
TABLE OF CONTENTS - ULisboarepositorio.ul.pt/bitstream/10451/1147/2/18315_ulsd_re282_MJD_Tese... ·...
169
1 TABLE OF CONTENTS Abbreviations list 5 Abstract 9 Resumo 11 Figure index 13 1. Introduction 19 1.1 Neurotrophins 19 1.1.1Neurotrophin synthesis 19 1.1.2 Neurotrophin receptors 20 1.1.3 Pathophysiological implications of BDNF actions 28 1.2 Adenosine 31 1.2.1 Adenosine synthesis 31 1.2.2 Adenosine receptors 33 1.2.3 Pathophysiological implications of adenosine actions 37 1.3 Hippocampus 41 2. Aims 45 3. Methods 49 3.1 Biological sample preparations 49 3.1.1 Hippocampal slices 49 3.1.2 Hippocampal homogenates 52 3.1.3 Neuronal cell cultures 52 3.1.4 Cytosolic protein isolation 54 3.2 Drugs and antibodies 55 3.2.1 Drugs 55 3.2.2 Antibodies 56 3.3 Techniques 57 3.3.1 Microelectrophysiological recordings 57 3.3.2 Western blotting 60 3.3.3 Binding assays 61 3.3.4 Apoptosis detection 62 3.4 Statistics 64 4. Results 65 4.1 Interplay between TrkB and adenosine A2A receptors in infant rats 65 4.1.1 Rationale 65
-
Upload
nguyentruc -
Category
Documents
-
view
221 -
download
0
Transcript of TABLE OF CONTENTS - ULisboarepositorio.ul.pt/bitstream/10451/1147/2/18315_ulsd_re282_MJD_Tese... ·...

1
TABLE OF CONTENTS Abbreviations list 5
Abstract 9
Resumo 11
Figure index 13
1. Introduction 19 1.1 Neurotrophins 19
1.1.1Neurotrophin synthesis 19 1.1.2 Neurotrophin receptors 20 1.1.3 Pathophysiological implications of BDNF actions 28
1.2 Adenosine 31 1.2.1 Adenosine synthesis 31 1.2.2 Adenosine receptors 33 1.2.3 Pathophysiological implications of adenosine actions 37
1.3 Hippocampus
41
2. Aims 45
3. Methods 49 3.1 Biological sample preparations 49
3.1.1 Hippocampal slices 49 3.1.2 Hippocampal homogenates 52 3.1.3 Neuronal cell cultures 52 3.1.4 Cytosolic protein isolation 54
3.2 Drugs and antibodies 55 3.2.1 Drugs 55 3.2.2 Antibodies 56
3.3 Techniques 57 3.3.1 Microelectrophysiological recordings 57 3.3.2 Western blotting 60 3.3.3 Binding assays 61 3.3.4 Apoptosis detection 62
3.4 Statistics 64 4. Results 65 4.1 Interplay between TrkB and adenosine A2A receptors in infant rats 65
4.1.1 Rationale 65

2
4.1.2 Pre-depolarisation induced by high K+ facilitates BDNF excitatory action on hippocampal synaptic transmission through TrkB receptors
65
4.1.3 Theta Burst stimulation paired with BDNF can elicit synaptic potentiation
69
4.1.4 Adenosine A2A receptor activation facilitates BDNF excitatory action on synaptic transmission
69
4.1.5 The excitatory action of BDNF is facilitated by the selective adenosine kinase inhibitor 5-iodotubercidin
73
4.1.6 The activation of the cAMP–PKA transducing system is a critical step in the excitatory action of BDNF
74
4.1.7 Discussion 78 4.2 Influence of age on BDNF modulation of hippocampal synaptic transmission: interplay with adenosine A2A receptors
83
4.2.1 Rationale 83 4.2.2 BDNF facilitates synaptic transmission in young adult rats in a “LTP like” process
83
4.2.3 Adenosine A2A receptors are involved in the excitatory action of BDNF in the hippocampus of young adult rats and aged rats
89
4.2.4 The density of TrkB full length receptors and adenosine A2A receptors is altered in older rats
94
4.2.5 Discussion 97 4.3 Influence of age on the BDNF modulation of long term potentiation: interplay with adenosine A2A receptors
101
4.3.1 Rationale 101 4.3.2 BDNF effect on LTP induced by θ-Burst stimulation (3 Bursts, 100Hz, 3 stimuli, separated by 200 ms).
102
4.3.3 BDNF effect on LTP induced by a weak θ-Burst stimulation (2 Bursts, 100Hz, 3 stimuli, separated by 200 ms)
111
4.3.4 Effects of a BDNF scavenger on LTP in hippocampal slices 113 4.3.5 Discussion 116
4.4 Influence of adenosine A2A receptors on the protective action of BDNF against apoptosis induced by the amyloid beta peptide
120
4.4.1 Rationale 120 4.4.2 Apoptosis induced by amyloid beta in primary cultures of rat neurons is prevented by BDNF and this protection is potentiated by adenosine A2A receptors activation.
121
4.4.3 Amyloid beta induced fragmentation of Pro-caspase-3 is prevented by BDNF in the presence of adenosine A2A receptors activation
126
4.4.4 Amyloid beta induced increase in caspase-3 activity is prevented by BDNF in the presence of adenosine A2A receptors activation
130

3
4.4.5 Discussion 132 5. Conclusions 135 6. Future perspectives 137 7. Acknowledgements 141 8. References 145

4

5
Abbreviations list Aβ – Amyloid beta peptide
AC – Adenylate cyclase
AD – Alzheimer’s disease
ADA – Adenosine deaminase
ADAC – Adenosine amine congenere
ADO – Adenosine
AK – Adenosine kinase
AMP – Adenosine 5’-monophosphate
AP5 – DL-2-amino-5-phosphonopentanoate
ATP – Adenosine 5’-triphosphate
BDNF – Brain-derived neurotrophic factor
Bmax – Maximal number of binding sites
cAMP – cyclic adenosine monophosphate
CGS 21680 – 4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuroanamidosyl)-9H-purin-2-
yl]amino]ethyl]benzenepropanoic acid
CNS – Central nervous system
CR – Cysteine-repeated
CREB – cAMP response element binding protein
DAG – Diacylglycerol
DbcAMP – Dibutyryl cyclic adenosine monophosphate
DG – Dentate Gyrus
DMEM – Dulbecco’s modified Eagle’s medium
DMSO – Dimethylsulphoxide
DPCPX- 1,3-Dipropyl-8- cyclopentylxanthine
DTT– 1,4-Dithiothreitol

6
EC – Entorhinal cortex
EDTA – Ethylenediaminetetraacetic acid
EGTA – Ethylene glycol-bis(2-aminoethyl ether)-N,N –tetraacetic acid
ER – Endoplasmic reticulum
ERK– extracellular signal-regulated protein kinases
FCT – Fundação para a ciência e a tecnologia
fEPSP – Field excitatory post-synaptic potential
GABA – γ-aminobutyric acid
GFAP – Glial fibrillary acidic protein
HBSS – Hank’s balanced salt solution
HD – Huntington’s disease
IP3 – Inositol 1,4,5-triphosphate
ITU – 5-Iodotubercidin
JNK – c-jun N-terminal kinases
KAc – Potassium acetate
Kd – Equilibrium dissociation constant
Ki – Equilibrium dissociation constant of the competitor
LDH – Lactate dehydrogenase
L-DOPA – (l-3,4-dihydroxyphenylalanine)
LRR– Leucine-rich repeats
LTP – Long-term potentiation
MAPK – mitogen-activated protein kinase
NF-KB – Nuclear factor KB
NGF – Nerve growth factor
NMDA– N-Methyl-D-aspartate
3-NP – 3-nitropropionic acid
NT – Neurotrophin

7
PBS – Phosphate buffered saline
PI3K – Phosphatydilinositol-3-kinase
PIP2 – Phosphatidylinositol-4,5-biphosphate
PKA – Protein kinase A
PKC – Protein kinase C
PLC – Phospholipase C
PLD – Phospholipase D
pNA – p-nitroanilide
SAH – S-adenosylhomocysteine
SAPK – Stress-activated protein kinases
SCH 58261 – 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1 ,2,4-triazolo
[1 ,5-c]pyrimidine
SDS - PAGE –Sodium dodecyl sulphate-polyacrylamide-gel electrophoresis
SEM – Standard error of the mean
TGN – Trans-golgi network
Tris – Tris-hydroxymethyl-aminomethane
Trk – Tropomyosin-related kinase
XAC – 1,3-dipropyl-8-phenylxanthine amine congener
ZM 241385 – 4-(2-[7-Amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-
ylamino]ethyl)phenol

8

9
Abstract
The brain-derived neurotrophic factor (BDNF) belongs to a group of signalling factors
that are essential for neuronal survival and differentiation, inducing modulation of cell
death events such as apoptosis. This neurotrophin also has synaptic regulatory
actions on basal synaptic transmission, including effects on plasticity such as long-
term potentiation (LTP).
The hippocampus is under neuromodulatory control of adenosine, which
through activation of inhibitory (A1) and excitatory (A2A) receptors fine-tunes the action
of neurotransmitters and neuromodulators. The hippocampus is a brain area where
both A2A and TrkB receptors are expressed. Since activation of A2A receptors can
acutely induce transactivation of BDNF TrkB receptors in cell culture, in the present
work it was evaluated how the activation of adenosine A2A receptors could influence
the actions of BDNF. The first aim of the present study was to investigate the
influence of adenosine A2A receptors on BDNF effects on hippocampal synaptic
transmission and the possible mechanisms involved. Secondly, the influence of age
on BDNF modulation of hippocampal synaptic transmission, as well as the interplay
between adenosine A2A receptors and BDNF TrKB receptors, was examined. As a
third objective, the BDNF actions on LTP during aging and the influence of adenosine
A2A receptors on these actions was also evaluated. Finally, the influence of the A2A
receptor activation on the neuroprotective action of BDNF on apoptosis was
investigated.
The acute excitatory action of BDNF on synaptic transmission in the
hippocampus of infant rats was found to be inducible by a depolarisation that is
dependent on adenosine A2A receptor activation, through a mechanism that requires
cyclic adenosine monophosphate (cAMP) formation and protein kinase A (PKA)

10
activity. Subsequently, for the first time a relationship between age-related changes in
the density of TrkB and A2A receptors as it concerns BDNF-induced enhancement of
synaptic transmission in the hippocampus was elucidated. In LTP studies, it was
observed that BDNF increases the magnitude of θ-Burst stimuli-induced LTP and that
this excitatory action is also dependent on A2A adenosine receptor activation.
Moreover, it was found that BDNF loses the ability to increase LTP in aged animals.
Finally, BDNF protected neurons from apoptosis induced by amyloid beta peptide
(Aβ) 25-35 and this protection was more evident when adenosine A2A receptors are
activated.
In conclusion, the results now presented demonstrate that activation of
adenosine A2A receptors facilitates the synaptic and neuroprotective actions of BDNF.

11
Resumo
O factor neurotrófico derivado do cérebro (BDNF, brain-derived neurotrophic factor)
pertence a um grupo de factores de sinalização essenciais para a sobrevivência e
diferenciação neuronal, modulando a morte celular por apoptose. Esta neurotrofina
regula quer a transmissão sináptica quer a plasticidade sináptica como é o caso da
potenciação de longa duração (LTP, long term potentiation).
O hipocampo encontra-se sob controlo da adenosina, que através da
activação dos seus receptores inibitórios (A1) e excitatórios (A2A) regula a acção de
neurotramissores e neuromoduladores. Uma vez que a activação dos receptores A2A
pode induzir transactivação dos receptores TrkB do BDNF em células em cultura, no
presente estudo avaliou-se se a activação dos receptores A2A poderia influenciar a
função do BDNF no hipocampo, área cerebral que expressa quer receptores A2A da
adenosina quer receptores TrkB para o BDNF. Primeiro avaliou-se a influência dos
receptores A2A na acção do BDNF sobre a transmissão sináptica no hipocampo e
possíveis mecanismos envolvidos. Seguidamente, foi avaliada a influência da idade
na modulação da transmissão sináptica induzida pelo BDNF, e a sua relação com os
receptores A2A da adenosina. Posteriormente estudaram-se as acções do BDNF na
LTP em função do envelhecimento e a influência exercida pelos receptors A2A da
adenosina. Finalmente, estudou-se a consequência da activação dos receptores A2A
da adenosina no efeito neuroprotector do BDNF, ou seja sobre a sua influência na
morte celular por apoptose.
Os resultados obtidos mostraram que a acção excitatória do BDNF aplicado
agudamente na transmissão sinaptica em hipocampo de ratos jovens pode ser
induzida por uma despolarização, e que este efeito é dependente da activação dos
receptores A2A da adenosina, através de um mecanismo que requer a formação de
monofosfato ciclico de adenosina (cAMP, cyclic adenosine monophosphate) e a

12
activação da cinase de proteínas tipo A (PKA, protein kinase A). Observou-se pela
primeira vez que, ao longo da idade, existe uma relação entre a alteração do efeito
do BDNF na transmissão sináptica e modificações nas densidades dos receptores
A2A da adenosina e TrkB de BDNF no hipocampo. Verificou-se também que o BDNF
aumenta a magnitude da LTP induzida por um estimulo θ-Burst, e que perde esta
acção em animais idosos. Estes efeitos são, igualmente, dependentes da activação
dos receptores A2A da adenosina. Finalmente, demonstrou-se que o BDNF protege
os neurónios da morte celular induzida pelo péptido beta amiloide (Aβ, amyloid beta
peptide) 25-35 e que esta protecção é mais evidente quando os receptores A2A se
encontram activados, deixando de se observar quando estes receptores são
bloqueados.
Em conclusão, os resultados apresentados na presente dissertação
demonstram que a activação dos receptores A2A da adenosina facilita a acção
sináptica e neuroprotectora do BDNF quando administrado agudamente.

13
Figure index
1. Background Figure 1.1.1 Structure of the rat BDNF gene and protein 21 Figure 1.1.2 Diagram illustrating the pathways of BDNF synthesis and
secretion
22 Figure 1.1.3 Representation of the domain structures of the Trk and
p75 receptors
24 Figure 1.1.4 TrK-mediated neurotrophin signalling 27 Figure 1.1.5 NGF administration directly to the brain 28 Figure 1.2.1 Adenosine chemical structure 31 Figure 1.2.2 Pathways of adenosine production, metabolism and
transport
32 Figure 1.2.3 Distribution of adenosine receptores (A1, A2A and human
A3) in brain regions.
35 Figure 1.2.4 Adenosine receptor-signalling pathways 36 Figure 1.3.1 Hippocampus in rat and human 43 Figure 1.3.2 Human hippocampus dissected free compared to a
specimen of Hippocampus leria
43 Figure 1.3.3 The hippocampal formation anatomy
44
3. Methods Figure 3.1.1 Hippocampal slices preparation 51 Figure 3.1.2 Resting chamber for hippocampal slices 52 Figure 3.3.1 Setup for extra-cellular microelectrophysiology recordings 57 Figure 3.3.2 Extracellular recordings in hippocampal slices
60
4. Results
Chapter 4.1 Figure 4.1.1 The averaged time course of changes in fEPSP slope
induced by application of BDNF alone 20 ng/ml or 100 ng/ml.
67 Figure 4.1.2 Pre-depolarisation induced by high K+ facilitates BDNF
excitatory action on hippocampal synaptic transmission through TrkB receptors.
68 Figure 4.1.3 Theta-Burst stimulation paired with BDNF can elicit
potentiation effect.
71

14
Figure 4.1.4 Adenosine A2A receptors activation facilitates BDNF excitatory action on synaptic transmission
72
Figure 4.1.5 The excitatory action of BDNF is facilitated by a selective adenosine kinase inhibitor, 5-iodotubercidin
75
Figure 4.1.6 The activation of the cAMP–PKA transducing system is a critical step for the excitatory action of BDNF
76
Figure 4.1.7 Presynaptic cAMP mimics the effect of high-K+, ITU, or CGS 21680
77
Figure 4.1.8 Mechanism of the facilitatory action of adenosine A2A receptor activation on the BDNF effects on hippocampal synaptic transmission in infant rats
80 Chapter 4.2
Figure 4.2.1 The action of BDNF on excitatory synaptic transmission varies in rats from different age groups
85
Figure 4.2.2 Comparison between the averaged effects of BDNF in rats from different age groups
86
Figure 4.2.3 A high concentration (100 ng/ml) of BDNF increases synaptic transmission in slices taken from old adult rats
87
Figure 4.2.4 BDNF facilitates synaptic transmission in young adult rats in a “LTP- like” process.
88
Figure 4.2.5 Involvement of adenosine A2A receptors in the excitatory action of BDNF in synaptic transmission
91
Figure 4.2.6 Involvement of adenosine A2A receptors in the excitatory action of BDNF in synaptic transmission
92
Figure 4.2.7 Pre-depolarisation induced by high K+ facilitates BDNF excitatory action on hippocampal synaptic transmission in slices taken from old adult (36-38 week-old) rats
93 Figure 4.2.8 Changes in the density of TrkB full length receptors in
different age groups
95 Figure 4.2.9 Specific binding of adenosine A2A receptors in the
hippocampus of rats from different age groups
96 Chapter 4.3
Figure 4.3.1 Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 4 week-old rats
103
Figure 4.3.2 Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 4 week-old rats is dependent on adenosine A2A receptors
104 Figure 4.3.3 Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced
LTP in slices taken from 10-16 week-old rats
105 Figure 4.3.4 Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced
LTP in slices taken from 10-16 week-old rats is dependent

15
LTP in slices taken from 10-16 week-old rats is dependent on adenosine A2A receptors
106
Figure 4.3.5
Absence of effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 36-38 week-old rats
108
Figure 4.3.6 Absence of effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 70-80 week-old rats
109
Figure 4.3.7 Age-related modulation of long-term potentiation (LTP) by BDNF
110
Figure 4.3.8 Absence of effect of the BDNF (20 ng/ml) on θ-Burst (2x3) induced LTP in slices taken from 36-38 week-old rats and 70-80 week-old rats
112 Figure 4.3.9 Effect of Trk-Fc (2 µg/ml) on θ-Burst (3x3) induced LTP in
slices taken from 38-38 week-old rats
114 Figure 4.3.10 Effect of the K252a (200 nM) on θ-Burst (3x3) induced
LTP in slices taken from 38-38 week-old rats
115 Chapter 4.4
Figure 4.4.1 Morphologic analysis of cells 123 Figure 4.4.2 Amyloid-beta (Aβ)-induced apoptosis in primary rat
neurons
123 Figure 4.4.3 Apoptosis induced by Amyloid-beta (Aβ) in primary rat
neurons is prevented by BDNF and CGS 21680
124 Figure 4.4.4 SCH 58261 reverts the neurprotection induced by BDNF 125 Figure 4.4.5 Processing of caspase-3 in primary rat neurons in the
absence of Amylod-beta
127 Figure 4.4.6 Amyloid-beta induced processing of caspase-3 in primary
rat neurons
128 Figure 4.4.7 Amyloid-beta induced processing of caspase-3 in primary
rat neurons, which is reduced by BDNF in the presence of adenosine A2A receptors activation
129 Figure 4.4.8 Activity of caspase-3 in primary rat neurons 130 Figure 4.4.9 Amyloid-beta induced enhancement of caspase-3 activity
in primary rat neurons is reduced by by BDNF in the presence of adenosine A2A receptors activation
131
5. Conclusions Figure 5.1 Cross talk between signalling pathways of adenosine A2A
receptors and TrkB receptors
139

16

17
The scientific content of the present thesis has been included in the publication of the
following original articles:
I. Diógenes M.J., Fernandes C.C., Sebastião A.M., Ribeiro J.A. (2004)
Activation of adenosine A2A receptor facilitates BDNF modultion of synaptic
transmission in hippocampal slices. J. Neurosci. 24, 2905-2913.
II. Diógenes M.J., Assaife-Lopes N., Pinto-Duarte A., Sebastião A.M., Ribeiro
J.A. (2007) Influence of age on BDNF modulation of hippocampal synaptic
transmission: interplay with adenosine A2A receptors. Hippocampus. 17, 577-
85.
The following chapters of this thesis are in preparation to be submitted as
manuscripts to international journals:
Chapter 4.3 “Influence of age on the BDNF modulation of long-term potentiation:
interplay with adenosine A2A receptors.”
Chapter 4.4 “Influence of the activation of adenosine A2A receptors on the
protective BDNF action against apoptosis induced by amyloid beta peptide.”
Other papers where the author of this thesis participate during her doctoral studies:
Pousinha P.A., Diógenes M.J., Ribeiro J.A., Sebastião A.M. (2006) Triggering of
BDNF facilitatory action on neuromuscular transmission by adenosine A2A
receptors. Neuroscience Letters 404, 143–147
Fontinha B.M., Diógenes M.J., Ribeiro J.A., Sebastião A.M. (2007) Enhancement
of long-term potentiation by BDNF requires adenosine A2A receptor activation by
endogenous adenosine. Neuropharmacology 54,924-933.

18

19
1. INTRODUCTION 1.1 NEUROTROPHINS
In 1953, Rita Levi-Montalcini and Victor Hamburger discovered that a mouse sarcoma
tumor implanted close to the spinal cord of developing chicks in ovo secreted a
soluble factor that induced the hypertrophy and fiber outgrowth of sympathetic
neurons. Later, this factor was isolated and named as nerve growth factor (NGF)
(Levi-Montalcini and Cohen, 1956). The discovery of this molecule opened a new field
in neurobiology research. After NGF identification, Barde and collaborators (1982)
isolated a different neurotrophin (NT), from a pig brain, designated as brain-derived
neurotrophic factor (BDNF). Since then other members of the family of NTs have
been identified in mammals namely, NT-3 and NT-4, and in fish NT-6 e NT-7 (Gotz et
al., 1994; Lai et al., 1998).
1.1.1 Neurotrophin synthesis All NTs are generated first as precursor proteins, pre-pro-neurotrophins
(approximately 240-260 amino acids long, Figure 1.1.1), which then are cleaved
intracellularly to mature proteins of 118-120 amino acids. The pre-mRNA sequence
directs the synthesis of the nascent protein in the endoplasmic reticulum (ER)
attached ribosomes, leading to the sequestration of the newly formed polypeptide
chain into the ER (see Lessmann et al., 2003). The signal peptide is cleaved off
immediately after sequestration in the ER. The resulting pro-neurotrophins can

20
spontaneously form non-covalently-linked homodimers in the ER. The pro-
neurotrophins in the ER then transit the Golgi apparatus most likely via intermediate
non-clathrin-coated transport vesicles and finally accumulate in the membrane stacks
of the trans-golgi network (TGN). There are three fates for intracellular pro-
neurotrophins: 1) intracellular cleavage followed by secretion, 2) secretion followed by
extracellular cleavage, or 3) secretion without subsequent cleavage. All of these
products serve as signalling molecules (see Lee et al., 2001).
Two different types of secretory vesicles may be generated (Figure 1.1.2),
filled with one or a combination of different neuropeptides. Thus, BDNF release can
occur through two different pathways: 1) a regulated pathway, where neurotrophins
are secreted in response to a stimuli or 2) a constitutive pathway, where these
molecules are spontaneously secreted. The secretory granules of the constitutive
pathway are smaller (50-100 nm in diameter) and independent of intracellular Ca2+
concentration elevation. Vesicles can fuse with the plasma membrane to release their
content in the absence of any specific triggering mechanisms. In the regulated
pathway, vesicles are large (100-300 nm in diameter) and their release depends on
intracellular Ca2+ concentration elevation. Secretion of neurotrophins can be regulated
by neuronal activity, potassium, and glutamate; this release is also dependent on the
stimulus-frequency (Lim et al., 2003). Interestingly, constitutive and regulated
secretion of BDNF can coexist in the same cell (see Lessman et al., 2003)
1.1.2 Neurotrophin receptors
Neurotrophins function by activating two distinct classes of transmembrane receptors:
the p75 neurotrophin receptor (p75NTR) and the Trk family of receptor tyrosine
kinases that includes TrkA, TrkB and TrkC. Unlike the non-selective p75NTR, which

21
has a similar affinity for all neurotrophins, each Trk receptor selectively binds a
different neurotrophin.
Figure 1.1.1- Structure of the rat BDNF gene and protein. (A) Each of the four 5’-exons (I–IV) includes its own promotor, and is combined with the 3’exon (V) to yield an mRNA (B) coding for pre-pro-BDNF. The short white stretch in exon V represents an alternative splice site, giving rise to two differentially spliced mRNA variants for each of the four transcripts. Each of these mRNAs is expressed in a tissue-specific and developmentally regulated manner. The gray portion in exon V codes for the pre-pro-BDNF protein. (C) Primary structure and sequence of the BDNF protein. The pre-sequence (18 aa, black) is cleaved off immediately after sequestration of the nascent protein into the ER. The mature BDNF protein (black) is excised from the pro-BDNF precursor by virtue of specific protein convertases residing either in the TGN or in immature secretory granules. (Adapted from Lessmann et al., 2003.)
A
B
C

22
Figure 1.1.2- Diagram illustrating the pathways of BDNF synthesis and secretion. ER represents the endoplasmic reticulum and TGN is the trans-golgi network. (Adapted from Lessmann et al., 2003.)
1.1.2.1 p75 receptor
p75NTR is a member of the tumor necrosis receptor family with an extracellular
domain that includes four cysteine-rich motifs, a single transmembrane domain and a
cytoplasmic domain, that includes a ‘death’ domain as represented in Figure 1.1.3
(Liepinsh et al., 1997; He and Garcia 2004).
All neurotrophins, including the pro-neurotrophins, can bind to p75NTR
(Figure1.1.3) (see Lu et al., 2005). This receptor can interact with several proteins
and forms multimeric receptor complexes that transmit important signals for regulating
neuronal survival and differentiation as well as synaptic plasticity (see e.g. Reichardt,
2006). P75NTR also regulates the responsiveness of Trk receptors to neurotrophins.
The presence of p75NTR enhances the specificity of TrkA and TrkB for their primary
ligands, NGF and BDNF, respectively (see Reichardt, 2006).

23
1.1.2.2 Trk receptors
The name TrK, from tropomyosin-related kinase, derives from the oncogene that
resulted in its discovery (Barbacid, 1991). The oncogene was discovered in a
carcinoma and it was found to consist in the first seven of eight exons of nonmuscle
tropomyosin fused to the transmembrane and cytoplasmic domains of a novel
tyrosine kinase. Consequently, the protooncogene was named tropomyosin-related
kinase (Trk), commonly referred as TrkA. The TrkB and TrkC genes were identified
because of their homology with TrkA. Seventeen years ago, it was demonstrated that
NGF binds to TrkA and that it activates the tyrosine kinase activity of the receptor
(Kaplan et al., 1991; Klein et al., 1990). Subsequently, TrkB was identified as the
receptor for BDNF and NT-4, and TrkC as the receptor for NT-3 (see for review
Huang and Reichardt, 2001; Bibel and Barde, 2000). Although NT-4 and BDNF bind
to the same receptor, several observations indicate that the biological effects
mediated by these neurotrophins are not identical, which may mean that neurons can
discriminate between these two molecules (see e.g. Lewin and Barde, 1996). The
extracellular domain of each Trk receptor consists of a cysteine-rich cluster, followed
by three leucine-rich repeats, another cysteine-rich cluster and two immunoglobulin-
like domains. Each receptor terminates with a cytoplasmic domain consisting of a
tyrosine kinase domain surrounded by several tyrosines that serve as
phosphorylation-dependent docking sites for cytoplasmic adaptors and enzymes.
The major site at which neurotrophins interact with these receptors is in the
membrane-proximal immunoglobulin-like domain (Figure 1.1.3).
Splicing generates isoforms of TrkB and TrkC that include comparatively
short cytoplasmic motifs without a tyrosine kinase domain; these receptors are
designated as truncated receptors. Different TrkB isoforms can be generated: T1, T2
and T-Shc. Expression of these non-kinase-containing isoforms has been shown to
inhibit productive dimerization of kinase-containing Trk receptors, thereby inhibiting

24
responses to neurotrophins (Eide et al. 1996). Moreover, recent data indicates that
some truncated isoforms help to regulate the expression of full-length TrkB receptors
(Haapasalo et al., 2002).
Figure 1.1.3- Representation of the domain structures of the Trk and p75 receptors. Extracellular domains of cysteine rich clusters (C), leucine-rich repeats (LRR) and immunoglobin-like domains (Ig) are illustrated. Trk and p75 have one transmembrane domain and one cytoplasmic tyrosine kinase domain. In both TrkA and TrkB, the Ig2 is the major ligand-binding interface. The p75 receptor is depicted on the far left. The extracellular domain of this receptor has four cysteine-rich motifs (CR) in which CR2 and CR3 are involved in the neurotrophin-binding interaction. (Adapted from Reichardt, 2006.)

25
1.1.2.3 Trk-mediated signalling pathways
The Trk receptors are activated specifically by the mature and not by the pro-forms of
the neurotrophin gene products (Lee et al., 2001). Binding of mature neurotrophins to
their specific Trk receptors induces dimerization and autophosphorylation at specific
tyrosine residues within intracellular domains. The generation of phosphotyrosine
residues in turn catalyses the formation of large signalling complexes. (Segal and
Greenberg, 1996). Phosphorylation at Y515 (in the case of TrkB receptors) creates
the binding site for Shc (Src homology 2/α-collagen-related protein), whereas
phosphorylation at Y816 (in case of TrkB receptors) forms the adaptor site for
Phospholipase C (PLC)γ as represented in Figure 1.1.4. The major signalling
pathways activated by the Trk receptors are; (1) phosphatydilinositol-3 kinase (PI3K);
(2) Ras-MAPK pathway and (3) PLCγ pathway, and their downstream effectors.
These include PI3K stimulation of Akt kinase, Ras stimulation of mitogen-activated
protein MAPK cascades and PLC-γ dependent generation of inositol 1,4,5-
triphosphate (IP3) and diacylglycerol (DAG), which results in mobilization of Ca2+
stores and activation of Ca2+ and DAG-regulated protein kinase (Segal and
Greenberg, 1996). These intracellular signalling pathways modulate gene expression
in a cell type-specific manner and are responsible for most of the effects of
neurotrophins related to neuronal growth, survival and differentiation, along with
synaptic transmission and plasticity.
Neurotrophins and activated Trk receptors are transported together in
endocytotic vesicles (see e.g. Lessman et al., 2003). Internalization and transport of
Trk receptors serve two functions: 1) to bring activated Trk receptors into proximity of
cell compartments, such a the nucleus, where signalling is required to enable specific
cellular responses, such as gene transcription and 2) to transport activated Trk
receptors to membrane compartments, where signalling effectors are concentrated
(see Reichardt, 2006). The classic retrograde route is destined to exert the long-

26
lasting effects of neurotrophins, which are, for example, mediated by new gene
transcription; but, there is also an additional anterograde targeting that occurs in the
axon towards synaptic terminals which is involved in the fast actions of neurotrophins
(see e.g. Lessmann et al., 2003). Mechanisms underlying the fast action of
neurotrophins include intracellular Ca2+ signalling, neuronal excitation, augmentation
of synaptic excitation by modulation of N-methyl-D-aspartate (NMDA) receptor activity
and control of synaptic inhibition through the regulation of the K+/Cl- cotransporter
KCC2. The fastest action of brain-derived neurotrophic factor and neurotrophin-4/5
occurs within milliseconds, and involves activation of TrkB and the opening of the Na+
channel Nav1.9. Through these rapid actions, neurotrophins orchestrate neuronal
activity, modulate synaptic transmission and produce instructive signals for the
induction of long-term changes in the efficacy of synaptic transmission (see
Kovalchuk et al., 2004).

27
Figure 1.1.4- TrK-mediated neurotrophin signalling. The activated signalling pathways mediate effects of neurotrophins on neuronal survival, differentiation, and gene expression as well as acute effects on synaptic transmission and plasticity, as indicated. Abbreviations represent the following: P, phosphorylation; Y, tyrosine residue; PI3K, phosphatidylinositol 3-kinase; AKT, AKT kinase; Ras, small GTP-binding protein Ras; PIP2, phosphatidylinositol 4,5-bisphosphate; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate. (Adapted from Blom and Konnerth, 2005).
P
P P
P
P P P
P
P
P
Shc PI3K AKT
MAPK Ras
1-PI3K pathway
2- Ras pathway
PLCγγγγ
3- PLC pathway
PIP2
DAG
IP3
Survival
Growth Differentiation
Synaptic transmission Plasticity
Ca2+
PKC
BDNF
Cytosol
Nucleus
Gene expression
+
Y515-Shc site
Y816-PLC site

28
1.1.3 Pathophysiological implications of BDNF actions
The first suggestion to use a neurotrophin for therapeutic purposes appeared in the
1980s, when NGF was the only known member of the NT family. This proposal was
based on the knowledge that in the CNS, NGF promotes the survival and function of
cholinergic neurons in the basal forebrain (Fisher et al., 1987). These neurons project
to the hippocampus and are believed to be important in the memory process, which is
specifically affected in patients with Alzheimer’s disease. Thus, in 1993, a group of
investigators from the Institute of Karolinska developed a way to administer NGF
directly to the brain of Alzheimer’s patients (Seiger et al., 1993). This method
consisted in a canule placed in the lateral ventricles connected to an infusion pump,
enabling administration of NGF to patients (Figure 1.1.5). However this route of
administration is troublesome since it requires neurosurgery.
Figure 1.1.5- NGF administration directly to the brain. NGF was delivered through a canule implanted in the lateral ventricle connected to an infunsion pump. (Adapted from Sieger et al., 1993.)

29
The discovery of other members of the neurotrophin family, in particular the
discovery of BDNF and the knowledge of its physiological functions, contributed to the
large number of studies correlating the pathogenesis of human neurodegenerative
disorders with alteration in BDNF actions. These changes can be a result of genetic
polymorphisms in the BDNF gene or in the genes for BDNF receptors (TrKB full
length, truncated, and P75 receptors), alterations in the levels of mRNA for BDNF,
modifications in levels of BDNF protein and changes in BDNF receptor densities.
Regarding modifications in the BDNF gene, mRNA for BDNF and protein
levels, a polymorphism in BDNF gene leading to a different BDNF protein (BDNFmet)
appears to alter susceptibility to neuropsychiatric disorders, such us Alzheimer’s
disease (AD) (Ventriglia et al., 2002), Parkinson’s disease (Momose et al., 2002),
depression (Sen, 2003), and bipolar disorders (Sklar et al., 2002; Neves-Pereira et
al., 2002). In 1991, Phillips and colleagues reported a selective reduction of BDNF
mRNA expression in the hippocampus of individuals with Alzheimer’s desease, which
was later, confirmed by others (Murray et al., 1994). Moreover, a decrease in BDNF
protein levels in the hippocampus has been associated with depression (Russo-
Neustadt et al., 2001; Chen et al., 2001a). Furthermore, a decrease in BDNF protein
levels has been reported in nigrostriatal dopamine regions in the brains of Parkinson’s
patients (Mogi et al., 1999, Parai et al., 1999). A decrease in the expression of the
BDNF protein in the hippocampus of people with dementia exhibiting lewy bodies
(Immamura et al., 2005) in patients with diabetic brain neuropathies (Nitta et al.,
2002) and in the striatum of individuals with Huntington’s disease (see Zuccato and
Cattaneo, 2007) was also reported. There is also evidence that brain BDNF content is
diminished in the cortex and the hippocampus of AD patients (Connor et al., 1997,
Ferrer et al., 1999).
Concerning modifications in TrkB receptors, a study demonstrated that a
mutation in the kinase domain of TrkB, that leads to impaired intracellular signalling is

30
associated with obesity and developmental delay (Yeo et al., 2004). On the other
hand, the expression of different TrkB splice variants is associated with cognitive
capacity, while over-expression of the kinase-containing form enhances memory and
learning in transgenic mice (Koponen et al., 2004); conversely, over-expression of the
TrkBT1 isoform in adult neurons impairs long-term memory (Saarelainen et al., 2000).
Interestingly, increased content of the TrkBT1 isoform and decreased TrkB
expression has been observed in AD patients (Ferrer et al., 1999).
Finally, genetic modification in the gene encoding P75NTR can be involved in
pathology. Polymorphisms in this gene can lead to an increased susceptibility to
depressive disorders (Kunugi et al., 2004).
The regulatory actions of neurotrophic factors in neuronal functions and in
response to neuronal injury, together with the evidence that changes in BDNF
signalling are associated with a wide variety of pathologies, led to the working
hypothesis that the delivery of BDNF to the brain could ameliorate those diseases. A
number of clinical trials involving the use of BDNF for neurodegenerative disorders
such as amyotrophic lateral sclerosis and diabetic neuropathy has been carried out in
the past decade. However, BDNF had minimal beneficial effects and produced side
effects such as pain and gastrointestinal symptoms (Thoenen and Sendtner, 2002).
The lack of promising results can be attributed to a short half-life for BDNF, a non-
controlled delivery of the neurotrophin, lack of controlled levels of BDNF at the site of
action and the presence of endogenous compensative processes that may regulate
BDNF levels. Therefore further basic studies are needed in order to better understand
BDNF actions in neurons, which may help in a better design of future clinical trials.

31
1.2 ADENOSINE
Adenosine (Figure 1.2.1) is a nucleoside formed when N9 of adenine (a purine) is
covalently bound to the C1’ position of ribose.
Adenosine is an ubiquitous homeostatic substance present in all cells, and it
is released from apparently all cells, including neurons and glia (see Ribeiro et al.,
2003). Adenosine plays a major role in the cardiovascular system, in the central
nervous system, in the gastrointestinal tract, in the immune system, in mast cell
degranulation, and asthma, as well as cell growth, proliferation and apoptosis (see
Schultle and Fredholm, 2003).
Figure 1.2.1 – Adenosine chemical structure. Adenosine contains adenine (in the blue square) and ribose (in the red square) attached by a C1’-N9 glycosideic bond.
1.2.1 Adenosine synthesis
Adenosine can be formed by the action of the enzyme endo-5’-nucleotidase on
adenosine monophosphate (AMP) (Phillis and Newsholme, 1979) and by the
hydrolysis of S-adenosylhomocysteine (SAH) (Nagata et al., 1984) catalysed by SAH
hydrolase. The intracellular concentration of adenosine at equilibrium is about 100 nM
(Meghji, 1992). This level is controlled by adenosine kinase (AK), which

32
phosphorylates adenosine to produce AMP, and by adenosine deaminase (ADA),
which catalyzes the formation of inosine (Figure 1.2.2). Adenosine is neither stored
nor released as a classical neurotransmitter since it does not accumulate in synaptic
vesicles, but is instead released from the cytoplasm into extracellular space through a
nucleoside transporter (see e.g. Ribeiro et al., 2003). Adenosine is released from the
presynaptic component, from the activated postsynaptic component or from non-
synaptic regions of neurons (see Cunha, 1997). The extracellular adenosine
concentration in all body fluids is rather constant under basal conditions (30-300 nM)
(see Schultle and Fredholm, 2003).
Figure 1.2.2- Pathways of adenosine production, metabolism and transport Abbreviations are as follows: ADA, adenosine deaminase; AK, adenosine kinase; es, equilibrative-sensitive nucleoside transporters; ei, equilibrative-insensitive nucleoside transporters; SAH, S-adenosyl homocysteine. (Adapted from Latini and Pedata, 2001.)
Equilibrative
nucleoside transporter ei es
ADENOSINE
Adenosine receptors activation

33
1.2.2 Adenosine receptors
Adenosine receptors have been intensively studied, and to the present date, four
different receptors have been cloned and designated as A1, A2A, A2B and A3 receptors.
These receptors are classified as seven transmembrane domain G-protein-coupled
receptors. The original receptor classification was based on the effect on adenosine
binding to the receptor affecting the cyclic AMP (cAMP) levels in different tissues, with
A1 decreasing and A2 increasing cAMP levels (van Calker et al., 1979; Londos et al.,
1980).
The adenosine A1 receptors are highly expressed in brain cortex, cerebellum,
hippocampus, and the dorsal horn of spinal cord (Figure 1.2.3) (see e.g. Fredholm et
al., 2001). These receptors are present at pre-, post- and non-synaptic sites. When
they are located pre-synaptically, they inhibit neurotransmitter release (Dunwiddie and
Hass, 1985) whereas post-synaptically and in neuronal cell bodies they inhibit calcium
influx through voltage-sensitive calcium channels, inhibit NMDA receptor and inhibit
potassium currents, leading to membrane hyperpolarization (reviewed in Fredholm et
al., 2005). A1 receptors are coupled to inhibitory G-proteins (Gi/Go) that lead to an
inhibition of the activity of the enzyme adenylate cyclase (Figure 1.2.4) (see Linden,
2001).
The A2A receptors are highly expressed postsynaptically in the striato-pallidal
GABAergic neurons and olfactory bulb (see e.g. Fredholm et al., 2001); they are also
expressed in hippocampus and cortex (Figure 1.2.3) (see e.g. Ribeiro et al., 2003),
where they have a predominant presynaptic localization (Rebola et al., 2005b).
Adenosine A2A receptors are mostly coupled to stimulatory G-proteins (Gs), which
consequently increase intracellular cAMP (Figure 1.2.4). In striatum, they are also
coupled to Golf (Corvol et al., 2001); and, in the hippocampus, there is evidence that
this receptor can be coupled to Gi/Go (Cunha et al., 1999), in addition to its usual

34
coupling to Gs proteins. Moreover, adenosine A2A receptors overexpressed in COS-7
cells were shown to couple to G15/16, but there is no evidence that this interaction
occurs in vivo (Offermanns and Simon, 1995). Investigating endogenously expressed
adenosine A2A receptors in human endothelial cells, Sexl and collaborators cautiously
proposed signalling via G12/13 proteins without providing direct experimental evidence
for this hypothesis (Sexl et al., 1997). Activation of the adenosine A2A receptors also
induces formation of inositol phosphates in COS-7 cells via pertuxis toxin-insensitive
Gα15 and Gα16 proteins (see Jacobson and Gao, 2006).
The A2B receptors display low levels of expression in the brain (Dixon et al.,
1996), however their expression is noted in astrocytes (van Calker et al., 1979). A2B
receptors are coupled to Gs proteins (Figure 1.2.4).
The A3 receptor is found frequently in peripheral tissues, in particular in mast
cells and testis (Schubert et al., 1994); and, it has apparent intermediate levels of
expression in the human cerebellum and hippocampus (Figure 1.2.3) and low levels
in the rest of the brain (see Fredholm et al., 2001). The affinity of this receptor for
adenosine (Ki=1000 nM) is considerably lower than the adenosine affinity of A1
receptors (Ki=10 nM) and A2A (Ki=30 nM). This receptor is usually coupled to
inhibitory G-proteins (Gi and Go) (Figure 1.2.4)(see Linden, 2001).

35
Figure 1.2.3 Distribution of adenosine receptores (A1, A2A and human A3) in brain regions. High levels of expression are indicated by larger font. (From Ribeiro et al., 2003.)

36
Figure 1.2.4- Adenosine receptor-signalling pathways. Activation of the A1 and A3 adenosine receptors inhibits adenylyl cyclase activity through activation of pertussis toxin-sensitive Gi proteins resulting in increased activity of phospholipase C (PLC). Activation of the A2A and A2B adenosine receptors increases adenylyl cyclase activity through activation of Gs proteins. Activation of the A2A receptor induces formation of inositol phosphates. A2B receptor-induced activation of PLC occurs via Gq proteins. All four subtypes of adenosine receptors can couple to mitogen-activated protein kinase (MAPK), giving them a role in cell growth, survival, death and differentiation. Abbreviations represent the folowing: CREB, cAMP response element binding protein; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PK, protein kinase; PLD, phospholipase D; NF-kB, nuclear factor-kB. (Adapted from Jacobson and Gao, 2006.)
A1
A3
A2A
A2B

37
1.2.3 Pathophysiological implications of adenosine actions
Adenosine has the ability to modulate the release of neurotransmitters and
neuromodulators. The adenosine receptors have a crucial function in the regulation of
the activity of other receptors that affect several biological functions such as
differentiation, synaptic transmission, plasticity and apoptosis. Thus, adenosine
receptors regulate the function of neuropeptide receptors, nicotinic autofacilitatory
receptors, metabotropic glutamate receptors, NMDA receptors and neurotrophic
factor receptors (see e.g Ribeiro et al., 2003).
Adenosine is apparently involved in many functions that can play a role in the
pathology of the nervous system. Therefore, the modifications of extracellular
adenosine levels or the pharmacological or molecular manipulation of adenosine
receptors will interfere with the action of other important molecules that regulate brain
functions. This may prove relevant in the treatment of several diseases, where
activation or inhibition of adenosine receptors through modulation of certain pathways
may change the fate of the diseases.
The pharmacological manipulation of adenosine has been suggested for the
treatment of several health conditions (see Ribeiro et al., 2003). Adenosine functions
as a natural sleep-promoting agent mostly through activation of A1 receptors
(Benington et al., 1995; Porkka-Heiskanen et al., 1997). It was suggested that
adenosine participates in resetting of the circadian clock by manipulation of
behavioral stats (Antle et al., 2001). Thus, it emerges that there exists a potential role
for adenosine-related compounds and of A1 receptor agonists as sleep promoters and
adenosine receptor antagonists as arousal stimulators. In addition, adenosine A1
receptor agonists have anxiolytic activity suggesting that drugs that facilitate
adenosine A1 receptor-mediated actions may be effective for the treatment of anxiety
(Jain et al., 1995; Florio et al., 1998). Adenosine A1 receptor antagonists have also

38
been proposed for the treatment of memory disorders (see Ribeiro et al., 2003).
Caffeine, which is an antagonist of adenosine receptors, has cognitive effects mostly
due to its ability to antagonize adenosine A1 receptors in the hippocampus and cortex,
the brain areas actively involved in cognition. Positive actions of caffeine on
information processing and performance might also be attributed to improvement of
behavioral routines, arousal enhancement and sensorimotor gating (Fredholm et al.,
1999). This is futher supported by the observation that theophylline, an adenosine
receptor antagonist, enhances spatial memory performance only during the light
period, which is the time of sleepiness in rats (Hauber and Bareiß, 2001). However,
some cognitive actions of caffeine were also described in relation to A2A receptors
antagonism (Chen et al., 2007).
In vivo and in vitro studies have demonstrated the neuroprotective role of
adenosine A1 receptor activation. Evaluating the action of hypoxia on synaptic
transmission in hippocampal slices, it has been shown that substances that are
released during hypoxia, such as GABA, acetylcholine, and even glutamate through
metabotropic receptors, may have a neuroprotective role; however, their action is
evident only when activation of adenosine A1 receptors is impaired, leading to the
proposal that adenosine A1 receptors play a pivotal role in response to hypoxia
(Sebastião et al., 2001).
The pharmacological manipulation of adenosine receptors has also been
evaluated in the context of Alzheimer’s disease treatment. Caffeine intake has been
associated with a significantly lower risk for AD (Maia and de Mendonça, 2002). In
recent studies with a model of AD in mice, chronic caffeine protected against
cognitive impairment and resulted in reduced brain levels of amyloid beta protein
(Arendash et al., 2006). The most widely used drugs to treat AD patients increase the
availability of acetylcholine in central cholinergic pathways by inhibiting the enzyme
acetylcholinesterase (Doody et al., 2001). Another strategy to promote cholinergic

39
transmission might be to activate adenosine A2A receptors, which facilitate
acetylcholine release, or to block adenosine A1 receptors, which inhibit acetylcholine
release (see Sebastião and Ribeiro, 1996). Thus, either adenosine A2A receptor
agonists or adenosine A1 receptor antagonists (or the combination of both) might be
useful as cognitive enhancers. However, the adenosine A2A receptor antagonist has
also been studied to improve cognition of AD patients (see Chen et al., 2007).
Furthermore, the pharmacologic manipulation of A2B-adenosine receptor have been
proposed as possible therapeutic agents for Alzheimer’s disease patients (Rosi et al.,
2003). AD is associated with glial activation and increased levels of pro-inflammatory
cytokines. Epidemiological results suggest that anti-inflammatory therapies can slow
the onset of AD (see Rojo et al., 2008). Adenosine, acting at A2A receptors, is an
effective endogenous anti-inflammatory agent that can modulate inflammation both in
the periphery and the brain (Mayne et al., 2001).
Moreover, antagonists of adenosine A2A receptors emerged as potential anti-
parkinsonian agents, based, in part, on the CNS distribution of the A2A receptors. A
key finding in this process is the co-localization and reciprocal antagonistic
interactions between A2A and D2 receptors in the striatum, initially observed in rats
(Ferré et al., 1999), but also described in humans (Díaz-Cabiale et al., 2001). A2A
receptor antagonism not only diminishes Parkinsonian-like muscle rigidity in rats but
also potentiates the effect of L-DOPA (Wardas et al., 2001), which may allow the use
of lower doses of L-DOPA and hence minimize or retard side effects and
tachyphylaxis caused by L-DOPA treatment in Parkinson’s disease.
In Huntington’s disease (HD) both adenosine A1 receptor agonists and
adenosine A2A receptor antagonists appear to exert neuroprotective actions. The
adenosine amine congener (ADAC), an adenosine A1 receptor agonist, attenuates the
striatal lesion, as well as the dystonia, induced in a rat model of HD by the
administration of the mitochondrial toxin 3-nitropropionic acid (3-NP) (Blum et al.,

40
2002). Since A2A receptors are mainly localized on the neurons, which degenerate
early in HD, and given their ability to stimulate glutamate outflow and inflammatory
gliosis, it was hypothesized that A2A receptors could be involved in the pathogenesis
of HD, thus A2A receptors antagonists could be neuroprotective.
In addition other pathologies such as schizophrenia, epilepsy, drug addiction,
pain, and control of ventilation can also be improved through the use of adenosine
receptor agonists and antagonists (see Ribeiro et al., 2003).

41
1.3 HIPPOCAMPUS
Buried deep within the medial temporal lobe of the human brain lies a group of many
million of neurons organized into a network quite different from that found anywhere
else in the nervous system. The hippocampal formation is a brain area consisting of
the dentate gyrus, hippocampus, subiculum, presubiculum, parasubiculum, and
entorhinal cortex (Andersen et al., 2007).
The hippocampus has played a central role in brain investigations. There are
several reasons for the interest in this specific brain area. It has a relatively simple
organization of principal cell layers coupled with the highly-organized laminar
distribution of its inputs. There is also increasing information concerning the
involvement of the hippocampus in several physiological processes, such as, memory
and in pathological processes, such as, epilepsy or AD. In addition, the hippocampus
is a brain area extremely susceptible to aging (see e.g., Smith, 1996), which can
contribute to several age related changes observed in older subjects. Moreover, the
basic layout of cells and fiber pathways is much the same in all mammals (Figure
1.3.1), although the volume of the hipocampus is about 10 times larger in monkeys
than in rats and 100 times larger in humans than in rats. Since the basic hippocampal
architecture is common in mammalian species (Andersen et al., 2007), the study of
hippocampal functions in rat models allows correlation to human hippocampal
processes.
The anatomist Giulio Cesare Aranzi was the first to coin the name
“hippocampus”, undoubtedly because of its similarity to the tropical fish Hippocampus
leria (Figure 1.3.2) (Andersen et al., 2007).
After the advent of microscopy, the hipocampus was deemed even more
impressive, with its characteristic, neatly regimented cellular arrangement. The
entorhinal cortex can be considered the first step in the intrinsic hippocampal circuit

42
because much of the neocortical input reaching the hippocampal formation does so
through the entorhinal cortex. Cells in the superficial layers of entorhinal cortex give
rise to axons that project, among other destinations, to the dentate gyrus and CA3
area. These projections from the entorhinal cortex to the dentate gyrus and CA3
form part of the major hippocampal input pathway called the perforant path. These
pathways are not reciprocal since dentate gyrus and CA3 do not project back to
entorhinal cortex. Neurons of entorhinal cortex also project to the CA1 field and to
the subiculum via the perforant and alvear pathways. Both the CA1 and the
subiculum project back to the deep layers of the entorhinal cortex (see Amaral and
Lavenex, 2007 and Figure 1.3.3).
The granule cells, which are the principal cells of the dentate gyrus, give rise
to axons called mossy fibers that connect with pyramidal cells of the CA3 field of the
hippocampus. However, CA3 cells do not project back to the granule cells. The
pyramidal cells of the CA3 field are in turn the source of major input to the CA1
hippocampal field (the Schaffer collateral axons). Again, CA1 neurons do not project
back to the CA3 field. The CA1 field projects to the subiculum, providing its major
excitatory input. Subiculum does not project back to CA1 field of the hippocampus
(see Amaral and Lavenex, 2007 and Figure 1.3.3).

43
Figure 1.3.1- Hippocampus in rat and human. Although there are some differences in certain regions, the most striking feature is the general similarity of hippocampus sctruture across phylogeny. A- Brain and hippocampus of the rat (adapted from Squire and Kandel, 2000). B- Brain and hippocampus of humans (adapted from Amaral and Lavenex, 2007).
Figure 1.3.2 Human hippocampus dissected free (left) compared to a specimen of Hippocampus leria (right). (Adapted from Andersen et al., 2007.)
A B

44
Figure 1.3.3 The hippocampal formation anatomy. The hippocampus forms a principally uni-directional network with input from the entorhinal cortex that forms connections with the dentate gyrus and CA3 pyramidal neurons via the perforant path. CA3 neurons also receive input from the DG via the mossy fibers. The dendrites of pyramidal cells in the CA1 regions form a thick band (stratum radiatum), where they receive synapses from Shaffer collaterals, the axons of pyramidal cells in the CA3 region (see Lopes da Silva and Arnolds, 1978). CA1 neurons also receive input directly from the perforant path and project axons into the subiculum. These neurons, in turn, send the main hippocampal output back to the entorhinal cortex, forming a loop. Abbreviations: EC, entorhinal cortex; DG, dentate gyrus; Pre, presubiculum; Para, parasubiculum and Sub, subiculum. (Adapted from Amaral and Lavenex , 2007.)

45
2. AIMS
As briefly reviewed in the Introduction, adenosine is a modulator of modulators; the
activation of its specific receptors can modify the action of other molecules interfering
with several biologic functions. In particular, adenosine A2A receptor activation
induces phosphorylation of TrkB receptors (Lee and Chao, 2001), the specific
receptor for the neurotrophin BDNF, which, is lacking in several neurodegenerative
diseases (see introduction 1.1.3). However the clinical use of BDNF is difficult and it
remains necessary to clarify if it can be administrated peripherally. Thus, it is crucial
to identify small molecules that can potentiate BDNF actions in the brain. Adenosine,
through A2A receptor activation, was considered as a good candidate. Therefore, the
first aim of this work was to understand if activation of adenosine A2A receptors
modulates TrkB receptor mediated actions in the hippocampus and to characterize
the possible mechanisms that underlie this cross talk.
Aging is an inevitable life event and the hippocampus is a brain area
extremely susceptible to aging (see e.g. Smith, 1996). Besides functional evidence for
age-related dysfunctions in this area (Lynch, 1998; Barnes, 2003; Gooney et al.,
2004), cellular and molecular mechanisms are also modified. A decrease in the
expression of brain-derived neurotrophic factor (BDNF) has been implicated in
neuronal death occurring with aging, as well as in some neurological disorders (Murer
et al., 2001). It is also known that the expression of the high-affinity receptor for
BDNF, TrkB, as well as TrkB mRNA expression, is decreased in aged rats (Silhol et
al., 2005) and in old humans (Webster et al., 2006). The neuromodulatory action of
adenosine and the expression of its membrane receptors also change with age
(Sebastião et al., 2000, Lopes et al., 1999a; Rebola et al., 2003).

46
Considering that the therapeutic potential for BDNF-based strategies is
greater in aged subjects, that the hippocampus is particularly vulnerable to aging, and
that A2A receptor-mediated actions are more evident in older subjects, it was
considered of interest to investigate how the BDNF actions in the hippocampus
change with age, as well as whether these actions may depend on adenosine A2A
receptors activation.
During normal aging, the brain and in particular the hippocampus suffers
changes that might contribute to age-related memory deficits. On the other hand, the
use of BDNF has been attempted in treating patients with AD, where memory is
dramatically affected. Since, both adenosine and BDNF can modulate long-term
potentiation (LTP), and LTP is considered the neuropsychological correlate of
synaptic plasticity and memory it was considered important to evaluate the influence
of A2A/TrkB receptor cross talk in a model of synaptic plasticity during the aging
process.
Finally, apoptosis is a type of cellular death present in the brain of
Alzheimer’s patients as a result of abnormal amyloid-beta deposition. It is currently
accepted that BDNF, acting through TrkB, protects neurons from apoptosis induced
by numerous agents. Moreover, adenosine can also modulate apoptosis in several
tissues including the brain. Thus, it is of vital importance to understand if the
activation of adenosine A2A receptors modulates the anti-apoptotic neuroprotective
role of BDNF.

47
In summary, the work now reported aims to identify and to understand the
functional consequences of the cross talk between adenosine A2A and TrkB
receptors, having the following specific objectives:
1- To understand whether activation of adenosine A2A receptors can modulate
BDNF action on synaptic transmission.
2- To evaluate age-related changes in synaptic transmission in the density of
A2A and TrkB receptors and in the cross talk between A2A and TrkB receptors.
3- To evaluate the age-related changes that could result from changes in the
cross talk between A2A and TrkB receptors, using an in vitro synaptic plasticity model,
LTP.
4- To evaluate the effect of the activation of A2A receptors on the
neuroprotective action of BDNF against apoptosis.

48

49
3. METHODS
3.1 BIOLOGIC SAMPLE PREPARATIONS
3.1.1 Hippocampal slices
3.1.1.1 Hippocampus
McIlwain and collaborators were pioneers in developing methods for ex vivo CNS
preparations to perform biochemistry studies. In 1957, Li and McIlwain published the
first electrophysiological study performed in cortex slices. In spite of these early
reports, these biological preparations were believed to maintain their normal
physiological properties only after the studies by Yamamoto and McIlwain (1966) and
Richards and McIlwain (1967), showing that hippocampal slices, sectioned
perpendicularly to the long axis of the hippocampus, maintained synaptic activity and
that the evoked responses were similar to those recorded in vivo. Since then, slices of
CNS are commonly used as experimental models in pharmacological, biochemical
and neurophysiological studies. The hippocampus is used extensively in these
techniques because of the unique laminated organization of neuronal pathways in this
area (see e.g. Andersen and Colingridge, 1971 and previous Chapter 1.3). The
arrangement of neurons in this brain structure allows it to be sectioned such that most
of the relevant circuitry is left intact. In this preparation, the cell bodies of the
pyramidal neurons lie in a single packed layer that is easily visualized.
Thus, the regular sequential arrangement of hippocampal neurons that
facilitates electrophysiological studies, the prominence of adenosine and BDNF
receptors in the hippocampus and the impact of aging and neurodegenerative

50
diseases on the hippocampus prompted us to choose this structure to perform the
studies here reported.
3.1.1.2 Hippocampus isolation and slice preparation
Male Wistar rats were decapitated after halothane anesthesia. The skull was exposed
by cutting the skin at the top of the head. The brain was removed as illustrated in
Figure 3.1.1 A,B as already described by others (e.g. Palkovits et al., 1983), and
placed into ice-cold Krebs’ solution (124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 26
mM NaHCO3, 1 mM MgSO4, 2 mM CaCl2, and 10 mM glucose) previously gassed
with 95% O2 and 5% CO2, pH 7.4. The cerebellum was cut off and discarded and the
cerebrum was bisected along the midline, separating the two hemispheres. The
hemispheres were placed with the medial surface facing up, as illustrated in Figure
3.1.1 C. The neocortex was peeled off toward the caudal surface, and the midbrain
was pulled away ventrally by pulling in opposite directions at the location marked by
the arrows, using small spatulas. At this point, the dentate surface of the
hippocampus was revealed. Care was taken not to touch the hippocampus with the
spatulas. The fornix was cut by pushing the point of the spatula into the brain at the
point indicated by the dotted line. Next, one spatula was inserted gently under the
fimbria and then further under the hippocampus (area of insertion indicated by the
large arrows). The hippocampus was flipped out of the brain by lifting and pushing on
the spatula, and then rotating the spatula tip around the long axis of the hippocampus.
Once the hippocampus was isolated (Figure 3.1.1 D), it was trimmed at the line
indicated by the two arrows, and the rest of the brain was pulled away from the
hippocampus. Slices (400 µm thick) were cut perpendicularly to the long axis of the
hippocampus (Figure 3.1.1 E,F) with a McIlwain tissue chopper and allowed to

51
recover for at least one hour in a resting chamber (Figure 3.1.2) in Krebs’ solution at
room temperature.
Figure 3.1.1- Hippocampal slices preparation. (A) Scissor introduction into the foramen magnum to remove the occipital bone. (B) Brain extraction. (C) Separation of the hemispheres and hippocampus isolation. (D) Isolated hippocampus. (E) Orientation used to prepare hippocampal slices for electrophysiological recordings. (F) Hippocampal slices. (Adapted from Palkovits and Brownstein, 1983).
A B C
F E D

52
Figure 3.1.2- Resting chamber for hippocampal slices. 1 is a support with a Teflon net to hold the slices; 2 is the inlet tub through which the gas mixture composed by 95% O2 and 5% CO2 is delivered and 3 represents the place where the support with a Teflon net (1) is located. (Adapted from Harvard apparatus catalog).
3.1.2 Hippocampal homogenates After isolation, the hippocampus was disrupted with a Teflon pestle in 0.32 M
sucrose-Tris pH 7.5 and supplemented with protease inhibitors (Complete; Roche
Applied Science, Mannheim, Germany).
3.1.3 Neuronal cell cultures Primary cultures of rat hippocampal or cortical neurons were prepared from 18- to 19-
day-old fetuses of Wistar rats as previously described (Brewer et al., 1993). Pregnant
rats were anesthetized with halothane and then decapitated. The fetuses were
collected in Hanks’ balanced salt solution (HBSS-1; Invitrogen, Grand Island, NY,
USA) and rapidly decapitated. After removal of meninges and white matter, the brain
1
2 3
1
2 3

53
cortex or hippocampus were collected in HBSS without Ca2+ and Mg2+ (HBSS-2;
Invitrogen, Grand Island, NY, USA). The cortex or hippocampus were then
mechanically fragmented, transferred to HBSS-2 solution containing 0.025% trypsin,
and incubated for 15 minutes at 37°C. Following trypsinization, the cells were washed
twice in HBSS-2 with 10% fetal bovine serum and resuspended in Neurobasal
medium (Invitrogen, Grand Island, NY, USA), supplemented with 0.5 mM L-glutamine,
25 µ M L-glutamic acid, and 2% B-27 supplement (Invitrogen, Grand Island, NY,
USA), and 12 mg/ml gentamicin. The cells were then plated on tissue culture plates
(2 x 106 cells/ml) precoated with poly-D-lysine and maintained at 37°C in a humidified
atmosphere of 5% CO2. On the third day, the medium was removed and replaced by
medium without B-27 supplement until the end of the experiment. All assays were
performed on cells cultured for 5 days. Cells were characterized by
immunofluorescence labelling, confirming that neuronal cultures contained < 5% of
glia.
3.1.3.1 Immunofluorescence characterization of cultured neuronal cells
The media was removed from the culture and cells were rinsed with phosphate
buffered saline (PBS) and the cells were then fixed for 30 minutes in
paraformaldehyde (4% in PBS). Cells were subsequently rinsed with PBS and
permeabilized with 0.2% Triton X-100 in PBS for 15 minutes. After washing with PBS,
blocking with 3% BSA in PBS for 30 minutes was performed. Cells were then
incubated overnight at 4°C with primary antibodies: mouse monoclonal antibody to
glial fibrillary acid protein (GFAP) (1:200; see table 3.3) and/or rabbit polyclonal
antibody to neurofilament (1:200; see table 3.3), with subsequent treatment with
secondary antibodies (anti rabbit IgG AlexaF488 and/or anti mouse IgG AlexaF568;
see table 3.3). Fluorescence was visualized with a 40 x (0.45 NA) using an inverted

54
fluorescence microscope Axiovert 135 TV (Zeiss) as previously described by others
(Rodrigues et al., 2002).
3.1.4 Cytosolic protein isolation Cells were homogenized in buffer A (composed by: 10 mM Tris pH 7.6, 5 mM MgCl,
1.5 mM KAc) supplemented with protease inhibitors and 2 mM 1,4-dithiothreitol
(DTT) as previously described with some modifications (Rodrigues et al., 2002).
Then, cells were centrifuged at 500 g for 10 minutes at 4°C. Supernatant was
recovered and the pellet was again resuspended in buffer A and centrifuged at 500 g
for 10 minutes at 4°C. The supernatants were combined and centrifuged at 3160 g for
10 minutes at 4°C. Pellets were discarded and supernatants containing cytosolic
proteins were rapidly frozen and stored at -20°C until used for experimentation.

55
3.2 DRUGS AND ANTIBODIES 3.2.1 Drugs
Table 3.2- Drugs used in the experimental work.
* Aliquots of stock solutions were kept frozen at –20º C until used. ŧ The concentration of DMSO added to the slices (0.001% v/v) was well bellow the concentration that influences glutamatergic synaptic transmission (0.02% v/v, Tsvyetlynska et al., 2005).
Abbreviation Designation Function Supplier Stock solution
ADA Adenosine deaminase (EC 3.5.4.4) Adenosine deamination promoting enzyme
Roche Diagnostics Corporation (Germany)
AP5 DL-2-amino-5-phosphonopentanoate NMDA receptor antagonist
Tocris (Bistol, UK)
Aβ 25-35 Amyloid beta fragment 25:35 Apoptosis inducer Bachem AG (Bubendorf, Switzerland)
100 mM in water
BDNF Brain-derived neurotrophic factor Neurotrophin Regeneron Pharmaceuticals (Tarrytown, NY)
Supplied in 1 mg/ml solution in 150 mM NaCl, 10 mM Na2PO4 buffer, 0.004% Tween 20*
CGS 21680 4-[2-[[6-Amino-9-(N-ethyl-β-D-ribofuranuronamidosyl)-9H-purin-2-yl]amino]ethyl]benzenepropanoic acid hydrochloride
Adenosine A2A receptor agonist
Tocris (Bistol, UK)
5 mM stock solutions in DMSO*ŧ
DbcAMP 5’-cAMP Tris salt, N6, 2’-o-dibutyryladenosine-3’:5’-cAMP
cAMP analogue Sigma (ST Luis, USA)
5 mM stock solutions in DMSO*ŧ
DPCPX 1,3-Dipropyl-8- cyclopentylxanthine Adenosine A1A receptor antagonist
Tocris (Bistol, UK)
5 mM stock solution in DMSO* ŧ
H-89 N-(2-[p-bromocinnamylamino] ethyl)-5-isoquinolinesulfonamide hydrochloride
Protein kinase A inhibitor
Sigma (ST Luis, USA)
5 mM stock solutions in DMSO*ŧ
ITU 5-iodotubercidin Adenosine kinase inhibitor
Sigma (ST Luis, USA)
5mM stock solution in DMSO*ŧ
[3H]ZM 241385
3H-4-(2-[7-amino-2-(2-furyl)-[1,2,4]triazolo[2,3-a][1,3,5]triazinylamino]ethyl)phenol
Tritiated adenosine A2A receptor antagonist
American Radiolabed Chemicals, Inc. (ST Luis, USA)
Supplied as a 36.5µ M solution in ethanol
K-252a (8R*,9S*,11S*)-(-)-9-hydroxy-9-methoxycarbony-8-methyl-2,3,9,10-tetrahydro-8,11-epoxy-1H,8H,11H-2,7b,11a-triazadibenzo(a,g)cycloocta(cde)trinden-1-one
Inhibitor of tyrosine protein kinase activity
Calbiochem (La Jolla, CA)
1 mM stock solution in DMSO*ŧ
TrkB-Fc Recombinant human TrkB/Fc chimera BDNF scavenger R&D Systems (Minneapolis, USA)
50 µg/ml stock solution in PBS with BSA 0.1% *
SCH 58261 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1 ,2,4-triazolo[1 ,5-c]pyrimidine
Adenosine A2A receptor antagonist
Tocris (Bistol, UK)
5 mM stock solution in DMSO* ŧ
XAC 8-4-[(2-aminoethyl)amino]carbonylmethyloxy phenylxanthine
Adenosine receptor antagonist
RBI (Natick, MA, USA)
ZM 241385 4-(2-[7-Amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol
Adenosine A2A receptor antagonist
Tocris (Bistol, UK)
5 mM stock solutions in DMSO*

56
3.2.2 Antibodies
Table 3.3- Antibodies used in the experimental work.
Description Supplier Dilution Use
Primary antibodies Mouse monoclonal antiboy to Adenosine A2A receptor Upstate Biotechnology
(Lake Placid, NY, USA) 1:500 Western Blotting
Mouse monoclonal antibody to TrkB receptor BD Transduction Laboratories (Mississauga, ON Canada)
1:1000 Western Blotting
Rabbit polyclonal antibody to beta-actin Abcam (Cambridge, UK)
1:10000 Western Blotting
Mouse monoclonal antibody to GFAP Abcam (Cambridge, UK)
1:200 Immunofluorescence
Rabbit polyclonal antibody to neurofilament 200 kD Chemicon (Temecula, CA,USA)
1:200 Immunofluorescence
Rabbit polyclonal antibody to caspase 3 Santa Cruz Biotechnology (Santa Cruz, CA, USA)
1:1000 Western Blotting
Secondary antibodies
Anti-mouse HRP Biorad laboratories (Hercules, CA, USA)
1:5000 Western Blotting
Anti-rabbit HRP Biorad laboratories (Hercules, CA, USA)
1:5000 Western Blotting
Anti-rabbit IgG AlexaF488 Invitrogen (Grand Island, NY, USA)
1:400 Immunofluorescence
Anti-mouse IgG AlexaF568 Invitrogen (Grand Island, NY, USA)
1:400 Immunofluorescence

57
3.3 TECHNIQUES 3.3.1 Microelectrophysiological recordings After 1 hour of functional and energetic recovery, slices were transferred to a
recording chamber for submerged slices and continuously superfused at 3 ml/min
with bathing solution gassed with 5% CO2 and 95% O2 at 31-32°C (Figure 3.3.1). The
drugs were added to this superfusion solution for experiments.
Recordings were obtained with an Axoclamp 2B amplifer and digitized (Axon
Instruments, Foster City, CA). Individual responses were monitored, and averages of
eight consecutive responses were continuously stored on a personal computer with
the LTP program (Anderson and Collingridge, 2001).
Figure 3.3.1- Setup for extra-cellular microelectrophysiology recordings. 1-reference electrode; 2-stimulation electrode; 3-recording electrode and 4-temperature sensor.
1
2
3
4
1
2
3
4

58
Field excitatory post-synaptic potentials (fEPSPs) were recorded (Figure
3.3.2C) through an extracellular microelectrode (4 M NaCl, 2–6 MΩ resistance)
placed in the stratum radiatum of the CA1 area (Figure 3.3.2A). Stimulation
(rectangular 0.1 ms pulses, once every 15 seconds) was delivered through a
concentric electrode placed on the Schaffer collateral-commissural fibers, in the
stratum radiatum near the CA3–CA1 border except as otherwise indicated. The
intensity of stimulus (80–200 µA) was initially adjusted to obtain a large fEPSP slope
with a minimum population spike contamination.
Alteration on synaptic transmission was evaluated as the % change in the
average slope of the fEPSP in relation to the average slope of the fEPSP measured
during the 10 minutes that preceded the addition of drugs as previously described
(Diógenes et al., 2004).
3.3.1.1 LTP induction and quantification
In LTP experiments, stimulation (rectangular 0.1 ms pulses, once every 10 seconds)
was delivered alternatively to two independent pathways through two bipolar
concentric electrodes placed on the Shaffer collateral/commissural fibers in the
stratum radiatum (Figure 3.3.2B). LTP was induced by θ-Burst with two or three trains
of 100 HZ, 3 stimuli, separated by 200 ms (Figure 3.3.2D).
The intensity of the stimulus was never changed during these induction
protocols. LTP was quantified as the % change in the average slope of the fEPSP
taken from 46 to 60 minutes after LTP induction in relation to the average slope of the
fEPSP measured during the 14 minutes that preceded the induction of LTP. In each
individual experiment the same LTP-inducing paradigm was delivered to each
pathway. One hour after LTP induction in one of the pathways, BDNF was added to

59
the superfusion solution and LTP was induced in the second pathway, no less than 30
minutes after BDNF perfusion and only after stability of fEPSP slope values was
observed for at least 10 minutes. The effect of BDNF upon LTP was evaluated by
comparing the magnitude of LTP in the first pathway in the absence of BDNF (control
pathway), with the magnitude of LTP in the second pathway in the presence of BDNF
(test pathway); each pathway was used as control or test on alternate days. To test
the modification of the effect of BDNF upon LTP, the modulatory drugs (agonist and
antagonist of adenosine A2A receptors, or the inhibitor of Trk receptors) were added to
the superperfusing bath at least 30 minutes before induction of LTP in the first
pathway and remained in the bath up to the end of the experiment. BDNF was added,
as usual, 60 minutes after induction of LTP in the first pathway. Thus, modulatory
drugs were present during both LTP-inducing periods, whereas BDNF was only
present during the second induction of LTP. This protocol allows the comparison
between the effect of BDNF upon LTP under different experimental conditions,
keeping the magnitude of LTP under the same drug condition, without BDNF in the
same slice as an internal control. When testing the effect of a drug upon LTP (rather
than the modulation of the BDNF effect on LTP), the drug was added to the bath 30
minutes before induction of LTP in the second pathway and the magnitude of the
resulting LTP was compared with that previously obtained (first pathway) in the
absence of the drug as previously described (Fontinha et al., 2008).

60
Figure 3.3.2- Extracellular recordings in hippocampal slices. Representation of the stimulation of one pathway (A) or two pathways (B) in a hippocampal slice to record field excitatory postsynaptic potentials (fEPSP) (C). Traces obtained after stimulation are composed of the stimulus artifact (1), followed by the presynaptic volley (2) and the fEPSP (3). A schematic representation of the stimulation paradigms used in plasticity experiments is represented in D.
3.3.2 Western blotting
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to
evaluate the level of the receptor TrKB immunolabeling as previously described
(Diógenes et al., 2007). Hippocampi were disrupted in sucrose-tris solution with a
S1
DG
CA3
CA1
S1
S2
DG
CA3
CA1
A- Stimulation of one pathawy
B- Stimulation of two pathawy
1 mV
5 ms
C- Traces recorded
D- θ- burst stimulation paradigms
1
2
3
I I I I I I I I I
I I I I I I
200 ms
3x3
2x3
100 Hz
S1
DG
CA3
CA1
S1
S2
DG
CA3
CA1
S1
S2
DG
CA3
CA1
A- Stimulation of one pathawy
B- Stimulation of two pathawy
1 mV
5 ms
C- Traces recorded
D- θ- burst stimulation paradigms
1
2
3
I I I I I I I I I
I I I I I I
200 ms
3x3
2x3
100 Hz

61
Teflon pestle. Hippocampal proteins (20 µg) were separated on 8% SDS-
polyacrylamide electrophoresis gels, and than transferred onto nitrocellulose
membranes. Immunoblots were incubated with 15% H2O2 for 15 min at room
temperature. After blocking with 5% milk solution, the blots were than incubated
overnight at 4°C with primary mouse monoclonal antibodies recognizing TrkB
receptors, adenosine A2A receptors, caspase-3 or β-actin; and, finally the membranes
were incubated with secondary antibodies conjugated with horseradish peroxidase for
3 hours at room temperature. The proteins of interest were detected using Super
SignalTM substrate (Pierce, Rockford, IL, USA). β-actin was used as a loading control.
Protein concentrations were quantified using the Bio-Rad protein assay according to
Bradford (Bradford, 1976).
3.3.3 Binding assays
[3H]ZM 241385 binding studies were performed as previously described (Palmer et
al., 1995), with some modifications. The experiments were performed with
hippocampal homogenates (90 µg protein per assay). After incubation with 2 U/mL
adenosine deaminase (ADA) for 30 minutes at 37°C, [3H]ZM 241385 binding was
carried out in an incubation solution containing 50 mM Tris-HCl buffer and 10 mM
MgCl2 (pH 7.4) for 60 minutes at room temperature, in a final volume of 200 µL.
Specific binding was calculated by subtraction of the nonspecific binding, defined in
the presence of 2 µM XAC. The reaction was stopped by addition of cold incubation
buffer and vacuum filtration through glass fiber filters (FilterMAT for receptor binding,
Skatron Instruments, Lier, Norway) using a semi-automatic cell harvester from
Skatron Instruments. The samples were transferred to scintillation vials and the
radioactivity of each sample measured using a liquid scintillation analyser (Tri Carb

62
2900TR, Perkin Elmer, Illinois, USA). Membrane protein content was quantified with
the Bio-Rad protein assay according to Bradford (Bradford, 1976).
3.3.4 Apoptosis detection
To induce apoptosis, rat neurons cultured for 4 days, were incubated with amyloid-β
peptide (Aβ) 25-35 (25 µ M) one hour after the administration of all drugs. 24 hours
after the induction of apoptosis by Aβ 25-35, cells were analyzed with the following
assays: hoescht staining for morphologic analysis, caspase activity assay and
caspase-3 cleavage detection as described previously by others (Rodrigues et al.,
2002).
3.3.4.1 Morphologic Analysis
Analysis of nuclear morphology was performed as described previously (Rodrigues et
al., 2002). Cell culture medium was gently removed from the cultured plates at the
end of the incubation period. Cells were fixed with 4% formaldehyde in PBS (pH 7.4)
for 10 minutes at room temperature, incubated with Hoechst dye 33258 (Sigma, St.
Luis, USA) at 5 µ g/mL in PBS for 5 minutes, washed with PBS, and mounted using
an aqueous mounting medium (Sigma, St. Luis, USA). Fluorescence was visualized
at 40x magnification (0.45NA) with an inverted fluorescence microscope Axiovert 135
TV (Zeiss). Fluorescent nuclei were independently scored by different observers and
categorized according to the condensation and staining characteristics of chromatin.
Normal nuclei showed noncondensed chromatin dispersed over the entire nucleus.
Apoptotic nuclei were identified by condensed chromatin, contiguous to the nuclear
membrane, as well as nuclear fragmentation of condensed chromatin. Five random

63
microscopic fields per sample were counted and mean values expressed as the
percentage of apoptotic nuclei in the counted cell population.
3.3.4.2 Pro-caspase-3 cleavage
To determine caspase-3 processing, cytosolic proteins were separated by 15% SDS-
PAGE. Blots were probed with primary rabbit polyclonal antibodies to caspase-3 at a
dilution of 1:1000, and subsequently incubated with horseradish peroxidase–
conjugated secondary anti-rabbit antibody. Finally, membranes were processed for
caspase-3 cleavage detection (32 KDa and 20 KDa) (Rodrigues et al., 2002).
3.3.4.3 Caspase activity assay
Caspase activation was evaluated in cytosolic protein extract as decribed previously
(Solá et al., 2003). Cells were harvested and homogenized in buffer A (previously
described in 3.1.4). General caspase-3-like activity was evaluated by enzymatic
cleavage of the chromophore p-nitroanilide (pNA) from the substrate N-acetyl-Asp-
Glu-Val-Asp (DEVD)-pNA. The proteolytic reaction was preceded in isolation buffer
containing 50 µ g of cytosolic protein and 50 µ M DEVD-pNA. The reaction mixtures
were incubated at 37° C for 1 hour, and the liberation of pNA determined by
measuring absorbance at 405 nm using a 96-well plate reader.

64
3.4 STATISTICS
The values presented are mean ± SEM of n experiments. To test the significance of
the effect of a drug versus control, a paired Student´s t-test was used. When making
comparisons from different set of experiments with control, a one-way variance
analysis (ANOVA) was used, followed by a Dunnett’s test. One-way variance analysis
(ANOVA) followed by a Bonferroni test was used when different sets of experiments
were compared between them. Values of p<0.05 were considered to be statistically
significant.

65
4. RESULTS
4.1 INTERPLAY BETWEEN TrkB AND ADENOSINE A2A RECEPTORS IN INFANT
RATS
4.1.1 Rationale Adenosine and adenosine agonists can induce TrkB phosphorylation through a
mechanism involving the adenosine A2A receptor (Lee and Chao, 2001). These data
together with the finding that adenosine A2A receptors and neurotrophin receptors
have a considerable overlap in their distribution (Lewin and Barde, 1996; Fredholm et
al., 2001), prompted the author to evaluate how endogenous activation of adenosine
A2A receptor could influence the action of BDNF on hippocampal synaptic
transmission. Because the excitatory action of BDNF on synaptic transmission at the
developing neuromuscular junction is facilitated by depolarisation (Boulanger and
Poo, 1999a) and depolarising conditions are able to induce adenosine release
(Pazzagli et al., 1993), it was also investigated how depolarising conditions influence
BDNF actions and how this could be related to adenosine receptor activation. Since
the actions of neurotrophins are expected to be more relevant at young ages, 3-week-
old animals were used.
4.1.2 Pre-depolarisation induced by high K+ facilitates BDNF excitatory action
on hippocampal synaptic transmission through TrkB receptors
As illustrated in Figure 4.1.1A, BDNF (20 ng/ml) when applied alone to the
hippocampal slices did not significantly influence the slope of fEPSP (n=7). Even at a
higher concentration (100 ng/ml), BDNF could not enhance synaptic transmission

66
(n=2) (Figure 4.1.1B). However, when applied to slices that had been shortly
depolarised (Figure 4.1.2A) with a pulse of high-K+ (10 mM) for 2 minutes, 46 minutes
before application of the neurotrophin, BDNF (20 ng/ml) caused a significant increase
in fEPSP slope (41 ± 9.8% increase; n=9; p<0.05). The depolarising pulse of high-K+
(10 mM) for 2 minutes, caused a transient enhancement of fEPSP slope, which
returned to the basal level, within 35 minutes after returning to the normal K+
concentration in the bath; in slices where BDNF was not applied (n=3), the fEPSP
slope remained within those basal levels for at least 60 minutes. To evaluate the type
of receptor involved in the excitatory action of BDNF on synaptic transmission, we
studied the effect of this neurotrophin in the presence of K252a, an inhibitor of Trk
receptors phosphorylation (Berg et al., 1992). This inhibitor was added to the
perfusion solution 30 minutes before the pulse of high-K+ and remained in the bath up
to the end of the experiment. The slices were therefore perfused with K252a (200 nM)
for 76 minutes before BDNF application. K252a (200 nM) was virtually devoid of effect
on synaptic transmission; and, as shown in Figure 4.1.2B, it abolished the excitatory
effect of BDNF (n =3; p<0.05).

67
Figure 4.1.1- The averaged time course of changes in fEPSP slope induced by application of BDNF alone 20 ng/ml (A) or 100 ng/ml (B). Values obtained in individual experiments at time 0 and 60 minutes are shown as a scatter representation in (A). The horizontal bars represent the application of BDNF. All values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 minutes immediately before BDNF application): -0.64 ± 0.05 mV/ms, n=7 (A), -0.70±0.03 mV/ms, n=2 (B).
A
B

68
Figure 4.1.2- Pre-depolarisation induced by high K+ facilitates BDNF excitatory action on hippocampal synaptic transmission through TrkB receptors. (Aa) illustrates the averaged time course of changes in fEPSP slope under pre-depolarisation (n=9). Slices were superfused with BDNF (20 ng/ml, corresponding to approximately 0.8 nM BDNF) 46 minutes after treatment with high-K+ (10 mM) for 2 minutes. In (Ab) are shown traces obtained in a representative experiment in (a); each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. In (B) is shown the averaged time course of the effect of BDNF (20 ng/ml) 46 minutes after treatment with high-K+ (10 mM) for 2 minutes in the presence of an inhibitor of TrkB receptors, K252a (200 nM), which was added to the slices 30 minutes before the pulse of high-K+ (n=3). Values obtained in individual experiments at time 0 and 60 minutes are shown as a scatter representation in (Aa). The arrows represent the beginning of the 2 minutes high-K+ application and the horizontal bars represent the application of the different drugs. All values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 minutes immediately before BDNF application): -0.57 ± 0.03 mV/ms (A), and -0.50 ± 0.10 mV/ms (B). Note that BDNF increased fEPSP slope only in pre-depolarised slices, an action prevented by the treatment with K252a.
Aa
Ab
B

69
4.1.3 Theta Burst stimulation paired with BDNF can elicit synaptic potentiation
The K+ depolarising stimulus could still be effective if applied after BDNF
administration. Indeed, when BDNF (20 ng/ml) was added to the slice at least
30 minutes before the K+ depolarising pulse (10 mM, 2 minutes) and remained in the
bath up to the end of the experiment, an increase (81 ± 2.7%; n= 2) in fEPSP slope
was observed (Figure 4.1.3), which is clearly different from what occurred when the
BDNF (20 ng/ml) was applied without the K+ pulse, or when the K+ pulse was applied
without BDNF (see above). Because the K+ pulse causes a broad depolarisation in all
cells, including glial cells and interneurons, we evaluated whether a focal
depolarisation delivered through the stimulation electrode placed at the synaptic
afferents (Schaffer collaterals) was able to elicit the BDNF effect. Weak theta Burst
stimulation (three Bursts of three pulses each at 100 Hz, delivered 100 ms apart) was
applied to slices that had been previously perfused with BDNF (20 ng/ml) for at least
10 minutes, and fEPSPs were increased by 91 ± 9.5% at 60 minutes after stimulus
(n=3, p<0.05 as compared with values before stimulation) (Figure. 4.1.3B); as
expected (de Mendonça and Ribeiro, 2000), the same stimulation applied in the
absence of BDNF caused a smaller and non-sustained increase in fEPSP slope (20 ±
16.1% increase 60 minutes after stimulation; n=3; p< 0.05).
4.1.4 Adenosine A2A receptors activation facilitates BDNF excitatory action on
synaptic transmission
To evaluate if adenosine A2A receptors activation could influence the action of BDNF
on synaptic transmission, the effect of a selective agonist of the adenosine A2A
receptor CGS 21680 (Jarvis et al., 1989) upon BDNF action was tested. CGS 21680
(10 nM) was added to the slices at least 30 minutes before BDNF application and was

70
virtually devoid of effect on fEPSP slope (Figure 4.1.4C). In the presence of CGS
21680 (10 nM), BDNF increased (Figure 4.1.4D) the slope of fEPSPs by 43 ± 6.7%
(n=6; p<0.05) in slices that had not been predepolarised by the high-K+ pulse.
Blockade of adenosine A2A receptors with the selective antagonist (Poucher et al.,
1995), ZM 241385 (50 nM), which was added 30 min before CGS 21680 (10 nM),
prevented this excitatory effect of BDNF (n =3; p<0.05 as compared with absence of
ZM 241385) (Figure 4.1.4D). Indeed, in the presence of ZM 241385 (50 nM), BDNF
(20 ng/ml) even decreased (26 ± 6.5%), rather than increased, fEPSP slope (Figure
4.1.4B). When added alone, ZM 241385 (50 nM) was virtually devoid of effect on
fEPSP slope (Figure 4.1.4A). To know whether the adenosine A2A receptor could play
a role in the excitatory action of BDNF observed after K+ depolarisation, the influence
of adenosine A2A receptor blockade upon the effect of BDNF was evaluated. In the
presence of the adenosine A2A receptor antagonist, ZM 241385 (50 nM), which was
applied for at least 30 minutes before the K+ pulse, the excitatory action of BDNF (20
ng/ml) was completely prevented (n=3; p<0.05) (Figure 4.1.4B). Under these
conditions BDNF (20 ng/ml) even decreased (17 ± 8.0%) fEPSP slope as it occurred
in the presence of CGS 21680 (10 nM) plus ZM 241385 (50 nM).

71
Figure 4.1.3-Theta-Burst stimulation paired with BDNF can elicit potentiation effect. (A) Shows the averaged time course of changes in fEPSP slope induced by BDNF when the high-K+ pulse was applied in the presence of the neurotrophin. In (Ba) is illustrated the averaged time course of changes in fEPSP slope induced by BDNF (20 ng/ml) after a theta-Burst stimulus (3 Bursts at 5 Hz, each composed of three pulses at 100 Hz) (n=3) () and the changes of fEPSP slope in the same conditions of stimulation but without administration of the neurotrophin (n=3) (). Slices were superfused with BDNF (20 ng/ml) at least 10 min before electrical stimulation. In (Bb) are shown traces obtained in representative experiments in (Ba); each trace is the average of eight consecutive responses obtained immediately before (1) and after (2) theta-Burst stimulation in the absence (n=3) () or in the presence (n=3) () of BDNF, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. The arrow represents the beginning of stimulation and the horizontal bars represent the application of the BDNF. All values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 minutes immediately before stimulation): -0.61 ± 0.07 mV/ms, n=2 (A), -0.70± 0.02 mV/ms, n=3 (Ba,), -0.66 ± 0.01 mV/ms, n=3 (Ba,).
A
Ba
Bb

72
Figure 4.1.4- Adenosine A2A receptors activation facilitates BDNF excitatory action on synaptic transmission. (A) ilustrates the averaged time course of changes in fEPSP slope induced by application of 50 nM ZM 241385 alone (n=3). (B) represents the averaged time course of the effect of BDNF after a depolarising stimulus of potassium in the presence of an A2A adenosine receptor antagonist (ZM 241385). Slices were superfused with BDNF (20ng/ml) 46 min after treatment with high-K+ (10 mM) for 2 min and in the presence of ZM 241385 (50 nM) which was added for at least 30 min before the potassium pulse. In (C) averaged time course of changes in fEPSP slope induced by application of 10 nM CGS 21860 alone (n=3) is ilustrated. In (Da), the averaged time course of the effect of BDNF (20 ng/ml) in the presence of the A2A receptor agonist, CGS 21680 (10 nM) () or in the presence of both CGS 21680 (10 nM) and the A2A receptor antagonist, ZM 241385 (50 nM) () are shown. CGS 21680 was applied at least 30 minutes before BDNF application ( and ) and ZM 241385 was applied 30 minutes before CGS 21680 application (). In (Db) are shown traces obtained in a representative experiment in (Da,), each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. The arrow represents the beginning of the 2 minutes high-K+ application and the horizontal bars represent drug application. All values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 min immediately before ZM 241385 application in (A), before BDNF application in (B and Da), or before CGS 21860 application in (C): -0.54 ± 0.090 mV/ms (A), -0.58 ± 0.06 mV/ms (B), -0.60 ± 0.09 mV/ms (C), -0.61 ± 0.01 mV/ms (Da,) and -0.53 ± 0.14 mV/ms (Db,). Note that activation of adenosine A2A receptors facilitates the excitatory action of BDNF on synaptic transmission, an action prevented by A2A receptor blockade.

73
4.1.5 The excitatory action of BDNF is facilitated by the selective adenosine
kinase inhibitor 5-iodotubercidin
The findings that a brief depolarisation by a pulse of high-K+ (10 mM) and subsequent
application of BDNF resulted in a significant increase in fEPSP slope, together with
previous findings that treatment of slices with high-K+ Krebs’ solution results in an
increase of adenosine release (Pazzagli et al., 1993), prompted the next question:
does an enhancement of the extracellular adenosine levels mimic the action of the
pulse of high-K+? A selective adenosine kinase inhibitor, 5-iodotubercidin (ITU), which
enhances the concentration of endogenous extracellular adenosine in hippocampal
slices (Pak et al., 1994) was used. The application of ITU (100 nM) caused a
decrease in synaptic transmission, which may be attributed to activation of adenosine
A1 receptors (the predominant adenosine receptors in the hippocampus) because it
was prevented by the DPCPX (50 nM), a specific adenosine A1 receptor antagonist
(Lohse et al., 1987), (Figure 4.1.5A).
In the experiments where the action of BDNF in the presence of ITU was
tested, the neurotrophin was applied when the full effect of ITU was achieved, and the
slope values of the fEPSPs recorded under these conditions were taken as 100%. As
shown in Figure 4.1.5B, perfusion of BDNF (20 ng/ml) under these conditions resulted
in a significant increase in fEPSP slope (44 ± 8.7% increase; n =3; p<0.05); this did
not occur when A2A receptors were blocked with ZM 241385 (50 nM) before ITU
application (n=3) (Figure 4.1.5B). When ZM 241385 (50 nM) was added only
50 minutes after ITU, BDNF (20 ng/ml) caused the expected enhancement (51%) of
synaptic transmission (one experiment). This may suggest that A2A receptors are
required to trigger the action of BDNF, but they do not need to remain activated
during the BDNF action to enhance synaptic transmission.

74
4.1.6 The activation of the cAMP–PKA transducing system is a critical step in
the excitatory action of BDNF
Because ITU and the pulse of high K+ mimicked the action of the adenosine A2A
receptor agonist, CGS 21680, in respect to its ability to trigger BDNF actions, and
because the activation of A2A receptors stimulates the formation of cAMP–PKA
transducing system (Fredholm et al., 2001), the next series of experiments were
designed to evaluate if a selective PKA inhibitor H-89 (Chijiwa et al., 1990) could
modify the action of BDNF. H-89 (1 µ M) was added for at least 30 minutes before K+
pulse application (Figure 4.1.6A), CGS 21680 perfusion (Figure 4.1.6B), or ITU
administration (Figure 4.1.6C) and remained in the bath up to the end of the
experiments. In all cases, H-89 abolished (p<0.05) the excitatory action of BDNF on
fEPSPs (n=3 for each experimental condition), but by itself H-89 was virtually devoid
of effect on fEPSPs (Figure 4.1.6D). To further evaluate the involvement of cAMP-
dependent PKA activity on the synaptic action of BDNF, experiments were designed
to test if a membrane-permeable cAMP analog, dibutyryl cAMP (dbcAMP) (Henion et
al., 1967), influences BDNF action. By itself dbcAMP (0.5 mM) was virtually devoid of
effect on fEPSP slope (Figure 4.1.7A), and it was added to the slices at least 30
minutes before BDNF application. In the presence of dbcAMP (0.5 mM), BDNF
caused a significant increase (49 ± 13.6%; n=4; p<0.05) in fEPSP slope (Figure
4.1.7B), an effect fully blocked by H-89 (1 µ M; n=2; p<0.05) (Figure 4.1.7B). In slices
predepolarised by a pulse of high-K+ (10 mM), dbcAMP caused only a small increase
in the fEPSP slope (17 ± 1.4%; n=2; p<0.05 as compared with dbcAMP alone),
suggesting that cAMP-mediated actions are slightly influenced by predepolarisation.

75
Figure 4.1.5- The excitatory action of BDNF is facilitated by a selective adenosine kinase inhibitor, 5-iodotubercidin. (A) Represents the averaged time courses of changes of fEPSP slope in the presence of 5-iodotubercidin (ITU, 100 nM) () and in the presence of both ITU (100 nM) plus DPCPX (50 nM) (), an A1 adenosine receptor agonist. Note that the application of DPCPX inhibited the decrease of synaptic transmission induced by ITU (n=3). (Ba,) shows the averaged time courses of changes in fEPSP slope induced by BDNF (20 ng/ml) in the presence of ITU (100 nM), (n=3) and (Ba,) in the presence of both ITU (100 nM) plus ZM 241385 (50 nM), an A2A adenosine receptor antagonist. The horizontal bars represent drug application. In (Bb) are shown traces obtained in a representative experiment in (Ba,). Each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. All values are mean ± SEM; 100% (averaged fEPSP slopes obtained during 10 minutes immediately before the ITU application in (A) or before BDNF application in (Ba)): -0.65 ± 0.06 mV/ms, n=3 (A,), -0.57 ± 0.03 mV/ms, n=3 (A,), -0.52 ± 0.02 mV/ms, n=3 (Ba,) and -0.50 ± 0.10 mV/ms, n=3 (Ba,).

76
Figure 4.1.6- The activation of the cAMP–PKA transducing system is a critical step for the excitatory action of BDNF. Averaged time course of changes in fEPSP slope induced by BDNF in the absence () and in the presence () of the PKA inhibitor H-89 (1 µM) are represented under the following experimental conditions: (A) after a pre-depolarising pulse of high-K+ (10 mM, 2 min); (B) in the presence of CGS 21680 (10 nM); (C) in the presence of 5-iodotubercidin (ITU; 100 nM). H-89 (1 µM) was added to the slices 30 minutes before the application of the K+ pulse (A), CGS 21680 (B), or ITU (C) and alone was virtually devoid of effect on fEPSP slope, as shown in D. In all panels the horizontal bars represent drug application, and the arrow in A represents the beginning of the 2 minutes high-K+ application. All values are mean ± SEM; 100% (averaged fEPSP slopes obtained at times -10–0): -0.57 ± 0.03 mV/ms, n=9 (A,), -0.48 ± 0.23 mV/ms, n=3 (A,), -0.61 ± 0.01 mV/ms, n=6 (B,), -0.47 ± 0.06 mV/ms, n=3 (B,), -0.50 ± 0.10 mV/ms, n=3 (C,), -0.43 ± 0.17 mV/ms, n=3 (C,), -0.59 ± 0.08 mV/ms, n=3 (D). Note that the PKA inhibition prevented the action of BDNF in all experimental conditions.
A
B
C
D

77
Figure 4.1.7- Presynaptic cAMP mimics the effect of high-K+, ITU, or CGS 21680. A shows the averaged time course of changes in fEPSP slope induced by application of dbcAMP (0.5 mM), and Ba shows the averaged time course of changes in fEPSP slope induced by BDNF (20 ng/ml) in the presence of dbcAMP (0.5 mM), either in the absence () and or in the presence () of the PKA inhibitor H-89 (1 µM). Slices were superfused with dbcAMP 30 minutes before BDNF application. Bb shows traces obtained in a representative experiment in Ba; each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. In all panels the horizontal bar represents the application of the different drugs. All values are mean ± SEM; 100% in A and B (averaged fEPSP slopes obtained at times -10–0: -0.70 ± 0.2, n=2 (A), -0.56 ± 1.60 mV/ms, n=4 (Ba,), and -0.64 ± 0.04 mV/ms, n=2 (Ba,).

78
4.1.7 Discussion
The present results show that the excitatory action of BDNF on hippocampal synaptic
transmission is facilitated by predepolarisation and that this effect is dependent on
adenosine A2A receptor activation. These actions were revealed by using a BDNF
concentration that on its own did not significantly modify the slope of fEPSPs and was
mediated by a receptor of the tyrosine kinase receptor family, because pretreatment
with the Trk receptor inhibitor K252a (Berg et al., 1992), prevented the excitatory
effect of this neurotrophin on fEPSPs. BDNF possesses a greater affinity for TrkB
receptors than for TrkA or TrkC receptors (Lewin and Barde, 1996) and, therefore, the
observed actions of BDNF are probably caused by activation of TrkB receptors, which
are present in the hippocampus (Klein et al., 1990; Lewin and Barde, 1996).
The findings that after the brief K+ depolarising pulse, BDNF could facilitate
synaptic transmission are in accordance with the results of Boulanger and Poo
(1999a) at developing neuromuscular synapses showing that presynaptic
depolarisation is a critical factor in causing the synaptic action of neurotrophins. This
could result from the following mechanisms: (1) the prepulse of K+ triggers secretion
of endogenous neurotrophin that could act synergistically with applied BDNF to
potentiate synapses; (2) potassium treatment could induce the expression of more
TrkB receptors, and (3) treatment with high-K+ induces release of adenosine
(Pazzagli et al., 1993), which could gate the excitatory action of BDNF on synaptic
transmission. The results obtained with the adenosine A2A selective agonist, CGS
21680 and with ITU, a selective inhibitor of adenosine kinase that enhances the
extracellular amount of adenosine (Pak et al., 1994), support this last hypothesis.
Thus, in the presence of CGS 21680, BDNF increased the slope of fEPSP, even in
slices that had not been predepolarised by the high-K+ pulse. Moreover the adenosine

79
A2A receptor antagonist ZM 241385 prevented the excitatory effects of BDNF in
conditions of predepolarisation, as well as in the presence of CGS 21680. Finally, ITU
was also able to unmask the BDNF excitatory action, as did CGS 21680 and the brief
K+ pulse, an action also prevented by the A2A atagonist ZM 241385.
There are two sources of extracellular adenosine: release of adenosine by
facilitated diffusion, through membrane adenosine transporters that are equilibrative
and bidirectional (Gu et al., 1995), and extracellular conversion of released adenine
nucleotides into adenosine through a series of ectoenzymes, the last one in the
cascade and the rate-limiting step for adenosine formation being ecto-5’-nucleotidase
(Zimmermann and Braun, 1999). ATP is co-stored with most neurotransmitters, and
therefore neuronal activity causes the release of ATP and extracellular formation of
adenosine in most brain areas, including the hippocampus (Wieraszko et al., 1989;
Cunha et al., 1996). Depolarisation by K+ predominantly induces the release of
adenosine as such (see Latini and Pedata, 2001). The intracellular adenosine levels
are kept low mainly because of the activity of adenosine kinase (see Arch and
Newsholme, 1978) and when this enzyme is inhibited (e.g., by ITU) there is a marked
increase in the release of adenosine (Pak et al., 1994). One may question why K+-
induced adenosine release did not cause an A1-receptor mediated decrease in
synaptic transmission, as ITU did. This most probably results from the time course of
A1-receptor mediated actions (Sebastião et al., 1990; Lupica et al., 1992): any fast
inhibitory actions, because of adenosine released during the 2 minutes application of
K+, should be masked below the transient excitation induced by depolarisation. In
contrast, activation of A2A receptors for a similar period of time might lead to long
lasting enhancement in intracellular cAMP caused by adenylate cyclase activation
(Fredholm et al., 2001), which could gate BDNF actions (Boulanger and Poo, 1999b).
In line with this possibility are the observations that the PKA inhibitor, H-89, prevented
the excitatory action of BDNF in the presence of K+ pulse, CGS 21680, or ITU.

80
Moreover in the presence of the cAMP analog dbcAMP, BDNF enhanced synaptic
transmission, an effect blocked by H-89.
Figure 4.1.8- Mechanism of the facilitatory action of adenosine A2A receptor activation on the BDNF hippocampal synaptic transmission effects on infant rats. The activation of adenosine A2A receptors by the selective agonist, CGS 21680 or through the increase of endogenous adenosine by the addition of the inhibitor of adenosine kinase, ITU or by the pulse of K+, facilitates the excitatory action of BDNF on hippocampal synaptic transmission. This mechanism is dependent on the transducing system cAMP/ PKA since its inhibition by H-89 blocks the BDNF excitatory action. The addition of an analogue of cAMP, dbcAMP, also facilitates the BDNF excitatory action. K252a, the inhibitor of Trk phosphorylation totally prevents BDNF effect on synaptic transmission.
It is known that cAMP can induce a variety of cellular processes in different
systems, including expression of mRNA for Trk receptors and neurotrophins in
primary astroglial cultures (Condorelli et al., 1994) and recruitment of TrkB receptors
to the plasma membrane of CNS neurons (Meyer-Franke et al., 1998). Cytosolic
cAMP can be positively modulated by depolarisation and synaptic activity (Ferrendelli
et al., 1980), and activation of cAMP signalling has been shown to potentiate
TrKB
Gs
AC
A2A
?
cAMP/PKA
BDNF
ITU
K+
ADO
CGS 21680
ZM 241385
(-)
(-)
(+)
(+)
(+)
(+)
H-89
K252a
(-)
dbcAMP(+)
synaptic transmission
TrKB
Gs
AC
A2A
?
cAMP/PKA
BDNF
ITU
K+
ADO
CGS 21680
ZM 241385
(-)
(-)
(+)
(+)
(+)
(+)
H-89
K252a
(-)
dbcAMP(+)
synaptic transmission

81
facilitatory actions of BDNF at the developing neuromuscular junction (Boulanger and
Poo, 1999b). A cAMP—PKA—CREB-dependent pathway is also involved in the
ability of adenosine A2A receptor agonists to potentiate nerve growth factor-induced
neurite outgrowth in cultured PC12 cells (Chen et al., 2002). The results now
described show that the interactions between neurotrophins and adenosine A2A
receptors can occur in CNS neurons that are not in culture, therefore not at
developing stage. The finding that blockade of adenosine A2A receptors prevents the
action of BDNF after a K+ predepolarising pulse, suggests that the predepolarisation
pulse per se is not an essential requisite to unmask BDNF excitatory effects,
eventually by a K+-induced increase in cAMP levels (Ferrendelli et al., 1980). In
contrast, activation of adenosine A2A receptors either by released adenosine or by a
selective agonist (CGS 21680) appears to be an essential step to trigger BDNF
excitatory action on synaptic transmission.
Clear excitatory actions of BDNF on hippocampal synaptic transmission have
been observed by some authors (Kang and Schuman, 1995, 1996), whereas others
reported minimal or no effects of this neurotrophin in the same brain area (Figurov et
al., 1996; Gottschalk et al., 1998). Whether this discrepancy results from different
levels of endogenous activation of adenosine A2A receptors, awaits further
investigation.
Neurotrophins have been suggested to have an important role in protecting
mature neurons from neuronal atrophy in the degenerating human brain. A decrease
in BDNF levels might be involved in neurodegenerative disorders such as Alzheimer’s
disease, Parkinson’s disease, Huntington’s disease (Connor and Dragunow, 1998), or
diabetic neuropathies (Nitta et al., 2002), making the use of the naturally occurring
neurotrophic factors very promising for treatment of these disorders. However, until
now the pharmacological administration of in vivo BDNF has not been easy. One of
the reasons is that these molecules are unable to cross the blood–brain barrier,

82
making invasive application strategies like intracerebroventricular infusion necessary.
Intravenous administration of BDNF has been attempted, but it involves a complex
molecular reformulation of the neurotrophin (Wu and Pardridge, 1999). The results
described in this chapter open a new prospective to potentiate BDNF actions on CNS
neurons, i.e., by co-activation of a specific type of adenosine receptors, the A2A
receptors. This possibility expands the pathophysiological implications of adenosine
receptor functioning in the brain (Ribeiro et al., 2003) and points toward new
strategies to interfere with neurotrophic factors in the therapeutics of some
neurodegenerative disease.
In summary, the results described in this chapter clearly indicate a way
to enhance the excitatory action of BDNF on synaptic transmission via
activation of adenosine A2A receptors, which can be achieved either by
enhancing adenosine release or by pharmacological manipulation of the A2A
receptors.

83
4.2 INFLUENCE OF AGE ON BDNF MODULATION OF HIPPOCAMPAL SYNAPTIC
TRANSMISSION: INTERPLAY WITH ADENOSINE A2A RECEPTORS
4.2.1 Rationale
As reported in chapter 4.1 (see also Diógenes et al., 2004), adenosine, through A2A
receptor activation, potentiates synaptic actions of brain-derived neurotrophic factor
(BDNF) in the hippocampus of infant (3-4 weeks) rats. Since A2A receptor mediated
actions are more relevant in older than in young rats and since the therapeutic
potential for BDNF-based strategies is greater in old subjects, the next step was
focused upon 1) the synaptic actions of BDNF, 2) the levels of TrkB receptors, and 3)
the levels of adenosine A2A receptors, in the hippocampus of three groups of adult
rats: young adult rats (10-16 weeks), an age where reproductive behavior is fully
established (Havenaar et al., 1993), old adult rats (36-38 weeks), the time where rats
get the third part of life expectancy (Havenaar et al., 1993), and aged rats (70-80
weeks old); a group of infant (3-4 weeks) rats was also used again for comparison.
4.2.2 BDNF facilitates synaptic transmission in young adult rats in a “LTP-like”
process
As previously observed (see chapter 4.1), BDNF (20 ng/ml) did not significantly
influence the slope of fEPSP when applied alone to hippocampal slices from infant
rats (3-4 weeks old) (n=7, Figure 4.2.1A and 4.2.2). However, in hippocampal slices
from young adult (10-16 week-old) rats, BDNF (20 ng/ml) significantly increased
fEPSP slope by 32 ± 5.3% (n=8, P<0.05, Figure 4.2.1B and 4.2.2). This excitatory
action of BDNF (20 ng/ml) was lost (n=9) in old adults (36-38 weeks, Figure 4.2.1C

84
and 4.2.2), though a higher BDNF concentration (100 ng/ml, Figure 4.2.3) increased
synaptic transmission by 18 ± 2.5%, n=5. Finally, in the aged group of rats (70-80
weeks), hippocampal synaptic transmission was enhanced by approximately 18 ±
4.4% (n=5; P<0.05, Figure 4.2.1D and 4.2.2) by BDNF (20 ng/ml).
The excitatory action of BDNF (20ng/ml) on synaptic transmission of young
adult rats (10-16 week-old) can be attributed to Trk receptor activation since it was
prevented (n=3; p<0.05, Figure 4.2.1B) by K252a (200 nM), an inhibitor of Trk
receptors phosphorylation (Berg et al., 1992). K252a (200 nM) was added to the
perfusion solution 30 minutes before BDNF administration and K252a alone was
devoid of effect on synaptic transmission (n=3).
Neurotrophins, and in particular BDNF, modulate long-term potentiation (LTP)
in the CA1 area of the hippocampus by presynaptically enhancing synaptic responses
to tetanic stimulation (Figurov et al., 1996; Gottschalk et al., 1998). N-methyl-D-
aspartate (NMDA) glutamate receptors play a key role in LTP induction but are not
required for LTP maintenance (Bliss and Collingridge, 1993). To evaluate if the
increase of synaptic transmission induced by BDNF in young adult rats was
dependent on NMDA receptor activation we studied the effect of BDNF in the
presence of AP5, an antagonist of NMDA receptors, at a concentration (50 µM)
known to prevent NMDA responses in the hippocampus (e.g. Sebastião et al., 2001).
As shown on Figure 4.2.4A, when AP5 (50 µM) was applied to hippocampal slices 30
minutes before BDNF (20 ng/ml), the neurotrophin failed to increase fEPSP slope
(n=4, p<0.05 as compared with absence of AP5). When AP5 (50 µ M) was added 60
minutes after BDNF (20 ng/ml) perfusion, therefore, after the full effect of BDNF on
synaptic transmission, it did not revert the action of the neurotrophin (n=3, Figure
4.2.4B). Therefore, as it occurs with LTP (Collingridge et al., 1983), NMDA receptors
are required for the induction of the excitatory action of BDNF on synaptic

85
Ba
C
Da
Bb Db
A
transmission but they do not need to remain activated during BDNF action. The AP5
(50 µ M) by it self, did not significantly change synaptic transmission (n=4).
Figure 4.2.1- The action of BDNF on excitatory synaptic transmission varies in rats from different age groups. In A-D are shown the averaged time courses of changes in fEPSP slope induced by application of 20 ng/ml (corresponding to ≈ 0.8 nM) BDNF to hippocampal slices taken from 3-4 week-old rats (A), 10-16 week-old rats (B), 36-38 week-old rats (C) and 70-80 week-old rats (D) in the absence (•) or in the presence () of the inhibitor of Trk phosphorylation, K252a (200 nM), which was applied at least 30 minutes before BDNF superfusion. The horizontal bars represent the application of the BDNF. Averaged fEPSPs (Bb and Db) obtained in a representative experiment in B and D are also shown below the respective panel; each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. In all panels the values are mean ± SEM; 100% represents the averaged fEPSP slopes recorded for 10 minutes immediately before BDNF, and were: -0.64 ± 0.05 mV/ms, n=7 (A), -0.73 ± 0.06 mV/ms, n=8 (B,•), -0.62 ± 0.04 mV/ms, n=3 (B,), -0.62 ± 0.04 mV/ms, n=9 (C) and -0.52 ± 0.05 mV/ms, n=5 (D).

86
Figure 4.2.2- Comparison between the averaged effects of BDNF in rats from different age groups. The ordinates shows the % change of fEPSP slope induced by BDNF (20 ng/ml) 50-60 minutes after its application to hippocampal slices taken from infant (3-4 weeks), young adult (10-16 weeks), old adult (36-38 weeks) and aged (70-80 weeks) rats, as indicated below each bar *p<0.05, **p<0.001 versus infants (one way ANOVA with the Dunnett’s correction).
Infants Young adult Old adult Aged
50
100
150
* **
fEP
SP
slo
pe
(% o
f ch
ang
e)

87
A
B
Figure 4.2.3- A high concentration (100 ng/ml) of BDNF increases synaptic transmission in slices taken from old adult rats. In A the averaged time courses of changes in fEPSP slope induced by application of 100 ng/ml BDNF to hippocampal slices taken from 36-38 week-old rats are shown. Averaged fEPSPs obtained in a representative experiment in A are shown in B; each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. The values in A are mean ± SEM; 100% represents the averaged fEPSP slopes recorded for 10 minutes immediately before BDNF, and were: -0.52 ± 0.05 mV/ms, n=5 (A).

88
A
B
Figure 4.2.4- BDNF facilitates synaptic transmission in young adult rats in a “LTP-like” process. A and B show the averaged time course of changes in fEPSP slope induced by BDNF (20 ng/ml) in hippocampal slices taken from young adult (10-16 week-old) rats; in the experiments shown in (A), the antagonist of NMDA receptors, AP5 (50 µM), was applied at least 30 minutes before BDNF application; whereas in the experiments shown in (B), AP5 was applied 60 minutes after BDNF, as indicated by the horizontal bars. All values are mean ± SEM; 100% (averaged fEPSP slopes recorded for 10 minutes immediately before BDNF: -0.55 ± 0.01 mV/ms, n=4 (A) and -0.52 ± 0.03 mV/ms, n=3 (B). Note that AP5 inhibited the induction but not the maintenance of the effect of BDNF.

89
4.2.3 Adenosine A2A receptors are involved in the excitatory action of BDNF in
the hippocampus of young adult rats and aged rats.
As shown previously (Chapter 4.1), to trigger an excitatory action of BDNF in infant
rats (3-4 weeks) it is necessary to co-activate adenosine A2A receptors. It was
therefore evaluated if the excitatory action of BDNF observed in young rats could also
be under the influence of adenosine A2A receptors. As illustrated in Figure 4.2.5A and
Figure 4.2.6, the A2A receptor selective antagonist, ZM 241385 (50 nM, added 30
minutes before BDNF) prevented the excitatory effect of BDNF (20ng/ml) in
hippocampal slices taken from young adult (10-16 weeks old) rats (n=5; p<0.05 as
compared with the absence of ZM 241385), suggesting that in this group of animals
the action of BDNF upon hippocampal synaptic transmission requires active
adenosine A2A receptors.
In the oldest group of rats (70-80 weeks) the small increase of the fEPSP
induced by application of the neurotrophin alone was also prevented by ZM 241385
(50 nM, n=3, Figure 4.2.5B and Figure 4.2.6, p<0.05 as compared with absence of
ZM 241385), which was applied 30 minutes before BDNF.
Considering that a pre-depolarisation pulse, by inducing adenosine release
and A2A receptor activation, can trigger a facilitatory action on synaptic transmission in
infant (3-4 weeks old) rats (Chapter 4.1), and that pre-depolarisation also triggers
BDNF actions in other preparations (Boulanger and Poo, 1999a; Pousinha et al.,
2006), it was now evaluated whether a similar facilitation of BDNF actions could occur
in the old adult (36-38 weeks) rats. As before, the slices were shortly depolarised by a
pulse of high-K+ (10 mM) for 2 minutes and BDNF was applied 46 minutes after the
pulse. As shown in Figure 4.2.7A, the depolarising pulse of high-K+ caused a transient
enhancement of fEPSP slope, which returned to the basal level, within 35 minutes
after returning to the normal K+ concentration in the bath. When BDNF was applied to

90
the pre-depolarised slices, it enhanced fEPSP slope by 18 ± 2.4% (n=4) (Figure
4.2.7A). This facilitatory action of BDNF upon synaptic transmission in slices from old
adult (36-38 weeks) rats was dependent on A2A receptor co-activation since it was
prevented by the adenosine A2A receptor selective antagonist, ZM 241385 (50 nM,
Figure 4.2.7B) (n= 3; p< 0.05).
In all age groups, ZM 241385 (50 nM), on its own, was virtually devoid of
effect on the EPSPs (n=3-5).

91
Figure 4.2.5- Involvement of adenosine A2A receptors in the excitatory action of BDNF in synaptic transmission. In (A,B) are shown the averaged time course of changes in synaptic transmission induced by application of BDNF (20 ng/ml) to hippocampal slices taken from young adult (10-16 weeks) and aged rats (70-80 weeks) in the absence (•), or the presence of the selective A2A receptor antagonist, ZM 241385 (50 nM) (), which was applied at least 30 minutes before BDNF. The horizontal bars represent time of BDNF application; 100% (averaged fEPSP slopes recorded for 10 minutes immediately before BDNF): -0.73 ± 0.06 mV/ms, n=8 (A,•), -0.51 ± 0.04 mV/ms, n=5 (A,), -0.52 ± 0.05 mV/ms, n=5 (B,•), -0.62 ± 0.08 mV/ms, n=3 (B,). The data in (A,•) is the same as shown in Figure 4.2.1B(•) and the data in (B,•) is the same as in Figure 4.2.1D(•).
A
B

92
Figure 4.2.6- Involvement of adenosine A2A receptors in the excitatory action of BDNF in synaptic transmission. The percentage of change of fEPSP slope (ordinates) induced by BDNF (20 ng/ml) 50-60 minutes after its application to the young adult and aged rats in the absence (filled bars) and presence (open bars) of ZM 241385 (50 nM) is shown. *p<0.05 versus the same age group in the absence of the A2A receptor antagonist.

93
Figure 4.2.7- Pre-depolarisation induced by high K+ facilitates BDNF excitatory action on hippocampal synaptic transmission in slices taken from old adult (36-38 weeks old) rats. In A are shown the averaged time courses of changes in fEPSP slope induced by application of BDNF (20 ng/ml), which was perfused 46 minutes after treatment with high-K+ (10 mM) for 2 minutes. Averaged fEPSPs (Ab) obtained in a representative experiment in A are shown; each trace is the average of eight consecutive responses obtained immediately before (1) and during (2) BDNF application, and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. B shows the averaged time course of the effect of BDNF (20 ng/ml) applied 46 minutes after treatment with high-K+(10 mM) for 2 minutes in the presence of the selective antagonist of adenosine A2A receptor, ZM 241385 (50 nM), which was added to the slices 30 minutes before the pulse of high-K+. In all panels, the arrows represent the beginning of the 2 minutes high-K+ application, and the horizontal bars represent the application of the different drugs. All values are mean ± SEM; 100% (averaged fEPSP slopes recorded for 10 minutes immediately before BDNF): -0.57 ± 0.04 mV/ms, n=4 (A), -0.55 ± 0.02 mV/ms, n=3 (B).
Aa
Ab
B

94
4.2.4 The density of TrkB full length receptors and adenosine A2A receptors is
altered in older rats
To evaluate if the changes in the BDNF excitatory effect on hippocampal slices taken
from animals with different ages could be related to changes in the number of TrkB
full-length receptors, the levels of this receptor in hippocampal homogenates taken
from the same animals used in electrophysiology were assessed by western blot. As
illustrated in Figure 4.2.8A and B, there was a small, though non-significant, increase
in TrkB receptor density in the hippocampus of young adult (10-16 weeks old) rats as
compared with TrkB receptor density in infant (3-4 weeks old, which was taken as
100% reference level) rats. TrkB receptor levels in the hippocampus of old adult (36-
38 weeks old) rats were significantly (p<0.05) reduced, and the decrease was even
more pronounced in the hippocampus from aged (70-80 weeks old) rats.
Since the excitatory action of BDNF on synaptic transmission is dependent on
adenosine A2A receptors activation, the density of A2A adenosine receptors in the
same animals was also evaluated. The existence of commercially available tritiated
ligands for A2A receptors allowed receptor binding assays, which are frequently
preferred for quantitative analysis of receptors. The A2A receptor antagonist [3H]ZM
241385 was used as ligand for A2A receptors (Palmer et al., 1995). Figure 4.2.9
shows the saturation isotherms for the binding of [3H]ZM 241385 to hippocampal
homogenates taken from the same hippocampi as those used in Western blot
analysis of TrkB receptors. It is evident that the specific [3H]ZM41385 binding is
higher in aged (70–80 weeks old) than in young adult (10–16 weeks old) rats, the
difference being more pronounced for higher ligand concentrations. The Bmax values
obtained by nonlinear regression analysis were 28 ± 7.7 fmol mg/protein (n = 5) for
the 10–16-week-old rats, and 67 ± 16 fmol mg/protein (n = 4) for the 70–80-week-old
rats (p<0.05). No significant differences in the Kd values were found (young adult: 1.7

95
± 1.1 nM, n = 5; aged: 2.4 ± 1.2 nM, n = 4, p>0.05) and a ZM 241385 concentration
near the Kd (2 nM) was therefore used in subsequent experiments. Binding of 2 nM
[3H]ZM 241385 was significantly higher in aged (70–80 weeks old, p<0.01) and old
adult (36–38 weeks, p<0.05) rats than in infant (3–4 weeks old) rats (Figure 4.2.9B).
DISCUSSION
Figure 4.2.8- Changes in the density of TrkB full length receptors in different age groups. In (A) are shown the Western blots of TrkB full length (FL) (145 KDa) receptors and for control purposes, β-actin protein (43 KDa), in homogenates of rat hippocampus taken from infant (3 weeks), young adult (10-16 weeks), old adult (36-38 weeks) and aged (70-80 weeks) rats, as indicated above each lane. In (B) are shown the averaged of TrkB FL receptor density (n=5-6). All values are mean ± SEM. 100% was taken as the density of TrkB (B) receptors in infant rats. *p<0.05 and **p<0.01 versus infants (one way ANOVA with Bonferroni correction). The hippocampus homogenates were taken from the same animals that were used in electrophysiology experiments.
A
B

96
Figure 4.2.9- Specific binding to adenosine A2A receptor receptors in the hippocampus of rats from different age groups. In (A) are shown saturation isotherms for the binding of the selective A2A receptor antagonist [3H]ZM 241385 to the hippocampal homogenates of young adult (,n=5 ) and aged (•,n=4) rats. In (B) is shown the comparison between the specific binding of 2 nM [3H]ZM 241385 to the hippocampal homogenates of infant (3-4 weeks), young adult (10-16 weeks), old adults (36-38 weeks) and aged (70-80 weeks) rats (n=3-5). In both panels the ordinates represent the specific binding of [3H]ZM 241385 obtained upon subtraction of the non-specific binding (determined in the presence of 2 µM XAC) from total binding. The specific binding of 2 nM [3H]ZM 241385 corresponded to 18 ± 1.9% of total binding for all age groups; no appreciable differences in the proportion of specific binding over total binding occurred among the different groups. All values are mean ± SEM. *p<0.05 versus infant rats; **p<0.01 versus infant rats; §p<0.01 versus young adults (one way ANOVA with Bonferroni correction). The hippocampus homogenates were taken from the same animals used in electrophysiology experiments, and from the same hippocampi as those used in western blot analysis.
A
B

97
4.2.5 Discussion
The present results show that the effect of BDNF on rat hippocampal synaptic
transmission changes according to the age of the animals and that this could be
related to changes induced by age in the density of TrkB and of adenosine A2A
receptors.
BDNF when applied alone to hippocampal slices from infant rats (3-4 weeks)
does not affect synaptic transmission, but it is able to do so when adenosine A2A
receptors are exogenously activated or when extracellular adenosine levels are
increased (see Chapter 4.1). This ability of A2A receptors to trigger a BDNF action in
infant rats requires cAMP formation and protein kinase A (PKA) activity (Chapter 4.1).
Moreover, in A2A receptor knockout mice BDNF is unable to facilitate hippocampal
synaptic transmission, reinforcing the idea that BDNF-induced modulation of synaptic
transmission requires functional adenosine A2A receptors (Tebano et al., 2008). These
new results show that when BDNF is applied to hippocampal slices taken from young
adult rats (10-16 weeks), it induces a significant increase in fEPSPs. This effect is
mediated by a tyrosine kinase receptor, since it was prevented by K252a, and it also
requires co-activation of adenosine A2A receptors, since it was abolished in the
presence of the selective A2A receptor antagonist, ZM 241385. Therefore, in spite of
not being necessary to exogenously activate A2A receptors to trigger a BDNF action in
young adult rats, A2A receptors are still required for the effect of BDNF on synaptic
transmission.
BDNF enhances glutamate release (Canas et al., 2004) and increases
activation (Levine et al., 1998) and phosphorylation of glutamate NMDA receptors
(Suen et al., 1997) in the hippocampus. By operating pre- and postsynaptic
mechanisms, BDNF also facilitates LTP induction (Figurov et al., 1996; Gottschalk et
al., 1998; Kang and Schuman, 1995; Xu et al., 2000). In the present work it was

98
observed that induction (but not the maintenance) of the excitatory action of BDNF
upon synaptic transmission is abolished by the NMDA receptor antagonist, AP5. It
thus appears that BDNF can by itself induce a long-lasting enhancement of synaptic
transmission that does not require high-frequency stimulation but involves, as LTP
does, NMDA receptor activation. This BDNF-induced “LTP-like” phenomenon is
clearly observed in young adult rats (10-16 weeks old) but disappears in old adult
rats. At this age (36-38 weeks old) animals have clearly lower density of TrkB
receptors and this may account for the loss of effect of BDNF on synaptic
transmission. Interestingly, after a pre-depolarising potassium pulse, a condition
known to increase extracellular adenosine levels (Pazzagli et al., 1993), the
neurotrophin increased synaptic transmission in an adenosine A2A receptor-
dependent manner, suggesting that in this age group an increase in the A2A receptor-
mediated adenosinergic tonus is able to trigger a BDNF action upon hippocampal
synaptic transmission.
A decrease in the amount of TrkB receptor with aging fits to previously
reported data (Silhol et al., 2005). The decrease was evident in old adult animals but
was much more pronounced in aged animals. Surprisingly, BDNF could enhance
synaptic transmission in aged (70-80 weeks old) animals. In this age group we could
observe: 1) a marked increase in Bmax value for A2A receptor binding, indicating
higher density in A2A receptors and 2) that the effect of BDNF on synaptic
transmission requires A2A receptors since it was prevented in the presence of the
selective antagonist of these receptors, ZM 241385. Taken together, these data
indicate that the ability of BDNF to enhance synaptic transmission in aged rats is
probably due to an increase in A2A receptor levels that partially compensate for the
marked loss of TrkB receptors. It is possible that critical levels of A2A and TrkB
receptors need to be present together to allow the facilitatory effect of BDNF on
synaptic transmission. However, concomitant changes in the transducing pathways

99
operated by A2A (Rebola et al., 2003) or TrkB receptors (Gooney et al., 2004), or in
the levels of endogenous extracellular adenosine (Cunha et al., 2001) or BDNF
(Narisawa-Saito and Nawa, 1996; Katoh-Semba et al., 1997; Croll et al., 1998) may
also account for a change in BDNF effects throughout age. Indeed, the changes in
TrkB and A2A receptors density in the hippocampus from infant (3-4 weeks old)
toyoung adult (10-16 weeks old) animals are very mild and probably do not account
entirely for the quantitative changes of the BDNF effect in the two age groups.
Unfortunately, the complexity of the transducing pathways operated by TrkB receptors
(Huang and Reihardt, 2003), or even A2A receptors (e.g. Sebastião and Ribeiro, 2000)
makes it difficult to directly address this issue.
The tonic action of adenosine results from a balance between inhibitory A1
and facilitatory A2A receptor-mediated actions. It is known that in the limbic cortex
there is a lower density of A1 receptors and a higher density of A2A receptors (Cunha
et al., 1995). These changes result in a decreased ability of A1 receptor agonists to
inhibit (Sebastião et al., 2000) and an enhanced efficiency of A2A receptors to
facilitate (Rebola et al., 2003) hippocampal synaptic transmission. Furthermore, the
A1/A2A cross talk also changes with age (Lopes et al., 1999b). How this A1/A2A
adenosine receptors interplay contributes to the observed age related changes in the
facilitatory effect of BDNF on synaptic transmission deserves further investigation.
After the pioneer publication by Kang and Schuman (1995) showing that
BDNF enhances synaptic transmission in the rat hippocampus, several papers
reported the absence of an effect of the neurotrophin on evoked EPSPs induced by
low frequency stimulation (e.g. Figurov et al., 1996; Gottschalk et al., 1998; Tanaka et
al., 1997). Methodological differences (e.g. flow rate, temperature, type of chamber)
may not be the sole reason for this discrepancy. Thus, Kang and Schumman (1995)
used young adult (≅ 50 days old) rats whereas the other studies used hippocampus
from developing (12-18 postnatal days old) rats. From the data now presented, where

100
the effect of BDNF was compared in the same experimental conditions in animals
from different ages, it is clear that at least two physiological variables that are not fully
independent may contribute to the variability of the effect of BDNF on fast excitatory
transmission in the hippocampus: age and adenosine through A2A receptor activation.
In summary, the present results show, for the first time, a relationship
between age-related changes in the density of TrkB receptors and adenosine
A2A receptors BDNF-induced enhancement of synaptic transmission in the
hippocampus. This interplay should be taken into consideration whenever
evaluating BDNF actions in nerve cells, and may prove relevant in the design of
BDNF-based therapeutic strategies.

101
4.3 INFLUENCE OF AGE ON THE BDNF MODULATION OF LONG TERM
POTENTIATION: INTERPLAY WITH ADENOSINE A2A RECEPTORS
4.3.1 Rationale
During normal aging and in some pathologies such as Alzheirmer’s disease, the
whole brain, and in particular the hippocampus, suffers changes that might contribute
to memory deficits. It is commonly accepted that the neurophysiological basis for
learning and memory involve modifications in the efficiency of synapses. Using
specific patterns of stimulation, it is possible to experimentally induce long-term
modifications in synaptic strength, for instance long-term potentiation (LTP) (Bliss and
Collingridge, 1993). Neurotrophins have a key role in LTP, in particular through
activation of TrkB receptors (Poo, 2001).
Besides functional evidence of dysfunctions in the hippocampus (Lynch,
1998; Barnes, 2003; Gooney et al., 2004), cellular and molecular mechanisms are
also modified during aging. A decrease in the expression of BDNF high-affinity
receptor, TrkB, in rats (Silhol et al., 2005, Diógenes et al., 2007, chapter 4.2) and also
in humans (Webster et al., 2006) has been reported. Furthermore, the
neuromodulatory action of adenosine and the expression of its receptors also change
with age. Thus, the inhibitory adenosine A1 receptor is less efficient in aged animals
(Sebastião et al., 2000), whereas the functioning and number of the adenosine A2A
receptors in the hippocampus is higher in aged rats (Lopes et al., 1999a; Rebola et
al., 2003, Diógenes et al., 2007, chapter 4.2 of this thesis).
Considering the relevance of the action of BDNF upon synaptic plasticity
phenomena and considering the change of TrkB and A2A receptors density upon
aging, the next step was developed to study the influence of BDNF upon LTP during
aging and the influence of adenosine A2A receptors on the effect of BDNF.

102
4.3.2 BDNF effect on LTP induced by θ-Burst stimulation (3 Bursts, 100Hz, 3
stimuli, separated by 200 ms).
As illustrated in Figure 4.3.1, when a θ-Burst (3 Burst of 3 stimuli each, 3x3) was
applied to hippocampal slices taken from 4 week-old rats, a very small LTP was
observed (12 ± 1.0% increasing in fEPSP slope, n=4). In contrast, when BDNF (20
ng/ml) was present, the same θ-Burst stimuli elicited a robust LTP (50 ± 5.1 %
increase in fEPSP slope, n=4, p<0.05). This facilitation of LTP induced by BDNF was
totally abolished when the adenosine A2A receptors were antagonized by the selective
antagonist, SCH 58261 (100 nM) (n=3, p<0.05, Figure 4.3.2). In these experiments
SCH 58261 was added to the superperfusing bath at least 30 minutes before
induction of LTP in the first pathway and remained in the bath up to the end of the
experiment, whereas BDNF was added 60 minutes after induction of LTP in the first
pathway and at least 30 minutes before induction of LTP in the second pathway. No
significant differences in the magnitude of the first LTP were found in the presence or
in the absence of SCH 58261 (100 nM, compare first and third columns of the Figure
4.3.2).

103
Aa Ab
Figure 4.3.1- Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 4 week-old rats. Aa shows averaged time courses changes of fEPSP slope induced by a θ-Burst (3x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL). The neurotrophin () was applied 60 minutes after induction of LTP in the first pathway (). The ordinates represents normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation (-0.61 ± 0.06 mV/ms, n= 4 () and -0.52 ± 0.06 mV/ms, n= 4 ()) and the abscissa represents the time of every recordings. The recordings obtained in representative experiments are shown in Ab; each recording is the average of eight consecutive responses obtained before (1) and 46-60 minutes after (2) LTP induction in the presence (upper panel) or in the absence of BDNF (lower panel), and is composed of the stimulus artifact, followed by the presynaptic volley and the fEPSP. All values are mean ± SEM.
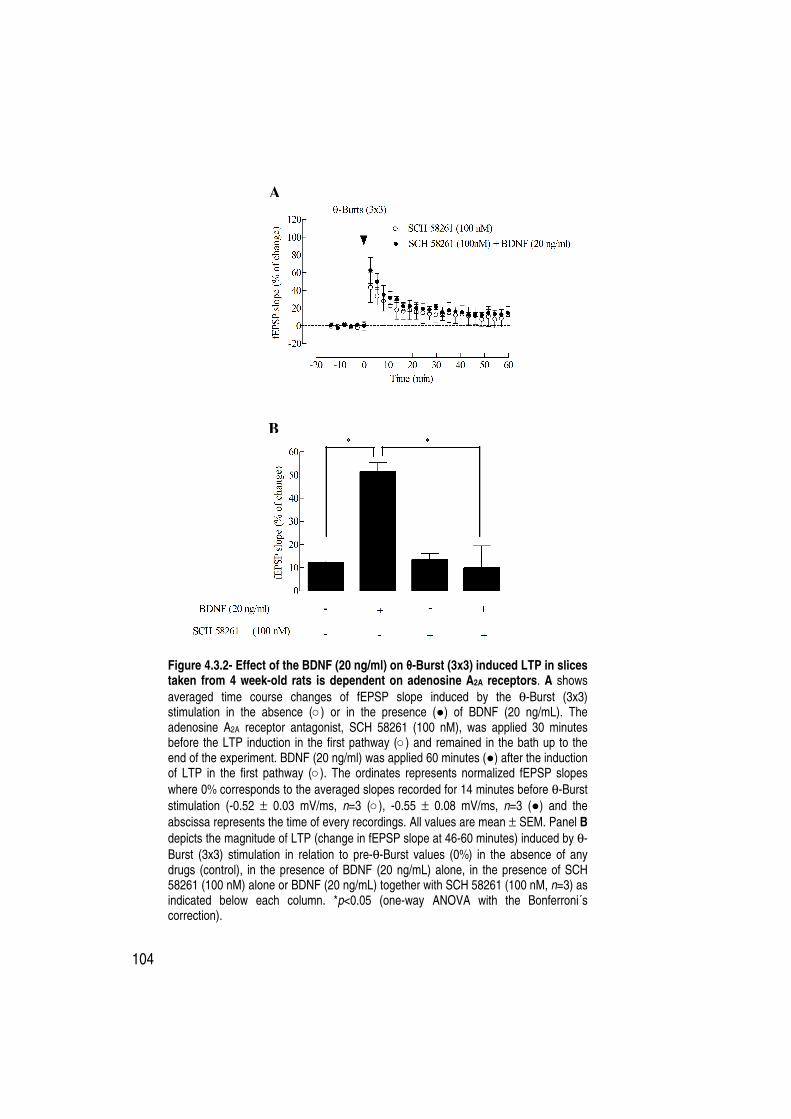
104
A
B
Figure 4.3.2- Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 4 week-old rats is dependent on adenosine A2A receptors. A shows averaged time course changes of fEPSP slope induced by the θ-Burst (3x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL). The adenosine A2A receptor antagonist, SCH 58261 (100 nM), was applied 30 minutes before the LTP induction in the first pathway () and remained in the bath up to the end of the experiment. BDNF (20 ng/ml) was applied 60 minutes () after the induction of LTP in the first pathway (). The ordinates represents normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation (-0.52 ± 0.03 mV/ms, n=3 (), -0.55 ± 0.08 mV/ms, n=3 () and the abscissa represents the time of every recordings. All values are mean ± SEM. Panel B depicts the magnitude of LTP (change in fEPSP slope at 46-60 minutes) induced by θ-Burst (3x3) stimulation in relation to pre-θ-Burst values (0%) in the absence of any drugs (control), in the presence of BDNF (20 ng/mL) alone, in the presence of SCH 58261 (100 nM) alone or BDNF (20 ng/mL) together with SCH 58261 (100 nM, n=3) as indicated below each column. *p<0.05 (one-way ANOVA with the Bonferroni´s correction).

105
A similar result was observed in slices taken from 10-16 week-old rats (Figure
4.3.3). Thus, the same θ-Burst stimulus (3x3) only induced a moderate LTP, (13 ±
1.8% increase in fEPSP slope, n=5) which in the presence of the BDNF (20 ng/ml)
was significantly higher (33 ± 3.2 % increase, n=5, p<0.05). In the presence of SCH
58261 (100 nM) the facilitation of LTP induced by the neurotrophin was prevented
(n=5 p<0.05, Figure 4.3.4). As before, SCH 58261 was added to the superperfusing
bath at least 30 minutes before induction of LTP in the first pathway and remained in
the bath up to the end of the experiment. No significant differences in the magnitude
of the first LTP were found in the presence or in the absence of SCH 58261 (100 nM,
compare first and third columns of the Figure 4.3.4).
Figure 4.3.3- Effect of BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 10-16 week-old rats. Averaged time course changes of fEPSP slope induced by a θ-Burst (3x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL). The neurotrophin () was applied 60 minutes after induction of LTP in the first pathway (). The ordinates represents normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation (-0.58 ± 0.06 mV/ms, n= 4 () and -0.57 ± 0.06 mV/ms, n= 4 ()) and the abscissa represents the time of every recordings. All values are mean ± SEM.

106
A
B
Figure 4.3.4- Effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 10-16 week-old rats is dependent on adenosine A2A receptors. A shows averaged time course changes of fEPSP slope induced by the θ-Burst (3x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL). The adenosine A2A receptor antagonist, SCH 58261 (100 nM), was applied 30 minutes before the LTP induction in the first pathway () and remained in the bath up to the end of the experiment. BDNF (20 ng/ml) was applied 60 minutes () after the induction of LTP in the first pathway (). The ordinates represents normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation (-0.54 ± 0.03 mV/ms, n=5 (), -0.63 ± 0.06 mV/ms, n=3 ()) and the abscissa represents the time of every recordings. All values are mean ± SEM. Panel B depicts the magnitude of LTP (change in fEPSP slope at 46-60 minutes) induced θ-Burst (3x3) stimulation in relation to pre-θ-Burst values (0%) in the absence of any drugs (control), in the presence of BDNF (20 ng/mL) alone, in the presence of SCH 58261 (100 nM) alone or BDNF (20 ng/mL) together with SCH 58261 (100 nM, n=5) as indicated below each column. *p<0.05 (one-way ANOVA with the Bonferroni´s correction).

107
As illustrated in Figure 4.3.5A, in slices taken from 36-38 week-old rats, the θ-Burst
stimulation (3x3) was able to induce LTP (37 ± 7.4 % increase in fEPSP slope, n=4).
In parallel, in slices from the same animals, BDNF (20 ng/ml) did not cause a further
significant enhancement of LTP (42 ± 7.0 %, n=4, p>0.05 as compared with the
absence of BDNF). Since the synaptic effects of BDNF are potentiated by adenosine
A2A receptors activation (Chapters 4.1 and 4.2), it was next studied if the agonist of
this receptor also potentiates the effect of BDNF on LTP. The presence of a selective
agonist of the adenosine A2A receptor, CGS 21680 (10 nM), did not trigger an effect of
the neurotrophin (20 ng/ml) upon LTP (n=3, Figure 4.3.5B). CGS 21680 was added
to the superperfusing bath at least 30 minutes before induction of LTP in the first
pathway and remained in the bath up to the end of the experiment.
Finally, in the oldest group of rats (70-80 wek old rats), θ-Burst stimulation
was able to induce LTP (35± 3.5 % increase in fEPSP slope, n=4, Figure 4.3.6), but
BDNF (20 ng/ml) did not induce any further significant increase in the magnitude of
LTP (37 ± 1.4% increase of fEPSP slpe, n=4 p>0.05, when compared with the
absence of BDNF Figure 4.3.6).
In Figure 4.3.7 are sumarized the results from the experiments with all age
groups, where it is clear that BDNF gradually looses the ability to facilitate LTP but it
also emerge that LTP is progressively larger with aging.

108
Figure 4.3.5- Absence of effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 36-38 week-old rats. A and B show the averaged time courses changes of fEPSP slope induced by a θ-Burst (3x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL). In panel B, the adenosine A2A receptor agonist, CGS 21680 (10 nM), was applied 30 minutes before the LTP induction in the first pathway () and remain in the bath up to the end of the experiment. BDNF (20 ng/ml) was applied 60 minutes () after the induction of LTP in the first pathway (). The ordinates represent normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation (-0.69 ± 0.07 mV/ms, n=4 (A,), -0.68 ± 0.05 mV/ms, n=4 (A,), -0.59 ± 0.07 mV/ms, n=3 (B,) and -0.59 ± 0.05 mV/ms, n=3 (B,)) and the abscissa represent the time of every recordings . All values are mean ± SEM.
A
B

109
Figure 4.3.6- Absence of effect of the BDNF (20 ng/ml) on θ-Burst (3x3) induced LTP in slices taken from 70-80 week-old rats. Averaged time course changes of fEPSP slope induced by a θ-Burst (3x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL). The neurotrophin () was applied 60 minutes after induction of LTP in the first pathway (). The ordinates represents normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation (-0.69 ± 0.07 mV/ms, n= 4 () and -0.68 ± 0.07 mV/ms, n= 4 ()) and the abscissa represent the time of every recordings. All values are mean ± SEM.

110
Figure 4.3.7- Age-related modulation of long-term potentiation (LTP) by BDNF. The bars represent the % change of fEPSP slope 46-60 minutes after LTP induction in the presence (black bars) and the absence (white bars) of BDNF (20 ng/ml). BDNF (20 ng/ml) significantly increases LTP magnitude in hippocampi taken from 4 (n=4) and 10-16 (n=5) week old rats (first and second black bars respectively) when compared to controls (first and second white bars). In slices taken from 36-38 (n=4) and 70-80 (n=4) week old, BDNF was not able to increase LTP (fourth and fifth black bars) magnitude compared to the control LTPs (fourth and fifth white bars). *P<0.05 (paired Student´s t test) as compared with absence of BDNF in the same experiments (adjacent white bar to the left).
0
10
20
30
40
50
60
Without BDNF
With BDNF (20 ng/ml)
10-16 36-384 70-80 Age (weeks)
*
*
fEP
SP
slo
pe (
% o
f ch
an
ge)

111
4.3.3 BDNF effect on LTP induced by a weak θ-Burst stimulation (2 Bursts,
100Hz, 3 stimuli, separated by 200 ms).
The observed reduction in the effect of BDNF upon LTP in aged animals could be due
to the age-related increase in the magnitude of LTP in the absence of BDNF, which
would therefore contribute to masking the effect of BDNF. Thus, the effect of BDNF in
LTP induced by a weaker θ-Burst stimulation (2 Bursts, 100Hz, 3 stimuli, separated
by 200 ms - 2x3) was tested. In these conditions, the magnitude of LTP (without
BDNF) attained in hippocampal slices taken from both 36-38 week-old and 70-80
week-old rats was lower, (14 ± 3.5 % and 31 ± 2.7 %, increase in fEPSP respectively,
Figure 4.3.8) as compared with LTP observed in the animals of the same age but
after the stronger θ-Burst (3x3) stimulation protocol (see 4.3.2).
BDNF was not able to increase LTP induced by the weak θ-Burst (2x3) in
slices taken from 36-38 week-old rats (Figure 4.3.8A) even when adenosine A2A
receptors were pharmacologically activated by CGS 21680 (10 nM) (n=5, P>0.05,
Figure 4.3.8.B). In slices taken from 70-80 week-old rats, BDNF was also unable to
increase the magnitude of LTP (31 ± 7.1% increase, n=3, Figure 4.3.8C).

112
Figure 4.3.8- Absence of effect of the BDNF (20 ng/ml) on θ-Burst (2x3) induced LTP in slices taken from 36-38 week-old rats and 70-80 week-old rats. Averaged time course changes of fEPSP slope induced by a θ-Burst (2x3) stimulation in the absence () or in the presence () of BDNF (20 ng/mL) in hippocampal slices taken from 36-38 week-old rats (A and B) or taken from 70-80 week-old rats (C). The neurotrophin () was applied 60 minutes after induction of LTP in the first pathway () in the absence (A and C) or in the presence (B) of CGS 21680 (10 nM). CGS 21680 was added to slice 30 minutes before induction of LTP in the first pathway and remained in the bath up to the end of the experiment. The ordinates represents normalized fEPSP slopes where 0% corresponds to the averaged slopes recorded for 14 minutes before θ-Burst stimulation: -0.51 ± 0.03 mV/ms, n=3 (A,), –0.50 ± 0.18 mV/ms, n=3 (A,), -0.58 ± 0.03 mV/ms, n=5 (B,), -0.54 ± 0.02 mV/ms, n=5 (B,), -0.66 ± 0.14 mV/ms, n=3 (C,), –0.56 ± 0.07 mV/ms, n=3 (C,) and the abscissa represent the time of every recordings. All values are mean ± SEM.
A B
C

113
4.3.4 Effects of a BDNF scavenger on LTP in hippocampal slices
In slices taken from 10-16 week-old rats, the magnitude of LTP after a θ-Burst (3x3) in
the presence of BDNF was similar to that observed on hippocampal slices taken from
36-38 week-old rats in the absence of the exogenous BDNF administration (Figure
4.3.7). This could suggest that the higher LTP in control conditions in older rats was
due to higher endogenous levels of BDNF. To test this hypotesis, hippocampal slices
from 36-38 week-old rats were treated with a BDNF scavenger, TrkB-Fc. Slices were
incubated as described by Patterson and collaborators (2001), submerged in either
gassed Krebs’ solution alone or in gassed Krebs’ solution containing 2 µ g/ml TrkB-Fc
at room temperature for 1–1.5 hours prior to use for electrophysiological experiments.
Slices were then moved to the interface-recording chamber and were stimulated
using the θ-Burst stimuli. The TrkB-Fc fusion protein (the ligand binding domain of
the native TrkB receptor coupled to the Fc fragment of human immunoglobulin)
scavenges unbound TrkB ligands (Shelton et al., 1995), such as the BDNF.
If extracellular BDNF levels in 36-38 week-old rats were higher and this was
the cause for a higher LTP, a lower LTP magnitude in the slices pre-treated with
TrkB-Fc would be expected. As shown in Figure 4.3.9, LTP magnitude in the
presence of Trk-Fc (2 µg/ml) was similar (n=3, p>0.05) to that observed without the
scavenger, suggesting that the age related increase of LTP magnitude may not be a
result of an increase of endogenous BDNF levels acting via TrkB receptors. However,
the slices were, as mentioned before, pretreated with TrkB-Fc for 1–1.5 hours prior to
use for electrophysiological experiments allowing the scavenging of the BDNF
available in that time. The slices were placed in the recording chamber continuously
perfused with Krebs’ solution, which probably removed the complex (BDNF)-(TrkB-
Fc) from the hippocampal slice. On the other hand, during the θ-Burst stimulation a
higher amount of BDNF can be released (Aicardi et al., 2004); since more TrkB-Fc

114
was not administerd, the activation of TrkB receptors could not be prevented because
of execess of BDNF. Thus, it was next tested the effect of the K252a, the inhibitor of
Trk phosphorylation, on LTP magnitude of hippocampal slices taken from 36-38
week-old rats.
Figure 4.3.9- Effect of Trk-Fc (2 µµµµg/ml) on θ-Burst (3x3) induced LTP in slices taken from 38-38 week-old rats. Averaged time course changes of fEPSP slope induced by a θ-Burst (3x3) stimulation. In the pathway represented as (), slices were pre-incubated for 1.5 hr with Trk-Fc (2 µg/ml). In the pathway represented by (), no drug was added to slices. The arrow represents θ-Burst stimulation. All values are mean ± SEM; 0% (averaged fEPSP slopes recorded for 14 minutes immediately before θ-Burst stimulation: -0.58 ± 0.06 mV/ms, n=5 () and –0.53 ± 0.02 mV/ms, n=3 ()).

115
A B
K252a was applied to slices 60 minutes after the induction of the LTP in the
first pathway and 30 minutes before LTP induction in second pathway. As illustrated
in Figure 4.3.10, in slices taken fom 36-38 week-old rats, K252a (200 nM) reduced
significantly the magnitude of the LTP (22 ± 2.8 %, n=3, p<0.05). K252a did not
influence the basal synaptic transmission as well as does not influence LTP in infant
rats (Fontinha et al., 2008). This suggests that the increase of LTP observed in older
rats comparing to the younger can be in part attributed to the activation of Trk
receptors as a result of the increase in BDNF levels during the θ-Burst stimulation.
Further experiments using animals for the different age groups are required for further
clarification of the ages related increase in LTP due to an age related enhancement of
BDNF release.
Figure 4.3.10- Effect of the K252a (200nM) on θ-Burst (3x3) induced LTP in slices taken from 38-38 week-old rats. A shows averaged time course changes of fEPSP slope induced by the θ-Burst (3x3) stimulation in the absence () or in the presence () of K252a (200 nM). K252a was applied 60 minutes () after the induction of LTP in the first pathway (). 0 % in the ordinates: -0.60 ± 0.03 mV/ms, n=3 (), -0.82 ± 0.02 mV/ms, n=3 (). All values are mean ± SEM. Panel B depicts the magnitude of LTP (change in fEPSP slope at 46-60 minutes) induced θ-Burst (3x3) stimulation in relation to pre-θ-Burst values (0%) in the absence or in the presence of K252a (200 nM). *p<0.05 (paired Student´s t-test).

116
4.3.5 Discussion The aim of the work described in this chapter was to study the effect of BDNF on LTP
induced by θ-Burst stimulus during aging and the influence of adenosine A2A receptor
activation on the BDNF effects.
In previous Chapters (4.1 an 4.2) it was observed that activation of adenosine
A2A receptors is required for the excitatory action of BDNF on hippocampal synaptic
transmission of young and aged rats; but the reduced ability of BDNF to increase
synaptic transmission in aged rats even when adenosine receptors are activated,
could be related to reduced TrkB receptor levels (Diógenes et al., 2007). These
observations lead to the question that was addressed in this chapter: Does the effect
of BDNF on LTP change upon aging and what is the involvement of adenosine A2A
receptors in this process?
In experiments performed using hippocampal slices from the two youngest
groups of rats (4 week-old and 10-16 week-old rats), a marked increase on LTP
induced by BDNF was observed. This effect was totally prevented when adenosine
A2A receptors were blocked. These results reinforce the conclusions from previous
chapters that the activation of adenosine A2A receptors is required for BDNF synaptic
excitatory actions, extending it to synaptic plasticity phenomena.
In slices taken from 36-38 and 70-80 weeks old rats, BDNF no longer
increased the magnitude of LTP even when adenosine A2A receptors were
pharmacologically activated. On the other hand, a marked increase was observed in
LTP magnitude in slices from 36-38 and 70-80 weeks old rats when compared with
LTP magnitude obtained in slices from young rats (4 or 10-16 week-old). To
understand if the effect of BDNF was masked by the increase in LTP magnitude, the
effect of BDNF upon LTP induced by a weaker θ-Burst stimulation was evaluated.
However, under these weak LTP induction conditions, BDNF was still devoid of effect.

117
In slices taken from 10-16 week-old rats, the magnitude of LTP induced by θ-
Burst in the presence of BDNF was similar to that observed in hippocampal slices
taken from 36-38 week-old rats in the absence of exogenous BDNF. This could
suggest that the higher LTP in control conditions in older rats was due to higher
endogenous levels of BDNF and a consequent increase in TrkB activation. The BDNF
scavenger, Trk-Fc, did not reduce LTP observed in older animals, suggesting that the
high LTP in aged animals is not due to an increase of extracellular BDNF levels.
However, it is important to be aware of some technical aspects of these experiments.
When Trk-Fc was tested, the slices were pre-incubated with this scavenger during 1.5
hours immediately before electrophysiological recordings. Thus, Trk-Fc scavenges
the available endogenous BDNF during that time and any molecules of Trk-Fc that
became loosely bound to the tissue would be expected to scavenge BDNF released
afterwards. The slices were then continuously perfused for electrophysiological
recordings. For economical reasons and in accordance with a previously reported
protocol (Patterson et al., 2001), no Trk-Fc was added to the perfusion solution.
However, it is known that θ-Burst induces BDNF release (Aicardi et al., 2004), which
facilitates LTP (Figurov and et al., 1996). If the amount of BDNF released during θ-
Burst stimulus is higher in aged then in young animals, the remaining Trk-Fc
molecules may not be sufficiently able to scavenge it. Thus, experiments were
performed using the inhibitor of Trk phosphorylation, K252a. In the presence of this
inhibitor, the LTP magnitude was reduced in aged animals, which may suggest that
the increase of LTP magnitude observed in aged rats may indeed be a consequense
of higher BDNF levels in these animals.
Figurov and collaborators (1996), studied θ-Burst induced LTP in the
hippocampus from Sprague-Dawley rats with ages between one to 2.5 week-old
postnatal days and a group of adult rats. They also found that exogenous BDNF
enhanced LTP in the neonatal hippocampus where endogenous BDNF level is low;

118
but, in the adult hippocampus, where endogenous BDNF is higher, the Trk-IgG
decreased LTP, suggesting that endogenous levels of this neurotrophin strongly
influenced LTP.
It is commonly accepted that in normal aging the hippocampus suffers
changes, which might contribute to memory deficits. Since LTP is related to learning
and memory (Lynch, 2004), the effect of aging on LTP is of great interest to
understand the age-related decline in cognitive function. However, the results relating
LTP and aging are controversial. The first reports indicated intact hippocampal LTP
induction at the Schaffer collateral-CA1 synapses in old rats (Landfield and Lynch,
1977; Landfield et al., 1978). On the other hand, further studies showed that age-
related deficits in cognitive function are accompanied by an impaired ability to sustain
LTP (see Barnes, 2003). An increase in LTP in slices from aged rats has ben
previously reported (Costenla et al., 1999).
What contributes to the changes here described in LTP, is a fundamental
question. It is known that 1) there is no loss of hippocampal CA1 pyramidal cells in old
rats (Rapp and Gallangher, 1996; Rasmussen et al., 1996), 2) most biophysical
properties of old pyramidal cells do not differ from those in young cells, and 3) there is
no change in spontaneous firing rates of single cells in the hippocampus of freely
behaving old rats comparing with young rats (see Barnes, 2003). However, 4) a deficit
in calcium regulation upon aging has been reported (Landfield and Lynch, 1977).
There are also results suggesting that an increase in microglia activation could be the
key factor for plasticity changes observed during aging (Griffin et al., 2006).
Additionally, changes in the density of NMDA subtype of glutamate receptors have
been discussed. With aging, declines in the expresion of mRNA for diferent NMDA
receptors subunits in rats (Magnusson, 2000) and in konkeys (Bai et al., 2004) were
already observed, as weel as, a decrease in the expression of several NMDA
subunits (Sonntag et al., 2000; Clayton et al., 2002). Yet, these changes do not

119
explain the now observed increase in LTP upon aging. But there are other points
which can, in fact, contribute to this increase in LTP: 1) a lower GABAergic inibition
observed in aged rat hippocampus (Potier et al., 1992; Billard et al., 1995); 2) a
possible increase in BDNF release or the release of other facilitatory substances upon
θ-Burst stimulation in aged animals; and 3) the increase of the levels of adenosine
A2A excitatory receptors in aged animals (Chapter 4.2). According to the results
observed with K252a, the second possibility seems to hold true, but this does not
exclude any of the others.
During the present work it was also observed that BDNF loses the ability to
increase LTP in old rats. The facilittatory effects of BDNF on plasticity have been
related to the activation of TrkB receptors (Figurov et al., 1996), and, TrkB receptor
density decreases with age (Chapter 4.2). Thus, it was postulated that BDNF would
be unable to increase LTP in aged rats. Since BDNF can activate both TrkB and p75
receptors and p75 receptor activation frequently has opposite effects to TrkB receptor
activation (Lu et al., 2005), further investigation needs to be done to understand the
role of p75 receptors in the age-related changes in LTP.
In summary, the work described here shows that BDNF increases θθθθ-
Burst induced-LTP in young and adult rats, that this facilitatory effect is
dependent on adenosine A2A receptor activation, that LTP magnitude increases
with the age of these animals and that this could be due to an increase of the
release of BDNF upon LTP induction, during aging and consequent increase in
TrkB receptor activation.

120
4.4 INFLUENCE OF ADENOSINE A2A RECEPTORS ON THE PROTECTIVE
ACTION OF BDNF AGAINST APOPTOSIS INDUCED BY THE AMYLOID BETA
PEPTIDE
4.4.1 Rational
Neurotrophins have been suggested to have an important role in protecting neurons
from neuronal atrophy in the degenerating human brain. A decrease in BDNF levels
might be involved in neurodegenerative disorders such as Alzheimer’s disease (AD)
(Connor and Dragunow, 1998), making the use of this molecule very promising for
treatment of these disorders. However, as discussed in Chapter 1.1.3, the
pharmacological administration of in vivo BDNF has not been easy since these
molecules are unable to cross the blood–brain barrier, making invasive application
strategies like intracerebroventricular infusion necessary. Intravenous administration
of BDNF has been attempted, but it involves a complex molecular reformulation of the
neurotrophin (Wu and Pardridge, 1999).
As showed in previous chapters, a way to potentiate BDNF actions on
hippocampal neurons is the co-activation of a specific type of adenosine receptors,
the A2A receptors. This could open a new strategy for the treatment of
neurodegenerative diseases by the use of drugs that can induce activation of A2A
receptors and consequently potentiate the neuroprotective effect of the endogenous
BDNF. However, this type of adenosine receptors can also interfere with apoptosis.
Thus, it was observed that the blockade rather than the activation of A2A receptors is
involved in neuroprotection (Dall’lgna et al., 2007). However, adenosine A2A receptor
stimulation also has been associated with decreased apoptosis in the amygdala
(Boucher et al., 2006) and it is known that endogenous adenosine via the A2A

121
receptor subtype may be important in mediating protection of sympathetic neurons
from apoptosis (Ramirez et al., 2004).
Taking into account that adenosine A2A receptors activation facilitates BDNF
actions on synaptic transmission (Chapter 4.1), that the contribution of this adenosine
receptor in apoptosis is controversial and that apoptosis can be a key factor for some
neuodegenerative diseases where BDNF may play a role, it was decided to evaluate:
1) the effect of the adenosine A2A receptors on apoptosis in cultured neurons, and 2)
the function of the A2A receptor activation on the neuroprotective action of BDNF. The
amyloid beta peptide (Aβ) was used as an apoptotic stimulus since Aβ accumulates
in the brain of AD patients (Finder and Glockshuber, 2007), where it seems to induce
apoptosis (Loo et al, 1993), which has been implicated as a potential cause for the
neuronal loss in AD (Shimohama, 2000).
4.4.2 Apoptosis induced by amyloid beta in primary cultures of rat neurons is
prevented by BDNF and this protection is potentiated by adenosine A2A
receptors activation.
Since all the work described in previous chapters of this thesis was carried out in
hippocampus, the initial apoptosis studies were performed separately in hippocampal
and cortical cultured neurons. The results obtained were similar in the two brain
regions and therefore all remaining experiments were performed in cortical neuron
cultures to increase the amount of available biological material and thus decrease the
number of animals required.
Neuronal cells were incubated in the presence of BDNF (20 ng/ml), of the
adenosine A2A receptor agonist CGS 21680 (10 nM), or both CGS 21680 (10 nM) and
the neurotrophin (20 ng/ml) for 24 hours. Whenever both drugs were added, CGS

122
21680 was applied 30 minutes before BDNF. The characteristic morphologic nuclear
changes associated with apoptosis were observed using Hoechst staining (Figure
4.4.1). As expected, few apoptotic cells were detected in control cultures as well as
when BDNF, CGS 21680 or both were present. In contrast, almost 31 ± 4.4% of
neurons exhibited condensed chromatin and nuclear fragmentation with formation of
apoptotic bodies after treatment with 25 µM of amyloid beta (Aβ) 23-35 for 24 hours
(p<0.05 as compared with control, Figure 4.4.2). To evaluate if increased cell death
could be prevented by BDNF and/or CGS 21680, neurons were pre-exposed to these
drugs for 1 hour before Aβ application (Figure 4.4.3). BDNF, added prior to Aβ 25-35,
reduced the percentage of apoptotic cells to 19 ± 1.6% (n=5). CGS 21680 reduced
apoptosis to 24 ± 2.1% (n=7, p>0.05, as compared with Aβ 25-35 alone). When
added together, BDNF and CGS 21680 fully prevented Aβ 25-35-induced apoptosis
reducing apoptotic cells to a value of 11 ± 1.2% (n=7, P<0.05 as compared with Aβ
25-35 alone), similar to that obtained in the absence of Aβ 25-35 or any other drug.
The effect of the selective antagonist of A2A receptors, SCH 58261 (100 nM)
was tested in two cultures. SCH 58261 (100 nM) applied 1 hour before Aβ 25-35 (25
µM) did not appreciably change the percentage of apoptotic cells when compared
with Aβ 25-35 alone (n=3, p>0.05, Figure 4.4.4). This antagonist of adenosine A2A
receptors, added 30 minutes before BDNF completely prevented the neuroprotective
effect of the neurotrophin (Figure 4.4.4A). As expected when applied 30 minutes
before CGS 21680 (10 nM), SCH 58261 (100 nM) also reverted the slight decrease of
apoptosis induced by the A2A receptor agonist (Figure 4.4.4B). Moreover, in
experiments where BDNF and CGS 21680 were added together, SCH 58268 fully
reverted the marked neuroprotection already observed (Figure 4.4.4C).

123
Figure 4.4.1- Morphologic analysis of cells. Cortical neurons were isolated and cultured for 4 days, subsequently they were incubated with no drugs (control), BDNF (20 ng/ml), CGS 21680 (10 nM) or both BDNF and CGS 21680 as indicated. In co-incubation experiments CGS 21680 was added 30 minutes before BDNF application. 24 hours after drugs administration, cells were fixed and then assessed for nuclear morphologic alterations characterized by condensed chromatin, fragmentation, and formation of apoptotic bodies using fluorescence microscopy with Hoechst staining as shown in A. In B the mean ± SEM values obtained in five to eight experiments are ilustrated. Dotted line the value obtained without any drug.
Figure 4.4.2 Amyloid-beta (Aβ)-induced apoptosis in primary rat neurons. Cortical neurons were isolated and cultured for 4 days. When Aβ 25-35 (25 µM) was added alone it remain in the cell cultures for 24h, afterward cells were fixed and then assessed for nuclear morphologic alterations characterized by condensed chromatin, fragmentation, and formation of apoptotic bodies using fluorescence microscopy with Hoechst staining as shown in A. In B the mean ± SEM values obtained in eight experiments are illustrated. *P<0.05 (paired Student´s t-test).
Aβ
25
-35
(2
5 µ
M)
A B
A B

124
Figure 4.4.3 Apoptosis induced by amyloid-beta (Aβ) in primary rat neurons is prevented by BDNF and CGS 21680. Cortical neurons were isolated and cultured for 4 days; subsequently, they were incubated with BDNF (20 ng/ml), CGS 21680 (10 nM) or both BDNF and CGS 21680 as indicated. In co-incubation experiments CGS 21680 was added 30 minutes before BDNF application. Aβ 25-35 (25 µM) was added 1 hour after BDNF and/or CGS 21680 and it remained in the cell cultures for 24 hours. Cells were then fixed and assessed for nuclear morphologic alterations characterized by condensed chromatin, fragmentation, and formation of apoptotic bodies using fluorescence microscopy with Hoechst staining as shown in A. Arrows indicate apoptotic cells. In B, the mean ± SEM values obtained in four to eight experiments are illustrated. *P<0.05 (one-way ANOVA with the Dunnett correction). Dotted line the value obtained without any drug.
A B

125
Figure 4.4.4- SCH 58261 reverts the neuroprotection induced by BDNF. In panel A, black bars illustrate the neuroprotective effect of BDNF upon Aβ 25-35-induced apoptosis (already showed in Figure 4.4.3), grey bars show the effect of SCH 58261, an antagonist of adenosine A2A receptors, in the presence of Aβ 25-35 (first grey bar, n=3) and on the effect of BDNF with Aβ 25-35 treatment (second gray bar, n=2). In panel B, black bars illustrate the effect of CGS 21680 upon Aβ 25-35-induced apoptosis (already showed in Figure 4.4.3), grey bars show the effect of SCH 58261, in the presence of Aβ 25-35 (first grey bar, n=3) and on the effect of CGS 21680 (second gray bar, n=2). In panel C, black bars illustrate the neuroprotective effect of BDNF in the presence of CGS 21680 upon Aβ 25-35-induced apoptosis (already showed in Figure 4.4.3), grey bars show the effect of SCH 58261 in the presence of Aβ 25-35 (first grey bar, n=3) and on the effect of BDNF plus CGS 21680 (second gray bar, n=2). *p<0.05 (one-way ANOVA with the Dunnett correction). Dottted lines represent the control values (without drugs).
A B
C

126
4.4.3 Amyloid beta induced fragmentation of pro-caspase-3 is prevented by BDNF in the presence of adenosine A2A receptor activation Mitochondria are central life and death regulator, which release cytochrome c, leading
to the activation of caspase-3, which subsequently cleaves downstream substrates.
Processed caspase-3 in the cytosol of neuronal cells, assessed by immunoblot
analysis, was therefore used as measure of the degree of apoptosis in cultured cells.
Proteolytically activated caspase-3 (20 KDa) was low in cultured neurons after
incubation without drugs (control), BDNF (20 ng/ml) and or CGS 21680 (10 nM)
(Figure 4.4.5). However, when Aβ 25-35 (25 µM) was added, a significant increase
(more than 2-fold) in caspase-3 formation (p<0.05 when compared with control cells)
was observed (Figure 4.4.6). The pre-incubation with BDNF (20 ng/ml) reduced the
formation of caspase-3 induced by Aβ 25-35, however the decrease did not reveal
statistical significance. A similar trend occurred when cells were incubated with the
A2A receptor agonist (CGS 21680, 10 nM, Figure 4.4.7). However, when cells were
incubated with BDNF (20 ng/ml) in the presence of CGS 21680 (10 nM), the caspase-
3 formation from pro-caspase-3 was significantly reduced to levels similar to the
control (without drugs) (P>0.05 when compared to control cells, Figure 4.4.7).

127
Figure 4.4.5 Processing of caspase-3 in primary rat neurons in the absence of amyloid-beta. Cortical neurons were isolated and cultured for 4 days before incubation as described in Methods. Cells were incubated either with vehicle (control, no aded drugs), BDNF (20 ng/ml), CGS 21680 (10 nM) or both BDNF and CGS 21680. In coincubation experiments CGS 21680 was added 30 minutes before BDNF application. Procaspase-3 (32 KDa) and active caspase-3 (20 KDa) expression were analysed using western blotting. In A representative Western blots of protein expression are shown for cytosolic protein fractions. The histograme B show mean ± SEM values of the ratio caspase 3/ pro-caspase- 3 for 3-5 independent experiments. Dotted line represents the control values (without drugs).
B
A
Co
ntr
ol
BD
NF
CG
S 2
16
80
BD
NF
+ C
GS
21
68
0
Pro-caspase 3
Caspase 3
32 KDa
20 KDa

128
Figure 4.4.6 Amyloid-beta induced processing of caspase-3 in primary rat neurons. Cortical neurons were isolated and cultured for 4 days before incubation as described in Methods. Cells were incubated either with vehicle (control) or with Aβ 25-35 (25 µM) for 24 hours. Procaspase-3 (32 KDa) and active caspase-3 (20 KDa), expression were analysed using Western blotting. Representative western blots of protein expression are shown for cytosolic protein fractions in A. The histograme B show mean ± SEM values of the ratio caspase 3/ pro-caspase- 3 for 3 independent experiments. *p<0.05 (paired Student´s t-test). Dotted line represents the control values (without drugs).
Co
ntr
ol
Aβ
25
-35
32 KDa
20 KDa
Pro-caspase 3
Caspase 3
B
A

129
Figure 4.4.7-Amyloid-beta induced processing of caspase-3 in primary rat neurons, which is reduced by BDNF in the presence of adenosine A2A receptors activation. Cortical neurons were isolated and cultured for 4 days before incubation as described in Methods. Cells were incubated with vehicle (values represented as the dotted line) or with Aβ 25-35 (25 µM) alone or together with BDNF (20 ng/ml), with CGS 21680 (10 nM) or with both BDNF and CGS 21680. In coincubation experiments CGS 21680 was added 30 minutes before BDNF application. Aβ 25-35 (25µM) was added 1 hour after BDNF and/or CGS 21689 and it remained in the cell cultures for 24 hours. Procaspase-3 (32 KDa) and active caspase-3 (20 KDa) expression was mesured using western blotting. Representative Western blots of protein expression are shown for cytosolic protein fractions in A. The histograme B shows mean ± SEM values of the ratio caspase 3/ pro-caspase- 3 for 3 independent experiments. *p<0.05 (one-way ANOVA with the Dunnett correction).
BD
NF
A
CG
S2
16
80
32 KDa
20 KDa Pro-caspase 3
Caspase 3
+ Aβ 25-35
BD
NF
+
C
GS
21
68
0
B

130
A B
4.4.4 Amyloid beta induced increase in caspase-3 activity is prevented by BDNF
in the presence of adenosine A2A receptors activation
Caspase-3-like activity was evaluated by enzymatic cleavage of the chromophore
p-nitroanilide (pNA) from the substrate N-acetyl-Asp-Glu-Val-Asp (DEVD)-pNA and
the formation of pNA was measured at 405 nm, as mentioned in Methods (Chapter
3.4).
Neither BDNF nor CGS 21680 alone or together influenced significantly the
activity of caspase 3 (Figure 4.4.8). Aβ 25-35 (25 µM) significantly increased
caspase-3 activity (p<0.05, n=4, Figure 4.4.8B). When either BDNF (20 ng/ml) or
CGS 21680 (10 nM) were added alone, a slight redution in the increase in caspase-3
activity induced by Aβ 25-35 (25µM) was noted (Figure 4.4.9). The increase in
caspase-3 activity caused by Aβ 25-35 (25 µM) was fully prevented when cells were
incubated with BDNF (20 ng/ml) together with CGS 21680 (10 nM) (Figure 4.4.8C).
Figure 4.4.8- Activity of caspase-3 in primary rat neurons. Cortical neurons were isolated and cultured for 4 days before incubation as described in Methods. Cells were incubated with either vehicle (control), BDNF (20 ng/ml), CGS 21680 (10 nM) or both BDNF and CGS 21680 as indicated. In coincubation experiments CGS 21680 was added 30 minutes before BDNF application (A). When Aβ 25-35 (25 µM) was added alone it remained in the cell cultures for 24 hours (B). Caspase activity was measured in cytosolic protein fractions as described in Methods. The histogrames shows mean ± SEM values of caspase-3 activity for 3-4 experiments are represented. *p<0.05 (paired Student´s t-test). Dotted line represents the control values (without drugs).

131
Figure 4.4.9- Aβ-induced enhancement of caspase-3 activity in primary rat neurons is reduced by BDNF in the presence of adenosine A2A receptor activation. Cortical neurons were isolated and cultured for 4 days before incubation as described in Methods. Cells were incubated with either vehicle (represented as the dotted line), or with Aβ 25-35 (25µM) alone or together with BDNF (20 ng/ml), CGS 21680 (10 nM) or both BDNF and CGS 21680. In coincubation experiments CGS 21680 was added 30 minutes before BDNF application. Aβ 25-35 (25µM) was added 1 hour after BDNF and/or CGS 21680 and it remained in the cell cultures for 24 hours. Caspase activity was measured in cytosolic protein fractions as described in Methods. These results are the mean ± SEM values of caspase-3 activity for 3 independent experiments. *p<0.05 (one-way ANOVA with the Dunnett correction).

132
4.4.5 Discussion
Treatment of rat cortical neurons with Aβ 25-35 resulted in high levels of apoptosis
showing the consequent elevated levels of pro-caspase-3 cleavage into caspase-3
and also an increase in its proteolytic activity. The main finding in the work described
in this chapter was that the molecular modifications resulting from an increase in
neuronal death by apoptosis were totally prevented when cells were incubated with
BDNF in the presence of a selective agonist of adenosine A2A receptors, CGS 21680.
In addition, when the antagonist of adenosine A2A receptors, SCH 58261, was added
to cells prior to the other drugs, it completetly reverted the protective affects of BDNF,
CGS 21680 or both BDNF and CGS 21680 added together. Therefore, A2A adenosine
receptors need to be activated to allow BDNF neuroprotection against apoptosis
induced by Aβ.
The accumulation of Aβ in the brain has been implicated as a potential cause
for the neuronal loss that occurs in AD (Shimohama, 2000). Cell death from Aβ-
induced toxicity is a complex process that is thought to involve a number of different
pathways, including oxidative stress, perturbation of calcium homeostasis,
mitochondrial dysfunction, and activation of caspases (Selkoe, 2001). It is generally
believed that Aβ peptides contribute significantly to the pathogenesis of AD, although
the mechanisms for this pathogenesis are not yet fully understood. Accordingly, it was
reported that central administration of Aβ 25-35 caused a memory impairment in
mice, assessed by a decrease in performance in both inhibitory avoidance and
spontaneous alteration tasks (Maurice et al., 1996; Dall’Igna et al., 2007).
Neurotrophins, after binding to their Trk receptors, activate two main signal
transduction pathways that regulate the cell death machinery (Segal and Greenberg,
1996, Kaplan and Miler, 2000). The primary pathway is the phosphatidylinositol 3-
kinase (PI3K)–Akt pathway, which suppresses cell death by inhibiting the apoptotic

133
activities of forkhead (Brunet et al., 1999) and BCL2 (B-cell leukaemia/ lymphoma 2)-
associated death protein (BAD)(del Pesos et al., 1997; Datta et al., 1997). The other
pathway is the mitogen-activated protein kinase (MAPK)–MEK (MAPK/ERK
extracellular signal regulated kinase) signalling cascade, which stimulates the activity
and/or expression of antiapoptotic proteins, including BCL2 and the transcription
factor cyclic AMP response element binding protein (CREB) (Riccio et al., 1999).
Adenosine is an ubiquitous homeostatic substance present in all cells, which
modulates several functions in the brain, including neuronal survival. However, the
actions of adenosine can be diverse depending on the cell type, the aggressiveness
of stimuli and the age of animals or cells. In neurons, Lee and Chao (2001) observed
by measuring lactate dehydrogenase (LDH) release, that activation of adenosine A2A
receptors rescues neurons from death induced by the absence of BDNF, in a TRK
receptor dependent way. In PC12 cell cultures, adenosine A2A receptor activation
antagonizes apoptosis due to serum deprivation (Huang et al., 2001) and also
rescues the impairement of neurite outgrowth when the NGF-evoked MAPK cascade
is suppressed (Chen et al., 2002). Recently it was shown that the adenosine A2A
receptor contributes to motoneuron survival by transactivating the TrkB receptors
(Wiese et al., 2007). In primary glial cultures from cerebral cortices, the activation of
adenosine A2A receptors increases NGF expression (Heese et al., 1997), which in
turn can exert its neuroprotective effects. Adenosine also has important roles in
peripheral nervous system where it has neuroprotective effects on neurotrophin-
deprived sympathetic neurons through A2A receptor activation, via a pathway that
requires ERK signalling (Ramirez et al., 2004).
During hypoxia or ischemic conditions, adenosine is released in large
amounts and can mediate cellular protection. This neuroprotection against ischemia
or hypoxia is mainly due to A1 receptor activation (Ribeiro et al., 2003). However, in
liver, administration of A2A receptor agonists before induction of ischemia can

134
attenuate post-ischemic apoptotic hepatic injury and thereby minimize liver injury
(Ben-Ari et al., 2005). On the other hand, A2A antagonists have been reported to
reduce hypoxic-ischemic neuronal injury (von Lubitz et al., 1995; Phillis, 1995) as well
as to block the neurotoxicity induced by Aβ in primary cultures of cerebellar granule
cells prepared from 8-day-old Wistar rats (Dall’Igna et al., 2003). In fact, the ability of
A2A agonists to enhance glutamate release (see Sebastião and Ribeiro, 1996)
together with the frequent finding that these receptors operate in an opposite way to
the A1 receptors, which are widely accepted as neuroprotective receptors, leads to a
frequent assumption that A2A receptors enhance cell death. As shown in the work now
reported, and in line with data mentioned above, A2A receptors enhance the
neuroprotective action of neurotophins and, even more importantly, the
neuroprotective actions of neurotrophins are fully lost when A2A receptors are
blocked. This clearly point towards a putative neuroprotective role of A2A receptors
whenever neurotrophic actions are required. Therefore, studies are required on the
consequences of therapy using specific A2A receptor antagonists in
neurodegenerative diseases, where neurotrophic factors play a beneficial role (see
e.g. Muller and Ferré, 2007).
In summary, as shown in the work presented in this chapter, activation
of adenosine A2A receptors did not intensify the apoptosis induced by Aβ. On
the contrary, adenosine A2A receptor activation enhanced the ability of BDNF to
decrease cell death, strengthening the idea that adenosine A2A receptor
agonists may be usefull in potentiating the BDNF therapeutic actions on AD.

135
5. CONCLUSIONS
The goal of the present study was to investigate functional aspects in the cross talk
between adenosine A2A receptors and the brain-derived neurotrophic factor (BDNF).
This study was started using hippocampal slices taken from infant rats (3-4
week-old rats) to understand if the activation of adenosine A2A receptors modulates
BDNF action actions in basal synaptic transmission and the possible mechanisms
that underlie this cross talk. We found that adenosine A2A receptor activation
facilitates the excitatory effect of BDNF on synaptic transmission and that this
facilitatory action of adenosine A2A receptor activation is due to the activation of the
cAMP/PKA transducing system (Chapter 4.1).
Since BDNF has been pointed out as a promising drug to treat
neurodegenerative diseases, such as Alzheimer’s disease, a pathology more frequent
in old people, it was imperative to study the influence of adenosine receptors
activation upon the effect of BDNF on synaptic transmission during aging. With the
present work it became clear that BDNF has distinct actions throughout the lifetime of
animals, that the effects of BDNF are also dependent on adenosine A2A receptors
activation and that these changes can be in part due to a decrease of TrkB receptor
levels and an increase of A2A receptor densities (Chapter 4.2).
In addition, it was considered essential to evaluate the influence of adenosine
A2A receptors upon the BDNF effect on LTP, a commonly accepted way to study the
neurophysiological basis of learning and memory, and to understand if A2A receptors
would influence the neuroprotection induced by BDNF. The main reason for this
approach was that in neurodegenerative pathologies, such as Alzheimer’s disease,
there is both a memory deficit and neuronal loss probably due to enhanced apoptosis
as a consequence of amyloid beta peptide deposition. It was observed that BDNF

136
increases LTP in young and adult rats, and that this facilitatory effect is dependent on
adenosine A2A receptor activation. It was also observed that LTP magnitude
increases with age and this may be due, in part, to an increase in BDNF release upon
stimulation. Finally, BDNF loses the capacity to increase LTP in aged animals, which
can be attributed to a decrease of TrkB receptors density.
As stated earlier, BDNF has been proposed to be used in the treatment of
Alzheimer’s disease patients, a pathology characterized by the deposition of amyloid
plaques containing the amyloid beta peptide, the formation of neurofibrillary tangles
with hyperphosphorylated tau protein and cell death. Thus we studied the
neuroprotective effect of BDNF against cellular death induced by amyloid beta
peptide and the involvement of adenosine A2A receptors in this neuroprotective action.
This study showed that activation of adenosine A2A receptors enhances the ability of
BDNF to prevent apoptosis cellular death in cultured neurons.
The chronic actions of neurotrophins, such as differentiation, growth and
maturation of CNS cells, are already well established. A new area of investigation was
recently opened with the finding that these molecules also have important acute
actions upon synaptic transmission. The work presented in this thesis contributes to a
more elucidated understanding of these acute neuronal actions of BDNF and
describes a way to potentiate these actions through the activation of adenosine A2A
receptors.

137
6. FUTURE PERSPECTIVES
The importance of administering neurotrophic factors, in particularly BDNF, to
Alzheimer’s patients has been pointed out several times throughout this thesis. The
discovery of a molecule that crosses the blood-brain barrier and that could potentiate
BDNF actions would be a great therapeutic breakthrough. The first hint of the
potential benefits of adenosine or adenosine A2A receptor agonists as drugs to
potentiate BDNF action obtained in 2001 by Lee and Chao (2001), who found that a
selective adenosine A2A agonist induced phosphorylation of the BDNF receptors,
TrkB. But it remained to be known if this molecular interplay would be relevant to the
actions of BDNF. Answering this question was the global objective of this work.
As reported here, A2A receptor activation is an obligatory step for the synaptic
and neuroprotective function of BDNF, and this A2A facilitatory action occurs in a
cAMP-PKA transducion signal-dependent way. In addition to the possibilities already
discussed about the manner in which the cAMP-PKA transducing system can
modulate TrkB actions, one could envision that this cross talk could also occur
between the signalling transdution pathways initiated by adenosine receptors and Trk
receptors. Thus, the work here reported opens new perspectives for the investigation
of common players involved in this interplay between adenosine and BDNF receptors.
A schematic representation of the relationship between pathways initiated by
activation of both A2A and TrkB receptors is presented in Figure 5.1. As mentioned in
detail in the introduction of this thesis, adenosine A2A receptors generally couple to Gs
proteins. In addition, in the striatum they are also coupled to Golf proteins (Corvol et
al., 2001) and, in the hippocampus, they can be coupled to Gi/Go proteins (Cunha et
al., 1999). Furthermore, adenosine A2A receptors, when overexpressed in COS-7
cells, were shown to couple to G15/16 proteins (Offermanns and Simon, 1995). In
human endothelial cells, the signalling via G12/13 proteins has been hypothezied (Sexl

138
et al., 1997). There is also evidence that adenosine A2A receptors can induce
formation of inositol phosphates via pertussis toxin-insensitive Gα15 and Gα16 (see
Jacobson and Gao, 2006). Therefore, the adenosine A2A receptor couple, via G
proteins, to an intricate network of signalling pathways, which enables adenosine to
communicate with cells in a complex manner. Initially, signalling pathways were
thought to be linear, starting with activation of receptors in the cellular membrane and
a subsequent activation of different steps including small signalling molecules, called
second messengers, protein kinases and transcription factors that regulate gene
expression. In the particular case of A2A receptors, this involves the cAMP and cAMP-
dependent protein kinase (PKA) pathway (in blue in the Figure 5.1). However,
adenosine A2A receptors can also be involved in the MAPK signalling and as weel as
in PI3K signalling (Figure 5.1 blue dotted line).
Regarding to Trk receptors, as described in the introduction, there are three
major pathways involved in the signalling mediated by these receptors: 1)
phosphotydilinositol-3 kinase (PI3K); 2) Ras-MAPK pathway and 3) PLCγ pathway,
and their downstream effectors (Figure 5.1 brown dotted line).
Therfore, signalling cascades resulting from the activation of A2A and TrkB
receptors have common steps (Figure 5.1 in green), the Ras-MAPK and PI3K
pathways. These are involved in survival, growth and differentiation as well as in
synaptic transmission and in plasticity (Blom and Konnerth, 2005). Since adenosine
A2A receptors activation potentiates BDNF actions on synaptic transmission, plasticity
and neuroprotection, I advance the hypothesis that one or both of these pathways are
involved in the cross talk between A2A and TrkB receptors.

139
Figure 5.1 Cross talk between signalling pathways of adenosine A2A receptors and TrkB receptors. Possible interaction between the signalling pathways of both receptors A2A and TrKB.
In conclusion, the key finding in this thesis is that adenosine A2A receptors activation
potentiates BDNF actions. Furthermore, as clearly shown throughout this work, the
actions of this neurotrophin upon both synaptic trasmission and neuronal death are
fully lost when A2A receptors are blocked. Therefore, agonists of adenosine A2A
receptors may prove to be of high therapeutic interest where there is the need to
increase BDNF actions in neurodegenerative diseases. In other words, in the early
stages of neurodegenerative diseases, where an enhancement of neurotrophic
factors is highly desirable, A2A receptor agonists can be an important therapeutic
option and antagonists should be avoided in order to allow beneficial neurotrophic
influences. On the other hand, in the late stages of neurodegenerative diseases, A2A
receptor antagonists can be advantageous to inhibit excitatory processes.
AC
cAMP
CREB
SrcRas
Raf
MEK
Erk
SOS
PI3K
AKT
PLCγ
DAG IP3
PKC Ca2+
Adenosine A2A receptors Trk receptors
Gαs
G12/13
PKA
Rac1/Cdc42
p38
G15/16
AC
cAMP
CREB
SrcRas
Raf
MEK
Erk
SOSSOS
PI3K
AKT
PLCγPLCγ
DAG IP3
PKC Ca2+
Adenosine A2A receptors Trk receptors
Gαs
G12/13
PKA
Rac1/Cdc42
p38
G15/16

140
In the future, investigation of how to promote the expression of TrkB
receptors in aged subjects and how to avoid eventual deleterious consequences of
the upregulation of neurotrophic actions (e.g. tumor promotion) deserves to be
investigated.

141
7.ACKNOWLEDGEMENTS
Em primeiro lugar gostaria de expressar o meu profundo agradecimento aos meus
orientadores, Professor Joaquim Alexandre Ribeiro e Professora Ana Sebastião. Ao
professor Alexandre Ribeiro, por quem tenho um enorme respeito e admiração, por
me ter recebido no seu laboratório, pelo incentivo e pelos ensinamentos muito
experientes que me deu. À Professora Ana Sebastião, por quem tenho uma grande
admiração e amizade, por todos ensinamentos, palavras de apoio, positivismo e
discussão científica. Gostaria de agradecer ao Professor Alexandre de Mendonça,
que sempre esteve disponível para qualquer esclarecimento e discussão científica
principalmente em temas relacionados com plasticidade sináptica.
Uma palavra de agradecimento vai para todos os colegas que ao longo destes anos
se tornaram amigos. Gostaria de dirigir um agradecimento especial aos que
partilharam discussão científica, experiências e artigos são eles a Catarina
Fernandes, o António, a Ana Rita, a Natalia, a Paula, a Luisa e o Bruno Fontinha. Ao
Bernardo, estudante que orientei no decorrer do último ano, pelo entusiasmo e
colaboração nas experiências de apoptose. Não posso deixar de expressar o meu
reconhecimento a todos os técnicos em particular à Alexandra, ao Sr. João e á
Elvira.
À Professora Cecília Rodrigues da Faculdade de Farmácia da Universidade de
Lisboa por me ter recebido no seu laboratório o que possibilitou a realização de parte
das experiências de apoptose bem como pela sua orientação na realização das
mesmas. Um agradecimento muito especial à Susana Solá e todos os estudantes do
grupo pela constante disponibilidade, simpatia e paciência.

142
Ao Professor Rui Silva, à Adelaide e à Sofia da Faculdade de Farmácia da
Universidade de Lisboa por toda a ajuda prestada aquando de implementação das
culturas primárias de neurónios.
À Professora Dora Brites por me ter dado a oportunidade de entrar no mundo da
ciência e de ter acompanhado os meus primeiros passos ainda como estudante de
Ciências Farmacêuticas na Faculdade de Farmácia da Universidade de Lisboa.
Desde esses primeiros passos já decorreram 12 anos.
Ao Professor Silva Carvalho e à Professora Isabel Rocha do Instituto de Fisiologia
pelo constante e indispensável apoio na cedência de espaço do biotério bem como
da colaboração preciosa de todas as pessoas que nele trabalham.
I also would like to thank Lori, for the English review of this thesis, suggestions, good
humor and friendship.
E finalmente, à minha família: aos meus pais por me terem incentivado a continuar e
a apostar na procura do conhecimento sabendo não ser muitas vezes o caminho
mais fácil; à minha irmã e ao Henrique por toda a paciência, boa disposição e apoio
incondicional.
À Lúcia, a minha filha, a quem dedico esta tese pelo seu sorriso e olhar que tantas
vezes foi o meu consolo e força para continuar em dias menos bons.
Um agradecimento às instituições que financiaram o trabalho desenvolvido. À
Fundação para a Ciência e a Tecnologia (FCT) da qual recebi uma bolsa de
doutoramento (SFRH/BD/12167/2003) e que financiou o trabalho de investigação

143
(POCTI/43634/99) bem como a Fundação Caloust Gulbenkian e à Regeneron que
gentilmente cedeu o BDNF.

144

145
8.REFERENCES
Aicardi G., Argilli E., Cappello S., Santi S., Riccio M., Thoenen H. and Canossa M.
(2004) Induction of long-term potentiation and depression is reflected by
corresponding changes in secretion of endogenous brain-derived neurotrophic
factor. Proc. Natl. Acad. Sci. USA. 101, 15788-15792.
Amaral D. and Lavenex P. (2007) The hippocampal formation in: The hippocampus
book ed. by Andersen P., Morris R., Amaral D., Bliss T. and O’Keefe J. Oxford,
NY, pp. 37-114.
Andersen P., Morris R., Amaral D., Bliss T. and O’Keefe J. (2007) The hippocampal
formation in: The hippocampus book ed. by Andersen P., Morris R., Amaral D.,
Bliss T. and O’Keefe J. Oxford, NY, pp. 3-8.
Anderson W.W. and Collingridge G.L. (2001) The LTP program: a data acquisition
program for online analysis of long-term potentiation and other synaptic events. J.
Neurosci. Methods 10, 71–83.
Antle M.C., Steen N.M. and Mistlberger R.E. (2001) Adenosine and caffeine modulate
circadian rhythms in the Syrian hamster. NeuroReport 12, 2901–2905.
Arch J.R. and Newsholme E.A. (1978) The control of the metabolism and the
hormonal role of adenosine. Essays Biochem. 14, 82–123.
Arendash G.W., Schleif W., Rezai-Zadeh K., Jackson E.K., Zacharia L.C., Cracchiolo
J.R., Shippy D. and Tan J. (2006) Caffeine protects Alzheimer's mice against
cognitive impairment and reduces brain beta-amyloid production. Neuroscience
142, 941-952.

146
Bai L., Hof P.R., Standaert D.G., Xing Y., Nelson S.E., Young A.B. and Magnusson
K.R. (2004) Changes in the expression of the NR2B subunit during aging in
macaque monkeys. Neurobiol. Aging 25, 201–208.
Barbacid M., Lamballe .F, Pulido D. and Klein R. (1991) The trk family of tyrosine
protein kinase receptors. Biochem. Biophys. Acta 1072, 115-127.
Barde Y.A., Edgar D. and Thoenen H. (1982) Purification of a new neurotrophic factor
from mammalian brain. EMBO J. 1, 549-553.
Barnes C.A (2003) Long-term potentiation and the aging brain. Philos. Trans. R. Soc.
Lond. B. Biol. Sci. 358, 765-72.
Ben-Ari Z., Pappo O., Sulkes J., Cheporko Y., Vidne B.A. and Hochhauser E. (2005)
Effect of adenosine A2A receptor agonist (CGS) on ischemia/reperfusion injury in
isolated rat liver. Apoptosis 10, 955–962
Benington J.H., Kodali S.K. and Heller H.C. (1995). Stimulation of A1 adenosine
receptors mimics the electroencephalographic effects of sleep deprivation. Brain
Res. 692, 79–85.
Berg M.M., Sternberg D.W., Parada L.F. and Chao M.V. (1992) K-252a inhibits nerve
growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase
activity. J. Biol. Chem. 267, 13–16.
Bibel M. and Barde Y.A. (2000) Neurotrophins: key regulators of cell fate and cell
shape in the vertebrate nervous system. Genes Dev. 14, 2919-2937.
Billard J.M., Lamour Y. and Dutar P. (1995) Decreased monosynaptic GABAB
mediated inhibitory postsynaptic potentials in hippocampal CA1 pyramidal cells in
the aged rat: pharmacological characterization and possible mechanisms. J.
Neurophysiol. 74, 539–546.

147
Bliss T.V. and Collingridge G.L. (1993) A synaptic model of memory: long-term
potentiation in the hippocampus. Nature 361, 31-39.
Blom R., Konnerth A. (2005) Neurotrophin-mediated rapid signalling in the central
nervous system: mechanisms and functions. Physiology 20,70-78.
Blum D., Gall D., Galas M.C., d’Alcantara P., Bantubungi K. and Schiffmann S.N.,
(2002) The adenosine A1 receptor agonist adenosine amine congener exerts a
neuroprotective effect against the development of striatal lesions and motor
impairments in the 3-nitropropionic acid model of neurotoxicity. J. Neurosci. 22,
9122–9133.
Boucher M., Wann B.P., Kaloustian S., Cardinal R., Godbout R. and Rousseau G.
(2006) Reduction of apoptosis in the amygdala by an A2A adenosine receptor
agonist following myocardial infarction. Apoptosis 11, 1067-1074.
Boulanger L. and Poo M. (1999a) Presynaptic depolarisation facilitates neurotrophin-
induced synaptic potentiation. Nat. Neurosci. 2, 346–351.
Boulanger L. and Poo M. (1999b) Gating of BDNF-induced synaptic potentiation by
cAMP. Science 284, 1982–1984.
Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem.
72, 248-254.
Brewer G.J., Torricelli J.R., Evege E.K. and Price P.J. (1993) Optimized survival of
hippocampal neurons in B27-supplemented neurobasal, a new serum-free
medium combination. J. Neurosci. Res. 35, 567- 576.
Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden
K.C., Blenis J. and Greenberg M.E. (1999) Akt promotes cell survival by
phosphorylating and inhibiting a forkhead transcription factor. Cell 96, 857–868.

148
Canas N., Pereira I.T., Ribeiro J.A. and Sebastião A.M. (2004) Brain-derived
neurotrophic factor facilitates glutamate and inhibits GABA release from
hippocampal synaptosomes through different mechanisms. Brain Res. 1016, 72-
78.
Chen B., Dowlatshahi D., MacQueen G.M., Wang J.F. and Young L.T. (2001a)
Increased hippocampal BDNF immunoreactivity in subjects treated with
antidepressant medication. Biol. Psychiatry 50, 260-265.
Chen H-C., Shih H-M. and Chern Y. (2002) Essential role of cAMP-response element-
binding protein activation by A2A adenosine receptors in rescuing the nerve
growth factor-induced neurite outgrowth impaired by blockage of the MAPK
cascade. J. Biol. Chem. 277, 33930–33942.
Chen J.F., Sonsalla P.K., Pedata F., Melani A., Domenici M.R., Popoli P., Geiger J.,
Lopes L.V. and de Mendonça A. (2007) Adenosine A2A receptors and brain injury:
broad spectrum of neuroprotection, multifaceted actions and "fine tuning"
modulation. Prog. Neurobiol. 83, 310-331.
Chen Z., Gibson T.B., Robinson F., Silvestro L., Pearson G., Xu B., Wright A.,
Vanderbilt C. and Cobb M.H. (2001b) MAP kinases Chem. Rev. 101, 2449-2476.
Chijiwa T., Mishima A., Hagiwara M., Sano M., Hayashi K., Inoue T., Naito K.,
Toshioka T., and Hidaka H. (1990) Inhibition of forskolin-induced neurite
outgrowth and protein phosphorylation by a newly synthesized selective inhibitor
of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-
isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol.
Chem. 265, 5267–5272.
Clayton D.A., Mesches M.H., Alavarez E., Bickford P. and Browning M.D. (2002) A
hippocampal NR2B deficit can mimic age-related changes in long-term

149
potentiation and spatial learning in the Fischer 344 rat. J. Neurosci. 22, 3628–
3637.
Collingridge G.L., Kehl S.J. and McLennan H. (1983) Excitatory amino acids in
synaptic transmission in the schaffer collateral-commissural pathway of the rat
hippocampus. J. Physiol. 334, 33-46.
Condorelli D.F., Dell’Albani P., Mudò G., Timmusk T. and Belluardo N. (1994)
Expression of neurotrophins and their receptors in primary astroglial cultures:
induction by cyclic AMP-elevating agents J. Neurochem. 63, 509-516.
Connor B, Young D, Yan Q, Faull RL, Synek B and Dragunow M (1997) Brain-derived
neurotrophic factor is reduced in Alzheimer's disease. Brain Res. Mol. Brain Res.
49, 71-81.
Connor B. and Dragunow M. (1998) The role of neuronal growth factors in
neurodegenerative disorders of the human brain. Brain. Res. Rev. 27, 1–39.
Corvol J.C., Studler J.M., Schonn J.S., Girault J.A. and Hervé D. (2001) G alpha(olf)
is necessary for coupling D1 and A2A receptors to adenylyl cyclase in the
striatum. J. Neurochem. 76, 1585-1588.
Costenla A.R., de Mendonça A. and Ribeiro J.A. (1999) Adenosine modulates
synaptic plasticity in hippocampal slices from aged rats. Brain Res. 851, 228–
234.
Croll S.D., Chesnutt C.R., Rudge J.S., Acheson A., Ryan T.E., Siuciak J.A., et al.
(1998). Co-infusion with a TrkB-Fc receptor body carrier enhances BDNF
distribution in the adult rat brain. Exp. Neurol. 152, 1–15.
Cunha R.A., Constantino M.C., Sebastião A.M. and Ribeiro J.A. (1995) Modification
of A1 and A2A adenosine receptor binding in aged striatum, hippocampus and
cortex of the rat. Neuroreport. 316, 1583-1588

150
Cunha R.A., Vizi E.S., Ribeiro J.A. and Sebastião A.M. (1996) Preferential release of
ATP and its extracellular catabolism as a source of adenosine upon high but not
low-frequency stimulation of rat hippocampal slices. J. Neurochem. 67, 2180–
2187.
Cunha R.A. (1997) Release of ATP and adenosine and formation of extracellular
adenosine in the hippocampus in: The role of adenosine in the nervous system,
ed. by Y. Okada, Elsevier Science, pp.135-142.
Cunha R.A., Constantino M.D. and Ribeiro J.A. (1999) G protein coupling of CGS
21680 binding sites in the rat hippocampus and cortex is different from that of
adenosine A1 and striatal A2A receptors Naunyn Schmiedebergs Arch.
Pharmacol. 359, 295-302.
Cunha R.A., Almeida T. and Ribeiro J.A. (2001) Parallel modification of adenosine
extracellular metabolism and modulatory action in the hippocampus of aged rats.
J. Neurochem. 76, 372-382.
Dall'Igna O.P., Porciúncula L.O., Souza D.O., Cunha R.A. and Lara D.R. (2003)
Neuroprotection by caffeine and adenosine A2A receptor blockade of β-amyloid
neurotoxicity. Br. J. Pharmacol. 138, 1207–1209.
Dall'Igna O.P., Fett P., Gomes M.W., Souza D.O., Cunha R.A. and Lara D.R. (2007)
Caffeine and adenosine A2A receptor antagonists prevent β-amyloid (25–35)-
induced cognitive deficits in mice. Exp. Neurology 203, 241–245.
Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. (1997)
Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death
machinery. Cell 91, 231–241.

151
de Mendonça A. and Ribeiro J.A. (2000) Long-term potentiation observed upon
blockade of adenosine A1 receptors in the hippocampus is N-methyl-D-aspartate
receptor dependent. Neurosci. Lett. 291, 81–84.
del Peso L., González-García M., Page C., Herrera R. and Nuñez G. (1997)
Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt.
Science 278, 687-689.
Díaz-Cabiale Z., Hurd Y., Guidolin D., Finnman U.B., Zoli M., Agnati L.F.,
Vanderhaeghen J.J., Fuxe K. and Ferré S. (2001) Adenosine A2A agonist CGS
21680 decreases the affinity of dopamine D2 receptors for dopamine in human
striatum. Neuroreport. 12, 1831-1834.
Diógenes M.J., Fernandes C.C., Sebastião A.M. and Ribeiro J.A. (2004) Activation of
adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of
synaptic transmission in hippocampal slices. J. Neurosci. 24, 2905-2913.
Diógenes M.J., Assaife-Lopes N., Pinto-Duarte A., Sebastião A.M., Ribeiro J.A.
(2007) Influence of age on BDNF modulation of hippocampal synaptic
transmission: interplay with adenosine A2A receptors. Hippocampus. 17, 577-585.
Dixon A.K., Gubitz A.K., Sirinathsinghji D.J., Richardson P.J. and Freeman T.C.
(1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br. J.
Pharmacol. 118, 1461–1468.
Doody R.S., Stevens J.C.and Beck C. (2001) Practice parameter: management of
dementia (an evidence-based review). Report of the Quality Standards
Subcommittee of the American Academy of Neurology. Neurology 56, 1154–
1166.

152
Dunwiddie T.V. and Hass H.L. (1985) Adenosine increases synaptic facilitation in the
in vitro rat hippocampus: evidence for a presynaptic site of action. J. Physiol. 369,
365-377.
Eide F.F., Vining E.R., Eide B.L., Zang K., Wang X.Y. and Reichardt L.F. (1996)
Naturally occurring truncated TrkB receptors have dominant inhibitory effects on
brain-derived neurotrophic factor signalling. J. Neurosci. 16, 3123–3129.
Ferrer I., Marín C., Rey M.J., Ribalta T., Goutan E., Blanco R., Tolosa E. and Martí E.
(1999) BDNF and full-length and truncated TrkB expression in Alzheimer’s
disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 58,
729-739.
Ferré S., Rimondini R., Popoli P., Reggio R., Pèzzola A., Hansson A.C., Andersson
A. and Fuxe K. (1999) Stimulation of adenosine A1 receptors attenuates
dopamine D1 receptor-mediated increase of NGFI-A, c-fos and jun-B mRNA
levels in the dopamine-denervated striatum and dopamine D1 receptor-mediated
turning behaviour. Eur. J. Neurosci. 11, 3884-3892.
Ferrendelli J.A., Blank A.C. and Gross R.A. (1980) Relationships between seizure
activity and cyclic nucleotide levels in brain. Brain Res. 200, 93–103.
Figurov A., Pozzo-Miller L.D., Olafsson P., Wang T. and Lu B. (1996) Regulation of
synaptic responses to high-frequency stimulation and LTP by neurotrophins in the
hippocampus. Nature (Lond) 381, 706–709.
Finder V.H. and Glockshuber R. (2007) Amyloid-beta aggregation. Neurodegener.
Dis. 4, 13-27.
Fischer W., Wictorin K., Björklund A., Williams L.R., Varon S. and Gage F.H. (1987)
Amelioration of cholinergic neuron atrophy and spatial memory impairment in
aged rats by nerve growth factor. Nature. 329, 65-68.

153
Florio C., Prezioso A., Papaioannou A. and Vertua, R. (1998) Adenosine A1 receptors
modulate anxiety in CD1 mice. Psychopharmacology (Berl.) 136, 311–319.
Fontinha B.M., Diógenes M.J., Ribeiro J.A. and Sebastião A.M. (2007) Enhancement
of long-term potentiation by BDNF requires adenosine A2A receptor activation by
endogenous adenosine. Neuropharmacology. In press.
Fredholm B.B., Bätig K., Holmén J., Nehlig A. and Zvartau E.E. (1999) Actions of
caffeine in the brain with special reference to factors that contribute to its
widespread use. Pharmacol. Rev. 51, 83–133.
Fredholm B.B., Ijzerman A.P., Jacobson K.A., Klotz K-N. and Linden J. (2001)
International union of pharmacology. XXV. Nomenclature and classification of
adenosine receptors. Pharmacol. Rev. 53, 527–552
Fredholm B.B., Chen J.-F. and Masino S.A. and Vaugeois J.-M. (2005) Actions of
adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annu.
Rev. Pharmacol. Toxicol. 45, 385–412.
Gooney M., Messaocine E., Maher F.O., Bramham C.R. and Lynch M.A. (2004)
BDNF-induced LTP in dentate gyrus is impaired with age: analysis of changes in
cell signalling events. Neurobiol. Aging 25, 1323-1331.
Gottschalk W., Pozzo-Miller L.D., Figurov A. and Lu B. (1998) Presynaptic modulation
of synaptic transmission and plasticity by brain-derived neurotrophic factor in the
developing hippocampus. J. Neurosci. 18, 6830-6839.
Gotz R., Koster R., Winkler C., Raulf F., Lottspeich F., Schartl M. and Tho H. (1994)
Neurotrophin-6 is a new member of the nerve growth factor family. Nature. 372,
266-269.

154
Griffin R., Nally R., Nolan Y., McCartney Y., Linden J. and Lynch M.A. (2006) The
age-related attenuation in long-term potentiation is associated with microglial
activation. J. Neurochem. 99, 1263–1272
Gu J.G., Foga I.O., Parkinson F.E. and Geiger J.D. (1995) Involvement of
bidirectional adenosine transporters in the release of L-[3H]adenosine from rat
brain synaptosomal preparations. J. Neurochem. 64, 2105–2110.
Haapasalo A., Sipola I., Larsson K., Akerman K.E., Stoilov P., Stamm S., Wong G.
and Castren E. (2002) Regulation of TRKB surface expression by brain-derived
neurotrophic factor and truncated TRKB isoforms. J. Biol. Chem. 277, 43160-
43167.
Hauber W., and Bareiß A. (2001) Facilitative effects of an adenosine A1/A2 receptor
blockade on spatial memory performance of rats: selective enhancement of
reference memory retention during the light period. Behav. Brain Res. 118, 43–
52.
Havenaar R., Meijer J.C., Morton D.B., Ritskes-Hoitinga and Zwart P. (1993) Biology
and husbandry of laboratory animals in: Principles of laboratory animal science. A
contribution to the humane use and care of animals and to the quality of
experimental results. Ed. by Van Zutphen L.F.M., Baumans V. and Beynen A.C.,
Elsevier, NY, pp.17-76.
He X.L. and Garcia K.C. (2004) Structure of nerve growth factor complexed with the
shared neurotrophin receptor p75. Science. 304, 870-875.
Heesea K., Bernd L.F., Bauerb J., and Ottena U. (1997) Nerve growth factor (NGF)
expression in rat microglia is induced by adenosine A2A-receptors, Neurosci. Let.
231, 83–86

155
Henion W.F., Sutherland E.W. and Posternak T.H. (1967) Effects of derivatives of
adenosine 3’,5’-phosphate on liver slices and intact animals. Biochem. Biophys.
Acta 148, 106–113.
Huang E.J. and Reichardt L.F. (2001) Neurotrophins: roles in neuronal development
and function. Annu. Rev. Neurosci. 24, 677-736.
Huang E.J. and Reihardt L.F. (2003) Trk receptors: roles in neuronal signal
transduction. Ann. Rev. Biochem. 72, 609-642.
Huang N-K., Lin Y-W., Huang C-L., Messing R.O. and Chern Y. (2001) Activation of
protein kinase A and atypical protein kinase C by A2A adenosine receptors
antagonizes apoptosis due to serum deprivation in PC12 cells. J. Biol. Chem.
276, 13838–13846
Jacobson K.A. and Gao Z.G. (2006) Adenosine receptors as therapeutic targets. Nat.
Rev. Drug Discov. 5, 247-264.
Jain N., Kemp N., Adeyemo O., Buchanan P. and Stone T.W. (1995) Anxiolytic
activity of adenosine receptor activation in mice. Br. J. Pharmacol. 116, 2127–
2133.
Jarvis M.F., Schulz R., Hutchison A.J., Do U.H., Sills M.A. and Williams M. (1989)
[3H]CGS 21680, a selective A2 adenosine receptor agonist, directly labels A2
receptors in rat brain. J. Pharmacol. Exp. Ther. 25, 888–893.
Jones P.A., Smith R.A. and Stone T.W. (1998) Protection against kainate-induced
excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience
85, 229–237
Kang H. and Schuman E.M. (1995) Long-lasting neurotrophin-induced enhancement
of synaptic transmission in the adult hippocampus. Science 267, 1658–1662.

156
Kang H. and Schuman E.M. (1996) A requirement for local protein synthesis in
neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406.
Kaplan D.R., Martin-Zanca D. and Parada L.F. (1991) Tyrosine phosphorylation and
tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature
350, 158-160.
Kaplan D.R.and Miller F.D. (2000) Neurotrophin signal transduction in the nervous
system. Curr. Opin. Neurobiol.10, 381–391.
Katoh-Semba R., Takeuchi I.K., Semba R. and Kato K. (1997) Distribution of brain-
derived neurotrophic factor in rats and its changes with development in the brain.
J. Neurochem. 69, 34-42
Klein R., Jing S.Q., Nanduri V., O'Rourke E. and Barbacid M. (1990) The trk proto-
oncogene encodes a receptor for nerve growth factor. Cell 65, 189-197.
Koponen E., Võikar V., Riekki R., Saarelainen T., Rauramaa T., Rauvala H., Taira T.,
and Castrén E. (2004) Transgenic mice overexpressing the full-length
neurotrophin receptor trkB exhibit increased activation of the trkB-PLCgamma
pathway, reduced anxiety, and facilitated learning. Mol. Cell Neurosci. 26, 166-
181.
Kovalchuk Y., Holthoff K. and Konnerth A. (2004) Neurotrophin action on a rapid
timescale. Curr. Opin. Neurobiol. 14, 558-563.
Kunugi H., Hashimoto R., Yoshida M., Tatsumi M. and Kamijima K. (2004) A
missense polymorphism (S205L) of the low-affinity neurotrophin receptor
p75NTR gene is associated with depressive disorder and attempted suicide Am.
J. Med. Genet. B. Neuropsychiatr. Genet. 129, 44-46.
Lai K.O., Fu W.Y., Ip FC. and Ip N.Y. (1998) Cloning and expression of a novel
neurotrophin, NT-7, from carp. Mol. Cell Neurosci. 11, 64-76.

157
Landfield P.W. and Lynch G. (1977) Impaired monosynaptic potentiation in in vitro
hippocampal slices from aged, memory-deficient rats. J. Gerontol. 32, 523-533
Landfield P.W., McGaugh J.L. and Lynch G. (1978) Impaired synaptic potentiation
process in the hippocampus of aged, memory deficient rats. Brain Res. 150, 85–
101.
Latini S. and Pedata F. (2001) Adenosine in the central nervous system: release
mechanisms and extracellular concentrations. J. Neurochem. 79, 463–484.
Lee F.S. and Chao M.V. (2001) Activation of Trk neurotrophin receptors in the
absence of neurotrophins. Proc. Natl. Acad. Sci. USA 98, 3555–3560.
Lee R., Kermani P., Teng K.K. and Hempstead B.L. (2001) Regulation of cell survival
by secreted proneurotrophins. Science 294, 1945-1948.
Lessmann V., Gottmann K. and Malcangio M. (2003) Neurotrophin secretion: current
facts and future prospects. Prog. Neurobiol. 69, 341-374.
Levi-Montalcini R. and Cohen S. (1956) In vitro and in vivo effects of a nerve growth-
stimulating agent isolated from snake venon. Proc. Natl. Acad. Sci. USA 42, 695-
699
Levine E.S., Crozier R.A., Black I.B. and Plummer M.R. (1998) Brain-derived
neurotrophic factor modulates hippocampal synaptic transmission by increasing
N-methyl-D-aspartic acid receptor activity. Proc. Natl. Acad. Sci. USA 95, 10235–
10239.
Lewin G.R. and Barde Y.A. (1996) Physiology of the neurotrophins. Annu. Rev.
Neurosci. 19, 289-317.
Li C.L. and McIlwIN H. (1957) Maintenance of resting membrane potentials in slices
of mammalian cerebral cortex and other tissues in vitro. J. Physiol. 139, 178-190.

158
Liepinsh E., Ilag L.L., Otting G. and Ibáñez C.F. (1997). NMR structure of the death
domain of the p75 neurotrophin receptor. Embo J. 16, 4999-5005.
Lim K.C., Lim S.T. and Federoff H.J. (2003) Neurotrophin secretory pathways and
synaptic plasticity. Neurobiol. Aging. 24,1135-1145.
Linden J. (2001) Molecular approach to adenosine receptors: receptor-mediated
mechanisms of tissue potection. Ann. Rev. Pharmacol. Toxicol. 41, 775-787.
Lohse M.J., Klotz K.N., Lindenborn-Fotinos J., Reddington M., Schwabe U. and
Olsson R.A. (1987) 8-Cyclopentyl-1,3-dipropylxanthine (DPCPX)- a selective high
affinity antagonist radioligand for A1 adenosine receptors. Naunyn
Schmiedebergs Arch. Pharmacol. 336, 204–210.
Londos C., Cooper D.M. and Wolff J. (1980) Subclasses of external adenosine
receptors. Proc. Natl. Acad. Sci. USA 77, 2551-2554.
Loo D.T., Copani A., Pike C.J., Whittemore E.R., Walencewicz A.J. and Cotman C.W.
(1993) Apoptosis is induced by β-amyloid in cultured central nervous system
neurons. Proc. Natl. Acad. Sci. U.S.A. 90, 7951-7955
Lopes da Silva F.H. and Arnolds D.E. (1978) Physiology of the hippocampus and
related structures. Annu. Rev. Physiol. 40, 185-216.
Lopes L.V., Cunha R.A. and Ribeiro J.A. (1999a) Increase in the number, G protein
coupling, and efficiency of facilitatory adenosine A2A receptors in the limbic
cortex, but not striatum, of aged rats. J. Neurochem. 73, 1733-1738.
Lopes L.V., Cunha R.A. and Ribeiro J.A. (1999b) Cross talk between A(1) and A(2A)
adenosine receptors in the hippocampus and cortex of young adult and old rats.
J. Neurophysiol. 82, 3196-3203.
Lu B., Pang P.T. and Woo N.H. (2005) The yin and yang of neurotrophins action. Nat.
Neurosci. 6, 603-614.

159
Lupica C.R., Proctor W.R. and Dunwiddie T-U. (1992) Presynaptic inhibition of
excitatory synaptic transmission by adenosine in rat hippocampus: analysis of
unitary fEPSP variance measured by whole-cell recording. J. Neurosci. 12, 3753–
3764.
Lynch M.A. (1998) Analysis of the mechanisms underlying the age-related impairment
in long-term potentiation in the rat. Rev. Neurosci. 9, 169-201.
Lynch M.A. (2004) Long-term potentiation and memory. Physiol Rev. 84:87-136.
Magnusson K.R. (2000) Declines in mRNA expression of different subunits may
account for differential effects of aging on agonist and antagonist binding to the
NMDA receptor. J. Neurosc. 20, 1666–1674.
Maia L. and de Mendonça A. (2002) Does caffeine intake protect from Alzheimer's
disease? Eur. J. Neurol. 9, 377-382
Mayne M., Fotheringham J., Yan H.J., Power C., Del Bigio M.R., Peeling J., Geiger
J.D. (2001). Adenosine A2A receptor activation reduces proinflammatory events
and decreases cell death following intracerebral hemorrhage. Ann. Neurol. 49,
727–735.
Maurice T., Lockhart B.P. and Privat A. (1996) Amnesia induced in mice by centrally
administrered beta-amyloid peptides involves cholinergic dysfunction. Brain Res.
706, 181-193.
McIIwain H., Buchel L. and Chesire J.D. (1951) The inorganic phosphate and
phosphocreatine of brain especially during metabolism in vitro. Biochem. J. 48,
12-20.
Meghji P.V. (1992) Adenosine production and metabolism in the: Adenosne in the
nervous system ed. by Stone T., Academinc Press, London, pp. 25-42.

160
Meyer-Franke A., Wilkinson G.A., Kruttgen A., Hu M., Munro E., Hanson M.G.,
Reichardt L.F. and Barres B.A. (1998) Depolarisation and cAMP elevation rapidly
recruit TrkB to the plasma membrane of CNS neurons. Neuron 21, 681–693.
Mogi M., Togari A., Kondo T., Mizuno Y., Komure O., Kuno S., Ichinose H. and
Nagatsu T. (1999) Brain-derived neurotrophic factor and nerve-growth factor
concentrations are decreased in the substantia nigra in Parkinson’s disease.
Neurosci. Lett. 270, 45–48.
Momose Y., Murata M., Kobayashi K., Tachikawa M., Nakabayashi Y., Kanazawa I.
and Toda T. (2002) Association studies of multiple candidate genes for
Parkinson's disease using single nucleotide polymorphisms. Ann. Neurol. 51,
133-136.
Müller C.E. and Ferré S. (2007) Blocking striatal adenosine A2A receptors: a new
strategy for basal ganglia disorders. Recent Patents CNS Drug Discov. 2, 1-21.
Murer M.G., Yan Q. and Raisman-Vozari R. (2001) Brain-derived neurotrophic factor
in the control human brain, and in Alzheimer’s disease and Parkinson’s disease.
Prog. Neurobiol. 63, 71-124.
Nagata H., Mimori Y., Nakamura S. and Kameyama M. (1984) Regional and
subcellular distribution in mammalian brain of the enzymes producing adenosine.
J. Neurochem. 42, 1001-1007.
Narisawa-Saito M. and Nawa H. (1996) Differential regulation of hippocampal
neurotrophins during aging in rats. J. Neurochem. 67, 1124– 1131.
Neves-Pereira M., Mundo E., Muglia P., King N., Macciardi F. and Kennedy J.L.
(2002)The brain-derived neurotrophic factor gene confers susceptibility to bipolar
disorder: evidence from a family-based association study. Am. J. Hum. Genet.
71, 651-655.

161
Nitta A., Murai R., Suzuki N., Ito H., Nomoto H., Katoh G., Furukawa Y. and Furukawa
S. (2002) Diabetic neuropathies in brain are induced by deficiency of BDNF.
Neurotoxico. Teratol. 24, 695–701.
Oberhammer F.A., Pavelka M., Sharma S., Tiefenbacher R., Purchio A.F., Bursch W.,
Schulte-Hermann R. (1992) Induction of apoptosis in cultured hepatocytes and in
regressing liver by transforming growth factor β1. Proc. Natl. Acad. Sci. USA 89,
5408-5412.
Offermanns S. and Simon M.I. (1995) G alpha 15 and G alpha 16 couple a wide
variety of receptors to phospholipase C. J. Biol. Chem. 270, 15175-15180.
Pak M.A., Haas H.L., Decking U.K.M. and Schrader J. (1994) Inhibition of adenosine
kinase increases endogenous adenosine and depresses neuronal activity in
hippocampal slices. Neuropharmacology 33, 1049–1053.
Palkovits M. and Brownstein M.J. (1983) Microdissection of brain areas by the punch
technique in: Brain microdissection techniques ed. by Cuello A. C. MeElsevier,
NY, pp. 1-37.
Palmer T.M., Poucher S.M., Jacobson K.A. and Stiles G.L. (1995) 125I-4-(2-[7-amino-
2-[2-furyl][1,2,4]triazolo[2,3-a][1,3,5]triazin-5-yl-amino]ethyl)phenol, a high affinity
antagonist radioligand selective for the A2A adenosine receptor. Mol. Pharmacol.
48, 970-974
Parain K., Murer M.G., Yan Q., Faucheux B., Agid Y., Hirsch E. and Raisman-Vozari
R. (1999) Reduced expression of brain-derived neurotrophic factor protein in
Parkinson's disease substantia nigra. Neuroreport 10, 557-561.
Patterson S.L., Pittenger C., Morozov A., Martin K.C., Scanlin H. Drake C. and Kandel
E.R. (2001) Some forms of cAMP-mediated long-lasting potentiation are

162
associated with release of BDNF and nuclear translocation of phospho-MAP
kinase. Neuron 32, 123–140.
Pazzagli M., Pedata F. and Pepeu G. (1993) Effect of K+ depolarisation, tetrodotoxin,
and NMDA receptor inhibition on extracellular adenosine levels in rat striatum.
Eur. J. Pharmacol. 234, 61–65.
Phillis E. and Newsholme E.A. (1979) Maximum activities, properties and distribution
of 5’-nucleotidase, adenosine kinase and adenosine deaminase in rat and human
brain. J. Neurochem. 33, 553-558.
Phillis J.W. (1995) The effects of selective A1 and A2A adenosine receptor antagonists
on cerebral ischemic injury in the gerbil. Brain Res. 705, 79– 84.
Phillips H.S., Hains J.M., Armanini M., Laramee G.R., Johnson S.A and Winslow
J.W. (1991) BDNF mRNA is decreased in the hippocampus of individuals with
Alzheimer’s disease. Neuron 7, 695-702.
Poo M.M. (2001) Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2, 24-32.
Porkka-Heiskanen T., Strecker R.E., Thakkar M., Bjorkum A.A., Greene R.W. and
McCarley R.W. (1997) Adenosine: a mediator of the sleep-inducing effects of
prolonged wakefulness. Science. 276, 1265-1268.
Potier B., Rascol O., Jazat F., Lamour Y. and Dutar P. (1992) Alterations in the
properties of hippocampal pyramidal neurons in the aged rat. Neuroscience 48,
793–806.
Poucher S.M., Keddie J.R., Singh P., Stoggall S.M., Caulkett P.W., Jones G. and Coll
M.G. (1995) The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2A
selective adenosine receptor antagonist. Br. J. Pharmacol. 115, 1096–1102.

163
Pousinha P.A., Diógenes M.J., Sebastião A.M. and Ribeiro J.A. (2006) Triggering of
BDNF facilitatory action on neuromuscular transmission by adenosine A2A
receptors. Neurosci. Lett. 404, 143-147.
Ramirez S.H., Fan S., Maguire C.A., Perry S., Hardiek K., Ramkumar V., Gelbard
H.A, Dewhurst S. and Maggirwar S.B. (2004) Activation of adenosine A2A receptor
protects sympathetic neurons against nerve growth factor withdrawal. J.
Neurosci. Res. 77, 258–269.
Rapp P.R. and Gallagher M.U. (1996) Preserved neuron number in the hippocampus
of aged rats with spatial learning deficits. Proc. Natl Acad. Sci. USA 93, 9926–
9930.
Rasmussen T., Schliemann T., Sorensen J.C., Zimmer J. and West M.J. (1996)
Memory impaired aged rats: no loss of principal hippocampal and subicular
neurons. Neurobiol. Aging 17, 143–147.
Rebola N., Sebastiao A.M., de Mendonça A., Oliveira C.R., Ribeiro J.A. and Cunha
R.A. (2003) Enhanced adenosine A2A receptor facilitation of synaptic transmission
in the hippocampus of aged rats. J. Neurophysiol. 90, 1295-1303.
Rebola N., Rodrigues R.J., Lopes L.V., Richardson P.J., Oliveira C.R. and Cunha
R.A. (2005a) Adenosine A1 and A2A receptors are co-expressed in pyramidal
neurons and co-localized in glutamatergic nerve terminals of the rat
hippocampus. Neuroscience. 133, 79-83.
Rebola N., Canas P.M., Oliveira C.R. and Cunha R.A. (2005b) Different synaptic and
subsynaptic localization of adenosine A2A receptors in the hippocampus and
striatum of the rat. Neuroscience. 132, 893-903
Reichardt L.F. (2006) Neurotrophin-regulated signalling pathways. Phil. Trans. R.
Soc. B 361, 1545– 564.

164
Ribeiro J.A., Sebastião A.M. and de Mendonça A. (2003) Adenosine receptors in the
nervous system: pathophysiological implications. Prog. Neurobiol. 68, 377-392.
Riccio A., Ahn S., Davenport C.M. Blendy J.A. and Ginty D. D. (1999) Mediation by a
CREB family transcription factor of NGF-dependent survival of sympathetic
neurons.Science 286, 2358–2361 (1999).
Richards C.D. and McIlwain H. (1967) Electrical responses in bain samples. Nature
215, 704-707.
Rojo L.E., Fernández J.A., Maccioni A.A., Jimenez J.M. and Maccioni R.B. (2008)
Neuroinflammation: implications for the pathogenesis and molecular diagnosis of
Alzheimer's disease. Arch. Med. Res. 39, 1-16.
Rodrigues C.M.P., Sóla S. and Brites D. (2002) Bilirubin induces apoptosis via the
mitochondrial pathway in developing rat brain neurons. Hepatology 35, 1186-
1196.
Rosi S., McGann K., Hauss-Wegrzyniak B. and Wenk G.L. (2003) The influence of
brain inflammation upon neuronal adenosine A2B receptors. J. Neurochem. 86,
220-227.
Russo-Neustadt A., Ha T., Ramirez R. and Kesslak J.P. (2001) Physical activity-
antidepressant treatment combination: impact on brain-derived neurotrophic
factor and behavior in an animal model. Behav. Brain Res. 120, 87-95
Saarelainen T., Pussinen R., Koponen E., Alhonen L., Wong G., Sirviö J. and Castrén
E. (2000) Transgenic mice overexpressing truncated trkB neurotrophin receptors
in neurons have impaired long-term spatial memory but normal hippocampal LTP.
Synapse. 38, 102-104.
Schulte G. and Fredholm B.B. (2003) Signalling from adenosine receptors to mitogen-
activated protein kinases. Cell Signal. 15, 813-827.

165
Sebastião A.M., Stone T.W. and Ribeiro J.A. (1990) The inhibitory adenosine receptor
at the neuromuscular junction and hippocampus of the rat: antagonism by 1,3,8-
substituted xanthines. Br. J. Pharmacol. 101, 453–459.
Sebastião A.M. and Ribeiro J.A. (1996) Adenosine A2 receptor-mediated excitatory
actions on the nervous system. Prog. Neurobiol. 48, 167-189.
Sebastião A.M. and Ribeiro J.A. (2000) Fine-tuning neuromodulation by adenosine.
Trends Pharmacol. Sci. 21, 341–346.
Sebastião A.M., Cunha R.A., de Mendonça A. and Ribeiro J.A. (2000) Modification of
adenosine modulation of synaptic transmission in the hippocampus of aged rats.
Br. J. Pharmacol. 131, 1629-1634.
Sebastião A.M., de Mendonça A., Moreira T. and Ribeiro J.A. (2001) Activation of
synaptic NMDA receptors by action potential-dependent release of transmitter
during hypoxia impairs recovery of synaptic transmission on reoxygenation. J.
Neurosci. 21, 8564-8571.
Segal R.A. and Greenberg M.E. (1996) Intracellular signalling pathways activated by
neurotrophic factors. Annu. Rev. Neurosci. 19, 463-489.
Seiger A, Nordberg A, von Holst H, Backman L, Ebendal T, Alafuzoff I, Amberla K,
Hartvig P, Herlitz A, Lilja A, et al (1993) Intracranial infusion of purified nerve
growth factor to an Alzheimer patient: the first attempt of a possible future
treatment strategy. Behav Brain Res. 57:255-61
Selkoe D.J. (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev.
Apr. 81, 741–66
Sen S., Nesse R.M., Stoltenberg S.F., Li S., Gleiberman L., Chakravarti A., Weder
A.B., Burmeister M. (2002) A BDNF coding variant is associated with the NEO

166
personality inventory domain neuroticism, a risk factor for depression.
Neuropsychopharmacology. 28, 397-401.
Sexl V., Mancusi G., Höller C., Gloria-Maercker E., Schütz W. and Freissmuth M.
(1997) Stimulation of the mitogen-activated protein kinase via the A2A-adenosine
receptor in primary human endothelial cells. J. Biol. Chem. 272, 5792-5799.
Shelton D.L., Sutherland J., Gripp J., Camerato T., Armkanini M.P., Phillips H.S.,
Carroll K., Spencer S.D., and Levinson A.D. (1995). Human trks: molecular
cloning, tissue distribution, and expression of extracellular domain
immunoadhesins. J. Neurosci. 15, 477–491.
Shimohama S. (2000) Apoptosis in Alzheimer’s disease—an update. Apoptosis 5, 9–
16
Silhol M., Bonnichon V., Rage F. and Tapia-Arancibia L. (2005). Age-related changes
in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the
hippocampus and hypothalamus in male rats. Neuroscience 132, 613-624.
Sklar P., Gabriel S.B., McInnis M.G., Bennett P., Lim Y.M., Tsan G., Schaffner S.,
Kirov G., Jones I., Owen M., Craddock N., DePaulo J.R. and Lander E.S. (2002)
Family-based association study of 76 candidate genes in bipolar disorder: BDNF
is a potential risk locus. Brain-derived neutrophic factor. Mol. Psychiatry. 7, 579-
593.
Smith M.A. (1996) Hippocampal vulnerability to stress and aging: possible role of
neurotrophic factors. Behav. Brain. Res. 78, 25-36.
Solá S., Castro R.E., Laires P.A., Steer C.J. and Rodrigues C.M.P. (2003)
Tauroursodeoxycholic acid prevents amyloid-β peptide–induced neuronal death
via a phosphatidylinositol 3-Kinase–dependent signaling pathway. Mol. Med. 9,
226-234.

167
Sonntag W.E., Benett S.A., Khan A.S., Thomton P.L., Xu X., Ingram R.L. and Brunso-
Bechtold J.K. (2000) Age and insulin-like growth factor-1 modulate N-methyl-d-
aspartate receptor subtype expression in rats. Brain Res. Bull. 51, 331–338.
Squire L.R. and Kandel E.R. (2000) Brain systems for declarative memory in:
Memory:From Mind to Molecules ed by Squire L.R. and Kandel E.R., Scientific
American Library, NY, pp 83-109.
Suen P-C., Wu K., Levine E.S., Mount H.T.J., Xu J-L., Lin S-Y. and Black I.B. (1997).
Brain-derived neurotrophic factor rapidly enhances phosphorylation of the
postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc. Natl. Acad. Sci. USA
94, 8191-8195.
Tanaka T., Saito H. and Matsuki N. (1997) Inhibition of GABAA synaptic responses of
brain-derived neurotrophic factor (BDNF) in rat hippocampus. J. Neurosci. 17,
2959-2966.
Tebano M.T., Martire A., Potenza R.L., Grò C., Pepponi R., Armida M., Domenici
M.R., Schwarzschild M.A., Chen J.F. and Popoli P. (2008) Adenosine A2A
receptors are required for normal BDNF levels and BDNF-induced potentiation of
synaptic transmission in the mouse hippocampus. J. Neurochem. 104, 279-286.
Thoenen H. and Sendtner M (2002) Neurotrophins: from enthusiastic expectations
through sobering experiences to rational therapeutic approaches. Nat. Neurosci.
Supp. 5, 1046-1050
Tsvyetlynska N.A., Hill R.H. and Grillner S. (2005) Role of AMPA receptor
desensitization and the side effects of DMSO vehicle on reticulospinal EPSPs
and locomotor activity. J. Neurophysiol. 94, 3951-3960.

168
van Calker D., Muller M. and Hamprecht B. (1979) Adenosine regulates via two
different types of receptors, the accumulation of cyclic AMP in cultured brain
cells. J. Neurochem. 33, 999-1005.
Ventriglia M., Bocchio Chiavetto L., Benussi L., Binetti G., Zanetti O., Riva M.A. and
Gennarelli M. (2002) Association between the BDNF 196 A/G polymorphism and
sporadic Alzheimer’s disease. Mol. Psychiatry 7, 136–137
von Lubitz, D., Lin, R., Jacobson, K. (1995) Cerebral ischemia in gerbils: effects of
acute and chronic treatment with adenosine A2A receptor agonist and antagonist.
Eur. J. Pharmacol. 287, 295–302.
Wardas J., Konieczny J. and Lorenc-Koci E. (2001). SCH 58261, an A2A adenosine
receptor antagonist, counteracts Parkinsonian-like muscle rigidity in rats.
Synapse 41, 160–171.
Webster M.J., Herman M.M., Kleinman J.E. and Shannon Weickert C. (2006) BDNF
and TrkB mRNA expression in the hippocampus and temporal cortex during the
human lifespan. Gene Exp. Patterns 6, 941-951.
Wenk G.L. and Barnes C.A. (2000) Regional changes in the hippocampal density of
AMPA and NMDA receptors across the lifespan of the rat. Brain Res., 885, 1–5.
Wieraszko A., Goldsmith G. and Seyfried T.N. (1989) Stimulation-dependent release
of adenosine triphosphate from hippocampal slices. Brain Res. 485, 244–250.
Wiese S., Jablonka S., Holtmann B., Orel N., Rajagopal R., Chao M.V. and Sendtner
M. (2007) Adenosine receptor A2A-R contributes to motoneuron survival by
transactivating the tyrosine kinase receptor TrkB. Proc. Natl. Acad. Sci. USA 104,
17210-17215.
Wu D. and Pardridge W.M. (1999) Neuroprotection with non-invasive neurotrophin
delivery to the brain. Proc. Natl. Acad. Sci. USA 96, 254–259.

169
Xu B., Gottschalk W., Chow A., Wilson R.I., Schnell E., Zang K., Wang D., Nicoll R.A.,
Lu B. and Reichardt L.F. (2000). The role of brain-derived neurotrophic factor
receptors in the mature hippocampus: modulation of long-term potentiation
through a presynaptic mechanism involving TrkB. J. Neurosci. 20, 6888-6897.
Yamamoto C. and McIIwain H. (1966) Potentials evoked in vitro in preparations from
the mammalian brain. Nature 210, 1055-1056.
Yeo G.S., Connie Hung C.C., Rochford J., Keogh J., Gray J., Sivaramakrishnan S.,
O'Rahilly S. and Farooqi I.S. (2004) A de novo mutation affecting human TrkB
associated with severe obesity and developmental delay. Nat. Neurosci. 7, 1187-
1189.
Zimmermann H. and Braun N. (1999) Ecto-nucleotidases: molecular structures,
catalytic properties, and functional roles in the nervous system. Prog. Brain Res.
120, 371–385.
Zuccato C. and Cattaneo E. (2007) Role of brain-derived neurotrophic factor in
Huntington’s disease. Prog. Neurobiol. 81, 294–330


















