
Studies on Islet Amyloid Polypeptide Aggregation: From Model ...
110
Linköping University Medical Dissertations No. 1254 Studies on Islet Amyloid Polypeptide Aggregation: From Model Organism to Molecular Mechanisms Sebastian W Schultz Department of Clinical and Experimental Medicine Linköping University, Sweden Linköping 2011
Transcript of Studies on Islet Amyloid Polypeptide Aggregation: From Model ...

Linköping University Medical Dissertations No. 1254 Studies on Islet Amyloid Polypeptide Aggregation:
From Model Organism to Molecular Mechanisms
Sebastian W Schultz
Department of Clinical and Experimental Medicine
Linköping University, Sweden
Linköping 2011

© Sebastian W Schultz Cover: Drosophila brain; green: cell nuclei of ventral lateral neurons, red: neuropil During the course of the research underlying this thesis, Sebastian W Schultz was enrolled in Forum Scientium, a multidisciplinary doctoral programme at Linköping University, Sweden. Printed by LiU-‐Tryck, Linköping, Sweden, 2011 ISBN 978-‐91-‐7393-‐099-‐4 ISSN 0345-‐0082

Der Weg ist das Ziel

Supervisor Gunilla T Westermark, Professor Department of Medical Cell Biology Uppsala University, Sweden Opponent Anne Simonsen, Associate Professor Department of Biochemistry University of Oslo, Norway

Preface
This thesis is based on the following papers, which are referred to in the text by their roman numerals:
I. Paulsson JF, Schultz SW, Kohler M, Leibiger I, Berggren PO, Westermark GT. Real-‐time monitoring of apoptosis by caspase-‐3-‐like protease induced FRET reduction triggered by amyloid aggregation. 2008, Exp Diabetes Res 2008: 865850.
A free, coloured version of this paper can be downloaded from: www.hindawi.com/journals/edr/2008/865850/
II. Schultz SW, Nilsson KP, Westermark GT. Drosophila melanogaster as a model system for studies of islet amyloid polypeptide aggregation. 2011, PLoS One 6:e20221.
A free, coloured version of this paper can be downloaded from: www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0020221
III. Schultz SW, Gu X, Rusten TE, Alenius M, Westermark GT. HIAPP and hproIAPP trigger selective autophagy and inhibit the neuro-‐protective effect of autophagy. Manuscript.

Abstract
The proper folding of a protein into its defined three-‐dimensional structure is one of the many fundamental challenges a cell encounters. A number of tightly controlled pathways have evolved to assist in the proper folding of a protein, but also to aid in the removal of misfolded proteins. Despite the presence of these pathways accumulation of misfolded proteins can still occur. Amyloid deposits consist of misfolded proteins with a characteristic highly ordered fibrillar structure that will exert affinity for the amyloid dye Congo red and has a unique X-‐ray diffraction pattern. Currently 27 different proteins have been identified as amyloid forming proteins in human, however the exact role of amyloid in the pathogenesis of the connected disease is most often unclear. Islet amyloid is made up of the beta cell derived hormone islet amyloid polypeptide (IAPP) and is associated with the development of type 2 diabetes. Propagation of IAPP-‐fibrils is believed to be one important cause of the pancreatic beta cell death detected in patients with type 2 diabetes. IAPP is a naturally occurring polypeptide hormone stored and secreted together with insulin. IAPP and insulin arise from posttranslational processing of their biological inactive precursors proIAPP and proinsulin. In addition to human, cat and monkey IAPP will form amyloid deposits in conditions resembling human type 2 diabetes. However, IAPP from mouse and rat do not form amyloid as a result of the differences in amino acid sequence. My main research goal was to establish a unique model system suitable to study the effects of proIAPP and IAPP aggregation. I selected Drosophila melanogaster due to its many suitable characteristics as a model organism and its superior genetic toolbox. I have demonstrated that over-‐expression of hproIAPP and hIAPP in the central nervous system (CNS) results in aggregate formation in the brain and neighbouring fat body. Consistent with previous studies, expression of mIAPP does not result in the formation of aggregates. To investigate the intracellular effects of hproIAPP and hIAPP aggregation on a specific population of neurons, we targeted the expression of these peptides specifically to 16 neurons in the brain, the pdf-‐neurons. These pdf-‐neurons are divided into 2 clusters of 8 cells per brain hemisphere. First I showed that expression of aggregation prone hIAPP and hproIAPP resulted in significant death of the 8 cells, whereas expression of mIAPP had no such effect. In efforts to pinpoint the mechanisms behind the observed cell death I demonstrated that hproIAPP and hIAPP both pass the ERs quality control for protein folding and that the initiated cell death does not occur through classical apoptosis. Instead, selective autophagy is activated by hIAPP and hproIAPP. This activation counteracts the usually neuro-‐protective effects of autophagy and contributes to cell death. Strikingly, I also showed that Aβ, the amyloid protein implicated in Alzheimer’s disease, does not exhibit any intracellular toxicity when expressed in pdf-‐cells. This supports the existence of separate toxic pathways for different amyloid proteins.

Popular scientific summary
Proteins are one of the building blocks of life. They are important for almost every process in the cell, e.g. forming a framework involved in cellular structure, activation of chemical reactions and mediating cell signals and cell interactions. However, proteins have to adopt a pre-‐defined three-‐dimensional fold, referred to as its native confirmation, in order to function. Because proteins are so important, cells have developed highly sophisticated and tightly controlled pathways used to assist their proper folding and to remove misfolded proteins. Despite quality control, accumulation of misfolded proteins can occur. Amyloidosis is a group of protein misfolding diseases. Hitherto, 27 different proteins have been identified as amyloid forming in man. Each amyloid protein is associated with a specific disease, but the exact role for amyloid in the pathogenesis of the illness is unclear. All amyloid deposits share certain characteristics, they have all affinity for amyloid specific dyes and methods providing high-‐resolution information reveal a highly ordered fibrillar structure. The protein I have been working on is the hormone islet amyloid polypeptide (IAPP) that together with insulin and glucagon participates in the regulation of blood glucose. IAPP can form amyloid in pancreas and this is associated with type 2 diabetes. After food intake the blood glucose concentration raises, which leads to release of insulin from beta cells in the pancreas. Insulin facilitates cellular uptake of sugar and thereby lowers the blood glucose concentration. Patients that suffer from type 2 diabetes cannot produce sufficient amounts of insulin and they develop chronic elevated blood sugar level. One reason for the decreased insulin secretion is the replacement of beta cells by IAPP-‐amyloid, and it is believed that islet amyloid is responsible for this cell reduction and contributes to insulin deficiency. One question that still remains to be answered is -‐ how does IAPP-‐amyloid mediate cell death? Since IAPP and insulin are produced by the same cells, death can be initiated from the inside or from the outside of the cell. For my work I have set up a new Drosophila melanogaster (fruit fly) model to study effects of aggregation of human IAPP and its precursor proIAPP. I have produced transgenic flies that secrete human IAPP or proIAPP and shown that expression of these proteins in the fly head results in aggregation (paper II). In paper III, I limited IAPP and proIAPP expression to a subset of 16 neurons, and showed that this caused cell death. The mechanism behind intracellular cell death was studied in detail and I was able to show that the autophagy (self-‐eating) pathway was selectively triggered by human IAPP and human proIAPP. Gained evidence indicates that activation of this self-‐eating (autophagy) pathway decreases the normal protective mechanism of this pathway and thereby contributes to cell death. I have included studies on Aβ, the protein that forms amyloid in patients with Alzheimer’s disease. Aβ expression in the 16 cells did not result in cell death. Instead, comparison of Aβ and IAPP/proIAPP expression revealed that amyloid proteins use different pathways to exhibit their toxicity.


TABLE OF CONTENTS ABBREVIATIONS ................................................................................................................. 1
INTRODUCTION ................................................................................................................... 3
PROTEIN FOLDING AND MISFOLDING ................................................................................................ 4
AMYLOID AND AMYLOIDOSIS .............................................................................................................. 5
History and definitions .............................................................................................................. 5
Amyloid and diseases ................................................................................................................. 6
Structure of amyloid ................................................................................................................... 8
Non-‐fibrillar components in amyloid deposits ................................................................. 9
Amyloid formation .................................................................................................................... 10
Toxic effects ................................................................................................................................. 11
Functional amyloid ................................................................................................................... 12
ISLET AMYLOID POLYPEPTIDE (IAPP) ........................................................................................... 13
General introduction ................................................................................................................ 13
Prohormone processing .......................................................................................................... 15
IAPP and type 2 diabetes ........................................................................................................ 17
IAPP fibril formation ................................................................................................................ 18
Transgenic animal models with hIAPP ............................................................................. 21
Aβ ......................................................................................................................................................... 22
Alzheimer’s disease ................................................................................................................... 22
Aβ and IAPP ................................................................................................................................. 23
DROSOPHILA MELANOGASTER AS MODEL SYSTEM ........................................................................ 25
History of Drosophila as model system ............................................................................. 25
Huge genetic toolbox: Gal4/UAS system ........................................................................... 26
Drosophila models for protein aggregation .................................................................... 28
MOLECULAR PATHWAYS CONNECTED TO PROTEIN MISFOLDING ............................................... 31
ER-‐stress and Unfolded protein response (UPR) ........................................................... 31
Apoptosis ....................................................................................................................................... 37
Autophagy .................................................................................................................................... 41
AIMS OF THE THESIS ....................................................................................................... 51

MATERIAL AND METHODS ............................................................................................ 53
WORKING WITH DROSOPHILA ......................................................................................................... 54
P-‐element insertion ................................................................................................................... 54
Survival assay .............................................................................................................................. 54
DETECTION METHODS ....................................................................................................................... 55
Immunofluorescence – tissue preparation ...................................................................... 55
Congo Red or pFTAA ................................................................................................................. 55
Image processing ....................................................................................................................... 56
RESULTS AND DISCUSSION ............................................................................................ 57
EXTRACELLULAR AMYLOID FORMATION INDUCES APOPTOSIS (PAPER I) ................................. 58
CHARACTERISATION OF A NEW DROSOPHILA MODEL FOR STUDIES OF IAPP AGGREGATION
(PAPER II) ............................................................................................................................................ 60
HPROIAPP AND HIAPP TRIGGER SELECTIVE AUTOPHAGY (PAPER III) ................................... 64
GENERAL DISCUSSION AND FUTURE PERSPECTIVES ........................................... 69
ACKNOWLEDGEMENTS ................................................................................................... 73
REFERENCES ....................................................................................................................... 77

1
Abbreviations Aβ amyloid-‐β peptide AD Alzheimer’s disease AGE advanced glycation end-‐products Alfy PI3P-‐binding autophagy-‐linked FYVE domain protein ApoE apolipoprotein E APP Aβ precursor protein ASK1 apoptosis signal regulation kinase-‐1 ATG autophagy-‐related genes ATF6 activating transcription factor-‐6 Bchs blue cheese Bcl-‐2 B cell lymphoma-‐2 BiP binding immunoglobulin protein CGRP calcitonin gene-‐related peptide CHOP C/EBP homologous protein CMA chaperone mediated autophagy CPE Carboxypeptidase E CRLR calcitonin-‐receptor-‐like-‐receptor CSF cerebrospinal fluid CT calcitonin CTR-‐2 calcitonin receptor 2 CVT cytosol-‐to-‐vacuole targeting EDEM ER degradation-‐enhancing α1,2-‐mannosidase like protein EM electron microscopy EOFAD early-‐onset FAD ER endoplasmic reticulum ERAD ER associated degradation ERAF ER associated folding ERdj ER-‐resident J-‐domains ERManI ER degradation α1,2-‐mannosidase I ESCRT endosomal sorting complex required for transport FAD familial form of Alzheimer’s disease FADD Fas-‐associated death domain GAGs Glycosaminoglycans GFP green fluorescent protein GS glycogen synthase GSK3α glycogen synthase 3α HDAC histone deacteylase HFNs human fetal neurons hIAPP human IAPP

2
HS heparin sulphate Hsc heat shock cognate Hsf1 heat shock factor-‐1 Hsp heat shock protein HSPG heparan sulphate proteoglycan HSR heat shock response Htt Huntingtin IAPP islet amyloid polypeptide IDE insulin degrading enzyme IRE1 inositol-‐requiring protein-‐1 JNK c-‐Jun N-‐terminal kinase LAMP lysosome-‐associated membrane type protein LC3 microtubule associated protein 1 light chain 3 mIAPP murine IAPP MVBs multivesicular bodies NEFA non-‐esterified fatty acids NFT neurofibrillary tangles NMR nuclear magnetic resonance OST oligosaccharyltransferase PAM peptidyl amidating monooxygenase PC prohormone convertase PD Parkinson’s disease PE phosphatidylethanolamine PERK protein kinase RNA-‐like ER kinase PI3K phosphatidylinositol 3-‐kinase PI3P phosphatidylinositol (3,4,5)-‐trisphosphate Poly-‐Q polyglutamine PS1 presenilin-‐1 RAMP receptor activity-‐modifying protein ROS reactive oxygen species SAP serum amyloid P SDS sodium dodecyl sulphate TNFR1 tumor necrosis factor receptor 1 TTR transthyretin TUNEL terminal deoxynucleotidyl transferase dUTP nick labelling UAS upstream activating sequence ULK Unc-‐51-‐like kinase UGGT UDP-‐glucose:glycoprotein glucosyltransferase UPR unfolded protein response UPRE unfolded protein response element UPS ubiquitin-‐proteasome system Xbp1 X-‐box binding protein-‐1 YFP yellow fluorescent protein

Introduction

Introduction
4
Protein folding and misfolding
One of the most fundamental processes in biology is the ability of a protein to fold into its defined three-‐dimensional structure. The function of a protein is tightly coupled to this defined conformation. Already in the 1950’s Anfinsen pointed out the relationship between the amino acid sequence of the enzyme ribonuclease and its functional conformation. This functional conformation could be destroyed by the addition of 8 M urea and the reducing agent β-‐mercaptoethanol but as soon as urea was removed and the protein re-‐oxidized, it reassembled into its native structure. The free energy gained in this assembly drives the refolding process [1]. As tribute to his work on ribonuclease Anfinsen was awarded the Nobel Prize in 1972. The native state of a protein is thought to be the most stable structure under physiological conditions. However it was for long not clear how this structure could be adopted and there was no reasonable explanation for the Levinthal paradox [2]. The basic concept introduced by Levinthal is that the search for the proper three-‐dimensional structure is a random “trial and error” event. If a protein of 100 amino acids had to try all of its putative conformations (each taking 10-‐11 seconds to find) the calculated time for this exceeds the age of our universe. However, from experiments we now know that folding occurs in the order of milliseconds to seconds. This time discrepancy is known as the Levinthal paradox [3]. Today, the current concept is that a polypeptides search for its native structure is following a “folding funnel” or “folding landscape” with the native structure as the lowest accessible point. Because, on average native-‐like interactions are more stable than non-‐native ones, not all possible conformations have to be tested, instead it is sufficient to test a small number of possible conformations. The shape of this energy landscape is encoded in the amino-‐acid sequence [4]. The crowded intracellular milieu with a protein concentration of 300-‐400 mg/ml complicates protein folding, since it increases the risk for undesirable interactions with other molecules [4,5]. A way to circumvent this problem is the engagement of folding catalysts and chaperones. They function either by accelerating slow folding steps or by protecting partially folded proteins from misfolding [6,7]. Despite all cellular efforts to optimize folding can protein misfolding occur. In fact, accumulation of misfolded proteins can have detrimental effects on the organism, and is indeed linked to many diseases, including amyloidosis. This dissertation deals with various aspects of misfolded proteins with focus on the amyloid forming islet amyloid polypeptide (IAPP), and the consequences that arise when cells are exposed to misfolded IAPP.

Introduction
5
Amyloid and amyloidosis
History and definitions
In 1854 the German physician Rudolph Virchow was the first to use the term amyloid (from Latin amylum = starch) to describe the macroscopic changes he found in some human organs after they had been treated with iodine and sulphuric acid [8]. At this time, this staining method was widely used by botanists to demonstrate cellulose [9]. Already five years later, Friedreich and Kekulé were able to show that amyloid isolated from the spleen was not “starch-‐like” material but instead it was mainly made up by protein [10]. With time, new staining methods evolved and in 1922 Bennhold introduced the cotton dye Congo red as a histological dye for amyloid [11]. In 1927 Divry and Florkin showed that Congo red emits green birefringence when observed in cross-‐polarized light [12]. A standardized Congo red staining protocol was introduced in 1962 and this is still in use [13,14]. The property of amyloid to emit green birefringence when stained with Congo red suggested a highly ordered structure, which was confirmed by Cohens and Calkins electron microscopy studies on amyloid fibrils. They showed that amyloid is made up of unbranched fibrils with a diameter of approximately 10 nm and undetermined length [15]. Further research revealed that all amyloid fibrils are made up of smaller sub-‐elements, named protofibrils, a finding that proved to be independent on the protein constituent of the amyloid [16]. X-‐ray diffraction analysis was used by Eanes at al. to define the well-‐ordered cross-‐β-‐sheet pattern of amyloid fibrils [17]. In order to be defined as amyloid, following criteria have to be fulfilled:
1. In vivo deposited material 2. Affinity for Congo red and presentation of green birefringence when
viewed in polarized light 3. The characteristic fibrillar structure when investigated with an electron
microscope 4. A specific X-‐ray diffraction pattern of the fibril
All stated criteria follow the consensus reached at the meeting of the Nomenclature Committee of the International Society of Amyloidosis in November 2006. During this meeting one previous characteristic of amyloid was actually revised. Due to the increasing evidence of intracellular amyloid, the definition of amyloid is no longer limited to extracellular material [18].

Introduction
6
Amyloid and diseases
Today, at least 27 different proteins have been identified to form amyloid in humans and the heterogeneous group of diseases associated with such deposits is referred to as amyloidosis [19]. Each type of amyloidosis is characterised by a distinct fibril protein [18]. Despite the common structural features of amyloid fibrils exhibit amyloid proteins only modest primary, secondary and tertiary structure homology [20,21]. Dependant on the amyloid distribution the disease is divided into localized and systemic amyloidosis. Amyloid that appears at a single site or in one tissue type is called localized amyloidoses. Typically, these deposits occur in close proximity of the amyloid protein expression site. Localized amyloidosis are often linked to ageing, e.g. Aβ deposition in Alzheimer’s disease or IAPP in type 2 diabetes. Amyloid diseases with deposits that affect several organs are referred to as systemic amyloidoses. The amyloid precursor in systemic amyloidosis is a plasma protein. Examples of systemic amyloidosis are reactive amyloidosis or secondary amyloidosis with protein AA deposits or AL-‐amyloidosis with light chain deposits [18].

Introduction
7
Table 1: Amyloid fibril proteins and their precursors in human [19].
Amyloid protein Precursor
Systemic (S), or localized (L)
Syndrome or involved tissue
AL Immunoglobulin light chain
S, L Primary Myeloma-‐associated
AH Immunoglobulin heavy chain
S, L Primary Myeloma-‐associated
Aβ2M β2-‐microglobulin S L?
Hemodialysis-‐associated Joints
ATTR Transthyretin S Familial Senile systemic
AA (Apo)serum AA S Secondary, reactive AApoAI Apolipoprotein AI S
L Familial Aorta, meniscus
AApoAII Apolipoprotein AII S Familial AApoAIV Apolipoprotein AIV S Sporadic, associated with ageing AGel Gelsolin S Familial (Finnish) ALys Lysozyme S Familial AFib Fibrinogen α-‐chain S Familial ACys Cystatin C S Familial ABri ABriPP S Familial dementia, British ALect2 Leukocyte chemotactic
factor 2 S Mainly kidney
ADan ADanPP L Familial dementia, Danish Aβ Aβ protein precursor
(AβPP) L Alzheimer’s disease, ageing
APrP Prion protein L Spongiform encephalopathies ACal (Pro)calcitonin L C-‐cell thyroid tumors AIAPP Islet amyloid
polypeptide (also called: amylin)
L Islets of Langerhans (type 2 diabetes) Insulinomas
AANF Atrial natriuretic factor L Cardiac atria APro Prolactin L Ageing pituitary
Prolactinomas AIns Insulin L Iatrogenic AMed Lactadherin L Senile aortic, arterial media AKer Kerato-‐epithelin L Cornea, familial ALac Lactoferrin L Cornea AOaap Odontogenic
ameloblast-‐associated protein
L Odontogenic tumors
ASemI Semenogelin I L Vesicula seminalis

Introduction
8
Structure of amyloid
The high-‐resolution structures of different in vitro assembled amyloid-‐like fibrils have been solved. The primary building block of the fibrils, the actual protein, gives rise to two, or more, β-‐strands that run perpendicular to the fiber axis. Amyloid fibrils are easily identified when viewed in an electron microscope [22]. The highly ordered, repetitive composition of the fibrils give rise to a characteristic X-‐ray diffraction pattern with an inter-‐β-‐strand distance of 4.7Å and a distance of 6-‐11Å between stacked β-‐sheets. Association of 2-‐6 protofilaments, each 2.5-‐3.5 nm in diameter, forms fibrils (see Figure 1). By twisting around one another along the fiber axis, these protofilaments contribute to the rigidity of the amyloid fibril [23]. Amyloid fibrils from the same protein are able to form different morphologies, depending on the surrounding conditions [24]. Solid-‐state NMR and EM images have supported the idea of structural polymorphism in amyloids [25,26]. Different local minima in the energy landscape of the unfolded amyloid protein are accounted for this diversity in vivo [27]. The structural heterogeneity of fibrils includes degree of twisting, the number of filaments per fibril, and the diameter or mass per length of the fibrils [25,26].
Figure 1: Structure of the amyloid fibril. The β-‐strands of the amyloid protein are stacked perpendicular to the fiber axis. The intermolecular distance of β-‐strands of neighbouring units is 4.7Å. Two to six protofilaments twist around each other and give rise to the mature amyloid fibril.

Introduction
9
Non-‐fibrillar components in amyloid deposits
The major amyloid constituent is the disease-‐specific fibril protein. In addition to this fibril protein other, non-‐fibrillar components are present, such as Glycosaminoglycans, Serum amyloid P (SAP) component and Apolipoprotein E (ApoE). Glycosaminoglycans (GAGs) are negatively charged heteropolysaccharides composed of repeating disaccharide units. The structure of the repeating disaccharide unit defines the five GAG classes, namely heparin/heparin sulphate (HS), chondroitin sulphate, dermatan sulphate, hyaluronan, and keratan sulphate. All GAGs except for hyaluronan are usually found covalently linked to a protein backbone and this complex is then called proteoglycan. In the light of amyloidogenesis are heparan sulphate and the heparan sulphate proteoglycan (HSPG) perlecan the best studied GAG and proteoglycan. Numerous in vitro experiments showed the potential of GAGs and HSPGs to promote fibril formation by increasing the β-‐sheet content of the amyloidogenic protein. It is also reported that HS is involved in processing of the amyloid precursor proteins and thereby influencing fibril formation kinetics and/or toxicity [28,29]. Experiments in animal models affirm an active role for HS in amyloidogenesis [30,31]. The interaction of GAGs and amyloid is a target for drug therapy [32,33,34]. Serum amyloid P component belongs to the pentraxin superfamily and binds amyloid fibrils in an calcium-‐dependent manner [35]. The binding of SAP to amyloid fibrils is suggested to prevent proteolysis of amyloid fibrils [36]. Due to its high and specific affinity, radiolabelled SAP is used to monitor amyloid deposits in a non-‐invasive manner [37]. Apolipoprotein E has been detected in association to numerous amyloid deposits, including IAPP derived islet amyloid and amyloid deposits of Alzheimer’s disease [38]. However, the exact role of ApoE in amyloidogenesis is unclear. Polymorphisms in the APOE gene, ε2, ε3, and ε4 strongly alter the likelihood of developing Alzheimer’s disease and cerebral amyloid angiopathy. It has been suggested that ApoE modulates Aβ metabolism and accumulation, although there are contradictive results on plaque density or number depending on the APOE genotype. Differential effects of APOE isoforms on lipid metabolism have been assigned a role in synaptic plasticity and neurodegeneration, independent of interactions with Aβ [39].
Introduction
10
Amyloid formation
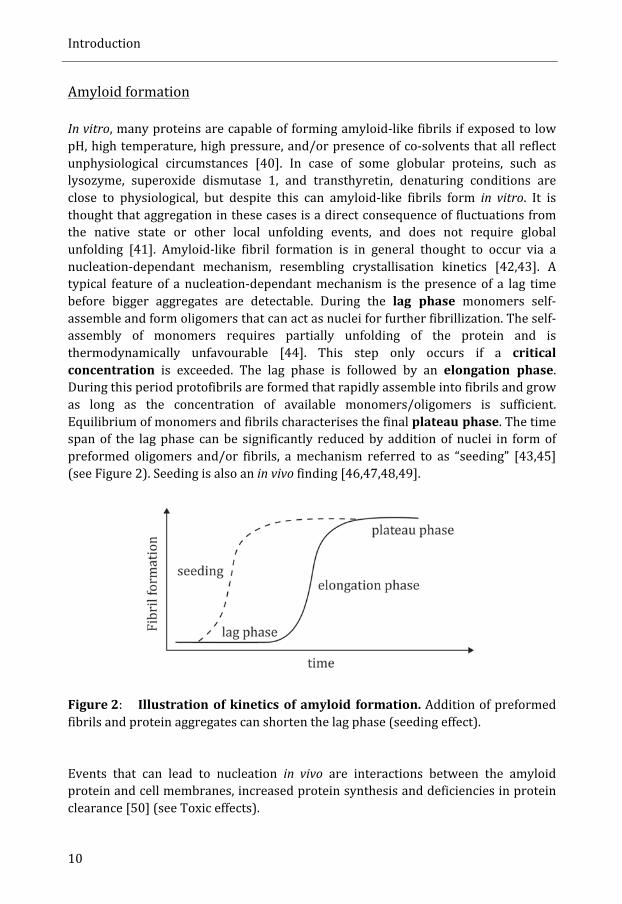
In vitro, many proteins are capable of forming amyloid-‐like fibrils if exposed to low pH, high temperature, high pressure, and/or presence of co-‐solvents that all reflect unphysiological circumstances [40]. In case of some globular proteins, such as lysozyme, superoxide dismutase 1, and transthyretin, denaturing conditions are close to physiological, but despite this can amyloid-‐like fibrils form in vitro. It is thought that aggregation in these cases is a direct consequence of fluctuations from the native state or other local unfolding events, and does not require global unfolding [41]. Amyloid-‐like fibril formation is in general thought to occur via a nucleation-‐dependant mechanism, resembling crystallisation kinetics [42,43]. A typical feature of a nucleation-‐dependant mechanism is the presence of a lag time before bigger aggregates are detectable. During the lag phase monomers self-‐assemble and form oligomers that can act as nuclei for further fibrillization. The self-‐assembly of monomers requires partially unfolding of the protein and is thermodynamically unfavourable [44]. This step only occurs if a critical concentration is exceeded. The lag phase is followed by an elongation phase. During this period protofibrils are formed that rapidly assemble into fibrils and grow as long as the concentration of available monomers/oligomers is sufficient. Equilibrium of monomers and fibrils characterises the final plateau phase. The time span of the lag phase can be significantly reduced by addition of nuclei in form of preformed oligomers and/or fibrils, a mechanism referred to as “seeding” [43,45] (see Figure 2). Seeding is also an in vivo finding [46,47,48,49].
Figure 2: Illustration of kinetics of amyloid formation. Addition of preformed fibrils and protein aggregates can shorten the lag phase (seeding effect). Events that can lead to nucleation in vivo are interactions between the amyloid protein and cell membranes, increased protein synthesis and deficiencies in protein clearance [50] (see Toxic effects).

Introduction
11
Toxic effects
In general, diseases associated with amyloid are of late onset and actual deposits have degenerative effects [45]. The role of amyloid in different diseases has been subject of discussion over a long period and during the last decade many new insights into structural properties of amyloid fibril precursor species have shed a new light on how to think about amyloid cytotoxicity. In 2006, the year this PhD thesis was initiated, it was believed that amyloid cytotoxicity is coupled to common mechanism independent of protein or peptide. Until then, several in vitro studies had shown that oligomeric species and/or protofibrils of several amyloid proteins were able to permeabilize cell membranes, resulting in cell dysfunction [51,52,53,54,55]. In the same year Cohen et al. were able to demonstrate in a C. elegans model that protofibrils of Aβ were toxic, whereas high molecular weight Aβ aggregates were not [56]. Today, oligomers are still seen as the major cause for cytotoxicity. Over the last few years there has been growing evidence for the concept that the same amyloidogenic peptide/protein can give rise to structurally different oligomers and structural distinct fibrils. This led to the proposal of an aggregation energy landscape with several local energy minima corresponding to distinguishable oligomeric states [50]. But toxicity is not only thought to be dependent on the structure of the oligomeric species but also on the biophysical and biochemical properties of the interacting membrane. Anionic surfaces (e.g. anionic phospholipid-‐rich liposomes, glycosaminoglycans) seem to play an important role as potent triggers for protein fibrillization. Also mature fibrils can be ascribed certain toxicity since the deposited amyloid can be massive and affect exchange of oxygen and nutrients. Moreover, mature fibrils might contribute to cytotoxicity by leakage of toxic oligomers [50]. When it comes to IAPP it is still unclear if toxic oligomers exist in vivo. In vitro, beta cell toxicity has been shown in the presence of freshly solubilized IAPP and this leads to activation of apoptosis [55,57,58]. On the other hand have different studies shown that even pre-‐formed IAPP fibrils induce beta cell death [59,60]. A recent study could show that there exists a significant relation between the amount of deposited islet amyloid and measured beta cell apoptosis. This latter study strongly suggests that islet amyloid deposition contributes to beta cell death [61]. The inhibitory effect of amyloid inhibitors on beta cell death further challenges the concept of toxic oligomers (reviewed in [62]). The oligomeric state might be transient, and this complicates the interpretation of the in vitro assays where cells are incubated with oligomers. If cells are incubated for longer times with oligomers, these oligomers might alter their structure and start fibrillization. So in order to be able to ascribe toxicity to oligomers it is crucial to make sure that these oligomers are stable. An alternative pathway for IAPP toxicity has been suggested by Engel et al.. In this model, IAPP binds to membranes, which results in fibril growth, significant changes of membrane curvature and will over time lead to physical breakage of the membrane. Notably, the kinetic profile of hIAPP fibril formation matched that of membrane leakage [63]. The model of membrane interaction as crucial step in

Introduction
12
mediating toxicity might be of general nature. Membranes can serve as a template that allow orientation of monomers in a way that favour aggregation [64]. In addition membrane interaction of amyloidogenic proteins can lead to increased local protein concentration and thereby catalyse aggregation [65]. Finally, it has been shown that membranes have the ability to alter the conformation of a protein and in this way induce aggregation [66,67]. Taken together results from different studies that all tried to identify toxic species of amyloidogenic proteins, it becomes clear that aggregation pathways have a major influence on how toxicity is mediated. Since these aggregation pathways not necessarily are the same for different amyloid-‐related peptides, we have to reconsider the concept that there exists a general mechanism that accounts for toxicity. In parallel to the attempt of identifying a toxic amyloid species, several groups have started to look at molecular pathways that might be altered upon protein aggregation and subsequent amyloid formation. Several pathways, such as autophagy, endoplasmic reticulum associated degradation (ERAD) and unfolded protein response (UPR), have been identified to be triggered upon protein aggregation (intra-‐ and extracellular) and a more detailed overview of our current knowledge how these pathways influence cell survival is given in a separate section of this introduction (see Molecular pathways connected to protein misfolding).
Functional amyloid
Since many, structurally unrelated proteins are capable of forming amyloid-‐like fibrils in vitro, it has been speculated that amyloid structures have been a prominent fold in early life [68]. In coherence with this speculation, the field of functional amyloid has evolved over the last decade. Originally it was hypothesised that some organisms have during evolution taken advantage of the widespread potential of proteins to fold in a stable, amyloid-‐like manner [69]. Today, several functional amyloid structures are reported in lower organisms, including curly and chaplins in bacteria [70,71], Sup32p and Ure2p in fungi [72,73], and chorion in insects [74]. In aplysia (sea slug) conversion of CBEP to an amyloid-‐like structure has been suggested to play a functional role in memory storage [75]. In humans Mα, a component of Pmel17, has been described to play a role as functional amyloid as it serves as template for melanin and thereby is involved in melanin polymerisation [76]. Maji et al. suggested in 2009 that peptide and protein hormones are stored in secretory granules in an amyloid like aggregation state [77]. Their hypothesis is based on different in vitro experiments in which they showed how 31 of 42 investigated protein hormones formed amyloid-‐like structures at pH 5.5 in the presence of heparin -‐ conditions that mimic the environment of secretory granules. In addition, they also investigated mouse pituitary tissue and were able to detect

Introduction
13
amyloid like structures. The proposed working model is that either a critical concentration in the Golgi per se and/or processing of prohormones can trigger amyloid formation. As a result, hormones can be packed in secretory granules at a highest density possible and even be stored over long periods due to high stability of the amyloid entity. At the same time, the secretory granules could serve as an “inert” membrane container protecting the cell of putative toxic effects of the formed amyloid. In this model, amyloid fibrils will be destabilized once they are released from the secretory granules and are exposed to pH 7.4 [77]. Unfortunately, neither insulin nor IAPP were part of the investigated protein hormones though. It is known however, that IAPP fibrils are extremely stable and generally need harsh conditions for depolymerisation [78]. It is questionable if secreted IAPP fibrils are able to dissolve once they are secreted from β-‐cells.
Islet amyloid polypeptide (IAPP)
General introduction
Eugene Opie reported in 1901 a hyaline substance to replace areas of the islets of Langerhans in autopsy material from a patient with type 2 diabetes [79]. Already in 1973 the characteristic interaction of extracellular amyloid fibrils with β-‐cell membranes was described [80]. But it was not until 1986 the amyloid protein was sequenced and for the first time fully characterised as 37 amino acid residue polypeptide [81,82]. This peptide was initially being called islet amyloid peptide (IAP) and later islet amyloid polypeptide (IAPP). Short after the very first description of IAPP, a second report was published describing the same polypeptide naming it diabetes associated peptide (DAP) and later amylin [83]. The gene for IAPP consists of 3 exons of which exon one is non-‐coding. It is situated on the short arm of chromosome 12 and has a promoter region similar to the promoter region of insulin [84,85,86,87]. IAPP belongs to the calcitonin gene peptide family together with calcitonin (CT), calcitonin gene-‐related peptide (CGRP), intermedin and adrenomedullin [88]. Sequence homology of hIAPP with CGRP-‐I and II is 43-‐46% and with human CT 20% [89,90]. IAPP is mainly expressed in the beta cells in the islets of Langerhans. Here, IAPP is stored in secretory granules together with insulin and those hormones are co-‐

Introduction
14
secreted upon stimulation [91,92,93]. The intra-‐granular concentration of IAPP is 1-‐4 mM and the insulin concentration is 10-‐40 times higher [94,95]. The plasma concentration of IAPP ranges between 2-‐10 pM [96]. Expression of IAPP has been found in mammals, avian, and the bony fish [94,97,98,99,100,101,102]. In rodents, expression of IAPP was also reported in delta cells in the islets of Langerhans, the gastrointestinal tract, in sensory neurons and in the central nervous system [103,104,105]. Over the years several different biological functions have been ascribed to IAPP. These functions include auto-‐ and paracrine effects in the islets of Langerhans, actions as a satiety peptide in the brain, antagonising insulin action in skeletal muscles and also a role in calcium homeostasis in regard to bone mass. Each of these different functions is briefly highlighted below. Auto-‐ and paracrine effects of IAPP are reported to regulate insulin secretion. Autocrine actions include a dual role for IAPP on insulin secretion. Transgenic mice that are deficient for IAPP show normal basal levels of circulating insulin and glucose. However, these knock-‐out mice have increased insulin responses and blood glucose elimination upon glucose administration when compared to wild type controls. It can be concluded that usually IAPP limits the degree of glucose-‐induced insulin secretion [106]. Studies about 5 years later gave a more differentiated picture of IAPPs role in insulin secretion. Akesson et al. detected a modest increase of basal insulin secretion in the presence of low IAPP concentrations (10-‐10 – 10-‐6 M) and physiological glucose concentrations (7 mM). In contrast, high IAPP concentrations (10-‐6 – 10-‐5 M) inhibited glucose stimulated (10 mM & 16.7 mM) insulin secretion [107]. In addition it has been shown that IAPP acts in a paracrine manner on alpha-‐ and delta-‐cells and suppresses glucagon and somatostatin release, respectively [107,108]. The observed inhibitory effect of IAPP on glucagon release was already seen at low concentrations (10-‐10 and 10-‐8 M) [107]. Today, IAPP has also been identified as a satiety hormone. This action was a matter of discussion but the identification of receptor activity-‐modifying proteins (RAMPs) was a major break-‐through [109,110]. McLatchie et al. showed that RAMPs, single-‐transmembrane-‐domain proteins, can bind to the Calcitonin-‐receptor-‐like receptor (CRLR). This binding and hence newly formed RAMP:CRLR complex has high affinities for substrates that do not bind CRLR alone. If any of the three RAMPs (RAMP-‐1, -‐2, or-‐3) binds to calcitonin receptor 2 (CTR-‐2), a class of receptors with affinity for IAPP is formed [109,111,112,113]. It is not clear if effects of IAPP in the brain are due to local expression in neurons or if IAPP crosses the blood-‐brain barrier [114]. Effects of IAPP on glycaemic control have also led to the development of pramlintide (symlin). Pramlintide is a hIAPP analogue with proline substitutions at position 25, 28, and 29. The proline substiutions abrogate the capacity to form amyloid fibrils.

Introduction
15
This hIAPP analogue is today an approved drug for use in conjugation with insulin therapy in patients with type 1 or type 2 diabetes. Furthermore reveal preliminary data a weight loss in obese patients with and without diabetes upon symlin intake [115,116,117]. A recent study in rats suggests a role for IAPP in maternal regulations and IAPP mRNA was up-‐regulated in the preoptic area of the hypothalamus of lactating dams [118]. IAPP also has been attributed a role in reducing pain [119,120]. In skeletal muscles IAPP has been found to inhibit insulin-‐stimulated incorporation of glucose into glycogen. The effect is described to occur via inhibition of glycogen synthase (GS) and activation of glycogen phosphorylase (GP), [121,122]. Insulin on the other hands stimulates dephosphorylation of GS thereby promoting glycogen synthesis. These effects of IAPP that are contrary to insulin action on skeletal muscles, are accounted for playing a role in developing insulin resistance [123]. Finally, I want to mention IAPPs effect on calcium homeostasis. Infusion of IAPP decreases circulating levels of calcium in humans [124]. Mice deficient for IAPP show a 50% reduction in bone mass when compared to wild-‐type littermates; an effect due to increased bone resorption mediated by IAPP [125].
Prohormone processing
Biological mature human IAPP derives from proteolytic cleavage of the 89 amino acid hormone preproIAPP. The first 22 amino acids account for the signal peptide and are cleaved off after entrance into the endoplasmic reticulum (ER) [126]. The remaining, 67 amino acid long, proIAPP enters the secretory pathway and there it is cleaved at its C-‐terminal and N-‐terminal site, giving rise to mature IAPP (see Figure 3) [126,127,128,129]. Processing of proIAPP is sequential and occurs first at the C-‐terminal site where prohormone convertase (PC) 1/3 cleaves at di-‐basic amino acid residues K50-‐R51 [128,130]. In the secretory granules PC2 removes the N-‐terminal flanking peptide processing after di-‐basic residues K10-‐R11 [129,130]. Notably, in absence of PC 1/3 is PC 2 capable to cleave at the C-‐terminal processing site. This redundancy does not work the other way round. Removal of the N-‐terminal flanking can solely be achieved by PC 2 [128]. Carboxypeptidase E (CPE) removes the dibasic residues lysine and arginine at the C-‐terminus of processed proIAPP. The exposed glycine is carboxyamidated by the peptidyl amidating monooxygenase (PAM) complex. Presence of active CPE is also necessary in order to facilitate processing at the N-‐terminal site by PC 2 [131].

Introduction
16
Both prohormone convertase are produced as precursor molecules themselves and have to undergo cleavage events in order to become fully active. PC 1/3 is first auto-‐catalytically cleaved at its N-‐terminus. This occurs already in the ER. In mature secretory granules PC 1/3 is additionally cleaved at the C-‐terminal. This site-‐specific maturation of PC 1/3 may explain the observed granule-‐specific processing by PC 1/3. At the same time, a partial activation of PC 1/3 in the late TGN was demonstrated and it was shown that C-‐terminal cleavage of proIAPP is already initiated in the TGN before entering secretory granules [130,132,133,134]. Sorting of PC2 starts in the ER where 7B2 binds to proPC 2 and enables relocalization of proPC 2 to the TGN. The binding of proPC 2 to 7B2 requires proper folding of proPC 2 [135,136]. ProPC 2 is finally cleaved in the secretory granule, a prerequisite to gain enzymatic function [134,137]. In mature IAPP, a disulphide bridge is present between cysteine 2 and 7 [94,138]. Several studies show that impaired processing of proIAPP influences fibril formation of IAPP [139,140,141,142,143]. The implications of incomplete processing of proIAPP on fibril formation are discussed below (see IAPP fibril formation).
Figure 3: Schematic drawing of prohormone processing. Both proIAPP and proinsulin are sequentially processed by the prohormone convertases 2 and 1/3.
The prohormone convertases that process proIAPP also sequentially cleave proinsulin into insulin. Initially PC 1/3 cleaves at two arginines at position 31 and 32 (R31-‐R32), separating the B-‐chain from the C-‐peptide [144,145]. PC 1/3 cleavage gives rise to the transient intermediate des-‐31,32 proinsulin. Thereafter, PC 2 removes the C-‐peptide from the A-‐chain after residues lysine 64 and arginine 65 [146]. Dibasic residues at the cleavage sites are removed by CPE [147].

Introduction
17
IAPP and type 2 diabetes
Diabetes is today classified into different types: type 1 and type 2 diabetes, maturity onset diabetes in young (MODY), ketosis prone diabetes (KPD), and latent autoimmune diabetes (LADA) [148]. Type 1 diabetes leads to insulin deficiency due to destruction of beta cells by an auto-‐immune reaction and usually debuts at a young age. The majority of individuals with diabetes suffer from type 2 diabetes [149]. Deposition of IAPP-‐derived islet amyloid is closely associated with type 2 diabetes. At autopsy, amyloid can be found in a great majority (up to 95%) of patients with type 2 diabetes [150,151,152]. Contrary, in healthy, age matched individuals, only 10-‐20% subjects show amyloid deposition in the pancreas, and the amyloid load found is much lower when compared to patients with type 2 diabetes [152,153]. Type 2 diabetes is a heterogeneous disease that is characterized by hyperglycaemia [154]. The prevalence for type 2 diabetes increases with age and the combination of several risk factors, such as genetic predisposition, physical inactivity, and obesity have been pointed out as determinants in developing this heterogeneous disease. An initial event in the pathogenesis of type 2 diabetes is peripheral insulin resistance, which will be compensated for by elevated insulin secretion from the pancreatic beta cells [155]. However, type 2 diabetes is not manifested as long as beta cells are able to keep insulin levels high enough. The transition of beta cells not being able to secrete sufficient amount of insulin and thereby leading to type 2 diabetes is referred to as “beta cell decompensation” or “beta cell failure” [156,157]. There is evidence that beta cell loss via increased apoptosis is important for the onset of type 2 diabetes [152,158]. One question that has been matter of debate over a long time concerns the actual role of islet amyloid in type 2 diabetes. Looking at a pancreatic section from a patient with type 2 diabetes where almost all islets are replaced by amyloid it is very tempting to conclude that amyloid most certainly will affect the amount of secreted insulin. However, today it is impossible to say if the formation of islet amyloid is a cause or a consequence of type 2 diabetes. The circumstance that not all patients with type 2 diabetes develop islet amyloid is most likely due to the multifactorial nature of the disease. It does not exclude the possibility that aggregation and fibrillization of IAPP might be of major importance in the development of type 2 diabetes in a considerable large subgroup of patients. The lack of in vivo techniques to track islet amyloid formation in humans leaves the question if islet amyloid deposition precedes type 2 diabetes unanswered [159]. Such chronological order has been observed in baboons though, where islet amyloid appeared before development of disease. In these animals the amount of amyloid correlated well with raised blood glucose levels, a good indicator for the progression of the disease [160]. In animals that “spontaneously” develop type 2 diabetes, i.e. monkey and cat, islet amyloid can be found. This is in contrast to animals like rats and mice, which do not spontaneously develop type 2 diabetes and neither deposit islet amyloid. As a matter

Introduction
18
of fact murine IAPP (mIAPP) lacks the capability to form amyloid fibrils (in vivo and in vitro) [161]. The reason for this can be found in the presence of three proline substitutions in mIAPP. All three prolines are situated between amino acid 20 and 29. Synthetic hIAPP 20-‐29 is extremely amyloidogenic, the corresponding murine not at all [162]. Proline is a known beta-‐sheet breaker and is absent in the primary sequence of hIAPP. However, transgenic mice expressing human IAPP (hIAPP) that were crossbred with ob/ob or Agoutivy mice respectively did develop islet amyloid in response to insulin resistance with hyperglycaemia and a type 2 diabetic phenotype [163,164]. Increased demands of insulin secretion also imply elevated production of IAPP since both hormones are co-‐secreted. It is possible that this rise in IAPP concentration initiates oligomerisation and IAPP fibril formation. Numerous in vivo studies have shown that IAPP fibril formation can cause beta cell death and some of these studies identified apoptosis as death mechanism [58,142,165,166,167]. Besides the presence of deposited islet amyloid are hyperproinsulinemia and elevated levels of circulating des 31-‐32 proinsulin hallmarks of type 2 diabetes [168]. As aberrant processing of proinsulin becomes more frequent, is also aberrant processing of proIAPP expected, a circumstance that is thought to accelerate formation of islet amyloid (see IAPP fibril formation). In Asian population a serine to glycine substitution at position 20 (S20G) has been reported and is associated with early onset of type 2 diabetes and increased risk for developing diabetes [169,170]. In vitro this mutation is more prone to form amyloid-‐like fibrils than the wild-‐type counterpart [171,172].
IAPP fibril formation
In order to understand more about the role of IAPP-‐derived amyloid in type 2 diabetic patients we have to find an answer to the question: “why does IAPP form amyloid in patients with type 2 diabetes?”. At the same time one can rephrase this fundamental question and ask: “which mechanisms prevent fibril formation of IAPP under normal conditions?”. Below, some of the results are presented that contain clues to these puzzling questions. The structure of the IAPP monomer is not determined. This is due to the circumstance that IAPP in an aqueous environment spontaneously aggregates into insoluble fibrils within a few hours. Structural data on IAPP in monomeric form are obtained by either analysing murine IAPP, the addition of SDS, or the binding of IAPP to a membrane or insulin. Results from several NMR experiments suggest IAPP to be an unfolded protein, however residues in the region 8-‐19 can dynamically adopt an α-‐helical structure [167,173,174].

Introduction
19
The formation of a N-‐terminal helix is thought to be stabilized by interaction with insulin [174]. More recently, three different conformational preferences for hIAPP in solution have been calculated using molecular simulations and infrared experiments. Regarding to this study, the most stable structure of hIAPP is an extended antiparallel β-‐hairpin with the turn region comprising residue 20-‐23. A slightly less stable structure has an α-‐helical segment spanning residues 9-‐17 and a short antiparallel β-‐sheet including residues 24-‐28 and 31-‐35. The least favourable conformation is a random coil structure [175]. The identification of several stable IAPP structures in solution is very interesting for several reasons. It has ben suggested that IAPP has to form monomeric β-‐hairpins that can aggregate and lead to fibril formation [176]. The structure described to be most stable contains such a β-‐hairpin and the turn region in this β-‐hairpin coincides with the turn region found in protofilaments of IAPP fibrils [177]. This could explain the fast aggregation of IAPP in vitro. It is known that insulin is found in the secretory granules together with IAPP. Even though those two hormones are found at different intra-‐granular localizations -‐ IAPP resides in the halo region of secretory granules, whereas insulin form a crystal in the core region – I want to mention some in vitro data from insulin-‐IAPP interaction studies as they exemplify how inhibition of IAPP aggregation could work [178]. In vitro insulin can inhibit fibril formation of IAPP [178,179,180]. Insulin is thought to interact with IAPP by keeping IAPP in its α-‐helical conformation [174]. Taken together these findings give rise to a model in which IAPP is very prone to form amyloid in absence of an inhibitor impeding the formation of the preferred β-‐hairpin structure. On the other hand, insulin can serve as inhibitor for IAPP fibril formation by stabilizing another naturally occurring structure of IAPP, in which IAPP forms an α-‐helix at its N-‐terminus. Parenthetically, I want to point out that this suggested structure of IAPP stabilized by insulin resembles very much the structure assigned for murine IAPP [175]. When looking closer at interactions of IAPP with insulin one can estimate the complexity of how the environment of beta cell secretory granules influences IAPP fibril formation and/or toxic effects of such events. As already mentioned, several studies have shown in vitro an inhibitory effect of insulin on IAPP fibrillization [178,179,180,181]. Brender et al. recently published results showing insulin to be capable of preventing fiber-‐dependant membrane disruption, but in this study insulin could neither prevent the formation of small oligomers on the membrane nor the initial phase of membrane disruption before fibrillogenesis [182]. Mice expressing hIAPP but not mIAPP (+hIAPP/-‐mIAPP) fed on a diet high on fat develop islet amyloid. In these mice intra-‐granular amyloid fibrils can be detected. This led to the assumption that IAPP initially forms amyloid intracellular in the secretory granules resulting in cell death. Once these cell have disappeared the degradation resistant amyloid will be found extracellularly and act as seed for further amyloid formation from exocytosed IAPP [142]. These in vivo data further

Introduction
20
highlight the importance of intra-‐granular events in promoting and inhibiting fibril formation. One important intra-‐granular event thought to influence fibril formation is the processing of proIAPP. The aberrant processing of proIAPP by PC 1/3 and PC 2 has been investigated in different cell lines with unique prohormone convertases expression profiles. Only cell lines in which proIAPP was not completely processed contained intracellular amyloid [143]. Our group also was able to detect unprocessed proIAPP in intra-‐cellular amyloid fibrils in vivo [142]. In addition, it has been reported that processing of proIAPP alters the binding capacity of heparan sulphate to the amyloidogenic protein. A binding site for heparan sulphate (HS) was identified in the N-‐terminal region of the prohormone – a region not present in processed IAPP [141]. Biophysical studies with unprocessed proIAPP and mature IAPP have supported these findings. This HS – proIAPP interaction might lead to local high concentrations of the amyloidogenic protein and initiate oligomerisation and subsequently fibril formation. Fibrils formed of proIAPP are competent to seed fibril formation of mature hIAPP [139,140]. Heparan sulphate synthesis is thought to occur in the Golgi, generally allowing for intra-‐granular HS-‐proIAPP interactions in vivo [183]. These results overlap with the above-‐described model of IAPP fibril formation at membranes being responsible for amyloid toxicity [63]. A direct interaction of IAPP with cell membranes has been suggested to locally increase IAPP concentrations and cause fibril formation. In all these models negatively charged lipids increase the propensity of membranes to interact with IAPP (reviewed in [167]). Even though IAPP fibrils can be found intracellular it is yet unclear where the primary step of IAPP aggregation occurs. Especially with regards to IAPP amyloid in connection to type 2 diabetes, I want to mention three extracellular factors that are associated with islet amyloid formation. Chronic hyperglycaemia leads to non-‐enzymatic glycation of proteins and is referred to as advanced glycation end-‐products (AGE) [184]. Non-‐enzymatic glycation occurs mainly on proteins with a low turnover rate such as haemoglobin and collagen. The short half-‐life of IAPP (approximately 30 min) therefore makes it unlikely that AGE-‐IAPP exists in vivo [185]. However, deposited islet amyloid might be glycated. In vitro, such AGE-‐IAPP amyloid-‐like fibrils are more prone to seed IAPP fibril formation that non-‐glycated IAPP amyloid-‐like fibrils [186]. These results argue for a role of hyperglycaemia-‐induced glycation in amyloid propagation rather than initiation of IAPP fibrillization. Patients with type 2 diabetes have elevated levels of non-‐esterified fatty acids (NEFAs) in plasma, and there is a linear correlation with levels of blood glucose [187,188]. In this context it is noteworthy that transgenic mice expressing human IAPP have to be fed a diet high in fat in order to develop islet amyloid. The mere over-‐expression of human IAPP in mice is not sufficient for islet amyloid formation [189]. When studied in vitro, NEFAs accelerate IAPP fibril formation without being

Introduction
21
incorporated into fibrils themselves [190]. But NEFAs might not just catalyse fibrillization of IAPP by direct interactions. The NEFAs palmitine and oleate dose-‐dependently induced IAPP expression in the murine pancreatic β-‐cell line MIN6 [191]. As mentioned above, several reports assign GAGs and HSPGs an important role in catalysing fibril formation of amyloidogenic proteins [31,183]. As HSPGs are a major component of extracellular matrix and basement membranes, one can speculate that secreted proIAPP can bind to proteoglycans present in the basement membranes, increase local IAPP concentration there and thereby facilitating fibril formation. This is supported by frequent occurrence of perivascular amyloid deposition [192,193].
Transgenic animal models with hIAPP
Today, in vivo-‐detection of human islet amyloid is not possible. Collected human material derived from either autopsy or from surgical resection does not allow for longitudinal studies. About 15 years ago several groups independently established transgenic mouse strains as models for islet amyloidogenesis. Mouse strains had to express hIAPP since mIAPP is not amyloidogenic. In most strains is the expression of hIAPP under control of the rat insulin I or II promoter [192,194,195,196]. In the model generated by Yagui et al. hIAPP expression is regulated by the human insulin promoter [197]. It soon became clear that the mere hIAPP overexpression did not lead to amyloid deposition despite elevated plasma concentrations of hIAPP (2-‐15 times). These mice were also normoglycaemic and normoinsulinemic [192,195,196,197]. However, one strain contained intracellular amyloid fibrils in the beta cell secretory granules and de Koning et al. found hIAPP in secondary lysosomes [192,197]. Transgenic mice that were hemizygous for hIAPP and treated with growth hormone and dexamethasone contained small intra-‐ and extracellular amyloid deposits [194]. When male mice from this strain were homozygous for hIAPP they spontaneously developed hyperglycaemia, diabetes with beta cell death and intra-‐ and extracellular amorphous IAPP aggregates by 8-‐14 weeks of age. After 20 weeks, the majority of the male mice had detectable amounts of islet amyloid. Deposits were often found in close vicinity to the nucleus and were neighboured by swollen mitochondria [198]. An interesting observation was done in the lab of Steven Kahn. When hIAPP transgenic mice were fed a diet high in fat, the mice became hyperglycaemic and in 80% of male and 11% of female mice older than 13 months were extensive amounts of amyloid found [199]. This emphasizes the importance of additional factors for IAPP fibrillization [195,199]. Oophorectomy in this strain did increase the occurrence of amyloid in female mice to 64%, suggesting a protective role of ovarian products on IAPP fibrillization [200]. Crossbreeding of hIAPP transgenic mice with insulin resistant traits also promoted amyloid formation and persistent hyperglycaemia [163,164]. A hIAPP strain deficient for mIAPP

Introduction
22
(+hIAPP/-‐mIAPP) developed islet amyloid when fed a diet high in fat [189]. In these mice fibrils made up of proIAPP were found in the halo region of the secretory granules [142]. Today not only mice but also rats are established that are transgenic for hIAPP. Rats transgenic for hIAPP (HIP-‐rat) that are hemizygous for hIAPP develop diabetes within 5-‐10 months, accompanied by the presence of extracellular amyloid, decreased beta cell mass, and increased beta cell apoptosis [201].
Aβ
Alzheimer’s disease
Alzheimer’s disease (AD) is the leading cause of dementia in the ageing population and clinical symptoms for AD include cognitive alterations, memory loss and behavioural changes. Neurodegeneration, initially characterized by synaptic injury and followed by neuronal loss has a causal role in the development of AD [202]. Histopathologically AD is hallmarked by the presence of amyloid-‐β (Aβ) containing plaques and neurofibrillary tangles (NFT), composed of hyper-‐phosphorylated forms of the microtubule-‐associated protein tau [203]. Almost 105 years have passed since Alois Alzheimer presented the first case of Alzheimer’s disease and extensive research has been undertaken since then in order to understand the mechanisms lying behind AD. By the time this text is written there are 58212 articles listed on Pubmed all in one way or the other dealing with AD. Still the precise mechanisms leading to neurodegeneration in AD are not completely clear. The majority of AD patients develop a sporadic form, with age as a main risk factor and disease onset between 60 and 70 years [204]. About 10-‐15% of patients have a genetically linked familial form of AD (FAD), with mutations in genes such as Aβ precursor protein (APP), tau and presenilin-‐1 (PS1). FAD patients often have an earlier onset of disease [202]. Many of the mutations found in patients with FAD lead to Aβ production or aggregation and hence Aβ has been ascribed an important role in the development of the disease. Today, this role is more defined in the amyloid cascade hypothesis that comprises deposition of Aβ as trigger for neuronal dysfunction and death in the brain. Aβ is a product of step-‐wise cleavage of APP and comes in different lengths, depending on the cleavage pattern of α-‐, β-‐, and γ-‐secretase [205]. Of these Aβ peptides, Aβ40 is the most abundant peptide but Aβ42 seems to be essential for initiating Aβ aggregation and is considered central to the amyloid cascade hypothesis [206]. The ratio of these two peptides measured in the cerebrospinal

Introduction
23
fluid (CSF) can be a useful measure to confirm the diagnosis of AD. The smaller the relative portion of Aβ1-‐42 is, the higher is the risk for developing the disease [207,208]. This counter-‐intuitive finding is explained by the idea that Aβ1-‐42 is trapped in amyloid deposits and therefore cannot transit from the brain to the CSF (“amyloid sink” hypothesis) [206]. Even though mechanisms leading to selective neuronal death are still debated, Aβ oligomers have emerged as potential culprit in causing neurodegeneration by interference with synaptic function [202,209]. Oligomers as toxic species also might explain the finding of most clinico-‐pathological studies, which fail to find a strong correlation between Aβ amyloid plaque burden and AD severity [210]. A mutation in APP (E693G), known as the arctic mutation, leads to early onset AD and the formed Aβ42 E22G peptide has shown to form increased amounts of protofibrils in vitro [211]. Mouse models transgenic for mutant APP in combinations with mutant PS1 (PS1 is part of the γ-‐secretase complex and involved in the proteolytic cleavage of APP), recapitulate several of the neuropathological characteristics, but it is notable that many of these mouse models don´t suffer neurodegeneration [212]. Lately, the second hallmark of AD, the presence of NFTs made up of hyper-‐phosphorylated tau has gained more attention again. The distribution and abundance of NFTs correlates well with clinical symptoms of AD [213]. Today a “tau axis hypothesis” is discussed in the Alzheimer field. Regarding to this model there are three different ways of how Aβ and tau toxicity interact with each other and thereby cause AD. In a hierarchical model is Aβ acting on tau (hyper-‐phosphorylation of tau due to Aβ amyloid formation), which in its turn mediates toxicity in neurons. In a second model is tau mediating Aβ toxicity. Synergistic toxic effects of tau and Aβ are summarized in a third model [203].
Aβ and IAPP
Aβ and IAPP form insoluble amyloid aggregates in AD and type 2 diabetes respectively. Several epidemiological studies have established a link between the two diseases, showing that patients with type 2 diabetes have an increased risk to develop AD [214] and vice versa [215,216]. Both proteins are thought to be dependant on proper degradation and clearance rate in order to avoid fibril formation and interestingly, insulin-‐degrading enzyme (IDE) degrades Aβ as well as IAPP [217,218]. In vivo and in vitro, Aβ and IAPP compete for IDE binding and can thereby influence the respective degradation efficiency [219,220]. Hence, modulating the clearance capacity of IDE has been discussed as beneficial in the treatment of AD and type 2 diabetes. Still the mechanistic basis for the correlation of AD and type 2 diabetes is unclear. Aβ and IAPP have 25% sequence homology and 50% sequence similarity (see Figure 4) [221]. This structural similarity prompted

Introduction
24
O’Nuallain et al. to test whether preformed fibrils of one of the two peptides (Aβ or IAPP) can serve as seed for the other peptide and accelerate fibril formation (“cross-‐seeding”). Interestingly, fibrils made up of Aβ1-‐40 or Aβ1-‐42 respectively have the same capacity in seeding IAPP fibrillization as preformed IAPP fibrils have. Fibrils made up of IAPP have only very poor seeding capacity on Aβ [221]. Therefore it is unlikely that circulating IAPP fibrils initiate Aβ aggregation in the brain. Jhamandas et al. have recently shown another connection between Aβ and IAPP. In their study in primary cultures of human fetal neurons (HFNs) they found Aβ to mediate toxicity via the IAPP receptor (CTR and RAMP3) and that down-‐regulation of this receptor has a neuro-‐protective effect [222].
Figure 4: Sequence alignment of IAPP and Aβ40. IAPP and Aβ40 share a 25% sequence identity (long bars) and 50% sequence homology (short bars) (adopted from [221]).

Introduction
25
Drosophila melanogaster as model system
History of Drosophila as model system
History has shown, that many results obtained from simple organisms like Drosophila melanogaster have invaluable impact on the understanding of biological correlations in vertebrates and as a matter of fact several biological concepts as we know them today would lack fundamental insights without research conducted in fruit flies. In the following section I want to list some selected examples of how Drosophila research helped us to get a better understanding of biology. Research in Drosophila started in 1908, when T.H. Morgan chose this model for his studies on heredity. In 1910 Morgan had found an eye phenotype (white eyes) that was sex-‐linked and he postulated the information for the phenotype to be found on the X-‐chromosome. Besides the eye colour, several other X-‐chromosome derived phenotypes, e.g. yellow body colour, vermilion eyes, miniature wings, were identified over the next years. In 1913 Sturtevant published a paper in which he put different genes in a linear order [223]. This and other results were the basis for a revolutionary chromosome theory of heredity that in 1933 was awarded the Nobel prize (reviewed in [224]). Notch mutations were first found in Drosophila and reported already in 1915. The systemic search for mutations causing similar phenotypes as Notch mutations led to the identification of the Notch signalling pathway. In the 1990s it became clear that the Notch pathway as it was described in Drosophila is conserved in vertebrates. The Notch pathway has a fundamental role in developmental neurobiology as it affects almost every aspect of neurogenesis and differentiation of neurons in vertebrates, both in the developing and adult brain (reviewed in [225]). Another Nobel prize awarded discovery came in 1927 when Muller was able to demonstrate the mutagenic effects of ionizing radiation on Drosophila [226]. The possibility of inducing mutations was expanded in 1968 when chemical mutagenesis with ethyl methane sulfonate was introduced to the scientific community, giving rise to a new set of experiments accelerating the functional identification of many new genes [227]. The systematic genome-‐wide mutation screen conducted by Nüsslein-‐Vollhard and Wieschaus led to the discovery of most major signalling pathways, including Hedgehog, Tumor growth factor-‐β , and

Introduction
26
Wingless. In 1995 Nüsslein-‐Vollhard and Wieschaus were awarded the Nobel prize in recognition for their work (reviewed in[225]). Even several insights into behaviour derive from discoveries made in Drosophila. Main regulators of circadian rhythm were first identified in flies and the isolation of the genes period, timeless and clock respectively, was an essential initiator for functionally dissecting the circadian rhythm that is conserved from flies to humans (reviewed in [225]). The identification of cAMP in learning and memory is another discovery first made in Drosophila [228]. Whole families of novel channels in vertebrates, such as transient receptor potential (TRP) channels, basic understandings of synaptic transmission are all based on discoveries first made in fruit flies (reviewed in [225]). In 2000 the genome of Drosophila melanogaster was published in a special edition of the Science magazine and today we know that 77% of human disease-‐related genes have a homologue in Drosophila and complicated molecular mechanisms, including apoptosis, autophagy, and unfolded protein response are widely conserved between human and fruit fly [229,230,231,232,233,234]. The Drosophila field working on diseases that are coupled to protein aggregation is still very young but has already shown to be of immense value (see “Drosophila models for protein aggregation”). One of many success stories includes research in flies related to Parkinson’s disease that has provided compelling evidence that parkin and PINK1 are components of a pathway that is involved in regulation of mitochondrial remodelling and that mitochondrial dysfunction is a cause of Parkinson’s disease (reviewed in [225]).
Huge genetic toolbox: Gal4/UAS system
The model organism Drosophila melanogaster offers a vast variety of genetical tools to investigate all sorts of scientific questions. This toolbox was expanded in 1993 when Andrea Brand and Norbert Perrimon published their ground-‐breaking paper introducing the Gal4/UAS system [235]. Until then it was only possible to manipulate gene expression in Drosophila by either driving the expression by a heat shock promoter or tissue specific promoters. The heat shock promoter approach allows for inducible expression, but also carries disadvantages like ubiquitous ectopic expression, basal expression from the heat shock promoter, and the fact that heat shock itself can induce altered phenotypes [236,237,238]. The use of tissue-‐specific promoters allows for targeted expression, but there are limitations in the availability of cloned and characterized promoters that can be used for this purpose. In addition, this technique does not allow for expression of genes coding for toxic products [235]. All these obstacles can be bypassed with the bipartite Gal4/UAS system. In this
Introduction
27
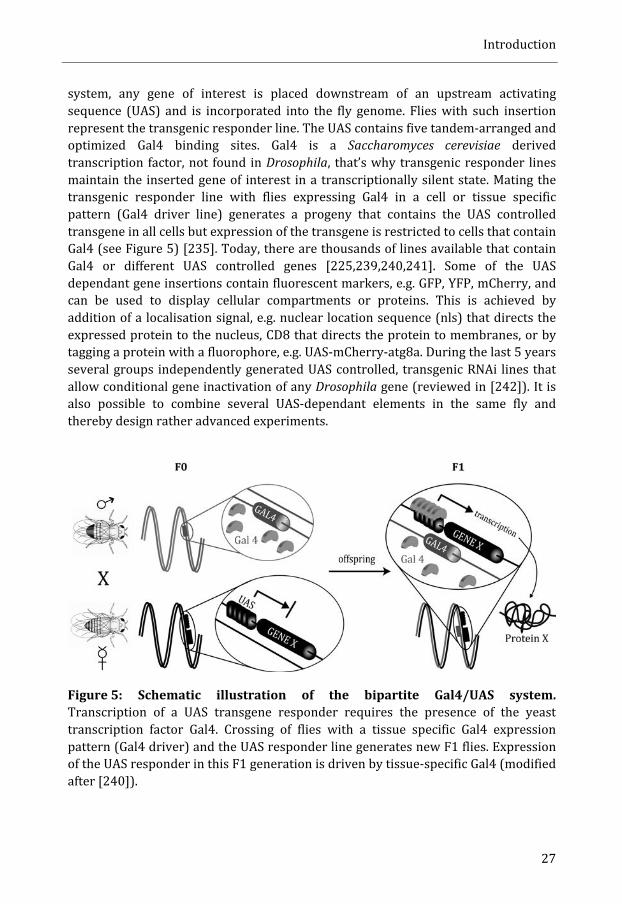
system, any gene of interest is placed downstream of an upstream activating sequence (UAS) and is incorporated into the fly genome. Flies with such insertion represent the transgenic responder line. The UAS contains five tandem-‐arranged and optimized Gal4 binding sites. Gal4 is a Saccharomyces cerevisiae derived transcription factor, not found in Drosophila, that’s why transgenic responder lines maintain the inserted gene of interest in a transcriptionally silent state. Mating the transgenic responder line with flies expressing Gal4 in a cell or tissue specific pattern (Gal4 driver line) generates a progeny that contains the UAS controlled transgene in all cells but expression of the transgene is restricted to cells that contain Gal4 (see Figure 5) [235]. Today, there are thousands of lines available that contain Gal4 or different UAS controlled genes [225,239,240,241]. Some of the UAS dependant gene insertions contain fluorescent markers, e.g. GFP, YFP, mCherry, and can be used to display cellular compartments or proteins. This is achieved by addition of a localisation signal, e.g. nuclear location sequence (nls) that directs the expressed protein to the nucleus, CD8 that directs the protein to membranes, or by tagging a protein with a fluorophore, e.g. UAS-‐mCherry-‐atg8a. During the last 5 years several groups independently generated UAS controlled, transgenic RNAi lines that allow conditional gene inactivation of any Drosophila gene (reviewed in [242]). It is also possible to combine several UAS-‐dependant elements in the same fly and thereby design rather advanced experiments.
Figure 5: Schematic illustration of the bipartite Gal4/UAS system. Transcription of a UAS transgene responder requires the presence of the yeast transcription factor Gal4. Crossing of flies with a tissue specific Gal4 expression pattern (Gal4 driver) and the UAS responder line generates new F1 flies. Expression of the UAS responder in this F1 generation is driven by tissue-‐specific Gal4 (modified after [240]).

Introduction
28
The Gal4 expression is temperature regulated and expression levels increase with raising temperature [240]. Other modifiers of the Gal4/UAS system include the temperature sensitive expression of the Gal4 inhibitor Gal80 and systems with hormone-‐induced Gal4 expression. The Gal4/UAS system can even be used for highly sophisticated “Mosaic Analysis with a Repressible Cell Marker” (MARCM) experiments, where directed mosaic clones are generated [243].
Drosophila models for protein aggregation
Drosophila melanogaster has become a popular model for studies of protein aggregation and for diseases associated with aggregated proteins, such as Alzheimer’s disease (Aβ and tau), familial amyloidotic polyneuropathy (transthyretin, TTR), Parkinson’s disease (α-‐synuclein), Huntington’s disease (poly-‐glutamine expanded huntingtin), and prion disease (HaPrP). Greve et al. published an Alzheimer’s disease (AD) model in which Aβ production is up-‐regulated by simultaneously overexpression of the human amyloid β-‐precursor protein APP, human β-‐secretase, and Drosophila presenilin (dPsn). The latter two proteins process APP into Aβ-‐peptide. These flies produced modest levels of Aβ40, Aβ42, an additional Aβ-‐peptide (δ-‐Aβ), and intracellular fragments of APP. Consequences of this overexpression were age-‐dependant neurodegeneration, formation of β-‐amyloid plaques and shortened life span. The neurodegeneration phenotype was enhanced in flies carrying mutations in dPsn that are associated with early-‐onset familial AD (EOFAD) [244]. In parallel, three groups independently generated transgenic Drosophila strains expressing human Aβ40, Aβ42, and Aβ42 E22G (arctic mutation). The produced peptides all contained an N-‐terminal signal peptide targeting the peptide for secretion [245,246,247]. Only the expression of either Aβ42 or Aβ42 E22G resulted in amyloid deposition in the fly brain. Aβ42 caused short-‐term memory defects, and at later stages, locomotor defects, age-‐dependant neurodegeneration and premature death. The arctic mutation in Aβ42 strongly enhanced all these phenotypes. Iijima et al. reported necrotic cell death as cause for neurodegeneration in their flies [245,247]. Expression of Aβ-‐peptides in the eye caused retinal degeneration [245,246]. These models set the stage for several follow up studies trying to understand more about the mechanisms underlying intra-‐neuronal Aβ42 accumulation and neurodegeneration [248]. In addition, these models were used in the search for new genetic and pharmacological modifiers preventing the phenotypes caused by Aβ42. Some of the approaches were to alter β-‐ and γ-‐secretase activity, enhance Aβ-‐degrading enzymes like neprilysin and insulin-‐degrading enzyme (IDE), or directly interact with Aβ aggregation, by feeding Congo red, or ligands that stabilize Aβ in an α-‐helical structure [248]. The addition of such ligands had protective effects and prolonged survival, increased locomotor activity,

Introduction
29
and reduced neurodegeneration [249]. Computational studies of predicted Aβ42 aggregation caused by different mutations were tested in Drosophila and revealed a strong relation between degree of neurotoxicity and the propensity of Aβ42 to form protofibrils [250]. In a genetic screen utilizing 1963 EP transposon insertions that allow Gal4 to up-‐ or down-‐regulate endogenous Drosophila genes, Cao et al. found 23 modifiers involved in the pathogenesis of AD. Some of the genes were involved in the secretory pathway, cholesterol homeostasis, copper transport, and ubiquitination/proteolysis (ubiquitin-‐conjugating enzyme E2Q and NEP 2). They also had some indication that the autophagy gene ATG1 is crucial in reducing Aβ42 toxicity. When transcription or chromatin remodelling was impaired, Aβ42 had more severe pathological effects [251]. In a genome wide expression analysis and complementary genetic screen on Aβ42 flies, several oxidative-‐stress related genes (e.g. ferritin and catalase) were identified to be potent suppressors of Aβ42 and Aβ42 E22G toxicity [252]. Ling et al. demonstrated an involvement of autophagy in Aβ42 expressing flies. In their hands, reduction of autophagy decreased toxicity while the opposite effect was seen upon autophagy induction and they suggested an autophagic-‐lysosomal injury to be involved in Aβ42 toxicity [253]. To look closer at the role for tau in AD were fly models created that express human wild-‐type or mutant forms of tau (R406W and V337M) that are associated with inherited frontotemporal dementia, and Parkinsonism linked to chromosome 17 (FTDP-‐17) [254,255]. The expression of tau in neurons reduced lifespan, caused neurodegeneration characterized by nuclear fragmentation and vacuole formation in neurons of the cortex and neuropil [255,256]. Expressed in the eye, tau evokes a “rough” phenotype [254]. Although tau expression mimics the phenotype of AD in several ways, the formation of neurofibrillary tangles (NFT) was not seen in these transgenic flies [255]. Flies expressing wild-‐type TTR, TTR V30M, and TTR L55P, two mutations associated with FAP have been created. Expression of any of the two mutant forms resulted in shortened lifespan, neurodegeneration, and attenuation of locomotor activity [257,258]. Recently, Pokrzywa et al. showed an uptake of circulating TTR by the fat body and subsequent storage of TTR as quasi-‐crystalline nanospherules as a mechanism to neutralize toxic species of TTR [259]. Research on Parkinson’s disease has made tremendous advancements due to Drosophila models [260]. Here, I will focus on the impact of aggregating α-‐synuclein. In 2000 Feany and Bender presented their results of flies with ectopic expression of wild-‐type α-‐synuclein, α-‐synuclein A30P and α-‐synuclein A53T. This ectopic expression led to loss of dopaminergic neurons, locomotor defects in adult flies, intra-‐neuronal inclusions and retinal degeneration [261]. These phenotypes were confirmed two years later, and the described phenotypes could be altered upon changes of chaperone activity. Increased levels of Hsp70 suppressed α-‐synuclein toxicity whereas the opposite was the case if chaperone activity was reduced [262].

Introduction
30
Polyglutamine (poly-‐Q)-‐expanded huntingtin is linked to Huntington’s disease and a fly model expressing poly-‐Q huntingtin was characterised by neuronal degeneration and nuclear inclusions. Age of onset and severity of neurodegeneration correlated with increases in poly-‐Q expansion length [263]. Changes in genes that regulate degradation of misfolded proteins, such as the E3 ligase CHIP (C-‐terminus of Hsc70 interacting protein) that also is responsible for targeting tau for ubiquitination and degradation, and genes related to apoptosis, and other signalling pathways have proven to strongly modify the degenerative effects of polyQ proteins. An effective strategy to prevent polyQ-‐expansion caused toxicity was the maintenance of acetylation levels by reduction of histone deacteylases (HDAC) (reviewed in [264]). One identified HDAC, HDAC6, has been shown to serve as molecular link between autophagy and the ubiquitin-‐proteasome system (UPS) [265]. Also several other factors, like phosphoinositide-‐dependant kinase-‐1 (PDK1), p70 ribosomal S6 kinase (S6K), and endosomal sorting complex required for transport (ESCRT) complexes, were identified to modify autophagy and thereby reduce polyQ mediated toxicity [266,267]. Drosophila as model system to study prion diseases has been more changeling than expected. Expression of wild-‐type PrP from Syrian hamster and different mutant forms of PrP, failed to induce neuropathology in flies and only very small amounts of mutant PrP was found to accumulate in fly brains. Flies producing mouse PrP P101L had signs of neurodegeneration, but contained no detergent-‐insoluble or proteinase K (PK)-‐resistant PrP conformers. Finally, the crossing of a Gal4 driver line optimized for high expression levels with a UAS-‐responder line transgenic for wild type PrP from Syrian hamster (HaPrP) generated a fly with neurons that underwent spongiform vacuolation. Guanidine-‐resistance of conformationally changed HaPrP and immunoreactivity with the conformation-‐dependant antibody 15B3 further confirmed the validity of this model. Only PK-‐resistant PrP was absent. Further studies with this model ascribed Hsp70 a protective role and suggested a mechanism of how cytosol-‐derived Hsc70 and HaPrP, which is secreted via the secretory pathway, can interact with each other. The idea is that in older flies expressing HaPrP, Hsp70 can colonize the lipid raft, the proposed site for PrP conversion (reviewed in [268]). Indeed, it has been shown that Hsc70 can move across membranous structures into organelles or into the extra-‐cellular space [269,270].

Introduction
31
Molecular pathways connected to protein misfolding
There is a continuously protein turnover in a cell and protein biogenesis is an error-‐prone process. Accumulation of damaged or misfolded proteins can disturb cellular homeostasis and provoke ageing and cytotoxicity. It is therefore necessary for cells to detect and counteract protein misfolding. This has led to the evolution of a compartment-‐specific quality control systems that can initiate cellular responses upon imbalances in protein folding [271]. The response mechanisms include refolding or degradation of misfolded proteins or the capability to undergo suicide if the process of misfolding is too excessive. In this chapter I will focus on the quality control system and response mechanisms in the ER, programmed cell death (apoptosis), and autophagy, a mechanisms shown to degrade misfolded protein aggregates.
ER-‐stress and unfolded protein response (UPR)
Secretory proteins are directly released into the ER after translation and roughly one quarter of the proteome that traverses the secretory pathway structurally matures in the ER [272]. In order to cope with these high demands has ER developed as a highly specialized and optimized compartment containing a high concentration of general chaperones and a quality control system that senses the folding state of present proteins [273]. An imbalance in ER load of unfolded and misfolded proteins and the capacity to cope with it is referred to as ER-‐stress [274]. But how can the ER sense the folding state of a residing protein? There are several systems for folding quality control in the ER. Glycoproteins become Asparagine (N)-‐linked glycosylated in the ER. Initially, oligosaccharyltransferase (OST) transfers glucose3-‐mannose9-‐N-‐acetylglucosamine2 (Glc3Man9GlcNAc2) to an acceptor sequence (N-‐X-‐S/T). The protein-‐bound oligosaccharide is processed in a cascade of reactions resulting in specific N-‐glycan structures that direct proteins for folding, export or degradation [275]. First, two glucoses are removed by glucosidase I and II generating a monoglucosylated (Glc1Man9GlcNAc2) glycoprotein, which is recognized by the lectins calreticulin and calnexin. Removal of the remaining glucose by glucosidase II results in the release of the glycoprotein from its interacting lectin. Correctly folded glycoproteins can then exit the ER. However, glycoproteins with an incorrect fold are re-‐glucosylated by UDP-‐glucose:glycoprotein glucosyltransferase (UGGT) and re-‐enter the on-‐off cycle with lectins until the glycoprotein has reached its native conformation or is targeted for degradation. Glucosylation plays an important role in

Introduction
32
quality control, as the added glucose acts as a tag for incomplete folded proteins. The glycoprotein can only exit this folding cycle if UGGT fails to re-‐glucosylate [273]. In vitro it could be shown that UGGT recognizes partially folded, molten-‐globule like conformations. Exposure of hydrophobic patches that usually are buried in the core of a folded protein, are important in the recognition of such incomplete folded structures by UGGT [276,277]. Removal of a specific mannose residue by the ER α1,2-‐mannosidase I (ERManI) leads to the recognition of the glycoprotein by the ER degradation-‐enhancing 1,2-‐mannosidase like protein (EDEM) and subsequent targeting for ERAD [273]. ERManI has a relative low catalytic activity and thereby sets a time window for productive glycoprotein folding [271]. The absence of an “N-‐X-‐S/T” acceptor sequence makes it unlikely that IAPP folding is monitored via this pathway. The ER has also developed an oligosaccharide independent control system in which binding of the immunoglobulin-‐binding protein (BiP) is crucial. BiP belongs to the Hsp70 chaperone family and is the most abundant ER-‐chaperone. The primary function of BiP is to assist protein folding and it is of no surprise that BiP is usually the first chaperone to bind a nascent polypeptide chain entering the ER. However, BiP is not only involved in ER associated folding (ERAF) but has also been suggested to target substrates for ERAD. Today it remains elusive how this distinction is done. However, some preliminary results point out ER-‐resident J-‐domains (ERdj) (the ER contains seven different ERdjs) to be involved in pathway selection. ERdjs catalyse hydrolysis of ATP by BiP, an important step in the on-‐off cycle of BiP and its substrate [278]. In general, it is speculated that extensive BiP binding of unfolded substrates is involved in ER-‐stress sensing (see below). Once ER-‐stress is sensed and induced, cells have different opportunities to counteract this imbalance. Cells can reduce the protein load entering the ER by lowering protein synthesis and/or increase ERs capacity to handle unfolded proteins. This requires targeted up-‐regulation of transcription of genes involved in ERAF and ERAD, e.g. chaperones. If ER-‐stress is too intensive and cannot be counteracted by ERAF and ERAD is cell death triggered. This minimizes the damage on organism level due to extensive production of misfolded proteins. Cellular reactions on ER-‐stress are embraced by the term unfolded protein response (UPR). There is no hierarchical order in how ER-‐stress is counteracted. Instead act three different arms of UPR in parallel, initiated by specific signal transducers, inositol-‐requiring protein-‐1 (IRE1), protein kinase RNA (PRK)-‐like ER kinase (PERK), and activating transcription factor-‐6 (ATF6) (see Figure 6) [274].
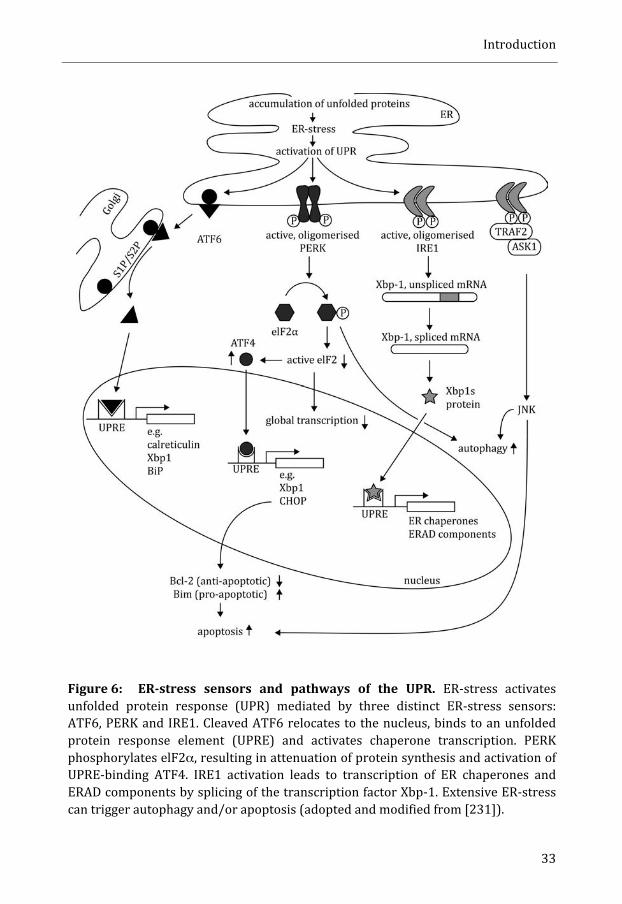
Introduction
33
Figure 6: ER-‐stress sensors and pathways of the UPR. ER-‐stress activates unfolded protein response (UPR) mediated by three distinct ER-‐stress sensors: ATF6, PERK and IRE1. Cleaved ATF6 relocates to the nucleus, binds to an unfolded protein response element (UPRE) and activates chaperone transcription. PERK phosphorylates elF2α, resulting in attenuation of protein synthesis and activation of UPRE-‐binding ATF4. IRE1 activation leads to transcription of ER chaperones and ERAD components by splicing of the transcription factor Xbp-‐1. Extensive ER-‐stress can trigger autophagy and/or apoptosis (adopted and modified from [231]).

Introduction
34
IRE1 is a transmembrane domain that resides in the ER. Activation of IRE1 and subsequent signalling requires its oligomerisation and is thought to be a direct consequence of ER-‐stress [279]. IRE1 possesses structural similarity to the peptide-‐binding domain of MHC complexes, suggesting a direct interaction of IRE1 with other proteins [280]. However, the crystal structure of IRE1 shows that the MHC-‐like binding groove is obstructed and not accessible for protein interactions [279]. This finding is in conflict with in vitro experiments showing direct interaction of IRE1 with unfolded proteins [281]. Hence, it is unclear if IRE1 oligomerisation is a direct outcome of binding to unfolded proteins. Alternatively, it has been suggested that BiP binds to IRE-‐1 and thereby prevents its oligomerisation. High amounts of unfolded proteins could then withdraw BiP from IRE-‐1 leading to IRE-‐1 oligomerisation. However, this model is challenged by BiP binding not to be essential for IRE1 regulation [280,282]. Also a hybrid model has been suggested, which requires both -‐ dissociation of BiP from, and binding of unfolded protein to IRE1 [274]. Once IRE1 oligomerises it becomes trans-‐autophosphorylated on its cytosolic side and activated [283]. The only substrate for IRE1 is mRNA of the transcription factor X-‐box binding protein-‐1 (Xbp1) [284,285]. Hence, IRE1 activity can be monitored by detection of spliced Xbp1. Only in its spliced form initiates Xbp1 transcription of several UPR genes, including chaperones that increase folding capacity, genes for lipid synthesis that result in ER-‐expansion, and ERAD proteins [274,286]. Phosphorylated IRE1 can also recruit Traf2 (tumour necrosis factor receptor-‐associated factor-‐2), a complex that has been linked to caspase-‐12 activation and cell death [287,288]. The cellular response to stress in the cytosol is termed heat shock response (HSR) and mediates gene expression of e.g. chaperones via the heat shock factor-‐1 (hsf1). It has been shown that activation of hsf1 can relief ER-‐stress and this led to the identification of several genes that are regulated by both, hsf1 and Xbp1. These results point towards a cross talk between UPR and HSR [289]. ER-‐stress can also result in oligomerisation and trans-‐autophosphorylation of PERK. This step is followed by phosphorylation of the α-‐subunit of eukaryotic translation initiation factor-‐2 (elF2α), which results in lower levels of active elF2. Low levels of active elF2 decrease general translation and thereby reduce the load from protein synthesis. In addition, low levels of active elF2 also lead to an increased translation of activating transcription factor 4 (ATF4). ATF4 triggers the transcription of genes involved in UPR, e.g. Xbp1 and CHOP (C/EBP homologous protein) [231,274]. The transmembrane protein ATF6 represents the third arm of UPR. Upon ER-‐stress is ATF 6 transported to the Golgi and cleaved by Golgi resident S1 and S2 proteases. The released cytosolic domain of ATF6 then transfers to the nucleus and activates transcription of Xbp1, calreticulin, and ER-‐chaperones such as BiP [231,285]. Expression of the unspliced form of Xbp1 alerts cells for further ER-‐stress responses [286]. The three different arms with their own signalling cascades interplay [274]. The basic principle of ERAD comprises the recognition and unfolding of misfolded

Introduction
35
proteins and their translocation over the ER membrane to the cytosol where these proteins are tagged with ubiquitin and are targeted for proteasomal degradation [290]. One important aspect of ERAD has to be kept in mind when discussing this mechanism in the context of amyloid prone proteins: even though the exact mechanism of retro-‐translocation of proteins from the ER to cytosol still is matter of debate, it is unlikely that larger aggregates can be transported this way. Indeed, FRAP (fluorescence recovery after photobleaching) experiments with a fluorescent ERAD substrate demonstrated that only non-‐aggregated populations were eliminated by ERAD [291]. Aggregated proteins can be isolated in the ER-‐associated compartment (ERAC) -‐ a subcompartment of the ER [292]. These subcompartments were found to be cleared by autophagy but the mechanistic details behind remain unclear [274]. Bernales et al. found autophagic removal of ER as response to UPR and termed this process ER-‐phagy [293]. This process has to occur in the absence of ubiquitination since the ER lacks the ubiquitin system [274]. Autophagy induction as a consequence of ER-‐stress occurs via both, IRE1 activation and subsequent JNK (c-‐Jun N-‐terminal Kinase) signalling, and elF2α phosphorylation by PERK [294]. If none of the above-‐mentioned actions help to ease ER-‐stress can UPR also initiate cell death. The exact link between ER-‐stress and apoptosis are still elusive, but some key signalling events have been identified. It is known that the ER-‐stress sensors PERK and IRE1 are involved in triggering apoptosis. PERK can activate CHOP (via ATF4), which in its turn down-‐regulates the anti-‐apoptotic factor B cell lymphoma-‐2 (Bcl-‐2) and up-‐regulates proapoptotic Bim (BH3-‐only member of the Bcl-‐2 family) [231]. As result of Bim and Bcl-‐2 modulation, mitochondria release cytochrome c, which leads to caspase 3 activation and apoptosis [295]. Drosophila melanogaster lacks an apparent CHOP homologue, but substrates of CHOP, such as Bcl-‐2 (in Drosophila DEBCL and BUFFY) can be found in fruit flies. There role of ER-‐stress induced apoptosis in Drosophila melanogaster is unclear though [231]. Activation of cell death by IRE1 involves complex-‐formation with Traf2 and ASK1 (apoptosis signal regulating kinase-‐1) and activation of JNK signalling [296]. This signalling pathway is independent of Xbp1 splicing and in vitro experiments suggest decreased Xbp1-‐splicing activity of IRE1 at late stages of ER-‐stress when apoptosis is induced [297]. A third connection between ER-‐stress and apoptosis has been found in caspase-‐12. Calcium activated calpains induce caspase 12 dependant procaspase-‐9 cleavage. Cleaved caspase 9 in its turn activates caspase 3 and thereby triggers apoptosis [231]. Caspase-‐12 deficient mice have been shown to have decreased levels of ER-‐stress induced apoptosis, but showed at the same time no alterations in apoptosis in response to other death stimuli [298,299]. The intensity of UPR activation seems to be critical for response selection upon ER-‐stress. In experiments with human cell lines, pro-‐apoptotic mRNAs were less stable when compared with mRNA of factors that facilitate protein folding and adaption. This difference was neutralized under conditions of strong and continuous ER-‐stress when pro-‐apoptotic factors prevailed. Hence programmed cell death was activated [300].

Introduction
36
The roles of UPR and ER-‐stress related responses in the context of IAPP are controversial. Expression of either mIAPP or hIAPP increased the amounts of BiP in 10 weeks old mice. Specific for hIAPP expressing mice was the increased occurrence of active Xbp1 (as detected with Western Blot), ubiquitin, active caspase-‐12 and CHOP, suggesting toxicity that might be due to ER-‐stress [301]. The same group also found high levels of CHOP in HiP rats and again this was interpreted as induction of apoptosis due to ER-‐stress [302]. Marchetti et al. found only modest signs of ER-‐stress in islets from individuals with type 2 diabetes. However, isolated islet from such patients showed signs of ER-‐stress in form of increased mRNA expression of BiP and Xbp1 when cultured with elevated glucose levels (11.1 mM/l). No ER-‐stress was detected at glucose levels of 5.5 mM/l [303]. Accumulation of BiP, spliced Xbp1, CHOP, and Bcl-‐2 was demonstrated in islets from human pancreas of individuals with type 2 diabetes. ER-‐stress was also detected in isolated islets from db/db mice [304]. Taking in account that these db/db mice are not transgenic for hIAPP, one might ask to which extent the observed ER-‐stress in diabetic patients really is due to IAPP aggregation and not to other abnormalities found in patients with type 2 diabetes. Treatment with exogenous hIAPP also has been reported to induce ER-‐stress [305]. Even though several of these models for type 2 diabetes concluded that ER-‐stress is initiated, the question remains unanswered if UPR is triggered by aggregation of hIAPP. A study performed by Hull et al. failed to find any involvement of ER-‐stress and questions if UPR is the pathway mediating toxic effects of hIAPP. In this study the authors looked at mRNA levels of BiP, CHOP, ATF4, and spliced Xbp1 in islets from human IAPP transgenic mice cultured at different glucose concentrations. As control they used islets from non-‐transgenic mice. There was no relative increase in mRNA levels of any investigated ER-‐stress marker in islets from hIAPP transgenic mice. However, ER-‐stress markers did increase in response to thapsigargin, a known ER-‐stress inducer. It was shown by immunohistochemistry that both BiP and activated Xbp1 were elevated in pancreas of mice fed a diet high in fat and in human pancreas from patients with type 2 diabetes. This staining was not influenced by the presence or absence of islet amyloid though. A finding that prompted the authors to discuss the possibility that other factors than IAPP aggregation, such as e.g. elevated levels of circulating NEFAs, have to be accounted for ER-‐stress associated with type 2 diabetes [306]. Immunohistochemical analysis of mice that were transgenic for hIAPP but lacked the gene for mIAPP (+hIAPP/mIAPP-‐) and were fed a diet high in fat failed to find an up-‐regulation of CHOP due to hIAPP expression. The antibodies used in this setup were the same as in the studies mentioned above (G. Westermark, unpublished results). These mice did develop amyloid though and even intra-‐granular amyloid fibrils were found [142]. A recent study by Gurlo et al. did not directly examine ER-‐stress in the context of hIAPP aggregation but rather investigated oligomer formation of hIAPP in islets of transgenic hIAPP mice and pancreas tissue of patients with insulinoma (with and without type 2 diabetes). Sections were taken from tumour-‐free pancreas providing material from individuals with and without type 2 diabetes. Oligomeric hIAPP

Introduction
37
species were identified in secretory granules in the beta cells from transgenic mice as well as beta cells from patients with type 2 diabetes [307]. The presence of such oligomers in secretory vesicles after the ER (only minor amounts were present in the ER and Golgi) further indicates that hIAPP can pass through the ER without being degraded by ERAD or causing UPR in such degree that apoptosis is triggered.
Apoptosis
It is of extreme importance for metazoans to control both cell survival and cell death. In this context, apoptosis (programed cell death) has a central role during development and homeostasis. Failure of apoptosis regulation has been linked to several diseases, e.g. cancer (too little apoptosis) and neurodegenerative diseases (removal of neurons due to apoptosis induction). Central hallmarks of apoptosis include caspase-‐activation, DNA fragmentation (often visualised by TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labelling)), membrane blebbing, and nuclear and cytoplasmic condensation (apoptotic bodies). There are two different pathways that can lead to the activation of programed cell death; one initiated by extracellular factors (extrinsic pathway), and the intrinsic pathway that is triggered upon intracellular death stimuli (reviewed in [229,308]). The extrinsic pathway is activated via a cell surface bound death receptor, such as the tumor necrosis factor receptor 1 (TNFR1) or the Fas receptor. Death receptors oligomerise upon binding of extracellular death ligands, e.g. TNFα or TNFβ. Death receptor oligomerisation leads to the binding of the adaptor protein FADD (Fas-‐associated death domain) forming a death-‐inducing signalling complex (DISC) at the plasma membrane. FADD further recruits procaspase-‐8 or -‐10, which are auto-‐activated upon this relocation. Once activated, the initiator caspase will cleave the activator caspase-‐3 or -‐7 and thereby set the stage for mobilization of DNase and induction of DNA fragmentation. Under normal conditions the DNase DFF40/CAD is tightly bound by DFF45/ICAD and thereby inhibited. Once caspase-‐3 or-‐7 are cleaved and activated they will degrade DFF45/ICAD and liberate DNase DFF40/CAD (reviewed in [229]). One central event of the intrinsic pathway is the modulation of members of the Bcl-‐2 protein family. Bcl-‐2 proteins can be divided into three classes; one class with anti-‐apoptotic function (Bcl-‐2 (the protein giving name to the whole family) and Bcl-‐xL) and two classes comprising pro-‐apoptotic proteins. These two subfamilies are classified as multi-‐domain subfamily (e.g. Bax and Bak) and BH-‐3 only subfamily (e.g. Bim, Bid, Bik). The anti-‐apoptotic effect of Bcl-‐2 or Bcl-‐xL can be ascribed to their interaction with the pro-‐apoptotic members of their family. Interaction occurs via binding to the BH3 domain. Apoptotic death signals (e.g. due to growth factor withdrawal or ER-‐stress) are associated with inhibition of anti-‐apoptotic members of the Bcl-‐2 family and/or activation of pro-‐apoptotic members of this protein family.

Introduction
38
As result of this complex interplay homo-‐oligomerise apoptosis-‐inducing Bcl-‐2 proteins and form pores in mitochondria leading to leakage of cytochrome c and Smac/DIABLO to the cytosol [229,309,310]. Cytochrome c forms a complex termed apoptosome together with the adaptor protein Apaf-‐1. [311]. The apoptosome cleaves caspase 9, which in its turn activates the effector caspase-‐3 and -‐7 and finally leads to the dismantling of the cell. All caspases are inactive unless they are cleaved proteolytically. Initiator caspases (caspase-‐8, -‐10, -‐9, and -‐2) are auto-‐activated under apoptotic conditions whereas effector caspases (caspase-‐3, -‐7, and -‐6) have to be cleaved by initiator caspases. There is a second mechanism of protection against caspase activity. The inhibitor of apoptosis (IAP) family negatively regulates caspases. In order to trigger apoptosis these IAPs have to be inhibited. Smac/DIABOLO (released together with cytochrome c from mitochondria) interacts and neutralizes IAPs (reviewed in [229]). The extrinsic and intrinsic pathways share some common players. Bid, a pro-‐apoptotic Bcl-‐2 family member is activated upon DISC formation (an early step in the extrinsic pathway) and induces mitochondrial release of pro-‐apoptotic factors [312,313]. The activation of caspase-‐3 is also a common feature of both signalling pathways (see Figure 7).
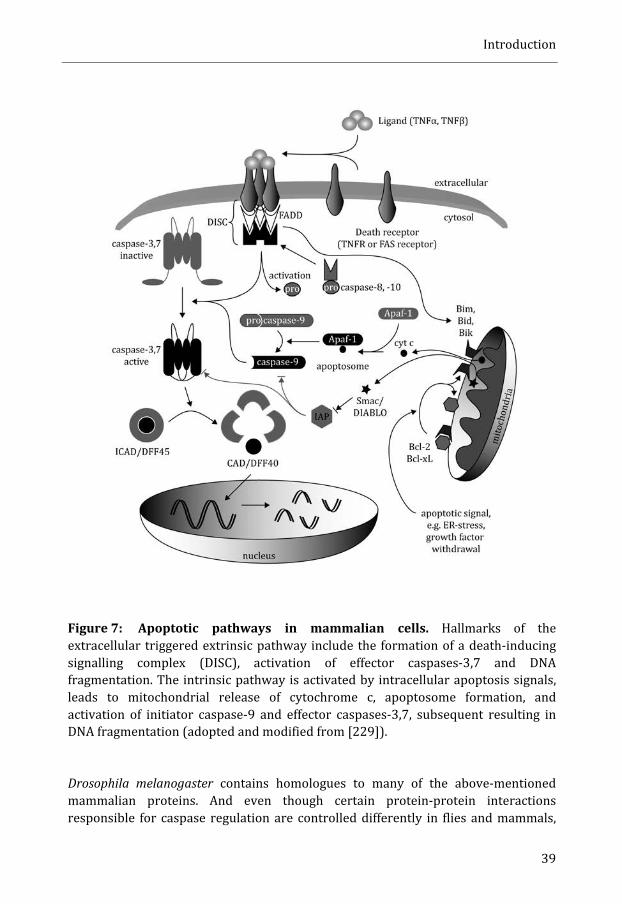
Introduction
39
Figure 7: Apoptotic pathways in mammalian cells. Hallmarks of the extracellular triggered extrinsic pathway include the formation of a death-‐inducing signalling complex (DISC), activation of effector caspases-‐3,7 and DNA fragmentation. The intrinsic pathway is activated by intracellular apoptosis signals, leads to mitochondrial release of cytochrome c, apoptosome formation, and activation of initiator caspase-‐9 and effector caspases-‐3,7, subsequent resulting in DNA fragmentation (adopted and modified from [229]).
Drosophila melanogaster contains homologues to many of the above-‐mentioned mammalian proteins. And even though certain protein-‐protein interactions responsible for caspase regulation are controlled differently in flies and mammals,

Introduction
40
there still is a big degree of conservation when it comes to the general control of the pathways. One example of different regulations includes inhibition of caspases by IAPs. In mammals is caspase-‐9 activity directly inhibited by XIAP. The Drosophila homologue DIAP1 shows no such direct inhibition of Dronc (the Drosophila caspase-‐9 homologue) but functions as E3 ligase recognizing Dronc and thereby targeting it for proteasomal degradation [229,314]. So even if the inhibitory effect of IAPs is different, the basic principle is the same: inactivation of caspases by IAPs, which only is countermanded by an apoptotic signal and subsequent Smac/DIABLO (Drosophila homologues: hid, grim, reaper, and sickle) release from the mitochondria. Drice is the Drosophila homologue of mammalian caspase-‐3, another important molecule in apoptosis. There is evidence that beta cells reduction in type 2 diabetic patients is due to increased apoptosis [152,315]. Several studies have suggested an apoptotic beta cell death as direct consequence of islet amyloid formation. IAPP has been shown to induce caspase-‐3 activation and JNK phosphorylation in a time-‐ and concentration dependant manner when added to RINm5F cells [316]. Addition of amyloidogenic IAPP to pancreatic beta cell lines did trigger apoptosis via caspase-‐8 and caspase-‐3 activation, whereas (25,28,29triprolyl)-‐IAPP (not capable of forming amyloid) did not have such effect [58]. The same group detected an up-‐regulation of Fas-‐associated death receptor in beta cells as response to hIAPP exposure [57]. Zraika et al. found an induction of oxidative stress and following production of reactive oxygen species (ROS) as response to IAPP presence in culturing medium. In their hands, this increase in oxidative stress did not mediate beta cell apoptosis in the short term. The authors speculated that ROS did accelerate amyloid formation via an unidentified feedback mechanism and thereby contribute to beta cell apoptosis [317]. The involvement of ROS in mediating toxicity is of interest in regard to a study recently published by Li et al.. They detected mitochondrial dysfunction accompanied by cytochrome c release, modulation of expression levels of Bcl-‐2 family members, and caspase activation in Ins-‐1 cells that were subjected to freshly dissolved hIAPP. These effects were not seen upon mIAPP exposure [318]. Sensitivity to IAPP amyloid formation depends on the cell type though. While beta cells are evidently vulnerable to islet amyloid and undergo apoptosis, were alpha-‐cells much more resistant to islet amyloid induced apoptosis in a study conducted in the lab of L. Marzban [319]. Also in vivo studies have shown increased beta cell apoptosis, both in transgenic mouse models and HIP rats [142,320]. Experiments conducted in baboons even showed a direct correlation between pancreatic islet amyloidosis severity and apoptosis [160]. Taken together, islet amyloid formation and/or presence are likely triggers of cell death. The performed studies often show induction of apoptosis upon extracellular hIAPP presence. An intracellular event leading to apoptosis could be the activation of CHOP upon ER-‐stress, however, results showing such CHOP involvement are contradictive (see ER-‐stress and unfolded protein response (UPR)). It therefore remains elusive if apoptosis even can be triggered by hIAPP while the protein still is intracellular.

Introduction
41
Autophagy
The term autophagy, which literally means “self-‐eating” (coined by Nobel Laureate Christian de Duve in 1963), comprises pathways that allow cells to digest cytosolic components via lysosomal degradation. Proteasome and autophagy-‐mediated degradation are the main cellular pathways for protein and organelle turnover and all eukaryotic cells can employ autophagy [321,322]. Today, three different classes of autophagy are distinguished: microautophagy, chaperone-‐mediated autophagy (CMA), and macroautophagy. Microautophagy is mainly studied in yeast (containing vacuoles instead of lysosomes) and consists of direct invagination of the vacuolar boundary membrane and budding of autophagic bodies into the vacuolar lumen [323]. Very little is known about molecular mechanisms underlying microautophagy in eukaryotic cells. However, a publication from Ana Maria Cuervos lab recently described a microautophagy-‐like process (named endosomal microautophagy, e-‐MI) in mammalian cells where soluble cytosolic proteins selectively were taken up by late endosomes/multivesicular bodies (MVBs). Cargo selection was dependent on the chaperone Hsc70 and electrostatic interactions with the endosomal membrane [324]. In CMA is cytosolic cargo selectively recognized by a complex of molecular chaperones, including Hsc70, bound by the lysosome-‐associated membrane type protein 2A (LAMP-‐2A) and taken up by the lysosome [325]. Some of the differences between CMA and e-‐MI are the binding to LAMP-‐2A and the requirement of protein unfolding in CMA [324]. The third common type of autophagy, macroautophagy (henceforth referred to as autophagy), is initiated by the formation of a cytosolic double membrane structure called the phagophore (also called isolation membrane). Cytosolic components are both selectively and non-‐selectively engulfed during growth of phagophores. Autophagosomes originate from the closure of phagophores and will subsequently fuse with lysosomes and thereby enable degradation of captured cytosolic constituents [294,326,327]. It is matter of intensive research on how the membrane of phagophores initially is formed. Results from several independent experiments gave rise to different models in which the ER, Golgi, or the outer membrane of mitochondria respectively is source of the initial phagophore double membrane [327,328]. Autophagy has early been identified as a cellular response mechanism to starvation, where resulting macromolecules from cytosolic bulk degradation can be recycled back to the cytosol and be reused. But autophagy has also been shown to turn over substrates in a selective manner in yeast, a pathway known as the cytoplasm-‐to-‐vacuole targeting (CVT) pathway [329]. In analogy, cargo selective degradation of aggregated proteins (aggrephagy [330]), mitochondria (mitophagy [331]), ribosomes (ribophagy [332]), peroxisomes (pexophagy [333]), endoplasmic reticulum (reticulophagy [293]) and many more have been reported for mammalian systems [334]. The role of aggrephagy will be further addressed later in this section.
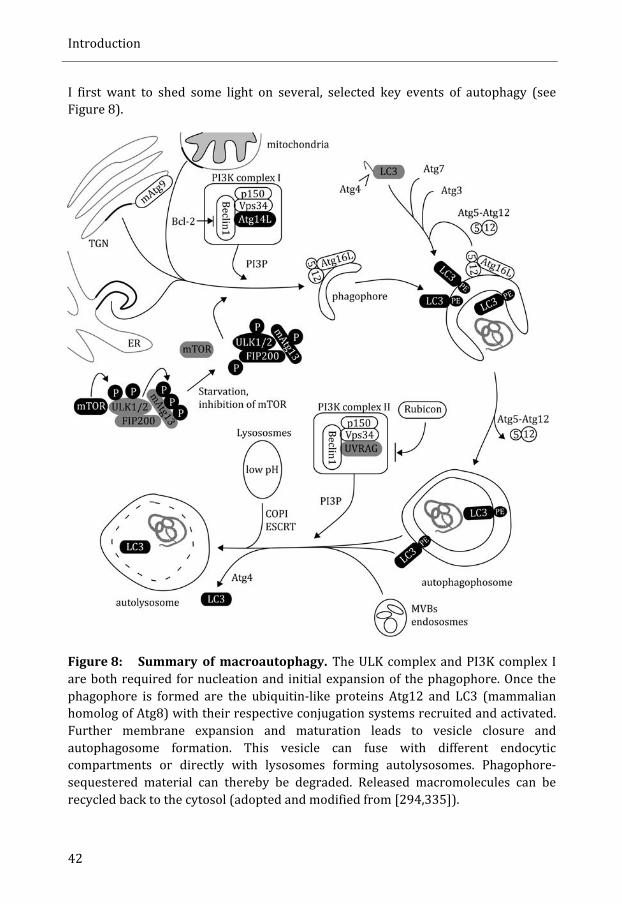
Introduction
42
I first want to shed some light on several, selected key events of autophagy (see Figure 8).
Figure 8: Summary of macroautophagy. The ULK complex and PI3K complex I are both required for nucleation and initial expansion of the phagophore. Once the phagophore is formed are the ubiquitin-‐like proteins Atg12 and LC3 (mammalian homolog of Atg8) with their respective conjugation systems recruited and activated. Further membrane expansion and maturation leads to vesicle closure and autophagosome formation. This vesicle can fuse with different endocytic compartments or directly with lysosomes forming autolysosomes. Phagophore-‐sequestered material can thereby be degraded. Released macromolecules can be recycled back to the cytosol (adopted and modified from [294,335]).

Introduction
43
Genetic screens in S. cerevisiae have led to the identification of numerous autophagy-‐related (ATG) genes and many homologs have been identified and characterized in higher eukaryotes [294]. In general, autophagy can be divided into three steps: 1) induction/nucleation; 2) expansion; and 3) maturation [335]. Basal levels of autophagy are low under normal conditions. However, intra-‐ and extracellular stress factors, such as starvation, ER-‐stress, hypoxia and pathogen invasion can induce autophagy [294]. One important regulator of autophagy is mammalian Target of Rapamycin (mTOR), a serine/threonine protein kinase that inhibits autophagy. Amino acid starvation negatively regulates mTOR and thereby induces autophagy [336,337]. Substrates of mTOR include the Unc-‐51-‐like kinase 1 (ULK1) and -‐2 (ULK2) (mammalian homologs of yeast Atg1), which form a complex with mammalian Atg13 (mATG13) and FIP200 (the focal adhesion kinase family-‐interacting protein of 200 kDa). Dephosphorylation of this complex (requiring inactivation of mTOR) induces autophagy [338]. As mentioned above, much is unknown about the site and exact mechanisms underlying initial phagophore double membrane formation. However, the class III phosphatidylinositol 3-‐kinase (PI3K) complex I, consisting of the class III PI3K Vps34 (vacuolar protein sorting 34), the Vps15-‐like serine/threonine kinase p150, Beclin1 (Atg6 in yeast), and Atg14L (also named Barkor) and its generation of phosphatidylinositol (3,4,5)-‐trisphosphate (PI3P) has been shown to be essential in the initial step of phagophore membrane formation. Bcl-‐2, which already was mentioned in the context of UPR and ER-‐stress, inhibits autophagy by binding and sequestering Beclin1 under nutrient-‐rich conditions. Atg14L has been assigned a pivotal role for this PI3K complex I once autophagy is activated as it recruits the complex to the site of phagophore formation, stimulates it’s activity and even interacts with proteins, such as LC3 (microtubule associated protein 1 light chain 3, the mammalian homolog of yeast atg8), that are crucial in later steps of autophagy. Elongation of the phagophore relies on the ubiquitin-‐like proteins Atg12 and Atg8/LC3 and their respective conjugation systems. Following the processing by a cysteine protease (Atg4) is Atg8/LC3 covalently linked to phosphatidylethanolamine (PE) through the action of Atg7 (E1 activating enzyme), Atg3 (E2 activating enzyme) and the E3 like Atg5-‐Atg12 complex The Atg5-‐Atg12 complex is coupled to the phagophore via Atg16L. Lipidation of Atg8/LC3 is a central step in autophagy. Once activated and lipid-‐conjugated is Atg8/LC3 localized to both sides of the phagophore. Atg4 removes only Atg8/LC3 residing on the cytosolic side of the autophagosome prior to autophagosome-‐lysosome/endosome fusion. It even has been reported that Atg8 can modulate the size of autophagosomes by influencing membrane curvature. Taken together, it is of no surprise that activation of Atg8/LC3 is widely used to monitor autophagy [294,339]. It has been proposed that stepwise fusion of autophagosomes with different endosomal populations account for maturation and culminates in the fusion with lysosomes, the organelle responsible for degradation – a model supported by several findings. Coat protein complex I (COPI), that is involved in ER-‐Golgi transport as well as in the maintenance of endosomal/lysosomal function, has been identified

Introduction
44
to be necessary for autophagy. Down-‐regulation of COPI coatomer subunits with siRNA in GFP-‐LC3 transfected cells resulted in the accumulation of GFP-‐LC3-‐positive autophagosomes. These autophagosomes were not fused with lysosomes. Loss of COPI was also accompanied by an accumulation of p62 and ubiquitinated proteins [340]. Impairment of ESCRT machinery has similar effects and results in defective autophagy [341]. Other known protein complexes of the endocytic pathway that also have been demonstrated to be involved in autophagosomes maturation include class III PI3K complex II (Vps34 together with p150 and Beclin1) with UVRAG as positive and Rubicon (RUN domain and cysteine-‐rich domain containing, Beclin1-‐interacting protein) as negative regulator, and the Vps-‐C complexes HOPS (homotypic vacuole fusion and protein sorting) and CORVET (class X core vacuole/endosome tethering) [342,343,344]. With the discovery of the PI3P-‐binding protein FYCO1 (FYVE and coiled-‐coil domain-‐containing protein 1) binding both LC3 and Rab7 (a late endosomal marker) there even is a molecular link between autophagy and the endocytic machinery. FYCO1 mediates microtubule plus end-‐directed transport of autophagosomes [345]. It has been shown before that the transport of autophagosomes along microtubules depends on dynein [346]. Selective autophagy in form of CVT has been known in yeast for a long time and has gained major attention in mammalian systems over the last years. Selectivity requires crucial, additional steps to the above described autophagy process: cargo has to be recognized by a specific receptor and must be delivered to the autophagic machinery. Here, I want to focus on selective degradation of aggregated proteins via the autophagy pathway (aggrephagy) and introduce essential players in tagging aggregating proteins, recognition of cargo and finally feeding aggregated proteins destined for degradation into the autophagy pathway (see Figure 9). Ubiquitin has been proven to be crucial when it comes to tag proteins that are determined for degradation but lately also in endocytosis, signal transduction and DNA repair [347]. Conjugation of ubiquitin depends on a complex reaction cascade requiring the enzymes E1 (activating ubiquitin), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin ligase). As result ubiquitin is covalently bound via an isopeptide bond between the C-‐terminal glycine of ubiquitin and the ε-‐amino group of a lysine residue of the substrate protein. The E3 ubiquitin ligase provides substrate specificity by recognizing its protein substrate and bringing it to the E2 ubiquitin conjugating enzyme. Cells contain several of E1, E2, and E3 enzymes providing cells with a great tool for primary selectivity for this signalling machinery [348]. Ubiquitin itself contains seven lysine residues and ubiquitin attached to itself forming a polyubiquitin tag. The best-‐characterised linkages occur via Lys48, targeting the substrate for proteasomal degradation, and via Lys63, which is preferred by ubiquitin-‐binding autophagy receptors. Furthermore, Lys63 ubiquitination has been reported to be a potent enhancer of inclusion formation and lead to substrate degradation via the autophagy/lysosome degradation pathway [347,349,350,351]. Also more atypical sites, such as Lys6 or Lys29, for polyubiquitination have been reported but the exact role of these ubiquitin chains is still poorly understood [352].
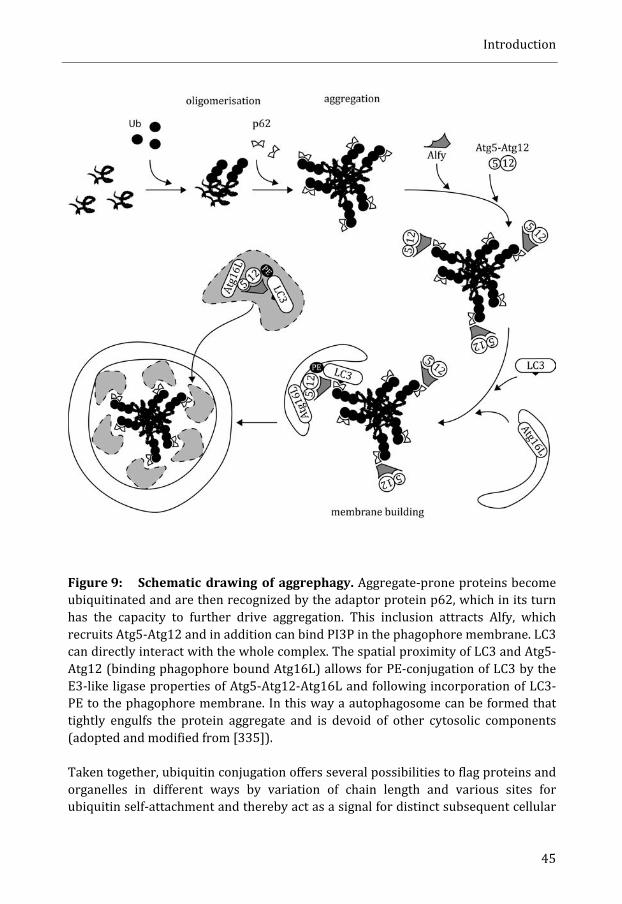
Introduction
45
Figure 9: Schematic drawing of aggrephagy. Aggregate-‐prone proteins become ubiquitinated and are then recognized by the adaptor protein p62, which in its turn has the capacity to further drive aggregation. This inclusion attracts Alfy, which recruits Atg5-‐Atg12 and in addition can bind PI3P in the phagophore membrane. LC3 can directly interact with the whole complex. The spatial proximity of LC3 and Atg5-‐Atg12 (binding phagophore bound Atg16L) allows for PE-‐conjugation of LC3 by the E3-‐like ligase properties of Atg5-‐Atg12-‐Atg16L and following incorporation of LC3-‐PE to the phagophore membrane. In this way a autophagosome can be formed that tightly engulfs the protein aggregate and is devoid of other cytosolic components (adopted and modified from [335]).
Taken together, ubiquitin conjugation offers several possibilities to flag proteins and organelles in different ways by variation of chain length and various sites for ubiquitin self-‐attachment and thereby act as a signal for distinct subsequent cellular

Introduction
46
processes. Molecular links between ubiquitinated proteins and autophagy were identified in form of the sequestosome marker SQSTM1/p62 and NBR1 (neighbour of BRCA1 gene). The conserved functional homolog for p62/NBR1 in Drosophila is Ref(2)p. Both, p62 and NBR1 contain an ubiquitin-‐associated (UBA) domain, allowing for interactions with ubiquitin, a LIR (LC3 interacting region) domain, and a N-‐terminal PB1 (Phox and Bem1p) domain that facilitates self-‐ and hetero-‐oligomerisation with other proteins [347,353,354]. p62 is necessary for the formation of protein aggregates that are degraded by autophagy [355,356,357]. Experiments in mice further proved the active role of p62 in protein aggregation and that these protein aggregates are targeted for autophagic degradation. Tissue specific autophagy deficiency (Atg7 knockout mice) led to p62 and ubiquitin positive inclusions in neurons and hepatocytes. However, formation of ubiquitin-‐positive inclusions was suppressed in p62 knockout mice, underscoring the aggregate-‐promoting role of p62 [358]. Due to its active involvement in autophagy serves p62 as useful marker for autophagic turnover [359]. Several diseases are associated with p62 containing ubiquitinated protein inclusions, such as Lewy bodies in PD, neurofibrillary tangles (AD), Huntingtin aggregates (HD) and Mallory bodies (alcoholic and non-‐alcoholic steatohepatitis). It remains to be clarified if these inclusions are a consequence of defective autophagy [330,360,361]. Very recently, Filimonenko et al. were able to identify Alfy (PI3P-‐binding Autophagy-‐linked FYVE domain protein) to be actively involved in autophagic degradation of polyglutamine expanded, aggregated proteins [362]. The 400 kDa protein Alfy usually resides in the nucleus decorating the nuclear membrane. The presence of ubiquitinated, aggregated proteins in the cytosol leads to relocalization of Alfy to these aggregates [363]. Alfy can directly interact with p62 and with Atg5 [357,362]. In vitro, Alfy is necessary to recruit atg5 to polyQ protein aggregates. In addition, Alfy scaffolds the Atg5-‐Atg12-‐Atg16L complex to p62-‐ and ubiquitin-‐positive polyQ inclusions [362]. The Atg5-‐Atg12-‐Atg16L complex on the other hand is important for LC3 lipidation [364]. Taken together, all these interactions allow for LC3 lipidation in close spatial proximity to ubiquitinated, aggregated proteins and explain the absence of other cytosolic components in aggregate filled autophagosomes [362]. Primary neurons expressing polyQ Htt (Huntingtin) had fewer poly inclusions upon ectopic Alfy expression. These results were confirmed in vivo with a Drosophila model where polyQ production provokes a phenotype that is due to toxicity. The outcome of this polyQ-‐mediated toxicity was much milder once bchs (blue cheese, the Drosophila homologue of Alfy) was co-‐expressed [362]. Reduced levels of bchs in mutant flies had opposite effects and led to shortened live span and extensive neurodegeneration [365]. It remains to be elucidated if Alfy directly recognizes ubiquitinated aggregates or if this interaction is mediated by p62 [330]. Polyubiquitination and p62 are not only required for selective autophagy of aggregated proteins. Aged, damaged mitochondria have to be turned over and the E3 ubiquitin ligase Parkin has been ascribed an important function in mitophagy. Parkin induces Lys63-‐linked polyubiquitination of mitochondrial substrates and thereby

Introduction
47
recruits p62, which in its turn mediates mitochondrial clustering [366]. There is contradicting data concerning whether mitophagy depends on p62 or not. Narendra et al. observed no decline in Parkin mediated mitophagy in MEFs (mouse embryonic fibroblasts) or HeLa cells upon p62 knockdown or siRNA depletion of p62 whereas Geisler et al saw a complete blockage of final clearance of damaged mitochondria once HeLa cells were treated with siRNA against p62 [366,367]. There also has been a report of p62 and ubiquitin dependent autophagy of peroxisomes (pexophagy) [368]. Over the years has autophagy been implicated in many neurodegenerative diseases, such as Huntington’s disease (HD), Parkinson’s disease (PD), Alzheimer’s disease (AD), or amyotrophic lateral sclerosis (ALS). All four are also associated with accumulation of protein aggregates [330]. Both, HD and PD have been shown to be connected to elevated autophagy and noteworthy, autophagy was only triggered by an aggregate prone mutant of huntingtin and not by the wild-‐type form. Cytosolic α-‐synuclein aggregates can be degraded by macroautophagy and CMA [369,370,371,372]. In ALS loss of motor neurons deprives patients of voluntary controlled muscle movements. The disease is associated with ubiquitinated, p62 positive protein inclusions of TDP-‐43 (TAR DNA binding protein 43) or SOD1 (superoxide dismutase 1) or rare mutations in a subunit of the ESCRT complex [373,374]. A defective ESCRT complex in its turn has been shown to result in autophagosome accumulation [341]. But also point mutations of the p150 subunit of dynactin resulting in defects in the transport machinery along microtubules have been implicated in ALS. Transport along microtubules is necessary for autophagosome-‐lysosome fusion and therefore crucial for functional autophagy [375,376]. Extensive alterations in macroautophagy can also be found in patients with AD. In an immuno-‐electron microscopy study on neocortical biopsies from AD patients, were autophagosomes, multivesicular bodies, multilamellar bodies, and cathepsin-‐containing autophagolysosomes the predominant organelles and occupied most of the cytosol of dystrophic neurites. Autophagy was seen in cell bodies with neurofibrillary pathology and was associated with a relative depletion of mitochondria and other organelles. It was speculated that the accumulations of immature autophagic vacuoles result from impaired transport to and fusion with lysosomes thereby hampering protective effects of autophagy [377]. Disruption of lysosomal proteolysis in primary mouse cortical neurons by inhibiting cathepsins, or by supressing lysosomal acidification, impaired transport of autolysosomes, endosomes and lysosomes and led to accumulations of these structures within dystrophic axonal swellings. Such a phenotype can also be seen in numerous mouse models of AD. The phenotype was not caused by general disruption of the axonal transport machinery, as mitochondria and cathepsin-‐lacking organelles were not influenced in their movements. Once lysosomal function was restored the axonal dystrophy was reversed [378]. Enhanced lysosomal cathepsin activity (achieved by

Introduction
48
genetically deletion of cystatin B, an endogenous lysosomal protease inhibitor) increased autophagy function and rescued the autophagy-‐lysosomal related pathology in the Alzheimer’s disease mouse model TgCRND8. This intervention prevented also behaviour defects, measured by learning and memory in fear conditioning and odour habituation tests [379]. Aβ42 also induced neurodegeneration mediated by age-‐dependant autophagy-‐lysosomal injury in a Drosophila model of Alzheimer’s disease [253]. Recently, the age dependence was shown to be of high importance as brain ageing is accompanied with increasing defects of the autophagy-‐lysosomal system and accumulation of dysfunctional autophagosomes and autolysosomes. As a consequence are intracellular membranes and organelles damaged. Such changes could be achieved in young Drosophila expressing Aβ42 and this raised the question if chronic deterioration of the autophagy-‐lysosomal system by Aβ42 simply accelerates brain ageing [380]. This concept is supported by a work done three years earlier with Drosophila. The decreased expression of autophagy genes was shown to be part of normal ageing, and disruption of the autophagy pathway reduced lifespan of flies. A genetically induced enhancement of autophagy extended the average survival and promoted resistance to oxidative stress, at the same time as age dependent accumulations of ubiquitinated proteins was reduced [381]. We are in the beginning of understanding the role of autophagy in pancreatic beta cells and its connections to type 2 diabetes. Beta cell specific disruption of Atg7 in transgenic mice led to reduced beta cell mass, accumulation of ubiquitin-‐ and p62-‐containing inclusions, swollen mitochondria and distended ER. Mice with depleted Atg7 in beta cells developed hypoinsulinaemia and hyperglycaemia. Beta cells from these mice were defective in glucose-‐induced Ca2+ increase. Taken together these results point to a physiological role of basal autophagy in the maintenance of structure, mass and function of pancreatic beta cells [382,383]. The formation of autophagosomes was also up-‐regulated in pancreatic beta cells of diabetic db/db and in non-‐diabetic high-‐fat-‐fed C57BL/6 mice [382]. Very little is known about the influence of IAPP aggregation on autophagy. In HIP rats an increased number of autophagosomes and presence of cytosolic p62-‐positive inclusions was detected when compared with wild-‐type rats. Already obesity per se up-‐regulated autophagy and might be an effect of increased protein burden as such animals developed insulin resistance that is compensated with higher insulin and IAPP production. However, hemizygous HIP rats exhibited a more prominent autophagy induction and it is tempting to ascribe this effect to the amyloidogenic character of hIAPP. Furthermore, when hIAPP was expressed in INS 832/13 cells was autophagy increased compared to cells expressing the non-‐amyloidogenic ratIAPP (rIAPP). The induction of autophagy had a cell protective effect since p62 RNA silencing in hIAPP cells led to increased cell death, while p62 over-‐expression ameliorated cell survival [384]. There is a complex entanglement between autophagy and cell death. The underlying mechanisms of this cross-‐regulation are largely unknown. It has been reported

Introduction
49
numerous times that cell death can be induced via autophagy (named autophagic, or type II, cell death) but autophagy has also been shown to be protective against cell death by inhibiting apoptosis [385,386,387,388]. Autophagic cell death is distinct from apoptosis and necrosis even though it sometimes shares common features [388]. Lately there have been reports from independent groups describing cell death in glioma cells and the midgut of developing Drosophila respectively that was dependant on autophagy but not on effector caspases or lysosomal damage [389,390]. In case of midgut cells in Drosophila this apoptosis-‐independent, autophagy-‐dependant, programmed cell death was seen despite the presence of high caspase activity in these cells [389].


Aims of the Thesis

Aims of the Thesis
52
The term amyloid was introduced to the scientific field more than 150 years ago, and already in the beginning of the 20th century was the association between islet amyloid and type 2 diabetes described. Since then, many researchers have spent time and efforts to understand more about amyloid and its relation to different diseases. In the past 5 years I have been a part of this research in order to increase our knowledge on islet amyloid polypeptide (IAPP). I have explored IAPP toxicity, and disclosed some of the cellular mechanisms that are activated by IAPP-‐aggregates. For this purpose I set different aims that I tried to achieve during this time:
• To set up a new tool that allows to monitor the activation of apoptosis in vitro (paper I).
• To identify which species of proIAPP or its metabolites is most toxic when
added to cells: solubilised protein, mature fibrils, or a mixture of preformed fibrils and solubilised protein (paper I).
• To establish Drosophila melanogaster as model system to study
aggregation/amyloid formation of proIAPP and IAPP in vivo (paper II and III).
• To determine in vivo which cellular mechanisms are altered upon intracellular presence of aggregation prone hproIAPP and hIAPP and to investigate if these mechanisms are involved in the described toxicity. The identification of such pathways allows us to understand more about initial events that might be involved in islet amyloid formation and subsequently lead to the manifestation of type 2 diabetes (paper III).

Material and Methods

54
Detailed description of materials and protocols for all methods used can be found in the papers and manuscript. Here I want highlight some of the pitfalls, advantages and disadvantages that should be taken into account.
Working with Drosophila
P-‐element insertion
At the time we generated transgenic Drosophila strains we decided for an approach with P-‐element mediated transformation [391]. Because the transgene insertion site is random there will be variations in the efficiency of gene transcription. In our case the gene of interest was inserted together with a mini-‐white gene that is responsible for red eye colour. Micro-‐injection was performed in a white mutant background, and red eyes can only be found in flies with successful P-‐element insertion. The intensity of red colour depends on the transcription rate of the mini-‐white gene and provides a hint of transcription rates for the adjacent gene of interest. An additional advantage with the random insertions is the possibility to combine transgenic lines with different insertion loci. It should be mentioned that random insertion can lead to disruption of Drosophila genes and have detrimental effects. When designing experiments it is therefore important to control for effects caused by P-‐element insertion. Hence, in paper II, we included transgenic hproIAPP flies that were not crossed with the used Gal4 driver line in our survival experiment. In recent years, a new technique has emerged that allows for gene-‐insertion at distinct, predefined gene loci. An advantage with this is the possibility to compare the effects of inserted genes that encode for different proteins. On the downside one loses the opportunity to randomly generate flies with different gene expression rates and recombine them.
Survival assay
We conducted numerous survival assays, and one obvious advantage of such assay is the fast read-‐out in form of dead or alive flies. For each survival assay I used 100-‐150 flies with 25 flies per vial kept at constant temperature and humidity with a 12 hours dark-‐light cycle. Despite these high demands I have experienced fluctuations between survival-‐assays performed at different time points. E.g. the median survival for hproIAPP expressing flies varied but was always significantly shorter than estimated for control flies. The reason for the pitfalls in survival-‐assay could be differences in food quality e.g. the surrounding relative humidity will influence the water content in food left to cool down after preparation. Flies can drown in wet and become dehydrated in dry food. Therefore we used mean survival data from a single

Material and Methods
55
experiment and used information from multiple experiments as a description of a trend.
Detection methods
Immunofluorescence – tissue preparation
Many results in this thesis are based on immunofluorescence. Material for these experiments was obtained in different ways. In several experiments heads from decapitated flies were embedded in Tissue-‐Tek® O.C.T.TM and sectioned. Several sections from different transgenic animals were placed on the same slide. This assures that antibody incubation was equal for all specimens placed on the same slide and this allows a better comparison of the results. Sections of fly heads allowed us to analyse antibody binding not only in the CNS but also in the surrounding tissue, such as the detection of hIAPP and hproIAPP in the head fat body. A drawback with the 10 μm thick head sections is the lack of three-‐dimensional information. Analysis of consecutive sections can partly circumvent this problem. However, analysis of dissected brains from Drosophila offers a more sophisticated way to obtain three-‐dimensional data. Counting LNvs in dissected brains is a more exact procedure and also less work–intensive compared to sections. A disadvantage with dissected brains can be encountered when quantifying absolute fluorescence signals. LNvs are located at different levels in the brain and the distance the fluorescent light has to penetrate varies and subsequently the absolute fluorescence. In addition, dissected whole brains exclude the opportunity to analyse surrounding tissue, such as the fat body.
Congo Red or pFTAA
The classic definition of amyloid includes affinity for the dye Congo red and the appearance of green birefringence when viewed in cross-‐polarized light. The fly head capsule has strong affinity for Congo red (the same applies to pFTAA) and results in green birefringence, which makes it difficult to detect small amyloid aggregates. Another drawback is that Congo red staining does not allow for parallel staining with fluorophore-‐conjugated antibodies, as these require mounting in water based mounting medium (e.g. 1:1 PBS/glycerol) that dissolve the Congo red binding. This is different from staining amyloid with pFTAA that is solubilised in PBS. pFTAA can be combined with immunofluorescence, and used for co-‐localisation studies. However, pFTAA does not solely define amyloid deposits instead, pFTAA can also bind to other highly ordered protein structures.

Material and Methods
56
Image processing
After image acquisition there are programs available for image processing that facilitates presentation of data. However, it is important to keep in mind that such powerful tools can provide misleading results when used frivolous. Especially quantification of reactivity requires a careful stringent approach and only images that are acquired under identical circumstances can be used.

Results and Discussion

Results and Discussion
58
Extracellular amyloid formation induces apoptosis (paper I)
This work introduces a novel detection assay for caspase-‐3-‐like activation in beta cells. The system was used to investigate the effects of proIAPP and the three putative processing metabolites N-‐IAPP, IAPP, and IAPP-‐C on beta cells. We compared the putative toxic effects exerted by mature amyloid fibrils, solubilised protein, and the process of fibril formation. The system is based on the measurement of fluorescence resonance energy transfer (FRET). We transfected murine Beta-‐TC-‐6 cells with a vector construct (pFRET2-‐DEVD) that codes for a fusion protein made up by enhanced cyan fluorescent protein (ECFP, emission wavelength 480nm) linked to enhanced yellow fluorescent protein (EYFP, emission wavelength 535nm) and established a stable cell line. The two fluorophores are linked by the caspase-‐3 specific cleavage site residues DEVD. When the two fluorophores are linked to each other is energy transferred from ECFP to EYFP via FRET. If caspase-‐3 is activated it will cleave the fusion protein at its DEVD site and the two fluorophores will disperse. As a result will FRET from ECFP to EYFP be lost in such cells as the energy transfer only occurs when the fluorophores are in very close spatial proximity. Loss of FRET can be used as a direct measurement of caspase-‐3 activity. The excitation wavelength in our experiments was 440 nm and emission was measured at 480 nm (ECFP) and 535 nm (EYFP) respectively. The more FRET occurs, the more light with 535 nm is emitted. Loss of FRET was monitored as a decrease in 535 nm/480 nm ratio. The 535 nm/ 480 nm ratio decreased within 4 hours from initially 2.2 to 1.2 upon addition of the apoptotic agent staurosporine. This method has several advantages compared to other apoptosis-‐detection assays: since apoptosis is a transient event it is of interest to follow the same cell population over time with several measurements. Staining methods such as TUNEL (detecting DNA fragmentation), the vybrant apoptosis detection kit or the apoptosis assay using Ac-‐DEVD-‐AMC are all restricted to one single time point and therefore do not offer time studies in the same way. The same is true for the frequently used MTT assay, which in fact is not a true apoptosis assay since it measures living cells. With the new method described one doesn’t have to consider factors such as cell density or rate of cell division since the number of cells does not influence the measured ratio of emitted light. This is not true for assays where absolute values, such as number of stained cells, are measured. A drawback of the here described conditions for the FRET assay is the relatively poor medium that limits cell survival to up to 12 hours. It would be desirable to define an assay condition that prolongs cell survival. We analysed in which form IAPP was capable of inducing apoptosis. Addition of solubilised synthetic IAPP did not activate caspase-‐3, but a combination of

Results and Discussion
59
solubilized IAPP with preformed IAPP fibrils induced caspase-‐3 activity. In a parallel experiment with a mixture of solubilized IAPP and preformed fibrils was fibril formation monitored with a ThT assay. Samples from different time points were investigated with an electron microscope, and fibrillation over time could be confirmed. Since extracellular fibril formation of hIAPP induced apoptosis, we asked if fibril formation of hproIAPP or the processing intermediates had similar effects or not. We therefore expressed recombinant peptides corresponding to proIAPP (recproIAPP), IAPP with the N-‐terminal flanking peptide (recN+IAPP); IAPP with the C-‐terminal flanking peptid (recIAPP+C) and fully processed IAPP (recIAPP). All peptides were expressed in E. coli as fusion protein with a N-‐terminal GST (glutathione S-‐transferase)-‐tag. The GST-‐tag was enzymatically removed, which led to spontaneous amyloid-‐like fibril formation of all recombinant peptides as confirmed with Cong red staining and TEM. Peptides were added to the cells as preformed amyloid-‐like fibrils, in monomeric form (50 μM), or in monomeric form (50 μM) mixed together with preformed synthetic hIAPP (30 nM) fibrils. Only the mixed solution led to induction of apoptosis and connected reduction in cell viability. There was no significant difference in the degree of cell death irrespective of which hproIAPP metabolite was added together with hIAPP-‐fibrills to the Beta-‐TC-‐6 cells. The cell viability after 12 hours compared to a negative control consisting of solubilized hIAPP without additional seeds, was as follows: recproIAPP: 51%, recN+IAPP: 54%, recIAPP: 28%, recIAPP+C: 41%. In general, apoptosis has been described to be responsible for the reduced beta cell mass found in patients with type 2 diabetes [152]. Islet amyloid formation and/or presence has been shown to induce apoptosis and therefore been suggested to lie behind the observed beta cell loss connected to type 2 diabetes [57,58,142,316,392]. In two different studies were apoptotic elements coupled to the extrinsic pathway, namely caspase-‐8 and FAS-‐associated death receptor signalling, identified [57,58]. It has previously been shown by our group that intracellular IAPP amyloid fibrils in beta cells of mice and humans contain proIAPP and/or incomplete processed proIAPP [142]. Several in vitro experiments have further strengthened the idea that aberrant processing of proIAPP might play a crucial role in amyloid formation and subsequent toxicity [139,140,141,143,393]. All together, the results presented in this study show that proIAPP and the processing intermediates contain the same cell toxic capacity as IAPP, and underline that the fibril propagation can have a central role in the beta cells reduction in type 2 diabetes.

Results and Discussion
60
Characterisation of a new Drosophila model for studies of IAPP aggregation (paper II)
This paper is the first description of a Drosophila melanogaster model for studies of the effects of IAPP and proIAPP aggregation. IAPP belongs to the calcitonin gene peptide family together with calcitonin, calcitonin gene-‐related peptide, intermedin and adrenomedullin [88]. Drosophila lacks nucleotide/amino acid sequence similarity to any of these genes/proteins making it plausible that observed effects of (pro)IAPP are due to its ectopic expression. When setting up this new IAPP model system we wanted to study the consequences of protein aggregation and we therefore included the non-‐amyloidogenic mouse IAPP (mIAPP) as control to all our experiments. The absence of propagation propensity for mIAPP and its sequence homology to human IAPP (hIAPP) makes it a perfect control that can help us to distinguish between effects caused mere protein over-‐expression and effects due to protein aggregation. Furthermore, mIAPP-‐expressing flies can be used as negative controls when staining with amyloid-‐specific dyes. Insertion of the Gal4 dependant transgenes into the Drosophila genome is random and it was important to make sure that this insertion itself does not introduce new phenotypes. Multiple transgenic lines were established. We chose three independent lines that were unique in their transgene insertion site for each transgene (hproIAPP, hIAPP, and mIAPP) and determined the mRNA levels as well as their survival when transgene expression was driven to the CNS (elavC155,Gal4). MRNA expression levels for each transgene were dependant on the insertion site but this did not significantly influence the survival. Neither hIAPP nor mIAPP expressing flies had a significant alteration in survival compared to control flies that only produced Gal4 in the CNS. However, hproIAPP expression had toxic effects and for two out of three transgenic lines, reduction of lifespan was significant when compared to control flies (p<0.02). Also the third line lived shorter than control flies but this effect did not reach statistical significance (p = 0.0577). Interestingly, the transgenic proIAPP line with the shortest lifespan had the lowest mRNA expression levels. The estimated mRNA levels in this hproIAPP line were most comparable to the expression levels detected for hIAPP and mIAPP transgenes and we therefore chose this hproIAPP line together with one mIAPP and one hIAPP transgene for all further experiments. However, the choice of Gal4 driver line and connected site of expression mattered for hproIAPP toxicity. Only when expressed in the CNS by elavC155,Gal4 led proIAPP to accelerated death of the whole organism. With this Gal4 driver was hproIAPPs relative toxicity (when compared to suitable control flies) persistent though and not influenced by increase or decrease in temperature. The observed toxicity for

Results and Discussion
61
hproIAPP was confirmed to be dependant on hproIAPP expression as transgenic hproIAPP flies lacking a Gal4 driver displayed no such shorter survival. If not otherwise stated was the expression of the respective transgenes for all subsequent experiments driven to CNS by elavC155,Gal4. Minor amounts of both hIAPP and hproIAPP could be detected by immunofluorescence at site of expression. But the majority of the respective proteins were found in the neighbouring head fat body. Unspecific cross-‐reactions of the used antibodies could be excluded, as Gal4 driver flies devoid of hproIAPP of hIAPP showed no such staining at neurons or in the fat body. Gal4 dependant expression of GFP with a nuclear localisation sequence did not lead to a GFP signal in the fat body when the same elavC155,Gal4-‐driver was used. This additional experiment allowed us to exclude the presence of Gal4 in the fat body elavC155,Gal4 driver flies (unpublished data). All expressed proteins contained a signal peptide determining the protein for secretion and we concluded that proteins found in the fat body were successfully secreted from neurons and finally delivered to or taken up by the fat body for storage and/or degradation. This finding is not very surprising taking in consideration that the fly fat body partly corresponds to the mammalian liver and is involved in immune responses. Flies expressing TTR in the eye also show accumulations of the protein in the fat body [259]. The amount of hproIAPP or hIAPP found within the humoral/CNS barrier differed depending on the age of the flies. Amyloid aggregation in man is related to age and we expected to find more hIAPP/hproIAPP in neurons from old flies. Surprisingly, this was not the case. Instead more hIAPP and hproIAPP was found in 5d and 15d young flies as compared to 40d old flies. This prompted us to investigate the levels of Gal4 over time. Expression of Gal4 dependant nlsGFP allowed us to follow the occurrence of Gal4. When comparing GFP intensity in 1, 5, 15 and 30d old flies it was obvious that green fluorescence was strongest at day 5, slight reduction occurred at day 15 and the signal was almost absent after 30 days. This decrease of Gal4 expression levels with age can explain the low amount of hproIAPP and hIAPP in neurons of old flies when compared to younger flies. At this stage we had shown toxicity due to pan-‐neuronal hproIAPP expression whereas hIAPP did not alter longevity. On the other hand we were not able to find any differences in protein abundance or tissue distribution. In human, proIAPP is processed at two sites by prohormone convertases 1/3 and 2 and Drosophila contains a homolog to PC2, amontillado. It can be speculated that processing of hproIAPP by amontillado could lead to intracellular imbalances that cause toxicity. However, processing of hproIAPP by amontillado is unlikely as shown by the presence of the N-‐terminal processing site of hproIAPP and the C-‐terminal flanking peptide of hproIAPP in the fat body. The presence of hproIAPP was verified by immunolabelling with antibodies specific for these two processing sites [143]. HproIAPP in the fat body has been produced and secreted from neurons in its unprocessed form.

Results and Discussion
62
The presence of hIAPP and hproIAPP within the humoral/CNS barrier raised the question whether the detected proteins are intra-‐ or extracellular. We designed flies that co-‐expressed Gal4-‐dependant nlsGFP, with the majority of GFP expressed in the nucleus and only a small portion of GFP leaking to the cytosol. With GFP as a cellular marker we could show that hproIAPP and hIAPP was found intracellular replacing the main portion of the cytosol. However, in several cases the proteins were also detected extracellular. As secreted protein can be expected to rapidly diffuse we concluded that detected extracellular proteins were due to protein aggregate depositions representing areas with high local protein concentrations. Extracellular deposits were often found at areas devoid of cell nuclei. Similar findings of intracellular IAPP replacing the cytosol can be done in hIAPP transgenic mice and there it associated with cell death caused by apoptosis [142]. To confirm the presence of amyloid like aggregates in flies sections of Drosophila heads were stained with amyloid specific dyes, such as Congo red and pFTAA (pentameric formic thiophene acetic acid) [394,395]. Both dyes exhibit high affinity for the chitin rich exoskeleton of Drosophila [396]. However, flies expressing hproIAPP and hIAPP both contained structures in the neurons, but more frequent in the fat body, with affinity for the dyes. This presence of pFTAA and Congo red staining besides the exoskeleton was absent in mIAPP expressing flies and control flies that only expressed Gal4. The observed, higher prevalence of pFTAA labelling compared to Congo red is probably due to pFTAAs capacity to bind pre-‐amyloid aggregates [397,398]. Co-‐staining of pFTAA with an IAPP-‐specific antibody confirmed that pFTAA positive structures were made up of hIAPP/hproIAPP. Several areas were only stained with the antibody but not with pFTAA. But all structures that stained with pFTAA also bound the IAPP-‐specific antibody. Staining with pFTAA was often found in close vicinity to nuclei. With transmission electron microscopy (TEM) we detected highly ordered, electron dense aggregates surrounding fat body nuclei. This ultra-‐structural analysis of the fat body revealed two distinct populations of aggregates; an ordered, electron dense and a less ordered, lighter structure. Both structures were found in hproIAPP and hIAPP expressing flies and were completely absent in flies producing mIAPP or Gal4 only. Even though we failed to stain these aggregates with an antibody, we are convinced that they are most likely represent hproIAPP or hIAPP. Absence of antibody labelling of tissue prepared for EM most likely is due to epitope blockage caused by fixation and embedding in epon. The detected aggregates were structurally distinct form aggregates found in TTR expressing flies [259]. HproIAPP and hIAPP expressing flies showed occasionally morphological changes of cell nuclei. This was manifested as an evenly dotted pattern that had replaced heterochromatin and euchromatin and points towards cell fragmentation and cell death. However, this was not accompanied with classic apoptotic hallmarks such as apoptotic bodies or nucleus shrinkage. Taken together this paper presents the characterisation of a novel Drosophila model that can be used for studies on IAPP aggregation. Both hproIAPP and hIAPP form

Results and Discussion
63
aggregates in this model but only hproIAPP exerts toxicity. This finding opens for further studies to understand more about mechanistic links between protein aggregation and toxicity. Clearly, protein aggregation per se is not always toxic and it remains to be elucidated which additional factors are important in determining the outcome in regards to cell/organism survival.

Results and Discussion
64
HproIAPP and hIAPP trigger selective autophagy (paper III)
We took advantage of the unique genetic toolbox offered by the Drosophila melanogaster system and investigated the effects hproIAPP and hIAPP have on several molecular pathways. The model as it is presented in paper II did not allow us to distinguish between responses triggered by extracellular protein deposition and reactions due to intracellular actions. We therefore modified our system in two ways: (i) expression of all transgenes was reduced to 8 ventral lateral neurons (LNvs) using a pdf-‐Gal4 driver line [399] and (ii) we included co-‐expression of Gal4 dependant GFP with a nuclear localization sequence (nlsGFP) if not stated different. This nlsGFP enabled us to monitor all 8 cells as they fluoresce green. Similar to the results with a pan-‐neuronal Gal4 driver (paper II) were hIAPP and hproIAPP present intra-‐ and extracellular. Comparing expression in brains of 1 day, 15 day, and 30 day old flies we detected a reduction of LNvs over time. A similar decrease in LNv number with age was observed for flies that additionally expressed mIAPP. However, the loss of green fluorescent LNvs was significantly higher for flies that expressed hproIAPP or hIAPP. There was no significant difference in toxicity for hproIAPP and hIAPP. We had previously shown that pan-‐neuronal expression of hproIAPP, but not hIAPP, shortens lifespan. However, the two proteins affect viability on a cellular level in similar ways. The loss of LNvs in hproIAPP and hIAPP expressing flies does not occur during development as no difference in LNv number was found in 1 day old flies expressing any of the transgenes, including mIAPP and control flies (nlsGFP only). Even though extracellular protein depositions were found, it is most likely that loss of cell number is due to intracellular events. Extracellular deposition can occur at any neuron that is close to LNvs. The chance of deposition surrounding LNvs is low compared to the probability of deposition next to any other neuron. Monitoring death of LNvs allowed us to distinguish between mIAPP and hproIAPP/hIAPP. This raised the questions about underlying mechanisms for this difference. Apoptosis has been reported to be involved in beta cell death of patients with type 2 diabetes [152]. Similar results are obtained from mouse models and in vitro studies investigating effects of IAPP aggregation [57,58,142,316,320]. However, we were not able to detect any sign of apoptosis in LNvs due to hproIAPP, hIAPP, mIAPP, or sole nlsGFP expression. Immunofluorescence with an antibody recognizing cleaved caspase-‐3 and TUNEL staining are both well-‐established markers for apoptosis but failed to stain LNvs in 15 day and 30 day old flies. This was irrespective of the studied transgene. Since apoptosis is a transient event we wanted to extend the window where caspase activity can be monitored and therefore made use of an in vivo caspase activity sensor, called Apoliner [400]. The basic idea of Apoliner resembles our previously used detection assay for caspase-‐3-‐

Results and Discussion
65
like activation (paper I). Apoliner terms a fusion protein of N-‐terminal mRFP and C-‐terminal eGFP. A synthetic caspase-‐3 cleavage site links both fluorophores. An N-‐terminal mCD8 transmembrane domain destines the fusion protein to membranes. Upon caspase-‐3 activity is this fusion protein cleaved and the two fluorophores separated. Separation of the two fluorophores can be detected as eGFP relocates to the nucleus due to the nuclear localization sequence preceding eGFP. Expression of Apoliner alone or together with hproIAPP, hIAPP, or mIAPP never resulted in such eGFP relocation. A genetic approach to investigate the involvement of apoptosis revealed similar results. Inhibition of apoptosis by the viral caspase inhibitor protein p35 did not revert cell death mediated by hproIAPP. Taken together, all results strongly suggest that cell death of LNvs is independent of apoptosis. Reports of apoptotic cell death of beta cells do not necessarily contradict our results as apoptosis still might be triggered by extracellular protein aggregates and/or amyloid fibrils. Actually, several reports show activity of members of the extrinsic pathway suggesting the initiation of apoptosis due to extracellular events [57,58]. Viral over-‐expression of hproIAPP has been shown to cause ER-‐stress, induce UPR and lead to cell death [301]. As our proteins are secreted and travel through the ER we considered the possibility that ER-‐stress caused cell death. One key event of ER-‐stress is splicing of Xbp1 mRNA, thereby activating this transcription factor [274]. We made use of a Gal4 dependant Xbp1-‐GFP reporter construct. In this construct, GFP mRNA is out of frame and not transcribed unless Xbp1 mRNA is spliced. Co-‐expression of hproIAPP, hIAPP or mIAPP in LNvs did not lead to Xbp1 splicing and subsequent GFP signal after 5, 15 or 30 days. This reporter did work though, as ER-‐stress was detected with Apoliner once RNAi against hsf was expressed. Hsf is a transcription factor known to play a crucial role in regulating the response to heat shock and misfolding proteins [401,402]. Therefore, it is of no surprise that lower hsf levels increased sensitivity for ER-‐stress. Additional experiments underscored our finding that neither hproIAPP nor hIAPP trigger ER-‐stress. The ER residing chaperone BiP (hsc70) is important in sensing ER-‐stress and inducing UPR but immunofluorescence showed no hsc70 accumulation in the ER. As low hsf levels reduced the capacity of ER to handle stress we tested the effects caused by co-‐expression of hproIAPP/hIAPP with RNAi against hsf. Staining with an IAPP specific antibody in such cells lead to accumulation of hIAPP/hproIAPP next to the nucleus and absence of extracellular protein. This result was interpreted to be a direct consequence of general down-‐regulation of protein synthesis, a possible outcome of mild UPR. Low hsf levels are responsible for the increased sensitivity of the ER to misfolded proteins. In conclusion, we can rule out the exhaustion of ER folding capacity evoked by hIAPP and/or hproIAPP expression as a cause for chronic ER-‐stress leading to UPR and subsequent cell death. The absence of ER-‐stress in our model is in accordance with findings from human pancreas of patients with type 2 diabetes and a hIAPP expressing mouse model [306]. Since neither apoptosis nor ER-‐stress could be accounted for cell death we asked which other known cell death pathways could be triggered by the presence of

Results and Discussion
66
amyloid prone peptides. Autophagy has emerged as a potential player in selective degradation of aggregated proteins and has been shown to have protective effects against neurodegeneration. We therefore quantified mRNA levels of ATG4, ATG8a, ATG8b and Ref(2)P (Drosophila homolog of p62) in flies expressing hproIAPP, hIAPP, or mIAPP in the whole CNS (elavGal4, C155-‐driver line). Only hproIAPP expressing flies had elevated levels of all of the investigated mRNAs. Noteworthy, only hproIAPP shortens lifespan when expressed in the whole CNS (paper II). The co-‐localization of antibodies recognizing Atg8a/Atg8b (Drosophila homolog of LC3) and Ref(2)P with protein aggregates made up of hIAPP or hproIAPP further strengthened the concept of autophagy involvement. An autonomously induction of autophagy in response to hIAPP and hproIAPP expression was again shown in LNvs. Expression of the fusion protein mCherry-‐Atg8a reports autophagic activity as cytoplasmic mCherry-‐Atg8a gets recruited to autophagosomal membranes and concentrates in autolysosomes upon autophagosome-‐lysosome fusion. Only hIAPP and hproIAPP, but not mIAPP, led to an accumulation of this autophagy marker in LNvs. In a next step, we were able to show that mCherry-‐Atg8a accumulation is not a consequence of defective autophagosome-‐lysosome fusion. We used the pH sensitive marker mCherry-‐GFP-‐Atg8a. Both fluorophores, GFP and mCherry, emit light from autophagosomes whereas only red signal (mCherry) can be seen from autolysosomes since GFP is unstable in the acidic environment of autolysosomes. Co-‐expression of this reporter with hproIAPP or hIAPP resulted only in red fluorescence consistent with an accumulation of autolysosomes. HproIAPP and hIAPP do not lead to defective autophagy. But is autophagy still involved in toxicity? To address this question we tested if genetic up-‐regulation of autophagy can hamper the increased death of hproIAPP and hIAPP expressing LNvs. We made use of a TOR-‐inhibitor (TOR-‐TED), which was co-‐expressed together with hproIAPP, hIAPP, mIAPP or only nlsGFP. Autophagy up-‐regulation completely restored survival of LNvs that expressed hIAPP and hproIAPP. Neuronal survival was now similar to control, and mIAPP expressing flies. Previously, autophagy has been ascribed a neuroprotective effect in aging by promoting cell survival and longevity through increased resistance to accumulation of ubiquitinated and oxidized proteins, and oxidative stress [381,403]. One can speculate that this protective effect is hampered if cells are challenged with aggregating hIAPP or hproIAPP. This hypothesis was tested by down-‐regulation of autophagy with RNAi against Atg8a and Atg8b. If hIAPP and hproIAPP exert toxicity by reducing protective effects of autophagy, down-‐regulation of autophagy should have similar effects as sole hIAPP or hproIAPP expression has. Counting the number of LNvs over time, we saw a significant decrease in cell viability once autophagy was down-‐regulated and survival rates dropped to levels that are comparable to hproIAPP and hIAPP producing cells. In addition, down-‐regulation of autophagy did not potentiate hIAPP or hproIAPPs toxicity. Taken together this indicates that hIAPP and hproIAPP expression per se already hampers the neuroprotective effects of autophagy.

Results and Discussion
67
Previously, it has been shown that Ref(2)P and ubiquitin positive protein aggregates accumulate in brains of aged Drosophila – this has been suggested to be a consequence of decreased autophagy capacity over time [356,404]. Ubiquitin is a tag known to direct proteins and organelles for degradation [405]. Ref(2)P is involved in promoting protein aggregation and the capacity of Ref(2)P to bind ubiquitin and Atg8 offers a molecular link between ubiquitinated substrates and the autophagy pathway [354,356,406,407]. As ubiquitin and Ref(2)P can deliver substrates to the autophagic machinery we asked if these two molecules are involved in the selective recognition of hproIAPP and hIAPP. Indeed, frequent ubiquitination of intracellular substrates was detected in cells expressing hproIAPP and hIAPP. No such ubiquitination pattern was present in cells producing non-‐aggregating mIAPP. We were able to identify Ref(2)P to be in involved in recognition of ubiquitin positive accumulations and targeting them for autophagic degradation. This conclusion is based on the finding that a decrease in autophagic capacity, mediated by RNAi against Atg8a and Atg8b, leads to accumulation of intracellular vesicles that stain for ubiquitin and Ref(2)P. Such vesicles were found in all cells with decreased Atg8a and Atg8b levels, however hIAPP and hproIAPP expression gave rise to a new population of such vesicles that were unique with their increased surface area. The smaller, common vesicles probably represent normal cellular substrates that are recognized by Ref(2)P and turned over by autophagy. We concluded that the new population consisting of enlarged vesicles is related to accumulation of hIAPP and hproIAPP. Lack of RNAi against Atg8a and Atg8b was enough to completely abolish the presence of these large ubiquitin and Ref(2)P containing vesicles. This further underlines the physiological role of autophagy in degrading these vesicles. We have shown for hIAPP and hproIAPP that autophagosomes were still capable to fuse with lysosomes. However, autophagic capacity was not sufficient to protect cells from accelerated cell death. For Aβ and Alzheimer’s disease it has been suggested that neurodegeneration is related to defective autophagosome-‐lysosme fusion, which can be observed in dystrophic dendrites and axons [408]. This points towards separate toxicity mechanisms for Aβ and hIAPP/hproIAPP. Expression of Aβ42 in the whole CNS shortens life span. This reduction in survival is even more prominent when Aβ42 contains the E22G mutation. However, LNvs that produce Aβ42, with and without E22G mutation, are not influenced in their viability. This demonstrates that Aβ42 can successfully exit the cells before exerting toxicity and further support our findings that the identity of the amyloid protein, and not the sole propensity to aggregate, determines the initiation of cell death. It remains to be elucidated which further characteristics of the amyloid protein are important in cell death initiation. Last but no least, our finding of extracellular ubiquitin and Ref(2)P aggregates at sites where we expected missing LNvs resembles a phenotype recently described in the pancreas of HIP rats. In these rats deposits contained p62 and ubiquitin and are suspected to be connected to beta cell death [384]. This phenotypic mimicry stresses the physiological relevance of findings from our Drosophila model.


General Discussion
and
Future Perspectives

General Discussion and Future Perspectives
70
“But you know, flies are not humans!?” is a reaction I faced several times over the last years when describing my research. And yes, without doubt there are numerous differences between fruit flies and humans. But does that necessarily mean that nature needs to find novel solutions for common problems during evolution? Most certainly not! As matter of fact, without research in flies we would not know as much about human biology as we do today. As history tends to repeat itself, there is no reason to doubt that research conducted in Drosophila melanogaster will continue to provide us with fundamental insights in human biology. So what can we learn from our Drosophila model? It is well described in the literature that IAPP is one of most amyloidogenic proteins in vitro. In an aqueous in vitro environment, synthetic and recombinant IAPP spontaneously forms amyloid-‐like fibrils. One would predict that over-‐expression of IAPP in the brain of Drosophila would result in large amounts of amyloid deposits, evoking a strong phenotype. However, the amounts of IAPP deposition and associated phenotype was less pronounced than expected. Reported phenotypes, such as shortened survival when expressing proIAPP in the whole brain, protein deposition in the fat body and loss of pdf-‐neurons that expressed hIAPP or hproIAPP were stable and reproducible, but not as distinct as the results from in vitro experiments suggest. This inconsistency between in vitro and in vivo properties also exists when comparing results from in vitro experiments with the human situation. Patients with type 2 diabetes develop islet amyloid with involved beta cell reduction, where the vast majority of individuals that do not suffer from the disease produce proIAPP and IAPP every day without deposition of islet amyloid. As type 2 diabetes is closely related to age, it is very likely that the involved amyloid phenotype also occurs later in life, further underscoring the fact that there must be cellular mechanisms to either preclude IAPP aggregation and/or to degrade IAPP aggregates. In order to fully comprehend the impact of amyloid formation it is crucial to identify mechanisms that are affected when IAPP aggregates, and to learn more about the chronological order regarding amyloid formation and cellular responses. Drosophila research with its genetic toolbox offers unequalled opportunities to address such questions in vivo. We were able to identify autophagy to be selectively triggered by hIAPP and hproIAPP over-‐expression and our results demonstrate that this activation neutralizes the protective effects of autophagy. It has previously been shown in mice that impaired autophagy in beta cells provokes phenotypes resembling those of type 2 diabetes [382,383]. First experiments in hIAPP transgenic rats confirm a role for IAPP in affecting the autophagy pathway [384]. These results emphasize the physiological relevance of our findings. It remains to be answered how autophagy is exactly triggered by hIAPP and hproIAPP. We took advantage of the non-‐

General Discussion and Future Perspectives
71
amyloidogenic properties of mIAPP and compared its impact with those of hproIAPP and hIAPP. We managed to point out several molecules, such as ubiquitin and p62, which are involved in the selective recognition of IAPP aggregates. IAPP is a secretory protein and it is of great interest to understand how cytosol resident ubiquitin and p62 can recognize protein misfolding that occurs in the secretory pathway. Can ubiquitin and p62 target complete secretory granules for degradation, and if so, what is the initial signal for such event? Or does protein misfolding in the secretory pathway lead to membrane disruption of e.g. ER, Golgi or secretory granules, followed by leakage of aggregates to the cytosol and subsequent recognition by selective autophagy? Jiminez et al. recently presented a poster demonstrating selective recruitment of the autophagic machinery upon damage of trafficking organelles. Damage of the Golgi apparatus, early and recycling endosomes all resulted in ubiquitin, p62/NBR1 and LC3 recruitment followed by up-‐regulated autophagy. As for hIAPP, Engel et al. described a model in which hIAPP is cytotoxic by its capacity to bind membranes resulting in fibril growth and significant changes of membrane curvature, which finally leads to physical breakage of the membrane [63]. Our results from paper I reveal that neither mature fibrils nor solubilised proteins were capable to induce apoptosis. However, caspase-‐3 was activated once we accelerated fibril formation by addition of small amounts of preformed fibrils to solubilised monomers of hproIAPP or any of its metabolites. This finding substantiates that the process of fibrillization is important in toxicity and is in accordance with the model suggested by Engel et al. [63]. From these results, a new model emerges in which hIAPP and hproIAPP can form intracellular aggregates that disrupt the surrounding organelle membrane. This starts a reaction cascade that involves activation of selective autophagy in order to degrade the hIAPP/hproIAPP aggregate and/or the dysfunctional organelle. Important roles for autophagy include general cytosol turnover, but also the protection of cells from accumulation of damaged organelles. Our experiments clearly showed an up-‐regulation of autophagy upon hIAPP and hproIAPP expression and that extensive autophagy activation hampers its normal protective effects. Insulin resistance leading to elevated insulin demand and simultaneous increase in IAPP production precedes type 2 diabetes. This intra-‐organelle concentration-‐increase could cause intracellular aggregation, organelle damage, and provoke autophagic breakdown. In short, this can protect cells from intracellular protein deposition, however if this state becomes chronic the autophagy machinery can get out of balance and not be sufficient to maintain cellular homeostasis, and eventually lead to cell death. Thus initiating a chain reaction where IAPP aggregates from dying beta cells are released to the extracellular space. There, preformed aggregates can act as seed for IAPP secreted from surrounding beta cells, which will lead to growth of islet amyloid deposits, thereby causing beta cell death and result in an incapacity to produce sufficient amounts of insulin. In this way hIAPP aggregation would play a major role in causing type 2 diabetes. Such a model leads to questions about the current treatment of type 2 diabetes by administrating drugs, e.g. sulfonylurea that

General Discussion and Future Perspectives
72
increase insulin production in order to meet elevated insulin demand. Instead, insulin treatment at an early stage of the disease together with arrangements to decrease insulin resistance, e.g. weight reduction, and increased amounts of exercise, should be considered as better option to reduce beta cell stress and to postpone a complete dependence for patients on external insulin administration. If extensive expression of hIAPP and hproIAPP is cytotoxic, why did only hproIAPP but not hIAPP shorten the survival of Drosophila when expressed in the whole brain? I think the answer can be found in the nature of the chosen Gal4 driver responsible for ectopic pan-‐neuronal expression. Gal4 production decreases rapidly with age and this decrease of hIAPP and hproIAPP production may be sufficient to avoid a collapse of the autophagy machinery. In this way neither hproIAPP nor hIAPP will exert intracellular toxicity. The observed shortening of lifespan is probably due to extracellular events. Such events could include hproIAPP to be secreted in a form that allowed further aggregation on surrounding cell membranes and thereby causing cell death. It also is possible that hproIAPP interacts with surrounding neurons differently than hIAPP, thus allowing for longer retention of hproIAPP in the brain before being cleared to the fat body. In the past, attempts have been made to identify general toxicity mechanisms for all amyloid proteins. Comparing the outcomes of Aβ expression with hIAPP and hproIAPP production, we were able to demonstrate that Aβ toxicity is not initiated intracellular. Cell death was triggered before the protein was secreted in cells producing hIAPP or hproIAPP. It will be interesting to see if Aβ still activates autophagy in the same way as hIAPP and hproIAPP do. This will give us clues about the relation of autophagy initiation and cell death. Since Aβ, as well as hIAPP and hproIAPP, can form amyloid, there clearly have to be additional factors that influence toxicity. Such factors might include the property to interact with membranes or the speed of aggregation. Both these factors can affect the site of initial fibril formation and fibril growth on membranes. It is conceivable that Aβ does not interact with membranes of organelles of the secretory pathway but with the extracellular part of the cell membrane. In this case could toxicity be initiated by the extracellular presence of Aβ. Future research will further dissect the molecular pathways affected by different amyloid proteins and clarify if amyloid deposits are a cause or consequence to its associated disease. This remains the crucial question to answer in the light of disease treatment.

Acknowledgements

Acknowledgements
74
I want to express my sincere gratitude to everybody who contributed to this thesis in one way or another. In particular I want to thank to: Gunilla Westermark, my supervisor. Firstly, thank you for introducing me to the field and giving me the opportunity to join your team. During my time in your lab I got the chance to learn a lot and I am grateful for all the help you provided to me. Your hard-‐working attitude and drive to fight a little bit harder combined with great generosity was a crucial support for my thesis. Mattias Alenius, after one evening of discussing research you soon became not only a mentor for me, but also a dear friend. I am glad that you were always willing to share you immense knowledge of research in general, and Drosophila in particular, with me. Many ideas in this thesis are a result of the discussions with you over the last years. Tor Erik Rusten, for your help in discussing results and designing good experiments to unravel some of the mysteries of autophagy. I really hope this thesis doesn’t put an end to our co-‐operation. Peter Nilsson, for our your collaboration and sharing your knowledge on LCPs. Stefan Klintström, for your unlimited commitment to Forum Scientium. It was a privilege to be part of Forum Scientium and to meet great PhD students from completely different fields. Per Westermark, you had potentially the biggest influence on the title of my thesis, as you were the person who named IAPP. It was always a great pleasure to meet you and to take part of your vast experience on amyloid research. Stefan Thor and his research group, for helping us getting started with our own fly research and later helping us to find solutions to get fly flood. Xiahong Gu, for joining our group and showing so much interest in Drosophila. I really enjoyed working together and wish you all the best with the continuation of this project! I can’t wait to read your thesis one day! Johan Paulsson, for taking me under your wing when I started in the group. I often think back to our work-‐intensive but great days and two fantastic conferences with you as a roommate. From you I learned that 4am might not be the best time to do Victor-‐measurements, but also to make sure not to burn my forehead skin…. Marie Oskarsson, for being such a wonderful co-‐worker and always being able to make me smile. You left a huge vacuum behind you when you moved to Uppsala. I wish you all the best for your future!

Acknowledgements
75
Sofia Nyström, for great discussions. Maybe not all of our discussions were worth the time and energy we put in to them, but I wouldn’t want to miss any one of them. I miss being able to turn around and get good advice from you. Jana Sponarova, for a great time together in our group. Even though I still prefer German to Czech beer, I am glad for all your effort to teach us more about the Czech Republic. Prague still is very high on my list and hope for your help the day I finally make it there… Marie-‐Louise Eskilsson, for your immense patience when sectioning fly heads. It is easy to say what you want to have, but sometimes extremely difficult to make it happen. You always succeeded… Aida, for being so extremely kind and always willing to help. All the small things you helped with when I was not in the lab made a huge difference for this work! Mildred, for contributing to the research presented in this thesis. It was fun working with you. Bengt-‐Arne Fredriksson, for your help with the microscope. I also want to thank all the other PhD students/postdocs that were not part of our group but still made a difference for me: Tobias, it was a great privilege for me to sit next to a person I had so much in common with – starting from being the only PhD student in your group left in Linköping, to having realized the huge advantages of Mac over Windows J. I think after so many years in the same office, water-‐skiing at your summerhouse and many parties we maybe should go for a camping trip… Åsa, Siri, and Cissi, unfortunately not all three of you were around during the last year. But I will miss being able to come to your office and discuss science or any of the other important issues that matter in life. A coffee break without you doesn’t feel complete. Without you Åsa, there would be so much good music I wouldn´t have had a clue exists. Cissi, you helped me to understand that Germany does not export as much music as Sweden does, and Siri, I can’t tell you how much I appreciate your sense of humour – even though it was not always an ego boost J. Ia, I always was fascinated by the energy you radiate. Gosia, for all our conversations on Drosophila and life in Sweden. You taught me a lot and I keep my fingers crossed that everything works out the way you wish for! Björn, for great training company. Go sub22! We both know that you can do it! Anita for sharing your wisdom with me. Good luck in Freiburg!

Acknowledgements
76
Peter Strålfors, Sven Hammarström, Mats Söderström, and all other members from level 12 (former and actual) for creating a nice working atmosphere. Anders, Calle, Jocke, Johan J, Johan O, Jonas, Lisa, Patiyan, Ryan, Ullis, I probably could fill a separate book listening all the great adventures, evenings, parties, conferences, and experiments (I never looked at 15ml the same way again) over the last years. And I think such a book couldn’t come close to describe how grateful I am for having been a PhD student the same time you were in Linköping. Maria, Pelle, Magdalena, Jason, Jenny, and Mattias, thank you so much for your friendship. I really hope we manage to increase the number of evenings discussing the big questions of life! Pelle and Maria, it is great to get those phone calls when you try to get me out climbing or invite me over for dinner. I am looking forward to be able to spend more time with you again. Magda and Jason, for all those great vacations, from ski mountaineering in Canada to paddling kayak in the Swedish archipelago. Even though there are thousands of kilometres between us now, so are you always close to me! Jenny and Mattias, for just being there – no matter when! Pernilla, for all the time we shared over the last years. I am deeply grateful for all your support and wish you all the best for your future! Christine, Ulrike, and Katharina, for being the best sisters a younger brother can ask for (at least after you all had left the house J). Katharina, you made sure to fill in for our parents when you thought life was getting a bit to easy for me J. But you also always took, and still take, time to listen to my problems. Ulrike, you always were, and still are, my role-‐model of how to interact with other people and you are an amazing mentor. Christine, you had a huge impact on me, and still have. To know that all of you always are there for me and support me whenever it is needed is an invaluable secureness that often allowed me to dare just a bit more. Meinen Eltern möchte ganz herzlich für all die Unterstützung danken. Ohne Euch und all die Werte die Ihr mir versucht habt zu übermitteln, hätte ich all das nie gewagt und erst recht nicht geschafft! Hanna, for all your support over the last months. All the laughs, adventures, and experiences we shared during this time gave me all the energy needed to finish my work. And now I can’t wait to discover the world together with you – and who knows, one day, after a great climb we might stand on top of “Presten”…

References

References
78
1. Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181: 223-230.
2. Levinthal C (1969) How to fold graciously. Mossbauer Spectroscopy in Biological Systems, Proceedings of a Meeting held at Allerton House, Monticello, Illinois: 22.
3. Karplus M (1997) The Levinthal paradox: yesterday and today. Folding & design 2: S69-75.
4. Dobson CM (2003) Protein folding and misfolding. Nature 426: 884-890. 5. Ellis RJ, Minton AP (2003) Cell biology: join the crowd. Nature 425: 27-28. 6. Gething MJ, Sambrook J (1992) Protein folding in the cell. Nature 355: 33-
45. 7. Richter K, Haslbeck M, Buchner J (2010) The heat shock response: life on
the verge of death. Molecular cell 40: 253-266. 8. Virchow R (1854) Zur Cellulose-Frage. Virchows Archiv A, Pathological
Anatomy and Histopathology 6: 416-426. 9. Aterman K (1976) A historical note on the iodine-sulphuric acid reaction of
amyloid. Histochemistry 49: 131-143. 10. Friedreich N, Kekulé A (1859) Zur Amyloidfrage. Virchows Archiv A,
Pathological Anatomy and Histopathology 16. 11. Bennhold H (1922) Eine spezifische Amyloidfärbung mit Kongorot. Münch
Med Wochenschr 69: 1537-1538. 12. Divry P, Florkin M (1927) Sur les propriétées optique de l'amyloide. CR Soc
Biol (Paris) 97: 1808-1810. 13. Puchtler H, Sweat F, Levine M (1962) On the binding of Congo red by
amyloid. J Histochem Cytochem 10: 355-364. 14. Westermark P (2005) Part I: Overview of Amyloidosis and Amyloid Proteins.
Amyloid proteins The beta sheet confirmation and disease, ed J D Sipe. 15. Cohen AS, Calkins E (1959) Electron microscopic observations on a fibrous
component in amyloid of diverse origins. Nature 183: 1202-1203. 16. Shirahama T, Cohen AS (1976) A brief review of the ultrastructure of
amyloid. Amyloidosis, eds J Marrink and M H van Rijswijk: 51-57. 17. Eanes ED, Glenner GG (1968) X-ray diffraction studies on amyloid
filaments. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 16: 673-677.
18. Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, et al. (2007) A primer of amyloid nomenclature. Amyloid 14: 179-183.
19. Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, et al. (2010) Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis 17: 101-104.
20. Kelly JW (1997) Amyloid fibril formation and protein misassembly: a structural quest for insights into amyloid and prion diseases. Structure 5: 595-600.
21. Lansbury PT, Jr. (1992) In pursuit of the molecular structure of amyloid plaque: new technology provides unexpected and critical information. Biochemistry 31: 6865-6870.
22. Sunde M, Blake C (1997) The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Advances in protein chemistry 50: 123-159.
23. Serpell LC, Sunde M, Benson MD, Tennent GA, Pepys MB, et al. (2000) The protofilament substructure of amyloid fibrils. Journal of molecular biology 300: 1033-1039.
24. Makin OS, Serpell LC (2005) Structures for amyloid fibrils. The FEBS journal 272: 5950-5961.

References
79
25. Fandrich M, Meinhardt J, Grigorieff N (2009) Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion 3: 89-93.
26. Greenwald J, Riek R (2010) Biology of amyloid: structure, function, and regulation. Structure 18: 1244-1260.
27. Wang L, Schubert D, Sawaya MR, Eisenberg D, Riek R (2010) Multidimensional structure-activity relationship of a protein in its aggregated states. Angewandte Chemie 49: 3904-3908.
28. Leveugle B, Ding W, Durkin JT, Mistretta S, Eisle J, et al. (1997) Heparin promotes beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Neurochemistry international 30: 543-548.
29. Scholefield Z, Yates EA, Wayne G, Amour A, McDowell W, et al. (2003) Heparan sulfate regulates amyloid precursor protein processing by BACE1, the Alzheimer's beta-secretase. The Journal of cell biology 163: 97-107.
30. Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, et al. (2005) In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proceedings of the National Academy of Sciences of the United States of America 102: 6473-6477.
31. Zhang X, Li JP (2010) Heparan sulfate proteoglycans in amyloidosis. Progress in molecular biology and translational science 93: 309-334.
32. Kisilevsky R, Szarek WA (2002) Novel glycosaminoglycan precursors as anti-amyloid agents part II. Journal of molecular neuroscience : MN 19: 45-50.
33. Hirschfield GM, Hawkins PN (2003) Amyloidosis: new strategies for treatment. The international journal of biochemistry & cell biology 35: 1608-1613.
34. Ariga T, Miyatake T, Yu RK (2010) Role of proteoglycans and glycosaminoglycans in the pathogenesis of Alzheimer's disease and related disorders: amyloidogenesis and therapeutic strategies--a review. Journal of neuroscience research 88: 2303-2315.
35. Pepys MB, Dyck RF, de Beer FC, Skinner M, Cohen AS (1979) Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clinical and experimental immunology 38: 284-293.
36. Tennent GA, Lovat LB, Pepys MB (1995) Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proceedings of the National Academy of Sciences of the United States of America 92: 4299-4303.
37. Hawkins PN (2002) Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis. Current opinion in nephrology and hypertension 11: 649-655.
38. Charge SB, Esiri MM, Bethune CA, Hansen BC, Clark A (1996) Apolipoprotein E is associated with islet amyloid and other amyloidoses: implications for Alzheimer's disease. The Journal of pathology 179: 443-447.
39. Verghese PB, Castellano JM, Holtzman DM (2011) Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet neurology 10: 241-252.
40. Stefani M, Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. Journal of molecular medicine 81: 678-699.
41. Chiti F, Dobson CM (2009) Amyloid formation by globular proteins under native conditions. Nature chemical biology 5: 15-22.
42. Jarrett JT, Lansbury PT, Jr. (1992) Amyloid fibril formation requires a chemically discriminating nucleation event: studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry 31: 12345-12352.
43. Jarrett JT, Lansbury PT, Jr. (1993) Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73: 1055-1058.

References
80
44. Kelly JW (1998) The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Current opinion in structural biology 8: 101-106.
45. Rochet JC, Lansbury PT, Jr. (2000) Amyloid fibrillogenesis: themes and variations. Current opinion in structural biology 10: 60-68.
46. Lundmark K, Westermark GT, Olsen A, Westermark P (2005) Protein fibrils
in nature can enhance amyloid protein A amyloidosis in mice: Cross-seeding as a disease mechanism. Proceedings of the National Academy of Sciences of the United States of America 102: 6098-6102.
47. Ranlov P (1967) The adoptive transfer of experimental mouse amyloidosis by intravenous injections of spleen cell extracts from casein-treated syngeneic donor mice. Acta pathologica et microbiologica Scandinavica 70: 321-335.
48. Ridley RM, Baker HF, Windle CP, Cummings RM (2006) Very long term studies of the seeding of beta-amyloidosis in primates. Journal of neural transmission 113: 1243-1251.
49. Westermark GT, Westermark P (2010) Prion-like aggregates: infectious agents in human disease. Trends in molecular medicine 16: 501-507.
50. Stefani M (2010) Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. The FEBS journal 277: 4602-4613.
51. Glabe CG (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiology of aging 27: 570-575.
52. Glabe CG, Kayed R (2006) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66: S74-78.
53. Quist A, Doudevski I, Lin H, Azimova R, Ng D, et al. (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proceedings of the National Academy of Sciences of the United States of America 102: 10427-10432.
54. Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, et al. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486-489.
55. Mirzabekov TA, Lin MC, Kagan BL (1996) Pore formation by the cytotoxic islet amyloid peptide amylin. The Journal of biological chemistry 271: 1988-1992.
56. Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A (2006) Opposing activities protect against age-onset proteotoxicity. Science 313: 1604-1610.
57. Zhang S, Liu H, Yu H, Cooper GJ (2008) Fas-associated death receptor signaling evoked by human amylin in islet beta-cells. Diabetes 57: 348-356.
58. Zhang S, Liu J, Dragunow M, Cooper GJ (2003) Fibrillogenic amylin evokes islet beta-cell apoptosis through linked activation of a caspase cascade and JNK1. The Journal of biological chemistry 278: 52810-52819.
59. Tomiyama T, Kaneko H, Kataoka K, Asano S, Endo N (1997) Rifampicin inhibits the toxicity of pre-aggregated amyloid peptides by binding to peptide fibrils and preventing amyloid-cell interaction. The Biochemical journal 322 ( Pt 3): 859-865.
60. Bhattacharya S, Latha JN, Kumresan R, Singh S (2007) Cloning and expression of human islet amyloid polypeptide in cultured cells. Biochemical and biophysical research communications 356: 622-628.
61. Jurgens CA, Toukatly MN, Fligner CL, Udayasankar J, Subramanian SL, et al. (2011) beta-cell loss and beta-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. The American journal of pathology 178: 2632-2640.
62. Zraika S, Hull RL, Verchere CB, Clark A, Potter KJ, et al. (2010) Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence? Diabetologia 53: 1046-1056.

References
81
63. Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, et al. (2008) Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci U S A 105: 6033-6038.
64. Knight JD, Miranker AD (2004) Phospholipid catalysis of diabetic amyloid assembly. Journal of molecular biology 341: 1175-1187.
65. Aisenbrey C, Borowik T, Bystrom R, Bokvist M, Lindstrom F, et al. (2008) How is protein aggregation in amyloidogenic diseases modulated by biological membranes? European biophysics journal : EBJ 37: 247-255.
66. Engel MF, Visser AJ, van Mierlo CP (2004) Conformation and orientation of a protein folding intermediate trapped by adsorption. Proceedings of the National Academy of Sciences of the United States of America 101: 11316-11321.
67. Norde W, Lyklema J (1991) Why proteins prefer interfaces. Journal of biomaterials science Polymer edition 2: 183-202.
68. Chernoff YO (2004) Amyloidogenic domains, prions and structural inheritance: rudiments of early life or recent acquisition? Current opinion in chemical biology 8: 665-671.
69. Fowler DM, Koulov AV, Balch WE, Kelly JW (2007) Functional amyloid--from bacteria to humans. Trends in biochemical sciences 32: 217-224.
70. Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, et al. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295: 851-855.
71. Claessen D, Rink R, de Jong W, Siebring J, de Vreugd P, et al. (2003) A novel class of secreted hydrophobic proteins is involved in aerial hyphae formation in Streptomyces coelicolor by forming amyloid-like fibrils. Genes & development 17: 1714-1726.
72. True HL, Lindquist SL (2000) A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407: 477-483.
73. Baxa U, Cheng N, Winkler DC, Chiu TK, Davies DR, et al. (2005) Filaments of the Ure2p prion protein have a cross-beta core structure. Journal of structural biology 150: 170-179.
74. Iconomidou VA, Vriend G, Hamodrakas SJ (2000) Amyloids protect the silkmoth oocyte and embryo. FEBS letters 479: 141-145.
75. Si K, Lindquist S, Kandel ER (2003) A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115: 879-891.
76. Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, et al. (2006) Functional amyloid formation within mammalian tissue. PLoS biology 4: e6.
77. Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, et al. (2009) Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325: 328-332.
78. Clark A, Cooper GJ, Lewis CE, Morris JF, Willis AC, et al. (1987) Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet 2: 231-234.
79. Opie EL (1901) The Relation Of Diabetes Mellitus to Lesions of the Pancreas. Hyaline Degeneration of the Islands Oe Langerhans. The Journal of experimental medicine 5: 527-540.
80. Westermark P, Grimelius L (1973) The pancreatic islet cells in insular amyloidosis in human diabetic and non-diabetic adults. Acta pathologica et microbiologica Scandinavica Section A, Pathology 81: 291-300.
81. Westermark P, Wernstedt C, Wilander E, Hayden DW, O'Brien TD, et al. (1987) Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A 84: 3881-3885.

References
82
82. Westermark P, Wernstedt C, Wilander E, Sletten K (1986) A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochemical and biophysical research communications 140: 827-831.
83. Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, et al. (1987) Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proceedings of the National Academy of Sciences of the United States of America 84: 8628-8632.
84. Christmanson L, Rorsman F, Stenman G, Westermark P, Betsholtz C (1990) The human islet amyloid polypeptide (IAPP) gene. Organization, chromosomal localization and functional identification of a promoter region. FEBS letters 267: 160-166.
85. Macfarlane WM, Campbell SC, Elrick LJ, Oates V, Bermano G, et al. (2000) Glucose regulates islet amyloid polypeptide gene transcription in a PDX1- and calcium-dependent manner. The Journal of biological chemistry 275: 15330-15335.
86. Macfarlane WM, Shepherd RM, Cosgrove KE, James RF, Dunne MJ, et al. (2000) Glucose modulation of insulin mRNA levels is dependent on transcription factor PDX-1 and occurs independently of changes in intracellular Ca2+. Diabetes 49: 418-423.
87. Mosselman S, Hoppener JW, Zandberg J, van Mansfeld AD, Geurts van Kessel AH, et al. (1988) Islet amyloid polypeptide: identification and chromosomal localization of the human gene. FEBS letters 239: 227-232.
88. Muff R, Born W, Lutz TA, Fischer JA (2004) Biological importance of the peptides of the calcitonin family as revealed by disruption and transfer of corresponding genes. Peptides 25: 2027-2038.
89. Wimalawansa SJ (1997) Amylin, calcitonin gene-related peptide, calcitonin, and adrenomedullin: a peptide superfamily. Critical reviews in neurobiology 11: 167-239.
90. Betsholtz C, Svensson V, Rorsman F, Engstrom U, Westermark GT, et al. (1989) Islet amyloid polypeptide (IAPP):cDNA cloning and identification of an amyloidogenic region associated with the species-specific occurrence of age-related diabetes mellitus. Experimental cell research 183: 484-493.
91. Lukinius A, Wilander E, Westermark GT, Engstrom U, Westermark P (1989) Co-localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic islets. Diabetologia 32: 240-244.
92. Westermark P, Wilander E, Westermark GT, Johnson KH (1987) Islet amyloid polypeptide-like immunoreactivity in the islet B cells of type 2 (non-insulin-dependent) diabetic and non-diabetic individuals. Diabetologia 30: 887-892.
93. Leffert JD, Newgard CB, Okamoto H, Milburn JL, Luskey KL (1989) Rat amylin: cloning and tissue-specific expression in pancreatic islets. Proceedings of the National Academy of Sciences of the United States of America 86: 3127-3130.
94. Nishi M, Sanke T, Nagamatsu S, Bell GI, Steiner DF (1990) Islet amyloid polypeptide. A new beta cell secretory product related to islet amyloid deposits. The Journal of biological chemistry 265: 4173-4176.
95. Hutton JC (1984) Secretory granules. Experientia 40: 1091-1098. 96. Percy AJ, Trainor DA, Rittenhouse J, Phelps J, Koda JE (1996) Development
of sensitive immunoassays to detect amylin and amylin-like peptides in unextracted plasma. Clinical chemistry 42: 576-585.
97. Betsholtz C, Christmanson L, Engstrom U, Rorsman F, Jordan K, et al. (1990) Structure of cat islet amyloid polypeptide and identification of amino acid residues of potential significance for islet amyloid formation. Diabetes 39: 118-122.

References
83
98. Fan L, Westermark G, Chan SJ, Steiner DF (1994) Altered gene structure and tissue expression of islet amyloid polypeptide in the chicken. Molecular endocrinology 8: 713-721.
99. Jordan K, Murtaugh MP, O'Brien TD, Westermark P, Betsholtz C, et al. (1990) Canine IAPP cDNA sequence provides important clues regarding diabetogenesis and amyloidogenesis in type 2 diabetes. Biochemical and biophysical research communications 169: 502-508.
100. Nishi M, Bell GI, Steiner DF (1990) Sequence of a cDNA encoding Syrian hamster islet amyloid polypeptide precursor. Nucleic acids research 18: 6726.
101. Nishi M, Chan SJ, Nagamatsu S, Bell GI, Steiner DF (1989) Conservation of the sequence of islet amyloid polypeptide in five mammals is consistent with its putative role as an islet hormone. Proceedings of the National Academy of Sciences of the United States of America 86: 5738-5742.
102. Westermark GT, Falkmer S, Steiner DF, Chan SJ, Engstrom U, et al. (2002) Islet amyloid polypeptide is expressed in the pancreatic islet parenchyma of the teleostean fish, Myoxocephalus (cottus) scorpius. Comparative biochemistry and physiology Part B, Biochemistry & molecular biology 133: 119-125.
103. Mulder H, Leckstrom A, Uddman R, Ekblad E, Westermark P, et al. (1995) Islet amyloid polypeptide (amylin) is expressed in sensory neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 15: 7625-7632.
104. Skofitsch G, Wimalawansa SJ, Jacobowitz DM, Gubisch W (1995) Comparative immunohistochemical distribution of amylin-like and calcitonin gene related peptide like immunoreactivity in the rat central nervous system. Canadian journal of physiology and pharmacology 73: 945-956.
105. Mulder H, Ekelund M, Ekblad E, Sundler F (1997) Islet amyloid polypeptide in the gut and pancreas: localization, ontogeny and gut motility effects. Peptides 18: 771-783.
106. Gebre-Medhin S, Mulder H, Pekny M, Westermark G, Tornell J, et al. (1998) Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochemical and biophysical research communications 250: 271-277.
107. Akesson B, Panagiotidis G, Westermark P, Lundquist I (2003) Islet amyloid polypeptide inhibits glucagon release and exerts a dual action on insulin release from isolated islets. Regulatory peptides 111: 55-60.
108. Wang F, Adrian TE, Westermark GT, Ding X, Gasslander T, et al. (1999) Islet amyloid polypeptide tonally inhibits beta-, alpha-, and delta-cell secretion in isolated rat pancreatic islets. The American journal of physiology 276: E19-24.
109. Christopoulos G, Perry KJ, Morfis M, Tilakaratne N, Gao Y, et al. (1999) Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Molecular pharmacology 56: 235-242.
110. McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, et al. (1998) RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393: 333-339.
111. Hay DL, Christopoulos G, Christopoulos A, Sexton PM (2004) Amylin receptors: molecular composition and pharmacology. Biochemical Society transactions 32: 865-867.
112. Muff R, Buhlmann N, Fischer JA, Born W (1999) An amylin receptor is revealed following co-transfection of a calcitonin receptor with receptor activity modifying proteins-1 or -3. Endocrinology 140: 2924-2927.

References
84
113. Tilakaratne N, Christopoulos G, Zumpe ET, Foord SM, Sexton PM (2000) Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. The Journal of pharmacology and experimental therapeutics 294: 61-72.
114. Banks WA, Kastin AJ (1998) Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19: 883-889.
115. Dunican KC, Adams NM, Desilets AR (2010) The role of pramlintide for weight loss. The Annals of pharmacotherapy 44: 538-545.
116. Riddle MC, Drucker DJ (2006) Emerging therapies mimicking the effects of amylin and glucagon-like peptide 1. Diabetes care 29: 435-449.
117. (2003) Pramlintide: (AC 137, AC 0137, Symlin, Tripro-Amylin). BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy 17: 73-79.
118. Dobolyi A (2009) Central amylin expression and its induction in rat dams. Journal of neurochemistry 111: 1490-1500.
119. Huang X, Yang J, Chang JK, Dun NJ (2010) Amylin suppresses acetic acid-induced visceral pain and spinal c-fos expression in the mouse. Neuroscience 165: 1429-1438.
120. Gebre-Medhin S, Mulder H, Zhang Y, Sundler F, Betsholtz C (1998) Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Brain research Molecular brain research 63: 180-183.
121. Deems RO, Deacon RW, Young DA (1991) Amylin activates glycogen phosphorylase and inactivates glycogen synthase via a cAMP-independent mechanism. Biochemical and biophysical research communications 174: 716-720.
122. Young AA, Mott DM, Stone K, Cooper GJ (1991) Amylin activates glycogen phosphorylase in the isolated soleus muscle of the rat. FEBS letters 281: 149-151.
123. Abaffy T, Cooper GJ (2004) GSK3 involvement in amylin signaling in isolated rat soleus muscle. Peptides 25: 2119-2125.
124. Gilbey SG, Ghatei MA, Bretherton-Watt D, Zaidi M, Jones PM, et al. (1991) Islet amyloid polypeptide: production by an osteoblast cell line and possible role as a paracrine regulator of osteoclast function in man. Clinical science 81: 803-808.
125. Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, et al. (2004) Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. The Journal of cell biology 164: 509-514.
126. Sanke T, Bell GI, Sample C, Rubenstein AH, Steiner DF (1988) An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J Biol Chem 263: 17243-17246.
127. Halban PA, Irminger JC (1994) Sorting and processing of secretory proteins. The Biochemical journal 299 ( Pt 1): 1-18.
128. Marzban L, Trigo-Gonzalez G, Zhu X, Rhodes CJ, Halban PA, et al. (2004) Role of beta-cell prohormone convertase (PC)1/3 in processing of pro-islet amyloid polypeptide. Diabetes 53: 141-148.
129. Wang J, Xu J, Finnerty J, Furuta M, Steiner DF, et al. (2001) The prohormone convertase enzyme 2 (PC2) is essential for processing pro-islet amyloid polypeptide at the NH2-terminal cleavage site. Diabetes 50: 534-539.
130. Marzban L, Trigo-Gonzalez G, Verchere CB (2005) Processing of pro-islet amyloid polypeptide in the constitutive and regulated secretory pathways of beta cells. Molecular endocrinology 19: 2154-2163.
131. Marzban L, Soukhatcheva G, Verchere CB (2005) Role of carboxypeptidase E in processing of pro-islet amyloid polypeptide in {beta}-cells. Endocrinology 146: 1808-1817.

References
85
132. Jutras I, Seidah NG, Reudelhuber TL, Brechler V (1997) Two activation states of the prohormone convertase PC1 in the secretory pathway. The Journal of biological chemistry 272: 15184-15188.
133. Zhou A, Mains RE (1994) Endoproteolytic processing of proopiomelanocortin and prohormone convertases 1 and 2 in neuroendocrine cells overexpressing prohormone convertases 1 or 2. The Journal of biological chemistry 269: 17440-17447.
134. Shennan KI, Taylor NA, Jermany JL, Matthews G, Docherty K (1995) Differences in pH optima and calcium requirements for maturation of the prohormone convertases PC2 and PC3 indicates different intracellular locations for these events. The Journal of biological chemistry 270: 1402-1407.
135. Muller L, Zhu X, Lindberg I (1997) Mechanism of the facilitation of PC2 maturation by 7B2: involvement in ProPC2 transport and activation but not folding. The Journal of cell biology 139: 625-638.
136. Zhu X, Lindberg I (1995) 7B2 facilitates the maturation of proPC2 in neuroendocrine cells and is required for the expression of enzymatic activity. The Journal of cell biology 129: 1641-1650.
137. Muller L, Cameron A, Fortenberry Y, Apletalina EV, Lindberg I (2000) Processing and sorting of the prohormone convertase 2 propeptide. The Journal of biological chemistry 275: 39213-39222.
138. Balasubramaniam A, Renugopalakrishnan V, Stein M, Fischer JE, Chance WT (1991) Syntheses, structures and anorectic effects of human and rat amylin. Peptides 12: 919-924.
139. Abedini A, Tracz SM, Cho JH, Raleigh DP (2006) Characterization of the heparin binding site in the N-terminus of human pro-islet amyloid polypeptide: implications for amyloid formation. Biochemistry 45: 9228-9237.
140. Meng F, Abedini A, Song B, Raleigh DP (2007) Amyloid formation by pro-islet amyloid polypeptide processing intermediates: examination of the role of protein heparan sulfate interactions and implications for islet amyloid formation in type 2 diabetes. Biochemistry 46: 12091-12099.
141. Park K, Verchere CB (2001) Identification of a heparin binding domain in the N-terminal cleavage site of pro-islet amyloid polypeptide. Implications for islet amyloid formation. The Journal of biological chemistry 276: 16611-16616.
142. Paulsson JF, Andersson A, Westermark P, Westermark GT (2006) Intracellular amyloid-like deposits contain unprocessed pro-islet amyloid polypeptide (proIAPP) in beta cells of transgenic mice overexpressing the gene for human IAPP and transplanted human islets. Diabetologia 49: 1237-1246.
143. Paulsson JF, Westermark GT (2005) Aberrant processing of human proislet amyloid polypeptide results in increased amyloid formation. Diabetes 54: 2117-2125.
144. Smeekens SP, Montag AG, Thomas G, Albiges-Rizo C, Carroll R, et al. (1992) Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proceedings of the National Academy of Sciences of the United States of America 89: 8822-8826.
145. Zhu X, Orci L, Carroll R, Norrbom C, Ravazzola M, et al. (2002) Severe block in processing of proinsulin to insulin accompanied by elevation of des-64,65 proinsulin intermediates in islets of mice lacking prohormone convertase 1/3. Proceedings of the National Academy of Sciences of the United States of America 99: 10299-10304.
146. Furuta M, Carroll R, Martin S, Swift HH, Ravazzola M, et al. (1998) Incomplete processing of proinsulin to insulin accompanied by elevation of Des-31,32 proinsulin intermediates in islets of mice lacking active PC2. The Journal of biological chemistry 273: 3431-3437.

References
86
147. Docherty K, Hutton JC (1983) Carboxypeptidase activity in the insulin secretory granule. FEBS letters 162: 137-141.
148. Leslie RD, Kolb H, Schloot NC, Buzzetti R, Mauricio D, et al. (2008) Diabetes classification: grey zones, sound and smoke: Action LADA 1. Diabetes/metabolism research and reviews 24: 511-519.
149. Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, et al. (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54 Suppl 2: S97-107.
150. Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, et al. (1988) Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes research 9: 151-159.
151. Westermark P (1972) Quantitative studies on amyloid in the islets of Langerhans. Ups J Med Sci 77: 91-94.
152. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, et al. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102-110.
153. Clark A, Charge SB, Badman MK, MacArthur DA, de Koning EJ (1996) Islet amyloid polypeptide: actions and role in the pathogenesis of diabetes. Biochem Soc Trans 24: 594-599.
154. Hoppener JW, Lips CJ (2006) Role of islet amyloid in type 2 diabetes mellitus. The international journal of biochemistry & cell biology 38: 726-736.
155. Stumvoll M, Goldstein BJ, van Haeften TW (2005) Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365: 1333-1346.
156. Kahn BB (1998) Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell 92: 593-596.
157. Larsson H, Ahren B (1996) Failure to adequately adapt reduced insulin sensitivity with increased insulin secretion in women with impaired glucose tolerance. Diabetologia 39: 1099-1107.
158. Rhodes CJ (2005) Type 2 diabetes-a matter of beta-cell life and death? Science 307: 380-384.
159. Westermark P (2011) Amyloid in the islets of Langerhans: Thoughts and some historical aspects. Ups J Med Sci 116: 81-89.
160. Guardado-Mendoza R, Davalli AM, Chavez AO, Hubbard GB, Dick EJ, et al. (2009) Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proceedings of the National Academy of Sciences of the United States of America 106: 13992-13997.
161. Westermark P, Andersson A, Westermark GT (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiological reviews 91: 795-826.
162. Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C (1990) Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proceedings of the National Academy of Sciences of the United States of America 87: 5036-5040.
163. Hoppener JW, Oosterwijk C, Nieuwenhuis MG, Posthuma G, Thijssen JH, et al. (1999) Extensive islet amyloid formation is induced by development of Type II diabetes mellitus and contributes to its progression: pathogenesis of diabetes in a mouse model. Diabetologia 42: 427-434.
164. Soeller WC, Janson J, Hart SE, Parker JC, Carty MD, et al. (1998) Islet amyloid-associated diabetes in obese A(vy)/a mice expressing human islet amyloid polypeptide. Diabetes 47: 743-750.
165. Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC (1999) The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 48: 491-498.

References
87
166. Lorenzo A, Razzaboni B, Weir GC, Yankner BA (1994) Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 368: 756-760.
167. Engel MF (2009) Membrane permeabilization by Islet Amyloid Polypeptide. Chem Phys Lipids 160: 1-10.
168. Porte D, Jr. (1991) Banting lecture 1990. Beta-cells in type II diabetes mellitus. Diabetes 40: 166-180.
169. Sakagashira S, Sanke T, Hanabusa T, Shimomura H, Ohagi S, et al. (1996) Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 45: 1279-1281.
170. Seino S (2001) S20G mutation of the amylin gene is associated with Type II diabetes in Japanese. Study Group of Comprehensive Analysis of Genetic Factors in Diabetes Mellitus. Diabetologia 44: 906-909.
171. Ma Z, Westermark GT, Sakagashira S, Sanke T, Gustavsson A, et al. (2001) Enhanced in vitro production of amyloid-like fibrils from mutant (S20G) islet amyloid polypeptide. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis 8: 242-249.
172. Sakagashira S, Hiddinga HJ, Tateishi K, Sanke T, Hanabusa T, et al. (2000) S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. The American journal of pathology 157: 2101-2109.
173. Nanga RP, Brender JR, Xu J, Hartman K, Subramanian V, et al. (2009) Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. Journal of the American Chemical Society 131: 8252-8261.
174. Wei L, Jiang P, Yau YH, Summer H, Shochat SG, et al. (2009) Residual structure in islet amyloid polypeptide mediates its interactions with soluble insulin. Biochemistry 48: 2368-2376.
175. Reddy AS, Wang L, Singh S, Ling YL, Buchanan L, et al. (2010) Stable and metastable states of human amylin in solution. Biophysical journal 99: 2208-2216.
176. Dupuis NF, Wu C, Shea JE, Bowers MT (2009) Human islet amyloid polypeptide monomers form ordered beta-hairpins: a possible direct amyloidogenic precursor. Journal of the American Chemical Society 131: 18283-18292.
177. Luca S, Yau WM, Leapman R, Tycko R (2007) Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry 46: 13505-13522.
178. Westermark P, Li ZC, Westermark GT, Leckstrom A, Steiner DF (1996) Effects of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS letters 379: 203-206.
179. Jaikaran ET, Nilsson MR, Clark A (2004) Pancreatic beta-cell granule peptides form heteromolecular complexes which inhibit islet amyloid polypeptide fibril formation. The Biochemical journal 377: 709-716.
180. Janciauskiene S, Eriksson S, Carlemalm E, Ahren B (1997) B cell granule peptides affect human islet amyloid polypeptide (IAPP) fibril formation in vitro. Biochemical and biophysical research communications 236: 580-585.
181. Andersson A, Bohman S, Borg LA, Paulsson JF, Schultz SW, et al. (2008) Amyloid deposition in transplanted human pancreatic islets: a conceivable cause of their long-term failure. Experimental diabetes research 2008: 562985.
182. Brender JR, Lee EL, Hartman K, Wong PT, Ramamoorthy A, et al. (2011) Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophysical journal 100: 685-692.

References
88
183. Stevens FJ, Kisilevsky R (2000) Immunoglobulin light chains, glycosaminoglycans, and amyloid. Cellular and molecular life sciences : CMLS 57: 441-449.
184. Furth AJ (1997) Glycated proteins in diabetes. British journal of biomedical science 54: 192-200.
185. Clodi M, Thomaseth K, Pacini G, Hermann K, Kautzky-Willer A, et al. (1998) Distribution and kinetics of amylin in humans. The American journal of physiology 274: E903-908.
186. Kapurniotu A, Bernhagen J, Greenfield N, Al-Abed Y, Teichberg S, et al. (1998) Contribution of advanced glycosylation to the amyloidogenicity of islet amyloid polypeptide. European journal of biochemistry / FEBS 251: 208-216.
187. Reaven GM, Chen YD (1988) Role of abnormal free fatty acid metabolism in the development of non-insulin-dependent diabetes mellitus. The American journal of medicine 85: 106-112.
188. Wyne KL (2003) Free fatty acids and type 2 diabetes mellitus. The American journal of medicine 115 Suppl 8A: 29S-36S.
189. Westermark GT, Gebre-Medhin S, Steiner DF, Westermark P (2000) Islet amyloid development in a mouse strain lacking endogenous islet amyloid polypeptide (IAPP) but expressing human IAPP. Molecular medicine 6: 998-1007.
190. Ma Z, Westermark GT (2002) Effects of free fatty acid on polymerization of islet amyloid polypeptide (IAPP) in vitro and on amyloid fibril formation in cultivated isolated islets of transgenic mice overexpressing human IAPP. Molecular medicine 8: 863-868.
191. Qi D, Cai K, Wang O, Li Z, Chen J, et al. (2010) Fatty acids induce amylin expression and secretion by pancreatic beta-cells. American journal of physiology Endocrinology and metabolism 298: E99-E107.
192. de Koning EJ, Hoppener JW, Verbeek JS, Oosterwijk C, van Hulst KL, et al. (1994) Human islet amyloid polypeptide accumulates at similar sites in islets of transgenic mice and humans. Diabetes 43: 640-644.
193. Narita R, Toshimori H, Nakazato M, Kuribayashi T, Toshimori T, et al. (1992) Islet amyloid polypeptide (IAPP) and pancreatic islet amyloid deposition in diabetic and non-diabetic patients. Diabetes research and clinical practice 15: 3-14.
194. Couce M, Kane LA, O'Brien TD, Charlesworth J, Soeller W, et al. (1996) Treatment with growth hormone and dexamethasone in mice transgenic for human islet amyloid polypeptide causes islet amyloidosis and beta-cell dysfunction. Diabetes 45: 1094-1101.
195. D'Alessio DA, Verchere CB, Kahn SE, Hoagland V, Baskin DG, et al. (1994) Pancreatic expression and secretion of human islet amyloid polypeptide in a transgenic mouse. Diabetes 43: 1457-1461.
196. Fox N, Schrementi J, Nishi M, Ohagi S, Chan SJ, et al. (1993) Human islet amyloid polypeptide transgenic mice as a model of non-insulin-dependent diabetes mellitus (NIDDM). FEBS letters 323: 40-44.
197. Yagui K, Yamaguchi T, Kanatsuka A, Shimada F, Huang CI, et al. (1995) Formation of islet amyloid fibrils in beta-secretory granules of transgenic mice expressing human islet amyloid polypeptide/amylin. European journal of endocrinology / European Federation of Endocrine Societies 132: 487-496.
198. Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, et al. (1996) Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proceedings of the National Academy of Sciences of the United States of America 93: 7283-7288.

References
89
199. Verchere CB, D'Alessio DA, Palmiter RD, Weir GC, Bonner-Weir S, et al. (1996) Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proceedings of the National Academy of Sciences of the United States of America 93: 3492-3496.
200. Kahn SE, Andrikopoulos S, Verchere CB, Wang F, Hull RL, et al. (2000) Oophorectomy promotes islet amyloid formation in a transgenic mouse model of Type II diabetes. Diabetologia 43: 1309-1312.
201. Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, et al. (2004) Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes 53: 1509-1516.
202. Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer's disease. Human molecular genetics 19: R12-20.
203. Ittner LM, Gotz J (2011) Amyloid-beta and tau--a toxic pas de deux in Alzheimer's disease. Nature reviews Neuroscience 12: 65-72.
204. Association As (2010) 2010 Alzheimer's disease facts and figures. Alzheimer's & dementia : the journal of the Alzheimer's Association 6: 158-194.
205. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, et al. (2011) Alzheimer's disease. Lancet 377: 1019-1031.
206. Perrin RJ, Fagan AM, Holtzman DM (2009) Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 461: 916-922.
207. Spies PE, Slats D, Sjogren JM, Kremer BP, Verhey FR, et al. (2010) The cerebrospinal fluid amyloid beta42/40 ratio in the differentiation of Alzheimer's disease from non-Alzheimer's dementia. Current Alzheimer research 7: 470-476.
208. Shoji M, Kanai M, Matsubara E, Tomidokoro Y, Shizuka M, et al. (2001) The levels of cerebrospinal fluid Abeta40 and Abeta42(43) are regulated age-dependently. Neurobiology of aging 22: 209-215.
209. Walsh DM, Selkoe DJ (2004) Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein and peptide letters 11: 213-228.
210. Rabinovici GD, Jagust WJ (2009) Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behavioural neurology 21: 117-128.
211. Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, et al. (2001) The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nature neuroscience 4: 887-893.
212. Philipson O, Lord A, Gumucio A, O'Callaghan P, Lannfelt L, et al. (2010) Animal models of amyloid-beta-related pathologies in Alzheimer's disease. The FEBS journal 277: 1389-1409.
213. Goedert M, Spillantini MG (2006) A century of Alzheimer's disease. Science 314: 777-781.
214. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, et al. (1999) Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 53: 1937-1942.
215. Gotz J, Ittner LM, Lim YA (2009) Common features between diabetes mellitus and Alzheimer's disease. Cellular and molecular life sciences : CMLS 66: 1321-1325.
216. Janson J, Laedtke T, Parisi JE, O'Brien P, Petersen RC, et al. (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53: 474-481.
217. Bennett RG, Hamel FG, Duckworth WC (2003) An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amylin-induced cytotoxicity, and increases amyloid formation in insulinoma cell cultures. Diabetes 52: 2315-2320.

References
90
218. Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, et al. (2004) Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. The American journal of pathology 164: 1425-1434.
219. Qiu WQ, Folstein MF (2006) Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer's disease: review and hypothesis. Neurobiology of aging 27: 190-198.
220. Shen Y, Joachimiak A, Rosner MR, Tang WJ (2006) Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443: 870-874.
221. O'Nuallain B, Williams AD, Westermark P, Wetzel R (2004) Seeding specificity in amyloid growth induced by heterologous fibrils. The Journal of biological chemistry 279: 17490-17499.
222. Jhamandas JH, Li Z, Westaway D, Yang J, Jassar S, et al. (2011) Actions of beta-amyloid protein on human neurons are expressed through the amylin receptor. The American journal of pathology 178: 140-149.
223. Sturtevant AH (1913) The Linnear Arrangement of Six Sex-linked Factors in Drosophila, asshown by Their Mode of Association. Journal of Experimental Zoology: 43-59.
224. Rubin GM, Lewis EB (2000) A brief history of Drosophila's contributions to genome research. Science 287: 2216-2218.
225. Bellen HJ, Tong C, Tsuda H (2010) 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nature reviews Neuroscience 11: 514-522.
226. Muller HJ (1927) Artificial Transmutation of the Gene. Science 66: 84-87. 227. Lewis EB, Bacher F (1968) Methods of feeding ethyl methane sulfonate
(EMS) to Drosophila males. Drosophila Information Service 43: 193. 228. McGuire SE, Deshazer M, Davis RL (2005) Thirty years of olfactory learning
and memory research in Drosophila melanogaster. Progress in neurobiology 76: 328-347.
229. Yan N, Shi Y (2005) Mechanisms of apoptosis through structural biology. Annual review of cell and developmental biology 21: 35-56.
230. Reiter LT, Potocki L, Chien S, Gribskov M, Bier E (2001) A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome research 11: 1114-1125.
231. Rasheva VI, Domingos PM (2009) Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis : an international journal on programmed cell death 14: 996-1007.
232. McPhee CK, Baehrecke EH (2009) Autophagy in Drosophila melanogaster. Biochimica et biophysica acta 1793: 1452-1460.
233. Zirin J, Perrimon N (2010) Drosophila as a model system to study autophagy. Seminars in immunopathology 32: 363-372.
234. Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. (2000) The genome sequence of Drosophila melanogaster. Science 287: 2185-2195.
235. Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401-415.
236. Petersen NS (1990) Effects of heat and chemical stress on development. Advances in genetics 28: 275-296.
237. Petersen NS, Mitchell HK (1987) The induction of a multiple wing hair phenocopy by heat shock in mutant heterozygotes. Developmental biology 121: 335-341.
238. Yost HJ, Petersen RB, Lindquist S (1990) RNA metabolism: strategies for regulation in the heat shock response. Trends in genetics : TIG 6: 223-227.

References
91
239. Pfeiffer BD, Jenett A, Hammonds AS, Ngo TT, Misra S, et al. (2008) Tools for neuroanatomy and neurogenetics in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 105: 9715-9720.
240. Duffy JB (2002) GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis 34: 1-15.
241. Jones WD (2009) The expanding reach of the GAL4/UAS system into the behavioral neurobiology of Drosophila. BMB reports 42: 705-712.
242. Perrimon N, Ni JQ, Perkins L (2010) In vivo RNAi: today and tomorrow. Cold Spring Harbor perspectives in biology 2: a003640.
243. Lee T, Luo L (2001) Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends in neurosciences 24: 251-254.
244. Greeve I, Kretzschmar D, Tschape JA, Beyn A, Brellinger C, et al. (2004) Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. The Journal of neuroscience : the official journal of the Society for Neuroscience 24: 3899-3906.
245. Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, et al. (2005) Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience 132: 123-135.
246. Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M (2004) A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Molecular and cellular neurosciences 26: 365-375.
247. Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, et al. (2004) Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America 101: 6623-6628.
248. Iijima-Ando K, Iijima K (2010) Transgenic Drosophila models of Alzheimer's disease and tauopathies. Brain structure & function 214: 245-262.
249. Nerelius C, Sandegren A, Sargsyan H, Raunak R, Leijonmarck H, et al. (2009) Alpha-helix targeting reduces amyloid-beta peptide toxicity. Proceedings of the National Academy of Sciences of the United States of America 106: 9191-9196.
250. Luheshi LM, Tartaglia GG, Brorsson AC, Pawar AP, Watson IE, et al. (2007) Systematic in vivo analysis of the intrinsic determinants of amyloid Beta pathogenicity. PLoS biology 5: e290.
251. Cao W, Song HJ, Gangi T, Kelkar A, Antani I, et al. (2008) Identification of novel genes that modify phenotypes induced by Alzheimer's beta-amyloid overexpression in Drosophila. Genetics 178: 1457-1471.
252. Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, et al. (2009)
Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer's disease. The European journal of neuroscience 29: 1335-1347.
253. Ling D, Song HJ, Garza D, Neufeld TP, Salvaterra PM (2009) Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One 4: e4201.
254. Jackson GR, Wiedau-Pazos M, Sang TK, Wagle N, Brown CA, et al. (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34: 509-519.
255. Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, et al. (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711-714.
256. Nishimura I, Yang Y, Lu B (2004) PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116: 671-682.

References
92
257. Berg I, Thor S, Hammarstrom P (2009) Modeling familial amyloidotic polyneuropathy (Transthyretin V30M) in Drosophila melanogaster. Neuro-degenerative diseases 6: 127-138.
258. Pokrzywa M, Dacklin I, Hultmark D, Lundgren E (2007) Misfolded transthyretin causes behavioral changes in a Drosophila model for transthyretin-associated amyloidosis. Eur J Neurosci 26: 913-924.
259. Pokrzywa M, Dacklin I, Vestling M, Hultmark D, Lundgren E, et al. (2010) Uptake of aggregating transthyretin by fat body in a Drosophila model for TTR-associated amyloidosis. PLoS One 5: e14343.
260. Botella JA, Bayersdorfer F, Gmeiner F, Schneuwly S (2009) Modelling Parkinson's disease in Drosophila. Neuromolecular medicine 11: 268-280.
261. Feany MB, Bender WW (2000) A Drosophila model of Parkinson's disease. Nature 404: 394-398.
262. Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295: 865-868.
263. Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, et al. (1998) Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21: 633-642.
264. Ambegaokar SS, Roy B, Jackson GR (2010) Neurodegenerative models in Drosophila: polyglutamine disorders, Parkinson disease, and amyotrophic lateral sclerosis. Neurobiology of disease 40: 29-39.
265. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447: 859-863.
266. Nelson B, Nishimura S, Kanuka H, Kuranaga E, Inoue M, et al. (2005) Isolation of gene sets affected specifically by polyglutamine expression: implication of the TOR signaling pathway in neurodegeneration. Cell death and differentiation 12: 1115-1123.
267. Rusten TE, Filimonenko M, Rodahl LM, Stenmark H, Simonsen A (2007) ESCRTing autophagic clearance of aggregating proteins. Autophagy 4.
268. Rincon-Limas DE, Casas-Tinto S, Fernandez-Funez P (2010) Exploring prion protein biology in flies: genetics and beyond. Prion 4: 1-8.
269. Goloubinoff P, De Los Rios P (2007) The mechanism of Hsp70 chaperones: (entropic) pulling the models together. Trends in biochemical sciences 32: 372-380.
270. Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nature reviews Neuroscience 6: 11-22.
271. Buchberger A, Bukau B, Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Molecular cell 40: 238-252.
272. Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125: 443-451.
273. Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nature reviews Molecular cell biology 4: 181-191.
274. Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular cell biology 8: 519-529.
275. Aebi M, Bernasconi R, Clerc S, Molinari M (2010) N-glycan structures: recognition and processing in the ER. Trends in biochemical sciences 35: 74-82.
276. Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ (2003) UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proceedings of the National Academy of Sciences of the United States of America 100: 86-91.

References
93
277. Caramelo JJ, Castro OA, de Prat-Gay G, Parodi AJ (2004) The endoplasmic reticulum glucosyltransferase recognizes nearly native glycoprotein folding intermediates. The Journal of biological chemistry 279: 46280-46285.
278. Maattanen P, Gehring K, Bergeron JJ, Thomas DY (2010) Protein quality control in the ER: the recognition of misfolded proteins. Seminars in cell & developmental biology 21: 500-511.
279. Zhou J, Liu CY, Back SH, Clark RL, Peisach D, et al. (2006) The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America 103: 14343-14348.
280. Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P (2005) On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America 102: 18773-18784.
281. Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, et al. (2007) Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. The Journal of cell biology 179: 75-86.
282. Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K (2004) A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. The Journal of cell biology 167: 445-456.
283. Papa FR, Zhang C, Shokat K, Walter P (2003) Bypassing a kinase activity with an ATP-competitive drug. Science 302: 1533-1537.
284. Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, et al. (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92-96.
285. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881-891.
286. Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, et al. (2003) A time-dependent phase shift in the mammalian unfolded protein response. Developmental cell 4: 265-271.
287. Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, et al. (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. The Journal of biological chemistry 276: 13935-13940.
288. Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, et al. (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 312: 572-576.
289. Liu Y, Chang A (2008) Heat shock response relieves ER stress. The EMBO journal 27: 1049-1059.
290. Meusser B, Hirsch C, Jarosch E, Sommer T (2005) ERAD: the long road to destruction. Nature cell biology 7: 766-772.
291. Fu L, Sztul E (2009) ER-associated complexes (ERACs) containing aggregated cystic fibrosis transmembrane conductance regulator (CFTR) are degraded by autophagy. European journal of cell biology 88: 215-226.
292. Huyer G, Longsworth GL, Mason DL, Mallampalli MP, McCaffery JM, et al. (2004) A striking quality control subcompartment in Saccharomyces cerevisiae: the endoplasmic reticulum-associated compartment. Molecular biology of the cell 15: 908-921.
293. Bernales S, McDonald KL, Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS biology 4: e423.
294. He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics 43: 67-93.

References
94
295. Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, et al. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Molecular cell 8: 705-711.
296. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, et al. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664-666.
297. Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, et al. (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318: 944-949.
298. Nakagawa T, Yuan J (2000) Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. The Journal of cell biology 150: 887-894.
299. Nakagawa T, Zhu H, Morishima N, Li E, Xu J, et al. (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98-103.
300. Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, et al. (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS biology 4: e374.
301. Huang CJ, Haataja L, Gurlo T, Butler AE, Wu X, et al. (2007) Induction of endoplasmic reticulum stress-induced beta-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. American journal of physiology Endocrinology and metabolism 293: E1656-1662.
302. Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, et al. (2007) High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 56: 2016-2027.
303. Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, et al. (2007) The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 50: 2486-2494.
304. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, et al. (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50: 752-763.
305. Casas S, Gomis R, Gribble FM, Altirriba J, Knuutila S, et al. (2007) Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes 56: 2284-2294.
306. Hull RL, Zraika S, Udayasankar J, Aston-Mourney K, Subramanian SL, et al. (2009) Amyloid formation in human IAPP transgenic mouse islets and pancreas, and human pancreas, is not associated with endoplasmic reticulum stress. Diabetologia 52: 1102-1111.
307. Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, et al. (2010) Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol 176: 861-869.
308. Richardson H, Kumar S (2002) Death to flies: Drosophila as a model system to study programmed cell death. Journal of immunological methods 265: 21-38.
309. Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nature reviews Cancer 2: 647-656.
310. Wang X (2001) The expanding role of mitochondria in apoptosis. Genes & development 15: 2922-2933.
311. Zou H, Li Y, Liu X, Wang X (1999) An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. The Journal of biological chemistry 274: 11549-11556.

References
95
312. Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491-501.
313. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481-490.
314. Ditzel M, Broemer M, Tenev T, Bolduc C, Lee TV, et al. (2008) Inactivation of effector caspases through nondegradative polyubiquitylation. Molecular cell 32: 540-553.
315. Butler AE, Janson J, Soeller WC, Butler PC (2003) Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes 52: 2304-2314.
316. Rumora L, Hadzija M, Barisic K, Maysinger D, Grubiic TZ (2002) Amylin-induced cytotoxicity is associated with activation of caspase-3 and MAP kinases. Biological chemistry 383: 1751-1758.
317. Zraika S, Hull RL, Udayasankar J, Aston-Mourney K, Subramanian SL, et al. (2009) Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 52: 626-635.
318. Li XL, Chen T, Wong YS, Xu G, Fan RR, et al. (2011) Involvement of mitochondrial dysfunction in human islet amyloid polypeptide-induced apoptosis in INS-1E pancreatic beta cells: An effect attenuated by phycocyanin. The international journal of biochemistry & cell biology 43: 525-534.
319. Law E, Lu S, Kieffer TJ, Warnock GL, Ao Z, et al. (2010) Differences between amyloid toxicity in alpha and beta cells in human and mouse islets and the role of caspase-3. Diabetologia 53: 1415-1427.
320. Matveyenko AV, Butler PC (2006) Beta-cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes 55: 2106-2114.
321. Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryotic cell 1: 11-21.
322. De Duve C (1963) The lysosome. Scientific American 208: 64-72. 323. Uttenweiler A, Schwarz H, Neumann H, Mayer A (2007) The vacuolar
transporter chaperone (VTC) complex is required for microautophagy. Molecular biology of the cell 18: 166-175.
324. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, et al. (2011) Microautophagy of cytosolic proteins by late endosomes. Developmental cell 20: 131-139.
325. Cuervo AM (2010) Chaperone-mediated autophagy: selectivity pays off. Trends in endocrinology and metabolism: TEM 21: 142-150.
326. Seglen PO, Gordon PB, Holen I (1990) Non-selective autophagy. Seminars in cell biology 1: 441-448.
327. Tooze SA, Yoshimori T (2010) The origin of the autophagosomal membrane. Nature cell biology 12: 831-835.
328. McEwan DG, Dikic I (2010) Not all autophagy membranes are created equal. Cell 141: 564-566.
329. Yorimitsu T, Klionsky DJ (2005) Autophagy: molecular machinery for self-eating. Cell death and differentiation 12 Suppl 2: 1542-1552.
330. Knaevelsrud H, Simonsen A (2010) Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett 584: 2635-2645.
331. Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nature reviews Molecular cell biology 12: 9-14.

References
96
332. Kraft C, Deplazes A, Sohrmann M, Peter M (2008) Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nature cell biology 10: 602-610.
333. Dunn WA, Jr., Cregg JM, Kiel JA, van der Klei IJ, Oku M, et al. (2005) Pexophagy: the selective autophagy of peroxisomes. Autophagy 1: 75-83.
334. Klionsky DJ, Cuervo AM, Dunn WA, Jr., Levine B, van der Klei I, et al. (2007) How shall I eat thee? Autophagy 3: 413-416.
335. Yamamoto A, Simonsen A (2010) The elimination of accumulated and aggregated proteins: A role for aggrephagy in neurodegeneration. Neurobiol Dis.
336. Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. The Journal of biological chemistry 273: 3963-3966.
337. Scott RC, Schuldiner O, Neufeld TP (2004) Role and regulation of starvation-induced autophagy in the Drosophila fat body. Developmental cell 7: 167-178.
338. Hara T, Takamura A, Kishi C, Iemura S, Natsume T, et al. (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. The Journal of cell biology 181: 497-510.
339. Simonsen A, Tooze SA (2009) Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. The Journal of cell biology 186: 773-782.
340. Razi M, Chan EY, Tooze SA (2009) Early endosomes and endosomal coatomer are required for autophagy. The Journal of cell biology 185: 305-321.
341. Rusten TE, Stenmark H (2009) How do ESCRT proteins control autophagy? Journal of cell science 122: 2179-2183.
342. Nickerson DP, Brett CL, Merz AJ (2009) Vps-C complexes: gatekeepers of endolysosomal traffic. Current opinion in cell biology 21: 543-551.
343. Lindmo K, Simonsen A, Brech A, Finley K, Rusten TE, et al. (2006) A dual function for Deep orange in programmed autophagy in the Drosophila melanogaster fat body. Experimental cell research 312: 2018-2027.
344. Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, et al. (2009) Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature cell biology 11: 468-476.
345. Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, et al. (2010) FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. The Journal of cell biology 188: 253-269.
346. Kimura S, Noda T, Yoshimori T (2008) Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell structure and function 33: 109-122.
347. Kirkin V, McEwan DG, Novak I, Dikic I (2009) A role for ubiquitin in selective autophagy. Molecular cell 34: 259-269.
348. Hershko A, Ciechanover A (1998) The ubiquitin system. Annual review of biochemistry 67: 425-479.
349. Welchman RL, Gordon C, Mayer RJ (2005) Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nature reviews Molecular cell biology 6: 599-609.
350. Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, et al. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Human molecular genetics 17: 431-439.
351. Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, et al. (2008) Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. The Journal of biological chemistry 283: 6783-6789.

References
97
352. Ikeda F, Dikic I (2008) Atypical ubiquitin chains: new molecular signals. 'Protein Modifications: Beyond the Usual Suspects' review series. EMBO reports 9: 536-542.
353. Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, et al. (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33: 505-516.
354. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, et al. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131-24145.
355. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, et al. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. The Journal of cell biology 171: 603-614.
356. Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, et al. (2008) Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol 180: 1065-1071.
357. Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, et al. (2010) p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6: 330-344.
358. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, et al. (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131: 1149-1163.
359. Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, et al. (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4: 151-175.
360. Kuusisto E, Salminen A, Alafuzoff I (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12: 2085-2090.
361. Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, et al. (2002) p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. The American journal of pathology 160: 255-263.
362. Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, et al. (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 38: 265-279.
363. Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, et al. (2004) Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. Journal of cell science 117: 4239-4251.
364. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, et al. (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Molecular biology of the cell 19: 2092-2100.
365. Finley KD, Edeen PT, Cumming RC, Mardahl-Dumesnil MD, Taylor BJ, et al. (2003) blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience 23: 1254-1264.
366. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090-1106.
367. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, et al. (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature cell biology 12: 119-131.
368. Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J (2008) Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proceedings of the National Academy of Sciences of the United States of America 105: 20567-20574.

References
98
369. Nagata E, Sawa A, Ross CA, Snyder SH (2004) Autophagosome-like vacuole formation in Huntington's disease lymphoblasts. Neuroreport 15: 1325-1328.
370. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, et al. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36: 585-595.
371. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278: 25009-25013.
372. Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305: 1292-1295.
373. Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, et al. (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67: 1074-1077.
374. Talbot K, Ansorge O (2006) Recent advances in the genetics of amyotrophic lateral sclerosis and frontotemporal dementia: common pathways in neurodegenerative disease. Human molecular genetics 15 Spec No 2: R182-187.
375. Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, et al. (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63: 724-726.
376. Rubinsztein DC (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443: 780-786.
377. Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, et al. (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of neuropathology and experimental neurology 64: 113-122.
378. Lee S, Sato Y, Nixon RA (2011) Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. The Journal of neuroscience : the official journal of the Society for Neuroscience 31: 7817-7830.
379. Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, et al. (2011) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain : a journal of neurology 134: 258-277.
380. Ling D, Salvaterra PM (2011) Brain aging and Abeta neurotoxicity converge via deterioration in autophagy-lysosomal system: a conditional Drosophila model linking Alzheimer's neurodegeneration with aging. Acta neuropathologica 121: 183-191.
381. Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, et al. (2008) Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4: 176-184.
382. Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, et al. (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell metabolism 8: 325-332.
383. Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, et al. (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell metabolism 8: 318-324.
384. Rivera JF, Gurlo T, Daval M, Huang CJ, Matveyenko AV, et al. (2011) Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic beta-cells: protective role of p62-positive cytoplasmic inclusions. Cell death and differentiation 18: 415-426.

References
99
385. Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, et al. (2005) Inhibition of macroautophagy triggers apoptosis. Molecular and cellular biology 25: 1025-1040.
386. Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, et al. (2008) To die or not to die: that is the autophagic question. Current molecular medicine 8: 78-91.
387. Tsujimoto Y, Shimizu S (2005) Another way to die: autophagic programmed cell death. Cell death and differentiation 12 Suppl 2: 1528-1534.
388. Chen Y, Klionsky DJ (2011) The regulation of autophagy - unanswered questions. Journal of cell science 124: 161-170.
389. Denton D, Shravage B, Simin R, Mills K, Berry DL, et al. (2009) Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Current biology : CB 19: 1741-1746.
390. Voss V, Senft C, Lang V, Ronellenfitsch MW, Steinbach JP, et al. (2010) The pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in malignant glioma. Molecular cancer research : MCR 8: 1002-1016.
391. Spradling AC, Rubin GM (1982) Transposition of cloned P elements into Drosophila germ line chromosomes. Science 218: 341-347.
392. Matveyenko AV, Butler PC (2006) Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR journal / National Research Council, Institute of Laboratory Animal Resources 47: 225-233.
393. Marzban L, Rhodes CJ, Steiner DF, Haataja L, Halban PA, et al. (2006) Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes 55: 2192-2201.
394. Aslund A, Sigurdson CJ, Klingstedt T, Grathwohl S, Bolmont T, et al. (2009) Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS chemical biology 4: 673-684.
395. Berg I, Nilsson KP, Thor S, Hammarstrom P (2010) Efficient imaging of amyloid deposits in Drosophila models of human amyloidoses. Nature protocols 5: 935-944.
396. Cohen E (1993) Chitin synthesis and degradation as targets for pesticide action. Archives of insect biochemistry and physiology 22: 245-261.
397. Hammarstrom P, Simon R, Nystrom S, Konradsson P, Aslund A, et al. (2010) A fluorescent pentameric thiophene derivative detects in vitro-formed prefibrillar protein aggregates. Biochemistry 49: 6838-6845.
398. Nilsson KP, Ikenberg K, Aslund A, Fransson S, Konradsson P, et al. (2010) Structural typing of systemic amyloidoses by luminescent-conjugated polymer spectroscopy. The American journal of pathology 176: 563-574.
399. Carolina R, Santiago W, María Fernanda C (2007) Neuronal death in <i>Drosophila</i> triggered by GAL4 accumulation. European Journal of Neuroscience 25: 683-694.
400. Bardet PL, Kolahgar G, Mynett A, Miguel-Aliaga I, Briscoe J, et al. (2008) A fluorescent reporter of caspase activity for live imaging. Proc Natl Acad Sci U S A 105: 13901-13905.
401. Hsu AL, Murphy CT, Kenyon C (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300: 1142-1145.
402. Morley JF, Morimoto RI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15: 657-664.
403. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, et al. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885-889.

References
100
404. Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, et al. (2011) p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 7: 572-583.
405. Clague MJ, Urbe S (2010) Ubiquitin: same molecule, different degradation pathways. Cell 143: 682-685.
406. Kirkin V, Lamark T, Johansen T, Dikic I (2009) NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5: 732-733.
407. Lamark T, Kirkin V, Dikic I, Johansen T (2009) NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8: 1986-1990.
408. Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer's disease-locating the primary defect. Neurobiology of disease 43: 38-45.





![November 2016 AHS Scottsdale Amylin...11/10/2016 1 Amylin (other names –islet amyloid polypeptide [IAPP], diabetes-associated peptide) Professor Debbie L Hay School of Biological](https://static.fdocuments.in/doc/165x107/5fa080ae893c796f1318561d/november-2016-ahs-scottsdale-amylin-11102016-1-amylin-other-names-aislet.jpg)












