
Supporting Information - Wiley-VCH file1 Arylamine-substituted Hexa-peri-hexabenzocoronenes: Facile...
20
Supporting Information for Angew. Chem. Int. Ed. 200460174 © Wiley-VCH 2004 69451 Weinheim, Germany
Transcript of Supporting Information - Wiley-VCH file1 Arylamine-substituted Hexa-peri-hexabenzocoronenes: Facile...

Supporting Information
for
Angew. Chem. Int. Ed. 200460174
© Wiley-VCH 2004
69451 Weinheim, Germany

1
Arylamine-substituted Hexa-peri-hexabenzocoronenes: Facile
Synthesis and Their Potential Applications as “Coaxial” Hole
Transport Materials Jishan Wu,a Martin Baumgarten,a Michael G. Debije,b John M. Warman,b Klaus
Müllen*, a
a Max-Planck Institut für Polymerforschung, Ackermannweg 10, D-55128, Mainz,
Germany
b IRI, Delft University of Technology, Mekelweg 15, 2629 JB Delft, the Netherlands
I. General
Unless otherwise noted, all starting materials were purchased from Aldrich, Acros,
ABCR, and used as received. THF and toluene for water-free reactions were refluxed
over potassium under argon and freshly distilled before use. Compound 10 [1], 15 [2] and
16 [3] were prepared according to the literatures.
1H NMR and 13C NMR spectra were recorded in deuterated solvents such as CD2Cl2,
C2D2Cl4 on a Bruker DPX 250 and Bruker DRX 500. Field desoption (FD) mass spectra
were obtained on a VG Instruments ZAB 2-SE-FPD. High resolution MALDI mass
spectra were recorded on a Bruker Reflex II-TOF Spectrometer using a 337 nm nitrogen
laser with TCNQ as matrix [4]. Elemental analysis was carried on a Foss Heraeus-Vario
EL. Differential scanning calorimetry (DSC) was measured on a Mettler DSC 30 with
heating and cooling rate of 10 K/min. A Zeiss Axiophot with a nitrogen flushed Linkam
THM 600 hot stage was used to characterize optical textures. 2-D X-ray diffraction of
oriented fibers was conducted using a rotating anode (Rigaku, 18kw) X-ray beam (CuKα,
pinhole collimation, double graphite monochromator) with the beam perpendicular to the
fiber axis and the 2D patterns were recorded by a CCD camera. Cyclic and differential

2
voltammetry of compounds 1-5 were recorded using a standard three-electrode cell: a Pt
disc as working electrode, a Pt wire as counter electrode and a nonaqueous AgNO3/Ag
electrode as reference. Normally, 1 mg/mL solution of 1-5 in 1,2-dichlorobenzene were
used and 0.1 M tetrabutyl ammonium hexafluorophosphorate (TBAPF6) was used as
supporting electrolyte. The electrochemical potentials in each measurements were
calibrated by an internal standard of ferrocenium/ferrocene(Fc+/Fc). The titration of
compounds 1-5 (about 1.0x10-5 M in dry dichloromethane) by oxidants such as SbCl5 and
THClO4 (about 1.0x10-3 M) [5] were conducted under argon in a quartz cuvette equipped
with a septum and recorded by a Perkin-Elmer Lambda 9 UV-vis-NIR
spectrophotometer. Electron spin resonance spectroscopy (ESR) were recorded on CW
X-band ESP 300E equipped with an NMR gauss meter (Brucker ER035), a frequency
counter (Brucker ER 041 XK) and a variable temperature control continues flow N2
cryostat (Brucker B-VT 2000).
The pulse-radiolysis time-resolved microwave conductivity technique (PR-TRMC), as
applied to the study of the conductive properties of discotic materials, has been described
fully elsewhere.[6] Briefly, roughly 30 mg powder samples are compressed into Perspex
sample holders which are insterted into a Ka-band (ca 30 GHz) microwave cell. A
uniform, micromolar concentration of charge carriers is produced in the sample by a 5
nanosecond pulse of 3 MeV electrons from a Van de Graaff accelerator. Any change in
the conductivity of the sample resulting from the formation of mobile charge carriers is
monitored (without the need of electrode contacts) as a decrease in the microwave power
reflected by the sample cell. The one-dimensional, intracolumnar charge carrier mobility
is determined from the end-of-pulse conductivity per unit dose, ∆σeop/D (Sm2/J), using
the relationship
Σµ1D = 3 ⋅∆σeop
D⋅
Ep
Wp (1)

3
In (1), Ep is the average energy deposited in eV per ionization event and Wp is the
probability that initially formed ion-pairs survive to the end of the pulse. The value of Ep
was taken to be 25 eV, and the values of Wp, which were all within the range 0.39 ± 0.11
for the present compounds, were calculated as described previously.[6] The factor of 3 in
(1) takes into account the fact that the organized columnar domains within the bulk
samples investigated are randomly orientated and that charge transport is expected to be
highly anisotropic and to occur almost exclusively along the axis of the macrocyclic
stacks. The mobility sum, Σµ = µ(+) + µ(-), is used in (1) since the PR-TRMC technique
does not allow the determination of the separate contributions of the positive and
negative charge carriers.
All the AFM measurements were made using tapping mode operation on a Nanoscope
IIIa Multimode scanning probe microscope from Digital Instruments (Santa Barbara, CA)
using an “E” and “J” vertical engage scanner. Tapping mode etched silicon probes (force
constant f0 = 320~350 KHz) with about 5~10 nm radiuses were used for imaging. The
solutions of compound 1-5 (3 mg/mL in chloroform) were spin-coated onto different
substrates such as silicon wafer, HOPG and quartz with a spin rate of 1000 rpm. The UV-
vis absorption spectra of the thin film were recorded on Perkin-Elmer Lambda 9
spectrophotometer at room temperature.

4
II. Synthesis and characterization
Compounds 1, 2 were synthesized by Buchwald-Hartwig coupling reactions between the
mono- (17) and para-dibromo HBC (18) [7], respectively, as shown in SI-scheme 1.
Br
RR
R
R R
NH
OCH3H3CO
Br
RR
Br
R R
NH
OCH3H3CO
1Pd2(dba)3-P(tert-Bu)3-NaOBut
toluene, 80°C
18
17
2Pd2(dba)3-P(tert-Bu)3-NaOBut
toluene, 80°C
R= n-C12H25
SI-scheme 1. Synthesis of compounds 1,2 by Pd-catalyzed Buchwald-Harwig coupling.
The synthesis of the D3h symmetry HBC building blocks such as 7 is a big challenge.
Several attempts to make appropriate C3 symmetry hexaphenylbenzene precursors
carrying functionalizable bromine or iodine sites have been done. For example, the
Co2(CO)8 catalyzed cyclotrimerization was carried out on unsymmetric tolane derivatives
19 and two structural isomers 20 (a+b), one desired 1,3,5-isomer (a) and another
undesired 1,2,4-isomer (b), were obtained (SI-scheme 2). However, several attempts to
separate these two isomers by column chromatography failed due to the small difference
in polarity. Regioregular cyclotrimerization of acetyl substituted arylenes with TiCl4 or

5
Br
R
Co2(CO)8
Br
BrBr
R
R R
R
BrBr
R
R Br
+
1,3,5-isomer 1,2,4-isomerR=n-C12H25 or
3
1920
a b SI-scheme 2. Attempted synthesis of C3-symmetric hexaphenylbenene derivatives by Co-
catalyzed cyclotrimerization.
SiCl4 is a powerful method to make 1,3,5-substituted benzene (C3 symmetry), and it is
noteworthy that such Lewis acid promoted condensation-trimerization was used in the
practical synthesis of C60 fullerene.[8] Herein, a similar precursor 23 was designed and
synthesized, which was made by nucleophilic substitution reaction[9] between carboxylic
chloride 21 and benzyl lithium-cuprates bromide 22 (SI-scheme 3). However, several
attempts to cyclotrimerize 23 with SiCl4 and TiCl4 under different conditions failed to
give the desired hexaphenylbenzene precursor 24. This failure is most probably due to the
steric hindrance arising from the additional phenyl ring. These drawbacks drive us to
change the idea: instead of making the hexaphenylbenzene precursors, we designed and
synthesized a tris-biphenylylbenzene 14 precursor and finally the D3h symmetry HBC
building block 7 carrying three active iodo groups was obtained as described in the main
text.

6
I
II
LiCuBr
ICl
OO
I
+(i) SiCl4
(ii) TiCl4
or
21
2223
24 SI-scheme 3. Attempted synthesis of the C3 -symmetric hexaphenylbenzene precursor 24
by SiCl4 mediated condensation-cyclotrimerization.
All the new compounds were well characterized by standard NMR and mass
spectroscopy, and elemental analysis and some data for compounds 1-5 was presented
below (SI-figure 1).
880 885 890 895 900 905 910 915 9200
1000
2000
3000
4000
5000
6000
7000
Inte
nsity
m/z
899.
45
I
I I
C42H15I3Mol. Wt.: 900.28
SI-figure 1(a). Isotope resolved MALDI mass spectrum of compound 7 (TCNQ as
matrix). The isotopic distribution is in good agreement with the simulated results,
indicating an exact loss of 12 hydrogens during the cyclodehydrogenation. Further
structural proof is the 1H NMR spectrum of soluble compound 3 (see below) which is
prepared from building block 7.

7
1-Di(4’-methoxyphenyl)amino-4,7,10,13,16-pentadodecyl-hexa-peri-hexabenzocoronene
(1) - a general procedure of Buchwald-Hartwig coupling reactions
200 mg mono bromo HBC 16, 38 mg di(4-methoxyphenyl) amine, 2.5 mg Pd2(dba)3,
0.91 mg tri-tert-butyl phosphorous (20mg/ml in toluene, 45.3 µL), 20 mg NaOBut, and
2.0 ml dry toluene were put together into a Schlenk tube in glove box. The mixture was
stirred at 80 °C overnight. After cooling down, the solvent was removed under vacuum
and the residue was purified by column chromatography (silica gel, PE/DCM=4:1 to
DCM) to afford 143 mg yellow powder (65%). FD MS (8 kV): m/z= 1592.10 (100%),
calcd.1591.50. 1H NMR (500 MHz, 413 K, C2D2Cl4): δ ppm 8.95 (br, Ar-H in HBC core,
8 H), 8.77 (s, Ar-H, HBC core, 2 H), 8.59 (s, Ar-H in HBC core, 2 H), 7.38 (d, J= 8.2 Hz,
Ar-H in –PhO-, 4H), 7.00 (d, J= 8.2 Hz, Ar-H in –PhO-, 4H), 3.88 (s, OCH3, 6H), 3.26
(t, J= 7.6 Hz, Ar-CH2-, 6H), 3.16 (t, J=7.5 Hz, 4H), 2.09~1.98 (m, Ar-CH2-CH2-C10H21,
10H), 1.65~0.86 (m, 105 H). 13C NMR (125 MHz, 302 K, C2D2Cl4): δ ppm 158.61,
155.51, 146.49, 145.17, 141.45, 140.27, 129.65, 129.28, 125.93~114.83 (m), 55.57,
36.91, 31.84, 29.78, 29.72, 29.64, 29.31, 22.62, 14.13. Elemental analysis: calcd.. C
87.55%, H 9.56 %, N 0.88 %, O 2.01%; found: C 87.42 %, H 9.70%.
1,10-Bis[di(4’-methoxyphenyl)amino]-4,7,13,16-tetradodecyl-hexa-peri-
hexabenzocoronene (2)
Following the procedure for compound 1, compound 2 was obtained in 76 % yield from
compound 17 after column chromatography (silica gel, PE/DCM= 7:3 to 2:1 and then
hot THF). FD MS (8 KV): m/z= 1651.17 (100%), calcd.1650.44 for C118H140N2O4. 1H
NMR (250 MHz, d8-THF/CS2=2:1): δ ppm 8.94 (s, 4H), 8.77 (s, 4H), 8.57 (s, 4H), 7.30
(d, J=8.8 Hz, 8H), 6.93 (d, J= 8.8Hz, 8H), 3.84 (s, -OCH3, 12H), 3.14 (t, J=7.6 Hz, Ar-
CH2-, 8H), 1.94 (m, Ar-CH2-CH2-C10H21, 8H), 1.56~0.85 (m, 84H). Elemental analysis:
calcd.: C 85.87%, H 8.55 %, N 1.70 %, O 3.88%; found: C 84.53%, H 8.80%.

8
8.
94
57
8.
77
24
8.
59
30
7.
38
97
7.
37
32
7.
00
77
6.
99
13
3.
87
93
3.
27
33
3.
25
81
3.
24
34
3.
17
94
3.
16
35
3.
15
01
2.
09
93
2.
08
53
2.
07
07
1.
99
99
1.
98
58
1.
97
18
1.
65
14
1.
63
80
1.
59
47
1.
58
07
1.
56
48
1.
54
83
1.
53
37
1.
52
09
0.
86
49
( p p m)0 . 01 . 02 . 03 . 04 . 05 . 06 . 07 . 08 . 09 . 01 0 . 0
N
C12H25C12H25
C12H25
C12H25 C12H25
OCH3
OCH3
Hb
HaHc
Hd
He
Hf
Hg
Hh
-OCH3 -CH2-CH2-C10H21-CH2-C11H23
N
C12H25C12H25
N
C12H25 C12H25
H3CO
H3CO
OCH3
OCH3
Ha
Hb
Hc
Hd
He-OCH3
abcde-h
abcde
**
*
*
1
2
SI-figure 1 (b). 1H NMR spectra of 1 (in D4-C2D2Cl4, 413 K) and 2 (d8-THF/CS2=2:1,
room temperature).

9
N
N
N
N
N
N
C8H17
C8H17
C8H17 C8H17
C8H17
C8H17
C8H17
C8H17
C8H17C8H17
C8H17
C8H17
Ha
Hb
Hc
HdHe
*4
12.0
00
12.6
68
25.2
0838
.033
27.4
83
99.9
00
133.
70
Inte
gral
8.88
90
7.62
957.
6149
7.25
737.
2408
7.02
187.
0065
1.71
98
1.39
031.
3695
0.79
11
(ppm)1.02.03.04.05.06.07.08.09.0
N
N
N
C8H17C8H17
C8H17
C8H17 C8H17
C8H17
Ha
Hb
Hc
HeHd
abc d e
*
**
3
ab cde
4
*
SI-figure 1 (c). 1H NMR spectrum of 3 in d8-THF/CS2=2:1 at room temperature, and 4 in
in D4-C2D2Cl4 at 373K.

10
2-Bromo-3’-(trimethylsilyl)biphenyl (11)
1.41 g 1-bromo-2-iodobenzene (9, 5 mmol), 1.07 g 3-(trimethylsilyl)-1-benzoboronic
acid (10, 5.5 mmol) and 144 mg Pd(PPh3)4 were dissolved in 35 ml toluene and then 25
ml 1M aqueous K2CO3 was added. The mixture was degassed by two “freeze-pump-
thaw” cycles and heated to reflux (95 °C in oil bath) for 20 h. After cooling down, the
mixture was poured into water and extracted by 100 ml toluene. The organic layer was
washed by water two times and dried over MgSO4. The solvent was removed under
vacuum and the residue was purified by column chromatography (silica gel, hexane) to
afford 1.42 g colorless liquid (93%). 1H NMR (250 MHz, CD2Cl2): δ ppm 7.72~7.20 (m,
Ar-H, 8H), 0.34 (s, TMS, 9H). 13C NMR (62.5 MHz, CD2Cl2): δ ppm 143.19, 140.73,
134.78, 133.51, 132.96, 131.81, 130.14, 129.14, 127.90, 127.71, 122.93, -1.02. Elemental
analysis: calcd. C 59.01%, H 5.61%, Br 26.17%, Si 10.39%; found: C 57.49%, H 5.75%.
3-(Trimethylsilyl)biphenyl-2’-boronic acid (12)
A solution of 1.2 g compound 11 (3.93 mmol) in 10 ml dry THF was cooled down to -78
°C in a dry-ice-acetone bath, and then 2.94 ml n-BuLi (1.6 M in hexane) was added
dropwise over 15 min. The mixture was stirred at -78 °C for 1.5 h and 494 mg
trimethoxylborane was added dropwise over 15 min. The mixture was allowed to
naturally warm to room temperature over night and then quenched by adding 10 ml
degassed hydrogen chloride solution (1M in water). The mixture was extracted by 40 ml
dichloromethane and the organic layer was washed with water two times, dried over
MgSO4. The filtrate after filtration was concentrated to 4 ml and 40 ml cold ethanol was
added to give white precipitate. The solid was collected, dried under vacuum( 1.0 g) and
submitted to next step without further purification. 1H NMR (250 MHz, CD2Cl2): δ ppm
7.76~7.11 (m, Ar-H), 0.31 (s, TMS).
1,3,5-tris[3’’-(trimethylsilyl)-2’-biphenyl]benzene (13)
180 mg 1,3,5-tribromobenzene, 850 mg compound 12, 61 mg Pd(PPh3)4, 1.45 g K2CO3,
15 ml toluene and 10 ml water were mixed together. The mixture was degassed by two
“freeze-pump-thaw” cycles and then heated to reflux overnight. After standard work-up
and purification by column chromatography (silica gel, PE/DCM=10:1), 250 mg white
solid was obtained (58%). FD MS (8 KV): m/z = 750.9, calcd.: 751.26 (M+) for

11
C51H54Si3. 1H NMR (250 MHz, CD2Cl2): δ ppm 7.55~6.72 (m, Ar-H, 24H), 0.27 (s,
TMS, 27H). 13C NMR (62.5 MHz, CD2Cl2): δ ppm 141.41, 141.26, 140.81, 140.74,
135.45, 131.80, 131.08, 130.68, 130.56, 130.32, 127.67, 127.59, -0.99. Elemental
analysis: calcd.: C 81.54%, H 7.25%, Si 11.22%; found: C 81.75%, H 7.11%.
1,3,5-Tri(3’’-iodo-2’-biphenyl)benzene (14)
200 mg compound 13 was dissolved in 60 ml chloroform. The solution was degassed by
bubbling argon for 20 min and then 1.6 ml iodine monochloride (1.0 M In
dichloromethane) was added. After stirring for 1 h, the reaction was quenched by adding
saturated aqueous sodium disulfite. The orgainic layer was washed by water three times
(40 mLx3) and dried over MgSO4. The solution was concentrated to 5 ml and then 50 ml
cold ethanol was added to precipitate the product. The solid was collected and dried
under vacuum to give 220 mg pure product (91%). FD MS (8KV): m/z= 912.5, calcd.
912.38 (M+) for C42H27I3. 1H NMR (250 MHz, CD2Cl2): δ ppm 7.65~6.78 (m, Ar-H,
27H). 13C NMR (62.5 MHz, CD2Cl2): δ ppm 144.30, 140.72, 140.29, 139.11, 138.97,
135.89, 130.88, 130.54, 130.34, 130.02, 128.34, 127.95, 94.17. Elemental analysis: calcd.
C 55.29%, H 2.98%, I 26.17%; found: C 54.78%, H 3.04%.
1, 7, 13-Triiodo-hexa-peri-hexabenzocoronene (7)
500 mg compound 14 was dissolved in 160 ml fresh dichloromethane. The solution was
degassed by bubbling argon for 20 min and then 3.2 g FeCl3 in 10 ml nitromethane was
added dropwise. Keeping the bubbling of argon through the reaction for 40 min and then
the reaction was quenched by 50 ml methanol. The yellow precipitate was collected,
washed by methanol and dried under vacuum to afford 370 mg yellow powder (75%).
MALDI-TOF MS (TCNQ as matrix): m/z= 899.45, calcd. 900.28 for C42H15I3. Elemental
analysis: calcd.: C 56.03%, H 1.68%, I 42.29%; found: C 57.96%, H 1.61%.
1, 7, 13-Tris[di(4-octylphenyl amino)] hexa-peri-hexabenzocoronene (3)
Following the procedure for compound 1, compound 3 was obtained in 24 % yield from
triiodo HBC 7 after column chromatography (silica gel, PE/DCM/CS2= 8:1:1 to 7:1:1).
FD MS (8 kV): m/z= 1699.1 (100%), calcd.1697.55 for C126H141N3. 1H NMR (250 MHz,
d8-THF/CS2=2:1): δ ppm 8.82 (s, 6H), 8.68 (d, J= 8.1 Hz, 6H), 7.86 (t, J= 7.8 Hz, 3H),

12
7.44 (d, J= 8,8 Hz, 12H), 7.32 (d, J= 8.8 Hz, 12H), 1.84 (br, Ar-CH2-, 12H), 1.47~1.29
(m, 48H), 0.92~0.85 (m, 52H).
Hexakis (4-iodo)-peri-hexabenzocoronene (8)
2 g of compound 15 (0.2862 mmol) was dissolved in 800 ml dichloromethane and the
solution was constantly sparged with argon. Then 8.03 g anhydrous iron (III) chloride in
20 ml nitromethane was added via a syringe. After 24 h the reaction was quenched by
methanol. The yellow precipitate was collected and washed by dichloromethane
methanol until the filtrate was colorless, dried under reduced pressure to give 1.62 g
insoluble product (82%). MALDI-TOF MS (TCNQ as matrix): m/z=1277.61 (100%).
Elemental analysis: calcd.: C 44.29% H 1.30% I 54.41%; Found: C 39.54%, H 0.90%.
Hexa-triarylamine substituted HBC 4
To a frame-dried 25 ml Schlenk flask was added 127.8 mg of compound 18 (0.10 mmol),
17.5 mg Pd(PPh3)4 (2.5 mol % per I), 5.7 mg of copper iodide (5.0 mol % per I), and 10
ml piperidine. The mixture was degassed by bubbling argon for 15 min, then 592 mg 16
was added. The solution was heated to 52 °C overnight, cooled to RT, and 20 ml
methanol was added. The yellow precipitate was collected, followed by column
chromatography (Al2O3, PE/DCM=4:1 to 7:3) to give 212 mg pure compound 4 (61%).
MALDI-MS (TCNQ as matrix): m/z=3473.08, calcd. 3473.21. 1H NMR (500 MHz,
C2D2Cl4, 373 K): δ ppm 8.89 (s, 12H), 7.62 (d, J=7.3 Hz, 12H), 7.25 (d, J= 8.2Hz, 24H),
7.01 (br, 36H), 1.71 (br, 24H), 1.39~0.79 (m, 180H). 13C NMR (125 MHz, C2D2Cl4,
302K): δ ppm 148.34, 145.06, 144.06, 133.36, 132.91, 132.46, 128.80, 128.77, 127.41,
126.98, 123.81, 123.30, 122.26, 121.87, 121.40, 120.95, 119.86, 118.92, 118.73, 118.60,
115.79, 114.45, 113.93, 113.29, 113.20, 113.00, 91.58, 89.35, 57.03, 38.05, 38.05,
32.24~30.32 (m), Elemental analysis: calcd.: C 89.22%, H 8.36 %, N 2.42 %; found: C
88.05%, H 8.44%.

13
III. Thermal behavior. Selected POM images of compounds 1, 2, and 3 were shown in SI-figure 2. Typical fan-
texture for 1 in a hexagonal liquid crystalline phase at 320 °C was observed.
(a) (b)
(c) SI-figure 2. POM textures of compounds 1 at 320 °C (a), 2 at 316 °C (b) and 3 at room
temperature(c) after slow cooling from the melts.

14
Some 2D WAXD patterns for compounds 1-5 at different temperature were shown in SI-
figure 3.
(a) (b)
SI-figure 3. 2D WAXD diagrams for compound 2 in the liquid crystalline phase at 217
°C (a), and compound 4 at room temperature (b).

15
IV. Cyclic and differential pulse voltammetry
0.0 0.2 0.4 0.6 0.8 1.0-0.6
-0.4
-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2
Cur
rent
µA
Potential V (vs AgNO3/Ag)0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1.08
00.
973
0.50
4
0.34
2
Cur
rent
µA
Potential V (vs AgNO3/Ag)
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0-2
-1
0
1
2
3
Cur
rent
µA
Potential V (vs AgNO3/Ag)0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4
0,2
0,3
0,4
0,5
0,6
0,7
0,8
1.41
6
1.23
0
0.71
6
0.51
2
Cur
rent
µA
Potential V (vs AgNO3/Ag)
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4-2
-1
0
1
2
3
4
5
0.76
6
0.90
8
0.58
9
Cur
rent
µA
Potential vs Ag+/Ag
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,80,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
1.24
41.
10
Cur
rent
Potential
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4
-1
0
1
2
3
4
0.59
3
0.84
1
1.07
0.91
9
0.72
1
Cur
rent
µA
Potential vs Ag+/Ag
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,80,0
0,1
0,2
0,3
0,4
0,5
0,6
Cur
rent
Potential
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6
-2
-1
0
1
2
3
4
Cur
rent
µA
Potential vs AgNO3/Ag0.0 0.5 1.0 1.5 2.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
Cur
rent
µA
Potential vs AgNO3/Ag
0.46
8
0.93
5
(c) 2-CV (d) 2-DPV
(e) 3-CV (f) 3-DPV
CVDPV DPV CV
(g) 4 (h) 5
(a) 1-CV (a) 1-DPV

16
SI-figure 4. Cyclic voltammograms (CV) and differential pulse voltammograms (DPV) of
compounds 1-5 in 1,2-dichlorobenzene and 0.1 M tetrabutylammonium
hexafluorophosphorate. Scan rate: 100 mV/s.

17
V. Morphology of the thin films.
a b
c d
e
SI-figure 5. Representative tapping mode AFM images of spin-coated thin films of
compounds 1 (a, height image), 2 (b, phase image), 3 (c, phase image), 4 (d, phase
image), and 5 (e, phase image) on quartz plates.
18
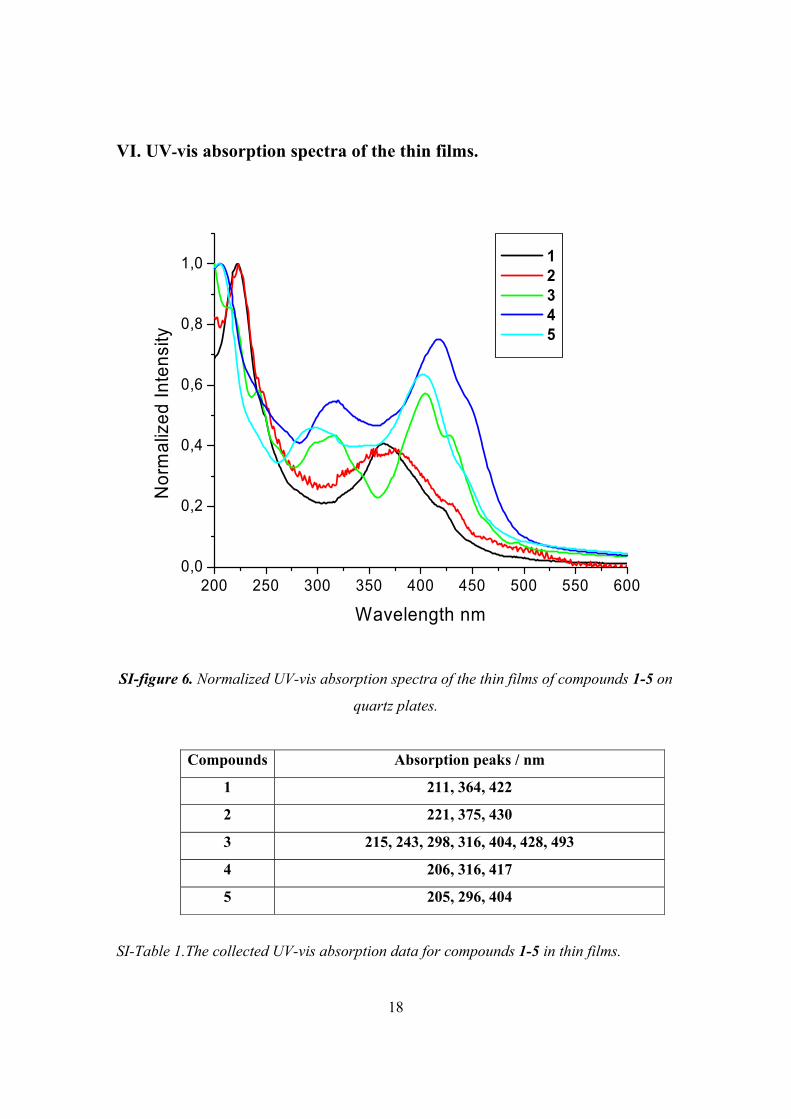
VI. UV-vis absorption spectra of the thin films.
200 250 300 350 400 450 500 550 6000,0
0,2
0,4
0,6
0,8
1,0
Nor
mal
ized
Inte
nsity
Wavelength nm
1 2 3 4 5
SI-figure 6. Normalized UV-vis absorption spectra of the thin films of compounds 1-5 on
quartz plates.
Compounds Absorption peaks / nm
1 211, 364, 422
2 221, 375, 430
3 215, 243, 298, 316, 404, 428, 493
4 206, 316, 417
5 205, 296, 404
SI-Table 1.The collected UV-vis absorption data for compounds 1-5 in thin films.

19
VII. References:
[1] C. L. Nesloney, J.W. Kelly, J. Org. Chem. 1996, 61, 3127-3137.
[2] J. A. Hyatt, Org. Prep. Proced. Int. 1991, 23, 460-463.
[3] J. Wu, M. D. Watson, K. Müllen, Angew. Chem. 2003, 115, 5487-5491; Angew.
Chem. Int. Ed. 2003, 42, 5329-5333.
[4] L. Przybilla, J. D. Brand, K. Yoshimura, J. Räder, K. Müllen, Anal. Chem. 2000, 72,
4591.
[5] Y. Murata, H. J. Shine, J. Org. Chem. 1969, 34, 3368-3372.
[6] a) P. G. Schouten, J. M. Warman, M. P. de Haas, C. F. van Nostrum, G. H. Gelinck,
R. J. M. Nolte, M. J. Copyn, J. W. Zwikker, M. K. Engel, M. Hanack, Y. H. Chang, W.
T. Ford J. Am. Chem. Soc. 1994, 116, 6880-6894; b) J. M. Warman, A. M. van de Craats
Mol. Cryst. Liq. Cryst. 2003, 396, 41-72.
[7] S. Ito, M. Wehmeier, J. D. Brand, C. Kübel, R. Epsch, J. P. Rabe, K. Müllen, Chem.
Eur. J. 2000, 6, 4327-4342.
[8] L. T. Scott, M. M. Boorum, B. McMahon, S. Hagen, J. Mack, J. Blank, H. Wegner, A.
de Meihere, Science 2002, 295, 1500-1503.
[9] S. C. Berk, M. C. P. Yeh, N. Jeong, P. Knochel, Organometallics 1990, 9, 3053-3064.


















