
Suppressor of Cytokine Signaling 2 Is a Feedback Inhibitor ... · PDF fileSuppressor of...
11
of May 24, 2018. This information is current as Dendritic Cells Activation in Human Monocyte-Derived Feedback Inhibitor of TLR-Induced Suppressor of Cytokine Signaling 2 Is a Horejs-Hoeck Gernot Posselt, Harald Schwarz, Albert Duschl and Jutta http://www.jimmunol.org/content/187/6/2875 doi: 10.4049/jimmunol.1003348 August 2011; 2011; 187:2875-2884; Prepublished online 15 J Immunol Material Supplementary 8.DC1 http://www.jimmunol.org/content/suppl/2011/08/15/jimmunol.100334 References http://www.jimmunol.org/content/187/6/2875.full#ref-list-1 , 24 of which you can access for free at: cites 51 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2011 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on May 24, 2018 http://www.jimmunol.org/ Downloaded from by guest on May 24, 2018 http://www.jimmunol.org/ Downloaded from
-
Upload
vuongthuan -
Category
Documents
-
view
219 -
download
1
Transcript of Suppressor of Cytokine Signaling 2 Is a Feedback Inhibitor ... · PDF fileSuppressor of...

of May 24, 2018.This information is current as
Dendritic CellsActivation in Human Monocyte-DerivedFeedback Inhibitor of TLR-Induced Suppressor of Cytokine Signaling 2 Is a
Horejs-HoeckGernot Posselt, Harald Schwarz, Albert Duschl and Jutta
http://www.jimmunol.org/content/187/6/2875doi: 10.4049/jimmunol.1003348August 2011;
2011; 187:2875-2884; Prepublished online 15J Immunol
MaterialSupplementary
8.DC1http://www.jimmunol.org/content/suppl/2011/08/15/jimmunol.100334
Referenceshttp://www.jimmunol.org/content/187/6/2875.full#ref-list-1
, 24 of which you can access for free at: cites 51 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2011 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Suppressor of Cytokine Signaling 2 Is a Feedback Inhibitor ofTLR-Induced Activation in Human Monocyte-DerivedDendritic Cells
Gernot Posselt, Harald Schwarz, Albert Duschl, and Jutta Horejs-Hoeck
Dendritic cells (DCs) are key players in initiating and directing the immune response. Therefore, their activation state and func-
tional differentiation need to be tightly controlled. The activating stimuli and their signaling networks have long been an area of
focus in DC research. Recent investigations have also shed light on the mechanisms of counterregulation and fine-tuning of DC
functions. One class of proteins involved in these processes is the family of suppressors of cytokine signaling (SOCS), whosemembers
were originally described as feedback inhibitors of cytokine-induced JAK/STAT signaling. Essential roles in DC function have been
assigned to SOCS1 and SOCS3. In this article, we show that SOCS2 also is involved in DC regulation. In human and in murine DCs,
SOCS2 is a highly TLR-responsive gene, which is expressed in a time-delayed fashion beginning 8 h after TLR ligation. Functionally,
silencing of SOCS2 in DCs results in hyperphosphorylation of STAT3 at later time points. As a consequence, SOCS2-deficient DCs
secrete increased amounts of the cytokines IL-1b and IL-10, both being transcriptional targets of STAT3. We propose a model in
which SOCS2 acts as a negative regulator of TLR-induced DC activation. The delayed expression of SOCS2 provides a mechanism
of late-phase counterregulation and limitation of inflammation-driving DC activity. The Journal of Immunology, 2011, 187:
2875–2884.
Dendritic cells (DCs) play important roles in immunerecognition of invading pathogens during infections.They bridge the two branches of innate and adaptive
immunity and are thought to be unique in their capacity to primenaive T cell responses. DCs act as sentinels, responding toevolutionary-conserved microbial structures as indicators of in-fection, using pattern recognition receptors. TLRs are the bestcharacterized group of pattern recognition receptors that recognizepathogen-associated molecular patterns such as LPS, peptidogly-cans, flagellin, lipoteichoic acid, or unmethylated CpG DNA, andthey can stimulate activation of the innate immune system (1, 2).As a result, DCs change their activation state, gaining immunos-timulatory capacity hallmarked by reinforced migratory homingto secondary lymphoid tissues, increased Ag processing and pre-sentation, expression of costimulatory molecules, and secretion ofproinflammatory cytokines. These activation-associated changesenable DCs to prime Ag-specific T cells and lead to the initiationof adaptive immune responses (3). The processes of immune ac-tivation are well understood; however, research on the mecha-
nisms of counterregulation and fine-tuning in the innate systemis still in its early stages. One class of molecules that is thoughtto be involved in negative regulation and/or modification of DC-activating signals contains the suppressors of cytokine signaling(SOCS) proteins (4).The family of SOCS proteins was originally described as
comprising feedback inhibitors of cytokine-induced JAK/STATsignaling and consists of eight members, namely, SOCS1-7 andcytokine-inducible Src homology 2 (SH2)-containing protein(CIS) (5). SOCS proteins share a tripartite structure with a centralSH2 domain flanked by a conserved C-terminal domain termedthe SOCS-box and an N-terminal region with low conservationexcept for an extended SH2 subdomain and, in SOCS1 andSOCS3, a kinase inhibitory region (KIR). This segmented archi-tecture reflects the functional repertoire of these proteins. SOCSproteins execute their function in three different ways. First,SOCS proteins bind competitively to their target proteins, thusdisplacing and sequestering the signaling intermediates away fromtheir respective receptors, thereby inhibiting downstream signal-ing. The common SH2 domain mediates binding to phospho-tyrosine residues, whereas the extended SH2 subdomain con-fines the substrate specificity (6). Second, the shared KIR motif ofSOCS1 and SOCS3 directly inhibits JAK activity, apparently byacting as a pseudosubstrate for the kinase (7). Third, SOCS pro-teins exert their inhibitory action, at least in part, by enhancingubiquitination and subsequent proteasomal depletion of receptorsor signal-transducing molecules (8). The degradation of interact-ing proteins is attributed to the SOCS-box, which interacts withand stabilizes complexes of ubiquitin ligases (9).SOCS1 and SOCS3 are induced as a consequence of TLR sig-
naling and are capable of modifying the functional properties ofAPCs (10–12). Regulatory functions in pathways other than cy-tokine signaling have been described. SOCS1 has been shown tointerfere with TLR signaling by proteasomal degradation of theMyD-like adapter molecule Mal (13). In addition, SOCS1 is ableto impede autocrine IFN stimulation and is thought to prevent
Division of Allergy and Immunology, Department of Molecular Biology, Paris Lo-dron University of Salzburg, 5020 Salzburg, Austria
Received for publication October 8, 2010. Accepted for publication July 11, 2011.
G.P. designed and performed research and wrote the manuscript; H.S. performedresearch; A.D. helped interpret data and revise the manuscript; J.H.-H. designedresearch and wrote the manuscript.
Address correspondence and reprint requests to Dr. Jutta Horejs-Hoeck, Division ofAllergy and Immunology, Department of Molecular Biology, Paris Lodron Universityof Salzburg, Hellbrunnerstrasse 34, 5020 Salzburg, Austria. E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: CIS, cytokine-inducible SH2-containing protein;DC, dendritic cell; GH, growth hormone; KIR, kinase inhibitory region; moDC,monocyte-derived DC; poly I:C, polyriboinosinic:polyribocytidylic acid; qRT-PCR,quantitative RT-PCR; RPLP0, large ribosomal protein P0; SH2, Src homology 2;siRNA, small interfering RNA; SOCS, suppressor of cytokine signaling.
Copyright� 2011 by The American Association of Immunologists, Inc. 0022-1767/11/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1003348
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

overshooting of immune activation (14). SOCS1 deficiency inAPCs results in hyperactivation and consecutive hyper-Th1 re-sponses (15).In contrast, SOCS3 deficiency furnishes DCs with a toleroge-
nic phenotype. In response to many gp130 cytokines, SOCS3-dependent signal termination limits the duration of STAT3 acti-vation. Opposed to that, IL-10 signaling is not sensitive to SOSC3inhibition and induces long-lasting phosphorylation of STAT3. Inthe absence of SOCS3, a wide array of gp130 cytokines is able toinduce prolonged STAT3 activation and to mimic IL-10 stimula-tion. As a consequence, these cytokines share the anti-inflam-matory properties of IL-10 (16–18).SOCS2 was originally described as a feedback inhibitor of the
growth hormone (GH)/insulin-like growth factor axis, which isreflected by the high growth phenotype of SOCS2-deficient mice(19). Surprisingly, SOCS2 transgenic mice also show an increasein body weight, suggesting a dual role for SOCS2 in the GHsignaling cascade. This is supported by in vitro experiments, inwhich only low-to-intermediate levels of SOCS2 show inhibitoryaction on GH signaling, whereas high levels of ectopic SOCS2expression even increase STAT5 activation in response to GHstimulation (20, 21). SOCS2 expression is induced by a number ofdifferent cytokines and hormones in many cell types (22). In ad-dition, dioxin and the lipid mediator lipoxin A4 have been de-scribed to stimulate SOCS2 expression in B cells and DCs,respectively, each of them dependent on aryl-hydrocarbon re-ceptor activation (23, 24). Based on forced expression, a regula-tory potential for SOCS2 has been suggested in several pathways;however, the physiological relevance of these results is contro-versial. Because SOCS2 lacks a KIR domain, the inhibitoryfunction of SOCS2 is dependent on competitive binding via itsSH2 domain and, even more important, on the proteasomal deg-radation of the proteins with which it interacts (8). Apart fromaltered GH signaling, SOCS2-deficient mice have also been shownto exhibit alterations in their immune system. They displayovershooting immune reactions in a model of toxoplasmosisbecause of a failure to counterregulate DC activation. SOCS2-dependent TRAF6 degradation in APCs was suggested as theunderlying mechanism (25). However, that study was recentlyretracted and the proposed mechanism remains in doubt (26).Nevertheless, the main conclusion that SOCS2 plays a regulatoryrole in DC activation appears to be well supported.In this study, we show that SOCS2 is induced as a consequence
of TLR stimulation in both human and murine DCs. Comparedwith SOCS1 and SOCS3, which are upregulated within 1 h afterLPS treatment, SOCS2 shows a time-delayed expression starting∼8 h poststimulation. Despite this delay, SOCS2 is a direct tar-get of TLR signaling, for its expression is robust under con-ditions of protein-synthesis inhibition. Silencing of SOCS2 in DCsleads to increased cytokine secretion, predominantly of cytokineswith a late and more sustained expression pattern, like IL-10and IL-1b. Cytokines with early and transient expression, suchas TNF-a or IL-6, were not affected. Moreover, elevated, IL-10–independent, STAT3 phosphorylation was observed in SOCS2-deficient DCs. In conclusion, our data suggest an inhibitoryfunction for SOCS2 in TLR ligand-induced DC activation.
Materials and MethodsAll studies involving human material were conducted in accordance withthe guidelines of the World Medical Association’s Declaration of Helsinki.
Generation of human monocyte-derived DCs
Monocyte-derived DCs (moDCs) were generated according to the slightlymodified standard protocol (27). In brief, PBMCs were isolated from buffy
coats from healthy donors (kindly provided by Transfusionsmedizin derParacelsus Medical University, Salzburg, Austria) over Ficoll-Paque PLUS(GE Healthcare, Uppsala, Sweden) by density gradient centrifugation,according to the manufacturer’s instructions. PBMCs were washed twicein RPMI 1640, and cells were allowed to adhere for 90 min. Adherentmonocytes were washed extensively with warm RPMI 1640 to remove allnonadherent cells and cultured for 7 d in DC medium (RPMI 1640; PAA,Pasching, Austria), 10% FCS (PAA), 2 mM L-glutamine, 100 U/ml peni-cillin, 100 mg/ml streptomycin, 50 mM 2-ME (all from Life TechnologiesLaboratories, Grand Island, NY), and stimulated with 50 ng/ml GM-CSFand 50 ng/ml IL-4 (generous gift from Novartis, Vienna, Austria). At day 3,cells were fed with 1 vol DC medium containing fresh cytokines. After 6 d,cells were harvested and replated in DC medium without cytokines. At thisstage, cells were phenotyped by flow cytometry and were routinely $90%CD1a+ CD14low.
Isolation of primary human blood DCs and CD4+CD45Ra+
naive T cells
CD1c+ DCs were isolated from CD19-depleted PBMCs using a BDCA1+
Kit (Miltenyi Biotech, Bergisch Gladbach, Germany), according to themanufacturer’s instructions. CD4+CD45RA+ T cells were isolated using theuntouched naive CD4 T cell isolation kit II (Miltenyi Biotech), according tothe manufacturer’s instructions. Isolated cells were phenotyped by FACS.
Generation of thioglycolate-induced mouse peritonealmacrophages
cDNAs from both untreated and LPS-stimulated, thioglycolate-inducedperitoneal macrophages were kindly provided by G. Schwamberger. Mac-rophages were obtained according to standard protocols. In brief, mice werei.p. injected with 1 ml 1% thioglycolate solution. After 4 d, mice were killedand peritoneal lavage cells were collected. Cells were plated in IMDM(PAA) with antibiotics for 3 h and washed extensively afterward. Theadherent fraction was used for further stimulation as indicated.
Generation of mouse bone marrow-derived DCs
Bone marrow-derived DCs were generated according to standard protocols.In brief, bone marrow cells were isolated from femurs of BALB/c mice(Charles River). Cells were differentiated in DCmedium supplemented with5% mouse GM-CSF–conditioned supernatant (mouse DC medium) for 9 d.At days 3 and 6, half of the medium was replaced with fresh mouse DCmedium. After 9 d, cells were sedimented and replated for further stim-ulation in mouse DC medium. Cells were routinely phenotyped for CD11cexpression by means of FACS analysis.
TLR ligands and cell culture reagents
Cells were stimulated with Escherichia coli LPS 055:B5 (Sigma-Aldrich),Pam3CSK4, FSL-1, Flagellin (all from InvivoGen, San Diego, CA), R848(Alexis Biochemicals, Lausen, Switzerland), immunostimulatory CpGoligodeoxynucleotide C274 (as described in Ref. 28, phosphorothioatemodified), and polyriboinosinic:polyribocytidylic acid (poly I:C) (Sigma-Aldrich). For the control of STAT3 phosphorylation in response to IL-10stimulation, cells were incubated with 25 ng/ml recombinant human IL-10(Immunotools, Friesoythe, Germany). Where indicated, experiments wereconducted in the presence of the IL-10R–blocking Ab anti-CDw210 (BDPharmingen) at 20 mg/ml for inhibition of autocrine/paracrine IL-10stimulation. The STAT3 inhibitor Stattic (6-nitrobenzo[b]thiophene-1,1-dioxide) was purchased from Calbiochem (Darmstadt, Germany).
ELISA
For each condition, 5 3 105 cells were plated per six wells in 1.5 ml DCmedium. Supernatants of DCs were collected and stored at 280˚C untilanalysis. Cytokine secretion was measured by using commercially avail-able ELISA kits for TNF-a, IL-6 (both from PeproTech, Eubio, Vienna,Austria), IL-12p70 (BD Pharmingen, Erembodegen, Belgium), IL-1b, andIL-10 (both from R&D Systems, Biomedica, Vienna, Austria). ELISAresults are shown as normalized secretion. Because of heterogeneous ab-solute values in different donors, LPS-stimulated secretion in control cellswas set as 100%.
Western blot analysis
Cells were sedimented, washed, and lysed either directly in 23 SDS samplebuffer (Bio-Rad, Vienna, Austria) or in ice-cold Nonidet P-40 lysis buffer(Invitrogen, Lofer, Austria) supplemented with 1 mM PMSF and CompleteMini proteinase inhibitor mixture (Roche) for 30 min. Nonidet P-40 lysateswere cleared of debris by centrifugation. Equal amounts of protein were
2876 SOCS2 CONTROLS TLR-INDUCED DC ACTIVATION
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
separated by 4–12% SDS-PAGE (Invitrogen) under reducing conditions,blotted on nitrocellulose membrane (Bio-Rad), and blocked with 5%nonfat dry milk (Carl-Roth, Karlsruhe, Germany) in TBS with 0.05%Tween 20. All Abs were purchased from Cell Signaling Technology(Danvers, MA) and used according to the manufacturer’s instructions.Western blot quantification was done with ImageJ software (NationalInstitutes of Health; http://rsb.info.nih.gov/ij/).
Quantitative real-time PCR
Quantitative RT-PCR (qRT-PCR) was done as described previously (29). Inbrief, total RNA was isolated with TRIzol (Invitrogen, Lofer, Austria) ora Nucleospin RNA II kit (Macherey-Nagel, Duren, Germany), according tothe manufacturer’s instructions. For qRT-PCR, 2 mg total RNA wasreverse-transcribed using RevertAid H Minus M-MuLV reverse tran-scriptase (MBI Fermentas, St. Leon-Roth, Germany), following the man-ufacturer’s protocol. qRT-PCR was performed in a Rotorgene 3000(Corbett Research) with ready-to-use 2x iQ SYBR Green Supermix (Bio-Rad). Primers were designed by using Vector NTI software (Invitrogen) toamplify targets ranging in size from 120 to 200 nucleotides with annealingtemperatures of 65˚C. Target specificity was assessed by product se-quencing and routine recording of melting curves. Sequences of the pri-mers are listed in Table I. mRNA content (x) was calculated using theformula x = 22Dct, where Dct represents the difference between the gene ofinterest and the reference gene, large ribosomal protein P0 (RPLP0). Thefold-change value represents the quotient of the mRNA content of theinduced sample and that of the corresponding noninduced sample.
Small interfering RNA and MLR
For knockdown experiments, we used the validated SOCS2 stealth RNAiDuopack (Invitrogen) and a scrambled sequence oligo with similar GCcontent as a control oligo. Cells were transfected with LipofectamineRNAiMax reagent (Invitrogen), according to the manufacturer’s guide-lines. In brief, at day 6 of differentiation, 5 3 105 cells were plated inantibiotic-free DC medium and transfected with 100 pmol/well smallinterfering RNA (siRNA). After overnight incubation, the medium wassupplemented with antibiotics and the cells were used for furtherexperiments. Transfection efficiency was routinely .90%, as assessedby flow cytometry with fluorescent control siRNA oligonucleotides(BlockIT; Invitrogen). Knockdown efficacy was analyzed by means ofqRT-PCR and Western blotting. For allogeneic MLRs, 1 3 105 naiveCD4+CD45RA+ T cells were cultured with DCs at a ratio of 30:1 (T cells/DCs) in 96-well, round-bottom plates. After 4 d of coculture, cells werepulsed with 1 mCi [3H]thymidine for 16 h. Cells were transferred onto
glass fiber filters (MACH III M cell harvester; TOMTEC), sealed withscintillation sheets (MeltiLex), and analyzed in a Microbeta 1450 scin-tillation counter (PerkinElmer). Analogous to the ELISA results,
Table I. qRT-PCR primer sequences
Organism Target Gene Strand Sequence
Human RPLP0 Forward 59-GGCACCATTGAAATCCTGAGTGATGTG-39Reverse 59-TTGCGGACACCCTCCAGGAAG-39
Human SOCS1 Forward 59-TTGGAGGGAGCGGATGGGTGTAG-39Reverse 59-AGAGGTAGGAGGTGCGAGTTCAGGTC-39
Human SOCS2 Forward 59-CCAAATCAACCAAAAAAAGTGACCATGAAGTCCTG-39Reverse 59-CGGGGATTGAGTTGCACCTGTATAGCATGATATTC-39
Human SOCS3 Forward 59-ATACTATACCTTCCTGTACCTGGGTGGATGGAGCG-39Reverse 59-TGAGTATGTGGCTTTCCTATGCTGGGTCCCTCT-39
Human SOCS4 Forward 59-TGGAATCACAACTTTAGCTTTGATGCACATGACCC-39Reverse 59-TGGTTCAAAGAACATACAGGCGCTTGGGTCC-39
Human SOCS5 Forward 59-AAACAGGCGTTTGGAATAGCTGCTGCAATGTAGTC-39Reverse 59-CACAAAATCATCCTGGGCATAGGAACAGATCCAAC-39
Human SOCS6 Forward 59GCTGAAAAAACTTGCAAAGCAAGGATGGTACTGGG-39Reverse 59-CGAACAAGAAAAGAACCATCTGGCACGTTTGC-39
Human SOCS7 Forward 59-TCTAAAGGAAGCGCAGCTCATTTCCAAACAGAAGC-39Reverse 59-TCTTCAAAGCTGGAGCTTGGCAACCAAATGC-39
Human TNFA Forward 59-CAAGCCTGTAGCCCATGTTG-39Reverse 59-GAGGTTGACCTTGGTCTGGTA-39
Human IL-1B Forward 59-GTACCTGAGCTCGCCAGTGA-39Reverse 59-TCGGAGATTCGTAGCTGGATG-39
Human IL-6 Forward 59-GTACATCCTTCGACGGCATCTC-39Reverse 59-GGCAAGTCTCCTCATTGAATC-39
Human IL-10 Forward 59-AGGGCACCCAGTCTGAGAACA-39Reverse 59-CGGCCTTGCTCTTGTTTTCAC-39
Mouse Rplp0 Forward 59-TGCACTCTCGCTTTCTGGAGGGTG-39Reverse 59-AATGCAGATGGATCAGCCAGGAAGG-39
Mouse Socs2 Forward 59-GCTCAGTCAAACAGGATGGTACTGGGGAAGTATG-39Reverse 59-TCTGAATTTCCCATCTTGGTACTCAATCCGCAGG-39
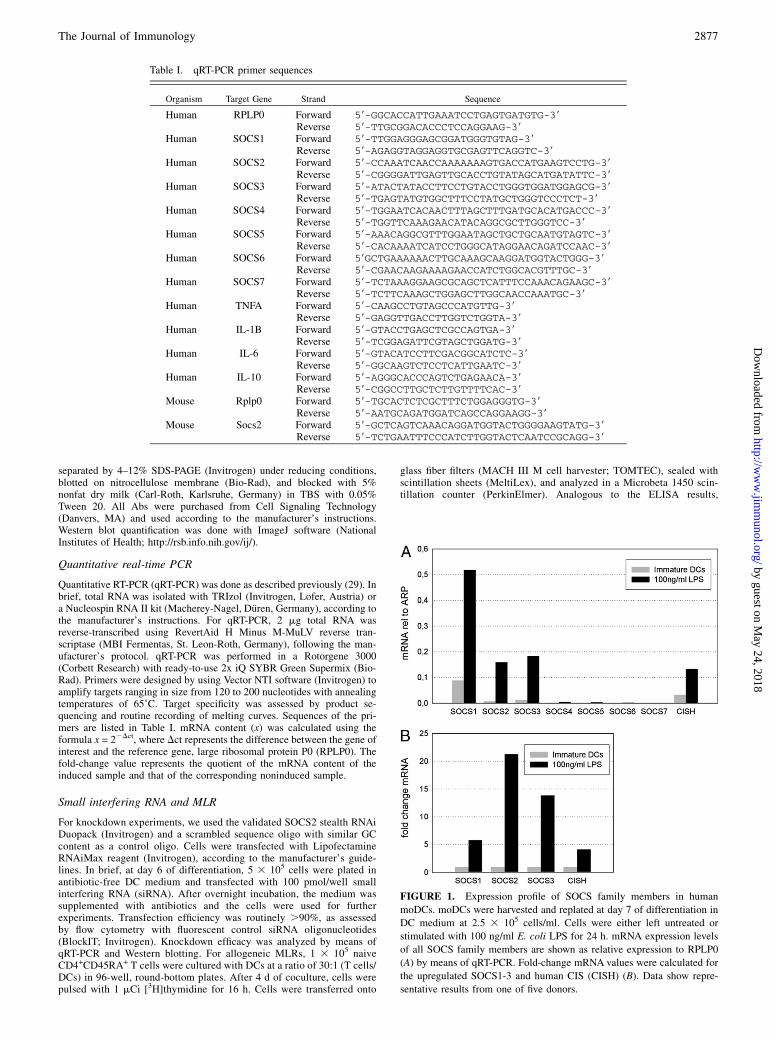
FIGURE 1. Expression profile of SOCS family members in human
moDCs. moDCs were harvested and replated at day 7 of differentiation in
DC medium at 2.5 3 105 cells/ml. Cells were either left untreated or
stimulated with 100 ng/ml E. coli LPS for 24 h. mRNA expression levels
of all SOCS family members are shown as relative expression to RPLP0
(A) by means of qRT-PCR. Fold-change mRNA values were calculated for
the upregulated SOCS1-3 and human CIS (CISH) (B). Data show repre-
sentative results from one of five donors.
The Journal of Immunology 2877
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

proliferation results are shown as normalized proliferation. Proliferationstimulated by LPS-treated control cells was set as 100%.
Flow cytometry
All FACS analyses were done on a FACS Canto II instrument (BectonDickinson) using FACS Diva software for acquisition and analysis. FACSAbs to human Ags (CD1a, CD14, CD40, CD80, CD86, HLA DR, PD-L1,PD-L2, CCR4, CCR7) were all purchased from BD Pharmingen. Stainingwas done according to standard procedures. Cells were then washed in PBS/FCS and fixed in 0.1% paraformaldehyde until analysis.
ResultsSOCS2 is expressed in APCs in response to TLR ligands
SOCS1, SOCS3, and CIS were described to be TLR-responsivegenes, and specific functions for these proteins in DCs havebeen established (10, 11, 13–16, 30). Therefore, we screenedhuman moDCs for expression of all SOCS family members (forprimer sequences, see Table I). In a long-term qRT-PCR kineticsexperiment, we observed that, in addition to SOCS1, SOCS3, andCIS, SOCS2 is a highly LPS-responsive gene. For SOCS4-7, nosignificant expression was detectable in LPS-treated DCs (Fig. 1).In contrast with SOCS1 and SOCS3, SOCS2 was upregulated ina delayed fashion, beginning ∼8 h after LPS stimulation andpeaking at 24 h after LPS treatment (Fig. 2A). To verify theseresults, we also analyzed the expression of SOCS2 at the proteinlevel, and the results fully agree with the data from the initial qRT-PCR screen: SOCS2 protein remained undetectable up to 8 h;thereafter, increasing protein expression was detectable, and
SOCS2 levels continued to accumulate up to 48 h (Fig. 2B). Toexclude potential contributions of residual impurities in our DCcultures, we also analyzed expression in CD1a+ FACS-sorted DCs(.99%) and obtained the same results (Supplemental Fig. 1).Because moDCs represent inflammation-induced DCs and do notshare all features with conventional DCs (31), we chose to isolateprimary CD1c+ blood DCs to confirm the results obtained with themoDCs (Fig. 3A, 3B).For further validation of our data, we also measured SOCS2
expression in murine APCs. LPS stimulation of mouse bonemarrow-derived DCs and thioglycolate-induced mouse peritonealmacrophages resulted in a significant upregulation of SOCS2mRNA levels. The time course follows a similar delayed expressionpattern (Fig. 3C, 3D). The delayed timing of SOCS2 expressionsuggested the possibility that the MyD88-independent TLR4pathway, which induces delayed activation of NF-kB and IFNregulatory factor 3 (32), could be responsible for SOCS2 ex-pression. To test this hypothesis, we investigated whether SOCS2expression is triggered solely by the TLR4 pathway or is equallyactivated by other TLRs that exclusively use the MyD88-dependent pathway. In addition to the TLR4 ligand LPS, wetested a whole panel of commercially available TLR ligands.moDCs were stimulated with Pam3CSK4 (for TLR1/2), FSL-1(TLR2/6), poly I:C (TLR3), flagellin (TLR 5), R848 (TLR7,TLR8), and type C CpG-containing oligonucleotides (28) (TLR9).SOCS2 expression was observed in response to stimulation ofTLR1/2, TLR2/6, TLR4, and TLR7/TLR8, albeit to varying
FIGURE 2. Time-course experiments show delayed expression of SOCS2. moDCs were harvested and replated at day 7 of differentiation in DC medium
at 2.5 3 105 cells/ml. Cells were stimulated with 100 ng/ml E. coli LPS for the indicated times. mRNA expression levels of SOCS1-3 are shown as relative
expression compared with RPLP0 by means of qRT-PCR and are representative of all donors analyzed throughout the study (A). Error bars indicate the SD
of two independent experiments for the same donor. B, DCs were stimulated with E. coli LPS as indicated, and total cell lysates were subjected to Western
blot analyses for SOCS2 and for STAT1 as a control for equal loading. All results are representative of five independent experiments.
2878 SOCS2 CONTROLS TLR-INDUCED DC ACTIVATION
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

extents (Fig. 4). These observations are in line with previousreports that human moDCs do not express TLR5, TLR7, andTLR9, but do express TLR1-4, TLR6, and TLR8 (33, 34). In-terestingly, TLR3 stimulation resulted in increased mRNA levelsbut failed to produce significant levels of SOCS2 protein. Takentogether, these results suggest that SOCS2 is a feedback moleculethat is induced by all TLRs that signal through the MyD88-dependent pathway, and that its expression is not reliant on thealternative signaling pathway of the LPS receptor TLR4. Notably,although SOCS2 is manifestly induced in all primary human andmouse APCs in response to TLR stimulation, we were not able toobserve induction of SOCS2 mRNA in response to LPS in any ofthe tested monocytic-like, macrophage-like, or DC-like cell lines(THP-1, Monomac-1; RAW, Jaws II). Therefore, primary humanmoDCs were used for all subsequent experiments.
SOCS2 is a direct downstream target of TLR ligation
Although delayed expression of SOCS2 has been repeatedly re-ported in other studies (35–37), the deferred induction raised thequestion whether SOCS2 is a direct downstream target of TLRsignaling or is stimulated via secreted factors in an autocrine/paracrine fashion. To exclude potential contributions of auto-crine cytokine stimulation, we treated immature DCs with dif-ferent candidate cytokines that are produced by activated DCsthemselves (IL-1, IL-6, IL-12, IL-10, TNF-a); however, none ofthe tested cytokines nor the combination of them was able to in-duce significant levels of SOCS2 mRNA in the absence of LPS
(data not shown). To further address this question, we stimulatedthe cells with LPS in the presence of the protein synthesis in-hibitor cycloheximide, and even though the time kinetics wasslightly changed, SOCS2 expression was robust in these settings(Supplemental Fig. 2). Because DCs are rather sensitive to cy-cloheximide treatment, we had to use a relatively low concen-tration of the drug (20 mM); nevertheless, protein synthesisinhibition was still efficient, as verified by inhibition of TNF-aproduction. Nevertheless, a potential contribution of preformedintracellular or membrane attached cytokines (38) cannot abso-lutely be excluded.
Silencing of SOCS2 in DCs leads to increased secretion ofIL-10 and IL-1b
To investigate the functional role of SOCS2 in DCs, we conductedRNA interference experiments. To avoid unintended adverse ef-fects, for example, cellular stress responses like the PKR/IFN re-sponse, we used chemically modified oligonucleotides applying theStealth RNAi technology. Silencing was performed with two dif-ferent oligos targeting SOCS2 mRNA to minimize the risk forboth sequence-dependent and off-target effects. Neither the SOCS2oligos nor the control oligo caused any DC maturation themselves,as determined by FACS analyses (CD83; CD86) and cytokineELISA (TNF-a), and showed no transcription of the type I IFNIFN-a2 compared with untransfected cells (data not shown). Si-lencing efficacy of the siRNA oligos on mRNA levels was ∼70%,as determined by qRT-PCR (data not shown); however, SOCS2protein was hardly detectable, as shown in Western blot analysis ofthe corresponding cell lysates (Fig. 5B). Analysis of the surfaceexpression of the DC activation-associated markers CD40, CD80,CD86, CD83, CCR4, and CCR7 failed to identify significant dif-ferences between SOCS2-silenced DCs and control cells 24 h afterLPS stimulation (data not shown). At the same time point, weanalyzed supernatants by ELISA and observed increased levelsof IL-1b (100% = 115 pg/ml) and IL-10 (100% = 26 pg/ml) in
FIGURE 3. Human primary DCs and murine APCs show SOCS2 ex-
pression in response to LPS. Human CD1c+ DCs (A, B) were isolated with
a BDCA-1 kit (Miltenyi), and 1 3 105 cells were stimulated for the in-
dicated times with 100 ng/ml E. coli LPS. Cells were then analyzed for
SOCS2 expression by qRT-PCR in relation to RPLP0. A, One donor
yielding enough cells provided the opportunity to perform a time-series
experiment. B, In total, three independent donors were analyzed, all
yielding similar results. C, cDNAs of thioglycolate-induced mouse peri-
toneal macrophages were analyzed for Socs2 expression in relation to
Rplp0. Results show mean and SD of three mice in three independent
experiments. D, cDNAs of murine bone marrow-derived DCs were ana-
lyzed for Socs2 expression in relation to Rplp0. Results show mean and SD
of two independent DC preparations.
FIGURE 4. SOCS2 expression is induced by MyD88-dependent TLRs.
moDCs were either left untreated or stimulated with LPS (100 ng/ml),
Pam3CSK (10 mg/ml), FSL-1 (1 mg/ml), poly I:C (10 mg/ml), flagellin (1
mg/ml), R848 (10 mg/ml), or CPG ODN (1 mg/ml). Cells were then ana-
lyzed for SOCS2 mRNA expression (upper panel) by qRT-PCR. In addi-
tion, total cells lysates of the same experiments were analyzed for SOCS2
protein expression by means of Western blot. Results show mean and SD of
three independent PCR experiments and one of three similar Western blots.
The Journal of Immunology 2879
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

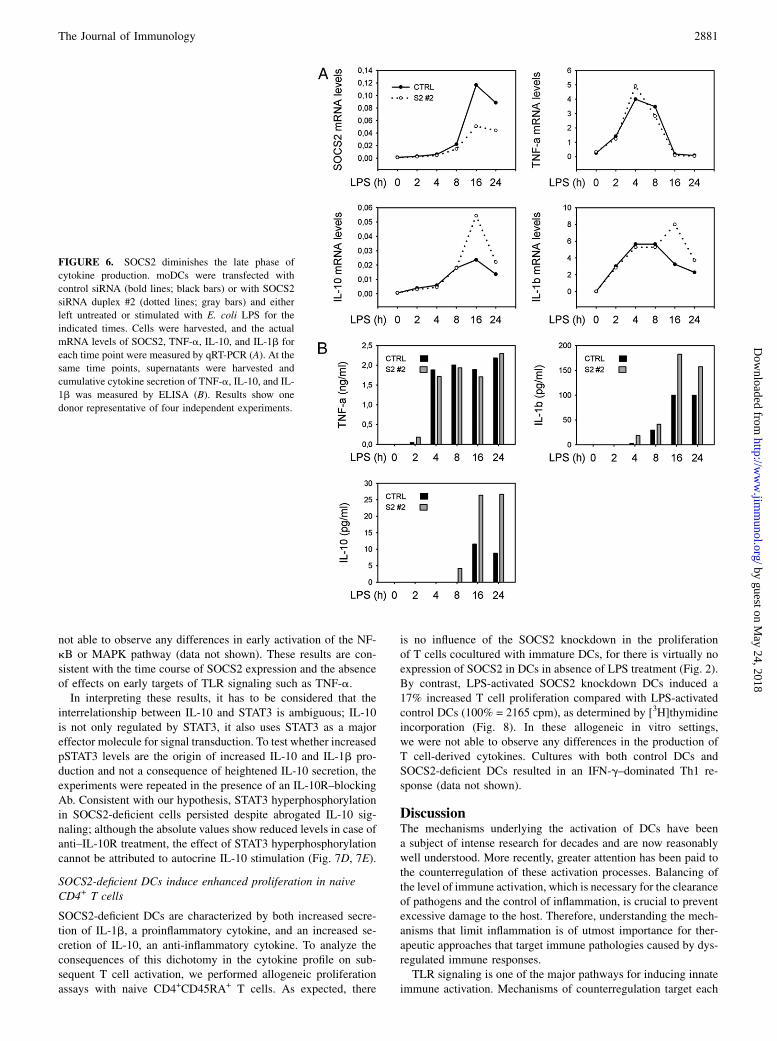
SOCS2-deficient DCs, whereas TNF-a (100% = 1.3 ng/ml) and IL-6 (100% = 1.4 ng/ml) levels remained unaffected (Fig. 5A). Fora better understanding of the observed changes in cytokine pro-duction, we conducted time-course experiments and measured theactual mRNA levels by qRT-PCR, as well as the cumulative cy-tokine secretion quantified by ELISA for each time point. Asreported previously, TNF-a and IL-6 showed an early-to-intermediate transcription profile (39) (data not shown), whichwas completely extinguished before substantial amounts ofSOCS2 were produced in control cells. In contrast, IL-1b and IL-10 showed delayed and sustained transcription. IL-1b and IL-10levels were increased after SOCS2 silencing, both at the tran-scriptional level and in terms of cumulative protein secretion (Fig.6). The observations corroborate our hypothesis that SOCS2specifically regulates time-delayed processes during DC matura-tion, whereas the early response, including IL-6 and TNF-a se-cretion, is not affected. In addition, a slight reduction of IL-12p70protein level was observed under SOCS2 knockdown, most likelybecause of heightened IL-10 secretion into the culture medium(40).
SOCS2-deficient DCs show hyperactivation of late-phaseSTAT3
The observed increased cytokine secretion with specificity for IL-10and IL-1b could indicate that SOCS2 targets a transcription factor
involved in regulating the expression of both cytokines. A prom-ising candidate was STAT3, because it was previously described toplay vital roles in the regulation of IL-6, IL-10, and IL-1b in re-sponse to LPS stimulation (41, 42). To test whether STAT3 acti-vation is necessary for the expression of these cytokines in humanmoDCs, we treated the cells with the STAT3-specific inhibitorcompound Stattic and found concentration-dependent repressionof IL-1b, IL-6, and IL-10 transcription, whereas TNF-a was notsensitive toward STAT3 inhibition (Supplemental Fig. 3).To analyze potential alterations of transcription factor activation,
we repeated the time-series experiments and subjected cell lysates toWestern blotting for analyses of phospho-STAT3, phospho-STAT1,phospho-ERK1/2, phospho-p38, and phospho-IkB. As hypothe-sized, significantly enhanced phosphorylation of STAT3 was ob-served, predominantly at late time points (Fig. 7A, 7B). Althoughdifferent donors showed slightly variant time kinetics of STAT3hyperphosphorylation, a robust effect was observed for all donorsat 24 h of LPS treatment (Fig. 7C). This also correlates with ourcytokine-secretion data, where significant enhanced production ofIL-1b and IL-10 is exclusively observed at later time points (Fig.6B). The absent effect of overloud STAT3 activation in SOCS2-silenced DCs on IL-6 production is explained by the differenttiming of SOCS2 and IL-6 transcription. As a matter of fact, IL-6transcription peaks and returns to basal levels before significantexpression of SOCS2 is detected (data not shown) (39). We were
FIGURE 5. Silencing of SOCS2 results
in increased secretion of IL-1b and IL-10.
moDCs were transfected with control siRNA
or with SOCS2 siRNA duplex S2 #1 and S2
#2, and either left untreated (black bars) or
stimulated with E. coli LPS for 24 h (gray
bars). Cytokine secretion was determined by
means of ELISA (A). Normalized secretion
was calculated with secretion of LPS-treated
control cells (CTRL-LPS) equals 100%.
Results represent mean and SD of four in-
dependent donors (*p , 0.05, **p , 0.005).
At the same time points, total cell lysates
were prepared and analyzed for SOCS2 and
STAT1 as controls for equal loading by
means of Western blot (B).
2880 SOCS2 CONTROLS TLR-INDUCED DC ACTIVATION
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
not able to observe any differences in early activation of the NF-kB or MAPK pathway (data not shown). These results are con-sistent with the time course of SOCS2 expression and the absenceof effects on early targets of TLR signaling such as TNF-a.In interpreting these results, it has to be considered that the
interrelationship between IL-10 and STAT3 is ambiguous; IL-10is not only regulated by STAT3, it also uses STAT3 as a majoreffector molecule for signal transduction. To test whether increasedpSTAT3 levels are the origin of increased IL-10 and IL-1b pro-duction and not a consequence of heightened IL-10 secretion, theexperiments were repeated in the presence of an IL-10R–blockingAb. Consistent with our hypothesis, STAT3 hyperphosphorylationin SOCS2-deficient cells persisted despite abrogated IL-10 sig-naling; although the absolute values show reduced levels in case ofanti–IL-10R treatment, the effect of STAT3 hyperphosphorylationcannot be attributed to autocrine IL-10 stimulation (Fig. 7D, 7E).
SOCS2-deficient DCs induce enhanced proliferation in naiveCD4+ T cells
SOCS2-deficient DCs are characterized by both increased secre-tion of IL-1b, a proinflammatory cytokine, and an increased se-cretion of IL-10, an anti-inflammatory cytokine. To analyze theconsequences of this dichotomy in the cytokine profile on sub-sequent T cell activation, we performed allogeneic proliferationassays with naive CD4+CD45RA+ T cells. As expected, there
is no influence of the SOCS2 knockdown in the proliferationof T cells cocultured with immature DCs, for there is virtually noexpression of SOCS2 in DCs in absence of LPS treatment (Fig. 2).By contrast, LPS-activated SOCS2 knockdown DCs induced a17% increased T cell proliferation compared with LPS-activatedcontrol DCs (100% = 2165 cpm), as determined by [3H]thymidineincorporation (Fig. 8). In these allogeneic in vitro settings,we were not able to observe any differences in the production ofT cell-derived cytokines. Cultures with both control DCs andSOCS2-deficient DCs resulted in an IFN-g–dominated Th1 re-sponse (data not shown).
DiscussionThe mechanisms underlying the activation of DCs have beena subject of intense research for decades and are now reasonablywell understood. More recently, greater attention has been paid tothe counterregulation of these activation processes. Balancing ofthe level of immune activation, which is necessary for the clearanceof pathogens and the control of inflammation, is crucial to preventexcessive damage to the host. Therefore, understanding the mech-anisms that limit inflammation is of utmost importance for ther-apeutic approaches that target immune pathologies caused by dys-regulated immune responses.TLR signaling is one of the major pathways for inducing innate
immune activation. Mechanisms of counterregulation target each
FIGURE 6. SOCS2 diminishes the late phase of
cytokine production. moDCs were transfected with
control siRNA (bold lines; black bars) or with SOCS2
siRNA duplex #2 (dotted lines; gray bars) and either
left untreated or stimulated with E. coli LPS for the
indicated times. Cells were harvested, and the actual
mRNA levels of SOCS2, TNF-a, IL-10, and IL-1b for
each time point were measured by qRT-PCR (A). At the
same time points, supernatants were harvested and
cumulative cytokine secretion of TNF-a, IL-10, and IL-
1b was measured by ELISA (B). Results show one
donor representative of four independent experiments.
The Journal of Immunology 2881
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

level of the signaling cascade and include soluble decoy receptorvariants (43), alternatively spliced adaptor molecules (MyD88s)(44, 45), and a kinase-inactive form of the IL-1R–associated ki-nase family (IRAK-M) (46). More recently, SOCS1 was shown tobe crucially involved in the regulation of TLR4 signaling, becauseit uses proteasomal depletion of the adaptor molecule MAL asa regulatory mechanism (13). Furthermore, SOCS3 is able to tagthe TRAF6/TAK1 complex for proteasomal degradation, therebylimiting signal duration (47). Whereas SOCS1 and SOCS3 havebeen extensively studied in immune cells, most studies of SOCS2addressed its function in GH/insulin-like growth factor-mediatedSTAT5 activation (22). It has been shown that SOCS2 binds tophospho-tyrosine residues of the GH receptor, and that STAT5inhibition is dependent on the SOCS-box (48). Crystal structuresshow interaction of the SOCS-box with elongin B and C, thusforming a ternary ubiquitin ligase complex able to mark inter-acting proteins for degradation (49).In this study, we showed that SOCS2 expression in DCs is in-
duced in direct response to TLR activation. Although time-delayedexpression appears to be a characteristic of SOCS2 (35–37), wedetermined that SOCS2 expression is independent of indirectautocrine stimulation, for it requires no de novo protein synthesis.Moreover, SOCS2 expression could be observed in response tomultiple TLR ligands, indicating that only MyD88-dependentsignaling is necessary for its transcription.Despite limited data pertaining to the function of SOCS2 in the
immune system, SOCS2 was recently described as the main me-diator of the anti-inflammatory capacities of both natural andaspirin-triggered lipoxins. Furthermore, SOCS2-deficient mice suf-fer from exuberant immune activation in a toxoplasmosis model(24, 25). Our data support the immune-dampening function ofSOCS2 in human DCs rather than a coactivating function in TLRsignaling, as suggested in a recent publication (50). In our ex-perimental setup, the early transcriptional targets of TLR acti-vation such as TNF-a or IL-6 remained unaffected; however,SOCS2-silenced DCs displayed increased secretion of IL-10 andIL-1b, two cytokines with a late and sustained expression patternand both transcriptional targets of STAT3 (41, 42). In allogeneicproliferation assays, we could show that the proinflammatoryactivity of IL-1b dominates over the IL-10 effect, for SOCS2-deficient DCs induce hyperproliferation in allogeneic naiveT cells (Fig. 8). Moreover, we were able to demonstrate that, inSOCS2-deficient DCs, the late activation of STAT3 is enhancedand is independent of autocrine IL-10 signaling, which suggests
FIGURE 8. LPS-induced, SOCS2-silenced DCs stimulate increased
proliferation in naive allogeneic T cells. For coculture experiments,
moDCs were transfected with control siRNA or with SOCS2 siRNA du-
plex #2 and transferred to 96-well flat-bottom plates. DCs were either left
untreated or stimulated with E. coli LPS for 8 h before the addition of 1 3105 MACS purified allogeneic T cells. [3H]thymidine incorporation was
measured in triplicates for each condition. Data represent mean and SD of
four independent experiments.
FIGURE 7. SOCS2-silenced DCs show augmented STAT3 activation. A
and B, moDCs were transfected with control siRNA or with SOCS2 siRNA
duplex #2 and either left untreated or stimulated with E. coli LPS for the
indicated times. Total cell lysates were subjected to Western blot analyses
of pSTAT3 (p-Tyr705), SOCS2, and ERK1/2 as control for equal loading
(A). Results were quantified in reference to ERK1/2 signals using ImageJ
software (B). Results are representative of three independent donors. C,
Mean and SD for 24-h LPS treatment (*p , 0.05, **p , 0.005) of three
donors are shown. D, moDCs were transfected with control siRNA or with
SOCS2 siRNA duplex #2 and either left untreated or stimulated with E.
coli LPS for 24 h in the presence of either an IL-10R–blocking Ab (aIL-
10R) or an isotype control. Cells were harvested, and total cell lysates were
analyzed for pSTAT3 and SOCS2. Blots were then reprobed with total
STAT3 Ab. E, Results from D were quantified and normalized for STAT3
levels. F, moDCs were preincubated with either an isotype control or aIL-
10R–blocking Ab (10 mg/ml) and stimulated with recombinant human IL-
10 (20 ng/ml) for 1 h. Total cell lysates were then analyzed for total and
phospho-STAT3 (pTyr705) by Western blot.
2882 SOCS2 CONTROLS TLR-INDUCED DC ACTIVATION
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

that SOCS2 targets an upstream kinase or signaling intermediateresponsible for STAT3 activation. It is unlikely that SOCS2-mediated degradation of SOCS3 protein (37) contributes to thiseffect, for it would lead to decreased levels of STAT3 activation.The proposed regulatory function of SOCS2 in STAT3 activationis in line with a recent study demonstrating that augmented STAT3activation correlates with hypermethylation-induced gene silenc-ing of SOCS1 and SOCS2 in ovarian and breast carcinomas (51).However, in opposition to a recent report showing hyperactivationof early MyD88-dependent and -independent TLR signaling (50),we were not able to detect any significant changes in activationof the NF-kB and ERK1/2 pathways (data not shown). Our ownobservations are corroborated by the delayed expression profile ofSOCS2, in which neither elevated SOCS2 mRNA nor protein wasdetectable earlier than 8 h after LPS treatment. The discrepanciesmight, at least in part, be explained by the use of different methodsfor siRNA transfection and the types of siRNA used. Furthermore,we suspect that the reduction in all parameters as observed by theprevious authors might be indicative of incomplete target speci-ficity in their silencing experiments. These results could also beattributed to siRNA- or electroporation-induced preactivation ofthe DCs and, therefore, an altered maturation response towardLPS treatment.In conclusion, our data provide evidence that SOCS2 is a
feedback inhibitor of TLR-induced DC maturation. The delayedexpression allows for a period of unrestricted immune activationfollowed by a phase of counterregulation and limitation of inflam-mation-driving activity. However, the molecular target that inter-connects SOCS2 expression with limited STAT3 activation, andconsequently limited cytokine secretion, remains elusive and needsto be addressed in further studies.
AcknowledgmentsWe thank Dr. Gunther Schwamberger for providing the mouse macrophage
cDNAs. We are indebted to Dr. Petra Desch (Laboratory for Immunolog-
ical and Molecular Cancer Research, Paracelsus Medical University,
Salzburg, Austria) for the sorting of CD1a+ DCs.
DisclosuresThe authors have no financial conflicts of interest.
References1. Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol.
4: 499–511.2. Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins
linking innate and acquired immunity. Nat. Immunol. 2: 675–680.3. Granucci, F., I. Zanoni, and P. Ricciardi-Castagnoli. 2008. Central role of den-
dritic cells in the regulation and deregulation of immune responses. Cell. Mol.Life Sci. 65: 1683–1697.
4. Yoshimura, A., T. Naka, and M. Kubo. 2007. SOCS proteins, cytokine signallingand immune regulation. Nat. Rev. Immunol. 7: 454–465.
5. Starr, R., T. A. Willson, E. M. Viney, L. J. Murray, J. R. Rayner, B. J. Jenkins,T. J. Gonda, W. S. Alexander, D. Metcalf, N. A. Nicola, and D. J. Hilton. 1997. Afamily of cytokine-inducible inhibitors of signalling. Nature 387: 917–921.
6. Babon, J. J., E. J. McManus, S. Yao, D. P. DeSouza, L. A. Mielke, N. S. Sprigg,T. A. Willson, D. J. Hilton, N. A. Nicola, M. Baca, et al. 2006. The structure ofSOCS3 reveals the basis of the extended SH2 domain function and identifies anunstructured insertion that regulates stability. Mol. Cell 22: 205–216.
7. Yasukawa, H., H. Misawa, H. Sakamoto, M. Masuhara, A. Sasaki, T. Wakioka,S. Ohtsuka, T. Imaizumi, T. Matsuda, J. N. Ihle, and A. Yoshimura. 1999. TheJAK-binding protein JAB inhibits Janus tyrosine kinase activity through bindingin the activation loop. EMBO J. 18: 1309–1320.
8. Piessevaux, J., D. Lavens, F. Peelman, and J. Tavernier. 2008. The many faces ofthe SOCS box. Cytokine Growth Factor Rev. 19: 371–381.
9. Kamura, T., S. Sato, D. Haque, L. Liu, W. G. Kaelin, Jr., R. C. Conaway, andJ. W. Conaway. 1998. The Elongin BC complex interacts with the conservedSOCS-box motif present in members of the SOCS, ras, WD-40 repeat, andankyrin repeat families. Genes Dev. 12: 3872–3881.
10. Dalpke, A. H., S. Opper, S. Zimmermann, and K. Heeg. 2001. Suppressors ofcytokine signaling (SOCS)-1 and SOCS-3 are induced by CpG-DNA andmodulate cytokine responses in APCs. J. Immunol. 166: 7082–7089.
11. Bode, J. G., A. Nimmesgern, J. Schmitz, F. Schaper, M. Schmitt, W. Frisch,D. Haussinger, P. C. Heinrich, and L. Graeve. 1999. LPS and TNFalpha induceSOCS3 mRNA and inhibit IL-6-induced activation of STAT3 in macrophages.FEBS Lett. 463: 365–370.
12. Stoiber, D., P. Kovarik, S. Cohney, J. A. Johnston, P. Steinlein, and T. Decker.1999. Lipopolysaccharide induces in macrophages the synthesis of the sup-pressor of cytokine signaling 3 and suppresses signal transduction in response tothe activating factor IFN-gamma. J. Immunol. 163: 2640–2647.
13. Mansell, A., R. Smith, S. L. Doyle, P. Gray, J. E. Fenner, P. J. Crack,S. E. Nicholson, D. J. Hilton, L. A. O’Neill, and P. J. Hertzog. 2006. Suppressorof cytokine signaling 1 negatively regulates Toll-like receptor signaling bymediating Mal degradation. Nat. Immunol. 7: 148–155.
14. Baetz, A., M. Frey, K. Heeg, and A. H. Dalpke. 2004. Suppressor of cytokinesignaling (SOCS) proteins indirectly regulate toll-like receptor signaling in in-nate immune cells. J. Biol. Chem. 279: 54708–54715.
15. Hanada, T., K. Tanaka, Y. Matsumura, M. Yamauchi, H. Nishinakamura,H. Aburatani, R. Mashima, M. Kubo, T. Kobayashi, and A. Yoshimura. 2005.Induction of hyper Th1 cell-type immune responses by dendritic cells lacking thesuppressor of cytokine signaling-1 gene. J. Immunol. 174: 4325–4332.
16. Lang, R., A. L. Pauleau, E. Parganas, Y. Takahashi, J. Mages, J. N. Ihle,R. Rutschman, and P. J. Murray. 2003. SOCS3 regulates the plasticity of gp130signaling. Nat. Immunol. 4: 546–550.
17. Yasukawa, H., M. Ohishi, H. Mori, M. Murakami, T. Chinen, D. Aki, T. Hanada,K. Takeda, S. Akira, M. Hoshijima, et al. 2003. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immu-nol. 4: 551–556.
18. Matsumura, Y., T. Kobayashi, K. Ichiyama, R. Yoshida, M. Hashimoto,T. Takimoto, K. Tanaka, T. Chinen, T. Shichita, T. Wyss-Coray, et al. 2007.Selective expansion of foxp3-positive regulatory T cells and immunosuppressionby suppressors of cytokine signaling 3-deficient dendritic cells. J. Immunol. 179:2170–2179.
19. Metcalf, D., C. J. Greenhalgh, E. Viney, T. A. Willson, R. Starr, N. A. Nicola,D. J. Hilton, and W. S. Alexander. 2000. Gigantism in mice lacking suppressor ofcytokine signalling-2. Nature 405: 1069–1073.
20. Favre, H., A. Benhamou, J. Finidori, P. A. Kelly, and M. Edery. 1999. Dualeffects of suppressor of cytokine signaling (SOCS-2) on growth hormone signaltransduction. FEBS Lett. 453: 63–66.
21. Greenhalgh, C. J., D. Metcalf, A. L. Thaus, J. E. Corbin, R. Uren, P. O. Morgan,L. J. Fabri, J. G. Zhang, H. M. Martin, T. A. Willson, et al. 2002. Biologicalevidence that SOCS-2 can act either as an enhancer or suppressor of growthhormone signaling. J. Biol. Chem. 277: 40181–40184.
22. Rico-Bautista, E., A. Flores-Morales, and L. Fernandez-Perez. 2006. Suppressorof cytokine signaling (SOCS) 2, a protein with multiple functions. CytokineGrowth Factor Rev. 17: 431–439.
23. Boverhof, D. R., E. Tam, A. S. Harney, R. B. Crawford, N. E. Kaminski, andT. R. Zacharewski. 2004. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces suppres-sor of cytokine signaling 2 in murine B cells. Mol. Pharmacol. 66: 1662–1670.
24. Machado, F. S., J. E. Johndrow, L. Esper, A. Dias, A. Bafica, C. N. Serhan, andJ. Aliberti. 2006. Anti-inflammatory actions of lipoxin A4 and aspirin-triggeredlipoxin are SOCS-2 dependent. Nat. Med. 12: 330–334.
25. Machado, F. S., L. Esper, A. Dias, R. Madan, Y. Gu, D. Hildeman, C. N. Serhan,C. L. Karp, and J. Aliberti. 2008. Native and aspirin-triggered lipoxins controlinnate immunity by inducing proteasomal degradation of TRAF6. J. Exp. Med.205: 1077–1086.
26. Machado, F. S., L. Esper, A. Dias, R. Madan, Y. Gu, D. Hildeman, C. N. Serhan,C. L. Karp, and J. Aliberti. 2009. Native and aspirin-triggered lipoxins controlinnate immunity by inducing proteasomal degradation of TRAF6. J. Exp. Med.206: 2573.
27. Sallusto, F., and A. Lanzavecchia. 1994. Efficient presentation of soluble antigenby cultured human dendritic cells is maintained by granulocyte/macrophagecolony-stimulating factor plus interleukin 4 and downregulated by tumor ne-crosis factor alpha. J. Exp. Med. 179: 1109–1118.
28. Duramad, O., K. L. Fearon, J. H. Chan, H. Kanzler, J. D. Marshall,R. L. Coffman, and F. J. Barrat. 2003. IL-10 regulates plasmacytoid dendriticcell response to CpG-containing immunostimulatory sequences. Blood 102:4487–4492.
29. Wirnsberger, G., D. Hebenstreit, G. Posselt, J. Horejs-Hoeck, and A. Duschl.2006. IL-4 induces expression of TARC/CCL17 via two STAT6 binding sites.Eur. J. Immunol. 36: 1882–1891.
30. Jackson, S. H., C. R. Yu, R. M. Mahdi, S. Ebong, and C. E. Egwuagu. 2004.Dendritic cell maturation requires STAT1 and is under feedback regulation bysuppressors of cytokine signaling. J. Immunol. 172: 2307–2315.
31. Geissmann, F., M. G. Manz, S. Jung, M. H. Sieweke, M. Merad, and K. Ley.2010. Development of monocytes, macrophages, and dendritic cells. Science327: 656–661.
32. Palsson-McDermott, E. M., and L. A. O’Neill. 2004. Signal transduction by thelipopolysaccharide receptor, Toll-like receptor-4. Immunology 113: 153–162.
33. Jarrossay, D., G. Napolitani, M. Colonna, F. Sallusto, and A. Lanzavecchia.2001. Specialization and complementarity in microbial molecule recognition byhuman myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 31: 3388–3393.
34. Visintin, A., A. Mazzoni, J. H. Spitzer, D. H. Wyllie, S. K. Dower, andD. M. Segal. 2001. Regulation of Toll-like receptors in human monocytes anddendritic cells. J. Immunol. 166: 249–255.
35. Adams, T. E., J. A. Hansen, R. Starr, N. A. Nicola, D. J. Hilton, andN. Billestrup. 1998. Growth hormone preferentially induces the rapid, transient
The Journal of Immunology 2883
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J. Biol.Chem. 273: 1285–1287.
36. Pezet, A., H. Favre, P. A. Kelly, and M. Edery. 1999. Inhibition and restoration ofprolactin signal transduction by suppressors of cytokine signaling. J. Biol. Chem.274: 24497–24502.
37. Tannahill, G. M., J. Elliott, A. C. Barry, L. Hibbert, N. A. Cacalano, andJ. A. Johnston. 2005. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signalingby accelerating SOCS3 degradation. Mol. Cell. Biol. 25: 9115–9126.
38. Quinones, M., S. K. Ahuja, P. C. Melby, L. Pate, R. L. Reddick, and S. S. Ahuja.2000. Preformed membrane-associated stores of interleukin (IL)-12 are a pre-viously unrecognized source of bioactive IL-12 that is mobilized within minutesof contact with an intracellular parasite. J. Exp. Med. 192: 507–516.
39. Langenkamp, A., M. Messi, A. Lanzavecchia, and F. Sallusto. 2000. Kinetics ofdendritic cell activation: impact on priming of TH1, TH2 and nonpolarizedT cells. Nat. Immunol. 1: 311–316.
40. De Smedt, T., M. Van Mechelen, G. De Becker, J. Urbain, O. Leo, and M. Moser.1997. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J.Immunol. 27: 1229–1235.
41. Benkhart, E. M., M. Siedlar, A. Wedel, T. Werner, and H. W. Ziegler-Heitbrock.2000. Role of Stat3 in lipopolysaccharide-induced IL-10 gene expression. J.Immunol. 165: 1612–1617.
42. Samavati, L., R. Rastogi, W. Du, M. Huttemann, A. Fite, and L. Franchi. 2009.STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Mol. Immunol.46: 1867–1877.
43. Iwami, K. I., T. Matsuguchi, A. Masuda, T. Kikuchi, T. Musikacharoen, andY. Yoshikai. 2000. Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 165: 6682–6686.
44. Burns, K., S. Janssens, B. Brissoni, N. Olivos, R. Beyaert, and J. Tschopp. 2003.Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the al-ternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4.J. Exp. Med. 197: 263–268.
45. Janssens, S., K. Burns, J. Tschopp, and R. Beyaert. 2002. Regulation ofinterleukin-1- and lipopolysaccharide-induced NF-kappaB activation by alter-native splicing of MyD88. Curr. Biol. 12: 467–471.
46. Kobayashi, K., L. D. Hernandez, J. E. Galan, C. A. Janeway, Jr., R. Medzhitov,and R. A. Flavell. 2002. IRAK-M is a negative regulator of Toll-like receptorsignaling. Cell 110: 191–202.
47. Frobøse, H., S. G. Rønn, P. E. Heding, H. Mendoza, P. Cohen, T. Mandrup-Poulsen, and N. Billestrup. 2006. Suppressor of cytokine Signaling-3 inhibitsinterleukin-1 signaling by targeting the TRAF-6/TAK1 complex. Mol. Endo-crinol. 20: 1587–1596.
48. Greenhalgh, C. J., E. Rico-Bautista, M. Lorentzon, A. L. Thaus, P. O. Morgan,T. A. Willson, P. Zervoudakis, D. Metcalf, I. Street, N. A. Nicola, et al. 2005.SOCS2 negatively regulates growth hormone action in vitro and in vivo. J. Clin.Invest. 115: 397–406.
49. Bullock, A. N., J. E. Debreczeni, A. M. Edwards, M. Sundstrom, and S. Knapp.2006. Crystal structure of the SOCS2-elongin C-elongin B complex definesa prototypical SOCS box ubiquitin ligase. Proc. Natl. Acad. Sci. USA 103: 7637–7642.
50. Hu, J., O. Winqvist, A. Flores-Morales, A. C. Wikstrom, and G. Norstedt. 2009.SOCS2 influences LPS induced human monocyte-derived dendritic cell matu-ration. PLoS ONE 4: e7178.
51. Sutherland, K. D., G. J. Lindeman, D. Y. H. Choong, S. Wittlin, L. Brentzell,W. Phillips, I. G. Campbell, and J. E. Visvader. 2004. Differential hyper-methylation of SOCS genes in ovarian and breast carcinomas. Oncogene 23:7726–7733.
2884 SOCS2 CONTROLS TLR-INDUCED DC ACTIVATION
by guest on May 24, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from


















