
Tamm-Horsfall protein-deficient thick ascending limbs ... · PDF filephil chemoattractant...
9
Tamm-Horsfall protein-deficient thick ascending limbs promote injury to neighboring S3 segments in an MIP-2-dependent mechanism Tarek M. El-Achkar, 1,2 Ruth McCracken, 1,2 Michael Rauchman, 1,2 Monique R. Heitmeier, 1 Ziyad Al-Aly, 2 Pierre C. Dagher, 3 and Xue-Ru Wu 4 1 Department of Medicine, Saint Louis University, St. Louis; 2 Department of Medicine, Saint Louis Veterans Affairs Medical Center, St. Louis, Missouri; 3 Department of Medicine, Indiana University, Indianapolis, Indiana; and 4 Departments of Urology and Pathology, New York University School of Medicine, New York, New York Submitted 20 October 2010; accepted in final form 6 January 2011 El-Achkar TM, McCracken R, Rauchman M, Heitmeier MR, Al-Aly Z, Dagher PC, Wu XR. Tamm-Horsfall protein- deficient thick ascending limbs promote injury to neighboring S3 segments in an MIP-2-dependent mechanism. Am J Physiol Renal Physiol 300: F999 –F1007, 2011. First published January 12, 2011; doi:10.1152/ajprenal.00621.2010.—Tamm-Horsfall protein (THP) is a glycoprotein expressed exclusively in thick ascending limbs (TAL) of the kidney. We recently described a novel protective role of THP against acute kidney injury (AKI) via downregulation of inflam- mation in the outer medulla. Our current study investigates the mechanistic relationships among the status of THP, inflammation, and tubular injury. Using an ischemia-reperfusion model in wild-type and THP/ mice, we demonstrate that it is the S3 proximal segments but not the THP-deficient TAL that are the main targets of tubular injury during AKI. The injured S3 segments that are surrounded by neutrophils in THP/ mice have marked overexpression of neutro- phil chemoattractant MIP-2 compared with wild-type counterparts. Neutralizing macrophage inflammatory protein-2 (MIP-2) antibody rescues S3 segments from injury, decreases neutrophil infiltration, and improves kidney function in THP/ mice. Furthermore, using immunofluorescence volumetric imaging of wild-type mouse kidneys, we show that ischemia alters the intracellular translocation of THP in the TAL cells by partially shifting it from its default apical surface domain to the basolateral domain, the latter being contiguous to the basolateral surface of S3 segments. Concomitant with this is the upregulation, in the basolateral surface of S3 segments, of the scav- enger receptor SRB-1, a putative receptor for THP. We conclude that TAL affects the susceptibility of S3 segments to injury at least in part by regulating MIP-2 expression in a THP-dependent manner. Our findings raise the interesting possibility of a direct role of basolaterally released THP on regulating inflammation in S3 segments. CXCL2; ischemia-reperfusion; uromodulin; acute kidney injury TAMM-HORSFALL PROTEIN (THP) is a unique glycoprotein be- cause it is exclusively expressed in the kidney, specifically in thick ascending limbs (TALs) and early distal tubules (4, 15, 29). Although this glycoprotein was discovered 60 years ago (37), its functions are still not fully understood (35). The association of THP with acute and chronic forms of renal disease, such as tubulointerstitial nephritis, familial juvenile hyperuricemic nephropathy, and more recently chronic kidney disease, argues for important regulatory functions of this gly- coprotein in maintaining normal function of renal tubules (4, 5, 14, 16, 19, 20, 35, 41). The role of THP in the setting of acute kidney injury (AKI) is even less well-understood. AKI remains a grave disease without a specific therapy and associated with increased risk of death (39). Experimental models of AKI such as ischemia- reperfusion injury (IRI) have been instrumental in understand- ing the complex pathogenesis of this disease. Indeed, AKI is a dynamic injury that involves epithelial and endothelial cell injury and alterations in various processes such vascular per- meability, oxidative stress, and inflammation (8, 9, 26). The role of inflammation in the pathophysiology of AKI involves various cells of the innate and adaptive immune response (18). Among these, neutrophils are believed to play a detrimental role in the ischemic insult (3, 18). Neutrophil infiltration of the injured site is mediated in part by the release of specific chemokines that function as powerful attractants. Macrophage inflammatory protein-2 (MIP-2) is part of the CXC chemokine family (CXCL-2) and is a major neutrophil chemoattractant (2, 34). This chemokine is thought to play an important role in neutrophil recruitment to the site of injury during AKI (22, 23, 40). Although MIP-2 expression is induced after ischemia- reperfusion (36), its specific tubular localization has not been well-defined in the context of AKI. We recently presented the first lines of evidence demonstrat- ing that, contrary to the traditional belief, THP is actually protective of renal tubules during AKI (12). Kidney injury, especially necrosis and neutrophil infiltration, is significantly aggravated in THP knockout mice compared with wild-type. We also showed that THP is not requisite for cast formation in the setting of AKI. The current studies were designed to address the mechanisms whereby THP protects the renal tu- bules in the outer medulla. We aimed to determine, using a THP knockout mouse model, the effect of THP deficiency on the specific type of tubule(s) involved in injury (TAL vs. S3) and the relationship to the neutrophil chemoattractant MIP-2. We therefore tested the hypothesis that THP-deficient TAL is more prone to injury by an augmented expression of MIP-2 that attracts neutrophils to the outer medulla. However, our results show that, instead of suspected THP-deficient TAL of the outer stripe (OS), necrosis predominantly affects S3 prox- imal segments. In addition, lack of THP led to an upregulation of MIP-2 in these S3 segments during AKI, and neutralizing MIP-2 rescued these tubules from injury. Our results indicate that a normal THP protects the kidney from acute injury by inhibiting a relay of inflammatory signaling between TAL and S3 segments. Our findings that ischemia increases ectopic THP expression on the basolateral surface and that putative THP receptors are present on S3 segments also suggest a direct THP-S3 link, a suggestion that warrants further validation. Address for reprint requests and other correspondence: T. M. El-Achkar, Div. of Nephrology, 111B JC, St. Louis VA Medical Center, 915 N. Grand, St. Louis, MO 63106 (e-mail: [email protected]). Am J Physiol Renal Physiol 300: F999–F1007, 2011. First published January 12, 2011; doi:10.1152/ajprenal.00621.2010. http://www.ajprenal.org F999 by 10.220.33.3 on September 21, 2017 http://ajprenal.physiology.org/ Downloaded from
Transcript of Tamm-Horsfall protein-deficient thick ascending limbs ... · PDF filephil chemoattractant...

Tamm-Horsfall protein-deficient thick ascending limbs promote injury toneighboring S3 segments in an MIP-2-dependent mechanism
Tarek M. El-Achkar,1,2 Ruth McCracken,1,2 Michael Rauchman,1,2 Monique R. Heitmeier,1 Ziyad Al-Aly,2
Pierre C. Dagher,3 and Xue-Ru Wu4
1Department of Medicine, Saint Louis University, St. Louis; 2Department of Medicine, Saint Louis Veterans Affairs MedicalCenter, St. Louis, Missouri; 3Department of Medicine, Indiana University, Indianapolis, Indiana; and 4Departmentsof Urology and Pathology, New York University School of Medicine, New York, New York
Submitted 20 October 2010; accepted in final form 6 January 2011
El-Achkar TM, McCracken R, Rauchman M, HeitmeierMR, Al-Aly Z, Dagher PC, Wu XR. Tamm-Horsfall protein-deficient thick ascending limbs promote injury to neighboring S3segments in an MIP-2-dependent mechanism. Am J Physiol RenalPhysiol 300: F999 –F1007, 2011. First published January 12, 2011;doi:10.1152/ajprenal.00621.2010.—Tamm-Horsfall protein (THP)is a glycoprotein expressed exclusively in thick ascending limbs(TAL) of the kidney. We recently described a novel protective role ofTHP against acute kidney injury (AKI) via downregulation of inflam-mation in the outer medulla. Our current study investigates themechanistic relationships among the status of THP, inflammation, andtubular injury. Using an ischemia-reperfusion model in wild-type andTHP�/� mice, we demonstrate that it is the S3 proximal segmentsbut not the THP-deficient TAL that are the main targets of tubularinjury during AKI. The injured S3 segments that are surrounded byneutrophils in THP�/� mice have marked overexpression of neutro-phil chemoattractant MIP-2 compared with wild-type counterparts.Neutralizing macrophage inflammatory protein-2 (MIP-2) antibodyrescues S3 segments from injury, decreases neutrophil infiltration, andimproves kidney function in THP�/� mice. Furthermore, usingimmunofluorescence volumetric imaging of wild-type mouse kidneys,we show that ischemia alters the intracellular translocation of THP inthe TAL cells by partially shifting it from its default apical surfacedomain to the basolateral domain, the latter being contiguous to thebasolateral surface of S3 segments. Concomitant with this is theupregulation, in the basolateral surface of S3 segments, of the scav-enger receptor SRB-1, a putative receptor for THP. We conclude thatTAL affects the susceptibility of S3 segments to injury at least in partby regulating MIP-2 expression in a THP-dependent manner. Ourfindings raise the interesting possibility of a direct role of basolaterallyreleased THP on regulating inflammation in S3 segments.
CXCL2; ischemia-reperfusion; uromodulin; acute kidney injury
TAMM-HORSFALL PROTEIN (THP) is a unique glycoprotein be-cause it is exclusively expressed in the kidney, specifically inthick ascending limbs (TALs) and early distal tubules (4, 15,29). Although this glycoprotein was discovered 60 years ago(37), its functions are still not fully understood (35). Theassociation of THP with acute and chronic forms of renaldisease, such as tubulointerstitial nephritis, familial juvenilehyperuricemic nephropathy, and more recently chronic kidneydisease, argues for important regulatory functions of this gly-coprotein in maintaining normal function of renal tubules (4, 5,14, 16, 19, 20, 35, 41).
The role of THP in the setting of acute kidney injury (AKI)is even less well-understood. AKI remains a grave diseasewithout a specific therapy and associated with increased risk ofdeath (39). Experimental models of AKI such as ischemia-reperfusion injury (IRI) have been instrumental in understand-ing the complex pathogenesis of this disease. Indeed, AKI is adynamic injury that involves epithelial and endothelial cellinjury and alterations in various processes such vascular per-meability, oxidative stress, and inflammation (8, 9, 26). Therole of inflammation in the pathophysiology of AKI involvesvarious cells of the innate and adaptive immune response (18).Among these, neutrophils are believed to play a detrimentalrole in the ischemic insult (3, 18). Neutrophil infiltration of theinjured site is mediated in part by the release of specificchemokines that function as powerful attractants. Macrophageinflammatory protein-2 (MIP-2) is part of the CXC chemokinefamily (CXCL-2) and is a major neutrophil chemoattractant (2,34). This chemokine is thought to play an important role inneutrophil recruitment to the site of injury during AKI (22, 23,40). Although MIP-2 expression is induced after ischemia-reperfusion (36), its specific tubular localization has not beenwell-defined in the context of AKI.
We recently presented the first lines of evidence demonstrat-ing that, contrary to the traditional belief, THP is actuallyprotective of renal tubules during AKI (12). Kidney injury,especially necrosis and neutrophil infiltration, is significantlyaggravated in THP knockout mice compared with wild-type.We also showed that THP is not requisite for cast formation inthe setting of AKI. The current studies were designed toaddress the mechanisms whereby THP protects the renal tu-bules in the outer medulla. We aimed to determine, using aTHP knockout mouse model, the effect of THP deficiency onthe specific type of tubule(s) involved in injury (TAL vs. S3)and the relationship to the neutrophil chemoattractant MIP-2.
We therefore tested the hypothesis that THP-deficient TALis more prone to injury by an augmented expression of MIP-2that attracts neutrophils to the outer medulla. However, ourresults show that, instead of suspected THP-deficient TAL ofthe outer stripe (OS), necrosis predominantly affects S3 prox-imal segments. In addition, lack of THP led to an upregulationof MIP-2 in these S3 segments during AKI, and neutralizingMIP-2 rescued these tubules from injury. Our results indicatethat a normal THP protects the kidney from acute injury byinhibiting a relay of inflammatory signaling between TAL andS3 segments. Our findings that ischemia increases ectopic THPexpression on the basolateral surface and that putative THPreceptors are present on S3 segments also suggest a directTHP-S3 link, a suggestion that warrants further validation.
Address for reprint requests and other correspondence: T. M. El-Achkar,Div. of Nephrology, 111B JC, St. Louis VA Medical Center, 915 N. Grand, St.Louis, MO 63106 (e-mail: [email protected]).
Am J Physiol Renal Physiol 300: F999–F1007, 2011.First published January 12, 2011; doi:10.1152/ajprenal.00621.2010.
http://www.ajprenal.org F999
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from
MATERIALS AND METHODS
Animal surgery. Animal experiments and protocols were approvedby the St. Louis VA Animal Care and Use Committee and conductedin conformity with the “Guiding Principles for Research InvolvingAnimals and Human Beings.” THP knockout animals (129/SvEvTHP�/�) and wild-type background strain have been characterizedin detail in previous publications (12, 24, 25). Animals were 8–10 wkold at the time of surgery. Ischemia-reperfusion surgery was aspreviously described (12, 17). In brief, mice were anesthetized withisoflurane and placed on a homeothermic table to maintain core bodytemperature at 37°C. Both renal pedicles were exposed via a midlineincision and occluded with micro aneurysm clamps for 22 minfollowed by 6- or 24-h reperfusion depending on the time pointstudied. This clamp time was chosen because of our previous workshowing that the corresponding injury was significant enough withoutcausing diffuse necrosis/or infarction of the outer medulla (12). Thisis especially relevant to our studies investigating the effect of THP onthe type of tubule injured. Sham surgery consisted of an identicalprocedure without application of the micro aneurysm clamps. Allanimals received subcutaneous analgesic (0.1 mg/kg bruprenorphine)at the end of surgery.
Serum creatinine was measured using capillary electrophoresis(28), performed at the UT Southwestern O’Brien Center of Excel-lence. Serum was obtained from maxillary vein bleeds before surgery(baseline) and 24 h after surgery (sham and IRI; n � 6 per strain perexperimental group).
Tissue harvesting and staining. Six or 24 hours after surgery,kidneys were perfusion-fixed in situ with 30 ml 4% paraformaldehydein PBS, pH 7.4; 100-�m vibratome sections for THP, MIP-2, andSR-B1 staining were obtained 24 h later. Alternatively, kidneys weredehydrated with increasing concentrations of ethanol and paraffin-embedded for histological studies using light microscopy as previ-ously described (10, 12).
We used the following primary antibodies for immunostaining: asheep polyclonal IgG for THP (AbD Serotec, cat. no. 8595-0054), agoat polyclonal IgG for MIP-2 (Santa Cruz Biotechnology, cat. no.Sc-26537), and a SR-B1 rabbit polyclonal antibody for SR-
B1(Abcam, cat. no. Ab396-100). The following conjugated secondaryantibodies were used: Alexa Fluor 555 donkey anti-sheep IgG andAlexa Fluor 647 chicken anti-goat IgG (both from Molecular Probes,cat. no. A21436 and A21469, respectively). Brush borders werestained with Oregon green-488 phalloidin (Molecular Probes, cat. no.O7466) and nuclei were stained with DAPI (Molecular Probes, cat.no. D21490). Negative controls were obtained by incubating tissuesfrom sham and ischemia animals with secondary antibodies in theabsence of primary antibodies.
Light microscopy and operational definitions for identification oftubular segments. Paraffin-embedded kidneys were sectioned at 4 �mand stained using periodic acid Schiff (PAS). This stain can differen-tiate proximal tubules in the outer medulla (predominantly S3 seg-ments, identified by the pink brush border) from nonproximal tubules(predominantly TAL, lacking any apical staining and appearing thin-ner). Nonnecrotic S3 tubules could still be identified after injury bythe pink brush-border remnant rim present at the apical area (see Fig.2B). Since we previously showed that necrosis, rather than apoptosis,is the predominant injury phenotype in THP knockout mice (12), wequantitated necrosis as an indicator of injury by tubule type. This wasaccomplished by counting tubules in the OS of the medulla (5 �20fields/kidney section) and grouping them into three types: S3, non-S3,and necrotic tubules. No necrotic tubules are expected in sham. Afterinjury, necrotic tubules are expected to appear, alongside a decrease inthe number of identifiable S3 or non-S3 tubules. Comparing thechanges in the tubule type percentages between sham and IRI inTHP�/� and THP�/� mice will identify the tubule type affected bynecrosis. Six animals per strain for each experimental group (shamTHP�/�, sham THP�/�, IRI THP�/�, IRI THP�/�) were used.Experiments were performed on coded sections, enabling blindedscoring.
Modified esterase staining specific for neutrophil detection usingthe naphthol AS-D chloroacetate esterase kit (Sigma, cat. no. 91C)was performed according to the manufacturer’s specifications. Neu-trophils were counted per intermediate power field (�20 magnifica-tion) and a general average was computed for each animal group (n �6 per strain for each experimental group).
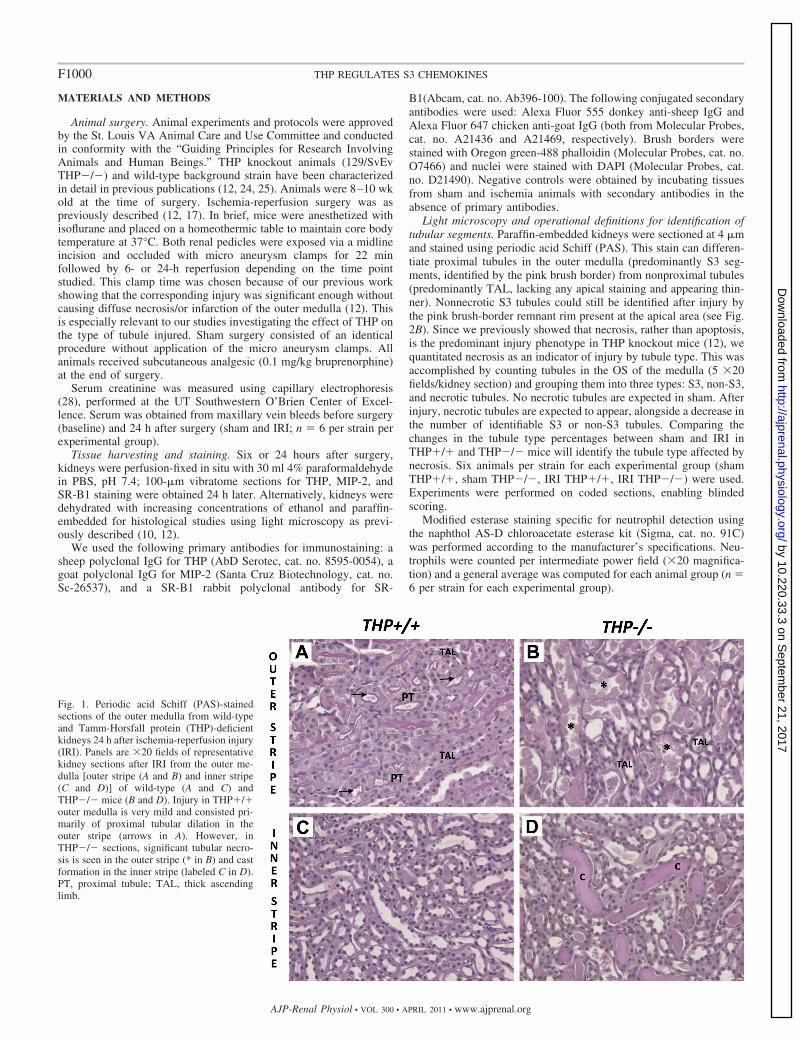
Fig. 1. Periodic acid Schiff (PAS)-stainedsections of the outer medulla from wild-typeand Tamm-Horsfall protein (THP)-deficientkidneys 24 h after ischemia-reperfusion injury(IRI). Panels are �20 fields of representativekidney sections after IRI from the outer me-dulla [outer stripe (A and B) and inner stripe(C and D)] of wild-type (A and C) andTHP�/� mice (B and D). Injury in THP�/�outer medulla is very mild and consisted pri-marily of proximal tubular dilation in theouter stripe (arrows in A). However, inTHP�/� sections, significant tubular necro-sis is seen in the outer stripe (* in B) and castformation in the inner stripe (labeled C in D).PT, proximal tubule; TAL, thick ascendinglimb.
F1000 THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from
Fluorescence microscopy. Vibratome sections (100-�m thickness)were stained for THP, MIP-2, or SR-B1 as previously described (11) andviewed under an Olympus Fluoview laser-scanning confocal microscopeavailable at St. Louis University. Images were collected under �20 or�60 magnification. Previously described fluorescence-based operationaldefinitions of kidney layers and structures were used to guide localizingproteins of interest within each kidney layer (11).
Three-dimensional reconstruction and volume rendering of THPexpression. Stacks of z-planes were collected at 0.4-�m intervals. Three-dimensional (3D) volume rendering was done using Voxx, a voxel-based3D imaging software available from the Indiana Center for BiologicalMicroscopy. Videos were produced in addition to snapshot imagesrepresenting views from different angles as previously described (11).
MIP-2 neutralization in THP�/� mice. THP�/� mice were sub-jected to IRI surgery. Following the release of the renal clamp, theyreceived an intraperitoneal injection of 100 �g of MIP-2 neutralizingantibody (n � 5; monoclonal rat anti-mouse MAB452, clone 40605,R&D Systems) or 100 �g of isotype IgG control (n � 5; also from R&DSystems). Mice were killed at 24 h following surgery. Serum creatinine
at baseline and 24 h after surgery was analyzed as above. Kidneys wereprocessed as described above to quantitate injury histologically by tubulartype and assess neutrophil infiltration as described above.
Statistical analysis. Values of each experimental group are reportedas means � SE. A chi square test was used to compare the differencein proportions and a two-tailed t-test was used to examine thedifference in means for continuous data. Statistical significance wasdetermined at the 0.05 significance level.
RESULTS
Renal IRI in THP�/� mice causes significant necrosis of S3segment and not TAL in the outer medulla. We studied theeffect of renal IRI on THP�/� and THP�/� mice 24 h afterIRI surgery. We focused on the OS of the outer medullabecause it is the main site of injury. As shown in Fig. 1,significant necrosis and cast formation were seen in THP�/�OS compared with THP�/�. This is consistent with ourprevious findings described recently (12).
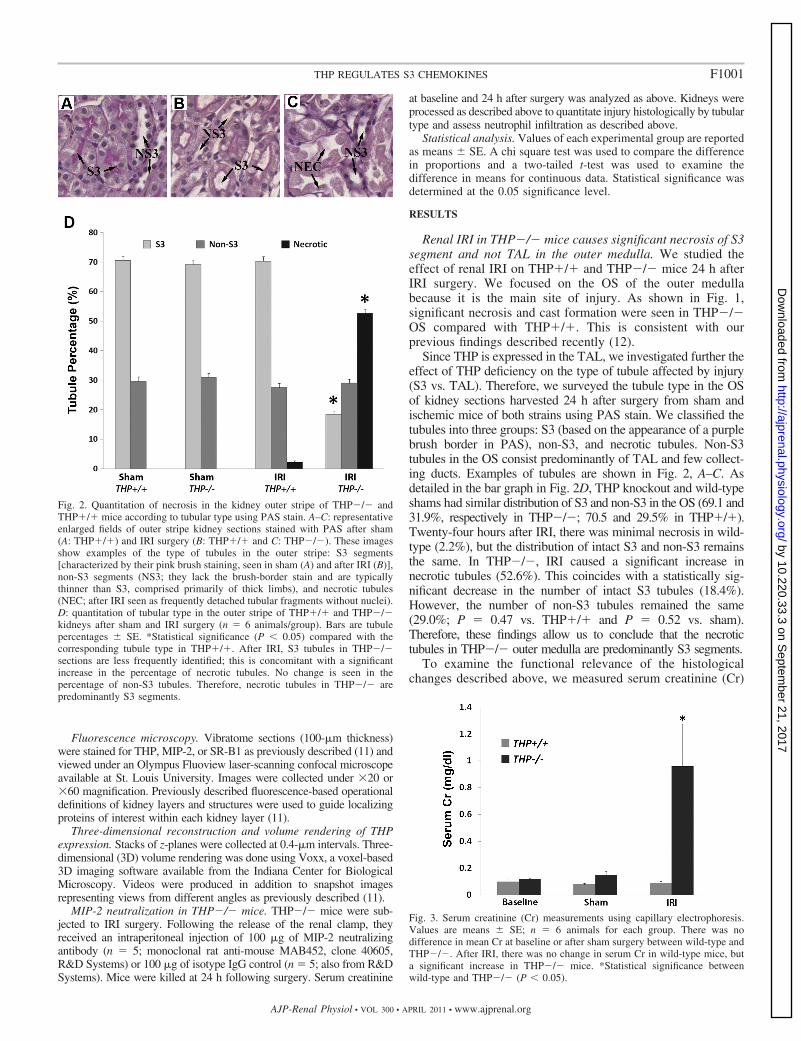
Since THP is expressed in the TAL, we investigated further theeffect of THP deficiency on the type of tubule affected by injury(S3 vs. TAL). Therefore, we surveyed the tubule type in the OSof kidney sections harvested 24 h after surgery from sham andischemic mice of both strains using PAS stain. We classified thetubules into three groups: S3 (based on the appearance of a purplebrush border in PAS), non-S3, and necrotic tubules. Non-S3tubules in the OS consist predominantly of TAL and few collect-ing ducts. Examples of tubules are shown in Fig. 2, A–C. Asdetailed in the bar graph in Fig. 2D, THP knockout and wild-typeshams had similar distribution of S3 and non-S3 in the OS (69.1 and31.9%, respectively in THP�/�; 70.5 and 29.5% in THP�/�).Twenty-four hours after IRI, there was minimal necrosis in wild-type (2.2%), but the distribution of intact S3 and non-S3 remainsthe same. In THP�/�, IRI caused a significant increase innecrotic tubules (52.6%). This coincides with a statistically sig-nificant decrease in the number of intact S3 tubules (18.4%).However, the number of non-S3 tubules remained the same(29.0%; P � 0.47 vs. THP�/� and P � 0.52 vs. sham).Therefore, these findings allow us to conclude that the necrotictubules in THP�/� outer medulla are predominantly S3 segments.
To examine the functional relevance of the histologicalchanges described above, we measured serum creatinine (Cr)
Fig. 2. Quantitation of necrosis in the kidney outer stripe of THP�/� andTHP�/� mice according to tubular type using PAS stain. A–C: representativeenlarged fields of outer stripe kidney sections stained with PAS after sham(A: THP�/�) and IRI surgery (B: THP�/� and C: THP�/�). These imagesshow examples of the type of tubules in the outer stripe: S3 segments[characterized by their pink brush staining, seen in sham (A) and after IRI (B)],non-S3 segments (NS3; they lack the brush-border stain and are typicallythinner than S3, comprised primarily of thick limbs), and necrotic tubules(NEC; after IRI seen as frequently detached tubular fragments without nuclei).D: quantitation of tubular type in the outer stripe of THP�/� and THP�/�kidneys after sham and IRI surgery (n � 6 animals/group). Bars are tubulepercentages � SE. *Statistical significance (P � 0.05) compared with thecorresponding tubule type in THP�/�. After IRI, S3 tubules in THP�/�sections are less frequently identified; this is concomitant with a significantincrease in the percentage of necrotic tubules. No change is seen in thepercentage of non-S3 tubules. Therefore, necrotic tubules in THP�/� arepredominantly S3 segments.
Fig. 3. Serum creatinine (Cr) measurements using capillary electrophoresis.Values are means � SE; n � 6 animals for each group. There was nodifference in mean Cr at baseline or after sham surgery between wild-type andTHP�/�. After IRI, there was no change in serum Cr in wild-type mice, buta significant increase in THP�/� mice. *Statistical significance betweenwild-type and THP�/� (P � 0.05).
F1001THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from
in wild-type and THP�/� mice at baseline and after sham or IRIsurgery. This was performed using capillary electrophoresis at theUT Southwestern O’Brien Center (Dallas, TX). Figure 3 showsthat there was no difference in serum Cr at baseline or aftersham surgery in mice from both strains. After IRI, THP�/�mice had a higher Cr compared with wild-type (0.96 � 0.3 vs.0.09 � 0.01 mg/dl, respectively; P � 0.05). This is consistentwith the injury seen in histology.
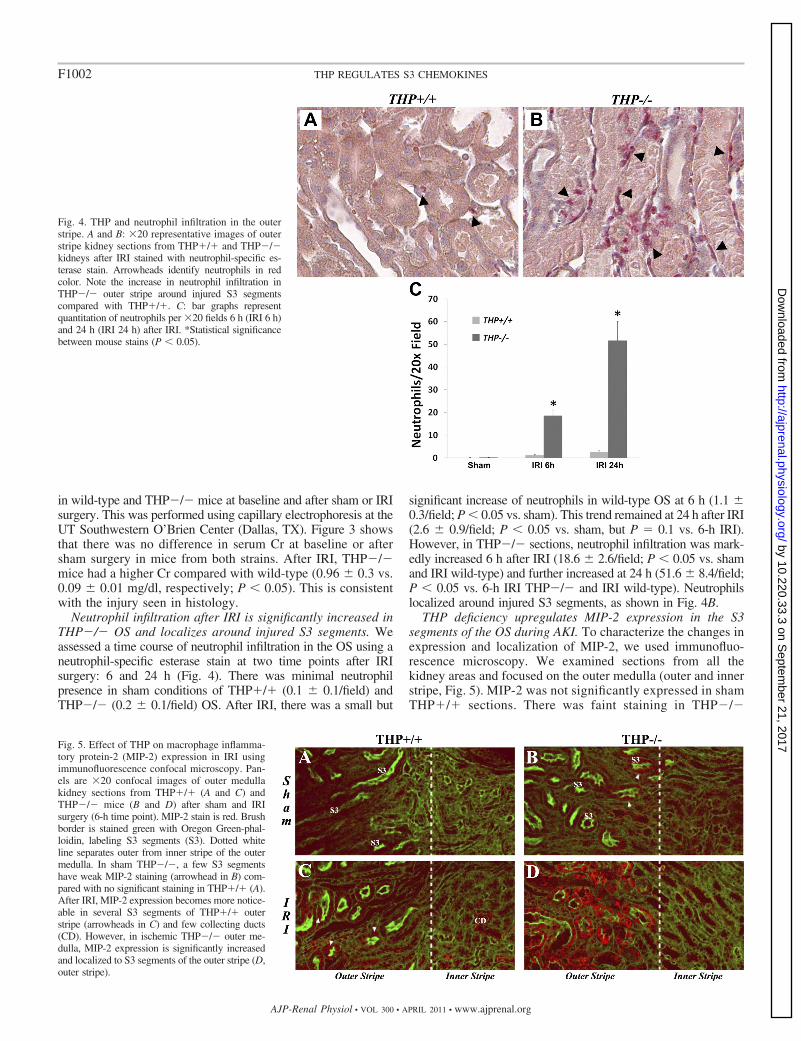
Neutrophil infiltration after IRI is significantly increased inTHP�/� OS and localizes around injured S3 segments. Weassessed a time course of neutrophil infiltration in the OS using aneutrophil-specific esterase stain at two time points after IRIsurgery: 6 and 24 h (Fig. 4). There was minimal neutrophilpresence in sham conditions of THP�/� (0.1 � 0.1/field) andTHP�/� (0.2 � 0.1/field) OS. After IRI, there was a small but
significant increase of neutrophils in wild-type OS at 6 h (1.1 �0.3/field; P � 0.05 vs. sham). This trend remained at 24 h after IRI(2.6 � 0.9/field; P � 0.05 vs. sham, but P � 0.1 vs. 6-h IRI).However, in THP�/� sections, neutrophil infiltration was mark-edly increased 6 h after IRI (18.6 � 2.6/field; P � 0.05 vs. shamand IRI wild-type) and further increased at 24 h (51.6 � 8.4/field;P � 0.05 vs. 6-h IRI THP�/� and IRI wild-type). Neutrophilslocalized around injured S3 segments, as shown in Fig. 4B.
THP deficiency upregulates MIP-2 expression in the S3segments of the OS during AKI. To characterize the changes inexpression and localization of MIP-2, we used immunofluo-rescence microscopy. We examined sections from all thekidney areas and focused on the outer medulla (outer and innerstripe, Fig. 5). MIP-2 was not significantly expressed in shamTHP�/� sections. There was faint staining in THP�/�
Fig. 4. THP and neutrophil infiltration in the outerstripe. A and B: �20 representative images of outerstripe kidney sections from THP�/� and THP�/�kidneys after IRI stained with neutrophil-specific es-terase stain. Arrowheads identify neutrophils in redcolor. Note the increase in neutrophil infiltration inTHP�/� outer stripe around injured S3 segmentscompared with THP�/�. C: bar graphs representquantitation of neutrophils per �20 fields 6 h (IRI 6 h)and 24 h (IRI 24 h) after IRI. *Statistical significancebetween mouse stains (P � 0.05).
Fig. 5. Effect of THP on macrophage inflamma-tory protein-2 (MIP-2) expression in IRI usingimmunofluorescence confocal microscopy. Pan-els are �20 confocal images of outer medullakidney sections from THP�/� (A and C) andTHP�/� mice (B and D) after sham and IRIsurgery (6-h time point). MIP-2 stain is red. Brushborder is stained green with Oregon Green-phal-loidin, labeling S3 segments (S3). Dotted whiteline separates outer from inner stripe of the outermedulla. In sham THP�/�, a few S3 segmentshave weak MIP-2 staining (arrowhead in B) com-pared with no significant staining in THP�/� (A).After IRI, MIP-2 expression becomes more notice-able in several S3 segments of THP�/� outerstripe (arrowheads in C) and few collecting ducts(CD). However, in ischemic THP�/� outer me-dulla, MIP-2 expression is significantly increasedand localized to S3 segments of the outer stripe (D,outer stripe).
F1002 THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from
kidneys localized to few S3 tubules. Six hours after IRI,MIP-2 staining was seen in few S3 segments in the OS ofTHP�/� mice and occasional collecting ducts in the innerstripe of these mice. However, MIP-2 staining in THP�/�was markedly increased after IRI and localized predomi-nantly to S3 segments of the outer medulla (Fig. 5D).Twenty-four hours after IRI, MIP-2 staining was compara-ble to the 6-h staining in wild-type and THP�/�, althoughsome staining was lost from S3 segments that underwentnecrosis (data not shown).
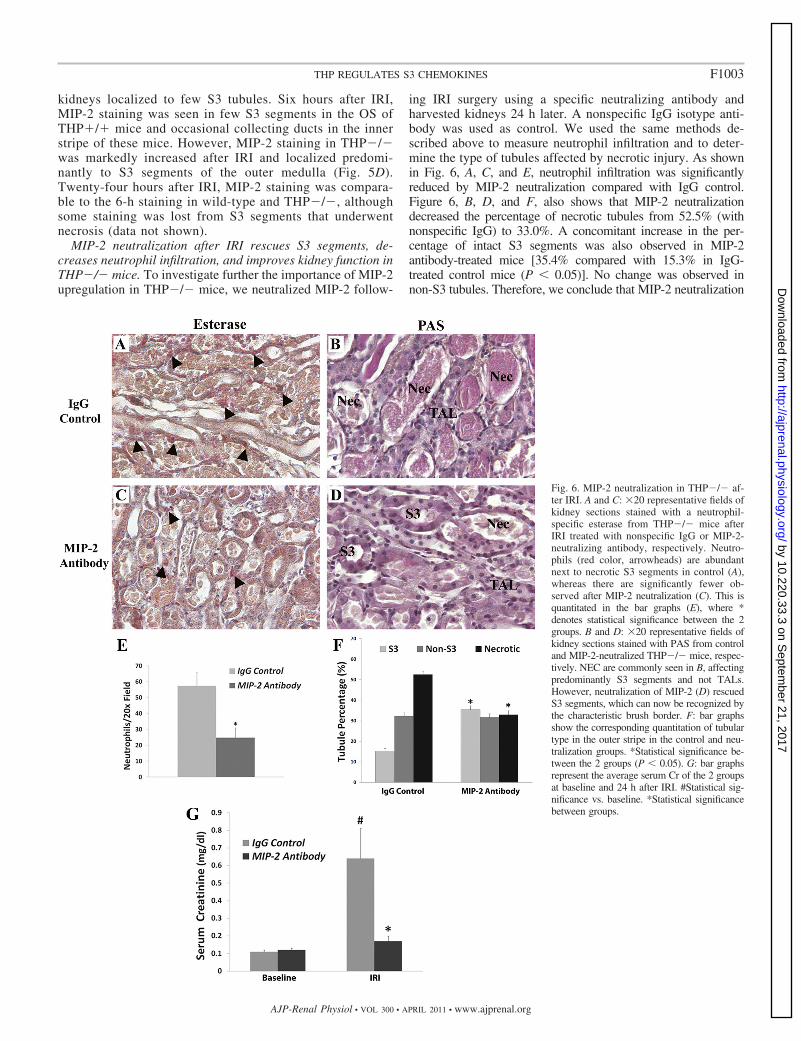
MIP-2 neutralization after IRI rescues S3 segments, de-creases neutrophil infiltration, and improves kidney function inTHP�/� mice. To investigate further the importance of MIP-2upregulation in THP�/� mice, we neutralized MIP-2 follow-
ing IRI surgery using a specific neutralizing antibody andharvested kidneys 24 h later. A nonspecific IgG isotype anti-body was used as control. We used the same methods de-scribed above to measure neutrophil infiltration and to deter-mine the type of tubules affected by necrotic injury. As shownin Fig. 6, A, C, and E, neutrophil infiltration was significantlyreduced by MIP-2 neutralization compared with IgG control.Figure 6, B, D, and F, also shows that MIP-2 neutralizationdecreased the percentage of necrotic tubules from 52.5% (withnonspecific IgG) to 33.0%. A concomitant increase in the per-centage of intact S3 segments was also observed in MIP-2antibody-treated mice [35.4% compared with 15.3% in IgG-treated control mice (P � 0.05)]. No change was observed innon-S3 tubules. Therefore, we conclude that MIP-2 neutralization
Fig. 6. MIP-2 neutralization in THP�/� af-ter IRI. A and C: �20 representative fields ofkidney sections stained with a neutrophil-specific esterase from THP�/� mice afterIRI treated with nonspecific IgG or MIP-2-neutralizing antibody, respectively. Neutro-phils (red color, arrowheads) are abundantnext to necrotic S3 segments in control (A),whereas there are significantly fewer ob-served after MIP-2 neutralization (C). This isquantitated in the bar graphs (E), where *denotes statistical significance between the 2groups. B and D: �20 representative fields ofkidney sections stained with PAS from controland MIP-2-neutralized THP�/� mice, respec-tively. NEC are commonly seen in B, affectingpredominantly S3 segments and not TALs.However, neutralization of MIP-2 (D) rescuedS3 segments, which can now be recognized bythe characteristic brush border. F: bar graphsshow the corresponding quantitation of tubulartype in the outer stripe in the control and neu-tralization groups. *Statistical significance be-tween the 2 groups (P � 0.05). G: bar graphsrepresent the average serum Cr of the 2 groupsat baseline and 24 h after IRI. #Statistical sig-nificance vs. baseline. *Statistical significancebetween groups.
F1003THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from
in THP�/� mice rescued S3 segments from injury. This was alsoreflected in measuring kidney function (Fig. 6G) where MIP-2neutralization prevented a rise in serum Cr 24 h after IRI com-pared with control (0.17 � 0.03 vs. 0.64 � 0.17 mg/dl, respec-tively; P � 0.05).
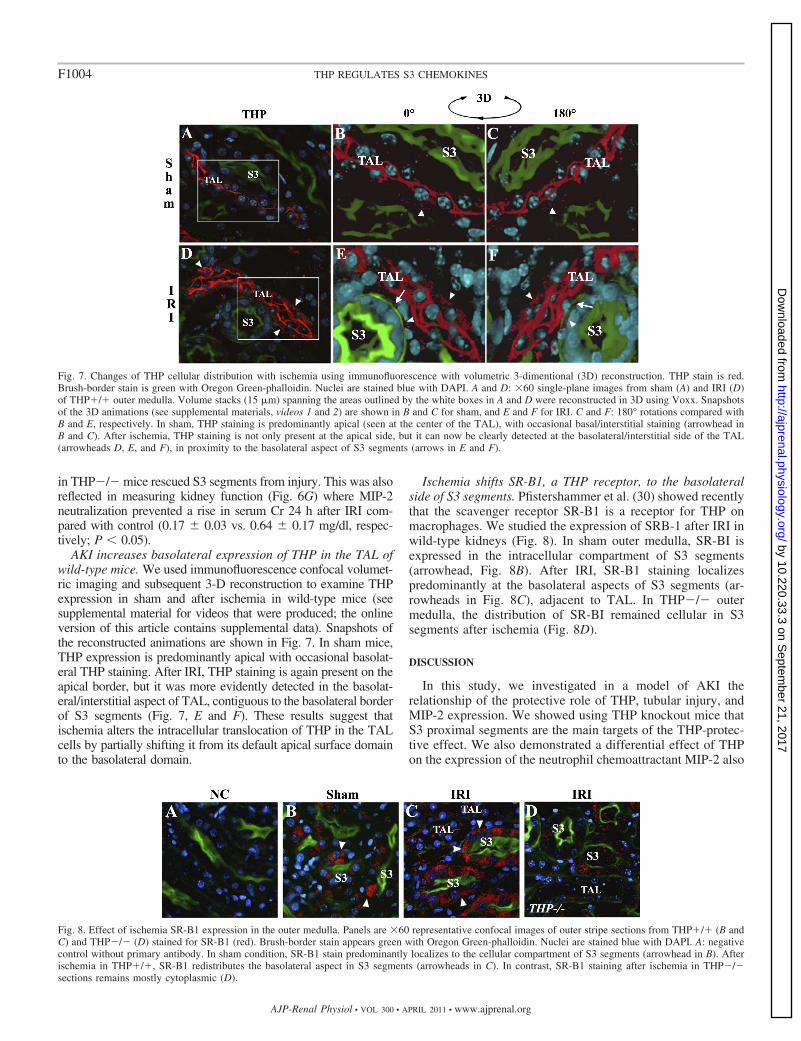
AKI increases basolateral expression of THP in the TAL ofwild-type mice. We used immunofluorescence confocal volumet-ric imaging and subsequent 3-D reconstruction to examine THPexpression in sham and after ischemia in wild-type mice (seesupplemental material for videos that were produced; the onlineversion of this article contains supplemental data). Snapshots ofthe reconstructed animations are shown in Fig. 7. In sham mice,THP expression is predominantly apical with occasional basolat-eral THP staining. After IRI, THP staining is again present on theapical border, but it was more evidently detected in the basolat-eral/interstitial aspect of TAL, contiguous to the basolateral borderof S3 segments (Fig. 7, E and F). These results suggest thatischemia alters the intracellular translocation of THP in the TALcells by partially shifting it from its default apical surface domainto the basolateral domain.
Ischemia shifts SR-B1, a THP receptor, to the basolateralside of S3 segments. Pfistershammer et al. (30) showed recentlythat the scavenger receptor SR-B1 is a receptor for THP onmacrophages. We studied the expression of SRB-1 after IRI inwild-type kidneys (Fig. 8). In sham outer medulla, SR-BI isexpressed in the intracellular compartment of S3 segments(arrowhead, Fig. 8B). After IRI, SR-B1 staining localizespredominantly at the basolateral aspects of S3 segments (ar-rowheads in Fig. 8C), adjacent to TAL. In THP�/� outermedulla, the distribution of SR-BI remained cellular in S3segments after ischemia (Fig. 8D).
DISCUSSION
In this study, we investigated in a model of AKI therelationship of the protective role of THP, tubular injury, andMIP-2 expression. We showed using THP knockout mice thatS3 proximal segments are the main targets of the THP-protec-tive effect. We also demonstrated a differential effect of THPon the expression of the neutrophil chemoattractant MIP-2 also
Fig. 7. Changes of THP cellular distribution with ischemia using immunofluorescence with volumetric 3-dimentional (3D) reconstruction. THP stain is red.Brush-border stain is green with Oregon Green-phalloidin. Nuclei are stained blue with DAPI. A and D: �60 single-plane images from sham (A) and IRI (D)of THP�/� outer medulla. Volume stacks (15 �m) spanning the areas outlined by the white boxes in A and D were reconstructed in 3D using Voxx. Snapshotsof the 3D animations (see supplemental materials, videos 1 and 2) are shown in B and C for sham, and E and F for IRI. C and F: 180° rotations compared withB and E, respectively. In sham, THP staining is predominantly apical (seen at the center of the TAL), with occasional basal/interstitial staining (arrowhead inB and C). After ischemia, THP staining is not only present at the apical side, but it can now be clearly detected at the basolateral/interstitial side of the TAL(arrowheads D, E, and F), in proximity to the basolateral aspect of S3 segments (arrows in E and F).
Fig. 8. Effect of ischemia SR-B1 expression in the outer medulla. Panels are �60 representative confocal images of outer stripe sections from THP�/� (B andC) and THP�/� (D) stained for SR-B1 (red). Brush-border stain appears green with Oregon Green-phalloidin. Nuclei are stained blue with DAPI. A: negativecontrol without primary antibody. In sham condition, SR-B1 stain predominantly localizes to the cellular compartment of S3 segments (arrowhead in B). Afterischemia in THP�/�, SR-B1 redistributes the basolateral aspect in S3 segments (arrowheads in C). In contrast, SR-B1 staining after ischemia in THP�/�sections remains mostly cytoplasmic (D).
F1004 THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from

in the S3 segments, which could explain the predisposition ofthese tubules to injury and neutrophil infiltration. This wasverified by neutralizing MIP-2 after IRI in THP�/� mice,which rescued S3 segments. The heightened inflammatorysignaling in S3 of THP�/� kidneys is consistent with ourprevious findings that THP alters the expression of the inflam-matory receptor Toll-like receptor 4 (TLR4) in these tubularsegments. Indeed, TLR4 is upstream from MIP-2 in the in-flammatory signaling axis in AKI (40). Therefore, THP derivedfrom the TAL is affecting the response of S3 segments toinjury. Our findings confirm the seminal proposition by Sa-firstein (32, 33) that the TAL is a key element in regulatingmolecular responses of neighboring tubular elements duringkidney injury.
How does THP in TAL regulate MIP-2 expression in neigh-boring S3 segments? The process is unlikely to be mediatedthrough the urinary luminal space because THP cannot accessS3 segments from that route. Since TAL and S3 segments runside-by-side in the OS, it is possible that a THP-dependentcross talk exists between TAL and S3 segments in the outermedulla. The concept of TAL-S3 cross talk in the medulla hasbeen previously suggested by others (13, 27, 32). A fewautocrine or paracrine factors have been proposed as potentialmediators, such as prostaglandins (PGE2), nitric oxide, kinins,and adenosine (13). However, data in the setting of AKI arevery sparse. Could THP itself be a major mediator of thistrans-tubular signaling?
Although THP is predominantly an apically secreted protein,its basolateral and interstitial expression has been well-de-scribed previously. In a physiological state, Bachmann et al.(4) showed using immunogold electron microscopy that theapical-to-basolateral distribution of THP within the TALs liesapproximately in a 2:1 ratio. More recently, Jennings et al. (16)also suggested a possible role for basolateral THP in thecontext of studying familial juvenile nephropathy. In injurymodels, previous studies suggest significant THP expression inthe interstitium (14, 35, 41). We recently showed that ische-mia-reperfusion promotes the interstitial presence of THParound S3 segments (12). In the current study, we furtherexpand these findings by showing that ischemia shifts THPexpression partially to the basolateral aspect of TAL, whichcould explain the presence of THP in the interstitium (throughan enhanced basolateral release). The expression by S3 tubulesof a putative receptor for THP (SRB-1) in an appropriatedistribution (at the basolateral aspect of S3) could support adirect role for THP on S3 segments. Figure 9 depicts workingmodels whereby a TAL-S3 tubular cross talk could be depen-dent on THP. Although our current and previous data (12)suggest a direct THP-S3 interaction (Fig. 9, model A), it is alsoimportant to consider, at this developing investigative stage,other alternative possibilities that do not necessarily involve adirect THP-S3 interaction; for example, the protective effectsof THP on S3 segments could be relayed through a secondarymessage produced from TAL (Fig. 9, model B). Indeed, thecurrent work underscores the importance of future investiga-tions to further elucidate the role of THP in mediating TAL-S3interactions in the outer medulla in maintaining homeostasisand during injury.
Inflammation is now believed to play a major role in thepathophysiology of AKI (8). In addition to involving variouscells from the innate and adaptive immune system, neutrophil
infiltration is a prominent feature of this response in murineAKI (7, 18). TLR signaling is at the core of the inflammatoryresponse and is believed to be activated in ischemic kidneyinjury (22, 40). These proteins recognize specific molecularpatterns of pathogens and select endogenous ligands and ini-tiate among others the NF-� and MAPKK signaling cascadesthat produce a myriad of cytokines and chemokines (1). Tu-bular epithelial cells are believed to be involved in the ensuinginflammatory response after AKI in part by activating TLRsignaling (TLR2 and TLR4) and the subsequent production ofcytokines such as TNF- and chemokines such as CXCL2(MIP-2) and MCP-1 (21–23, 38, 40). We focused on MIP-2 inthis manuscript because of the significant neutrophil infiltrationaround S3 segments seen after injury in THP�/� medulla andour previous findings of TLR4 upregulation specifically inthese segments (12).
MIP-2 is part of the CXC chemokine family and is a majorneutrophil chemoattractant (2, 34). MIP-2 is expressed in thekidney (31), and induced with injury (22, 36, 40), but itsspecific tubular localization has not been well-defined. The roleof MIP-2 in the recruitment of neutrophils after kidney injuryhas been previously studied by Miura and colleagues (23),where neutralization of MIP-2 by specific antibodies in amouse model of ischemia-reperfusion reduced neutrophil in-filtration and attenuated kidney injury (23). Our current datafurther extend these studies, by showing that the renalepithelial MIP-2 expression after injury localizes primarilyto S3 segments and is regulated by THP. Since we previ-ously showed that THP alters the expression of TLR4 inthese S3 segments, we speculate that THP regulates aTLR4-MIP-2 signaling axis in S3 segments, which couldadd up to be an important determinant of neutrophil infil-tration and susceptibility to kidney injury. Future studies areneeded to understand why TAL, independent of THP, isinherently more resistant to neutrophil-induced injury com-pared with S3 segments.
In addition to our studies in AKI, the use of THP knockoutmice has been instrumental in uncovering the multifaceted roleof THP as a defense agent against stone formation (24) andbladder infections (6, 25). Indeed, THP�/� mice show an
Fig. 9. Possible mechanisms of THP-mediated TAL-S3 tubular cross talk. Aand B: conceptual models illustrating possible mechanisms of THP-mediatedcross talk between S3 segments and TAL. A: possibility where basolateral/interstitial THP released from TAL could act directly on S3 segments.B: alternative explanation where THP affects the TAL, which in turn releasesa secondary mediator that acts on S3 tubules. Another possible “hybrid”mechanism (not depicted here) could involve THP in the interstitium withouta direct effect on S3, but rather through an effect on interstitial or endothelialcells.
F1005THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from

inability to clear inoculated bladder infections. However, it isimportant to note that these mice do not develop spontaneousurinary tract infections (6, 25). Therefore, it is unlikely thatsuperimposed kidney infections in our IRI model in THP�/�mice could explain the augmented inflammatory response andthe increased susceptibility to kidney injury.
In summary, we showed data that the protective effect ofTHP in AKI involves primarily S3 segments of the outermedulla. THP has a differential effect on the expression of theneutrophil chemoattractant MIP-2 in these S3 segments,thereby affecting neutrophil infiltration. Our findings raise thepossibility of a direct role of basolaterally released THP ondownregulating inflammation in S3 segments, a hypothesis thatwarrants further investigation. We propose that THP is amediator of tubular cross talk aimed at stabilizing the outermedulla in the face of injury.
ACKNOWLEDGMENTS
The authors acknowledge J. Lucas from UT Southwestern O’Brien Centerfor assistance with capillary electrophoresis.
GRANTS
This work was supported by a Carl Gottschalk Award from the AmericanSociety of Nephrology to T. M. El-Achkar.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
1. Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol 4:499–511, 2004.
2. Anders HJ, Vielhauer V, Schlondorff D. Chemokines and chemokinereceptors are involved in the resolution or progression of renal disease.Kidney Int 63: 401–415, 2003.
3. Awad AS, Rouse M, Huang L, Vergis AL, Reutershan J, Cathro HP,Linden J, Okusa MD. Compartmentalization of neutrophils in the kidneyand lung following acute ischemic kidney injury. Kidney Int 75: 689–698,2009.
4. Bachmann S, Koeppen-Hagemann I, Kriz W. Ultrastructural localiza-tion of Tamm-Horsfall glycoprotein (THP) in rat kidney as revealed byprotein A-gold immunocytochemistry. Histochemistry 83: 531–538, 1985.
5. Bachmann S, Mutig K, Bates J, Welker P, Geist B, Gross V, Luft FC,Alenina N, Bader M, Thiele BJ, Prasadan K, Raffi HS, Kumar S.Renal effects of Tamm-Horsfall protein (uromodulin) deficiency in mice.Am J Physiol Renal Physiol 288: F559–F567, 2005.
6. Bates JM, Raffi HM, Prasadan K, Mascarenhas R, Laszik Z, MaedaN, Hultgren SJ, Kumar S. Tamm-Horsfall protein knockout mice aremore prone to urinary tract infection: rapid communication. Kidney Int 65:791–797, 2004.
7. Bolisetty S, Agarwal A. Neutrophils in acute kidney injury: not neutralany more. Kidney Int 75: 674–676, 2009.
8. Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatorydisease? Kidney Int 66: 480–485, 2004.
9. Devarajan P. Update on mechanisms of ischemic acute kidney injury. JAm Soc Nephrol 17: 1503–1520, 2006.
10. El-Achkar TM, Huang X, Plotkin Z, Sandoval RM, Rhodes GJ,Dagher PC. Sepsis induces changes in the expression and distribution ofToll-like receptor 4 in the rat kidney. Am J Physiol Renal Physiol 290:F1034–F1043, 2006.
11. El-Achkar TM, Plotkin Z, Marcic B, Dagher PC. Sepsis induces anincrease in thick ascending limb Cox-2 that is TLR4 dependent. Am JPhysiol Renal Physiol 293: F1187–F1196, 2007.
12. El-Achkar TM, Wu XR, Rauchman M, McCracken R, Kiefer S,Dagher PC. Tamm-Horsfall protein protects the kidney from ischemicinjury by decreasing inflammation and altering TLR4 expression. Am JPhysiol Renal Physiol 295: F534–F544, 2008.
13. Gobe GC, Johnson DW. Distal tubular epithelial cells of the kidney:potential support for proximal tubular cell survival after renal injury. Int JBiochem Cell Biol 39: 1551–1561, 2007.
14. Howie AJ, Brewer DB. Extratubular deposits of Tamm-Horsfall proteinin renal allografts. J Pathol 139: 193–206, 1983.
15. Hoyer JR, Sisson SP, Vernier RL. Tamm-Horsfall glycoprotein: ultra-structural immunoperoxidase localization in rat kidney. Lab Invest 41:168–173, 1979.
16. Jennings P, Aydin S, Kotanko P, Lechner J, Lhotta K, Williams S,Thakker RV, Pfaller W. Membrane targeting and secretion of mutanturomodulin in familial juvenile hyperuricemic nephropathy. J Am SocNephrol 18: 264–273, 2007.
17. Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reducesapoptosis and protects renal function in the setting of ischemic injury. JClin Invest 108: 1291–1298, 2001.
18. Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury.Nephron Exp Nephrol 109: e102–e107, 2008.
19. Kottgen A, Glazer NL, Dehghan A, Hwang SJ, Katz R, Li M, Yang Q,Gudnason V, Launer LJ, Harris TB, Smith AV, Arking DE, Astor BC,Boerwinkle E, Ehret GB, Ruczinski I, Scharpf RB, Ida Chen YD, deBoer IH, Haritunians T, Lumley T, Sarnak M, Siscovick D, BenjaminEJ, Levy D, Upadhyay A, Aulchenko YS, Hofman A, Rivadeneira F,Uitterlinden AG, van Duijn CM, Chasman DI, Pare G, Ridker PM,Kao WH, Witteman JC, Coresh J, Shlipak MG, Fox CS. Multiple lociassociated with indices of renal function and chronic kidney disease. NatGenet 41: 712–717, 2009.
20. Kumar S. Mechanism of injury in uromodulin-associated kidney disease.J Am Soc Nephrol 18: 10–12, 2007.
21. Lech M, Avila-Ferrufino A, Allam R, Segerer S, Khandoga A, Krom-bach F, Garlanda C, Mantovani A, Anders HJ. Resident dendritic cellsprevent postischemic acute renal failure by help of single Ig IL-1 receptor-related protein. J Immunol 183: 4109–4118, 2009.
22. Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ,Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S.Renal-associated TLR2 mediates ischemia/reperfusion injury in the kid-ney. J Clin Invest 115: 2894–2903, 2005.
23. Miura M, Fu X, Zhang QW, Remick DG, Fairchild RL. Neutralizationof Gro alpha and macrophage inflammatory protein-2 attenuates renalischemia/reperfusion injury. Am J Pathol 159: 2137–2145, 2001.
24. Mo L, Huang HY, Zhu XH, Shapiro E, Hasty DL, Wu XR. Tamm-Horsfall protein is a critical renal defense factor protecting against calciumoxalate crystal formation. Kidney Int 66: 1159–1166, 2004.
25. Mo L, Zhu XH, Huang HY, Shapiro E, Hasty DL, Wu XR. Ablation ofthe Tamm-Horsfall protein gene increases susceptibility of mice to bladdercolonization by type 1-fimbriated Escherichia coli. Am J Physiol RenalPhysiol 286: F795–F802, 2004.
26. Nath KA, Norby SM. Reactive oxygen species and acute renal failure.Am J Med 109: 665–678, 2000.
27. Neuhofer W, Beck FX. Cell survival in the hostile environment of therenal medulla. Annu Rev Physiol 67: 531–555, 2005.
28. Paroni R, Fermo I, Cighetti G, Ferrero CA, Carobene A, Ceriotti F.Creatinine determination in serum by capillary electrophoresis. Electro-phoresis 25: 463–468, 2004.
29. Peach RJ, Day WA, Ellingsen PJ, McGiven AR. Ultrastructural local-ization of Tamm-Horsfall protein in human kidney using immunogoldelectron microscopy. Histochem J 20: 156–164, 1988.
30. Pfistershammer K, Klauser C, Leitner J, Stockl J, Majdic O, Weich-hart T, Sobanov Y, Bochkov V, Saemann M, Zlabinger G, SteinbergerP. Identification of the scavenger receptors SREC-I, Cla-1 (SR-BI), andSR-AI as cellular receptors for Tamm-Horsfall protein. J Leukoc Biol 83:131–138, 2008.
31. Roche JK, Keepers TR, Gross LK, Seaner RM, Obrig TG. CXCL1/KCand CXCL2/MIP-2 are critical effectors and potential targets for therapyof Escherichia coli O157:H7-associated renal inflammation. Am J Pathol170: 526–537, 2007.
32. Safirstein R. Gene expression in nephrotoxic and ischemic acute renalfailure. J Am Soc Nephrol 4: 1387–1395, 1994.
33. Safirstein R, Megyesi J, Saggi SJ, Price PM, Poon M, Rollins BJ,Taubman MB. Expression of cytokine-like genes JE and KC is increasedduring renal ischemia. Am J Physiol Renal Fluid Electrolyte Physiol 261:F1095–F1101, 1991.
34. Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors,and renal disease: from basic science to pathophysiologic and therapeuticstudies. J Am Soc Nephrol 11: 152–176, 2000.
35. Serafini-Cessi F, Malagolini N, Cavallone D. Tamm-Horsfall glycopro-tein: biology and clinical relevance. Am J Kidney Dis 42: 658–676, 2003.
F1006 THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from

36. Stroo I, Stokman G, Teske GJ, Raven A, Butter LM, Florquin S,Leemans JC. Chemokine expression in renal ischemia/reperfusion injury ismost profound during the reparative phase. Int Immunol 22: 433–442, 2010.
37. Tamm I, Horsfall FL Jr. Characterization and separation of an inhibitorof viral hemagglutination present in urine. Proc Soc Exp Biol Med 74:106–108, 1950.
38. Thurman JM, Lenderink AM, Royer PA, Coleman KE, Zhou J,Lambris JD, Nemenoff RA, Quigg RJ, Holers VM. C3a is required forthe production of CXC chemokines by tubular epithelial cells after renalishemia/reperfusion. J Immunol 178: 1819–1828, 2007.
39. Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology andoutcomes of acute kidney injury. Clin J Am Soc Nephrol 3: 844–861,2008.
40. Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alex-ander SI, Sharland AF, Chadban SJ. TLR4 activation mediateskidney ischemia/reperfusion injury. J Clin Invest 117: 2847–2859,2007.
41. Zager RA, Cotran RS, Hoyer JR. Pathologic localization of Tamm-Horsfall protein in interstitial deposits in renal disease. Lab Invest 38:52–57, 1978.
F1007THP REGULATES S3 CHEMOKINES
AJP-Renal Physiol • VOL 300 • APRIL 2011 • www.ajprenal.org
by 10.220.33.3 on Septem
ber 21, 2017http://ajprenal.physiology.org/
Dow
nloaded from

















