
Synthesis and Characterization of Polyimides: …ncl.csircentral.net/835/1/th1780.pdf · Synthesis...
227
Synthesis and Characterization of Polyimides: Membrane Material for Gas Permeation and Polymer Electrolyte for Fuel Cell A thesis submitted to the UNIVERSITY OF PUNE for the degree of DOCTOR OF PHILOSOPHY in CHEMISTRY by Sandeep S. Kothawade Research Guide Dr. Ulhas K. Kharul Polymer Science and Engineering Division National Chemical Laboratory Pune 411 008, INDIA December 2009
Transcript of Synthesis and Characterization of Polyimides: …ncl.csircentral.net/835/1/th1780.pdf · Synthesis...

Synthesis and Characterization of Polyimides:
Membrane Material for Gas Permeation
and Polymer Electrolyte for Fuel Cell
A thesis submitted to the UNIVERSITY OF PUNE
for the degree of DOCTOR OF PHILOSOPHY
in
CHEMISTRY
by
Sandeep S. Kothawade
Research Guide Dr. Ulhas K. Kharul
Polymer Science and Engineering Division National Chemical Laboratory
Pune 411 008, INDIA
December 2009

Dedicated to
My Family
Their patience, understanding, support and most importantly love has made this possible.

Acknowledgement I think, I am one of the luckiest and hence thankful to GOD, for being contoured
myself in the beautiful and fathomless world of CHEMISTRY, though accidentally. This doctoral work is nothing but my contribution for understanding the myriad intricate in science which may render the living for betterment.
There are so many people, whose support, encouragement and inspiration are very much obligatory to accomplish major achievements in life, especially, if it involves the elements of fulfilling one’s cherish dreams. For me, this thesis is such an important destiny and I am indeed indebted to lot of people for their well wishes and blessings for completing this journey.
Among the various people, I would like to take this opportunity first to Dr U. K. Kharul, my research supervisor, whose inspiration, encouragement, and continual guidance and not to forget timely pressurization, has lead me to bring my dream to reality. I owe big gratitude for his guidance and support for some of the past years. Dr. Kharul not only guided me in the scientific problem but also in my personnel life, shaping it for professional as well as personnel matching. Hence I am in debt to him.
I would also like to offer my sincere admiration to Dr. S. P. Vernekar, for all his help, support, suggestion and moral advice during the course of this study. Dr .Vernekar had been my immediate advisor in the initial stages of my research work. Infact, he acted as a window for my entrance in the research. I especially enjoyed his style of mentoring students from which I have benefited tremendously in my personal and professional growth.
I also would like to thank my research co-supervisor Dr. K. Vijaymohanan, whose advice especially in the fuel cell characterization has been valuable during the course of this study.
My thanks are duly acknowledged to CSIR, New Delhi for valuable support in the form of a Senior Research Fellowship.
I wish to place on record my sincere thanks to Dr. S. Sivaram, Director, National Chemical Laboratory for allowing me to carry out my research work at this prestigious Institute. I am grateful to Dr. B. D. Kulkarni, Deputy Director, NCL and Dr. M. G. Kulkarni, Head, PSE Division, for providing the infrastructure and facilities for my research work and allowing me to access the facilities in the division. I am thankful to NMR facility, Elemental Analysis Group, Glass Blowing and Workshop groups for their technical support. I also wish to thank the Library, Administrative and other supporting staff at NCL.
I was really an auspicious student for getting the rare opportunity in the membrane group to interact with all the students working in divergent research areas. I truly enjoyed the valuable discussions with Harshada, Yogesh Chendake, Santosh, Manoj Achalpurkar, Prasad, Sundar, Nazarul, Yogesh Bhole, Pankaj and Anita. I am thankful to Harshada who really put all the possible ways for completing my thesis work, especially in the last stages.
I owe special thanks to Mr. A. S. Patil, who assisted me in an acquirement of experimental skills at my embryonic stage of research career. I am also thankful to Dr. P. P. Wadgoankar, Dr. A. K. Lele, Dr. Guruswamy, Dr. Premanth, Dr. B. B. Idage, Dr. (Mrs.) S. B. Idage, Dr. C. V. Avadhani, Dr. R. P. Singh, Mr. K. G. Raut, Dr.(Mrs.) A. N.

Bote, Dr. S. Shreekumar, Dr. C. V. Rode, Dr. R. S. Khisti, Mr. S. K. Menon, Dr. N. N. Chavan, Dr. S. S. Mahajan, Dr. M. B. Sabne, Dr. S.D. Patil, Dr. Badigar, Dr. (Mrs.) J. P. Jog, Dr. (Mrs.) Garnaik, Mrs. D. A. Dhoble, Dr. B. D. Sarwade, Dr. A. S. Jadhav Dr. B. M. Shinde, Dr. T. P. Mohandas, Dr. P. G. Sukhla, Dr. I. S. Mulla, Dr. C. Ramesh, Mr. Borkar, Mr. Kiran Pandhare and Dr. Saini for their valuable help and cooperation during my research stay in NCL.
Contribution from one of closet friends, Liladhar Bagad and his family can not be forgotten. I feel proud to have friends like Ravi Potrekar, Arvind and Mahesh who motivated at my every stage during this research sojourn. They not only contributed my happiness but also shared sorrows. I can not forget all my lab members Rupesh, Nahire, Alkesh, Ritesh, Manoj Patil, Shubhangi, Mrunal, Soumya, Prerana, Pradnya, Bhavana, Anurag, Kailas and Yogesh with whom I spent nice time and thankful to them for their co-operation. I would like to mention special thanks to Mr. Soraj Singh for his assistance during my work in the membrane group.
I also extend my thank my friends Kannann, Vikas, JP, Wasif, Rakesh, Girish, Hamid, Ravi Sonawane, Ram, Narkhede, Sanjay Desale, Ajit Deore, Prashant Sonar, Mallikarjuna, Vivek, Sony, Arun, Vijay, Pandurang, Anjana, Mukesh, Sunil, Dnyaneshwar, Jadab, Niranjan, Bhaskar, Bhalchandra, Trupti, Harsha, Mahima, Mukta, Vishal, Pinak, Ausutosh and Satish Patil for their support and co-operation.
I find no words to express my feelings for my Father and Mother, whose moral support, love and constant encouragements have helped me to complete this journey. Their patience and sacrifice are always a main source of my inspiration and will remain throughout my life, motivating me to pursue still higher goals. I would like to thanks my Sister, Jijaji, Ashu and little Vedant for their love and prayer. I do not have any word to thank Bapu Mama, Mr. Balasaheb, Shri. Morankar Tatya, Bhushan, Dr. Tushar, Dr. Vivek, Pravin, Nilesh, Nayana, Jeetendra, Mr. Bhamare, sister-in-law, brother-in-law, mother-in-law, father-in-law, Mihir, Abhineet and Shreya for their constant encouragement.
Last, but certainly not least, I would like to acknowledge deep thanks to my wife, Ashwini. She has taken care of every problem, every possible responsibility without a complaint to help me focus on finishing my dissertation. Thank you Ashwini! I am also thankful to my sweet and cute daughter; Anwesha…..! her smiling has really been the catalyst for expediting this doctoral work.
The thesis would be useful, if my efforts are ever to be of any use for the welfare of mankind and the advancement of science.
Finally, I genuflect before the GOD (‘Shree Swami Samarth’) for giving me an opportunity to have wondered in such a nice world of polymer science.
Sandeep Subhash Kothawade

Certificate of the Guide
Certified that the work incorporated in the thesis entitled “Synthesis and
Characterization of Polyimides: Membrane Material for Gas Permeation and
Polymer Electrolyte for Fuel Cell” submitted by Sandeep Subhash Kothawade was
carried out under our supervision. Such material as has been obtained from other sources
has been duly acknowledged in this thesis.
December, 2009 Dr. U. K. Kharul Dr. K. Vijayamohanan Pune (Research Guide) (Research Co-Guide)

Declaration by the Candidate
I declare that the thesis entitled “Synthesis and Characterization of Polyimides:
Membrane Material for Gas Permeation and Polymer Electrolyte for Fuel Cell” is
my own work conducted under the supervision of Dr. U. K. Kharul, at Polymer Science
and Engineering Division, National Chemical Laboratory, Pune. I further declare that to
the best of my knowledge, this thesis does not contain any part of work, which has been
submitted for the award of any degree either of this University or any other University
without proper citation.
December 2009 Research Student Pune (Sandeep S. Kothawade)
Senior Research Fellow
Polymer Science and Engineering Division National Chemical Laboratory
Pune – 411 008, India.

Contents
Description
Page No.
Abstract……………………………………………………………………. i
List of abbreviations …………………………………………………….. iii
List of Tables……………………………………………………………… v
List of Schemes …………………………………………………………… vii
List of Figures…………………………………………………………….
viii
Chapter 1
Introduction and literature survey
1.1 Polyimides ………………………………………………………………… 1
1.2 A brief history of polyimides …………………………………………….. 1
1.3 Synthesis of polyimides…………………………………………………… 2
1.3.1 Polycondensation of a diamine and a dianhydride ………………… 2
1.3.1.1 Two-step method via poly(amic acid) intermediate………. 3
1.3.1.1.a Role of monomers……………………………….. 3
1.3.1.1.b Role of solvent ………………………………….. 4
1.3.1.1.c Transformation of poly(amic acid) to polyimide…. 5
(i) Chemical imidization of poly(amic acid)….. 6
(ii) Thermal Imidization of poly(amic acid)…… 6
1.3.1.2 One-step or one pot method ………………………………... 7
1.4 Properties and applications of polyimides…………………………………. 7
1.5 Gas Permeation in polyimides………………………………..……………. 9
1.5.1 Application of membrane based gas separation……..…………… 11
1.5.2 Theoretical consideration……………………………..……………. 12
1.5.3 Physical parameters affecting gas permeation………..……………. 15
1.6 Sulfonated polyimides as polymer electrolyte membrane (PEM) materials 19
1.6.1 Synthesis of sulfonated polyimides (SPIs) …………..……………. 19
I

1.6.1.1 SPI based on sulfonated diamine (SDA) as monomers…… 20
1.6.1.2 SPI based on naphthalic type dianhydrides (PNI)………… 23
1.6.2 Membrane preparation of SPIs …………………..………………. 24
1.7 Other polymers as polymer electrolyte membrane (PEM) materials 24
1.7.1 Perfluorosulfonic acid (PFSA) membranes…………………..…… 24
1.7.2 Polymers with sulfonic acid group…………………..……………. 25
1.7.2.1 Post-sulfonation of polymers …………………..………… 26
1.7.2.2 Sulfonated polymers and copolymers from sulfonated monomers …………………..……………………………..
27
1.8 Fuel Cells…………………..……………………………………………… 29
1.8.1 Types of Fuel cells (FC) …………………………………………... 30
1.8.2 Evolution and major aspects of PEMFC…………………………… 32
1.8.3 Advantages and functioning of PEMFC…………………………… 32
1.8.4 Criteria for PEM materials ………………………………………… 33
1.9 Aims and objectives…………..…………………………………………… 36
1.10 Organization of the thesis…………..………………………………………
36
Chapter 2
Monomer synthesis and characterization
2.1 Introduction…………..……………………………………………………. 39
2.2 Experimental………..…………………………………………………….. 41
2.2.1 Materials…..……………………………………………………… 41
2.2.2 Analytical methods………………………………………………. 41
2.2.3 Synthesis of PDNB, PDAB and SPDAB based on unsubstituted phenol………..…………………………………………………….
42
2.2.3.1 Synthesis of 1-phenoxy-2,4-dinitrobenzene (PDNB)……. 42
2.2.3.2 Synthesis of 1-phenoxy-2,4-diaminobenzene (PDAB)……. 43
2.2.3.3 Synthesis of 4-sulfonic-1-phenoxy-2,4-diaminobenzene (SPDAB) ………..…………………………………………
43
2.2.4 Synthesis of dinitro precursors and diamines based on CDNB and substituted phenols ..…………………………………………..
44
2.2.4.1 Synthesis of 2-methyl-1-phenoxy-2,4-dinitro benzene (2MPDNB) ..……………………………………………..
44
II

2.2.4.2 Synthesis of 2methyl-1-phenoxy-2,4-diamino benzene (2MPDAB) ) ..……………………………………………
45
2.2.4.3 Synthesis of 4-methyl-1-phenoxy-2,4-dinitro benzene (4MPDNB) .……………………………………………
45
2.2.4.4 Synthesis of 4-methyl-1-phenoxy-2,4-diamino benzene (4MPDAB) .……………………………………………
46
2.2.4.5 Synthesis of 2,6-dimethyl-1-phenoxy-2,4-dinitro benzene (2,6DMPDNB) ………………………………..
46
2.2.4.6 Synthesis of 2,6-dimethyl-1-phenoxy-2,4-diamino benzene (2,6DMPDAB) ………………………………..
46
2.2.4.7 Synthesis of 4’-t-butyl-1-phenoxy-2,4-dinitro benzene (4tBPDNB) .……………………………………………..
47
2.2.4.8. Synthesis of 4’-t-butyl-1-phenoxy-2,4-diamino benzene (4tBPDAB) .……………………………………………..
47
2.3 Results and Discussion……………………………………………………. 48
2.3.1 Reaction of CDNB with unsubstituted phenols …….…………….. 48
2.3.1.1 Synthesis and characterization of PDNB and PDAB…….. 48
2.3.1.2 Synthesis and characterization of SPDAB………………. 51
2.3.2 Reaction of CDNB with substituted phenols……………………… 52
2.3.2.1 Synthesis and characterization of 2MPDNB and 2MPDAB.……………………………………………..
52
2.3.2.2 Synthesis and characterization of 4MPDNB and 4MPDAB.……………………………………………..
55
2.3.2.3 Synthesis and characterization of 26DMPDNB and 26DMPDAB.……………………………………………..
58
2.3.2.4 Synthesis and characterization of 4tBPDNB and 4tBPDAB.……………………………………………..
60
2.4 Conclusions .……………………………………………………………….. 63
Chapter 3
Polyimide synthesis and their characterization
3.1 Introduction.……………………………………………………………….. 64
3.2 Materials .…………………………………………………………………. 65
3.3 Polymer Synthesis…………………………………………………………. 66
3.3.1 Polyimides (PIs) and copolyimides (CPIs) based on PDAB ……… 66
3.3.1.1 Synthesis of PIs……………………………………………. 66
3.3.1.2 Preparation of dense membrane………………………….. 67
III

3.3.2 Sulfonated polyimides (SPIs) based on SPDAB…………………. 68
3.3.2.1 Synthesis of SPI…………………………………………… 68
3.3.2.2 Synthesis of polybenzimidazoles (PBIs) ………………… 69
3.3.2.3 Preparation of dense membrane based on sulfonated PIs and their proton exchange…………………………………
70
3.3.2.4 Preparation of dense membrane based on PBIs…………… 71
3.3.2.5 Preparation of blend membranes based on SPI and PBI….. 71
3.3.3 Polyimides based on alkyl substituted phenoxy diamines…………. 72
3.3.3.1 Synthesis of polymers…………………………………… 72
3.3.3.2 Preparation of dense membranes………………………… 73
3.4 Characterization of polymers…………………………………………….. 73
3.5 Results and discussion……………………………………………………. 74
3.5.1 Synthesis and physical characterization of polyimides based on PDAB ……………………………………………………………..
74
3.5.1.1 Synthesis………………………………………………….. 74
3.5.1.2 Elemental analysis………………………………………… 75
3.5.1.3 Solubility and solution viscosity………………………….. 75
3.5.1.4 FT-IR analysis…………………………………………….. 78
3.5.1.5 Thermal Properties of Polymers…………………………... 78
3.5.1.6 Wide-angle X-ray diffraction (WAXD) ………………… 81
3.5.2 Synthesis and physical characterization of sulfonated polyimides (SPIs) …………………………………………………
82
3.5.2.1 Synthesis ………………………………………………… 82
3.5.2.2 FT-IR analysis……………………………………………. 84
3.5.2.3 Solubility and solution viscosity………………………….. 85
3.5.2.4 Thermal Properties of Polymers…………………………... 88
3.5.2.5 WAXD Analysis………………………………………….. 90
3.5.3 Preparation and physical characterization of blend membranes based on SPIs and PBI-BuI…………………………………………
91
3.5.3.1 Membrane preparation…………………………… 92
3.5.3.2 FT-IR analysis of blend membranes………………………. 92
3.5.3.3 Thermal analysis of blend membranes……………………. 95
3.5.4 Synthesis and physical characterization of polyimides based on various alkyl substituted diamines……………………………….
97
IV

3.5.4.1 Synthesis………………………………………………….. 97
3.5.4.2 Elemental analysis ………………………………………... 99
3.5.4.3 FT-IR analysis…………………………………………….. 100
3.5.4.4 Solubility and solution viscosity…………………………. 100
3.5.4.5 Thermal Properties of Polymers………………………….. 102
3.5.4.6 WAXD analysis………………………………………….. 105
3.6 Conclusions………………………………………………………………… 106
Chapter 4
Structure-gas permeability correlation in polyimides
4.1 Introduction………………………………………………………………. 108
4.1.1 Literature on PIs containing pendant groups: A brief review ……. 109
4.1.2 Gas permeation in aromatic polymers with methyl and t-butyl group as substituent………………………………………………………..
110
4.1.3 Effect of moisture on CO2 permeation properties…………………… 111
4.2 Experimental section………………………………………………………. 112
4.2.1 PI Synthesis and membrane preparation …………………………. 112
4.2.2 Measurement of gas permeability………………………………… 114
4.3 Results and discussion ……………………………………………………. 115
4.3.1 Assessing capability of O-Ph group incorporation: Gas Permeability of PIs based on 1-phenoxy-2,4-diamino benzene (PDAB)……
116
4.3.2 Substitution of alkyl group on O-Ph of diamine moiety ………….. 119
4.3.2.1 Chain packing density and rigidity …………………… 119
4.3.2.2 Permeability and selectivity ………………………….. 122
4.3.2.3 Effect of dianhydride………………………………….. 125
4.3.2.4 Effect of alkyl substituent…………………………….. 125
4.3.3 Gas permeability of sulfonated polyimides (SPI) ………………… 129
4.3.3.1 Gas permeability in dry state………………………….. 129
4.3.3.2 Permeability with humidified gas ……………………. 131
4.3.4 Gas permeability of blend membranes ……………………………. 132
4.5 Conclusions………………………………………………………………… 134
V

Chapter 5
Investigation of polyimides towards their applicability as PEM
materials
5.1 Introduction ………………………………………………………………. 136
5.2 Experimental ……………………………………………………………… 138
5.2.1 Syntheses of polymer electrolytes ………………………………… 138
5.2.1.1 Synthesis of Sulfonated Polyimide (SPI) ………………… 138
5.2.1.2 Synthesis of polybenzimidazole (PBI) …………………… 139
5.2.2 Membrane preparation ……………………………………………. 139
5.2.3 Determination of water uptake (WU) capacity of membranes……. 140
5.2.4 Determination of Phosphoric acid uptake of membranes …………. 140
5.2.5 Determination of Ion exchange capacity (IEC) of membranes……. 140
5.2.6 Determination of Hydrolytic and Oxidative stability of membranes 141
5.2.7 Determination of Proton conductivity……………………………. 142
5.3 Results and Discussion…………………………………………..….….…. 143
5.3.1 Membrane preparation…………………………………………….. 143
5.3.2 Phosphoric acid doping ……………………………………………. 144
5.3.3 Water uptake (WU) capacity ……………………………………… 146
5.3.4 Ion exchange capacity (IEC) ……………………………………… 148
5.3.5 Hydrolytic stability………………………………………….……. 153
5.3.6 Oxidative stability…………………………………………..….…. 155
5.3.7 Proton conductivity measurements …………………………..….… 156
5.3.7.1 Proton conductivity of sulfonated polyimides (SPIs)….. 158
5.3.7.2 Proton conductivity of blends constituting SPI and PBI- BuI…………………………………………..….….……..
162
(A) Proton conductivity of blend membranes [SPI(H)/PBI-BuI(a/b)] ………………..….….……..
162
(B) Proton conductivity of polymer electrolyte membranes doped with 12 M H3PO4……………..….….……..
164
(C) Proton conductivity of blend membranes and PBI- BuI doped with 1M H2SO4 and 1M H3PO4……………..…
173
5.4 Conclusions…………………………………………..….….……..….…. 181
VI

VII
Conclusions 183
eferences…………………………………………..….……………… 187
List of Publications……………………………..….……………… 204
Chapter 6
R

i
Abstract The present dissertation constitutes synthesis and characterization of polyimides
(PI) having a pendant flexible phenoxy group and their investigations as membrane
materials for gas permeation and polymer electrolytes for fuel cell.
Accordingly, monomers (aromatic diamines) were designed in such a fashion that
the resultant PIs comprise pendant phenoxy group as a side chain exhibiting improved
solubility without sacrificing their thermal properties and became the prime objective of
this thesis. Polyimides were prepared from these synthesized aromatic diamines with
commercial dianhydrides such as 4,4′-oxydiphthalic dianhydride (ODPA), 4,4′-
(hexafluoroisopropylidene)diphthalic anhydride (6FDA), 1,4,5,8-naphthalene
tetracarboxylic dianhydride (NTDA), etc. by using single step solution polycondensation
and imidization in meta-cresol as the solvent.
In Chapter 1, a brief literature survey on synthesis of PIs with an emphasis on
polycondensation and applications of PIs are given. Gas permeation in PIs and membrane
based gas separation is discussed, followed by theoretical consideration and physical
parameters affecting gas permeation. A literature reports on sulfonated PIs with an
importance on side-chain-type PIs and other polymers as PEMFC is also discussed. This
chapter ends with history and types of fuel cell covering criteria for PEM.
Chapter 2 presents a detailed study on synthesis and characterization of
monomers viz., aromatic meta-phenylene diamines containing phenoxy group, to be used
in the synthesis of polyimides. Diamines were prepared by condensing 1-chloro-2,4-
dinitrobenzene with various phenols. In this way, a detailed discussion on synthesis and
characterization of dinitro precursors and their corresponding diamines viz., 1-phenoxy-
2,4-diamino benzene (PDAB), 2methyl-1-phenoxy-2,4-diamino benzene (2MPDAB),
4-methyl-1-phenoxy-2,4-diamino benzene (4MPDAB), 2,6-dimethyl-1-phenoxy-2,4-
diamino benzene (2,6DMPDAB), 4’-t-butyl-1-phenoxy-2,4-diamino benzene
(4tBPDAB) and 4-sulfonic-1-phenoxy-2,4-diaminobenzene (SPDAB) is given.
Chapter 3 presents a detailed study on synthesis of PIs based on monomers
mentioned in Chapter 2, their characterizations by FT-IR, solubility, solution viscosity,
thermal properties and WAXD analysis. Preparation and characterization of blend

ii
membranes based on sulfonated polyimide (SPI) with polybenzimidazole is also
discussed.
Chapter 4 constitutes investigations towards effects of bulk and site of alkyl
substituent on pendant phenoxy group in PIs on their pure gas permeability (using He, H2,
Ar, N2, O2, CH4 and CO2). Crucial physical properties of these polyimides are correlated
with gas permeability. Alkyl substituent on the pendant phenoxy group at para position is
more effective than at ortho substitution for increasing gas permeability in PIs. Structural
variation in diamine moiety greatly affects chain packing and gas permeability in ODPA
based PIs than that of 6FDA based PIs. SPIs in salt form as well in sulfonic acid form
were also evaluated for H2, O2 and CO2 permeation with and without humidifying feed
gas. Permeability coefficient for H2, O2 and CO2 were also evaluated for blends
membranes.
Chapter 5 describes proton conductivity of SPIs (in imidazolium and
triethylammonium sulfonate form as well as in –SO3H form) and blends of SPI and PBI
measured at various temperatures. Physico-chemical properties of SPIs and blend
membranes influencing proton conductivity such as water uptake capacity, IEC,
hydrolytic stability and oxidative stability were also evaluated. Proton conductivity in
humid and anhydrous conditions with varying temperature was also measured. NTDA
based SPIs in –SO3H exhibited higher ion exchange capacity (IECs) and proton
conductivity while in salt form hydrolytic and oxidative stability was improved. Detailed
investigation on proton conductivity of H3PO4 doped blend membranes based on SPI
with polybenzimidazole reveals the transportation of proton both by–SO3H belonging to
SPI and H3PO4. Detailed analysis done by electrochemical impedance spectroscopy (EIS)
of proton conductivity of blends membrane on doping with 12 M H3PO4 in anhydrous
condition and with 1M H3PO4 in humid atmosphere at various temperatures is discussed.
While same blend membranes after converting in pure –SO3H form exhibited
comparatively lower proton conductivity as compared with SPIs in 100 % humidification
with increase in temperature.
Chapter 6 summarizes the results and describes salient conclusions of the
investigations reported in this thesis.

iii
List of Abbreviations CDNB 1-chloro-2,4-dinitrobenzene PDNB 1-phenoxy-2,4-dinitrobenzene PDAB 1-phenoxy-2,4-diaminobenzene SPDAB 4-sulfonic-1-phenoxy-2,4-diaminobenzene 2MPDNB 2-methyl-1-phenoxy-2,4-dinitro benzene 2MPDAB 2methyl-1-phenoxy-2,4-diamino benzene 4MPDNB 4-methyl-1-phenoxy-2,4-dinitro benzene 4MPDAB 4-methyl-1-phenoxy-2,4-diamino benzene 2,6DMPDNB 2,6-dimethyl-1-phenoxy-2,4-dinitro benzene 2,6DMPDAB 2,6-dimethyl-1-phenoxy-2,4-diamino benzene 4tBPDNB 4’-t-butyl-1-phenoxy-2,4-dinitro benzene 4tBPDAB 4’-t-butyl-1-phenoxy-2,4-diamino benzene ODPA 4,4′-Oxydiphthalic dianhydride BPDA 3,3′,4,4′-biphenyltetracorboxylic dianhydride BTDA 3,3′,4,4′-benzophenone tetracarboxylic dianhydride 6FDA 4,4′-(hexafluoroisopropylidene)diphthalic anhydride PMDA pyrromellitic dianhydride NTDA 1,4,5,8-naphthalene tetracarboxylic dianhydride ODA 4,4′-oxydianiline DAB 3,3′-diaminobenzedine IPA isophthalic acid BuI 5-tert-butyl isophthalic acid TEA Triethyl amine Im Imidazole TEAH+ Triethylammonium cation ImH+ Imidazolium cation PI Polyimide SPI Sulfonated polyimide CPI Copolyimide PBI Polybenzimidazole PDAB-ODPA PI based on diamine ‘PDAB’ and dianhydride ‘ODPA’ PDAB-6FDA PI based on diamine ‘PDAB’ and dianhydride ‘6FDA’ PDAB-BPDA PI based on diamine ‘PDAB’ and dianhydride ‘BPDA’ PDAB-BTDA PI based on diamine ‘PDAB’ and dianhydride ‘BTDA’ PDAB-PMDA PI based on diamine ‘PDAB’ and dianhydride ‘PMDA’ ODA-ODPA PI based on diamine ‘ODA’ and dianhydride ‘ODPA’ CPI-0595 CPI based on 5 mole% PDAB, 95 mole% ODA with ODPA CPI-2575 CPI based on 25 mole% PDAB, 75 mole% ODA with ODPA CPI-5050 CPI based on 50 mole% PDAB, 50 mole% ODA with ODPA SPDAB•TA-N SPI based on SPDAB and NTDA in triethylammonium
sulfonate salt form

iv
SPDAB•Im-N SPI based on SPDAB and NTDA in imidazolium sulfonate salt form
SPDABI-N SPI based on SPDAB and NTDA in –SO3H form obtained from SPDAB•TA-N
SPDABII-N SPI based on SPDAB and NTDA in –SO3H form obtained from SPDAB•Im-N
SPDAB•TA-O SPI based on SPDAB and ODPA in triethylammonium sulfonate salt form
SPDAB-O SPI based on SPDAB and ODPA –SO3H form obtained from SPDAB•TA-O
PBI-I Polybenzimidazole based on DAB and IPA PBI-BuI Polybenzimidazole based on DAB and BuI SPI(TEA)/PBI-BuI(a/b)
Blends with “a” wt% of SPDAB•TA-N and “b” wt% of PBI-BuI, where ‘TEA’ denotes SPI in triethylammonium sulfonate salt form
SPI(H)/PBI-BuI(a/b)
Blends with “a” wt% of SPDAB•TA-N and “b” wt% of PBI-BuI, where ‘H’ represents proton form of SPI
SPI(PA)/PBI-BuI(a/b)
Blends with “a” wt% of SPDAB•TA-N and “b” wt% of PBI-BuI, where ‘PA’ denotes acid doping with H3PO4
SPI(SA)/PBI-BuI(a/b)
Blends with “a” wt% of SPDAB•TA-N and “b” wt% of PBI-BuI, where ‘SA’ denotes acid doping with H2SO4
4′MPDAB-ODPA PI based on diamine ‘4′MPDAB’ and dianhydride ‘ODPA’ 4′MPDAB-6FDA PI based on diamine ‘4′MPDAB’ and dianhydride ‘6FDA’ 2′MPDAB-ODPA PI based on diamine ‘2′MPDAB’ and dianhydride ‘ODPA’ 2′MPDAB-6FDA PI based on diamine ‘2′MPDAB’ and dianhydride ‘6FDA’ 2′,6′DMPDAB-ODPA
PI based on diamine ‘2′,6′DMPDAB’ and dianhydride ‘ODPA’
2′,6′DMPDAB -6FDA
PI based on diamine ‘2′,6′DMPDAB’ and dianhydride ‘6FDA’
4′tBPDAB-ODPA PI based on diamine ‘4′tBPDAB’ and dianhydride ‘ODPA’ 4′tBPDAB-6FDA PI based on diamine ‘4′tBPDAB’ and dianhydride ‘6FDA’ MEA Membrane Electrode Assembly PEMFC Polymer Electrolyte Membrane Fuel Cell DSC Differential Scanning Calorimetry TGA Thermo Gravimetric Analysis IEC Ion Exchange Capacity IDT Initial Decomposition Temperature SD Degree of Sulfonation CTC Charge transfer complex WAXD Wide angle X-ray diffraction

v
List of Tables
Table
No. Description
Page
No.
1.1 General aspects of fuel cells……………………………………………… 31
3.1 Elemental analysis of PDAB based polyimides………………………….. 75
3.2 Solubility of PDAB based polyimides and copolyimides………………... 77
3.3 Physical properties of PDAB based polyimides and copolyimides……… 79
3.4 Solubility of SPIs in various solvents……………………………………. 87
3.5 Thermal properties of SPIs………………………………………………. 90
3.6 Thermal properties of blend membranes…………………………………. 96
3.7 Elemental analysis of PIs based on alkyl susbtituted phenoxy diamines… 99
3.8 Solvent solubility of polyimides based on alkyl substituted phenoxy diamines…………………………………………………………………..
101
3.9 Physical properties of polyimides based on alkyl substituted phenoxy diamines…………………………………………………………………..
104
4.1 Polymers investigated for gas permeation……………….......................... 113
4.2 Physical properties of PIs based on PDAB………………......................... 116
4.3 Gas permeability (P) and selectivity of PIs based on PDAB…………….. 117
4.4 Physical properties of polyimides based on diamines possessing substituted o-phenoxy side chain…………………………………………
120
4.5 Gas permeability (P) of polyimides based on alkyl substituted phenoxy group in meta-PDA……………………………………………………….
123
4.6 Gas permselectivity () of polyimides based on alkyl substituted phenoxy group in meta-PDA……………………………………………..
124
4.7 Gas permeability (P) and selectivity () of SPIs under dry conditions….. 129
4.8 Permeability (P) and selectivity () of SPIs under humidified gas conditions ………………………………………………………………...
131
4.9 Gas Permeability and selectivity of the blend membranes………………. 133
5.1 PEM and their abbreviation……………………………………………… 139
5.2 Physical properties of membranes based on SPIs, PBIs and their blends... 145

vi
5.3 Determination of benzimidazole and sulfonic acid group ratio in blends.. 148
5.4 Proton conductivity of blend membranes in humidified conditions……... 163
5.5 Change in RΩ for the blends and PBIs with temperature………………… 167
5.6 Change in Rf for the blends and PBIs with temperature…………………. 171
5.7 RΩ values of blend membranes and PBI-BuI in 1M H2SO4……………… 174
5.8 RΩ values of blend membranes and PBI-BuI in 1M H3PO4……………… 177

vii
List of Schemes
Scheme 2.1 Synthetic route to PDNB, PDAB and SPDAB
Scheme 2.2 Synthesis of dinitro precursors and diamines based on alkyl substituted
phenols
Scheme 3.1 Synthetic route to PDAB based PIs
Scheme 3.2 Synthetic route to copolyimides
Scheme 3.3 Schematic presentation for the synthesis of sulfonated polyimides (SPIs)
Scheme 3.4 Synthetic scheme to Polybenzimidazoles
Scheme 3.5 Synthetic route to PIs based on alkyl substituted phenoxy diamines.
Scheme 5.1 Probable exchange of protons in 12M H3PO4 doped blend membranes

viii
List of Figures
Figure
No. Description
Page
No.
1.1 General structures of aromatic polyimides………………………………. 1
1.2 Two step synthesis of polyimides………………………………………... 3
1.3 Major breakthroughs in membrane based gas separation technology
(1850-2000)……………………………………………………………….
12
1.4 Representation of gas permeation mechanisms in polymeric membranes 13
1.5 Main-chain-type sulfonated diamine…………………………………….. 21
1.6 Side-chain-type sulfonated diamines…………………………………….. 22
1.7 Various structures of naphthalenic dianhydride monomers……………… 23
1.8 A general structure of commercially available PFSA membranes………. 25
1.9 Sulfonation of PEEK and PEES ……………………………………… 26
1.10 Sulfonation reaction of copolymers……………………………………… 27
1.11 Sulfonated-PES from sulfonated (4,4'-dichlorodiphenylsulfone)
monomer………………………………………………………………….
27
1.12 Schematic of PEMFC…………………………………………………… 33
2.1 Number of available sites in common aromatic diamine and dianhydride 39
2.2 FT-IR spectra of PDNB and PDAB ……………………………………... 48
2.3 1H NMR spectrum of PDNB……………………………………………... 49
2.4 1H NMR spectrum of PDAB……………………………………………... 50
2.5 FT-IR spectrum of SPDAB……………………………………………..... 51
2.6 1H NMR spectrum of SPDAB……………………………………………. 52
2.7 FT-IR spectra of 2MPDNB and 2MPDAB……………………………... 53
2.8 1H NMR spectrum of 2MPDNB………………………………………… 54
2.9 1H NMR spectrum of 2MPDAB………………………………………… 55
2.10 FT-IR spectra of 4MPDNB and 4MPDAB……………………………... 56
2.11 1H NMR spectrum of 4'MPDNB………………………………………… 56

ix
2.12 1H NMR spectrum of 4'MPDAB………………………………………… 58
2.13 FT-IR spectra of 2,6DMPDNB and 2,6DMPDAB …………………… 58
2.14 1H NMR spectrum of 2,6DMPDNB……………………………............. 60
2.15 1H NMR spectrum of 2,6DMPDAB……………………………............. 60
2.16 FT-IR spectra of 4tBPDNB and 4tBPDAB…………………………….. 61
2.17 1H NMR spectrum of 4tBPDNB………………………………………… 62
2.18 1H NMR spectrum of 4tBPDAB………………………………………… 62
3.1 IR spectra of PDAB based PIs and CPIs…………………………………. 78
3.2 DSC of PDAB based polyimides and copolyimides……………………... 80
3.3 TGA of PDAB based PIs and CPIs………………………………………. 81
3.4 WAXD spectra of PDAB-ODPA and PDAB-6FDA…………………….. 81
3.5 FT-IR spectra of SPIs…………………………………………………….. 85
3.6 Variation in reduced viscosity of SPIs with concentration………………. 88
3.7 TGA of SPIs……………………………………………………………… 90
3.8 WAXD spectra of SPIs…………………………………………………... 91
3.9 FT-IR spectra of blends in the sulfonic acid form……………………….. 94
3.10 TGA of blends…………………………………………………………… 96
3.11 IR spectra of polyimides based on alkyl substituted phenoxy diamines 100
3.12 DSC curves of polyimides based on alkyl substituted phenoxy diamines 103
3.13 TGA curves of polyimides based on alkyl substituted phenoxy diamines 105
3.14 WAXD spectra of polyimides based on alkyl substituted phenoxy
diamines…………………………………………………………………..
105
4.1 Gas permeation Equipment………………………………………………. 115
4.2 Lengthening of side chain due to substitution of alkyl groups at para
position……………………………………………………………………
128
4.3 Widening of the side chain due to substitution of alkyl group at ortho
position……………………………………………………………………
128

x
5.1 Structures of cations in salt form………………………………………… 149
5.2 Dissociation of SPIs in aqueous medium………………………………… 151
5.3 Schematic representation of the SPIs showing relative distance of
sulfonic acid group form the imide linkage………………………………
153
5.4 Schematic representation of the apparatus used for proton conductivity
measurement……………………………………………………………...
157
5.5 A Nyquist plot for the impedance measurements ………………………….. 158
5.5 B Randles equivalent circuit used for fitting this data……………………… 158
5.6 Impedance spectra of SPIs and Nafion-117 at 50 C…………………….. 158
5.7 Proton conductivity of SPIs with temperature at 100% RH……………... 159
5.8 Arrhenius plot of proton conductivity of SPI with temperature at 100%
RH………………………………………………………………………..
162
5.9 Proton conductivity of SPIs doped with 12M H3PO4 against temperature 165
5.10 Variation of proton conductivity of blend membranes and PBI-BuI
doped with 12 M H3PO4 at different temperature………………………...
168
5.11 Impedance spectra of blend membranes doped with 12M H3PO4……….. 169
5.12 Comparative EIS spectra of blends and PBIs doped with 12M H3PO4 at
100 °C……………………………………………………………………
172
5.13 Arrhenius plot of proton conductivity of 12 M H3PO4 doped membranes
against temperature……………………………………………………….
172
5.14 Variation of proton conductivity of blend membranes [SPI(SA)/PBI-
BuI(a/b)] and PBI-BuI doped with 1M H2SO4 against temperature…….
174
5.15 Impedance spectra of blend membranes and PBI-BuI doped with in 1 M
H2SO4……………………………………………………………………..
176
5.16 Variation of proton conductivity of blend membranes and PBI-BuI
doped with 1M H3PO4 with temperature………………………………….
178
5.17 Arrhenius plot of proton conductivity of 1M H3PO4 doped membranes
with temperature………………………………………………………….
179
5.18 Impedance spectra of blend membranes and PBI-BuI doped with 1M
H3PO4…………………………………………………………………….
180

Chapter 1
Introduction and literature survey
1.1 Polyimides
Polyimides (PIs) are the condensation polymers derived mainly from organic
diamines or their derivatives and dianhydrides. They possess heterocyclic five or six
membered imide linkage in their chain repeat unit (Figure 1.1).
Figure 1.1 General structures of aromatic polyimides
The importance of six membered-imide linkage came in the picture due its
relative high chemical stability in acidic environment as compared to five membered
analogues in the emerging advanced technology such as polymer electrolyte membrane
fuel cells (PEMFC) (Genies, 2001a; Yin, 2006a). The excellence of aromatic PIs as gas
separation materials can be credited to their physical properties (Kim, 1988; Stern, 1989;
Sroog, 1991; Robeson, 2008). Considerable research has been accelerated towards
aromatic PIs for their use in pioneering fields such as aerospace, liquid crystal displays,
automotive components, microelectronics, gas separation membrane materials, PEMFC,
etc. (Takekoshi 1990; Sroog, 1991; de Abajo, 1999; Ding, 2007; Marestin, 2008).
The present chapter briefly describes history of polyimides, their synthetic routes,
physical characterizations, applications and scope of aromatic polyimides as membrane
material in gas permeation and PEMFC. Since the invention of aromatic PI was laid on
the five-membered imide ring, these PIs will be discussed in the beginning, followed by
PIs with six-membered ring.
1.2 A brief history of polyimides
Retrospection in PIs informs their first synthesis in 1908 by Bogert and Renshaw
(1908). In their invention, they observed elimination of water instead of melting upon
heating the relatively stable organic compound, 4-aminophthalic anhydride and

Chapter 1 Introduction and literature survey
2
concluded the formation of “polymolecular imide”. The development of modern
polyimides can be linked to the outcome of use of the polycarboxylic acids prepared from
oxidation of a major petroleum component, polymethyl benzenes. At the embryonic stage
of polyimides, aliphatic diamines in salt form were prepared specially for making Nylons
(polyamides). Aliphatic diamines were condensed with polycarboxylic acids such as
tetracarboxylic aromatic acid or their derivatives; typically pyrromellitic dianhydride
(PMDA) (Edward, 1959a and 1959b). The obtained aliphatic-aromatic polyimides could
only be melt-processed if aliphatic diamines used had either linear long-chain or
relatively shorter branched chain. Later on, implementation of less basic aromatic
diamines following two stage synthesis through intermediate poly(amic acid) formation
drove the development in polyimides. Commercial polyimides such as Pyralin and
Kapton H followed these routes. With the advancement in aromatic PIs, it was further
observed that such a fully aromatic system constituted CTC because of the interaction
between electron withdrawing cyclic imide rings and electron donating π-cloud of phenyl
rings. Such interactions make them densely packed and hence intractable (Chen, 2008;
Zhang, 2007). However, excellent mechanical properties, outstanding oxidative stability,
extraordinary non-flammability and good electrical properties have made them more
useful in advanced technologies (Ghosh, 1996; Li, 2007a) and they emerged as high
performance polymers.
1.3 Synthesis of polyimides
Various synthetic methods such as polycondensation via ester derivatives of
poly(amic acids), polycondensation of dianhydrides and diisocynates, transition metal
catalyzed polycondensation of diamines and polymerization of cycloaddition reaction to
prepare polyimides have been explored (Mathew, 2001; Shingte, 2006). Most widely
used polycondensation methods are briefly discussed below.
1.3.1 Polycondensation of a diamine and a dianhydride
This is one of the most widely adopted methods for the synthesis of polyimides
(Sroog, 1991; Volksen, 1994). It can be carried out in two ways, viz., two-step method
via poly(amic acid) intermediate and one-step or one pot method.

Chapter 1 Introduction and literature survey
3
1.3.1.1 Two-step method via poly(amic acid) intermediate
A successful route to the synthesis of high molecular weight polyimide was first
described by Endrey et. al. in 1962 and successively by Du Pont patents (Mathew, 2001;
Shingte, 2006). In this method, the synthesis was conducted in two stages in a suitable
solvent. First, a soluble poly(amic acid) was prepared; which was then converted to the
desired polyimide by imidization as exemplified in Figure 1.2.
Figure 1.2 Two step synthesis of polyimides
Various factors such as structure of monomers, type of solvent, imidization
method, etc. play an important role in the formation of high molecular weight polyimide
as described below.
1.3.1.1.a Role of monomers
Poly(amic acid) (PAA) is formed by reversible nucleophilic substitution reaction.
Herein, nucleophilic attack of a diamine takes place at one of the carbonyl carbons of an
anhydride moiety followed by its opening to carboxylate functionality. This is followed
by intra-proton abstraction (-NH2-) from the attacked diamine leading to the formation of
amic acid in the first stage. This reaction is an equilibrium reaction, favored by the high
concentration of monomers, reactivity of each monomeric unit, basicity of the solvent
and low to moderate temperature (Cassidey, 1982). In general, diamine is dissolved in a
solvent at the beginning of polymerization, followed by solid dianhydride addition to the
solution of diamine (Dine-Hart, 1967; Johnson, 1972; Kumar, 1981). Since poly(amic
acid) formation is a nucleophilic substitution reaction, consequently the major boosters
for such a reaction are high nucleophilicity of the amino nitrogen atom of diamine and
greater electrophilicity of the carbonyl groups belonging to dianhydride. In other words,

Chapter 1 Introduction and literature survey
4
more basic character of the diamine and the enhanced electronic affinity (EA) in
dianhydride result in a higher reaction rate.
The Lewis acid-base concept can also be well correlated in poly(amic acid)
formation. Diamine acts as a Lewis base and dianhydride as a Lewis acid. Consequently,
the larger electron pair donation ability of amino group in diamine which is associated
with pKa and the more acceptance nature of the carbonyl group in dianhydride correlating
with EA should favor higher forward reaction rate of poly(amic acid) formation (Zubkov,
1981). This phenomenon can be exemplified with comparative reactivity of PMDA,
BPDA, ODPA and BTDA (Koton, 1984; Cornelius, 2000). Higher EA value in BTDA is
attributed to the bridged electron withdrawing carbonyl carbon, while the lower EA value
in ODPA is due to the bridged electron donating ether linkage. BPDA with absence of
such bridged functionality demonstrates EA between BTDA and ODPA. In addition,
placement of such functionality with respective to carbonyl becomes more effective as
para>ortho>meta (Pine, 1987; Cornelius, 2000). In diamines, similar correlation
between pKa with their reactivity is found (Zubkov, 1981; Cornelius, 2000). Higher
values of pKa show enhanced reactivity, since basicity of these diamines gets increased.
Embodiment of electron donating functionality in diamines increases their nucleophilic
nature. Such diamines therefore show higher basicity with increased pKa values executing
rapid polymerization for the same period of polymerization as compared with diamines
with lower basicity and pKa values.
Molecular weight of poly(amic acid) is affected by the sequence of monomer
addition. This is due to the solubility difference of monomers in polymerization solvent.
In general, complete dissolution of diamine is followed by dianhydride addition. Diamine
can be rapidly dissolved in the solvent at room temperature, while dianhydride consumes
more time for dissolution in diamine solution. This sequential procedure confers high
molecular weight of poly(amic acid). Orwoll et. al. (1981) attributed this phenomenon to
the interfacial condensation of crystalline dianhydride and diamine.
1.3.1.1.b Role of solvent
More polar as well as basic solvents enhance the rate of poly(amic acid)
formation (Vygodskii, 1977). Most of the polar aprotic solvents with basic nature such as

Chapter 1 Introduction and literature survey
5
DMF, DMAc, NMP etc. are utilized for preparation of poly(amic acid). Such types of
solvents have ability to effectively interact as well as solvate the propagating poly(amic
acid). In addition, the heat evolved during such acid-base interactions drives the reaction
in forward direction. In order to achieve high molecular weight for poly(amic acid),
removal of water from the starting monomers as well as reaction medium becomes
important. Presence of water in the solvent affects reaction kinetics of poly(amic acid)
formation as it can hydrolyze poly(amic acid) (Bower, 1963; Sroog, 1965).
Probability of hydrogen bonding of hydroxyl group belonging to –COOH with
neighboring chain of poly(amic acid) leads to increased viscosity of poly(amic acid)
solution (Bryant, 2006). As a result, nucleophilic attack of diamine also becomes
prominent on the carbonyl carbon of anhydride moiety due to hydrogen bonding with
carbonyl oxygen leading to its enhanced electrophilicity. This phenomenon can be
considered as autocatalytic type of behavior (Kaas, 1981). However, autocatalysis is not
observed in polar aprotic basic solvents (DMF, DMAc) (Kuzenetsove, 2000; Bryant,
2006). This can be attributed to the better solvation and association with acidic carboxyl
group of propagating poly(amic acid) by these basic solvents, thus isolating them from
interaction with anhydride as well as amic acids (Kuzenetsove, 2000; Bryant, 2006;
Hasanain, 2008). Hence benzoic acid is added in such amide type basic solvents in order
to enhance the rate of poly(amic acid) formation. Poly(amic acid) can also be prepared in
polar protic solvents like m-cresol (Mathew, 2001). This enhances the rate of nucleophilic
substitution reaction in amide formation. Carbonyl groups of an anhydride become highly
electrophilic after their protonation, as a result nucleophilic attack of diamine becomes
feasible. High molecular weight polyimide has been synthesized in acidic solvents such
as salicylic acid (Hasanain, 2008) and melt of benzoic acid (Kuzenetsove, 2000).
1.3.1.1.c. Transformation of poly(amic acid) to polyimide
Properties of polyimides depend on an extent of imidization of intermediate
poly(amic acid) (PAA). Presence of amic acid in PI significantly affects thermal,
hydrolytic stability and other properties. Imidization is achieved by cyclodehydration of
amic acid, which is accomplished by two methods as described below.

Chapter 1 Introduction and literature survey
6
(i) Chemical imidization of poly(amic acid)
Chemical imidization is carried out by external dehydrating agents. Type of
solvent, temperature, concentration of amic acid, ratio of dehydrating agent (usually
acetic anhydride) to the catalyst organic base, catalyst structure and its base strength
influence the chemical imidization process (Endrey, 1965a and 1965b; Kailani, 1998a;
Kailani, 1998b). Since the reagent used for dehydration lowers the activation energy for
ring closure, transforming poly(amic acid) to polyimide becomes feasible at relatively
lower temperature. A combination of external dehydrating agents and a catalyst in dipolar
aprotic solvents are also used (Endrey, 1965a). Dehydrating reagents such as acetic
anhydride or anhydrides of other acids and the catalysts such as tertiary amines, viz;
triethylamine (TEA), pyridine etc. are chosen (Kailani, 1998a; Kailani, 1998b). The
advantage of chemical imidization over thermal imidization is that former reveals faster
conversion to polyimides. However, in chemical imidization process, imide formation is
accompanied by isoimide formation. The presence of water in the solvent also affects the
kinetics of imidization (Kailani, 1998a). The chemical structure as well as base strength
of the catalyst also plays the dominant role in chemical imidization process. Imidization
kinetics is well investigated by various researchers (Dickinson, 1992; Kailani, 1992;
1998a; 1998b; Kim, 1993; Yu, 1997)
(ii) Thermal Imidization of poly(amic acid)
In this process, imidization is accomplished by gradual heating of poly(amic acid)
in the solid state to 250-350 °C (Volksen, 1994; Mathew, 2001; Shingate, 2006). In
thermal imidization, removal of the condensate produced during cyclodehydration
becomes an indispensable step. The simultaneous water removal, which is a byproduct,
moves imidization in the forward direction. In the solid state, cyclodehydration of
poly(amic acid) is difficult. Hence, high viscous solution of poly(amic acid) is also
utilized, as residual solvent plays the significant role. It is said that use of dipolar aprotic
solvents speeds up cyclization since they can efficiently solvate poly(amic acid) chains as
well as offer favorable conformation of the orthoamic acid to cyclize (Kreuz, 1966;
Bryant, 2006). In addition, being weak Lewis bases, they are able to accept protons
during cyclization. It is reported that various factors such as film thickness, molecular

Chapter 1 Introduction and literature survey
7
mobility, amount of residual solvent, physical state of the poly(amic acid), conformations
of each macromolecule, degree of imidization, degradation of poly(amic acid) chains, and
curing reaction of intermolecular imide link formation at higher temperature (350 °C)
significantly affect the kinetics, in particular for solid phase imidization (Kim, 1993). Use
of various aromatic acids is known to accelerate the imidization in solid state (Lavrov,
1977 and 1980).
1.3.1.2 One-step or one pot method
In this process, polymerization is typically carried out in high boiling solvent such
as m-cresol, p-chlorophenol, nitrobenzene, benzonitrile etc.; as well as dipolar aprotic
amide solvents or a mixture of these at 180 °C, where imidization proceeds rapidly
(Dotcheva, 1994; Mathew, 2001; Bryant, 2006). Those polyimides which are not likely to
precipitate out in high boiling polymerizing solvent at higher temperature can only be
synthesized by this method. Solvents like toluene, o-dichlorobenzene are typically used
to eliminate the water formed azeotropically. One of the advantages on following this one
step method is that amic acid and isoimide are not present in the formed polymer. Thus,
polyimides formed by this method possess different physical properties than that of
formed by conventional two-step process. Obstruction in direct processing of polyimides
into final end application (films and coatings) due to lower polymerization concentration
as well as use of carcinogen solvents and requirement of long polymerization time
(> 18 h) are some of the drawbacks of this technique (Hasanain, 2008).
The most favorable solvent used for this high temperature solution
polymerization is m-cresol, indicating favorable imidization catalysis in acidic media.
Kuznetsov et. al. (2000) and Hasanain et. al. (2008) prepared polyimides by this method
in acidic solvents viz; melt of benzoic acid and salicylic acid. Built up of high molecular
weight polymer within a short period of time (1-2 h) was demonstrated.
1.4 Properties and applications of polyimides
Polyimides usually have high mechanical properties, excellent thermal stability,
high chemical resistance, high Tg and good oxidative stability. These polymers also have
film forming properties and low dielectric constant. Owing to these properties, they find

Chapter 1 Introduction and literature survey
8
wide applications in different fields such as aerospace, liquid crystal displays (LCD),
automotives, gas separation membranes, dielectric, microelectronics, etc. Since the
introduction of first commercial polyimides (Kapton) by Du Pont in 1960 (Sroog, 1991;
de Abajo, 1999), polyimides have been explored in innumerable fields (Mittal, 2001).
Kapton was especially made as the heat resistant polymer for applications in space and
military requirements. In microelectronics, a major demand for insulating materials with
lower dielectric constants relative to the silicon dioxide (ε = 3.9–4.2) are needed and
polyimides containing, in particular, perfluoroalkyl groups are the best suited for this
application (de Abajo, 1999). Fluorinated PI in the fully imidized state have also proved
better performance in the field of optical waveguides. The presence of fluorine containing
linkage such as hexafluoroisopropylidene, in the main chain not only increases the
solubility and Tg, but also decreases the chain packing and charge transfer complexes.
This is favorable for many applications. The low thermal expansion coefficient, good
adhesion property, high modulus and tough ductile mechanical properties of polyimides
are also the other driving forces for their implementation as insulators in microelectronics
(Hedrick, 1999a and 1999b).
Polyimides possess excellent adhesion to a number of substrates including
metals, enabling their composites build-up feasible. Such composites can further raise the
mechanical properties to a superior level, which includes high flexural modulus and
compressive strength, outstanding dimensional stability under loads, aerodynamic
properties, low fatigue, corrosion resistance making useful in the engineering field like
aerospace. Polyimides are also used as biomaterials for cardiovascular medical
applications (Wang, 2007a). NASA developed the thermoset polyimide PMR-15 for the
specific application as a matrix resin for graphite fiber resin composite in the aerospace
industry. Later on Ciba-Geigy also developed another type of thermoset PI having end
capped allylnorbornenedicarboxyl anhydride (Takekoshi, 1990). Fire retardancy in
polyimides can be improved by incorporation of phosphorous containing organic
compounds (Lee, 1995). They showed improvement in solubility of these PIs and
adhesive strength to titanium. PI coating having excellent heat as well as chemical
resistance is of specially interest in electronic devices as the stress buffer of
semiconductor and interlayer dielectrics. Fabrication of PI in thin film form has

Chapter 1 Introduction and literature survey
9
successfully been applied in organic electroluminescent diode (OLED) as emissive layer
and hole transporting layer.
Deposition of metals like Co(II), Pd, Pt, Ag, Au, etc. in the poly(amic acid) stage
followed by thermal curing leads to surface conductive polyimide films (Rancourt, 1987;
Bergemeister, 1990; Biswas, 1994). PIs have also been used as substrate material of
electrode panels in the crystallization of protein under an electric field (Al-Haq, 2007).
Another important application of PI is for separation of gases. PIs with wide
structural variations have been studied as membrane material for gas separation. In
consistent with present trend, the present thesis also describes synthesis and application
of alkyl substituted PIs as membrane material for gas separation. In this context, a brief
overview on gas separation is given below.
1.5 Gas Permeation in polyimides
The beauty of aromatic polyimides as membrane material for gas permeation lies
in the rigidity of the imide ring, their high mechanical and thermal properties (Koros,
1988; de Abajo, 1999). Several reports are available on structure-gas permeation relation
of polyimide, conveying effects of monomer structure modification on gas permeability
of resulting PIs (Hirayama, 1996a; Koros 1988; Langsam, 1993; Stern, 1989, 1990 and
1993; Tsujita, 2003; Ding, 2007).
Polyimides exhibit charge transfer complex (CTC) formation (Matsumoto, 1993b)
resulting in poor processability and lower gas permeation properties. Hence various
efforts on structural modification in PIs leading to improved processability as well as gas
permeability have been reported (Stern, 1989; Tanaka, 1992a; Shao, 2005; Xiao, 2007;
Qiu, 2006). These include structural variations in the diamine or dianhydride molecule
leading to reduced molecular order and torsional mobility (Kim, 1988; Tanaka, 1992b;
Matsumoto, 1993a and 1993b). Incorporation of -C(CF3)2- linkage in a polyimide is one
of the ways to improve solubility of PI in common organic solvents with increase in
permeability and appreciable permselectivity. The -C(CF3)2- linkage inhibits rotation of
the neighbouring phenyl ring leading to increased rigidity of polymer chain, thus
enhancing the permselectivity (Chung, 2001). Presence of this linkage in a dianhydride

Chapter 1 Introduction and literature survey
10
moiety was shown to be more effective towards governing permeation properties than in
the diamine (Matsumoto, 1993b).
To develop a better gas separation material, various approaches comprise
designing of proper chemical structure of monomers, facile modifications of either
monomers or polymers, embodiment of bulky substituents on the precursor monomers,
making meta-linked PIs than para-linked, lowering of interchain interactions, enhancing
rigidity of the PIs, etc. Among these, one of the widely followed efforts is incorporation
of voluminous substituent on phenyl rings of PIs. Such substituent if located at ortho to
an imide linkage not only reduces the interchain interactions by increasing free volume
but it also increases Tg and hence chain stiffness by restricting rotation around C-N bond
of phenyl and imide rings (Takekoshi, 1990; Tanaka, 1992b; Hsiao, 1998a and 1998b;
Liu, 1999; Mittal, 2001; Qiu, 2006). Thus, enhancement in both, permeability due to
bulky substituent and permselectivity due to increased stiffness can be accomplished.
Structure-property correlations in gas permeation suggests that polymers exhibiting high
gas permeability tend to lower the permselectivity or vice versa (Robeson, 2008).
Architecture of polymers leading to increased inter-segmental distance with decreased
intra-segmental mobility should offer both, high permeability and selectivity. PIs having -
C(CF3)2- linkage in their backbone are typical examples of such behavior (Matsumoto,
1993b; Chung, 2001). Being bulkier, -C(CF3)2- reduces the interchain interactions with
increased free volume and thus cerates spaces for gas diffusion conferring high
permeation (Kim, 1988; Tanaka, 1992a; Matsumoto, 1993b). 6FDA based PIs can
therefore reveal higher permselectivity and comparable permeability with other polymers,
deviating the general trade-off relationship between permeability and permselectivity
(Lin, 2000; Liu, 2001). Furthermore, it has been reported that meta-isomers of PIs lead to
decrease in permeability and an increase in permselectivity as compared with their para-
isomer analogues due to the difference in the intrasegmental mobility (Stern, 1989;
Coleman, 1990; Mi, 1993). This also confers more bent structure in main chain of meta-
isomers of PIs (Ding, 2007).
Side chain substitution could also be one of the methods for polyimide structural
architecture towards enhancement of gas permeation in polyimides. However,
embodiment of bulky groups by flexible linkages like C-O-C can be advantageous than

Chapter 1 Introduction and literature survey
11
that of by C-C since the former offers more sub-group mobility. In addition, depending
on such sub-group mobility, permeability coefficient of gases can be varied. Qiu et. al.
(2007) prepared PIs by modifying the dianhydride BPDA with substitution of various
pendant flexible phenoxy groups and reported increase in permeability than that of PIs
based on unsubstituted BPDA. Authors also revealed improvements in gas-separation
performance as evidenced by their permeability values lie closer to the Robeson’s 1991
upper bound limit (Robeson, 1991).
1.5.1 Application of membrane based gas separation
Application of membranes encompasses separation of H2 from CO, CH4 and N2,
acid gas separation from natural gas, gas dehydration, removal of volatile organic
compounds (VOC) from air, O2 or N2 enrichment of air, etc. Because of simplicity and
low capital investment, membrane system competes with adsorptive and cryogenic
separations. Monsanto established the first commercial application based on membrane
gas separation by separating H2 from N2 in ammonia pure gas streams (Baker, 2004;
Koros, 2000). Recovery of H2 gas from waste gases produced at various refinery
operations finds major application for hydrogen-selective membrane (Bollinger, 1982).
Palladium alloy membrane demonstrated above 99.9% purity of H2 (McBride, 1965).
Today, various polymeric membranes such as polyimides (Ube, Praxair), polyaramide
(Medal) or brominated polysulfone (Permea) are used for hydrogen membrane gas
separation. Gas separation is well acknowledged in the separation of air into its
component, i.e. O2 or N2 enrichment. Oxygen separation membranes with a separation
factor of 4-6 and oxygen permeability around 250 Barrers is desired for effective
commercialization (Koros, 2000). Air containing 30-40% O2 is useful in enhanced
combustion and biochemical processes, while for medical purposes, O2 enrichment is
valuable. Purified N2 above 90% facilitates inert atmosphere, being useful in blanketing
fuel storage tanks and pipelines for diminishing fire hazards. In order to avoid spoilage of
food during transport and storage, N2-enriched air is used. In the recent times, green
house effect has built up the tremendous pressure over membrane technologist for
separation of CO2 from effluent streams. Membrane based gas separation is competitive
over conventional technologies for CO2 capture (Scholes, 2008). For the specific

Chapter 1 Introduction and literature survey
12
application of CO2 capture from the flue gas, a CO2/N2 selectivity of >70 and a minimum
CO2 permeability of 100 Barrers (a permeance of 1000 GPU) are required for the
economic operation (Hirayama, 1999). In the future, market would eventually expand
with an economical opportunity for the membrane community.
The following Figure 1.3 represents major breakthroughs in gas separation
technology from the year 1850 to 2000.
.
Figure 1.3 Major breakthroughs in membrane based gas separation technology (1850-
2000) (Baker, 2000)
1.5.2 Theoretical consideration
Gas permeation in polymeric membranes can take place based on one of the three
general transport mechanisms (Koros, 1993): Knudsen-diffusion, molecular sieving or
solution-diffusion, which can be schematically represented as shown in Figure 1.4.

Chapter 1 Introduction and literature survey
13
Figure 1.4 Representation of gas permeation mechanisms in polymeric membranes (Koros, 1993)
When gaseous mixture is allowed to diffuse through a porous membrane into a
region of lower pressure, lower molecular weight components are permeated
preferentially, as they diffuse more rapidly. When the pore size is much smaller than the
mean free path of gas molecules, the gases diffuse independently by Knudsen diffusion.
The diffusivity is proportional to the pore size of membrane and average molecular
velocity of gas molecules, which varies inversely with the square root of the molecular
weight. Kundsen diffusion of gas in cylindrical pores is (Gilron, 2002) given by;
M
RT8
3
dD p
K
where, dp is nominal pore diameter, R is universal gas constant, T is absolute temperature
and is DK is gas diffusivity. For standard gas separation applications, though membranes
following Kundsen diffusion are not commercially attractive due to low selectivities,
some specialty applications are known, e.g. membrane reactors (Koros, 1993).
The molecular sieving separation mechanism is based primarily on much higher
diffusion rates of the smallest molecules. Diffusion coefficient for this mechanism can be
expressed as (Gilron, 2002):
RT
EexpDD MS,a0
MSMS
where, 0MSD is pre-exponential factor and MS,aE is an energy of activation for molecular
sieving. The sorption level difference may be important for similarly sized penetrants like
O2 and N2. The membranes are based on either ultramicroporous carbon or glass hollow
fiber membranes. This mechanism received attention since higher productivities and

Chapter 1 Introduction and literature survey
14
selectivities than solution-diffusion membranes are known (Koros, 1993). Some of these
membranes are commercialized for producing enriched hydrogen streams at high
pressure from mixed hydrocarbon (Acharya, 2000). Fragility and fouling by condensable
gas has made this mode of separation unacceptable for commercialization.
The more common model used to explain and predict gas permeation through
non-porous polymers is the solution-diffusion model. It is based on both solubility and
diffusivity (mobility) of gases into the membrane matrix. The diffusivity selectivity
favors the smallest molecule, while solubility selectivity favors the most condensable
molecule. It states that permeability coefficient P is the product of diffusivity coefficient
D (a kinetic term) and solubility coefficient S (a thermodynamic term).
DSP
If the permeability of gas pairs; PA and PB is known then the selectivity of a
polymer to gas A relative to another gas B can be expressed in terms of an ideal
selectivity, αAB, defined by the relation,
αAB = PA/ PB = DA/ DB SA/ SB
The ratio DA/DB is termed as mobility selectivity, which is favored for smallest
molecule; while the ratio SA/SB is solubility selectivity and is favored for molecules
having high condensability. Selectivity in rubbery polymers is generally favored by
solubility selectivity, while in case of glassy polymers; diffusivity selectivity dominates
to determine overall selectivity. Both, rubbery and glassy polymer can be effectively
utilized in gas separation applications. High solubility of acidic gases such as CO2 and
H2S at low partial pressure as compared with CH4 with low diffusivity and solubility
entails use of rubbery polymers offering a good selectivity of former gases over CH4
(Chatterjee, 1997). On the other hand, diffusive selectivity is favored by glassy polymers.
For example, diffusive selectivity of N2 over pentane of 100 000 is favored in poly(vinyl
chloride) (Baker, 2000).
Gas solubility (S) mainly depends upon penetrant condensability, polymer-
penetrant interactions and polymer morphology (i.e. crystallinity, orientation, fractional
free volume, etc.). Gas diffusivity depends on the ability of a gas molecule to undergo
diffusive jumps, which occur when thermally driven, random, cooperative polymer
segmental dynamics form transient gaps, those are large enough to accommodate

Chapter 1 Introduction and literature survey
15
penetrant, in the immediate vicinity of the gas molecule (Ghosal, 1994). Thus, diffusion
coefficient D decreases with increases in size of the penetrant. In addition, it also depends
on flexibility of the polymer backbone, available free volume, etc. Mobility selectivity
for a given gas pair increases as the polymer matrix becomes more and more rigid. The
mobility selectivity is dominant for most glassy polymers. Hence, the transport of smaller
molecules is favored. On the other hand, solubility of gases generally increases with
molecular size, because the intermolecular forces between the gas and the polymer
increase. Most rubbers show low mobility selectivity due to their flexible polymer chains,
but their ability to separate gases is dominated by their solubility selectivity (Nunes,
2001).
Usually, polymers with high permeability tend to have low selectivity and vice
versa (trade-off relationship). To make membrane-based separations more competitive,
membrane material design which would have both, high permeability and high
permselectivity is envisaged. Variation in chemical structure of the polymer allows
control over its permeation properties. In view of this, it becomes essential to address
physical properties of polymers as well as of penetrants affecting the permeation
behavior.
1.5.3 Physical parameters affecting gas permeation
i) Chain packing density
The d-spacing (average intersegmental distance) and fractional free volume (Vf or
FFV) reveals the chain packing density in polymers. Larger d-spacing as well as FFV
indicates more openness structure of the polymer matrix. Free volume (Vf) is simply the
volume of the polymer mass not occupied by the molecules themselves. In other words, it
is the fraction of the total polymer specific volume that is not occupied by polymer
molecules. It can be calculated as follows
Vf = V- Vo
where, V is the polymer specific volume and Vo is the occupied volume by the
polymer chains at 0° K. Vo can be calculated from the relation Vo = 1.3 Vw, where Vw is
Van der Waals volume and is calculated by group contribution method (Van Krevelen,
1997).

Chapter 1 Introduction and literature survey
16
ii) Chain and subgroup mobility
Structural alterations which inhibit chain packing while simultaneously inhibiting
rotational motion about flexible linkages on polymer backbone tend to increase
permeability while maintaining or increasing selectivity (Coleman, 1990). Inhibition of
the segmental and sub-segmental mobility can be judged by glass transition (Tg) or sub-
Tg temperatures. Typical methods to characterize torsional mobility include dynamic
mechanical relaxation spectroscopy, differential scanning calorimetry, dipole relaxation
spectroscopy and nuclear magnetic resonance (Ghosal, 1994).
iii) Polarity
Since polarity leads to net forces of attraction between macromolecules, dsp and
Vf in polar polymers tend to be smaller. Gases such as CO2, which has a quadrupole
moment, are in general, more soluble in polar polymers (Hirayama, 1999). Presence of
polar groups like bromo, chloro, nitro, sulfonic/carboxylic acid group and their salts, etc.
generally lead to a decrease in permeability and increase in selectivity. This was observed
for various types of polymers such as poly(phenylene oxide) (Bhole, 2005), polysulfone
(Ghosal, 1992), polyarylate (Murugandam, 1987; Houde, 1995, Bhole, 2007 etc.)
iv) Crystallinity
Crystalline regions in polymers typically preclude penetrant solubility (Ghosal,
1994) and thus are impermeable regions. They act to increase the tortuosity of the path
followed by penetrant molecules through a polymer. They may also restrict segmental
mobility in the non-crystalline regions of the polymer. Both effects tend to reduce gas
diffusivity. Thus, crystallinity in gas permeation membrane materials is generally
undesirable.
v) Crosslinking
Diffusion coefficients typically decrease with increasing crosslink density, since
crosslinking reduces polymer segmental mobility (Crank, 1956) as demonstrated in case
of vulcanization of natural rubber (Barrer, 1948). On the other hand, crosslinking can be
taken into benefit for reducing plasticization effects by gases like CO2 (Wind, 2003).

Chapter 1 Introduction and literature survey
17
vi) Molecular Weight
In low molecular weight polymers, a large contribution to segmental mobility
comes from chain ends, which are less constrained by chain connectivity requirements
and are therefore more mobile (Freeman, 1990). As polymer molecular weight increases,
concentration of chain ends decreases and, in turn, the polymer free volume decreases. As
a result, penetrant diffusivity decreases with increasing molecular weight.
vii) Penetrant condensability
Gas permeability increases due to the increased condensability of the gas. Gas
critical temperature (Tc), normal boiling point (Tb), or Lennard-Jones force constant
( /) are all measures of condensability that correlate well with the solubility
coefficients in a polymer. For example, in most polymers, CO2 (Tc= 31°C) is more
soluble than CH4 (Tc= -82.1°C), O2 (Tc= -118.4°C) or N2 (Tc= -147°C) (Bird, 2002).
viii) Penetrant size and shape
In general, a diffusion coefficient decreases with increasing penetrant size and is
also sensitive to penetrant shape. The diffusivity of linear penetrant molecules such as
CO2 is higher than the diffusivities of spherical molecules of equivalent molecular
volume. The sorption coefficient also increases with molecular diameter, because large
molecules are normally more condensable than smaller ones. Hence permeability
increases for the penetrants with high diffusion and sorption coefficient.
ix) Penetrant – polymer interactions
Gas solubility is sensitive to specific interactions between gas and polymer
molecules. Gases such as CO2 has a quadrupole moment and more soluble in polar
polymers. The interaction energy , between a quadrupole and a dipole is given by
(Ghosal, 1994),
Tr
Q8
22

Chapter 1 Introduction and literature survey
18
where, μ is the dipole moment, Q is the quadrupole moment, r is the distance between the
dipolar and quadrupolar moieties, is Boltzmann's constant and T is the absolute
temperature. The strength of interaction between a quadrupole and a dipole increases
rapidly as the dipole or quadrupole moment increases or as the separation between the
quadrupole and dipole decreases.
x) Temperature
The temperature dependence of the permeation coefficient can be expressed as,
P = Po exp(-EP/RT)
where Po is preexponential constant (equal to SoDo) and Ep is the activation energy of
permeation, which is equal to the algebraic sum of Ed and ΔHs. The diffusivity is
generally a stronger function of temperature., than the solubility coefficient (i.e. Ed >
ΔHs). As a result, gas permeability usually increases with temperature. Diffusivity
selectivity is affected with temperature since molecules having highest kinetic diameter
possess higher activation energy and therefore their diffusivity increases. Diffusion of
small molecules, a thermally activated process can be expressed as.
D=Do exp(-ED/RT)
where, ED is the activation energy for diffusion and Do is a constant.
Gas solubility increases with its condensability factor and is governed by van’t
Hoff relation as (Robeson, 1994)
S=So exp(ΔHs / RT)
where So is preexponential and ΔHs is the partial molar enthalpy of sorption.
xi) Pressure
The gas solubility, diffusivity and, in turn, permeability may vary appreciably
with pressure of penetrant in contact with the polymer changes. The gas solubility
decreases with increasing pressure, which is typically observed in the permeation of
glassy polymers to penetrants such as CO2. With increasing pressure, diffusivity is
enhanced.
Polyimides can be easily modified by use of diamines of different chemical
structures to modify the properties to suit targeted applications. Wide varieties of

Chapter 1 Introduction and literature survey
19
modified PIs with different chemical structures are reported in the literature. Sulfonated
PIs (SPIs) is one such modification, projected as possible candidate for application as
membrane material for fuel cells (PEMFC). Presently, significant research work is being
conducted to study the electrochemical and other properties of SPIs of different
structures. Since, one of the objectives of present dissertation is to synthesize novel SPI
containing sulfonic acid group in side chain and study general and electrochemical
properties of this SPI to assess possible application as membrane material for polymer
electrolyte for fuel cell, a general literature survey on sulfonated PIs is given below.
1.6 Sulfonated polyimides as polymer electrolyte membrane (PEM)
materials
Ability of forming numerous sulfonated monomer chemical structures for
aromatic polyimides widens their breadth in PEMFC application. High susceptibility of
conventional five membered imide rings towards hydrolysis under severe acidic
environment makes them brittle demoting their application in PEMFC (Faure, 1997;
Genies, 2001a; Fang, 2002). On the other hand, SPIs with six membered imide rings,
based on 1,4,5,8-naphthalene tetracarboxylic dianhydride (NTDA) have proved to be
stable towards acid hydrolysis, promoting them to be used in PEMFC (Fang, 2002; Yin,
2004 and 2006a). Group of Mercier (Genies, 2001a and 2001b; Marestin, 2008),
McGrath (Gunduz, 2001; Einsla, 2004; 2005a and 2005b), Okamoto (Fang, 2002; Yin,
2006a), Watanabe (Miyatake, 2003; Asano, 2006), Holdcraft (Rodgers, 2006; Savard,
2008) and Zhang (Li, 2007b; 2007c and 2008) have diligently worked towards the
synthesis of six membered SPI in PEMFC application.
1.6.1 Synthesis of sulfonated polyimides (SPIs)
General method for the synthesis of SPIs is by sulfonating parent PI in suitable
solvents using a sulfonating agents such as SO3, oleum or chlorosulfonic acid. Sulfonated
PIs precipitate out from solvents making it difficult to control the degree of sulfonation.
Thus, SPIs with known degree of sulfonation are usually prepared by the
polycondensation of a dianhydride with sulfonated diamine (SDA).

Chapter 1 Introduction and literature survey
20
1.6.1.1 SPI based on sulfonated diamine (SDA) as monomers
SDA possess both basic (–NH2) and acidic groups (–SO3H), form zwitter ion and
thus become insoluble in organic solvents. To make it soluble, protection of –SO3H
group by an organic base such as triethyl amine (TEA) is usually done. In general, SPIs
are prepared by polycondensation reaction between SDA and dianhydrides in m-cresol as
the solvent (Genies 2001a; Fang, 2002). In the first step, SDA in its triethylammonium
sulfonate form is prepared and after its complete dissolution in the solvent, equimolar
quantity of dianhydride is added followed by the addition of benzoic acid. As explained
earlier, the role of benzoic acid is to catalyze the polymerization reaction. CPIs are
prepared with varying ratio of SDA to the non-sulfonated diamine. Li et. al (2008)
synthesized random as well as block CPI from hexamethylene diamine with varying ratio
of sulfonated and non-sulfonated dianhydrides.
Among the different types of CPIs, synthetic procedure for block CPI is quite
different and is followed in two steps. In the first step, a block of SPI with desired
molecular weight is prepared by condensing SDA with a dianhydride in the desired ratio.
In the second step, this block was reacted with a non-sulfonated diamine and a
dianhydride (Genies, 2001a; Li, 2008). While, for the statistical random CPI synthesis, all
the reactants can be mixed at once in the beginning of polymerization (Li, 2008).
Most of the literature on SPIs constitute polycondensation of SDA with
dianhydride; NTDA. Though, recent literature reveals synthesis of SPI involving large
number of SDA, most of them have been synthesized in the laboratory and the least are
commercially available including DABSA, DMBDSA, DSDSA and BDSA as shown in
Figure 1.5. Genies et al. (2001a) synthesized sulfonated diamine; 4,4’-diamino-biphenyl
2,2’-disulphonic acid (BDSA). Its copolymers with various non-sulfonated diamines in
different ratio with either ODPA or NTDA were prepared. They observed that phthalic-
based SPI based on ODPA was highly prone to the hydrolysis leading to chain scission.
On the other hand, naphthalic-based SPI prepared from NTDA demonstrated good
performance as membrane material in fuel cell environment (Genies, 2001a and 2001b).
Fang et al. (2002) synthesized ODADS and its SPI and compared PEM performance
between these SPIs and BDSA-based SPIs (Genies, 2001a). The former revealed better
hydrolytic stability than the latter. In BDSA-type SPI, steric hindrance due to two

Chapter 1 Introduction and literature survey
21
sulfonic groups resists the rotation of two neighboring phenyl rings of BDSA resulting in
induced strain with decreased hydrolytic stability (Fang, 2002; Yin, 2006a).
Figure 1.5 Main-chain-type sulfonated diamine
With advancement towards better PEM, Okamoto proved that SPI based on
highly basic diamines possessed better hydrolytic stability. Due to this basicity, “main-
chain-type” SPIs were further classified into “Type 1” and “Type 2”. In “Type 1” the
sulfonic group is directly attached to amino-phenyl ring, while “Type 2” consisted of
bonding of sulfonic group to the phenyl ring other than amino-phenyl ring as shown in
the Figure 1.3 (Yin, 2006a). Authors stated that “Type 1” SPIs have lower hydrolytic
stability, since their basicity is decreased due to the electron withdrawing effect of
sulfonic group resulting in more facile nature of carbon-nitrogen bond of imide group. In
contrast, in case of “Type 2” SPIs, there is no such decrease in electron density on the

Chapter 1 Introduction and literature survey
22
amino-phenyl ring. In addition, the electron density around the nitrogen atom of imide
moiety is increased due to the electron donor effect of amino-phenyl ring, becoming
imide linkage less vulnerable for the nucleophilic attack of water molecule. The net result
is the improved hydrolytic stability for the “Type 2” SPIs.
With further design for SDA monomers, “side-chain-type” SPIs derived from
these diamines have gathered major attention. The various structures of such SDA are
shown in Figure 1.6. These types of SPIs can form micro-phase separation structures,
similar as in case of Nafion. Such phase morphology not only facilitates the proton
transport but also improves the mechanical strength needed for PEM after water swelling.
In other words, positioning of –SO3H group away from the imide moiety restricts the
migration of proton in hydrophilic ion-rich domains only. Thus “side-chain-type” SPIs
constitute imide linkages in hydrophobic domain, while the sulfonic group in the
hydrophilic domains. Since imide hydrolysis is an acid-catalytic reaction, restriction of
proton mobility away from the polymer main chain can alleviate the hydrolysis of imide
ring. Moreover, phase separation increases the proton conductivity in side-chain-type
SPIs (Yin, 2004 and 2006a).
Figure 1.6 Side-chain-type sulfonated diamines

Chapter 1 Introduction and literature survey
23
1.6.1.2 SPI based on naphthalic type dianhydrides (PNI)
The dense structures due to the planar and symmetric nature of NTDA make PNI
more intractable than the five-membered ring (phthalic) polyimides. Thus, solubility of
PNIs is limited only to strong acids (sulfuric, methylsulfuric, polyphosphoric) (Rusanov,
1994). It should be noted that the step wise approach for the synthesis of PNI becomes
more difficult due to the low electrophilicity of the six membered anhydride as compared
with the five membered anhydride moieties (Rusanov, 1994). Hence, efforts were
diverted towards the synthesis of organic soluble PNI. The modification in NTDA
structure such as introduction of “hinge” groups between two fragments of naphthalic
anhydride (facilitating the bond rotation around the main chain axis) and embodiment of
bulky group substituents can improve PNI solubility significantly (Rusanov, 1994). It
was also said that structural modifications in NTDA to ease solvent solubility. The
following types of bis(naphthalic anhydride) were used for making SPIs (Figure 1.7).
Figure 1.7 Various structures of naphthalenic dianhydride monomers
Li et al. (2008) proposed that use of BNTDA as compared with NTDA for
making SPI should be more advantageous as it forms non-coplanar binaphthyl rings,
separating their electronic conjugation. As a result, imide linkage possesses higher
electron density and thus becomes less vulnerable for the nucleophilic attack of water
molecule. Li et al. (2008) further sulfonated this BNTDA to SBTDA and observed the
enhancement in water uptake and proton conductivity due to the increased free volume
and kinked non-coplanar structure of SBTDA. Chen et al. (2006) synthesized SPIs based
on more reactive dianhydride KDNTDA. These SPIs showed tough mechanical
properties, high-reduced viscosity (0.8 to 4.5 dL.g-1), good solubility in proton form and
reasonable proton conductivity at their lower IECs.

Chapter 1 Introduction and literature survey
24
Blends of SPIs with other polymers are demonstrated in the literature, as reviewed
separately in Chapter 5.
1.6.2 Membrane preparation of SPIs
Membrane preparation of SPIs in pure sulfonic acid form is difficult since they
are usually insoluble in common organic solvents. After synthesizing SPI, they are first
isolated in the salt form (triethylammoium sulfonate form) by precipitating in a non-
solvent like acetone. Subsequently, this SPI is purified by reprecipitation to remove traces
of polymerizing solvent and catalyst. Generally, membranes of SPI in salt form are
prepared and then they are acidified by immersing in acid to get the membrane in –SO3H
form for the application as PEM for fuel cell.
1.7 Other polymers as polymer electrolyte membrane (PEM)
materials
Polymers containing strong acid groups such as sulfonic acid are capable of
conducting protons in presence of water. Basic polymers such as polybenzimidazole
(PBI) on doping with acids such as phosphoric acid or sulfuric acid are also used (Wang,
1996). Sulfonated polymers can be further divided into fluorinated and non-fluorinated
polymers. A brief literature survey on these polymers is given below.
1.7.1 Perfluorosulfonic acid (PFSA) membranes
PEMFC accomplished the major breakthrough in late 1960s, when Dupont
developed the membrane based on perfluorinated ionomer; Nafion with superior
chemical and mechanical stability. It is perfluorosulfonic acid ionomer consisting of
Teflon like backbone with sulfonic acid located at the terminal of the side chain. The
presence of C-F backbone offers several advantages. Its high C-F bond strength (441 kJ/
mol) improves its stability in severe redox environment as well as at higher potential
(~1.0 V at cathode) than the low C-H bond strength (356 kJ/ mol). The peculiar
arrangement of hydrophobic Teflon backbone in main chain and hydrophilic sulfonic
acid (–SO3¯H+) in the side chain through flexible ether linkage organizes the phase
morphology so as to separate the hydrophobic and hydrophilic regions. Because of such

Chapter 1 Introduction and literature survey
25
ionic groups, large quantity of water can be imbibed in the polymer matrix making
Nafion a dilute acid in solid state.
An important parameter of the Nafion is its equivalent weight (EW). Nafion is
commercially available with trade name as Nafion 117, Nafion 115, Nafion 1110, Nafion
112, etc. The first two digits represents the EW as 1100, while the remaining digits show
its thickness in mil. The chemical structure of Nafion is shown as in Figure 1.8.
Being perfluorinated polymer backbone, the characteristic properties of these
membranes such as chemical, thermal and mechanical are elevated. However, high
production cost, low proton conductivity at reduced humidity and/ or high temperature,
high permeability for H2, O2 and MeOH, CO poisoning of platinum catalyst due to low
operational temperature of these membranes have limited their widespread commercial
application in PEMFC (Rikukawa, 2000; Rusanov, 2005). Therefore, efforts have been
taken for the development of alternative membranes with fulfillment of the desired
properties for PEMFC, as shown in Figure 1.8. Flemion, Aciplex and Dow membranes
have been commercialized by Asahi Glass, Asahi Kasei Chemical Co. and Dow
chemicals, respectively (Figure 1.8).
Figure 1.8 A general structure of commercially available PFSA membranes
In PEMFC, the oxidation reduction rate (ORR) occurring at cathode is sluggish,
which being further suppressed by low operating temperature. Thus, synthesizing novel
non-fluorinated solid electrolyte addressing various drawbacks as given above becomes
necessary.
1.7.2 Polymers with sulfonic acid group
These polymers can be synthesized by either sulfonating the high molecular
weight aromatic polymers (post-sulfonation) or preparing the polymers from monomers
containing sulfonic acid groups, as elaborated below.

Chapter 1 Introduction and literature survey
26
1.7.2.1 Post-sulfonation of polymers
Various polymers including poly(1,4-phenylenes), poly(1,4-oxyphenylenes),
poly(ether ether ketone) (PEEK), poly(arylene ether sulfone) (PEES),
poly(phenylqunixalines), poly(phthalazinones), poly(imides), poly(benzimidazole), etc.
can be sulfonated by using sulfonating agents such as concentrated sulfuric acid, oleum,
chlorosulfonic acid, SO3, a mixture of methanesulfonic acid and conc. H2SO4 and acetyl
sulfonate (Roziere, 2003; Gao, 2003a and 2003b; Hickner, 2004; Liu, 2007; Mader,
2008). The feasibility of easy scale up due to the simple procedure and availability of
inexpensive commercial high molecular weight thermoplastic polymers make post
sulfonation more attractive. For instance, several researchers have studied sulfonation of
the commercial Victrex (PEEK) (Hickner, 2004; Liu, 2007, etc.). However, drawbacks
such as degradation of polymers while using strong acidic sulfonating agents
(chlorosulfonic acid or fuming sulfuric acid), uneven distribution of sulfonic acid groups
and difficulties in optimization of degree of sulfonation (DS) can cause the rough
surfaces, crosslinking and excessive swelling in the post-sulfonated PEEK (Liu, 2007;
Shin, 2004; Wu, 2006). Moreover, DS also determines the solvent solubility of sulfonated
polymers. DS depends on the sulfonation time, temperature and sulfonating agents. The
irregular placements of sulfonic acid groups can induce the stress formation when
hydrated, making membrane cracking more feasible. PEEK and PEES confer sulfonation
only to a phenylene ring attached to an electron donating ether linkage while restricting to
phenylene ring that of electron withdrawing carbonyl and sulfone moieties as shown in
following Figure 1.9.
Figure 1.9 Sulfonation of PEEK and PEES (Pienemann, 2008)
These sulfonic groups are less acidic being situated ortho or para to electron-
donating substituents and become more acidic when located meta to the electron
withdrawing moiety (–CO– and –SO2–). The latter sulfonated polymer becomes more
stable towards thermal de-sulfonation (Shin, 2004; Hamrock, 2006; Wu, 2006; Li,

Chapter 1 Introduction and literature survey
27
2007c). Desired or controllable DS in post-sulfonation can be achieved by sulfonating
copolymers. Copolymers based on monomers with capability of different ease of
sulfonation are synthesized first and then post sulfonated (Liu, 2007). Thus, the design of
copolymers is done in such a fashion that some structures could not be sulfonated easily
and letting degree of sulfonation to be dependant on feed ratio of such monomers as
exemplified in the following Figure 1.10.
Figure 1.10 Sulfonation reaction of copolymers (Pienemann, 2008)
1.7.2.2 Sulfonated polymers and copolymers from sulfonated monomers
This method comprises the direct polymerization using sulfonated monomers.
This technique enables copolymer synthesis with desired degree of sulfonation and hence
IEC values. The synthesis of sulfonated monomer was reported by Robeson and
Matzener (1983), whose primary interest was to check at its flame retarding additive
property (Peinemann, 2008). Ueda et al. (1993) successfully demonstrated synthesis,
characterization and purification of the sulfonated (4,4'-dichlorodiphenylsulfone)
monomer and its polymer (sulfonated PES) by condensation polymerization route as
shown in Figure 1.11.
Figure 1.11 Sulfonated-PES from sulfonated (4,4'-dichlorodiphenylsulfone) monomer
Wang et al. (1998) added another analogous halogenated sulfonated monomer
(5,5’-carbonylbis(2-fluorobenzenesulfonate)), which could offer various chemical
structural alteration in the community of PAEKs. Difluorinated monomers though are
expensive, they are highly reactive than the analogues dichlorinated monomers towards
nucleophilic aromatic substitution reaction (Lau, 1989). With the progressive shift of

Chapter 1 Introduction and literature survey
28
synthesizing sulfonated polymer, scientists soon started focusing on the sulfonation of
important raw materials of the above mentioned HPP polymers such as dihalogenated
monomers including aromatic diols (Gao, 2004 and 2005) as well. Synthesis of
disulfonated dihalogenated monomers is documented (Shin, 2004; Einsala, 2005b)
The position of sulfonic acid group in phenyl ring becomes convincing when the
enhanced acidic character is observed due to the location in the deactivated phenyl ring as
compared with its location at ortho or para to electron donating groups (activated phenyl
groups) (Shin, 2004; Hamrock, 2006; Li, 2007b and Li, 2007c). As a result, stability
towards desulfonation is improved in the former case than in the later case. On the other
hand, the access of sulfonation to such deactivated phenyl ring by post-sulfonation route
become insupportable by mild sulfonating agents as observed in the case of poly(4-
benzoyl)-1,4-phenylene [(PBP)] (Rikukawa, 2000).
The general method for making block copolymers includes the presynthesis of
hydrophilic and hydrophobic oligomers containing reactive functional end groups and
later pursuing their copolymerization (Hu, 2009). The existence of better phase separated
morphology in block copolymers than random copolymers is due to the better
agglomeration of hydrophilic sulfonic acid in the former due to the electrostatic
interactions in ion pairs (Elabd, 2006). The hydrophilic domain transports protons while
the hydrophobic domain provides morphological stability as well as prevents dissolution
in water. Such system is highly resembled with the morphology of PFSA polymers.
Properties of the PEM can be aligned with adjustment of either the length of hydrophilic
or hydrophobic segments. For the copolymers with similar chemical structure, physical
properties such as glass transition temperature, solvent solubility, IEC values, oxidative
and hydrolytic stability, proton conductivity, etc. of the block copolymers behave
significantly different as compared with random copolymers (Ding, 2001; Elabd, 2006;
Hu, 2009).
From the above review, it is evident that efforts are mainly directed to develop
sulfonated hydrocarbon polymer as alternative to expensive Nafion type polymers.
Sulfonated PI is one of such alternatives. The objective of the present dissertation is to
study electrochemical and other properties of new SPI containing sulfonic acid group in
side chain for their application as PEMFC. Thus, it is essential to know about fuel cell. A

Chapter 1 Introduction and literature survey
29
brief account of history and evolution of fuel cell, types of fuel cell, functioning of fuel
cell, evaluation and other aspects of PEMFC, functioning of PEMFC and prerequisites
for a polymer electrolyte membrane is given below.
1.8 Fuel Cells
The birth of fuel cell (FC) concept goes to Sir William Grove who in 1839 first
achieved electrical energy from a chemical reaction of oxygen and hydrogen with
platinum as catalyst (Grove, 1839; Bacon, 1969; Appleby, 1990). In 1893, Friedrich
Wilhelm Ostwald paid attention on the theoretical understanding of fuel cell operation
(Boudghene Stambouli, 2002). The technology revolution happened during World War II
in 1930s, witnessing the birth of many technologies including alkaline fuel cell (AFC)
invented by the British researcher F.T. Bacon for the reliable source of power for Royal
Navy submarines. In 1959, Bratt and Whiteny licensed this idea of AFC in Appollo
spacecrafts by using higher concentration (~85%) of alkali electrolytes (Perry, 2002). The
long hibernation in fuel cell research for commercialization was strongly shaken in
1950s-60s when scientists in National Aeronautics and Space Agency (NASA), USA had
been looking for power upcoming to manned space flights (Gemini program). Shortly,
the collaborative work of General Electric and NASA’s researchers lead to the
development of new type fuel cell; proton exchange membrane fuel cell (PEMFC). The
research in fuel cell areas had to be accelerated in late 20th century due to the tremendous
pressure built up to alleviate the greenhouse effect and global warming concerns.
Demand for energy has been growing with increase in population and living
standards. The concurrent consciousness about environmental cost of energy has entailed
various governments of nations, organizations and industries to find the new ways of
accessing the expansion of energy sources. Efforts have been performed for generating
plentiful energy sources, which have also been progressively shifting from solids (wood,
coal) to liquids (petroleum) to gas (LNG, natural gas). This process is called
“decarbonization”, implying progressive shift to fuels with increasing hydrogen to carbon
ratio (Devezas, 2008). Logically, the impending logical progression in this evolution is
the use of only hydrogen as a fuel with complete elimination of carbon. Advent energy
sources technologies would therefore largely dependant on hydrogen in the coming

Chapter 1 Introduction and literature survey
30
decades (Tseng, 2005; Penner, 2006; Dincer, 2007). Fuel cells use hydrogen as the feed,
are electrochemical devices which directly converts chemical energy into electrical
energy. This one step process has higher efficiency than internal combustion engine
(ICE) having multi step processes and lower efficiency (Wright, 2004). The conventional
popular ICE follows the Carnot cycle adiabatically as well as isothermally with 40-50%
theoretical efficiency. In contrast, fuel cells are operated isothermally with more than
80% (Barbir, 2005) efficiency provided by hydrogen as the fuel, as follows.
H2 + ½ O2→ H2O [ΔH° = -286.0 kJ.mole-1 and ΔG°= -237.2 kJ.mole-1]
Efficiency (%) = [ΔG° / ΔH°] = 83.0
The fuel cells produce only water, heat and electricity by electrochemical reaction
isothermally. Moreover, absence of moving parts with resultant low noise operation in
fuel cells eliminate complexity in FC systems making them more adaptable towards
simpler designing as well as benignant to environment.
1.8.1 Types of Fuel cells (FC)
Classification of FC primarily depends on their operation at specified temperature
as well as the types of ion conductive electrolyte used. Five major types of FC have been
demonstrated. These include Proton Exchange Membrane Fuel Cell (PEMFC), Alkaline
Fuel Cell (AFC), Phosphoric Acid Fuel Cell (PAFC), Molten Carbonate Fuel Cell
(MCFC) and Solid Oxide Fuel Cell (SOFC) (Barbir, 2005), as shown in Table 1.1.
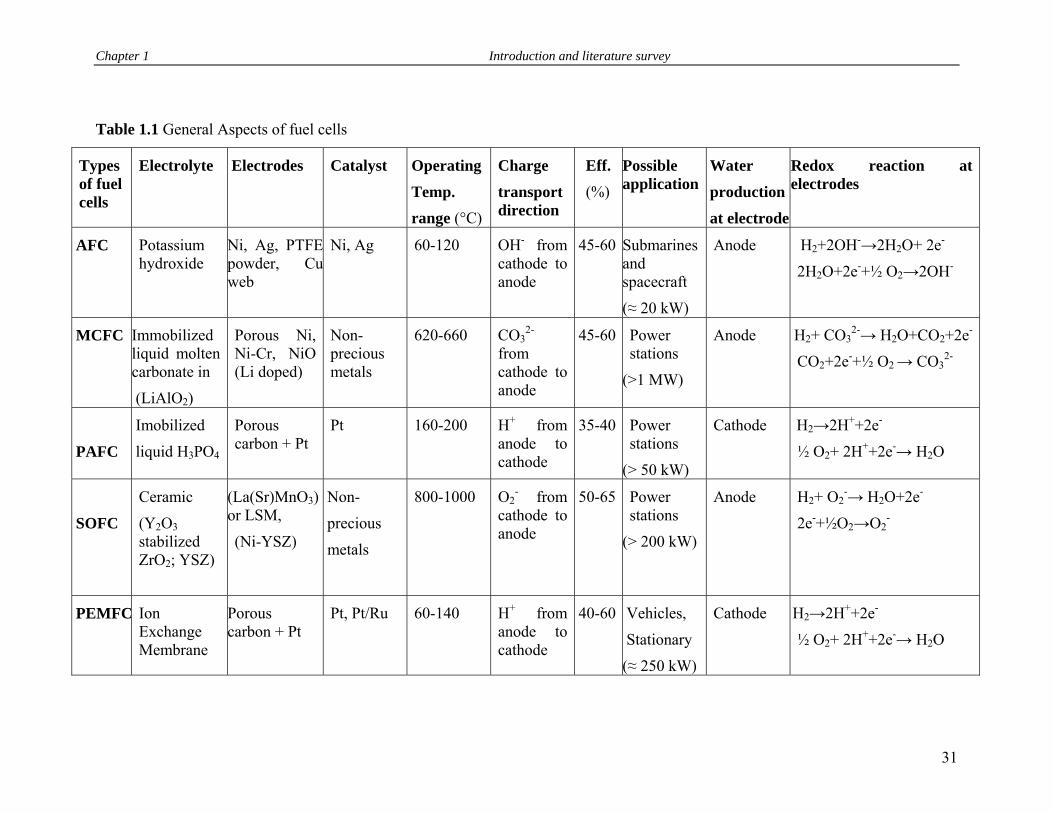
Chapter 1 Introduction and literature survey
31
Table 1.1 General Aspects of fuel cells
Types of fuel cells
Electrolyte Electrodes
Catalyst
Operating
Temp.
range (°C)
Charge
transport direction
Eff.
(%)
Possible application
Water
production
at electrode
Redox reaction at electrodes
AFC
Potassium hydroxide
Ni, Ag, PTFEpowder, Cuweb
Ni, Ag 60-120 OH- from cathode to anode
45-60
Submarines and spacecraft
(≈ 20 kW)
Anode H2+2OH-→2H2O+ 2e-
2H2O+2e-+½ O2→2OH-
MCFC
Immobilized liquid molten carbonate in
(LiAlO2)
Porous Ni, Ni-Cr, NiO (Li doped)
Non-precious metals
620-660 CO32-
from cathode to anode
45-60 Power stations
(>1 MW)
Anode H2+ CO32-→ H2O+CO2+2e-
CO2+2e-+½ O2 → CO32-
PAFC
Imobilized
liquid H3PO4
Porous carbon + Pt
Pt 160-200 H+ from anode to cathode
35-40 Power stations
(> 50 kW)
Cathode H2→2H++2e-
½ O2+ 2H++2e-→ H2O
SOFC
Ceramic
(Y2O3 stabilized ZrO2; YSZ)
(La(Sr)MnO3) or LSM,
(Ni-YSZ)
Non-
precious
metals
800-1000 O2- from
cathode to anode
50-65 Power stations
(> 200 kW)
Anode H2+ O2-→ H2O+2e-
2e-+½O2→O2-
PEMFC
Ion Exchange Membrane
Porous carbon + Pt
Pt, Pt/Ru 60-140 H+ from anode to cathode
40-60 Vehicles,
Stationary
(≈ 250 kW)
Cathode H2→2H++2e-
½ O2+ 2H++2e-→ H2O

Chapter 1 Introduction and literature survey
32
1.8.2 Evolution and major aspects of PEMFC
Thomas Grubb and Leonard Niedrach in General Electric (GE) company were
working on fuel cell and ended up into a new type of fuel cells, referred as Grubb-
Niedrach fuel cell (Grubb, 1959; Niedrach, 1964; Beuscher, 2005). At the same time,
NASA had also been in search for building up a compact electricity generator for use on
manned space missions. Recognizing the potential in Grubb-Niedrach fuel cell, a team of
GE and NASA generated a new type of fuel cell; polymer electrolyte membrane fuel cell
(PEMFC) (Perry, 2002). Utilization of this PEMFC in NASA’s Gemini space program
demonstrated the first successful implementation and commercialization of fuel cell
technology. This PEMFC constituted polystyrene sulfonic acid as the membrane material
for proton transportation and platinum as the electrocatalyst (Rikukawa, 2000). The high
loading of platinum catalyst, poor oxidative stability of polystyrene sulfonic acid
membrane after certain period leading to degradation and low attainable power density
raised the question of its practicality. PEMFC accomplished the first major breakthrough
in late 1960s, when Dupont developed the membrane based on perfluorinated ionomer,
Nafion with superior chemical and mechanical stability (Rikukawa, 2000).
1.8.3 Advantages and functioning of PEMFC
PEMFC has progressively become more and more attractive due to its low
operating temperature, quick start up and load following convenient way of electrolyte
handling, low corrosion and system robustness due to solid electrolyte and its
compactness. It also generates higher specific power (W/kg) and power density (W/cm2)
than other types of fuel cells. Hence wide application range of power as portable,
stationary and automotive applications is possible. Furthermore, larger heat generated at
cathode when greater number of stacks are used, can be effectively utilized. The basic
design of single PEMFC stack is shown in Figure 1.12. Fabrication of PEMFC comprises
polymer electrolyte membrane (PEM) sandwiched by catalyst coating from both the
sides, constituting membrane electrode assembly (MEA). This is the ‘heart’ of the
PEMFC. PEM not only transports the proton from anode to cathode but it also acts as
barrier for unoxidized H2 and unreduced O2 from passing through.

Chapter 1 Introduction and literature survey
33
Figure 1.12 Schematic of PEMFC (Rusanov 2005)
Protons generated by oxidation of H2 (H2 → 2H+ + 2e-) at the anode migrate
towards cathode through the membrane. PEM also serves as barrier for evolved electrons.
The cathode is fed with O2, which is reduced by the electrons reached through
external circuit with the help of catalyst, resulting in production of water, heat and
electricity.
At Cathode, ½ O2 + 2H+ + 2e- → H2O + heat + electricity (Ecathode = 1.23 V)
This electrochemical half-cell reaction at cathode is kinetically sluggish.
1.8.4 Criteria for PEM materials
PEM materials can be broadly categorized as polymer containing –SO3H
functionality and others based on acid-base polyelectrolyte complexes. These two types
have different properties and their own mechanism of proton transport. Their requisite
properties can be described as given below.
For the –SO3H group containing polymers, high proton transport at low humidity,
strong acidic functional groups with higher dissociation constant and lower pKa constant,
lower IEC values (< 2) coupled with better thermal, oxidative and hydrolytic stability in
acidic medium are required. In addition, such PEM materials should exhibit low electro
osmotic water drag, low swelling, better connectivity of proton conducting channels,
almost zero electron conductivity, separation of hydrophilic and hydrophobic domains
with lower interfacial tension are also importanat criteria. PEM should not contain any

Chapter 1 Introduction and literature survey
34
pores and should have minimum H2 and O2 permeability. It should confer excellent
mechanical strength in hydrated condition, at elevated temperature (≈150 °C), high
flexibility particularly in dry state and high CO tolerance.
In order to be capable for membrane electrode assembly (MEA) fabrication,
membrane must possess good adherence with porous gas diffusion electrodes. Hu et al.
(2009) reported higher through-plane proton conductivity (conductivity in thickness
direction of membrane) than in-plane conductivity (conductivity in plane direction) and
lower in-plane swelling for better adhesion as well as mechanical stability of resulting
MEA. It should also be environmental friendly with low production cost for effective
commercialization.
PEM, containing –SO3H group conduct proton only in hydrated condition by
vehicular mechanism, limiting the operation of PEMFC at < 100 °C. This entails
permanent humidification system with auxiliary devices and making PEMFC expensive.
Hence replacement of water with another species capable of proton transportation at
higher temperature is being explored. In this context, azole group containing heterocyclic
polymers like PBI while doping with phosphoric acid looks attractive due to its high CO
tolerance, non-volatile nature of phosphoric acid, low metahanol-fuel crossover,
operation performance up to 200 °C, almost zero osmotic drag coefficient and
comparable proton conductivity (Li, 2003 and 2009; Mader, 2008; Potrekar, 2009a).
Operating PEMFC at higher temperature confers fast electrode kinetics with less cathode
flooding. Operating PEMFC above 100 °C becomes simplified due to the presence of
water in single gas phase. Though PBI doped with amphoteric acid like phosphoric acid
is emerging as PEMFC for high temperature application, it carries some drawbacks.
These include poor solubility of pristine PBI in common organic solvents, leaching of
phosphoric acid from the membrane, unestablished long term durability, effect of on-off
cycle, etc. Large efforts are being made to overcome these drawbacks.
Future efforts towards increasing acid retention capability and acid content in PBI
are being made. Incorporation of bulky groups improves solvent solubility of PBI as well
as acid uptake due to the increased free volume with disruption of rigid structure in PBI.
However, bulky group with additional basic sites in PBI can not only increase acid base
interaction but also the acid retention capacity with higher proton conductivity (Kulkarni,

Chapter 1 Introduction and literature survey
35
2008; Kim, 2009). PBI with high molecular weight is desirable since it improves the
mechanical and chemical stability (Lobato, 2007). Functionalization, polymer blending
and doping with different electrolytes have also been adopted towards improving
performance of PBI as PEM material (Mader, 2008). Presence of basic benzimidazole
group in PBI offers sites for interaction with polymers having acidic moieties and
creating favorable environment for miscible blending. Blends of PBI are prepared in
order to increase thermal stability, acid uptake and proton conductivity (Mader, 2008);
which can be accomplished by blending with sulfonated polymer or addition of inorganic
filler. Swelling and IEC of sulfonated polymer can be reduced by blending with PBI
(Wysick, 2005; Mader, 2008). Hasiotis et al. (2001a and 2001b) reported higher
conductivity of PBI/PSF blends than the acid doped PBI (Daletou, 2009). Blends of PBI
with PSF copolymer containing pyridine units showed good oxidative stability (Daletou,
2005). High acid doping deteriorates mechanical strength of PBI film. In this context,
addition of inorganic additives looks attractive, since with increasing proton conductivity,
mechanical properties can also be greatly improved (Mader, 2008).
Polytriazole (PT) and polyoxadiazole (POD) also belong to azole group
heterocyclic polymers and on doping with acids, they can conduct proton in absence of
water. Potrekar et al. (2009b) synthesized PBI tethered with N-phenyl 1,2,4-triazole and
reported maximum proton conductivity of 1.8 X 10-2 S/cm at 175 °C. Zaidi et al. (2000)
demonstrated high proton conductivity of phosphoric acid doped POD, comparable with
that of Nafion. Subbaraman et al. (2007) reported 4,5-dicyano-1H-[1,2,3]-triazole as a
viable proton transport facilitator. Zhou et al. (2005) proved that poly(4-vinyl-1H-1,2,3-
triazole) possessed higher conductivity with significant improvement in the
electrochemical stability than poly-(4-vinylimidazole) suggesting the significance of
triazole as proton conductor. Since 1H-1,2,3-triazole (pKa1 =1.17 and pKa2 = 9.26)
possess higher acidic character than imidazole (pKa1 = 7.18 and pKa2 = 14.52)
(Gagliano, 1989), solid electrolyte based on the former can offer significantly improved
proton conductivity.

Chapter 1 Introduction and literature survey
36
1.9 Aims and objectives
Aim of this thesis was to synthesize polyimides (PI) containing side chain
substituent and assess their applicability in two promising areas, as a gas separation
membrane material and as a polymer electrolyte membrane for fuel cell. The side chain
incorporation in PI was anticipated to be better feasible with modification of amine
moiety of PI than that of anhydride moiety.
Developing a viable synthesis methodology of systematically architected
diamines, which would allow requisite architecture of diamine structure was the primary
objective of the work. Polymerization of these structurally different side chain containing
diamines with chosen dianhydrides, characterization of their physical properties,
membrane preparation and their evaluation as gas separation membrane material or as
polymer electrolyte membrane material was thought to be the path of this thesis work.
These objectives can be further split as
a) Synthesis of a diamine, viz., 1-phenoxy-2,4-diaminobenzene (PDAB), which
contains a phenoxy substituent on the m-phenylene diamine. This was thought to be
achieved by condensation of 1-chloro-2,4-dinitrobenzene and phenol followed by
reduction of nitro groups. Polymerization of PDAB with selected dianhydrides, physical
property evaluation of formed polymers and their permeability determination would set a
guideline towards further structural modification of the diamine.
b) Substitution of bulky alkyl groups on the aromatic ring of phenoxy linkage
belonging to PDAB, their polymerization with select dianhydrides, physical property
determination of formed polyimides, membrane preparation and evaluation of permeation
properties using pure gases.
c) Sulfonation of PDAB, polymerization with selected dianhydrides, their blend
preparation with PBI and proton conductivity evaluations.
1.10 Organization of the thesis
The present thesis provides synthesis, characterization and application of side-
chain-type polyimides for gas permeability and polymer electrolyte for fuel cell. It has
been presented in following chapters.

Chapter 1 Introduction and literature survey
37
Chapter 1. Introduction and literature survey
This chapter presents a brief literature overview of synthesis and applications of
polyimides. Properties of PIs as membrane material, particularly in gas permeation and
PEMFC are illustrated. Scope and objective of the work is presented at the end of this
chapter.
Chapter 2. Monomer synthesis and characterization
This chapter presents synthesis of various diamines used in the present
investigation. These diamines were designed so that the formed polyimides exhibit side
chain phenoxy groups. All diamines were synthesized from condensation of a single
precursor, 1-chloro-2,4-dinitro benzene. It was condensed with various phenols
possessing systematic structural variation. Characterizations of precursors and resulting
diamines were performed by usual structural elucidation methods, viz., FT-IR, 1H NMR,
elemental analysis and melting point determination.
Chapter 3. Polyimide synthesis and their characterization
This chapter presents synthesis of polyimides by single step solution
polycondensation method. All polyimides investigated possessed side chain oxy-phenyl
ring possessing chosen substitutes either for gas permeation or for PEM material
investigation. Physical characterizations of these PIs (requisite for gas permeation and
PEM material) are presented. Blend membranes of sulfonated polyimides with
polybenzimidazole (PBI) for PEM analysis are also described.
Chapter 4. Structure-gas permeability correlation in polyimides
This chapter presents investigation of gas permeability of PIs based on diamines
possessing alkyl substitution on pendant phenoxy group. It presents physical properties,
those are known to affect gas permeation properties and their correlation with
permeability, of pure gases (He, H2, Ar, N2, O2, CH4 and CO2). Gas permeation in SPIs as
well as blend membranes (using dry as well as humidified gases) is also investigated.

Chapter 1 Introduction and literature survey
38
Chapter 5. Investigation of polyimides towards their applicability as PEM
materials
Evaluation of side-chain-type SPIs and blend of SPI with PBI as PEM material is
discussed in this chapter. Physical properties influencing proton conductivity such as
water uptake capacity, phosphoric acid uptake, ion exchange capacity (IEC), hydrolytic
stability and oxidative stability were evaluated and proton conductivity analysis measured
by electrochemical impedance spectroscopy (EIS) is discussed.
Chapter 6. Conclusions
This chapter presents conclusions with major results obtained.

Chapter 2
Monomer synthesis and characterization
2.1 Introduction
Tailor made structural variations in polymers to achieve desired properties is
usually based on designing a suitable structure of their monomers. In case of polyimide
structural architecture, polycondensation of a diamine with a dianhydride has played the
major contribution. Between diamines and dianhydrides, synthesis of novel diamines
have always surpassed over dianhydrides. This could be attributed to their relative ease of
synthesis, comparatively lower number of synthetic steps, higher stability towards
moisture, etc. In addition, precursors of diamines, viz., dinitro compounds are stable at
ambient conditions and therefore can easily be stored. These can be reduced to diamines
by a suitable reducing agent viz., Fe/HCl, Sn/HCl, LiAlH4, Co2(CO)8 or H2 in presence of
Raney Ni, Pt or Pd/C at the time of utilization (Scriven, 1988; Kabalka, 1991; Huldlicky,
1996; Lee, 2004). Conversely, dianhydrides need to be stored in dry conditions, as they
are highly susceptible to hydrolysis and need to be sublimed before polymerization.
Aromatic diamines offer more number of sites for substitution than the analogous
aromatic dianhydrides. For instance, between, p-phenylene diamine (p-PDA) and
pyrromellitic dianhydride (PMDA), the latter has only two sites for modification as
compared to four sites in the former (Figure 2.1). Aromatic diamines also exhibit more
number of structural isomers than that of analogous aromatic dianhydrides. For instance,
in phenylene diamines, three isomers, viz., p-PDA, m-PDA and o-PDA are possible. On
the other hand, in a dianhydride like PMDA, no structural isomer is possible.
Figure 2.1 Number of available sites in common aromatic diamine and dianhydride
Though phenylene diamines are used in synthesis of polyimides possessing high
tensile strength and heat resistance, these polymers are not easily processable. It is

Chapter 2 Monomer synthesis and characterization
40
necessary to synthesize PIs based on structurally modified diamines to fine-tune polymer
properties. Such diamines can be prepared by introducing flexible and/or bulky pendant
substituents on phenyl ring. This improves solubility/processability of resulting polymers
while retaining thermal stability, in comparison to PI based on diamine monomers
incorporating flexible groups in the main chain (Yang, 1993 and 1994). Substitutions on
phenyl ring of a diamine moiety disrupt chain packing, renders amorphous nature and
lead to improved solubility (St.Clair, 1976; Takekoshi, 1990). Bulky group substitution
with aromatic nature would become more attractive since they not only reduce chain
interactions, but also improve solubility without sacrificing thermal properties (de Abajo,
1999; Kim, 2002; Qiu, 2005). Among phenylene diamines, m–phenylene diamines induce
kinks and disrupt the symmetry due to meta-linkage in resulting polymer (Takekoshi,
1990; Abajo, 1999; Kim, 2002). As a result, amorphous nature is rendered to such PIs
leading to improved solubility.
Diamines are generally prepared either by ammonolysis route or by the reduction
of dinitro compounds, dioximes or dinitriles. In first route, functional group like halogen
(Muhlbauer, 1968), hydroxyl (MacKenzie, 1958), keto or aldehyde (Habermann, 1979) is
replaced by amino group. Reports indicate that synthesis of amines by reduction route use
reagents like (i) metal and acid (Fe, Zn or Sn along with HCl, H2SO4, CH3COOH or
HCOOH), (ii) metal and alkali (Zn or Fe in alkaline solutions), (iii) sulfides in alkaline
solutions (sodium sulfide or disulfide, ammonium sulfide, polysulfides or hydrosulfides),
(iv) sodium hydrosulfite in alkaline solution, (v) metal hydrides like LiAlH4, LiH, or
NaBH4 and (vi) amalgams (March, 1992). The substrates used for reduction include nitro
compounds, amides, oximes, isocyanates, isothiocyanates, azides, N-nitroso compounds,
azo, azoxy or hydrazo compounds (Rylander, 1967; March, 1992). The reagents
mentioned above produce large amounts of salts as by-products posing environmental
issues. The alternative route, catalytic hydrogenation, is thought to be the most efficient
route for synthesis of diamines and can be carried out using gaseous hydrogen (Rylander,
1967) or using hydrogen donors like NH2-NH2 with HCOOH (transfer hydrogenation) in
presence of a catalyst (Sivanandaiah, 1985). The catalysts used for transfer hydrogenation
include Raney nickel, platinum and palladium black. Hydrogenation using gaseous H2 is
more effective and requires shorter reaction times. Extensive studies have been carried

Chapter 2 Monomer synthesis and characterization
41
out on different types of catalysts, pressures, temperatures, solvents, reactant
concentrations and mode of reactions. A number of patents are available on the synthesis
of aliphatic (Williams, 1968), cycloaliphatic (Barkdoll, 1953; Arthur, 1967) and aromatic
diamines (Reynolds, 1951; Gonzalez, 1970; Bhutani, 1976) by catalytic hydrogenation.
Present Chapter deals with synthesis of diamines possessing meta linkage and
bulky substituent in the form of phenyl ether. This phenyl ether ring was also substituted
with alkyl groups to elevate bulky characteristics, desirous in gas separation application.
2.2 Experimental
This section describes synthesis of dinitro precursors, respective diamines and
their characterizations. Aromatic diamines possessing meta-linkage and flexible ether
(Ar–O–Ar') side group were prepared in two steps. In the first step, 1-chloro-2,4-
dinitrobenzene (CDNB) was condensed with various phenols to form dinitro precursors.
These precursors were subsequently reduced to diamines by heterogeneous catalysis
method using H2 in presence of Pd/C in a high pressure Parr reactor as described in the
following sections.
2.2.1 Materials
Phenol, o-cresol, p-cresol, 1-chloro-2,4-dinitrobenzene (CDNB), triethylamine
(TEA), potassium carbonate (K2CO3), anhydrous sodium sulfate (Na2SO4), benzoic acid,
calcium chloride (CaCl2), sodium hydroxide (NaOH), conc. HCl, conc. H2SO4 and
various solvents of AR grade [chloroform, methanol, ethanol, acetone, pet ether, ethyl
acetate and N,N-dimethylformamide (DMF)] were procured from S.D. Fine Chemicals,
(India). 4-t-Butylphenol and 2,6-dimethyl phenol were procured from Aldrich Chemicals
(USA). 4-t-Butylphenol was used as received, while other phenols were purified by
vacuum distillation prior to use. Oleum (60%) was procured from Dharmasi Morarji
Chemical Co. Ltd., Ambernath (India). Ultra-pure water was obtained using a Millipore
Milli-Q water purification system.
2.2.2 Analytical methods
Elemental analysis of diamines and their precursors was performed on Elementar
Vario-EL (Germany). FT-IR spectra were recorded on a Perkin Elmer 16 PC FT-IR

Chapter 2 Monomer synthesis and characterization
42
spectrophotometer (MA, USA) at ambient temperature. KBr pellet containing powdered
sample (~10 mg in 90 mg KBr) was prepared and vacuum dried at 60 C for 24 h prior to
use. In case of compounds having melting point (Mp) lower than 60 C, KBr pellets were
dried in vacuum oven at room temperature for 24 h. 1H NMR spectra were recorded on a
Bruker AC-200 (Switzerland). Depending on the solubility, samples were either
dissolved in CDCl3 with tetramethylsilane (TMS) as an internal reference or in dimethyl
sulfoxide (DMSO-d6). Melting point was determined by using BUCHI Melting Point B-
540 equipment (Switzerland)
Two sets of diamines were prepared. In the first set, the unsubstituted phenol was
reacted with CDNB to obtain dinitro precursor (PDNB). This was subsequently reduced
to obtain PDAB. This diamine was further sulfonated in view of applicability of resulting
polyimides as proton exchange membrane materials. In the second set of diamines, alkyl
substituted phenols were reacted with CDNB and reduced to obtain diamines.
2.2.3 Synthesis of PDNB, PDAB and SPDAB based on unsubstituted phenol
Scheme 2.1 given below describes the structure and synthetic route followed.
NO2
NO2
Cl
Et3N,
Reflux,48 h
NO2
NO2
OHH2, Pd/C
65 oC, 700 psi,4.5 h.
NH2
NH2
+
O
PDABPDNB
SO3 (60%),H2SO4
0-80 oC, 4-6 h
NH2
NH2
SO3H
SPDAB
OO
Acetone
Scheme 2.1 Synthetic route to PDNB, PDAB and SPDAB
2.2.3.1 Synthesis of 1-phenoxy-2,4-dinitrobenzene (PDNB)
A 250 mL single neck round bottom flask, equipped with a condenser and a
CaCl2 guard tube was charged with 9.411 g (0.1 mol) of phenol, 10.1 g (0.1 mol) of TEA,
20.25 g (0.1 mol) of CDNB and 150 mL of acetone. The mixture was refluxed for 48 h.
Solvent was distilled off and the residue was dissolved in CHCl3. This solution was

Chapter 2 Monomer synthesis and characterization
43
washed with 5 % HCl, 2 % aq. NaOH, followed by water, till neutral to pH. The solution
was dried over anhydrous Na2SO4, filtered and solvent was removed to obtain the crude
product. It was recrystallized from methanol to obtain pale yellow crystals of PDNB.
Yield: 24.1 g (90%), Mp: 75 C.
Elemental Analysis, (C12H8N2O5): Calculated: C, 55.4%; H, 3.1%; N, 10.8%. Found: C,
55.5%; H, 3.1%; N, 10.6%.
Figure 2.2 and 2.3 show FT-IR and 1H NMR spectra of PDNB, respectively.
2.2.3.2 Synthesis of 1-phenoxy-2,4-diaminobenzene (PDAB)
A Parr reactor (250 mL) was charged with 10 g (0.03846 mol) of 1-phenoxy-2,4-
dinitrobenzene (PDNB), 70 mL of methanol and 0.3 g of 5 % Pd/C. It was heated at 65
C under 700 psi of H2. When the absorption of H2 was complete, the contents were
cooled to room temperature and discharged after the pressure was released. The time
required for this reduction process was ~4.5 h. The solution was then filtered to remove
the catalyst. The filtrate was treated with activated charcoal and solvent was distilled off.
Obtained crude product was recrystallized from methanol to offer faint brownish shiny
crystals of PDAB. It was finally dried in vacuum oven for a day at room temperature and
stored in the desiccators under N2 atmosphere to prevent its oxidation.
Yield: 6.7 g (87 %), Mp: 60-61 C.
Elemental Analysis, (C12H12N2O): Calculated: C, 72.0%; H, 6.0%; N, 14.0%. Found: C,
2.0%; H, 6.4%; N, 14.0%.
Figure 2.2 and 2.4 show FT-IR and 1H NMR spectra of PDAB, respectively.
2.2.3.3 Synthesis of 4-sulfonic-1-phenoxy-2,4-diaminobenzene (SPDAB)
A three-necked 50 ml round-bottom flask equipped with a mechanical stirrer, a
dropping funnel, CaCl2 guard tube and gas inlet was purged with dry N2 and charged with
3.0 mL of conc. H2SO4 (95 %). The flask was cooled to 0 C in an ice bath followed by
slow addition of finely crushed powder of 5.0 g (25 mmol) of diamine (PDAB) and 7.0
mL of 95 % conc. H2SO4 while stirring. After ensuring complete dissolution of PDAB in
conc. H2SO4, 10.0 mL of fuming H2SO4 (60% SO3) was slowly added at 0 C for 30 min.
The temperature of the reaction mixture was elevated at the ambient and kept stirring for

Chapter 2 Monomer synthesis and characterization
44
30 min. This was followed by heating at 80 C for 1 h. The reaction mixture was
subsequently cooled to the ambient and poured onto crushed ice. This acidic solution was
then slowly neutralized with saturated solution of aq. NaOH at 0 C, till precipitation of
crude SPDAB. The crude product was filtered and added to 100 mL of deionised water at
0 C. It was dissolved as its sodium salt by slow addition of 2% aq. NaOH solution.
Obtained solution was filtered, followed by slow addition of 5% HCl at 0 C till
occurrence of brownish precipitate of pure SPDAB. The product was filtered, thoroughly
washed with cold water till neutral to pH, followed by acetone wash and dried at 60 C
under reduced pressure for a day.
Yield: 3.8 g (65 %), Mp: 330 C.
Elemental Analysis, (C12H12N2O4S): Calculated: C, 51.42%; H, 4.28%; N, 10.00%; S,
11.42%. Found: C, 51.08%; H, 4.29%; N, 9.75%; S, 11.13%.
FT-IR and 1H NMR spectra of SPDAB are given in Figure 2.5 and 2.6,
respectively.
2.2.4 Synthesis of dinitro precursors and diamines based on CDNB and
substituted phenols
Scheme 2.2 given below describes the structure and synthetic route followed.
Scheme 2.2 Synthesis of dinitro precursors and diamines based on alkyl substituted phenols
2.2.4.1 Synthesis of 2-methyl-1-phenoxy-2,4-dinitro benzene (2MPDNB)
A 500 mL single-necked round bottom flask, equipped with a condenser and a
CaCl2 guard tube was charged with 21.62 g (0.2 mol) of o-cresol, 16.58 g (0.24 mol) of

Chapter 2 Monomer synthesis and characterization
45
dry K2CO3 and 150 mL of DMF. The reaction mixture was stirred at ambient temperature
for 30 minutes. To this solution, 40.51 g (0.2 mol) of CDNB dissolved in 150 mL of
DMF was added and the mixture was heated for 12 h at 120 ºC. After cooling, the
resulting solution was poured in stirred water (500 mL). Obtained slurry was filtered and
solid was washed with water till neutral to pH. The crude product (2MPDNB) was
purified by recrystallization in methanol to yield orange crystals.
Yield: 41.0 g (75 %), Mp: 90 C.
Elemental Analysis, (C13H10N2O5); Calculated: C, 56.94%; H, 3.68%; N, 10.22%. Found:
C, 56.93%; H, 3.90%; N, 10.37%.
Figure 2.7 and 2.8 show FT-IR and 1H NMR spectra of 2MPDNB, respectively.
2.2.4.2 Synthesis of 2methyl-1-phenoxy-2,4-diamino benzene (2MPDAB)
2MPDAB was obtained by reducing 10 g 2MPDNB in the Parr reactor using
similar procedure described in the Section 2.2.3.2. The crude product was purified by
recrystallization in methanol, conferring faint brown crystals.
Yield: 5.8 g (74 %), Mp: 78 C.
Elemental Analysis, (C13H10N2O): Calculated: C, 72.87%; H, 6.59%; N, 13.07%. Found:
C, 72.66%; H, 6.61%; N, 13.03%.
Figure 2.7 and 2.9 show FT-IR and 1H NMR spectra of 2MPDAB, respectively.
2.2.4.3 Synthesis of 4-methyl-1-phenoxy-2,4-dinitro benzene (4MPDNB)
A 500 mL single-necked round bottom flask, equipped with a reflux condenser
and a CaCl2 guard tube, was charged with 21.62 g (0.2 mol) of p-cresol, 27.8 ml (0.2
mol) of TEA and 150 mL of acetone and stirred at ambient temperature for 30 minutes.
To this solution, 40.51 g (0.2 mol) of CDNB dissolved in 150 mL of acetone was added
and the reaction mixture was refluxed for 48 h. Acetone was distilled off to obtain crude
product, 4MPDNB, with green-yellowish color. It was dissolved in 500 mL of CHCl3
and washed with 5% HCl, 2% aq. NaOH and finally with water (till neutral to pH),
sequentially. The CHCl3 solution was dried over anhydrous Na2SO4, filtered and solvent
distilled off to recover crude product. It was purified by recrystallization in methanol and
dried in vacuum oven at 60 C for a day.

Chapter 2 Monomer synthesis and characterization
46
Yield: 45.2 g (82 %), Mp: 91 C.
Elemental Analysis, (C13H10N2O5): Calculated: C, 56.94%; H, 3.68%; N, 10.22%. Found:
C, 56.53%; H, 3.51%; N, 10.6%.
Figure 2.10 and 2.11 show FT-IR and 1H NMR spectra of 4MPDNB,
respectively.
2.2.4.4 Synthesis of 4-methyl-1-phenoxy-2,4-diamino benzene (4MPDAB)
4MPDAB was obtained by reducing 10 g of 4MPDNB in the Parr reactor using
similar procedure described in the Section 2.2.3.2.
Yield: 6.2 g (80 %), Mp: 59 C.
Elemental Analysis, (C13H10ON2): Calculated: C, 72.87%; H, 6.59%; N, 13.07%. Found:
C, 72.31%; H, 6.58%; N, 12.96%.
Figure 2.10 and 2.12 show FT-IR and 1H NMR spectra of 4MPDAB,
respectively.
2.2.4.5 Synthesis of 2,6-dimethyl-1-phenoxy-2,4-dinitro benzene (2,6DMPDNB)
2,6DMPDNB was synthesized using 24.43 g (0.2 mol) of 2,6-dimethylphenol
following the procedure described in Section 2.2.4.1. Obtained product could not be
purified by recrystallization using common organic solvents or their mixtures. Therefore,
purification was done by washing the obtained crude product with cold methanol. The
purified product was vacuum dried at 60 C for a day. Its analysis is given below.
Yield: 40.6 g (70 %), Mp: 121 C.
Elemental Analysis, (C14H12N2O5): Calculated: C, 58.33%; H, 4.20%; N, 9.72%. Found:
C, 58.35%; H, 4.55%; N, 9.74%.
Figure 2.13 and 2.14 show FT-IR and 1H NMR spectra of 2,6DMPDNB,
respectively.
2.2.4.6 Synthesis of 2,6-dimethyl-1-phenoxy-2,4-diamino benzene (2,6DMPDAB)
2,6DMPDAB was obtained by reducing 10 g of 2,6DMPDNB in a Parr reactor
using similar procedure described in the Section 2.2.3.2. The crude product was

Chapter 2 Monomer synthesis and characterization
47
recrystallized in the mixture of pet ether: ethyl acetate (6:1). The analysis performed is as
given below.
Yield: 5.4 g (68 %), Mp: 157 C.
Elemental Analysis, (C14H16N2O): Calculated: C, 73.66%; H, 7.06%; N, 12.27%. Found:
C, 74.12%; H, 7.50%; N, 11.59%.
Figure 2.13 and 2.15 show FT-IR and 1H NMR spectra of 2,6DMPDAB,
respectively.
2.2.4.7 Synthesis of 4’-t-butyl-1-phenoxy-2,4-dinitro benzene (4tBPDNB)
In a 1 L single neck round bottom flask, 27.64 g (0.2 mol) of 4-t-butylphenol and
40.50 g (0.2 mol) of CDNB was reacted by using 20.2 g (0.2 mol) of TEA in 500 mL of
acetone, following the similar procedure as given in Section 2.2.4.3. The crude product
was recrystallized in methanol to obtain needle shaped yellowish crystals. Its analysis
was as given below.
Yield: 55.0 g (87 %), Mp: 108 C.
Elemental Analysis, (C16H16N2O5): Calculated: C, 60.76%; H, 5.10%; N, 8.86%. Found:
C, 60.90%; H, 5.27%; N, 8.88%.
Figure 2.16 and 2.17 show FT-IR and 1H NMR spectra of 4tBPDNB,
respectively.
2.2.4.8 Synthesis of 4’-t-butyl-1-phenoxy-2,4-diamino benzene (4tBPDAB)
This was synthesized using 5 g of 4tBPDNB and 70 ml of methanol following
the similar procedure as described in the Section 2.2.3.2. The crude product was purified
by recrystallization in water: methanol mixture (1:1). The obtained crystals were floppy
in nature and had silver shining.
Yield: 3.1 g (76 %), Mp: 97 C.
Elemental Analysis, (C16H20N2O): Calculated: C, 74.97%; H, 7.86%; N, 10.93%. Found:
C, 74.57%; H, 7.77%; N, 10.92%.
Figure 2.16 and 2.18 show FT-IR and 1H NMR spectra of 4tBPDAB,
respectively.
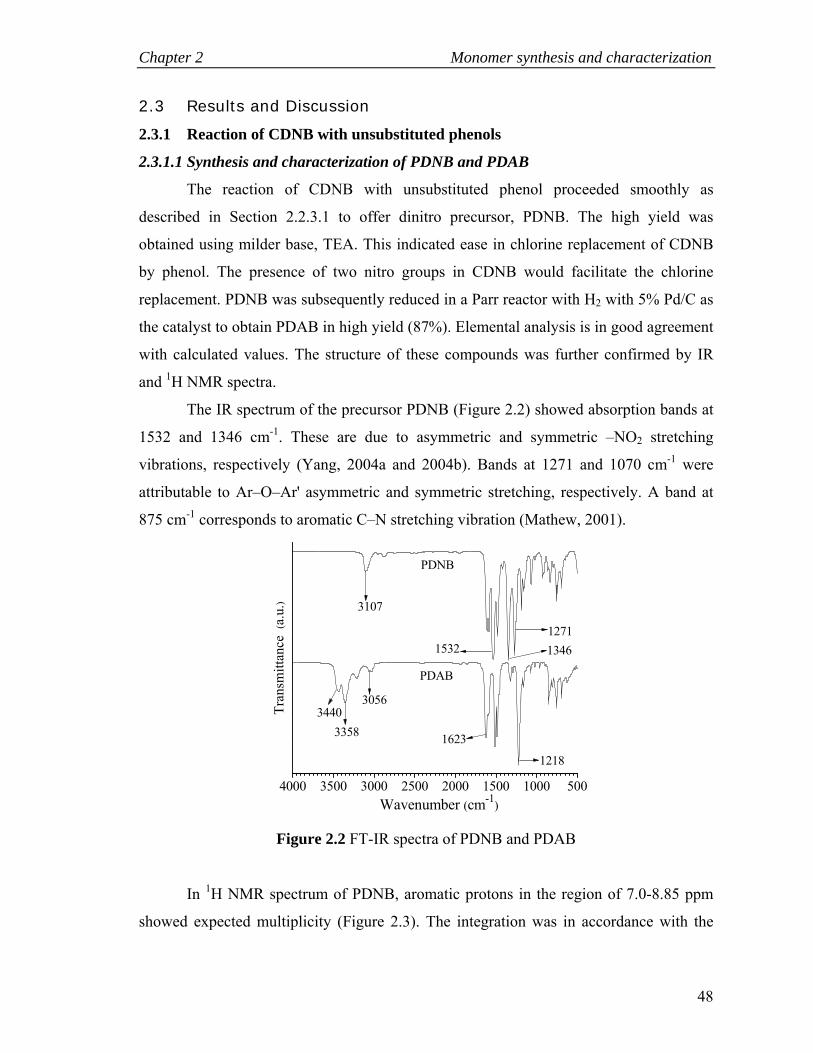
Chapter 2 Monomer synthesis and characterization
48
2.3 Results and Discussion
2.3.1 Reaction of CDNB with unsubstituted phenols
2.3.1.1 Synthesis and characterization of PDNB and PDAB
The reaction of CDNB with unsubstituted phenol proceeded smoothly as
described in Section 2.2.3.1 to offer dinitro precursor, PDNB. The high yield was
obtained using milder base, TEA. This indicated ease in chlorine replacement of CDNB
by phenol. The presence of two nitro groups in CDNB would facilitate the chlorine
replacement. PDNB was subsequently reduced in a Parr reactor with H2 with 5% Pd/C as
the catalyst to obtain PDAB in high yield (87%). Elemental analysis is in good agreement
with calculated values. The structure of these compounds was further confirmed by IR
and 1H NMR spectra.
The IR spectrum of the precursor PDNB (Figure 2.2) showed absorption bands at
1532 and 1346 cm-1. These are due to asymmetric and symmetric –NO2 stretching
vibrations, respectively (Yang, 2004a and 2004b). Bands at 1271 and 1070 cm-1 were
attributable to Ar–O–Ar' asymmetric and symmetric stretching, respectively. A band at
875 cm-1 corresponds to aromatic C–N stretching vibration (Mathew, 2001).
4000 3500 3000 2500 2000 1500 1000 500
1623
3107
3056
1271
13461532
PDAB
PDNB
Tra
nsm
ittan
ce (
a.u.
)
Wavenumber (cm-1)
3358
3440
1218
Figure 2.2 FT-IR spectra of PDNB and PDAB
In 1H NMR spectrum of PDNB, aromatic protons in the region of 7.0-8.85 ppm
showed expected multiplicity (Figure 2.3). The integration was in accordance with the

Chapter 2 Monomer synthesis and characterization
49
number of protons. The extreme downfield appearance of two aromatic protons, Ha and
Hb belonging to the nitro-phenyl ring was due to the highly electron withdrawing nature
of – NO2 groups. Between these two protons, proton Ha resonated at the downfield region
since it has been flanked by electron withdrawing two –NO2 groups. The proton Hc
appeared in the relative upfield region due to its meta position to –NO2 groups.
Moreover, to its ortho position, Ar–O–Ar' linkage would be in the conjugation effect.
Aromatic protons corresponding to the phenyl ring of phenoxy group showed their
expected multiplicity and integration in the region of 7.0-7.55 ppm as shown in Figure
2.3. The sharp melting point of PDNB at 74.3-75.1 °C indicated its good purity.
Figure 2.3 1H NMR spectrum of PDNB
The IR spectrum of PDAB (Figure 2.2) showed the characteristic bands of amino
groups at 3358 and 3293 cm-1 (Yang, 2004a and 2004b). These were due to asymmetric
and symmetric N-H stretching, respectively. Band at 1489 cm-1 corresponds to carbon-
carbon stretching of aromatic ring. A band at 1218 cm-1 was due to the ether linkage,
while the band at 1275 cm-1 was due to the C–N stretching (Mathew, 2001). The structure
of diamine was further confirmed by the disappearance of the bands at 1532 and 1346

Chapter 2 Monomer synthesis and characterization
50
cm-1 due to asymmetric and symmetric stretching vibrations of –NO2 group of the
precursor PDNB.
The 1H NMR spectrum of the PDAB (Figure 2.4) showed multiple peaks in the
range of 6.0-7.3 ppm. This upfield shift of the aromatic protons in comparison to PDNB
confirmed the reduction of PDNB. Protons of the amino-phenyl ring shifted to upfield as
compared to protons of the phenoxy ring due to the activated amino-phenyl ring (electron
donating nature of –NH2 groups by mesomeric +R effect). Among the protons of amino-
phenyl ring, proton Hc appeared at the downfield since it was located meta to both –NH2
groups. It is interesting to note that, proton Ha resonated at the downfield region than
proton Hb, despite being flanked by electron donating –NH2 groups. Protons He,e’
corresponding to phenoxy group shifted to downfield region due to its meta location to
the ether linkage. It also appeared as the triplet. The remaining protons Hd,d’ and Hf of the
phenoxy group appeared in the region of 6.85 -7.05 ppm with expected multiplicity and
integration. The signal appearing at the upfield region 3.5 ppm was corresponding to the
primary aromatic amine protons. PDAB showed the sharp melting at 61 °C. Elemental
analysis is also in good agreement with calculated values.
Figure 2.4 1H NMR spectrum of PDAB

Chapter 2 Monomer synthesis and characterization
51
2.3.1.2 Synthesis and characterization of SPDAB
A new diamine, 4-sulphonic-1-phenoxy-2,4-diaminobenzene (SPDAB), was
synthesized by sulfonating PDAB (Scheme 2.1) as described in Section 2.2.3.3. After
dissolving in 95% H2SO4 solution, the amino-phenyl ring became deactivated due to the
transformation of meta-linked electron donating –NH2 groups to the electron withdrawing
ammonium sulfonate groups (–NH3+HSO4
–). Thus, electrophilic substitution by SO3H
group preferentially occurred at para position of pendant phenoxy ring in PDAB.
The IR spectrum of sulfonated diamine; SPDAB is shown in Figure 2.5.
Characteristic bands at 3409 cm-1 and 3308 cm-1 corresponded to asymmetric and
symmetric –N–H stretching, respectively. The bands at 1229 cm-1 and 1308 cm-1 were
due to the Ar–O–Ar' linkage and C–N stretching, respectively. Various peaks
corresponding to –SO2 stretching were observed at 1168, 1089 and 1026 cm-1.
4000 3500 3000 2500 2000 1500 1000 500
3409
3308
12291026
Tra
nsm
itta
nce
(a.u
.)
Wavenumber (cm-1)
SPDAB
1308
Figure 2.5 FT-IR spectrum of SPDAB
The 1H NMR spectrum of SPDAB (Figure 2.6) showed aromatic proton signals in
the range of 5.83-7.52 ppm. The aromatic protons corresponding to the amino-phenyl
ring (Ha, Hb and Hc) appeared in the upfield region due to the activation of this phenyl
ring by electron donating nature of–NH2 groups. While, the remaining aromatic protons
corresponding to the pendant mono sulfophenoxy moiety showed two doublets (Hd,d’ and
He,e’) at 6.75 and 7.52 ppm, respectively. This confirmed the attachment of sulfonic acid

Chapter 2 Monomer synthesis and characterization
52
group, to para position of phenoxy moiety. The appearance of signals at two different
chemical shifts (4.7 and 4.5 ppm) in the upfield region was peculiar of the meta-amino
groups and suggested their different basicity. Its melting point was 329-331 °C.
Figure 2.6 1H NMR spectrum of SPDAB
Observed values of elemental analysis for PDNB, PDAB and SPDAB matched
well with the calculated values. This along with FT-IR and 1H NMR analysis confirmed
respective structure of these compounds.
2.3.2 Reaction of CDNB with substituted phenols
The rational for the design of attempted alkyl substituted diamine monomers was
to investigate effects of systematic structural variations on gas permeability of polyimides
based on these diamines. Four novel diamines viz., 2-methyl-1-phenoxy-2,4-
diaminobenzene (2MPDAB), 4-methyl-1-phenoxy-2,4-diaminobenzene (4MPDAB),
26-dimethyl-1-phenoxy-2,4-diaminobenzene (2,6DMPDAB) and 4-t-butyl-1-
phenoxy-2,4-diaminobenzene (4tBPDAB) were synthesized in two steps as described in
experimental Section 2.2.4.
2.3.2.1 Synthesis and characterization of 2MPDNB and 2MPDAB
Alkyl substituted phenol such as o-cresol could not be reacted with CDNB at low
temperature in acetone. So, high boiling polar solvent, DMF, was used for the reaction.

Chapter 2 Monomer synthesis and characterization
53
Anhydrous K2CO3 was used as acid acceptor. The compound 2MPDNB was obtained in
high yield (75 %). 2MPDNB was subsequently reduced in a Parr reactor with H2 with
5% Pd/C as the catalyst to obtain 2MPDAB in good yield (74%). Calculated and
observed values of elemental analysis for 2MPDNB and 2MPDAB matched well. The
purity of these compounds reflected in their sharp melting points (Section 2.2.4).
Structures of these compounds were further confirmed by FT-IR and 1H NMR analysis.
In the FT-IR spectrum of 2MPDNB, absorption bands appearing at 3058 and
3109 cm-1 were due to aromatic C–H stretching (Figure 2.7). The sharp C–H stretching
band corresponding to the methyl group was observed at 2858 and 2926 cm-1. Occurrence
of bands at 1609 and 1477 cm-1 was due to the presence of –C=C– aromatic stretching.
The strong band at 1271 and a weak band at 1067 cm-1 were due to C–O–C stretching of
asymmetric and symmetric type, respectively. The strong band at 748 cm-1 was due to
ortho-substitution in the benzene ring (Silverstein, 2006). Bands appearing at 1539 and
1346 cm-1 were due to asymmetric and symmetric –NO2 stretching, respectively.
4000 3500 3000 2500 2000 1500 1000 500
1272
33543440
11861620
1228
2922
1346
1539
751
748
31092926
2'MPDAB
Wavenumber (cm-1)
Tra
nsm
itanc
e (a
.u.)
2'MPDNB
2858
Figure 2.7 FT-IR spectra of 2MPDNB and 2MPDAB
The 1H NMR spectrum of 2MPDNB is shown in Figure 2.8. The aromatic
protons appeared in the range of 6.84-8.8 ppm. Among them, protons in the vicinity of
electron withdrawing –NO2 groups, protons Ha and Hb were shifted to the downfield

Chapter 2 Monomer synthesis and characterization
54
region. On the other hand, proton Hc of the same phenyl ring containing nitro groups
appeared in upfield. This could be attributed to its meta position to the –NO2 moieties as
well as ortho to the electron donating effect of Ar–O–Ar' linkage. The signals of
remaining aromatic protons appeared between the positions of protons Ha and Hc in the
range of 6.84-8.8 ppm with expected multiplicity and integration. The sharp singlet in the
upfield region at 2.20 ppm belongs to the methyl protons.
Figure 2.8 1H NMR spectrum of 2MPDNB
In case of FT-IR spectrum of 2MPDAB, absence of peak of –NO2 stretching
(Figure 2.7) indicated complete conversion of nitro to amine functionality. Characteristic
absorption of N–H stretching appeared in the region 3300-3500 cm-1. The asymmetric
and symmetric stretching occurred at 3440 and 3354 cm-1, respectively. The strong
absorption peak near 1620 cm-1 could be assigned to the N–H bending. The Ar–O–Ar'
stretching peak shifted to lower frequency at 1228 cm-1 as compared to higher frequency
at 1271 cm-1 in case of 2MPDNB. This could be due to electron donating effect of amino
groups. The strong band due to C–N stretching was observed at 1186 cm-1. The
absorption bands appearing between 2923 and 2862 cm-1 correspond to the asymmetric
and symmetric stretching of –CH3 groups, respectively.
The 1H NMR spectrum of 2MPDAB is shown in the Figure 2.9. Aromatic
protons appeared in the range of 6.06-7.2 ppm. Among them, protons of meta-amino-
phenyl ring appeared in the upfield region due to activation of the phenyl ring by electron

Chapter 2 Monomer synthesis and characterization
55
donating –NH2 groups. The signal of proton Hb appeared in the form of doublet of a
doublet, since it was ortho to the proton Hc and meta to the proton Ha. The proton Ha
showed a doublet at 6.17 ppm due its meta coupling with proton Hc. Among the aromatic
protons of methyl substituted phenoxy ring, proton He and proton Hf appeared as a triplet
at 7.07 and 6.92 ppm, respectively. While proton Hd and proton Hg appeared as doublet at
6.7 and 7.2 ppm, respectively. Protons of the –NH2 appeared in the upfield region at 3.4
ppm as a broad peak, while the signal of methyl protons appeared at 2.35 ppm.
Figure 2.9 1H NMR spectrum of 2MPDAB
2.3.2.2 Synthesis and characterization of 4MPDNB and 4MPDAB
Both compounds, 4MPDNB and 4MPDAB were synthesized in good yields by
the procedure described in experimental part (Section 2.2.4). Besides, well matched
calculated and observed values of elemental analysis, the structure of these compounds
was confirmed by, FT-IR and 1H NMR analysis.
The FT-IR spectra of the 4MPDNB and 4MPDAB are shown in Figure 2.10. In
the case of 4MPDNB, the aromatic C–H stretching band appeared at 3107 cm-1, while
the asymmetric and symmetric methyl C–H stretching vibrations were observed at 2923
and 2855 cm-1, respectively. The aromatic –C=C– stretching bands were observed at
1600 and 1533 cm-1. The strong band at C–O–C stretching was observed at 1270 cm-1.
The para substitution in 4MPDNB could be further confirmed by the appearance of

Chapter 2 Monomer synthesis and characterization
56
strong band at 831 cm-1 (Silverstein, 2006). The asymmetric and symmetric −NO2
stretching frequencies appeared at 1534 and 1348 cm-1 in 4MPDNB, respectively.
The 1H NMR spectrum of 4MPDNB is shown in the Figure 2.11. The aromatic
protons appeared in the range of 7.02-8.82 ppm. The order as well as positions of
aromatic protons Ha and Hb of the meta-nitro-phenyl ring was similar as in the case of
2MPDNB (Figure 2.8). While, the proton Hc at 7.02 ppm was merged with that of the
protons Hd,d’ of the para-methyl substituted phenoxy ring. Protons He,e’ were located at
7.28 ppm. The sharp singlet at 2.20 ppm was corresponding to the methyl protons.
4000 3500 3000 2500 2000 1500 1000 500
3107
1226
1622 856 821
1503
2923
2921
33633444
8311348
4'MPDAB
4'MPDNB
Tra
nsm
itan
ce (
a.u.
)
Wavenumber (cm-1)
1534
3024
Figure 2.10 FT-IR spectra of 4MPDNB and 4MPDAB
Figure 2.11 1H NMR spectrum of 4'MPDNB

Chapter 2 Monomer synthesis and characterization
57
In 4MPDAB, −NO2 stretching peaks of 4MPDNB at 1534 and 1348 cm-1
disappeared (Figure 2.10) and the new peaks at 3444 and 3363 cm-1 corresponding to the
respective asymmetric and symmetric –N–H stretching appeared, authenticating the
reduction of –NO2 to –NH2 (Yang, 2004a and 2004b). The bending vibrations of –N–H
appear at 1622 cm-1. The sharp band appearing at 1226 cm-1 was due to the C–O–C
stretching, which shifted to lower wave number (Mathew, 2001; Yang, 2002). The
appearance of bands at ~1505 cm-1 corresponding to the aromatic –C=C–stretching
proved that aromatic ring had not been affected by hydrogenation. The –C–H stretching
bands of methyl group were observed at 2861 and 2921 cm-1, while that of aromatic
region appeared at 3024 and 3213 cm-1. The strong peaks appeared at 821 and 856 cm-1
validated the para-substitution in 4MPDAB (Silverstein, 2006). The intense
characteristic peak in the region of 821-856 cm-1 in 4MPDNB and 4MPDAB confirmed
the para-substitution of –NH2 and –CH3 groups to the Ar–O–Ar' ether linkage.
The 1H NMR spectrum of 4MPDAB is shown in the Figure 2.12. Aromatic
protons appeared in the upfield region of 6.07 - 7.05 ppm as compared to same protons
appearing at in the range of 7.02-8.82 ppm for its dinitro precursor, 4MPDNB.
Moreover, the signal obtained at 3.25 ppm corresponding to –NH2 group could confirm
the reduction of nitro to amine. The protons Ha, Hb and Hc corresponding to the meta-
amino-phenyl ring appeared at 6.16, 6.07 and 6.72 ppm, respectively. The remaining
protons viz., Hd,d’ and He,e’ of the para-methyl substituted phenoxy ring appeared at 6.82
and 7.05 ppm, respectively. The methyl protons appeared at 2.3 ppm.
The calculated values of elemental analysis were in good agreement with the
observed values, as given in Section 2.2.4.1.

Chapter 2 Monomer synthesis and characterization
58
Figure 2.12 1H NMR spectrum of 4'MPDAB
2.3.2.3 Synthesis and characterization of 26DMPDNB and 26DMPDAB
The compound 26DMPDNB could be obtained in good yields by synthetic
procedure described in experimental section 2.2.4.5. However, this compound could not
be crystallized from common solvents. So, the compound was purified by washing of
crude compound with cold methanol. The purity of the compound was ascertained by
sharp melting point and well matched observed and calculated elemental analysis values,
while the structure was further confirmed by FT-IR and 1H NMR spectral analysis.
4000 3500 3000 2500 2000 1500 1000 500
1215 119233383437 1537
162128482918 772792
775 742
1271
1344
1537
2924
Tra
nsm
itanc
e (a
.u.)
Wavenumber (cm-1)
2',6'DMPDAB
2',6'DMPDNB
2861
Figure 2.13 FT-IR spectra of 2,6DMPDNB and 2,6DMPDAB

Chapter 2 Monomer synthesis and characterization
59
The FT-IR spectra of the 26DMPDNB and 26DMPDAB are shown in Figure
2.13. The band observed at 3108 cm-1 in 26DMPDNB was due to the aromatic –C–H
stretching. Bands at 2861 and 2924 cm-1 were due to the symmetric and asymmetric
stretching of two –CH3 groups. The band displayed at 1607 and 1477 cm-1 were due to
the –C=C– aromatic stretching. The strong band at 1271 cm-1 was due to C–O–C
stretching. Two sharp shoulder peaks at 775 and 742 cm-1 could be ascribed to the
presence of ortho substitution in benzene ring. Bands appearing at 1537 and 1344 cm-1
were corresponding to the asymmetric and symmetric –NO2 stretching, which
subsequently disappeared in the respective diamine (26DMPDAB) after hydrogenation,
confirming complete reduction. In addition, the asymmetric and symmetric stretching of
primary aromatic amine occurred at 3437 and 3338 cm-1. In 26DMPDAB, N–H bending
was observed at 1621 cm-1. The sharp bands appearing at 1215 and 1192 cm-1 were due
to the C–N and C–O–C stretching, respectively. The bands at 2918 and 2848 cm-1
informed the presence of methyl groups. Two sharp shoulder peaks at 792 and 772 cm-1
could be ascribed to the presence of ortho substitution in benzene ring.
The 1H NMR spectra of 26DMPDNB and 26DMPDAB are shown in the Figure
2.14 and Figure 2.15, respectively. In 2,6DMPDNB, aromatic protons were observed in
the range of 6.70-8.86 ppm, while for 2,6DMPDAB, they were located in the range of
5.88-7.05 ppm with expected multiplicity and integration. It was noticed that, among all
the dinitro precursors as well as diamines, proton Hc in 2,6DMPDNB and
2,6DMPDAB had acquired the most upfield region. The donating effect of dimethyl
substituted phenoxy moiety was responsible for this effect. Elemental analysis indicated
good match between experimental and theoretically calculated values.

Chapter 2 Monomer synthesis and characterization
60
Figure 2.14 1H NMR spectrum of 2,6DMPDNB
Figure 2.15 1H NMR spectrum of 2,6DMPDAB
2.3.2.4 Synthesis and characterization of 4tBPDNB and 4tBPDAB
Both 4tBPDNB and 4tBPDAB were obtained in good yield (Section 2.2.4). Due
to low solubility of t-butyl phenol and 4tBPDNB, the quantity of solvent required for the
reaction was higher. 4tBPDNB was obtained as needle shaped yellowish crystals on
crystallization from methanol, while 4tBPDAB was crystallized from water: methanol
(1:1) mixture as silver shining crystals. Elemental analysis of both the compounds
matched well with calculated values.
The FT-IR spectra of the 4tBPDNB and 4tBPDAB are shown in Figure 2.16. In
4'tBPDNB, the aromatic C–H stretching was observed at 3110 cm-1. Sharp bands at 2965
and 2872 cm-1 were due to the asymmetric and symmetric C–H stretching of t-butyl

Chapter 2 Monomer synthesis and characterization
61
group, respectively. The aromatic –C=C– stretching band was observed at 1610 cm-1. The
strong band of C–O–C stretching was observed at 1272 cm-1. The asymmetric and
symmetric –NO2 stretching was observed at 1540 and 1345 cm-1, respectively. The sharp
and strong band at 835 cm-1 indicated the presence of para substitution in 4tBPDNB.
In 4tBPDAB, appearance of bands at 3367 and 3417 cm-1 was due to asymmetric
and symmetric –N–H stretching, respectively (Figure 2.16). The appearance of –N–H
bending at 1619 cm-1 and disappearance of –NO2 stretching peaks (1540 and 1345 cm-1)
revealed the complete hydrogenation of dinitro precursor to diamine. The sharp bands
appearing in the spectra of 4tBPDAB at 1235 and 1212 cm-1 were due to the C–N and
C–O–C stretching, respectively. The intense asymmetric and symmetric C–H stretching
corresponding to t-butyl group were detected at 2960 and 2867 cm-1. The band at 3039
cm-1 was due to the aromatic C–H stretching. The strong peak at 838 cm-1 was
attributable to the presence of para-substitution in 4tBPDAB.
4000 3500 3000 2500 2000 1500 1000 500
1235
1364
1619 838
1212
1272835
1345
1540
2867
29603367
3417
2872
Tra
nsm
itanc
e (a
.u.)
Wavenumber (cm-1)
4'tBPDAB
4'tBPDNB
2965
Figure 2.16 FT-IR spectra of 4tBPDNB and 4tBPDAB
The 1H NMR spectra of 4tBPDNB and 4tBPDAB are shown in the Figure 2.17
and 2.18, respectively. The placement of aromatic protons was observed to be similar
with that of 4MPDNB and 4MPDAB. In 4tBPDNB, aromatic protons were observed in
the range of 7.05-8.82 ppm while that of 4tBPDAB was in the range of 6.05-7.27 ppm
with expected multiplicity and integration. Protons of t-butyl group were shifted to
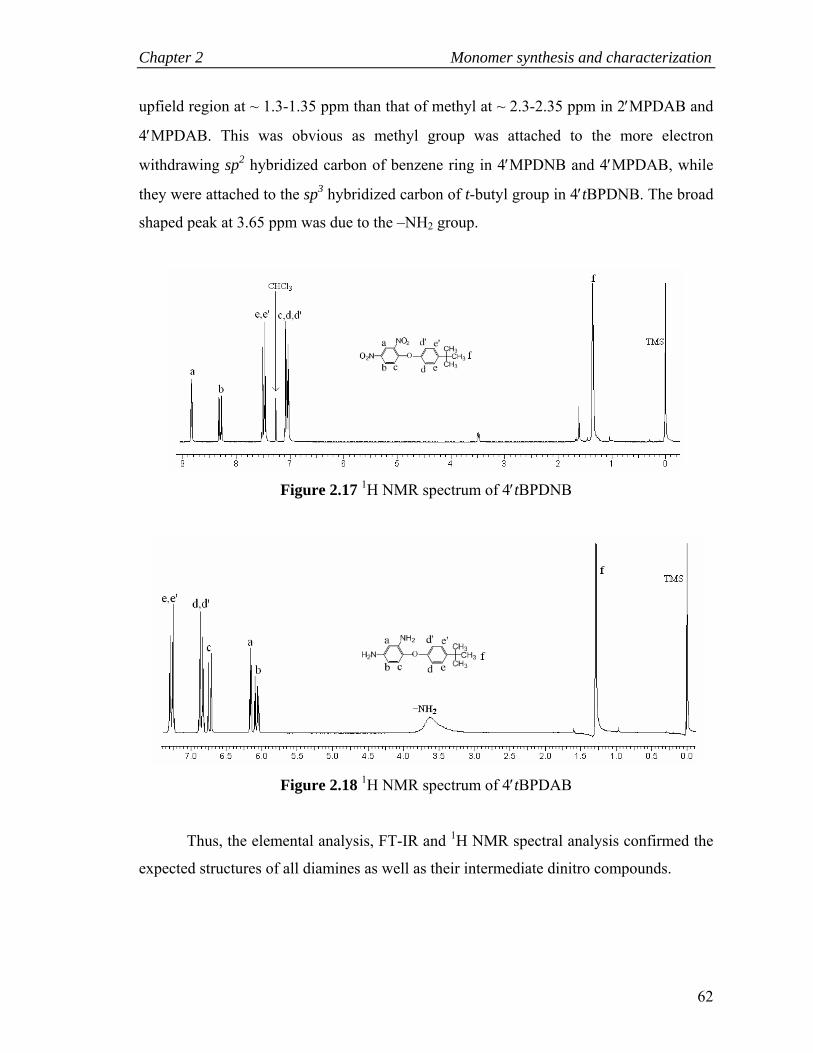
Chapter 2 Monomer synthesis and characterization
62
upfield region at ~ 1.3-1.35 ppm than that of methyl at ~ 2.3-2.35 ppm in 2MPDAB and
4MPDAB. This was obvious as methyl group was attached to the more electron
withdrawing sp2 hybridized carbon of benzene ring in 4MPDNB and 4MPDAB, while
they were attached to the sp3 hybridized carbon of t-butyl group in 4tBPDNB. The broad
shaped peak at 3.65 ppm was due to the –NH2 group.
Figure 2.17 1H NMR spectrum of 4tBPDNB
Figure 2.18 1H NMR spectrum of 4tBPDAB
Thus, the elemental analysis, FT-IR and 1H NMR spectral analysis confirmed the
expected structures of all diamines as well as their intermediate dinitro compounds.

Chapter 2 Monomer synthesis and characterization
63
2.4 Conclusions
1. Various diamines, namely, 1-phenoxy-2,4-diaminobenzene (PDAB), 4'-sulphonic-1-
phenoxy-2,4-diaminobenzene (SPDAB), 4'-methyl-1-phenoxy-2,4-diaminobenzene
(4'MPDAB), 2'-methyl-1-phenoxy-2,4-diaminobenzene (2'MPDAB), 2',6'-dimethyl-
1-phenoxy-2,4-diaminobenzene (2',6'DMPDAB) and 4'-t-butyl-1-phenoxy-2,4-
diaminobenzene (4'tBPDAB) were synthesized by simple condensation reaction.
These were designed with specifically aiming application of PIs based on them; either
as PEM or gas separation membrane materials.
2. The reaction pathway for the synthesis of these diamines was same, wherein first step
was etherification followed by reduction of –NO2 group to –NH2 using H2 Pd/C.
3. Simple, cheap and easily available phenols were successfully utilized for the
synthesis of these novel diamines.
4. Condensation of para-substituted phenols with CDNB could be carried out at low
temperature using acetone as the solvent and TEA as an acid acceptor. The ortho-
substituted phenols required high temperature and these reactions were conducted in
DMF using K2CO3 as the base.
5. The duration of etherification reactions was longer in the case of organic base (TEA),
as compared to the inorganic base (K2CO3).
6. Synthesis of all the monomers were architectured in such a fashion that the resultant
diamine would be placed at meta-position to each other and simultaneously possess
phenoxy ring at 4-position.
7. All the monomers, except SPDAB could be easily crystallized.
8. Melting point of the dinitro precursors was in the order PDNB < 2'MPDNB ≈
4'MPDNB < 4'tBPDNB. In case of the diamines, the following order of increasing
Mp was observed: SPDAB > 2',6'DMPDAB > 4'tBPDAB > 2'MPDAB > 4'MPDAB ≈
PDAB.
9. The monomers as well as their dinitro precursors were characterized by FT-IR and 1H
NMR, elemental analysis and melting point determination. The FT-IR and 1H NMR
confirmed their structures, while elemental analysis and melting point conveyed their
good purity.

Chapter 3
Polyimide synthesis and their characterization
3.1 Introduction
Aromatic polyimides (PI) represent a major class of high performance polymers
with excellent thermal, chemical, mechanical, dielectric properties and good permeation.
Some of their important application areas include alignment layers for liquid crystal
display devices, interlayer dielectrics for semiconductor devices, membrane material for
gas separation, proton exchange membrane for fuel cell (PEMFC) applications, etc.
However, difficulty in their processing due to the strong interchain attractions is an issue.
Therefore, industrious efforts are devoted to synthesize processable PIs.
Attempts to understand the structure-property relationship in polyimides by
systematically changing the diamine and dianhydride moiety have been numerous. As a
result of these studies, several criteria have been proposed to obtain polyimides with
improved solubility. These include (1) introduction of aliphatic or other kinds of flexible
linkages that reduce chain stiffness, (2) introduction of bulky side chain substituents,
which help in the separation of the polymer chains and hinder molecular packing and
crystallization, (3) use of enlarged monomers containing angular bonds, which suppress
coplanar structures, (4) use of 1,3-substituted instead of 1,4-substituted monomers, which
lower regularity and molecular ordering and (5) copolymerization of two or more
diamines or dianhydrides (de Abajo, 1999).
The incorporation of ether groups or other flexible linkages into the main chain
generally leads to an improvement in solubility (de Abajo, 1999; Liaw, 2001), whereas
the glass transition temperatures (Tg) and thermal stability are lowered significantly
(Tamai, 1996). On the other hand, introduction of bulky groups into the polymer main
chain or the attachment of bulky pendant groups like phenyl along the backbone helps in
retaining the thermal properties, while providing a good solubility due to decrease in
packing density and crystallinity (Jeong, 1994; Liou, 1998). Pendant ether and thio
groups are also known to improve the solubility (Kakimoto, 1988). Incorporation of
meta-linkages with pendant aromatic ring can significantly improve the solubility of

Chapter 3 Polymer synthesis and their characterization
65
polyimides (Tsuda, 2000). It was expected that suitable combination of these structural
modifications would minimize the trade off between the processability and thermal
properties of aromatic polyimides and provide PI with desired properties for specific
application.
The objective of this thesis was to synthesize polyimides based on phenyl ether
substituted meta-phenylene diamines and assessment of their performance, both as gas
permeation and proton exchange membrane (PEM) materials. With this as an aim,
present chapter describes synthesis of various PIs based on systematically designed
monomers as described in Chapter 2. It also describes blending of sulfonated polyimide
(SPI) with polybenzimidazoles (PBI) with specific aim of using acid-base blend
membranes as PEM materials.
3.2 Materials
Monomers with defined structural variations, viz.; 1-phenoxy-2,4-
diaminobenzene (PDAB), 4′-sulphonic-1-phenoxy-2,4-diaminobenzene (SPDAB), 4′-
methyl-1-phenoxy-2,4-diaminobenzene (4′MPDAB), 2′-methyl-1-phenoxy-2,4-
diaminobenzene (2′MPDAB), 2′,6′-dimethyl-1-phenoxy-2,4-diaminobenzene
(2′,6′DMPDAB) and 4′-t-butyl-1-phenoxy-2,4-diaminobenzene (4′tBPDAB) were
synthesized as described in the experimental section of Chapter 2. Select dianhydrides,
monomers and reagents viz.; 4,4′-Oxydiphthalic dianhydride (ODPA), 3,3′,4,4′-
biphenyltetracorboxylic dianhydride (BPDA), 3,3′,4,4′-benzophenone tetracarboxylic
dianhydride (BTDA), 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA),
pyrromellitic dianhydride (PMDA), 1,4,5,8-naphthalene tetracarboxylic dianhydride
(NTDA), 4,4′-oxydianiline (ODA), 3,3′-diaminobenzedine (DAB), isophthalic acid
(IPA), 5-tert-butyl isophthalic acid (BuI) and imidazole were purchased from Aldrich
chemicals, USA. All dianhydrides and a diamine, ODA were purified by sublimation
prior to use. m-Cresol was procured form S.D. Fine Chemicals, (India) and vacuum
distilled prior to use. Other solvents viz., chloroform (CHCl3), sym-tetrachloroethane
(TCE), methanol, acetone, N,N-dimethylacetamide (DMAc), N,N-dimethylformamide
(DMF), dimethyl sulfoxide (DMSO), nitrobenzene (Nitro-Bz), triethylamine (TEA),
benzoic acid, N-methyl-2-pyrrolidone (NMP), conc. sulfuric acid (H2SO4), conc.

Chapter 3 Polymer synthesis and their characterization
66
hydrochloric acid (HCl), polyphosphoric acid (PPA), ortho-phophoric acid (H3PO4) and
tetrahydrofuran (THF) (all AR grade) were procured from S.D. Fine Chemicals, (India)
and used as received. Ultra-pure water was obtained from a Millipore Milli-Q water
purification system.
3.3 Polymer Synthesis
Syntheses of polymers are categorized into three sections, depending on the
diamine used.
(I) Polyimides (PIs) and copolyimides (CPIs) based on 1-phenoxy-2,4-diaminobenzene
(PDAB) with various dianhydrides,
(II) Sulfonated polyimides (SPIs) based on SPDAB with two select dianhydrides,
NTDA and ODPA; their blend membranes with PBIs,
(III) Polyimides based on alkyl substituted phenoxy diamines (2'MPDAB, 4'MPDAB,
4'tBPDAB and 2',6'DMPDAB).
3.3.1 Polyimides (PIs) and copolyimides (CPIs) based on PDAB
3.3.1.1 Synthesis of PIs
Polyimides based on PDAB with various dianhydrides were synthesized by one
step polymerization process as shown in Scheme 3.1.
Scheme 3.1 Synthetic route to PDAB based PIs
A 50 mL three necked round bottom flask, equipped with a mechanical stirrer, N2
gas inlet and a guard tube was charged with 2.002 gm (0.01 mol) of PDAB and 10 mL of
m-cresol. This mixture was stirred for 15 minutes under a stream of N2 at room

Chapter 3 Polymer synthesis and their characterization
67
temperature to dissolve the diamine. To the formed solution, 0.01 mol of dianhydride and
15 mL of m-cresol were added. The temperature was raised to 200 C and maintained for
12-24 h, depending on the dianhydride used. Water formed during the imidization was
removed continuously with a stream of N2. The resultant viscous solution was cooled and
poured into 300 mL of stirred methanol to precipitate the polymer. It was filtered, washed
several times with methanol and dried at 100 C for 24 h. The polymer was further
purified by dissolution in either TCE or DMAc, followed by precipitation in methanol.
After filtration, it was dried at 60 °C under vacuum for a week. Abbreviation for polymer
synthesized (Scheme 3.1) is based on the monomer abbreviation as used in Chapter 2.
The polymer synthesized from oxydianiline (ODA) and 4,4′-oxydiphthalic dianhydride
(ODPA) is abbreviated as ODA-ODPA.
Copolyimides based on the mixture of diamines, PDAB and ODA in a ratio of
5:95, 25:75 and 50:50 with ODPA were prepared using similar procedure described
above. The polymer designation is given in Scheme 3.2 below.
Scheme 3.2 Synthetic route to copolyimides
3.3.1.2 Preparation of dense membrane
The dense membranes of PDAB-ODPA were prepared by casting its 3 wt %
tetrachloroethane (TCE) solution at 60 C on a flat glass surface under dry atmosphere.
Though, it is soluble in chloroform, TCE was used as the solvent for membrane casting

Chapter 3 Polymer synthesis and their characterization
68
since those prepared with chloroform as the solvent exhibited wavy surface. The TCE
casted membrane was transparent and tough in nature. In the case of PDAB-6FDA based
membrane preparation, the solvent used was chloroform and casting was done at 40 C.
For all the remaining polyimides and copolyimides (PDAB-BPDA, PDAB-BTDA, CPI-
0595, CPI-2575, CPI-5050) dense membranes were prepared using their DMAc solution
and casting was done at 80 C. After initial evaporation of the solvent, formed film was
peeled off and dried in a vacuum oven at 60 C for 1 week in order to remove residual
solvent. In case of membranes casted from DMAc, vacuum oven temperature was 80 °C.
The complete removal of solvent was confirmed by DSC.
3.3.2 Sulfonated polyimides (SPIs) based on SPDAB
3.3.2.1 Synthesis of SPI
SPIs based on SPDAB with two different dianhydrides, NTDA and ODPA, were
prepared as follows (Scheme 3.3).
Scheme 3.3 Schematic presentation for the synthesis of sulfonated polyimides (SPIs)
To a 50 mL three-necked round bottom flask equipped with a mechanical stirrer,
N2 gas inlet and a guard tube, were added 1.4 g (5.0 mmol) of 4'-sulphonic-1-phenoxy-
2,4-diaminobenzene (SPDAB), 10.0 mL of m-cresol and 0.6 g (6.0 mmol) of TEA or
0.408 gm (6.0 mmol) of imidazole while stirring. After SPDAB was completely
dissolved, 1.34 g (5.0 mmol) of NTDA and 0.866 g (7.1 mmol) of benzoic acid were

Chapter 3 Polymer synthesis and their characterization
69
added, followed by addition of 15 mL of m-cresol at ambient under N2 atmosphere. The
mixture was heated gradually to 200 C and stirred further at this temperature for 8 h, in
case of imidazole as the base, or 24 h in case of TEA as the base. Water formed during
the imidization was continuously removed with a stream of N2. The resultant viscous
solution was cooled to ambient and poured onto stirred acetone. In case of use of
imidazole as the base, the solution became highly viscous within 8 h at 200 C. Hence,
additional 18 mL of m-cresol was added and then precipitated. Obtained fibrous polymer
was stirred in an excess of acetone to remove m-cresol, followed by drying at 80 C in
the vacuum oven for a day. After drying, polymer was further purified by dissolving in
DMAc and re-precipitating in acetone. The purified SPI was dried at 80 C in vacuum
oven for 3 days. The polymer obtained using triethyl amine as the base is designated as
SPDAB•TA-N; while polymer synthesized with imidazole as the base is designated as
SPDAB•Im-N.
Similar procedure was followed for the synthesis of SPI based on ODPA and
SPDAB. In this case, only TEA was used as the base. Polymerization was carried out for
24 h. This polymer was abbreviated as SPDAB•TA-O.
3.3.2.2 Synthesis of polybenzimidazoles (PBIs)
Polybenzimidazoles used for blending with sulfonated PIs were synthesized as
follows (Scheme 3.4).
Scheme 3.4 Synthetic scheme to Polybenzimidazoles

Chapter 3 Polymer synthesis and their characterization
70
Polybenzimidazoles were synthesized by solution polycondensation method using
PPA, which acts as a solvent as well as condensation agent (Iwakura, 1964; Saegusa,
1997). Typically, a 250 mL three-necked flask equipped with a mechanical stirrer, N2
inlet and CaCl2 drying tube was charged with 60 g of PPA and 2 g (0.0093 mol) of DAB
and temperature was raised to 140 ºC. After complete dissolution, 0.0093 mol of
dicarboxylic acid (isophthalic acid or 5-tert-butyl isophthalic acid) was added and the
temperature was slowly raised to 170 ºC and maintained for 5 h under constant flow of
nitrogen. The temperature was further raised to 200 ºC and maintained for 12 h. After
completion of the reaction, temperature was lowered and the reaction mixture was poured
into the stirred water. The precipitated polymer was crushed and thoroughly washed with
water till it was neutral to pH. The polymer was then kept overnight in 10% aqueous
Na2CO3 for 24 h, washed with water until neutral to pH and soaked in acetone for 16 h to
extract water. The dried polymer (100 ºC, 3 days) was further purified by dissolving in
DMAc, removing undissolved material, if any, by centrifugation at 3000 rpm for 3 h and
reprecipitation from stirred water. The polymer was finally dried at 60 °C for 24 h and
then in vacuum oven at 100 °C for 3 days. The polymer based on isophthalic acid is
designated as PBI-I; while the one based on t-butyl isophthalic acid is designated as PBI-
BuI.
3.3.2.3 Preparation of dense membrane based on sulfonated PIs and their proton
exchange
Dense membranes of the SPIs in salt form (SPDAB•TA-N, SPDAB•Im-N and
SPDAB•TA-O) were prepared by casting their DMAc solutions (~ 3 wt %) on a flat glass
surface, under dry conditions at 80 C for 24 h. The formed film was peeled off and
soaked in deioinised water for a day at 60 C and then vacuum dried at 60 C for a week.
In this way, dense membranes with average thickness of 40 μm and 120 μm were
prepared for the gas permeation and proton conductivity measurements, respectively. To
measure proton conductivity, proton exchange treatment was done by immersing these
membranes into 1 N hydrochloric acid solution for a day, followed by thorough washing
with deionised water. The proton exchanged membranes based on SPDAB•TA-N,
SPDAB•Im-N and SPDAB•TA-O were abbreviated as SPDABI-N, SPDABII-N and

Chapter 3 Polymer synthesis and their characterization
71
SPDAB-O, respectively. Thus, after HCl treatment, these membranes are in sulfonic acid
form.
3.3.2.4 Preparation of dense membrane based on PBIs
Dense membranes were prepared by solution casting method under identical
conditions using 3% (w/v) polymer solution in DMAc. The solution was prepared by
stirring at 80 C for 14–18 h under dry atmosphere. The solution after centrifugation at
3000 rpm (to remove undissolved particles, if any) was poured on to the flat glass surface
and heated at 80 C for ~18 h under inert atmosphere. Formed film was peeled off and
soaked in water at 60 C for 3 days in order to remove traces of DMAc. Such films were
finally dried in vacuum oven at 100 C for a week and used for subsequent analysis.
3.3.2.5 Preparation of blend membranes based on SPI and PBI
SPI-PBI blend membranes were prepared in their, 30:70, 40:60, 50:50 weight
ratio. The blend membranes based on PBI-I were found to disintegrate and were not
pursued further. PBI-BuI was dissolved in DMAc by stirring at 80 C for 10 h while
SPDAB•TA-N was dissolved in DMAc by stirring at ambient for 2 h. SPI solution was
subsequently added to the DMAc solution of PBI-BuI at 60 C and stirred for 1 h. The
dense blend membranes were obtained by casting this mixed solution at 80 C on a flat
glass surface under dry atmosphere for 24 h in an oven. Formed film was peeled off and
dried in a vacuum oven at 80 C for 3 days in order to remove residual solvent. The
average thickness of membranes for proton conductivity measurement was ≈120 μm and
for the gas permeation analysis, it was 40 μm. The blend membrane constituting SPI in
triethylammonium salt form was subsequently converted sulfonic acid form by
immersing it in 1 N HCl for 48 h at ambient conditions. On the other hand, membranes
based on PBI-BuI were quite stable and maintained their integrity. The membranes thus
obtained in the sulfonic acid form were thoroughly washed with deionised water,
followed by soaking in water for 24 h. These membranes were kept in a vacuum oven at
80 C for three days. The membranes were again kept in the deionised water for 24 h
prior to their proton conductivity measurements.

Chapter 3 Polymer synthesis and their characterization
72
3.3.3 Polyimides based on alkyl substituted phenoxy diamines
3.3.3.1 Synthesis of polymers
The synthetic route to these PIs is as shown below in Scheme 3.5. The
abbreviations for polymers are based on monomers used.
Scheme 3.5 Synthetic route to PIs based on alkyl substituted phenoxy diamines
Polyimides (PIs) based on alkyl substituted phenoxy diamines (2'MPDAB,
4'MPDAB, 4'tBPDAB and 2',6'DMPDAB) were prepared by the single step solution
polycondensation in m-cresol. Typically, 0.01 mol of alkyl substituted diamine and
10 mL of m-cresol were added to a 50 mL three-necked round bottom flask equipped
with a mechanical stirrer, N2 gas inlet and a guard tube. This solution was stirred for 15-
20 minutes under a stream of N2 at ambient condition to dissolve the diamine. To this
solution 0.01 mol of dianhydride (either ODPA or 6FDA) was added, followed by the
addition of 10 mL m-cresol. The temperature was raised gradually to 200 C and
maintained for 8-24 h. For 2',6'DMPDAB-based polyimides, highly viscous solution was
formed within 8 h. Hence, 20 mL of m-cresol was added before precipitating in methanol.
While in case of other polyimides, it took 15-24 h to complete the polymerization. This
was also dependant on the dianhydride used. Between ODPA and 6FDA reacted with the
same diamine, the former took lesser time than the latter for the polymerization. Water

Chapter 3 Polymer synthesis and their characterization
73
formed during the imidization was removed continuously with a stream of N2. The
resulting polymer solution was cooled and poured onto 500 ml of stirred methanol. The
precipitated polymer was washed several times with methanol and dried at 80 C for a
day in vacuum oven. It was further purified by dissolving in chlorinated solvents (ODPA-
based PI in TCE and 6FDA-based PI in chloroform), reprecipitating in methanol and
finally drying in vacuum oven at 60 C for a week in order to ensure complete removal of
the solvent.
3.3.3.2 Preparation of dense membranes
The dense membranes of polyimides based on ODPA were prepared by solution
casting from 3 wt % sym-tetrachloroethane (TCE) solution at 70 C on a flat glass surface
under dry atmosphere. In case of polyimides based on 6FDA, the solvent used was
chloroform and the membrane preparation was done at 40 C. After initial evaporation of
the solvent, formed film was peeled off and dried in a vacuum oven at 70 C for a week
in order to remove residual solvent. Absence of solvent was confirmed by DSC.
3.4 Characterization of polymers
Elemental analysis of polymers was obtained using Elementar Vario-EL
(Germany). The IR spectra were recorded on a Perkin Elmer 16 PC FT-IR
spectrophotometer. Inherent viscosity of PDAB based PIs was determined at 0.5 g.dL-1
concentration in DMF (ODA-ODPA in m-cresol and CPI-0595 in TCE) at 35 C
(± 0.2 C) using an Ubbelohde viscometer. Intrinsic viscosity [] of polyimides based on
alkyl substituted phenoxy diamines was determined in TCE at 35 C. The inherent as
well as reduced viscosity of the SPIs was measured under isothermal condition at 35 C
in the thermostat in DMAc with varying concentrations. Inherent viscosity of PBI-I and
PBI-BuI was determined using conc. H2SO4 solution (0.2 g.dL-1). Efflux time of the
dilute polymer solutions and the pure solvent was recorded by a digital timer (Q&Q,
Japan CBM Corp., Japan) having a least count of 1 milisecond. Time measurements were
continued till three consistent readings were obtained and then averaged. Solubility of PIs
in various solvents was determined with 0.5 % (w/v) concentration. Thermogravimetric

Chapter 3 Polymer synthesis and their characterization
74
analysis (TGA) was performed on Perkin Elmer TGA-7 (TA instruments, USA) in N2
atmosphere at a heating rate of 10 C. min-1. The glass transition temperature (Tg) of all
polymers was measured on DSC Q-10 (TA instruments, USA) in N2 atmosphere at a
heating rate of 10 C. min-1.
The density () of polyimides used for gas permeation was obtained in the film
form by floatation method using aq. K2CO3 solution. Based on density measurement,
solubility parameter [=(Ecoh/ Vsp )1/2] was estimated by the group contribution method
(Van Kravelen, 1997), where Vsp is the specific free volume (ratio of the molecular
weight of the repeat unit and measured density), Vo is the occupied volume calculated
from the correlation Vo = 1.3Vw, (Vw is the Van der Waals volume calculated from
Bondi’s group contribution method). Ecoh is the cohesive energy of the polymer and Vsp is
the molar volume of the polymer. The wide-angle Χ-ray diffraction (WAXD) spectra of
the dry membrane samples were obtained at ambient using Rigaku Dmax 2500
diffractometer (Tokyo, Japan). Samples were scanned in the 2θ range of 4-40° at the scan
rate of 2°.min-1 with a Cu-Kα radiation source. The amorphous peak maxima on each X-
ray scattering profile was ascribed to the average intersegmental distance of polymer
chains, and the d-spacing was calculated by substitution of the scattering value (2) of the
peak into the Bragg’s equation, n= 2d Sin(), where, n=1 and =1.54 Å for Cu-K
radiation.
3.5 Results and discussion
3.5.1 Synthesis and physical characterization of polyimides based on PDAB
3.5.1.1 Synthesis
New polyimides based on PDAB containing pendant phenoxy group with various
aromatic dianhydrides, viz., ODPA, BTDA, BPDA, 6FDA and PMDA were prepared by
one step high temperature solution polymerization in m-cresol, as described in the
experimental Section 3.3.1.1. All the polymerization reactions proceeded homogeneously
without gelation or precipitation, except for the PMDA based polyimide; which
precipitated from the solution during polymerization. This could be probably due to the
rigid and closed packed structure of PDAB-PMDA. It is known that PMDA based
polyimides are rigid (de Abajo, 1999; Liu, 2005). The polymers were obtained by

Chapter 3 Polymer synthesis and their characterization
75
pouring the resultant viscous polymer solution into methanol with vigorous stirring. The
copolyimides of PDAB and ODA with ODPA containing 5, 25 and 50 mol % PDAB
were synthesized with a similar procedure.
Dense membranes of these polymers, with varying thickness prepared by solution
casting method and tough films were obtained. For FT-IR spectral analysis, dense
membranes of relative lower thickness (~10 μm) were prepared. These polymers were
characterized by elemental analysis, FT-IR spectroscopy, WAXD, viscosity measurement
and thermal analysis as elaborated below.
3.5.1.2 Elemental analysis
The elemental analysis for polyimides based on PDAB is shown in the following
Table 3.1. The observed values of elemental analysis were in good agreement with the
calculated values, except for the PDAB-PMDA. Due to partial solubility of PDAB-
PMDA at high concentration (> 2 wt%) even in DMF, it could not be purified. This could
have resulted in larger difference in calculated and observed values of elemental analysis.
Table 3.1 Elemental analysis of PDAB based polyimides
Element analysis Polymer
abbreviation
Formula and molecular weight of
chain repeat unit (CRU)
% C % H % N
Calculated 70.89 2.95 5.90 PDAB-ODPA
(C28H14N2O6)n
(474) Observed 70.59 3.49 5.57
Calculated 71.18 2.30 4.60 PDAB-6FDA
(C31H14N2O5F6)n
(608) Observed 72.05 2.72 4.07
Calculated 73.36 3.05 6.11 PDAB-BPDA
(C28H14N2O5)n
(458) Observed 71.68 3.00 5.85
Calculated 71.60 2.88 5.76 PDAB-BTDA
(C29H14N2O6)n
(486) Observed 71.49 3.60 4.80
Calculated 69.10 2.61 7.32 PDAB-PMDA
(C22H10N2O5)n
(382) Observed 67.50 3.22 5.64
3.5.1.3 Solubility and solution viscosity
The solubility of the PDAB based polyimides and copolyimides was determined
at a 0.5% concentration in common organic solvents at the ambient temperature, as

Chapter 3 Polymer synthesis and their characterization
76
shown in Table 3.2. Polyimides based on 6FDA and ODPA were soluble in many
solvents, including chlorinated solvents. The high solubility of the 6FDA-based
polyimides was attributed to the presence of the –C(CF3)2– linkage, which is known to
increase the free volume and decrease the intermolecular interaction between the polymer
chains (Stern, 1989). Similarly, polyimides prepared from the ODPA dianhydride showed
good solubility in polar aprotic solvents such as NMP, DMF, DMAc, nitrobenzene, m-
cresol and chlorinated solvents. Polyimides based on BTDA (PDAB-BTDA) and BPDA
(PDAB-BPDA) were soluble at the ambient temperature only in m-cresol and on heating
in polar aprotic solvents such as DMF, DMAc and nitrobenzene, possibly because of
their rigid structure. The polyimide based on PMDA (PDAB-PMDA) was soluble in
DMF and m-cresol only on heating. The polyimide based on ODA and ODPA (ODA-
ODPA) was insoluble in all solvents except m-cresol, and that too on heating. Thus, the
solubility of ODA- and ODPA-based polyimides could be enhanced by the incorporation
of PDAB as evidenced by the good solubility of the copolyimides in many solvents. All
these copolyimides were soluble in TCE at the ambient temperature. The presence of the
pendant phenoxy group on asymmetrically (meta) linked PDAB could also be responsible
for the better solubility observed for these polyimides. The pendant phenoxy group with a
flexible ether linkage could disrupt the chain packing in the polymer backbone to ease
solvent solubility.
The inherent viscosity of polyimides and copolyimides is given in Table 3.3. The
inherent viscosities of PDAB-ODPA, PDAB-6FDA, PDAB-BTDA, PDAB-BPDA, CPI-
2575, and CPI-5050 were measured in DMF, whereas the viscosities of ODA-ODPA and
CPI-0595 were determined in m-cresol and TCE, respectively, as they were not soluble in
DMF. The viscosities of the polyimides based on PDAB ranged from 0.33 to 0.48 dL.g-1
for homopolyimides and from 0.38 to 1.16 dL.g-1 for copolyimides (Table 3.3). The ODA
(structural isomer of PDAB) based polyimide showed higher viscosity than the PDAB-
based polyimide obtained from the same dianhydride (ODPA).
The lower viscosity exhibited by PDAB based PIs could be attributed to the
relative lower basic character due to the meta-substituted amino groups and the steric
hindrance created by the bulky substitution of phenoxy moiety, ortho to one of the amine
groups. This phenomenon is supported by the fact that among m-, o- and p-PDA, m-PDA

Chapter 3 Polymer synthesis and their characterization
77
has the highest oxidation potential. Thus, it has least tendency to loose an electron pair,
resulting in lower basic character (Li, 2002). In PDAB, the electron donating ether group
should enhance the basicity of amino groups and being located ortho and para- to amino
groups, it’s donating ability would be even more effective (Clayden, 2001). Such effect
of enhanced basicity of aromatic diamine by electron donating ether linkage is well
known (Yin, 2004). However, in PDAB, the increased basicity of amine group by
electron donating ether linkage (Ar-O-Ar') should counteract by the steric hindrance of
bulky phenoxy ring. At higher temperature, the feasibility of rotation of flexible phenoxy
group would be more, creating steric hindrance towards its reactivity with dianhydride
molecule. Thus, PDAB could be considered as the sterically hindered diamine, which is
known to react slowly (Grubb, 1999). In contrast to PDAB, ODA has two amino-phenyl
ring bridged by electron donating ether linkage (Ar-O-Ar'), which enhances basicity of
amino groups. In addition, in ODA, there is no substitution to the ortho position of –NH2
groups making it sterically free. It has also been reported in the literature that ODA has
higher pKa (5.20) than that of mPDA (4.80) (Zubkov, 1981). Thus, this may be one of the
reasons for PI based on ODA exhibiting higher viscosity than PDAB, due to high basicity
of ODA compared to PDAB.
Table 3.2 Solubility of PDAB based polyimides and copolyimides
Polymer CHCl3 DMF DMSO NMP Nitro-Bz m- Cresol DMAc THF TCE
PDAB-ODPA ++ ++ – ++ ++ ++ ++ – ++
PDAB-6FDA ++ ++ – ++ ++ ++ ++ – ++
PDAB-BTDA – + – ± + ++ + – –
PDAB-BPDA – + – + + ++ + – –
PDAB-PMDA – + – – – + – – –
ODA-ODPA – – – – – + – – –
CPI-0595 – – – – – + – – ++
CPI-2575 + + – ± – + + – ++
CPI-5050 + + – + + + + – ++
++: soluble at room temperature; +: soluble on heating at 80 C; ±: partially soluble; –: insoluble.

Chapter 3 Polymer synthesis and their characterization
78
3.5.1.4 FT-IR analysis
FT-IR spectra of polyimides are shown in Figure 3.1. The characteristic
absorption bands of the 5 membered imide ring appeared at 1785 cm-1 and 1720 cm-1.
They correspond to asymmetric and symmetric C=O stretching, respectively. A band at
1350 cm-1 was due to C−N stretching. The imide ring deformations appeared at 1100 cm-
1 and 720 cm-1. A band at 1240 cm-1 was due to ether group of diamine. The complete
imidization of polymers was confirmed by the absence of peaks at 1650, 1534, 1712 cm-1
(acid C=O) and 3250 cm-1 (N−H and O−H groups) of amic acid (Mathew, 2001).
Thus, elemental and FT-IR analysis confirm the formation of polyimides
containing pendant phenoxy groups.
Figure 3.1 IR spectra of PDAB based PIs and CPIs
3.5.1.5 Thermal Properties of Polymers
High thermal stability is one of the important physical properties of PI. The
thermal behavior of present PIs and copolyimides was studied by TGA and DSC. Results

Chapter 3 Polymer synthesis and their characterization
79
are summarized in Table 3.3. The DSC curves of these polyimides are shown in Figure
3.2. All polyimides and copolyimides had a high Tg (> 248 C). The increase in Tg
generally corresponded to an increase in the rigidity of the dianhydride monomer like
6FDA.
Table 3.3 Physical properties of PDAB based polyimides and copolyimides
Polymer Diamine Dianhydrideinh
(dL.g-1)
Tg
(C )
IDT
(C)
T10
(C)
Char yield
( %)
PDAB-
ODPA PDAB ODPA 0.47a 256 580 608 59.02
PDAB-
6FDA PDAB 6-FDA 0.43a 281 537 551 52.15
PDAB-
BTDA PDAB BTDA 0.34a 264 553 584 59.76
PDAB-
BPDA PDAB BPDA 0.32a 248 555 594 58.65
PDAB-
PMDA PDAB PMDA ---- 250 543 564 56.46
ODA-ODPA ODA ODPA 1.91b 269 576 618 55.00
CPI-0595 5% PDAB + 95%ODA ODPA 0.38c 259 571 627 53.41
CPI-2575 25% PDAB + 75% ODA ODPA 1.16a 272 606 622 56.14
CPI-5050 50% PDAB + 50% ODA ODPA 0.57a 273 590 633 57.60
a: 0.5g/dL in DMAc at 35 C, b: 0.5g/dL in m-Cresol at 35 C, c: 0.5g/dL in TCE at 35 C
It is evident that, bridging group (–O–, –C=O) between the two phenyl rings
present in the dianhydrides like ODPA and BTDA, facilitating the bond rotation would
decrease the Tg. Among these, all PDAB based polyimides with different dianhydrides, in
particular the one based on 6FDA showed the highest Tg because of the polarizable
hexafluoroisopropylidene group, which inhibited the molecular motions. It has also been
reported that bulky –C(CF3)2– linkage restricts the segmental mobility of the polymer

Chapter 3 Polymer synthesis and their characterization
80
main chain (Stern, 1989). The Tg of copolyimides synthesized by addition of ODA to
PDAB were marginally higher than Tg of the PDAB based homopolymer.
The TGA thermograms of the polyimides and copolyimides are shown in Figure
3.3. All synthesized polyimides showed an initial decomposition temperature (IDT)
above 500 C and T10 values in the range of 551-633 C, thus exhibiting high thermal
stability. However, among all the polyimides, the IDT of PDAB-6FDA was the lowest,
despite the high C–F bond strength (441 kJ/mol) (Van Krevelen, 1997). It is known that
the voluminous size of the CF3 retards the free rotation around its own axis resulting in
high torsional strain as well as gain in high conformational energy (Hougham, 1999;
Yang, 2004c). It leads to the low thermal stability of CF3 group, (Lee, 2006) which is lost
in the form of a CF3 radical (Yang, 2004c). Because of the strong electron affinity of F
atom, the C-C bond energy in CF3-C-CF3 becomes lower (Ren, 2004). Hence, polymer
with –C(CF3)2– linkage demonstrated low thermal stability, which was exhibited by the
lowest weight residue of 6FDA-based polyimide in the series. All polyimides showed a
char yield greater than 50 %.
Figure 3.2 DSC of PDAB based polyimides and copolyimides

Chapter 3 Polymer synthesis and their characterization
81
Figure 3.3 TGA of PDAB based PIs and CPIs
3.5.1.6 Wide-angle X-ray diffraction (WAXD)
Crystallinity in a polymer has significant effect on solubility, thermal behaviors
and other properties of the polymers. WAXD patterns of present PIs revealed their
complete amorphous nature (Figure 3.4). This observation was reasonable because the
presence of the non-coplanar conformation of the 1-phenoxy-2,4-diamino benzene might
have reduced the intermolecular forces between polymer chains. Amorphous nature of
these polymers is also reflected in good solubility of these polymers in solvents. The high
d-spacing value of PDAB-6FDA (5.82 Ǻ) than that of PDAB-ODPA (4.23 Ǻ) was due to
the bulky nature of –C(CF3)2– linkage, which could reduce the interchain interaction.
5 10 15 20 25 30 35 40
Inte
nsity
(a.
u.)
2
PDAB-ODPA
PDAB-6FDA
Figure 3.4 WAXD spectra of PDAB-ODPA and PDAB-6FDA

Chapter 3 Polymer synthesis and their characterization
82
Thus, having confirmed the good solubility, film forming property and thermal
stability of polyimides containing pendant phenoxy group based on PDAB, the study was
further extended to look for possible application of these polymers as PEMFC by
introducing –SO3H group in pendant phenoxy moiety. Most of the sulfonated polyimides
(SPIs), studied as PEMFC bears –SO3H group in the main chain. Much work has not
been reported on effect of –SO3H groups in flexible side chain of PI on performance of
these polymers as polymer electrolytes in fuel cell. So, in the present work, we
synthesized novel polyimides containing –SO3H groups in flexible pendant phenoxy in
side chain, based on SPDAB.
Much work has also not been reported on effect of pendant phenoxy group in PI
on gas permeability. Therefore study of PDAB based PIs was also extended for
application of PIs in gas permeability field. In view of this, we decided to synthesize
novel diamines in such a fashion that their resultant PIs would contain alkyl substituted
phenoxy group as a side chain. So the present dissertation also comprises synthesis of
novel PIs based on alkyl substituted pendant phenoxy group in view of investigations of
these PIs towards gas permeability.
3.5.2 Synthesis and physical characterization of sulfonated polyimides (SPIs)
3.5.2.1 Synthesis
New SPIs containing pendant sulfophenoxy group were prepared by one step high
temperature solution polycondensation of SPDAB and NTDA in m-cresol with benzoic
acid as the catalyst. Protection of sulfonic acid group in SPDAB was done with two
different organic bases; TEA and imidazole, separately, as described in the experimental
Section 3.3.2.1. In presence of TEA as the base, all reactions proceeded homogeneously
without gelation. Whereas, polymerization of SPDAB with NTDA by using imidazole as
the base resulted in viscous solution within 8 h. This phenomenon could be attributed to
the catalytic role of unreacted excess imidazole (1.2 moles of SPDAB), which is
amphoteric in nature having pKa 7.1 (Clayden, 2001; Lv, 2008).
Six-membered ring anhydride exhibits lower reactivity towards an amine group
due to its strain free and hence more stable form than strained and more reactive five-
membered ring analogues (Sek, 1992; Sek, 1995). Hence, use of catalyst becomes an

Chapter 3 Polymer synthesis and their characterization
83
essential step for such dianhydrides in order to enhance their electrophilicity. It has also
been reported that the addition of organic acids such as benzoic acid and organic
heterocyclic base such as isoquinoline, quinoline or imidazole during polymerization
increases viscosity of the PI (Sek, 1992). In the present condensation polymerization
based on NTDA, benzoic acid was added along with both of the organic bases, TEA or
imidazole. A short polymerization period of 8 h required for the polymerization with
SPDAB in presence of imidazole, as compared to 24 h period in presence of TEA, infers
catalytic activity of imidazole. The unreacted imidazole, remaining after salt formation
with SPDAB (0.2 mole) can form the imidazole-amic acid derivative of NTDA. In this
case, excess of imidazole first activates NTDA turning it to more electrophilic followed
by nucleophilic attack of imidazolium salt of SPDAB forming amic-acid derivative. In
other words, imidazole hydrolyzes six-membered anhydride moiety to imidazole-amic
acid derivative, which reacts faster with SPDAB forming poly(amic-acid) in the first step.
In this step imidazole is removed owing to its good leaving group capacity and thus,
molecular weight could be built up by propagating poly(amic-acid) with regeneration of
imidazole. Such condensation reactions of amide and imide formation from carboxylic
acids and dianhydride with amine, promoted and catalyzed by imidazole, its derivative
and carbonyl diimidazole (CDI) have been reported (Kingston, 1969; Jung, 1995;
Baldwin, 1996; Eastmond, 2002). Jung et al. (1995, 1996) has reported the synthesis of
polyimides with imidazole blocked isocyantes. Imidazole and its derivatives have been
used as the catalyst for various condensation reactions like acetylation of carbohydrates
(Tiwari, 2005), ester synthesis (Morton, 1988; Hirao, 2003; Hirose, 2003), peptide
synthesis (Nakashima, 1987) and curing of an epoxide group (Klaren, 1973). Lin et al.
(2006) prepared polybenzoxazines by using imidazole as the catalyst.
It should also be noted that, imidazole not only accelerates the poly(amic-acid)
formation, but it can also further catalyze imide formation. The similar catalytic activity
by benzimidazole has also been reported in the imidization of polyamic acids to
polyimides (Nelson, 1988). It is reported that imide formation could be substantially
enhanced with imidazole blocked isocynates (Wenzel, 1987; Helmer-Metzmann, 1989).
Thus, rapid polymerization with increasing viscosity of SPDAB•Im-N in the
polymerizing solvent (m-cresol) can be attributed to the catalytic role of imidazole.

Chapter 3 Polymer synthesis and their characterization
84
Whereas, another base; TEA only neutralizes sulfonic acid group to enhance solubility of
SPDAB in m-cresol, without any catalytic activity.
The polymer in fiber form was obtained by adding polymer solution in acetone. It
could not be precipitated in methanol (conventional non-solvent for PI) properly from m-
cresol solution (Mathew, 2001). It was observed that polymer solution of SPDAB•Im-N
became too viscous to be precipitated in acetone at room temperature, even after pouring
additional m-cresol, indicating formation of high molecular weight polymer.
Dense membranes of SPIs in salt form (triethylammonium sulfonate and
imidazolium sulfonate) were prepared by casting their 3 wt% solution onto clean glass
substrate at 80 C for 24 h. These formed reddish (SPDAB•TA-N), dark greenish
(SPDAB•Im-N) and faint greenish (SPDAB•TA-O) colored tough films on casting from
their DMAc solution. Dense membrane of imidazolium sulfonate salt form (SPDAB•Im-
N), possessed wavy nature. Membranes in their triethylammonium sulfonate
(SPDAB•TA-N) as well as imidazolium sulfonate (SPDAB•Im-N) salt forms were
converted to the corresponding –SO3H form (SPDABI-N and SPDABII-N, respectively)
by immersing in 1N HCl for 24 h at ambient followed by soaking in deionised water for
24 h at ambient and then dried. In case of proton conductivity measurements, these
membranes were equilibrated with water for at least 24 h at ambient, prior to
measurements.
3.5.2.2 FT-IR analysis
The formation of SPI was confirmed by FT-IR spectroscopy (Figure 3.5). The
presence of sulfonic acid moiety was confirmed by broad band around 1200-1250 cm-1.
Asymmetric and symmetric S=O stretching occurred at 1123 cm-1 and 1032 cm-1,
respectively; which showed maximum intensity due to more polar –SO3¯ group in
SPDAB•Im-N and SPDAB•TA-N than their respective sulfonic acid form (SPDABI-N
and SPDABII-N) (Yin, 2004; Hu, 2006). The bands corresponding to carbonyl
asymmetric and symmetric stretching of naphthalic based imides were observed at 1714
cm-1 and 1682 cm-1 (Li, 2008), while that of phthalic based imides at 1780 cm-1 and 1722
cm-1 (Genies, 2001b), respectively. The stretching and out of phase bending bands of

Chapter 3 Polymer synthesis and their characterization
85
C−N−C of naphthalic based imides were observed at 1344 cm-1 and 768 cm-1, while
corresponding bands for phthalic based imides were observed at 1368 cm-1 and 746 cm-1.
In SPDAB•Im-N, characteristic peaks in the range 2600-3200 cm-1 could be
assigned to the presence of imidazolium ring and its strong aromatic C−H stretching
vibrations occurred at 3154 and 2988 cm-1 (Dong, 2006; Iimori, 2007) and out of plane
bending band at 901 cm-1 (Cordes de N.D.; 1968). While, -N-H out of plane bending
vibration occurred at 631 cm-1. In addition, the intensity of the peak at 1450 cm-1 was
maximum due to the, corresponding added C=N conjugation with C=C bond present in
imidazolium ring. Bands appeared at 1066 cm-1 and 1054 cm-1 corresponded to the -N-C-
H frequency in the salt membrane SPDAB•TA-N and SPDAB•Im-N, respectively, and
subsequently disappeared after treatment of acid (1M HCl) in SPDABI-N and SPDABII-
N, hence confirming the complete exchange of proton.
4000 3500 3000 2500 2000 1500 1000 500
SPDAB-O
SPDABII-N
SPDABI-N
SPDABIm-N
SPDAB·TA-N
SPDAB·TA-O
Tra
nsm
ittan
ce (
%)
Wavenumber (cm-1)
Figure 3.5 FT-IR spectra of SPIs
3.5.2.3 Solubility and solution viscosity
The solubility of SPIs was determined for 0.5 % concentration on stirring for a
day at ambient and then at 80 °C for 24 h. These SPIs revealed excellent solubility in
organic solvents such as DMAc, DMF, DMSO, NMP and m-cresol. They were also
soluble in conc. H2SO4 (Table 3.4). SPDAB•TA-O and SPDAB•TA-N could readily be
dissolved in these solvents. SPDAB•Im-N also exhibited good solubility in these

Chapter 3 Polymer synthesis and their characterization
86
solvents, however, the relative time for dissolution of SPDAB•Im-N was higher (4-24 h).
In addition, it was dissolved in DMAc and NMP only on heating at 80 °C. This could be
attributed to the higher anticipated molecular weight.
In general, NTDA based SPIs in their sulfonic acid form are known to exhibit
poor organic solvent solubility (Yin, 2006a). However, in the present case, NTDA based
SPI in sulfonic acid form also exhibited good solvent solubility at ambient, except for the
partial solubility in NMP and insolubility in m-cresol. The improved solubility may be
due to asymmetric side-chain substitution of bulkier pendant sulfophenoxy group as well
as formation of kink structure by the presence of meta-imido moiety.
It is interesting to note that SPDAB•Im-N got precipitated from DMAc solution
of 0.5% concentration on increasing the temperature above 95 °C and redissolved on
lowering the temperature below 95 °C. This procedure was repeated 3 times and same
phenomenon of dissolution below and precipitation above 95 °C temperature was
noticed. Thus, 0.5% concentration solution of SPDAB•Im-N in DMAc exhibited the
critical solution temperature (CST) phenomenon. Investigation of this phenomenon for
different concentration revealed that the phenomenon was observed at and above 0.5%
concentration till 3% concentration. At 0.25% concentration, this CST phenomenon was
not observed. In case of SPDAB•TA-N solution in DMAc, such phenomenon at various
concentration was not observed. A peculiar flat structure and aromatic nature of
imidazolium cation in SPDAB•Im-N can play the major role for such CST phenomenon.
Such phenomenon is observed in ionic liquids (IL) (Anthony, 2001; Crosthwaite, 2005;
Domanska, 2006) based on alkyl substituted imidazolium cation. It is therefore possible
to have accommodated such planar structure of imidazolium cation in the available free
volume between the polymer chains in SPDAB•Im-N, resulting in close packing, hence
requiring more time for dissolution in the above mentioned solvents. It should be noted
that, gel formation was observed for SPDAB•Im-N at the end of polymerization in m-
cresol, probably due to build up of high molecular weight resulting in decrease in
solubility. Gelation of SPDAB•Im-N in m-cresol and precipitation in DMAc could be
attributed to the difference of interaction induced by aromatic imidazolium cation with
aromatic solvent m-cresol and non-aromatic solvent DMAc. The electrostatic or π-π
interaction of aromatic imidazole with π-cloud of m-cresol could have been increased

Chapter 3 Polymer synthesis and their characterization
87
with either increasing concentration or molecular weight (Domanska, 2007). As a result,
gelation was observed instead of precipitation. While in DMAc, there were no such
interactions, besides on increasing temperature as well as concentration, there might have
been close contact ion-pairs of imidazolium rings resulting in precipitation. Such
phenomenon has been observed in case of IL, which become more soluble in aromatic
solvents than non-aromatic solvents of equivalent molecular weight and polarity
(Blanchard, 2001).
Table 3.4 Solubility of SPIs in various solvents
++: soluble at room temperature, +: soluble on heating at 80 C, +: partially soluble on heating at 80 C, – : insoluble.
The reduced viscosity of the SPI was determined in DMAc at different
concentration with Ubbelohde viscometer at 35 °C. From the Figure 3.5, it is observed
that reduced viscosity increases non-linearly with lowering concentration, indicating the
typical polymer ionomer characteristics for these SPIs (Contois, 1955; Lundberg, 1982;
Hara, 1989; Campos, 1997). This anomalous viscosity behavior can be attributed to
coalescing of polymer ionomer molecules at high concentration due to increased intra or
intermolecular ionic association. While on dilution, these aggregated molecules go away
from each other due to greater electrostatic repulsion among them, which leads to
expansion of the polymer chain and hence displaying increased viscosity (Hara, 1989;
Campos, 1997). The higher viscosity of the SPDAB•Im-N than SPDAB•TA-N could
either be ascribed to the probable formed high molecular weight due to rapid
polymerization or discrepancy in solute-solvent interaction owing to different nature of
Polymer DMAc DMF DMSO NMP m-cresol CHCl3 Conc.
H2SO4 THF
SPDAB•TA-N ++ ++ ++ ++ ++ – ++ –
SPDAB •Im-N + ++ ++ + ++ – ++ –
SPDAB I-N + ++ ++ + – – ++ –
SPDAB II-N + ++ ++ + – – ++ –
SPDAB•TA-O ++ ++ ++ ++ ++ – ++ –
SPDAB-O ++ ++ ++ ++ ++ – ++ –

Chapter 3 Polymer synthesis and their characterization
88
pendant imidazolium and triethylammonium salt form. The viscosity of SPDABI-N and
SPDABII-N were measured after acidifying SPDAB•TA-N and SPDAB•Im-N membrane,
respectively. A slight higher viscosities of SPDABII-N with concentration as compared to
SPDABI-N may be assigned to higher viscosity of parent SPDAB•Im-N (Figure 3.6). It
should be noted that, the molecular weight of the SPDABII-N could have been further
increased by increasing period of polymerization to 24 h for SPDAB•Im-N, i.e. similar to
period for SPDAB•TA-N, by using more solvent (m-cresol). Thus, it may be concluded
that polymerization of SPI with imidazole as base, can increase viscosity of SPI
compared to that of TEA.
0.1 0.2 0.3 0.4 0.5
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
2.2
2.4
2.6
Conc. (g/dL)
Red
uced
vis
cosi
ty
SPDABIm-N SPDABTA-N SPDAB
II-N
SPDABI-N
SPDABTA-O SPDAB-O
Figure 3.6 Variation in reduced viscosity of SPIs with concentration
3.5.2.4 Thermal Properties of Polymers
Thermal stability of SPIs was investigated by TGA in N2 atmosphere at a heating
rate of 10 C. min-1 and the results are summarized in Table 3.5. Samples were taken in
film form, except for SPDAB-O, which was in powder form. These samples exhibited
water loss at 50-100 °C. It could be seen that SPIs exhibited typical two-step degradation
(Figure 3.7). The initial decomposition temperature (IDT) was ~260 °C, attributable to
desulfonation in the side chain. The SPI in sulfonic acid form viz., SPDABI-N and
SPDABII-N showed the similar two step degradation pattern with residue 44.5 % and
44.9 %, respectively. Moreover, these two curves were almost superimposable with each

Chapter 3 Polymer synthesis and their characterization
89
other. It could thus be deduced that, after acid hydrolysis of the corresponding
membranes in salt form, resulting membranes in sulfonic acid form SPDABI-N and
SPDABII-N yielded the same type of polymers. Among all the SPIs, the IDT for
SPDAB•Im-N occurred at the highest temperature (348 °C) and ~68 °C higher than its
analogous SPDAB•TA-N, indicating stronger interaction between the pendant sulfonic
and imidazolium moiety. In addition, aromatic nature of imidazolium ring could also
elevate the thermal stability. On the other hand, SPDAB•TA-N membrane (in
triethylammonium salt form) exhibited lower IDT as well as char yield (23.82 %) than
the acidified membrane SPDABI-N and SPDABII-N. Same pattern of lower IDT and char
yield of SPIs in triethylammonium salt form than that of their respective sulfonic acid
form has also been observed earlier (Einsla, 2004). Same was the pattern for SPIs based
on ODPA. Thermal stability for six membered imides decreases in the order
SPDAB•Im-N > SPDABI-N ≈ SPDABII-N > SPDAB•TA-N. In conclusion, between the
polymerization of SPI with a base (TEA or imidazole), the latter not only improved the
IDT, but also increased the char yield as compared with the former demonstrating its fire
retardant tendency. The second stage weight loss occurring above 500 °C in SPIs
represented the decomposition in their main chain. SPI containing five membered imide
ring based on ODPA (SPDAB•TA-O) decompose at lower temperature compared to SPI
with six membered imide ring.
Attempts were made to determine the glass transition temperature (Tg) of these
SPIs by DSC at a heating rate of 10 °C min-1 in N2 atmosphere. However, when the
samples were scanned in a temperature range of 50−350 °C, within which, no obvious
glass transition could be detected. This indicated the stronger interaction among the
chains of SPI induced by the pedant sulfonated moiety. In NTDA based SPI, presence of
rigid planar naphthalo–imide ring in main chain would further assist in increasing the
glass transition temperature. It had also been observed that splitting-off sulfonic group
(cleavage of CAr−SO3H bond) occured around 260 °C as explained earlier in TGA.
Furthermore, Tg of these SPIs could also be raised by their increased rigidity due to an
alternate ortho substituted bulkier sulfophenoxy group, which had an ability to restrict the
bond rotation between phenyl ring and nitrogen (Ar–N bond) in main chain. Considering
all these aspects, the apparent Tg of polymer appears to be above the decomposition

Chapter 3 Polymer synthesis and their characterization
90
temperature (~260 °C), which could not be located. It also implied that free volume made
available by pendant bulkier short side chain sulfophenoxy group, was not sufficient to
lower Tg below the temperature corresponding to splitting-off sulfonic acid group i.e IDT
(260 °C). Some of the SPIs have also been reported, which do not show glass transition
(Yasuda, 2005; Rodgers, 2006).
Table 3.5 Thermal properties of SPIs
Degradation temperature (C) Polymer
I II Residue (%)
SPDAB•TA-N 272 512 23.82
SPDAB•Im-N 340 555 40.87
SPDABI-N 295 526 44.91
SPDABII-N 302 518 44.55
SPDAB•TA-O 262 402 15.14
SPDAB-O 288 520 27.36
150 300 450 600 750 9000
10
20
30
40
50
60
70
80
90
100
SPDABII-NSPDABTA-N
SPDABI-N
SPDABIm-N
SPDATA-O
SPDAB-O
Wei
ght
loss
(%
)
Temperature (C)
Figure 3.7 TGA of SPIs
3.5.2.5 WAXD Analysis
Dry form of the membrane samples were used for WAXD analysis except for
SPDAB-O, which was analyzed in powder form. Initially salt form of the membrane
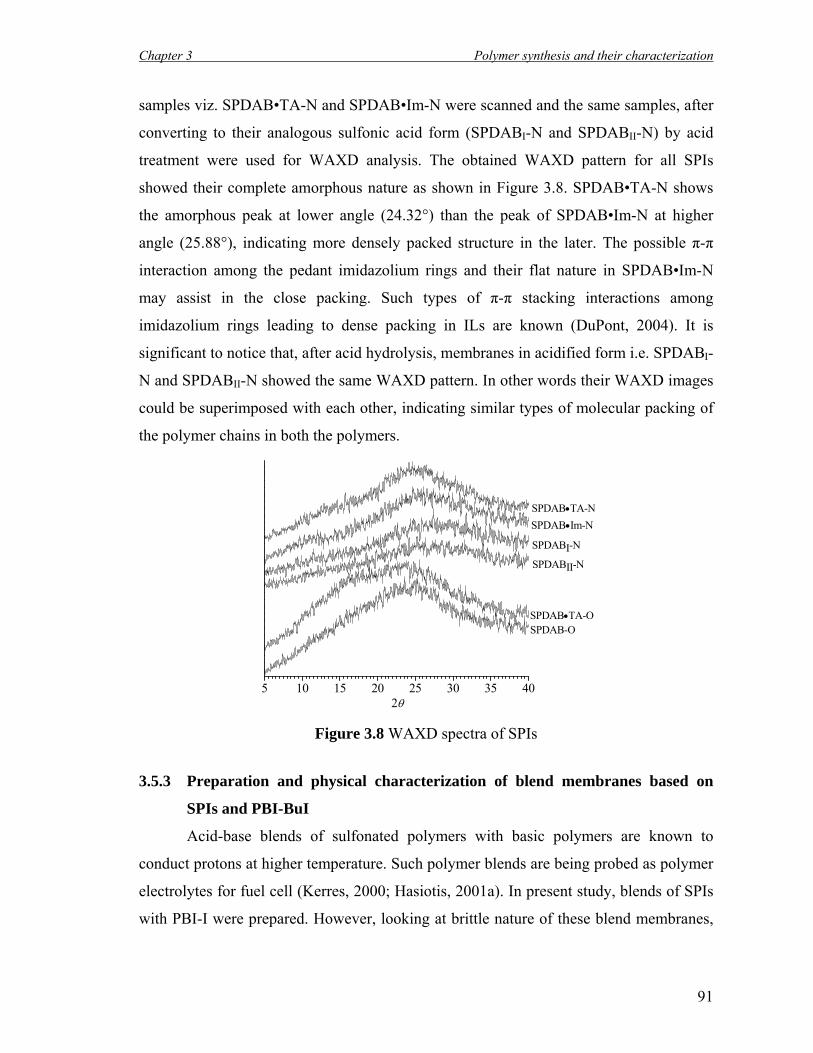
Chapter 3 Polymer synthesis and their characterization
91
samples viz. SPDAB•TA-N and SPDAB•Im-N were scanned and the same samples, after
converting to their analogous sulfonic acid form (SPDABI-N and SPDABII-N) by acid
treatment were used for WAXD analysis. The obtained WAXD pattern for all SPIs
showed their complete amorphous nature as shown in Figure 3.8. SPDAB•TA-N shows
the amorphous peak at lower angle (24.32°) than the peak of SPDAB•Im-N at higher
angle (25.88°), indicating more densely packed structure in the later. The possible π-π
interaction among the pedant imidazolium rings and their flat nature in SPDAB•Im-N
may assist in the close packing. Such types of π-π stacking interactions among
imidazolium rings leading to dense packing in ILs are known (DuPont, 2004). It is
significant to notice that, after acid hydrolysis, membranes in acidified form i.e. SPDABI-
N and SPDABII-N showed the same WAXD pattern. In other words their WAXD images
could be superimposed with each other, indicating similar types of molecular packing of
the polymer chains in both the polymers.
5 10 15 20 25 30 35 40
SPDAB-OSPDABTA-O
SPDABII-N
SPDABIm-N
SPDABI-N
SPDABTA-N
2
Figure 3.8 WAXD spectra of SPIs
3.5.3 Preparation and physical characterization of blend membranes based on
SPIs and PBI-BuI
Acid-base blends of sulfonated polymers with basic polymers are known to
conduct protons at higher temperature. Such polymer blends are being probed as polymer
electrolytes for fuel cell (Kerres, 2000; Hasiotis, 2001a). In present study, blends of SPIs
with PBI-I were prepared. However, looking at brittle nature of these blend membranes,

Chapter 3 Polymer synthesis and their characterization
92
blends of SPI and PBI-BuI were prepared with objective to study proton conduction of
these blend membranes.
3.5.3.1 Membrane preparation
Blend membranes were prepared from SPI and polybenzimidazole (PBI) in
different weight ratio in order to evaluate their performance as material for PEMFC. SPI
in triethylammonium sulfonate (TEA) form (SPDAB•TA-N) and PBI (inherent viscosity
of 1.8) based on DAB and IPA were initially attempted to prepare blend membranes.
However, membranes cast from them were brittle, though homogeneous solution of these
polymers in different weight ratio could be made. Hence we replaced PBI-I by PBI-BuI.
It was observed that, though flexible blend membranes based on SPDAB•TA-N and PBI-
BuI could be made, the membranes containing more than 50 wt% of SPDAB•TA-N
became brittle.
Thus, blend membranes based on SPI and PBI-BuI with a weight ratio 50:50,
40:60 and 30:70 were prepared and named as SPI(TEA)/PBI-BuI(a/b), where a and b
represents weight percentage of SPI and PBI-BuI, respectively, while ‘TEA’ denotes SPI
in triethylammonium sulfonate form. In order to evaluate the performance of these blend
membranes as polymer electrolyte membrane (PEM) for fuel cell, they were transformed
into sulfonic acid form after treating with 1N HCl for 24 h followed by washing with
deionised water. These membranes were named as SPI(H)/PBI-BuI(a/b). Here, ‘H’
represents proton form of SPI. The blend membranes doped by 12 M H3PO4 were named
as SPI(PA)/PBI-BuI(a/b), where ‘PA’ denotes the exchange of triethylamine by
phosphoric acid protons.
3.5.3.2 FT-IR analysis of blend membranes
Figure 3.9 shows the FTIR spectra of SPDABI-N, PBI-BuI and their respective
blends. In SPDABI-N, bands at 1715 cm-1 and 1680 cm-1 correspond to carbonyl
asymmetric and symmetric stretching, respectively present in naphthalic imide group.
The characteristic absorption bands at 1252, 1180 and 1030 cm-1 are associated with the
stretching of the sulfonate groups. In, PBI-BuI the sharp bands in the region 3380-3430
cm-1 are assigned to the isolated -N-H stretching of the imidazole ring. However there is

Chapter 3 Polymer synthesis and their characterization
93
no observable sharp band within 3100-3140 cm-1 corresponding to the associated -N-H
stretching which is usually present in the conventional PBI based on IPA (Wang, 2007b;
Kumbharkar, 2009). This implies that, bulky t-butyl group in PBI-BuI has lower
interchain interactions (Kumbharkar, 2006). In PBI-BuI, two distinct bands at 2962 cm-1
and 2872 are due to the asymmetric and symmetric stretching of the –C–H bonds of t-
butyl group, respectively. The peaks obtained in the range between 1650 and 1500 cm-1
are the characteristics of the benzimidazole rings (Musto, 1993).
It can be seen that the peak at 1626 cm-1 corresponding to -C=N stretching of the
imidaozle ring in PBI-BuI was shifted to higher frequency at 1634-1636 cm-1 in the
respective blend, implying the involvement benzimidazole ring interaction. The peak at
the 804 cm-1 corresponding to the five-member heterocyclic ring in PBI-BuI shifts to
higher wavenumber 807, 809 and 810 cm-1 with decrease in PBI-BuI content. This was
obvious, since with increase in relative sulfonic content, quantitative interaction between
five membered heterocyclic benzimidazole ring and SPI would be more. Such
observation was also made in the blend prepared from sulfonated PEEK (SPEEK) and
PBI, where a peak of −C=N stretching at 1625 cm-1 and five membered imidazole peak at
806 cm-1 shifts to 1645 and 814 cm-1, respectively (Zhang, 2008), indicating the
interaction between SPEEK and PBI. However, it should be noted that, shift of these
peaks was relatively lower in the present blend system. The characteristic bands of
benzimidazole are seen at 1625 cm-1 (C=N stretching) and aromatic C=C stretching at
1602 cm-1 of the benzene ring. The peak at 3634 cm-1 in PBI-BuI can be assigned to the
O−H stretching of absorbed water. Brooks et al. (1993) and Li et al. (2004) have also
mentioned the occurrence of the same peak at 3620 cm-1, which disappeared on heating at
120 ºC. Among the blends made, the appearance of this peak at 3621 cm-1 can be
observed only in case of SPI (H)/PBI-BuI (30/70) due to high content of PBI-BuI.
The peak corresponding to imidazole ring vibration at 1284 cm-1 (Musto, 1993) is
intensified with increase in PBI-BuI content in the blend. The peak at 1344 cm-1
corresponding to the O=S=O asymmetric stretching shifts to a lower wavenumber at
1340, 1339 and 1338 cm-1 with decrease in SPI content indicating its involvement in the
interaction for blend formation. The characteristic intensity of the asymmetric and
symmetric imide ring at 1715 and 1680 cm-1, respectively has neither been intensified nor

Chapter 3 Polymer synthesis and their characterization
94
shifted, suggesting that imide ring might not have participated for interactions with PBI-
BuI. So the probable H-bonding may be involved between imidazole and sulfonic acid
moiety. Similarly, with increase in PBI-BuI content the same heterocyclic ring vibration
at 700 cm-1 is shifted to 692, 693 and 697 cm-1. The peak at 1032 due to SO3¯ in SPI(H)/
PBI-BuI(50/50) has been intensified than parent SPI and remaining blend system also.
This can be attributed to the more polarized nature of SO3¯ group due to the more
dissociated form of sulfonic acid group into SO3¯ and H+. This observation is reasonable
as in SPI(H)/PBI-BuI(40/60) and SPI(H)/PBI-BuI(30/70) blend system, the number of
sulfonic groups available for H-bonding becomes less as compared with SPI(H)/PBI-
BuI(50/50).
It is interesting to note that in the respective blend a change in shift of the
corresponding frequency pertaining to particular moiety interactions was within the range
3-10 cm-1, suggesting the either medium or weak interaction. This can be attributed to the
presence of pendant sulfonic acid group in the side chain of NTDA based SPI as well as
the presence of pendant bulky t-butyl group in the main chain of PBI, which would
screen the imidazole moiety from interaction, leading to weak intermolecular H-bonding.
This can be further justified by the absence of associated H-bonding occurring in the
range of 3100-3140 cm-1 in PBI-BuI (Kumbharkar, 2009) and present in PBI-I (Musto,
1989). The shift in specific bands of both components of polymers in blends indicates the
specific interaction between the groups, a characteristic of miscible blend formation.
3500 3000 2500 2000 1500 1000 500
SPI(H)/ PBI-BuI(30/70)
SPI(H)/ PBI-BuI(40/60)
SPI(H)/ PBI-BuI(50/50)
SPDABI-N
PBI-BuI
Tra
nsm
ittan
ce (
%)
Wavenumber (cm-1)
Figure 3.9 FT-IR spectra of blends in the sulfonic acid form

Chapter 3 Polymer synthesis and their characterization
95
3.5.3.3 Thermal analysis of blend membranes
The thermal stability of the blend membrane was measured by TGA. The blend
membranes exhibited 2 step degradation processes as shown in Figure 3.10. The initial
little weight loss at ~100 C is due to the loss of absorbed moisture. The first weight loss
corresponds to the thermal splitting of sulfonic group either in salt form with TEA or free
sulfonic acid group form. The second weight loss is due to the decomposition of main
chain backbone of the polymer components viz; SPI and basic polymer, PBI-BuI,
respectively.
It is interesting to note that, there is noticeably improved thermal stability of blend
membranes in sulfonic acid form viz; SPI(H)/PBI-BuI(a/b), than their counterpart in
triethylammonium salt form viz; SPI(TEA)/PBI-BuI(a/b). The splitting of –SO3H of SPI
(H)/PBI-BuI(a/b) blend system occurs above 400 C compared to 250 C for SPI(TEA)/
PBI-BuI(a/b), indicating the thermal splitting of –SO3H stability was better for blend
membrane in sulfonic acid form. This result indicated that acidic –SO3H is involved in
the interaction with the imidazole of PBI-BuI. While comparing the degradation behavior
among the blend system, 10 % weight loss for SPI(H)/PBI-BuI(a/b) blend system occurs
at a higher temperature in the range of 443-480 C than SPI(TEA)/PBI-BuI(a/b) blend
system (258-327 C) as shown in the Table 3.6. Thus, result proves that free sulfonic acid
group (–SO3H) is more liable for ionic interaction than in triethylammonium sulfonate
salt form with the imidazole ring of the basic polymer; PBI-BuI. In this way,
improvement in the thermal –SO3H splitting of stability can be distinctly accomplished
by the blending of SPI with the basic polymers such as PBI-BuI. The specific acid-base
interactions in the blend as well as the extraordinary thermal stability of the PBI-BuI
(Kumbharkar, 2006) assist in the improvement of thermal stability of the blend.
We endeavored for determining the glass transition temperature (Tg) of these
blend membranes by DSC at a heating rate of 10 °C min-1 in N2 atmosphere, however no
apparent glass transition could be seen. This could be attributed to the probable stronger
interaction between the sulfonic acid moieties of SPIs with the bezimidazole ring PBI-
BuI. Because of this segmental mobility of the polymeric chains in the resultant blend
membrane can be reduced. The Tg of these polymers is probably higher than the
decomposition temperature of sulfonic acid groups, hence it could not be detected by

Chapter 3 Polymer synthesis and their characterization
96
DSC. Moreover, the glass transition temperature for the parent polymers PBI-BuI as well
as SPIs, could also not be detected.
Table 3.6 Thermal properties of blend membranes
Polymer T10 Residue (%)
SPDAB•TA-N 270.4 23.8
SPDABI-N 309.2 44.9
PBI-I 649.8 66.9
PBI-BuI 564.5 64.9
SPI(TEA)/PBI-BuI(30/70) 257.9 49.8
SPI(H)/PBI-BuI(30/70) 451.6 60.5
SPI(TEA)/PBI-BuI(40/60) 326.8 55.3
SPI(H)/PBI-BuI(40/60) 431.5 58.8
SPI(TEA)/PBI-BuI(50/50) 320.2 54.5
SPI(H)/PBI-BuI(50/50) 443.2 55.2
50 150 250 350 450 550 650 750 85010
20
30
40
50
60
70
80
90
100
6 6'
5
4
3'
5'
4'
32
1
Wei
ght
Los
s (
%)
Temperature (C)
1 . PBI-I2 . PBI-BuI3 . SPI(TEA)/PBI-BuI(50/50)3'. SPI(H)/PBI-BuI(50/50)4 . SPI(TEA)/PBI-BuI(40/60)4'. SPI(H)/PBI-BuI(40/60)5 . SPI(TEA)/PBI-BuI(30/70)5'. SPI(H)/PBI-BuI(30/70)6 . SPDABTA-N6'. SPDAB
I-N
Figure 3.10 TGA of blends
Thus, SPI and PBI-BuI form miscible blends having high thermal stability in
sulfonic acid form.

Chapter 3 Polymer synthesis and their characterization
97
3.5.4 Synthesis and physical characterization of polyimides based on various alkyl
substituted phenoxy diamines
The aromatic PIs with alkyl substituents in different position of pendant phenoxy
ring were synthesized with the objective to study effects of alky substituents on the gas
permeability. PIs were substituted with methyl groups at ortho, para and 2,6-positions;
while bulky t-butyl group was situated at para position to the ether linkage. These PIs
were synthesized by condensing corresponding diamine with ODPA and 6FDA. These
two dianhydrides were chosen in order to compare the effect between the presence of
flexible (C-O-C) link with bulk and rigid (–C(CF3)2–) groups in the main chain of the
resultant PIs on their gas permeation properties. The purpose of utilizing these two
commercially available dianhydrides, for the preparation of present PIs lies in excellent
gas permeation performance of PIs based on these dianhydrides (Stern, 1989; Li, 1997).
6FDA based PIs have well been acknowledged in gas permeation field due to their
intrinsic high permeability as well as selectivity. Moreover, 6FDA improves solvent
solubility, even in chlorinated organic solvents such as TCE and chloroform (de Abajo,
1999; Tamai, 2001).
3.5.4.1 Synthesis
Aromatic polyimides were prepared by the classical one-step solution
polycondensation technique at 200 °C in m-cresol. Their preparation involved the
addition of solid dianhydride to the diamine solution in m-cresol at the ambient condition,
followed by heating at 200 °C. Water formed during the process of imidization was
removed continuously with the flush of N2, to obtain high viscosity polymer. All
reactions proceeded homogeneously without gelation or precipitation in m-cresol.
Depending on the reactivity of diamine and dianhydride, the polymerization time varied
between 8-24 h. The diamine 2',6'DMPDAB being more basic, gave polymer with high
viscosity within 8 h. The comparative short time for the 2',6'DMPDAB based PI can be
credited to its increased basicity as compared with the other diamines. It is well known
that basicity of a diamine can be increased with the embodiment of electron donating
substituents (Yin, 2004 and 2006a; Marestin, 2008) and hence such diamines become
more reactive. Such phenomenon of increased basicity in the amine group by the ortho-

Chapter 3 Polymer synthesis and their characterization
98
substituted propoxy linkage has been observed by Yin et al. (2004). In the present case,
basicity of 2',6'DMPDAB is enhanced due to the strong electron donating effect of the
ortho-dimethyl substituted phenoxy group. The presence of these two –CH3 groups at
2,6-position of phenoxy ring bestowed two advantages. First, being ortho substituted to
the ether linkage, electron donating ability of the oxygen atom in the ether linkage was
boosted due to their +I effect. Second, the rotation of phenoxy ring and meta-amino
phenyl ring around the Ar-O-Ar’ linkage could become more energetic due to the steric
hindrance of the ortho-substitution of two –CH3 groups in the former and amino
functionality in the later. As a result, phenyl rings of 2',6'DMPDAB become coplanar to
each other. Hence, ortho located -NH2 group of meta-amino phenyl ring (present at the
vicinity of methyl disubstituted phenoxy group) experiences less steric hindrance of the
bulky dimethyl substituted phenoxy group resulting in its enhanced reactivity with
dianhydride. This observation can be proved by the 1H-NMR of 2',6'DMPDAB (Figure
2.15), wherein the proton Hc of meta-amino phenyl ring (ortho to dimethyl substituted
phenoxy group) shifts towards upfield as compared with the other diamines. This
indicates stronger electron donating ability of dimethyl substituted phenoxy group in
2',6'DMPDAB as compared with the other alkyl substituted phenoxy group of other
diamines. In this way, more basic character of 2',6'DMPDAB results in its PI with
increased viscosity in the lesser duration.
Between ODPA and 6FDA based PIs with the same diamine, the former took less
time than the latter for the polymerization, despite having high electron affinity (EA) of
6FDA. However, the polymerization duration for 2',6'DMPDAB with either ODPA or
6FDA was the same. Polymers were isolated by precipitating their viscous solution into
methanol with vigorous stirring. The yield of polymers thus obtained was high (> 93 %).
Dense membranes of these polymers (40-50 μm thick), were prepared by solution
casting method. For ODPA based PIs, dense membranes were prepared from TCE, while
for 6FDA based PI, chloroform was used. In case of FT-IR spectral analysis, dense
membranes of relatively lower thickness (~10 μm) were prepared, while for density
measurements, films of comparatively higher thickness (~100 μm) were prepared.

Chapter 3 Polymer synthesis and their characterization
99
Obtained polymers were characterized by elemental analysis, FT-IR spectroscopy,
solvent solubility, WAXD, viscosity measurements and thermal analysis. The physical
properties investigated of these PIs are given in Table 3.7, Table 3.8 and Table 3.9.
3.5.4.2 Elemental analysis
The elemental analysis calculated for PIs based on alkyl substituted diamines are
shown in Table 3.7. Observed elemental analysis values of these PIs showed good
agreement with the calculated values.
Table 3.7 Elemental analysis of PIs based on alkyl susbtituted phenoxy diamines
Element analysis Polymer
Formula and
Molecular weight
% C % H % N % F
Calculated 71.31 3.30 5.74 2MPDAB-ODPA
(C29H16O6N2)n
(488)n Observed 70.42 3.80 6.19
Calculated 61.75 2.59 4.50 18.31 2MPDAB-6FDA
(C32H16O5N2F6)n
(622)n Observed 61.38 2.45 4.71 17.19
Calculated 71.31 3.30 5.74 4MPDAB-ODPA
(C29H16O6N2)n
(488)n Observed 70.75 3.71 6.12
Calculated 61.75 2.59 4.50 18.31 4MPDAB-6FDA
(C32H16O5N2F6)n
(622)n Observed 61.08 3.01 3.96 17.39
Calculated 71.71 3.61 5.58 2',6'DMPDAB-ODPA
(C30H18O6N2)n
(502.48)n Observed 71.25 3.89 5.86
Calculated 62.27 2.85 4.40 17.91 2',6'DMPDAB-6FDA
(C30H18O5N2F6)n
(636.5)n Observed 62.24 2.69 4.72 16.18
Calculated 72.45 4.18 5.28 4tBPDAB-ODPA
(C32H22O6N2)n
(530)n Observed 71.09 4.82 5.13
Calculated 63.26 3.34 4.22 17.15 4tBPDAB-6FDA
(C35H22O5N2F6)n
(664)n Observed 62.89 3.57 3.88 15.67

Chapter 3 Polymer synthesis and their characterization
100
3.5.4.3 FT-IR analysis
IR spectra of present alkyl substituted polyimides (Figure 3.11) showed
characteristic absorption bands of the 5 membered imide ring at 1779-1784 cm-1 and
1722-1727 cm-1 (corresponding to asymmetric and symmetric –C=O stretching),
respectively and 1360-1365 cm-1 (C–N stretching). Imide ring deformation peaks
appeared at 1100-1106 and 720-745 cm-1 (Mathew, 2001). Absence of peaks at 1650 cm-1
(–C=O of amic acid) and 3250 cm-1 (N–H and O–H groups) confirmed complete
imidization. In case of 6FDA based polyimides, C–F multiple stretching bands appeared
at 1100-1300 cm-1 (Jeong, 2001) and the C–O–C stretching appeared at 1240-1250 cm-1
(Mathew, 2001).
3500 3000 2500 2000 1500 1000
2'MPDAB-ODPA
2'MPDAB-6FDA
4'MPDAB-ODPA
2',6'DMPDAB-ODPA
4'tBPDAB-6FDA
4'tBPDAB-ODPA
2',6'DMPDAB-6FDA
4'MPDAB-6FDA
Tra
nsm
itta
nce
(a.u
.)
Wavenumber (cm-1)
Figure 3.11 IR spectra of polyimides based on alkyl substituted phenoxy diamines
Thus, elemental analysis and FT-IR spectral analysis indicate formation of alkyl
substituted PIs of desired structures.
3.5.4.4 Solubility and solution viscosity
The solvent solubility of polyimides was determined for 0.5 % concentration in
common organic solvents (Table 3.8). All PIs showed appreciable solubility at ambient
temperature, which could be advantageous in membrane formation. Excellent solubility
could be attributed to the presence of pendant phenoxy group with alkyl substitution
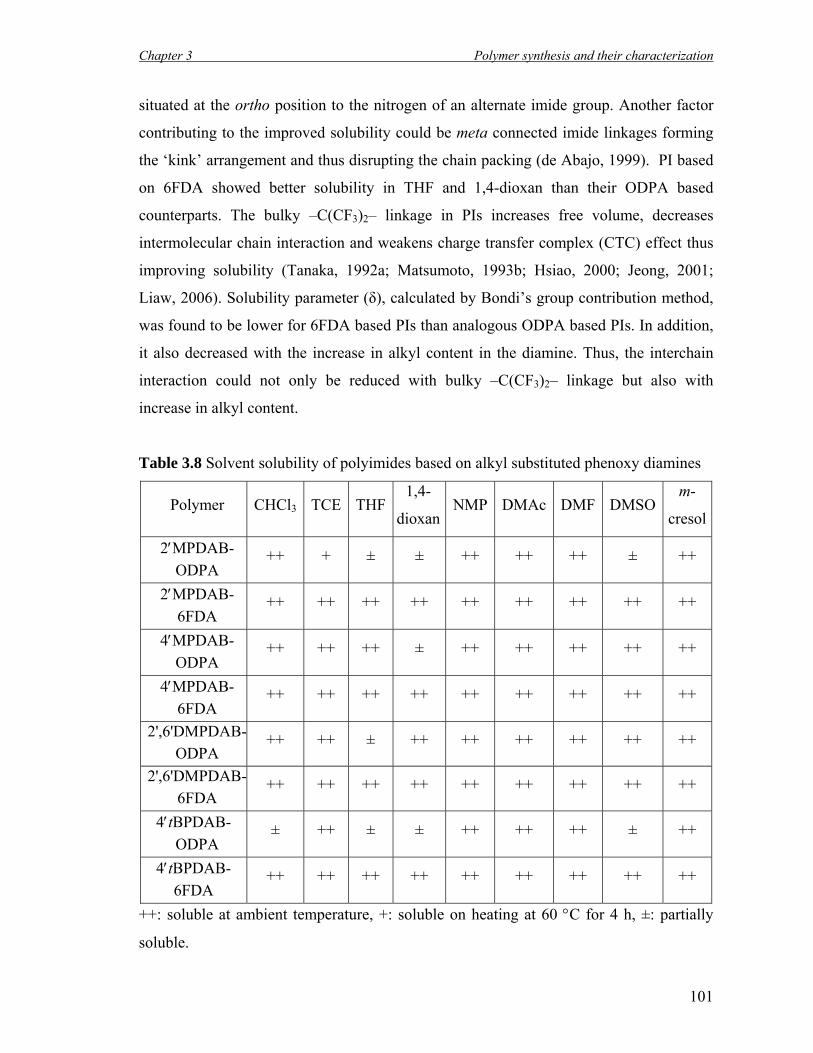
Chapter 3 Polymer synthesis and their characterization
101
situated at the ortho position to the nitrogen of an alternate imide group. Another factor
contributing to the improved solubility could be meta connected imide linkages forming
the ‘kink’ arrangement and thus disrupting the chain packing (de Abajo, 1999). PI based
on 6FDA showed better solubility in THF and 1,4-dioxan than their ODPA based
counterparts. The bulky –C(CF3)2– linkage in PIs increases free volume, decreases
intermolecular chain interaction and weakens charge transfer complex (CTC) effect thus
improving solubility (Tanaka, 1992a; Matsumoto, 1993b; Hsiao, 2000; Jeong, 2001;
Liaw, 2006). Solubility parameter (δ), calculated by Bondi’s group contribution method,
was found to be lower for 6FDA based PIs than analogous ODPA based PIs. In addition,
it also decreased with the increase in alkyl content in the diamine. Thus, the interchain
interaction could not only be reduced with bulky –C(CF3)2– linkage but also with
increase in alkyl content.
Table 3.8 Solvent solubility of polyimides based on alkyl substituted phenoxy diamines
Polymer CHCl3 TCE THF1,4-
dioxanNMP DMAc DMF DMSO
m-
cresol
2MPDAB-
ODPA ++ + ± ± ++ ++ ++ ± ++
2MPDAB-
6FDA ++ ++ ++ ++ ++ ++ ++ ++ ++
4MPDAB-
ODPA ++ ++ ++ ± ++ ++ ++ ++ ++
4MPDAB-
6FDA ++ ++ ++ ++ ++ ++ ++ ++ ++
2',6'DMPDAB-ODPA
++ ++ ± ++ ++ ++ ++ ++ ++
2',6'DMPDAB-6FDA
++ ++ ++ ++ ++ ++ ++ ++ ++
4tBPDAB-
ODPA ± ++ ± ± ++ ++ ++ ± ++
4tBPDAB-
6FDA ++ ++ ++ ++ ++ ++ ++ ++ ++
++: soluble at ambient temperature, +: soluble on heating at 60 C for 4 h, ±: partially
soluble.

Chapter 3 Polymer synthesis and their characterization
102
The intrinsic viscosities [η] of these polyimides were in the range of 0.33-0.55
dL.g-1 in TCE, which was high enough to offer strong and tough films. The intrinsic
viscosity increased in the order for diamines 2'MPDAB ≈ 4'MPDAB < 4tBPDAB <
2',6'DMPDAB.
3.5.4.5 Thermal Properties of Polymers
Thermal properties of all polyimides were evaluated by DSC (Figure 3.12) and
TGA (Figure 3.13) and results are summarized in Table 3.9. These polymers exhibit high
Tg in the range of 265-315 C. It is observed that ODPA based PI of a particular diamine
shows lower Tg than the corresponding 6FDA based analogue, owing to the presence of
flexible ether linkage in ODPA. The voluminous –C(CF3)2– linkage in 6FDA would
impose limitations on free rotation around the CF3CCF3 bond leading to restricted
torsional motion of neighboring phenyl rings. Such increase in Tg due to inhibited
segmental motion is well known (Matsumoto, 1993b; Coleman, 1994). Tg values are
dependant on structure of diamine also. In the present series of PIs based on either ODPA
or 6FDA, Tg of PI increased in the order of 4'MPDAB ≈ 4'tBPDAB < 2'MPDAB <
2',6'DMPDAB. This indicated that methyl substitutions at 2 and 6 positions on the
pendant phenoxy ring in 2',6'DMPDAB based PI would hinder the rotational motion
around phenoxy group the most, followed by ortho-substituted 2'MPDAB based PI;
while the least hindrance by the substitution at 4-position, irrespective of its bulk
(4'MPDAB and 4'tBPDAB based PIs). This also signifies that position of alkyl
substituent is more crucial in depicting the rotational barrier than the bulk of substituent.
It is interesting to compare the trend of Tg between PI based on two dianhydrides
for different diamines. In case of ODPA based PI with different diamines Tg decreases in
the order PDAB (256 C) < 4'MPDAB (267 C) < 4tBPDAB (270 C) < 2'MPDAB
(282 C) < 2',6'DMPDAB (311 C). While, for 6FDA based PIs with different diamines,
Tg decreased in the order 4'MPDAB (278 C) < PDAB (281 C) ≈ 4tBPDAB (282 C) <
2'MPDAB (289 C) < 2',6'DMPDAB (313 C). This indicates that, in case of ODPA
based PIs, chemical structure of diamine plays the dominant role in governing the Tg
while for 6FDA based PIs, diamine-structural effect becomes prevailing only when they
have ortho-substitution.

Chapter 3 Polymer synthesis and their characterization
103
Thermal stability of the PIs was evaluated by the TGA (Figure 3.12) in N2
atmosphere at a heating rate of 10 C. min-1 and the results are summarized in Table 3.9.
These PIs show IDT in the range 473-515 C and 10% weight loss in the range 495-
545 C. It is interesting to look at the char yield data of these PIs. PI based on diamines
such as 2',6'DMPDAB and 2'MPDAB exhibited lower thermal stability and hence lower
weight residue than PI based on 4tBPDAB and 4'MPDAB. This could be assigned to the
increased rotational barrier of the pendant phenoxy group and hence inducing high
torsional strain due to the ortho-substitution of methyl groups on the pendant phenoxy
moiety. It should also be noted that, char yield of the 6FDA based PIs is lower than the
ODPA based PIs. This could be assigned to the loss of CF3 radical in the former case as
explained earlier in the Section 3.5.1.5.
100 125 150 175 200 225 250 275 300 325 350
4'MPDAB-ODPA
4'tBPDAB-ODPA
2',6'DMPDAB-ODPA
2',6'DMPDAB-6FDA
4'tBPDAB-6FDA
4'MPDAB-6FDA
2'MPDAB-6FDA
Hea
t F
low
(a.
u.)
Temperature (°C)
2'MPDAB-ODPA
Figure 3.12 DSC curves of polyimides based on alkyl substituted phenoxy diamines

Chapter 3 Polymer synthesis and their characterization
104
Table 3.9 Physical properties of polyimides based on alkyl substituted phenoxy diamines
Polymer []
(dL.g-1)
dsp
(Å)
(cal.cm-3)1/2
(g.
cm-3)
Tg
(C)
IDT
(C)
T10%
(C)
Char yield
at 900 C
(%)
2MPDAB-
ODPA 0.34 4.23 12.22 1.402 282 478 496 50.8
2MPDAB-
6FDA 0.32 5.70, 3.38 10.97 1.404 289 493 500 45.0
4MPDAB-
ODPA 0.38 4.53 12.20 1.404 267 515 545 54.0
4MPDAB-
6FDA 0.30 6.08, 3.62 10.97 1.403 278 486 543 53.3
2',6'DMPDA
B-ODPA 0.50 4.36 12.04 1.375 311 473 494 53.0
2',6'DMPDA
B-6FDA 0.55 6.29, 3.46 10.87 1.387 313 477 494 45.4
4tBPDAB-
ODPA 0.47 4.18 11.69 1.333 270 504 530 55.4
4tBPDAB-
6FDA 0.45 5.32, 3.52 10.57 1.335 281 506 540 54.5
[]: Intrinsic viscosity using TCE at 35 C.
: Density determined by floatation method at 40 C using aq. K2CO3 solutions.
dsp: Average interchain distance from WAXD spectra.
: Solubility parameter (Van Krevelen, 1997).
Tg: Glass transition temperature measured by DSC at a heating rate of 10 C.min-1.
T5%: Onset of 5% weight loss on TGA thermograms at a heating rate of 10 C.min-1
under N2 atmosphere.
Thus, Tg values and TGA analysis indicate high thermal stability of these polymers
suitable for gas separation even at higher temperature.

Chapter 3 Polymer synthesis and their characterization
105
100 200 300 400 500 600 700 800 90040
50
60
70
80
90
1008
7
6
5
1. 2'MPDAB-ODPA2. 2'MPDAB-6FDA3. 4'MPDAB-ODPA4. 4'MPDAB-6FDA5. 2',6'DMPDAB-ODPA6. 2',6'DMPDAB-6FDA7. 4'tBPDAB-ODPA8. 4'tBPDAB-6FDA
4
3
21
Wei
ght
Los
s (%
)
Temperature (C)
Figure 3.13 TGA curves of polyimides based on alkyl substituted phenoxy diamines
3.5.4.6 WAXD analysis
All these polyimides were amorphous in nature as could be seen from their
WAXD spectra (Figure 3.14). 6FDA based PI shows two amorphous peaks suggesting
that there could be two types of predominant chain packing. In ODPA based polyimides,
the average intersegmental spacing (dsp) varies marginally with the variation of alkyl
content in their diamine moiety. 6FDA based alkyl substituted PIs show higher d-spacing
at lower angle corresponding to d-spacing 5.32-6.29 Å, which is normally obtained for
6FDA based PI.
5 10 15 20 25 30 35 40
2',6'DMPDAB-6FDA
2',6'DMPDAB-ODPA
4'tBPDAB-6FDA
4'MPDAB-6FDA
4'MPDAB-ODPA
2'MPDAB-6FDA
2'MPDAB-ODPA
Inte
nsit
y (
a.u.
)
2
4'tBPDAB-ODPA
Figure 3.14 WAXD spectra of polyimides based on alkyl substituted phenoxy diamines

Chapter 3 Polymer synthesis and their characterization
106
3.6 Conclusions
1. A diamine containing pendant phenoxy group, 1-phenoxy-2,4-diaminobenzene
(PDAB) and polyimides based on this diamine with PMDA, ODPA, 6FDA, BTDA
and BPDA were synthesized and characterized. These PI showed good solvent
solubility in common organic solvents. The low viscosity of the homopolyimides can
be attributed to the bulky phenoxy group substitution at ortho position of the diamine,
which reduced the reactivity of one amino group due to steric hindrance. Thermal
properties of all the PIs are high as evidenced by their high Tg (> 248 C) and high
IDT (> 500 C). PDAB-6FDA showed high d-spacing, high density, high Tg but low
IDT.
2. Novel side chain type sulfonated polyimides (SPIs) based on a new diamine, 4'-
sulfonic,1-phenoxy-2,4-diaminobenzene (SPDAB) with dianhydrides viz., NTDA and
ODPA were synthesized and characterized. Imidazole as a base catalyses
polymerization of NTDA with PDAB with high viscosity in short period compared
with TEA as a base. SPIs, both in their salt form (SPDAB•TA-N and SPDAB•Im-N)
and sulfonic acid form (SPDABI-N and SPDABII-N), exhibited good solvent
solubility in polar aprotic solvents like DMF, DMAc, NMP and DMSO. The critical
solution temperature (CST) phenomenon is observed for SPDAB•Im-N in DMAc.
The low decomposition temperature in the range 270-350 °C of these polyimides
shows their fair thermal stability. Among these SPIs, thermal stability for six
membered imide moieties decreased in the order SPDAB•Im-N > SPDABI-N ≈
SPDABII-N > SPDAB•TA-N. These polymers did not reveal any glass transition.
Being ionomer, SPIs shows increase in their reduced viscosity with lowering in
concentration. WAXS pattern of SPIs reveals their complete amorphous nature. Blend
membranes of SPI and PBI-I are brittle in nature. Whereas, SPI and PBI-BuI form
flexible blends below 50 wt% of SPI in blend composition. Miscibility of these
blends was confirmed by FT-IR analysis. Blend membranes in sulfonic acid form
[SPI(H)/PBI-BuI(a/b)] exhibit higher thermal stability (443-480 C) than that of in
salt form [SPI(TEA)/PBI-BuI, 258-327 C]. Blend membrane did not exhibit any
glass transition.

Chapter 3 Polymer synthesis and their characterization
107
3. Side chain type PIs with alkyl substituted pendant phenoxy moiety with two
dianhydrides, ODPA and 6FDA, were successfully prepared and characterized.
2',6'DMPDAB based PI shows higher polymerization rate. FT-IR spectra of these PIs
show their complete imidization. These PIs reveal good solvent solubility in common
organic solvents. Solubility parameter (δ) decreases with increase in alkyl content.
Alkyl substitution at ortho position of pendant phenoxy group increases Tg than at
para-position. 6FDA based PIs shows higher Tg as compared with ODPA based PIs.
These PIs exhibit high thermal stability with IDT > 473 °C. PIs based on
2',6'DMPDAB exhibits the highest intrinsic viscosity. All these PIs are amorphous in
nature and have good film forming property.

Chapter 4
Structure-gas permeability correlation in polyimides
4.1 Introduction
Polyimides have been well recognized as gas separation membrane material and
couple of reviews are available on their structure – permeation property relationship
(Tsujita, 2003; Ding, 2007; Xiao, 2009). Effects of variation in diamine moiety
(Langsam, 1993; Stern, 1993; Tanaka, 1995; Hirayama, 1996a; Li, 1997; Liu, 1999; Xu,
2003; Qiu, 2005 and 2006) as well as dianhydride moiety of polyimide on gas permeation
and related physical properties are reported (Stern, 1989; Tanaka, 1992a, 1992b and
1995; Coleman, 1994; Qiu, 2006 and 2007). The incorporation of
hexafluoroisopropylidene moiety in diamine (Stern, 1989; Tanaka, 1992a; Coleman,
1994) or dianhydride (Langsam, 1993; Stern, 1993; Hirayama, 1996a and 1996b; Xu,
2003) is known to improve permeation properties of resulting polyimide. It is known that
better benefits of incorporation of this moiety in dianhydride can be drawn than its
incorporation in diamine (Matsumoto, 1993b). This can be attributed to an enhanced
electron affinity in dianhydride because of more electron withdrawing nature of
-C(CF3)2- linkage. This results in greater reduction in CTC complex with an increase in
free volume and gas permeability in the PIs on dianhydrides having -C(CF3)2- linkage.
(Matsumoto, 1993b). A study on effect of various pendant groups on permeation
properties and other physical properties of resulting polyimide would be very useful
towards structural design of membrane material for gas separation.
The focus of present thesis is on polyimides obtained from diamines containing
oxyphenyl linkage as pendant group and their application as membrane material for PEM
and gas separation. Following section briefly discusses literature reports on gas
permeation properties of polyimides containing pendant groups. Some of the polymers
from present work which were evaluated as PEM materials were also investigated for
their CO2 and related gas permeability under dry and humidified gas conditions. Thus,
literature reports investigating CO2 gas permeation properties under humid atmosphere
are also reviewed at the end.

Chapter 4 Structure-gas permeability correlation in polyimides
109
4.1.1 Literature on PIs containing pendant groups: A brief review
One of the ways to develop processible polyimides (PIs) for targeted application
comprises substitution of bulky pedant groups on its backbone. Such substituent reduces
interchain interactions with increase in free volume necessitated for the penetration of
solvent molecules, leading to better solubility and thus, processability. Substitution of
phenyl group on PI backbone is well reported (Korshak, 1969; Imai, 1984; Akutsu, 1994;
Spiliopoulous, 1997 and 1998; Mikroyannidis, 1999a and 1999b; Xu, 2003; Morikawa,
2005 and 2006; Qiu, 2005 and 2007; Ghaemy, 2009). Increasing the Tg along with
attainment of solubility in PIs, can be achieved if such embodied group is able to
decrease the segmental mobility (Takekoshi, 1990; Liu, 1999; Morikawa, 2005 and 2006;
Qiu, 2006). This phenomenon was attributed to the decrease in rotational flexibility
around the diphenyl ether unit due to the embodied phenyl substituents (Morikawa 2005
and 2006). Introduction of bulky groups at ortho position of the imide nitrogen can
restrict the rotation around C-N bonds of the two neighboring phenyl rings in PIs and
thus is known to decrease the intra-segmental mobility, which leads to increase in chain
rigidity and Tg (Takekoshi, 1990; Liu, 1999; Hsiao, 1998b). This phenomenon can be
beneficial in improving gas permeability of polyimides while retaining permselectivity
(Qiu, 2006).
Efforts encompassing bulky group substitutions in PIs to increase chain rigidity
with better processability are known to be beneficial towards improving gas permeation
properties of polyimides. If the bulky substituent is phenyl or aromatic ring, it also offers
sites for electrophilic substitution. Tanaka et al. (1992b) substituted a number of methyl
groups at various ortho positions in mPDA and pPDA and observed an increase in FFV
with number of methyl groups. Gas permeability increased due to decreased activation
energy for diffusion with increasing FFV. Li et al. (1997) also observed increase in FFV
and gas permeability for ODPA based PIs with number of methyl groups being located at
ortho positions in mPDA. The Tg of these PIs was increased due to the restricted local
mobility of chain segments arisen from ortho substitution of methyl groups to an imide
linkage.

Chapter 4 Structure-gas permeability correlation in polyimides
110
Liu et al. (1999) substituted three –CH3 groups on the phenyl ring of m-PDA
(3MPDA) and four –CH3 groups on the phenyl ring of p-PDA (4MPDA). The gas
permeability of resulting PIs based on BTDA and 6FDA was investigated. PIs based on
4MPDA revealed higher FFV and gas permeability than that of 3MPDA analogs. 6FDA
based PIs could be placed above the upper bound relationships for O2/N2. Qiu et al.
(2006) reported PIs with isomeric substitution of bulkier -CF3 group at ortho and meta to
the amine moieties of 1,4-bis(4-aminophenoxy)benzene and observed almost same
permselectivity with improved permeability as well as higher Tg for ortho isomers. This
indicated the effectiveness of ortho substitution at imide nitrogen in PIs for improvement
of permeability while retaining gas permselectivity.
Incorporation of bulky groups at ortho position to an imide linkage restricts the
free rotation around C-N bond, leading to stiffening of polymer backbone and increase in
Tg. This strategy can be helpful, since with increase in permeability, the stiffened polymer
backbone can resist the decrease in selectivity to some extent. Similar approach has been
proved in 6FDA based PIs, wherein the presence of bulky -C(CF3)2- in main chain
increases permeability with appreciable selectivity (Coleman, 1990; Matsumoto, 1993b;
Chung, 2001 ).
4.1.2 Gas permeation in aromatic polymers with methyl and t-butyl
group as substituent
In the present work, selection of alkyl group (methyl and t-butyl) as a substituent
on the aromatic ring of oxyphenyl linkage of diamine monomer is based on promises of
these groups in elevating gas permeation properties in various family of polymers as
briefly discussed below.
Attractive gas permeation properties by methyl group substitution are
demonstrated in polysulfone (McHattie, 1991a and 1992), polycarbonates (Murugandam,
1987; Zolandz, 1992; Koros, 1993), polyarylates (Chen, 1993), polyimides (Yamamoto,
1990; Li, 1997), etc. In polyarylate, polysulfone and polycarbonate, the methyl group
substitution on one of their monomer, i.e. bisphenol-A resulted in general increase in
permeability with marginal or no reduction in selectivity of different gas pairs. The
methyl group substitution on acid moiety of polyarylate (Pixton, 1995) also led to

Chapter 4 Structure-gas permeability correlation in polyimides
111
increase in permeability with marginal reduction in selectivity. In case of polyimide, as
stated above, methyl group substitution on m-PDA resulted in increase in permeability
and decrease in selectivity than polyimide based on unsubstituted m-PDA, obtained from
same dianhydride (Yamamoto, 1990; Li, 1997). The substitution by t-butyl group on the
monomer level generally increase permeability of the resulting polymer, coupled with
lower selectivity, in comparison to the polymer based on unsubstituted monomer. This
was exemplified by t-butyl group substitution in acid moiety of polyarylate, which led to
3-6.5 times increase in permeability and 6-25% decrease in selectivity (Pixton, 1995).
Bhole et al. (2007) reported that gas permeation properties of polyarylate containing t-
butyl group appear near the Robeson upper bond curve. Substitution of t-butyl group on
acid moiety of polybenzimidazole led to 10-40 times increase in gas permeability,
compared to PBI based on unsubstituted isophthalic acid (Kumbharkar, 2006).
4.1.3 Effect of moisture on CO2 permeation properties
Various hydrophilic polymers, especially ion-exchange membrane have proved to
be selective towards CO2 (Pellegrino, 1988 and 1995; Quinn 1997a and 1997b). These
membranes are known to be plasticized by H2O and show increase in permeation. The
permselectivity is increased due to the selective transport of polar gases like CO2, H2S
over non-polar gases like H2, N2 and CH4. Current CO2-selective membrane transports
CO2 either by solution-diffusion mechanism or by facilitated transport mechanism. The
latter mechanism is known to reveal high permeability with high permselectivity (Zhang,
2002, Zou, 2006).
Reports on enhanced permeability of CO2 with increase in the water content are
known (Quinn 1997a and 1997b; Zou, 2006). Matsuyama et al. (1996) reported the
facilitated transport based separation of CO2 in a cation exchange membrane, containing
protonated amine as a carrier for CO2. Increase in water content decreased the
crystallinity and energy of diffusion of the gases. Increased diffusivity of the gases due to
the enhanced flexibility or plasticization of the membrane is observed by Kim et al.
(2004). Okamoto et al. (1996) reported the increase in polarity of the membrane with
humidification and observed the enhanced solubility selectivity of the polar CO2 over
non-polar N2. Increase in water content in the membrane or having functional group

Chapter 4 Structure-gas permeability correlation in polyimides
112
results in the increased volume fraction of the ionic network, which significantly
increases gas permeability (Pellegrino, 1988).
Present chapter deals with investigation of gas permeation properties in selected
polyimides based on m-phenylene diamine possessing oxyphenyl pendant group.
Initially, gas permeability in PIs based on PDAB (1-phenoxy-2,4-diamono benzene) and
select dianhydrides (ODPA and 6FDA) were investigated. Encouraged by promising
results of these polymers, the phenyl ring belonging to oxyphenyl moiety of PDAB was
substituted by selected alkyl groups that are bulky in nature and polymerized with two
dianhydrides (ODPA and 6FDA). Effects of bulk and position of substitution of alkyl
group in oxyphenyl ring of diamine and dianhydride used for the polymer preparation on
physical and gas permeation properties of resulting PIs is discussed. Some of the
polymers possessing polar pendant groups (sulfonated PIs) in their backbone and blends
of PI with PBI were also investigated for their gas permeability.
4.2 Experimental section
4.2.1 PI Synthesis and membrane preparation
Synthesis of diamines with desired structural variations aiming at application of
resulting PIs for gas permeation is already described in Chapter 3. Diamines synthesized
for this purpose were PDAB, 2′MPDAB, 4′MPDAB, 2′,6′DMPDAB and 4′tBPDAB.
Polymerization of these diamines with ODPA and 6FDA was performed (Section 3.3.1.1
and 3.3.3.1). Sulfonated polyimides based on SPDAB with dianhydrides ODPA and
NTDA were also investigated for gas permeation under dry as well as humidified gas
conditions. Blends based on SPI and PBI-BuI were also investigated for permeation of
H2, O2 and CO2, in view of their importance as polymer electrolyte membrane for fuel
cell (PEMFC). Synthesis, purification, characterization of physical properties and
membrane preparation in dense film form based on these polymers is already discussed in
Chapter 3.
Details of various polyimides used for gas permeation analysis and their
abbreviation are given below in the Table 4.1.

Chapter 4 Structure-gas permeability correlation in polyimides
113
Table 4.1 Polymers investigated for gas permeation
Polyimide Sr. No.
Diamine Dianhydride Abbreviation used
1 PDAB ODPA PDAB-ODPA
2 PDAB 6FDA PDAB-6FDA
3 2′MPDAB ODPA 2′MPDAB-ODPA
4 2′MPDAB 6FDA 2′MPDAB-6FDA
5 4′MPDAB ODPA 4′MPDAB-ODPA
6 4′MPDAB 6FDA 4′MPDAB-6FDA
7 2′,6′DMPDAB ODPA 2′,6′DMPDAB-ODPA
8 2′,6′DMPDAB 6FDA 2′,6′DMPDAB-6FDA
9 4′tBPDAB ODPA 4′tBPDAB-ODPA
10 4′tBPDAB 6FDA 4′tBPDAB-6FDA
11 SPDAB (as triethylammonium
sulfonate) NTDA SPDAB•TA-N
12 SPDAB (as imidazolium
sulfonate) NTDA SPDAB•Im-N
13 SPDAB in –SO3H form NTDA SPDABI-N
14 SPDAB (as triethylammonium
sulfonate) ODPA SPDAB•TA-O
Other polymers investigated
15 Polybenzimidazole based on 5-tert-butyl
isophthalic acid (BuI) PBI-BuI
16 Blends of SPI (–SO3H form) and PBI in ‘a’ and
‘b’ weight percent, respectively SPI(H)/PBI-BuI(a/b)

Chapter 4 Structure-gas permeability correlation in polyimides
114
4.2.2 Measurement of gas permeability
Gas permeability of dense membranes was measured using pure gases at upstream
gas pressure of 10 atm and at 35 C using variable volume method (Stern, 1963); while
maintaining permeate side at the ambient pressure. A schematic representation of
permeation equipment is shown in Figure 4.1. Design of the permeation cell, component
specifications, material of construction, etc. are discussed elsewhere (Houde, 1991). One
end of the feed side of the cell was connected through valve V1 to feed gas cylinder outlet
and a pressure gauge (0-550 psi range). The valve V2 was used to control the feed
pressure. On the permeate side of the permeation cell, a calibrated borosilicate glass
capillary containing a small mercury slug (~ 4-6 mm in length) was connected. The
membrane cell assembly was kept in a water bath maintained at 35 + 0.1 C. Pure gases,
viz., He, H2, Ar, N2, O2 with 99.9% purity were obtained from Inox Air Products Ltd.,
India, while CH4 and CO2, with purity of 99.995 % were obtained from Air liquid, USA.
Displacement of the mercury slug was monitored against time using cathetometer. The
permeability (P) of a gas was calculated using the following equation
ptA76
.C.Fd7.14P
where,
d = distance traveled by the mercury slug (cm),
F.C. = flow meter constant (volume of the gas per unit length (cm3/cm)),
= thickness of the membrane (cm),
A = effective membrane area (cm2),
t = time (sec) and
∆p = pressure difference across the membrane (psi).
The permeation measurements were repeated with at least 3 different membrane
samples prepared under identical conditions and the data averaged. Variation in the
permeability measurement was upto 15% for different gases studied. The variation was
dependant on type of the gas. It was lesser for lighter gas like He and was higher for

Chapter 4 Structure-gas permeability correlation in polyimides
115
heavier gas like N2 and CH4. The unit of permeability (P) for different gases is expressed
in Barrer, where, 1 Barrer = 10-10 cm3 (STP) cm/ cm2 s cm Hg. The ideal selectivity (α =
P1/P2) of various gas pairs was calculated by taking a ratio of pure gas permeability.
Figure 4.1 Gas permeation Equipment
Gas permeability in some of the polymers of interest (sulfonated polymers) was
also studied under humidified conditions. In these cases, the feed gas was passed through
a chamber filled with water, prior to its entry in the permeation cell. The membranes used
for such study were initially humidified by immersing in deionised water for a day,
before mounting in the permeation cell.
4.3 Results and discussion
Gas permeation properties of PDAB based polyimides are discussed initially as
the base (unsubstituted O-Ph) case. This is followed by the gas permeability
investigations of PIs containing O-Ph, substituted with alkyl groups. At the end, gas
permeation properties of sulfonated polyimides and their blends with PBI-BuI are

Chapter 4 Structure-gas permeability correlation in polyimides
116
discussed. Physical properties of these polyimides though are given in Chapter 3, some
of these (those are known to affect gas permeation) are reproduced in this chapter for
convenience.
4.3.1 Assessing capability of O-Ph group incorporation: Gas permeability of PIs
based on 1-phenoxy-2,4-diamino benzene (PDAB)
To investigate the effect of side chain phenoxy group substituted at the 4-position
in PDAB, gas permeation properties of two polyimides of PDAB with ODPA and 6FDA
were investigated at 35C and 10 atm upstream pressure. Dense membranes of these
polymers of thickness ~ 40 m prepared by solution casting method were used. Though,
both the polymers, viz., PDAB-ODPA and PDAB-6FDA were soluble in chloroform,
TCE was used as the solvent for casting PDAB-ODPA membrane; since this membrane
prepared with chloroform as the solvent exhibited wavy surface. The TCE casted
membrane was transparent and tough in nature and was able to withstand applied gas
pressure in the permeation cell. Dense membrane of PDAB-6FDA was casted using
chloroform as the solvent. The physical properties of these polymers are reproduced in
Table 4.2, while gas permeability and selectivity of various gas pairs in these polyimide
membranes is given in Table 4.3.
Table 4.2 Physical properties of PIs based on PDAB
Property PDAB-ODPA PDAB-6FDA
(g/cm3 ) 1.354 1.416
dsp (Å) 4.23 5.82
FFV 0.1425 0.1728

Chapter 4 Structure-gas permeability correlation in polyimides
117
Table 4.3 Gas permeability (P) and selectivity of PIs based on PDAB
Property PDAB-ODPA PDAB-6FDA
P(He) 6.9 58
P(H2) 6.1 53
P(O2) 0.3 5.8
P(Ar) 0.12 1.96
P(N2) 0.045 1.44
P(CH4) 0.017 0.44
P(CO2) 1.26 16.8
(He/N2) 164 41
(He/CH4) 406 131
(O2/N2) 7 4.1
(CO2/N2) 30 12
(CO2/CH4) 74 37
It was observed that the permeability coefficient (P) of 6FDA based polyimide for
gases studied were considerably higher than that of ODPA based polyimide. The
fractional free volume as well as d-spacing as given in Table 4.2 of 6FDA based
polyimide was higher than that of ODPA based polyimide; which led to higher
permeability of all gases in PDAB-6FDA. Such increase in permeability as a result of
lowering in packing density is known in case of polyimides containing -C(CF3)2- group
(Stern, 1989). Incorporation of this group restricts intra-segmental mobility of polymer
chain resulting in increase chain stiffness as evidenced by the higher Tg of 6FDA based
polyimide, PDAB-6FDA, as compared to that of ODPA based polyimide, PDAB-ODPA.

Chapter 4 Structure-gas permeability correlation in polyimides
118
If permeability difference for individual gases in PDAB-ODPA and PDAB-6FDA is
considered, the permeability in PDAB-6FDA was higher for bigger molecules like N2
and CH4, than that of smaller molecules like He and H2. This phenomenon can be
attributed to the availability of the free volume for diffusion and the kinetic diameter of
the penetrants (Stern, 1989; Koros, 1993). In other words, effect of incorporation of
hexafluoroisopropylidene group in increasing permeability coefficient was less
significant for smaller gases like He and H2, than for larger gases like N2 and CH4; in
view of the fact that smaller gases can diffuse easily through the interstitial spaces (Xu,
2003). PDAB-ODPA exhibited excellent selectivity for various gas pairs. The potential
for incorporation of pendant phenoxy linkage in polyimide was depicted by the O2/N2
selectivity of 7, high He based selectivity combined with high He or H2 permeability and
CO2/CH4 selectivity of 74. Observed high permeability of He and H2 in PDAB-6FDA (58
and 53 barrer, respectively), coupled with appreciable selectivity of He/CH4 (131) and
CO2/CH4 (37) in PDAB-6FDA depicted usefulness of the phenoxy group substitution in
improving permeation properties.
It could be worth comparing permeation properties of present polyimides with
that of reported in the literature, exhibiting structural similarities. The gas permeability of
present polyimides was higher than that of respective polyimides based on an
unsubstituted m-phenylenediamine (m-PDA) with either ODPA (Li, 1997) or 6FDA
(Yamamoto, 1990). This is attributable to the introduction of pendant phenoxy group in
diamine moiety of present polyimides. This increase in permeability however was found
to be lower than that obtained by the introduction of methyl group at the same position
(Li, 1997). This could be ascribed to the flat nature of phenyl ring, which is known to
reduce permeability and increase selectivity in other families of polymers like polyarylate
(Houde, 1995), as compared to their methyl substituted analogs. It is interesting to note
that the presence of flexible -O- linkage of phenoxy group in the PDAB did not adversely
affect the selectivity performance in comparison to polyimides based on methyl
substituted m-PDA and respective dianhydride. Owing to such promises, it was thought
to substitute phenoxy linkage by alkyl groups like methyl and t-butyl at appropriate

Chapter 4 Structure-gas permeability correlation in polyimides
119
positions and to investigate gas permeability of resulting polyimides, as elaborated in the
following section.
4.3.2 Substitution of alkyl group on O-Ph of diamine moiety
It was thought to employ the capability of O-Ph group of PDAB towards
accommodation of alkyl groups (methyl and t-butyl) by substitution at ortho and / or para
positions to phenoxy linkage for altering gas permeation properties of resulting
polyimides. Thus, four novel diamines, viz., 2′MPDAB, 4′MPDAB, 2′,6′DMPDAB and
4′tBPDAB were synthesized and polymerized with two selected dianhydrides, ODPA and
6FDA. These two dianhydrides were kept same as used for the polymerization with
PDAB, so that the effect of substitution of alkyl group on gas permeation properties can
be investigated.
4.3.2.1 Chain packing density and rigidity
The preparation of polyimides using alkyl substituted phenoxy diamines and two
dianhydrides, ODPA and 6FDA is already described in Chapter 3. These polymers were also
evaluated for physical properties those are known to affect gas permeation. These are
reproduced in following Table 4.4 for ease. It could be seen that dsp and FFV, which are
representative of openness of polymer matrix are higher for 6FDA based PI than ODPA
based PI based on the same diamine. Similar property variations were observed for density
and Tg. Increase in Tg in 6FDA based PIs can be attributed to the bulky nature of -C(CF3)2-
linkage, restricting the segmental mobility of the polymer main chain (Stern, 1989). It has
been reported that density of halogenated polymer becomes higher than that of non-
halogenated polymers such as polycarbonate (Hellums, 1989), polyarylate (Kharul, 1994),
polysulfone (McHattie, 1992), etc. 6FDA based PIs shows lower values of solubility
parameter () than that of ODPA based PIs for the same analogous diamine, indicating the
reduced molecular interaction and more free volume in the former than that of latter.
Moreover, the bulky nature of -C(CF3)2- linkage and the steric effect induced by the -CF3
group would disturb the efficient packing of the polymer chains leading to increase in FFV.
In this way, for the polyimides based on same diamine, 6FDA based PIs shows higher dsp,
FFV and Tg with lower values of as shown in Table 4.4.

Chapter 4 Structure-gas permeability correlation in polyimides
120
Table 4.4 Physical properties of polyimides based on diamines possessing substituted o-
phenoxy side chain
Polymer dsp
(Å) FFV
(cal.cm-3)1/2
Tg
(C)
PDAB-ODPA 4.23 0.1425 12.06 256
PDAB-6FDA 5.82 0.1728 11.03 281
2'MPDAB-ODPA 4.23 0.0998 12.22 282
2'MPDAB-6FDA 5.70, 3.38 0.168 10.97 289
4'MPDAB-ODPA 4.53 0.0985 12.20 267
4'MPDAB-6FDA 6.08, 3.62 0.167 10.97 278
2',6'DMPDAB-ODPA 4.36 0.102 12.04 311
2',6'DMPDAB-6FDA 6.29, 3.46 0.155 10.87 313
4'tBPDAB-ODPA 4.18 0.112 11.69 270
4'tBPDAB-6FDA 5.32, 3.52 0.178 10.57 281
The comparison of packing density parameters in a series of ODPA based
polymers and different diamines indicates that methyl substitution lowered the packing
density in comparison to unsubstituted PDAB-ODPA, irrespective of its position of
substitution, either ortho- or para- to the phenoxy linkage. Though 4'MPDAB-ODPA
exhibited slightly higher dsp than that of 2'MPDAB-ODPA, both of them exhibited
similar FFV. It is known that for similar kinds of substitution, d-spacing obtained by
WAXD analysis may not necessarily show the same variation as that of fractional free
volume. For example, Wilks et al. (2006) reported the increasing d-spacing in a series of
polynorbornene (PNBs) with increasing alkyl side chain lengths such as methyl, butyl
and hexyl groups. However FFV was found to be decreased with increasing alkyl side

Chapter 4 Structure-gas permeability correlation in polyimides
121
chain lengths of PNBs. This anomalous trend in FFV was attributed to the filling up of
the increased spaces with increasing side chain length. Pinnau et al. (2004) also observed
decrease in FFV for poly(4-methyl-1-pentyne) with increasing the side chain length by
either one or two methyl groups. Substitution of two methyl groups at 2 and 6 position of
phenoxy linkage in 2',6'DMPDAB-ODPA did not loosen chain packing further as that of
methyl substitution cases. This could be attributed to accommodation of second methyl
group in the available free space, as also observed previously (Matsumoto, 1993b). Such
accommodation of bulk of a small methyl group in available free space is also known in
case of dimethylbisphenol-A based polysulfone (McHattie, 1991), polycarbonate
(Schmidhauser, 1990) and polyarylate (Pixton, 1995; Bhole, 2003).
In case of 6FDA based polymers obtained from different alkyl substituted
diamines, the effect of substitution on chain packing density can be better viewed in
terms of FFV values of these polyimides, rather than their d-spacing. Their WAXD
spectra though showed amorphous nature, they exhibited multiple peaks indicating at
least two types of chain packing arrangement (Figure 3.14 in Chapter 3). Such multiple
peaks were observed previously for polymers containing halogen, (Pixton, 1995;
Langsam, 1993) or polymers possessing pendant phenyl ring, as observed in cases
polystyrene (Mitchell, 1984) and poly(2,6-diphenyl-1,4-phenylene oxide) (Wrasidlo,
1971). In these polymers, the higher dsp value corresponds to the average chain spacing
while the lower dsp value corresponds to the loose “stacks” of pendant phenyl rings on
adjacent polymer chains (Pixton, 1995). In case of 6FDA based polyimide, occurrence of
two d-spacing values was assigned to the probable helix structure (Matsumoto et al.,
1993c).
A close look at FFV of present polyimides based on 6FDA showed a general
decrease in FFV after the methyl substitution in case of 2'MPDAB-6FDA, 4'MPDAB-
6FDA and 2'6'MPDAB-6FDA in comparison to the parent case of PDAB-6FDA. This is
indicative of the fact that in case of methyl substitution, the bulk of the added methyl
group is accommodated in the available space created by inherent nature of 6FDA;
leading to a reduction in available FFV. Such decrease in FFV of polymer with addition
of small alkyl group like methyl has already been discussed. On the other hand, the FFV
of 4'tBPDAB-6FDA is slightly higher than the parent case of PDAB-6FDA. Thus, bulk

Chapter 4 Structure-gas permeability correlation in polyimides
122
of the added t-butyl group could further enhance the FFV, though to a small extent. FFV
enhancement by t-butyl group in ODPA based PI (in comparison to base case of PDAB-
ODPA) was much higher than in case of FDA based PI. This is indicative of the fact that
6FDA is responsible in a major way for governing the chain packing density than the
diamine counterpart; while in case of ODPA based polyimides, diamine part was
predominantly responsible for governing chain packing density.
The chain rigidity as evident from the glass transition temperature indicated that in
case of ODPA based polyimides, alkyl substituent led to considerable increase in the Tg in
comparison to the base case of PDAB-ODPA. This increase was by 11-55 °C by methyl
and t-butyl substituents at various positions. On the other hand, comparison in case of
6FDA based PIs indicated that Tg was marginally changed by alkyl substituent irrespective
of its nature or position. This could be because 6FDA by itself made polymer chains
enough rigid, (as evident from increase in Tg of PDAB-ODPA from 256 °C to 281 °C for
PDAB-6FDA); so that the addition of alkyl group had little effect in enhancing chain
rigidity further.
4.3.2.2 Permeability and selectivity
The gas permeability and selectivity results of polyimides having alkyl
substitution at the pendant phenoxy moiety are summarized in Table 4.5 and Table 4.6,
respectively. In case of a particular diamine, PI based 6FDA shows higher permeability
than its ODPA based counterpart. Though, the variation in permeability seems to be
dependant upon the structure of dianhydride and diamine, position, bulk and number of
alkyl substituent on the pendant phenoxy group also play a role, as explained in the
following sections.

Chapter 4 Structure-gas permeability correlation in polyimides
123
Table 4.5 Gas permeability (P) of polyimides based on alkyl substituted phenoxy group in meta-PDA
Gas
Permeability
2'MPDAB
-ODPA
4'MPDAB
-ODPA
2',6'DMPDAB
-ODPA
4'tBPDAB
-ODPA
2'MPDAB
-6FDA
4'MPDAB
-6FDA
2',6'DMPDAB
-6FDA
4'tBPDAB
-6FDA
P(He) 5.4 8.8 11.8 15.8 40.1 46.1 77.0 78.0
P(H2) 5.3 7.5 10.0 16.7 35.7 43.8 81.5 78.0
P(Ar) 0.11 0.17 0.34 0.53 1.04 1.52 3.8 3.7
P(N2) 0.077 0.12 0.21 0.32 0.77 1.15 2.8 2.5
P(O2) 0.32 0.53 0.85 1.32 3.47 4.25 10.9 10.8
P(CH4) 0.028 0.047 0.13 0.17 0.26 0.53 1.01 1.3
P(CO2) 1.06 2.3 3.6 5.0 12.8 14.6 38.0 36.7

Chapter 4 Structure-gas permeability correlation in polyimides
124
Table 4.6 Gas permselectivity () of polyimides based on alkyl substituted phenoxy group in meta-PDA
Gas
Permeability
2'MPDAB
- ODPA
4'MPDAB
-ODPA
2',6'DMPDAB
-ODPA
4'tBPDAB
-ODPA
2'MPDAB
-6FDA
4'MPDAB
-6FDA
2',6'DMPDAB
-6FDA
4'tBPDAB
-6FDA
(He/H2) 1.02 1.18 1.18 0.95 1.12 1.05 0.95 1.0
(He/N2) 69.5 74.0 56.0 49.4 52.0 40.0 28.0 31.4
(He/CH4) 190.0 185.0 89.0 93.0 152.0 86.0 76.0 61.0
(H2/CH4) 187.0 158.0 75.0 98.0 135.0 82.0 80.0 61.0
(Ar/N2) 1.43 1.42 1.62 1.66 1.35 1.32 1.36 1.48
(O2/N2) 4.1 4.5 4.0 4.1 4.5 3.7 4.0 4.35
(CO2/N2) 13.7 19.6 17.0 15.8 16.5 12.7 13.7 14.8
(CO2/CH4) 37.5 49.0 27.0 30.0 48.3 27.3 37.5 28.7
(N2/CH4) 2.7 2.5 1.6 1.9 2.9 2.2 2.7 2.0

Chapter 4 Structure-gas permeability correlation in polyimides
125
4.3.2.3 Effect of dianhydride
PI based on 6FDA showed higher permeability (4.6 to 13.3 times) and lower
selectivity (0.04 to 0.53 times) than its ODPA based counterpart for the same analogous
diamines (Table 4.5 and Table 4.6). It has been well reported that 6FDA based PIs show
higher permeability and lower selectivity than that of other dianhydrides such as ODPA,
BTDA, PMDA, BPDA etc. with similar diamine structure (Koros, 1988; Tanaka, 1992;
Stern, 1993; Matsumoto, 1993b; Hirayama, 1996a; Li, 1996; Wang 2005; Ding 2007).
The major focus of this work is on the alkyl substitution on dianhydride moiety of PI.
Effects of dianhydride could also be elaborated along with effect of alkyl substituent, as
given below.
4.3.2.4 Effect of alkyl substituent
In case of ODPA based polyimides with alkyl substituent, the permeability
increases in an order: 2'MPDAB-ODPA < 4'MPDAB-ODPA < 2',6'DMPDAB-ODPA <
4'tBPDAB-ODPA (Table 4.5). It is observed that substitution of alkyl group on the
pendant phenoxy moiety of PI based on ODPA led to increase in permeability, except in
case of 2'MPDAB-ODPA. In this case; permeability of O2, N2 and CH4 increased while
that of He, H2, Ar and CO2 was decreased compared to base case of PDAB-ODPA.
4'MPDAB-ODPA showed increase in permeability compared to PDAB-ODPA and
2'MPDAB-ODPA. This shows that –CH3 group substitution on pendant phenoxy group at
para position is more effective in increasing the permeability than that of substitution of
this group at ortho position. There is an increase in permeability for 4'MPDAB-ODPA by
1.42 to 2.17 times and 1.23 to 2.78 times with respect to 2'MPDAB-ODPA and PDAB-
ODPA, respectively. In spite of the increase in permeability, selectivity of various gas
pairs for both the methyl group substituted PIs are almost similar (except a small
lowering in (H2/CH4) and a increase in CO2 based selectivities for para substituted PI).
With substitutions of two –CH3 groups at ortho positions of pendant phenoxy group in
2',6'DMPDAB-ODPA, the permeability further increased by 1.89 to 4.60 times and 1.33
to 2.77 times than that of 2'MPDAB-ODPA and 4'MPDAB-ODPA, respectively. This
led to a large decrease in He and H2 based selectivities. Comparatively, CO2 based
selectivities were lowered to a smaller extent.

Chapter 4 Structure-gas permeability correlation in polyimides
126
Among the alkyl substituted PIs, incorporation of bulky t-butyl group on the
pendant phenoxy group (4'tBPDAB-ODPA) showed increase in permeability by 1.31 to
6.01 times than that of the base case. Though 4'tBPDAB-ODPA exhibited lowest d-
spacing (4.18 Ǻ), it revealed the highest FFV (0.112) with lowest value of solubility
parameter (11.69) (Table 4.4). The later parameter is an indicative of the reduced
interchain interactions and decrease in packing density, accounting for its higher
permeability. Considering the voluminous size of t-butyl group (73.6 Ǻ3) than that of
methyl (22.7 Ǻ3) (Langsam, 1993), the decreased interchain packing in the resultant PIs
by the embodiment of former group would be more than that by the later. Increased
permeability by incorporation of bulky t-butyl group in PIs (Kim, 2002 and 2005) and
polyarylates (Pixton, 1995; Bhole, 2007) are known.
An interesting result is seen between 4'tBPDAB-ODPA and 2',6'DMPDAB-
ODPA (Table 4.5 and 4.6). Former exhibits better selectivity performance despite its
increase in permeability by a factor of 1.31 to 1.67 than that of latter. 4'tBPDAB-ODPA
also exhibits highest Ar/N2 selectivity (1.66) and its O2/N2 selectivity (4.1) is close as
compared to other ODPA based PIs having alkyl substituent.
In case of 6FDA based polyimides, the permeability increases in an order
2'MPDAB-6FDA < 4'MPDAB-6FDA < 2',6'DMPDAB-6FDA ≈ 4'tBPDAB-6FDA.
Substitution of a single –CH3 group at either ortho or para (2'MPDAB-6FDA and
4'MPDAB-6FDA) on pendant phenoxy group demonstrated decrease in permeability and
increase in selectivity of different gas pairs than that of the unsubstituted analogue
(PDAB-6FDA); except for the increase in CH4 permeability in 4'MPDAB-6FDA. On the
other hand, such –CH3 group substitution at para position in case of ODPA based PIs
(4'MPDAB-ODPA) led to increase in permeability significantly (Table 4.5). In other
words, dianhydride was predominantly responsible for governing permeation. This can be
attributed to the voluminous –C(CF3)2– linkage existing in 6FDA based PIs, which by
itself causes the looser intersegmental packing. An incorporation of a –CH3 group on the
phenoxy group in diamine can be accommodated in the large free volume created by –
C(CF3)2– linkage. In other words, a single –CH3 group substitution in 6FDA based PIs
pays no role to disruption of chain packing; while there is an effective disruption in chain
packing by –CH3 group substitution (particularly at para position) in ODPA based PIs. A

Chapter 4 Structure-gas permeability correlation in polyimides
127
decrease in permeability by –CH3 group in PIs substitution has been observed by
Matsumoto et al. (1993b). Similar effect of decrease in permeability by –CH3 group
substitution in polyarylates (Pixton, 1995; Kharul 1998; Bhole, 2003), polysulfone
(McHattie; 1991) and polycarbonate (Schmidhauser, 1990) was also noted.
2'MPDAB-6FDA exhibits better selectivity for all the gas pairs, except Ar/N2
(Table 4.6). This is in good agreement with the general trend of polymers exhibiting
lower permeability and higher selectivity. With an embodiment of second –CH3 group at
ortho position on pendant phenoxy group (2',6'DMPDAB-6FDA) permeability further
increases. However, permeability by incorporating t-butyl group in 4'tBPDAB-6FDA is
almost same as that of 2',6'DMPDAB-6FDA, despite their large difference in
intrasegmental mobility (difference in Tg). 2',6'DMPDAB-6FDA can be considered as the
most rigid PIs as evidenced by its lowest intrasegmental mobility (highest Tg: 313 °C)
since the two ortho substituted –CH3 groups on pendant phenoxy group should sterically
hinder the rotation of pendant phenoxy group. It has been observed in PIs that
modifications conferring increased stiffness and a decrease in packing density result in an
increase in permeability without loss of selectivity (Hoehn, 1980; Kim, 1988; Kulkarni,
1994; Pixton, 1995). In 4'tBPDAB-6FDA, voluminous t-butyl group being substituted at
para position not only increases the side chain length but also confers more rotational
freedom to the pendant phenoxy group. Such architecture should reduce the interchain
interactions significantly contributing to an increase in FFV and hence rendering higher
permeability. No considerable difference in selectivity between 2',6'DMPDAB-6FDA
and 4'tBPDAB-6FDA is observed (Table 4.6). It should be noticed that, 4'tBPDAB based
PIs exhibits highest permeability despite its lowest d-spacing values indicating that apart
from average intersegmental distance some sort of other scattering features has been
reflected by their X-ray spectra. Such phenomenon with lower d-spacing values and
higher permeability has also been observed in polyarylates having t-butyl group as
reported by Pixton et al. (1995). However, the lowest density and highest FFV exhibited
by 4'tBPDAB based PIs further support the higher distance between their chains
accounting their higher permeability.
Substituting alkyl group at the para-position as compared with at ortho-position
bestows some advantages for increasing the gas permeation. In both the cases, there is an

Chapter 4 Structure-gas permeability correlation in polyimides
128
increase in the volume of the resultant bulky side group with the substitution as shown in
the Figure 4.2 and Figure 4.3.
Figure 4.2 Lengthening of side chain due to substitution of alkyl groups at para position
The probability of reduction of interchain interaction due to the lengthening of the
side chain with an increase in free volume in para substitution case may result in lower
intermolecular hindrance to rotation of torsion mobility of main chain facilitating
intrasegmental mobility. This phenomenon can be evidenced by the lower Tg of the
ODPA based PIs with alkyl substituent at para-position than that of at ortho-position
(Table 4.4). This phenomenon leads to increase in permeability of various gases (Table
4.5). This effect of increasing the side chain length is more pronounced in ODPA based
PIs. In 6FDA based PIs, permeability increases in the order 4'MPDAB-6FDA < PDAB-
6FDA < 4'tBPDAB-6FDA indicating that added bulk of substituent in 4'MPDAB-6FDA
is accommodated in the free volume created by -C(CF3)2- linkage.
Form the Figure 4.3 below, it can be seen that the increase in volume of the side
chain substituent due to alkyl substitution at ortho position does not increase the chain
length (as in the case of para substituent) but increases the width of the side chain.
Figure 4.3 Widening of the side chain due to substitution of alkyl group at ortho position
Xu et al. (2003) reported that the relative rigid and longer side substituents impart
a higher level of ring torsion leading to increase in permeability. In the present case, alkyl
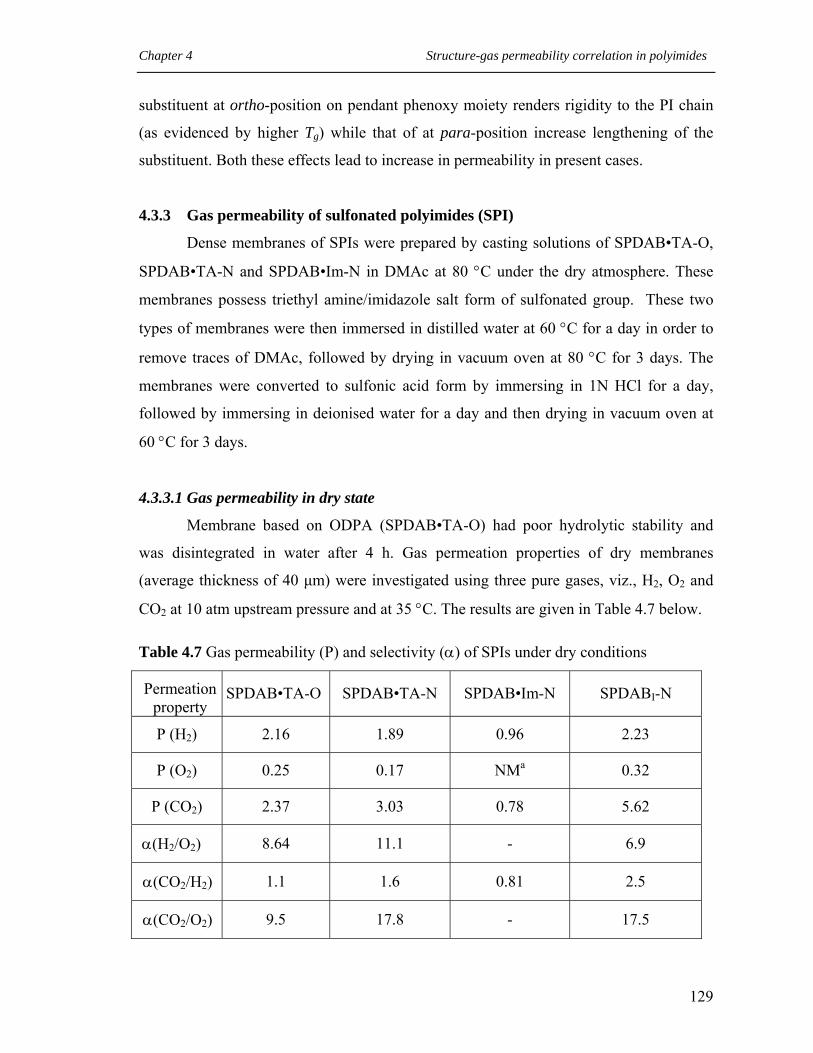
Chapter 4 Structure-gas permeability correlation in polyimides
129
substituent at ortho-position on pendant phenoxy moiety renders rigidity to the PI chain
(as evidenced by higher Tg) while that of at para-position increase lengthening of the
substituent. Both these effects lead to increase in permeability in present cases.
4.3.3 Gas permeability of sulfonated polyimides (SPI)
Dense membranes of SPIs were prepared by casting solutions of SPDAB•TA-O,
SPDAB•TA-N and SPDAB•Im-N in DMAc at 80 C under the dry atmosphere. These
membranes possess triethyl amine/imidazole salt form of sulfonated group. These two
types of membranes were then immersed in distilled water at 60 C for a day in order to
remove traces of DMAc, followed by drying in vacuum oven at 80 C for 3 days. The
membranes were converted to sulfonic acid form by immersing in 1N HCl for a day,
followed by immersing in deionised water for a day and then drying in vacuum oven at
60 C for 3 days.
4.3.3.1 Gas permeability in dry state
Membrane based on ODPA (SPDAB•TA-O) had poor hydrolytic stability and
was disintegrated in water after 4 h. Gas permeation properties of dry membranes
(average thickness of 40 μm) were investigated using three pure gases, viz., H2, O2 and
CO2 at 10 atm upstream pressure and at 35 C. The results are given in Table 4.7 below.
Table 4.7 Gas permeability (P) and selectivity () of SPIs under dry conditions
Permeation property
SPDAB•TA-O SPDAB•TA-N SPDAB•Im-N SPDABI-N
P (H2) 2.16 1.89 0.96 2.23
P (O2) 0.25 0.17 NMa 0.32
P (CO2) 2.37 3.03 0.78 5.62
(H2/O2) 8.64 11.1 - 6.9
(CO2/H2) 1.1 1.6 0.81 2.5
(CO2/O2) 9.5 17.8 - 17.5

Chapter 4 Structure-gas permeability correlation in polyimides
130
Permeability of H2 and O2 in 4,4′-Oxydiphthalic dianhydride (ODPA) based
polymer, SPDAB•TA-O was higher than that of naphthalene dicarboxylic dianhydride
based polymers (SPDAB•TA-N, SPDAB•Im-N). SPDAB•TA-O has higher structural
flexibility due to ether linkage in its dianhydride moiety, while naphthalene dianhydride
based polymer SPDAB•TA-N is anticipated to be rigid. This difference in rigidity could
be responsible for the observed difference in gas permeability. Between SPDAB•TA-N
and SPDAB•Im-N, the former showed higher permeation than that of the later. This can
be attributed to the peculiar planar structure of the imidazolium cation, rendering close
chain packing.
In SPDAB•TA-N, the salt form has tetrahedral geometry for three ethyl groups of
Et3N+–, vis-à-vis the flat nature of imidazolium cation (Im+) in SPDAB•Im-N. Flat or
planar structures are known to be accommodated in the available free volume of the
polymer as compared with tetrahedral structure like Et3N+–, resulting in decrease in
permeability. Such a phenomenon is explained earlier in Section 4.3.1. In the WAXD
spectra of SPDAB•TA-N and SPDAB•Im-N (Figure 3.8 of Chapter 3), the latter shows
amorphous peak maxima at higher angle (25.88°) than that of lower angle (24.32°) in the
former (Section 3.5.2.5 of Chapter 3). Though this difference is small, it is indicative of
the close-packed structure in SPDAB•Im-N. Flat structure and increase in aromatic
character due to the imidazolium cation in SPDAB•Im-N may also lead to the π-π
interaction between the imidazolium cation and other aromatic phenyl rings, resulting in
close packing. Such types of π-π interactions between imidazolium rings and other
aromatic structures are known (DuPont, 2004; Jiang, 2009).
CO2 permeability through these polymers varied in the order as SPDAB•Im-N <
SPDAB•TA-O < SPDAB•TA-N < SPDABI-N. It is interesting to note that all these
polymers (except SPDAB•Im-N) exhibit higher CO2 permeability than that of H2.
SPDABI-N exhibited the highest CO2/H2 selectivity among the series. Conversely, in
most of the glassy polymers, smaller gas, H2 usually shows higher permeability than that
of CO2. This dominance of penetrant size diffusivity in glassy state leads to CO2/H2
selectivity of < 1 (or H2/ CO2 selectivity of > 1). The observed behavior of higher
permeation for CO2 in present polymers reflects that this functionality (-SO3H or its salt)
leads to higher CO2 solubility, enhancing its permeation than that of smaller gas H2.
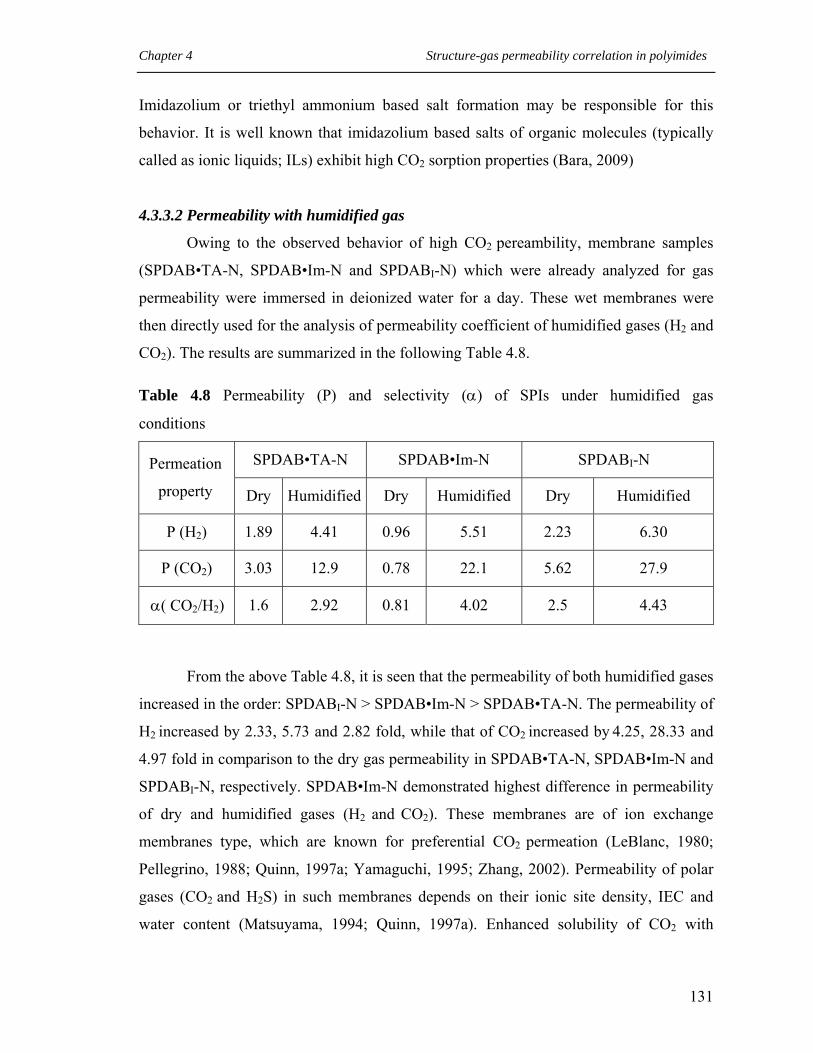
Chapter 4 Structure-gas permeability correlation in polyimides
131
Imidazolium or triethyl ammonium based salt formation may be responsible for this
behavior. It is well known that imidazolium based salts of organic molecules (typically
called as ionic liquids; ILs) exhibit high CO2 sorption properties (Bara, 2009)
4.3.3.2 Permeability with humidified gas
Owing to the observed behavior of high CO2 pereambility, membrane samples
(SPDAB•TA-N, SPDAB•Im-N and SPDABI-N) which were already analyzed for gas
permeability were immersed in deionized water for a day. These wet membranes were
then directly used for the analysis of permeability coefficient of humidified gases (H2 and
CO2). The results are summarized in the following Table 4.8.
Table 4.8 Permeability (P) and selectivity () of SPIs under humidified gas
conditions
SPDAB•TA-N SPDAB•Im-N SPDABI-N Permeation
property Dry Humidified Dry Humidified Dry Humidified
P (H2) 1.89 4.41 0.96 5.51 2.23 6.30
P (CO2) 3.03 12.9 0.78 22.1 5.62 27.9
( CO2/H2) 1.6 2.92 0.81 4.02 2.5 4.43
From the above Table 4.8, it is seen that the permeability of both humidified gases
increased in the order: SPDABI-N > SPDAB•Im-N > SPDAB•TA-N. The permeability of
H2 increased by 2.33, 5.73 and 2.82 fold, while that of CO2 increased by 4.25, 28.33 and
4.97 fold in comparison to the dry gas permeability in SPDAB•TA-N, SPDAB•Im-N and
SPDABI-N, respectively. SPDAB•Im-N demonstrated highest difference in permeability
of dry and humidified gases (H2 and CO2). These membranes are of ion exchange
membranes type, which are known for preferential CO2 permeation (LeBlanc, 1980;
Pellegrino, 1988; Quinn, 1997a; Yamaguchi, 1995; Zhang, 2002). Permeability of polar
gases (CO2 and H2S) in such membranes depends on their ionic site density, IEC and
water content (Matsuyama, 1994; Quinn, 1997a). Enhanced solubility of CO2 with

Chapter 4 Structure-gas permeability correlation in polyimides
132
increase in IEC and water uptake of cation-exchange membrane is known (Pellegrino,
1988; Quinn, 1995 and 1997a; Matsuyama, 1994 and 1999). The IEC in present
membranes increases in the order SPDAB•TA-N (0.39) < SPDAB•Im-N (1.49) <
SPDABI-N (1.92), while the water content increases in the order SPDAB•Im-N (30%) <
SPDAB•TA-N (39%) < SPDABI-N (49%), (Table 5.2 in Chapter 5). Since SPDABI-N
shows both, higher IEC (1.92 mequv/g) and water content (48.9%), both these parameters
would lead to enhanced interaction with CO2; leading to its enhanced permeability.
Highly swollen state would also have its effect on increased permeability, as the polymer
chains are more apart than from dry state and are more dynamic due to surrounded water
(plasticization effect), facilitating diffusion process.
The transport of humidified CO2 can be considered as the facilitated types of
transport. The observed difference in the selectivity, α (CO2/H2) in dry and humidified
state conveys the significance of humidified gas conditions. The largest jump in
permeability was shown by SPDAB•Im-N; which could be the effect of imidazolium
cation. Imidazolium based ionic liquids are well appreciated for high CO2 sorption (Tang,
2005; Bara, 2009). These membranes even in dry state were selective for CO2 over H2.
This indicates that these types of polyimides can be further tuned to manipulate
diffusivity to draw an ultimate advantage of enhanced CO2 interactions. Quantification of
gas sorption and diffusion analysis would have led to better understanding towards
effects of these modifications on the crucial permeation properties, which could not be
attempted in present investigation.
4.3.4 Gas permeability of blend membranes
Dense membranes of blends of sulfonated PI and polybenzimidazole (PBI-BuI)
were prepared as discussed earlier in Section 3.3.2.5 of Chapter 3. Membranes had an
average thickness of 40 μm. These membranes were soaked in distilled water at 60 C for
a day in order to remove traces of DMAc and then acidified in 1N HCl in order to convert
them from triethylammonium sulfonate to sulfonic acid form. This was followed by
rinsing with deionized water and drying in vacuum oven at 80 C for 3 days. These
membranes were abbreviated as SPI(H)/PBI-BuI(a/b), where ‘H’ stands for sulfonic acid
form of sulfonated PI, while ‘a’ and ‘b’ denotes the weight percentage of SPI and PBI-
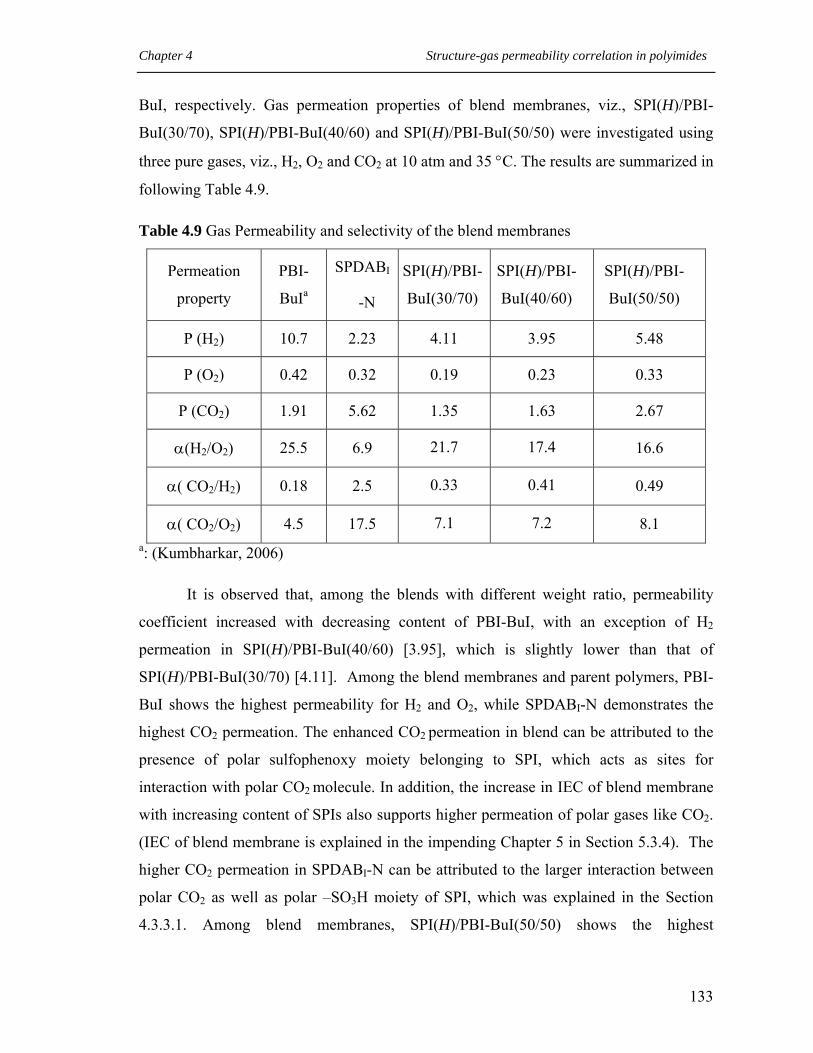
Chapter 4 Structure-gas permeability correlation in polyimides
133
BuI, respectively. Gas permeation properties of blend membranes, viz., SPI(H)/PBI-
BuI(30/70), SPI(H)/PBI-BuI(40/60) and SPI(H)/PBI-BuI(50/50) were investigated using
three pure gases, viz., H2, O2 and CO2 at 10 atm and 35 C. The results are summarized in
following Table 4.9.
Table 4.9 Gas Permeability and selectivity of the blend membranes
Permeation
property
PBI-
BuIa
SPDABI
-N
SPI(H)/PBI-
BuI(30/70)
SPI(H)/PBI-
BuI(40/60)
SPI(H)/PBI-
BuI(50/50)
P (H2) 10.7 2.23 4.11 3.95 5.48
P (O2) 0.42 0.32 0.19 0.23 0.33
P (CO2) 1.91 5.62 1.35 1.63 2.67
(H2/O2) 25.5 6.9 21.7 17.4 16.6
( CO2/H2) 0.18 2.5 0.33 0.41 0.49
( CO2/O2) 4.5 17.5 7.1 7.2 8.1 a: (Kumbharkar, 2006)
It is observed that, among the blends with different weight ratio, permeability
coefficient increased with decreasing content of PBI-BuI, with an exception of H2
permeation in SPI(H)/PBI-BuI(40/60) [3.95], which is slightly lower than that of
SPI(H)/PBI-BuI(30/70) [4.11]. Among the blend membranes and parent polymers, PBI-
BuI shows the highest permeability for H2 and O2, while SPDABI-N demonstrates the
highest CO2 permeation. The enhanced CO2 permeation in blend can be attributed to the
presence of polar sulfophenoxy moiety belonging to SPI, which acts as sites for
interaction with polar CO2 molecule. In addition, the increase in IEC of blend membrane
with increasing content of SPIs also supports higher permeation of polar gases like CO2.
(IEC of blend membrane is explained in the impending Chapter 5 in Section 5.3.4). The
higher CO2 permeation in SPDABI-N can be attributed to the larger interaction between
polar CO2 as well as polar –SO3H moiety of SPI, which was explained in the Section
4.3.3.1. Among blend membranes, SPI(H)/PBI-BuI(50/50) shows the highest

Chapter 4 Structure-gas permeability correlation in polyimides
134
permeability which can be attributed to the increased concentration of bulky pendant
polar sulfophenoxy group due to the increased content of SPI. Such bulky pendant group
should increase the available free volume for the diffusion of gases.
One of the interesting features in the permselectivity of the blend membranes is
that permselectivity for H2/O2 decreases while that of CO2 over H2 and O2 increases with
increasing content of SPI. Thus, our attempt to make blends of polybenzimidazole (PBI-
BuI) with sulfonated PI has improved the selectivity performance of polar gases such as
over CO2 over H2 and O2 than that of pristine PBI-BuI.
3.5 Conclusions
1) For the analogous diamine, 6FDA based PI shows higher dsp, Tg and permeability
coefficient than that of ODPA based PIs. Though the increase in permeability was by
4.6 to 13.3 times, the decrease in selectivity was just by a factor of 0.04 to 0.53.
2) In case of ODPA based PIs permeability increases in the order PDAB-ODPA ≈
2'MPDAB-ODPA < 4'MPDAB-ODPA < 2',6'DMPDAB-ODPA < 4'tBPDAB-ODPA.
In 6FDA based PIs permeability increases in the order 2'MPDAB-6FDA <
4'MPDAB-6FDA < PDAB-6FDA < 2',6'DMPDAB-6FDA ≈ 4'tBPDAB-6FDA. In
ODPA based PIs, better selectivity performance is shown by PDAB-ODPA while in
6FDA based PIs 2'MPDAB-6FDA demonstrates the best selectivity.
3) Though d-spacing value for substitution of voluminous t-butyl group on pendant
phenoxy group in PIs (4'tBPDAB-ODPA and 4'tBPDAB-6FDA) is low; these
polymers demonstrated the highest permeability, which is in tune of variation in FFV.
This indicated that the packing density is better expressed in terms of FFV for these
polymers.
4) Between the PIs based on isomeric diamines 2'MPDAB and 4'MPDAB (with either
ODPA or 6FDA), the later exhibited lower Tg, higher permeability and lower
selectivity.
5) In case of ODPA based PIs, chain packing and gas permeability is largely affected by
the structural variation in diamine moiety. Conversely, in case of PIs based on 6FDA,
the dependence of structural variation in diamine moiety on gas permeability was

Chapter 4 Structure-gas permeability correlation in polyimides
135
largely suppressed by the effects of hexafluoroisopropylidene (–C(CF3)2– ) linkage of
dianhydride moiety.
6) 4'tBPDAB-ODPA shows higher permeability without any appreciable loss of
selectivity as compared with 2',6'DMPDAB-ODPA. In addition, selectivity
performance of the former is higher for some of the gas pairs than of latter.
4'tBPDAB-6FDA and 2',6'DMPDAB-6FDA exhibits almost same permeability and
no significant difference of selectivity is observed between them also.
7) In case of SPIs, though humidification of both gases and membranes of SPIs lead to
an increase in permeability selectivity also increases. This effect is more pronounced
in CO2, indicating the importance of SPIs in CO2 separation.
8) In blend membranes, permeability of CO2 increases with increasing content of SPIs.
In addition, permselectivity for H2/O2 decreases while that of CO2 over H2 and O2
increases with increasing content of SPI.

Chapter 5
Investigation of polyimides towards their applicability
as PEM materials
5.1 Introduction
Since inception of fuel cell almost half century ago, perfluorinated sulfonic acid
copolymer, Nafion, is symbolized as the current state-of-the-art PEMs due to its high
proton conductivity, excellent hydrolytic and redox stability (Roziere, 2003; Souzy,
2005; Hamrock, 2006). However, inherent drawbacks of Nafion, particularly their high
cost (explained in Section 1.7.1 of Chapter 1) encouraged researchers to look for alternate
polymers containing sulfonic acid groups. This resulted in synthesis and evaluation of
various novel hydrocarbon polymers containing sulfonic acid group and acid-base
polymer electrolytes as PEM material for fuel cells.
Among these, sulfonated polyimides (SPIs) prepared particularly from various
sulfonated diamines (SDAs), have become the cynosure of all attention due to their
proven ability as PEM (Guo, 2002; Fang, 2002; Asano, 2006). Besides, the ease of
chemical structural modifications in SPI conferring low manufacturing cost, enhanced
thermal and mechanical properties make them more attractive (Yin, 2006a; Marestin,
2008). However, under severe acidic environment of PEMFC, conventional five
membered-imide linkage is susceptible towards hydrolysis (Genies, 2001a; Fang, 2002;
Alvarez-Gallego, 2007). Attention towards SPIs with organic soluble six membered-
imide linkages has therefore been favored; as they possess better thermal, hydrolytic and
chemical stability (Fang, 2002; Marestin, 2008). Side-chain-type SPIs are being paid
more attention since they hold better option for the above mentioned disadvantages
(Fang, 2006; Hu, 2007; Yin, 2006a; Savard, 2008). Infact, SPIs can be categorized into
two main types, as main-chain-type and side-chain-type, which is already described in
Section 1.6.1 of Chapter 1.
Being a zwitter ion, sulfonated diamine (SDA) is insoluble in organic solvents.
Therefore, protection of –SO3H group of SDA by an organic base; triethyl amine (TEA)
is frequently followed in order to dissolve it in organic solvent (m-cresol). Subsequently,

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
137
SDA in its triethylammonium sulfonate form is then condensed with six-membered
dianhydride to synthesize resultant SPI (Genies, 2001a; Fang, 2002; Lee, 2004; Li,
2007c; Savard, 2008; Chhabra, 2009). In water media, this resultant SPI in the salt form
comprises Bronsted conjugate acid-base pair of mobilized triethylammonium cation with
immobilized sulfonate anion tethered to either main chain or side chain. Subsequent
acidification of SPI in the salt form confers SPI with free sulfonic acid group, behaving
as proton exchange membrane (PEM) in fuel cell. However, utilization of myriad organic
bases in place of TEA can constitute different skeleton of Bronsted conjugate acid-base
pair in SPI and in-situ polymer-in-salt electrolyte can thus be generated. The present
chapter focuses on such type of PEM.
Polymer blending is one of the adaptable techniques to develop new PEMs for
tailor made application for PEMFC, which combine the excellent properties of more than
one existing polymer (Kerres, 2000 and 2002; Deimede, 2002). However, the
development of new useful blends is limited by the immiscibility of many polymer pairs
of interest. Polymers can only be miscible when there is a negative free energy of mixing.
ΔGmix = ΔHmix-T. ΔSmix
However, considering the high molecular weight of the polymers, ΔSmix becomes
negligible. Thus only the deciding factor for the negative free energy of mixing of blend
remains ΔHmix. In other words the mixing must be exothermic, entitling the blend
component with specific interaction, which range from strongly ionic to weak and non-
bonding interaction such as hydrogen bonding, ion-dipole, dipole-dipole and donor-
acceptor interaction. As a result, presence of ionic moieties, which in particular capable
of specific interaction in either or both the polymer blend components, can tune blending
towards miscible blend formation. We, therefore, determined to make the blends from
functional group containing polymers like sulfonated polyimides with PBI. In the former,
presence of acidic sulfonic acid group should interact favorably with the basic
benzimidazole group of the latter. Though, several reports were available on blends of
polyimides and PBI (Guerra, 1988; Jaffe, 1994; Ahn, 1997; Lee, 1999), no reports, till
dates were available on blends of side-chain-type SPI and PBI to the best of our
knowledge.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
138
Aromatic polybenzimidazole (PBI) is a basic heterocyclic polymer having
excellent thermal stability with outstanding high glass transition temperature, high
chemical resistance, low gas permeability and excellent mechanical strength (Buckley,
1988; Kulkarni, 2008; Kumbharkar, 2009). Such basic polymer when doped with
amphoteric acid, which both donates and accepts the proton, leads to proton migration by
Grotthus mechanism (Bouchet, 2001; Ma, 2004). Additionally, proton migration becomes
more feasible at higher temperature as the energy of activation (Ea) necessitated for the
proton transportation becomes less. Another approach is to make acid-base polymer
blend through the formation of hydrogen bridging and base protonation, reducing the
swelling behavior as well as improving the proton conductivity, thermal stability and
mechanical flexibility. Blends of PBI with different sulfonated polymers have already
been used in PEMFC (Kerres, 1999; 2000 and 2004; Hasiotis, 2001b; Kosmala, 2002;
Wysick, 2005). However, blending of PBI with SPI is advantageous as apart from
interaction between sulfonic acid group and basic imidazole group, imide group also
interacts with benzimidazole groups assisting formation of miscible blends (Guerra,
1988; Ahn, 1997; Pu, 2005).
Thus in the present Chapter, our study is focused on investigation of membranes
prepared from SPI containing flexible sulfophenoxy group in the side chain, described
earlier in Section 3.3.2 of Chapter 3. In addition, blends of this polymer with PBI based
on 5-tert-butyl isophthalic acid (BuI) were also prepared for the application as PEM
material.
5.2 Experimental
5.2.1 Syntheses of polymer electrolytes
5.2.1.1 Synthesis of Sulfonated Polyimide (SPI)
SPIs were synthesized from the novel sulfonated diamine viz., 4'-sulphonic-1-
phenoxy-2,4-diaminobenzene (SPDAB) and dianhydrides such as ODPA and NTDA.
NTDA based SPI was synthesized using two different organic bases viz., TEA and
imidazole while for ODPA based SPI, only TEA was used as the organic base as given in
the Section 3.3.2.1 of Chapter 3.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
139
5.2.1.2 Synthesis of polybenzimidazole (PBI)
PBI based on 3,3’-diaminobenzedine (DAB) with aromatic dicarboxylic acids,
viz., isophathalic acid (IPA) and 5-tert-butyl isophthalic acid (BuI) were synthesized as
explained earlier in the Section 3.3.2.2 of Chapter 3.
5.2.2 Membrane preparation
Dense membranes of SPIs, PBIs and blend of these polymers were prepared as
described previously in Section 3.3.2.3, Section 3.3.2.4 and Section 3.3.2.5, respectively
of Chapter 3.
Details of various PEM and their abbreviation are given below in the following
Table 5.1.
Table 5.1 PEM and their abbreviation
Sr. No.
PEM used Abbreviation used
1 Polybenzimidazole based on isophthalic acid (IPA) PBI-I 2 Polybenzimidazole based on 5-tert-butyl isophthalic acid (BuI) PBI-BuI
3 SPI based on SPDAB and NTDA in triethylammonium sulfonate form
SPDAB•TA-N
4 SPI based on SPDAB and NTDA in –SO3H form after acidification of SPDAB•TA-N in 1N HCl, followed by washing with deionised water
SPDABI-N
5 SPI based on SPDAB and NTDA in imidazolium sulfonate form SPDAB•Im-N 6 SPI based on SPDAB and NTDA in –SO3H form after
acidification of SPDAB•Im-N in 1N HCl, followed by washing with deionised water
SPDABII-N
7 SPI based on SPDAB and ODPA in triethylammonium sulfonate form
SPDAB•TA-O
8 Blends prepared from SPI (SPDAB•TA-N) and PBI (PBI-BuI), where ‘a’ and ‘b’ denotes weight percentage of SPDAB•TA-N and PBI-BuI, respectively, while ‘TEA’ shows SPI in triethylammonium sulfonate form
SPI(TEA)/PBI-BuI(a/b)
9 Corresponding blend, after acidification of SPI(TEA)/PBI-BuI(a/b) in 1N HCl followed by washing in deionised water, where ‘H’ denotes SPI in –SO3H form
SPI(H)/PBI-BuI(a/b)
10 Corresponding blend, after doping of SPI(TEA)/PBI-BuI(a/b) with either 1M or 12M H3PO4
SPI(PA)/PBI-BuI(a/b)
11 Corresponding blend, after doping of SPI(TEA)/PBI-BuI(a/b) with 1M H2SO4
SPI(SA)/PBI-BuI(a/b)

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
140
5.2.3 Determination of water uptake (WU) capacity of membranes
Initially, membranes of 5 X 5 cm2 size and thickness of ~ 50 μm were kept in a
vacuum oven at 80 C for 48 h to ensure the constant weight of the dry membrane (Wd).
It was then immersed in deionised water for 48 h at ambient condition. The membranes
were then taken out and wiped instantly by a tissue paper to remove only the adhered
water present on the membrane surface. Afterwards, the wet membranes were weighed
(Ww) and thus, water imbibed inside the membrane could be determined. The precise WU
capacity was subsequently calculated with the following equation,
WU = (Ww - Wd / Wd) X 100 (%)
A balance with 0.1 mg accuracy was used and at least three measurements were
conducted under identical conditions and the values reported are the average of these
measurements.
5.2.4 Determination of phosphoric acid uptake of membranes
Membranes of 4 X 3 cm2 size based on parent polymers and blends were first
dried in a vacuum oven at 80 C for 3 days and then weighed (Wi). These membranes
were then doped by immersing in 12 M H3PO4 for 3 days. Doped membranes were then
wiped out with tissue paper to remove acid adhered on the membrane surface followed by
drying in vacuum oven at 80 C for 4 days. After taking out the sample form vacuum
oven, it was immediately weighed (Wf). The acid uptake by the membrane was
subsequently calculated with the following equation.
Acid uptake = [(Wf -Wi)/ Wi] X 100 (%)
5.2.5 Determination of Ion exchange capacity (IEC) of membranes
The ion exchange capacity (IEC) of PEM was determined by using classical acid-
base titration method. The membranes were dried in a vacuum oven at 80 C for 48 h,
then immediately weighed (Wdry) followed by soaking in 2 M NaCl solution to exchange
the available H+ with Na+. The exchange was performed in the shaking water bath for a
day at ambient temperature. The H+ released in the solution was titrated with
standardized 0.01 N NaOH (against oxalic acid) solution with phenolphthalein as an
indicator. At least five membrane samples of size 5 X 3 cm2 having comparatively lower

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
141
thickness of ~20-25 μm were used. Afterward, separate titrations for these membranes
were performed and the results of ion exchange capacities of three samples averaged.
The following equation was used to determine the IEC.
dry
NaOH
W
N V IEC
where, V is the volume of standardized NaOH in mL and NNaOH is the standardized
normality of 0.01N NaOH.
In case of blend PEM, the theoretical IEC was calculated with the following
equation (Zhao, 2007).
Blend
BuI-PBIBuI-PBISPISPIBlend W
W IEC W IEC IEC
where, IECBlend is the theoretical IEC of the PEM blend
WBlend = Actual weight of the PEM blend in dry state
IECSPI = IEC of the SPI
i.e. theoretical value of IEC of SPDABI-N (1.95 mequv/g)
WSPI = Actual weight of the SPI in WBlend = [ WBlend * (% SPI)] / 100
IECPBI-BuI = IEC of the PBI-BuI considered same as of PBI-I i.e. 0.02 mequv/g (Kerres,
2000)
WPBI-BuI = Actual weight of the PBI-BuI in WBlend = [ WBlend * (% PBI-BuI)] / 100
5.2.6 Determination of hydrolytic and oxidative stability of membranes
Hydrolytic stability of the PEM samples having rectangular size 4 X 1 cm2 and
thickness ~40-50 µm was evaluated by immersing them in deionised water at 80 C. The
water bath was adjusted at a lower speed of 30 rpm for continuous shaking. Hydrolytic
stability was then determined by recording the immersion period required for breaking
the film when bent slightly, due to the loss of flexibility. If the blend films retained their
shape even after folding, it was then said to be stable. The observed duration of these
experiments were in the range of few hours to a week, depending on the stability of the
resultant PEM.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
142
Oxidative stability was tested by immersing membrane strips of the same size as
mentioned above, into Fenton’s reagent (3% H2O2 containing 3 ppm FeSO4) at 80 C.
The stability towards oxidation was evaluated by recording the elapsed immersion time
when the membrane had started breaking (τ1) and dissolved completely (τ2).
5.2.7 Determination of proton conductivity
Proton conductivity was measured with AC impedance spectroscopy (Autolab
PGSTAT 30 with FRA software) over a frequency range of 1-106 Hz. The proton
conductivity measurements were performed for the samples of SPIs, Nafion-117 and the
blends prepared from SPI and PBI-BuI at various ratios in the humidified conditions
(100% RH) at various temperatures. These membranes were fully hydrated in deionised
water for 24 h prior to conductivity measurements. In case of 1M H2SO4 and 1M H3PO4
doped membranes, samples were directly used after taking out from the solution of acid
at this concentration (1M) and the proton conductivity measurement were performed at
100% RH and at various temperature. For the membranes doped with 12 M H3PO4,
measurements were performed in the temperature range of room temperature to 175 C in
anhydrous condition. Proton conductivity of PBI-I doped with 12M H3PO4 was also
evaluated for comparison.
A round-shaped membrane sample with diameter of 2.0 cm and thickness 120 μm
was placed between the two stainless steel electrodes, which were inserted in a
cylindrical closed glass chamber, wrapped around with a resistance heater and equipped
with a feed back temperature controller. A 200 mL of deionised water was placed at the
bottom of the cylinder in order to maintain the humidifying atmosphere. The AC
impedance measurement was performed in 100 % relative humidity (RH) at the desired
temperature. The proton conductivity (σ) of membranes was measured using the
following equation.
σ = L/( RΩ*A)
where, L is the thickness of the membrane in cm, RΩ is the membrane resistance obtained
from the high frequency intercept of imaginary component (Z", Y-axis) on the real
component (Z', X-axis) and A is the area of circular stainless steel electrodes in cm2.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
143
5.3 Results and Discussion
5.3.1 Membrane preparation
Dense membranes based on the parent SPI, PBI and their blends were cast from
their respective DMAc solution. The solution concentration was 3% (w/v). PBI solutions
were centrifuged in order to remove the undissolved particles. While in case of SPI, their
solutions were filtered through filter paper prior to casting in order to remove undissolved
particles, if any. In order to maintain moisture free environment in the casting oven, the
oven temperature was adjusted at 80 C and fused CaCl2 was kept inside it, 2 h before the
casting procedure. All prepared dense membranes had a uniform surface except for the
membranes belonging to SPDAB•Im-N, which had a wavy surface. These membranes,
after peeling off from the glass surface were then immersed in water for a day at 60 C in
order to remove the traces of DMAc, followed by vacuum drying at 80 C for 3 days. It
was observed that membrane of SPDAB•TA-O was broken within a span of 4 h after
keeping in water at 60 C. This could be assigned to the lower hydrolytic stability of the
five membered imide groups due to the induced ring strain (Genies, 2001a). Hence only
SPIs membrane based on NTDA having strain free six-membered imide groups were
investigated for further polymer electrolyte membrane (PEM) characterizations.
Membranes viz., SPDAB•TA-N and SPDAB•Im-N were converted to respective sulfonic
acid form (SPDABI-N and SPDABII-N) by keeping the membranes in 1N HCl and then in
deionised water for 24 h each at ambient conditions.
Blend membranes were prepared by mixing solutions of SPDAB•TA-N (SPI in
triethylammonium sulfonate form) and PBI-BuI in DMAc in different ratio. The
concentration of each solution was 3% (w/v), while percentage of weight ratio of
SPDAB•TA-N to PBI-BuI was 50:50, 40:60 and 30:70. These blend membranes were
abbreviated as SPI(TEA)/PBI-BuI(a/b), where ‘a’ and ‘b’ denotes weight percentage of
SPDAB•TA-N and PBI-BuI, respectively, while ‘TEA’ shows SPI in triethylammonium
sulfonate form. These membranes were also soaked in distilled water for a day at 60 C
to remove the traces of DMAc, followed by vacuum drying at 80 C for 3 days. These
blend membranes were subsequently used for doping with different acids for proton
conductivity measurements. They were transformed into sulfonic acid form after treating
with 1N HCl for 24 h followed by washing with deionised water and abbreviated as

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
144
SPI(H)/PBI-BuI(a/b), where ‘H’ denotes SPI in –SO3H form. SPI(TEA)/PBI-BuI(a/b),
when doped with either 1M or 12M H3PO4 were abbreviated as SPI(PA)/PBI-BuI(a/b)
and for 1M H2SO4 doping, they were named as SPI(SA)/PBI-BuI(a/b).
5.3.2 Phosphoric acid doping
Pure PBI is an insulator having an intrinsic conductivity of about 10-12 S.cm-1 at
25 C (Xing, 2000; Bouchet, 2001; Ma, 2004b). It is known that, doping of PBI with
strong acids (H3PO4 and H2SO4) bestows the proton conductivity in the range of 10-2 -10-
3 S.cm-1 (Glipa, 1999; Bouchet, 2001). PBI is generally doped with 85-88% phosphoric
acid (16.2 M H3PO4). In our case, doping of membranes of SPI and blends with 88%
phosphoric (16.2 M H3PO4) acids were either disintegrated or dissolved hence they were
doped in 12 M H3PO4. Doping level was defined as the weight percentage of phosphoric
acid present in per gram of the polymer membrane. Duration of doping was kept for 3
days, followed by drying in a vacuum oven at 80 C for the same period, after wiping out
adhered acid with tissue paper. In order to remove the water completely from the acid
doped membrane, these doped membranes were kept at 100 C for 3 days in vacuum
oven. However, on following this procedure, doped blend membranes based on PBI-BuI
and doped SPI membranes were broken. In order to compare the proton conductivity of
blend membrane in anhydrous condition, both PBI-I and PBI-BuI were also doped with
12 M H3PO4.
The acid uptake for SPDAB•TA-N and SPDAB•Im-N is nearly same, being 67.7
wt% and 65.3 wt%, respectively as shown in Table 5.2. This is probably due to exchange
of organic cations (triethylammoium and imidazolium cation) of both polymer
membranes with H+ released from phosphoric acid. In this way, the resultant polymer
could absorb same quantity of acid using 12 M H3PO4 conferring same chemical structure
with –SO3H group. The acid uptake of SPI can be attributed to the presence of N-
heterocycle in the imide linkage as well as –O– in the side chain belonging to Ar-O-Ar'
linkage, which can establish H-bonding with H3PO4. In addition, oxygen belonging to -
SO3H has also capability for H-bonding with H3PO4.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
145
Table 5.2 Physical properties of membranes based on SPIs, PBIs and their blends
Oxidative stability
(h) Polymer IEC
(mequiv/g)
WUc
(%)
12 M H3PO4
uptake (%)
Hydrolytic
Stabilityf
(h) τ 1 τ2
SPDABI-N 1.92 48.90 --- 11 3.5 5
SPDABII-N 1.86 46.40 --- 11.5 3.5 5
SPDAB•TA-N 0.39 39.30 67.7 > 168 10 21
SPDAB•Im-N 1.49 30.40 65.3 > 168 16 62
SPI(H)/PBI-
BuI(50/50) 0.89 27.70 94.7e 153 82 > 168
SPI(H)/PBI-
BuI(40/60) 0.66 26.77 100.3e > 168 114 > 168
SPI(H)/PBI-
BuI(30/70) 0.48 22.32 109.4e > 168 > 168 > 168
PBI-BuI 0.02a 12.30d 165.2 Stable NM NM
PBI-I 0.02b 20.40d 150.5 Stable 30 ming ---
IEC: Ion exchange capacity measured by titration method in mequiv/g a: Determined in the same way as that of PBI-I b: Kerres, 2000 c: Water uptake capacity in wt % d: Kumbharkar, 2009 e: 12M H3PO4 uptake of blend membranes having been abbreviated as SPI(PA)/PBI- BuI(a/b) (Table 5.1) f: Elapsed time when membrane was broken while bending g: Li, 2007b τ 1: Elapsed time when membrane was broken τ2 : Elapsed time when membrane had been completely dissolved NM: Not measured
The membranes of PBI-I and PBI-BuI were also doped with 12 M H3PO4 for 3
days. PBI-BuI exhibited higher acid uptake of ~165.2 wt % compared to 150.5 wt % of
PBI-I (Table 5.2). This can be attributed to comparatively higher initial free volume in
PBI-BuI, wherein more number of H3PO4 along with water molecules can be imbibed. In
addition, larger swelling of PBI-BuI on doping with H3PO4 also supports its higher acid
uptake. Bulky t-butyl group in PBI-BuI, restricts efficient packing polymer chains as that

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
146
of PBI-I. This results in the reduction of inter-molecular H-bonding leading to reduced
interchain interactions as well as increase in initial fractional free volume (0.3393
cm3/cm3) in PBI-BuI as compared with PBI-I (0.3096 cm3/cm3) (Kumbharkar, 2006).
This observation can also be supported from the lower N-H group density in PBI-BuI
(8.24%) than PBI-I (9.74%). In case of blend membranes, the acid uptake is in the range
of 94-110 wt% and increases with PBI-BuI content. Thus, acid uptake decreases in the
order PBI-BuI > PBI-I > SPI(PA)/PBI-BuI(30/70) > SPI(PA)/PBI-BuI(40/60) >
SPI(PA)/PBI-BuI(50/50) > SPDAB•TA-N ≈ SPDAB•Im-N. This is attributed to the
decreased content of PBI in the membrane.
5.3.3 Water uptake (WU) capacity
Since proton conductivity as well as the mechanical strength of PEM comprising
sulfonated polymers, is highly dependant on water content, determination of their WU
becomes an indispensable criterion. High water content in the membrane though increases
the proton conductivity, it deteriorates the mechanical strength of PEM (Roziere, 2003).
Hence sufficient amount of WU commensurate with proton conductivity of magnitude 0.1
S/cm is desirable for better performance in PEM. From Table 5.2, it is observed that WU
of SPIs decreases in the order SPDABI-N ≈ SPDABII-N > SPDAB•TA-N > SPDAB•Im-N.
Water uptake of SPDABI-N and SPDABII-N is 48.9 % and 46.4 %, respectively. The
reported WU for different SPIs is in the range of 21-250 % (Fang, 2002; Fang, 2006; Yin,
2006a; Savard, 2008). The WU of present SPIs is thus at the lower level when compared to
WU of reported SPIs. This can be mainly attributed to the lower IEC (<2) and presence of
a single sulfonated moiety per chain repeating unit (CRU) of the present SPIs. It should be
noted that most of the reported SPIs used as PEM are in the copolymer form since
homopolymers are either soluble in water or show high swelling with poor mechanical
resistance (Guo, 2002; Fang, 2006).
It is interesting to compare the water uptake between SPDAB•TA-N (39.3%) and
SPDAB•Im-N (30.4%). The latter shows lower water uptake than the former despite its
higher IEC. In general, WU increases with higher IEC conferring either high swelling or
even dissolution in water of the membranes (Guo, 2002; Fang, 2006; Yin, 2006a). This
anomalous behavior of SPDAB.Im-N can be assigned to the peculiar structural

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
147
arrangement of pendant imidazolium ring in SPDAB•Im-N. The planar and aromatic
structure of the imidazolium ring is thought to be well accommodated in the available free
volume of the polymer chains. On the other hand, the relative reduction between interchain
interaction due to tetrahedrally attached three alkyl (ethyl) groups at the side chain end in
SPDAB•TA-N may result in increased free volume as compared with that of planar
structure of imidazolium ring in SPDAB•Im-N. As a result more number of water
molecules can be harbored in the available free volume of former than that of latter. This
observation can be supported from the permeability of H2 and CO2 explained earlier in
Section 4.3.3 in Chapter 4, wherein the permeability of these gases are lower in
SPDAB•Im-N than SPDAB•TA-N in dry state.
Water uptake of SPI is also governed by the chemical structure of SDA. SDA
having angled or non-linear monomer leads to the greater chain entanglements in the
resulting polymer structure, thereby offering more resistance to swelling and increasing
proton concentration (Rodgers, 2006). The present SPDAB is a non-linear SDA, with
asymmetric substitution of sulfophenoxy moiety on meta-amino phenyl ring, which acts as
a ‘kink’ in the resulting structure of SPI. Hence present SPIs would be expected to exhibit
larger number of entanglements. In addition, absence of any flexible linkage (C-O-C) in
main chain of polyimide as well as the planar and rigid structure of the naphthalo-diimide
structure (Gianolio, 2000) would further offer strong resistance towards the reorganization
of individual polymer chains in the wet condition. Consequently, these SPIs offer strong
resistance towards water swelling resulting in decrease in WU with better mechanical
strength in wet conditions. Hence these SPIs, being homopolymer are stable in water. IEC
and water uptake in membrane form of SPDAB•TA-O and its acidified form polymer
(SPDAB-O) could not be measured due to lower stability of these polymer membranes in
water, arisen from strained five-membered imide ring as also observed previously (Genies,
2001a).
WU capacity of the blend membrane in sulfonic acid form is shown in Table 5.2.
Initially, the blend membranes SPI(TEA)/ PBI-BuI(a/b) was placed in 1N HCl for 48 h in
order to regenerate –SO3H of SPI, followed by washing with deionised water at ambient
[abbreviated as SPI(H)/PBI-BuI(a/b)]. The blend membranes SPI(H)/PBI-BuI(a/b)
exhibits water uptake capacity in the range of 22.3% -27.70%, which is lower than parent
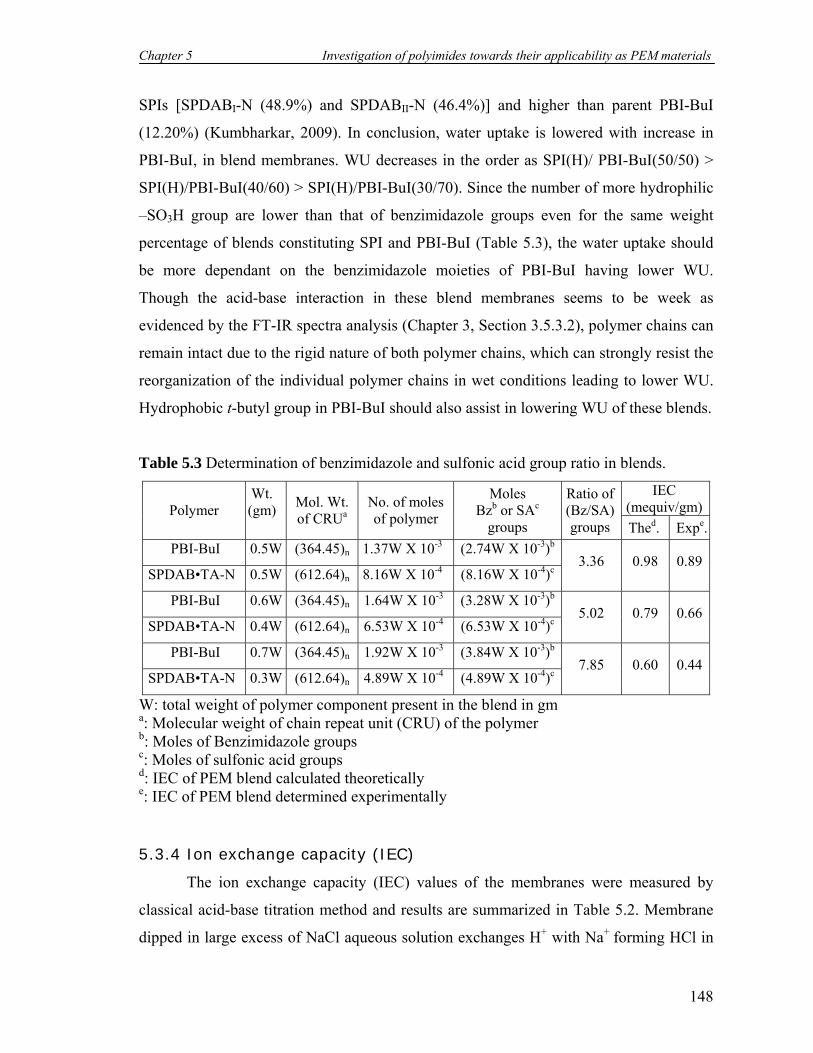
Chapter 5 Investigation of polyimides towards their applicability as PEM materials
148
SPIs [SPDABI-N (48.9%) and SPDABII-N (46.4%)] and higher than parent PBI-BuI
(12.20%) (Kumbharkar, 2009). In conclusion, water uptake is lowered with increase in
PBI-BuI, in blend membranes. WU decreases in the order as SPI(H)/ PBI-BuI(50/50) >
SPI(H)/PBI-BuI(40/60) > SPI(H)/PBI-BuI(30/70). Since the number of more hydrophilic
–SO3H group are lower than that of benzimidazole groups even for the same weight
percentage of blends constituting SPI and PBI-BuI (Table 5.3), the water uptake should
be more dependant on the benzimidazole moieties of PBI-BuI having lower WU.
Though the acid-base interaction in these blend membranes seems to be week as
evidenced by the FT-IR spectra analysis (Chapter 3, Section 3.5.3.2), polymer chains can
remain intact due to the rigid nature of both polymer chains, which can strongly resist the
reorganization of the individual polymer chains in wet conditions leading to lower WU.
Hydrophobic t-butyl group in PBI-BuI should also assist in lowering WU of these blends.
Table 5.3 Determination of benzimidazole and sulfonic acid group ratio in blends.
IEC (mequiv/gm)
Polymer Wt. (gm)
Mol. Wt. of CRUa
No. of moles of polymer
Moles Bzb or SAc
groups
Ratio of
(Bz/SA) groups Thed. Expe.
PBI-BuI 0.5W (364.45)n 1.37W X 10-3 (2.74W X 10-3)b
SPDAB•TA-N 0.5W (612.64)n 8.16W X 10-4 (8.16W X 10-4)c3.36 0.98 0.89
PBI-BuI 0.6W (364.45)n 1.64W X 10-3 (3.28W X 10-3)b
SPDAB•TA-N 0.4W (612.64)n 6.53W X 10-4 (6.53W X 10-4)c 5.02 0.79 0.66
PBI-BuI 0.7W (364.45)n 1.92W X 10-3 (3.84W X 10-3)b
SPDAB•TA-N 0.3W (612.64)n 4.89W X 10-4 (4.89W X 10-4)c 7.85 0.60 0.44
W: total weight of polymer component present in the blend in gm a: Molecular weight of chain repeat unit (CRU) of the polymer b: Moles of Benzimidazole groups c: Moles of sulfonic acid groups d: IEC of PEM blend calculated theoretically e: IEC of PEM blend determined experimentally
5.3.4 Ion exchange capacity (IEC)
The ion exchange capacity (IEC) values of the membranes were measured by
classical acid-base titration method and results are summarized in Table 5.2. Membrane
dipped in large excess of NaCl aqueous solution exchanges H+ with Na+ forming HCl in

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
149
the aqueous solution. This formed acid is then titrated with standardized 0.01 N NaOH
using phenolphthalein as an indicator. The IEC values obtained from titration of the
acidified membranes of SPIs viz; SPDABI-N (1.92) and SPDABII-N (1.86) are in good
agreement with the theoretical value (1.95). This implied that almost all the protons from
sulfonic acid groups are exchangeable. Thus, there are no concealed sulfonic acidic sites
present in these polymers and all the protons from sulfonic acid groups should take part
in the proton conduction through the well connected hydrophilic segments.
While comparing the IEC between the SPDAB•TA-N and SPDAB•Im-N, the later
shows almost more than 3 times IEC value (1.49) than the former (0.39). These values
are lower than their corresponding sulfonic acid form [SPDABI-N (1.92) and SPDABII-N
(1.86)]. This discrepancy in IEC values can be better explained on the basis of their
cationic structural formation and dissociation in aqueous medium. In SPDAB•Im-N, an
imidazolium cation contains three hydrogen atoms belonging to carbon attached through
covalent bonds (C2, C4 and C5 as shown in the following Figure 5.1) while the remaining
two hydrogen of the nitrogen are in more detachable form. Among hydrogen atoms of
carbons, C2 is the most acidic (Dupont, 2004) and hence can be considered to be
exchangeable. Thus due to the presence of more number of exchangeable hydrogens in
imidazolium cation as compared with only one in triethylammonium salt form, the
former exhibited higher IEC value than the latter.
Imidazolium Triethyl ammonium cation (ImH+) cation (TEAH+)
Figure 5.1 Structures of cations in salt form
Different IEC of these salt type membranes can also be better illustrated by the
comparison of their acidic nature of cation, dissociation and solvation of ion pairs in
aqueous media. Since IEC was measured by using 0.01N NaOH, the acidic nature of
cations should be one of the major contributors. Consequently, in aqueous medium,

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
150
SPDAB•Im-N could be more dissociated due to better solvation of an imidazolium cation
(ImH+) due to its higher acidic nature than that of triethylammonium hydronium ion
(TEAH+). Higher acidic nature of ImH+ may be attributed to the better stabilization of its
conjugate base imidazole due to its aromaticity as well as better solubility in aqueous
media after loosing its proton. While in case of TEAH+, capacity of proton donation is
less since the conjugate base TEA is less soluble in water. It is known that, TEA has only
5.5% solubility in water whereas; imidazole is completely soluble in water. It should be
noted that, TEA (pKa=10.75) (Kailani, 1988b) is more basic than imidazole (pKa= 7.1)
(Clayden, 2001; Lv, 2008) hence the conjugate Bronsted acid; imidazolium cation
(ImH+) should be more acidic than triethylammonium cation (TEAH+). Infact,
SPDAB•Im-N and SPDAB•TA-N can be considered as protic neutral salt type
membranes due to acidic nature of ImH+ and TEAH+ cations. Reports of acidic nature of
ImH+ and TEAH+ cations are known (MacFarlane, 2006). Protic ionic liquids based on
these cations have well been reported (Belieres, 2007; Greaves, 2008). Acid catalyzed
reactions based on such type of acidic cations have also been reported. Moreover, ionic
liquids containing Bronsted acidic nature of dialkyimidazolium cations forming a salt
with a side chain containing –SO3H group have been demonstrated as acid catalysis in
many conventional reactions such as Friedal-Crafts reaction (Qiao, 2004; Du, 2005),
esterification (Fraga-Dubreuil; 2002, Du, 2005) and etherification (Cole, 2002; Du,
2005). Poor solvation of TEAH+ in water can also be attributed to its hydrophobic three
ethyl groups. Though SPDAB•TA-N reveals higher water uptake than SPDAB•Im-N,
TEAH+ is expected to be poor solvated than ImH+ due to the hydrophobic three ethyl
groups. This phenomenon may confer larger dissociation of SPDAB•Im-N than that of
SPDAB•TA-N in aqueous media. In other words, dissociation constant for imidazolium
sulfonate is more as compared with triethylammonium sulfonate as shown in the Figure
5.2. This can also be supported from the delocalization of positive charge on the
imidazolium cation due to its aromatic character, which is effective for the improvement
in the degree of salt dissociation in SPDAB•Im-N. While in case of SPDABI-N and
SPDABII-N, -SO3H group is completely dissociated with complete exchange of H+. In
this way, IEC values increase in the order SPDABI-N ≈ SPDABII-N > SPDAB•Im-N >
SPDAB•TA-N.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
151
Figure 5.2 Dissociation of SPIs in aqueous medium
where,
K1D is the dissociation constant of SPI in triethylammonium sulfonate form
K2D is the dissociation constant of SPI in imidazolium sulfonate form
KD is the dissociation constant of SPI in –SO3H form and (K1D < K2
D < KD)
In case of blend membranes, the membranes prepared in TEA form were first
converted to sulfonic acid form and then IEC measured. IEC values decreases in the
order SPI(H)/PBI-BuI(50/50) > SPI(H)/PBI-BuI(40/60) > SPI(H)/PBI-BuI(30/70) as
shown in the Table 5.2. This suggests that IEC values increase with SPI content. Since
the present blend system were prepared on the percentage of weight basis and not on the
mole basis hence we thought that it was necessary to dig out the actual number of
sulfonic acid groups available for proton exchange. If this exact number of sulfonic acid
groups can be calculated, it would shed more light on the IEC values of blends. To
calculate exact number of moles of sulfonic acid, moles of SPI in TEA form was
considered instead of sulfonic acid form as SPI was taken in the TEA salt form while
preparing blends. From the Table 5.3, it can be seen that number of sulfonic acid groups
present in the blend is less as compared with the number of benzimidazole moieties even
for the same weight percentage of the polymer. This discrepancy is attributed to the
larger difference in molecular weight of chain repeat unit of respective polymers.
Moreover, there is only a single sulfonic acid group in the CRU of SPI as compared with
the two imidazole groups in the PBI-BuI. This also implies that blend prepared with
equimolar quantities of SPI and PBI would also contain two fold of imidazole moieties
than that of sulfonic acid moieties. Increase in benzimidazole to sulfonic acid ratio should
raise acid-base interactions since more number of benzimidazole moieties available per

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
152
sulfonic acid group. In other words with increasing PBI-BuI content in the blend number
of –SO3H group governing IEC should be decreased. This statement can be supported
from the fact that percentage difference between theoretical IEC and experimentally
determined IEC decreases with decreasing PBI-BuI content i.e. SPI(H)/PBI-BuI(30/70)
(26.67%) < SPI(H)/PBI-BuI(40/60) (16.5%) < SPI(H)/PBI-BuI(50/50) (9.7%).
It is evident that the PEM exhibiting high IEC implies the existence of more
number of free sulfonic acid groups. From the Table 5.3, it is observed that IEC of blend
membranes determined experimentally is less than that of their theoretical values. This
implies that some of the –SO3H groups have not participated in the H+ exchange process
indicating their involvement in other interactions which can probably be none other than
acid-base interactions. However, the difference in IECs is not too high suggesting that
most of the sulfonic acid groups have labile H+. In case of the IEC’s of blend system
constituting acid-base interactions, protons can not become exchangeable to the solution
to be titrated if acidic sulfonic group forms the strong interactions with the basic groups
resulting in larger difference between theoretical and experimental IEC values (Kosmala,
2002). In the present blend component, this difference is not too high, indicating
moderate or weak acid-base interactions. This observation is reasonable, as the presence
of t-butyl group can shield the imidazole group in the main chain of PBI-BuI and thus can
resist such interactions. Another factor contributing to such weak functional interactions
is the rigidity of the main chain of these polymers. Though SPI has flexible sulfophenoxy
group, it lacks such flexibility in its main chain, which can further resist the aligning of
polymer main chains favorable for strong interactions with benzimidazole groups of PBI.
PBI and polyimide comprise extreme rigid structures in their main chain. Polymer blends
constituting extreme rigid structure in their main chain sterically hinder the interactions
of functional groups (Wang, 2008). In other words, functional groups accessibility for
enthalpic interactions required for the miscibility of polymers becomes less. Since the
entanglement required for functional group association become less in such rigid
structures, the probability of forming the interactions necessary for miscibility also
becomes little (Cote, 1994). Radmard et al. (2001) also reported the reduction in
intermolecular hydrogen bonding in the blend with increase in chain rigidity of polyether.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
153
IEC of present blend system can therefore be attributed to the weakened acid-base
interactions arising from the chain rigidity of PBI-BuI and SPI.
5.3.5 Hydrolytic stability
Table 5.2 lists the hydrolytic stability of PEM membrane. It was measured by
immersing the membranes in water at 80 °C and determining the period required for
breaking the film. SPIs viz., SPDABI-N and SPDABII-N could maintain their hydrolytic
stability up to 11 h and 11.5 h, respectively. After this duration, membranes became
brittle and remained un-dissolved in water. Some of the reported SPIs (homopolymers
based on SDA with NTDA) displayed lower hydrolytic stability in the range of few
seconds to 5h and became water soluble (Fang, 2002; Yin, 2006a). Comparatively the
present SPIs displayed an improved hydrolytic stability. This can be attributed to the
lower IEC (< 2 mequv/g) due to the presence of flexible sulfophenoxy group having
single sulfonic group per CRU and comparatively low water uptake (46-49%). In SPIs,
sulfonic acid group is at the para position of the pendant –OPh ring, thus located away
from the functional imide groups. Consequently, the increased distance between the
imide ring and sulfonic acid group (as compared in main-chain-type SPIs) has an ability
to alleviate the acid-hydrolysis of imide linkage. This can be better represented by
schematic representation as shown in the following Figure 5.3.
(a) Main-chain-type SPI (b) Side-chain-type SPI
Figure 5.3 Schematic representation of the SPIs showing relative distance of sulfonic
acid group form the imide linkage
(a) –SO3H and imide moieties become nearer in main-chain type-SPI (Fang, 2002)
(b) –SO3H and imide moieties go away from each other (e.g. SPDABI-N and
SPDABII-N)

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
154
Moreover, in hydrated state, region with large content of water is present to the
proximity of sulfonic group only i.e. hydrophilic ion-rich domains which are away from
the imide group in the main chain. Consequently, hydrolytic stability of SPDABI-N and
SPDABII-N should increase. Thus, side-chain-type SPI membranes seems to be less
vulnerable for hydrolysis, consequently exhibiting better hydrolytic stability than main-
chain-type ones (Yin, 2004 and 2006a; Fang, 2006; Savard, 2008). It has been mentioned
that hydrolytic stability is subjective towards chemical structure, basicity of the SDA,
IEC, water uptake and flexible linkages (C-O-C) in the main chain (Yin, 2004; Marestin,
2008).
However, the brittleness induced after 11 h in SPDABI-N and SPDABII-N can be
ascribed to the extreme rigid structure of the main chain. As explained earlier in section
5.3.3, these SPIs therefore offer strong resistance towards swelling since SPDAB is an
angled or non-linear type monomer. Though these SPIs have flexible ether linkage in the
side chain, they lack flexible linkages in main chain, which is prerequisite for the
relaxation and orientation of polymer molecules in water medium (Yin, 2006b). This
would further impart brittleness in these membranes. In addition, ortho substitution of
pendant sulfophenoxy group present to alternative imide linkages restrict the rotation of
neighboring phenyl rings attached to C-N bond resulting in reduced segmental mobility
and hence enhancement in rigidity of SPI. Consequently, hydrolytic stability of these
SPIs is decreased (Rodgers, 2006; Savard, 2008).
SPIs in salt form viz; SPDAB•TA-N and SPDAB•Im-N reveal better results and
can maintain their hydrolytic stability even after dipping these membranes in deionised
water for a week at 80 C. This can be attributed to their lower IEC and water uptake
values than SPI in sulfonic acid form (SPDABI-N ≈ SPDABII-N). However, enhanced
basicity of the SPDAB in salt form could also corroborate for their increased hydrolytic
stability (Yin, 2004). In fact, sulfonic acid group is electron withdrawing, however in the
salt form (triethylammoium sulfonate and imidazolium sulfonate), its electron
withdrawing capacity is appeased, as a result the oxygen in the C-O-C linkage becomes
relatively electron donating towards amino phenyl ring resulting in increased basicity of
the –NH2 groups. Such phenomenon has also been proved for the sulfopropoxy linkage
(Yin, 2004). Consequently, electron density is enhanced in the carbonyl carbon of

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
155
naphthalo-imido ring and became less vulnerable by the oxygen atom of water molecule.
In this way, hydrolysis of naphthalo-imido ring can be alleviated in salt form as
compared with sulfonic acid form. Though, we didn’t continue our investigation after a
period of a week (168 h), between SPDAB•TA-N and SPDAB•Im-N, the later might have
shown increased hydrolytic stability than former. Since hydrolysis of imide linkage is
nothing but the reverse reaction of polymerization (Yin, 2006b), the increased
polymerization rate in SPDAB•Im-N would probably further lessen its imide hydrolysis.
As explained earlier, strained five membered ODPA based SPI revealed lower hydrolytic
stability than strain free six-membered NTDA based SPI counterpart (Genies, 2001a).
The evaluation of hydrolytic stability for the blend membranes reveals improved
hydrolytic stability than that of SPIs. This can be due to their lower water uptake
capacity. Besides, hydrolytic stability improves with increasing content of base
component (PBI-BuI). It was observed that, blend membrane SPI(H)/PBI-BuI(50/50) was
broken after 6 days (153 h) while the remaining blend membranes SPI(H)/PBI-
BuI(40/60) and SPI(H)/PBI-BuI(30/70) could maintain their hydrolytic stability even
after 7 days. This can be attributed to low percentage of SPI which is more prone to
hydrolysis as compared with high content of better hydrolytically stable PBI-BuI.
5.3.6 Oxidative stability
Membrane stability of SPIs to oxidation was studied by determining the elapsed
time for the membrane to disintegrate and/or its complete dissolution into Fenton’s
reagent (3% H2O2 and 3 ppm FeSO4) at 80 C. Oxidative stability decreases in the order
SPDAB•Im-N > SPDAB•TA-N > SPDABI-N ≈ SPDABII-N (Table 5.2). In other words,
oxidative stability increases with decreasing WU of SPIs. This observation is reasonable
as the highly oxidizing generated radical species (OH and OOH) attack favorably in the
hydrophilic region. It should be noted that the present SPI exhibits oxidative stability for
longer duration as compared with reported sulfonated homopolyimides in same
composition of Fenton’s reagent (Miyatake, 2004; Zhou, 2005). Thus, our attempt to put
hydrophilic sulfonated moiety away from main chain has resulted in improved oxidative
stability. Among these SPIs, the effect of side chain sulfophenoxy group on prolonging
the dissolving time in Fenton’s reagent is more pronounced in SPDAB•Im-N (Table 5.2)

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
156
due to its peculiar structure. The planar and aromatic nature of this cation can effectively
be accommodated in the available free volume of polymer chains which could restrict
water molecules from being imbibed (explained earlier in Chapter 4). In addition,
imidazolium cation, being aromatic becomes more stable to the radical attack as
compared with the cation TEAH+ in SPDAB•TA-N. However, it should be noted that the
presence of flexible electron donating ether linkage (Ar-O-Ar') reduces the oxidative
stability of SPIs as it is known to be susceptible to breaking by electrophilic hydroxyl
radicals (•OH and •OOH) (Hubner, 1999; Lee, 2007).
In the blend membrane same trend of increasing oxidative stability with decrease
of water uptake is observed i.e. oxidative stability increases in the order SPI(H)/PBI-
BuI(30/70) > SPI(H)/PBI-BuI(40/60) > SPI(H)/PBI-BuI(50/50) (Table 5.2). Thus,
increase in SPI content decreases the oxidative stability or vice versa since hydrophilic
content increases with SPI content. The probable acid-base interactions in blend, though
week should also increase the oxidative stability as it prevents membrane from excessive
water swelling.
5.3.7 Proton conductivity measurements
Proton conductivity (σ) of the PEM was measured by a two-probe impedance
technique consisting of two stainless steel plates of diameter 2.0 cm. A circular
membrane sample of diameter 2.0 cm having thickness in the range of 100-150 micron
was placed in between the two SS electrodes. Proper ohmic contact was ensured by
spring action. The impedance measurements were taken in the range of 1 to 106 Hz with a
10 mV rms amplitude at 10 points per decade. Proton conductivity of SPIs and blends of
the type SPI(H)/PBI-BuI(a/b) was measured in 100% RH at various temperatures. Proton
conductivity of PBI-BuI and blends after doping separately with 1M H2SO4 and 1M
H3PO4 were also measured in 100% RH at various temperatures. Proton conductivity of
PBI-I, PBI-BuI, blends and SPIs viz., SPDAB•Im-N and SPDAB•TA-N after doping with
12M H3PO4 was performed at dry condition and at various temperatures ranging from 30
to 175 °C. The schematic representation of the apparatus used for proton conductivity
measurement is shown in the Figure 5.4.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
157
Figure 5.4 Schematic representation of the apparatus used for proton conductivity
measurement
Proton conductivity of the polymer electrolyte membrane (PEM) was calculated
from the plots obtained from electrochemical impedance measurements. The following
Figure 5.5 elucidates the general pattern of impedance spectra of (PEM) sandwiched
between two stainless electrodes at various temperatures and at ambient pressure. The
results are presented in Nyquist plot comprising a plot of imaginary impedance (Z") at Y-
axis against the real impedance (Z') on X-axis (Figure 5.5 A). It can be seen that
semicircle arc can be generated from high-frequency region to the low frequency region.
The intercept of imaginary component on X-axis in the high frequency domain represents
the ohmic resistance (RΩ) of the PEM while the behavior of Z" and Z' in the low
frequency domain represents the mass transport related to the diffusion of H+ (Rf)
through the PEM. We used the basic Randles equivalent circuit as shown in (Figure 5.5

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
158
B) for comparing our results as it describes the different process related to membrane
conductivity in a simple manner.
Figure 5.5 A) Nyquist plot for the impedance measurements
B) Randles equivalent circuit used for fitting this data.
where, RΩ denotes the ohmic resistance between the electrode and membrane, Rf denotes
the charge transfer resistance for diffusion of H+ through PEM and Cdl is the capacitance.
Proton conductivity was calculated by inserting the value of RΩ in the following equation.
σ = L/( RΩ*A)
where, L is the thickness of the membrane in cm, RΩ is the membrane resistance obtained
from the impedance measurements and A is the area of the circular membrane in cm2.
5.3.7.1 Proton conductivity of sulfonated polyimides (SPIs)
Figure 5.6 Impedance spectra of SPIs and Nafion-117 at 50 C

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
159
Figure 5.6 represents the Nyquist plots measured for SPIs and Nafion-117 at
50 C in 100 % RH. Figure 5.7 represents the temperature dependant proton conductivity
(σ) of sulfonated polyimides and Nafion-117 at 100% RH.
30 40 50 60 70 800.0
4.0x10-3
8.0x10-3
1.2x10-2
1.6x10-2
2.0x10-2
2.4x10-2
2.8x10-2
Temperature (oC)
Con
d. (S
/cm
)
Nafion-117 SPDAB
I-N
SPDABII-N
SPDABIm-N SPDABTA-N
Figure 5.7 Proton conductivity of SPIs with temperature at 100% RH
In case of membranes of SPDABI-N, SPDABII-N and Nafion-117, proton
conductivity linearly increases from RT to 80 C and then further decreases sharply
above 80 C. While for N-SPDAB•TA and SPDAB•Im-N it increases from RT to 50 and
60 C, respectively and then further decreases with increase in temperature. At a
particular temperature, proton conductivity increases in the order SPDAB•TA-N <
SPDAB•Im-N < SPDABII-N < SPDABI-N < Nafion-117. For example, conductivity at
50 C for SPDAB•TA-N, SPDAB•Im-N, SPDABII-N, SPDABI-N and Nafion-117 is 1.35
X 10-3, 3.91 X 10-3, 1.27 X 10-2, 1.41 X 10-2 and 2.22 X 10-2 S/cm, respectively. In SPIs,
the trend of proton conductivity can be well matched with IEC which increases as
SPDAB•TA-N < SPDAB•Im-N < SPDABII-N < SPDABI-N. The high conductivity of
Nafion-117 is attributable to the higher acidity (pKa,-6) (Chikvaidze, 2007; Sartori, 2009)
of the –CF2–SO3H group as compared with –Ar–SO3H group (pKa, 2.70) (Mauritz,
2004). Besides, in the swollen state, the phase separated morphology in Nafion-117

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
160
retains a bulk amount of water inside the hydrophilic domain due to the agglomeration of
side chain hydrophilic –SO3H groups. As a result inter-sulfonate group separation
becomes less with fine connectivity of hydrated domains, lower hydrophobic-hydrophilic
interface and better proton transport performance (Kreuer, 2004). Hydrocarbon based
sulfonated polymers seldom form such separated morphology. The present SPIs contain
rigid main chain backbone with flexible pendant sulfophenoxy group as a short side
chain. Hence reorganization of individual polymer main chains becomes difficult in wet
conditions with less probable agglomeration of -SO3H. This concept has earlier been well
explained in Section 5.3.3. Hence, SPDABI-N and SPDABII-N exhibits lower proton
conductivity than Nafion-117. However, proton conductivity of the order of 10-2-10-3
S.cm-1 of SPDABI-N and SPDABII-N, in spite of their comparatively lower IEC (<2) and
optimum water uptake (46-49%) is worth noting. The lower proton conductivity of
SPDABII-N as compared with SPDABI-N can be attributed to its wavy nature due to
which better surface contact of electrodes with PEM (SPDABII-N) could not have been
achieved.
It should be noted that proton conductivity is significantly dependant on IEC than
on water uptake. In general, proton conductivity increases with water uptake (Rodgers,
2006). However, for better proton conduction in sulfonic acid based polymers, not only
the higher water uptake but larger IEC i.e. actual number of exchangeable protons is also
an indispensable factor (Xing, 2006). It is interesting to look at the proton conductivity
between salt membranes viz; SPDAB•Im-N and SPDAB•TA-N which can be considered
as PEM having protic neutral salts, i.e., imidazolium sulfonate and triethylammonium
sulfonate, respectively. Former reveals higher IEC and lower water uptake than that of
latter and has been thoroughly described in Section 5.3.3 and 5.3.4. Transport of protons
through such types of protic neutral salts has been reported (Noda, 2003; Nakamoto,
2007). Despite lower water uptake of SPDAB•Im-N than SPDAB•TA-N, former shows
higher proton conductivity than that of latter. One of the possible reasons for this
phenomenon can be attributed to its higher IEC (1.49) than SPDAB•TA-N (0.39). In
addition, more acidic nature, better cationic stability and solvation of imidazolium cation
(ImH+) in water should also enhance the proton conductivity of SPDAB•Im-N. However,
conduction in these salt membranes is due to the ionic transport or by proton transport

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
161
remains ambiguous. It is reported that conductivity of SPI in proton form is 300 times
larger as compared with triethylammonium form (Nakayama, 2003; Ueda, 2007). Cornet
et al. (2000) studied the transport of different sizes of ammonium ion for SPIs in neutral
form and observed the decrease in ionic conductivity with increase in ionic size. Few
others have observed that SPI in -SO3H form exhibited higher conductivity than that of
SPIs in neutral form due to the lower mobility of cations in the latter as compared with
higher mobility of H+ in the former (Cornet, 2000; Marestin, 2008). Takahashi et al.
(1976) reported amine based-protonic acid system wherein complexes of
triethylenediamine and hexamethylenetriamine with H2SO4 demonstrated proton
conductivity.
Higher conductivity of SPDAB•Im-N may also be attributed to the higher ability
of ImH+ to form H-bonding with water molecules due to more number of hydrogen atoms
in it. Higher dissociation of SPDAB•Im-N (K2D > K1
D, Figure 5.2) leads to the better
solvation of ImH+ in aqueous media facilitating H-bonding with water molecules. While
in TEAH+, possibility of H-bonding becomes less due to the three hydrophobic ethyl
groups and lower dissociation of SPDAB•TA-N, which has previously been explained in
Section 5.3.4. Furthermore, TEA (pKa=10.75) (Kailani, 1988b) is more basic than
imidazole (pKa= 7.1) (Clayden, 2001; Lv, 2008), and therefore the conjugate Bronsted
acid-imidazolium cation (ImH+) formed in the aqueous media should be more acidic than
triethylammonium cation (TEAH+) resulting in increased conductivity for SPDAB•Im-N.
However, if the surface of the membrane of SPDAB•Im-N could be smooth instead of
wavy like that of SPDAB•TA-N, conductivity of the former could have also been
increased.
We have also calculated the energy of activation (Ea) for the proton conductivity
of SPIs in -SO3H form and compared this with that of Nafion-117 from RT to 80 C.
Within this temperature range, the conductivity follows the following Arrhenius equation.
RT
Eao exp
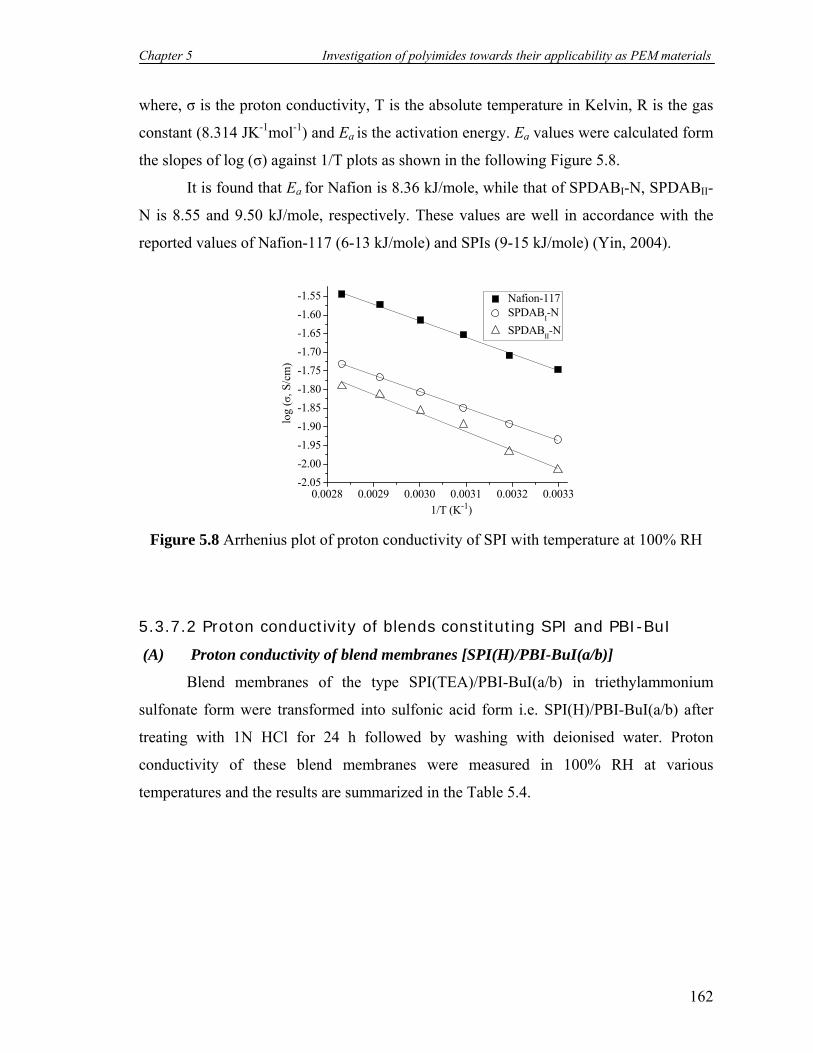
Chapter 5 Investigation of polyimides towards their applicability as PEM materials
162
where, σ is the proton conductivity, T is the absolute temperature in Kelvin, R is the gas
constant (8.314 JK-1mol-1) and Ea is the activation energy. Ea values were calculated form
the slopes of log (σ) against 1/T plots as shown in the following Figure 5.8.
It is found that Ea for Nafion is 8.36 kJ/mole, while that of SPDABI-N, SPDABII-
N is 8.55 and 9.50 kJ/mole, respectively. These values are well in accordance with the
reported values of Nafion-117 (6-13 kJ/mole) and SPIs (9-15 kJ/mole) (Yin, 2004).
0.0028 0.0029 0.0030 0.0031 0.0032 0.0033-2.05
-2.00
-1.95
-1.90
-1.85
-1.80
-1.75
-1.70
-1.65
-1.60
-1.55 Nafion-117 SPDAB
I-N
SPDABII-N
1/T (K-1)
log
(S
/cm
)
Figure 5.8 Arrhenius plot of proton conductivity of SPI with temperature at 100% RH
5.3.7.2 Proton conductivity of blends constituting SPI and PBI-BuI
(A) Proton conductivity of blend membranes [SPI(H)/PBI-BuI(a/b)]
Blend membranes of the type SPI(TEA)/PBI-BuI(a/b) in triethylammonium
sulfonate form were transformed into sulfonic acid form i.e. SPI(H)/PBI-BuI(a/b) after
treating with 1N HCl for 24 h followed by washing with deionised water. Proton
conductivity of these blend membranes were measured in 100% RH at various
temperatures and the results are summarized in the Table 5.4.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
163
Table 5.4 Proton conductivity of blend membranes in humidified conditions
Proton conductivity (σ) at 100 % RH (S.cm-1) Temp.
(C) SPI(H)/PBI-BuI(50/50) SPI(H)/PBI-BuI(40/60) SPI(H)/PBI-BuI(30/70)
30 2.25 X 10-5 1.34 X 10-5 2.93X 10-5
40 3.31 X 10-5 1.44 X 10-5 2.99 X 10-5
50 3.54 X 10-5 1.53 X 10-5 3.22 X 10-5
60 3.82 X 10-5 1.74 X 10-5 3.19 X 10-5
70 2.03 X 10-5 1.42 X 10-5 2.58 X 10-5
80 1.77 X 10-5 1.31 X 10-5 2.03 X 10-5
90 1.48 X 10-5 1.26 X 10-5 1.91 X 10-5
100 1.14 X 10-5 1.01 X 10-5 1.87 X 10-5
The proton conductivity obtained is in the range of 10-5 S.cm-1, which is lower
than that of SPIs. It is observed that conductivity initially increases with increase in
temperature till temperature reaches up to 50 or 60 C and decreases with further increase
in temperature. These blend membranes exhibit lower proton conductivity in the order of
10-5 S.cm-1 in 100% RH. This may be attributed to the low content of SPI in blends.
However, from Table 5.3, it can be observed that number of –SO3H groups are almost
more than 3 times lower than that of the benzimidazole groups for the blend constituting
same weight percentage of SPI and PBI-BuI and number of –SO3H groups further
decrease with increase in PBI-BuI percentage. Thus, for a single –SO3H group more than
three benzimidazole moieties are present and it reaches to almost 7.85 in the case of
SPI(H)/PBI-BuI(30/70). Since –SO3H group are the major contributors for H+ in the
humidified atmosphere, its decreasing concentration should lower the proton
conductivity. It has been reported that, for the blends of sulfonated PEEK and PBI, that
the conductivity decreases with increase in PBI content also (Zhang, 2008). Proton
conductivity of the order of 10-2 S.cm-1, for the blends with maximum 20 wt% of PBI has
been reported. In the present case PBI content is more than 50 wt%. We have already
explained (Section 5.3.4 and Section 3.5.3.2 of Chapter 3) that these blends form weak
interaction of –SO3H and benzimidazole functionality resulting in exchangeable H+ from
–SO3H group of SPI responsible for proton transport in aqueous medium. It is known

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
164
that, proton transports in sulfonated polymer is by vehicular mechanism, i.e. through the
displacement of hydronium ion (H3O+) (Mauritz, 2004) and for better transportation of
H+ excellent connectivity of hydrophilic channels i.e. –SO3H group is a vital. Hydrophilic
channels formation requires sufficiently high content of sulfonic acid groups and water.
However in the present blends, understudy, low content of sulfonic acid groups and low
water uptake due to the presence of hydrophobic t-butyl group in PBI-BuI, adversely
affect formation of hydrophilic channels, which explains observed proton conductivity in
the blends.
Considering these facts, we therefore expected that, rather than proton
transportation as hydronium ion (H3O+), these blend membranes may show better
performance by acid-base interactions or hopping mechanism. Hence we decided to dope
these membranes with acids. Proton conductivity of these blend membranes doped with
12M H3PO4 in anhydrous condition and 1M H3PO4 in the humidified conditions (100%
RH), at various temperatures was measured.
(B) Proton conductivity of polymer electrolyte membranes doped with 12 M H3PO4
Proton conductivity (σ) of membranes doped with 12 M H3PO4 was measured at
various temperatures in anhydrous condition. Membranes viz., SPIs (SPDAB•TA-N and
SPDAB•Im-N), polybenzimidazoles (PBI-I and PBI-BuI) and blends [SPI(PA)/PBI-
BuI(a/b)] were doped with 12 M H3PO4 and evaluated for proton conductivity
measurements.
(I) Proton conductivity of SPIs (SPDAB•TA-N and SPDAB•Im-N) doped with 12 M
H3PO4
Proton conductivity (σ) of SPDAB•TA-N and SPDAB•Im-N, doped with 12M
H3PO4 was measured at various temperatures (Figure 5.9). It can be seen that proton
conductivity increases with temperature till 100 C and then it decreases. At room
temperature proton conductivity of SPDAB•TA-N is 6.20 X 10-4 S.cm-1, which increases
to 2.66 X 10-3 S.cm-1 at 100 C and decreases further continuously with increase in
temperature. The same pattern of proton conductivity is observed in SPDAB•Im-N.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
165
40 60 80 100 120 140 160 180
5.0x10-4
1.0x10-3
1.5x10-3
2.0x10-3
2.5x10-3
3.0x10-3
Temp. (OC)
Con
d. (,
S/c
m)
SPDAB·TA-N SPDAB·Im-N
Figure 5.9 Proton conductivity of SPIs doped with 12M H3PO4 against temperature
In general, acid doped basic polymers, such as PBI, transports proton by a
hopping or Grotthus mechanism. In H3PO4 doped PBI, the basic imidazole abstracts the
proton from doped acid generating H2PO4¯ as a conjugate base, transporting proton
through acid-base moieties. However, in SPI imide linkage possess N-heterocycle, which
is a much weaker base than imidazole group due to the strong electron withdrawing effect
of its neighboring two carbonyl groups. As a result, in H3PO4 doped SPI, extent of
dissociation of H3PO4 generating H2PO4¯ becomes less therefore transport of proton by
Grotthus mechanism is doubtful. Pu et al. (2005) reported the H3PO4 doped polyimide as
anhydrous PEM and mentioned proton transport through H-bonding. The present SPI
doped with H3PO4 can also transport through such H-bonding as the carbonyl group, the
oxygen attached through pendant sulfophenoxy group and oxygen belonging to –SO3H
group can form H-bonding with the acid H3PO4. Besides, –SO3H group present in SPI
can also contribute to the proton conductivity. However, at higher temperature although
proton conductivity decreases, it is still higher than that of lower temperatures viz., RT
and 50 C. This can be attributed to the increased dissociation of H3PO4 as well as –SO3H

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
166
group at higher temperature than that of at lower temperature which can enhance the
proton conductivity. In addition, these membranes were doped with lower concentration
of H3PO4 (12M) and dried in a vacuum oven at 80 C hence complete removal of water
can also be not ascertained. As a result, role of water in proton conduction can also be not
neglected. In summary, doped H3PO4, –SO3H group and presence of small concentration
of conjugate base H2PO4¯ and water can assist in proton transportation.
It should be noted that, SPDAB•Im-N shows lower proton conductivity than that
of SPDAB•TA-N despite having same phosphoric acid uptake (≈65-68%). This
difference of proton conductivity can be attributed to the wavy nature of membrane of
SPDAB•TA-N, because of which proper electrode and electrolyte contact could not have
been achieved. The same decrease in proton conductivity in case of SPDABII-N than that
of SPDABI-N due to the wavy nature of the former was observed and explained
previously in Section 5.3.7.1.
(II) Proton conductivity of PBIs (PBI-I and PBI-BuI) and blend membranes
[SPI(PA)/PBI-BuI(a/b)] doped with 12M H3PO4
Proton conductivity of blend membranes of the type SPI(PA)/PBI-BuI(a/b) and
PBIs; PBI-BuI and PBI-I doped with 12M H3PO4 were measured in anhydrous conditions
at various temperatures. Membrane resistance (RΩ) is shown in Table 5.5 while proton
conductivity trend with temperature is seen Figure 5.10. It can be seen that, proton
conductivity is in the range of 2.07 X 10-4 to 1.12 X 10-2 S/cm, which varies with both
acid uptake and temperature. For all the membranes, it increases from room temperature
to 150 °C and further decreases at 175 °C. For example SPI(PA)/PBI-BuI(30/70) shows
proton conductivity of 5.78 X 10-4 S/cm at room temperature which increases to 9.86 X
10-3 S/cm at 150 °C and further decreases to 4.97 X 10-3 S/cm at 175 °C (Figure 5.10).
The decrease in proton conductivity at 175 °C can be attributed to the self-dehydration of
phosphoric acid at this temperature to the less conductive pyrophosphoric acid (Lobato,
2007). In general, for the H3PO4 acid doped membranes such as PBI, proton conductivity
increases with the uptake of phosphoric-acid due to the increase in proton carriers (Ma,
2004b).

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
167
Table 5.5 Change in RΩ for the blends and PBIs with temperature
RΩ in ohms Temp.
( °C) SPI(PA)/PBI-
BuI(50/50)
SPI(PA)/PBI-
BuI(40/60)
SPI(PA)/PBI
-BuI(30/70) PBI-BuI PBI-I
RT 27.51 13.1 10.75 5.89 13
50 8.25 5.53 3.97 2.24 3.8
75 2.8 1.80 1.67 0.89 1.84
100 1.35 1.11 1.06 0.68 1.65
125 0.89 0.86 0.76 0.57 1.54
150 0.76 0.76 0.63 0.52 1.45
175 1.28 1.03 1.25 1.11 1.65
From the Table 5.5, it can be seen that membrane resistance (RΩ) decreases from
RT to 150 °C and further increases at 175 °C. It is also noticed that, among all
membranes, PBI-BuI shows the least values for RΩ at all temperatures indicating its
higher proton conductivity. For example, as shown in Figure 5.10, proton conductivity of
PBI-BuI at RT is 9.86 X 10-4 S/cm which increases till 150 °C to 1.12 X 10-2 S/cm and
further decreases to 5.23 X 10-3 S/cm at 175 °C. On the other hand, PBI-I demonstrates
the highest RΩ, particularly above the temperature 75 °C. In other words, blend
membrane also show higher proton conductivity than PBI-I despite having their lower
uptake of H3PO4. From the Table 5.2, it is seen that acid uptake of PBI-I is 150.4 wt%
and that of PBI-BuI is 165.2 wt%. While that of blend membranes is in the range of 94.7
to 109.4 wt% only. Considering H3PO4 uptake, the maximum number of moles of acid
taken per chain repeating unit (CRU) for PBI-I and PBI-BuI is 4.7 and 6.2, respectively.
Though the membranes were heated at 80 °C under reduced pressure, the complete
removal of water can not be ascertained. As a result, moles of acid uptake for these PBIs
should definitely be lower than these values (4.7 and 6.2).
Both acid uptake and RH were found to have significantly effect on proton
conductivity of PBI doped with H3PO4 (Wainright, 1995; Li, 2001; He, 2003; Ma,
2004b). Proton conductivity increases with increase in acid uptake and RH (Fontella,
1998). Ma et al. (2004a) obtained proton conductivity value of 4.7 X 10-3 S/cm at 5% RH

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
168
and 5.9 X 10-2 S/cm at 30% RH at 150 °C temperature. Li et al. (2004) also observed a
higher proton conductivity value with an increase in RH at a H3PO4 doping level of 5.6
moles per CRU of PBI-I.
40 60 80 100 120 140 160 1800.0
2.0x10-3
4.0x10-3
6.0x10-3
8.0x10-3
1.0x10-2
Con
d. (,
S/c
m)
SPI(PA)/PBI-BuI(50/50) SPI(PA)/PBI-BuI(40/60) SPI(PA)/PBI-BuI(30/70) PBI-BuI PBI-I
Temp. (OC)
Figure 5.10 Variation of proton conductivity of blend membranes and PBI-BuI doped
with 12 M H3PO4 at different temperature
From the Table 5.2, it can be clearly seen that, acid uptake decreases in the order
PBI-BuI (165.2 wt%) > PBI-I (150.5 wt%) > SPI(PA)/PBI-BuI(30/70) (109.4 wt%) >
SPI(PA)/PBI-BuI(40/60) (100.3 wt%) > SPI(PA)/PBI-BuI(50/50) (94.7 wt%). In the
present study, it is also observed that, proton conductivity increases sharply from RT to
75 °C while increase in proton conductivity from 75 °C to 150 °C is smooth. This
behavior is particularly observed in case of PBI-I (Figure 5.10). Since we doped the
membranes with lower concentration of H3PO4 (12 M) and dried the membrane in
vacuum oven below 100 °C i.e. at 80 °C, presence of water in the membrane can not be
neglected. Hence, we attribute the proton transportation to be governed by water as well
as H3PO4. In other words, in the temperature range from RT to 75 °C, transportation of
proton could be by both vehicular and Grotthus mechanism. While above the 75 °C, role
of water for proton transportation becomes insignificant while that of H3PO4 becomes
prominent resulting in proton transportation by Grotthus mechanism. The pattern of

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
169
proton conductivity of the present blend membranes doped with H3PO4 can be better
illustrated by electrochemical impedance spectroscopy (EIS) analysis as discussed below
(Figure 5.11).
0 1500 3000 4500 60000
1000
2000
3000
4000
5000
0 20 40 60 800
7
14
21
28
35
175 oC
150 oC125 oC
100 oC
75 oC 50 oC
RT
Z'
Z"
SPI(PA)/PBI-BuI(50/50) in 12M H3PO4
Z''
Z'
75 oC
50 oC
RT
0 1000 2000 3000 4000
0
1000
2000
3000
4000
5000
0 20 40 600
4
8
12
16
20
175 oC
150 oC
125 oC
100 oC
75 oC 50 oC
RT
Z"
Z'
Z''
Z'
SPI(PA)/PBI-BuI(40/60) in 12M H3PO4
75 oC
50 oC
RT
0 500 1000 1500 20000
250
500
750
1000
1250
0 12 24 36 48 600
4
8
12
16
20
Z"
Z'
175 oC
150 oC
125 oC
50 oC75 oC100 oC
RT
50 oC
RT
Z''
Z'
SPI(PA)/PBI-BuI(30/70) in 12M H3PO4
75 oC
0 200 400 600 800 1000
0
300
600
900
1200
1500
0 50 100 1500
25
50
75
Z"
Z'175 oC
150 oC
125 oC
75 oC
100 oC
RT50 oC PBI-BuI in 12M H3PO4
Z''
Z'
75 oC
50 oC
RT
0 200 400 600 800 10000
500
1000
1500
2000
0 10 20 300
10
20
30
40
Z"
Z'
100 oC75 oC
50 oC
125 oC 175 oC
150 oC
RT
PBI-I in 12M H3PO4
Z''
Z'
125 oC
100 oC
75 oC
50 oC
RT
Figure 5.11 Impedance spectra of blend membranes doped with 12M H3PO4
Although 12 M H3PO4 uptake of blend membranes was lower than that of PBI-I,
the former shows the least value for RΩ, in particular above 75 °C (Table 5.5). This can
be attributed to the probable participation of H+ from –SO3H group belonging to SPI in
the proton conductivity of the blend membrane. It is possible to have proton donation
from the –SO3H group of SPI to the imidazole or conjugate base of (H2PO4-) in the blend
components resulting in decrease in RΩ. In the present case, we have determined the

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
170
conductivity in anhydrous conditions, hence only the water present in 12 M H3PO4
should be considered. It is known that water promotes dissociation of H3PO4 (Ma,
2004a). In the PBIs (PBI-I and PBI-BuI), H3PO4 can only be dissociated by both, basic
PBI and water. However, PBI is more basic, it contributes to the larger dissociation of
H3PO4 than water as shown in following equation.
Proton transportation in PBI is due to the H3PO4, H2PO4¯ and water. On the other
hand, in case of blend system, along with H3PO4, sulfonic group can also be dissociated
due to PBI and water. In addition, the conjugate base formed –SO3¯ can also take the H+
from H3PO4 while that of another conjugate base H2PO4¯ can also take up H+ from –
SO3H of SPI in the blend. This can be better illustrated as follows (Scheme 5.1).
Scheme 5.1 Probable exchange of protons in 12M H3PO4 doped blend membranes.
Moreover, the difference in pKa of –SO3H group of SPI and 12M H3PO4 should
also govern the proton conductivity in blend membranes. Nevertheless, from the Table
5.3, the number of benzimidazole groups is almost more than 3 times higher than that of
–SO3H group even for the same weight percentage of blend. Hence proton conductivity
would be largely dependant on benzimidazole moieties, which is known to transport it by
hopping mechanism. In this way, in blend membranes proton conductivity is contributed
by H3PO4, –SO3H group, H2O, H3O+, H2PO4¯, –SO3¯, PBI and PBI-H+ species. At higher
temperature, i.e. above 75 °C role of H2O and H3O+ diminishes due to loss of water.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
171
Table 5.6 Change in Rf for the blends and PBIs with temperature
Temp.
( °C)
SPI(PA)/PBI-
BuI(50/50)
SPI(PA)/PBI-
BuI(40/60)
SPI(PA)/PBI-
BuI(30/70) PBI-BuI PBI-I
RT -- --- 3769.25 -- >10k
50 2702.75 4061.47 830.03 1960.76 >10k
75 597.2 506.2 195.33 189.11 >10k
100 86.65 73.89 54.94 52.32 >10k
125 50.11 52.14 23.24 44.43 >10k
150 38.24 26.24 17.37 34.48 >10k
175 45.72 38.97 29.75 39.89 >10k
From the Table 5.6 and the above Figure 5.11, it can be seen that with increasing
PBI-BuI content in the blend, Rf decreases in particular in the range of 75-175 °C. In case
of PBI-I, Rf values are too large hence they are not discussed in detail. Since the acid
uptake would be commensurate with the PBI-BuI content in the blend and at higher
H3PO4 uptake the proton diffusion happens with the least resistant path resulting in more
facile proton conduction. As a result, semicircle with least Rf can be obtained in 12M
H3PO4 blend membrane, which is shown by SPI(PA)/PBI-BuI(30/70) blend. It is worth to
mention that, though phosphoric acid uptake is lower for blend membrane than that of
PBI-I, the blend membrane shows a semicircle with less diameter than that of PBI-I. This
indicates the active participation of the –SO3H moiety in the proton transportation from
one of the blend components; SPI. It is also noticed that with increasing temperature, Rf
decreases till 150 °C i.e. proton conductivity increases. This is obvious as the diffusion of
the proton would become faster at higher temperatures. However, at the highest
temperature (175 °C) measured, the Rf increases indicating the conversion of phosphoric
acid to more resistive pyro phosphoric acid that results in reduced conductivity and
charge transport at this temperature. (Lobato, 2007).

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
172
0 15 30 45 60 75 900
10
20
30
Z''
Z'
SPI(PA)/PBI-BuI(50/50)
SPI(PA)/PBI-BuI(40/60)
SPI(PA)/PBI-BuI(30/70)
PBI-BuI
PBI-I
Figure 5.12 Comparative EIS spectra of blends and PBIs doped with 12M H3PO4 at 100 °C
From the above Figure 5.12, it can be seen that, at 100 °C, among the 12 M
H3PO4 doped membranes, PBI-BuI shows the semicircle with least diameter indicating
lower Rf value. With increasing SPI content however the diameter of the semicircle
increases demonstrating higher hindrance for proton transport. This fact can be supported
from the lower uptake of phosphoric acid with increasing SPI content coupled with
dehydration of the membrane that effectively removes the -SO3H moieties from the
channel network (OR) contributing towards proton transport.
0.0024 0.0026 0.0028 0.0030 0.0032
-1.2
-0.9
-0.6
-0.3
0.0
0.3
0.6
SPI(PA)/PBI-BuI(50/50) SPI(PA)/PBI-BuI(40/60) SPI(PA)/PBI-BuI(30/70) PBI-BuI PBI-I
1/T (K-1)
log
[(S
/cm
) X
T(K
)]
Figure 5.13 Arrhenius plot of proton conductivity of 12 M H3PO4 doped membranes
against temperature

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
173
For 12M H3PO4 doped membranes, we used the following Arrhenius equitation in
order to determine the temperature dependence of proton conductivity.
RT
E
T
A
RT
E aa expexp0
Figure 5.13 shows log (σT) against 1/T curves for 12M H3PO4 doped membrane
for temperatures from 30 °C to 150 °C. It is observed that proton conductivity follows the
non-Arrhenius behavior as inferred from the non-linear curves. This may be attributed to
the enhanced segmental mobility of the polymer chains after doping with acid as well as
with increase in temperature.
In all the blends, benzimidazole group content is more than sulfonic acid groups
on molar basis (Table 5.3). In these blends benzimidazole groups are not neutralized by
sulfonic acid groups and they may not contribute to proton conductivity without doping.
On doping with high molar concentration (12M) of H3PO4, H3PO4 acts as vehicle for H+
transport by Grotthus mechanism in an anhydrous condition. To understand, H+ behavior
with a low acid content of blends sufficient to neutralize free benzimidazole groups,
study was conducted to determine the proton conductivity by doping with low
concentration of two acids; 1M H3PO4 and 1M H2SO4. Proton conductivity of these blend
membranes were measured particularly in 100% RH since low proton conductivity in
anhydrous condition due to lower concentration of these acids was expected.
(C) Proton conductivity of blend membranes and PBI-BuI doped with 1M H2SO4
and 1M H3PO4
Blend membranes were separately dipped in 1M H2SO4 and 1M H3PO4 for three
days and then directly used for the proton conductivity analysis with 100% RH at various
temperatures.
(i) Proton conductivity of blend membranes [SPI(SA)/PBI-BuI(a/b)] and PBI-BuI
doped with 1M H2SO4
The results of the membrane resistance (RΩ) are summarized in Table 5.7. Plot of
proton conductivity against temperature can be seen from the Figure 5.14.
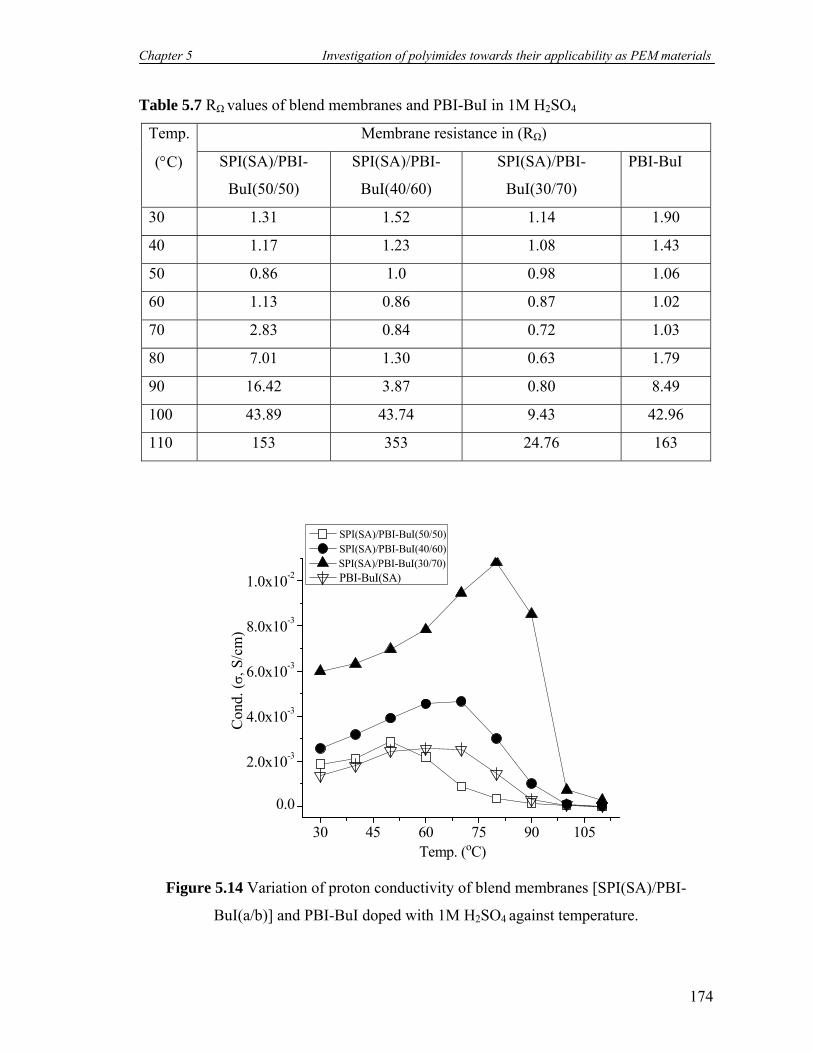
Chapter 5 Investigation of polyimides towards their applicability as PEM materials
174
Table 5.7 RΩ values of blend membranes and PBI-BuI in 1M H2SO4
Membrane resistance in (RΩ) Temp.
(C) SPI(SA)/PBI-
BuI(50/50)
SPI(SA)/PBI-
BuI(40/60)
SPI(SA)/PBI-
BuI(30/70)
PBI-BuI
30 1.31 1.52 1.14 1.90
40 1.17 1.23 1.08 1.43
50 0.86 1.0 0.98 1.06
60 1.13 0.86 0.87 1.02
70 2.83 0.84 0.72 1.03
80 7.01 1.30 0.63 1.79
90 16.42 3.87 0.80 8.49
100 43.89 43.74 9.43 42.96
110 153 353 24.76 163
30 45 60 75 90 105
0.0
2.0x10-3
4.0x10-3
6.0x10-3
8.0x10-3
1.0x10-2
Con
d. (S
/cm
)
SPI(SA)/PBI-BuI(50/50) SPI(SA)/PBI-BuI(40/60) SPI(SA)/PBI-BuI(30/70) PBI-BuI(SA)
Temp. (oC)
Figure 5.14 Variation of proton conductivity of blend membranes [SPI(SA)/PBI-
BuI(a/b)] and PBI-BuI doped with 1M H2SO4 against temperature.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
175
From the above Table 5.7, RΩ decreases with increase in temperature up to a
certain temperature and there after it increases. As the PBI-BuI content increases, RΩ
decreases becoming less with increasing temperature. Thus, maximum proton
conductivity obtained at higher temperature for the blend membrane having higher
content of PBI-BuI. From the above Figure 5.14, among the blend membranes
conductivity becomes higher at elevated temperature with increasing content of PBI-BuI.
For example, at 70, 80 and 90 °C conductivity decreases in the order SPI(SA)/PBI-
BuI(30/70) > SPI(SA)/PBI-BuI(40/60) > SPI(SA)/PBI-BuI(50/50). Thus, at 90 °C
conductivity values for SPI(SA)/PBI-BuI(30/70), SPI(SA)/PBI-BuI(40/60) and
SPI(SA)/PBI-BuI(50/50) are 8.52 X 10-3, 1.01 X 10-3 and 1.50 X 10-4 S/cm, respectively.
This result shows that at higher temperature the presence of benzimidazole groups
dominates proton conduction, which can possibly arise due to the exchange of proton
from H2SO4 to the benzimidazole moieties. However, PBI-BuI shows lower proton
conductivity than that of blend membranes, in particular for SPI(SA)/PBI-BuI(40/60) and
SPI(SA)/PBI-BuI(30/70) from RT to 100 °C indicating the optimum quantity of SPI in
the blend for better proton conductivity after being doped with 1M H2SO4. In other words
our attempt to make the blends for enhancing proton conductivity, is revealed, these
blends was doped with 1M H2SO4. However, at further higher temperatures (100 and 110
°C) proton conductivity suddenly decreases. This can possibly due to the loss of water
indicating that water is an essential medium for transportation of protons. The results are
better illustrated from the Nyquist EI plots that describe the trend.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
176
0 200 400 600 800 10000
200
400
600
800
1000
1200
0.5 1.0 1.5 2.0 2.5 3.00
1
2
3
4
5
6
70 oC
60 oC
50 oC
40 oC
RTZ''
Z'
SPI(SA)/PBI-BuI(50/50) in 1M H2SO4
100 oC
90 oC
80 oC
70 oC
60 oC
50 oC40 oC
RT
Z''
Z' 0 200 400 600 800 1000 1200 14000
500
1000
1500
2000
2500
0.8 1.0 1.2 1.4 1.60
1
2
3
Z''
Z'
80 oC
70 oC 60 oC
50 oC
40 oC
RT
60 oC
50 oC
40 oC
Z''
SPI(SA)/PBI-BuI(40/60) in 1M H2SO4
100 oC
90 oC
80 oC
70 oC
RT
Z'
0 50 100 150 200 250 300 350 4000
200
400
600
800
1000
1200
1400
0.6 0.8 1.0 1.2 1.40.0
0.6
1.2
1.8
2.4
3.0
Z''
Z'
90 oC
80 oC70 oC
60 oC
50 oC
40 oCRT
SPI(SA)/PBI-BuI(30/70) in 1M H2SO4
Z''
Z'
100 oC
80 oC
70 oC
50 oC
40 oCRT
0 100 200 300 400 500 6000
200
400
600
800
1000
1200
1400
1600
1.0 1.2 1.4 1.6 1.8 2.00.0
0.4
0.8
1.2
1.6
2.0
Z''
Z'
80 oC
70 oC60 oC
50 oC40 oC
RT
PBI-BuI in 1M H2SO4
Z''
Z'
100 oC
90 oC
50 oC
70 oC
60 oC
80 oC
40 oC RT
Figure 5.15 Impedance spectra of blend membranes and PBI-BuI doped with in 1 M
H2SO4
From above Figure 5.15, it is seen that only SPI(SA)/PBI-BuI(50/50) blend
membrane having higher SPI content gives the semicircle at high temperatures, which
indicates the charge transport of controlled behavior (proton, Rf). Increasing PBI-BuI
content in blends results in the disappearance of semicircle which indicates a less facile
transport of proton. However, on the other hand, increasing the temperature also
improves the proton transport that results in a semicircle at low frequency range. In case
of SPI(SA)/PBI-BuI(50/50), the semicircle can only be observed at higher temperature
(70, 80 and 90 °C) and diameter of which become less at 90 °C indicating the facile
diffusion of proton at these temperatures. However, at this temperature (90 °C), RΩ
increases with rapid decrease in proton conductivity. At further higher temperature (100
°C), not only RΩ increases but the diameter of the semicircle is also enhanced indicating
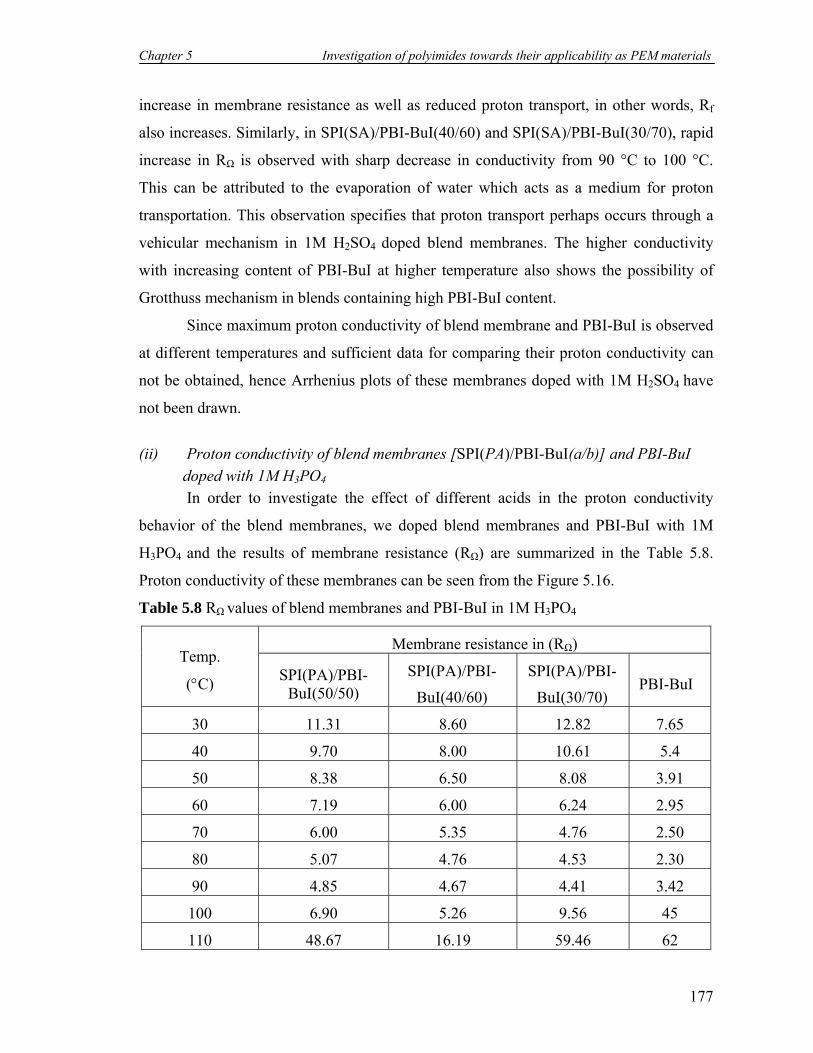
Chapter 5 Investigation of polyimides towards their applicability as PEM materials
177
increase in membrane resistance as well as reduced proton transport, in other words, Rf
also increases. Similarly, in SPI(SA)/PBI-BuI(40/60) and SPI(SA)/PBI-BuI(30/70), rapid
increase in RΩ is observed with sharp decrease in conductivity from 90 °C to 100 °C.
This can be attributed to the evaporation of water which acts as a medium for proton
transportation. This observation specifies that proton transport perhaps occurs through a
vehicular mechanism in 1M H2SO4 doped blend membranes. The higher conductivity
with increasing content of PBI-BuI at higher temperature also shows the possibility of
Grotthuss mechanism in blends containing high PBI-BuI content.
Since maximum proton conductivity of blend membrane and PBI-BuI is observed
at different temperatures and sufficient data for comparing their proton conductivity can
not be obtained, hence Arrhenius plots of these membranes doped with 1M H2SO4 have
not been drawn.
(ii) Proton conductivity of blend membranes [SPI(PA)/PBI-BuI(a/b)] and PBI-BuI doped with 1M H3PO4
In order to investigate the effect of different acids in the proton conductivity
behavior of the blend membranes, we doped blend membranes and PBI-BuI with 1M
H3PO4 and the results of membrane resistance (RΩ) are summarized in the Table 5.8.
Proton conductivity of these membranes can be seen from the Figure 5.16.
Table 5.8 RΩ values of blend membranes and PBI-BuI in 1M H3PO4
Membrane resistance in (RΩ) Temp.
(C) SPI(PA)/PBI-
BuI(50/50)
SPI(PA)/PBI-
BuI(40/60)
SPI(PA)/PBI-
BuI(30/70) PBI-BuI
30 11.31 8.60 12.82 7.65
40 9.70 8.00 10.61 5.4
50 8.38 6.50 8.08 3.91
60 7.19 6.00 6.24 2.95
70 6.00 5.35 4.76 2.50
80 5.07 4.76 4.53 2.30
90 4.85 4.67 4.41 3.42
100 6.90 5.26 9.56 45
110 48.67 16.19 59.46 62

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
178
From the Table 5.8, among the blend membranes, membrane resistance (RΩ)
decreases with increase in temperature till 90 °C in 100% RH. However, RΩ increases
with a sharp increase in resistance from 100 °C to 110 °C. While comparing the values of
RΩ of blend membrane with that of PBI-BuI, latter shows lesser RΩ up to 90 °C. This
behavior is exactly the reverse of results seen in membrane doped with 1M H2SO4, where
PBI-BuI exhibits higher RΩ than that of blend membrane. This shows that presence of –
SO3H group in the blend improves the proton conductivity as compared with that of PBI-
BuI on doping with 1M H2SO4. While, opposite behavior is observed when membranes
are doped with 1M H3PO4. It should also be noted that, comparing RΩ between
membranes doped with 1M H2SO4 and 1M H3PO4, former shows much lower RΩ than
latter. This can be due to higher dissociation constant of the H2SO4 (pKa: -3) than H3PO4
(pKa: 2.15). As a result, availability of [H+] is more in 1M H2SO4 than that of 1M H3PO4,
resulting in more number of protonated benzimidazole moieties on doping with former
than that of latter. Proton conductivity of these membranes doped with 1M H3PO4 is
shown in the following Figure 5.16.
30 40 50 60 70 80 90 100 1100.0
5.0x10-4
1.0x10-3
1.5x10-3
2.0x10-3
SPI(PA)/PBI-BuI(50/50) SPI(PA)/PBI-BuI(40/60) SPI(PA)/PBI-BuI(30/70) PBI-BuI
Temp. (oC)
Con
d. (S
/cm
)
Figure 5.16 Variation of proton conductivity of blend membranes and PBI-BuI doped
with 1M H3PO4 with temperature

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
179
From the Figure 5.16, it is seen that PBI-BuI shows higher proton conductivity
than that of blend membranes, in particular from RT to 90 °C. For example proton
conductivity at 60 °C for PBI-BuI, SPI(PA)/PBI-BuI(30/70), SPI(PA)/PBI-BuI(40/60)
and SPI(PA)/PBI-BuI(50/50) is 1.58 X 10-3, 8.02 X 10-4, 5.12 X 10-4 and 3.95 X 10-4
S/cm, respectively. In this case, conductivity increases with increase in PBI-BuI content,
being highest for PBI-BuI. This is because of the increase in protonated benzimidazole
moieties. This also indicates that PBI-BuI is the main contributor for proton
transportation while SPI plays the supporting role by efficiently interconnecting the
channels with –SO3H groups after doping with 1M H3PO4. This further supports the fact
that proton transport between basic benzimidazole and acidic H3PO4 is presumably by
exchange which suggests a Grotthus type mechanism. However, conductivity decreases
above 90 °C, demonstrating strong dependence on water also.
0.0027 0.0028 0.0029 0.0030 0.0031 0.0032 0.0033
-3.6
-3.5
-3.4
-3.3
-3.2
-3.1
-3.0
-2.9
-2.8
-2.7
log
(,
S/c
m)
1/T (K-1)
SPI(PA)/PBI-BuI(50/50) SPI(PA)/PBI-BuI(40/60) SPI(PA)/PBI-BuI(30/70) PBI-BuI
Figure 5.17 Arrhenius plot of proton conductivity of 1M H3PO4 doped membranes with temperature
We have attempted to investigate the proton conductivity behavior of blend
membranes on doping with 1M H3PO4, by using following Arrhenius equation
RT
Eao exp

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
180
From the above Figure 5.17, it is observed that PBI-BuI and SPI(PA)/PBI-
BuI(30/70) shows a slight deviation from Arrhenius type of behavior for proton transport
when doped with 1M H3PO4. While the blends SPI(PA)/PBI-BuI(40/60) and
SPI(PA)/PBI-BuI(50/50) show Arrhenius type of behavior with Ea values of 10 and 14
kJ/mole, respectively. This indicates that with increase in SPI content, proton
conductivity is also governed by sulfonic acid group content in the blend even when
doped with 1M H3PO4. In other words, with increasing SPI content in the blend, it shows
Arrhenius behavior.
Following Figures 5.18 represents EIS spectra of the blend membranes when
doped with 1M H3PO4, which can shed more light on proton transportation.
0 200 400 600 800 10000
300
600
900
1200
1500
1800
4 6 8 10 120
4
8
12
16
20
Z''
Z'
90 oC
100 oC
80 oC
70 oC
60 oC
50 oC
40 oC
RT
RT
100 oC
90 oC
80 oC
70 oC
60 oC
50 oC
40 oC
SPI(PA)/PBI-BuI(50/50) in 1M H3PO
4
Z''
Z'0 100 200 300 400 500 600 700
0
300
600
900
1200
5 6 7 8 90
2
4
6
8
10
Z''
Z'
100 oC
90 oC
80 oC
70 oC
60 oC
50 oC
40 oC
RT
110 oC
100 oC
90 oC
80 oC
40 oC
70 oC
60 oC
SPI(PA)/PBI-BuI(40/60) in 1M H3PO
Z''
Z'
50 oCRT
0 200 400 600 800 1000 1200 14000
200
400
600
800
1000
1200
1400
4 6 8 10 12 140
4
8
12
16
20
100 oC
90 oC
80 oC
70 oC
60 oC
50 oC
40 oC
RT
Z''
Z'
100 oC
90 oC
80 oC
70 oC
60 oC
50 oC
40 oC
RT
SPI(PA)/PBI-BuI(30/70) in 1M H3PO4
Z''
Z' 0 250 500 750 1000 12500
300
600
900
1200
1500
1800
2 3 4 5 6 7 80.0
0.6
1.2
1.8
2.4
3.0
Z''
Z'
90 oC
80 oC 70 oC
60 oC
50 oC
40 oC
RT
PBI-BuI in 1M H3PO4
Z''
Z'
110 oC
100 oC
90 oC
80 oC
70 oC
60 oC
50 oC
40 oC
RT
Figure 5.18 Impedance spectra of blend membranes and PBI-BuI doped with 1M H3PO4

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
181
Among membranes doped with 1M H3PO4 (Figure 5.18), PBI-BuI shows the least
RΩ at all temperatures as seen in the Table 5.8. Among 1M H3PO4 doped blend
membranes the decrease in RΩ shifts at higher temperature with increase in PBI-BuI
content. This can be attributed to the higher benzimidazole content which can take up the
proton from the phosphoric acid. In addition, in these blend membranes, with increase in
temperature, the diameter of the semicircle keeps decreasing indicating more facile
transport of proton at higher temperatures (Figure 5.18). In these blend membranes least
value of RΩ is obtained at 90 °C. From RT to 90 °C, RΩ decreases gradually and increases
suddenly at 100 °C. This indicates that water is an essential medium for proton
transportation perhaps by vehicular mechanism.
From Tables 5.7 and 5.8 and Figure 5.15 and 5.18, it could be seen that, 1M
H2SO4 doped blend membranes exhibit lower value of RΩ as compared with that of 1M
H3PO4 doped membranes. This indicates that the former has less membrane resistance
than the latter. However, the semicircle region is clear, in particular at higher temperature
in 1M H3PO4 doped blend membranes, indicating enhanced proton transport. On doping
these membranes either with 1M H2SO4 or 1M H3PO4, both types of mechanism i.e. by
vehicular and Grotthus for proton conductivity have been observed.
5.4 Conclusions 1. Water uptake (WU) of SPIs decreases in the order SPDABI-N ≈ SPDABII-N >
SPDAB•TA-N > SPDAB•Im-N while incase of blend membranes WU decreases in
the order as SPI(H)/PBI-BuI(50/50) > SPI(H)/PBI-BuI(40/60) > SPI(H)/PBI-
BuI(30/70). In other words WU increase with SPI content and decreases with PBI-
BuI content.
2. Ion exchange capacity (IEC) decreases in the order SPDABI-N > SPDABII-N >
SPDAB•Im-N > SPDAB•TA-N. In case of blend membranes, it decreases in the order
as SPI(H)/PBI-BuI(50/50) > SPI(H)/PBI-BuI(40/60) > SPI(H)/PBI-BuI(30/70).
3. For sulfonated polyimides, hydrolytic stability as well as oxidative stability is higher
for SPIs in salt form (SPDAB•TA-N and SPDAB•Im-N) as compared with SPI in –
SO3H form (SPDABI-N and SPDABII-N). While incase of blend membranes
hydrolytic and oxidative stability increases with increase in PBI-BuI content.

Chapter 5 Investigation of polyimides towards their applicability as PEM materials
182
4. Proton conductivity (σ) of SPIs increases in the order SPDAB•TA-N < SPDAB•Im-N
< SPDABII-N < SPDABI-N < Nafion-117. SPDAB•Im-N exhibits higher proton
conductivity, IEC and lower water uptake than that of SPDAB•TA-N. Blend
membranes with SPI in –SO3H form [SPI(H)/PBI-BuI(a/b)] exhibits much lower
proton conductivity in the order of 10-5 S/cm.
5. PBI-BuI membrane, on doping with 1M H2SO4, show lower conductivity than blend
membranes, while on doping with 1M H3PO4, it exhibits higher proton conductivity
than blend membranes.
6. In case of 12 M H3PO4 doped membranes, highest conductivity was exhibited by PBI-
BuI. Among the blend membranes, conductivity increases with increase in PBI-BuI
content. Above 75 C, blend membrane exhibits higher proton conductivity than that
of PBI-I.
7. For the 12M H3PO4 doped membranes, H3PO4 uptake decreases in the order PBI-BuI
> PBI-I > SPI(PA)/PBI-BuI(30/70) > SPI(PA)/PBI-BuI(40/60) > SPI(PA)/PBI-
BuI(50/50) > SPDAB•TA-N ≈ SPDAB•Im-N.
8. In case of membranes doped with 12 M H3PO4, blend membrane, SPI(PA)/PBI-
BuI(30/70) exhibits proton conductivity almost equivalent to that of PBI-BuI even
though it is having lower phosphoric acid uptake. This is especially important as
membranes with lower acid uptake are expected to be more robust than that with
higher acid uptake.

Chapter 6
Conclusions
Successful methodology was developed for the synthesis of diamines containing
pendant phenoxy group which would allow synthesis of
PIs with systematic structure architecture. 1-phenoxy-2,4-diamino benzene (PDAB) was
synthesized as a primary monomer of this family. Substitution of bulky group was done
on aromatic ring of oxyphenyl (phenoxy) ring of PDAB. This methodology yielded four
types of diamines having methyl and t-butyl substitution on phenoxy group. In this way,
novel diamines viz., 2methyl-1-phenoxy-2,4-diamino benzene (2MPDAB), 4-methyl-
1-phenoxy-2,4-diamino benzene (4MPDAB), 2,6-dimethyl-1-phenoxy-2,4-diamino
benzene (2,6DMPDAB) and 4’-t-butyl-1-phenoxy-2,4-diamino benzene (4tBPDAB)
were succefully syntesized and characterized. Sulfonated diamine, viz., 4-sulfonic-1-
phenoxy-2,4-diaminobenzene (SPDAB) was synthesized by sulfonation of PDAB using a
simple methodology to offer quantitative substitution of –SO3H moiety at para-position
of aromatic ring at oxyphenyl (phenoxy) linkage.
Polycondensation of a diamine (PDAB) with selected dianhydrides by one step
solution condensation technique yielded polyimides with flexible pendant phenoxy
group, which were characterized for requisite physical properties. This methodology was
subsequently followed for the polycondensation of other diamines with commercial
dianhydrides (ODPA, 6FDA and NTDA). All these PIs exhibited good solvent solubility,
high Tg (> 250 °C) and high thermal stability (IDT > 500 C). PI based on 6FDA (PDAB-
6FDA) showed high d-spacing, high density, high Tg but low IDT compared to PI based
on ODPA. Copolyimides (CPIs) of PDAB and ODA in different ratios were prepared.
These CPIs also showed good solubility and high thermal stability. Results indicate that
incorporation of pendant phenoxy group in PIs enhance solvent solubility without
sacrificing thermal stability.
Polyimides with alkyl substituted phenoxy groups as a side chain were
successfully synthesized with two select dianhydrides (ODPA and 6FDA). Intrinsic
viscosity of these polyimides was in the range of 0.30-0.55 dL/g and dense membranes

Chapter 6 Conclusions
184
could be cast in chlorinated solvents. Solubility parameter of PIs decreases with increase
in alkyl content. Polyimides based on 6FDA showed better solubility, higher Tg, lower
decomposition temperature, lower solubility parameter and higher d-spacing than that of
ODPA based polyimides. Structural isomers of PIs based on diamines, viz., 4'MPDAB
and 2'MPDAB with either ODPA or 6FDA revealed different d-spacing. Polyimides with
–CH3 substitution at ortho position in pendant phenoxy group exhibited high Tg. WAXS
pattern revealed their amorphous nature. While, 6FDA based PIs disclosed two humps in
their corresponding WAXS spectra, indicating their two types of chain packing.
Gas permeability investigations of these PIs reveal that 6FDA based PIs exhibit
higher permeability than that of ODPA based PIs obtained from same diamine. Increase
in permeability was by 4.6 to 13.3 times, while decrease in selectivity was by a factor of
0.04 to 0.53, confirming the importance of –C(CF3)2– linkage in gas permeability of PIs.
In ODPA based PIs, better selectivity is shown by PDAB-ODPA, while in 6FDA based
PIs 2'MPDAB-6FDA demonstrates the best selectivity for various gas pairs. In the case
of ODPA based PIs, permeability for different gases increase in an order PDAB-ODPA ≈
2'MPDAB-ODPA < 4'MPDAB-ODPA < 2',6'DMPDAB-ODPA < 4'tBPDAB-ODPA. In
6FDA based PIs, permeability increases in the order 2'MPDAB-6FDA < 4'MPDAB-
6FDA < PDAB-6FDA < 2',6'DMPDAB-6FDA ≈ 4'tBPDAB-6FDA. Intrasegmental
mobility (Tg) largely governs gas permeation in these PIs. Alkyl substituent on the
pendant phenoxy group at para position is more effective than at ortho substitution for
increasing permeability in PIs. As a result, PIs based on isomeric diamines 2'MPDAB
and 4'MPDAB (with either ODPA or 6FDA), the later exhibited lower Tg, higher
permeability, but lower selectivity. Though d-spacing for substitution of voluminous t-
butyl group on pendant phenoxy group in PIs (4'tBPDAB-ODPA and 4'tBPDAB-6FDA)
is low; these polymers demonstrated the highest permeability, which is in tune of
variation in FFV. Between 4'tBPDAB-ODPA and 2',6'DMPDAB-ODPA, former reveals
better selectivity for some of the gas pairs. On the other hand, 4'tBPDAB-6FDA and
2',6'DMPDAB-6FDA exhibits almost same permeability and no significant difference in
selectivity. In ODPA based PIs, structural variation in diamine moiety greatly affects
chain packing and thus gas permeability. On the other hand in 6FDA based PIs,
dependence of structural variation in diamine moiety on gas permeability was largely

Chapter 6 Conclusions
185
suppressed by the effects of–C(CF3)2– linkage of 6FDA. In case of SPIs, feed gas
humidification lead to an increase in permeability as well as selectivity. This effect is
more pronounced in CO2 based permeation properties. In blend membranes, permeability
of CO2 increases with increasing content of SPIs.
Polyimides containing sulfonic acid group in pendant phenoxy were synthesized
by polycondensation of SPDAB with NTDA. Polycondensation in presence of imidazole
is faster than TEA. These SPIs in corresponding salt form as well as in –SO3H form
revealed good solubility in polar aprotic solvents like DMF, DMAc and DMSO and their
reduced viscosity increased with decreasing concentration in the solution of DMAc, thus
showing a typical ionomer behavior. Tough and flexible films of these SPIs in their
corresponding salt form could be cast from their DMAc solution and on converting these
membranes into their respective –SO3H form. NTDA based SPIs could retain their
mechanical strength while ODPA based SPI became brittle. Thermal stability of SPIs
decreased in the order SPDAB•Im-N > SPDABI-N ≈ SPDABII-N > SPDAB•TA-N and
they did not show any glass transition. SPDAB•Im-N showed some interesting features
such as critical solution temperature (CST) phenomenon, high thermal stability, delayed
time for dissolution in solvents and close packing from its WAXD spectrum. Blends
prepared from PBI-I with SPDAB•TA-N are brittle while that of with PBI-BuI become
flexible below 50 wt% of SPDAB•TA-N. These blends are miscible as supported by their
FT-IR spectra.
Physicochemical characterizations and proton conductivity analysis of SPIs as
well as blend membranes as PEM material was evaluated. It is observed that water uptake
and IEC of SPIs in –SO3H form (SPDABI-N and SPDABII-N) is higher than that of their
salt forms (SPDAB•Im-N and SPDAB•TA-N). While reverse is the trend for hydrolytic
and oxidative stability. Proton conductivity (σ) of SPIs decreases in the order Nafion-117
> SPDABI-N > SPDABII-N > SPDAB•Im-N > SPDAB•TA-N. Proton conductivity of the
order of 10-2-10-3 S.cm-1 of SPIs in –SO3H form in spite of their comparatively lower IEC
(<2) and optimum water uptake (46-49%) indicates their importance as PEM material.
Arrhenius type of behavior is observed for SPIs in –SO3H form with Ea in the range of 8-
9.5 for kJ/mole. These values matched well with reported ones for other SPIs and
sulfonated hydrocarbon polymers. The lower time for the polymerization of SPDAB•Im-

Chapter 6 Conclusions
186
N and higher proton conductivity, higher hydrolytic as well as oxidative stability, higher
IEC and lower water uptake than that of SPDAB•TA-N indicate the effectual of SPIs in
an imidazolium sulfonate salt form.
In case of blend membranes [SPI(H)/PBI-BuI(a/b)] as PEM material, water
uptake and IEC values increase with SPI content and decrease with PBI-BuI content.
While hydrolytic stability and oxidative stability of such blend membranes show the
reverse trend. 12M H3PO4 uptake of blend membranes increases with increasing PBI-BuI
content, as anticipated. Their proton conductivity also increases with PBI-BuI content,
which being highest for one of their components i.e. 12M H3PO4 doped PBI-BuI at all
temperatures. Proton conductivity of 12M H3PO4 doped membranes was observed to
follow non-Arrhenius behavior. This implies importance of blend membranes while
doping with 12M H3PO4 despite of their low acid doping level. This is especially
important as membranes with lower acid uptake are expected to be more robust than that
with higher acid uptake. Though, proton conductivity of 1M H3PO4 doped membranes in
100% RH is found to be lower than that of membranes doped with 12M H3PO4 in
anhydrous condition, however proton conductivity of the former in the range of 10-3-10-4
S.cm-1 at such lower concentration (1M) of acid is worth noting.

187
References
Acharya M. and Foley H. C., AIChE Journal, 46, 5, 2000, 911.
Ahn T. K., Kim M., and Choe S., Macromolecules, 30, 1997, 3369.
Akutsu F., Kataoka T., Shimizu H., Naruchi K. and Miura M., Macromol. Rapid
Commun., 15, 1994, 411.
Al-Haq M. I., Lebrasseur E., Tsuchiya H. and Torri T., Crystall. Rev., 13, 1, 2007, 29.
Alvarez-Gallego Y., Nunes S. P., Lozano A. E., de la Campa J. G. and de Abajo J.,
Macro. Rap. Comm., 28, 2007, 616.
Anthony J. L., Maginn E.J. and Brennecke J. F., J. Phys. Chem. B, 105, 2001, 10942.
Appleby A. J., J. Power Sources, 29, 1990, 3.
Arthur W. J., U. S. Pat., 3347917, 1967.
Asano N., Aoki M., Suzuki S., Miyatake K., Uchida H. and Watanabe M., J. Am. Chem.
Soc., 128, 2006, 1762.
Bacon F. T., Electrochimica Acta., 14, 1969, 569.
Baker R. W., Membrane Technology and Application, 2nd Edition, John Wiley and Sons,
Ltd., England, 2004.
Baldwin B. W., Hirose T. and Wang Z. H., Chem. Commun., 1996, 2669.
Bara J. E., Carlisle T. K., Gabriel C. J., Camper D., Finotello A., Gin D. L. and Noble R.
D., Ind. Eng. Chem. Res., 48, 2009, 2739.
Barbir F., PEM Fuel Cells: Theory and Practice, Elsevier Academic Press, Chapter 2,
2005.
Barkdoll A. E., England D. C., Gray H. W., Kirk W. J. and Whiltman G. M., J. Am.
Chem. Soc., 75, 1953, 1156.
Barrer R. M. and Skirrow G., J. Polym. Sci., 3, 1948, 549.
Beliers J.-P. and Angell C. A., J. Physc. Chem. B, 111, 2007, 4926.
Bergemeister J. J., Rancourt J. D., Taylor L. T., Chem. Mater., 2, 1990, 640.
Beuscher U., Cleghorn S. J. C. and Johnson W. B., Int. J. Energy Res., 29, 2005, 1103.
Bhole Y. S. and Kharul U.K., Polym. Int., 52, 2003, 1474.
Bhole Y. S., Karadkar P. B. and Kharul U. K., J. Polym. Sci., Part B: Polym. Phys., 45,
2007, 3156.

188
Bhole Y. S., Kharul U. K., Somani S. P. and Kumbharkar S.C., Eur. Polym. J., 41, 2005,
2461.
Bhutani S. K., U. S. Pat., 3935264, 1976.
Biswas M. and Mukherjee A., Adv. Polym. Sci., 115, 1994, 90.
Blanchard L. A. and Brennecke J. F., Ind. Eng. Chem., 40 (1), 2001, 287.
Bogert M. T. and Renshaw R. R., J. Am. Chem. Soc., 30, 1908, 1135.
Bollinger W. A., MacLean D. L. and Narayan R. S., Chem. Eng. Prog., 78, 1982, 27.
Bouchet R., Miller S., Duclot M. and Souquet J. L., Solid State Ionics, 145, 2001, 69.
Boudghene Stambouli A. and Traversa E., Renew. Sust. Energy Rev., 6, 2002, 433.
Bower G. M. and Frost L. W., J. Polym. Sci., Part A: Polym. Chem., 1, 1963, 3135.
Brooks N. W., Duckett R. A., Rose J., Ward I. M. and Clements J., Polymer, 34 (19),
1993, 4038.
Bryant R. G., Polyimides, Kirk-Othmer Encyclopedia of Chemical Technology, Vol. 20,
page no. 268, John Wiley and Sons, Inc. 2006.
Buckley A., Stuetz D.E. and Serad G.A., Encycl. Polym. Sci. Eng., 11, 1987, 572.
Campos A., Garcia R., Suner I. and Figueruelo J. E., Polymer, 38 (12), 1997, 3011.
Cassidey P. E., Fawcett, N. C., “Kirk-Othmer Encyclo. Chem. Technol.”, John Wiley and
Sons, New York, 18, 1982, 704.
Chatterjee G., Houde A. A. and Stern S. A., J. Membr. Sci., 135, 1997, 99.
Chen C., Huang X. and Qin W., J. Macromol. Sci., Part B: Phys., 47, 2008, 109.
Chen N., Tien C.-F., Patton S. M., Langsam M. and Burgoyne W.F., Polym. Mater. Sci.
Eng., 69, 1993, 161.
Chen X., Yin Y., Tanaka K., Kita H. and Okamoto K.-I., High Perform. Polym., 18,
2006, 637.
Chhabra P. and Choudhary V. Eur. Polym. J., 45, 2009, 1467.
Chikvaidze G., Gabrusenoks J., Kleperis J. and Vaivars G., Functional Materials and
Nanotechnologies, J. Phys., Conference Series, 93, 2007, 012026, 1.
Chung T. S., Lin W.-H. and Vora R. H., J. Appl. Polym. Sci., 81, 2001, 3552.
Clayden J., Greeves N., Warren S. and Wothers P., Organic Chemistry, Oxford
University Press, New York, 2001.

189
Cole A. C., Jensen J. L., Ntai I., Tran K. L. T., Weaver K. J., Forbes D. C. and Davis J.
H. Jr., J. Am. Chem. Soc., 124, 2002, 5962.
Coleman M. R. and Koros W. J., J. Membr. Sci., 50, 1990, 285.
Coleman M. R. and Koros W. J., J. Polym. Sci., Part B: Polym. Phy., 32, 1994, 1915.
Contois L. L. and Trementozzi Q. A., J. Polym. Sci., XVIII, 1955, 479.
Cordes De N. D. M. and Walter J. L., Spectrochimica Acta, 24A, 1968, 237.
Cornelius C. J., Ph. D. Dissertation, Virginia Polytechnic Institute and State University,
2000.
Cornet N., Diat O., Gebel G., Jousse F., Marsacq D., Mercier R. and Pineri M, J. New
Mater. Electrochem. Syst., 3, 2000, 33.
Cote P. and Brisson J., Macromolecules, 27, 1994, 7329.
Crank J., “The mathematics of diffusion”, Clarendon Press, 1956.
Crosthwaite J. M, Aki S. N.V. K, Maginn E. J. and Brennecke J. F., Fluid Phase
Equilibria, 228, 2005, 303.
Daletou M. K., Gourdoupi N. and Kallitsis J. K., J. Membr. Sci., 252, 2005, 115.
Daletou M. K., Kallitsis J. K., Voyiatzis G. and Neophytidesa S. G., J. Membr. Sci., 326,
2009, 76.
de Abajo J. and de la Campa J. G., Adv. Polym. Sci., 140, 1999, 23.
Deimede V., Voyiatzis G. A. Kallitsis J. K., Qingfeng L. and Bjerrum N. J.,
Macromolecules, 33, 2002, 7609.
Devezas T., LePoire D., Matias J. C.O. and Silva A.M.P., Features, 40, 2008, 1.
Dickinson P. R. and Sung C. S. P., Macromolecules, 25, 1992, 3758.
Dincer I., Int. J. Energy Res., 31, 2007, 29.
Dine-Hart R. A. and Wright W. W., J. Appl. Polym. Sci., 11, 1967, 609.
Ding J., Chuy C. and Holdcroft S., Chem. Mater., 13, 7, 2001, 2231.
Ding M., Prog. Polym. Sci., 32, 2007, 623.
Domanska U., Thermochimica Acta., 448, 2006, 19.
Domanska U., Zolek-Tryznowska Z. and Krolikowski M., J. Chem. Eng. Data, 52, 2007,
1872.
Dong K., Zhang S., Wang D. and Yao X., J. Phys. Chem. A, 110 (31), 2006, 9775.
Dotcheva D., Klapper M. and Mullen K ., Macromol. Chem. Phys., 195, 1994, 1905.

190
Du Z., Li Z., Guo S., Zhang J., Zhu L. and Deng Y., J. Physc. Chem. B, 109, 2005,
19542.
Dupont J., J. Braz. Chem. Soc., 15, 3, 2004, 341.
Eastmond G. C., Gibas M., Pacynko W. F. and Paprotny J., J. Membr. Sci., 207, 2002,
29.
Edward, W. M. and Robinson, I. M., U. S. Pat., 2867609, 1959a.
Edward, W. M. and Robinson, I. M., U. S. Pat., 2880230, 1959b.
Einsla B. R., Hong Y.-T., Kim S. Y., Wang F., Gunduz N. and McGrath J. E., J. Polym.
Sci. Part A: Polym. Chem., 42, 2004, 862.
Einsla B. R., Kim S. Y. Hickner M. A., Hong Y.-T., Hill M. L., Pivovar B. S. and
McGrath J. E., J. Membr. Sci., 255, 2005a, 141.
Einsla B. R., Ph. D. Dissertation, Virginia Polytechnic Institute and State University,
Blacksburg, Virginia, 2005b.
Elabd Y. A., Napadensky E., Walker C. W. and Winey K. I., Macromolecules, 39, 2006,
399.
Endrey A. L., U. S. Pat., 3179631, 1965a.
Endrey A. L., U. S. Pat., 3179633, 1965b.
Fang J., Guo X., Harada S., Watari T., Tanaka K., Kita H. and Okamoto K.,
Macromolecules, 35, 2002, 9022.
Fang J., Guoa X., Xua H. and Okamoto K., J. Power Sources, 159, 2006, 4.
Faure S., Cornet N., Gebel G., Mercier R., Pineri M. and Sillion B. Proceedings of
Second International Symposium on New Materials for Fuel Cell and Modern
Battery Systems, Montreal, Canada, July 6–10, 1997, p. 818.
Fontanella J. J., Wintersgill M. C., Wainright J. S., Savinell R. F. and M. Litt,
Electrochim. Acta., 43 (10-11), 1998, 1289.
Fraga-Dubreuil J., Bourahla K., Rahmouni M., Bazureau J. P. and Hamelin J., Catal.
Commun., 3, 2002, 185.
Freeman B. D., Bokobza L., Sergot P., Monnerie L. and De Schryver F. C.,
Macromolecules, 23, 1990, 2566.
Gagliano A. R., Knowlton R. C. and Byers L. D., J. Org. Chem., 54, 1989, 5247.

191
Gao Y., Robertson G. P., Guiver M. D. and Jian X., J. Polym. Sci., Part A: Polym.
Chem., 41, 2003a, 497.
Gao Y., Robertson G. P., Guiver M. D., Jian X., Mikhailenko S. D., Wang K. and
Kaliagaine S., J. Polym. Sci., Part A: Polym. Chem., 41, 2003b, 2731.
Gao Y., Robertson G. P., Guiver M. D., Mikhailenko S. D., Li X. and Kaliagaine S.,
Macromolecules, 37, 2004, 6748.
Gao Y., Robertson G. P., Guiver M. D., Mikhailenko S. D., Li X. and Kaliagaine S.,
Macromolecules, 38, 2005, 3237.
Genies C., Mercier R., Sillion B., Cornet N., Gebel G. and Pineri M., Polymer, 42,
2001a, 359.
Genies C., Mercier R., Sillion B., Petiaud R., Cornet N., Gebel G. and Pineri M.,
Polymer, 42, 2001b, 5097.
Ghaemy M. and Alizadeh R., Eur. Polym. J., 45, 2009, 1681.
Ghosal K. and Chern R. T., J. Membr. Sci., 72, 1992, 91.
Ghosal K. and Freeman B. D., Polym. Adv. Tech., 5, 1994, 673.
Ghosh M. K. and Mittal K. L., Polyimides: Fundamentals and Applications, Marcel
Dekker, New York, 1996.
Gianolio D. A., Segismundo J. M. and Mclaughlin L.W., Nucleic Acids Research, 10,
2000, 2128.
Gilron J. and Soffer A., J. Membr. Sci., 209, 2002, 339.
Glipa X., Bonnet B., Mula B., Jones D. J. and Roziere R., J. Mater. Chem., 9, 1999,
3045.
Gonzalez R. A., U. S. Pat., 3499034, 1970.
Greaves T. L. and Drummond C. J., Chem. Rev., 108, 2008, 206.
Grove W. R., Phil. Mag. Ser., 3, 14, 1839, 127.
Grubb T. L., Ulery V. L., Smith T. J., Tullos G. L., Yagci H., Mathias L. J. and Langsam
M., Polymer, 40, 1999, 4279.
Grubb W. T., U. S. Pat., 2913511, 1959.
Guerra G., Choe S., Williams D. J., Karasz F. E. and MacKnight W. J., Macromolecules,
21, 1988, 231.

192
Gunduz N., Ph. D. Dissertation, Virginia Polytechnic Institute and State University,
Blacksburg, VA, 2001.
Guo X., Fang J., Watari T., Tanaka K., Kita H. and Okamoto K., Macromolecules, 35,
2002, 6707.
Habermann C. E., U. S. Pat., 4153581, 1979.
Hamrock S. J. and Yandrasits M. A., J. Macrom. Sci., Part C: Polym. Rev., 46, 2006,
219.
Hara M., Wu J. and Lee A. H, Macromolecules, 22, 1989, 754.
Hasanain F. and Wang Z. Y., Polymer, 49, 2008, 831.
Hasiotis C., Deimede V. and Kontoyannis C., Electrochimica Acta., 46, 2001b, 2401.
Hasiotis C., Qingfeng L., Deimede V., Kallitsis J. K., Kontoyannis C. G. and Bjerrum N.
J., J. Electrochem. Soc., 148 (5), 2001a, A513.
He R., Li Q., Xiao G. and Bjerrum N. J., J. Membr. Sci., 226, 2003, 169.
Hedrick J. L., Carter K. R., Labadie J. W., Miller R .D., Volksen W., Hawker C. J., Yoon
D. Y., Russell T. P., McGrath J. E. and Briber R. M., Adv. Polym. Sci., 141,
1999a, 1.
Hedrick J. L., Labadie J. W., Volksen W. and Hilborn J. G., Adv. Polym. Sci., 147,
1999b, 61.
Hellums M. W., Koros W. J., Husk G. R. and Paul D. R., J. Membr. Sci., 1989, 46, 93.
Helmer-Metzmann F., Ballauff M., Schulz R. C. and Wegner G., Makromol. Chem., 190,
1989, 985.
Hickner M. A., Ghassemi H., Kim Y. S., Einsla B. R. and McGrath J. E., Chem. Rev.,
104, 2004, 4587.
Hirao T., Santhitikul S., Takeuchi H., Ogawa A. and Sakurai H., Tetrahedron, 59, 2003,
10147.
Hirayama Y., Kase Y., Tanihara N., Sumiyama Y., Kusukia Y. and Haraya K., J. Membr.
Sci., 160, 1999, 87.
Hirayama Y., Yoshinaga T., Kusuki Y., Ninomiya K., Sakakabira T. and Tamari T., J.
Membr. Sci., 111, 1996a, 169.
Hirayama Y., Yoshinaga T., Kusuki Y., Ninomiya K., Sakakabira T. and Tamari T., J.
Membr. Sci., 111, 1996b, 183.

193
Hirose T., Kopek B. G., Wang Z. H., Yusa R. and Baldwin B. W., Tetrahedron Letters,
44, 2003, 1831.
Hoehn H. and Richter J. W., U.S. Patent Reissue, 30, 1980, 351.
Houde A. Y., Kulkarni S. S., Kharul,U. K., Charati C. G. and Kulkarni M. G., J. Membr.
Sci., 103, 1995, 167.
Hougham G., Cassidy P. E., Jones K. and Davidson T., Fluoropolymers 2: Properties,
Eds., Plenum Press, New York, 1999, p 233.
Hsiao S. H. and Huang P. C., J. Polym. Sci., Part A: Polym. Chem., 36, 1998a, 1649.
Hsiao S.-H, Yang C.-P and Lo T.-K., J. Polym. Res., 5,3, 1998b, 193.
Hsiao S.-H., Yang C.-P. and Chen S.-H., J. Polym. Sci. Part A: Polym. Chem., 38, 2000,
1551.
Hu Z., Yin Y., Chen S., Yamada O., Tanaka K., Kita H. and Okamoto K. I., J. Polym.
Sci. Part A: Polym. Chem., 44, 2006, 2862.
Hu Z., Yin Y., Kita H., Okamoto K.-I., Suto Y., Wang H. and Kawasato H., Polymer, 48,
2007, 1962.
Hu Z., Yin Y., Yaguchi K., Endo N., Higa M. and Okamoto K.-I., Polymer, 50, 2009,
2933.
Hubner G. and Roduner E., J. Mater. Chem., 9, 1999, 409.
Hudlicky M., Reductions in Org. Chem., 2nd Ed., ACS Monograph, 1996.
Iimori T., Iwahashi T., Kanai K., Seki K., Sung J., Kim D., Hamaguchi H. and Ouchi Y.
J. Phys. Chem. B, 111 (18), 2007, 4860.
Imai Y., Maldar N. N. and Kakimoto M-A., J. Polym. Sci., Polym. Chem. Ed., 22, 1984,
2189.
Iwakura Y., Uno K. and Imai Y., J. Polym. Sci. Part A: Polym. Chem., 2 (6), 1964, 2605.
Jaffe M., Chen P., Choe E. W., Chung T. S. and Makhija S., Adv. Poly. Sci., 117, 1994,
298.
Jeong H.-J., Kobayashi A., Kakimoto M. and Imai Y., Polym. J., 26 (3), 1994, 373.
Jeong K. U., Kim J.-J. and Yoon T.-H., Polymer, 42, 2001, 6019.
Jiang P., Acta Cryst., E65, 2009, o2178.
Johnston N. J., J. Polym. Sci., Part A-1, 10, 1972, 2727.
Jung J. C. and Park S. B., J. Polym. Sci. Part A: Polym. Chem., 34, 1996, 357.

194
Jung J. C. and Park S. B., Polym. Bulletin, 35, 1995, 423.
Kaas R. L., J. Polym. Sci., Part A: Polym. Chem. Ed., 19, 1981, 2255.
Kabalka G. W. and Varma R. S. Comprehensive Organic Synthesis, Eds.; Pergamon:
Oxford, Vol. 8, 1991, p 363.
Kailani M. H. and Sung C. S. P., Macromolecules, 31, 1998a, 5771.
Kailani M. H. and Sung C. S. P., Macromolecules, 31, 1998b, 5779.
Kailani M. H., Sung C. S. P. and Huang S. J., Macromolecules, 25, 1992, 3751.
Kakimoto M.-A., Yoneyama M. and Imai Y., J. Polym. Sci. Part A: Polym. Chem., 26,
1988, 149.
Kerres J., Tang C. M. and Graf C., Ind. Eng. Chem. Res., 43, 2004, 4571.
Kerres J., Ullrich A., Häring T., Baldauf M., Gebhardt U. and Preidel W., J. New Mat.
Electrochem. Systems., 3, 2000, 229.
Kerres J., Ullrich A., Meier F. and Haring T., Solid State Ionics, 125, 1999, 243.
Kerres J., Zhang W., Jörissen L. and Gogel V., J. New. Mat. Electrochem. Systems, 5,
2002, 97.
Kharul U. K. and Kulkarni S. S., Bull. Mater. Sci., 17 (6), 1994, 1071.
Kharul U. K., Kulkarni S. S., Kulkarni M. G., Houde A.Y. and Charati S. G., Polymer, 39
(10), 1998, 2011.
Kim M.-J., Park Y.-I., Youm K.-H. and Lee K.-H., J. Appl. Polym. Sci., 91, 2004, 3225.
Kim S.-K., Kim T.-H., Jung J.-W. and Lee J.-C., J. Polym. Sci., Part A: Polym. Chem.,
50, 2009, 3495.
Kim T. H., Koros W. J., Husk G. R. and O’brien K. C., J. Membr. Sci., 37, 1988, 45.
Kim Y. J., Glass T. E., Lyle G. D. and McGrath J. E., Macromolecules, 26, 1993, 1344.
Kim Y.-H., Ahn S.-K., Kim H. S. and Kwon S.-K., J. Polym. Sci., Part A: Polym. Chem.,
40, 2002, 4288.
Kim Y.-H., Kim H.-S. and Kwon S.-K., Macromolecules, 38, 2005, 7950.
Kingston B. H. M., Garey J. J. and Hellwig W. B, Anal. Chem., 41 (1), 1969, 86.
Klaren Cornelis H. J. and Krak H., U. S. Pat., 3756984, 1973.
Koros W. J. and Fleming G. K., J. Membr. Sci., 83, 1993, l.
Koros W. J. and Mahajan R., J. Membr. Sci., 175, 2000, 181.

195
Koros W. J., Fleming G. K., Jordan S. M., Kim T. H. and Hoehn H. H., Prog. Polym.
Sci., 13, 1988, 339.
Korshak V. V., Rusanov A. L., Izv Akad Nauk SSSr Ser Khim, 10, 1969, 2418.
Korshak V. V., Rusanov A. L., J. Macromol. Sci.: Part C: Polym. Rev., 21, 2, 1981, 272.
Kosmala B. and Schauer J., J. Appld. Polym. Sci., 85, 2002, 1118.
Koton M. M., Kudryavtsev V. V. and Svetlichny V. M., Polyimides: Synthesis,
Characterization and Applications 1 & 2. Ed. Mittal K. L., Plenum, New York,
1984.
Kreuer K. D., Peddison S. J., Spohr E. and Schuster M., Chem. Rev. 2004, 104, 4637.
Kreuz J. A., Endrey A. L., Gay F. P. and Sroog C. E., J. Polym. Sci., Part A-1: 4, 1966,
2607.
Kulkarni M., Potrekar R., Kulkarni R. A. and Vernekar S. P., J. Polym. Sci., Part A:
Polym. Chem., 46, 2008, 5776.
Kulkarni S. S., Bull. Mater. Sci., 17 (7), 1994, 1307.
Kumar D., J. Polym. Sci., Polym. Chem. Ed., 19, 1981, 795.
Kumbharkar S. C., Islam Md. N., Potrekar R. A. and Kharul U. K., Polymer, 50, 2009,
1403.
Kumbharkar S. C., Karadkar P. B. and Kharul U.K.., J. Membr. Sci., 286, 2006, 161.
Kuzenetsove A. A., High Perform. Polym., 12, 2000, 445.
Langsam M. and Burgoyne W.F., J. Polym. Sci., Part A: Polym. Chem., 31, 1993, 909.
Lau K. S. Y. and Dougherty T. K., U. S. Pat., 4827054, 1989.
Lavrov S. V., Ardashnikov A. Ya., Kardash I. Ye. and Pravednikov A. N., Polym. Sci.,
USSR, 19, 5, 1977, 1212.
Lavrov S. V., Talankina O. B., Vorob'ev V. D., Izumnikov A. L., Kardash I. E. and
Pravednikov A. N., Polym. Sci. USSR (Engl Transl.), 22, 1980, 2069.
LeBlanc O. H., Ward W. J., Matson S. L. and Kimura S. G., J. Membr. Sci., 6, 1980, 339.
Lee C. W., Kwak S. M. and Yoon T. H., Polymer, 47, 2006, 4140.
Lee C., Sundar S., Kwon J. Han H., J. Polym. Sci., Part A: Polym. Chem., 42, 2004,3612.
Lee H.-Y. and An M., Bull. Korean Chem. Soc., 25, 11, 2004, 1717.
Lee J. K. and Kerres J., J. Membr. Sci, 294, 2007, 75.
Lee S., Lee J. G., Lee H. and Choe S., Macromolecules, 32 , 1999 , 5961.

196
Lee Y. J., Gungor A., Yoon T. H., McGrath J. E. The Journal of Adhesion, 55, 1, 1995,
165.
Li N., Cui Z., Zhang S. and Li S., J. Polym. Sci. Part A: Polym. Chem., 46, 2008, 2820.
Li N., Cui Z., Zhang S. and Xing W., J. Membr. Sci, 295, 2007c, 148.
Li N., Cui Z., Zhang S., Li S. and Zhang F., J. Power Sources, 172, 2007b, 511.
Li Q., He R. H., Jensen J. O., Bjerrum N. J., Chem. Mater., 2003, 15, 4896.
Li Q., He R., Berg R.W., Hjuler H. A. and Bjerrum N. J., Solid State Ionics, 168, 2004,
177.
Li Q., Hjuler H.A., Bjerrum N. J., J. Appl. Electrochem., 31, 2001, 773.
Li Q., Jensen J. O., Savinell R. F. and Bjerrum N. J., Prog. Polym. Sci., 34, 2009, 449.
Li W., Zhang S., Chen G. and Zhang Q., Polymer, 48, 2007a, 3082.
Li X.-G., Huang M.-R., Duan W. and Yang Y.-L., Chem. Rev., 102, 2002, 2925.
Li Y., Ding M. and Xu J., Macromol. Chem. Phys., 198, 1997, 2769.
Li Y., Wang X., Ding M. and Xu J., J. Appl. Polym. Sci., 61, 1996, 741.
Liaw D.-J., Huang C.-C. and Chen W.-H., Polymer, 47, 2006, 2337.
Liaw D.-J., Liaw B.-Y., Hus P.-N. and Hwang C.-Y., Chem. Mater., 13, 2001, 1811.
Lin C. H., Cai S. X., Leu T. S., Hwang T. Y. and Lee H. H., J. Polym. Sci. Part A:
Polym. Chem., 44, 2006, 3454.
Lin W.-H., Vora R. H. and Chung T.-S., J. Polym. Sci., Part B: Polym. Phys., 38, 2000,
2703.
Liou G.-S., J. Polym. Sci. Part A: Polym. Chem., 36, 1998, 1937.
Liu B., Hu W., Matsumoto T., Jiang Z. and Ando S., J. Polym. Sci. Part A: Polym.
Chem., 43, 2005, 3018.
Liu B., Robertson G. P., Kim D. S., Guiver M. D., Hu W. and Jiang Z., Macromolecules,
40, 2007, 1934.
Liu S. L., Wang R., Liu Y., Chng M. L. and Chung T. S., Polymer, 42, 2001, 8847.
Liu Y., Pan C., Ding M. and Xu J., Polym. Int., 48, 1999, 832.
Lobato J., Canizares P., Rodrigo M. A., Linares J. J. and Aguilar J. A., J. Membr. Sci.,
306, 2007, 47.
Lundberg D. and Phillips R. R., J. Polym. Sci.: Polym. Phys. Ed., 20, 1982, 1143.
Lv J., Wang K. Y. and Chung T. S., J. Membr. Sci., 310, 2008, 557.

197
Ma Y., Ph. D. Dissertation, Case Western Reserve University, USA, 2004b.
Ma Y., Wainright J., Litt M. and Savinella R., J. Electrochemical Soc., 151 (1), 2004, A8.
MacFarlane D. R., Pringle J. M., Johansson K. M., Forsyth S. A. and Forsyth M., Chem.
Commun., 2006, 1905.
Mackenize G. F., U. S. Pat., 2861995, 1958.
Mader J., Xiao L., Schmidt T. J. and Benicewicz B. C., Adv. Polym. Sci., 216, 2008, 63.
March J., “Advnced Organic Chemistry: Reactions, Mechanisms and Structure”, 3rd edn.,
Wiley Eastern Ltd., New Delhi, 1992.
Marestin C., Gebel G., Diat O. and Mercier R., Adv. Polym. Sci., 216, 2008, 185.
Mathew J. S., Ph. D. Dissertation, University of Pune, Pune, India, 2001.
Matsumoto K. and Xu P., J. Appl. Polym. Sci., 47, 1993c, 1951.
Matsumoto K. and Xu P., J. Membr. Sci., 81, 1993b, 23.
Matsumoto K., Xu P. and Nishikimi T., J. Membr. Sci., 81, 1993a, 15.
Matsuyama H., Terada A., Nakagawara T., Kitamura Y. and Teramoto M., J. Membr.
Sci., 163, 1999, 221.
Matsuyama H., Teramoto M. and Iwai K., J. Membr. Sci., 93, 1994, 237.
Matsuyama H., Teramoto M., Sakakura H. and Iwai K., J. Membr. Sci., 117, 1996, 251.
Mauritz K. A. and Moore R. B., Chem. Rev., 104, 2004, 4535.
McBride R.B. and McKinley D.L., Chem. Eng. Prog., 61, 1965, 81.
McHattie J. S., Koros W. J. and Paul D.R., Polymer, 32, 1991, 840.
McHattie J. S., Koros W. J. and Paul D.R., Polymer, 33, 1992, 1701.
Mi Y., Stern S. A. and Trohalaki S., J. Membr. Sci., 77, 1993, 41.
Mikroyannidis J. A. and Tsivgoulis G. M., J. Polym. Sci., Part A: Polym. Chem., 37,
1999a, 3646.
Mikroyannidis J. A., Polymer, 40, 1999b, 3107.
Mitchell G. R. and Windle A. H., Polymer, 25, 1984, 906.
Mittal K.L., Polyimides and Other High Temperature Polymers: Synthesis,
Characterization and Applications, Vol. 1, VSP publication, Netherlands, 2001.
Miyatake K., Zhou H., Matsuo T., Uchida H. and Watanabe M.., Chem. Commun., 2003,
368.

198
Miyatake K., Zhou H., Matsuo T., Uchida H. and Watanabe M., Macromolecules, 37,
2004, 4961.
Morikawa A., Furukawa T. and Moriyama Y., High Perform. Polym., 18, 2006, 593.
Morikawa A., Furukawa T. and Moriyama Y., Polym. J., 37, 10, 2005, 759.
Morton R. C., Mangroo D. and Gerber G. E., Can. J. Chem., 66, 1988, 1701.
Muhlbauer H. G., U. S. Pat., 3394186, 1968.
Muruganandam N., Koros W. J. and Paul D. R., J. Polym. Sci., Part B: Polym. Phys., 25,
1987, 1999.
Musto P., Karasz F. E. and MacKnight W. J., Polymer, 30, 1989, 1012.
Musto P., Karasz F. E. and MacKnight W. J., Polymer, 34 (14), 1993, 2934.
Nakamoto H., Noda A., Hayamizu K., Hayashi S., Hamaguchi H. and Watanabe H., J.
Phys. Chem. C., 111 (3), 2007, 1541.
Nakashima T., Topics in Current Chemistry, 139, 1987, 57.
Nakayama T., Watari T., Tanaka K., Kita H. and Okamoto K., Trans. Mater. Res. Soc.
Jpn., 28, 2003, 777.
Nelson A., Guerra G., Williams D. J., Karasz F. E., MacKnight W. J., J. Appl. Polym.
Sci., 36, 1988, 243.
Niedrach L. W., Fuel cell, U.S. Patent 3,134,697, General Electric Corporation, 1964.
Noda A., Susan A. B. H., Kudo K., Mitsushima S., Hayamizu K. and Watanabe M., J.
Phys. Chem. B., 107, 2003, 4024.
Nunes S.P. and Peinemann K.V., “Membrane Technology in Chemical Industry”, Wiley-
VCH, 2001.
Okamoto K. I., Yasugi N., Kawabata T., Tanaka K. and Kita H., Chem. Lett., 1996, 613.
Orwoll R. A., Clair T. L. St., Dobbs K. D., J. Polym. Sci., Polym. Phys. Ed., 19, 1981,
1385.
Peinemann K -V. and Nunes S. P., Membranes for Energy Conversion, Vol. 2, WILEY-
VCH Verlag GmbH & Co. KGaA, Weinheim, 2008.
Pellegrino J. and Kang Y. S., J. Membr. Sci., 99, 1995, 163.
Pellegrino J. J., Nassimbene R. and Noble R. D., Gas Sep. and Purf., 2, 1988,126.
Penner S. S., Energy, 31, 2006, 33.
Perry M. L. and Fuller T. F., J. Electrochem. Soc., 149,7, 2002, S59.

199
Pine S. H., Organic Chemistry, 5th Ed., McGraw-Hill Book Company, New York, United
States, 1987.
Pinnau I., He Z. and Morisato A., J. Membr. Sci., 241, 2004, 363.
Pixton M. R. and Paul D. R., Macromolecules, 28, 1995, 8277.
Potrekar R. A., Kulkarni M. P., Kulkarni R. A. and Vernekar S. P., J. Polym. Sci., Part A:
Polym. Chem., 47, 2009b, 2289.
Potrekar R. A., Ph. D. Dissertation, University of Pune, Pune, India, 2009a.
Pu H., Liu Q., Qiao L. and Yang Z., Polym. Eng. Sci., 45, 10, 2005, 1395.
Qiao K. and Yokoyama C., Chem. Lett., 33, 4, 2004, 472.
Qiu Z. and Zhang S., Polymer, 46, 2005, 1693.
Qiu Z., Chen G., Zhang Q. and Zhang S., Eur. Polym. J., 43, 2007, 194.
Qiu Z., Wang J., Zhang Q., Zhang S., Ding M., Gao L., Polymer, 47, 2006, 8444.
Quinn R. and Laciak D. V., J. Membr. Sci., 131, 1997a, 49.
Quinn R., Laciak D. V. and Pez Z. P., J. Membr. Sci., 104, 1995, 139.
Quinn R., Laciak D. V. and Pez Z. P., J. Membr. Sci., 131, 1997b, 61.
Radmard B. and Dadmun M. D., Polymer, 42, 2001, 1591.
Rancourt J. D. and Taylor L .T., Macromolecules, 20, 1987,790.
Ren L., Fu W., Luo Y., Lu H., Jia D., Shen J., Pang B. and Ko T. M., J. Appl. Polym.
Sci., 91, 2004, 2295.
Reynolds G. A., J. Am. Chem. Soc., 73, 1951, 4996.
Rikukawa M. and Sanui K., Prog. Polym. Sci., 25, 2000, 1463.
Robeson L. M., Burgoyne W. F., Langsam M., Savoca A. C. and Tien C. F., Polymer, 35,
1994, 4970.
Robeson L. M., J. Membr. Sci., 320, 2008, 390.
Robeson L. M., J. Membr. Sci., 62, 1991, 165.
Robeson L.M. and Matzner M., U. S. Pat., 4380598, 1983.
Rodgers M., Sang Y. and Holdcroft S., Eur. Polym. J., 42, 2006, 1075.
Roziere J. and Jones D. J., Annu. Rev. Mater. Res., 33, 2003, 503.
Rusanov A. L., Adv. Polym. Sci., 111, 1994, 115.
Rusanov A. L., Likhatchev D., Kostoglodov P. V., Müllen K. and Klapper M ., Adv.
Polym. Sci., 179, 2005, 83.

200
Rylander P. N., “Catalytic hydrogenation over Platinum Metals”, Academic Press, New
York, 1967, 168.
Saegusa Y., Horikiri M. and Nakurmura S., Macrom. Chem. and Phys., 198 (2), 1997,
619.
Sartori G. and Maggi R., Advances in Friedal-Crafts Acylation Reactions: Catalytic and
Green Processes, 2009, CRC press, Taylor and Francis Group, Boca Raton, FL.
Savard O., Peckham T. J., Yang Y. and Holdcroft S., Polymer, 49, 2008, 4949.
Schmidhauser J. C. and Longley K. L., J. Appl. Polym. Sci., 39, 1990, 2083.
Scholes C.A., Kentish S.E. and Stevens G.W., Recent Patents on Chem. Eng., 1, 2008,
52.
Scriven Eric F. V. and Turnbull K., Chem. Rev., 88, 2, 1988, 297.
Sek D., Pijet P. and Wanic A., Polymer, 33 (1), 1992, 190.
Sek D., Wanic A. and Schab-Balcerzak E., J. Polym. Sci. Part A: Polym. Chem., 33,
1995, 547.
Shao L., Chung T.-S., Goh S. H. and Pramoda K. P., J. Membr. Sci., 267, 2005, 78.
Shin C. K., Maier G. and Scherer G. G., J. Membr. Sci., 245, 2004, 163.
Shingate R. D., Ph D. Dissertation, University of Pune, Pune, India, 2006.
Silverstein R. M. and Webter F. X., Spectroscopic Identification of Organic Compounds,
6th edition, Wiley India Pvt. Ltd., New Delhi-110002, 2006.
Sivanandaiah K. M., Gurusiddappa S. and Gowda D. C., Indian J. Chem., 24B, 11, 1985,
1185.
Souzy R. and Ameduri B., Prog. Polym. Sci., 30, 2005, 644.
Spiliopoulos I. K. and Mikroyannidis J. A., Polymer, 38 (11), 1997, 2733.
Spiliopoulos I. K., Mikroyannidis J. A. and Tsivgoulis G. M., Macromolecules, 31, 1998,
522.
Sroog C. E., Endrey A. L., Abramo S. V., Berr C. E., Edwards W. M. and Olivier K. L.,
J. Polym. Sci., Part A, 3, 1965, 1373.
Sroog C. E., Prog. Polym. Sci., 16, 1991, 561.
St. Clair T. L., St. Clair A. K., Smith E. N., Polym. Prep.,17, 1976, 359.
Stern S. A., Vaidyanathan R. and Pratt J. R., J. Membr. Sci., 49, 1990, 1.
Stern S. A., Liu Y., Feld W. A., J. Polym. Sci., Part B: Polym. Phy., 31, 1993, 939.

201
Stern S. A., Mi Y., Yamamoto H. and Clair A. K. St., J. Polym. Sci., Part B: Polym. Phy.,
27, 1989, 1887.
Subbaraman R., Ghassemi H. and Zawodzinski Jr., T. A., J. Am. Chem. Soc. 129, 2007,
2238.
Takahashi T., Tanase S., Yamamoto O. and Yamauchi S., J. Solid State Chemistry, 17,
1976, 353.
Takekoshi T., Adv. Polym. Sci., 94, 1990, 1.
Tamai S., Kamada J., Goto K. and Yamaguchi A., High Perform. Polym., 13, 2001, 173.
Tamai S., Yamaguchi A. and Ohta M., Polymer, 37 (16), 1996, 3683.
Tanaka K., Kita H., Okano M. and Okamoto K.-I., Polymer, 33 (3), 1992a, 585.
Tanaka K., Okano M., Toshino H., Kita H. and Okamoto K.-I., J. Polym. Sci., Part B:
Polym. Phys., 30, 1992b, 907.
Tanaka K., Osada Y., Kita H. and Okamoto K.-I., J. Polym. Sci., Part B: Polym. Phy., 33,
1995, 1907.
Tang J., Tang H., Sun W., Radosz M. and Shen Y., J. Polym. Sci. Part A: Polym. Chem.,
43, 2005, 5477.
Tiwari P., Kumar R., Maulik P. R. and Misra A. K., Eur. J. Org. Chem., 2005, 4265.
Tseng P., Lee J. and Friley P., Energy, 30, 2005, 2703.
Tsuda Y., Kawauchi T., Hiyoshi N. and Mataka S., Polym. J., 32 (7), 2000, 594.
Tsujita Y., Prog. Polym. Sci., 28, 2003, 1337.
Ueda M., Nakamura K., Tanaka K., Kita H. and Okamoto K., Sensors and Actuators B,
127, 2007, 463.
Ueda M., Toyato H., Ouchi T., Sugiyama J.-I, Yonetake K., Masuko T. and Teramoto T.
J. Polym. Sci., Part A: Polym. Chem., 31, 1993, 853.
Van Krevelen D. W., Properties of Polymers, Elsevier Science B. V.: Amsterdam, The
Netherlands, 1997.
Volksen W., Adv. Polym. Sci., 117, 1994, 111.
Vygodskii Ya. S., Spirina T. N., Nechayev P. P., Chudina L. I., Zaikov G. Ye., Korshak
V. V., and Vinogradova S. V., Polym. Sci. USSR, 19(7), 1977, 1738.
Wainright J. S., Wang J., Weng D., Savinell R. F. and Litt M., J. Electrochem Soc., 142,
1995, L121.

202
Wang D-A., Adv. Polym. Sci., 209, 2007a, 179.
Wang F., Chen T. and Xu J., Macromol. Chem. Phys., 199, 1998, 1421.
Wang J. T., Savinell R. F., Wainright J., Litt M. and Yu H., Electrochimica Acta., 41, 2,
1996, 193.
Wang P., Liu B. and Zheng S., J. Macromol. Sci., Part B: Physics, 47, 2008, 800.
Wang Y., Goh S. H. and Chung T. S., Polymer, 48, 2007b, 2901.
Wang Y.-C., Huang S.-H., Hu C.-C., Li C.-L., Lee K.-R., Liaw D.-J. and Lai J.-Y., J.
Membr. Sci., 248, 2005, 15.
Wenzel M., Ballauff M. and Wegner G., Makromol. Chem., 188, 1987, 2865.
Wilks B. R., Won J. C., Ludovice P. J., Rezac M. E., Meakin P. and Hill A. J., J. Polym.
Sci., Part B: Polym. Phy., 44, 2006, 215.
Williams R. A., U. S. Pat., 3398195, 1968.
Wind J. D., Staudt-Bickel C., Paul D. R. and Koros W. J., Macromolecules, 36, 2003,
1882.
Wrasidlo W., Macromolecules, 4, 1971, 642.
Wright S. E., Renew. Energy, 29, 2004, 179.
Wu S., Qiu Z., Zhang S., Yang X., Yang F. and Li Z., Polymer, 47, 2006, 6993.
Wycisk R., Lee J. K. and Pintauro P. N., J. Electrochem. Soc., 152 (5), 2005, A892.
Xiao S., Huang Robert Y. M. and Feng X., Polymer, 48, 2007, 5355.
Xiao Y., Low B. T., Hosseini S. S., Chung T. S. and Paul D. R., Prog. Polym. Sci., 34,
2009, 561.
Xing D. and Kerres J., J. New Mat. Electro. Syst., 9, 2006, 51.
Xing O. and Savadogo O., Electrochem. Comm., 2, 2000, 697.
Xu J. W., Chng M. L., Chung T. S., He C. B. and Wang R., Polymer, 44, 2003, 4715.
Yamaguchi T., Boetje L. M., Koval C. A., Noble R. D. and Bowman C. N., Ind. Eng.
Chem. Res., 34, 1995, 4071.
Yamamoto H., Mi Y., Stern S. A. and Clair A. K. St., J. Polym. Sci., Part B: Polym. Phy.,
28, 1990, 2291.
Yang C.-P. and Chen W.-T., Macromolecules, 26, 1993, 4865.
Yang C.-P. and Hsiao F.-Z., J. Polym. Sci., Part A: Polym. Chem., 42, 2004a, 2272.
Yang C.-P. and Lin J.-H., J. Polym. Sci., Part A: Polym. Chem., 32, 1994, 423.

203
Yang C.-P., Hsiao F.-Z. and Hsu M-F., J. Polym. Sci., Part A: Polym. Chem., 40, 2002,
524.
Yang C.-P., Su Y.-Y. and Wu K.-L., J. Polym. Sci., Part A: Polym. Chem., 42, 2004b,
424.
Yang S.-J., Lee C., Jang W., Kwon J., Sundar S. and Han H., J. Polym. Sci. Part B:
Polym. Phys., 42, 2004c, 4293.
Yasuda T., Miyatake K., Hirai M., Nanasawa M. and Watanabe M., J. Poly. Sci.: Part A:
Poly. Chem., 43, 2005, 4439.
Yin Y., Chen S., Guo X., Fang J., Tanaka K., Kita H. and Okamoto K., High Perform.
Polym., 18, 2006b, 617.
Yin Y., Fang J., Watari T., Tanaka K., Kita H. and Okamoto K. I., J. Mater. Chem., 14,
2004, 1062.
Yin Y., Yamada O., Tanaka K. and Okamoto K.-I., Polym. J., 38 (3), 2006a, 197.
Yu J.-W. and Sung C. S. P., Macromolecules, 30, 1997, 1845
Zaidi S. M. J., Chen S. F., Mikhailenko S. D. and Kaliaguine S., J. New Mater.
Electrochem. Syst., 3, 2000, 27.
Zhang H., Li X., Zhao C., Fu T., Shi Y. and Na H., J. Membr. Sci., 308, 2008, 66.
Zhang Q., Chen G. and Zhang S., Polymer, 48, 2007, 2250.
Zhang Y., Wang Z., Wang S. C., Desalination, 145, 2002, 385.
Zhao C, Wang Z., Bi D., Lin H., Shao K., Fu T., Zhong S. and Na H., Polymer, 48, 2007,
3090.
Zhou Z., Li S., Zhang Y., Liu M. and Li W., J. Am. Chem. Soc., 127, 2005, 10824.
Zolandz L. R. and Fleming G. K., Membrane Handbook, Ho W.S.W., Sirkar K.K. (eds.)
van Nostrand Reinhold, 1992.
Zou J. and Winston Ho W. S., J. Membr. Sci., 286, 2006, 310.
Zubkov V. A., Koton M. M., Kudryavtsev V. V. and Svetlichnyi V. M., J. Organic
Chem. of the USSR, 17 (8), 1981, 1501.

204
List of Publications
S. S. Kothawade, M. P. Kulkarni, U. K. Kharul, A.S. Patil, S. P. Vernekar, “Synthesis, Characterization and Gas Permeability of Aromatic Polyimides Containing Pendant Phenoxy Group” J. Appl. Polym. Sci., 108, 2008, 3881.
S. S. Kothawade, U. K. Kharul, K. Vijayamohanan, S. P. Vernekar, “Novel polyimides containing pendant sulfophenoxy group as membrane material for polymer electrolyte fuel cell (PEFC) and gas permeation” to be communicated.
S. S. Kothawade, S. P. Vernekar, U. K. Kharul, “Structure–gas permeability co-relationship in aromatic polyimides having alkyl substituted pendant phenoxy group” to be communicated.
S. S. Kothawade, U. K. Kharul, K. Vijayamohanan, S. P. Vernekar, “Blend membranes based on side-chain-type sulfonated polyimide and polybenzimidazole (PBI) for polymer electrolyte fuel cell (PEFC) and gas permeation” to be communicated.
S. S. Kothawade, N. Ramgir, T. Gopakumar, U. K. Kharul, K. Vijayamohanan, S. P. Vernekar, “An improved surface functionalized polymer electrolyte membrane for planar fuel cells” to be communicated.
Presentations in conferences
S. S. Kothawade, N. S. Ramgir, M. P. Kulkarni, R. A. Potrekar, S. P. Vernekar, K. Vijayamohanan, A Paper titled “Comparison of surface functionalized HDPE, block Copolymers of Styrene- Propylene-Butadiene and PVDF polymer Membranes for Planar Fuel Cell Applications” presented in National Seminar on Fuel-to-Fuel Cells, Indian Institute of Chemical Technology, December 4-5, 2003, Hyderabad, Andhra Pradesh, India.
S. S. Kothawade, N. S. Ramgir, S. P. Vernekar, K. Vijayamohanan, A Paper titled “Temperature dependent impedance studies of Nafion-117 membrane for PEMFC applications” presented in National seminar on Membrane Science and Technology: Challenges and Opportunities, February 12-13, 2004, Regional Research Laboratory, Jorhat, Assam, India.
S. S. Kothawade, N. Ramgir, B. Kakade, A. B. Gaikwad, S.P.Vernekar, I. S. Mulla, K. Vijayamohanan, A poster titled “Temperature Dependent Impedance Studies of Nafion-117 and Sulfonated Polyimide Membranes for PEMFC Applications” presented in National Seminar on Fuel cells-Materials, Systems and Accessories Sept. 25-26, 2003, Ambernath (MS), India.
S. S. Kothawade, M. P. Kulkarni, U. K. Kharul, A.S. Patil, S. P. Vernekar, A poster titled “Synthesis, Characterization and Gas Permeability of Aromatic Polyimides Containing Pendant Phenoxy Group” presented in ‘MACRO 2006’ December 17-20, 2006, held at National Chemical Laboratory, Pune, India.


















