
The Biosynthesis of Sialic Acids - Journal of Biological ...The Biosynthesis of Sialic Acids ... was...
12
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 237, No. 5, May 1962 Printed in 7J.S.A The Biosynthesis of Sialic Acids LEONARD WARREN AND HERBERT FELSENFELD* From the National Institute of Arthritis and Metabolic Diseases, National Institutes of Health, United States Public Health Service, Bethesda 14, Maryland (Received for publication, December 4, 1961) The sialic acids (1) have in common a 9 carbon carboxylic acid, neuraminic acid (Fig. l), which may be regarded as a con- densation product of a 3 carbon and a 6 carbon unit. Neu- raminic acid itself is not found naturally, but several N- and O-substituted derivatives are widely distributed in nature (1). Chemical (2-4) and enzymic (5-7) degradations of N-acetyl- neuraminic acid have shown that this substance is composed of pyruvic acid and an N-acetylhexosamine. Working with neuraminic acid aldolase, an enzyme first described by Heimer and Meyer (5), Comb and Roseman (6) made the important observation that the N-acetylhexosamine component of N-acetylneuraminic acid is N-acetyl-n-mannos- amine. This compound was a product of the reversible N-ace- tylneuraminic acid aldolase-catalyzed reaction: N-Acetylneuraminic acids N-acetyl-n-mannosamine + pyruvate NAN1 has been synthesized chemically from oxaloacetate and N-acetyl-n-glucosamine (8, 9) or N-acetyl-n-mannosamine (10) in basic solutions. At the high pH of the reaction medium em- ployed, N-acetyl-n-glucosamine is effective because it is, in part, racemized to the actual reactant, N-acetyl-n-mannosamine (6). Decarboxylation of the fl-carboxyl group of oxaloacetate accom- panies the condensation reactions. In previous communications (11, 12) we have described a new enzyme system which is responsible for the biosynthesis of N-acetylneuraminic acid in extracts of rat liver and bovine sub- maxillary gland. Pyruvic acid is inactive in this system, whereas it is a participant in the NAN-aldolase catalyzed-reaction. The equilibrium of the latter reaction favors degradation of NAN (6, 7), whereas the over-all biosynthetic reaction process cat- alyzed by this new enzyme system permits almost quantitative conversion of the precursors, phosphoenolpyruvate and N-ace- tyl-n-mannosamine to the final product, N-acetylneuraminic acid. As previously reported (13), the over-all biosynthetic Reaction 4 is catalyzed by three enzymes, N-acetylmannosamine kinase,* * Present address, Department of Pharmacology, Yale Uni- versity, New Haven, Connecticut. 1The abbreviations used are: NAN, N-acetylneuraminic acid; NAN-g-P, N-acetylneuraminic acid g-phosphate; NAN-aldolase, N-acetylneuraminic acid aldolase. 2 In accordance with the nomenclature proposed by the Com- mission on Enzymes of the International Union of Biochemistry, the svstematic name of the kinase is ATP: N-acetamido-2- deox;n-mannose phosphotransferase and that for the condensing enzyme is phosphoenolpyruvate: N-acetamido-2-deoxy-n-man- nose-6-phosphate ligase. Since the mechanism of dephosphor- ylation of N-acetylneuraminic acid g-phosphate is not known (see discussion), no name is proposed for this enzyme at the pres- ent time. a condensing enzyme, and a dephosphorylating enzyme. These have been partially purified and shown to catalyze Reactions 1, 2, and 3, respectively. N-Acetyl-n-mannosamine + ATP Mg++K+ (1) N-acetyl-D-mannosamine-6-P + ADP N-Acetyl-n-mannosamine-6-P + Hz0 GSH, KCN Mg++ + phosphoenolpyruvate N-acetylneuraminic-9-P + Pi (2) Mg++ N-Acetylneuraminic acid-g-P + Hz0 3 (3) N-acetylneuraminic acid + Pi N-Acetyl-n-mannosamine + phosphoenolpyruvate + ATP + 2 Hz0 -+ N-acetylneuraminic acid + ADP + 2 Pi (4) This paper describes the isolation, purification, and properties of the enzymes involved, which have been isolated from rat liver and bovine submaxillary gland, and the identification of the products of the reactions. Recently Roseman and his co-workers have described a similar series of enzymes (14, 15). EXPERIMENTAL PROCEDURE Materials and Method-s ATP, GTP, CTP, and UTP were the products of the Pabst Laboratories. The tricyclohexylamine salt of phosphoenol- pyruvate was purchased from Mann Research Laboratories, Inc. Glycolaldehyde phosphate acetal, dicyclohexylammonium salt, was obtained from the California Corporation for Biochemical Research. For conversion to glycolaldehyde phosphate, a solu- tion of the latter was treated with Dowex 50-H+ at room tem- perature overnight (16). Bentonite was purchased from the Fisher Scientific Company and South Carolina kaolin (“Dixie clay”) from the R. T. Vanderbilt Company. DEAE-cellulose was obtained from Distillation Products, Inc. n-Mannosamine was synthesized by the method of Kuhn and Kirschenlohr (17) and was separated from its epimer n-glucosamine by fractional crystallization from HCl. [a]; -4.7” (c = 5% in 1 N HCl) (17). n-Mannosamine labeled in carbon 1 was made with Cl4- cyanide in the synthesis. Acetylation of the amino group of n-mannosamine and n-galactosamine was carried out according to the method of Roseman and Ludoweig (18). CX4-N-acetyl-n- mannosamine was made by the same method with the use of Cr4-acetic anhydride. The Cr4-acetic anhydride and NaCl4N employed were purchased from Volk Radio-Chemical Company. N-Acetylneuraminic acid was isolated and crystallized from human plasma protein by the procedure of Svennerholm (19). 1421 by guest on April 10, 2020 http://www.jbc.org/ Downloaded from
Transcript of The Biosynthesis of Sialic Acids - Journal of Biological ...The Biosynthesis of Sialic Acids ... was...

THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 237, No. 5, May 1962
Printed in 7J.S.A
The Biosynthesis of Sialic Acids
LEONARD WARREN AND HERBERT FELSENFELD*
From the National Institute of Arthritis and Metabolic Diseases, National Institutes of Health, United States Public Health Service, Bethesda 14, Maryland
(Received for publication, December 4, 1961)
The sialic acids (1) have in common a 9 carbon carboxylic acid, neuraminic acid (Fig. l), which may be regarded as a con- densation product of a 3 carbon and a 6 carbon unit. Neu- raminic acid itself is not found naturally, but several N- and O-substituted derivatives are widely distributed in nature (1). Chemical (2-4) and enzymic (5-7) degradations of N-acetyl- neuraminic acid have shown that this substance is composed of pyruvic acid and an N-acetylhexosamine.
Working with neuraminic acid aldolase, an enzyme first described by Heimer and Meyer (5), Comb and Roseman (6) made the important observation that the N-acetylhexosamine component of N-acetylneuraminic acid is N-acetyl-n-mannos- amine. This compound was a product of the reversible N-ace- tylneuraminic acid aldolase-catalyzed reaction:
N-Acetylneuraminic acids N-acetyl-n-mannosamine + pyruvate
NAN1 has been synthesized chemically from oxaloacetate and N-acetyl-n-glucosamine (8, 9) or N-acetyl-n-mannosamine (10) in basic solutions. At the high pH of the reaction medium em- ployed, N-acetyl-n-glucosamine is effective because it is, in part, racemized to the actual reactant, N-acetyl-n-mannosamine (6). Decarboxylation of the fl-carboxyl group of oxaloacetate accom- panies the condensation reactions.
In previous communications (11, 12) we have described a new enzyme system which is responsible for the biosynthesis of N-acetylneuraminic acid in extracts of rat liver and bovine sub- maxillary gland. Pyruvic acid is inactive in this system, whereas it is a participant in the NAN-aldolase catalyzed-reaction. The equilibrium of the latter reaction favors degradation of NAN (6, 7), whereas the over-all biosynthetic reaction process cat- alyzed by this new enzyme system permits almost quantitative conversion of the precursors, phosphoenolpyruvate and N-ace- tyl-n-mannosamine to the final product, N-acetylneuraminic acid.
As previously reported (13), the over-all biosynthetic Reaction 4 is catalyzed by three enzymes, N-acetylmannosamine kinase,*
* Present address, Department of Pharmacology, Yale Uni- versity, New Haven, Connecticut.
1 The abbreviations used are: NAN, N-acetylneuraminic acid; NAN-g-P, N-acetylneuraminic acid g-phosphate; NAN-aldolase, N-acetylneuraminic acid aldolase.
2 In accordance with the nomenclature proposed by the Com- mission on Enzymes of the International Union of Biochemistry, the svstematic name of the kinase is ATP: N-acetamido-2- deox;n-mannose phosphotransferase and that for the condensing enzyme is phosphoenolpyruvate: N-acetamido-2-deoxy-n-man- nose-6-phosphate ligase. Since the mechanism of dephosphor- ylation of N-acetylneuraminic acid g-phosphate is not known (see discussion), no name is proposed for this enzyme at the pres- ent time.
a condensing enzyme, and a dephosphorylating enzyme. These have been partially purified and shown to catalyze Reactions 1, 2, and 3, respectively.
N-Acetyl-n-mannosamine + ATP Mg++K+
(1) N-acetyl-D-mannosamine-6-P + ADP
N-Acetyl-n-mannosamine-6-P
+ Hz0 GSH, KCN
Mg++
+ phosphoenolpyruvate
N-acetylneuraminic-9-P + Pi (2)
Mg++ N-Acetylneuraminic acid-g-P + Hz0 3
(3) N-acetylneuraminic acid + Pi
N-Acetyl-n-mannosamine + phosphoenolpyruvate + ATP + 2 Hz0 -+ N-acetylneuraminic acid + ADP + 2 Pi (4)
This paper describes the isolation, purification, and properties of the enzymes involved, which have been isolated from rat liver and bovine submaxillary gland, and the identification of the products of the reactions. Recently Roseman and his co-workers have described a similar series of enzymes (14, 15).
EXPERIMENTAL PROCEDURE
Materials and Method-s
ATP, GTP, CTP, and UTP were the products of the Pabst Laboratories. The tricyclohexylamine salt of phosphoenol- pyruvate was purchased from Mann Research Laboratories, Inc. Glycolaldehyde phosphate acetal, dicyclohexylammonium salt, was obtained from the California Corporation for Biochemical Research. For conversion to glycolaldehyde phosphate, a solu- tion of the latter was treated with Dowex 50-H+ at room tem- perature overnight (16). Bentonite was purchased from the Fisher Scientific Company and South Carolina kaolin (“Dixie clay”) from the R. T. Vanderbilt Company. DEAE-cellulose was obtained from Distillation Products, Inc. n-Mannosamine was synthesized by the method of Kuhn and Kirschenlohr (17) and was separated from its epimer n-glucosamine by fractional crystallization from HCl. [a]; -4.7” (c = 5% in 1 N HCl) (17). n-Mannosamine labeled in carbon 1 was made with Cl4- cyanide in the synthesis. Acetylation of the amino group of n-mannosamine and n-galactosamine was carried out according to the method of Roseman and Ludoweig (18). CX4-N-acetyl-n- mannosamine was made by the same method with the use of Cr4-acetic anhydride. The Cr4-acetic anhydride and NaCl4N employed were purchased from Volk Radio-Chemical Company. N-Acetylneuraminic acid was isolated and crystallized from human plasma protein by the procedure of Svennerholm (19).
1421
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

1422 Biosynthesis of Sialic Acids Vol. 237, No. 5
I HO-F-COOH
0 HC-H
HO-h-H 9 HI
H3C-C-NY-H
-C-H
H-h-OH
H-b-OH
bi20H
FIG. 1. N-Acetylneuraminic acid
TABLE I Summary of purijication of condensing enzyme from
extract of bovine submaxillary gland I
ml &ml units/mg
1. Original extract. . 100 22 0.003 6.60 2. Acetone (33 to 52%). 20 12.8 0.020 5.12 3. Ammonium sulfate (0 to 56%). 10 4.0 0.072 2.88 4. DEAE-cellulose.. 15 0.27 0.284 1.16
Inorganic phosphate was measured by the method of Lowry and Lopez (20). Total phosphate was measured by the method of Fiske and SubbaRow after digestion (21). Proteins were determined by the method of Lowry et al. (22), and reducing sugar by the Park-Johnson assay (23).
N-Acetylneuraminic acid g-phosphate and N-acetylneuraminic acid were measured by the thiobarbituric acid assay (24) with crystalline N-acetylneuraminic acid isolated from human plasma protein as a standard. The thiobarbituric acid chromo- gen was extracted into 3 ml rather than 4.3 ml of cyclohexanone. Optical densities were determined with a Beckman DU spectro- photometer with a microcell attachment (l-cm light path). N-Acetylneuraminic acid was also determined by the orcinol, Ehrlich, resorcinol, and diphenylamine tests (25-27). Infrared spectra3 were determined on 1-mg samples pressed in 600 mg of KBr with a Perkin-Elmer model 21 spectrophotometer.
Assays
N-Acetylmunnosamine Kinase: Assay l-This assay was based upon Reaction 4, the biosynthesis of NAN from phosphoenol- pyruvate, N-acetyl-D-mannosamine, and ATP. The kinase was assayed in the presence of excess condensing and dephosphorylat- ing enzyme. This assay was employed in the purification of fractions from rat liver. Since there was an excess of the dephos- phorylating enzyme both in the kinase and condensing enzyme fractions of rat liver, the final product measured was free NAN. For routine assays vessels contained the following in a volume of 0.3 ml: Tris-chloride buffer pH 7.6, 30 pmoles; ATP, 1 pmole; phosphoenolpyruvate, 0.5 pmole; N-acetylmannosamine, 0.17 pmole; MgC$, 6 pmoles; KCl, 10 pmoles; DPN, 0.02 pmole; TPN, 0.02 pmole. After 2 hours of incubation at 37” the entire vessel contents were assayed by the thiobarbituric acid method
3 We wish to thank Mr. H. K. Miller (NIH) for determining the infrared spectra.
(24). Synthesis of 0.01 pmole of NAN led to an increase of optical density of 0.193 at 549 rnl.c. A unit of enzyme activity is defined as the amount of enzyme that leads to the synthesis of 1 pmole of NAN per hour under the conditions of the assay. In the presence of an excess of condensing enzyme (0.05 unit dialyzed enzyme), the amount of NAN synthesized was propor- tional to the amount of kinase present.
Assay 2-A second assay for the kinase was that used by Leloir, Cardini, and Olavarria (28), in which sugars that are phosphorylated by kinase in the presence of ATP are precipi- tated, and the disappearance of sugar from the supernatant solu- tion is measured. Incubation vessels contained in a volume of 0.2 ml: Tris-chloride buffer, pH 7.6, 30 pmoles; N-acetylmannos- amine, 0.25 pmole; MgC12, 5 pmoles; enzyme, and where indi- cated ATP, 1 pmole. After 2 hours of incubation at 37”, 0.3 ml of a solution of 10% zinc sulfate and 0.3 ml of 0.3 M barium hy- droxide were added, and the tubes were centrifuged. An assay for N-acetylhexosamine (29) was carried out on 0.4 ml of super- natant solution. The difference in the optical density values of the vessels with and without ATP is a measure of the N-acetyl- hexosamine phosphorylated. In some experiments the amount of reducing sugar (23) remaining in the barium-zinc supernatant solution was used to measure the extent of phosphorylation.
Condensing Enzyme: Assay l-The condensing enzyme of liver was assayed in the presence of excess kinase, 0.05 unit, in the system described in Assay 1 for N-acetylmannosamine kinase (over-all reaction).
Assay 2-A second assay for the condensing enzyme was based upon the conversion of N-acetylmannosamine 6-phosphate and phosphoenolpyruvate to NAN or N-acetylneuraminic acid 9- phosphate (Reaction 2). Purification of the condensing enzyme from bovine submaxillary gland was followed by this method. In this assay N-acetylmannosamine 6-phosphate, 0.07 pmole; phosphoenolpyruvate, 0.5 pmole; MgC12, 1 pmole; reduced glutathione 2.5 pmoles; KCN, 1 pmole; Tris-chloride buffer, pH 7.6, 30 pmoles; and enzyme, in a volume of 0.20 ml, were incu- bated for 1 hour at 37”. The thiobarbituric acid assay for free N-acetylneuraminic acid was carried out on the entire vessel contents. Synthesis of product was linear with respect to time and enzyme concentration in the assay.
As purification of the condensing enzyme proceeded and the dephosphorylating enzyme was removed (Table I, Steps 3 and 4) the product of the reaction was NAN-g-P instead of NAN. Absence of dephosphorylating enzyme was detected by heating the reaction vessels after their regular l-hour incubation for 1 minute at loo”, adding 1 unit of semen phosphomonoesterase* and 50 pmoles of sodium acetate, pH 5.2, and further incubating for 30 minutes before assay by the thiobarbituric acid method. Since NAN-g-P has a molar extinction coefficient that is 62% that of NAN, treatment of vessels containing NAN-g-P with phosphomonoesterase resulted in an increase of color in the thiobarbituric acid assay compared to vessels not treated with phosphomonoesterase. This procedure allowed a correct esti- mation of condensing enzyme activity.
Assay for N-Acetyl-n-mannosamine CT-Phosphate-N-Acetyl-D-
mannosamine 6-phosphate was quantitated by measuring the maximal quantity of NAN formed in the presence of excess
4 Kindly supplied by Dr. L. A. Heppel. One unit of enzyme (0.3 pg of protein) can dephosphorylate 1 pmole of 5’-AMP per hour at pH 5.2.
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from
May 1962 L. Warren and H. Felsenfeld 1423
phosphoenolpyruvate and condensing and dephosphorylating enzymes. Samples of N-acetyl-n-mannosamine 6-phosphate of 0.005 to 0.03 pmole were incubated with 0.5 pmole of phospho- enolpyruvate, 1.0 pmole of MgCL, 30 pmoles of Tris-chloride buffer, pH 7.6, and 0.1 unit of rat liver-condensing enzyme for 2 hours at 37”. The thiobarbituric acid assay was carried out on the entire vessel contents. An optical density of 0.193 at 549 rnp was observed when 0.01 pmole of N-acetyl-n-mannosamine 6-phosphate was present originally.
RESULTS
N-Acetylneuraminic Acid, Product of Over-all Reaction--In
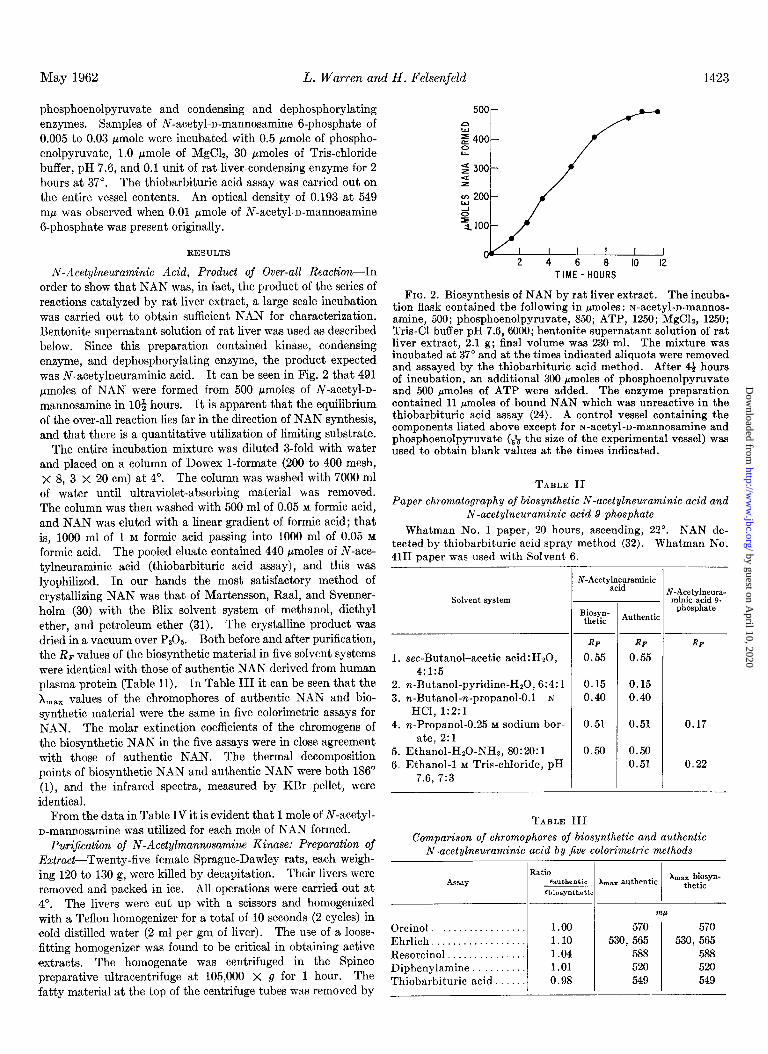
order to show that NAN was, in fact, the product of the series of reactions catalyzed by rat liver extract, a large scale incubation was carried out to obtain sufficient NAN for characterization. Bentonite supernatant solution of rat liver was used as described below. Since this preparation contained kinase, condensing enzyme, and dephosphorylating enzyme, the product expected was N-acetylneuraminic acid. It can be seen in Fig. 2 that 491 pmoles of NAN were formed from 500 pmoles of N-acetyl-n- mannosamine in lO$ hours. It is apparent that the equilibrium of the over-all reaction lies far in the direction of NAN synthesis, and that there is a quantitative utilization of limiting substrate.
The entire incubation mixture was diluted 3-fold with water and placed on a column of Dowex I-formate (200 to 400 mesh, X 8, 3 X 20 cm) at 4”. The column was washed with 7000 ml of water until ultraviolet-absorbing material was removed. The column was then washed with 500 ml of 0.05 M formic acid, and NAN was eluted with a linear gradient of formic acid; that is, 1000 ml of 1 M formic acid passing into 1000 ml of 0.05 M
formic acid. The pooled eluate contained 440 I.rrnoles of N-ace- tylneuraminic acid (thiobarbituric acid assay), and this was lyophilized. In our hands the most satisfactory method of crystallizing NAN was that of Martensson, Raal, and Svenner- holm (30) with the Blix solvent system of methanol, diethyl ether, and petroleum ether (31). The crystalline product was dried in a vacuum over P,Os. Both before and after purification, the RF values of the biosynthetic material in five solvent systems were identical with those of authentic NAN derived from human plasma protein (Table II). In Table III it can be seen that the x max values of the chromophores of authentic NAN and bio- synthetic material were the same in five calorimetric assays for NAN. The molar extinction coefficients of the chromogens of the biosynthetic NAN in the five assays were in close agreement with those of authentic NAN. The thermal decomposition points of biosynthetic NAN and authentic NAN were both 186’ (l), and the infrared spectra, measured by KBr pellet, were identical.
From the data in Table IV it is evident that 1 mole of N-acetyl- D-mannosamine was utilized for each mole of NAN formed.
Puti$cation of N-Acetylmannosamine Kinase: Preparation of Extract-Twenty-five female Sprague-Dawley rats, each weigh- ing 120 to 130 g, were killed by decapitation. Their livers were removed and packed in ice. All operations were carried out at 4”. The livers were cut up with a scissors and homogenized with a Teflon homogenizer for a total of 10 seconds (2 cycles) in cold distilled water (2 ml per gm of liver). The use of a loose- fitting homogenizer was found to be critical in obtaining active extracts. The homogenate was centrifuged in the Spinco preparative ultracentrifuge at 105,000 X g for 1 hour. The fatty material at the top of the centrifuge tubes was removed by
OY, 2 4 6 8 IO 12
TIME-HOURS
FIG. 2. Biosynthesis of NAN by rat, liver extract. The incuba- tion flask contained the following in pmoles : N-acetyl-n-mannos- amine, 500; phosphoenolpyruvate, 850; ATP, 1250; MgCL, 1250; Tris-Cl buffer pH 7.6,6000; bentonite supernatant solution of rat liver extract, 2.1 g; final volume was 230 ml. The mixture was incubated at 37” and at the times indicated aliquots were removed and assayed by the thiobarbituric acid method. After 4+ hours of incubation, an additional 300 rmoles of phosphoenolpyruvate and 500 pmoles of ATP were added. The enzyme preparation contained 11 rmoles of bound NAN which was unreactive in the thiobarbituric acid assay (24). A control vessel containing the components listed above except for N-acetyl-n-mannosamine and phosphoenolpyruvate (& the size of the experimental vessel) was used to obtain blank values at the times indicated.
TABLE II Paper chromatography of biosynthetic N-acetylneuraminic acid and
N-acetylneuraminic acid g-phosphate
Whatman No. 1 paper, 20 hours, ascending, 22’. NAN de- tected by thiobarbituric acid spray method (32). Whatman No. 41H paper was used with Solvent 6.
Solvent system
1. set-Butanol-acetic acid:HzO, 4:1:5
2. n-Butanol-pyridine-HZO, 6:4: 1 3. n-Butanol-n-propanol-0.1 N
HCI, 1:2:1 4. n-Propanol-0.25 M sodium bor-
ate, 2:l 5. Ethanol-HzO-NHa, 80:20: 1 6. Ethanol-l M Tris-chloride, pH
7.6, 7:3
-
-
N-Acety$raminic
Biosyn- thetic Authentic
RF RF
0.55 0.55
0.15 0.15 0.40 0.40
0.51 0.51
0.50 0.50 0.51
I-Acetylneura- minic acid 9-
phosphate
RF
0.17
0.22
TABLE III Comparison of chromophores of biosynthetic and authentic
N-acetylneuraminic acid by jive calorimetric methods /
Assay X,,, authentic Xmsx biosyn- thetic
Orcinol ............... 1.00 Ehrlich ................ 1.10 Resorcinol ............. 1.04 Diphenylamine ........ 1.01 Thiobarbituric acid. ... 0.98
+w 570 570
530, 565 530, 565 588 588 520 520 549 549
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

1424 Biosynthesis of Sialic Acids Vol. 237, No. 5
TABLE IV
Formation of N-aeelylneuraminic acid from C14-N-acetyl-D-mannosamine
The incubation mixture contained in micromoles: N-acetyl-n- mannosamine, 0.57, 139,000 c.p.m.; phosphoenolpyruvate, 3; ATP, 5; Tris-chloride buffer, pH 7.6, 150; N-acetyl-r-mannosamine kinase (rat liver), 4 mg; condensing enzyme (rat liver), 3 mg; Hz0 to 2.8 ml. After 5 hours of incubation at 37”, the protein was precipitated with 3 volumes of ethanol and centrifuged. The supernatant solution was evaporated to dryness, and the residue was taken up in water and applied to a column of Dowex l-acetate, 200 to 400 mesh, 1 X 8 cm. The column was washed with 60 ml of water and then eluted with a linear gradient of acetic acid, 100 ml of 3 M acetic acid into 100 ml of water. The yield of NAN was 0.54 pmoles by the thiobarbituric acid test. The Cl4 specific activity of the NAN in seven vessels over the elution peak varied from 239,000 to 250,000 c.p.m. per pmole with a standard deviation zt1300 c.p.m. per pmole.
Substrate
Substrate Product
CWN-Acetyl-n-mannosamine . 242 ) 000 243,000
suction and discarded, and the supernatant solution collected. The volume of this extract was approximately 190 ml.
Bentonite Step-To l-ml aliquots of the extract in separate tubes were added 30, 40, 50, and 60 mg of bentonite. The solutions were stirred, left in an ice bath for 10 minutes, and then centrifuged at 5000 X g in an angle centrifuge at 4” for 10 min- utes. The minimal quantity of bentonite per ml which removed the red color of the extract, leaving a yellow supernatant solu- tion, was employed for the preparation of the remainder of the extract. This amount was usually 50 mg per ml of extract. The bentonite supernatant solution was approximately 10% less in volume than the original liver extract. It has been observed that large losses of enzyme activity occurred with bentonite treatment if extracts were made from livers weighing less than 6 g each.
DEAE-Cellulose Chromatography-An aliquot of 50 ml of the bentonite supernatant solution was added to a column of DEAE- cellulose (1.5 x 16 cm) in the chloride form which had been washed with distilled water. The extract was placed on the column. The column was then washed with 50 ml of cold dis- tilled water, and lo-ml fractions were collected. A large quan- tity of protein, containing the condensing enzyme free of kinase, passed through the column. A linear gradient was then begun
TABLE V
Summary of purifLcation of N-acetylmannosamine kinase from extract of rat liver
, I I Fraction
1. Original extract. 2. Bentonite supernatant
solution 3. DEAE-column eluate 4. Zinc precipitate. 5. Cobalt precipitate.
ml m/ml units/mg 55 50.0 0.029
50 22.6 0.057 64.5 126 3.8 0.109 52.2
15 6.4 0.400 38.4 10 3.0 0.900 27.0
79.8
with 200 ml of 0.02 M NaCl in 0.001 M Tris-chloride, pH 7.6, in the mixing chamber and 200 ml of 0.5 M NaCl in 0.001 M Tris- chloride, pH 7.6, in the reservoir. The kinase was eluted at approximately 0.25 M NaCl.
Zinc Precipitation-The tubes containing the kinase activity as determined by Assay 1 were pooled (126-ml volume) and 2 pmoles of zinc acetate were added per ml of solution. A precipi- tate formed immediately and after 10 minutes it, was removed by centrifugation in an angle centrifuge at 5000 X g for 15 minutes. The precipitate, containing the kinase, was dissolved in a buffer containing 0.01 M disodium EDTA, 0.05 M Tris-chloride, pH 7.6, and 0.05 M KCI. The final volume of the solution was 15 ml. EDTA was necessary for solubilization of the precipitate.
Cobalt Precipitation-To each milliliter of this solution were added 7 pmoles of cobalt chloride, and the solution was allowed to stand at, 0”. B precipitate formed slowly, and after 20 to 30 minutes the precipitate containing the kinase was removed by centrifugation and then dissolved in 10 ml of the Tris-EDTA- KC1 buffer.
This enzyme preparation was 38fold purified and contained approximately 34% of the original activity (Table V). It con- tained no NAN-aldolase or pyrophosphatase. Approximately half of the preparations contained no N-acetylglucosamine kinase. It was completely free of condensing enzyme activity.
Preparations of Condensing Enzyme. (1) Rat Liver-This en- zyme was difficult to purify from rat liver although it could be readily separated from the kinase. The enzyme passed through the column of DEAE-cellulose, chloride form, and was not ame- nable to purification by other types of columns, by gels, ethanol, ammonium sulfate, or by isoelectric precipitation. Activity did not, survive more than a few freezings and thawings, and at 4”, activity was lost within a week. Salts, EDTA, and sulfhydryl compounds did not stabilize the enzyme.
Stable preparations of condensing enzyme free of kinase were prepared by treating the bentonite supernatant solution of rat liver extract with South Carolina Kaolin (“Dixie clay”). To each milliliter of bentonite supernatant solution were added 75 mg of Dixie clay. The mixture was kept at 0” for 10 minutes and then centrifuged at 8000 X g for 15 minutes. The clear supernatant solution was dialyzed against a large excess of cold distilled water (20 ml to 4 liters) for approximately 15 hours un- til a very heavy white precipitate formed, which was removed by centrifugation. The supernatant solution contained an enzyme which was 2-fold purified (specific activity, 0.06) from the origi- nal extract with 7Oa/, of the original activity. This preparation was stable for at least 3 months at, -14”; it contained no kinase but did contain dephosphorylating enzyme. It, was used rou- tinely, in excess, for the assay of the kinase.
(2) Bovine Submaxillary Gland-Condensing enzyme was also obtained from bovine submaxillary gland. Fresh gland contains large quantities of mucin which makes fractionation difficult. Since a slow destruction of mucin takes place during storage, aged frozen glands (1 year at -14”) were employed.
Preparation of Extract-Sixty grams of frozen gland were minced and placed in a Waring Blendor at 4” with 120 ml of distilled water. After blending for 30 seconds, the homogenate was strained through a double thickness of gauze. The liquid that passed through and that which was pressed out, of the gauze were pooled and centrifuged for 45 minutes at 105,000 X g in the Spinco preparative ultracentrifuge (No. 40 rotor). The solid fatty material at the tops of the centrifuge tubes was removed
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

May 1962 L. Warren and H. Felsenfeld 1425
by aspiration, and the supernatant solution remaining was de- canted and pooled. The supernatant solution could be stored for several months at -14” with no loss of activity.
Acetone Fractionation-All procedures were performed at - 14”. To 100 ml of the extract were added 50 ml of acetone (precooled to - 14”) ; the mixture was stirred and immediately centrifuged for 5 minutes at 8000 X g at -14”. The precipitate was dis- carded. To the supernatant solution were added 58 ml of ace- tone, and after brief stirring, the mixture was centrifuged at 8000 X g for 5 minutes. This precipitate (33-52 y0 acetone) was allowed to drain for 60 seconds and was then dissolved in 20 ml of cold 0.05 M Tris-chloride buffer, pH 7.6, containing 0.05 M
KCI. Ammonium Su2fate Step (0 to 56% Fraction)-To 18 ml of the
enzyme solution at 0” were added 25.5 ml of a saturated solution (4”) of (NH&S04 (recrystallized from 0.001 M EDTA), pH 7.0. After standing for 10 minutes, the mixture was centrifuged at 8000 X g for 15 minutes at 4” and the precipitate was dissolved in 10 ml of Tris-chloride-KC1 buffer. The solution was dialyzed against two changes of 0.05 M Tris-acetate buffer of 4 liters each, pH 7.6, and potassium acetate, 0.05 M, for 4 hours at 4”.
DEAE-Cellulose Chromatography-Nine milliliters of the am- monium sulfate fraction were diluted to 18 ml with 0.02 M Tris- HCl, pH 7.6, and were placed on a column of DEAE-cellulose (1.5 X 11 cm) which had been previously equilibrated with 0.003 M Tris-HCl, pH 7.6, and 0.001 M EDTA. After washing the column with 50 ml of a solution containing 0.001 M EDTA and 0.003 M Tris-chloride, pH 7.6, the enzyme was eluted with a linear gradient of KC1 and NaCl, with 100 ml of 0.25 M KCl, 0.25 M NaCl, 0.001 M EDTA, 0.02 M Tris-HCl, pH 7.6, that passed into a mixing chamber containing 100 ml of 0.01 M KCI, 0.01 M NaCI, 0.001 M EDTA, and 0.02 M Tris-HCl, pH 7.6. Fractions of 5 ml were collected. The condensing enzyme was eluted in two or three tubes, beginning with tube twelve. The enzyme was concentrated by lyophilization with 10 to 20% loss of activity.
The most purified enzyme preparations were 95-fold purified with an 18% recovery of units. Approximately half of the am- monium sulfate preparations (Step 3) contained no NAN-g-P-de- phosphorylating activity. Fractions from the DEAE column were free of this activity. A summary of the purification is pre- sented in Table I. Units of enzyme activity are the same as for the kinase enzyme preparation.
Phosphorylation of N-Acetyl-o-mannosamine (Reaction 1). Formation, Isolation, and Characterization of Product, N-Acetyl- o-mannosamine B-Phosphate-In a typical incubation (13) the following materials were incubated for 3 hours at 37”: N-Acetyl- n-mannosamine, 15 pmoles; ATP, 40 pmoles; magnesium acetate, 200 pmoles; 8 units of liver kinase; Tris acetate buffer, pH 7.6, 300 pmoles in a volume of 12 ml. At the end of the incuba- tion the vessel was heated for 3 minutes at loo”, and the de- natured protein was removed by centrifugation. The super- natant solution was diluted 3-fold with water and placed on a column of DEAE-cellulose in the hydroxyl form (2 x 14 cm). After washing with 50 ml of water at 4”, N-acetylmannosamine 6-phosphate was eluted by lithium chloride (linear gradient, 150 ml of 0.15 N LiCl passing into 150 ml of water). In a pre- vious report (13), it was erroneously stated that N-acetyl-n- mannosamine B-phosphate was eluted with HCl rather than LiCl. Fractions of 5 ml were collected. N-Acetylmannosamine 6-phosphate was eluted in a volume of approximately 40 ml after
the passage of 200 ml of solution. Eluates were assayed for product by determinations of N-acetylhexosamine (29)) Cl4 radioactivity, or by enzymic conversion to NAN, as described in “Materials and Methods.” The vessels containing N-acetyl- mannosamine g-phosphate were pooled and lyophilized. The dry white residue was dissolved in 1 ml of water and 30 pmoles of barium acetate were added, followed by 4 ml of absolute ethanol. After 3 hours at 0”, the precipitate was collected by centrifugation, and dried and stored in a vacuum over Pz06. Yields of 10 to 13 pmoles of product were obtained.
In more recent experiments 500+mole batches of N-acetyl- mannosamine g-phosphate have been obtained in correspond- ingly larger incubation mixtures. The incubation mixtures were deproteinized as above, and the product was purified on columns of Dowex 1-formate (1.5 X 15 cm; 200 to 400 mesh; 8% cross- linked) with a linear gradient of formic acid (300 ml of 3 M formic acid into 300 ml of water). N-Acetylmannosamine g-phosphate was eluted in the last 100 ml of the run and was precipitated as its barium salt, as above. Elution was carried out at 4’.
As previously reported (13), analysis of the product purified by means of a column of DEAE-cellulose proved it to be N- acetylmannosamine 6-phosphate. It did not absorb ultraviolet light. With N-acetylmannosamine 6-phosphate with a 84 specific activity of 12,000 c.p.m. per pmole as standard of 1.00, 1.08 pmoles of NAN could be formed enzymically from 1 pmole of N-acetylmannosamine B-phosphate. One micromole of the material contained 0.98 mole of phosphate as determined by the Fiske - SubbaRow method after digestion (21). When 1.00 pmole was incubated with 5 units of purified semen phospho- monoesterase in 0.05 M sodium acetate, pH 5.2, for 1 hour, 1.02 pmoles of inorganic phosphate (20) were released from a bound form. Less than 0.01 mole of inorganic phosphate was present per mole of product before treatment with semen phospho- monoesterase.
The material, 0.01 to 0.05 per mole, obtained after treatment with semen phosphomonoesterase was incubated with 20 pmoles of periodic acid, in a volume of 0.4 ml of 2.5 N H2S04 for 30 minutes at 37” and then assayed for formaldehyde formation (33); 0.92 mole of formaldehyde was generated per mole of orig- inal N-acetylmannosamine 6-phosphate only after enzymic dephosphorylation had taken place. This strongly suggested that the phosphate was attached to the hydroxyl group on car- bon 6. To verify this, the periodate oxidation products of the phosphorylated intermediate (1 pmole of intermediate to 20 pmoles of periodic acid, pH 4.5, 20 hours, room temperature) were placed on Whatman No. 43 H paper and chromatographed for 18 hours at room temperature in a solvent consisting of ter- tiary butanol and water, 80:20, containing 4% picric acid (34, 35). A material containing phosphate, located by the Hanes- Isherwood spray procedure (34), with an RF of 0.42, cochromato- graphed with authentic glycolaldehyde phosphate (35). This material could only have come from carbon atoms 5 and 6 of N-acetylmannosamine 6-phosphate.
Since the purified material could reduce ferricyanide (23)) there was no substitution on carbon atom 1. As previously reported the reducing power of N-acetylmannosamine g-phosphate was found to be 47% that of N-acetylmannosamine. One pmole of the dephosphorylated intermediate had 1.07 reducing groups compared to an authentic N-acetyl-n-mannosamine standard.
The absorption spectrum and the molar extinction coefficient (E = 16,100) of the product in the assay of Reissig, Strominger,
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Biosynthesis of Sialic Acids Vol. 237, No. 5
TABLE VI
Paper chromatography of N-acetyl-n-mannosamine 6-phosphate
Ascending chromatography on Whatman No. 1 paper for 20 hours at room temperature. N-Acetyl-n-mannosamine 6-phos- phate was located by quinine dip for phosphate (38), spray for N-acetylhexosamine (39), and for phosphate (34). The area containing the intermediate was also cut out and extracted with water. To the solution thus obtained were added phosphoenol- pyruvate, rat liver condensing enzyme, and Tris-chloride buffer, pH 7.6, as described in “Materials and Methods.” Upon incuba- tion at 37”, enzymic conversion to NAN was observed.
Solvent system RF
n-Butanol-acetic acid:HsO, 4:1:5.........................
ethanol-l M Tris-chloride, pH 0.14
7.6, 65:35. _. _. n-Propanol-0.25 M sodium bor-
0.43
ate, 2:1....................... 0.58 Ethanol-acetic acid, 65:35. 0.63
and Leloir (29) with an g-minute boiling in alkaline borate (36) were identical with those of N-acetylmannosamine. This in- dicates that there was no substitution on carbon atoms 3 or 4 of the hexosamine (37).
The Morgan-Elson spray method (38) was employed for locat- ing N-acetylhexosamines on paper (Table VI). N-Acetylhex- osamines substituted at carbon 1 do not react (39). However, the biosynthetic, N-acetyl-n-mannosamine 6-phosphate, yielded a characteristic purple spot.
Table VI gives the RF values of N-acetylmannosamine 6-phos- phate in four solvent systems. The material was eluted from the paper and converted to NAN under appropriate conditions, described under “Materials and Methods.” N-Acetylmannos- amine g-phosphate traveled as a single band toward the positive pole during electrophoresis with Whatman No. 3MM paper, 450 volts, 0.5 M ammonium formate buffer, pH 3.5, 6) hours. Under these conditions N-acetylmannosamine g-phosphate traveled 10 cm, whereas picric acid and uridine diphosphate- N-acetylglucosamine markers traveled 13 cm. The material in the band was identified by Cl4 radioactivity as well as by the methods described in the paper chromatographic studies.
N-Acetyl-n-mannosamine g-phosphate and its enzymically dephosphorylated product were chromatographed on Schleicher and Schuell No. 598 paper (ascending 22” for 24 hours). Whereas N-acetylmannosamine 6-phosphate remained very close to the origin on borate-treated paper (40)) the phosphomonoesterase- treated material cochromatographed with authentic N-acetyl- n-mannosamine, RF 0.10, in a solvent consisting of n-butanol, pyridine, and water (6:4:3) (7,40). The RF value of N-acetyl- n-glucosamine was 0.22. The authentic samples of N-acetyl- hexosamines employed were incubated with semen phos- phomonoesterase before chromatography to detect possible interconversion; none was found. A second solvent also showed that the dephosphorylated material wa N-acetyl-n-mannosa- mine. In butanol, acetic acid, and water (4:1:5) the chro- matogram was run for 26 hours, beyond the point at which the solvent had reached the edge of the paper. The RF of N-acetyl- n-mannosamine was 2.2 times that of N-acetyl-n-glucosamine.
Specijkity and Requirements of N-Acetylmannosamine Kinase- The most purified kinase preparations of liver were unable to
phosphorylate n-glucosamine, n-galactosamine, and their N- acetyl derivatives, n-mannosamine, n-glucose, n-ribose, n-galac- tose, or n-mannose, when followed by Assay 2 for kinase. It appears that the kinase is specific for N-acetyl-n-mannosamine and that the enzyme can be called N-acetyl-n-mannosamine kinase. GTP, UTP, and CTP would not substitute for ATP. Magnesium ions were essential for the reaction even with crude extracts of liver. Under the conditions of the assay, the optimal concentration was 0.015 M. Manganous, ferric, cobaltous, cupric, zinc, and aluminum ions in concentrations ranging from 0.001 M to 0.02 M could not substitute for magnesium ions. The latter 4 ions precipitated the enzyme. Potassium ions, although not essential, stimulated phosphorylation maximally, 35%, at a concentration of 0.04 M.
Equilibrium-In experiments in which N-acetyl-n-mannos- amine was incubated with magnesium ions, kinase, and a a-fold excess of ATP, all the substrate was phosphorylated and no detectable free N-acetylmannosamine remained as measured by the assay of Leloir, Cardini, and Olavarria (28). When the reaction was allowed to proceed to completion, with C14-N- acetyl-n-mannosamine as substrate, 142,000 c.p.m. per pmole, chromatography of the reaction mixture revealed no detectable N-acetylmannosamine. As with other kinase reactions, the equilibrium of this reaction is such as to favor strongly the phos- phorylation of the 6 hydroxyl group of N-acetyl-n-mannosamine.
Stoichiometry-For every mole of N-acetyl-n-mannosamine converted to N-acetyl-n-mannosamine 6-phosphate, 1 mole of ADP was formed (Table VII). No inorganic phosphate was formed during the course of the reaction.
TABLE VII
Stoichiometry of N-acetyl-D-mannosamine kinase reaction
The incubation mixture contained in a volume of 36.5 ml; N- acetyl-o-mannosamine, 100 Mmoles; ATP, 200 Mmoles; MgC12, 450 pmoles; enzyme (zinc precipitate), 35 mg; Tris-acetate, pH 7.6, 1000 pmoles. The mixture was incubated for 8 hours at 37”. It was then heated for 3 minutes at 100” to inactivate the kinase and centrifuged to remove protein. To measure the amount of N-acetylmannosamine 6-phosphate formed, aliquots of 0.01 ml were incubated with phosphoenolpyruvate and condensing en- zyme of rat, liver, kinase free, for varying periods of time until maximal conversion of intermediate to free N-acetylneuraminic acid occurred. Disappearance of N-acetyl-r-mannosamine was measured by the method of Leloir, Cardini, and Olavarria (28). ADP was measured by placing an aliquot of 0.5 ml of the heated incubation mixture on a column of Dowex l-formate, 7 X 0.7 cm, and eluting with a linear gradient of formic acid from 0 to 5 M.
The eluted ADP was pooled and measured by its ultraviolet light absorption. It, cochromatographed on paper with authentic ADP in three solvent systems: (a) ethanol-l M Tris-chloride buffer, pH 7.6, 65:35, RF 0.23; (b) isobutyric acid-O.5 N NH3, 100:60 (RF 0.50) ; (c) sodium phosphate, 0.1 M, pH 7.8, containing 600 g of (NH,)&‘Oa per 1000 ml, in an atmosphere of n-propanol (Rp 0.17).
/moles pmoles
Initial. 100.0 200.0 Final. 24.4 127.8* A -75.6 -72.2*
* Calculated from the ADP value.
pmozes /.lmoles
0 0 76.0 72.2
+76.0 +72.2
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

May 1962 L. Warren and H. FeEsenjeld 1427
Characteristics of Einase Reaction-The pH optimum meas- ured by Assay 2 lies between 7.6 and 8.0. The K, values for N-acetyl-n-mannosamine and ATP under the conditions of Assay 2 were 2.3 X 1W4 M and 1.6 X 10V3 M, respectively, as calculated from Lineweaver-Burk plots (41).
Condensation of N-Acetyl-o-mannosamine B-Phosphate and Phosphoenolpyruvate (Reaction 2). Formation, Isolation, dnd Characterization of N-Acetylncuraminic acid 9-phosphate-Incu- bation of N-acetylmannosamine 6-phosphate and phosphoenol- pyruvate with partially purified condensing enzyme from bovine submaxillary gland led to the accumulation of N-acetylneura- minic acid g-phosphate (Reaction 2). A typical incubation and purification of NAN-g-P on a column of Dowex 1-formate have been reported previously (13).
Paper chromatography (Table II) and electrophoresis with Whatman No. 3MM paper, at 450 volts, and 0.5 M ammonium formate, pH 3.5, for 6 hours, revealed a single spot that con- tained radioactivity and phosphate (34) and was reactive in the thiobarbituric acid assay. When NAN-g-P was treated with semen phosphomonoesterase, the produce cochromatographed with authentic NAN in Solvents 1, 4, and 6 of Table II.
An analysis was made on NAN-g-P which had been enzy- mically formed from N-acetyl-n-mannosamine 6-phosphate, labeled with Cl4 in the first carbon. The specific activity of the substrate was 137,000 c.p.m. per pmole, and the same specific activity was used as a standard in the analysis of the product. On this basis, 1 mole of the product, NAN-g-P, contained 1.00 mole of phosphate. No inorganic phosphate was detectable (20). Semen phosphomonoesterase released 1.05 molar equiva- lents of inorganic phosphate, and 0.95 molar equivalent of form- aldehyde was released upon periodate oxidation (33) only after treatment of NAN-g-P with phosphomonoesterase. No form- aldehyde was released without prior exposure to phosphomono- esterase. Further evidence that the phosphate was on the 9 hydroxyl group was that a material cochromatographing with glycolaldehyde phosphate (35) was formed upon periodate oxi- dation of the N-acetylneuraminic acid phosphate product. Methods of analysis were the same as those described for N- acetylmannosamine B-phosphate. Enzymic dephosphorylation was carried out by incubating 0.5 pmole of NAN-g-P with 3 units of purified semen phosphomonoesterase in 0.05 M sodium acetate buffer, pH 5.2. The final volume was 0.3 ml and the incubation lasted 14 hours at 37”.
Biosynthesized sialic acids can be degraded by the NAN- aldolase of Clostridium perfringens. Enzymically dephospho- rylated NAN-O-P, as well as NAN formed by liver, was incubated with a crude preparation of NAN-aldolase from Clostridium perfringens, ATCC 10543 (7). Incubation vessels contained 0.5 pmole of NAN, 3 mg of protein, and 30 pmoles of potassium phosphate buffer, pH 7.2, in a final volume of 0.3 ml. After 1 hour of incubation at 37”, only 12% of the original NAN re- mained. After removal of protein by heating, the incubation mixtures were chromatographed in two solvent systems which separate N-acetyl-o-glucosamine from N-acetyl-n-mannosamine, as described in the section on the characterization of N-acetyl- n-mannosamine B-phosphate. A material which cochromato- graphed with authentic N-acetyl-n-mannosamine was the only N-acetylhexosamine formed upon enzymic degradation of NAN. Authentic N-acetyl-n-glucosamine and N-acetyl-n-mannosamine, incubated with clostridial extract, were employed as controls. These were unaltered by the clostridial preparation.
The molar extinction coefficient in the thiobarbituric acid assay (24) increased from 35,400 to 57,000 upon removing the phosphate of NAN-g-P by semen phosphomonoesterase. Max- imal absorption occurred at 549 rnp with both compounds. Roseman et al. (15) have reported that the molar extinction coefficient of NAN-g-P is 78% that of NAN in another modifi- cation of the thiobarbituric acid assay (42). NAN-g-P gave the characteristic color in the orcinol and direct Ehrlich (25) assays for NAN.
NAN-g-P was reducible with sodium borohydride, indicating that the 2-position is not blocked (43). After 0.05 pmole of NAN-g-P or NAN was incubated with 1 mg of sodium boro- hydride at room temperature for 30 minutes in 0.2 ml of 0.05 M Tris, pH 7.6, the reaction was terminated by the addition of 0.02 ml of acetone. The product was unreactive in the thio- barbituric acid and orcinol assays, presumably because of the reduction of the keto group at carbon 2 of NAN and NAN-g-P. Weissbach and Hurwitz (43) have reported that borohydride reduction of the analogous compound 2-keto-3-deoxy-7-phos- phoheptonic acid also leads to the disappearance of reactivity in the thiobarbituric acid reaction.
Xpeci$city of Condensing Enzyme-N-Acetyl-n-glucosamine, N-acetyl-n-glucosamine 6-phosphate, iv-acetyl-n-galactosamine, and their deacylated forms as well as n-mannosamine, n-ribose 5-phosphate, n-glucose 6-phosphate, and n-mannose 6-phosphate could not substitute for N-acetyl-n-mannosamine 6-phosphate. In studies of the over-all reaction by crude rat liver extracts, UDP-N-acetylglucosamine could replace N-acetylmannosamine, presumably because of an epimerase which converts UDP-N- acetyl-n-glucosamine to N-acetyl-n-mannosamine (4446). In the course of investigation it was observed that some fractions could catalyze NAN synthesis from N-acetylmannosamine but not from UDP-N-acetylglucosamine. Since the reverse was never found, it was concluded that N-acetyl-n-mannosamine was the more immediate precursor of NAN. The biosynthesis of sialic acid by the rat liver system serves as a convenient assay method for measurement of this epimerase.
An analogue of N-acetyl-n-mannosamine, 3-acetamido-3- deoxy-o-mannose,5 was inactive in the over-all rat liver system and did not inhibit synthesis of NAN when present in the same concentration as N-acetyl-n-mannosamine or N-acetyl-n-man- nosamine 6-phosphate.
Pyruvate, lactate, and oxaloacetate could not substitute for phosphoenolpyruvate. Under conditions described in Assay 1, more than 80% of the oxaloacetate remained unaltered after a l-hour incubation with condensing enzyme from liver or bovine submaxillary gland. Oxaloacetate was measured by the method of Rosenthal (47).
Cofactor Requirements and Inhibitors of Condensing Reaction- As the purification of the condensing enzyme from bovine sub- maxillary gland proceeded, it was found that activity was com- pletely lost in all fractions unless boiled crude extracts were present in the incubation vessel. The ammonium sulfate frac- tion (Table I, Step 3) was almost inactive without heated extract (Fig. 3). It has been found that three factors, i.e. mag- nesium ions, KCN, and sulfhydryl compounds such as gluta- thione, could fully replace the heated extract. The dependence of NAN-BP synthesis on these factors is illustrated in Fig. 3. The optimal concentrations of these factors are also shown in
5 Kindly supplied by Dr. H. H. Baer.
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

1428 Biosynthesis of Sialic Acids Vol. 237, No. 5
S z 0.025
d 9 0-
1.0 2.0 3.0 MOLARITY X IO-’
FIG. 3. Cofactor requirements for the synthesis of NAN-g-P (Reaction 2). Each vessel contained in a volume of 0.2 ml: N- acetyl-n-mannosamine 6-phosphate, 0.15rmole; phosphoenolpyru- vate, 0.5 pmole; condensing enzyme 0.2 mg (ammonium sulfate fraction). and Tris-Cl buffer nH 7.6.30 umoles. Curve A-Vessels contained 0.005 M KCN: O.Oli5 M glutathione. MgClz concentra- tion as indicated. Curve B-Vessels contained 0.0125 M gluta- thione: 0.005 M MgCls . KCN concentration as indicated. Curve C-Vessels contained 0.005 M KCN: 0.005 M MgC12. Glutathione concentration as indicated. Incubation for 1 hour at 37”. Thio- barbituric acid assay was performed on the entire contents of the vessels (24).
Fig. 3. Magnesium ions could not be replaced by manganous or cobaltous ions. Zinc, aluminum, and cuprous ions at a con- centration of 10v3 M resulted in a complete inhibition of syn- thetic activity of a system activated by 5 X 10m3 M MgC12. EDTA at its optimal concentration (1.5 X 10m4 M), was only 57 y. as effective as potassium cyanide at its optimal concentra- tion (1.25 x 10e2 M). Mercaptoethanol and homocysteine at a concentration of 1.5 x 10m2 M could replace glutathione. The enzyme reaction was inhibited 80% by 5 x 10-a M p-hydroxy- mercuribenzoate in an incubation mixture containing 1.25 X 10e2 M glutathione. The cofactor requirements of the enzyme and the nature of the inhibitory substances that have been described strongly suggest that the condensing enzyme of bovine submaxillary gland contains sulfhydryl groups which are essen . tial for activity. Sulfhydryl compounds had no apparent effect on the condensing enzyme of liver. DPN and TPN did not stimulate the purified bovine submaxillary gland enzyme, but they did show an erratic stimulation of the crude rat liver sys- tem (11).
The synthesis of 2-keto-3-deoxyheptonic acid by crude puri- fied extracts of Escherichiu coli is stimulated by sulfhydryl com- pounds and by DPN (43, 48). Srinivasan and Sprinson (49) have also reported that mercaptoethanol stimulates the syn- thesis of 2-keto-3-deoxy-7-phosphoheptonic acid from erythrose 4-phosphate and phosphoenolpyruvate by a go-fold purified enzyme from E. coli.
Stoichiometry and Equilibrium of Condensing Reaction-From Table VIII it can be seen that 1 mole of N-acetyl-n-mannos- amine g-phosphate and 1 mole of phosphoenolpyruvate were converted to equimolar quantities of NAN-g-P and Pi according to Reaction 2. The NAN-g-P formed had a molar extinction coefficient that was 62% that of NAN. By analogy, the ex- tinction coefficient of 2-keto-3-deoxy-7-phosphoheptonic acid is 25 ye lower than the corresponding nonphosphorylated com- pound with the thiobarbituric acid assay (43). After incubation of the biosynthesized NAN-g-P with semen phosphomonoes- terase, the molar extinction increased to that of NAN and an
equimolar amount of phosphate (20) was released. It was ap- parent that the preparation of condensing enzyme that was employed for biosynthesis of NAN-O-P contained no dephos- phorylating activity. This preparation was also tested with the NAN-O-P that had been used for analysis and was found to be devoid of dephosphorylating activity. The equilibrium of Re- action 2 lies far in the direction of synthesis of NAN-g-P, since the reaction proceeded until no iir-acetyl-n-mannosamine 6-phos- phate remained in the presence of 3 times as much phospho- enolpyruvate.
Attempts to reverse Reaction 2 have been unsuccessful. In these experiments NAN-g-P, excess inorganic phosphate, co- factors, and condensing enzyme were incubated and then assayed by the thiobarbituric acid assay. No destruction of NAN-g-P was observed.
Characteristics of Condensing Reaction-The pH optimum of the reaction was 7.6 in Tris buffer, and it fell off sharply at higher and lower pH values. At pH 7.0 and 8.0, the activity was ap- proximately 65% that at 7.6. Under the conditions described in Fig. 4, the K, value for phosphoenolpyruvate was 6.5 X 1W5 M and for N-acetyl-n-mannosamine 6-phosphate it was 2.5 X 10m4 M (41).
Dephosphorylation of N-Acetylneuraminic Acid g-Phosphate (Reaction S)-Crude extracts of rat liver and bovine submaxillary gland which were capable of forming NAN-O-P also contained dephosphorylating activity. By purifying extracts of bovine submaxillary gland, condensing enzyme was prepared which was free of dephosphorylating activity (Table I, Steps 3 and 4). The dephosphorylating activity was found in the supernatant solution of the 0 to 56% ammonium sulfate precipitate (Table I, Step 3) and could be precipitated with a further addition to the 0 to 56% supernatant solution of ammonium sulfate to 65% saturation. From Table IX it can be seen that the enzyme required glutathione and magnesium ions for activity. In Table X the amount of inorganic phosphate released was equivalent to the amount of NAN formed from NAN-g-P. This was meas- ured by an increase of molar extinction coefficient from 35,400 for NAN-g-P to 57,000 for NAN in the thiobarbituric acid assay.
TABLE VIII
Stoichiometry of reaction catalyzed by condensing enzyme iv-Acetyl-n-mannosamine g-phosphate, 0.4 pmole; phospho-
enolpyruvate, 1.25 pmoles; MgC12, 7 pmoles; KCN, 10 pmoles; glutathione, 25 pmoles; Tris-chloride, pH 7.6, 50 pmoles; and condensing enzyme, ammonium sulfate fraction, 3 mg in a final volume of 1.27 ml were incubated at 37”. Similar vessels without enzyme served as controls. Aliquots (0.04 ml) of the complete incubation mixture were assayed by the thiobarbituric acid method every 30 minutes. After 2 hours, further synthesis did not take place, and the reaction was terminated at 23 hours.
pmJle pmole pmole pmole Initial. 0.40 1.22 0 0 Final. . . 0.00 0.80 0.43 0.45
-0.40 -0.42 +0.43 +0.45
a Measured by the assay of Reissig, Strominger, and Leloir (29). b Measured by the enzymic method of Kornberg and Pricer (50). c Molar extinction coefficient of NAN-g-P is 62’% that of NAN. d Measured by the Lowry-Lopez method (20).
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Equilibrium of this reaction appears to lie far in the direction of dephosphorylation. The reaction is being investigated fur- ther at the present time.
DISCUSSION
The enzyme system that synthesizes NAN-S-P and NAN, by the reactions described here, appear to be of wide occurrence in nature. The system is found in mammals (11, 12, 15), bacteria, and in the ovary of the sea urchin, Arbacia punctuluta.6
The condensing enzyme system differs from NAN-aldolase. Although NAN can be formed in the presence of NAN-aldolase, N-acetyl-n-mannosamine, and pyruvate, the equilibrium favors degradation of NAN (7). In contrast, pyruvate is inactive in the condensing enzyme system, and the reaction with phospho- enolpyruvate strongly favors synthesis of NAN-g-P. It is also pertinent that NAN-aldolase is found in relatively large quan- tity in Clostridium perfringens (7) and Vibrio cholercze (5), which do not appear to contain sialic acids.
Specificity studies of the kinase permit the name N-acetyl-n- mannosamine kinase to be applied to the enzyme. It is note- worthy that the enzyme can be partially purified by precipita- tion with relatively small amounts of cobaltous and zinc ions (Table V) . This newly described enzyme has also been partially purified from rat liver by Ghosh and Roseman (14). N-Acetyl- mannosamine B-phosphate can also be formed from N-acetyl- glucosamine g-phosphate by an epimerase which has been de- scribed in bacteria (51).
Recently, Roseman et al. (15) have described the biosynthesis of NAN-g-P by partially purified extracts of pig submaxillary gland. The conversion of N-acetyl-n-mannosamine 6-phosphate and N-glycolyl-n-mannosamine 6-phosphate to the correspond- ing sialic acid g-phosphate was observed. Whether or not there is a separate enzyme for the biosynthesis of each form is not known.
The biosynthesis of NAN-S-P is analogous to the biosynthesis of 2-keto-3-deoxy-7-phosphoheptonic acid (43,48,49) and 2-keto- 3-deoxy-8-phosphooctonic acid (52) by bacterial systems. The synthesis of NAN-g-P is the first of this class of reactions to be described in mammalian tissues. In these reactions phospho- enolpyruvate condenses with a sugar phosphate, with removal of the phosphate of phosphoenolpyruvate, probably by a con- certed mechanism as suggested by Srinivasan and Sprinson (49).
It appears that in each of the three reactions described in this paper the equilibrium is such that the reaction proceeds to the right, as written, until one of the substrates is used up. The thermodynamics of the condensing reaction has been discussed by Srinivasan and Sprinson (49). In the biosynthesis of 2-keto- 3-deoxy-7-phosphoheptonic acid described by these authors, heptonic acid-7-phosphate synthesis was favored and the reaction could not be reversed, as is the case in the reaction de- scribed here. As in the formation of the heptonic acid 7-phos- phate, synthesis of NAN-g-P is strongly favored, not only be- cause the phosphate of phosphoenolpyruvate is eliminated, but because the condensation product cyclizes to form a pyranose ring. In those systems containing dephosphorylating activity the reaction is “pulled” even further in the direction of synthesis of NAN.
A partially specific phosphatase has been described which cleaves the phosphate of 2-keto-3-deoxy-7-phosphoheptonic acid
May 1962 L. Warren and H. Felsenfeld 1429
PHOSPHOENOLPYRUVATE N-ACETYL-D-MANNOSAMINE-
'4 (SyMOLES/ML.) 8 (S=pidoLES/MC.XlOi
FIG. 4. Dependence of NAN-O-P synthesis on the concentration of N-acetyl-n-mannosamine 6-phosphate and phosphoenolpyru- vate. Each vessel contained, in a volume of 0.2 ml, KCN, 1 pmole; GSH, 2.5 pmoles; MgC12, 1 pmole; condensing enzyme (ammonium sulfate fraction) 0.2 mg; and Tris-Cl buffer, pH 7.6, 30 pmoles. Curves A and C-O.2 pmole of N-acetyl-D-mannos- amine B-phosphate and concentrations of phosphoenolpyruvate as indicated. Curves B and D-0.5 pmole of phosphoenolpyruvate and concentrations of N-acetylmannosamine 6-phosphate as in- dicated. Incubation was for 1 hour at 37”. Thiobarbituric acid assay was performed on the entire contents of the vessels.
TABLE IX
Requirement for magnesium ions and glutathione for dephosphorylation of NAN&P
The complete vessel contained, in a volume of 0.2 ml: NAN-g-P, 0.02 pmole, measured by Cl4 specific activity and by thiobarbituric acid assay; MgC12,l pmole; glutathione, 2.5pmoles; and 56 to 65yo ammonium sulfate fraction of bovine submaxillary gland extract, 0.1 mg. The mixture was incubated 14 hours at 37” and assayed by the thiobarbituric acid assay.
Omission NAN-9-P dephosphorylation
None......................... Enzyme ...................... Mg++ ......................... Glutathione .................. %++a glutathione ............
m/moles
20 0 5 5 0
TABLE X
Stoichiometry of dephosphorylation of NAN-g-P
The experimental details are the same as in Table IX, except that 0.054 pmole of NAN-g-P was used.
P j releaseda I
NAN-9-P dephosphorylatedb /
pm&
0.057 I 0.054
a Phosphate release was measured by the method of Lowry and Lopez (20).
b NAN-B-P dephosphorylation was followed by an increase in molar extinction coefficient from 35,400 to 57,000 in the thio-
6 L. Warren, unpublished observations. barbituric acid assay. See text.
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

1430 Biosynthesis of Sialic Acids Vol. 237, No. 5
UDP-N-Acetylglucosamine
DIAGRAM 1 Metabolism of N-acetylneuraminic acid
U,,/ (44-W I N-Acetylmannosamine (llAzP14)
’
N-acetylmannosamine 6-phosphate
pyruvate
(5,7)
phosphoenolpyruvate (13, 15)
mucopolysac- charides
tose gangliosides,
1 (13) etc.
aialidaso(l)r -NAN ~
(48, 49). Although it is probable that there is an analogous phosphatase for NAN-g-P, an alternative explanation may be that the enzyme is a mutase converting NAN-g-P to a very labile NAN-Z-P. Examination of molecular models of NAN-g-P do not exclude this possibility. The mutase could form an inter- mediate activated at the proper site for insertion of the NAN moiety into glycoproteins, but in the system in vitro would hy- drolyze nonenzymically and would appear to be a dephosphoryl- ation.
The necessity for a loose homogenizer should be emphasized. We have found that activity can be completely eliminated by vigorous homogenization. This inhibition may be related to the observation that the addition of particulate matter of rat liver (nuclear, mitochondrial, or microsomal fractions) to an active liver system causes complete inhibition of synthesis of NAN. The site and mechanism of inhibition are unknown. Perhaps synthesis is prevented by the release of phosphatases which de- stroy N-acetylmannosamine g-phosphate and ATP. Our ex- perience has been similar to that of Bucher (53), who in 1953 observed that the use of tight fitting homogenizers or prolonged grinding prevented the formation of cholesterol from acetate by extracts of rat liver.
At the present time our knowledge of the metabolism of N- acetylneuraminic acid and its precursors may be summarized in Diagram 1. A number of other derivatives of N-acetyl- neuraminic acid such as cytidine monophospho-N-acetylneura- minic acid (54)) N, 0-diacetylneuraminic acid, and triacetyl- neuraminic acid (55) have been described. These are not included in the scheme because the reactions that link these compounds to those shown are unknown at the present time.
SUMMARY
Three reactions involved in the biosynthesis of N-acetyl- neuraminic acid have been studied in rat liver and bovine sub- maxillary gland preparations.
In the first reaction, N-acetyl-n-mannosamine is converted to N-acetyl-n-mannosamine 6-phosphate. Adenosine triphosphate, K+, and Mg++ ions are required. The enzyme that catalyzes the reaction, N-acetyl-n-mannosamine kinase, has been partially purified.
The second reaction, the condensation of phosphoenolpyruvate with N-acetyl-n-mannosamine g-phosphate, results in the forma- tion of N-acetylneuraminic acid g-phosphate and inorganic phosphate. The reaction requires Mg++ ions, KCN, and a
sulfhydryl compound such as glutathione for activity. The condensing enzyme that catalyzes this reaction has been partially purified from bovine submaxillary gland.
The condensing enzyme from bovine submaxillary gland has been freed of a third activity which is responsible for the de- phosphorylation of N-acetylneuraminic acid g-phosphate to form N-acetylneuraminic acid and inorganic phosphate. This enzyme requires Mgft ions and glutathione for activity.
N-Acetyl-n-mannosamine B-phosphate and N-acetylneura- minic acid g-phosphate have been isolated and characterized.
The equilibrium of all three reactions strongly favors the formation of N-acetylneuraminic acid.
Acknowledgment-The authors would like to acknowledge the invaluable technical assistance of Miss C. W. Spearing.
REFERENCES
1. GOTTSCHALK, A., The chemistry and biology of sialic acids and related substances, Cambridge University Press, New York, 1960, p. 88.
2. GOTT&;ALK, A., Yale J. Biol. and Med., 28,525 (1956). 3. KUHN. R.. AND BROSSMER. R.. Chem. Ber.. 89. 2471 (1956). 4. ZILL&N,‘F., AND GLICK, ‘ikl. k., Naturwi~sen~chafte~, 43; 536
(1956). 5. HEIMER, R., AND MEYER, K., Proc. Natl. Acad. Sci., U. S.,
42, 728 (1956). 6. COMB, D. G., AND ROSEMAN, S., J. Am. Chem. Sot., 80, 497
(1958); 80, 3166 (1958). 7. COMB, D. G., AND ROSEMAN, S., J. Biol. Chem., 236, 2529
(1960). 8. CORNFORTH, J.W., FIRTH, M.E., AND GOTTSCHALK, A., Bio-
<hem. J., 68.57 (1958). 9. BRUQ, J., AND PAERLS, G. P., Nature, 182, 1159 (1958).
10. CARROLL, P. M., AND CORNFORTH, J. W., Biochim. et Biophys. Acta, 39, 161 (1960).
11. WARREN, L., AND FELSENFELD, H., Biochem. and Biophys. Research Communs., 4, 232 (1961).
12. WARREN, L., AND FELSENFELD, H., Federation Proc., 20, 80 1961.
13. WARREN, L., AND FELSENFELD, H., Biochem. and Biophys. Research Communs., 6, 185 (1961).
14. GHOSH, S., AND ROSEMAN, S., Proc. Natl. Acad. Sci., U. S., 47,955 (1961).
15. ROSEMAN, S., JOURDIAN, G. W., WATSON, D., AND ROOD, R., Proc. Natl. Acad. Sci., U. S., 47,958 (1961).
16. BALLOU, C. E., Arch. Biochem. Biophys., 78, 328 (1958). 17. KUHN, R., AND KIRSCHENLOHR, W., Liebigs Ann. Chem., 800,
115 (1956). 18. ROSEMAN, S., AND LUDOWEIG, J., J. Am. Chem. Sot., 76, 301
(1954).
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

May 1962 L. Warren and H. Felsenfeld 1431
19. SVENNERHOLM, L., Acta Sot. Med. Upsaliensis, 61, 75 (1956). 37. JEANLOZ, R. W., AND TREMEGE, M., Federation Proc., 16, 282 20. LELOIR, L. F., AND CARDINI, C. E., in S. P. COLOWICK AND (1956).
N. 0: KAPLAN (Editors), Methods in enzymology, vol. III, 38. R~REM, E. S., Nature, 183, 1739 (1959). Academic Press. Inc.. New York. 1957. v. 845. 39. PARTRIDGE. S. M.. Biochem. J.. 42, 283 (1948).
21. FISKE, C. H., AND S&AROW, Y’., J. ‘&ol. Chem., 66, 375 40. CARDINI, d. E., AND LELOIR, L. k., J.‘BioZ: Chem., 226, 317 (1925). (1957).
22.
23.
LOWRY, 0. H., ROGEBROUGH, N. J., FARR, A. L., AND RANDALL, R. J., J. Biol. Chem., 193, 265 (1951).
PARK, J. T., AND JOHNSON, M. J., J. Biol. Chem., 161, 149 (1949).
24. 25.
WARREN, L., J. Biol. Chem., 234,197l (1959). WERNER, I., AND ODIN, L., Acta Sot. Med. Upsaliensis, 67,
230 (1952).
41. LINEWEAVER, H., AND BURK, D., J. Am. Chem. Sot., 66, 658 (1934).
42. AMINOFF, D., Virology, 7, 355 (1959); Biochem. J., 81, 384 (1961).
43. WEISSBACH, A., AND HURWITZ, J., J. BioZ. Chem., 234, 706 (1959).
26. 27.
SVENNERHOLM, L., Biochim. et Biophys. Acta, 24,604 (1957). AYALA, W., MOORE, L. V., AND HESS, E. L., J. CZin. Invest.,
30, 781 (1951).
44. CARDINI, C. E., AND LELOIR, F., J. BioZ. Chem., 226,317 (1957). 45. COMB, D. G., AND ROSEMAN, S., Biochim. et Biophys. Acta,
29, 653 (1968). 46. GLASER. L.. Biochim. et BioDhus. Acta. 41, 534 (1960).
28.
29.
LELOIR, L. F., CARDINI, C. E., AND OLAVARRIA, J. M., Arch. Biochem. Biophys., 74, 84 (1958).
REISSIG, J. L., STROMINGER, J. L., AND LELOIR, L. F., J. BioZ. Chem., 217, 959 (1955).
ROSENTHAL, S. M., J. BioZ. *C&m., 179, 1235 (19‘49). ’ HURWITZ, J., AND WEISSBACH, A., J. Biol. Chem., 234, 710
(1959).
30.
31.
MARTENSSON, E., RAAL, A., AND SVENNERHOLM, L., Biochim. et Biophys. Acta, 30, 124 (1958).
BLIX, G., LINDBERG, E., ODIN, L., AND WERNER, I., Acta Sot. Med. Upsaliensis, 61, 1 (1956).
WARREN, L., Nature, 186, 237 (1960). MACFADYEN, D. A., J. BioZ. Chem., 168, 107 (1945). HANES, C. W., AND ISHERWOOD, F. A., Nature, 164,1107 (1949). LORING, H. S., LEVY, L. W., Moss, L. K., AND PLOESER, J.
M., J. Am. Chem. Sot., 78, 3724 (1966).
47. 48.
49.
50.
SRINIVASAN, P. R., AND SPRINSON, D. B., J. BioZ. Chem., 234, 716 (1959).
KORNBERG, A., AND PRICER, W. E., JR., J. BioZ. Chem., 193, 481 (1951).
51.
32. 33. 34. 35.
ROSEMAN, S., HAYES, F., AND GHOSH, S., Federation PTOC., 19, 85 (1960).
52. LEVIN, D. H., AND RACKER, E., Arch. Biochem. Biophys., 79, 396 (1959).
63. 54.
BUCHER, N. L. R., J. Am. Chem. Sot., 76, 498 (1953). COMB, D. G., SHIMIZU, F., AND ROSEMAN, S., J. Am. Chem.
Sot., 81, 6513 (1959). 36. MALEY, F., AND LARDY, H. A., J. Am. Chem. Sot., 78, 1393 55. BLIX, G., AND LINDBERO, E., Acta Chem. &and., 14, 1809
(1956). (1960).
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from

Leonard Warren and Herbert FelsenfeldThe Biosynthesis of Sialic Acids
1962, 237:1421-1431.J. Biol. Chem.
http://www.jbc.org/content/237/5/1421.citation
Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/237/5/1421.citation.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on April 10, 2020
http://ww
w.jbc.org/
Dow
nloaded from


















