
Symmetry in order-disorder changes of molecular clusters
6
Symmetry in order-disorder changes of molecular clusters Ana Proykova, 1 Dessislava Nikolova, 1 and R. Stephen Berry 2 1 Faculty of Physics, University of Sofia, 5 James Bourchier Blvd., Sofia-1126, Bulgaria 2 Department of Chemistry, the University of Chicago, Chicago, Illinois 60637 ~Received 23 April 2001; revised manuscript received 28 September 2001; published 7 February 2002! Clusters of octahedral AF 6 molecules transform when cooled from a body-centered-cubic, orientationally disordered phase to a monoclinic, orientationally ordered phase in a two-step process. Recently, the higher- temperature transition has been shown to be a finite-system counterpart of a first-order transition. Here, with the help of symmetry-adapted rotator functions, we show that the lower-temperature transition is continuous. With our analytical model, we predict a temperature of 27 K for this transition, for a 59-molecule cluster of TeF 6 , consistent with the ;30-K result from canonical Monte Carlo calculations. DOI: 10.1103/PhysRevB.65.085411 PACS number~s!: 36.40.Ei, 61.50.2f, 64.70.Kb I. INTRODUCTION Free clusters of ;100 or more octahedral molecules of SF 6 ~Refs. 1 and 2! and TeF 6 ~Ref. 3! solidify in body- centered-cubic ~bcc! structures with molecules disordered orientationally. A further decrease of temperature produces a phase transition to a more orientationally ordered phase with a lattice structure of a lower, monoclinic symmetry, as dif- fraction experiments have shown. 4–7 Numerical experiments show analogous structural transformations. 8–12 Our previous molecular-dynamics simulations 12,13 indicated two steps in the ordering structural transition. First, when the solid cluster is cooled from near freezing, it undergoes a transition from a bcc orientationally disordered phase to a partially ordered monoclinic phase. The two phases coexist dynamically, 14,15 indicative of the small-system counterpart of a first-order phase transition. A second transition, monoclinic to mono- clinic but from partially ordered to completely ordered ori- entations, occurs with a further temperature decrease. There has been no clear indication of the nature of this second transition: the absence of coexisting phases is a nec- essary but not sufficient condition to claim that this is a continuous transition. In fact, it might be a weak first-order transition, distorted by the presence of a symmetry-breaking surface. In order to understand the two-step solid-solid transfor- mation induced by temperature changes, we develop a mi- croscopic, symmetry-based description of the ordering of the molecular centers of mass and then of the axes of symmetry, in terms of point group theory. 16 Such a description makes it possible to calculate the relative contributions of various in- teractions to the total potential of the cluster, and thereby to infer the dominant mechanism responsible for a specific tran- sition. The methodology was developed 17 to describe the rotation-rotation interactions in C 60 and translation-rotation in KCN. 18 A broader aim is to determine the nature and origins of the temperature-driven solid-solid transformations of clusters of rigid molecules ~intramolecular vibrations are neglected!, and to gain insights into the nature of second- order and weak first-order transitions. Solid ~structural! changes in a cluster take place when some distortion develops in active constituent groups, either individually or collectively. The distortion is typically char- acterized by a change in the lattice symmetry. A change in a suitable order parameter 19 describes the relaxation of the sys- tem in terms of its broken symmetry. The Landau theory gives a qualitative description of continuous phase transi- tions if the order parameter is a scalar and the so-called mar- ginal spatial dimension is exceeded. 20 Being a mean-field theory, it does not allow a detailed description of the various interactions and their relative contribution to the total poten- tial at different temperatures. It is instructive to implement a microscopic description to elucidate the phase changes in small systems, and then to connect them to Landau theory for the corresponding bulk matter. Here, we adopt a specific model 17 to describe the dynamic orientational disorder of the high-temperature phase, and the transition to the orientationally ordered monoclinic structure of clusters of octahedral molecules. The model is applicable to any cluster composed of 59 molecules or more. This num- ber of molecules ensures that at least one primitive cell for the high-T structure ~bcc! is in a proper surrounding de- manded by the group symmetry operations for a lattice. Solid-solid transitions of free nanoclusters tend to initiate on the surface. 9,13,21 The current analysis assumes that in the vicinity of phase transitions molecules change their immedi- ate neighborhood from disordered to more ordered structures when the temperature decreases. With a decrease of the ki- netic energy, the orientational dependence of the interacting potential becomes relatively more and more pronounced. Hence, with cooling, there is a relative increase in the angle- dependent interactions that lead to a lower-temperature phase change. In Secs. II and III we consider only the lattice symmetry of the volume, with the ~interior! molecules treated as having all the neighbors required by the corresponding point group of a crystal. Surface effects are included in Sec. IV in the application example by explicit calculation of binding ener- gies: the surface molecules have larger amplitudes of motion around the lattice points, and are of course less strongly bound than those in the interior. Another alternative to treat- ing the surface explicitly would be to use continuous sym- metry measures 22,23 or a perturbation approach such as ‘‘near symmetry groups.’’ 24 The model for the orientation order-disorder transforma- tion of molecules in a given surrounding symmetry is de- scribed in Sec. II. We use rotator functions 25 to compute the PHYSICAL REVIEW B, VOLUME 65, 085411 0163-1829/2002/65~8!/085411~6!/$20.00 ©2002 The American Physical Society 65 085411-1
Transcript of Symmetry in order-disorder changes of molecular clusters

PHYSICAL REVIEW B, VOLUME 65, 085411
Symmetry in order-disorder changes of molecular clusters
Ana Proykova,1 Dessislava Nikolova,1 and R. Stephen Berry2
1Faculty of Physics, University of Sofia, 5 James Bourchier Blvd., Sofia-1126, Bulgaria2Department of Chemistry, the University of Chicago, Chicago, Illinois 60637
~Received 23 April 2001; revised manuscript received 28 September 2001; published 7 February 2002!
Clusters of octahedral AF6 molecules transform when cooled from a body-centered-cubic, orientationallydisordered phase to a monoclinic, orientationally ordered phase in a two-step process. Recently, the higher-temperature transition has been shown to be a finite-system counterpart of a first-order transition. Here, withthe help of symmetry-adapted rotator functions, we show that the lower-temperature transition is continuous.With our analytical model, we predict a temperature of 27 K for this transition, for a 59-molecule cluster ofTeF6, consistent with the;30-K result from canonical Monte Carlo calculations.
DOI: 10.1103/PhysRevB.65.085411 PACS number~s!: 36.40.Ei, 61.50.2f, 64.70.Kb
of
ds
wiif
te
re
enori-
thna
ein
ormthetitin
a
ndn
d
enth-in
s-rysi-ar-
usn-
t as inory
thereblem-for-
tehedi-urese ki-tinged.le-
hase
rygupher-tionglyat-m-ar
a-e-
I. INTRODUCTION
Free clusters of;100 or more octahedral moleculesSF6 ~Refs. 1 and 2! and TeF6 ~Ref. 3! solidify in body-centered-cubic~bcc! structures with molecules disordereorientationally. A further decrease of temperature producephase transition to a more orientationally ordered phasea lattice structure of a lower, monoclinic symmetry, as dfraction experiments have shown.4–7 Numerical experimentsshow analogous structural transformations.8–12 Our previousmolecular-dynamics simulations12,13 indicated two steps inthe ordering structural transition. First, when the solid clusis cooled from near freezing, it undergoes a transition frombcc orientationally disordered phase to a partially ordemonoclinic phase. The two phases coexist dynamically,14,15
indicative of the small-system counterpart of a first-ordphase transition. A second transition, monoclinic to moclinic but from partially ordered to completely ordered oentations, occurs with a further temperature decrease.
There has been no clear indication of the nature ofsecond transition: the absence of coexisting phases is aessary but not sufficient condition to claim that this iscontinuous transition. In fact, it might be a weak first-ordtransition, distorted by the presence of a symmetry-breaksurface.
In order to understand the two-step solid-solid transfmation induced by temperature changes, we develop acroscopic, symmetry-based description of the ordering ofmolecular centers of mass and then of the axes of symmin terms of point group theory.16 Such a description makespossible to calculate the relative contributions of variousteractions to the total potential of the cluster, and therebyinfer the dominant mechanism responsible for a specific trsition. The methodology was developed17 to describe therotation-rotation interactions in C60 and translation-rotationin KCN.18 A broader aim is to determine the nature aorigins of the temperature-driven solid-solid transformatioof clusters of rigid molecules~intramolecular vibrations areneglected!, and to gain insights into the nature of seconorder and weak first-order transitions.
Solid ~structural! changes in a cluster take place whsome distortion develops in active constituent groups, eiindividually or collectively. The distortion is typically characterized by a change in the lattice symmetry. A change
0163-1829/2002/65~8!/085411~6!/$20.00 65 0854
ath-
rad
r-
isec-
rg
-i-ery,
-ton-
s
-
er
a
suitable order parameter19 describes the relaxation of the sytem in terms of its broken symmetry. The Landau theogives a qualitative description of continuous phase trantions if the order parameter is a scalar and the so-called mginal spatial dimension is exceeded.20 Being a mean-fieldtheory, it does not allow a detailed description of the variointeractions and their relative contribution to the total potetial at different temperatures. It is instructive to implemenmicroscopic description to elucidate the phase changesmall systems, and then to connect them to Landau thefor the corresponding bulk matter.
Here, we adopt a specific model17 to describe the dynamicorientational disorder of the high-temperature phase, andtransition to the orientationally ordered monoclinic structuof clusters of octahedral molecules. The model is applicato any cluster composed of 59 molecules or more. This nuber of molecules ensures that at least one primitive cellthe high-T structure ~bcc! is in a proper surrounding demanded by the group symmetry operations for a lattice.
Solid-solid transitions of free nanoclusters tend to initiaon the surface.9,13,21The current analysis assumes that in tvicinity of phase transitions molecules change their immeate neighborhood from disordered to more ordered structwhen the temperature decreases. With a decrease of thnetic energy, the orientational dependence of the interacpotential becomes relatively more and more pronouncHence, with cooling, there is a relative increase in the angdependent interactions that lead to a lower-temperature pchange.
In Secs. II and III we consider only the lattice symmetof the volume, with the~interior! molecules treated as havinall the neighbors required by the corresponding point groof a crystal. Surface effects are included in Sec. IV in tapplication example by explicit calculation of binding enegies: the surface molecules have larger amplitudes of moaround the lattice points, and are of course less stronbound than those in the interior. Another alternative to treing the surface explicitly would be to use continuous symetry measures22,23or a perturbation approach such as ‘‘nesymmetry groups.’’24
The model for the orientation order-disorder transformtion of molecules in a given surrounding symmetry is dscribed in Sec. II. We use rotator functions25 to compute the
©2002 The American Physical Society11-1

ianmitn
nsllrote
Fs
ed
otio
ith
nth
e
e
i
-one
th
n
a-atnf-
ys-is
sfr
tions
the
ar.
ties
redan
g-
PROYKOVA, NIKOLOVA, AND BERRY PHYSICAL REVIEW B 65 085411
various contributions to the interaction potential describedSec. III, and apply the microscopic model to predict the trsition temperature of a cluster in Sec. IV. The computed teperature from the analytical theory is in good agreement wthe computer experiments performed with Monte Carlo amolecular-dynamics methods. The theory presented hereveals the origin of the two-step order-disorder phase tration in molecular clusters, and shows that clusters as sma59-molecule aggregates of octahedral molecules have perties which can be correlated with these of the bulk marial.
II. A SINGLE MOLECULEAT A SPECIFIC MOLECULAR SITE
In order to take into account the symmetries of the A6molecule and of the molecular site in the cluster we uconcepts from the theory of orientationally disordersubstances.26,16We consider that the AF6 molecules withOhsymmetry are rigid, and are located either in anOh localenvironment at elevated temperatures or in aC2h local envi-ronment below the transition temperature. The locationeach molecule is specified by its center of mass. Orientadisorder characterizes the high-temperature phase.
We refer to~i! a space~laboratory! axis systemXYZfixedat the cluster’s center of mass,~ii ! a nonrotating systemX8Y8Z8 parallel toXYZ but with its origin translating withthe molecular center of mass, and~iii ! a rotating, body-fixedsystemxyz. For rigid molecules, these axes coincide wthe principal axes of inertia.
At the first stage, we consider molecular translation arotation separately. Molecular translation is described asmotion of the molecular centers of mass in theXYZ coordi-nates. Thexyz orientation in theXYZ system is specified bythe three Euler anglesv5(a,b,g), wherea (0<a<2P)andb (0<b<P) are the ordinary polar coordinates of thz axis in theXYZsystem, andg (0<g<2P) is an angle inthe xy plane measuring the clockwise rotation about thzaxis.
We describe molecular sites and molecular orientationorientationally disordered high-T crystals in terms ofsymmetry-adapted rotator functions16 S(V), V5(u,w),used by James and Keenan27 to describe solid methane, having a tetrahedral symmetry. These angles, the conventicolatitude and azimuth, describe the orientation of a fixvector in the molecule. For a tetrahedral~octahedral, as well!symmetry and a rigid molecule, one can choose one ofbonds as the fixed vector.
The orientational changes at acubic site are expressed iterms of site-symmetry-adapted functions16
Slt~V!5 (
m52 l
l
Ylm~V!a l
mt , ~1!
wheret5(G, G, p, r) indicates the irreducible representtions G of the groupG, p distinguishes representations thoccur more than once, andr denotes the rows of a giverepresentation;Yl
m(V) are the spherical harmonics; the coe
08541
n--hdre-i-asp--
e
fn
de
n
ald
e
ficients a lm are tabulated.16 The functionsSl
t(V) form acomplete basis inV-space if all l 8s are included in thesum @Eq. ~1!#.
A. Molecular axes aligned with the cluster’s center of mass
Let an octahedral molecule have an initial orientationv inwhich the molecular axes coincide with the laboratory stem axes. The resulting orientational density distributionexpanded in terms of spherical harmonicsYl
m(V) in thesame way as in Eq.~1!. The molecular symmetry requirethat l 50,4,6, . . . , andonly certain linear combinations oYl
m occur. For each allowedl, we determine the moleculasymmetry-adapted functionsSl
l(V), wherel refers to theidentity representation of the cubic groupOh .16 The l 54manifold of the symmetry-adapted functions reduces toA1g ,Eg , T1g , andT2g .
At low temperatures, clusters of AF6 molecules adopt amonoclinic structureC2h .3 The l 54 manifold of thesymmetry-adapted functions reduces to the representa5Ag and 4Bg underG[C2h . Ag is the representation whichbelongs toOh andC2h .
B. Arbitrary orientation of a molecule at its site
For a molecule in an arbitrary orientation, rotated atvwith respect to the orientation discussed in Sec. II A,symmetry-adapted function becomes
R~v!Sll~Vb!5 (
k52 l
l
(m52 l
l
Ylm~Vs!Dl
mk~v!a lkl , ~2!
with R(v) the rotator operator andDlmk(v) the Wigner ma-
trices;Vb andVs determine the polar angle in the molecul~body! and laboratory~space! systems, respectively. In Eq~2! we put the spherical harmonicsYl
m(Vs), determined fromthe inverse of equation~1!,
R~v!Sll~Vb!5 (
t,m,kSl
t~Vs!~a lmt!Dl
mk~v!a lkl
5(t
Slt~Vs!D l
tl~v!, ~3!
where
D ltl~v!5 (
k52 l
l
(m52 l
l
~a lmt!Dl
mk~v!a lkl ~4!
are the rotator functions defined by the symmetry properof the moleculea l
kl and of the sitea lmt . Equation~3! relates
the symmetry-adapted functionsSll(Vb) for the body~mo-
lecular! system to the symmetry-adapted functionsSlt(Vs)
for the system—a cluster with a specific symmetry.The rotator function’s average valueD l
tl(v) is zero in thedisordered phase and nonzero in the orientationally ordephase. This property makes it suitable to be chosen asorder parameterfor the solid-solid transformations, as sugested by James and Keenan for solidCD4 ( l 53).27
1-2

ci
si
t
weigs
ce
-
on
e
r-
es,
y
otal
SYMMETRY IN ORDER-DISORDER CHANGES OF . . . PHYSICAL REVIEW B65 085411
III. INTERMOLECULAR POTENTIAL EXPANSIONIN TERMS OF ROTATOR FUNCTIONS
So far we have considered a single molecule at a spesite. The orientational configuration ofN molecules in thecluster is given by D l
tl(v(n))5D ltl(n) where n
51,2, . . . ,N, labels each molecule’s center at its site potion rn . The total potentialV is the sum overN molecules ofinteractions between two moleculesn andn8, each of whichcan be written as a sum of atom-atom potentials10
V5 (n,n8
N
(n,n8
Na
V~n,n;n8,n8!, ~5!
where (n,n) labels thenth atom in thenth molecule at sitern , with Na atoms. The atom-atom potentialV(n,n;n8n8)depends on the distancer nn8 between the atomsn and n8.The position of thenth atom in thenth molecule with respecto the space system is given by
R~n,dn!5rn1rnn1u~n!,
with u(n) the displacement of thenth molecule from itsequilibrium site positionrn ; the vectorrnn@dn,Vn(n)# is de-termined by the lengthdn and the orientationVn(n) in thespace system. For a rigid molecule,dn is equal to the bondlength.
In order to determine the translation-rotation couplingneed to expand the pair potential by considering a nonrlattice. Expansion of Eq.~5! in terms of the displacementu(n) yields a sum over terms ofpth order,
V~n,n8!5 (p50
`
(n,n8
1
~p! !Vi 1••• i p
(p) ~r nn8!@ui 1~n!
2ui 1~n8!#•••@ui p
~n!2ui p~n8!#, ~6!
with the notation
rnn85uR~n,dn!2R~n8,dn!u5ur ~n!2r ~n8!1u~n!2u~n8!
1rnn2rn8n8u
Vi 1••• i p
(p) ~r nn8!5]pV~r nn8!
]@u~n!2u~n8!# i 1•••]@u~n!2u~n8!# i p
.
~7!
The coefficientsV(p) contain the orientational dependenof the molecules at the sitesn and n8. We expand them interms ofSl
t @Eq. ~1!#. In the following we writeSm(n) forSl
t(Vn), wherem[m(t,l ):
Vi 1••• i p
(p) ~r nn8!5 (mm8
ci 1••• i pmm8(p)
~n,n8!Sm~n!Sm8~n8!. ~8!
The coefficientsci 1••• i pmm8(p) (n,n8) are determined by inte
grating the pairwise potential over the possible orientatiof r nn and r nn8 :
08541
fic
-
id
s
ci 1••• i pmm8(p)
~n,n8!
5E dVnE dVn8Vi 1••• i p
(p) ~n,n8!Sm~n!Sm8~n8!.
~9!
Now we substituteV(p) from Eq. ~8! into Eq. ~6!, and intro-duce the molecular form factorgl
l :
V~n,n8!5(p
(mm8
1
p!ci 1••• i pmm8
p~n,n8!gl
lgl 8l Dm~n!Dm8~n8!
3@ui 1~n!2ui 1
~n8!#•••@ui p~n!2ui p
~n8!#. ~10!
The molecular form factor is defined for allowedl asgll
5(n50Na Sl
l@Vn(n)# if the molecular axes coincide with thspace axes or as(nSm(n)5gl
lDm(v) if the molecule is ro-tated at an anglev. Molecular and site symmetry consideations restrict the number of terms in the sums in Eq.~10!.
A. Rotation-rotation interaction
The valuep50 corresponds to a rigid lattice~no motionsof the molecular centers of mass!. This case yields onlyrotation-rotation interaction between molecules, withmÞ(0,0) andm8Þ(0,0):
V(0)~n,n8!5 (mm8
cmm80
~n,n8!gllgl 8
l Dm~n!Dm8~n8!.
~11!
The total rotation interaction is the sum over all moleculVRR5(n,n8
N V(0)(n,n8).The matrix of rotation-rotation interactions is defined b
Jmm8~n,n8!5cmm8(0)
~n,n8!gllgl 8
l , ~12!
where the interaction matricescmm8(0) (n,n8) follow from Eq.
~9!. Their structure depends on the symmetry ofSm and onthe relative position (n,n8) of two interacting molecules on alattice with symmetry specified byt.
To calculate the transition temperature we need the tfield VR acting on the molecule at siter (n). The zeroth ap-proximation treats spherically symmetric moleculesm85(0,0) acting on a moleculen with mÞ(0,0) on a rigidlattice p50:
V(0)~n,n8!u l 8505(m
cm(0)~n,n8!gl
lg0lDm~n!.
Settingm85(0,0) yields
Sm85S0A1g5~4p!21/2 and g05Na~4p!21/2.
The coefficientscm(0)(n,n8) become
cm(0)~n,n8!5
1
A~4p!E dVnE dVn8V
(0)~n,n8!Sm~n!.
1-3

q.
ac-du
ed
ss
ica
roaewinath
lar-an-d
uc-ithtry.s isow-
duc--
igid,and
urald aesare
swe
ms
heics
is-samee.
PROYKOVA, NIKOLOVA, AND BERRY PHYSICAL REVIEW B 65 085411
Let us denote the interaction matricescm(0)(n,n8)
weighted with the molecular factorsgll andg0
l by yaR , where
a is an index for (l ,A1g ,p,r):
yaR5(
n8ca
(0)~n,n8!gllg0
l . ~13!
The crystal field acting on the moleculen is
VR~n!5(a
yaRDa~n!. ~14!
The rotator functionsD lA1g(n) in Eq. ~14! are cubic func-
tions.
B. Translation-rotation and translation-translation interaction
The pair translation-rotation interaction follows from E~10! for p51, mÞ(0,0), andm85(0,0):
VTR~n,n8!5V(1)~n,n8!5(im
cim(1)~n,n8!gl
lg0lDm~n!
3@ui~n!2ui~n8!#. ~15!
The sum over all molecules yields the total bilinear intertion VTR5(n,n8V
TR(n,n8). The translation-rotation interaction is caused by the change of the orientational potentialto the displacement of nearest neighbors.
The pair translation-translation interaction is obtainfrom Eq. ~10! for p52, m85(0,0), andm5(0,0):
VTT~n,n8!5V(2)~n,n8!5 (i 1 ,i 2
1
2ci 1i 2
(2) ~n,n8!g0lg0
l
3@ui 1~n!2ui 1
~n8!#@ui 2~n!2ui 2
~n8!#.
~16!
This gives a total translation-translation interactionVTT
5(n,n8VTT(n,n8). Now the total potential separates:
V5VR1VTT1VTR1VRR. ~17!
Equation ~17! may be expanded with higher order termwhich might become important in some structural phatransitions.28
IV. APPLICATION OF THE ROTATOR FUNCTIONSTO STRUCTURAL TRANSITIONS
IN TELLURIUM HEXAFLUORIDE CLUSTERS
As an example, we have applied this group-theoretapproach to determine the nature and factors of Eq.~17!,stimulating the phase changes of a cluster of 59 TeF6 mol-ecules, and we have compared the results to those fsimulations.10–12 Let us recall that 59 molecules ensure ththe interior @Eq. ~15!# molecules in the cluster all have theight neighbors required for a bcc structure. In Fig. 1show 12 interior molecules with their orientations: one isthe center; it has a surrounding cube of eight molecules,three of the six molecules are located in the bodies of
08541
-
e
,e
l
mt
e
nde
neighboring cubes. The data are taken from molecudynamics simulations14 at constant temperature. This ismolecular configuration just below the first solid-solid trasition, taking the cluster from orientationally disorderebody-centered-cubic to a partially ordered monoclinic strture. ‘‘Partially’’ means that the molecules are aligned wrespect to only one of the three molecular axes of symmeThe black and dark gray molecules are almost aligned, athe case with white and light gray molecules. Further, belthis temperature,;76 K, the displacements of the molecular center of masses couple to the orientation changes, ining rotation-translation interaction. With a still further decrease of the temperature, the cluster becomes almost rthe molecular centers undergo very small displacements,rotation-rotation interaction dominates the second structtransition, which consists of continuous rearrangement anfull ordering of the molecular axes of symmetry. The valuof the relevant contributions to the interaction potentialsgiven in a following paragraph.
We compare the rotation-rotation contributions@Eq. ~11!#,with their translation-rotation counterparts@Eq. ~15!#, for theinteraction of a TeF6 molecule with its nearest neighborlocated at cubic and at monoclinic sites. For this purpose,determine the crystal field@Eq. ~14!#, and the rotational ma-trix @Eq. ~12!#. The CERN Library29 is used to compute theelements of Eq.~12!.
Here we give as example the values of the first two terof the expansion. For octahedral TeF6, the form factors areg05(Na)(4p)21/251.98, g451.29 in anOh environmentandg453.88 in aC2h environment.
The l 50 value ofS is (4p)21/2. The l 54 manifold ofthis system reduces to representationsA1g , Eg , T1g , andT2g underG[Oh and to representations 5Ag and 4Bg underG[C2h .
For the normalized functionS4t we find in Oh ,
FIG. 1. The interior 12 molecules of 59-molecule cluster, in tvicinity of a phase transition found in the molecular-dynamsimulation~Ref. 10!. The arrows indicate the space (u,f) orienta-tion of one of the molecular axes of symmetry. It is seen that dtances between the molecular centers of masses are almost thebut still somewhat different: in the bulk the behavior is the samThis is the reason we call such materials ‘‘plastic.’’
1-4
ee
tm
-osurs.ld
-
ni
c
ladhee
erera
uln
ly
delas
er-
u-e
r
l
-thein
iththe
t
by
cep-
SYMMETRY IN ORDER-DISORDER CHANGES OF . . . PHYSICAL REVIEW B65 085411
[email protected],m50;0.457,m564# for Oh ,A1g,1,1,
[email protected],m50;20.541,m564# for Oh ,Eg,1,1,
[email protected],m562# for Oh ,Eg,2,1,
a4mt5@2 i0.663,m561;2 i0.25,
m563] for Oh ,T1g,1,1,
a4mt5@6 i0.663,m561;2 i0.25,
m563] for Oh ,T1g,2,1,
a4mt5@6 i0.707,m564# for Oh ,T1g,3,1,
a4mt5@6 i0.663,m563;6 i0.25,
m561] for Oh ,T2g,1,1
[email protected],m563;60.25,m561# for Oh ,T2g,2,1,
a4mt5@6 i0.707,m562# for Oh ,T2g,3,1.
In a C2h environment, all coefficientsat are 1 for the five-fold representationAg (m50,62,64) and for the fourfoldrepresentationBg (m561,63).
The contribution to the rotational-rotational matrix is thlargest for l 5 l 854. In a cubic symmetry environment thmatrix is quasidiagonal, while inC2h surrounding the matrixis diagonal~only one-dimensional representations!.
We correlateOh andC2h in two steps: first, we pass fromOh to D4h , and then use the inferred table forD4h to go onto C2h .16 Another two-step correlation is also possible:Oh toD3d to C2h . Different possibilities are realized in differenstructures observed in the experiments with telluriuclusters3 at low temperatures. In a recent paper,30 structuralstudies of ferroelectric PbZr12xTixO3 showed the metastability of different phases as a result of changes in the comption x of Ti in the substance, which is a confirmation of ogeneral prediction based on the symmetry consideration
The coefficientsyR, necessary to compute the crystal fie@Eq. ~14!#, are calculated from Eq.~9! with p50. The cubicsymmetry value isyOh
R 527.98 in the approximation includ
ing only the nearest eight neighbors. For the monoclistructure,C2h , this coefficient isyC2h
R 520.61.
In the approximation of only nearest-neighbor interations, we determine the energy per molecule: inOh , therotation-rotation interaction energy is 4 meV and the transtion rotation is 0.3 meV. InC2h , these values are 0.18 an0.002 meV, respectively. The conclusion is that ttranslation-rotation interaction can be neglected in the ording of molecules on monoclinic sites, so that the lowtemperature transition is entirely driven by rotational ording, and that the transition is continuous even in smsystems. However, on cubic sites, motions of the moleccenters of mass are large enough, and the transitioorientation-displacive first-order-like~with two local minimain the free energy!, at least in small systems. The high
08541
i-
c
-
-
r---llaris
degenerate state of anOh molecule in anOh environment isresolved by a distortion of the cluster structure if the morequires rigid molecules. Thus we can think of the processa Jahn-Teller effect that distorts the cluster from its localOhsymmetry toC2h via D3d or D4h . The large value of therotation-rotation interaction leads to a distinct partial ording of the molecules.11 The transition fromOh to C2h isresolved by invoking the representation ofEg in D4h ~or inD3d), equivalent to a splitting of a degenerate mode.
Having the interactions and the total field, we can calclate the free energyF of each phase as a function of throtator functions considered as order parameters,25
F50.5(q
†1x0211F@ J#‡dm~q!dm8~2q!, ~18!
whereF( J) anddm(q) are the Fourier images of the rotatomatrix J andDm(v), respectively; 1ˆ is the 333 unit matrix;and x0[xT21 is the single-molecule orientationasusceptibility,17
x5Z21~g4A1g!2E exp@2VR~v!/T#@Dm~v!#2dv, ~19!
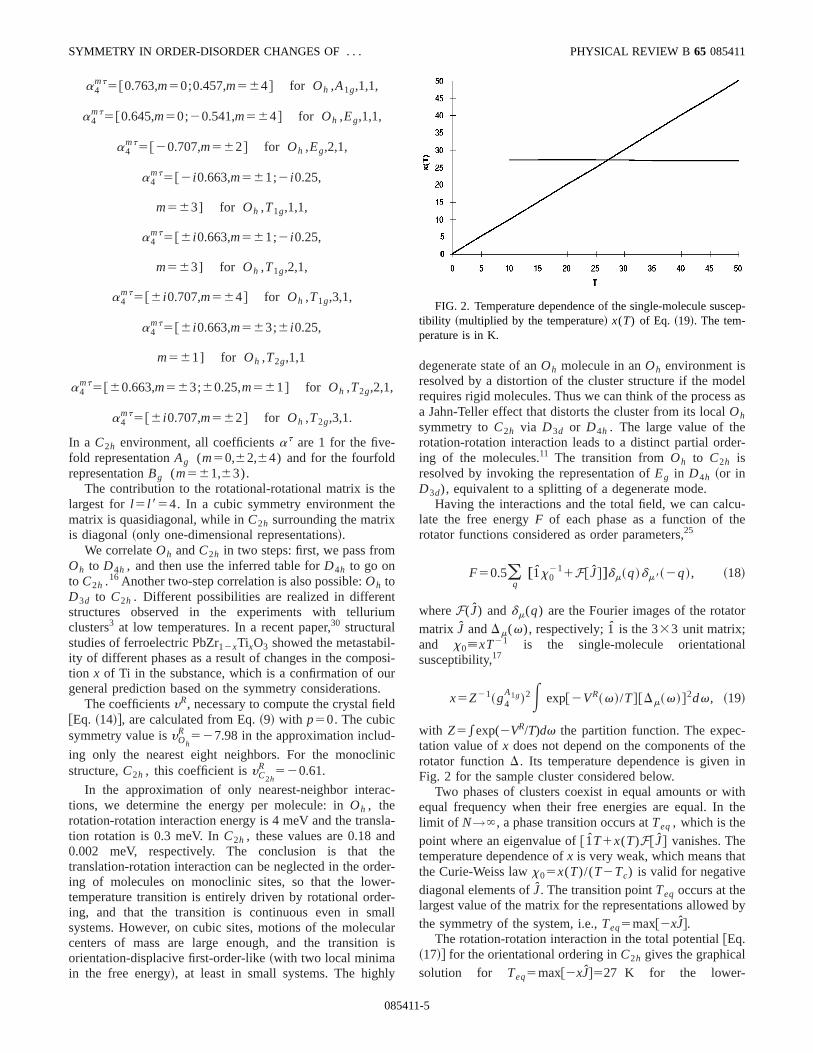
with Z5*exp(2VR/T)dv the partition function. The expectation value ofx does not depend on the components ofrotator functionD. Its temperature dependence is givenFig. 2 for the sample cluster considered below.
Two phases of clusters coexist in equal amounts or wequal frequency when their free energies are equal. Inlimit of N→`, a phase transition occurs atTeq , which is thepoint where an eigenvalue of@ 1T1x(T)F@ J# vanishes. Thetemperature dependence ofx is very weak, which means thathe Curie-Weiss lawx05x(T)/(T2Tc) is valid for negativediagonal elements ofJ. The transition pointTeq occurs at thelargest value of the matrix for the representations allowedthe symmetry of the system, i.e.,Teq5max@2xJ#.
The rotation-rotation interaction in the total potential@Eq.~17!# for the orientational ordering inC2h gives the graphicalsolution for Teq5max@2xJ#527 K for the lower-
FIG. 2. Temperature dependence of the single-molecule sustibility ~multiplied by the temperature! x(T) of Eq. ~19!. The tem-perature is in K.
1-5

e
in
ribe
in-s ofnts
it. Innd
ln-asetheing
gy
SA
nt.
nse
PROYKOVA, NIKOLOVA, AND BERRY PHYSICAL REVIEW B 65 085411
temperature transition~Fig. 2!. We have implemented thcubic rotator functionsD l
A1g in calculating the crystal field@Eq. ~14!#. This prediction of the transition temperature isa good agreement with the results (;30 K) from the ca-nonical Monte Carlo simulations,31 ~Fig. 3!, and molecular-
FIG. 3. Caloric curve from canonical Monte Carlo simulatioof a 59-molecule TeF6 cluster; 1 comp. unit is about 1 eV. Thcalculation uncertainties are 0.02–0.05 comp. units.
e
J.
yS.
e
hy
/
m
m
in
08541
dynamics calculations.10 This means that the cubic rotatofunctions@Eq. ~14!# are suitable order parameters to descrthe mechanism of structural phase changes.
V. CONCLUSION
To summarize, we have shown how the near-neighbortermolecular interactions of a cluster can be cast in termlocal molecular and site symmetry in a manner that accoufor the multistep phase changes that these clusters exhibparticular, we have shown how translation-rotation arotation-rotation interactions enter into theOh-D4h-2@D3d#transition of TeF6 to yield a phase change with two locafree-energy minima for the small system, but only rotatiorotation interactions enter into the lower-temperature phchange from partial to complete orientational ordering ofmolecules on a monoclinic lattice, a change that, accordto all indications, involves only a single local free-enerminimum.
ACKNOWLEDGMENTS
The research was completed under the NATO CLG~PST CLG 976363!5437. Support by Grant No. 3270 fromthe Scientific Fund at the University of Sofia and a grafrom the National Science Foundation are acknowledged
f
tt.
.
p.
. E.
1J. Farges, M. F. de Feraudy, B. Raoult, and G. J. Torchet, J. ChPhys.78, 5067~1983!.
2J. B. Maillet, A. Boutin, S. Buttefey, F. Calvo, and A. H. Fuchs,Chem. Phys.109, 329 ~1998!.
3L. S. Bartell, L. Harsami, and E. J. Valente, inThe Physics andChemistry of Small Clusters, Vol. 158 ofNATO Advanced StudInstitute, Series B: Physics, edited by P. Jena, B. K. Rao, andN. Khanna~Plenum, Richmond, VA, 1987!, p. 37.
4J. Farges, M. F. de Feraudy, B. Raoult, and G. J. Torchet, J. ChPhys.84, 3491~1986!.
5B. Raoult, J. Farges, M. F. de Feraudy, and G. Torchet, Z. PD: At., Mol. Clusters12, 85 ~1989!.
6L. S. Bartell, F. J. Dulls, and B. Chunko, J. Phys. Chem.95, 6481~1991!.
7T. S. Dibble and L. S. Bartell, J. Phys. Chem.96, 8603~1992!.8S. Xu and L. S. Bartell, Z. Phys. D: At., Mol. Clusters26, 364
~1993!.9S. Xu and L. S. Bartell, Z. Phys. D: At., Mol. Clusters31, 117
~1994!.10A. Proykova and R. S. Berry, Z. Phys. D: At., Mol. Clusters40,
215 ~1997!.11R. A. Radev, A. Proykova, and R. S. Berry, http://www.ijc.com
articles/1998v1/36.12R. A. Radev, A. Proykova, Feng-Yin Li, and R. S. Berry, J. Che
Phys.109, 3596~1998!.13A. Proykova, R. Radev, Feng-Yin Li, and R. S. Berry, J. Che
Phys.110, 3887~1999!.14A. Proykova, S. Pisov, R. Radev, I. Daykov and R. S. Berry,
Proceedings of the Second Workshop on Nanoscience, edited byE. Balabanova~Heron Press, Sofia, 2001!.
m.
m.
s.
.
.
15A. Proykova, S. Pisov and R. S. Berry, J. Phys. Chem.115,8583~2001!.
16C. J. Bradley and A. P. Cracknell,The Mathematical Theory oSymmetry in Solids~Clarendon, Oxford, 1972!.
17K. H. Michel, J. R. Copley, and D. N. Neumann, Phys. Rev. Le68, 2929~1992!.
18K. H. Michel and J. M. Rowe, Phys. Rev. B32, 5818~1985!; K.H. Michel and J. M. Rowe,ibid. 32, 5827~1985!.
19L. D. Landau and M. E. Lifshitz,Statistical Physics~Pergamon,New York, 1969!.
20E. Brezin, J. C. le Guillou, and J. Zinn-Justin, inPhase Transi-tions and Critical Phenomena, edited by C. Domb and M. SGreen~Academic Press, New York, 1976!, Vol. VI, p. 125.
21A. Proykova and R. S. Berry, Eur. Phys. J. D9, 445 ~1999!.22H. Zabrodsky, S. Peleg, and D. Avnir, J. Am. Chem. Soc.114,
7843 ~1992!.23M. Pinsky and D. Avnir, Inorg. Chem.37, 5575~1998!.24P. R. Bunker,Molecular Symmetry and Spectroscopy~Academic
Press, New York, 1979!, Chap. 11.25R. M. Lynden-Bell and K. H. Michel, Rev. Mod. Phys.66, 721
~1994!.26F. Seitz, Modern Theory of Solids~McGraw Hill, New York,
1940!.27H. M. James and T. A. Keenan, J. Chem. Phys.31, 12 ~1959!.28V. L. Ginzburg, A. P. Levanyuk, and A. A. Sobyanin, Phys. Re
57, 151 ~1980!.29CERN Program Library~CERN, Geneva, Switzerland, 1995!.30B. Noheda, D. E. Cox, G. Shirane, R. Guo, B. Jones, and L
Cross, Phys. Rev. B63, 014 103~2000!.31P. Michailov, M. thesis, University of Sofia, 1998.
1-6


















