns, B. Echtenacher, and D.N. Mannel. 2005.
Improved resistance to bacterial superinfection in mice by
treatment with macrophage migration inhibitory factor. Infect
Immun 73:6488-6492.
23. Gregory, J.L., M.T. Leech, J.R. David, Y.H. Yang, A. Dacumos, and
M.J. Hickey. 2004. Reduced leukocyte-endothelial cell interactions
in the inflamed microcirculation of macrophage migration inhibitory
factor-deficient mice. Arthritis Rheum 50:3023-3034.
24. Mikulowska, A., C.N. Metz, R. Bucala, and R. Holmdahl. 1997.
Macrophage migration inhibitory factor is involved in the

References – 99
pathogenesis of collagen type II-induced arthritis in mice. J
Immunol 158:5514-5517.
25. Leech, M., C. Metz, L. Santos, T. Peng, S.R. Holdsworth, R. Bucala,
and E.F. Morand. 1998. Involvement of macrophage migration
inhibitory factor in the evolution of rat adjuvant arthritis. Arthritis
Rheum 41:910-917.
26. Santos, L., P. Hall, C. Metz, R. Bucala, and E.F. Morand. 2001. Role
of macrophage migration inhibitory factor (MIF) in murine antigen-
induced arthritis: interaction with glucocorticoids. Clin Exp
Immunol 123:309-314.
27. Ichiyama, H., S. Onodera, J. Nishihira, T. Ishibashi, T. Nakayama,
A. Minami, K. Yasuda, and H. Tohyama. 2004. Inhibition of joint
inflammation and destruction induced by anti-type II collagen
antibody/lipopolysaccharide (LPS)-induced arthritis in mice due to
deletion of macrophage migration inhibitory factor (MIF). Cytokine
26:187-194.
28. Leech, M., D. Lacey, J.R. Xue, L. Santos, P. Hutchinson, E.
Wolvetang, J.R. David, R. Bucala, and E.F. Morand. 2003.
Regulation of p53 by macrophage migration inhibitory factor in
inflammatory arthritis. Arthritis Rheum 48:1881-1889.
29. Schurigt, U., C. Pfirschke, I.M. Irmler, M. Huckel, M. Gajda, T.
Janik, R. Baumgrass, J. Bernhagen, and R. Brauer. 2008.
Interactions of T helper cells with fibroblast-like synoviocytes: Up-
regulation of matrix metalloproteinases by macrophage migration
inhibitory factor from both Th1 and Th2 cells. Arthritis Rheum
58:3030-3040.
30. Leech, M., C. Metz, P. Hall, P. Hutchinson, K. Gianis, M. Smith, H.
Weedon, S.R. Holdsworth, R. Bucala, and E.F. Morand. 1999.
Macrophage migration inhibitory factor in rheumatoid arthritis:
evidence of proinflammatory function and regulation by
glucocorticoids. Arthritis Rheum 42:1601-1608.
31. Onodera, S., H. Tanji, K. Suzuki, K. Kaneda, Y. Mizue, A. Sagawa,
and J. Nishihira. 1999. High expression of macrophage migration
inhibitory factor in the synovial tissues of rheumatoid joints.
Cytokine 11:163-167.
32. Libby, P. 2002. Inflammation in atherosclerosis. Nature 420:868-
874.
33. Burger-Kentischer, A., H. Goebel, R. Seiler, G. Fraedrich, H.E.
Schaefer, S. Dimmeler, R. Kleemann, J. Bernhagen, and C. Ihling.
2002. Expression of macrophage migration inhibitory factor in
different stages of human atherosclerosis. Circulation 105:1561-
1566.
34. Pan, J.H., G.K. Sukhova, J.T. Yang, B. Wang, T. Xie, H. Fu, Y.
Zhang, A.R. Satoskar, J.R. David, C.N. Metz, R. Bucala, K. Fang,
D.I. Simon, H.A. Chapman, P. Libby, and G.P. Shi. 2004.
Macrophage migration inhibitory factor deficiency impairs
atherosclerosis in low-density lipoprotein receptor-deficient mice.
Circulation 109:3149-3153.

References – 100
35. Burger-Kentischer, A., H. Gobel, R. Kleemann, A. Zernecke, R.
Bucala, L. Leng, D. Finkelmeier, G. Geiger, H.E. Schaefer, A.
Schober, C. Weber, H. Brunner, H. Rutten, C. Ihling, and J.
Bernhagen. 2006. Reduction of the aortic inflammatory response in
spontaneous atherosclerosis by blockade of macrophage migration
inhibitory factor (MIF). Atherosclerosis 184:28-38.
36. Morand, E.F., M. Leech, and J. Bernhagen. 2006. MIF: a new
cytokine link between rheumatoid arthritis and atherosclerosis. Nat
Rev Drug Discov 5:399-410.
37. Ray, A., and L. Cohn. 1999. Th2 cells and GATA-3 in asthma: new
insights into the regulation of airway inflammation. J Clin Invest
104:985-993.
38. Rossi, A.G., C. Haslett, N. Hirani, A.P. Greening, I. Rahman, C.N.
Metz, R. Bucala, and S.C. Donnelly. 1998. Human circulating
eosinophils secrete macrophage migration inhibitory factor (MIF).
Potential role in asthma. J Clin Invest 101:2869-2874.
39. Baugh, J.A., S. Chitnis, S.C. Donnelly, J. Monteiro, X. Lin, B.J.
Plant, F. Wolfe, P.K. Gregersen, and R. Bucala. 2002. A functional
promoter polymorphism in the macrophage migration inhibitory
factor (MIF) gene associated with disease severity in rheumatoid
arthritis. Genes Immun 3:170-176.
40. Lue, H., A. Kapurniotu, G. Fingerle-Rowson, T. Roger, L. Leng, M.
Thiele, T. Calandra, R. Bucala, and J. Bernhagen. 2006. Rapid and
transient activation of the ERK MAPK signalling pathway by
macrophage migration inhibitory factor (MIF) and dependence on
JAB1/CSN5 and Src kinase activity. Cell Signal 18:688-703.
41. Mitchell, R.A., C.N. Metz, T. Peng, and R. Bucala. 1999. Sustained
mitogen-activated protein kinase (MAPK) and cytoplasmic
phospholipase A2 activation by macrophage migration inhibitory
factor (MIF). Regulatory role in cell proliferation and glucocorticoid
action. J Biol Chem 274:18100-18106.
42. Fukuzawa, J., J. Nishihira, N. Hasebe, T. Haneda, J. Osaki, T.
Saito, T. Nomura, T. Fujino, N. Wakamiya, and K. Kikuchi. 2002.
Contribution of macrophage migration inhibitory factor to
extracellular signal-regulated kinase activation by oxidative stress
in cardiomyocytes. J Biol Chem 277:24889-24895.
43. Kleemann, R., A. Hausser, G. Geiger, R. Mischke, A. Burger-
Kentischer, O. Flieger, F.J. Johannes, T. Roger, T. Calandra, A.
Kapurniotu, M. Grell, D. Finkelmeier, H. Brunner, and J.
Bernhagen. 2000. Intracellular action of the cytokine MIF to
modulate AP-1 activity and the cell cycle through Jab1. Nature
408:211-216.
44. Lue, H., M. Thiele, J. Franz, E. Dahl, S. Speckgens, L. Leng, G.
Fingerle-Rowson, R. Bucala, B. Luscher, and J. Bernhagen. 2007.
Macrophage migration inhibitory factor (MIF) promotes cell
survival by activation of the Akt pathway and role for CSN5/JAB1
in the control of autocrine MIF activity. Oncogene 26:5046-5059.

References – 101
45. Hudson, J.D., M.A. Shoaibi, R. Maestro, A. Carnero, G.J. Hannon,
and D.H. Beach. 1999. A proinflammatory cytokine inhibits p53
tumor suppressor activity. J Exp Med 190:1375-1382.
46. Mitchell, R.A., H. Liao, J. Chesney, G. Fingerle-Rowson, J. Baugh,
J. David, and R. Bucala. 2002. Macrophage migration inhibitory
factor (MIF) sustains macrophage proinflammatory function by
inhibiting p53: regulatory role in the innate immune response. Proc
Natl Acad Sci U S A 99:345-350.
47. Jung, H., H.A. Seong, and H. Ha. 2008. Critical role of cysteine
residue 81 of macrophage migration inhibitory factor (MIF) in MIF-
induced inhibition of p53 activity. J Biol Chem 283:20383-20396.
48. Roger, T., J. David, M.P. Glauser, and T. Calandra. 2001. MIF
regulates innate immune responses through modulation of Toll-like
receptor 4. Nature 414:920-924.
49. Shi, X., L. Leng, T. Wang, W. Wang, X. Du, J. Li, C. McDonald, Z.
Chen, J.W. Murphy, E. Lolis, P. Noble, W. Knudson, and R. Bucala.
2006. CD44 is the signaling component of the macrophage
migration inhibitory factor-CD74 receptor complex. Immunity
25:595-606.
50. Sun, H.W., J. Bernhagen, R. Bucala, and E. Lolis. 1996. Crystal
structure at 2.6-A resolution of human macrophage migration
inhibitory factor. Proc Natl Acad Sci U S A 93:5191-5196.
51. Suzuki, M., H. Sugimoto, A. Nakagawa, I. Tanaka, J. Nishihira, and
M. Sakai. 1996. Crystal structure of the macrophage migration
inhibitory factor from rat liver. Nat Struct Biol 3:259-266.
52. Muhlhahn, P., J. Bernhagen, M. Czisch, J. Georgescu, C. Renner, A.
Ross, R. Bucala, and T.A. Holak. 1996. NMR characterization of
structure, backbone dynamics, and glutathione binding of the
human macrophage migration inhibitory factor (MIF). Protein Sci
5:2095-2103.
53. Subramanya, H.S., D.I. Roper, Z. Dauter, E.J. Dodson, G.J. Davies,
K.S. Wilson, and D.B. Wigley. 1996. Enzymatic ketonization of 2-
hydroxymuconate: specificity and mechanism investigated by the
crystal structures of two isomerases. Biochemistry 35:792-802.
54. Nishihira, J., T. Kuriyama, M. Sakai, S. Nishi, S. Ohki, and K.
Hikichi. 1995. The structure and physicochemical properties of rat
liver macrophage migration inhibitory factor. Biochim Biophys Acta
1247:159-162.
55. Delano, W. 2002. The PyMOL Molecular Graphics System. In San
Carlos, CA, USA.
56. Kleemann, R., A. Kapurniotu, R.W. Frank, A. Gessner, R. Mischke,
O. Flieger, S. Juttner, H. Brunner, and J. Bernhagen. 1998.
Disulfide analysis reveals a role for macrophage migration
inhibitory factor (MIF) as thiol-protein oxidoreductase. J Mol Biol
280:85-102.
57. Burger-Kentischer, A., D. Finkelmeier, M. Thiele, J. Schmucker, G.
Geiger, G.E. Tovar, and J. Bernhagen. 2005. Binding of JAB1/CSN5

References – 102
to MIF is mediated by the MPN domain but is independent of the
JAMM motif. FEBS Lett 579:1693-1701.
58. Kamir, D., S. Zierow, L. Leng, Y. Cho, Y. Diaz, J. Griffith, C.
McDonald, M. Merk, R.A. Mitchell, J. Trent, Y. Chen, Y.K. Kwong,
H. Xiong, J. Vermeire, M. Cappello, D. McMahon-Pratt, J. Walker,
J. Bernhagen, E. Lolis, and R. Bucala. 2008. A leishmania ortholog
of macrophage migration inhibitory factor modulates host
macrophage responses. J Immunol 180:8250-8261.
59. Rosengren, E., S. Thelin, P. Aman, C. Hansson, L. Jacobsson, and
H. Rorsman. 1997. The protein catalysing the conversion of D-
dopachrome to 5,6-dihydroxyindole is a phenylpyruvate
tautomerase (EC 5.3.2.1). Melanoma Res 7:517-518.
60. Rosengren, E., R. Bucala, P. Aman, L. Jacobsson, G. Odh, C.N.
Metz, and H. Rorsman. 1996. The immunoregulatory mediator
macrophage migration inhibitory factor (MIF) catalyzes a
tautomerization reaction. Mol Med 2:143-149.
61. Lubetsky, J.B., A. Dios, J. Han, B. Aljabari, B. Ruzsicska, R.
Mitchell, E. Lolis, and Y. Al-Abed. 2002. The tautomerase active
site of macrophage migration inhibitory factor is a potential target
for discovery of novel anti-inflammatory agents. J Biol Chem
277:24976-24982.
62. Swope, M., H.W. Sun, P.R. Blake, and E. Lolis. 1998. Direct link
between cytokine activity and a catalytic site for macrophage
migration inhibitory factor. Embo J 17:3534-3541.
63. Onodera, S., K. Kaneda, Y. Mizue, Y. Koyama, M. Fujinaga, and J.
Nishihira. 2000. Macrophage migration inhibitory factor up-
regulates expression of matrix metalloproteinases in synovial
fibroblasts of rheumatoid arthritis. J Biol Chem 275:444-450.
64. Senter, P.D., Y. Al-Abed, C.N. Metz, F. Benigni, R.A. Mitchell, J.
Chesney, J. Han, C.G. Gartner, S.D. Nelson, G.J. Todaro, and R.
Bucala. 2002. Inhibition of macrophage migration inhibitory factor
(MIF) tautomerase and biological activities by acetaminophen
metabolites. Proc Natl Acad Sci U S A 99:144-149.
65. Winner, M., J. Meier, S. Zierow, B.E. Rendon, G.V. Crichlow, R.
Riggs, R. Bucala, L. Leng, N. Smith, E. Lolis, J.O. Trent, and R.A.
Mitchell. 2008. A Novel, Macrophage Migration Inhibitory Factor
Suicide Substrate Inhibits Motility and Growth of Lung Cancer
Cells. Cancer Res 68:7253-7257.
66. Odh, G., A. Hindemith, A.M. Rosengren, E. Rosengren, and H.
Rorsman. 1993. Isolation of a new tautomerase monitored by the
conversion of D-dopachrome to 5,6-dihydroxyindole. Biochem
Biophys Res Commun 197:619-624.
67. Aroca, P., J.C. Garcia-Borron, F. Solano, and J.A. Lozano. 1990.
Regulation of mammalian melanogenesis. I: Partial purification and
characterization of a dopachrome converting factor: dopachrome
tautomerase. Biochim Biophys Acta 1035:266-275.
68. Bjork, P., P. Aman, A. Hindemith, G. Odh, L. Jacobsson, E.
Rosengren, and H. Rorsman. 1996. A new enzyme activity in human

References – 103
blood cells and isolation of the responsible protein (D-dopachrome
tautomerase) from erythrocytes. Eur J Haematol 57:254-256.
69. Sugimoto, H., M. Taniguchi, A. Nakagawa, I. Tanaka, M. Suzuki,
and J. Nishihira. 1999. Crystal structure of human D-dopachrome
tautomerase, a homologue of macrophage migration inhibitory
factor, at 1.54 A resolution. Biochemistry 38:3268-3279.
70. Nishihira, J., M. Fujinaga, T. Kuriyama, M. Suzuki, H. Sugimoto,
A. Nakagawa, I. Tanaka, and M. Sakai. 1998. Molecular cloning of
human D-dopachrome tautomerase cDNA: N-terminal proline is
essential for enzyme activation. Biochem Biophys Res Commun
243:538-544.
71. Sonesson, B., E. Rosengren, A.S. Hansson, and C. Hansson. 2003.
UVB-induced inflammation gives increased d-dopachrome
tautomerase activity in blister fluid which correlates with
macrophage migration inhibitory factor. Exp Dermatol 12:278-282.
72. Coleman, A.M., B.E. Rendon, M. Zhao, M.W. Qian, R. Bucala, D.
Xin, and R.A. Mitchell. 2008. Cooperative regulation of non-small
cell lung carcinoma angiogenic potential by macrophage migration
inhibitory factor and its homolog, D-dopachrome tautomerase. J
Immunol 181:2330-2337.
73. Schatz, G., and B. Dobberstein. 1996. Common principles of protein
translocation across membranes. Science 271:1519-1526.
74. Devillers-Thiery, A., T. Kindt, G. Scheele, and G. Blobel. 1975.
Homology in amino-terminal sequence of precursors to pancreatic
secretory proteins. Proc Natl Acad Sci U S A 72:5016-5020.
75. Palade, G. 1975. Intracellular aspects of the process of protein
synthesis. Science 189:347-358.
76. Kleizen, B., and I. Braakman. 2004. Protein folding and quality
control in the endoplasmic reticulum. Curr Opin Cell Biol 16:343-
349.
77. Lee, M.C., E.A. Miller, J. Goldberg, L. Orci, and R. Schekman. 2004.
Bi-directional protein transport between the ER and Golgi. Annu
Rev Cell Dev Biol 20:87-123.
78. Wiertz, E.J., D. Tortorella, M. Bogyo, J. Yu, W. Mothes, T.R. Jones,
T.A. Rapoport, and H.L. Ploegh. 1996. Sec61-mediated transfer of a
membrane protein from the endoplasmic reticulum to the
proteasome for destruction. Nature 384:432-438.
79. Helenius, A., and M. Aebi. 2001. Intracellular functions of N-linked
glycans. Science 291:2364-2369.
80. Keller, P., and K. Simons. 1997. Post-Golgi biosynthetic trafficking.
J Cell Sci 110 ( Pt 24):3001-3009.
81. Lee, M.C., and E.A. Miller. 2007. Molecular mechanisms of COPII
vesicle formation. Semin Cell Dev Biol 18:424-434.
82. Orci, L., M. Ravazzola, P. Meda, C. Holcomb, H.P. Moore, L. Hicke,
and R. Schekman. 1991. Mammalian Sec23p homologue is
restricted to the endoplasmic reticulum transitional cytoplasm. Proc
Natl Acad Sci U S A 88:8611-8615.

References – 104
83. Bannykh, S.I., N. Nishimura, and W.E. Balch. 1998. Getting into
the Golgi. Trends Cell Biol 8:21-25.
84. Bednarek, S.Y., L. Orci, and R. Schekman. 1996. Traffic COPs and
the formation of vesicle coats. Trends Cell Biol 6:468-473.
85. Waters, M.G., and S.R. Pfeffer. 1999. Membrane tethering in
intracellular transport. Curr Opin Cell Biol 11:453-459.
86. Kee, Y., J.S. Yoo, C.D. Hazuka, K.E. Peterson, S.C. Hsu, and R.H.
Scheller. 1997. Subunit structure of the mammalian exocyst
complex. Proc Natl Acad Sci U S A 94:14438-14443.
87. Loh, E., and W. Hong. 2004. The binary interacting network of the
conserved oligomeric Golgi tethering complex. J Biol Chem
279:24640-24648.
88. Shorter, J., M.B. Beard, J. Seemann, A.B. Dirac-Svejstrup, and G.
Warren. 2002. Sequential tethering of Golgins and catalysis of
SNAREpin assembly by the vesicle-tethering protein p115. J Cell
Biol 157:45-62.
89. Barr, F.A. 2002. The Golgi apparatus: going round in circles?
Trends Cell Biol 12:101-104.
90. Losev, E., C.A. Reinke, J. Jellen, D.E. Strongin, B.J. Bevis, and B.S.
Glick. 2006. Golgi maturation visualized in living yeast. Nature
441:1002-1006.
91. Matsuura-Tokita, K., M. Takeuchi, A. Ichihara, K. Mikuriya, and A.
Nakano. 2006. Live imaging of yeast Golgi cisternal maturation.
Nature 441:1007-1010.
92. Alvarez, C., H. Fujita, A. Hubbard, and E. Sztul. 1999. ER to Golgi
transport: Requirement for p115 at a pre-Golgi VTC stage. J Cell
Biol 147:1205-1222.
93. Kooy, J., B.H. Toh, J.M. Pettitt, R. Erlich, and P.A. Gleeson. 1992.
Human autoantibodies as reagents to conserved Golgi components.
Characterization of a peripheral, 230-kDa compartment-specific
Golgi protein. J Biol Chem 267:20255-20263.
94. Burkhard, P., J. Stetefeld, and S.V. Strelkov. 2001. Coiled coils: a
highly versatile protein folding motif. Trends Cell Biol 11:82-88.
95. Nelson, D.S., C. Alvarez, Y.S. Gao, R. Garcia-Mata, E. Fialkowski,
and E. Sztul. 1998. The membrane transport factor TAP/p115 cycles
between the Golgi and earlier secretory compartments and contains
distinct domains required for its localization and function. J Cell
Biol 143:319-331.
96. Allan, B.B., B.D. Moyer, and W.E. Balch. 2000. Rab1 recruitment of
p115 into a cis-SNARE complex: programming budding COPII
vesicles for fusion. Science 289:444-448.
97. Sapperstein, S.K., D.M. Walter, A.R. Grosvenor, J.E. Heuser, and
M.G. Waters. 1995. p115 is a general vesicular transport factor
related to the yeast endoplasmic reticulum to Golgi transport factor
Uso1p. Proc Natl Acad Sci U S A 92:522-526.
98. Nakamura, N., M. Lowe, T.P. Levine, C. Rabouille, and G. Warren.
1997. The vesicle docking protein p115 binds GM130, a cis-Golgi
matrix protein, in a mitotically regulated manner. Cell 89:445-455.

References – 105
99. Diao, A., L. Frost, Y. Morohashi, and M. Lowe. 2008. Coordination
of Golgin Tethering and SNARE Assembly: GM130 BINDS
SYNTAXIN 5 IN A p115-REGULATED MANNER. J Biol Chem
283:6957-6967.
100. Short, B., A. Haas, and F.A. Barr. 2005. Golgins and GTPases,
giving identity and structure to the Golgi apparatus. Biochim
Biophys Acta 1744:383-395.
101. Puthenveedu, M.A., and A.D. Linstedt. 2001. Evidence that Golgi
structure depends on a p115 activity that is independent of the
vesicle tether components giantin and GM130. J Cell Biol 155:227-
238.
102. Puthenveedu, M.A., and A.D. Linstedt. 2004. Gene replacement
reveals that p115/SNARE interactions are essential for Golgi
biogenesis. Proc Natl Acad Sci U S A 101:1253-1256.
103. Sohda, M., Y. Misumi, S. Yoshimura, N. Nakamura, T. Fusano, S.
Sakisaka, S. Ogata, J. Fujimoto, N. Kiyokawa, and Y. Ikehara.
2005. Depletion of vesicle-tethering factor p115 causes mini-stacked
Golgi fragments with delayed protein transport. Biochem Biophys
Res Commun 338:1268-1274.
104. Hughes, R.C. 1999. Secretion of the galectin family of mammalian
carbohydrate-binding proteins. Biochim Biophys Acta 1473:172-185.
105. Flieger, O., A. Engling, R. Bucala, H. Lue, W. Nickel, and J.
Bernhagen. 2003. Regulated secretion of macrophage migration
inhibitory factor is mediated by a non-classical pathway involving
an ABC transporter. FEBS Lett 551:78-86.
106. Rubartelli, A., F. Cozzolino, M. Talio, and R. Sitia. 1990. A novel
secretory pathway for interleukin-1 beta, a protein lacking a signal
sequence. Embo J 9:1503-1510.
107. Nickel, W. 2003. The mystery of nonclassical protein secretion. A
current view on cargo proteins and potential export routes. Eur J
Biochem 270:2109-2119.
108. Lippincott-Schwartz, J., L.C. Yuan, J.S. Bonifacino, and R.D.
Klausner. 1989. Rapid redistribution of Golgi proteins into the ER
in cells treated with brefeldin A: evidence for membrane cycling
from Golgi to ER. Cell 56:801-813.
109. Orci, L., M. Tagaya, M. Amherdt, A. Perrelet, J.G. Donaldson, J.
Lippincott-Schwartz, R.D. Klausner, and J.E. Rothman. 1991.
Brefeldin A, a drug that blocks secretion, prevents the assembly of
non-clathrin-coated buds on Golgi cisternae. Cell 64:1183-1195.
110. Nickel, W. 2005. Unconventional secretory routes: direct protein
export across the plasma membrane of mammalian cells. Traffic
6:607-614.
111. Charras, G.T., C.K. Hu, M. Coughlin, and T.J. Mitchison. 2006.
Reassembly of contractile actin cortex in cell blebs. J Cell Biol
175:477-490.
112. Tournaviti, S., S. Hannemann, S. Terjung, T.M. Kitzing, C.
Stegmayer, J. Ritzerfeld, P. Walther, R. Grosse, W. Nickel, and O.T.
Fackler. 2007. SH4-domain-induced plasma membrane

References – 106
dynamization promotes bleb-associated cell motility. J Cell Sci
120:3820-3829.
113. Denny, P.W., S. Gokool, D.G. Russell, M.C. Field, and D.F. Smith.
2000. Acylation-dependent protein export in Leishmania. J Biol
Chem 275:11017-11025.
114. Schafer, T., H. Zentgraf, C. Zehe, B. Brugger, J. Bernhagen, and W.
Nickel. 2004. Unconventional secretion of fibroblast growth factor 2
is mediated by direct translocation across the plasma membrane of
mammalian cells. J Biol Chem 279:6244-6251.
115. Temmerman, K., A.D. Ebert, H.M. Muller, I. Sinning, I. Tews, and
W. Nickel. 2008. A direct role for phosphatidylinositol-4,5-
bisphosphate in unconventional secretion of fibroblast growth factor
2. Traffic 9:1204-1217.
116. Zehe, C., A. Engling, S. Wegehingel, T. Schafer, and W. Nickel.
2006. Cell-surface heparan sulfate proteoglycans are essential
components of the unconventional export machinery of FGF-2. Proc
Natl Acad Sci U S A 103:15479-15484.
117. Blott, E.J., and G.M. Griffiths. 2002. Secretory lysosomes. Nat Rev
Mol Cell Biol 3:122-131.
118. Andrei, C., C. Dazzi, L. Lotti, M.R. Torrisi, G. Chimini, and A.
Rubartelli. 1999. The secretory route of the leaderless protein
interleukin 1beta involves exocytosis of endolysosome-related
vesicles. Mol Biol Cell 10:1463-1475.
119. Mambula, S.S., and S.K. Calderwood. 2006. Heat shock protein 70
is secreted from tumor cells by a nonclassical pathway involving
lysosomal endosomes. J Immunol 177:7849-7857.
120. Gardella, S., C. Andrei, D. Ferrera, L.V. Lotti, M.R. Torrisi, M.E.
Bianchi, and A. Rubartelli. 2002. The nuclear protein HMGB1 is
secreted by monocytes via a non-classical, vesicle-mediated
secretory pathway. EMBO Rep 3:995-1001.
121. Andrei, C., P. Margiocco, A. Poggi, L.V. Lotti, M.R. Torrisi, and A.
Rubartelli. 2004. Phospholipases C and A2 control lysosome-
mediated IL-1 beta secretion: Implications for inflammatory
processes. Proc Natl Acad Sci U S A 101:9745-9750.
122. Raposo, G., H.W. Nijman, W. Stoorvogel, R. Liejendekker, C.V.
Harding, C.J. Melief, and H.J. Geuze. 1996. B lymphocytes secrete
antigen-presenting vesicles. J Exp Med 183:1161-1172.
123. Zitvogel, L., A. Regnault, A. Lozier, J. Wolfers, C. Flament, D.
Tenza, P. Ricciardi-Castagnoli, G. Raposo, and S. Amigorena. 1998.
Eradication of established murine tumors using a novel cell-free
vaccine: dendritic cell-derived exosomes. Nat Med 4:594-600.
124. Johnstone, R.M., A. Mathew, A.B. Mason, and K. Teng. 1991.
Exosome formation during maturation of mammalian and avian
reticulocytes: evidence that exosome release is a major route for
externalization of obsolete membrane proteins. J Cell Physiol
147:27-36.

References – 107
125. Keller, S., M.P. Sanderson, A. Stoeck, and P. Altevogt. 2006.
Exosomes: from biogenesis and secretion to biological function.
Immunol Lett 107:102-108.
126. Gomes, C., S. Keller, P. Altevogt, and J. Costa. 2007. Evidence for
secretion of Cu,Zn superoxide dismutase via exosomes from a cell
model of amyotrophic lateral sclerosis. Neurosci Lett 428:43-46.
127. Cheng, C.F., J. Fan, M. Fedesco, S. Guan, Y. Li, B. Bandyopadhyay,
A.M. Bright, D. Yerushalmi, M. Liang, M. Chen, Y.P. Han, D.T.
Woodley, and W. Li. 2008. Transforming growth factor alpha
(TGFalpha)-stimulated secretion of HSP90alpha: using the receptor
LRP-1/CD91 to promote human skin cell migration against a
TGFbeta-rich environment during wound healing. Mol Cell Biol
28:3344-3358.
128. MacKenzie, A., H.L. Wilson, E. Kiss-Toth, S.K. Dower, R.A. North,
and A. Surprenant. 2001. Rapid secretion of interleukin-1beta by
microvesicle shedding. Immunity 15:825-835.
129. Dean, M. 2005. The genetics of ATP-binding cassette transporters.
Methods Enzymol 400:409-429.
130. Davidson, A.L., and J. Chen. 2004. ATP-binding cassette
transporters in bacteria. Annu Rev Biochem 73:241-268.
131. Hamon, Y., M.F. Luciani, F. Becq, B. Verrier, A. Rubartelli, and G.
Chimini. 1997. Interleukin-1beta secretion is impaired by inhibitors
of the Atp binding cassette transporter, ABC1. Blood 90:2911-2915.
132. Zhou, X., T. Engel, C. Goepfert, M. Erren, G. Assmann, and A. von
Eckardstein. 2002. The ATP binding cassette transporter A1
contributes to the secretion of interleukin 1beta from macrophages
but not from monocytes. Biochem Biophys Res Commun 291:598-
604.
133. Daryadel, A., R.F. Grifone, H.U. Simon, and S. Yousefi. 2006.
Apoptotic neutrophils release macrophage migration inhibitory
factor upon stimulation with tumor necrosis factor-alpha. J Biol
Chem 281:27653-27661.
134. Nishino, T., J. Bernhagen, H. Shiiki, T. Calandra, K. Dohi, and R.
Bucala. 1995. Localization of macrophage migration inhibitory
factor (MIF) to secretory granules within the corticotrophic and
thyrotrophic cells of the pituitary gland. Mol Med 1:781-788.
135. Waeber, G., T. Calandra, R. Roduit, J.A. Haefliger, C. Bonny, N.
Thompson, B. Thorens, E. Temler, A. Meinhardt, M. Bacher, C.N.
Metz, P. Nicod, and R. Bucala. 1997. Insulin secretion is regulated
by the glucose-dependent production of islet beta cell macrophage
migration inhibitory factor. Proc Natl Acad Sci U S A 94:4782-4787.
136. Eickhoff, R., B. Wilhelm, H. Renneberg, G. Wennemuth, M. Bacher,
D. Linder, R. Bucala, J. Seitz, and A. Meinhardt. 2001. Purification
and characterization of macrophage migration inhibitory factor as a
secretory protein from rat epididymis: evidences for alternative
release and transfer to spermatozoa. Mol Med 7:27-35.

References – 108
137. Park, E.K., H.S. Jung, H.I. Yang, M.C. Yoo, C. Kim, and K.S. Kim.
2007. Optimized THP-1 differentiation is required for the detection
of responses to weak stimuli. Inflamm Res 56:45-50.
138. Engling, A., R. Backhaus, C. Stegmayer, C. Zehe, C. Seelenmeyer,
A. Kehlenbach, B. Schwappach, S. Wegehingel, and W. Nickel.
2002. Biosynthetic FGF-2 is targeted to non-lipid raft microdomains
following translocation to the extracellular surface of CHO cells. J
Cell Sci 115:3619-3631.
139. Bradford, M.M. 1976. A rapid and sensitive method for the
quantitation of microgram quantities of protein utilizing the
principle of protein-dye binding. Anal Biochem 72:248-254.
140. Fingerle-Rowson, G., P. Koch, R. Bikoff, X. Lin, C.N. Metz, F.S.
Dhabhar, A. Meinhardt, and R. Bucala. 2003. Regulation of
macrophage migration inhibitory factor expression by
glucocorticoids in vivo. Am J Pathol 162:47-56.
141. Shorter, J., and G. Warren. 1999. A role for the vesicle tethering
protein, p115, in the post-mitotic stacking of reassembling Golgi
cisternae in a cell-free system. J Cell Biol 146:57-70.
142. Bikfalvi, A., S. Klein, G. Pintucci, and D.B. Rifkin. 1997. Biological
roles of fibroblast growth factor-2. Endocr Rev 18:26-45.
143. Nugent, M.A., and R.V. Iozzo. 2000. Fibroblast growth factor-2. Int
J Biochem Cell Biol 32:115-120.
144. Powers, C.J., S.W. McLeskey, and A. Wellstein. 2000. Fibroblast
growth factors, their receptors and signaling. Endocr Relat Cancer
7:165-197.
145. Huang, S., R.I. Endo, and G.R. Nemerow. 1995. Upregulation of
integrins alpha v beta 3 and alpha v beta 5 on human monocytes
and T lymphocytes facilitates adenovirus-mediated gene delivery. J
Virol 69:2257-2263.
146. Prebeck, S., H. Brade, C.J. Kirschning, C.P. da Costa, S. Durr, H.
Wagner, and T. Miethke. 2003. The Gram-negative bacterium
Chlamydia trachomatis L2 stimulates tumor necrosis factor
secretion by innate immune cells independently of its endotoxin.
Microbes Infect 5:463-470.
147. Bulut, Y., E. Faure, L. Thomas, H. Karahashi, K.S. Michelsen, O.
Equils, S.G. Morrison, R.P. Morrison, and M. Arditi. 2002.
Chlamydial heat shock protein 60 activates macrophages and
endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-
dependent pathway. J Immunol 168:1435-1440.
148. Mpiga, P., S. Mansour, R. Morisset, R. Beaulieu, and M.
Ravaoarinoro. 2006. Sustained interleukin-6 and interleukin-8
expression following infection with Chlamydia trachomatis serovar
L2 in a HeLa/THP-1 cell co-culture model. Scand J Immunol
63:199-207.
149. Arjona, A., H.G. Foellmer, T. Town, L. Leng, C. McDonald, T. Wang,
S.J. Wong, R.R. Montgomery, E. Fikrig, and R. Bucala. 2007.
Abrogation of macrophage migration inhibitory factor decreases

References – 109
West Nile virus lethality by limiting viral neuroinvasion. J Clin
Invest 117:3059-3066.
150. Esumi, N., M. Budarf, L. Ciccarelli, B. Sellinger, C.A. Kozak, and G.
Wistow. 1998. Conserved gene structure and genomic linkage for D-
dopachrome tautomerase (DDT) and MIF. Mamm Genome 9:753-
757.
151. Atsumi, T., Y.R. Cho, L. Leng, C. McDonald, T. Yu, C. Danton, E.G.
Hong, R.A. Mitchell, C. Metz, H. Niwa, J. Takeuchi, S. Onodera, T.
Umino, N. Yoshioka, T. Koike, J.K. Kim, and R. Bucala. 2007. The
proinflammatory cytokine macrophage migration inhibitory factor
regulates glucose metabolism during systemic inflammation. J
Immunol 179:5399-5406.
152. Miller, E.J., J. Li, L. Leng, C. McDonald, T. Atsumi, R. Bucala, and
L.H. Young. 2008. Macrophage migration inhibitory factor
stimulates AMP-activated protein kinase in the ischaemic heart.
Nature 451:578-582.
153. Wu, S.P., L. Leng, Z. Feng, N. Liu, H. Zhao, C. McDonald, A. Lee,
F.C. Arnett, P.K. Gregersen, M.D. Mayes, and R. Bucala. 2006.
Macrophage migration inhibitory factor promoter polymorphisms
and the clinical expression of scleroderma. Arthritis Rheum
54:3661-3669.
154. Baugh, J.A., and R. Bucala. 2002. Macrophage migration inhibitory
factor. Crit Care Med 30:S27-S35.
155. Lue, H., R. Kleemann, T. Calandra, T. Roger, and J. Bernhagen.
2002. Macrophage migration inhibitory factor (MIF): mechanisms of
action and role in disease. Microbes Infect 4:449-460.
156. Bozza, F.A., R.N. Gomes, A.M. Japiassu, M. Soares, H.C. Castro-
Faria-Neto, P.T. Bozza, and M.T. Bozza. 2004. Macrophage
migration inhibitory factor levels correlate with fatal outcome in
sepsis. Shock 22:309-313.
157. Lehmann, L.E., U. Novender, S. Schroeder, T. Pietsch, T. von
Spiegel, C. Putensen, A. Hoeft, and F. Stuber. 2001. Plasma levels
of macrophage migration inhibitory factor are elevated in patients
with severe sepsis. Intensive Care Med 27:1412-1415.
158. Beishuizen, A., L.G. Thijs, C. Haanen, and I. Vermes. 2001.
Macrophage migration inhibitory factor and hypothalamo-pituitary-
adrenal function during critical illness. J Clin Endocrinol Metab
86:2811-2816.
159. Sprong, T., P. Pickkers, A. Geurts-Moespot, J. van der Ven-
Jongekrijg, C. Neeleman, M. Knaup, D. Leroy, T. Calandra, J.W.
van der Meer, F. Sweep, and M. van Deuren. 2007. Macrophage
migration inhibitory factor (MIF) in meningococcal septic shock and
experimental human endotoxemia. Shock 27:482-487.
160. Takasuka, N., K. Matsuura, S. Yamamoto, and K.S. Akagawa. 1995.
Suppression of TNF-alpha mRNA expression in LPS-primed
macrophages occurs at the level of nuclear factor-kappa B
activation, but not at the level of protein kinase C or CD14
expression. J Immunol 154:4803-4812.

References – 110
161. Sacco, R.E., S.K. Nibbelink, M.J. Baarsch, M.P. Murtaugh, and M.J.
Wannemuehler. 1996. Induction of interleukin (IL)-1beta and IL-8
mRNA expression in porcine macrophages by lipopolysaccharide
from Serpulina hyodysenteriae. Infect Immun 64:4369-4372.
162. Seemann, J., E.J. Jokitalo, and G. Warren. 2000. The role of the
tethering proteins p115 and GM130 in transport through the Golgi
apparatus in vivo. Mol Biol Cell 11:635-645.
163. Plutner, H., A.D. Cox, S. Pind, R. Khosravi-Far, J.R. Bourne, R.
Schwaninger, C.J. Der, and W.E. Balch. 1991. Rab1b regulates
vesicular transport between the endoplasmic reticulum and
successive Golgi compartments. J Cell Biol 115:31-43.
164. Peter, F., C. Nuoffer, S.N. Pind, and W.E. Balch. 1994. Guanine
nucleotide dissociation inhibitor is essential for Rab1 function in
budding from the endoplasmic reticulum and transport through the
Golgi stack. J Cell Biol 126:1393-1406.
165. Puig, O., F. Caspary, G. Rigaut, B. Rutz, E. Bouveret, E. Bragado-
Nilsson, M. Wilm, and B. Seraphin. 2001. The tandem affinity
purification (TAP) method: a general procedure of protein complex
purification. Methods 24:218-229.
166. Sriskandan, S., and D.M. Altmann. 2008. The immunology of sepsis.
J Pathol 214:211-223.
167. Whitman, C.P. 2002. The 4-oxalocrotonate tautomerase family of
enzymes: how nature makes new enzymes using a beta-alpha-beta
structural motif. Arch Biochem Biophys 402:1-13.
168. Smit, M.J., P. Verdijk, E.M. van der Raaij-Helmer, M. Navis, P.J.
Hensbergen, R. Leurs, and C.P. Tensen. 2003. CXCR3-mediated
chemotaxis of human T cells is regulated by a Gi- and
phospholipase C-dependent pathway and not via activation of
MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood 102:1959-1965.
169. Kinseth, M.A., C. Anjard, D. Fuller, G. Guizzunti, W.F. Loomis, and
V. Malhotra. 2007. The Golgi-associated protein GRASP is required
for unconventional protein secretion during development. Cell
130:524-534.

111
Curriculum vitae
Persönliche Daten
Geburtsdatum 20. November 1979
Geburtsort Freiburg im Breisgau
Familienstand ledig
Staatsangehörigkeit deutsch
Bildungsweg
08/90 – 06/99 Gymnasium
Goethe-Gymnasium, Freiburg
Abschluss: Abitur
10/99 – 09/01 Grundstudium Biologie
Albert-Ludwig Universität, Freiburg
Abschluss: Vordiplom
04/02 – 08/05 Hauptstudium Biologie
RWTH Aachen, Aachen
Abschluss: Diplom
Hauptfächer: Molekularbiologie, Bioinformatik
Nebenfächer: Biotechnologie, Mikrobiologie,
Informatik
10/04 – 09/06 Diplomarbeit
Institut für Biochemie und Molekulare Zellbiologie
der RWTH Aachen unter Betreuung von Prof.
Bernhagen
Titel: Charakterisierung der Bindung von Cyclinen
an das Zytokin MIF und seine Phosphorylierung
durch CDKs
01/06 – heute Promotion
Institut für Biochemie und Molekulare Zellbiologie
der RWTH Aachen unter Betreuung von Prof.
Bernhagen in Kooperation mit Yale University
School of Medicine (USA) unter der Anleitung von
Prof. Bucala

112
Auszeichnungen
10/01 – 03/02 Förderung des Deutschen Akademischen
Austauschdienst
01/06 – heute Promotionsstipendium der Studienstiftung des
deutschen Volkes
Publikationen
M. Merk, J. Baugh, S. Zierow, L. Leng, U. Pal, S. Lee, A. Ebert, Y. Mizue,
J. Trent, R. Mitchell, W. Nickel, P. Kavathas, J. Bernhagen, and R. Bucala
(2008). The Golgi-associated Protein p115 mediates the secretion on
Macrophage Migration Inhibitory Factor (MIF). (In Revision Mol Biol Cell)
D. Kamir, S. Zierow, L. Leng, Y. Cho, Y. Diaz, J. Griffith, C. McDonald, M.
Merk, R.A. Mitchell, J. Trent, Y. Chen, Y.K. Kwong, H. Xiong, J.
Vermeire, M. Cappello, D. McMahon-Pratt, J. Walker, J. Bernhagen, E.
Lolis, and R. Bucala. 2008. A leishmania ortholog of macrophage
migration inhibitory factor modulates host macrophage responses. J
Immunol 180:8250-8261.


















