
Targeting Tumor-Associated Fibroblasts for Therapeutic ... · Therapeutics, Targets, and Chemical...
14
Therapeutics, Targets, and Chemical Biology Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors Lei Miao 1 , Qi Liu 1,2 , C. Michael Lin 1 , Cong Luo 1,3 , Yuhua Wang 1 , Lina Liu 1 , Weiyan Yin 2 , Shihao Hu 4 , William Y. Kim 6 , and Leaf Huang 1,5 Abstract The off-target distribution of anticancer nanoparticles to fibro- blasts creates a barrier to the effective treatment of desmoplastic tumors. However, we hypothesized that this nanoparticle detri- ment might be exploited to target the expression of secreted cytotoxic proteins from tumor-associated fibroblasts (TAF) as an anticancer strategy. In addressing this hypothesis, plasmids encoding the secretable TNF-related factor sTRAIL were loaded into lipid-coated protamine DNA complexes and administered by infusion in a murine xenograft model of human desmoplastic bladder carcinoma. Three doses were sufficient to generate approximately 70% of TAFs as sTRAIL-producing cells. sTRAIL triggered apoptosis in tumor cell nests adjacent to TAFs. Fur- thermore, it reverted residual fibroblasts to a quiescent state due to insufficient activation, further compromising tumor growth and remodeling the microenvironment to favor sec- ond-wave nanotherapy. We confirmed the efficacy of this strategy in an orthotopic xenograft model of human pancreatic cancer, where the desmoplastic stroma is well known to be a major barrier to the delivery of therapeutic nanoparticles. Collectively, our results offer a proof of concept for the use of nanoparticles to modify TAFs as an effective strategy to treat desmoplastic cancers. Cancer Res; 77(3); 719–31. Ó2016 AACR. Introduction The enhanced permeation and retention (EPR) effect describes the increased intratumoral accumulation and cellular uptake of therapeutic nanoparticles in oncology, demonstrating promising preclinical responses (1). Unfortunately, the early promise of several therapeutic nanoparticles has failed to translate clinically (2). One of the major mechanisms is the heterogeneous uptake of drugs or nanoparticles in neighboring stromal cells (3). Tumor- associated macrophages (TAM) are a major off-target depletion site for nanoparticles (2, 4). In addition, in tumors with desmo- plastic stroma, tumor-associated fibroblasts (TAF) that wrap around blood vessels constitute another barrier for tumor-specific nanoparticle delivery (1, 5). Off-target distribution of therapeutic nanoparticles can result in adverse effects (6). Depletion of stromal cells has been proposed to circumvent stroma-induced adverse effects and improve tumor cells' capture of therapeutic agents (7, 8). However, this strategy has many limitations. First, it runs the risk of eliminating stromal components needed for tissue homeostasis, paradoxically facilitat- ing metastasis (9). We also found that TAFs damaged by a nano- formulation of cisplatin could produce survival factors, such as Wnt16, to support the proliferation of tumor cells (5). In addition, significant heterogeneity appears to be in the type of stroma within tumors, with some stroma being tumor-suppressive and some protumorigenic (10). Stromal components can produce small proteins (e.g., cytokines) secreted in situ, which can bypass stromal cells barriers and bind avidly to targeted cells causing overexpression of their receptors (11). As cytokines can modulate tumor growth, we sought to engineer an in situ stromal depot capable of secreting cytotoxic cytokine-like proteins, as an alternative stroma modulat- ing strategy, to constrain the growth of desmoplastic tumors. The best way to generate this theoretical stromal depot is gene therapy, as it allows the proteins to be produced locally at higher rates and more quantities than through systemic delivery of recombinant proteins (12). While the off-target delivery of therapeutic nanoparticles traditionally compromises the efficacy of tumor-specific treatments, this phenomenon can be exploited to specifically deliver genes to stroma cells, thus providing the basis for in situ synthesis and secretion. As macrophages and fibroblasts are the major off-target sites in desmoplastic tumors, they are excellent candidates for in situ reprogramming. However, expression of plasmids in macrophages is limited by the macro- phages' natural enzymes for plasmid degradation (13). In addi- tion, the regeneration of macrophage or other circulating mono- cytes limits the persistency of gene expression (1). Therefore, TAFs, as a locally recruited cell population, may serve as a more suitable protein-producing reservoir. TNF-related apoptosis-inducing ligand (TRAIL) efficiently induces apoptosis in a wide range of tumor cells while sparing normal cells, making it an ideal candidate for cancer therapy (14). Full-length TRAIL is a transmembrane protein lacking a leader 1 Division of Molecular Pharmaceutics and Center for Nanotechnology in Drug Delivery, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina. 2 UNC & NCSU Joint Department of Biomedical Engineering, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina. 3 Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, China. 4 Department of Pharmaceutics, China Pharmaceutical University, Nanjing, China. 5 UNC Lineberger Comprehen- sive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina. 6 Department of Medicine, Division of Hematology and Oncology, UNC Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina. Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/). L. Miao and Q. Liu contributed equally to this article. Corresponding Author: Leaf Huang, E-mail: [email protected] doi: 10.1158/0008-5472.CAN-16-0866 Ó2016 American Association for Cancer Research. Cancer Research www.aacrjournals.org 719 on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866
Transcript of Targeting Tumor-Associated Fibroblasts for Therapeutic ... · Therapeutics, Targets, and Chemical...

Therapeutics, Targets, and Chemical Biology
Targeting Tumor-Associated Fibroblasts forTherapeutic Delivery in Desmoplastic TumorsLei Miao1, Qi Liu1,2, C. Michael Lin1, Cong Luo1,3, Yuhua Wang1, Lina Liu1,Weiyan Yin2,Shihao Hu4,William Y. Kim6, and Leaf Huang1,5
Abstract
The off-target distribution of anticancer nanoparticles to fibro-blasts creates a barrier to the effective treatment of desmoplastictumors. However, we hypothesized that this nanoparticle detri-ment might be exploited to target the expression of secretedcytotoxic proteins from tumor-associated fibroblasts (TAF) as ananticancer strategy. In addressing this hypothesis, plasmidsencoding the secretable TNF-related factor sTRAIL were loadedinto lipid-coated protamineDNA complexes and administered byinfusion in a murine xenograft model of human desmoplasticbladder carcinoma. Three doses were sufficient to generateapproximately 70% of TAFs as sTRAIL-producing cells. sTRAIL
triggered apoptosis in tumor cell nests adjacent to TAFs. Fur-thermore, it reverted residual fibroblasts to a quiescent statedue to insufficient activation, further compromising tumorgrowth and remodeling the microenvironment to favor sec-ond-wave nanotherapy. We confirmed the efficacy of thisstrategy in an orthotopic xenograft model of human pancreaticcancer, where the desmoplastic stroma is well known to be amajor barrier to the delivery of therapeutic nanoparticles.Collectively, our results offer a proof of concept for the use ofnanoparticles to modify TAFs as an effective strategy to treatdesmoplastic cancers. Cancer Res; 77(3); 719–31. �2016 AACR.
IntroductionThe enhanced permeation and retention (EPR) effect describes
the increased intratumoral accumulation and cellular uptake oftherapeutic nanoparticles in oncology, demonstrating promisingpreclinical responses (1). Unfortunately, the early promise ofseveral therapeutic nanoparticles has failed to translate clinically(2). One of themajormechanisms is the heterogeneous uptake ofdrugs or nanoparticles in neighboring stromal cells (3). Tumor-associated macrophages (TAM) are a major off-target depletionsite for nanoparticles (2, 4). In addition, in tumors with desmo-plastic stroma, tumor-associated fibroblasts (TAF) that wraparound blood vessels constitute another barrier for tumor-specificnanoparticle delivery (1, 5).
Off-target distribution of therapeutic nanoparticles can result inadverse effects (6). Depletion of stromal cells has been proposed tocircumvent stroma-induced adverse effects and improve tumor
cells' capture of therapeutic agents (7, 8). However, this strategyhas many limitations. First, it runs the risk of eliminating stromalcomponents needed for tissue homeostasis, paradoxically facilitat-ing metastasis (9). We also found that TAFs damaged by a nano-formulation of cisplatin could produce survival factors, such asWnt16, to support the proliferation of tumor cells (5). In addition,significant heterogeneity appears to be in the type of stromawithintumors, with some stroma being tumor-suppressive and someprotumorigenic (10). Stromal components can produce smallproteins (e.g., cytokines) secreted in situ, which can bypass stromalcellsbarriers andbindavidly to targeted cells causingoverexpressionof their receptors (11). As cytokines canmodulate tumorgrowth,wesought to engineer an in situ stromal depot capable of secretingcytotoxic cytokine-like proteins, as an alternative stroma modulat-ing strategy, to constrain the growth of desmoplastic tumors.
The best way to generate this theoretical stromal depot is genetherapy, as it allows the proteins to be produced locally at higherrates and more quantities than through systemic deliveryof recombinant proteins (12). While the off-target delivery oftherapeutic nanoparticles traditionally compromises the efficacyof tumor-specific treatments, this phenomenon can be exploitedto specifically deliver genes to stroma cells, thus providing thebasis for in situ synthesis and secretion. As macrophages andfibroblasts are the major off-target sites in desmoplastic tumors,they are excellent candidates for in situ reprogramming. However,expression of plasmids in macrophages is limited by the macro-phages' natural enzymes for plasmid degradation (13). In addi-tion, the regeneration of macrophage or other circulating mono-cytes limits thepersistency of gene expression (1). Therefore, TAFs,as a locally recruited cell population, may serve as amore suitableprotein-producing reservoir.
TNF-related apoptosis-inducing ligand (TRAIL) efficientlyinduces apoptosis in a wide range of tumor cells while sparingnormal cells, making it an ideal candidate for cancer therapy (14).Full-length TRAIL is a transmembrane protein lacking a leader
1Division of Molecular Pharmaceutics and Center for Nanotechnology in DrugDelivery, UNC Eshelman School of Pharmacy, University of North Carolina atChapel Hill, Chapel Hill, North Carolina. 2UNC & NCSU Joint Department ofBiomedical Engineering, University of North Carolina at Chapel Hill, Chapel Hill,North Carolina. 3Department of Pharmaceutics, School of Pharmacy, ShenyangPharmaceutical University, Shenyang, China. 4Department of Pharmaceutics,China Pharmaceutical University, Nanjing, China. 5UNC Lineberger Comprehen-sive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NorthCarolina. 6Department of Medicine, Division of Hematology and Oncology, UNCLineberger Comprehensive Cancer Center, University of North Carolina atChapel Hill, Chapel Hill, North Carolina.
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
L. Miao and Q. Liu contributed equally to this article.
Corresponding Author: Leaf Huang, E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-16-0866
�2016 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 719
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

sequence for extracellular secretion (12). Its apoptotic effects arelimited to cells near the plasmid-transfected cells, compromisingthe therapeutic efficacy (15). Thereby, a secretable form of TRAIL(sTRAIL) was engineered. sTRAIL consists of an extracellulardomain of TRAIL fused with an NH2-terminal extracellulardomain of Flt3L, a ligand for flt3 tyrosine kinase receptor thataids in secretion (16). It was then necessary to utilize the off-targetdistribution of nanoparticles to target sTRAIL-containing nano-particles toward fibroblasts, seeking to make them tumor-inhib-itive in desmoplastic tumors. The utilization of TRAIL-resistantlow-proliferating fibroblasts as a gene-producing reservoir hastwo important advantages: (i) allowing a comparatively long geneexpression compared with sensitive tumor cells and (ii) main-taining the stroma cell components for tissue homeostasis.
To confirm this proof of concept, a stroma–vessel desmoplasiamodel was established by coinoculating UMUC3 bladder cancercells with NIH3T3 fibroblasts. Lipid-coated protamine DNA com-plexes (LPD) were developed and utilized for encapsulatingsTRAIL plasmids. The fibroblasts' expression of sTRAIL and theapoptosis of neighboring tumor cells were assessed. The conceptwas further evaluated with a clinically relevant, TRAIL-sensitive,orthotopic desmoplastic PDACmodel of BXPC3. As expected, thein situ expression of sTRAIL by fibroblasts induced potent tumorinhibition. However, residual TAFs unexpectedly reverted to qui-escence, presumably due to the death of neighboring tumor cells.This led to the remodelingof tumormicroenvironment (TME) andprovides a new paradigm for a second-wave nanoparticle therapy.
Materials and MethodsCell lines, animals, and antibodies
The mouse embryonic fibroblast cell lines NIH3T3 and thehuman lung fibroblasts MRC-5 were purchased from University ofNorth Carolina (UNC) Tissue Culture Facility. The human bladdertransitional cell line UMUC3 was provided by Dr. William Kim(University of NorthCarolina at Chapel Hill, Chapel Hill, NC). Thehuman pancreatic cancer BXPC3-Luc2 was purchased from Perki-nElmer. UMUC3 andNIH3T3weremaintained inDMEM (Invitro-gen), supplemented with 10% FBS (Sigma) or 10% bovine calfserum (Sigma). BXPC3-Luc2 cells were cultured in full RPMI1640medium (Invitrogen), while MRC-5 cells were cultured in fullaMEM (Invitrogen). Cell lines were authenticated by Dr. WilliamKim'sgroupandUNCTissueCultureFacilityusing the short tandemrepeat (STR) profiling method. Female nudemice 6- to 8-week-oldwere obtained from and raised by the University of North Carolinaanimal facility. All animal handling procedures were approved bythe University of North Carolina at Chapel Hill's InstitutionalAnimal Care and Use Committee. Primary and secondary antibo-dies used for Western blot analysis, flow cytometry, immunofluo-rescence staining, and IHC staining are listed in SupplementaryTable S1.
Preparation and characterization of LPDLPDs were prepared through a stepwise self-assembly process
basedonprevious protocols (17). Briefly, 1,2-dioleoyl-3-trimethyl-ammonium-propane (DOTAP) and cholesterol (1:1, mol/mol)were dissolved in chloroform, and the solvent was removed.The lipid film was then hydrated with distilled water to make thefinal concentration of 10 mmol/L cholesterol and DOTAP. Then,the liposome was extruded through 200-nm and 100-nm polycar-bonate membranes (Millipore) to form 70–100 nm unilamellar
liposomes. The LPD polyplex cores were formulated by mixing140 mL of 36 mg protamine in 5% glucose with equal volume of50 mg plasmid in 5% glucose. The mixture was incubated at roomtemperature for 10 minutes and then 60-mL cholesterol/DOTAPliposomes (10 mmol/L each) were added. Post-insertion of 15%DSPE-PEG and DSPE-PEG-AA was performed at 60�C for 15minutes. The size and surface charge of the nanoparticles weredetermined by a Malvern ZetaSizer Nano series. Transmissionelectron microscopy (TEM) images were acquired using a JEOL100 CX II TEM (JEOL).
Construction of sTRAIL and TRAIL plasmidThe p-sTRAIL–containing genes encoding a Flt3L leader
sequence, isoleucine zipper, the extracellular domain of TRAIL,followedby an internal ribosome entry site (IRES), andGFPunderaCMVpromoter were previously constructed and provided byDr.Shawn Hingtgen (University of North Carolina at Chapel Hill,Chapel Hill, NC). To establish the p-TRAIL construct, the sTRAILsequence was cleaved from the p-sTRAIL vector via digesting withXhoI/BamHI (New England Biolabs). The full-length TRAIL cDNAwas amplifiedbyPCRusing a sense primer containing theXhoI site(50-CAGCCTCGAGCGACCATGGCTATGATGGAGGTC-30) andan antisense primer containing the BamHI site (50-CAGCG-GATCCTTAGCCAACTAAAAAGGCCCCG-30). The amplifiedDNA was digested with XhoI/BamHI, and inserted into theXhoI/BamHI site of the preremoved p-sTRAIL construct. Theinsertion was confirmed by double digestion and PCR. Thesequence was verified using Applied Biosystems 3730xl GeneticAnalyzers.
Tumor growth inhibitionThe UMUC3/NIH3T3 model was established as previously
reported with little modification (18, 19). In brief, UMUC3 cells(5 � 106) and NIH 3T3 cells (2.5 � 106) were subcutaneouslycoinoculated into the right flank of mice with Matrigel (BDBiosciences). Treatments were initiated on the 11th day whentumor sizes reached approximately 500 mm3. Mice were thenrandomized into 4 groups (n�7 per group) as follows: Untreatedgroup (PBS), GFP LPD, TRAIL LPD, and sTRAIL LPD. Intravenousinjections were performed every other day for a total of 4 doses of50 mg plasmid/mouse. Tumor volume (1/2 � length � length �width) was measured every day with a digital caliper (ThermoFisher Scientific) and body weight was also recorded. The desmo-plastic BXPC3-Luc2 model was established by orthotopic injec-tionof 1�106 cells into the tail of the pancreas. Injections of LPDswere started 15days after inoculation anddosed every 2days, for atotal of 4 times. Tumor growth wasmonitored using IVIS KineticsOptical System (Perkin Elmer) twice a week. The increases intumor volumes were calculated as the radiance of the intensities.
Flow cytometry analysisTo study the cell population that took up nanoparticles within
tumors, mice were injected with DiI-labeled LPD (0.1 mg/kg DiI)and were sacrificed at determined time postintravenous injection.Fresh tumor tissues were dissociated with 1 mg/mL collagenase(Invitrogen), 1 mg/mL hyaluronidase (Sigma), and 200 mg/mLDNAase (Invitrogen) in DMEM/2% FBS for 40 minutes to gen-erate a single-cell suspension. The fibroblasts were pretransfectedwith GFP. Leukocytes were stained with APC-conjugated CD45antibody. The cells were then subjected to flow cytometry analysis
Miao et al.
Cancer Res; 77(3) February 1, 2017 Cancer Research720
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

after washing. The ratios of DiI-labeled nanoparticles distributedin different cell populations were then calculated.
To analyze the expression of IRES GFP in fibroblasts and othercells within the bulk tumor, tumor tissues were collected aftersingle-dose or multiple-dose treatments. Tumor tissues weredissociated, and the ratio of GFP-expressed fibroblasts (RFP-fibroblasts in UMUC3/3T3 model or aSMA-positive fibroblastsin BXPC3 model) in the dissociated cells was analyzed by flowcytometry on a BD FACS Aria instrument (Beckon Dickinson).
To quantify the expression of TRAIL and TAF markers in RFP-fibroblasts of the UMUC3/3T3 model, the dissociated cells weresorted using MoFlo XDP (Beckman Coulter), and the collectedfibroblasts and other cells were processed through RNA extract,DNA reversion, and qPCR analysis.
Statistical analysisStatistical analysis was undertaken using Prism 5.0c Software. A
two-tailed t test or a one-way ANOVA was performed whencomparing two groups or more than two groups, respectively.Statistical significance was defined as P < 0.05. Data are shown asmean � SD.
Additional methodsDetailed methodology including materials, gene transfection,
cell viability assay, quantitative real-time PCR (qPCR) assay,ELISA, Western blot analysis, immunofluorescence, and IHC,etc., are described in the Supplementary Methods.
ResultsIdentification of fibroblasts as themajor off-target cells for LPDuptake within a stroma–vessel desmoplastic tumor model
LPD nanoparticles were prepared according to the establishedprotocols with few adjustments (5). Aminoethylanisamide was
conjugated onto the surface of nanoparticles as a ligand for cellsoverexpressing the sigma receptor (including tumor cells andTAFs). A secondary amine 2 carbons away from the amide carbonwas added to the aminoethylanisamide structure (DSPE-PEG-SP2-AA) to ensure enhanced binding affinity and specificity accordingto the previous report (20, 21). The final nanoparticles wereapproximately 70 nm in diameter, with a surface charge of approx-imately 25mV(Supplementary Table S2). TEM images confirm thesize of LPD and indicate its spherical shape and homogenousdistribution (Supplementary Fig. S1).DiI (0.5%)was incorporatedinto the lipid membrane as an in vivo tracker of LPD.
A stroma–vessel type (a common stroma architecture in desmo-plasia) desmoplastic tumor model was generated from simulta-neous subcutaneous inoculation of UMUC3 bladder cancer cellsalong with NIH3T3 fibroblasts (UMUC3/3T3; Fig. 1A). Histopa-thology demonstrates an anatomic vicinity between blood vesselsand TAFs within the UMUC3/3T3 xenografts (Fig. 1A and Sup-plementary Fig. S2).DiI-labeledLPDreachedUMUC3/3T3 tumorswithin 10 hours of intravenous injection, and plateaued over 48hours (Supplementary Fig. S1C). Consistent with other nanopar-ticles of similar size, liver and spleen were the major LPD-accu-mulatingorgans. Flow cytometrywas performed todetermine LPDaccumulation in various cell populations within the tumor. Stableexpression of GFP and fluorophore-conjugated antibody againstmouse CD45 defined fibroblasts and leukocytes populations,respectively. Results showed that approximately 27% of the cellswithin the bulk tumor were fibroblasts (GFP positive) whileapproximately 16% were CD45þ leukocytes. The majority ofremaining cells, as shown in a previous study, were tumor cells(Fig. 1B; ref. 5). More than approximately 60%of fibroblasts (GFPpositive) took up LPD at 10 hours postintravenous injection,accounting for approximately 65% of the total nanoparticle-asso-ciated cells (Fig. 1CandD,Supplementary Fig. S3).Despite gradual
Figure 1.
Cell populations that take up LPD in the stroma–vessel type desmoplastic tumors.A, The schematic architecture of stroma–vessel tumors. Immunofluorescence (IF)and hematoxylin and eosin (H&E; adjacent section of immunofluorescence) images (right) show the histology of a stroma–vessel tumor model: UMUC3/3T3.Examination revealed nests of tumor cells (yellow dotted circles in the inset magnified images, labeled as T), surrounded by fibrotic components (filled by aSMA-positive myofibroblasts, red) between them. CD31þ blood vessels (cyan) were embedded in the interstitium near myofibroblasts; almost no vessels were observedinside the nests of tumor cells. B, Flow cytometry gating of the cell populations in the UMUC3/3T3-GFP tumors. C, Flow cytometry histograms of thepercentage of cells that took up DiI-labeled LPD in each cell population at determined time points. D, Quantitation of the percentage of DiI-positive cellsin each population (based on the flow data), n ¼ 4.
In Situ Generation of Tumor-Suppressive Fibroblasts
www.aacrjournals.org Cancer Res; 77(3) February 1, 2017 721
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866
clearance or degradation of the fluorescent nanoparticles, approx-imately 20% of fibroblasts still remained DiI positive 72 hoursafter injection. In contrast, only approximately 20% of the CD45þ
leucocytes initially tookupDiI LPD. Furthermore, less than10%oftumor cells took up nanoparticles during any of the time pointsassayed. These analyses ultimately indicate that fibroblasts are themajor off-target cells responsible for LPD uptake in the stroma–vessel desmoplastic tumors.
In vitro transfection of fibroblasts with sTRAIL in LPD inducesapoptosis of neighboring tumor cells
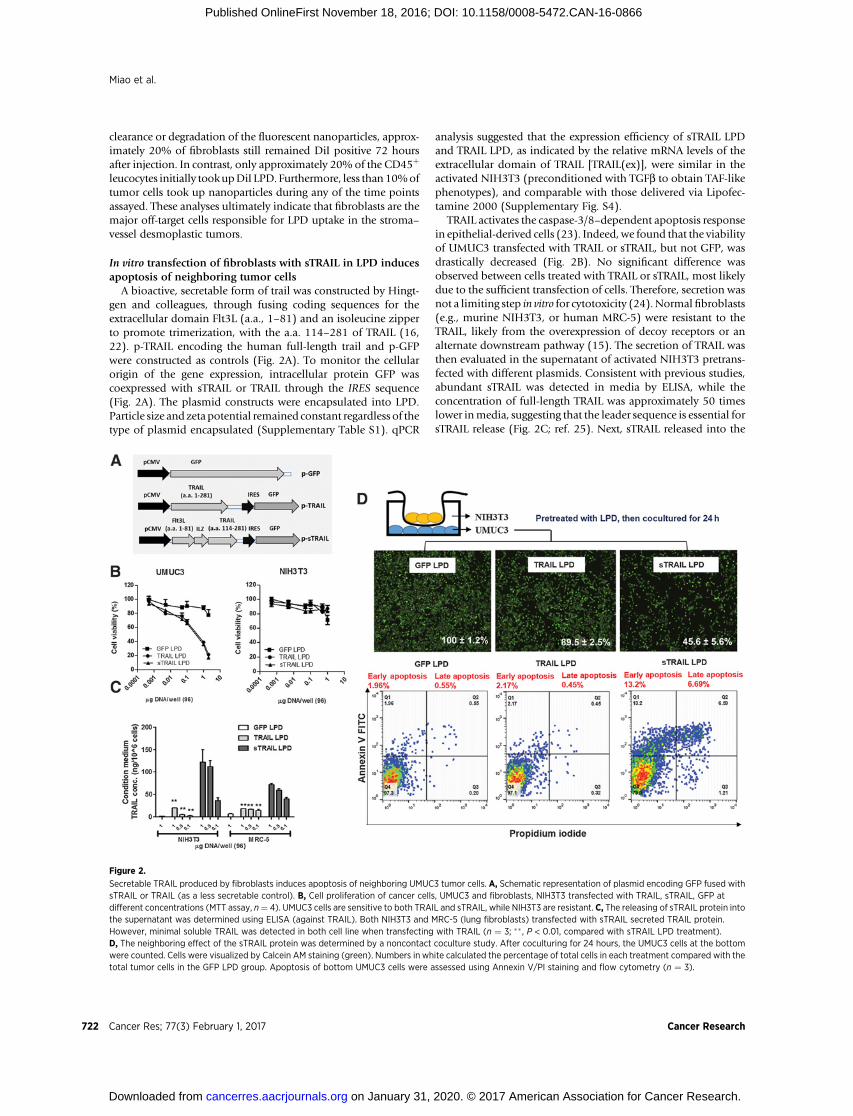
A bioactive, secretable form of trail was constructed by Hingt-gen and colleagues, through fusing coding sequences for theextracellular domain Flt3L (a.a., 1–81) and an isoleucine zipperto promote trimerization, with the a.a. 114–281 of TRAIL (16,22). p-TRAIL encoding the human full-length trail and p-GFPwere constructed as controls (Fig. 2A). To monitor the cellularorigin of the gene expression, intracellular protein GFP wascoexpressed with sTRAIL or TRAIL through the IRES sequence(Fig. 2A). The plasmid constructs were encapsulated into LPD.Particle size and zeta potential remained constant regardless of thetype of plasmid encapsulated (Supplementary Table S1). qPCR
analysis suggested that the expression efficiency of sTRAIL LPDand TRAIL LPD, as indicated by the relative mRNA levels of theextracellular domain of TRAIL [TRAIL(ex)], were similar in theactivated NIH3T3 (preconditioned with TGFb to obtain TAF-likephenotypes), and comparable with those delivered via Lipofec-tamine 2000 (Supplementary Fig. S4).
TRAIL activates the caspase-3/8–dependent apoptosis responsein epithelial-derived cells (23). Indeed, we found that the viabilityof UMUC3 transfected with TRAIL or sTRAIL, but not GFP, wasdrastically decreased (Fig. 2B). No significant difference wasobserved between cells treated with TRAIL or sTRAIL, most likelydue to the sufficient transfection of cells. Therefore, secretion wasnot a limiting step in vitro for cytotoxicity (24). Normal fibroblasts(e.g., murine NIH3T3, or human MRC-5) were resistant to theTRAIL, likely from the overexpression of decoy receptors or analternate downstream pathway (15). The secretion of TRAIL wasthen evaluated in the supernatant of activated NIH3T3 pretrans-fected with different plasmids. Consistent with previous studies,abundant sTRAIL was detected in media by ELISA, while theconcentration of full-length TRAIL was approximately 50 timeslower inmedia, suggesting that the leader sequence is essential forsTRAIL release (Fig. 2C; ref. 25). Next, sTRAIL released into the
Figure 2.
Secretable TRAIL produced by fibroblasts induces apoptosis of neighboring UMUC3 tumor cells. A, Schematic representation of plasmid encoding GFP fused withsTRAIL or TRAIL (as a less secretable control). B, Cell proliferation of cancer cells, UMUC3 and fibroblasts, NIH3T3 transfected with TRAIL, sTRAIL, GFP atdifferent concentrations (MTT assay, n¼ 4). UMUC3 cells are sensitive to both TRAIL and sTRAIL, while NIH3T3 are resistant. C, The releasing of sTRAIL protein intothe supernatant was determined using ELISA (against TRAIL). Both NIH3T3 and MRC-5 (lung fibroblasts) transfected with sTRAIL secreted TRAIL protein.However, minimal soluble TRAIL was detected in both cell line when transfecting with TRAIL (n ¼ 3; �� , P < 0.01, compared with sTRAIL LPD treatment).D, The neighboring effect of the sTRAIL protein was determined by a noncontact coculture study. After coculturing for 24 hours, the UMUC3 cells at the bottomwere counted. Cells were visualized by Calcein AM staining (green). Numbers in white calculated the percentage of total cells in each treatment compared with thetotal tumor cells in the GFP LPD group. Apoptosis of bottom UMUC3 cells were assessed using Annexin V/PI staining and flow cytometry (n ¼ 3).
Miao et al.
Cancer Res; 77(3) February 1, 2017 Cancer Research722
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

culture media was assayed for biological activity (SupplementaryFig. S5). The growthmedia forUMUC3were replacedwith culturesupernatants from NIH3T3 cells transfected with sTRAIL, TRAIL,or GFP. The culture supernatant containing sTRAIL, but notTRAIL, exerted a significant cytotoxic effect on UMUC3. Theneighboring effect was further confirmed by nondirect contactcoculture (Fig. 2D). UMUC3 (bottom layer) was cocultured withactivated fibroblasts (top chamber) preloaded with differentplasmids. The total cell number was significantly lower in thesTRAIL coculture group. Furthermore, approximately 13.2% earlyapoptosis and approximately 6.7% late apoptosis were observedin the sTRAIL coculture group as compared with other treatmentgroups (Fig. 2D). All together, these data verify the prerequisitesfor in situ gene modification of fibroblasts: (i) fibroblasts areresistant to both sTRAIL and TRAIL, while tumor cells are sensitiveto them; (ii) sTRAIL can be efficiently released within the super-natant; and (iii) the neighboring effect thereby occurs instantly.
Secreted TRAIL induces superior antitumor efficacy in thestroma–vessel desmoplastic bladder cancer model
The efficacy of systemically delivered sTRAIL was evaluatedusing theUMUC3/3T3model. Treatment beganwhen tumor sizes
reached 500 mm3, allowing the stroma–vessel structure to form.Equal amounts of plasmids in LPDs were intravenously injectedinto mice. As shown in Fig. 3A, the tumor growth in mice treatedwith sTRAIL LPD was significantly inhibited compared with othergroups. GFP-LPD exhibited a slight antitumor effect, likely fromthe nonspecific induction of inflammatory cytokines from thecargos andDNA backbones (26). To demonstrate the neighboringeffect induced from sTRAIL, the full-length TRAIL LPDwas admin-istered as a less secretable control. As expected, full-length TRAILshowedminimal antitumor efficacy compared with sTRAIL. qPCRanalysis of the mRNA expression using primers (SupplementaryFig. S4; Supplementary Table S3) for TRAIL(ex) suggested com-parable expression of sTRAIL and TRAIL in tumors treated with 4doses of LPDs (Fig. 3B). This, together with the in vitro transfectiondata (Supplementary Fig. S4), which indicated a similar transfec-tion efficiency between these 2 plasmids, dismissed the possibilitythat a difference in expression levels between sTRAIL and TRAILcould affect the antitumor effect. Notably, both TRAIL and sTRAILLPD induced >7 times greater expression of TRAIL(ex) comparedwith the PBS-treated group with a baseline level of endogenousTRAIL. In addition, the expression of mRNA persisted at least 4days after the endpoint dose, suggesting relatively long gene
Figure 3.
Intravenous injection of sTRAIL LPD leads to the expression of sTRAIL in fibroblasts in situ, inhibiting stroma–vessel UMUC3/3T3 tumor growth. A, Tumor inhibitioncurve of mice bearing desmoplastic UMUC/3T3 tumors. Mice were treated with PBS, GFP LPD, TRAIL LPD, or sTRAIL LPD (50 mg plasmid/mice) for 4 times(n ¼ 6–8; � , P < 0.05; ��, P < 0.01; ��� , P < 0.001, in the legend, compared with PBS group; in the data, compared with each time point of the TRAIL LPD group).B, qPCR quantitation of relative mRNA levels of TRAIL or sTRAIL in the treated tumors. The primers simultaneously for the extracellular domain of TRAILand sTRAIL were used for the detection (n ¼ 6; �� , P < 0.01; n.s, no significant difference, compared with PBS group). C–F, Immunofluorescence staining of GFP(green), RFP fibroblasts (red), and cell nuclei (DAPI, blue) at indicated time points after treatments on cryo-tumor (UMUC3/3T3-RFP) tissues collected. Resultsshowed that the majority of expression of GFP fusion protein colocalized with the RFP-labeled fibroblasts.G, Flow cytometry analysis of GFP's association with RFPfibroblasts at indicated time points in the dissociated cells from the collected tumor tissues (n¼ 4). The association of GFP in fibroblasts increased dose dependently.H,One day after three doses of sTRAIL LPD, the RFP-labeled fibroblasts were sorted by flow cytometry. sTRAIL mRNA level in the RFP-labeled fibroblasts and otherunlabeled cells were analyzed and compared with the GFP LPD–treated group (n ¼ 4, �� , P < 0.01). The inset chart indicates gating of the RFP fibroblasts.
In Situ Generation of Tumor-Suppressive Fibroblasts
www.aacrjournals.org Cancer Res; 77(3) February 1, 2017 723
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

expression profiles. qPCR assay using primers specific for sTRAILconfirms the expression of sTRAIL plasmid (Supplementary Fig.S6). With a lower baseline level compared with TRAIL(ex)mRNA,the relative sTRAIL production was approximately 200 timeshigher relative to the control. The expression and secretion ofTRAIL protein were further examined in the UMUC3/3T3 xeno-grafts. Dissociated cells from the treated tumors were cultured andthe conditioned media (secretomes) were measured by ELISA forTRAIL protein (both normal TRAIL and sTRAIL). Indeed, tumorsfrom sTRAIL LPD–treated animals secreted more TRAIL proteinthan any other groups including the ones treated with TRAIL LPD(Supplementary Fig. S7). Results indicate the strong potency ofgene transfection andprotein secretionbyusingLPD, and suggest apromising therapeutic outcome of sTRAIL LPD in treating thedesmoplastic UMUC3/3T3 tumors (Fig. 3A).
The sTRAIL LPDs were delivered to and expressed in fibroblastsin situ
Next, we assessed the locoregional expression of sTRAIL indifferent cell populations within the tumors. As the intracellularprotein GFP was fused with sTRAIL through an IRES sequence, thecells that expressed GFP represented cells that secrete sTRAIL.Moreover, to visualize the fibroblasts, RFP-expressing NIH3T3cells were coinoculated with UMUC3 cells. Notably, the xenograftdeveloped from UMUC3 cell lines alone had minimal to noendogenous fibroblasts (19). Therefore, the RFP fibroblasts con-stituted themajority offibroblast populations in theUMUC3/3T3-RFPmodel. As shown in Fig. 3C–F, mice treated with a single doseof sTRAIL LPD exhibited moderate GFP expression, exclusivelylocalized within the RFP fibroblasts. The strongest GFP expressionwas elicited after 4 doses of nanoparticles. The expression of GFPwas quantitatively confirmed using flow cytometry (Fig. 3G).Consistently, overall expression of GFP increased dose dependent-ly, but the majority of expression was limited to RFP fibroblasts.
These data confirmed that fibroblasts were the major reservoirfor in situ generation of LPD-delivered proteins. This was mostlikely due to off-target distribution of nanoparticles and relativelyhigh and stable expression of genes in fibroblasts compared withother off-target cell populations (e.g.,macrophages). Notably, theexpression of GFP in other cells were observed two days after theendpoint dose (Fig. 3F and G), suggesting that a portion ofnanoparticles had overcome the fibroblast-elicited barriers andentered into the tumor nest. However, the expression of GFP inthis groupof cells decreaseddramatically 4days after the endpointinjection while the expression in fibroblasts remained constant(Fig. 3G). There are two hypothesized mechanisms related tothese observations: (i) the tumor cells may internalize the nano-particles but undergo apoptosis immediately, or (ii) the infiltrat-ing leukocytes take up the nanoparticles, but cells escaped into thecirculate afterwards. Either cell population demonstrated tran-sient expression of the genes compared with local TRAIL-resistantfibroblasts, confirmingfibroblasts as themost suitable engineeredreservoir. The expression levels of sTRAIL mRNA were furtherassayed in the RFPfibroblasts sorted from tumorswith three dosesof sTRAIL or GFP LPD (Fig. 3H). As expected, only the fibroblaststreated with sTRAIL LPD elicited the synthesis of sTRAIL mRNA.
Neighboring effect unveiled the apoptotic effect of TRAIL inthe stroma–vessel desmoplastic bladder cancers
The distribution of apoptotic cells was then examined using aterminal deoxynucleotidyl transferase dUTP nick end labeling
(TUNEL) assay on the UMUC3/3T3 model (using wild-type unla-beledfibroblasts). TAFswere simultaneously visualizedby stainingwith aSMA. Only a trace amount of TUNEL-positive cells wasobserved after single dose of sTRAIL LPD, the majority of whichwas localized near aSMA-positive TAFs (Fig. 4A). The apoptoticarea grows around aSMA-positive TAFs as the doses increase.Sections from different tumors after each dose were then analyzedto quantify the average distances between apoptotic cells and thenearest aSMA-positive TAFs. Consistent with the fluorescenceimages, the distribution radius of apoptotic cells increases withdose and over time (Fig. 4B). The data substantiate the claim that aneighboring effect of fibroblasts is indeed present in fibroblastsin situ, facilitated by diffusion, and amplifiedwith escalated dosingschedules. The apoptotic assay was also performed in other treat-ment groups after the endpoints (Fig. 4C). As expected, minimalapoptosis was observed in the PBS and GFP-LPD group, whereas asmall amount of TUNEL-positive nuclei were observed in groupstreated with TRAIL LPD. We hypothesized that the proteolyticallycleaved extracellular domain of the full-length TRAIL inducedapoptosis of neighboring tumor cells, or a paucity of nanoparticlesdiffused through theTAF layer, inducing the synthesisof TRAIL andapoptosis in neighboring tumor cells, thus explaining the limitedapoptotic cells observed in the TRAIL LPD near TAFs. In compar-ison, an extensive amount of apoptotic cells was observed in thesTRAIL LPD group. In addition, the residual fibroblasts (especiallyTAFs) were clustered, sparing any tumor nest structure. Potent,well-dispersed apoptosis along with this disordered and clusteredfibroblast structure suggested a tumor microenvironment lessstructurally and functionally capable of growth and progression,thus verifying the superior antitumor activity of sTRAIL LPDcompared with other treatments.
Apoptosis of neighboring tumor cells induced by sTRAIL LPDcauses reprogramming of residual fibroblasts, facilitating thedelivery of second-wave therapeutic nanoparticles
We next examined the function of residual fibroblasts. Thelevels of collagen, a major extracellular matrix (ECM) protein,were assessed after multiple sTRAIL treatments in mice bearingUMUC3/NIH3T3 (27). Unexpectedly, the collagen contentdecreased approximately 3-fold compared with other treatmentgroups (Fig. 5A and B). Collagen level within TME under sTRAILLPD treatment was also monitored (Supplementary Fig. S8).Results indicated a gradual reduction of collagen level in responseto sTRAIL treatment. Reductions were also observed on otherproteins unique to fibroblast activation of functional significancein the TME, including fibronectin and hepatocyte growth factor(HGF; Fig. 5C; refs. 5, 9, 28). In addition, fibroblast activationmarkers, aSMA and fibroblast activation protein alpha (FAPa)decreased by approximately 90% and approximately 84% (com-pared with total RFP fibroblasts), respectively (Fig. 5C; refs. 5, 9,29). These data suggested that residual TAFs were shifted from anactivated to a quiescent state. However, the proteins (i.e., FAPaand fibronectin) described above are not exclusively secreted byfibroblasts (25, 30). To further confirm the state shift of fibro-blasts, we sorted the RFP fibroblasts from the dissociated cellscollected from tumors after 3 doses of sTRAIL LPD. Indeed, wefound themRNA level ofCOL1A1 (collagen) andACTA2 (aSMA)in sTRAIL-treated fibroblasts decreased approximately 2- to 5-foldcompared with untreated fibroblasts (Fig. 5D). Meanwhiletumors treated with full-length TRAIL or GFP-LPD failed to affectdesmoplasia, eliminating any possibility of TRAIL directly
Miao et al.
Cancer Res; 77(3) February 1, 2017 Cancer Research724
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

inducing fibroblast reprogramming (Fig. 5A–D). As it is com-monly accepted that the majority of TAFs are transdifferentiatedfrom resident fibroblasts in response to TGFb (31), the down-stream portions of TGFb signaling, including pSMAD2 and plas-minogen activator inhibitor type 1 (PAI-1) were examined (29,31, 32). Indeed, data revealed a decreased level of pSmad2 nucleistaining and an inhibition of the transcriptional activation ofPAI-1 in fibroblasts of sTRAIL-treated tumors (Fig. 5D and E).Again, the data support the reprogramming of TAFs (9).
We next assessed the response of intratumoral blood vessels tothe normal stroma restoration and the neoplastic cell loss. Boththe vascular density and vessel diameter were observed andquantified in mice bearing UMUC3/3T3 xenografts (Fig. 5F;Supplementary Fig. S9A and S9B). There was no significantincrease in vessel density associated with any of the treatments,suggesting no obvious angiogenesis effects. In fact, the vesseldensity decreased in some sTRAIL- or TRAIL-treated mice, likelydue to the inhibition of the proangiogenic factor, VEGF, throughreduced mRNA expression. Yet this effect was not statisticallysignificant (33). Although, sTRAIL, not TRAIL LPD, profoundlyincreased the blood vessel diameter and vessel lumen size (Fig. 5F;Supplementary Fig. S9). This finding indicates that sTRAIL LPD
could effectively decompress the intratumoral vasculatures (34),leading to the normalization of blood vessels. Normalized vas-culatures are often characterized by uncompressed vessels withgreater pericyte coverage (23, 35). Indeed, the loosely attached orabsent pericytes in tumor vessels were replaced with compact,normalized pericytes after sTRAIL LPD treatment (SupplementaryFig. S9G). Furthermore, these morphologic changes were accom-panied by functional changes, that is, the increased tumor oxy-genation, as indicated by the rapid downregulation of the hyp-oxia-inducible factor 1 alpha (HIF 1a; ref. 24) after sTRAIL LPDtreatment (Supplementary Fig. S9D; refs. 35, 36). The normali-zation of blood vessels and restoration of normal stroma wouldultimately lead to reduction of the interstitial fluidic pressure asindicated by studies reported elsewhere (23, 35).
As normalized blood vessels with reduced IFP and hypoxiawere characterized and reported, it was then questioned whetherthe remodeled TME would ultimately increase the accumulation,penetration, and efficacy of second-wave nanocarriers, whichdescribes an additional course of chemotherapy following TMEmodulation (37). Lipid-coated cisplatin nanoparticles (LPCnanoparticles, �30 nm) were previously developed in our labo-ratory andused for the tumor accumulation studyherein.Detailed
Figure 4
Fibroblasts (in situ) that secreted TRAIL induced the apoptosis of neighboring tumor cells. A, Immunofluorescence staining of aSMA and TUNEL from tumortissues (paraffin-embedded sections) after dose escalation of sTRAIL LPD NPs. Dashed lines indicate edge of the tumor nests. B, Quantification of relativeapoptotic cells' distance from the nearest aSMA-positive fibroblasts after different doses of sTRAIL LPD nanoparticles (NP). Numbers on top indicate the furthestdistance of apoptotic cells to aSMA after different treatments. C, Paraffin-embedded tissues sections from UMUC3/3T3 tumors 2 days after final treatmentswere stained for aSMA (red, TAFs) and TUNEL (green, apoptosis). The percentage of aSMA coverage was quantified using ImageJ and is presented as numbers inwhite on the left corner of each panel. Well-structured tumor nests are highlighted in the images. sTRAIL LPD NPs lead to the disruption of tumor nests andremodeling of the tumor microenvironment.
In Situ Generation of Tumor-Suppressive Fibroblasts
www.aacrjournals.org Cancer Res; 77(3) February 1, 2017 725
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

Figure 5.
sTRAIL LPD induces the reprogramming of residual fibroblasts and remodeling of TME, facilitating the delivery and antitumor effect of a second-wavenanoformulated cisplatin. A, Masson trichrome staining of the collagen after endpoint treatments. B, Quantification of the trichrome staining (n ¼ 5; �� , P < 0.01,compared with PBS group). C, Western blot analysis of the TME markers mainly secreted by TAFs and TAF markers. Intensities of each ECM protein werecalculated by comparing with the RFP-transfected fibroblasts and are shown (right). Protein/RFP ratios in the PBS group were set as 1 (n ¼ 3; � , P < 0.05;�� , P < 0.01, compared with the PBS group). D, mRNA levels of TAF markers in the sorted RFP-fibroblasts after sTRAIL treatment (n ¼ 5; � , P < 0.05; ���, P < 0.01,compared with the PBS group). E, IHC staining of aSMA and pSMAD2 in the adjacent sections of PBS and sTRAIL LPD groups. Red dotted circles, tumor nests;yellow dotted circles, fibroblasts. F, IHC staining of blood vessel (CD31, brown) after different treatments. sTRAIL LPD induces decompression of blood vessels.Red arrows, the decompressed vessels. Percentage of blood vessel coverage and percentage of normalized blood vessels were quantified using ImageJand is shown (right; n ¼ 4; ���, P < 0.001). G, Dosing schedule of the second-wave chemotherapy. H, ICP-MS analysis of cisplatin accumulation after a singledose of cisplatin NP (LPC NP) in mice pretreated with sTRAIL LPD or without pretreatment (n ¼ 5; ��� , P < 0.001). I, Tumor inhibition curve of LPC NP (cisplatin,1.9 mg/kg) after treating the tumors with sTRAIL LPD (n ¼ 5; � , P < 0.05). J, Fluorescence images of the intratumoral distribution of DiI-labeled LPC NP afterpretreating tumors with sTRAIL LPD. Blood vessels were stained with CD31 (cyan). Numbers in white indicate the average percent of DiI-positive cells in theselected views. Yellow arrows in magnified images demonstrate the extravasation of DiI-NP from the blood vessels. NP, nanoparticle.
Miao et al.
Cancer Res; 77(3) February 1, 2017 Cancer Research726
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

characterizations of the nanoparticles have been described by Guoand colleagues (38). Indeed, pretreatment of the tumors withsTRAIL LPD led to an approximately 2-fold increase of cisplatinretention 24 hours after a single-dose intravenous injection ofcisplatin nanoparticles (Fig. 5G and H). The intratumoral distri-bution of cisplatin nanoparticles were visualized using DiI-labeledcisplatin nanoparticles. As shown inFig. 5J, normalizationof bloodvessels after sTRAIL treatment efficiently improved the extravasa-tion and penetration of small nanoparticles. To demonstrate thepossible therapeutic benefits of the two-wave therapy, mice werefirst pretreated with 3 doses of sTRAIL LPD, as shown in Fig. 5G.Results herein suggest that the two-wave therapy significantlyinhibited anddelayed the tumor growthmore than singlemodalitytherapy (Fig. 5I). In conclusion, the data suggest residualfibroblastsand TMEafter sTRAIL treatment are remodeled, benefiting not onlythe sTRAIL-mediated antitumor efficacy, but also the delivery of asecond-wave chemotherapy.
In situ engineering of pancreatic stellate cells with sTRAIL LPDnanoparticles shows promising antitumor efficacy in anorthotopic desmoplastic pancreatic cancer BXPC3
To evaluate whether the abovementioned findings could berecapitulated in tumors that are clinically known to have extreme-ly high desmoplastic fibrosis, the antitumor efficacy of sTRAILLPD was assessed on mice bearing the human pancreatic adeno-carcinoma BXPC3. BXPC3 are stroma–vessel type desmoplastictumors characterized with nests of tumor cells surrounded byvessel-embedded fibrotic tissues (Supplementary Fig. S10;refs. 39–41). The cultured BXPC3 had greater sensitivity to TRAILcomparedwith other pancreatic cancer cell lines (42). To visualizetumor growth in vivo, BXPC-3 cells were stably transfected withluciferase vector (BXPC3-Luc2). BXPC3-Luc2 was orthotopicallyinjected into the tail of the pancreas. The dosing schedule ofsTRAIL LPD is presented in Fig. 6A. Tumor volume correlated fromthe number of photons emitted from the tumor were assessed(Fig. 6B) and quantified (Fig. 6C). Results demonstrated thatsTRAIL LPD, but not other treatment groups, effectively inhibitedtumor growth. Furthermore, of all mice treated with differentregimens, only sTRAIL treatment significantly improved the over-all survival. Mean survival time (MST) was increased to 65 days ascompared with other treatment groups (43, 50, and 53 days forPBS, GFP LPD, and TRAIL LPD groups, respectively, Fig. 6D),conveying a potent therapeutic effect and a prolonged survivaleffect. Major organs were imaged for metastasis and results werepresented in Supplementary Fig. S11. Results demonstrated thatsTRAIL LPD treatment also efficiently ameliorated tumor metas-tasis (primarily to liver and lung). To verify that the hypothesisregarding in situ engineering of fibroblasts occurred in the BXPC3model, the expression of GFP (the IRES-GFP from sTRAIL andTRAIL LPD or GFP from GFP LPD) in fibroblasts and other cellswas examined. Indeed, more than approximately 25% of fibro-blasts expressed GFP, accounting for approximately 40% of thetotal GFP-expressing cells (Fig. 6D). In addition, only approxi-mately 8% of the CD45þ leucocytes expressed GFP (Supplemen-tary Fig. S12). This again supports the claim thatfibroblasts are theprimary engineered population for sTRAIL secretion. To verifythat fibroblasts induced neighboring effect and the remodeling ofTME, the post-treatment apoptosis and collagen level are shownin Fig. 6E–G. Consistently, an increased amount of TUNEL-positive cells was localized near fibroblasts. The overall level ofcollagen in the non-scar tissue area decreased. Heterogeneities,
particularly the extensive extracellular matrix with few viable cells(the scar tissue; ref. 43) was primarily observed in the sTRAIL LPDtumors (Supplementary Fig. S13) due to the efficient eliminationof tumor cells by sTRAIL. Normalization of blood vasculaturesand reduction of tumor hypoxia were also found in the sTRAILLPD tumors (Supplementary Fig. S14). Once again, the resultsclearly demonstrate that in situ engineering of fibroblasts benefitsanticancer therapy in stroma–vessel desmoplastic tumors.
Toxicity evaluation for the different treatments and bloodchemistry analysis
Toxicologic evaluation demonstrated little to no noticeablemorphologic changes in organs where LPD nanoparticles aredistributed (e.g., liver and spleen; Supplementary Fig. S14A andS14B). The serum biochemical value analysis demonstrated thatthe sTRAIL treatment group had no liver (aspartate aminotrans-ferase and alanine aminotransferase) or kidney (creatinine andblood urea nitrogen) toxicity caused by tumor progression (Sup-plementary Table S4). One possible mechanism is that althoughliver is the major site for nanoparticle accumulation, approxi-mately 65% of the nanoparticles were actually trapped in CD68-positive Kupffer cells with low plasmid expression (Supplemen-tary Fig. S14D–S14F; ref. 44). Tumors were the main plasmidtransfection and protein synthesis reservoir (Supplementary Fig.S14D and S14E). In addition, hematology study showed nosignificant bone marrow suppression in sTRAIL-treated micecompared with the control groups, suggesting the treatment didnot cause anemia (Supplementary Fig. S14B).
DiscussionDespite recent advances in nanotherapeutics, efficacy against
desmoplastic tumors, including pancreatic cancer and advancedurothelial carcinoma, has not changed in decades (9). In part, thedense stromal barrier captures nanoparticles, preventing themfrom reaching the tumor (1, 19). Given the large amount ofnanoparticles delivered to fibroblasts, we hypothesized that wecould take advantage of this natural property of bladder andpancreatic cancers and target cancer treatment throughfibroblasts.Inspired by fibroblast's ability to secrete tumor supportive cyto-kines to neighboring tumor cells (45, 46), modification of fibro-blasts to secrete tumor-suppressive cytokines through gene deliv-ery with nanoparticles was proposed in the current article. The insitu engineering of fibroblasts harnesses the location of fibroblastsbetween blood vessels and tumor cells, bypassing major cellularbarriers for nanoparticle delivery; subsequently converting fibro-blasts from a tumor-supporting role to a tumor depletion center.
The choice of the secretable tumor-suppressive factor shouldnot be understated. TRAIL is a highly selective, tumor apoptosis–inducing cytokine. The resistance of mesenchymal stroma cells,especially fibroblasts to TRAIL, was a conceivable mechanism forthe clinical failure of TRAIL (15). However, this feature demon-strates fibroblasts as a durable synthesis reservoir for TRAIL, withprolonged expression compared with other TRAIL-sensitive cells.In reality, TRAIL-secreting human mesenchymal stem cells havebeen used for prolonged delivery of TRAIL in glioma (47). Toachieve the original hypothesis, TRAIL was fused with a leadersequence into a bioactive secretable form (sTRAIL). Despite thecomparable cytotoxicity observed in vitro with both sTRAIL andTRAIL plasmid, only sTRAIL encapsulated in LPD-induced super-ior antitumor efficacy in desmoplastic tumors. Consistent with
In Situ Generation of Tumor-Suppressive Fibroblasts
www.aacrjournals.org Cancer Res; 77(3) February 1, 2017 727
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

Figure 6.
Intravenous administration of sTRAIL LPD inhibited the orthotopic desmoplastic BXPC3 tumor growth and remodeled the tumor microenvironments. A, Dosingschedule of sTRAIL treatment on BXPC3-Luc2. B, IVIS images of BXPC3-Luc2 tumor after different treatments (n ¼ 5). C, Tumor inhibition curve of BXPC3(n ¼ 6–10; � , P < 0.05 compared with PBS group). D, The survival proportions of the treated groups. Median survival time (MST) is presented in the inset.Data shown as mean � SD, n ¼ 6–8. ��� , P < 0.001. E, Flow cytometry analysis of GFP's association with aSMA-positive fibroblasts 2 days after the thirdinjection of the LPD (n ¼ 4; � , P < 0.05; �� , P < 0.01). F, Immunofluorescence staining of aSMA and TUNEL assay from BXPC3-Luc2 tumor tissues after differenttreatments. G, Masson trichrome staining for collagen from the BXPC3-Luc2 tumors after different treatments. Heterogeneities are observed in the sTRAILLPD groups. Scar tissue (with few cell structures) is observed. H, The quantification of collagen levels based on non-scar area (n ¼ 4–5; � , P < 0.05).
Miao et al.
Cancer Res; 77(3) February 1, 2017 Cancer Research728
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

the in situ fibroblast engineering hypothesis, we found themajority of sTRAIL was expressed within fibroblasts within 3doses of sTRAIL LPD. The penetration of sTRAIL protein isanother concern for the in situ engineering. Compared withmost mAbs, the trimerized sTRAIL with a smaller molecularweight offered rapid diffusion. The current work illustrated thatapoptotic tumor cells induced by sTRAIL LPD can be observed500 mm away from the nearest fibroblast (Fig. 4). Comparedwith the average diameter (400 mm) of tumor nests in theUMUC3/3T3 model, the depth of penetration was undoubtedlysufficient to induce potent efficacy.
The apoptosis of tumor cells destroyed the nest structure,keeping fibroblasts as the major population of remaining cells.A quiescent characteristic marked by a reduction in ECM proteinsynthesis and decreased TAFs marker expression in the residualfibroblasts was found only after sTRAIL treatment. Downregula-tion of pSMAD2 in fibroblasts suggested this process may bemediated by TGFb (31, 48). Although further mechanistic studiesshould be conducted, the original hypothesis stated the apoptosisof neighboring tumor cells reciprocally reprograms TAFs due toinsufficient TGFb signaling activation. To verify this hypothesis,an experiment was conducted in vitro consisting of a noncontactcoculture of 3T3 cells secreting sTRAIL with UMUC3 tumor cells.Indeed, we found significant correlation between decreased fibro-blast aSMAmarkers and increased tumor cell apoptosis (Supple-
mentary Fig. S15). Support for this hypothesis can also be foundin that pSMAD was also downregulated in apoptotic tumor cellsin vivo, which was mediated through autocrine signaling of TGFb.In addition to the apoptotic tumor cells, the apoptosis-inducedrecruitment of proinflammatory cells responsible for TAF repro-gramming would be an additional source of the stroma remodel-ing. This particular mechanism will be evaluated in the futurestudies. In addition, the phenotype of fibroblasts in TRAIL treat-ment group remained constant, demonstrating TRAIL had min-imal direct effect on fibroblasts.
Reverting of TAFs has a dual benefit. First, the cellular andstructural changes of the stroma resulted from "normalized"fibroblasts are reported to exert tumor-suppressive forces andsignals, potentially inhibiting tumor growth (9, 49). Second, thereduction of the fibrotic content, which decompresses the intra-tumoral vasculature, creates a window for the second-wavenanotherapy (9, 35). Such expectations were presented both intheory and in the current study. Treatment with sTRAIL LPDsignificantly enhanced the delivery, retention, penetration, andefficacy of additional cisplatin nanoparticles.
Moreover, the potency of sTRAIL LPD monotherapy, observedon a hypovascular orthotopic pancreatic carcinoma (BXPC3)further verified the feasibility of this in situ engineering approach.The result is promising, as only a small population of therapeuticnanoparticles have shown efficacy against PDACs.
B
A
Systemic deliveryNPs Target to TAFs
In situ engineering of tumor-suppressive fibroblasts
TAFs in vicinity to blood vesselHigh binding affinity
Protamine
Remodel TMENP penetraton
Apoptosis
Reversion
TG
F-ββ
suppressive
ExtracellularMatrix
Tumorcells
TAFs
Therapeutic NP(e.g., cisplatin NP)
sTRAIL protein
(Second wave)
(First wave)
NFsp-sTRAIL
p-sTRAIL
p-sTRAIL
sTRAIL LPD
DSPE-PEG
Aminoethylanisamide(high binding to TAFs)
C
Figure 7.
Proposedmechanism of targeting TAFs for in situ engineering.A, Plasmid encoding secretable TRAIL protein is condensedwith protamine and further encapsulatedinto PEGylated liposomes coating with aminoethylanisamide-targeting motif (LPD). Diagram of the p-sTRAIL LPD is shown. B, LPD is systemically deliveredto tumor region and then extravasated from blood vessel due to the EPR effect. In most desmoplastic tumors, a thick layer of fibroblasts wraps around the bloodvessel. TAFs are the major cells taken up the targeted LPD. C, sTRAIL protein is synthesized by TAFs and diffuses to the neighboring tumor cells. Apoptotictumor cells reciprocally failed to activate local fibroblasts, reverting the TAFs to normal fibroblasts (NF). Normal fibroblasts can suppress tumor growth on one end,remodel the TME, and increase the penetration of a second-wave chemotherapeutic NPs on the other. Collectively, this multi-wave therapy can induce potentgrowth inhibition of the desmoplastic tumors. NP, nanoparticle.
In Situ Generation of Tumor-Suppressive Fibroblasts
www.aacrjournals.org Cancer Res; 77(3) February 1, 2017 729
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

Collectively, a novel regimen for the treatment of desmoplastictumors was developed by utilizing their off-target uptake of nano-particles to our benefit (Fig. 7). Three advantages highlight thesophistication of this approach: (i) the traditionally problematicbinding site barrier was used to induce potent apoptosis within thetumor nest with only a single conventional gene therapy agent; (ii)the fibroblasts in situwere not only engineered to secrete a cytotoxicprotein but also mechanistically reprogrammed to be tumor sup-pressive in a feedback fashion; and (iii) finally, reprogramming offibroblasts paved the way for coupling signal-dependent stromalreprogramming with tumor-directed cytotoxic nanoparticles andperhaps immunologic drugs, offering a new paradigm in thetreatment of desmoplastic tumors. Furthermore, fibroblasts couldbe engineered to produce various cytokines, orchestrating thesuppressive microenvironment to achieve a more sustained anti-tumor response. In the real clinical cases, the ECMcontentmay varyamong different individuals, as well as the ratio of stroma cells/tumor cells. All these factors would affect the potency of nanopar-ticles delivery to fibroblasts and the efficacy of protein productionin fibroblasts. Therefore, with regard to future clinical application,the combination of our conceptual strategy with individualizedtherapy would be more promising.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L. Miao, C.M. Lin, Y. Wang, L. Liu, L. HuangDevelopment of methodology: L. Miao, C.M. Lin, W.Y. Kim
Acquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): L. Miao, Q. Liu, C.M. Lin, C. Luo, S. HuAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): L. Miao, Q. Liu, C.M. Lin, W. Yin, W.Y. KimWriting, review, and/or revision of the manuscript: L. Miao, Q. Liu, C.M. Lin,W.Y. KimAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): L. Miao, C. Luo, Y. WangStudy supervision: L. Huang
AcknowledgmentsWe thank Dr. ShawnHingtgen (UNC) for providing the sTRAIL plasmid. We
acknowledge UNC Animal Histopathology Core and UNC TranslationalPathology Laboratory for tissue embedding, IF, and IHC staining. We thankUNC-CH Genome Core for gene sequencing of sTRAIL and TRAIL. We alsoappreciate the help of UNC Flow Cytometry Core, the ICP-MS Core and UNCCHANL for cell sorting, platinum quantification, and TEM imaging. Weappreciate Dr. Yi Zhao (UNC) andDr. Sai An's (UNC) help with themanuscriptrevision.
Grant SupportThe work was supported by NIH grants CA149363, CA151652, and
CA149387, and by the North Carolina Biotech Center Institutional SupportGrant2005-IDG-1016.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received March 29, 2016; revised September 26, 2016; accepted October 21,2016; published OnlineFirst November 18, 2016.
References1. Ernsting MJ, Hoang B, Lohse I, Undzys E, Cao P, Do T, et al. Targeting of
metastasis-promoting tumor-associated fibroblasts and modulation ofpancreatic tumor-associated stroma with a carboxymethylcellulose-doce-taxel nanoparticle. J Control Release 2015;206:122–30.
2. Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al.Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun 2015;6:8692.
3. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery ofnanomedicine to desmoplastic tumors. J Control Release 2015;219:192–204.
4. Roode LE, BrightonH,BoT, Perry JL, ParrottMC,Kersey F, et al. Subtumoralanalysis of PRINT nanoparticle distribution reveals targeting variationbased on cellular and particle properties. Nanomedicine 2016;12:1053–62.
5. Miao L, Wang Y, Lin CM, Xiong Y, Chen N, Zhang L, et al. Nanoparticlemodulation of the tumormicroenvironment enhances therapeutic efficacyof cisplatin. J Control Release 2015;217:27–41.
6. Smith NR, Baker D, Farren M, Pommier A, Swann R, Wang X, et al. Tumorstromal architecture can define the intrinsic tumor response to VEGF-targeted therapy. Clin Cancer Res 2013;19:6943–56.
7. LeBeau AM, Brennen WN, Aggarwal S, Denmeade SR. Targeting the cancerstroma with a fibroblast activation protein-activated promelittin protoxin.Mol Cancer Ther 2009;8:1378–86.
8. Murakami M, Ernsting MJ, Undzys E, Holwell N, Foltz WD, Li SD.Docetaxel conjugate nanoparticles that target alpha-smooth muscleactin-expressing stromal cells suppress breast cancer metastasis. CancerRes 2013;73:4862–71.
9. ShermanMH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. VitaminD receptor-mediated stromal reprogramming suppresses pancreatitis andenhances pancreatic cancer therapy. Cell 2014;159:80–93.
10. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al.Virtual microdissection identifies distinct tumor- and stroma-specific sub-types of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78.
11. De Palma M, Lewis CE. Macrophage regulation of tumor responses toanticancer therapies. Cancer Cell 2013;23:277–86.
12. Yoo J, Choi S, Hwang KS, Cho WK, Jung CR, Kwon ST, et al. Adeno-associated virus-mediated gene transfer of a secreted formof TRAIL inhibitstumor growth and occurrence in an experimental tumor model. J GeneMed 2006;8:163–74.
13. Zhang X, Edwards JP, Mosser DM. The expression of exogenous genes inmacrophages: obstacles and opportunities. Methods Mol Biol 2009;531:123–43.
14. Voortman J, Resende TP, Abou El HassanMA, Giaccone G, Kruyt FA. TRAILtherapy in non-small cell lung cancer cells: sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib.MolCancer Ther2007;6:2103–12.
15. O'Leary L, van der Sloot AM, Reis CR, Deegan S, Ryan AE, Dhami SP, et al.Decoy receptors block TRAIL sensitivity at a supracellular level: the role ofstromal cells in controlling tumour TRAIL sensitivity. Oncogene 2015;35:1261–70.
16. Shah K, Tung CH, Yang K, Weissleder R, Breakefield XO. Inducible releaseof TRAIL fusion proteins from a proapoptotic form for tumor therapy.Cancer Res 2004;64:3236–42.
17. Wang Y, Xu Z, Guo S, Zhang L, Sharma A, Robertson GP, et al. Intravenousdelivery of siRNA targeting CD47 effectively inhibits melanoma tumorgrowth and lung metastasis. Mol Ther 2013;21:1919–29.
18. Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with preciseratiometric co-loading and co-delivery of gemcitabine monophosphateand cisplatin for treatment of bladder cancer. Adv Funct Mater 2014;24:6601–11.
19. Zhang J, Miao L, Guo S, Zhang Y, Zhang L, Satterlee A, et al. Synergisticanti-tumor effects of combined gemcitabine and cisplatin nanoparticlesin a stroma-rich bladder carcinoma model. J Control Release 2014;182:90–6.
20. Fatin-Rouge N, Starchev K, Buffle J. Size effects on diffusion processeswithin agarose gels. Biophys J 2004;86:10.
Cancer Res; 77(3) February 1, 2017 Cancer Research730
Miao et al.
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

21. Dasargyri A, Hervella P, Christiansen A, Proulx ST, Detmar M, Leroux JC.Findings questioning the involvement of Sigma-1 receptor in the uptake ofanisamide-decorated particles. J Control Release 2016;224:229–38.
22. Hingtgen S, Ren XH, Terwilliger E, Masson M, Weissleder R, Shah K.Targeting multiple pathways in gliomas with stem cell and viral deliveredS-TRAIL and Temozolomide. Mol Cancer Ther 2008;7:3575–85.
23. Chono S, Li SD, Conwell CC, Huang L. An efficient and low immunos-timulatory nanoparticle formulation for systemic siRNA delivery to thetumor. J Control Release 2008;131:64–9.
24. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche incancer progression. J Cell Biol 2012;196:395–406.
25. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al.Tumour micro-environment elicits innate resistance to RAF inhibitorsthrough HGF secretion. Nature 2012;487:500–4.
26. Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T, et al.Metformin reduces desmoplasia in pancreatic cancer by reprogrammingstellate cells and tumor-associated macrophages. PLoS One 2015;10:e0141392.
27. UngefrorenH, KruseML, Trauzold A, Roeschmann S, Roeder C, Arlt A, et al.FAP-1 in pancreatic cancer cells: functional and mechanistic studies on itsinhibitory role in CD95-mediated apoptosis. J Cell Sci 2001;114(Pt 15):2735–46.
28. Novak M, Leonard MK, Yang XH, Kowluru A, Belkin AM, Kaetzel DM.Metastasis suppressor NME1 regulates melanoma cell morphology, self-adhesion and motility via induction of fibronectin expression. Exp Der-matol 2015;24:455–61.
29. Hawinkels LJ, PaauweM, Verspaget HW, Wiercinska E, van der Zon JM, vander Ploeg K, et al. Interactionwith colon cancer cells hyperactivates TGF-betasignaling in cancer-associated fibroblasts. Oncogene 2014;33:97–107.
30. Zhu Y, YinWL, Ba YF, Tian L, Gu ZQ, ZhangMS, et al. Transforming growthfactor-1 promotes the transcriptional activation of plasminogen activatorinhibitor type 1 in carcinoma-associated fibroblasts. Mol Med Rep 2012;6:1001–5.
31. Cantarella G, Risuglia N, Dell'eva R, Lempereur L, Albini A, Pennisi G, et al.TRAIL inhibits angiogenesis stimulated by VEGF expression in humanglioblastoma cells. Br J Cancer 2006;94:1428–35.
32. Griffon-EtienneG, Boucher Y, BrekkenC, SuitHD, JainRK. Taxane-inducedapoptosis decompresses blood vessels and lowers interstitial fluid pressurein solid tumors: clinical implications. Cancer Res 1999;59:3776–82.
33. Jain RK. Normalization of tumor vasculature: an emerging concept inantiangiogenic therapy. Science 2005;307:58–62.
34. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization forcancer and other angiogenic diseases. Nat Rev Drug Discov 2011;10:417–27.
35. Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, andtumor glucose metabolism to improve radiotherapy efficacy. Clin CancerRes 2012;18:5585–94.
36. Zhang X,Dong Y, Zeng X, Liang X, Li X, TaoW, et al. The effect of autophagyinhibitors on drug delivery using biodegradable polymer nanoparticles incancer treatment. Biomaterials 2014;35:1932–43.
37. Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, KamounWS, et al. Normalization of tumour blood vessels improves the delivery ofnanomedicines in a size-dependent manner. Nat Nanotechnol 2012;7:383–8.
38. Guo S,Wang Y,Miao L, Xu Z, Lin CM, Zhang Y, et al. Lipid-coated Cisplatinnanoparticles induce neighboring effect and exhibit enhanced anticancerefficacy. ACS Nano 2013;7:9896–904.
39. CabralH,Matsumoto Y,Mizuno K, ChenQ,MurakamiM, KimuraM, et al.Accumulation of sub-100 nm polymeric micelles in poorly permeabletumours depends on size. Nat Nanotechnol 2011;6:815–23.
40. Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, et al. Improve-ment of cancer-targeting therapy, using nanocarriers for intractable solidtumors by inhibition of TGF-beta signaling. Proc Natl Acad Sci U S A2007;104:3460–5.
41. MengH,ZhaoY,Dong J, XueM, Lin YS, Ji Z, et al. Two-wave nanotherapy totarget the stroma and optimize gemcitabine delivery to a humanpancreaticcancer model in mice. ACS Nano 2013;7:10048–65.
42. Matsuzaki H, Schmied BM, Ulrich A, Standop J, Schneider MB, Batra SK,et al. Combination of tumor necrosis factor-related apoptosis-inducingligand (TRAIL) and actinomycin D induces apoptosis even in TRAIL-resistant human pancreatic cancer cells. Clin Cancer Res 2001;7:407–14.
43. Kimura J, Ono HA, Kosaka T, Nagashima Y, Hirai S, Ohno S, et al.Conditionally replicative adenoviral vectors for imaging the effect ofchemotherapy on pancreatic cancer cells. Cancer Sci 2013;104:1083–90.
44. Kircheis R, Schuller S, Brunner S, Ogris M, Heider KH, Zauner W, et al.Polycation-basedDNA complexes for tumor-targeted gene delivery in vivo.J Gene Med 1999;1:111–20.
45. De Vlieghere E, Verset L, Demetter P, Bracke M, De Wever O. Cancer-associated fibroblasts as target and tool in cancer therapeutics and diag-nostics. Virchows Arch 2015;467:367–82.
46. Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: adiabolic liaisondriving cancer progression.CancerMetastasis Rev 2012;31:195–208.
47. Kim SM, Lim JY, Park SI, Jeong CH, Oh JH, Jeong M, et al. Gene therapyusing TRAIL-secreting human umbilical cord blood-derived mesenchymalstem cells against intracranial glioma. Cancer Res 2008;68:9614–23.
48. Islam SS, Mokhtari RB, El Hout Y, Azadi MA, Alauddin M, Yeger H, et al.TGF-beta1 induces EMT reprogramming of porcine bladder urothelial cellsinto collagen producing fibroblasts-like cells in a Smad2/Smad3-depen-dent manner. J Cell Commun Signal 2014;8:39–58.
49. BissellMJ,HinesWC.Whydon'twe getmore cancer? Aproposed role of themicroenvironment in restraining cancer progression. Nat Med 2011;17:320–9.
www.aacrjournals.org Cancer Res; 77(3) February 1, 2017 731
In Situ Generation of Tumor-Suppressive Fibroblasts
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866

2017;77:719-731. Published OnlineFirst November 18, 2016.Cancer Res Lei Miao, Qi Liu, C. Michael Lin, et al. Desmoplastic TumorsTargeting Tumor-Associated Fibroblasts for Therapeutic Delivery in
Updated version
10.1158/0008-5472.CAN-16-0866doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2016/11/18/0008-5472.CAN-16-0866.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/77/3/719.full#ref-list-1
This article cites 49 articles, 14 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/77/3/719.full#related-urls
This article has been cited by 2 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/77/3/719To request permission to re-use all or part of this article, use this link
on January 31, 2020. © 2017 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst November 18, 2016; DOI: 10.1158/0008-5472.CAN-16-0866


















